 Parth M. Patel1
Parth M. Patel1 Margaret R. Connolly1
Margaret R. Connolly1 Taylor M. Coe1
Taylor M. Coe1 Anthony Calhoun1,2
Anthony Calhoun1,2 Franziska Pollok1,3
Franziska Pollok1,3 James F. Markmann1,4Lars Burdorf1,2Agnes Azimzadeh1,2
James F. Markmann1,4Lars Burdorf1,2Agnes Azimzadeh1,2 Joren C. Madsen1,2Richard N. Pierson III1,2*
Joren C. Madsen1,2Richard N. Pierson III1,2*- 1Department of Surgery, Center for Transplantation Sciences, Massachusetts General Hospital and Harvard Medical School, Boston, MA, United States
- 2Department of Surgery, Division of Cardiac Surgery, Massachusetts General Hospital and Harvard Medical School, Boston, MA, United States
- 3Department of Anesthesiology, University Hospital Hamburg-Eppendorf, Hamburg, Germany
- 4Department of Surgery, Division of Transplantation, Massachusetts General Hospital and Harvard Medical School, Boston, MA, United States
The recent dramatic advances in preventing “initial xenograft dysfunction” in pig-to-non-human primate heart transplantation achieved by minimizing ischemia suggests that ischemia reperfusion injury (IRI) plays an important role in cardiac xenotransplantation. Here we review the molecular, cellular, and immune mechanisms that characterize IRI and associated “primary graft dysfunction” in allotransplantation and consider how they correspond with “xeno-associated” injury mechanisms. Based on this analysis, we describe potential genetic modifications as well as novel technical strategies that may minimize IRI for heart and other organ xenografts and which could facilitate safe and effective clinical xenotransplantation.
Introduction
Xenotransplantation has historically been studied by scientists and physicians as an appealing potential solution to numerous medical ailments. The earliest documented xenotransplant is the 17th century animal blood transfusion into humans (1, 2). Solid organ xenotransplantation studies began in the mid 19th century with the development of vascular anastomotic techniques and ex vivo machine perfusion (3). The current emphasis on using porcine xenografts in humans arose in the 1990s, and stimulated development of pig-to-non-human primate pre-clinical models (4–6). Simultaneously, genetic modification of pigs by nuclear deoxyribonucleic acid (DNA) microinjection coupled with advances in in vitro fertilization of large mammals raised optimism that genetic engineering of source pigs could help overcome the formidable immunological barriers identified in early solid organ xenotransplantation efforts (7). While these efforts have generated remarkable progress over the last 25 years, recent evidence points to a particularly important role for ischemia reperfusion injury in the initial graft dysfunction often observed following whole organ xenotransplantation.
The concept of “ischemia” has been known for millennia; originating from the ancient Greek term “ischaimos,” meaning to restrain blood. However, the idea that reperfusion, rather than the ischemic event itself, triggers many of the adverse consequences associated with ischemia is relatively new, enabled by Alexis Carrell’s development of vascular surgical, organ transplant, and organ perfusion techniques that allowed restoration of flow to an ischemic limb or organ (5, 6, 8–11). Effects of reperfusion after acute myocardial infarction (MI) and associated with ischemic limb, kidney, and liver revascularization were first studied at a mechanistic level in the early 1970s (12–16). Critical ischemia reperfusion injury (IRI) pathways were discovered or better understood based on work in the 1980’s, which defined key roles for integrins, selectins, and complement, as well as involvement of diverse cell death pathways (12–20). In parallel, IRI and techniques of ischemia reperfusion injury minimization (IRIM) have been studied extensively in solid organ allotransplantation (21–24). Herein, we review the literature regarding the mechanisms of IRI following allotransplantation in order to better understand how they may correspond with known mechanisms of xenograft injury. We identify applications of IRIM techniques, as well as genetic and pharmacologic approaches, that could facilitate effective clinical use of organ xenografts.
Mechanisms of IRI
Understanding the mechanisms that mediate IRI and IRI-driven cell death is critical to developing strategies to avoid it. IRI ranges in severity from minor and transient to severe and life-threatening, reflecting variable degrees of cell and organ damage which manifest at the molecular, cellular, tissue, and whole organism levels. Table 1 and Figure 1 highlight the critical molecular mechanisms involved while Figure 2 shows how the systemic mechanisms interact. Each of the broad categories of IRI mechanisms considered below interact extensively to propagate the individual damage caused by the different mechanisms.
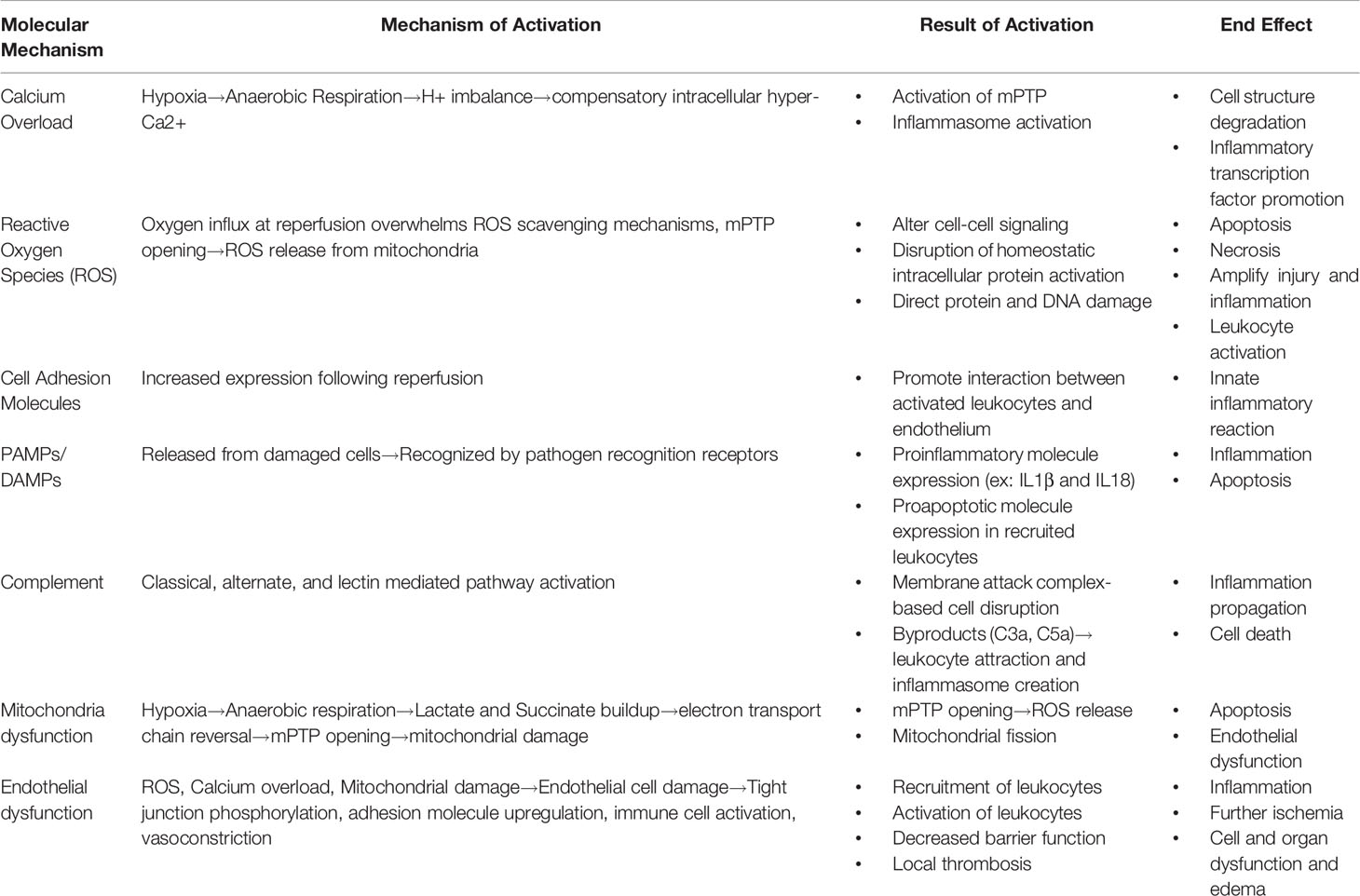
Table 1 Enumeration of the various molecular mechanisms at play during ischemia reperfusion injury, their various mechanisms of activation, how their activation propagates injury, and the end effect of the activation.
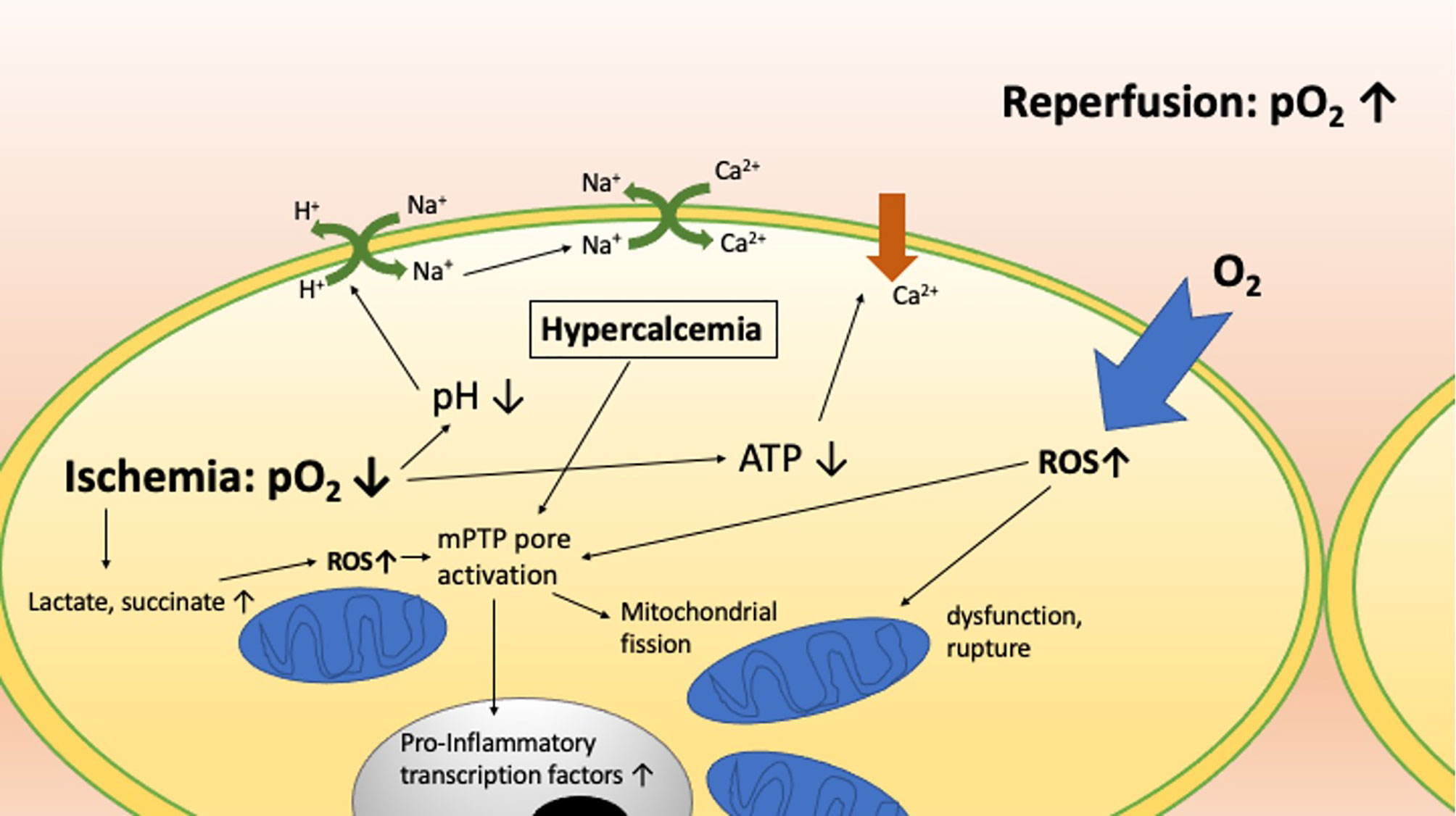
Figure 1 The molecular mechanisms involved in ischemia reperfusion injury and how they interact with one another.
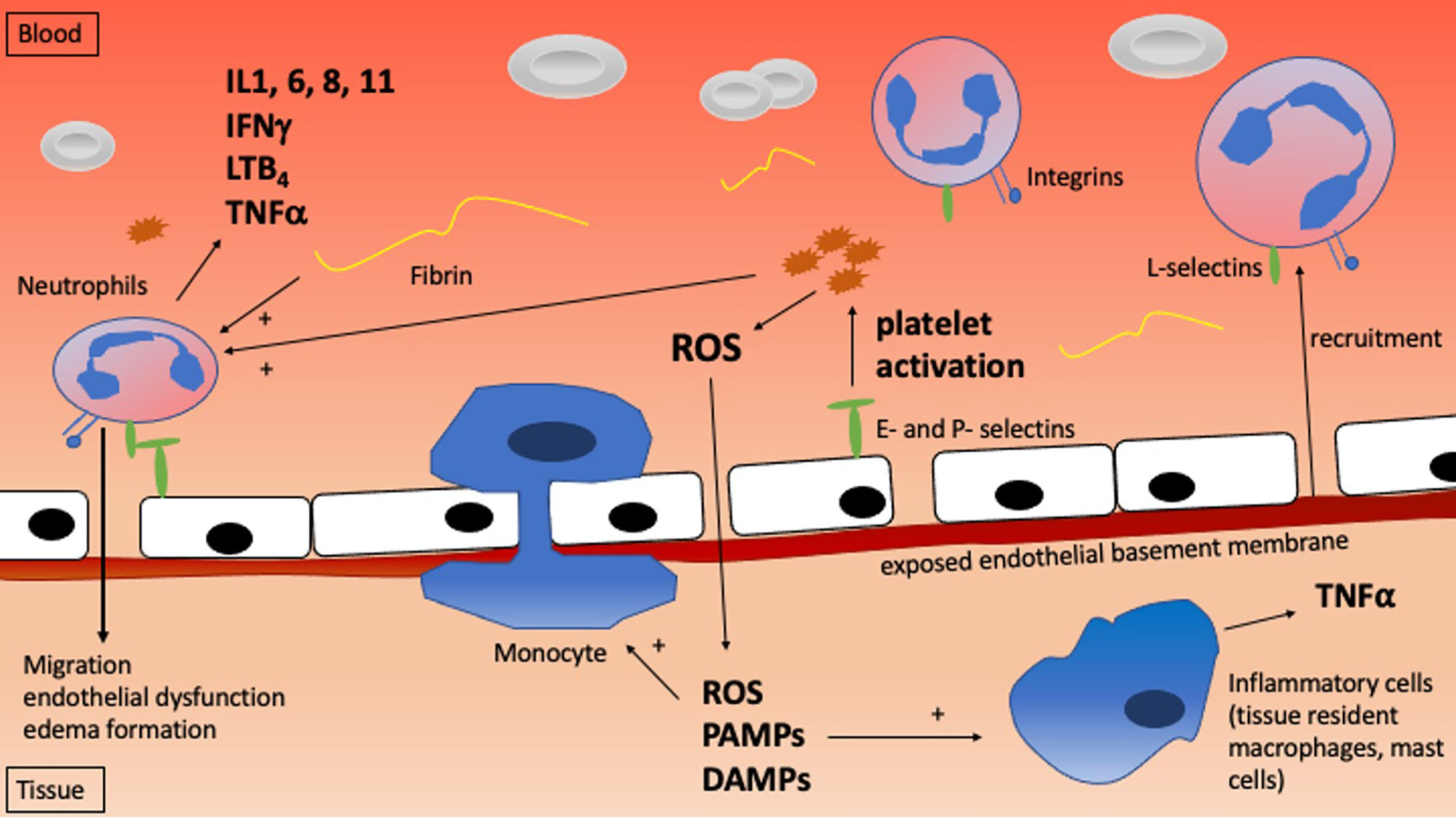
Figure 2 The systemic mechanisms involved with ischemia reperfusion injury.
Allo- and xenotransplant IRI presumably share some of these mechanisms. However, the relatively short cold ischemia intervals typical of experimental xenotransplant research studies would almost never be associated with graft dysfunction in clinical practice or in preclinical allograft models (25, 26). Recent evidence suggests that IRI mechanisms may be amplified by xeno-rejection mechanisms, including recipient innate anti-pig antibodies, interspecies complement dysregulation, coagulation cascade incompatibilities, and perhaps by other features of the profound innate xeno-immune response (25, 27).
Innate Immune Cellular and Molecular Mechanisms of IRI
Intracellular Calcium Overload/Calcium Paradox
Ischemia induces anaerobic respiration, which results in intracellular lactate and acid accumulation which decreases intracellular pH. Membrane Na+/H+ exchange pump activity increases to mitigate this (28–32). Intracellular hypernatremia then leads to an increase in cell membrane Na+/Ca++ exchanger activity, increasing the intracellular calcium levels (28–33). Ischemia also causes loss of adenosine triphosphate (ATP), which leads to dysfunctional intracellular organelle and cell membrane ATP-dependent calcium pumps, further exacerbating calcium overload (29, 31, 33). Upon reperfusion, the extracellular acid is washed out, increasing the proton gradient across the cell membrane and resulting in accumulation of cytosolic calcium (13, 28). This sequence of events was first observed by Zimmerman et al. in rat hearts; he termed this the ‘calcium paradox’ because although calcium was thought to be necessary for cardiac myocyte function, its reintroduction was detrimental during the reperfusion phase (34). This cytosolic hypercalcemia with immediate return of normal intracellular pH activates the mitochondrial permeability transition pore (mPTP), calpains, and calcium-dependent kinases which lead to lethal cell structure degradation, uric acid formation with inflammasome activation, inflammatory cytokine formation, and inflammatory transcription factor promotion (31, 35, 36). The fluctuations in pH and the ‘calcium paradox’ are particularly important in kidney, liver, and heart IRI given the importance of pH and calcium balance in normal cell and organ function (24, 31, 37–39).
Reactive Oxygen Species
Reactive oxygen species (ROS) such as superoxide, hydroxyl radicals, peroxides, and singlet oxygen play a significant role in IRI (23, 24, 40, 41). Low levels of ROS are a physiologic mechanism of cell-to-cell signaling (42). The double bond in molecular oxygen provides a critical energy source that is normally harnessed by the mitochondrial oxidation-reduction system where the byproducts of converting oxygen and carbon-bound hydrogen molecular species to carbon dioxide and water maintains cellular oxygenation and reduction homeostasis (42, 43). Reperfusion causes a massive oxygen influx that generates significant quantities of free radicals (28). ROS generation in excess of physiologic ROS scavenging mechanisms coupled with acidosis leads to mitochondrial dysfunction and eventual rupture. ROS-mediated mitochondrial injury combined with mPTP opening driven by cytosolic hypercalcemia leads to an additional burst of ROS release from mitochondria (31, 44, 45). This bolus of ROS without functional protective scavenging molecules exceeds the capacity of ROS-degrading enzymes, and disrupts cellular function through three main pathways: 1) alteration of cell-cell signaling, 2) disruption of the oxygenation/reduction balance causing alteration of intracellular proteins, and 3) direct damage to cell proteins and molecules such as DNA (31, 42, 43). These three general mechanisms of damage can lead to cell death via apoptosis or necrosis (46).
ROS and oxidative stress are particularly important in allotransplantation (23, 24, 37, 47, 48). In lung allografts, ROS formed consequent to adenosine A2A receptor activation trigger activation of invariant NKT (iNKT) cells, leading to epithelial and endothelial damage (49–53). ROS are one of the initial mediators of damage to hepatocytes and liver sinusoidal cells during liver allograft reperfusion (31). The heart is also particularly sensitive to ischemia and subsequent oxidative stress during reperfusion secondary to its high oxygen demand and rapid exhaustion of intracellular ATP stores with brief periods of normothermic ischemia (38, 39, 48, 54, 55).
In the context of xenotransplantation, ROS are also presumed to play a role in xenotransplantation ischemia reperfusion injury (XIRI). In vitro data from rat livers perfused with human blood showed that hyperacute rejection requires reactive oxygen species, with additional primary or secondary contributions from leukocytes and complement activation (56). Additionally, hydrogen peroxide and extracellular calcium have been shown to strongly induce cell adhesion molecule expression on porcine islet xenografts, presumably amplifying injury, inflammation, and islet xenograft attrition (57).
Cell Adhesion Molecules
Reperfusion following ischemia leads to increased expression of cell adhesion molecules including selectins and integrins on the endothelial surface of the injured tissue (58, 59). Upon reperfusion, endothelial cells and leukocytes activated by ROS and other pathways interact through these adhesion molecules via ligand proteins such as P-selectin glycoprotein ligand and sialyl LewisX (60, 61). This interaction is the basis of the innate inflammatory reaction associated with IRI in allotransplantation. Blocking selectin and integrin interactions in pre-clinical allotransplant models reduces both tissue damage and elaboration of pro-inflammatory chemokines into the circulation (59, 62).
