 Dongwei Xu
Dongwei Xu Yizhu Tian
Yizhu Tian Qiang Xia
Qiang Xia Bibo Ke
Bibo Ke
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 April 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.682736
This article is part of the Research Topic Nucleic Acid-Associated Inflammation View all 22 articles
Liver diseases represent a major global health burden accounting for approximately 2 million deaths per year worldwide. The liver functions as a primary immune organ that is largely enriched with various innate immune cells, including macrophages, dendritic cells, neutrophils, NK cells, and NKT cells. Activation of these cells orchestrates the innate immune response and initiates liver inflammation in response to the danger signal from pathogens or injured cells and tissues. The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway is a crucial signaling cascade of the innate immune system activated by cytosol DNA. Recognizing DNA as an immune-stimulatory molecule is an evolutionarily preserved mechanism in initiating rapid innate immune responses against microbial pathogens. The cGAS is a cytosolic DNA sensor eliciting robust immunity via the production of cyclic GMP-AMPs that bind and activate STING. Although the cGAS-STING pathway has been previously considered to have essential roles in innate immunity and host defense, recent advances have extended the role of the cGAS-STING pathway to liver diseases. Emerging evidence indicates that overactivation of cGAS-STING may contribute to the development of liver disorders, implying that the cGAS-STING pathway is a promising therapeutic target. Here, we review and discuss the role of the cGAS-STING DNA-sensing signaling pathway in a variety of liver diseases, including viral hepatitis, nonalcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD), primary hepatocellular cancer (HCC), and hepatic ischemia-reperfusion injury (IRI), with highlights on currently available therapeutic options.
Liver disease presents a globally recognized health threat with a mortality rate of 2 million deaths per year worldwide (1). It often occurs in response to hepatocyte injury caused mainly by the hepatitis B and C virus, alcohol abuse, bile duct damage, nonalcoholic fatty liver disease (NAFLD) or nonalcoholic steatohepatitis (NASH) (2–4). Hepatic inflammation is a critical player in triggering liver diseases. During the initial event of hepatic inflammation, innate immune cells, such as macrophages, neutrophils, natural killer (NK) cells, and NKT cells recognize cell damage or invading pathogens with intracellular-expressed pattern recognition receptors (PRRs) at the cell surface. PRRs detect distinct evolutionarily conserved structures on pathogens, termed pathogen-associated molecular patterns (PAMPs), and trigger innate inflammatory responses by activating a multitude of intracellular signaling pathways (5). Indeed, the innate immune system depends on PRRs, including the cyclic GMP-AMP (cGAMP) synthase (cGAS) and its downstream effector stimulator of interferon genes (STING), inflammasomes, and Toll-like receptors (TLRs) that recognize PAMPs and coordinate antimicrobial defense (6–9). PRRs also recognize a plethora of damage-associated molecular patterns (DAMPs), such as nucleic acids of uncontrolled death of host cells, to further activate the innate immune system, contributing to inflammatory diseases and cancer (10, 11). Therefore, aberrant nucleic acid recognition has emerged as a critical host defense mechanism mediated by cytosolic nucleic acid sensors.
DNA generally resides within the nucleus and mitochondria of eukaryotic cells. Aberrant presence of DNA in the cytoplasm from cellular damage or infection elicits robust immunity leading to activation of type I interferon-stimulated genes (ISGs) that confer increased susceptibility to the pathogens and promote host survival (12). The most robust response following DNA stimulation is initiated by cyclic GMP-AMP (cGAMP) synthase (cGAS), which is activated upon binding to double-stranded DNA (dsDNA) (13). cGAS is a critical cytosolic DNA sensor that catalyzes the synthesis of cGAMP from ATP and GTP and activates type I interferons (IFNs) through the endoplasmic reticulum (ER)-resident adaptor protein STING (13, 14), which subsequently activates the transcription factors NF-kB and IFN regulatory factor (IRF) 3 via the TANK-binding kinase 1 (TBK1) (13, 14). Besides, the binding of cGAS to DNA is irrespective of DNA sequence (15). Thus, self-DNA from the mitochondria or nucleus could also act as the cGAS ligand to activate the cGAS-STING pathway in triggering inflammatory responses (16). Recent studies suggested that endogenous cGAS was tightly tethered in the nucleus and prevented its autoreactivity against self-DNA (17–19). The structural basis for inhibiting cGAS by chromatin was verified via cryo-electron microscopy by other studies (20, 21). Moreover, cGAS was reported to inhibit homologous recombination-mediated DNA repair and promote genome destabilization, micronucleus generation, and cell death under conditions of genomic stress via a STING-independent manner (18). These findings indicate that activation of the cGAS-STING pathway by exogenous or endogenous DNA may contribute to the development of various human diseases. Here, we provide an overview of the cGAS-STING pathway in immunity. Moreover, we summarize and discuss the role of the cGAS-STING DNA pathway in a variety of liver diseases. Finally, we highlight current or prospective therapeutic strategies targeting the pathway.
DNA is a crucial DAMP that is recognized by innate immune receptors and triggers intracellular signaling cascades (22). dsDNA is primed by damaged mitochondria, dying cells, DNA damage, genomic instability, bacteria, DNA viruses, and retroviruses (12, 23, 24). DNA viruses can induce type I interferon production through activation of the STING pathway (25). Emerging evidence demonstrated that cGAS was required to trigger innate immune response during HIV and other retrovirus infections (26). The cGAS consists of a critical catalytic domain, C-terminal nucleotidyltransferase (NTase) domain, which is composed of two structural lobes with the active site (7). dsDNA activates cGAS by forming 2:2 cGAS-dsDNA complexes (27, 28). The stabilized structure modulates the catalytic domain’s rearrangement to transform GTP and ATP to cGAMP through induction of a conformational change in the C-terminal domain (13, 27, 28). cGAMP is an endogenous second messenger with a high affinity for STING (29). The binding of cGAMP to STING promotes STING translocation to the Golgi apparatus and activates TBK1, which phosphorylates STING and IRF3 transcription factor (13). The activated IRF3 enters the nucleus and triggers the production of type I IFNs, leading to the expression of IFN-stimulated genes (7, 30). STING can also recruit IκB kinase (IKK), which in turn catalyzes the phosphorylation of the nuclear factor-κB (NF-κB) inhibitor IκBα. IκBα phosphorylation accelerates nucleus translocation of NF-κB to promote transcription of target inflammatory cytokines (16). In addition, the N-terminal domain is also responsible for the maintenance of the liquid phase dsDNA and cGAS (31, 32). DNA binding to cGAS promotes the formation of liquid-like droplets, which facilitates cGAS activation via augmented cGAS liquid phase separation and enzyme activity (31). These findings demonstrate the multivalent interactions between DNA and the binding domain of cGAS in activating innate immune signaling (Figure 1).
