 Marc-André Limoges
Marc-André Limoges Subburaj Ilangumaran
Subburaj Ilangumaran Sheela Ramanathan
Sheela Ramanathan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 May 2021
Sec. T Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.679739
Overview: Long-term survival of T lymphocytes in quiescent state is essential to maintain their cell numbers in secondary lymphoid organs and in peripheral circulation. In the BioBreeding diabetes-prone strain of rats (BB-DP), loss of functional GIMAP5 (GTPase of the immune associated nucleotide binding protein 5) results in profound peripheral T lymphopenia. This discovery heralded the identification of a new family of proteins initially called Immune-associated nucleotide binding protein (IAN) family. In this review we will use ‘GIMAP’ to refer to this family of proteins. Recent studies suggest that GIMAP proteins may interact with each other and also be involved in the movement of the cellular cargo along the cytoskeletal network. Here we will summarize the current knowledge on the characteristics and functions of GIMAP family of proteins.
In the BioBreeding diabetes-prone strain of rats (BB-DP), the recessive lyp mutation causes a profound loss of T lymphocytes in secondary lymphoid organs (1). Positional cloning of the gene responsible for the lymphopenic phenotype in the BB-DP rats independently by two groups led to the discovery of a family of proteins that are conserved in vertebrates (2, 3). The lyp allele arises from a frame shift mutation within the GTPase domain of the immune associated nucleotide binding protein 5 (Gimap5) gene, resulting in a truncated protein lacking 223 amino acids at the C-terminus (2, 3). GIMAP5 is a member of the GIMAP family that are implicated in immune functions in mammals (4). Initially this family of proteins was named IAN, for ‘immune associated nucleotide binding’ proteins, as they were predominantly expressed in the cells of the hematopoietic system and contained domains that can bind to GDP/GTP. In this review we will summarize our current knowledge of the structure and functions of GIMAP proteins, many of which are implicated in the development and maintenance of lymphocytes (Table 1). All Gimap genes are clustered within a short locus in the genome. The human