Pathogen and Damage Associated Molecular Proteins
Cell damage triggered by the ‘calcium paradox’ and/or ROS elaboration triggers the release of multiple pro-inflammatory mediators and cytokines including chemotactic cytokines (chemokines), as well as activation of complement cascade proteins. The most potent proinflammatory mediators are broadly classified as damage-associated molecular proteins (DAMPs), such as various adenine phosphate nucleotides (ATP, ADP, and AMP). Heat shock proteins (HSPs) are also released. HSPs are produced during oxidative stress to ensure proper folding of other proteins and act as DAMPs (63, 64). DAMPs differ from bacterial- and virally derived pathogen associated molecular proteins (PAMPs) because they are endogenous molecules that are normally carefully prevented from release into the circulation. Like PAMPs, DAMPs are recognized by pattern recognition receptors (PRRs) such as Toll Like Receptors (TLRs). Among PRRs, DAMPs are particularly efficient in activating NLRp3 (65–68). TLRs have been generally associated with both immediate inflammatory responses (such as those implicated in IRI), and with later kidney fibrosis, thought consequent to amplifying inflammation with secondary recruitment of T and B cells (37). TLR2 and TLR4 are of particular interest as PRRs mediating DAMP-driven injury because genetic deletion of one or both decreases severity of kidney IRI in a mouse model (69). When PRRs recognize PAMPs and DAMPs, there is additional upregulation of a variety of transcription factors, most importantly NF-κB, which increase the expression of proinflammatory and pro-apoptotic molecules and proteins within the recruited immune cells (31, 66, 70–73). NLRp3 activation leads to assembly of a caspase 1 activating platform called the inflammasome with subsequent release of active forms of IL1β and IL18 to promote the sterile inflammatory response characteristic of IRI (66, 70, 71). For example, formation of the inflammasome has been demonstrated in association with myocardial damage during heart IRI (66, 74–76). The NF-κB pathway and the inflammasome are critical mediators common to the multiple upstream pro-inflammatory pathways within the systemic sterile inflammatory response that is characteristic of IRI (77).
Complement
Each of the three complement pathways (classical, driven by antibody binding and Fc-mediated complement activation; alternative, triggered by spontaneous unknown mechanisms; and lectin-mediated) can participate in IRI (31, 78, 79). Each pathway leads to formation of the C5b-9 ‘membrane attack complex’, the final common complement activation pathway which physically disrupts the cell membrane, overwhelming the ability of ion channels to maintain membrane polarization and osmolar gradients, resulting in cell death (31). Upstream byproducts of complement activation such as C5a and C3a are chemoattractants for neutrophils and macrophages which can lead to tissue inflammation (68, 72). Additionally, C3a and C5a can bind a variety of G protein coupled receptors as well as TLR2, which trigger NF-κB activation and the formation of the inflammasome (80, 81). Complement activation and deposition during reperfusion plays a role in IRI after acute MI, as well as in acute peripheral artery occlusion and in allografts, particularly kidney and liver (82–84). Accordingly, inhibiting complement activation has shown therapeutic benefit in animal models of each of these conditions (82–88). Clinical translation, however, has been slow and remains in preclinical and early-stage clinical trials (89–91).
In wild type pig-to-non-human primate (NHP) xenotransplantation, complement activation by the direct pathway (antibody-driven complement activation) is a central mechanism contributing to the phenomenon of hyperacute rejection (HAR) (92). Prolific complement activation associated with HAR was mitigated by two key genetic engineering accomplishments. First, human complement pathway regulatory protein (hCPRP)-transgenic pigs were created to enhance pig endothelial cell (EC) protection from human complement-mediated injury. Later, the porcine Gal-α1,3Galactosyl transferase gene was knocked out (GalTKO) to eliminate the predominant carbohydrate antigen recognized by human-anti-pig antibodies (93–95). Addition of one or more hCPRP’s to GalTKO further decreased the incidence of initial graft dysfunction by constraining amplification of the antibody-mediated classical complement cascade, inhibiting formation of the membrane attack complex, and better preventing endothelial injury relative to either hCPRP or GalTKO alone (96, 97).
Mitochondrial Dysfunction
Mitochondrial dysfunction plays an important role in IRI and subsequent cell death. Mitochondrial dysfunction starts during ischemia, with accumulation of lactate and succinate from anaerobic respiration damaging molecules in the electron transport chain (98). Upon reperfusion, the accumulated succinate leads to electron transport chain reversal and ROS formation (98). This reduces the mitochondrial membrane potential leading to mitochondrial Ca2+ bursts, and mPTP opening, as discussed above (98). mPTP opening causes structural damage in the form of mitochondrial swelling and is a key step in irreversible cell injury after IRI, which has been especially well-studied in heart IRI (13, 44, 45, 55, 98, 99). While ischemia and the associated lactate accumulation decreases intracellular pH and prevents mPTP opening, reperfusion rapidly returns the intracellular pH to normal, contributing to mPTP opening, particularly when combined with increased intracellular ROS and calcium (13, 100–103). The open mPTP allows H+ to enter the inner mitochondrial matrix, further uncoupling the electron transport chain and decreasing ATP production (104–106). Decreased ATP and increased mitochondrial ROS secondary to mPTP opening can lead to mitochondrial fission, a canonical step in initiating apoptosis (13, 31, 107). Mitochondrial fission also results in dysfunction of other cell organelles, particularly in endothelial cells, leading to endothelial dysfunction, seen in both IRI and xenograft injury (108).
Endothelial Dysfunction
Healthy endothelial cells are essential to maintain organ homeostasis and represent the first barrier between donor tissue parenchyma and recipient immune cells. Normal endothelium facilitates nutritive blood flow and waste clearance by preventing non-physiologic platelet adhesion and regulating local coagulation cascade activation (31). IRI disrupts multiple normal EC functions. Inflammatory cytokines, chemokines, proteases, histamine, and other proinflammatory mediators cause EC tight junction phosphorylation and internalization as well as calcium dependent myosin light chain phosphorylation, which leads to cytoskeletal contraction and loss of both paracellular and transcellular vascular barrier function (31, 109–113). Dysregulation of sphingosine-1-phospate, an endothelial tight cell junction protein, has been noted to play a significant role in IRI, most thoroughly studied in lung endothelial IRI (114).
Dying or dead cells release the ROS created after reperfusion. ROS enhance adhesion between leukocytes and endothelial cells by means of upregulation or activation of selectins and integrins, including ICAM-1, P-selectin, JAM-A, JAM-C, and PECAM-1 (31, 115–121). Similarly, multiple matrix metalloproteinases (MMPs) such as MMP9, are inducible gelatinases that disrupt liver sinusoidal endothelial cells and promote leukocyte adhesion via changes to PECAM-1 (122). Macrophages and mast cells are then activated, releasing chemoattractants such as TNFα, other proinflammatory cytokines, platelet activating factor (PAF), and the lipid mediator LTB4 (31). These mediators amplify recruitment, adhesion, and activation of neutrophils, which release chemokines and cytokines that further propagate a pro-inflammatory response and recruit other immune cells to the site of injury (112). Liver sinusoidal endothelial cells are normally protective against inflammation by their expression of transcription factors KLF2 and NRF2, but IRI damages these cells, which results in degradation of the anti-inflammatory transcription factors (41, 123, 124). In addition to elaboration of pro-inflammatory cytokines, expression of angiogenic growth factors such as VEGF is increased during IRI, and contributes to increased endothelial permeability during IRI of the lung, heart, and liver but not kidney (125–134).
ECs, damaged by pro-inflammatory cytokines and ROS, decrease production of endothelial nitric oxide (NO), a powerful vasodilator, which therefore results in vasoconstriction (135, 136). The decrease in EC NO release, coupled with upregulation of cell adhesion molecules, promotes platelet activation and adhesion, creating a prothrombotic local environment (137, 138). This leads to microthrombi formation, production of transcription factors for inflammatory mediators, and upregulation of costimulatory proteins such as CD28-B7 and ICOS-ICOSL that further propagate inflammation and cell/tissue death. They also contribute to amplification of adaptive immune responses to ‘non-self’ antigens, whether allo or xeno (117, 139, 140). Hypoxia inducible factor 1 (HIF-1) normally protects endothelial barrier function and vasodilatory function through modulation of VEGF and nitric oxide, but its dysregulation during ischemia is a significant factor in IRI (132, 141, 142).
In addition to endothelial injury, in all organs, but particularly in the lungs, epithelial injury increases levels of the receptor for advanced glycation end products (RAGE). HMGB1 is a traditionally intracellular protein that regulates a variety of nuclear functions, however upon ischemia induced necrosis, it becomes extracellular and acts as a DAMP. HMGB1 by itself or bound to inflammatory cytokines binds to membrane RAGE and becomes endocytosed. HMGB1 and/or its accompanying molecule then signal TLR4 which leads to activating the NF-κB pathway (143–145).
As in allotransplants performed in the setting of preformed anti-donor antibody (ABO mismatch, presensitized), in xenotransplantation, endothelial damage is initiated by pre-formed antibodies, in this case directed against pig carbohydrate and other antigens, that bind to the xenograft endothelium upon reperfusion, and are a critical initiator of XIRI. The pre-formed anti-pig antibodies activate complement and trigger Fc-mediated inflammatory cell adhesion and activation, amongst other inflammatory responses that damage the endothelium (146). Endothelial activation and consequent loss of thromboregulatory, anti-inflammatory, and anticoagulant functions propagates an already robust inflammatory response secondary to prolonged systemic inflammation in xenograft recipients (SIXR), which has been implicated as the culprit explaining the apparent requirement for an expanded spectrum of immunosuppressive treatments in successful xenotransplantation experiments (27).
Of particular note, the inflammatory mediators associated with SIXR are similar to those associated with XIRI (27). The importance of these inflammatory mediators in XIRI is highlighted by the especially robust response of immune cells to cross-species inflammatory mediators (147). French et al. explored this concept in IL8 and neutrophil interactions in an ex vivo pig-to-human lung xenotransplant model and in vivo pig-to-baboon lung xenotransplant model (147). These studies demonstrated that compared to allo- and autotransplantation, the xenotransplant model had significant elevation of baboon IL8 as early as four hours post-reperfusion (147). Additionally, human neutrophil adhesion was more robust when the neutrophil was activated by pig IL8 compared to human IL8 (147). Human TNFα activation of pig aortic endothelial cells also resulted in greater neutrophil adhesion compared to the neutrophil adhesion seen with human TNFα activation of human aortic endothelial cells (147).
Physiologically inappropriate formation or propagation of clot is also a feature shared between IRI and xenograft rejection due to hypoxia and inflammatory cytokine-induced upregulation of tissue factor production from endothelial cells (148). Tissue factor is a procoagulant enzyme that activates the extrinsic coagulation cascade. Intrinsic factor activation has also been implicated in the procoagulable state associated with IRI, especially in xenotransplantation (149). The procoagulable state associated with antibody binding and complement activation is amplified by a disturbance of the homeostatic balance between endothelial plasminogen activator inhibitor-1 to tissue plasminogen activator (150). As a consequence, transgenic expression of human tissue factor pathway inhibitor (hTFPI) in pigs is being studied to limit this coagulation dysregulation (151).
Dysregulated coagulation across species is known to contribute to xenograft injury, and has been addressed by expressing human coagulation pathway regulatory proteins (e.g., thrombomodulin, hTBM; endothelial protein C receptor, hEPCR; tissue factor pathway inhibitor, hTFPI) on pig endothelium (152). Xenografts from pigs with GTKO and complement regulatory transgenes, with or without additional coagulation pathway regulatory proteins, are the longest survivors to date in pig-to-NHP xenotransplantation, suggesting that the additional expression of one or more human coagulation pathway regulatory molecules may protect xenografts from IRI (25, 153, 154). Ultimately, genetic modification of the pig endothelium to prevent XIRI may prove to be critical in enabling successful clinical xenotransplantation (96, 155–157).
Cell Death Mechanisms
Cell death occurs by four basic means: necrosis, apoptosis, autophagy, and necroptosis (31).
Necrosis
Necrosis is an uncontrolled or uncoordinated form of cell death generally triggered by extrinsic toxic exposures that damages the cell and disrupts biochemical functions essential to cell survival.
Apoptosis
Apoptosis is considered “programmed” cell death, and is often normal, coordinated, and beneficial, but may also occur in response to harmful stimuli. It is a controlled process of removing and recycling cells without causing inflammation or harming the organism. It occurs as the membrane blebs, shrinking the cell, while the nucleus collapses, chromatin condenses, and DNA fragments, until the remains are engulfed by macrophages. This process is mediated by a cascade of caspase proteases that result in cleavage of intracellular substrates. There are intrinsic and extrinsic pathways to initiate the caspase cascade. Importantly, apoptosis can be initiated by hypoxia via the intrinsic (mitochondrial) pathway, a key component of ischemic injury, as well as by ROS from reperfusion (158).
Autophagy
Autophagy, or “eating of self” is similar to apoptosis in that is also a noninflammatory process. As the cell begins to die, it degrades its own components via lysosomes (159). This is initiated by formation of a phagophore around intracellular contents to create an autophagosome, which fuses with the lysosome for degradation (159). This process is seen in both phases of IRI (160). ULK1 is a key component in autophagy, and is activated when the cell is depleted of nutrients and energy in ischemia (158).
Necroptosis
Necroptosis is an intermediate phenomenon, an amalgam of mechanisms that overlap between coordinated and uncoordinated cell death (31). It is essentially a “programmed” form of necrosis, triggered by RIPK3, seen in response to viral or intracellular bacterial infections and inflammatory diseases. This contrasts with apoptosis in that caspases are uninvolved and that the result is disorganized leakage of intracellular contents, including DAMPs, triggering innate and adaptive immune responses and causing inflammation (161). This is important in IRI, and RIPK3-deficient mice are protected from IRI (162).
Cell Death in Xenotransplantation
Cell death mechanisms in xenotransplantation are starting to be studied and apoptosis has been identified as one of the critical pathways (27, 163). Inhibition of the NF-κB pathway has been shown to lead to decreased porcine endothelial cell death/apoptosis in an in vitro model of xenotransplantation, whereas endothelial cell apoptosis has been shown to lead to xenograft rejection (164). These studies, however, reviewed the impact of SIXR leading to apoptosis, and were not specifically evaluating the role of cell death in XIRI (164).
Systemic Mechanisms of IRI
Innate Immune System
Neutrophils
Leukocytes are recruited by the local release of inflammatory chemokines and cytokines, by endothelial expression of selectins, integrins, and bound antibodies, by products of clot formation such as platelets and fibrin, and by exposed basement membrane secondary to endothelial cell retraction (165). Subsequently, neutrophils release additional inflammatory mediators including IL1, IL6, IL8, IL11, IFNγ, TNFα, LTB4, as well as proteases, and monocyte chemotactic factor 1 (23, 112). IL6, IL8, and TNFα have all been associated with xenograft injury and failure in multiple models (166–168). The selectin-dependent, complement-driven migration of neutrophils into the parenchyma, endothelial dysfunction, parenchymal edema, and numerous other processes have been implicated in the condition of “No-Reflow”. No-Reflow is a clinical finding of lack of perfusion to the microvasculature despite reperfusion at the tissue level following a prolonged ischemic event. It has been established to be associated with IRI in particular of the heart and limb (65, 169–173). Blocking cell adhesion molecules, particularly selectins and integrins, disrupting chemokine and cytokine release, as well as complement inhibition has each been shown to attenuate IRI and prevent or reduce the severity of “No-Reflow” (174–178).
Macrophages and Mast Cells
Tissue resident macrophages are activated by ROS, PAMPs, and DAMPs (179). Activated macrophages release TNFα, which leads to upregulation of NF-κB and other transcription factors throughout the reperfused organ or tissue (31, 180). The resident macrophages of the liver, Kupffer cells, are activated by DAMPs as well as by hepatocyte damage and release of the proinflammatory ligand HMGB1 (181, 182). Kupffer cell activation can further exacerbate hepatocyte injury by promoting neutrophil recruitment and upregulating NF-κB (31, 182–185). Similarly, resident lung macrophages are activated during ischemia reperfusion and enhance epithelial and endothelial dysfunction and inflammation by the TLR pathway (77, 186–189). ROS, TNFα, IL17, and C-X-C motifs are released from the injured epithelium, activated macrophages, and iNKT cells (51, 143, 186, 190–192). These are powerful pro-inflammatory cytokines and chemokines that recruit leukocytes (143, 186, 190–192). ROS, complement, LTB4, and other molecular mediators of IRI activate mast cells as well (193). Mast cells are subendothelial and reside in nearly all tissues. Mast cell activation leads to release of various monoamines, proteases, TNFα, and other inflammatory mediators that result in edema, inflammation, and local hemorrhage (193).
Mast cells and resident tissue macrophages also play a significant role in xenograft dysfunction, and may aggravate XIRI (155). Burdorf et al. demonstrated in an ex vivo lung perfusion xenograft model that blocking thromboxane and histamine receptors, mediators released by peri-endothelial mast cells, led to a blunting of initial rise in pulmonary vascular resistance, decreased pulmonary edema, and delayed loss of vascular barrier function after reperfusion (155).
Platelets
During IRI, platelet activation occurs through a multitude of receptors including platelet collagen receptor, adenosine diphosphate receptor, glycoprotein-IIb/IIIa (GPIIb/IIIa), P-selectin, and G-proteins. Additionally, complement byproduct-mediated, histamine-mediated, and thromboxane-mediated pathways exist which lead to platelet activation (194–197). Activated platelets release ROS, serotonin, and platelet activating factors, as well as interact with leukocytes and endothelial cells, propagating sterile inflammation, coagulation, and IRI (194).
In addition to these processes, Fc receptor mediated platelet activation and complement mediated platelet activation are particularly critical in xenotransplantation (198). The interaction between the complement system, coagulation-fibrinolytic system, and platelets leads to thromboinflammation. In the long run this leads to consumptive coagulopathy and is a major barrier in xenotransplantation (198). These processes have been traditionally associated with SIXR however each of the component pieces leading to thromboinflammation are also activated during XIRI (198).
Adaptive Immune System
The adaptive immune system has been found to play an important role in IRI (37, 199–203). Dendritic cells, which are stimulated by ROS and DAMPs, are pivotal to triggering and amplifying activation of adaptive immunity (37). However, it is clear that the adaptive immune response not only propagates injurious mechanisms in IRI but can also down-regulate IRI (37). CD4+ and CD8+ T cells secrete inflammatory cytokines and chemokines such as IL1, TNFα, and IL17 to recruit other cells (37). CD8+ cells amplify IRI independent of the T-cell receptor and antigen specific T-cell functions (204). IgM and IgG produced by B-cells also contribute to IRI by ligating complement, neutrophils, macrophages, and platelets to xenoantigens in the graft and after release of cells and cell fragments into the circulation (37). The role of T-regulatory cells is an active avenue of research and they are thought to provide anti-inflammatory regulatory function in reducing IRI (37, 205, 206). NK and NKT cells are also thought to play a role in IRI, specifically in xenotransplantation where they have been shown to adhere to and damage the endothelium because of absence of self-recognition molecules (CD47, HLA-E), similar to a mechanism found in IRI-damaged or neoplastic cells (207).
Brain Death
Brain dead donors constitute a significant portion of the allograft donor pool, including more than 50% of kidneys, 85% of livers and lungs, and almost all hearts (208). Brain death induces a sterile inflammatory response that has been well-characterized, and activates many of the same pathways that mediate IRI (209). These pathways include pro-inflammatory cytokine release, endothelial injury, coagulation pathway dysregulation, and enhanced leukocyte adhesion and migration in the lung and other tissues (210–213). Additionally, neuropeptides, such as neuropeptide Y, calcitonin gene related peptide, and substance P, released during brain death, propagate sterile inflammation within the donor (209, 214). Brain death also causes dysregulation of the hypothalamic-pituitary-adrenal axis resulting in decreased adrenocorticotropic hormone and cortisol (215–217). There is also a rapid depletion of antidiuretic hormone resulting in diabetes insipidus, and thyroid stimulating hormone resulting in central hypothyroidism and subsequent decrease in serum T3 and T4 (215–217). These changes have significant electrolyte and hemodynamic consequences throughout the brain death period requiring special attention and treatment so as to preserve the donor organs (215–217). The injurious consequences of each of these “sterile inflammation” pathways differs from organ to organ, as exemplified by varying behavior of different organs from individual donors (209). Sterile inflammatory mediators elaborated in association with brain death “prime” transplanted organs for a ‘second hit’, such as IRI (218). For renal allografts, where there is a robust reference group of case-matched controls, allografts from brain dead donors demonstrate a higher incidence of primary graft dysfunction and reduced graft survival relative to living donors when all other variables are taken into account such as graft ischemic time, HLA mismatch, and immunosuppression regimen (219, 220). The brain death priming process is blunted in organs procured from donors after cardiac death (DCD). The effects and mechanisms of IRI from DCD organ procurement, and the competing risk of extended warm ischemic time between cardiac death and organ flush in situ, still need to be elucidated (40, 221–226). With regards to known mechanisms contributing to IRI, xenotransplantation has the advantage of avoiding the “brain death priming” phenomenon seen in allotransplantation.
Organ-Specific Ischemia Reperfusion Injury Minimization (IRIM)
Specific organ allografts have unique properties that make them more or less susceptible to different mechanisms of IRI (23, 24, 40, 41, 66, 165). In allotransplantation, there is now significant research on organ-specific mechanisms contributing to IRI, and IRIM strategies (23, 24, 40, 41, 66, 165, 227). Current research in IRIM for heart, lungs, kidney, and liver allografts include reducing the effects of ROS, and directly or indirectly inhibiting inflammatory mediators, complement, immune cell platelet adhesion and activation, and maintaining, preserving, or restoring endothelial barrier and vasoregulatory functions (23, 41, 50, 52, 114, 190, 227–265). Promising findings in reduction of lung IRI have included adenosine A2A receptor activation reducing microvascular permeability and lung injury, early growth response 1 (Egr1) deletion reducing neutrophil infiltration, C3a receptor antagonist decreasing cell injury and inflammation, carbon monoxide in cold flush reducing inflammatory mediators and cellular infiltrate, and nitric oxide to reduce pulmonary arterial pressures, inflammation, and apoptosis (50, 52, 190, 266–269).