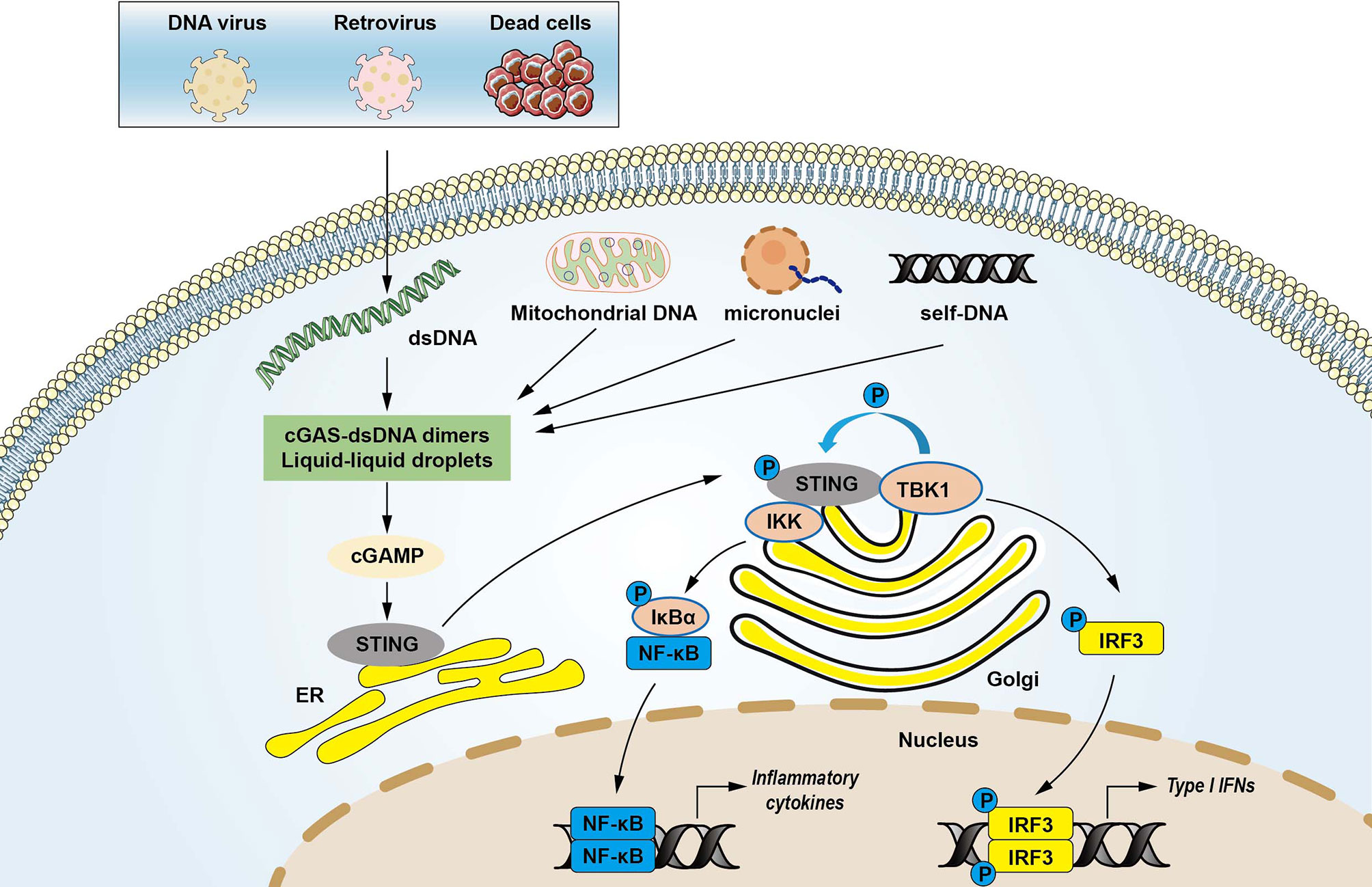
Figure 1 The cytosolic DNA-sensing cGAS-STING pathway in innate immunity. Cyclic GMP-AMP synthase (cGAS) is a protein, which detects various cytosolic dsDNA, including viral DNA, damaged self-DNA released by dying cells, micronuclei, and mitochondrial origins. dsDNA activates cGAS via forming cGAS-dsDNA in 2:2 complexes. Mitochondrial damage and the release of mitochondrial DNA (mtDNA) in the cytosol also activates cGAS. The interactions of cGAS with DNA induce the formation of the liquid droplets through a phase transition, in which cGAS exerts its catalytic role to create the second messenger cGAMP that stimulates the stimulator of interferon genes (STING) at the endoplasmic reticulum (ER). STING then translocates from the ER to Golgi compartments and recruits kinases such as TANK-binding kinase 1 (TBK1) and IκB kinase (IKK), which in turn catalyzes the phosphorylation of IFN regulatory factor 3 (IRF3) and the nuclear factor-κB (NF-κB) inhibitor IκBα. Phosphorylated IRF3 translocates to the nucleus to activate transcription of genes encoding type I interferons and other inflammatory genes. IκBα phosphorylation accelerates nucleus translocation of NF-κB to promote transcription of target inflammatory cytokines, leading to activating inflammatory responses.
Hepatitis B virus (HBV) and Hepatitis C virus (HCV) infections remains a major public health problem in the 21st century with over 300 million people worldwide affected, despite the implementation of various therapeutics (33, 34). HBV is an enveloped partially double-stranded DNA virus (35). HBV infection of human hepatocytes leads to acute and chronic hepatitis, which remarkably increases the risk of liver cirrhosis and hepatocellular carcinoma (HCC) (33, 36). The role of innate immune response in the HBV natural infection process remains unclear and controversial (37, 38). Accumulating data suggested that HBV can escape from recognition by the innate system (39–42). Lacking strong innate immune responses may also account for the convenient transformation of HBV infections to chronic HBV hepatitis (43). Other studies have identified that HBV-derived dsDNA fragments (44), viral genomic relaxed circular (RC) DNA (45), and naked HBV genome (46) could activate the innate antiviral immune responses. As a critical DNA cytosolic DNA sensor, the role of the cGAS-STING pathway during HBV infection has been investigated by several research groups (42, 44, 46–48). Recent studies demonstrated that both primary murine hepatocytes and primary human hepatocytes (PHH) failed to produce type I IFN in response to the foreign DNA in the cytosol or HBV infection due to the lack of STING expression in these hepatocytes (42). However, the hepatoma cell line HepG2 showed an innate immune response after HBV infection since STING expression was observed (42). The lack of DNA-sensing signaling impaired the hepatocytes’ ability to control HBV but induction of STING in vivo reduced viral gene expression and replication in hepatocytes (42), suggesting that the absence of the intracellular DNA-sensing pathway dampens the innate immune response against HBV infection in hepatocytes. These results were further validated by another in vitro study, which showed that increased STING expression exhibited resistance to HBV infection whereas disruption of STING expression depressed IFN response and enhanced HBV transcription activity in human immortalized hepatocyte NKNT-3 cells (49). Thus, the STING pathway is essential for modulating susceptibility to HBV.
Interestingly, another study suggested that the packaged HBV genome evaded recognition by innate immune cells during natural infection, while naked HBV genomic rcDNA was sensed in a cGAS-dependent manner in human hepatoma cell line HepG2-NTCP (46). Moreover, HBV infection could inhibit the cGAS expression and function in cell culture and humanized liver chimeric mice by downregulating the cGAS-related gene MB21D1, a classic member of IFN-stimulated genes (ISGs) (46). However, HBV-derived dsDNA can also induce the innate immune response by expressing high levels of cGAS in human hepatoma Li23 cells (44). Activation of the cGAS-STING pathway induced ISG56, one of the antiviral genes mediated by type I IFNs, and inhibited HBV assembly (44). Moreover, activation of the cGAS-STING pathway by dsDNA or cGAMP significantly depressed HBV replication in vitro and in vivo (48). A recent study revealed that HBV DNAs but not RNAs in the viral particles were immunostimulatory and sensed by the cGAS-STING pathway in HepG2 cells (47). HBV rcDNA triggered the hepatocyte response, whereas HBV infection did not suppress the DNA-sensing pathway but can evade the surveillance of the cGAS-STING mediated immune response (47). Indeed, activation of cGAS or STING with pharmaceutical treatment induced IFN response and inhibited viral replication in HBV-infected human hepatoma cells and immortalized mouse hepatocytes (50, 51). As an essential part of the innate immune system, Kupffer cells, which are macrophages residing in the liver, may also contribute to detecting foreign DNA and induction of inflammatory response by phagocytosis during HBV infection. Unlike PHH, the Kupffer cells certainly have intact DNA-sensor signaling, as they exhibit significantly enhanced cGAS-STING pathway levels after HBV infection (41, 42). Pharmaceutical activation of STING by 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in macrophages could remarkably inhibit hepatocyte HBV replication in mice (50). Although Kupffer cells are positive regulators of antiviral immunity during HBV infection (37), the HBV core has been known to activate TLR2 on Kupffer cells leading to inhibition of HBV-specific T cell response by producing IL-10 (52). Genetic knocking out of TLR2 or pharmaceutical depletion of Kupffer cells resulted in a stronger antiviral immune response (52). Another study suggested that instead of promoting liver inflammation, Kupffer cells can inhibit immune response by removing apoptotic hepatocytes during HBV infection (53). These conflicting results on the role of the cGAS-STING pathway in hepatocytes and Kupffer cells during HBV infection suggest that more investigation is needed to clarify the underlying mechanism. Thus, it would be interesting to explore the cGAS-STING signaling role in other innate immune cell types in HBV infection.