GIMAP cluster, spanning about 500kb on chromosome 7, contains seven functional genes and one pseudogene (4). In mice and rats the Gimap genes are present as a tight cluster within a 150kb region on chromosome 6 and 4, respectively (2, 3). Gimap-like genes have been identified in angiosperms, corals, nematodes and in snails wherein they are implicated in protection from infections (56–59). The observed homology between GIMAP proteins and the plant avrRpt2 induced gene 1 (AIG1) might have resulted from convergent evolution of the AIG1 domain (4, 60). In fact, initial homology searches identified Arpt1 in Arabhdiposis thaliana (61) as the closest homolog of the mammalian Gimap family proteins.
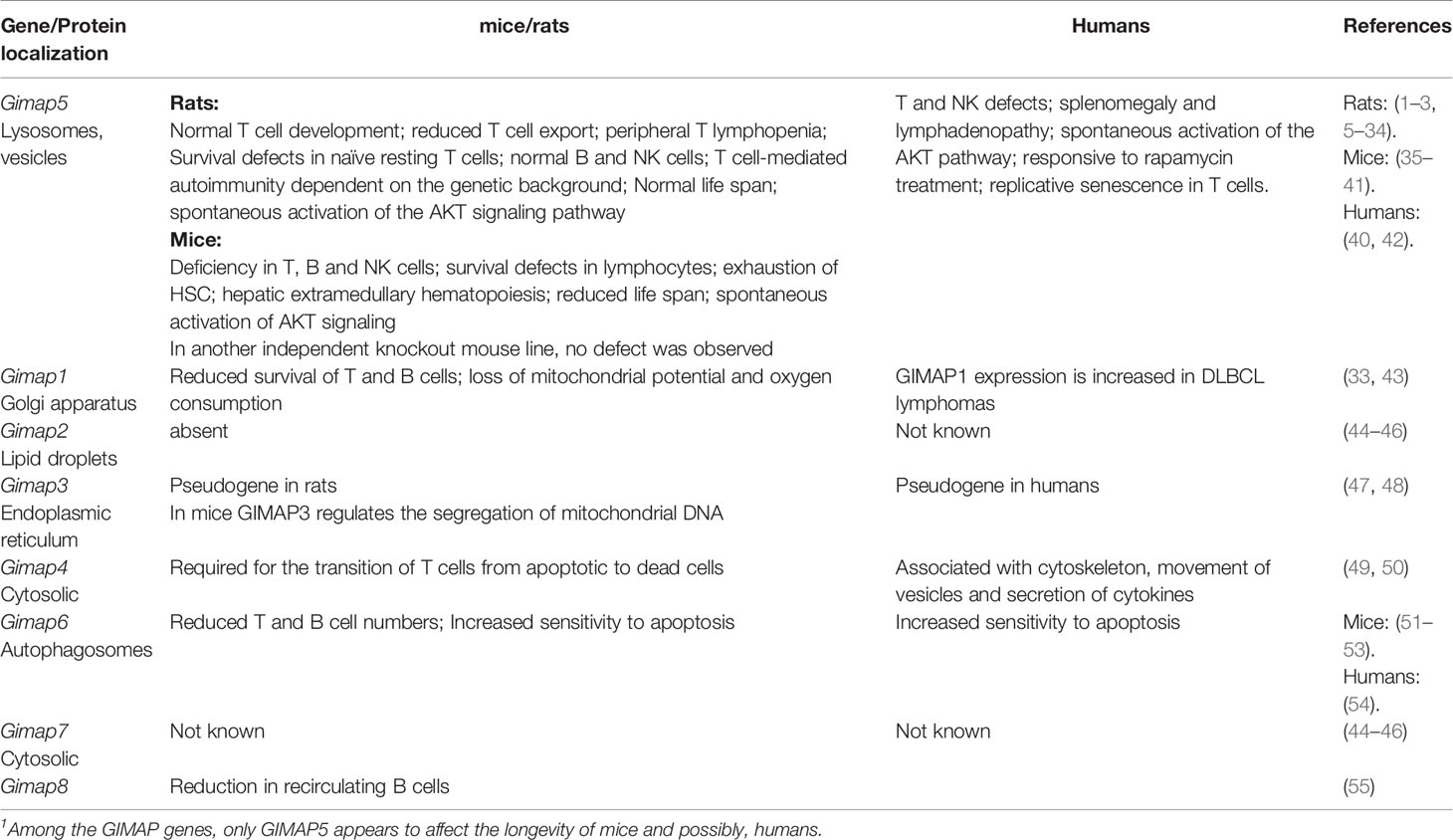
Table 1 Phenotype of deficiency in GIMAP genes.
The GIMAP family consists of putative small GTP-or/and ATP binding proteins that are conserved among vertebrates (Figure 1). Among the GIMAP family proteins, sequence similarity is restricted to the N-terminus, which contains a guanine nucleotide-binding domain (4, 62). All GIMAP family members harbor the AIG1 domain containing a GTP-binding motif, that is referred to as GIMAP GTPase domain comprised of the five G motifs G1, G2, G3, G4, and G5, which are involved in nucleotide binding. It is thought that the GIMAP GTPase domains may be functional and that their activity may be controlled by homo/hetero dimerization (44–46, 63). However, only some GIMAPs have been shown to bind GDP/GTP or to hydrolyze GTP. GIMAP4 is the first member reported to bind GDP and GTP and exhibit GTPase activity (63). Further studies have shown that GIMAP2 and GIMAP5 can bind GTP with high affinity but cannot hydrolyze it on their own (44). GIMAP7 can stimulate its own GTPase activity and promote GTP hydrolysis by GIMAP2 (44). Besides the GTPase domain, all GIMAP proteins contain a helical segment that folds back on to the GTPase domain and may mediate interaction with partner proteins. In addition, GIMAP1, GIMAP3, and GIMAP5 contain a transmembrane hydrophobic domain at the C-terminus that have been shown to mediate membrane anchoring and target them to intracellular organelles (64). In the next sections each of the GIMAP family member is discussed in detail, starting with the founding member GIMAP5.