Certain IRIM targets and treatments are common between multiple organs. Mesenchymal stem cell (MSC) treatment at the time of procurement, or during ex vivo perfusion before implantation, is a novel area of research in IRIM. MSCs are collected from bone marrow, amniotic tissue, or the umbilical cord, as their extracellular vesicles include apoptotic bodies, which were hypothesized to interact with other cells to quiet the inflammation seen in IRI (227, 228, 270). These have been administered by culturing the cells (with or without separation of extracellular vesicles) and adding to a perfusate such as Steen solution in ex vivo perfusion, as an intravenous infusion to the recipient at the time of reperfusion, or bronchoscopically (227, 239–241, 244, 246, 270–273).
MSC treatment has been found to be broadly protective of IRI in the heart, lungs, and kidney in preclinical models (23, 227, 239–241, 246, 270–274). It has been proposed that MSCs may have their protective effect by three general actions: paracrine secretion of soluble factors, increasing cell to cell interactions via microtubules, and secretion of vesicles containing proteins and nucleic acids (275). There is currently one phase 1 clinical trial using MSCs in kidney transplantation; early results have shown safety of MSC infusion in these patients (276). We expect the number of these studies to increase as has been done for in other disease processes that have trialed MSC therapy (277–279). Similarly, use of inhaled pharmacologic agents, specific inhaled anesthetic, or either inhaled or perfusate dissolved gasses such as nitric oxide, hydrogen sulfide, and carbon monoxide have been used in preclinical models to mitigate IRI of the lungs, liver, and kidneys (266–269, 280–284).
Lung
In lung transplantation, primary graft dysfunction (PGD) is closely associated with duration of cold and warm ischemia, and is usually attributed to IRI (285, 286). Ex vivo lung perfusion (EVLP) for normothermic organ preservation and rehabilitation is currently in use clinically and has been found to potentially reduce IRI (287–289). EVLP was introduced to the clinic in 2011 for high-risk lungs (290). Preclinical studies in a porcine model have shown that EVLP may reduce IRI by decreasing the insult on the endothelium or by reducing the inflammatory cytokine load (291–293). Controlling reperfusion by means of reversing oxygen debt in the absence of mechanical stress/strain and in the absence of platelets and neutrophils are also beneficial attributes of EVLP (291, 292). When comparing tissue cytokine levels of EVLP lungs versus cold static preserved lungs from biopsies obtained two hours post implantation, EVLP lung tissue had significantly lower concentrations of IL1β, IL18, and IFNγ. This indicates that ischemia minimization is associated with reduced elaboration of pro-inflammatory cytokines. In this study, the potential IRI reduction may have been secondary to decreasing the injury mechanisms associated with cold storage and subsequent reperfusion (291). EVLP has the potential to significantly reduce IRI by improving organ preservation by reducing or preventing oxygen debt and provides a platform for delivering preventive and/or adjunctive reparative treatments based on defined IRI mechanisms.
Kidney
In kidney transplantation, PGD encompasses both the relatively common delayed graft function (DGF), with recovery of glomerular filtration rate in days or weeks after transplant, as well as primary non-function, which does not recover. Renal allograft PGD is closely associated with duration of cold and warm ischemia, and is usually attributed to IRI (24). Inhibition of apoptosis, TLR signaling, and complement activation/injury are the IRIM strategies supported by strong preclinical and clinical evidence base, and are closest to clinical practice. QPI-1002 is an siRNA that temporarily inhibits p53 (37, 274). p53 inhibition in renal tubular cells has been shown to reduce apoptosis (37). QPI1002 was successful in reducing incidence and severity of DGF in phase I and II trials, and is currently in a phase III clinical trial to reduce DGF in kidney transplant patients (NCT0261096) (37, 294). An anti-TLR2 monoclonal antibody (Tomaralimab, Opsona Therapeutics Ltd, Dublin, Ireland) is currently in phase II clinical trials for DGF reduction (37, 262). Recombinant C1 inhibitors (C1-INH) are being evaluated in multiple clinical trials, with interim reports suggesting equivocal effects and other studies showing reduced severity of DGF (37). Complement Receptor 1 (CR1) is a protein found on the surface of many different cell types that downregulates C3 convertase (24). Mirococept (APT070, Inflazyme Pharmaceuticals, Richmond, BC, Canada) is a CR1 with a membrane anchor currently in phase III clinical trials for preventing IRI in kidneys (295). Orthosteric inhibition of leukocyte integrin CD11b/CD18 to diminish kidney IRI is an active area of research in a non-human primate model (296). Hypothermic (1-8°C) ex vivo perfusion is a long-established technology, and is a widely used method in many organ procurement organizations based on evidence of improved outcomes, including reduced DGF for deceased donor kidneys from high-risk donors or with expected prolonged cold ischemic times, intending to reduce IRI (253). More recently, normothermic blood based ex vivo perfusion has been shown to reduce the incidence of DGF in kidney allografts and this protocol is now in phase II clinical trials with primary outcomes being the incidence and nature of renal allograft DGF (ISRCTN15821205) (297, 298).
Liver
Liver PGD (early allograft dysfunction [EAD] or primary nonfunction [PNF]) is generally ascribed to IRI (299). Machine perfusion (MP) has been studied most extensively in preclinical models to combat IRI, paving the way for clinical trials (22, 300–312). The three main techniques of MP that have become clinically applicable include hypothermic MP, hypothermic oxygenated MP, and normothermic MP (NMP). Each technique aims to mitigate IRI by reducing standard cold storage time and associated ischemic injury. In recent trials, the use of MP has been shown to reduce histologic bile duct injury and decrease post-operative hepatocellular enzyme release, both surrogates for IRI (22, 302, 304). Of interest, the first ischemia-free liver transplant was performed in China, where NMP was initiated in the donor and continued without interruption until graft revascularization in the recipient (313). This technique is hypothesized to have reduced IRI given minimal hepatocyte necrosis and apoptosis seen on post-reperfusion graft histology, low levels of inflammatory cytokine release by immunohistochemical staining and quantitative real-time polymerase chain reaction, and low post-reperfusion hepatocellular enzyme levels.
Heart
Heart allograft primary graft dysfunction has been linked to IRI and manifests as global biventricular dysfunction – ranging from diastolic dysfunction in milder cases to progressively depressed systolic function in its more severe presentation (314). PGD after cardiac transplantation remains a significant clinical problem despite decades of research (315). Targeting inflammatory cytokines may be a new and promising avenue for IRIM. Tocilizumab (Genentech/Roche, San Francisco, CA, USA), an anti-IL6R monoclonal antibody, has proven efficacious in a clinical trial in reducing myocardial inflammation after acute myocardial infarction (316). It is currently being investigated in the heart transplant setting with study endpoints being donor specific antibodies, acute cellular rejection, antibody mediated rejection, hemodynamic compromise secondary to clinical rejection, death, or transplantation in the first year (NCT03644667). Antibodies against IL1R and TNFα reduce associated heart IRI in preclinical models but have not yet been evaluated in clinical trials (317, 318). The availability of clinically approved anti-IL1R and anti-TNFα antibodies (Anakinra, Swedish Orphan Biovitrum AB, Stockholm, Sweden and Etanercept, AMGEN, Thousand Oaks, CA, USA) make these avenues especially enticing. Heart IRIM via normothermic ex vivo perfusion has clinical approval and is currently available (319–322). On the horizon for cardiac allotransplantation is hypothermic ex vivo perfusion. Nilsson et al. compared hypothermic ex vivo perfusion to cold static storage in a recent phase II clinical trial and showed safety and efficacy of the circuit in humans (323). Ex vivo heart perfusion limits IRI by minimizing cold static storage time and thus minimizing organ ischemia.
Cardiac reanimation after DCD procurement is being used to expand the transplant donor pool (319–322). Optimizing the initial reperfusate of the heart contributed to the success of heart DCD (40). The DCD heart can be procured in two ways: direct procurement and preservation (DPP) and normothermic regional perfusion (NRP) (40). In DPP an emergent sternotomy is performed after declaration of death and the heart is perfused with cardioplegia solution during dissection. The heart is then explanted, connected, and reanimated with the normothermic ex vivo perfusion device. Throughout travel to the recipient’s facility the heart is perfused with normothermic oxygenated blood procured from the donor. The principles of the cardioplegia strategy for DPP and IRIM both aim to reoxygenate the myocardium, flush out ROS, replete ATP stores, and restore calcium homeostasis (40). In NRP, once death is declared, a sternotomy is performed, the head vessels are clamped, and the body is resuscitated via central or peripheral extracorporeal life support. The heart is then assessed in situ and if it is functionally normal, it is dissected, arrested, removed, and placed on the ex vivo perfusion device.
Discussion
IRI in Xenotransplantation
If shown to be dependably successful, organ xenotransplantation from pigs would provide an abundant and ethically acceptable alternative to human donors as a source of organs for transplantation that would be available on demand. Over the past 20 years, pigs have been genetically modified to address known xenograft injury mechanisms driven mainly by preformed antibody, complement activation, and coagulation pathway dysregulation. Long-term, life-supporting xenograft survival has recently been achieved using organs from several of these pig strains and transplanting them into non-human primates, the best available model for porcine-to-human xenotransplantation (4, 324). Three components of conventional allograft IRI that we believe are particularly critical to XIRI are complement activation, sterile inflammation, and endothelial activation (27, 146, 325, 326). However, heart xenografts of multiple different phenotypes and even pig hearts with three key xeno gene modifications (GalTKO.hCPRP.hTBM) were unable to dependably support life in vivo in the hands of multiple experienced investigators. Only with XIRI minimization were Langin et al. able to achieve consistent xenograft survival more than one day (25, 327, 328).
XIRI is a critical barrier to clinical xenotransplantation, but it affects xeno-organs to different degrees (25, 153, 329, 330). The xeno-heart seems to require robust XIRI minimization technique to avoid perioperative cardiac xenograft dysfunction (PCXD) (25, 153, 328). Additionally, Shah et al. showed that liver xenotransplant recipients suffer lethal coagulopathy, and Watanabe et al. showed that lung xenotransplantation is characterized by significant endothelial dysfunction and loss of vascular barrier function (alveolar hemorrhage) (329, 330). The mechanistic basis for the vulnerability of hearts and other organ xenografts to IRI may be related to xeno-specific mechanisms. For example, Khalpey et al. found that pig endothelial cells lose expression of ecto-5’ nucleotidase when exposed to human and non-human primate blood. This leads to loss of a key producer of extracellular cryoprotective, antithrombotic, and immunosuppressive adenosine which would put the porcine organ at increased risk of IRI (331). Porcine organ vulnerability may be due to this or a yet undiscovered peculiarity of pig physiology.
Ischemia Reperfusion Injury Minimization in Xenotransplantation
Complement and coagulation pathway regulatory molecules, self-recognition receptors that inhibit cell-mediated injury, and constitutive expression of anti-inflammatory transgenes that have been incorporated into porcine donors to address xeno-specific injury mechanisms are likely to play an important role in minimizing IRI (Table 2) (156, 157, 332). Preventing detrimental complement activation, endothelial dysfunction, and reducing the cross species inflammatory response will be critical for future research into XIRI minimization. Langin et al. and Mohiuddin et al. have used pigs that express complement regulatory protein (hCD46) and pretreat their heart xenotransplant recipients with complement inhibitors. However, Langin reported that those interventions along with additional expression of human thrombomodulin, did not prevent initial xenograft dysfunction (IXD) (25, 333). Thus, while expression of these two human complement and coagulation pathway regulatory proteins would be expected to be protective for GalTKO hearts in baboons, ischemia minimization was necessary and sufficient, at least when anti-IL6R, anti-IL-1R, and anti-TNFα were also administered.

Table 2 Summary of the promising mechanisms of ischemia reperfusion injury minimization currently being used in in vivo and in vitro models.
The relative contribution of complement, coagulation, and inflammation to XIRI remains unknown. Xenograft injury and allograft IRI are both associated with pro-inflammatory cytokines (168, 316–318, 334–336). Whether the IL6, IL1, and TNFα pathways and complement and coagulation pathway dysregulation contribute significantly to IXD of heart xenografts has not been addressed and could be studied by omitting these interventions from the regimen, or by including these agents when using hearts without the hCD46 or hTBM genetic modifications.
As described by Langin et al. in the orthotopic pig-to-baboon heart xenotransplant model, organ preservation with machine perfusion is the most obvious and readily available approach to IRIM. In their initial experiments, standard cold static ischemia was associated with IXD in all but one of four consecutive xenografts. In contrast, when the hearts were perfused with cold (8°C) oxygenated cardiac STEEN solution (XVIVO, Gothenburg, Sweden), a hyperkalemic, hyperosmotic, blood-containing cardiac preservation solution developed by the Steen group, no IXD was reported for 9 consecutive subjects (25, 337). The perfusate was administered continuously to the donor heart prior to explant and either continuously or intermittently, at 20-minute intervals, throughout implant. Their perfusate contained hormones, inotropes, and anesthetic agents based on 20 years of work in the field. Delivery of an oxygenated perfusate in an arrested heart allowed for restoration of oxygen supply with minimal consumption; avoiding ischemia minimized ROS production. The continual circulation of an electrolyte and pH balanced solution likely minimized ATP depletion, mPTP opening, and calcium and other ion flux. The absence of platelets and leukocytes and their associated pro-inflammatory cytokine and lipid arachidonic acid-derived mediators may have also helped in preventing IRI. The pretreatment of the recipient with a cytokine antibody cocktail may have blocked the effects associated with release of these inflammatory mediators at initial xenograft reperfusion. These multiple different treatments, in combination, were sufficient to prevent graft dysfunction; whether they had direct or indirect effects to inhibit significant endothelial dysfunction, complement activation, and significant initial inflammation remains to be determined.
Normothermic ex vivo perfusion is used clinically in allotransplantation with good effect, particularly in liver and heart transplantation (338, 339). There are no current studies demonstrating the effects of normothermic ex vivo perfusion in xenotransplantation. Normothermic machine perfusion shortens cold ischemia while supporting the full organ metabolism allowing for assessment of injury and function. Hypothermic and subnormothermic machine perfusion have also been shown to enhance mitochondrial function and replenish cellular energy stores (338). Lastly, ischemia-free organ transplantation requires normothermic machine perfusion and completely abolishes ischemia. Given that there is no component of trauma or brain death with xenotransplantation, and ideally no question of organ viability before procurement, the role of ex vivo/machine perfusion and ischemia minimization in xenotransplantation require further investigation for all organs, and may be critical to safe, successful clinical translation.
Other strategies in XIRI minimization include the use of mesenchymal stem cells (MSCs). MSCs are hypothesized to mitigate IRI through their anti-inflammatory, pro-immunomodulatory, and tissue repair characteristics. Dayan et al. utilized a myocardial infarction model in sheep with treatment with human MSCs, showing improvement in myocardial perfusion at one month in the treated group without adverse events (270, 273). Qiang et al. utilized a canine model of cardiopulmonary bypass-induced IRI and treatment with human amniotic MSCs which led to mitigation of IRI as evidence by decreased levels of TNFα and IL8 and increased levels of IL10 relative to dogs not treated with MSCs (211).
Significant work has shown that activating the protective NRF2/heme oxygenase-1 (HO-1) pathway may reduce oxidative and inflammatory stress associated with IRI (340, 341). Bach et al. demonstrated the importance of HO-1 in a mouse-to-rat cardiac xenograft model, noting increased expression of HO-1 in xenografts with long-term survival (342, 343). In vitro work involving transgenic expression of human HO-1 into pigs has shown a decrease in reactivity of pig fibroblasts to oxidative stress as well as human TNFα, therefore this gene has been added in genetically engineered pigs (344). The separate effect of HO-1 on organ behavior in xenotransplantation has not yet been studied. Of note, delivery of an ROS scavenger may also reduce oxidative stress and attenuate XIRI, but further work is needed (345, 346).
Limiting cross-species coagulation dysregulation may help minimize XIRI. Anticoagulant genes should diminish coagulation pathway dysregulation and blunt inflammation. CD39 expression should improve ADP and AMP catabolism, reducing platelet and EC activation, and promote elaboration of anti-inflammatory adenosine by endogenous CD73, thus inhibiting both physiologically inappropriate clotting and inflammation (347). Of note, pigs are relatively deficient in CD73, perhaps accounting in part for the pig’s particular IRI susceptibility. Human endothelial protein C receptor (hEPCR) transgenesis along with hTBM and hTFPI are all likely to reduce dysregulated coagulation, and EPCR, interacting with PPAR-1, upregulates potentially critical anti-inflammatory intracellular mechanisms (348, 349). Including one or more of these genetic modifications in the pig construct may reduce the need for ischemia minimization strategies to reduce IRI and its consequences. Parenthetically, addressing porcine vWF-human GP1b abnormal interaction (blocking Fab; humanizing pvWF by genetic engineering) reduces, but does not prevent, platelet sequestration seen in heart, kidney, liver, and lung models (350).
Molecular mechanisms of IRI mentioned previously such as the calcium paradox, mitochondrial dysfunction, and mPTP have not been studied in the context of xenotransplantation but may be presumed to be similarly important. Ex vivo perfusion should minimize activation of these pathogenic mechanisms since the cellular electrochemical and energy imbalances associated with ischemia and reperfusion are reduced or prevented prior to transplantation.
Immune cell adhesion and activation also play a role in XIRI. Using genetic engineering to express self-recognition molecules on the porcine endothelium may help attenuate XIRI. Damaged or neoplastic cells lose expression of self-recognition receptors such as CD47 and HLA-E, resulting in activation of innate immune cell scavenging by neutrophils and monocyte/macrophage lineage cells (CD47), NK cells, and other myeloid cells (HLA-E) (351, 352). Similarly, pig cells that lack human CD47 and HLA-E are susceptible to phagocytosis or cytolysis by these recipient effector leukocyte populations. In response, CD47 has been expressed in pigs, and was associated with prolonged survival in a porcine-NHP lung xenotransplant model (329). Incorporation of hHLA-E into the porcine endothelium also suppresses NK cell and macrophage activation (351).
Selectin antagonism is effective in decreasing IRI in MI, peripheral arterial, and allograft IRI models. It seems logical that selectin blockade will similarly attenuate XIRI by decreasing the rolling and adhesion of neutrophils, although this has not yet been confirmed. Further work specifically for XIRI needs to be done in models where other xeno-driven cell adhesive interactions are addressed, allowing this specific question to be answered (146).
While complement inhibition and coagulation regulation have been mentioned with respect to genetic engineering, these mechanisms can be targeted by other methods. Soluble hTBM or direct thrombin inhibitors can be administered, and should work synergistically to membrane bound hTBM to decrease activation of the coagulation cascade (353). IL4 activates the PI3K/Akt pathway in endothelial cells and has been shown to be protective of complement-mediated endothelial cell damage in xenotransplantation in vitro models. IL4 transfection with or without ex vivo perfusion, may be another avenue for protection from complement mediated IRI injury (354).
In addition to the innate immune system, altering how the adaptive immune system responds to the xenograft may attenuate XIRI by use of conventional immunosuppressive agents or other approaches. Artificial antigen presenting cells have been used in oncology to stimulate anti-tumor specific T-cell responses, and could in principle be engineered to regulate how the recipient immune system responds to the pig antigen (355).
Conclusion
IRI and its consequences for initial allograft organ function are well known to exert a critical negative influence on the initial and long-term outcomes of allotransplantation (23, 24, 41, 315). Minimizing IRI by ex vivo organ perfusion has allowed expansion of the donor pool in allotransplantation, and specifically enabled safer use of DCD organs. In addition, there is considerable evidence that IRI minimization by machine perfusion of ‘marginal’ kidneys, lungs, and livers is associated with improved allotransplant outcomes (288, 319, 356). To the extent that the major injury mechanisms are shared between xenograft injury and allograft IRI, including complement activation, endothelial dysfunction, and a profound inflammatory response, it is logical that genetic engineering modifications targeting these mechanisms should also attenuate XIRI (27, 146, 147, 155, 325, 326). Evidence to date indicates that XIRI is particularly critical in cardiac xenotransplantation. Efficacy of ischemia minimization in other xeno organs has not yet been evaluated (25, 153, 329, 330). By analogy with allograft experience, minimizing IRI either with ex vivo perfusion, or by targeting shared, overlapping injury mechanisms, may be critical for clinical translation. We suspect that using a combination of both approaches may offer critical advantages through redundancy to improve safety, maximize the odds of initial success with xenotransplantation in the short term, and the potential long-term benefits. With the advances of genetic modification, cytokine inhibition along with ischemia minimization via ex vivo organ perfusion has enabled a breakthrough in cardiac xenotransplantation. It remains to be seen which features are critical, and whether any drug treatments to the recipient can be eliminated using pigs with genes that constrain IRI mechanisms, like HO-1, HLA-E, hCD47. If not, continued progress in our understanding of XIRI mechanisms is likely to contribute to significant additional progress in pig-to-non-human primate XIRI minimization and xenotransplantation preclinical outcomes, providing hope for future clinical translation (25, 153, 329, 330).