HCV infection, followed by liver failure, liver cirrhosis, and HCC, is considered one of the most common causes of liver transplantation in Western countries (54). Evading innate and adaptive immune responses is the primary mechanism for HCV to defeat host immune surveillance and responses. The mechanism underlying HCV regulaton of host interferon response has been investigated for years. Several studies revealed that casein kinase II (CK2) was required for HCV core protein-mediated modulation (55) and served as a critical regulator in controlling IFN response. Activation of CK2 inhibited retinoic acid-inducible gene I (RIG-I)-mediated immune response, whereas disruption of CK2 promoted STING-mediated TBK1 activation and triggered IFN-β immune defense against HCV infection (56, 57). In addition, the hepatitis C virus non-structural 4B (HCV-NS4B) protein, an essential component of viral replication, was found to directly and specifically bind to STING and block the STING-Cardif interaction, contributing to potent inhibition of RIG-I-medicated IRF-3 phosphorylation and IFN-β (58). HCV-NS4B was also found to impair the interaction of STING and TBK1 (59, 60). These findings suggest that the STING-mediated immune defense mechanism contributes to host antiviral immune response.
Recently, it was reported that the delivery of synthetic cGAMP agonist for activation of the cGAS-STING pathway remarkably inhibited the HBV replication by inducing IFN production in the HBV-infected mouse model (48). The therapeutic drugs combined with an effective vaccine have shown high efficacy in eliminating viral hepatitis (61). As an HBV or HCV vaccine adjuvant, administration of STING agonists can induce a robust immune response via up-regulation of cytokines and chemokines, which may restrain tolerance in patients with chronic viral hepatitis (62, 63). Collectively, the interaction between the cGAS-STING pathway mediated innate immune response and HBV in hepatocytes and macrophages during natural infection is still elusive and controversial. Much more work is needed to investigate the molecular mechanisms underlying the role of the cGAS-STING pathway in HBV and HCV infection. These studies may provide a novel therapeutic approach for viral hepatitis.
Nonalcoholic fatty liver disease (NAFLD) is characterized by a series of diseases ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), subsequent cirrhosis, and even hepatocellular carcinoma. Currently, NAFLD is increasing globally, and the prevalence of NAFLD is about 25% (64). NAFLD is becoming the most common cause of chronic liver disease and the leading cause of liver failure requiring liver transplantation in western countries (65). However, there is no safe and effective therapy for patients with NASH due to the pathogenesis of NASH not being fully understood.
It is well known that the innate immune system, especially macrophages, plays an essential role in the development of hepatic steatosis to NASH (66). During the past years, numerous reports have identified the vital role of the cGAS-STING signaling pathway in NASH progression by regulating innate immune activation. Metabolic stress, such as a high-fat diet (HFD), could activate cGAS and the STING-IRF3-mediated inflammatory response (67). By contrast, STING deficiency mitigated HFD-induced adipose tissue inflammation, obesity, insulin resistance, and glucose intolerance (67). Disruption of either STING or IRF3 significantly attenuated free fatty acid (FFA)-induced inflammatory response, lipid accumulation, and hepatocellular apoptosis through regulation of the nuclear factor κB (NF-κB) signaling pathway (68). As lipotoxicity appears to be the central driver in NASH progression by oxidative stress and ER stress (69), lipotoxic activation of TBK1, a downstream of cGAS-STING kinase, is also crucial for the control of the NASH development (70). Recently, mitochondrial DNA (mtDNA) released from injured hepatocytes has been recognized as an endogenous DAMP, which activates the cGAS-STING pathway and promotes hepatic inflammation through release of cytokines in NASH (71), suggesting that cytosolic mtDNA sensed by the cGAS-STING signaling is key to trigger innate immune response in NASH progression. Several studies have indicated that human and murine hepatocytes did not express STING protein (42, 71, 72). However, increased STING expression was observed in Kupffer cells in patients with NASH (72). Myeloid-specific STING induced TGF-β1 and activated hepatic stellate cells (HSCs), which promoted NASH progression, whereas disruption of myeloid STING alleviated hepatic inflammation, steatosis, and liver fibrosis in a mouse model of HFD or methionine and choline-deficient diet (MCD)-induced NASH (72), suggesting that activation of STING regulates macrophage function and augments hepatic lipid accumulation, profibrotic gene expression, and proinflammatory responses in NASH (Figure 2). Moreover, a study in liver samples from 98 patients with NAFLD revealed that STING expression in Kupffer cells and monocyte-derived macrophages (MoMFs) was correlated with hepatic inflammation and fibrosis in human NAFLD (73). These findings indicate that activation of the cGAS-STING pathway in macrophages is critical in NASH progression.
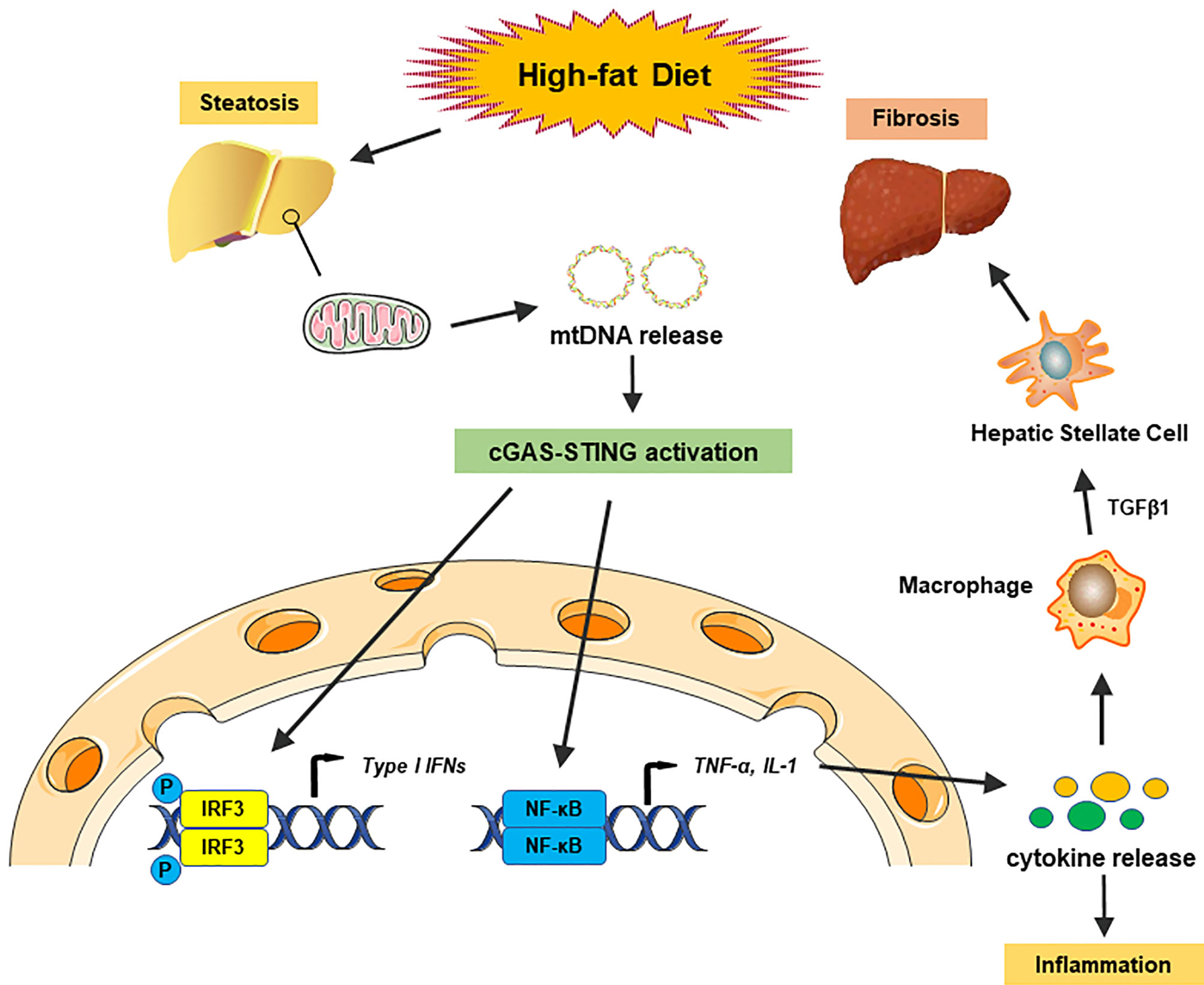
Figure 2 The cGAS-STING pathway in nonalcoholic fatty liver disease. A high-fat diet (HFD) causes steatosis, which induces mitochondrial stress damage in hepatocytes and subsequent releases of mitochondrial DNA (mtDNA) into the cytosol. Cytosolic mtDNA is recognized as an endogenous DAMP, which activates the cGAS-STING pathway and induces the IRF3 signaling to promote transcription of type I IFNs. Activation of the cGAS-STING pathway also induces the NF-κB signaling to produce proinflammatory cytokines, which triggers hepatic inflammatory responses. Moreover, proinflammatory cytokines activate macrophage function and produce TGF-β1, which activates hepatic stellate cells (HSCs) and promotes liver fibrosis in NASH.