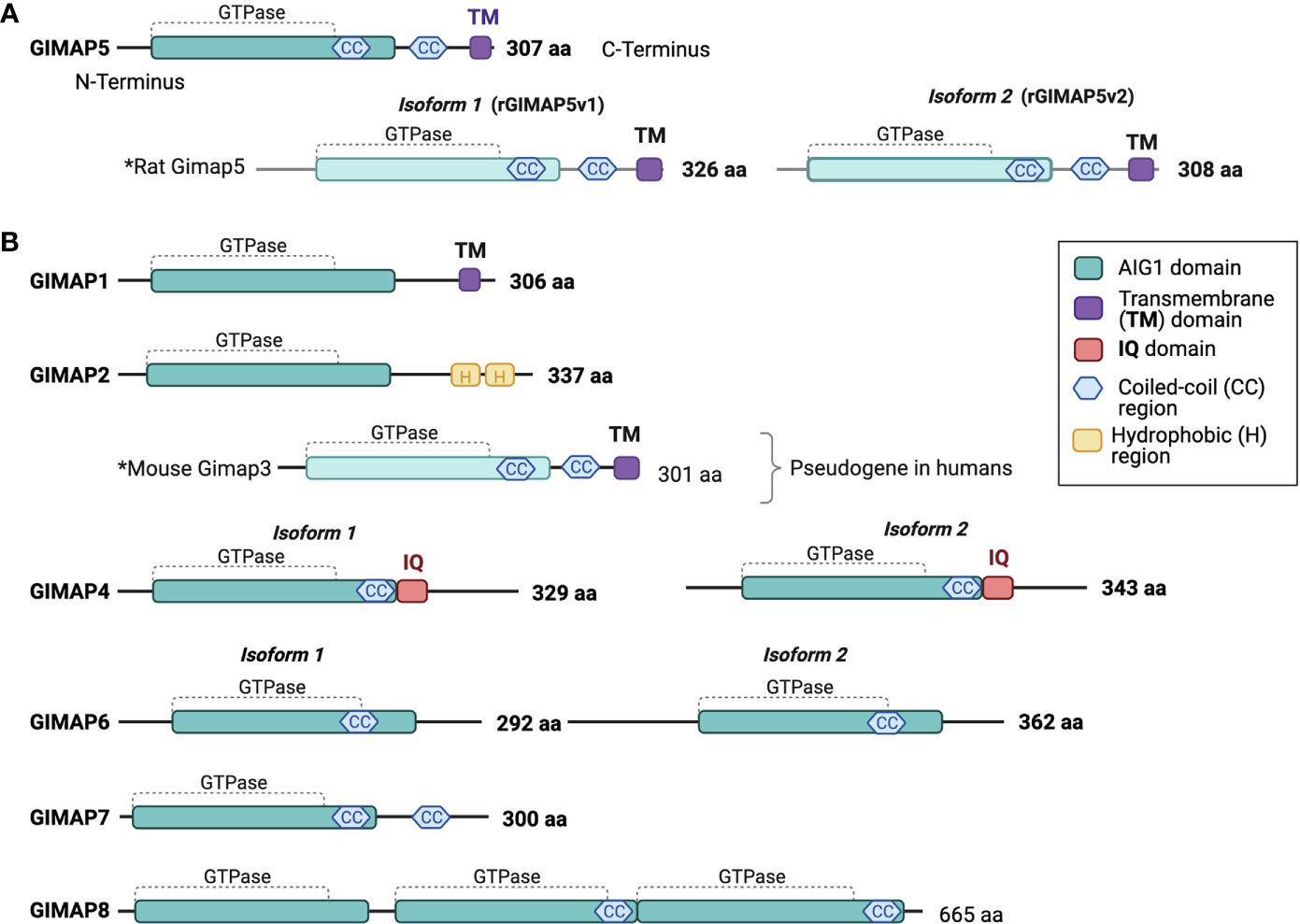
Figure 1 Human GIMAP family protein structure. Predicted structural domains of human GIMAP family members. (A) The 307-amino acid long human GIMAP5 protein contains the GIMAP GTPase domain, coiled-coil (CC) regions and a C-terminal transmembrane (TM) domain. Rat GIMAP5 protein is found in two isoforms that differ in length (GIMAP5v1 and GIMAP5v2). Both retain the CC regions and the TM domain. (B) Predicted structures of other human GIMAP family proteins and mouse GIMAP3, as the latter is a pseudogene in humans. The calmodulin interaction domain (IQ) is unique to both GIMAP4 isoforms. The drawings are not to scale.