Author Contributions
PP: Performed majority of background research and writing of the paper. MC and TC: Participated in background research and writing of the paper. AC, FP, JFM, LB, AA, and JCM: Participated in writing of the paper. RP: Participated in concept development and writing of the paper. All authors contributed to the article and approved the submitted version.
Funding
RP: NIH RO1 AI153612 (PI: RP) CRISPR-Modified Cardiac Xenograft Transplantation. FP: German Heart Foundation (Deutsche Herzstiftung e.V.).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ATP, adenosine triphosphate; CR1, complement receptor 1; DAMPs, damage-associated molecular proteins; DGF, delayed graft function; DNA, deoxyribonucleic acid; DPP, direct procurement and preservation; DCD, donors after cardiac death; EAD, early allograft dysfunction; EC, endothelial cell; EVLP, ex vivo lung perfusion; GalTKO, gal-α1,3galactosyl transferase gene was knocked out; GPIIb/IIIa, glycoprotein-IIb/IIIa; HO-1, heme oxygenase-1; hCPRP, human complement pathway regulatory protein; hEPCR, human endothelial protein C receptor; hTFPI, human tissue factor pathway inhibitor; HAR, hyperacute rejection; HIF-1, hypoxia inducible factor 1; IXD, initial xenograft dysfunction; iNKT, invariant NKT; IRI, ischemia Reperfusion Injury; IRIM, ischemia reperfusion injury minimization; MP, machine perfusion; MSC, mesenchymal stem cell; MMPs, metalloproteinases; mPTP, mitochondrial permeability transition pore; MI, myocardial infarction; NO, nitric oxide; NHP, non-human primate; NMP, normothermic Machine perfusion; NRP, normothermic regional perfusion; PAMPs, pathogen associated molecular proteins; PRRs, pattern recognition receptors; PCXD, perioperative cardiac xenograft dysfunction; PAF, platelet activating factor; PGD, primary graft dysfunction; PNF, primary nonfunction; ROS, reactive oxygen species; RAGE, receptor for advanced glycation end products; SIXR, systemic inflammation in xenograft recipients; TLRs, toll like receptors; XIRI, xenotransplantation ischemia reperfusion injury.
References
1. Aristizabal AM, Caicedo LA, Martínez JM, Moreno M, J Echeverri G. Clinical Xenotransplantation, a Closer Reality: Literature Review. Cir Esp. Feb (2017) 95(2):62–72. doi: 10.1016/j.ciresp.2016.12.008
2. Roux FA, Saï P, Deschamps JY. Xenotransfusions, Past and Present. Xenotransplantation (2007) 14(3):208–16. doi: 10.1111/j.1399-3089.2007.00404.x
3. Cyon E. Über Den Einfluss Der Temperaturänderungen Auf Zahl, Dauer Und Stärke Der Herzschläge. Berichte Über Die Verhandlungen Der Königlich Sächsischen Gesellschaft Der Wissenschaften Zu Leipzig, Mathematisch-Physische Classe1866 (1866) pp. 256–306.
4. Lu T, Yang B, Wang R, Qin C. Xenotransplantation: Current Status in Preclinical Research. Front Immunol (2019) 10:3060. doi: 10.3389/fimmu.2019.03060
5. Cooper DKC, Ekser B, Tector AJ. A Brief History of Clinical Xenotransplantation. Int J Surg (2015) 23(Pt B):205–10. doi: 10.1016/j.ijsu.2015.06.060
6. Carrel A. The Transplantation of Organs: A Preliminary Communication. 1905 [Classical Article]. Yale J Biol Med (2001) 74(4):239–41.
7. Camsonne R, Pottier D, Bustany P. Xenografts: Clinical Trials and Perspectives. Presse Med May (1998) 27(18):865–8.
8. Carrel A. La Technique Operatoire Des Anastomosis Vasculaires Et La Transplnatation Des Visceres. Lyon Med (1902) 1:859–64.
9. Carrel A. On The Permanent Life Of Tissues Outside Of The Organism. J Exp Med May (1912) 15(5):516–28. doi: 10.1084/jem.15.5.516
10. Carrel A. Technique and Remote Results of Vacular Anastomosis. Surg Gynecol Obstret (1912) 11:246–54.
11. Carrel A, Lindbergh CA. The Culture Of Whole Organs. Science (1935) 81(2112):621–3. doi: 10.1126/science.81.2112.621
12. Heyndrickx GR, Millard RW, McRitchie RJ, Maroko PR, Vatner SF. Regional Myocardial Functional and Electrophysiological Alterations After Brief Coronary Artery Occlusion in Conscious Dogs. J Clin Invest Oct (1975) 56(4):978–85. doi: 10.1172/JCI108178
13. Ferrari R, Balla C, Malagù M, Guardigli G, Morciano G, Bertini M, et al. Reperfusion Damage- A Story of Success, Failure, and Hope. Circ J Jan (2017) 81(2):131–41. doi: 10.1253/circj.CJ-16-1124
14. Jennings RB, Sommers HM, Smyth GA, Flack HA, Linn H. Myocardial Necrosis Induced by Temporary Occlusion of a Coronary Artery in the Dog. Arch Pathol (1960) 70:68–78.
15. Summers WK, Jamison RL. The No Reflow Phenomenon in Renal Ischemia. Lab Invest (1971) 25(6):635–43.
16. Chait LA, May JW, O’Brien BM, Hurley JV. The Effects of the Perfusion of Various Solutions on the No-Reflow Phenomenon in Experimental Free Flaps. Plast Reconstr Surg (1978) 61(3):421–30. doi: 10.1097/00006534-197803000-00017
17. McCord JM. Oxygen-Derived Free Radicals in Postischemic Tissue Injury. N Engl J Med (1985) 312(3):159–63. doi: 10.1056/NEJM198501173120305
18. Yong K, Khwaja A. Leucocyte Cellular Adhesion Molecules. Blood Rev (1990) 4(4):211–25. doi: 10.1016/0268-960x(90)90001-9
19. Lachmann PJ. Biological Functions of the Complement System. Biochem Soc Trans (1990) 18(6):1143–5. doi: 10.1042/bst0181143
20. Searle J, Kerr JF, Bishop CJ. Necrosis and Apoptosis: Distinct Modes of Cell Death With Fundamentally Different Significance. Pathol Annu (1982) 17 Pt 2:229–59.
21. Wernly B, Mirna M, Rezar R, Prodinger C, Jung C, Podesser BK, et al. Regenerative Cardiovascular Therapies: Stem Cells and Beyond. Int J Mol Sci (2019) 20(6). doi: 10.3390/ijms20061420
22. Nasralla D, Coussios CC, Mergental H, Akhtar MZ, Butler AJ, Ceresa CDL, et al. A Randomized Trial of Normothermic Preservation in Liver Transplantation. Nature (2018) 557(7703):50–6. doi: 10.1038/s41586-018-0047-9
23. Laubach VE, Sharma AK. Mechanisms of Lung Ischemia-Reperfusion Injury. Curr Opin Organ Transplant (2016) 21(3):246–52. doi: 10.1097/MOT.0000000000000304
24. Salvadori M, Rosso G, Bertoni E. Update on Ischemia-Reperfusion Injury in Kidney Transplantation: Pathogenesis and Treatment. World J Transplant (2015) 5(2):52–67. doi: 10.5500/wjt.v5.i2.52
25. Längin M, Mayr T, Reichart B, Michel S, Buchholz S, Guethoff S, et al. Consistent Success in Life-Supporting Porcine Cardiac Xenotransplantation. Nature (2018) 564(7736):430–3. doi: 10.1038/s41586-018-0765-z
26. D’Alessandro C, Golmard JL, Barreda E, Laali M, Makris R, Luyt CE, et al. Predictive Risk Factors for Primary Graft Failure Requiring Temporary Extra-Corporeal Membrane Oxygenation Support After Cardiac Transplantation in Adults. Eur J Cardiothorac Surg (2011) 40(4):962–9. doi: 10.1016/j.ejcts.2011.01.064
27. Li J, Hara H, Wang Y, Esmon C, Cooper DKC, Iwase H. Evidence for the Important Role of Inflammation in Xenotransplantation. J Inflammation (Lond) (2019) 16:10. doi: 10.1186/s12950-019-0213-3
28. Sanada S, Komuro I, Kitakaze M. Pathophysiology of Myocardial Reperfusion Injury: Preconditioning, Postconditioning, and Translational Aspects of Protective Measures. Am J Physiol Heart Circ Physiol (2011) 301(5):H1723–41. doi: 10.1152/ajpheart.00553.2011
29. Baines CP. The Mitochondrial Permeability Transition Pore and Ischemia-Reperfusion Injury. Basic Res Cardiol (2009) 104(2):181–8. doi: 10.1007/s00395-009-0004-8
30. Baines CP. The Molecular Composition of the Mitochondrial Permeability Transition Pore. J Mol Cell Cardiol (2009) 46(6):850–7. doi: 10.1016/j.yjmcc.2009.02.007
31. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell Biology of Ischemia/Reperfusion Injury. Int Rev Cell Mol Biol (2012) 298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7
32. Murphy E, Steenbergen C. Ion Transport and Energetics During Cell Death and Protection. Physiol (Bethesda) (2008) 23:115–23. doi: 10.1152/physiol.00044.2007
33. Talukder MA, Zweier JL, Periasamy M. Targeting Calcium Transport in Ischaemic Heart Disease. Cardiovasc Res (2009) 84(3):345–52. doi: 10.1093/cvr/cvp264
34. Zimmerman AN, Hülsmann WC. Paradoxical Influence of Calcium Ions on the Permeability of the Cell Membranes of the Isolated Rat Heart. Nature (1966) 211(5049):646–7. doi: 10.1038/211646a0
35. Contreras L, Drago I, Zampese E, Pozzan T. Mitochondria: The Calcium Connection. Biochim Biophys Acta (2010) 1797(6-7):607–18. doi: 10.1016/j.bbabio.2010.05.005
36. Croall DE, Ersfeld K. The Calpains: Modular Designs and Functional Diversity. Genome Biol (2007) 8(6):218. doi: 10.1186/gb-2007-8-6-218
37. Nieuwenhuijs-Moeke GJ, Pischke SE, Berger SP, Sanders JSF, Pol RA, Struys MMRF, et al. Ischemia and Reperfusion Injury in Kidney Transplantation: Relevant Mechanisms in Injury and Repair. J Clin Med (2020) 9(1). doi: 10.3390/jcm9010253
38. Miyamae M, Camacho SA, Weiner MW, Figueredo VM. Attenuation of Postischemic Reperfusion Injury Is Related to Prevention of [Ca2+]m Overload in Rat Hearts. Am J Physiol (1996) 271(5 Pt 2):H2145–53. doi: 10.1152/ajpheart.1996.271.5.H2145
39. Lemasters JJ, Bond JM, Chacon E, Harper IS, Kaplan SH, Ohata H, et al. The pH Paradox in Ischemia-Reperfusion Injury to Cardiac Myocytes. EXS (1996) 76:99–114. doi: 10.1007/978-3-0348-8988-9_7
40. White CW, Messer SJ, Large SR, Conway J, Kim DH, Kutsogiannis DJ, et al. Transplantation of Hearts Donated After Circulatory Death. Front Cardiovasc Med (2018) 5:8. doi: 10.3389/fcvm.2018.00008
41. Gracia-Sancho J, Casillas-Ramírez A, Peralta C. Molecular Pathways in Protecting the Liver From Ischaemia/Reperfusion Injury: A 2015 Update. Int J Mol Sci (2015) 129: (4):345–62. doi: 10.1042/cs20150223
42. Lima B, Forrester MT, Hess DT, Stamler JS. S-Nitrosylation in Cardiovascular Signaling. Circ Res Mar (2010) 106(4):633–46. doi: 10.1161/CIRCRESAHA.109.207381
43. Go YM, Park H, Koval M, Orr M, Reed M, Liang Y, et al. A Key Role for Mitochondria in Endothelial Signaling by Plasma Cysteine/Cystine Redox Potential. Free Radic Biol Med Jan (2010) 48(2):275–83. doi: 10.1016/j.freeradbiomed.2009.10.050
44. Lee HL, Chen CL, Yeh ST, Zweier JL, Chen YR. Biphasic Modulation of the Mitochondrial Electron Transport Chain in Myocardial Ischemia and Reperfusion. Am J Physiol Heart Circ Physiol (2012) 302(7):H1410–22. doi: 10.1152/ajpheart.00731.2011
45. Perrelli MG, Pagliaro P, Penna C. Ischemia/reperfusion Injury and Cardioprotective Mechanisms: Role of Mitochondria and Reactive Oxygen Species. World J Cardiol (2011) 3(6):186–200. doi: 10.4330/wjc.v3.i6.186
46. Shen HM, Liu ZG. JNK Signaling Pathway Is a Key Modulator in Cell Death Mediated by Reactive Oxygen and Nitrogen Species. Free Radic Biol Med (2006) 40(6):928–39. doi: 10.1016/j.freeradbiomed.2005.10.056
47. Porteous MK, Lee JC. Primary Graft Dysfunction After Lung Transplantation. Clin Chest Med (2017) 38(4):641–54. doi: 10.1016/j.ccm.2017.07.005
48. Zweier JL, Flaherty JT, Weisfeldt ML. Direct Measurement of Free Radical Generation Following Reperfusion of Ischemic Myocardium. Proc Natl Acad Sci USA (1987) 84(5):1404–7. doi: 10.1073/pnas.84.5.1404
49. Yang Z, Sharma AK, Linden J, Kron IL, Laubach VE. CD4+ T Lymphocytes Mediate Acute Pulmonary Ischemia-Reperfusion Injury. J Thorac Cardiovasc Surg (2009) 137(3):695–702; discussion 702. doi: 10.1016/j.jtcvs.2008.10.044
50. Sharma AK, Laubach VE, Ramos SI, Zhao Y, Stukenborg G, Linden J, et al. Adenosine A2A Receptor Activation on CD4+ T Lymphocytes and Neutrophils Attenuates Lung Ischemia-Reperfusion Injury. J Thorac Cardiovasc Surg (2010) 139(2):474–82. doi: 10.1016/j.jtcvs.2009.08.033
51. Sharma AK, Mulloy DP, Le LT, Laubach VE. NADPH Oxidase Mediates Synergistic Effects of IL-17 and TNF-α on CXCL1 Expression by Epithelial Cells After Lung Ischemia-Reperfusion. Am J Physiol Lung Cell Mol Physiol (2014) 306(1):L69–79. doi: 10.1152/ajplung.00205.2013
52. Sharma AK, LaPar DJ, Stone ML, Zhao Y, Mehta CK, Kron IL, et al. NOX2 Activation of Natural Killer T Cells Is Blocked by the Adenosine A2A Receptor to Inhibit Lung Ischemia-Reperfusion Injury. Am J Respir Crit Care Med (2016) 193(9):988–99. doi: 10.1164/rccm.201506-1253OC
53. Ferrari RS, Andrade CF. Oxidative Stress and Lung Ischemia-Reperfusion Injury. Oxid Med Cell Longev (2015) 2015:590987. doi: 10.1155/2015/590987
55. Hausenloy DJ, Yellon DM. The Mitochondrial Permeability Transition Pore: Its Fundamental Role in Mediating Cell Death During Ischaemia and Reperfusion. J Mol Cell Cardiol (2003) 35(4):339–41. doi: 10.1016/s0022-2828(03)00043-9
56. Ngo BT, Beiras-Fernandez A, Hammer C, Thein E. Hyperacute Rejection in the Xenogenic Transplanted Rat Liver Is Triggered by the Complement System Only in the Presence of Leukocytes and Free Radical Species. Xenotransplantation (2013) 20(3):177–87. doi: 10.1111/xen.12035
57. Lee S, Ha IS, Kim JH, Park KS, Han KH, Kim SH, et al. Hydrogen Peroxide-Induced VCAM-1 Expression in Pancreatic Islets and Beta-Cells Through Extracellular Ca2+ Influx. Transplantation (2008) 86(9):1257–66. doi: 10.1097/TP.0b013e318188ab04
58. Andonian S, Coulthard T, Smith AD, Singhal PS, Lee BR. Real-Time Quantitation of Renal Ischemia Using Targeted Microbubbles: In-Vivo Measurement of P-Selectin Expression. J Endourol (2009) 23(3):373–8. doi: 10.1089/end.2008.0229
59. Palazzo AJ, Jones SP, Anderson DC, Granger DN, Lefer DJ. Coronary Endothelial P-Selectin in Pathogenesis of Myocardial Ischemia-Reperfusion Injury. Am J Physiol (1998) 275(5):H1865–72. doi: 10.1152/ajpheart.1998.275.5.H1865
60. Zarbock A, Müller H, Kuwano Y, Ley K. PSGL-1-Dependent Myeloid Leukocyte Activation. J Leukoc Biol (2009) 86(5):1119–24. doi: 10.1189/jlb.0209117
61. Mulligan MS, Lentsch AB, Miyasaka M, Ward PA. Cytokine and Adhesion Molecule Requirements for Neutrophil Recruitment During Glycogen-Induced Peritonitis. Inflammation Res (1998) 47(6):251–5. doi: 10.1007/s000110050326
62. Martinez-Mier G, Toledo-Pereyra LH, McDuffie JE, Warner RL, Ward PA. P-Selectin and Chemokine Response After Liver Ischemia and Reperfusion. J Am Coll Surg (2000) 191(4):395–402. doi: 10.1016/s1072-7515(00)00360-4
63. Jin C, Cleveland JC, Ao L, Li J, Zeng Q, Fullerton DA, et al. Human Myocardium Releases Heat Shock Protein 27 (HSP27) After Global Ischemia: The Proinflammatory Effect of Extracellular HSP27 Through Toll-Like Receptor (TLR)-2 and TLR4. Mol Med (2014) 20:280–9. doi: 10.2119/molmed.2014.00058
64. Szyller J, Bil-Lula I. Heat Shock Proteins in Oxidative Stress and Ischemia/Reperfusion Injury and Benefits From Physical Exercises: A Review to the Current Knowledge. Oxid Med Cell Longev (2021) 2021:6678457. doi: 10.1155/2021/6678457
65. Hausenloy DJ, Yellon DM. Myocardial Ischemia-Reperfusion Injury: A Neglected Therapeutic Target. J Clin Invest (2013) 123(1):92–100. doi: 10.1172/JCI62874
66. Toldo S, Quader M, Salloum FN, Mezzaroma E, Abbate A. Targeting the Innate Immune Response to Improve Cardiac Graft Recovery After Heart Transplantation: Implications for the Donation After Cardiac Death. Int J Mol Sci (2016) 17(6):958–82. doi: 10.3390/ijms17060958
67. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular Danger Signals Guide Neutrophils to Sites of Sterile Inflammation. Science (2010) 330(6002):362–6. doi: 10.1126/science.1195491
68. Matzinger P. The Danger Model: A Renewed Sense of Self. Science (2002) 296(5566):301–5. doi: 10.1126/science.1071059
69. Rusai K, Sollinger D, Baumann M, Wagner B, Strobl M, Schmaderer C, et al. Toll-Like Receptors 2 and 4 in Renal Ischemia/Reperfusion Injury. Pediatr Nephrol (2010) 25(5):853–60. doi: 10.1007/s00467-009-1422-4
70. Arslan F, de Kleijn DP, Pasterkamp G. Innate Immune Signaling in Cardiac Ischemia. Nat Rev Cardiol (2011) 8(5):292–300. doi: 10.1038/nrcardio.2011.38
71. Toldo S, Abbate A. The NLRP3 Inflammasome in Acute Myocardial Infarction. Nat Rev Cardiol (2018) 15(4):203–14. doi: 10.1038/nrcardio.2017.161
72. Liu L, Fang C, Fu W, Jiang B, Li G, Qin L, et al. Endothelial Cell-Derived Interleukin-18 Released During Ischemia Reperfusion Injury Selectively Expands T Peripheral Helper Cells to Promote Alloantibody Production. Circulation (2020) 141(6):464–78. doi: 10.1161/CIRCULATIONAHA.119.042501
73. Luo JL, Kamata H, Karin M. IKK/NF-kappaB Signaling: Balancing Life and Death–a New Approach to Cancer Therapy. J Clin Invest (2005) 115(10):2625–32. doi: 10.1172/JCI26322
74. Bracey NA, Beck PL, Muruve DA, Hirota SA, Guo J, Jabagi H, et al. The Nlrp3 Inflammasome Promotes Myocardial Dysfunction in Structural Cardiomyopathy Through Interleukin-1β. Exp Physiol (2013) 98(2):462–72. doi: 10.1113/expphysiol.2012.068338
75. Sandanger Ø, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV, et al. The NLRP3 Inflammasome Is Up-Regulated in Cardiac Fibroblasts and Mediates Myocardial Ischaemia-Reperfusion Injury. Cardiovasc Res (2013) 99(1):164–74. doi: 10.1093/cvr/cvt091
76. Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, et al. Inflammasome Activation of Cardiac Fibroblasts Is Essential for Myocardial Ischemia/Reperfusion Injury. Circulation (2011) 123(6):594–604. doi: 10.1161/CIRCULATIONAHA.110.982777