The hepatic inflammatory response has a fundamental role in NASH progression. Activation of STING induces the IRF3 and NF-κB pathways, and produces various inflammatory cytokines (74). It was reported that global knockout (KO) of IRF3 were significantly reduced liver injury, steatosis, and inflammation (75). However, another study showed that disrupted IRF3 resulted in increased insulin resistance and liver inflammation in HFD-induced NAFLD (76). Indeed, STING activated the innate immune response and contributed to the NASH progression in an NF-κB dependent manner (71). Moreover, IRF3 KO mice showed higher fasting glycemia and higher body weight (76), which was not consistent with the model of HFD-fed STING-deficient mice (71). STING might regulate glucose levels but not body weight in an IRF3-dependent manner. Further studies are needed to elucidate the underlying mechanism. Therefore, targeting STING to inhibit innate immune activation could provide a novel approach to managing NAFLD and NASH in patients.
Alcohol-related liver disease (ALD) affects more than 150 million people worldwide. It is the second most common indication for liver transplantation due to ALD-induced cirrhosis (77). Liver failure by ALD accounts for approximately half of liver cirrhosis-associated deaths in the United States (78). A previous study found that ER stress-induced IRF3 activation in the liver was associated with ER adaptor protein STING in the acute ALD model (79). IRF3 deficiency ameliorated hepatocyte apoptosis and the inflammatory responses in an ethanol-feeding mouse model (79). Alcohol-feeding remarkably increased cytoplasmic mtDNA release, resulting in activating the cGAS-IRF3 signaling (80). Activation of IRF3 by cGAS drove liver inflammation and injury in both alcohol-exposed hepatocytes and the neighboring parenchyma through a gap junction intracellular communication pathway (80). RNA-seq analysis of ALD patients showed that the cGAS-IRF3 pathway was positively associated with disease severity (80). Thus, cGAS, STING, and IRF3 are crucial determinants in the pathogenesis of ALD and potential therapeutic targets in ALD (Figure 3).
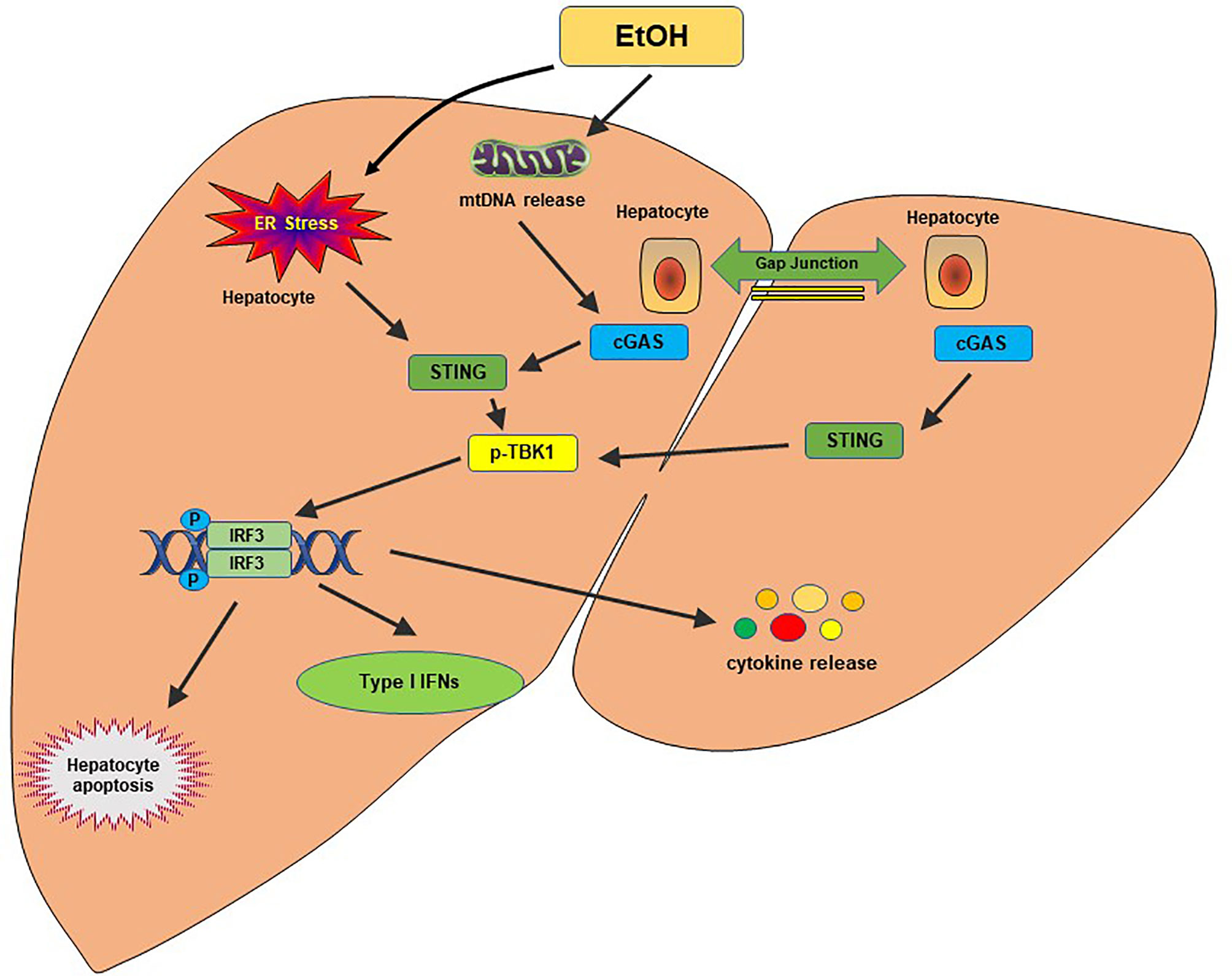
Figure 3 The cGAS-STING pathway in alcohol-related liver disease. Alcohol-induced ER stress and mtDNA release activate the STING pathway. STING facilitates IRF3 phosphorylation by TBK1, which results in the translocation of IRF3 into the nucleus, where it induces transcription of type I IFNs. A gap junction intracellular communication pathway between alcohol-exposed hepatocytes and the neighboring parenchyma also contributes to the IRF3 activation by cGAS. Activation of IRF3 could trigger hepatocyte apoptosis, type I IFN response and produce proinflammatory cytokines, leading to hepatic inflammation and injury.
Hepatocellular carcinoma (HCC) is the most common primary liver cancer and is the second leading cause of cancer-related death in the world (81). Despite the availability of multiple therapeutic approaches for the early stage of HCCs, including surgical liver resection, liver transplantation, and percutaneous ablation, most patients are diagnosed at relatively advanced stages with fewer treatment options and a poor prognosis (82). Recently, cancer immunotherapy has emerged as an effective therapy for various types of cancers (83). Accumulating evidence demonstrates the vital role of the innate immune system in liver cancer immunosurveillance and immunotherapy (84). During tumorigenesis, tumor cell death and genome instability could lead to abnormal localization of genomic DNA in the cytosol and micronuclei formation (16, 85). As a solid tumor, the hypoxic microenvironment inside the HCC tumor can also induce cancer cell necrosis, which promotes the release of mitochondrial DNA (mtDNA) (86, 87). These exogenous and endogenous cytosolic DNA are subsequently recognized by the immune cells, resulting in activation of the innate immune response. Emerging studies show that cGAS also detects tumor-derived DNA, initiating antitumor immunity in some cancers (88). Indeed, the cGAS-STING pathway plays an essential role in HCC progression. It was reported that low levels of STING in tumor tissues were associated with poor prognosis in HCC patients (89). Activation of the cGAS-STING pathway augmented immune cell infiltration in HCC tissues (90). The cGAS-STING pathway members also displayed strong associations with immune markers involved in clinical stages, pathological grades, and overall survival in patients with HCC (90), suggesting that the cGAS-STING pathway members could be used as potential prognostic biomarkers in patients with HCC. In a mouse model of mutagen-induced HCC, STING deficiency reduced phosphorylated-STAT1, autophagy, and cleaved caspase 3 levels but accelerated tumor progression, with increased numbers of large tumors at advanced stages. In contrast, treatment with a cyclic dinucleotide (CDN) STING agonist promoted cell death, autophagy, and IFN responses in HCC (91). Notably, CDN treatment markedly reduced tumor size and the number of HCC in mice (91). These findings indicate STING is a promising therapeutic target for the treatment of HCC.