A spontaneous mutation in an outbred colony of Wistar rats was associated with the development of autoimmune type 1 diabetes (T1D) (5). Incidentally these rats were also lymphopenic. Further inbreeding resulted in the establishment of the strain of BB-DP rats (1, 6–8). Genetic studies mapped the lymphopenia phenotype to the lyp locus on chromosome 4 (9). In 2002, two groups independently identified a frameshift mutation within the Gimap5 gene as being responsible for this lymphopenic phenotype (2, 3). In BB-DP rats, lymphopenia is restricted to the T cell compartment with a 5-10 fold reduction in the number of mature T cells in secondary lymphoid organs (10). This lymphopenia is more severe in the CD8+ T cell compartment than in the CD4+ T cell compartment (11, 12). Thus, most studies characterizing the function of GIMAP5 in T cells have been carried out on the CD4+ T cell subset. Mature CD4+ T cells, characterized by the expression of the RT6 marker in the rats, were almost completely absent in the secondary lymphoid organs of BB-DP rats (13, 14). In vitro studies have shown that the few surviving T cells in BB-DP rats are not fully functional. Purified BB-DP rat CD4+ T cells stimulated with mitogens or anti-CD3/CD8 antibodies displayed impaired proliferative response (15). In vivo, the decreased functionality of the BB-DP rat T cells was attributed to the increased nitric oxide (NO) production by macrophages (16). Bone-marrow chimeras showed that the increased NO production by macrophages of the BB-DP rats was secondary to the T lymphopenia, as correcting the latter decreased NO production (15). In addition, the lyp mutation affects the development of regulatory T cells, γδ T cells and intra-epithelial lymphocytes (iIELs) in the intestinal mucosa (17, 18). The development and survival of NK cells and B lymphocytes are not affected by the lyp mutation in BB-DP rats (genotype: Gimap5lyp/lyp) (1). An important paradox in BB-DP rats that aroused much scientific investigation in the 80s and 90s was the T cell dependency of autoimmune T1D development despite severe lymphopenia. Depletion of the few CD4+ or CD8+ T cells, but not NK cells, prevented T1D development in BB-DP rats (19, 20), indicating that the remaining lymphocytes are the crucial mediators of disease.
During T cell development, hematopoietic precursors with restricted multipotency enter the thymic cortex from the bone marrow and undergo a series of developmental changes that are demarcated by specific phenotypic characteristics associated with their commitment towards the T cell lineage (65). Until the rearrangement of the T cell antigen receptor (TCR) genes, thymocytes do not express CD4 or CD8 co-receptors and are referred to as double negative (DN) thymocytes. TCR rearrangement upregulates the expression of both CD4 and CD8 co-receptors, allowing these double positive (DP) cells to undergo positive selection (66, 67). Subsequent to TCR signaling, DP cells lose one of the two co-receptors, depending on their ability to recognize MHC class I or class II molecules, and become CD4 or CD8 single positive (SP) cells that transit to the thymic medulla where T cells with high affinity to self-antigens are deleted by the negative selection process. The surviving SP thymocytes undergo further maturation before exiting to the periphery as recent thymic emigrants (RTE). RTE undergo additional maturation in the periphery to become long-lived naïve T cells. Naïve T cell survival is maintained in the periphery by constant low-level interactions with the self MHC:peptide complexes (MHC:p) (68). Upon encounter with the cognate antigen, the TCR-stimulated T cell clones undergo proliferative expansion and initiate the adaptive immune response. Some of the antigen-specific T cells undergo reprogramming to become long-lived memory T cells that constantly patrol the tissues. Both naïve and memory T cells rely on cues from TCR- MHC:p interactions and cytokines such as IL-7 and IL-15 for their survival and homeostasis in the periphery.
Even though the proportion of DN, DP and SP subsets are comparable between control and BB-DP rats, the reduced thymic output in BB-DP rats (1, 21, 22) suggested that the lymphopenic phenotype caused by Gimap5lyp/lyp genotype manifests during the later stages of T cell development in the thymus. In line with this observation, transcripts for Gimap5 are detected at higher levels starting from DP stage of T cell development in normal rats (23). Some groups have observed a decrease in the frequency of immature CD8+ SP thymocytes (24–26). Nevertheless, SP thymocytes from Gimap5lyp/lyp rats undergo accelerated apoptosis in vitro (1, 25–27).