77. Cantu E, Lederer DJ, Meyer K, Milewski K, Suzuki Y, Shah RJ, et al. Gene Set Enrichment Analysis Identifies Key Innate Immune Pathways in Primary Graft Dysfunction After Lung Transplantation. Am J Transplant (2013) 13(7):1898–904. doi: 10.1111/ajt.12283
78. Weiser MR, Williams JP, Moore FD, Kobzik L, Ma M, Hechtman HB, et al. Reperfusion Injury of Ischemic Skeletal Muscle Is Mediated by Natural Antibody and Complement. J Exp Med May (1996) 183(5):2343–8. doi: 10.1084/jem.183.5.2343
79. Le Bas-Bernardet S, Tillou X, Branchereau J, Dilek N, Poirier N, Châtelais M, et al. Bortezomib, C1-Inhibitor and Plasma Exchange do Not Prolong the Survival of Multi-Transgenic GalT-KO Pig Kidney Xenografts in Baboons. Am J Transplant (2015) 15(2):358–70. doi: 10.1111/ajt.12988
80. Lin Z, Lin H, Li W, Huang Y, Dai H. Complement Component C3 Promotes Cerebral Ischemia/Reperfusion Injury Mediated by TLR2/Nfκb Activation in Diabetic Mice. Neurochem Res (2018) 43(8):1599–607. doi: 10.1007/s11064-018-2574-z
81. Triantafilou M, Hughes TR, Morgan BP, Triantafilou K. Complementing the Inflammasome. Immunology (2016) 147(2):152–64. doi: 10.1111/imm.12556
82. Damman J, Daha MR, van Son WJ, Leuvenink HG, Ploeg RJ, Seelen MA. Crosstalk Between Complement and Toll-Like Receptor Activation in Relation to Donor Brain Death and Renal Ischemia-Reperfusion Injury. Am J Transplant (2011) 11(4):660–9. doi: 10.1111/j.1600-6143.2011.03475.x
83. Núñez K, Thevenot P, Alfadhli A, Cohen A. Complement Activation in Liver Transplantation: Role of Donor Macrosteatosis and Implications in Delayed Graft Function. Int J Mol Sci (2018) 19(6):1750–71. doi: 10.3390/ijms19061750
84. Danobeitia JS, Djamali A, Fernandez LA. The Role of Complement in the Pathogenesis of Renal Ischemia-Reperfusion Injury and Fibrosis. Fibrogenesis Tissue Repair (2014) 7:16. doi: 10.1186/1755-1536-7-16
85. Panagiotou A, Trendelenburg M, Osthoff M. The Lectin Pathway of Complement in Myocardial Ischemia/Reperfusion Injury-Review of Its Significance and the Potential Impact of Therapeutic Interference by C1 Esterase Inhibitor. Front Immunol (2018) 9:1151. doi: 10.3389/fimmu.2018.01151
86. Mercurio D, Piotti A, Valente A, Oggioni M, Ponstein Y, Van Amersfoort E, et al. Plasma-Derived and Recombinant C1 Esterase Inhibitor: Binding Profiles and Neuroprotective Properties in Brain Ischemia/Reperfusion Injury. Brain Behav Immun (2021). doi: 10.1016/j.bbi.2021.01.002
87. Li C, Patel K, Tu Z, Yang X, Kulik L, Alawieh A, et al. A Novel Injury Site-Natural Antibody Targeted Complement Inhibitor Protects Against Lung Transplant Injury. Am J Transplant (2020). doi: 10.1111/ajt.16404
88. Kusakabe J, Hata K, Tamaki I, Tajima T, Miyauchi H, Wang Y, et al. Complement 5 Inhibition Ameliorates Hepatic Ischemia/reperfusion Injury in Mice, Dominantly via the C5a-Mediated Cascade. Transplantation (2020) 104(10):2065–77. doi: 10.1097/TP.0000000000003302
89. Smith EF, Griswold DE, Egan JW, Hillegass LM, Smith RA, Hibbs MJ, et al. Reduction of Myocardial Reperfusion Injury With Human Soluble Complement Receptor Type 1 (BRL 55730). Eur J Pharmacol (1993) 236(3):477–81. doi: 10.1016/0014-2999(93)90487-3
90. Sommer W, Tudorache I, Kühn C, Avsar M, Salman J, Ius F, et al. C1-Esterase-Inhibitor for Primary Graft Dysfunction in Lung Transplantation. Transplantation (2014) 97(11):1185–91. doi: 10.1097/TP.0000000000000034
91. Huang E, Vo A, Choi J, Ammerman N, Lim K, Sethi S, et al. Three-Year Outcomes of a Randomized, Double-Blind, Placebo-Controlled Study Assessing Safety and Efficacy of C1 Esterase Inhibitor for Prevention of Delayed Graft Function in Deceased Donor Kidney Transplant Recipients. Clin J Am Soc Nephrol (2020) 15(1):109–16. doi: 10.2215/CJN.04840419
92. Platt JL, Fischel RJ, Matas AJ, Reif SA, Bolman RM, Bach FH. Immunopathology of Hyperacute Xenograft Rejection in a Swine-to-Primate Model. Transplantation (1991) 52(2):214–20. doi: 10.1097/00007890-199108000-00006
93. Ekser B, Ezzelarab M, Hara H, van der Windt DJ, Wijkstrom M, Bottino R, et al. Clinical Xenotransplantation: The Next Medical Revolution? Lancet (2012) 379(9816):672–83. doi: 10.1016/S0140-6736(11)61091-X
94. Carrington CA, Richards AC, van den Bogaerde J, Tucker AW, White DJ. Complement Activation, Its Consequences, and Blockade by Gene Transfer. World J Surg (1997) 21(9):907–12. doi: 10.1007/s002689900325
95. Shimizu A, Hisashi Y, Kuwaki K, Tseng YL, Dor FJ, Houser SL, et al. Thrombotic Microangiopathy Associated With Humoral Rejection of Cardiac Xenografts From Alpha1,3-Galactosyltransferase Gene-Knockout Pigs in Baboons. Am J Pathol (2008) 172(6):1471–81. doi: 10.2353/ajpath.2008.070672
96. Burdorf L, Stoddard T, Zhang T, Rybak E, Riner A, Avon C, et al. Expression of Human CD46 Modulates Inflammation Associated With GalTKO Lung Xenograft Injury. Am J Transplant (2014) 14(5):1084–95. doi: 10.1111/ajt.12673
97. Ramírez P, Montoya MJ, Ríos A, García Palenciano C, Majado M, Chávez R, et al. Prevention of Hyperacute Rejection in a Model of Orthotopic Liver Xenotransplantation From Pig to Baboon Using Polytransgenic Pig Livers (CD55, CD59, and H-Transferase). Transplant Proc (2005) 37(9):4103–6. doi: 10.1016/j.transproceed.2005.09.186
98. Marin W, Marin D, Ao X, Liu Y. Mitochondria as a Therapeutic Target for Cardiac Ischemia−Reperfusion Injury (Review). Int J Mol Med (2021) 47(2):485–99. doi: 10.3892/ijmm.2020.4823
99. Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu Rev Pharmacol Toxicol (2017) 57:535–65. doi: 10.1146/annurev-pharmtox-010715-103335
100. Di Lisa F, Kaludercic N, Carpi A, Menabò R, Giorgio M. Mitochondria and Vascular Pathology. Pharmacol Rep (2009) 61(1):123–30. doi: 10.1016/s1734-1140(09)70014-3
101. Ong SB, Gustafsson AB. New Roles for Mitochondria in Cell Death in the Reperfused Myocardium. Cardiovasc Res (2012) 94(2):190–6. doi: 10.1093/cvr/cvr312
102. Qian T, Nieminen AL, Herman B, Lemasters JJ. Mitochondrial Permeability Transition in pH-Dependent Reperfusion Injury to Rat Hepatocytes. Am J Physiol (1997) 273(6):C1783–92. doi: 10.1152/ajpcell.1997.273.6.C1783
103. He L, Lemasters JJ. Regulated and Unregulated Mitochondrial Permeability Transition Pores: A New Paradigm of Pore Structure and Function? FEBS Lett (2002) 512(1-3):1–7. doi: 10.1016/s0014-5793(01)03314-2
104. Griffiths EJ, Halestrap AP. Mitochondrial non-Specific Pores Remain Closed During Cardiac Ischaemia, But Open Upon Reperfusion. Biochem J (1995) 307(Pt 1):93–8. doi: 10.1042/bj3070093
105. Halestrap AP. What Is the Mitochondrial Permeability Transition Pore? J Mol Cell Cardiol (2009) 46(6):821–31. doi: 10.1016/j.yjmcc.2009.02.021
106. Baines CP. The Cardiac Mitochondrion: Nexus of Stress. Annu Rev Physiol (2010) 72:61–80. doi: 10.1146/annurev-physiol-021909-135929
107. Chen L, Knowlton AA. Mitochondria and Heart Failure: New Insights Into an Energetic Problem. Minerva Cardioangiol (2010) 58(2):213–29.
108. Giedt RJ, Yang C, Zweier JL, Matzavinos A, Alevriadou BR. Mitochondrial Fission in Endothelial Cells After Simulated Ischemia/Reperfusion: Role of Nitric Oxide and Reactive Oxygen Species. Free Radic Biol Med (2012) 52(2):348–56. doi: 10.1016/j.freeradbiomed.2011.10.491
109. Alexander JS, Elrod JW. Extracellular Matrix, Junctional Integrity and Matrix Metalloproteinase Interactions in Endothelial Permeability Regulation. J Anat (2002) 200(6):561–74. doi: 10.1046/j.1469-7580.2002.00057.x
110. Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular Mechanisms of Endothelial Hyperpermeability: Implications in Inflammation. Expert Rev Mol Med (2009) 11:e19. doi: 10.1017/S1462399409001112
111. Mehta D, Malik AB. Signaling Mechanisms Regulating Endothelial Permeability. Physiol Rev (2006) 86(1):279–367. doi: 10.1152/physrev.00012.2005
112. Rodrigues SF, Granger DN. Role of Blood Cells in Ischaemia-Reperfusion Induced Endothelial Barrier Failure. Cardiovasc Res (2010) 87(2):291–9. doi: 10.1093/cvr/cvq090
113. Terao S, Yilmaz G, Stokes KY, Russell J, Ishikawa M, Kawase T, et al. Blood Cell-Derived RANTES Mediates Cerebral Microvascular Dysfunction, Inflammation, and Tissue Injury After Focal Ischemia-Reperfusion. Stroke (2008) 39(9):2560–70. doi: 10.1161/STROKEAHA.107.513150
114. Okazaki M, Kreisel F, Richardson SB, Kreisel D, Krupnick AS, Patterson GA, et al. Sphingosine 1-Phosphate Inhibits Ischemia Reperfusion Injury Following Experimental Lung Transplantation. Am J Transplant (2007) 7(4):751–8. doi: 10.1111/j.1600-6143.2006.01710.x
115. Cooper D, Russell J, Chitman KD, Williams MC, Wolf RE, Granger DN. Leukocyte Dependence of Platelet Adhesion in Postcapillary Venules. Am J Physiol Heart Circ Physiol (2004) 286(5):H1895–900. doi: 10.1152/ajpheart.01000.2003
116. Ishikawa M, Cooper D, Arumugam TV, Zhang JH, Nanda A, Granger DN. Platelet-Leukocyte-Endothelial Cell Interactions After Middle Cerebral Artery Occlusion and Reperfusion. J Cereb Blood Flow Metab (2004) 24(8):907–15. doi: 10.1097/01.WCB.0000132690.96836.7F
117. Gawaz M. Role of Platelets in Coronary Thrombosis and Reperfusion of Ischemic Myocardium. Cardiovasc Res (2004) 61(3):498–511. doi: 10.1016/j.cardiores.2003.11.036
118. Kokura S, Wolf RE, Yoshikawa T, Ichikawa H, Granger DN, Aw TY. Endothelial Cells Exposed to Anoxia/Reoxygenation Are Hyperadhesive to T-Lymphocytes: Kinetics and Molecular Mechanisms. Microcirculation (2000) 7(1):13–23. doi: 10.1111/j.1549-8719.2000.tb00739.x
119. Nourshargh S, Krombach F, Dejana E. The Role of JAM-A and PECAM-1 in Modulating Leukocyte Infiltration in Inflamed and Ischemic Tissues. J Leukoc Biol (2006) 80(4):714–8. doi: 10.1189/jlb.1105645
120. Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, et al. The Junctional Adhesion Molecule JAM-C Regulates Polarized Transendothelial Migration of Neutrophils In Vivo. Nat Immunol (2011) 12(8):761–9. doi: 10.1038/ni.2062
121. Proebstl D, Voisin MB, Woodfin A, Whiteford J, D'Acquisto F, Jones GE, et al. Pericytes Support Neutrophil Subendothelial Cell Crawling and Breaching of Venular Walls In Vivo. J Exp Med (2012) 209(6):1219–34. doi: 10.1084/jem.20111622
122. Kato H, Kuriyama N, Duarte S, Clavien PA, Busuttil RW, Coito AJ. MMP-9 Deficiency Shelters Endothelial PECAM-1 Expression and Enhances Regeneration of Steatotic Livers After Ischemia and Reperfusion Injury. J Hepatol (2014) 60(5):1032–9. doi: 10.1016/j.jhep.2013.12.022
123. Russo L, Gracia-Sancho J, García-Calderó H, Marrone G, García-Pagán JC, García-Cardeña G, et al. Addition of Simvastatin to Cold Storage Solution Prevents Endothelial Dysfunction in Explanted Rat Livers. Hepatology (2012) 55(3):921–30. doi: 10.1002/hep.24755
124. Gracia-Sancho J, Villarreal G, Zhang Y, Yu JX, Liu Y, Tullius SG, et al. Flow Cessation Triggers Endothelial Dysfunction During Organ Cold Storage Conditions: Strategies for Pharmacologic Intervention. Transplantation (2010) 90(2):142–9. doi: 10.1097/TP.0b013e3181e228db
125. Abraham D, Taghavi S, Riml P, Paulus P, Hofmann M, Baumann C, et al. VEGF-A and -C But Not -B Mediate Increased Vascular Permeability in Preserved Lung Grafts. Transplantation (2002) 73(11):1703–6. doi: 10.1097/00007890-200206150-00003
126. Taghavi S, Abraham D, Riml P, Paulus P, Schäfer R, Klepetko W, et al. Co-Expression of Endothelin-1 and Vascular Endothelial Growth Factor Mediates Increased Vascular Permeability in Lung Grafts Before Reperfusion. J Heart Lung Transplant (2002) 21(5):6003. doi: 10.1016/s1053-2498(01)00346-1
127. Krenn K, Klepetko W, Taghavi S, Lang G, Schneider B, Aharinejad S. Recipient Vascular Endothelial Growth Factor Serum Levels Predict Primary Lung Graft Dysfunction. Am J Transplant (2007) 7(3):700–6. doi: 10.1111/j.1600-6143.2006.01673.x
128. Krenn K, Klepetko W, Taghavi S, Paulus P, Aharinejad S. Vascular Endothelial Growth Factor Increases Pulmonary Vascular Permeability in Cystic Fibrosis Patients Undergoing Lung Transplantation. Eur J Cardiothorac Surg (2007) 32(1):35–41. doi: 10.1016/j.ejcts.2007.04.006
129. Diamond JM, Porteous MK, Cantu E, Meyer NJ, Shah RJ, Lederer DJ, et al. Elevated Plasma Angiopoietin-2 Levels and Primary Graft Dysfunction After Lung Transplantation. PloS One (2012) 7(12):e51932. doi: 10.1371/journal.pone.0051932
130. van der Heijden M, van Nieuw Amerongen GP, Koolwijk P, van Hinsbergh VW, Groeneveld AB. Angiopoietin-2, Permeability Oedema, Occurrence and Severity of ALI/ARDS in Septic and Non-Septic Critically Ill Patients. Thorax (2008) 63(10):903–9. doi: 10.1136/thx.2007.087387
131. Salama M, Andrukhova O, Hoda MA, Taghavi S, Jaksch P, Heinze G, et al. Concomitant Endothelin-1 Overexpression in Lung Transplant Donors and Recipients Predicts Primary Graft Dysfunction. Am J Transplant (2010) 10(3):628–36. doi: 10.1111/j.1600-6143.2009.02957.x
132. Tsurui Y, Sho M, Kuzumoto Y, Hamada K, Akashi S, Kashizuka H, et al. Dual Role of Vascular Endothelial Growth Factor in Hepatic Ischemia-Reperfusion Injury. Transplantation (2005) 79(9):1110–5. doi: 10.1097/01.tp.0000161627.84481.5e
133. Andersson L, Scharin Täng M, Lundqvist A, Lindbom M, Mardani I, Fogelstrand P, et al. Rip2 Modifies VEGF-Induced Signalling and Vascular Permeability in Myocardial Ischaemia. Cardiovasc Res (2015) 107(4):478–86. doi: 10.1093/cvr/cvv186
134. Kanellis J, Paizis K, Cox AJ, Stacker SA, Gilbert RE, Cooper ME, et al. Renal Ischemia-Reperfusion Increases Endothelial VEGFR-2 Without Increasing VEGF or VEGFR-1 Expression. Kidney Int (2002) 61(5):1696–706. doi: 10.1046/j.1523-1755.2002.00329.x
135. Zhang C, Wu J, Xu X, Potter BJ, Gao X. Direct Relationship Between Levels of TNF-Alpha Expression and Endothelial Dysfunction in Reperfusion Injury. Basic Res Cardiol (2010) 105(4):453–64. doi: 10.1007/s00395-010-0083-6
136. Gourdin MJ, Bree B, De Kock M. The Impact of Ischaemia-Reperfusion on the Blood Vessel. Eur J Anaesthesiol (2009) 26(7):537–47. doi: 10.1097/EJA.0b013e328324b7c2
137. Lindemann S, Klingel B, Fisch A, Meyer J, Darius H. Increased Platelet Sensitivity Toward Platelet Inhibitors During Physical Exercise in Patients With Coronary Artery Disease. Thromb Res (1999) 93(2):51–9. doi: 10.1016/s0049-3848(98)00155-8
138. Pigazzi A, Heydrick S, Folli F, Benoit S, Michelson A, Loscalzo J. Nitric Oxide Inhibits Thrombin Receptor-Activating Peptide-Induced Phosphoinositide 3-Kinase Activity in Human Platelets. J Biol Chem (1999) 274(20):14368–75. doi: 10.1074/jbc.274.20.14368
139. Khandoga A, Biberthaler P, Enders G, Axmann S, Hutter J, Messmer K, et al. Platelet Adhesion Mediated by Fibrinogen-Intercelllular Adhesion Molecule-1 Binding Induces Tissue Injury in the Postischemic Liver In Vivo. Transplantation (2002) 74(5):681–8. doi: 10.1097/00007890-200209150-00016
140. de Ramon L, Guiteras J, Guiteras R, Cruzado JM, Grinyó JM, Torras J. The Costimulatory Pathways and T Regulatory Cells in Ischemia-Reperfusion Injury: A Strong Arm in the Inflammatory Response? Int J Mol Sci (2018) 19(5):1283–97. doi: 10.3390/ijms19051283
141. Zhao X, Jin Y, Li H, Wang Z, Zhang W, Feng C. Hypoxia-Inducible Factor 1 Alpha Contributes to Pulmonary Vascular Dysfunction in Lung Ischemia-Reperfusion Injury. Int J Clin Exp Pathol (2014) 7(6):3081–8.
142. Shen B, Mei M, Pu Y, Zhang H, Liu H, Tang M, et al. Necrostatin-1 Attenuates Renal Ischemia and Reperfusion Injury via Meditation of HIF-1α/Mir-26a/TRPC6/PARP1 Signaling. Mol Ther Nucleic Acids (2019) 17:701–13. doi: 10.1016/j.omtn.2019.06.025
143. Sharma AK, LaPar DJ, Zhao Y, Li L, Lau CL, Kron IL, et al. Natural Killer T Cell-Derived IL-17 Mediates Lung Ischemia-Reperfusion Injury. Am J Respir Crit Care Med (2011) 183(11):1539–49. doi: 10.1164/rccm.201007-1173OC
144. Weber DJ, Gracon AS, Ripsch MS, Fisher AJ, Cheon BM, Pandya PH, et al. The HMGB1-RAGE Axis Mediates Traumatic Brain Injury-Induced Pulmonary Dysfunction in Lung Transplantation. Sci Transl Med (2014) 6(252):252ra124. doi: 10.1126/scitranslmed.3009443
145. Andersson U, Yang H, Harris H. Extracellular HMGB1 as a Therapeutic Target in Inflammatory Diseases. Expert Opin Ther Targets (2018) 22(3):263–77. doi: 10.1080/14728222.2018.1439924
146. Laird CT, Hassanein W, O’Neill NA, French BM, Cheng X, Fogler WE, et al. P- and E-Selectin Receptor Antagonism Prevents Human Leukocyte Adhesion to Activated Porcine Endothelial Monolayers and Attenuates Porcine Endothelial Damage. Xenotransplantation (2018) 25(2):e12381. doi: 10.1111/xen.12381
147. French BM, Sendil S, Sepuru KM, Ranek J, Burdorf L, Harris D, et al. Interleukin-8 Mediates Neutrophil-Endothelial Interactions in Pig-to-Human Xenogeneic Models. Xenotransplantation (2018) 25(2):e12385. doi: 10.1111/xen.12385
148. Verrier E. The Microvascular Cell and Ischemia-Reperfusion Injury. J Cardiovasc Pharmacol (1996) 27(Suppl 1):S26–30. doi: 10.1097/00005344-199600001-00007
149. Jakobs FM, Davis EA, White T, Sanfilippo F, Baldwin WM. Prolonged Discordant Xenograft Survival by Inhibition of the Intrinsic Coagulation Pathway in Complement C6-Deficient Recipients. J Heart Lung Transplant (1998) 17(3):306–11.