Immunotherapy has been rapidly expanded as a novel option in the treatment of advanced HCC. Data from the early stage of clinical trials with PD-1/PD-L1 therapy have suggested promising results with encouraging survival and safety data in HCC patients (92). While some therapeutic benefits have been reported with immune checkpoint blockade therapy, the low efficacy of immunotherapy remains a significant challenge in HCC treatment. Several studies have revealed that STING-deficient mice are less responsive to immunotherapy (93, 94). A combination treatment of cGAMP with PD-L1 inhibitor has shown a more potent antitumor effect in a xenograft model (95), indicating that stimulation of the cGAS-STING pathway may improve immunotherapeutic efficacy for the treatment of HCC. Further studies are needed to elucidate the crosstalk between the cGAS-STING and PD-1/PD-L1 pathway in antitumor immunity against HCC.
Liver ischemia-reperfusion injury (IRI), an innate immunity-dominated local sterile inflammatory response (96), is a significant cause of hepatic dysfunction and failure in liver transplantation (97). Oxidative and ER stress are important factors in the pathogenesis of hepatic IRI. IR-induced stress activates liver macrophages (Kupffer cells) to generate reactive oxygen species (ROS), leading to sterile inflammation in the liver (98). ROS, an endogenous ‘danger’ signal released from necrotic and stressed cells, triggers toll-like receptor 4 (TLR4) or NLRP3 inflammasome-driven innate immune response in ischemic livers (98–101). ROS can induce oxidative mitochondrial damage, resulting in mtDNA leaks into the cytosol (102). The mtDNA is recognized by the DNA sensor cGAS and activates STING, which triggers an innate immune response (103). Recent studies showed that mtDNA release from hepatocytes was significantly increased during liver IRI (104). Increased mtDNA induced STING activation in macrophages after liver IRI, whereas disruption of STING reduced NLRP3 activation and proinflammatory mediators in mtDNA-stimulated macrophages from aged mice (105).
Moreover, another study showed that IR-induced stress in hepatocytes promoted cGAS expression but they did not express STING under oxidative stress conditions (106). Interestingly, cGAS global knockout (KO) mice displayed increased IR-induced liver injury compared to the wild-type or STING-deficient mice. Disruption of cGAS in hepatocytes augmented cell death and apoptosis but reduced autophagy induction in response to oxidative stress (106), suggesting that cGAS regulates hepatic autophagy in a STING-independent manner during liver IRI. Indeed, the tissue-specific roles and regulatory mechanisms of the cGAS-STING pathway remain mostly elusive. As liver macrophages, including resident Kupffer cells and infiltrated bone marrow-derived macrophages, are a major player in innate immune responses in the pathogenesis of liver IRI (98, 99, 107), it is also unclear how the cGAS-STING pathway influences the interplay between hepatocytes and innate immune cells in liver IRI. Further studies will be needed to elucidate the coordination and orchestration of these IR-stressed cells regulated by the cGAS-STING pathway.
It is now clear that innate immunity plays a central role in the pathogenesis of liver diseases. The innate immune response may drive the progression of liver disease and contribute to liver damage, fibrosis, cirrhosis, and even HCC. The cGAS-STING pathway functions as a direct innate immune sensor of cytosolic DNA. While self-DNA sensor cGAS can recognize cellular or tissue damages, excessive activation of the cGAS-STING pathway triggers liver inflammation and subsequent disease. Studies on the cGAS-STING pathway in liver diseases have led to a better understanding of the role of the innate immune response in the development of liver inflammation and injury. New findings involved in regulating the cGAS–STING pathway will allow us to identify the essential molecules as potential therapeutic targets for liver diseases. Indeed, the cGAS–STING pathway is a dual-edged sword. Transient activation of this pathway shows an antitumor and antiviral effect, but persistent activation may promote inflammation-driven tumorigenesis (108). cGAS-STING dependent DNA-sensing of micronuclei in tumor cells can stimulate tumor metastasis due to chromosomal instability (109). However, tumor-derived cGAMP triggered natural killer (NK) cell response and inhibited tumor growth by activating the STING pathway (110). Although the STING agonists have shown promising results in HBV/HCV infection and HCC therapy in disease models (50, 51, 91, 111), more preclinical studies and early-stage clinical trials are needed to verify these encouraging survival and safety data.
The current research on the cGAS-STING signaling pathway in liver diseases has revealed only ‘tip of the iceberg’. Further studies on tissue-specific roles of the cGAS-STING pathway with other DNA sensing pathways in liver inflammation and injury are critical. They may provide new insights into the mechanism of therapy for liver diseases.
BK was responsible for the conception of the review. DX wrote the first draft of the manuscript and YT produced ideas for the figures. QX and BK participated in scientific discussion. All authors contributed to the article and approved the submitted version.
This work was supported by the NIH grants R01AI139552, R21AI146742, R21AI112722, and R21AI115133.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ALD, alcoholic liver disease; cGAMP-AMP, cyclic guanosine monophosphate-adenosine monophosphate; cGAS, cyclic GMP-AMP synthase; DAMPs, damage-associated molecular patterns; dsDNA, double-stranded DNA; ER, endoplasmic reticulum; HCC, hepatocellular cancer; IFNs, type I interferons; IRF3, IFN regulatory factor 3; IRI, ischemia/reperfusion injury; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PAMPs, pathogen-associated molecular patterns; RIG-1, retinoic acid-inducible gene I; ROS – reactive oxygen species; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; IKK, IκB kinase; NF-κB, nuclear factor-κB.
1. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of Liver Diseases in the World. J Hepatol (2019) 70:151–71. doi: 10.1016/j.jhep.2018.09.014
2. Nakamoto Y, Kaneko S. Mechanisms of Viral Hepatitis Induced Liver Injury. Curr Mol Med (2003) 3:537–44. doi: 10.2174/1566524033479591
3. Gao B, Tsukamoto H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology (2016) 150:1704–9. doi: 10.1053/j.gastro.2016.01.025
4. Younossi ZM, Bernstein D, Shiffman ML, Kwo P, Kim WR, Kowdley KV, et al. Diagnosis and Management of Primary Biliary Cholangitis. Am J Gastroenterol (2019) 114:48–63. doi: 10.1038/s41395-018-0390-3
5. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
6. Ma Z, Damania B. The Cgas-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe (2016) 19:150–8. doi: 10.1016/j.chom.2016.01.010
7. Hopfner KP, Hornung V. Molecular Mechanisms and Cellular Functions of cGAS-STING Signalling. Nat Rev Mol Cell Biol (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x
8. Akira S, Takeda K. Toll-Like Receptor Signalling. Nat Rev Immunol (2004) 4:499–511. doi: 10.1038/nri1391
9. Kanneganti TD. The Inflammasome: Firing Up Innate Immunity. Immunol Rev (2015) 265:1–5. doi: 10.1111/imr.12297
10. Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic Acid Recognition by the Innate Immune System. Annu Rev Immunol (2011) 29:185–214. doi: 10.1146/annurev-immunol-031210-101340
11. Wu J, Chen ZJ. Innate Immune Sensing and Signaling of Cytosolic Nucleic Acids. Annu Rev Immunol (2014) 32:461–88. doi: 10.1146/annurev-immunol-032713-120156
12. Riley JS, Tait SW. Mitochondrial DNA in Inflammation and Immunity. EMBO Rep (2020) 21:e49799. doi: 10.15252/embr.201949799
13. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science (2013) 339:786–91. doi: 10.1126/science.1232458
14. Chen Q, Sun L, Chen ZJ. Regulation and Function of the cGAS-STING Pathway of Cytosolic DNA Sensing. Nat Immunol (2016) 17:1142–9. doi: 10.1038/ni.3558
15. Li T, Chen ZJ. The cGAS-cGAMP-STING Pathway Connects DNA Damage to Inflammation, Senescence, and Cancer. J Exp Med (2018) 215:1287–99. doi: 10.1084/jem.20180139
16. Motwani M, Pesiridis S, Fitzgerald KA. DNA Sensing by the cGAS-STING Pathway in Health and Disease. Nat Rev Genet (2019) 20:657–74. doi: 10.1038/s41576-019-0151-1
17. Volkman HE, Cambier S, Gray EE, Stetson DB. Tight Nuclear Tethering of cGAS is Essential for Preventing Autoreactivity. Elife (2019) 8:1–21. doi: 10.7554/eLife.47491
18. Jiang H, Xue X, Panda S, Kawale A, Hooy RM, Liang F, et al. Chromatin-Bound cGAS is an Inhibitor of DNA Repair and Hence Accelerates Genome Destabilization and Cell Death. EMBO J (2019) 38:e102718. doi: 10.15252/embj.2019102718
19. Gentili M, Lahaye X, Nadalin F, Nader GPF, Puig Lombardi E, Herve S, et al. The N-Terminal Domain of Cgas Determines Preferential Association With Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep (2019) 26:2377–93.e2313. doi: 10.1016/j.celrep.2019.01.105
20. Michalski S, de Oliveira Mann CC, Stafford CA, Witte G, Bartho J, Lammens K, et al. Structural Basis for Sequestration and Autoinhibition of cGAS by Chromatin. Nature (2020) 587:678–82. doi: 10.1038/s41586-020-2748-0
21. Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, et al. Structural Basis for the Inhibition of cGAS by Nucleosomes. Science (2020) 370:455–8. doi: 10.1126/science.abd0237
22. Paludan SR. Activation and Regulation of DNA-driven Immune Responses. Microbiol Mol Biol Rev (2015) 79:225–41. doi: 10.1128/MMBR.00061-14
23. Jackson SP, Bartek J. The DNA-damage Response in Human Biology and Disease. Nature (2009) 461:1071–8. doi: 10.1038/nature08467
24. Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature (2017) 548:461–5. doi: 10.1038/nature23449
25. Ishikawa H, Ma Z, Barber GN. STING Regulates Intracellular DNA-mediated, Type I Interferon-Dependent Innate Immunity. Nature (2009) 461:788–92. doi: 10.1038/nature08476
26. Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, et al. Cyclic GMP-AMP Synthase is an Innate Immune Sensor of HIV and Other Retroviruses. Science (2013) 341:903–6. doi: 10.1126/science.1240933
27. Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, et al. The Cytosolic DNA Sensor cGAS Forms an Oligomeric Complex With DNA and Undergoes Switch-Like Conformational Changes in the Activation Loop. Cell Rep (2014) 6:421–30. doi: 10.1016/j.celrep.2014.01.003
28. Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, et al. Cyclic GMP-AMP Synthase is Activated by Double-Stranded DNA-induced Oligomerization. Immunity (2013) 39:1019–31. doi: 10.1016/j.immuni.2013.10.019
29. Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, et al. Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages is an Endogenous High-Affinity Ligand for STING. Mol Cell (2013) 51:226–35. doi: 10.1016/j.molcel.2013.05.022
30. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science (2013) 339:826–30. doi: 10.1126/science.1229963
31. Du M, Chen ZJ. DNA-Induced Liquid Phase Condensation of cGAS Activates Innate Immune Signaling. Science (2018) 361:704–9. doi: 10.1126/science.aat1022
32. Xie W, Lama L, Adura C, Tomita D, Glickman JF, Tuschl T, et al. Human cGAS Catalytic Domain has an Additional DNA-binding Interface That Enhances Enzymatic Activity and Liquid-Phase Condensation. Proc Natl Acad Sci USA (2019) 116:11946–55. doi: 10.1073/pnas.1905013116
33. Nguyen MH, Wong G, Gane E, Kao JH, Dusheiko G. Hepatitis B Virus: Advances in Prevention, Diagnosis, and Therapy. Clin Microbiol Rev (2020) 33:1–38. doi: 10.1128/CMR.00046-19
34. Ward JW, Hinman AR. What Is Needed to Eliminate Hepatitis B Virus and Hepatitis C Virus as Global Health Threats. Gastroenterology (2019) 156:297–310. doi: 10.1053/j.gastro.2018.10.048
35. Datta S, Chatterjee S, Veer V, Chakravarty R. Molecular Biology of the Hepatitis B Virus for Clinicians. J Clin Exp Hepatol (2012) 2:353–65. doi: 10.1016/j.jceh.2012.10.003
36. McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of Hepatocellular Carcinoma. Hepatology (2020) 73(S1):4–13. doi: 10.1002/hep.31288
37. Maini MK, Gehring AJ. The Role of Innate Immunity in the Immunopathology and Treatment of HBV Infection. J Hepatol (2016) 64:S60–s70. doi: 10.1016/j.jhep.2016.01.028
38. Ferrari C. HBV and the Immune Response. Liver Int (2015) 35 Suppl 1:121–8. doi: 10.1111/liv.12749
39. Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic Analysis of the Host Response to Hepatitis B Virus Infection. Proc Natl Acad Sci USA (2004) 101:6669–74. doi: 10.1073/pnas.0401771101
40. Fletcher SP, Chin DJ, Ji Y, Iniguez AL, Taillon B, Swinney DC, et al. Transcriptomic Analysis of the Woodchuck Model of Chronic Hepatitis B. Hepatology (2012) 56:820–30. doi: 10.1002/hep.25730
41. Cheng X, Xia Y, Serti E, Block PD, Chung M, Chayama K, et al. Hepatitis B Virus Evades Innate Immunity of Hepatocytes But Activates Cytokine Production by Macrophages. Hepatology (2017) 66:1779–93. doi: 10.1002/hep.29348
42. Thomsen MK, Nandakumar R, Stadler D, Malo A, Valls RM, Wang F, et al. Lack of Immunological DNA Sensing in Hepatocytes Facilitates Hepatitis B Virus Infection. Hepatol (Baltimore Md) (2016) 64:746–59. doi: 10.1002/hep.28685
43. Gehring AJ, Protzer U. Targeting Innate and Adaptive Immune Responses to Cure Chronic HBV Infection. Gastroenterology (2019) 156:325–37. doi: 10.1053/j.gastro.2018.10.032
44. Dansako H, Ueda Y, Okumura N, Satoh S, Sugiyama M, Mizokami M, et al. The Cyclic GMP-AMP synthetase-STING Signaling Pathway is Required for Both the Innate Immune Response Against HBV and the Suppression of HBV Assembly. FEBS J (2016) 283:144–56. doi: 10.1111/febs.13563