Homeostatic expansion of T cells present in the periphery can compensate for reduced thymic export and can almost fully restore the T cell numbers under conditions of lymphopenia (69). However, the peripheral T lymphopenia in the Gimap5lyp/lyp rats suggests that the homeostatic expansion may also be compromised by GIMAP5 deficiency or, alternately, that the expanding cells are unable to survive and persist in the secondary lymphoid organs. To address this issue, we thymectomized control and Gimap5lyp/lyp rats and labeled the cycling cells with bromodeoxyuridine (27). Whereas only 5-10% T cells incorporated the label in control rats with a full T cell compartment, almost 100% of the T cells in Gimap5lyp/lyp rats had incorporated BrDU during the same period. Despite this increased T cell cycling in the periphery, Gimap5lyp/lyp rats fail to restore their T cells numbers in the periphery. Follow up of the BrDU-labeled cells during the chase period suggested that most of them were lost from the secondary lymphoid organs in Gimap5lyp/lyp rats while they were present in control rats. The complete loss of the BrDU-labeled cells from the secondary lymphoid organs of Gimap5lyp/lyp rats indicated that the progeny of the cycling cells was unable to persist and survive and that the homeostatic pressure maintains the few surviving cells in a perpetual cycling phase. Reconstitution of the lymphopenic BB-DP rats with splenocytes from syngenic, diabetes-resistant (BB-DR) rats that carry the wildtype Gimap5 allele eliminated the cycling of endogenous BB-DP T cells that were eventually lost from the periphery.
Paradoxically, the few cells that persist in the secondary organs of Gimap5lyp/lyp rats have been shown to be activated by their cognate antigen and incorporated into the pool of recycling cells (27). As there are no TCR transgenics available for rats, the antigen specificity of the peripheral T cell pool was assessed using allogenic T cells from Wistar Furth (WF) background as lymphopenic BB-DP rats could reject allogenic T cells (27). To determine whether antigen-reactive cells were inducted into the pool of recirculating cells, the BB-DP rats were thymectomized following the rejection of WF T cells. One month after thymectomy, the antigen-exposed rats were still capable of rejecting the allogenic T cells. However, in the absence of prior exposure, thymectomized rats were unable to eliminate the allogenic cells. Additional experiments showed that RTEs had a narrow window of one week after thymic exit in order to be ‘rescued’ by TCR stimulation. These experiments helped resolve the paradox of T cell mediated autoimmunity in BB-DP rats. In an appropriate genetic background these cycling Gimap5lyp/lyp T cells recognize self-antigens and induce autoimmune diseases. For example, in the BB-DP rats these cycling T cells recognize islet antigens and induce T1D. In the PVG background the lyp mutation contribute to eosinophilic inflammatory bowel disease (70) whereas in Lewis rats experimental autoimmune encephalomyelitis (EAE) becomes aggravated (71).
In humans, transcripts for GIMAP5 can be detected in peripheral blood T cells but not in B cells (62). Human GIMAP5 was initially identified in 2001 as the Oar-2 clone from a Jurkat-derived cDNA library that could confer resistance to gamma-radiation and okadaic acid (OA)-induced apoptosis (72). In 2003, the protein was identified as Irod (inhibitor of radiation- and OA- induced death) (73). Overexpression of Oar-2 conferred protection in a CaMKII dependent manner in Jurkat cells (73). Regulation of sensitivity to OA may be species-specific, as no significant alterations in protein phosphatase activity was observed in Gimap5lyp/lyp rat T cells (28).
To better understand the functions of Gimap5, three groups generated mice with inactivating mutations in Gimap5 (35–37). Two lines of Gimap5-deficient mice generated by the groups of H. Weiler and K. Hoebe exhibited comparable phenotype (35, 36). The Weiler laboratory generated Gimap5-/- mice by replacing the Gimap5 exon 2 and a part of exon 3 with the neomycin cassette (36). The Hoebe group inactivated Gimap5 through ENU mediated mutagenesis to generate the Sphinx (Gimap5sph/sph) line of mice (35). The latter carried a point mutation G38C that can abrogate the binding of GTP/GDP to a site that is conserved in RAS family of GTPases. Unlike in rats, where the defect caused by the lyp allele is restricted to T cells, absence of functional GIMAP5 leads to a paucity of peripheral T, B and NK cells in Gimap5-/- and in Gimap5sph/sph mice. However, positive and negative selection of T cells was not altered by the absence of GIMAP5 (35). In addition to T and B lymphopenia, both lines of mice show exhaustion of hematopoietic stem cells (HSC) and hepatic extramedullary hematopoiesis that is independent of T and B lymphocytes, as Rag1-/-Gimap5-/- mice also show the same phenotype (35, 38). In contrast to the above two mouse strains, Gimap5-/- mice developed by the Takahama group did not exhibit any T cell survival defects (37). Reasons for this discrepancy remains unclear. The Sphinx mice have also been reported to develop intestinal inflammation (35, 39).