150. Kalady MF, Lawson JH, Sorrell RD, Platt JL. Decreased Fibrinolytic Activity in Porcine-to-Primate Cardiac Xenotransplantation. Mol Med Sep (1998) 4(9):629–37. doi: 10.1007/BF03401762
151. Jung SH, Hwang JH, Kim SE, Young Kyu K, Park HC, Lee HT. The Potentiating Effect of hTFPI in the Presence of Hcd47 Reduces the Cytotoxicity of Human Macrophages. Xenotransplantation (2017) 24(3). doi: 10.1111/xen.12301
152. Azimzadeh AM, Kelishadi SS, Ezzelarab MB, Singh AK, Stoddard T, Iwase H, et al. Early Graft Failure of GalTKO Pig Organs in Baboons Is Reduced by Expression of a Human Complement Pathway-Regulatory Protein. Xenotransplantation (2015) 22(4):310–6. doi: 10.1111/xen.12176
153. Kim SC, Mathews DV, Breeden CP, Higginbotham LB, Ladowski J, Martens G, et al. Long-Term Survival of Pig-to-Rhesus Macaque Renal Xenografts Is Dependent on CD4 T Cell Depletion. Am J Transplant (2019) 19(8):2174–85. doi: 10.1111/ajt.15329
154. Singh AK, Chan JL, DiChiacchio L, Hardy NL, Corcoran PC, Lewis BGT, et al. Cardiac Xenografts Show Reduced Survival in the Absence of Transgenic Human Thrombomodulin Expression in Donor Pigs. Xenotransplantation (2019) 26(2):e12465. doi: 10.1111/xen.12465
155. Burdorf L, Harris D, Dahi S, Laird C, Zhang T, Ali F, et al. Thromboxane and Histamine Mediate PVR Elevation During Xenogeneic Pig Lung Perfusion With Human Blood. Xenotransplantation (2019) 26(2):e12458. doi: 10.1111/xen.12458
156. Mohiuddin MM, Singh AK, Corcoran PC, Hoyt RF, Thomas ML, Ayares D, et al. Genetically Engineered Pigs and Target-Specific Immunomodulation Provide Significant Graft Survival and Hope for Clinical Cardiac Xenotransplantation. J Thorac Cardiovasc Surg (2014) 148(3):1106–13; discussion 1113-4. doi: 10.1016/j.jtcvs.2014.06.002
157. Iwase H, Ekser B, Hara H, Phelps C, Ayares D, Cooper DK, et al. Regulation of Human Platelet Aggregation by Genetically Modified Pig Endothelial Cells and Thrombin Inhibition. Xenotransplantation (2014) 21(1):72–83. doi: 10.1111/xen.12073
158. Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, et al. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol Biochem (2018) 46(4):1650–67. doi: 10.1159/000489241
159. Glick D, Barth S, Macleod KF. Autophagy: Cellular and Molecular Mechanisms. J Pathol (2010) 221(1):3–12. doi: 10.1002/path.2697
160. Ma S, Wang Y, Chen Y, Cao F. The Role of the Autophagy in Myocardial Ischemia/Reperfusion Injury. Biochim Biophys Acta (2015) 1852(2):271–6. doi: 10.1016/j.bbadis.2014.05.010
161. Linkermann A, Green DR. Necroptosis. N Engl J Med (2014) 370(5):455–65. doi: 10.1056/NEJMra1310050
162. Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller JO, et al. Two Independent Pathways of Regulated Necrosis Mediate Ischemia-Reperfusion Injury. Proc Natl Acad Sci USA (2013) 110(29):12024–9. doi: 10.1073/pnas.1305538110
163. Holzknecht ZE, Kuypers KL, Plummer TB, Williams J, Bustos M, Gores GJ, et al. Apoptosis and Cellular Activation in the Pathogenesis of Acute Vascular Rejection. Circ Res (2002) 91(12):1135–41. doi: 10.1161/01.res.0000046236.20251.fa
164. Shimizu A, Meehan SM, Kozlowski T, Sablinski T, Ierino FL, Cooper DK, et al. Acute Humoral Xenograft Rejection: Destruction of the Microvascular Capillary Endothelium in Pig-to-Nonhuman Primate Renal Grafts. Lab Invest (2000) 80(6):815–30. doi: 10.1038/labinvest.3780086
165. Boros P, Bromberg JS. New Cellular and Molecular Immune Pathways in Ischemia/Reperfusion Injury. Am J Transplant (2006) 6(4):652–8. doi: 10.1111/j.1600-6143.2005.01228.x
166. Gao H, Liu L, Zhao Y, Hara H, Chen P, Xu J, et al. Human IL-6, IL-17, IL-1β, and TNF-α Differently Regulate the Expression of Pro-Inflammatory Related Genes, Tissue Factor, and Swine Leukocyte Antigen Class I in Porcine Aortic Endothelial Cells. Xenotransplantation (2017) 24(2). doi: 10.1111/xen.12291
167. Gao H, Zhang Q, Chen J, Cooper DKC, Hara H, Chen P, et al. Porcine IL-6, IL-1β, and TNF-α Regulate the Expression of Pro-Inflammatory-Related Genes and Tissue Factor in Human Umbilical Vein Endothelial Cells. Xenotransplantation (2018) 25(5):e12408. doi: 10.1111/xen.12408
168. Iwase H, Liu H, Li T, Zhang Z, Gao B, Hara H, et al. Therapeutic Regulation of Systemic Inflammation in Xenograft Recipients. Xenotransplantation (2017) 24(2). doi: 10.1111/xen.12296
169. Liu NB, Wu M, Chen C, Fujino M, Huang JS, Zhu P, et al. Novel Molecular Targets Participating in Myocardial Ischemia-Reperfusion Injury and Cardioprotection. Cardiol Res Pract (2019) 2019:6935147. doi: 10.1155/2019/6935147
170. Yellon DM, Hausenloy DJ. Myocardial Reperfusion Injury. N Engl J Med Sep (2007) 357(11):1121–35. doi: 10.1056/NEJMra071667
171. Ge L, Zhou X, Ji WJ, Lu RY, Zhang Y, Zhang YD, et al. Neutrophil Extracellular Traps in Ischemia-Reperfusion Injury-Induced Myocardial No-Reflow: Therapeutic Potential of DNase-Based Reperfusion Strategy. Am J Physiol Heart Circ Physiol (2015) 308(5):H500–9. doi: 10.1152/ajpheart.00381.2014
172. Barzyc A, Łysik W, Słyk J, Kuszewski M, Zarębiński M, Wojciechowska M, et al. Reperfusion Injury as a Target for Diminishing Infarct Size. Med Hypotheses (2020) 137:109558. doi: 10.1016/j.mehy.2020.109558
173. Cowled PA, Khanna A, Laws PE, Field JB, Varelias A, Fitridge RA. Statins Inhibit Neutrophil Infiltration in Skeletal Muscle Reperfusion Injury. J Surg Res Aug (2007) 141(2):267–76. doi: 10.1016/j.jss.2006.11.021
174. Atkinson C, He S, Morris K, Qiao F, Casey S, Goddard M, et al. Targeted Complement Inhibitors Protect Against Posttransplant Cardiac Ischemia and Reperfusion Injury and Reveal an Important Role for the Alternative Pathway of Complement Activation. J Immunol (2010) 185(11):7007–13. doi: 10.4049/jimmunol.1001504
175. Farmer DG, Anselmo D, Da Shen X, Ke B, Carmody IC, Gao F, et al. Disruption of P-Selectin Signaling Modulates Cell Trafficking and Results in Improved Outcomes After Mouse Warm Intestinal Ischemia and Reperfusion Injury. Transplantation (2005) 80(6):828–35. doi: 10.1097/01.tp.0000174337.53658.b0
176. Chamoun F, Burne M, O’Donnell M, Rabb H. Pathophysiologic Role of Selectins and Their Ligands in Ischemia Reperfusion Injury. Front Biosci (2000) 5:E103–9. doi: 10.2741/chamoun
177. Rabb H, Ramirez G, Saba SR, Reynolds D, Xu J, Flavell R, et al. Renal Ischemic-Reperfusion Injury in L-Selectin-Deficient Mice. Am J Physiol (1996) 271(2 Pt 2):F408–13. doi: 10.1152/ajprenal.1996.271.2.F408
178. Connolly ES, Winfree CJ, Prestigiacomo CJ, Kim SC, Choudhri TF, Hoh BL, et al. Exacerbation of Cerebral Injury in Mice That Express the P-Selectin Gene: Identification of P-Selectin Blockade as a New Target for the Treatment of Stroke. Circ Res (1997) 81(3):304–10. doi: 10.1161/01.res.81.3.304
179. Mosser DM, Edwards JP. Exploring the Full Spectrum of Macrophage Activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
180. Meldrum DR. Tumor Necrosis Factor in the Heart. Am J Physiol (1998) 274(3):R577–95. doi: 10.1152/ajpregu.1998.274.3.R577
181. Li X, Wu Y, Zhang W, Gong J, Cheng Y. Pre-Conditioning With Tanshinone IIA Attenuates the Ischemia/Reperfusion Injury Caused by Liver Grafts via Regulation of HMGB1 in Rat Kupffer Cells. BioMed Pharmacother (2017) 89:1392–400. doi: 10.1016/j.biopha.2017.03.022
182. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding Cell Death Signals in Liver Inflammation. J Hepatol (2013) 59(3):583–94. doi: 10.1016/j.jhep.2013.03.033
183. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective Depletion of Macrophages Reveals Distinct, Opposing Roles During Liver Injury and Repair. J Clin Invest (2005) 115(1):56–65. doi: 10.1172/JCI22675
184. Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, et al. Hepatic Ischemia/Reperfusion Injury Involves Functional TLR4 Signaling in Nonparenchymal Cells. J Immunol (2005) 175(11):7661–8. doi: 10.4049/jimmunol.175.11.7661
185. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The Nuclear Factor HMGB1 Mediates Hepatic Injury After Murine Liver Ischemia-Reperfusion. J Exp Med (2005) 201(7):1135–43. doi: 10.1084/jem.20042614
186. Fiser SM, Tribble CG, Long SM, Kaza AK, Cope JT, Laubach VE, et al. Lung Transplant Reperfusion Injury Involves Pulmonary Macrophages and Circulating Leukocytes in a Biphasic Response. J Thorac Cardiovasc Surg (2001) 121(6):1069–75. doi: 10.1067/mtc.2001.113603
187. de Perrot M, Liu M, Waddell TK, Keshavjee S. Ischemia-Reperfusion-Induced Lung Injury. Am J Respir Crit Care Med (2003) 167(4):490–511. doi: 10.1164/rccm.200207-670SO
188. Johnston LK, Rims CR, Gill SE, McGuire JK, Manicone AM. Pulmonary Macrophage Subpopulations in the Induction and Resolution of Acute Lung Injury. Am J Respir Cell Mol Biol (2012) 47(4):417–26. doi: 10.1165/rcmb.2012-0090OC
189. Naidu BV, Krishnadasan B, Farivar AS, Woolley SM, Thomas R, Van Rooijen N, et al. Early Activation of the Alveolar Macrophage Is Critical to the Development of Lung Ischemia-Reperfusion Injury. J Thorac Cardiovasc Surg (2003) 126(1):200–7. doi: 10.1016/s0022-5223(03)00390-8
190. Yamamoto S, Yamane M, Yoshida O, Waki N, Okazaki M, Matsukawa A, et al. Early Growth Response-1 Plays an Important Role in Ischemia-Reperfusion Injury in Lung Transplants by Regulating Polymorphonuclear Neutrophil Infiltration. Transplantation (2015) 99(11):2285–93. doi: 10.1097/TP.0000000000000783
191. Zhao M, Fernandez LG, Doctor A, Sharma AK, Zarbock A, Tribble CG, et al. Alveolar Macrophage Activation Is a Key Initiation Signal for Acute Lung Ischemia-Reperfusion Injury. Am J Physiol Lung Cell Mol Physiol (2006) 291(5):L1018–26. doi: 10.1152/ajplung.00086.2006
192. Somers J, Ruttens D, Verleden SE, Vandermeulen E, Piloni D, Wauters E, et al. Interleukin-17 Receptor Polymorphism Predisposes to Primary Graft Dysfunction After Lung Transplantation. J Heart Lung Transplant (2015) 34(7):941–9. doi: 10.1016/j.healun.2015.03.009
193. He Z, Ma C, Yu T, Song J, Leng J, Gu X, et al. Activation Mechanisms and Multifaceted Effects of Mast Cells in Ischemia Reperfusion Injury. Exp Cell Res (2019) 376(2):227–35. doi: 10.1016/j.yexcr.2019.01.022
194. Schanze N, Bode C, Duerschmied D. Platelet Contributions to Myocardial Ischemia/Reperfusion Injury. Front Immunol (2019) 10:1260. doi: 10.3389/fimmu.2019.01260
195. Lupia E, Del Sorbo L, Bergerone S, Emanuelli G, Camussi G, Montrucchio G. The Membrane Attack Complex of Complement Contributes to Plasmin-Induced Synthesis of Platelet-Activating Factor by Endothelial Cells and Neutrophils. Immunology (2003) 109(4):557–63. doi: 10.1046/j.1365-2567.2003.01692.x
196. Chueh TH, Cheng YH, Chen KH, Chien CT. Thromboxane A2 Synthase and Thromboxane Receptor Deletion Reduces Ischaemia/Reperfusion-Evoked Inflammation, Apoptosis, Autophagy and Pyroptosis. Thromb Haemost (2020) 120(2):329–43. doi: 10.1055/s-0039-3400304
197. Nosal R. Antiplatelet and Antileukocyte Effects of Cardiovascular, Immunomodulatory and Chemotherapeutic Drugs. Cardiovasc Hematol Agents Med Chem (2006) 4(3):237–61. doi: 10.2174/187152506777698317
198. Zhou H, Hara H, Cooper DKC. The Complex Functioning of the Complement System in Xenotransplantation. Xenotransplantation (2019) 26(4):e12517. doi: 10.1111/xen.12517
199. Hsu J, Krishnan A, Lee SA, Dodd-O JM, Kim BS, Illei P, et al. Cd3. J Thorac Cardiovasc Surg (2019). doi: 10.1016/j.jtcvs.2019.09.188
200. Ysebaert DK, De Greef KE, De Beuf A, Van Rompay AR, Vercauteren S, Persy VP, et al. T Cells as Mediators in Renal Ischemia/Reperfusion Injury. Kidney Int (2004) 66(2):491–6. doi: 10.1111/j.1523-1755.2004.761_4.x
201. de Perrot M, Young K, Imai Y, Liu M, Waddell TK, Fischer S, et al. Recipient T Cells Mediate Reperfusion Injury After Lung Transplantation in the Rat. J Immunol (2003) 171(10):4995–5002. doi: 10.4049/jimmunol.171.10.4995
202. Fiorina P, Ansari MJ, Jurewicz M, Barry M, Ricchiuti V, Smith RN, et al. Role of CXC Chemokine Receptor 3 Pathway in Renal Ischemic Injury. J Am Soc Nephrol (2006) 17(3):716–23. doi: 10.1681/ASN.2005090954
203. Rabb H. The T Cell as a Bridge Between Innate and Adaptive Immune Systems: Implications for the Kidney. Kidney Int (2002) 61(6):1935–46. doi: 10.1046/j.1523-1755.2002.00378.x
204. Rabb H, Daniels F, O’Donnell M, Haq M, Saba SR, Keane W, et al. Pathophysiological Role of T Lymphocytes in Renal Ischemia-Reperfusion Injury in Mice. Am J Physiol Renal Physiol (2000) 279(3):F525–31. doi: 10.1152/ajprenal.2000.279.3.F525
205. Waldmann H, Graca L, Cobbold S, Adams E, Tone M, Tone Y. Regulatory T Cells and Organ Transplantation. Semin Immunol (2004) 16(2):119–26. doi: 10.1016/j.smim.2003.12.007
206. Ferrer IR, Hester J, Bushell A, Wood KJ. Induction of Transplantation Tolerance Through Regulatory Cells: From Mice to Men. Immunol Rev (2014) 258(1):102–16. doi: 10.1111/imr.12158
207. Inverardi L, Samaja M, Motterlini R, Mangili F, Bender JR, Pardi R. Early Recognition of a Discordant Xenogeneic Organ by Human Circulating Lymphocytes. J Immunol (1992) 149(4):1416–23.
208. Hassanain M, Simoneau E, Doi SA, Aljiffry M, Aloraini A, Madkhali A, et al. Trends in Brain-Dead Organ Donor Characteristics: A 13-Year Analysis. Can J Surg (2016) 59(3):154–60. doi: 10.1503/cjs.007415
209. Barklin A. Systemic Inflammation in the Brain-Dead Organ Donor. Acta Anaesthesiol Scand (2009) 53(4):425–35. doi: 10.1111/j.1399-6576.2008.01879.x
210. McKeating EG, Andrews PJ, Signorini DF, Mascia L. Transcranial Cytokine Gradients in Patients Requiring Intensive Care After Acute Brain Injury. Br J Anaesth (1997) 78(5):520–3. doi: 10.1093/bja/78.5.520
211. Baldwin AS. The NF-Kappa B and I Kappa B Proteins: New Discoveries and Insights. Annu Rev Immunol (1996) 14:649–83. doi: 10.1146/annurev.immunol.14.1.649
212. Tesfamariam B, DeFelice AF. Endothelial Injury in the Initiation and Progression of Vascular Disorders. Vascul Pharmacol (2007) 46(4):229–37. doi: 10.1016/j.vph.2006.11.005
213. Mertes PM, el Abassi K, Jaboin Y, Burtin P, Pinelli G, Carteaux JP, et al. Changes in Hemodynamic and Metabolic Parameters Following Induced Brain Death in the Pig. Transplantation (1994) 58(4):414–8. doi: 10.1097/00007890-199408270-00004
214. Barklin A, Theodorsson E, Tyvold SS, Larsson A, Granfeldt A, Sloth E, et al. Alteration of Neuropeptides in the Lung Tissue Correlates Brain Death-Induced Neurogenic Edema. J Heart Lung Transplant (2009) 28(7):725–32. doi: 10.1016/j.healun.2009.04.008
215. Cooper DK, Novitzky D, Wicomb WN. The Pathophysiological Effects of Brain Death on Potential Donor Organs, With Particular Reference to the Heart. Ann R Coll Surg Engl (1989) 71(4):261–6.
216. Tanriverdi F, Schneider HJ, Aimaretti G, Masel BE, Casanueva FF, Kelestimur F. Pituitary Dysfunction After Traumatic Brain Injury: A Clinical and Pathophysiological Approach. Endocr Rev (2015) 36(3):305–42. doi: 10.1210/er.2014-1065
217. Pouw IS, Drop SL, Ladée-Levy JV, Slijper FM. Pseudo- and Central Precocious Puberty Due to Adrenogenital Syndrome. Tijdschr Kindergeneeskd (1986) 54(3):77–83.