45. Cui X, Clark DN, Liu K, Xu XD, Guo JT, Hu J. Viral DNA-Dependent Induction of Innate Immune Response to Hepatitis B Virus in Immortalized Mouse Hepatocytes. J Virol (2016) 90:486–96. doi: 10.1128/JVI.01263-15
46. Verrier ER, Yim SA, Heydmann L, El Saghire H, Bach C, Turon-Lagot V, et al. Hepatitis B Virus Evasion From Cyclic Guanosine Monophosphate-Adenosine Monophosphate Synthase Sensing in Human Hepatocytes. Hepatology (2018) 68:1695–709. doi: 10.1002/hep.30054
47. Lauterbach-Rivière L, Bergez M, Mönch S, Qu B, Riess M, Vondran FWR, et al. Hepatitis B Virus DNA is a Substrate for the Cgas/STING Pathway But is Not Sensed in Infected Hepatocytes. Viruses (2020) 12:1–21. doi: 10.3390/v12060592
48. He J, Hao R, Liu D, Liu X, Wu S, Guo S, et al. Inhibition of Hepatitis B Virus Replication by Activation of the cGAS-STING Pathway. J Gen Virol (2016) 97:3368–78. doi: 10.1099/jgv.0.000647
49. Dansako H, Imai H, Ueda Y, Satoh S, Shimotohno K, Kato N. High-Level Expression of STING Restricts Susceptibility to HBV by Mediating Type III IFN Induction. FASEB Bioadv (2019) 1:67–80. doi: 10.1096/fba.1022
50. Guo F, Han Y, Zhao X, Wang J, Liu F, Xu C, et al. STING Agonists Induce an Innate Antiviral Immune Response Against Hepatitis B Virus. Antimicrob Agents Chemother (2015) 59:1273–81. doi: 10.1128/AAC.04321-14
51. Guo F, Tang L, Shu S, Sehgal M, Sheraz M, Liu B, et al. Activation of Stimulator of Interferon Genes in Hepatocytes Suppresses the Replication of Hepatitis B Virus. Antimicrob Agents Chemother (2017) 61:1–15. doi: 10.1128/AAC.00771-17
52. Li M, Sun R, Xu L, Yin W, Chen Y, Zheng X, et al. Kupffer Cells Support Hepatitis B Virus-Mediated Cd8+ T Cell Exhaustion Via Hepatitis B Core Antigen-TLR2 Interactions in Mice. J Immunol (2015) 195:3100–9. doi: 10.4049/jimmunol.1500839
53. Sitia G, Iannacone M, Aiolfi R, Isogawa M, van Rooijen N, Scozzesi C, et al. Kupffer Cells Hasten Resolution of Liver Immunopathology in Mouse Models of Viral Hepatitis. PloS Pathog (2011) 7:e1002061. doi: 10.1371/journal.ppat.1002061
54. Moradpour D, Grakoui A, Manns MP. Future Landscape of Hepatitis C Research - Basic, Translational and Clinical Perspectives. J Hepatol (2016) 65:S143–55. doi: 10.1016/j.jhep.2016.07.026
55. Foka P, Dimitriadis A, Kyratzopoulou E, Giannimaras DA, Sarno S, Simos G, et al. A Complex Signaling Network Involving Protein Kinase CK2 is Required for Hepatitis C Virus Core Protein-Mediated Modulation of the Iron-Regulatory Hepcidin Gene Expression. Cell Mol Life Sci (2014) 71:4243–58. doi: 10.1007/s00018-014-1621-4
56. Du M, Liu J, Chen X, Xie Y, Yuan C, Xiang Y, et al. Casein Kinase II Controls TBK1/IRF3 Activation in IFN Response Against Viral Infection. J Immunol (Baltimore Md 1950) (2015) 194:4477–88. doi: 10.4049/jimmunol.1402777
57. Sun Z, Ren H, Liu Y, Teeling JL, Gu J. Phosphorylation of RIG-I by Casein Kinase II Inhibits its Antiviral Response. J Virol (2011) 85:1036–47. doi: 10.1128/JVI.01734-10
58. Nitta S, Sakamoto N, Nakagawa M, Kakinuma S, Mishima K, Kusano-Kitazume A, et al. Hepatitis C Virus NS4B Protein Targets STING and Abrogates RIG-I-mediated Type I Interferon-Dependent Innate Immunity. Hepatol (Baltimore Md) (2013) 57:46–58. doi: 10.1002/hep.26017
59. Ding Q, Cao X, Lu J, Huang B, Liu Y-J, Kato N, et al. Hepatitis C Virus NS4B Blocks the Interaction of STING and TBK1 to Evade Host Innate Immunity. J Hepatol (2013) 59:52–8. doi: 10.1016/j.jhep.2013.03.019
60. Yi G, Wen Y, Shu C, Han Q, Konan KV, Li P, et al. Hepatitis C Virus NS4B can Suppress Sting Accumulation to Evade Innate Immune Responses. J Virol (2016) 90:254–65. doi: 10.1128/JVI.01720-15
61. Shahzad A, Köhler G. Inactivated Polio Vaccine (IPV): A Strong Candidate Vaccine for Achieving Global Polio Eradication Program. Vaccine (2009) 27:5293–4. doi: 10.1016/j.vaccine.2009.06.106
62. Landi A, Law J, Hockman D, Logan M, Crawford K, Chen C, et al. Superior Immunogenicity of HCV Envelope Glycoproteins When Adjuvanted With cyclic-di-AMP, a STING Activator or Archaeosomes. Vaccine (2017) 35:6949–56. doi: 10.1016/j.vaccine.2017.10.072
63. Ito H, Kanbe A, Hara A, Ishikawa T. Induction of Humoral and Cellular Immune Response to HBV Vaccine can be Up-Regulated by STING Ligand. Virology (2019) 531:233–9. doi: 10.1016/j.virol.2019.03.013
64. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat Med (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
65. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD Disease Burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the Period 2016-2030. J Hepatol (2018) 69:896–904. doi: 10.1016/j.jhep.2018.05.036
66. Kazankov K, Jorgensen SMD, Thomsen KL, Moller HJ, Vilstrup H, George J, et al. The Role of Macrophages in Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Nat Rev Gastroenterol Hepatol (2019) 16:145–59. doi: 10.1038/s41575-018-0082-x
67. Mao Y, Luo W, Zhang L, Wu W, Yuan L, Xu H, et al. Sting-Irf3 Triggers Endothelial Inflammation in Response to Free Fatty Acid-Induced Mitochondrial Damage in Diet-Induced Obesity. Arterioscler Thromb Vasc Biol (2017) 37:920–9. doi: 10.1161/ATVBAHA.117.309017
68. Qiao JT, Cui C, Qing L, Wang LS, He TY, Yan F, et al. Activation of the STING-IRF3 Pathway Promotes Hepatocyte Inflammation, Apoptosis and Induces Metabolic Disorders in Nonalcoholic Fatty Liver Disease. Metabolism (2018) 81:13–24. doi: 10.1016/j.metabol.2017.09.010
69. Neuschwander-Tetri BA. Hepatic Lipotoxicity and the Pathogenesis of Nonalcoholic Steatohepatitis: The Central Role of Nontriglyceride Fatty Acid Metabolites. Hepatology (2010) 52:774–88. doi: 10.1002/hep.23719
70. Cho CS, Park HW, Ho A, Semple IA, Kim B, Jang I, et al. Lipotoxicity Induces Hepatic Protein Inclusions Through TANK Binding Kinase 1-Mediated p62/sequestosome 1 Phosphorylation. Hepatology (2018) 68:1331–46. doi: 10.1002/hep.29742
71. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. STING-Mediated Inflammation in Kupffer Cells Contributes to Progression of Nonalcoholic Steatohepatitis. J Clin Invest (2019) 129:546–55. doi: 10.1172/JCI121842
72. Luo X, Li H, Ma L, Zhou J, Guo X, Woo S-L, et al. Expression of STING is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology (2018) 155:1971–84. doi: 10.1053/j.gastro.2018.09.010
73. Wang X, Rao H, Zhao J, Wee A, Li X, Fei R, et al. STING Expression in Monocyte-Derived Macrophages is Associated With the Progression of Liver Inflammation and Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Lab Invest (2020) 100:542–52. doi: 10.1038/s41374-019-0342-6
74. Barber GN. STING: Infection, Inflammation and Cancer. Nat Rev Immunol (2015) 15:760–70. doi: 10.1038/nri3921
75. Petrasek J, Dolganiuc A, Csak T, Nath B, Hritz I, Kodys K, et al. Interferon Regulatory Factor 3 and Type I Interferons are Protective in Alcoholic Liver Injury in Mice by Way of Crosstalk of Parenchymal and Myeloid Cells. Hepatology (2011) 53:649–60. doi: 10.1002/hep.24059
76. Wang X-A, Zhang R, She Z-G, Zhang X-F, Jiang D-S, Wang T, et al. Interferon Regulatory Factor 3 Constrains Ikkβ/Nf-κb Signaling to Alleviate Hepatic Steatosis and Insulin Resistance. Hepatol (Baltimore Md) (2014) 59:870–85. doi: 10.1002/hep.26751
77. Wong RJ, Singal AK. Trends in Liver Disease Etiology Among Adults Awaiting Liver Transplantation in the United States, 2014-2019. JAMA Netw Open (2020) 3:e1920294. doi: 10.1001/jamanetworkopen.2019.20294