Homeostatic survival of naive T cells requires two essential signals, one provided by the cytokine interleukin-7 (IL-7) and the other by MHC:self-peptide complexes that engage the TCR (74). Signals delivered via the IL-7 receptor and the TCR impact the classical pathway that maintains quiescence in most cell types involving LKB1 and AMPK (75, 76). AMPK, the energy sensor activated by an elevated AMP/ATP ratio, inhibits the mTORC1 complex by activating its suppressor, the TSC1/2 complex (77, 78). TCR engagement triggers the activation of LCK and ZAP70 that phosphorylate many substrates including the scaffolding protein LAT, resulting in the formation of multi-molecular signaling complex at the plasma membrane (79) (Figure 2). Activation of the PI3K/AKT signaling pathway downstream of TCR signaling phosphorylates TSC1/2 complex thereby releasing RHEB GTPase from suppression to activate the mTORC1 complex (80). mTORC1 promotes translation and protein synthesis by activating 70-kDa ribosomal S6 kinase (S6K1) and releasing the repressor protein 4E-BP1 from the translation initiation factor eIF-E4. Thus, in the absence of functional LKB1, AMPK or the TSC1/2 complex, T cell quiescence is lost (75–78). We observed that Gimap5-deficient T cells showed normal AMPK activation and mitochondrial respiration but displayed defects in IL-7 signaling, proximal TCR signaling manifested as reduced phosphorylation of ZAP70 and LAT, T cell calcium response and constitutive activation of AKT and the mTORC1 pathway (28–31) (Figure 2). Molecular mechanisms by which GIMAP5 impacts TCR and IL-7R signaling pathways remain to be elucidated.
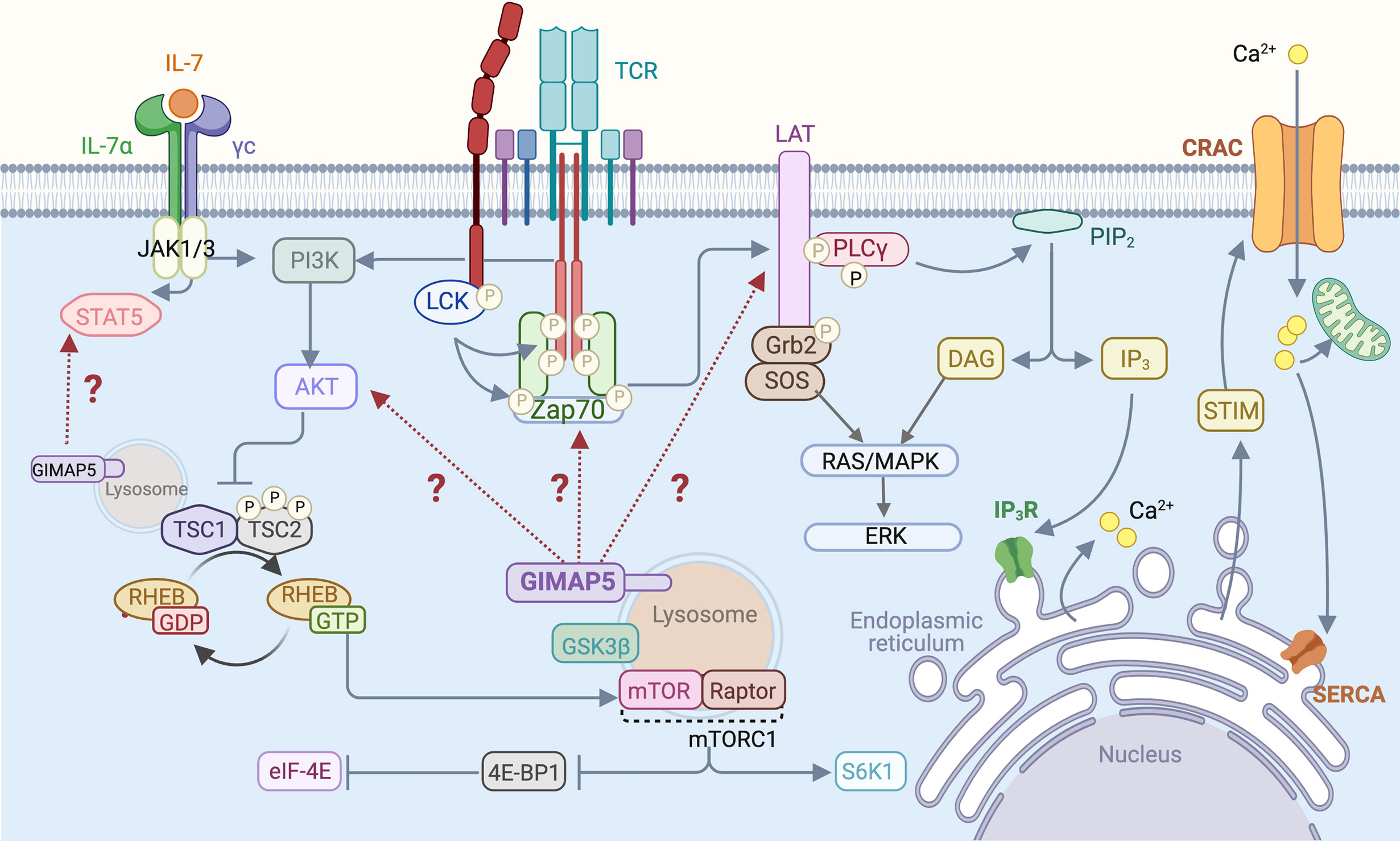