218. Michel SG, Madariaga MLL, LaMuraglia GM, Villani V, Sekijima M, Farkash EA, et al. The Effects of Brain Death and Ischemia on Tolerance Induction Are Organ-Specific. Am J Transplant (2018) 18(5):1262–9. doi: 10.1111/ajt.14674
219. Terasaki PI, Cecka JM, Gjertson DW, Takemoto S. High Survival Rates of Kidney Transplants From Spousal and Living Unrelated Donors. N Engl J Med (1995) 333(6):333–6. doi: 10.1056/NEJM199508103330601
220. Hariharan S, Johnson CP, Bresnahan BA, Taranto SE, McIntosh MJ, Stablein D. Improved Graft Survival After Renal Transplantation in the United States, 1988 to 1996. N Engl J Med (2000) 342(9):605–12. doi: 10.1056/NEJM200003023420901
221. Xu J, Sayed BA, Casas-Ferreira AM, Srinivasan P, Heaton N, Rela M, et al. The Impact of Ischemia/Reperfusion Injury on Liver Allografts From Deceased After Cardiac Death Versus Deceased After Brain Death Donors. PloS One (2016) 11(2):e0148815. doi: 10.1371/journal.pone.0148815
222. Weiss S, Kotsch K, Francuski M, Reutzel-Selke A, Mantouvalou L, Klemz R, et al. Brain Death Activates Donor Organs and Is Associated With a Worse I/R Injury After Liver Transplantation. Am J Transplant (2007) 7(6):1584–93. doi: 10.1111/j.1600-6143.2007.01799.x
223. Foley DP, Fernandez LA, Leverson G, Anderson M, Mezrich J, Sollinger HW, et al. Biliary Complications After Liver Transplantation From Donation After Cardiac Death Donors: An Analysis of Risk Factors and Long-Term Outcomes From a Single Center. Ann Surg (2011) 253(4):817–25. doi: 10.1097/SLA.0b013e3182104784
224. Mateo R, Cho Y, Singh G, Stapfer M, Donovan J, Kahn J, et al. Risk Factors for Graft Survival After Liver Transplantation From Donation After Cardiac Death Donors: An Analysis of OPTN/UNOS Data. Am J Transplant (2006) 6(4):791–6. doi: 10.1111/j.1600-6143.2006.01243.x
225. Inci I. Donors After Cardiocirculatory Death and Lung Transplantation. J Thorac Dis (2017) 9(8):2660–9. doi: 10.21037/jtd.2017.07.82
226. De Oliveira NC, Osaki S, Maloney JD, Meyer KC, Kohmoto T, D'Alessandro AM, et al. Lung Transplantation With Donation After Cardiac Death Donors: Long-Term Follow-Up in a Single Center. J Thorac Cardiovasc Surg (2010) 139(5):1306–15. doi: 10.1016/j.jtcvs.2010.02.004
227. Stone ML, Zhao Y, Robert Smith J, Weiss ML, Kron IL, Laubach VE, et al. Mesenchymal Stromal Cell-Derived Extracellular Vesicles Attenuate Lung Ischemia-Reperfusion Injury and Enhance Reconditioning of Donor Lungs After Circulatory Death. Respir Res (2017) 18(1):212. doi: 10.1186/s12931-017-0704-9
228. Inci I, Zhai W, Arni S, Hillinger S, Vogt P, Weder W. N-Acetylcysteine Attenuates Lung Ischemia-Reperfusion Injury After Lung Transplantation. Ann Thorac Surg (2007) 84(1):240–6; discussion 246. doi: 10.1016/j.athoracsur.2007.03.082
229. De Perrot M, Sekine Y, Fischer S, Waddell TK, McRae K, Liu M, et al. Interleukin-8 Release During Early Reperfusion Predicts Graft Function in Human Lung Transplantation. Am J Respir Crit Care Med (2002) 165(2):211–5. doi: 10.1164/ajrccm.165.2.2011151
230. Sayah DM, Mallavia B, Liu F, Ortiz-Muñoz G, Caudrillier A, DerHovanessian A, et al. Neutrophil Extracellular Traps Are Pathogenic in Primary Graft Dysfunction After Lung Transplantation. Am J Respir Crit Care Med (2015) 191(4):455–63. doi: 10.1164/rccm.201406-1086OC
231. Sharma AK, LaPar DJ, Stone ML, Zhao Y, Kron IL, Laubach VE. Receptor for Advanced Glycation End Products (RAGE) on iNKT Cells Mediates Lung Ischemia-Reperfusion Injury. Am J Transplant (2013) 13(9):2255–67. doi: 10.1111/ajt.12368
232. Sternberg DI, Gowda R, Mehra D, Qu W, Weinberg A, Twaddell W, et al. Blockade of Receptor for Advanced Glycation End Product Attenuates Pulmonary Reperfusion Injury in Mice. J Thorac Cardiovasc Surg (2008) 136(6):1576–85. doi: 10.1016/j.jtcvs.2008.05.032
233. Schossleitner K, Habertheuer A, Finsterwalder R, Friedl HP, Rauscher S, Gröger M, et al. A Peptide to Reduce Pulmonary Edema in a Rat Model of Lung Transplantation. PloS One (2015) 10(11):e0142115. doi: 10.1371/journal.pone.0142115
234. Mallavia B, Liu F, Sheppard D, Looney MR. Inhibiting Integrin αvβ5 Reduces Ischemia-Reperfusion Injury in an Orthotopic Lung Transplant Model in Mice. Am J Transplant (2016) 16(4):1306–11. doi: 10.1111/ajt.13605
235. Stone ML, Sharma AK, Zhao Y, Charles EJ, Huerter ME, Johnston WF, et al. Sphingosine-1-Phosphate Receptor 1 Agonism Attenuates Lung Ischemia-Reperfusion Injury. Am J Physiol Lung Cell Mol Physiol (2015) 308(12):L1245–52. doi: 10.1152/ajplung.00302.2014
236. Ibrahim M, Wang X, Puyo CA, Montecalvo A, Huang HJ, Hachem RR, et al. Human Recombinant Apyrase Therapy Protects Against Canine Pulmonary Ischemia-Reperfusion Injury. J Heart Lung Transplant (2015) 34(2):247–53. doi: 10.1016/j.healun.2014.09.034
237. Sugimoto S, Lin X, Lai J, Okazaki M, Das NA, Li W, et al. Apyrase Treatment Prevents Ischemia-Reperfusion Injury in Rat Lung Isografts. J Thorac Cardiovasc Surg (2009) 138(3):752–9. doi: 10.1016/j.jtcvs.2009.04.049
238. Gao W, Zhao J, Kim H, Xu S, Chen M, Bai X, et al. α1-Antitrypsin Inhibits Ischemia Reperfusion-Induced Lung Injury by Reducing Inflammatory Response and Cell Death. J Heart Lung Transplant (2014) 33(3):309–15. doi: 10.1016/j.healun.2013.10.031
239. Lu W, Si YI, Ding J, Chen X, Zhang X, Dong Z, et al. Mesenchymal Stem Cells Attenuate Acute Ischemia-Reperfusion Injury in a Rat Model. Exp Ther Med (2015) 10(6):2131–7. doi: 10.3892/etm.2015.2806
240. Tian W, Liu Y, Zhang B, Dai X, Li G, Li X, et al. Infusion of Mesenchymal Stem Cells Protects Lung Transplants From Cold Ischemia-Reperfusion Injury in Mice. Lung (2015) 193(1):85–95. doi: 10.1007/s00408-014-9654-x
241. La Francesca S, Ting AE, Sakamoto J, Rhudy J, Bonenfant NR, Borg ZD, et al. Multipotent Adult Progenitor Cells Decrease Cold Ischemic Injury in Ex Vivo Perfused Human Lungs: An Initial Pilot and Feasibility Study. Transplant Res (2014) 3(1):19. doi: 10.1186/2047-1440-3-19
242. Hashimoto K, Kim H, Oishi H, Chen M, Iskender I, Sakamoto J, et al. Annexin V Homodimer Protects Against Ischemia Reperfusion-Induced Acute Lung Injury in Lung Transplantation. J Thorac Cardiovasc Surg (2016) 151(3):861–9. doi: 10.1016/j.jtcvs.2015.10.112
243. Kim H, Zhao J, Zhang Q, Wang Y, Lee D, Bai X, et al. δv1-1 Reduces Pulmonary Ischemia Reperfusion-Induced Lung Injury by Inhibiting Necrosis and Mitochondrial Localization of Pkcδ and P53. Am J Transplant (2016) 16(1):83–98. doi: 10.1111/ajt.13445
244. Martens A, Ordies S, Vanaudenaerde BM, Verleden SE, Vos R, Van Raemdonck DE, et al. Immunoregulatory Effects of Multipotent Adult Progenitor Cells in a Porcine Ex Vivo Lung Perfusion Model. Stem Cell Res Ther (2017) 8(1):159. doi: 10.1186/s13287-017-0603-5
245. Lin H, Chen M, Tian F, Tikkanen J, Ding L, Andrew Cheung HY, et al. α1-Anti-Trypsin Improves Function of Porcine Donor Lungs during Ex-Vivo Lung Perfusion. J Heart Lung Transplant (2018) 37(5):656–66. doi: 10.1016/j.healun.2017.09.019
246. Park J, Kim S, Lim H, Liu A, Hu S, Lee J, et al. Therapeutic Effects of Human Mesenchymal Stem Cell Microvesicles in an Ex Vivo Perfused Human Lung Injured With Severe. Thorax (2019) 74(1):43–50. doi: 10.1136/thoraxjnl-2018-211576
247. Zhou J, Chen J, Wei Q, Saeb-Parsy K, Xu X. The Role of Ischemia-Reperfusion Injury in Early Hepatic Allograft Dysfunction. Liver Transplantation (2020) 26(8):1034–48. doi: 10.1002/lt.25779
248. Dar WA, Sullivan E, Bynon JS, Eltzschig H, Ju C. Ischaemia Reperfusion Injury in Liver Transplantation: Cellular and Molecular Mechanisms. Liver Int (2019) 39(5):788–801. doi: 10.1111/liv.14091
249. Czigany Z, Lurje I, Schmelzle M, Schöning W, Öllinger R, Raschzok N, et al. Ischemia-Reperfusion Injury in Marginal Liver Grafts and the Role of Hypothermic Machine Perfusion: Molecular Mechanisms and Clinical Implications. J Clin Med (2020) 9(3):846. doi: 10.3390/jcm9030846
250. Barzegar M, Kaur G, Gavins FNE, Wang Y, Boyer CJ, Alexander JS. Potential Therapeutic Roles of Stem Cells in Ischemia-Reperfusion Injury. Stem Cell Res (2019) 37:101421. doi: 10.1016/j.scr.2019.101421
251. Moers C, Smits JM, Maathuis MH, Treckmann J, van Gelder F, Napieralski BP, et al. Machine Perfusion or Cold Storage in Deceased-Donor Kidney Transplantation. N Engl J Med (2009) 360(1):7–19. doi: 10.1056/NEJMoa0802289
252. Moers C, Pirenne J, Paul A, Ploeg RJ, Group MPTS. Machine Perfusion or Cold Storage in Deceased-Donor Kidney Transplantation. N Engl J Med (2012) 366(8):770–1. doi: 10.1056/NEJMc1111038
253. Gill J, Dong J, Eng M, Landsberg D, Gill JS. Pulsatile Perfusion Reduces the Risk of Delayed Graft Function in Deceased Donor Kidney Transplants, Irrespective of Donor Type and Cold Ischemic Time. Transplantation (2014) 97(6):668–74. doi: 10.1097/01.TP.0000438637.29214.10
254. Kelpke SS, Chen B, Bradley KM, Teng X, Chumley P, Brandon A, et al. Sodium Nitrite Protects Against Kidney Injury Induced by Brain Death and Improves Post-Transplant Function. Kidney Int (2012) 82(3):304–13. doi: 10.1038/ki.2012.116
255. Hill P, Shukla D, Tran MG, Aragones J, Cook HT, Carmeliet P, et al. Inhibition of Hypoxia Inducible Factor Hydroxylases Protects Against Renal Ischemia-Reperfusion Injury. J Am Soc Nephrol (2008) 19(1):39–46. doi: 10.1681/ASN.2006090998
256. Imamura R, Moriyama T, Isaka Y, Namba Y, Ichimaru N, Takahara S, et al. Erythropoietin Protects the Kidneys Against Ischemia Reperfusion Injury by Activating Hypoxia Inducible Factor-1alpha. Transplantation (2007) 83(10):1371–9. doi: 10.1097/01.tp.0000264200.38926.70
257. Grenz A, Osswald H, Eckle T, Yang D, Zhang H, Tran ZV, et al. The Reno-Vascular A2B Adenosine Receptor Protects the Kidney From Ischemia. PloS Med (2008) 5(6):e137. doi: 10.1371/journal.pmed.0050137
258. Land W, Schneeberger H, Schleibner S, Illner WD, Abendroth D, Rutili G, et al. The Beneficial Effect of Human Recombinant Superoxide Dismutase on Acute and Chronic Rejection Events in Recipients of Cadaveric Renal Transplants. Transplantation (1994) 57(2):211–7. doi: 10.1097/00007890-199401001-00010
259. Danilovic A, Lucon AM, Srougi M, Shimizu MH, Ianhez LE, Nahas WC, et al. Protective Effect of N-Acetylcysteine on Early Outcomes of Deceased Renal Transplantation. Transplant Proc (2011) 43(5):1443–9. doi: 10.1016/j.transproceed.2011.02.020
260. Xue C, Liu Y, Li C, Li Y, Yang T, Xie L, et al. Powerful Protection Against Renal Ischemia Reperfusion Injury by T Cell-Specific NF-κb Inhibition. Transplantation (2014) 97(4):391–6. doi: 10.1097/01.TP.0000438622.89310.95
261. Lepper PM, Bals R. On the Edge: Targeting Toll-Like Receptor 2 in Ischemia/Reperfusion Injury. Circ Cardiovasc Interv (2012) 5(2):146–9. doi: 10.1161/CIRCINTERVENTIONS.112.968495
262. Reilly M, Miller RM, Thomson MH, Patris V, Ryle P, McLoughlin L, et al. Randomized, Double-Blind, Placebo-Controlled, Dose-Escalating Phase I, Healthy Subjects Study of Intravenous OPN-305, a Humanized Anti-TLR2 Antibody. Clin Pharmacol Ther (2013) 94(5):593–600. doi: 10.1038/clpt.2013.150
263. Gueler F, Rong S, Gwinner W, Mengel M, Bröcker V, Schön S, et al. Complement 5a Receptor Inhibition Improves Renal Allograft Survival. J Am Soc Nephrol (2008) 19(12):2302–12. doi: 10.1681/ASN.2007111267
264. Kondo M, Tahara A, Hayashi K, Abe M, Inami H, Ishikawa T, et al. Renoprotective Effects of Novel Interleukin-1 Receptor-Associated Kinase 4 Inhibitor AS2444697 Through Anti-Inflammatory Action in 5/6 Nephrectomized Rats. Naunyn Schmiedebergs Arch Pharmacol (2014) 387(10):909–19. doi: 10.1007/s00210-014-1023-z
265. Powell JT, Tsapepas DS, Martin ST, Hardy MA, Ratner LE. Managing Renal Transplant Ischemia Reperfusion Injury: Novel Therapies in the Pipeline. Clin Transplant (2013) 27(4):484–91. doi: 10.1111/ctr.12121
266. Cheng Q, Patel K, Lei B, Rucker L, Allen DP, Zhu P, et al. Donor Pretreatment With Nebulized Complement C3a Receptor Antagonist Mitigates Brain-Death Induced Immunological Injury Post-Lung Transplant. Am J Transplant (2018) 18(10):2417–28. doi: 10.1111/ajt.14717
267. Kohmoto J, Nakao A, Sugimoto R, Wang Y, Zhan J, Ueda H, et al. Carbon Monoxide-Saturated Preservation Solution Protects Lung Grafts From Ischemia-Reperfusion Injury. J Thorac Cardiovasc Surg (2008) 136(4):1067–75. doi: 10.1016/j.jtcvs.2008.06.026
268. Takashima S, Koukoulis G, Inokawa H, Sevala M, Egan TM. Inhaled Nitric Oxide Reduces Ischemia-Reperfusion Injury in Rat Lungs From Non-Heart-Beating Donors. J Thorac Cardiovasc Surg (2006) 132(1):132–9. doi: 10.1016/j.jtcvs.2006.02.032
269. Yamashita H, Akamine S, Sumida Y, Inoue M, Sawada T, Nagayasu T, et al. Inhaled Nitric Oxide Attenuates Apoptosis in Ischemia-Reperfusion Injury of the Rabbit Lung. Ann Thorac Surg (2004) 78(1):292–7. doi: 10.1016/j.athoracsur.2003.12.025
270. Qiang Y, Liang G, Yu L. Human Amniotic Mesenchymal Stem Cells Alleviate Lung Injury Induced by Ischemia and Reperfusion After Cardiopulmonary Bypass in Dogs. Lab Invest (2016) 96(5):537–46. doi: 10.1038/labinvest.2016.37
271. Nakajima D, Watanabe Y, Ohsumi A, Pipkin M, Chen M, Mordant P, et al. Mesenchymal Stromal Cell Therapy During Ex Vivo Lung Perfusion Ameliorates Ischemia-Reperfusion Injury in Lung Transplantation. J Heart Lung Transplant (2019) 38(11):1214–23. doi: 10.1016/j.healun.2019.07.006
272. Korkmaz-Icöz S, Li S, Hüttner R, Ruppert M, Radovits T, Loganathan S, et al. Hypothermic Perfusion of Donor Heart With a Preservation Solution Supplemented by Mesenchymal Stem Cells. J Heart Lung Transplant (2019) 38(3):315–26. doi: 10.1016/j.healun.2018.12.003
273. Dayan V, Sotelo V, Delfina V, Delgado N, Rodriguez C, Suanes C, et al. Human Mesenchymal Stromal Cells Improve Cardiac Perfusion in an Ovine Immunocompetent Animal Model. J Invest Surg (2016) 29(4):218–25. doi: 10.3109/08941939.2015.1128997
274. Benoit SW, Devarajan P. Acute Kidney Injury: Emerging Pharmacotherapies in Current Clinical Trials. Pediatr Nephrol (2018) 33(5):779–87. doi: 10.1007/s00467-017-3695-3
275. Oliva J. Therapeutic Properties of Mesenchymal Stem Cell on Organ Ischemia-Reperfusion Injury. Int J Mol Sci (2019) 20(21):5511–32. doi: 10.3390/ijms20215511
276. Sun Q, Huang Z, Han F, Zhao M, Cao R, Zhao D, et al. Allogeneic Mesenchymal Stem Cells as Induction Therapy Are Safe and Feasible in Renal Allografts: Pilot Results of a Multicenter Randomized Controlled Trial. J Transl Med (2018) 16(1):52. doi: 10.1186/s12967-018-1422-x
277. Krześniak AM, Radzimowski K, Stolarczyk A. Comparison of the Treatment Results of Knee Osteoarthritis Using Adipose Tissue Mesenchymal Stromal Cells Derived Through Enzymatic Digestion and Mechanically Fragmented Adipose Tissue. Med (Baltimore) (2021) 100(9):e24777. doi: 10.1097/MD.0000000000024777
278. Chung JW, Chang WH, Bang OY, Moon GJ, Kim SJ, Kim SK, et al. Efficacy and Safety of Intravenous Mesenchymal Stem Cells for Ischemic Stroke. Neurology (2021) 96(7):e1012–23. doi: 10.1212/WNL.0000000000011440
279. Lanzoni G, Linetsky E, Correa D, Messinger Cayetano S, Alvarez RA, Kouroupis D, et al. Umbilical Cord Mesenchymal Stem Cells for COVID-19 Acute Respiratory Distress Syndrome: A Double-Blind, Phase 1/2a, Randomized Controlled Trial. Stem Cells Transl Med (2021) 10(5):660–73. doi: 10.1002/sctm.20-0472
280. Macdonald P, Mundy J, Rogers P, Harrison G, Branch J, Glanville A, et al. Successful Treatment of Life-Threatening Acute Reperfusion Injury After Lung Transplantation With Inhaled Nitric Oxide. J Thorac Cardiovasc Surg (1995) 110(3):861–3. doi: 10.1016/S0022-5223(95)70125-7
281. Rea RS, Ansani NT, Seybert AL. Role of Inhaled Nitric Oxide in Adult Heart or Lung Transplant Recipients. Ann Pharmacother (2005) 39(5):913–7. doi: 10.1345/aph.1E228
282. George TJ, Arnaoutakis GJ, Beaty CA, Jandu SK, Santhanam L, Berkowitz DE, et al. Inhaled Hydrogen Sulfide Improves Graft Function in an Experimental Model of Lung Transplantation. J Surg Res (2012) 178(2):593–600. doi: 10.1016/j.jss.2012.06.037
283. Lang JD, Teng X, Chumley P, Crawford JH, Isbell TS, Chacko BK, et al. Inhaled NO Accelerates Restoration of Liver Function in Adults Following Orthotopic Liver Transplantation. J Clin Invest (2007) 117(9):2583–91. doi: 10.1172/JCI31892
284. Khajuria A, Tay C, Shi J, Zhao H, Ma D. Anesthetics Attenuate Ischemia-Reperfusion Induced Renal Injury: Effects and Mechanisms. Acta Anaesthesiol Taiwan (2014) 52(4):176–84. doi: 10.1016/j.aat.2014.10.001
285. den Hengst WA, Gielis JF, Lin JY, Van Schil PE, De Windt LJ, Moens AL. Lung Ischemia-Reperfusion Injury: A Molecular and Clinical View on a Complex Pathophysiological Process. Am J Physiol Heart Circ Physiol (2010) 299(5):H1283–99. doi: 10.1152/ajpheart.00251.2010
286. Porteous MK, Diamond JM, Christie JD. Primary Graft Dysfunction: Lessons Learned About the First 72 H After Lung Transplantation. Curr Opin Organ Transplant (2015) 20(5):506–14. doi: 10.1097/MOT.0000000000000232
287. Fumagalli J, Rosso L, Gori F, Morlacchi LC, Rossetti V, Tarsia P, et al. Early Pulmonary Function and Mid Term Outcome in Lung Transplantation After Ex Vivo Lung Perfusion- a Single-Center, Retrospective, Observational, Cohort Study. Transpl Int (2020) 20(5):506–14. doi: 10.1111/tri.13606
288. Chakos A, Ferret P, Muston B, Yan TD, Tian DH. Ex-Vivo Lung Perfusion Versus Standard Protocol Lung Transplantation-Mid-Term Survival and Meta-Analysis. Ann Cardiothorac Surg (2020) 9(1):1–9. doi: 10.21037/acs.2020.01.02
289. Slama A, Schillab L, Barta M, Benedek A, Mitterbauer A, Hoetzenecker K, et al. Standard Donor Lung Procurement With Normothermic Ex Vivo Lung Perfusion: A Prospective Randomized Clinical Trial. J Heart Lung Transplant (2017) 36(7):744–53. doi: 10.1016/j.healun.2017.02.011
290. Cypel M, Yeung JC, Liu M, Anraku M, Chen F, Karolak W, et al. Normothermic Ex Vivo Lung Perfusion in Clinical Lung Transplantation. N Engl J Med (2011) 364(15):1431–40. doi: 10.1056/NEJMoa1014597
291. Charles EJ, Huerter ME, Wagner CE, Sharma AK, Zhao Y, Stoler MH, et al. Donation After Circulatory Death Lungs Transplantable Up to Six Hours After Ex Vivo Lung Perfusion. Ann Thorac Surg (2016) 102(6):1845–53. doi: 10.1016/j.athoracsur.2016.06.043
292. Shafaghi S, Mortaz E, Abbasi Dezfuli A, Godarzi H, Sheikhy K, Ansari Aval Z, et al. Normothermic Ex Vivo Lung Perfusion in Brain-Dead Donors Reduces Inflammatory Cytokines and Toll-Like Receptor Expression. Iran J Allergy Asthma Immunol (2016) 15(5):340–54.