78. Schwartz JM, Reinus JF. Prevalence and Natural History of Alcoholic Liver Disease. Clinics liver Dis (2012) 16:659–66. doi: 10.1016/j.cld.2012.08.001
79. Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, et al. Sting-IRF3 Pathway Links Endoplasmic Reticulum Stress With Hepatocyte Apoptosis in Early Alcoholic Liver Disease. Proc Natl Acad Sci USA (2013) 110:16544–9. doi: 10.1073/pnas.1308331110
80. Luther J, Khan S, Gala MK, Kedrin D, Sridharan G, Goodman RP, et al. Hepatic Gap Junctions Amplify Alcohol Liver Injury by Propagating Cgas-Mediated IRF3 Activation. Proc Natl Acad Sci USA (2020) 117:11667–73. doi: 10.1073/pnas.1911870117
81. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
82. Forner A, Reig M, Bruix J. Hepatocellular Carcinoma. Lancet (2018) 391:1301–14. doi: 10.1016/S0140-6736(18)30010-2
83. Callahan MK, Postow MA, Wolchok JD. Targeting T Cell Co-receptors for Cancer Therapy. Immunity (2016) 44:1069–78. doi: 10.1016/j.immuni.2016.04.023
84. Ruf B, Heinrich B, Greten TF. Immunobiology and Immunotherapy of HCC: Spotlight on Innate and Innate-Like Immune Cells. Cell Mol Immunol (2020) 18(1):112–127. doi: 10.1038/s41423-020-00572-w
85. Yum S, Li M, Chen ZJ. Old Dogs, New Trick: Classic Cancer Therapies Activate Cgas. Cell Res (2020) 30:639–48. doi: 10.1038/s41422-020-0346-1
86. Li W, Li Y, Siraj S, Jin H, Fan Y, Yang X, et al. Fun14 Domain-Containing 1-Mediated Mitophagy Suppresses Hepatocarcinogenesis by Inhibition of Inflammasome Activation in Mice. Hepatology (2019) 69:604–21. doi: 10.1002/hep.30191
87. Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D, et al. Hypoxia Induced HMGB1 and Mitochondrial DNA Interactions Mediate Tumor Growth in Hepatocellular Carcinoma Through Toll-like Receptor 9. J Hepatol (2015) 63:114–21. doi: 10.1016/j.jhep.2015.02.009
88. Wang Y, Luo J, Alu A, Han X, Wei Y, Wei X. cGAS-STING Pathway in Cancer Biotherapy. Mol Cancer (2020) 19:136. doi: 10.1186/s12943-020-01247-w
89. Bu Y, Liu F, Jia Q-A, Yu S-N. Decreased Expression of TMEM173 Predicts Poor Prognosis in Patients With Hepatocellular Carcinoma. PloS One (2016) 11:e0165681. doi: 10.1371/journal.pone.0165681
90. Qi Z, Yan F, Chen D, Xing W, Li Q, Zeng W, et al. Identification of Prognostic Biomarkers and Correlations With Immune Infiltrates Among cGAS-STING in Hepatocellular Carcinoma. Biosci Rep (2020) 40:1–17. doi: 10.1042/BSR20202603
91. Thomsen MK, Skouboe MK, Boularan C, Vernejoul F, Lioux T, Leknes SL, et al. The cGAS-STING Pathway is a Therapeutic Target in a Preclinical Model of Hepatocellular Carcinoma. Oncogene (2020) 39:1652–64. doi: 10.1038/s41388-019-1108-8
92. Xu W, Liu K, Chen M, Sun JY, McCaughan GW, Lu XJ, et al. Immunotherapy for Hepatocellular Carcinoma: Recent Advances and Future Perspectives. Ther Adv Med Oncol (2019) 11:1758835919862692. doi: 10.1177/1758835919862692
93. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep (2015) 11:1018–30. doi: 10.1016/j.celrep.2015.04.031
94. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING Activation of Tumor Endothelial Cells Initiates Spontaneous and Therapeutic Antitumor Immunity. Proc Natl Acad Sci USA (2015) 112:15408–13. doi: 10.1073/pnas.1512832112
95. Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, et al. cGAS is Essential for the Antitumor Effect of Immune Checkpoint Blockade. Proc Natl Acad Sci USA (2017) 114:1637–42. doi: 10.1073/pnas.1621363114
96. Ke B, Shen XD, Kamo N, Ji H, Yue S, Gao F, et al. Beta-Catenin Regulates Innate and Adaptive Immunity in Mouse Liver Ischemia-Reperfusion Injury. Hepatology (2013) 57:1203–14. doi: 10.1002/hep.26100
97. Ito T, Naini BV, Markovic D, Aziz A, Younan S, Lu M, et al. Ischemia-Reperfusion Injury and its Relationship With Early Allograft Dysfunction in Liver Transplant Patients. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surgeons (2020) 21(2):614–25. doi: 10.1111/ajt.16219
98. Yue S, Zhu J, Zhang M, Li C, Zhou X, Zhou M, et al. The Myeloid Heat Shock Transcription Factor 1/Beta-Catenin Axis Regulates NLR Family, Pyrin Domain-Containing 3 Inflammasome Activation in Mouse Liver Ischemia/Reperfusion Injury. Hepatology (2016) 64:1683–98. doi: 10.1002/hep.28739
99. Lu L, Yue S, Jiang L, Li C, Zhu Q, Ke M, et al. Myeloid Notch1 Deficiency Activates the RhoA/ROCK Pathway and Aggravates Hepatocellular Damage in Mouse Ischemic Livers. Hepatology (2018) 67:1041–55. doi: 10.1002/hep.29593
100. Jin Y, Li C, Xu D, Zhu J, Wei S, Zhong A, et al. Jagged1-mediated Myeloid Notch1 Signaling Activates HSF1/Snail and Controls NLRP3 Inflammasome Activation in Liver Inflammatory Injury. Cell Mol Immunol (2020) 17:1245–56. doi: 10.1038/s41423-019-0318-x
101. Li C, Sheng M, Lin Y, Xu D, Tian Y, Zhan Y, et al. Functional Crosstalk Between Myeloid Foxo1-beta-catenin Axis and Hedgehog/Gli1 Signaling in Oxidative Stress Response. Cell Death Differ (2020). doi: 10.1038/s41418-020-00695-7
102. Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, et al. Mitochondrial Damage Causes Inflammation Via Cgas-STING Signaling in Acute Kidney Injury. Cell Rep (2019) 29:1261–73.e1266. doi: 10.1016/j.celrep.2019.09.050
103. West AP, Shadel GS. Mitochondrial DNA in Innate Immune Responses and Inflammatory Pathology. Nat Rev Immunol (2017) 17:363–75. doi: 10.1038/nri.2017.21
104. Xu D, Chen L, Chen X, Wen Y, Yu C, Yao J, et al. The Triterpenoid CDDO-imidazolide Ameliorates Mouse Liver Ischemia-Reperfusion Injury Through Activating the Nrf2/HO-1 Pathway Enhanced Autophagy. Cell Death Dis (2017) 8:e2983. doi: 10.1038/cddis.2017.386
105. Zhong W, Rao Z, Rao J, Han G, Wang P, Jiang T, et al. Aging Aggravated Liver Ischemia and Reperfusion Injury by Promoting STING-mediated NLRP3 Activation in Macrophages. Aging Cell (2020) 19:e13186. doi: 10.1111/acel.13186
106. Lei Z, Deng M, Yi Z, Sun Q, Shapiro RA, Xu H, et al. cGAS-mediated Autophagy Protects the Liver From Ischemia-Reperfusion Injury Independently of STING. Am J Physiol Gastrointest Liver Physiol (2018) 314:G655–67. doi: 10.1152/ajpgi.00326.2017
107. Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver Ischemia and Reperfusion Injury: New Insights Into Mechanisms of Innate-Adaptive Immune-Mediated Tissue Inflammation. Am J Transplant (2011) 11:1563–9. doi: 10.1111/j.1600-6143.2011.03579.x
108. Kwon J, Bakhoum SF. The Cytosolic Dna-Sensing Cgas-STING Pathway in Cancer. Cancer Discovery (2020) 10:26–39. doi: 10.1158/2159-8290.CD-19-0761
109. Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal Instability Drives Metastasis Through a Cytosolic DNA Response. Nature (2018) 553:467–72. doi: 10.1038/nature25432
110. Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-Derived Cgamp Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity (2018) 49:754–763 e754. doi: 10.1016/j.immuni.2018.09.016
Keywords: DNA sensor, cyclic GMPAMP synthase, stimulator of interferon genes, Innate immunity, inflammation, liver diseases
Citation: Xu D, Tian Y, Xia Q and Ke B (2021) The cGAS-STING Pathway: Novel Perspectives in Liver Diseases. Front. Immunol. 12:682736. doi: 10.3389/fimmu.2021.682736
Received: 19 March 2021; Accepted: 19 April 2021;
Published: 29 April 2021.
Edited by:
Vincenzo Torraca, University of London, United KingdomReviewed by:
Chenhe Su, Wistar Institute, United StatesCopyright © 2021 Xu, Tian, Xia and Ke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bibo Ke, YmtlQG1lZG5ldC51Y2xhLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.