Figure 2 T cell signaling pathways that are influenced by GIMAP5. Following TCR stimulation by MHC:peptide complex or by Ab-mediated TCR cross-linking, LCK phosphorylates CD3 zeta chains and ZAP70, resulting in the phosphorylation of LAT that acts as a scaffold for downstream signaling molecules such as PLCγ. Activation of the PI3K/AKT signaling pathway downstream of TCR phosphorylates and inhibits the TSC1/2 complex, relieving repression of the mTORC1 kinase and leading to activation of downstream signaling events. IL-7 signaling stimulates STAT5 and also activates the PI3K/AKT pathway. GIMAP5 deficiency in rat and mouse T cells compromises proximal TCR signaling characterized by reduced Tyr phosphorylation of ZAP70 and LAT, but results in constitutive activation of AKT and mTORC1. GIMAP5 deficient T cells also display reduced IL-7-induced STAT5 phosphorylation. It is unclear how GIMAP5 impacts the TCR and IL-7R signaling pathways and regulates AKT activity (29, 34, 40–42).
The TCR signalosome recruits and activates phospholipase Cγ (PLCγ), which hydrolyzes the membrane bound phosphatidylinositol 4,5 bisphosphate (PIP2) to generate inositol 1,4,5-triphosphate (IP3) and diacyl glycerol (DAG) (Figure 2). IP3 binds to its receptor IP3R on the endoplasmic reticulum (ER) and triggers Ca2+ release from the ER store, resulting in a conformational change in the ER-localized STIM1 protein (81, 82) (Figure 3). This event relays a signal to open the Ca2+ release-activated Ca2+ channel (CRAC) on the plasma membrane, inducing the capacitative Ca2+ entry from the extracellular milieu (83, 84). CRAC channels consisting of the ORAI proteins are the major store-operated channels in T lymphocytes (85, 86). TCR stimulation by antigen induces sustained Ca2+ influx via CRAC channels leading to T cell proliferation (87).