293. Stone JP, Ball AL, Crichley W, Yonan N, Liao Q, Sjöberg T, et al. Ex Vivo Lung Perfusion Improves the Inflammatory Signaling Profile of the Porcine Donor Lung Following Transplantation. Transplantation (2020) 104(9):1899–905. doi: 10.1097/TP.0000000000003338
294. Demirjian S, Ailawadi G, Polinsky M, Bitran D, Silberman S, Shernan SK, et al. Safety and Tolerability Study of an Intravenously Administered Small Interfering Ribonucleic Acid (siRNA) Post On-Pump Cardiothoracic Surgery in Patients at Risk of Acute Kidney Injury. Kidney Int Rep (2017) 2(5):836–43. doi: 10.1016/j.ekir.2017.03.016
295. Kassimatis T, Qasem A, Douiri A, Ryan EG, Rebollo-Mesa I, Nichols LL, et al. A Double-Blind Randomised Controlled Investigation Into the Efficacy of Mirococept (APT070) for Preventing Ischaemia Reperfusion Injury in the Kidney Allograft (EMPIRIKAL): Study Protocol for a Randomised Controlled Trial. Trials (2017) 18(1):255. doi: 10.1186/s13063-017-1972-x
296. Dehnadi A, Benedict Cosimi A, Neal Smith R, Li X, Alonso JL, Means TK, et al. Prophylactic Orthosteric Inhibition of Leukocyte Integrin CD11b/CD18 Prevents Long-Term Fibrotic Kidney Failure in Cynomolgus Monkeys. Nat Commun (2017) 8:13899. doi: 10.1038/ncomms13899
297. Nicholson ML, Hosgood SA. Renal Transplantation After Ex Vivo Normothermic Perfusion: The First Clinical Study. Am J Transplant (2013) 13(5):1246–52. doi: 10.1111/ajt.12179
298. Hosgood SA, Saeb-Parsy K, Wilson C, Callaghan C, Collett D, Nicholson ML. Protocol of a Randomised Controlled, Open-Label Trial of Ex Vivo Normothermic Perfusion Versus Static Cold Storage in Donation After Circulatory Death Renal Transplantation. BMJ Open (2017) 7(1):e012237. doi: 10.1136/bmjopen-2016-012237
299. Neves DB, Rusi MB, Diaz LGG, Salvalaggio P. Primary Graft Dysfunction of the Liver: Definitions, Diagnostic Criteria and Risk Factors. Einstein (São Paulo) (2016) 14(4):567–72. doi: 10.1590/s1679-45082016rw3585
300. Kim J, Zimmerman MA, Hong JC. Emerging Innovations in Liver Preservation and Resuscitation. Transplant Proc (2018) 50(8):2308–16. doi: 10.1016/j.transproceed.2018.03.080
301. Van Rijn R, Van Leeuwen OB, Matton APM, Burlage LC, Wiersema-Buist J, Van Den Heuvel MC, et al. Hypothermic Oxygenated Machine Perfusion Reduces Bile Duct Reperfusion Injury After Transplantation of Donation After Circulatory Death Livers. Liver Transplantation (2018) 24(5):655–64. doi: 10.1002/lt.25023
302. Van Rijn R, Van Den Berg AP, Erdmann JI, Heaton N, Van Hoek B, De Jonge J, et al. Study Protocol for a Multicenter Randomized Controlled Trial to Compare the Efficacy of End-Ischemic Dual Hypothermic Oxygenated Machine Perfusion With Static Cold Storage in Preventing Non-Anastomotic Biliary Strictures After Transplantation of Liver Gra. BMC Gastroenterol (2019) 19(1). doi: 10.1186/s12876-019-0956-6
303. van Leeuwen OB, de Vries Y, Fujiyoshi M, Nijsten MWN, Ubbink R, Pelgrim GJ, et al. Transplantation of High-Risk Donor Livers After Ex Situ Resuscitation and Assessment Using Combined Hypo- and Normothermic Machine Perfusion: A Prospective Clinical Trial. Ann Surg (2019) 270(5):906–14. doi: 10.1097/sla.0000000000003540
304. Ravikumar R, Jassem W, Mergental H, Heaton N, Mirza D, Perera MTPR, et al. Liver Transplantation AfterEx VivoNormothermic Machine Preservation: A Phase 1 (First-In-Man) Clinical Trial. Am J Transplantation (2016) 16(6):1779–87. doi: 10.1111/ajt.13708
305. Jomaa A, Gurusamy K, Siriwardana PN, Claworthy I, Collier S, De Muylder P, et al. Does Hypothermic Machine Perfusion of Human Donor Livers Affect Risks of Sinusoidal Endothelial Injury and Microbial Infection? A Feasibility Study Assessing Flow Parameters Sterility Sinusoidal Endothelial Ultrastructure (2013) 45(5):1677–83. doi: 10.1016/j.transproceed.2013.01.011
306. Henry SD, Nachber E, Tulipan J, Stone J, Bae C, Reznik L, et al. Hypothermic Machine Preservation Reduces Molecular Markers of Ischemia/Reperfusion Injury in Human Liver Transplantation. Am J Transplant (2012) 12: (9):2477–86. doi: 10.1111/j.1600-6143.2012.04086.x
307. Ghinolfi D, Rreka E, De Tata V, Franzini M, Pezzati D, Fierabracci V, et al. Pilot, Open, Randomized, Prospective Trial for Normothermic Machine Perfusion Evaluation in Liver Transplantation From Older Donors. Liver Transplantation (2019) 25(3):436–49. doi: 10.1002/lt.25362
308. Czigany Z, Schöning W, Ulmer TF, Bednarsch J, Amygdalos I, Cramer T, et al. Hypothermic Oxygenated Machine Perfusion (HOPE) for Orthotopic Liver Transplantation of Human Liver Allografts From Extended Criteria Donors (ECD) in Donation After Brain Death (DBD): A Prospective Multicentre Randomised Controlled Trial (HOPE ECD-DBD). BMJ Open (2017) 7(10):e017558. doi: 10.1136/bmjopen-2017-017558
309. Bral M, Gala-Lopez B, Bigam D, Kneteman N, Malcolm A, Livingstone S, et al. Preliminary Single Centre Canadian Experience of Human Normothermic Ex Vivo Liver Perfusion: Results of a Clinical Trial. Am J Transplant (2016) 17:1071–1080. doi: 10.1111/ajt.14049
310. Guarrera JV, Henry SD, Samstein B, Odeh-Ramadan R, Kinkhabwala M, Goldstein MJ, et al. Hypothermic Machine Preservation in Human Liver Transplantation: The First Clinical Series. Am J Transplantation (2010) 10(2):372–81. doi: 10.1111/j.1600-6143.2009.02932.x
311. Dutkowski P, Schlegel A, de Oliveira M, Müllhaupt B, Neff F, Clavien P-A. HOPE for Human Liver Grafts Obtained From Donors After Cardiac Death. J Hepatol (2014) 60(4):765–72. doi: 10.1016/j.jhep.2013.11.023
312. Mergental H, Perera MTPR, Laing RW, Muiesan P, Isaac JR, Smith A, et al. Transplantation of Declined Liver Allografts Following Normothermic Ex-Situ Evaluation. Am J Transplant (2016) 16: (11):3235–45. doi: 10.1111/ajt.13875
313. He X, Guo Z, Zhao Q, Ju W, Wang D, Wu L, et al. The First Case of Ischemia-Free Organ Transplantation in Humans: A Proof of Concept. Am J Transplant (2018) 18(3):737–44. doi: 10.1111/ajt.14583
314. Singh SSA, Dalzell JR, Berry C, Al-Attar N. Primary Graft Dysfunction After Heart Transplantation: A Thorn Amongst the Roses. Heart Fail Rev (2019) 24(5):805–20. doi: 10.1007/s10741-019-09794-1
315. Subramani S, Aldrich A, Dwarakanath S, Sugawara A, Hanada S. Early Graft Dysfunction Following Heart Transplant: Prevention and Management. Semin Cardiothorac Vasc Anesth (2020) 24(1):24–33. doi: 10.1177/1089253219867694
316. Kleveland O, Kunszt G, Bratlie M, Ueland T, Broch K, Holte E, et al. Effect of a Single Dose of the Interleukin-6 Receptor Antagonist Tocilizumab on Inflammation and Troponin T Release in Patients With Non-ST-Elevation Myocardial Infarction: A Double-Blind, Randomized, Placebo-Controlled Phase 2 Trial. Eur Heart J (2016) 37(30):2406–13. doi: 10.1093/eurheartj/ehw171
317. Yang M, Chen J, Zhao J, Meng M. Etanercept Attenuates Myocardial Ischemia/Reperfusion Injury by Decreasing Inflammation and Oxidative Stress. PloS One (2014) 9(9):e108024. doi: 10.1371/journal.pone.0108024
318. Zhu J, Huang J, Dai D, Wang X, Gao J, Han W, et al. Recombinant Human Interleukin-1 Receptor Antagonist Treatment Protects Rats From Myocardial Ischemia-Reperfusion Injury. BioMed Pharmacother (2019) 111:1–5. doi: 10.1016/j.biopha.2018.12.031
319. Ardehali A, Esmailian F, Deng M, Soltesz E, Hsich E, Naka Y, et al. Ex-Vivo Perfusion of Donor Hearts for Human Heart Transplantation (PROCEED II): A Prospective, Open-Label, Multicentre, Randomised Non-Inferiority Trial. Lancet (2015) 385(9987):2577–84. doi: 10.1016/S0140-6736(15)60261-6
320. Chan JL, Kobashigawa JA, Reich HJ, Ramzy D, Thottam MM, Yu Z, et al. Intermediate Outcomes With Ex-Vivo Allograft Perfusion for Heart Transplantation. J Heart Lung Transplant (2017) 36(3):258–63. doi: 10.1016/j.healun.2016.08.015
321. Page A, Messer S, Large SR. Heart Transplantation From Donation After Circulatory Determined Death. Ann Cardiothorac Surg (2018) 7(1):75–81. doi: 10.21037/acs.2018.01.08
322. Messer S, Page A, Berman M, Colah S, Dunning J, Pavlushkov E, et al. First to 50: Early Outcomes Following Heart Transplantation at Royal Papworth Hospital From Donation After Circulatory Determined Death (DCD) Donors. J Heart Lung Transplant (2019) 12:1311–8. doi: 10.1016/j.healun.2019.01.090
323. Nilsson J, Jernryd V, Qin G, Paskevicius A, Metzsch C, Sjöberg T, et al. A Nonrandomized Open-Label Phase 2 Trial of Nonischemic Heart Preservation for Human Heart Transplantation. Nat Commun (2020) 11(1):2976. doi: 10.1038/s41467-020-16782-9
324. Cooper DK, Gollackner B, Sachs DH. Will the Pig Solve the Transplantation Backlog? Annu Rev Med (2002) 53:133–47. doi: 10.1146/annurev.med.53.082901.103900
325. Ezzelarab MB, Ekser B, Azimzadeh A, Lin CC, Zhao Y, Rodriguez R, et al. Systemic Inflammation in Xenograft Recipients Precedes Activation of Coagulation. Xenotransplantation (2015) 22(1):32–47. doi: 10.1111/xen.12133
326. Bongoni AK, Kiermeir D, Jenni H, Wünsch A, Bähr A, Ayares D, et al. Activation of the Lectin Pathway of Complement in Pig-to-Human Xenotransplantation Models. Transplantation (2013) 96(9):791–9. doi: 10.1097/TP.0b013e3182a3a52b
327. Knosalla C. Success for Pig-to-Baboon Heart Transplants. Nature (2018) 564(7736):352–3. doi: 10.1038/d41586-018-07419-5
328. Längin M, Reichart B, Steen S, Sjöberg T, Paskevicius A, Liao Q, et al. Cold Non-Ischemic Heart Preservation With Continuous Perfusion Prevents Early Graft Failure in Orthotopic Pig-to-Baboon Xenotransplantation. Xenotransplantation (2021) 28(1):e12636. doi: 10.1111/xen.12636
329. Watanabe H, Sahara H, Nomura S, Tanabe T, Ekanayake-Alper DK, Boyd LK, et al. GalT-KO Pig Lungs Are Highly Susceptible to Acute Vascular Rejection in Baboons, Which may be Mitigated by Transgenic Expression of Hcd47 on Porcine Blood Vessels. Xenotransplantation (2018) 25(5):e12391. doi: 10.1111/xen.12391
330. Shah JA, Patel MS, Elias N, Navarro-Alvarez N, Rosales I, Wilkinson RA, et al. Prolonged Survival Following Pig-To-Primate Liver Xenotransplantation Utilizing Exogenous Coagulation Factors and Costimulation Blockade. Am J Transplant (2017) 17(8):2178–85. doi: 10.1111/ajt.14341
331. Khalpey Z, Yuen AH, Kalsi KK, Kochan Z, Karbowska J, Slominska EM, et al. Loss of Ecto-5’nucleotidase From Porcine Endothelial Cells After Exposure to Human Blood: Implications for Xenotransplantation. Biochim Biophys Acta (2005) 1741(1-2):191–8. doi: 10.1016/j.bbadis.2005.03.008
332. Sykes M, Sachs DH. Transplanting Organs From Pigs to Humans. Sci Immunol (2019) 4(41):1–13. doi: 10.1126/sciimmunol.aau6298
333. Mohiuddin MM, Singh AK, Corcoran PC, Thomas Iii ML, Clark T, Lewis BG, et al. Chimeric 2C10R4 Anti-CD40 Antibody Therapy Is Critical for Long-Term Survival of GTKO.hCD46.hTBM Pig-to-Primate Cardiac Xenograft. Nat Commun (2016) 7:11138. doi: 10.1038/ncomms11138
334. Jordan SC, Choi J, Kim I, Wu G, Toyoda M, Shin B, et al. Interleukin-6, A Cytokine Critical to Mediation of Inflammation, Autoimmunity and Allograft Rejection: Therapeutic Implications of IL-6 Receptor Blockade. Transplantation (2017) 101(1):32–44. doi: 10.1097/TP.0000000000001452
335. Weigt SS, Palchevskiy V, Belperio JA. Inflammasomes and IL-1 Biology in the Pathogenesis of Allograft Dysfunction. J Clin Invest (2017) 127(6):2022–9. doi: 10.1172/JCI93537
336. Zhao Y, Cooper DKC, Wang H, Chen P, He C, Cai Z, et al. Potential Pathological Role of Pro-Inflammatory Cytokines (IL-6, TNF-α, and IL-17) in Xenotransplantation. Xenotransplantation (2019) 26(3):e12502. doi: 10.1111/xen.12502
337. Steen S, Paskevicius A, Liao Q, Sjöberg T. Safe Orthotopic Transplantation of Hearts Harvested 24 Hours After Brain Death and Preserved for 24 Hours. Scand Cardiovasc J (2016) 50(3):193–200. doi: 10.3109/14017431.2016.1154598
338. Boteon YL, Afford SC. Machine Perfusion of the Liver: Which Is the Best Technique to Mitigate Ischaemia-Reperfusion Injury? World J Transplant (2019) 9(1):14–20. doi: 10.5500/wjt.v9.i1.14
339. Koerner MM, Ghodsizad A, Schulz U, El Banayosy A, Koerfer R, Tenderich G. Normothermic Ex Vivo Allograft Blood Perfusion in Clinical Heart Transplantation. Heart Surg Forum (2014) 17(3):E141–5. doi: 10.1532/HSF98.2014332
340. Sun W, Wang Z, Sun M, Huang W, Wang Y. Aloin Antagonizes Stimulated Ischemia/Reperfusion-Induced Damage and Inflammatory Response in Cardiomyocytes by Activating the Nrf2/HO-1 Defense Pathway. Cell Tissue Res (2021) 3:735–44. doi: 10.1007/s00441-020-03345-z
341. Shen Y, Liu X, Shi J, Wu X. Involvement of Nrf2 in Myocardial Ischemia and Reperfusion Injury. Int J Biol Macromol (2019) 125:496–502. doi: 10.1016/j.ijbiomac.2018.11.190
342. Soares MP, Lin Y, Anrather J, Csizmadia E, Takigami K, Sato K, et al. Expression of Heme Oxygenase-1 can Determine Cardiac Xenograft Survival. Nat Med (1998) 4(9):1073–7. doi: 10.1038/2063
343. Öllinger R, Pratschke J. Role of Heme Oxygenase-1 in Transplantation. Transpl Int (2010) 23(11):1071–81. doi: 10.1111/j.1432-2277.2010.01158.x
344. Yeom HJ, Koo OJ, Yang J, Cho B, Hwang JI, Park SJ, et al. Generation and Characterization of Human Heme Oxygenase-1 Transgenic Pigs. PloS One (2012) 7(10):e46646. doi: 10.1371/journal.pone.0046646
345. Kim MJ, Hwang YH, Kim YH, Lee DY. Immunomodulation of Cell-Penetrating Tat-Metallothionein for Successful Outcome of Xenotransplanted Pancreatic Islet. J Drug Targeting (2017) 25(4):350–9. doi: 10.1080/1061186X.2016.1258704
346. Yokoyama I, Negita M, Liu DG, Nagasaka T, Kobayashi T, Hayakawa A, et al. Prevention of Free-Radical Induced Apoptosis by Induction of Human Recombinant Cu, Zn-SOD in Pig Endothelial Cells. Transpl Int (2002) 15(5):220–5. doi: 10.1007/s00147-002-0394-0
347. Knosalla C, Yazawa K, Behdad A, Bodyak N, Shang H, Bühler L, et al. Renal and Cardiac Endothelial Heterogeneity Impact Acute Vascular Rejection in Pig-to-Baboon Xenotransplantation. Am J Transplant (2009) 9(5):1006–16. doi: 10.1111/j.1600-6143.2009.02602.x
348. Kim H, Hawthorne WJ, Kang HJ, Lee YJ, Hwang JI, Hurh S, et al. Human Thrombomodulin Regulates Complement Activation as Well as the Coagulation Cascade in Xeno-Immune Response. Xenotransplantation (2015) 22(4):260–72. doi: 10.1111/xen.12173
349. Harris DG, Quinn KJ, French BM, Schwartz E, Kang E, Dahi S, et al. Meta-Analysis of the Independent and Cumulative Effects of Multiple Genetic Modifications on Pig Lung Xenograft Performance During Ex Vivo Perfusion With Human Blood. Xenotransplantation (2015) 22(2):102–11. doi: 10.1111/xen.12149
350. Burdorf L, Riner A, Rybak E, Salles II, De Meyer SF, Shah A, et al. Platelet Sequestration and Activation During GalTKO.hCD46 Pig Lung Perfusion by Human Blood Is Primarily Mediated by GPIb, GPIIb/IIIa, and Von Willebrand Factor. Xenotransplantation (2016) 23(3):222–36. doi: 10.1111/xen.12236
351. Maeda A, Kawamura T, Ueno T, Usui N, Eguchi H, Miyagawa S. The Suppression of Inflammatory Macrophage-Mediated Cytotoxicity and Proinflammatory Cytokine Production by Transgenic Expression of HLA-E. Transpl Immunol (2013) 29(1-4):76–81. doi: 10.1016/j.trim.2013.08.001
352. Chang CL, Wu CC, Hsu YT, Hsu YC. Immune Vulnerability of Ovarian Cancer Stem-Like Cells Due to Low CD47 Expression Is Protected by Surrounding Bulk Tumor Cells. Oncoimmunol (2020) 9(1):1803530. doi: 10.1080/2162402X.2020.1803530
353. Miwa Y, Yazaki S, Iwamoto M, Suzuki S, Iwasaki K, Haneda M, et al. Functional Difference Between Membrane-Bound and Soluble Human Thrombomodulin. Transplantation (2015) 99(4):702–9. doi: 10.1097/TP.0000000000000571
354. Black SM, Grehan JF, Rivard AL, Benson BA, Wahner AE, Koch AE, et al. Porcine Endothelial Cells and Iliac Arteries Transduced With AdenoIL-4 Are Intrinsically Protected, Through Akt Activation, Against Immediate Injury Caused by Human Complement. J Immunol (2006) 177(10):7355–63. doi: 10.4049/jimmunol.177.10.7355
355. Bruns H, Bessell C, Varela JC, Haupt C, Fang J, Pasemann S, et al. CD47 Enhances Ex Vivo Functionality of Artificial Antigen-Presenting Cells. Clin Cancer Res (2015) 21(9):2075–83. doi: 10.1158/1078-0432.CCR-14-2696
Keywords: xenotranplantation, ischemia reperfusion (I/R) injury, ex vivo perfusion, initial xenograft dysfunction, ischemia reperfusion injury mechanisms, ischemia reperfusion injury minimization
Citation: Patel PM, Connolly MR, Coe TM, Calhoun A, Pollok F, Markmann JF, Burdorf L, Azimzadeh A, Madsen JC and Pierson RN III (2021) Minimizing Ischemia Reperfusion Injury in Xenotransplantation. Front. Immunol. 12:681504. doi: 10.3389/fimmu.2021.681504
Received: 16 March 2021; Accepted: 12 August 2021;
Published: 09 September 2021.
Edited by:
Niels Olsen Saraiva Camara, University of São Paulo, BrazilReviewed by:
Yi Wang, Second Affiliated Hospital of Hainan Medical University, ChinaMatthias Längin, Klinikum der Universität München, Germany
Copyright © 2021 Patel, Connolly, Coe, Calhoun, Pollok, Markmann, Burdorf, Azimzadeh, Madsen and Pierson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard N. Pierson III, UlBJRVJTT05AbWdoLmhhcnZhcmQuZWR1