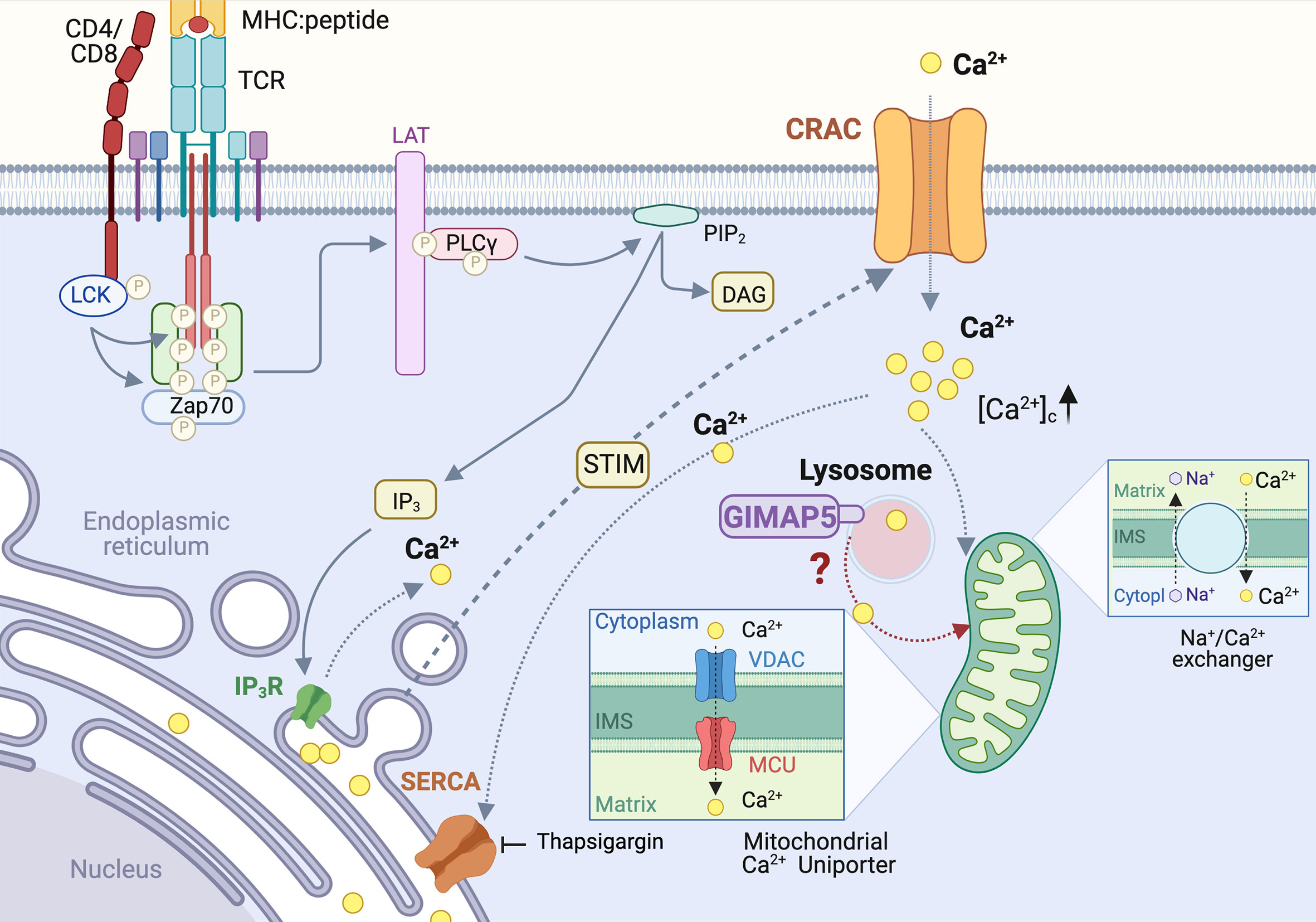
Figure 3 Regulation of Calcium homeostasis in rat T cells by GIMAP5. TCR-induced PLCγ activation leads to cleavage of plasma membrane-associated phosphatidylinositol 4,5 bisphosphate (PIP2) to generate inositol 1,4,5-triphosphate (IP3). IP3 binds to its receptor IP3R on the endoplasmic reticulum (ER) and triggers Ca2+ release from the ER store, resulting in a conformational change in the ER-localized STIM1 protein. This event relays a signal to open the Ca2+ release-activated Ca2+ channel (CRAC) on the plasma membrane, inducing the capacitative Ca2+ entry. The rising concentration of cytosolic calcium ([Ca2+]c) activates the Ca2+ uniporter on the mitochondrial membrane to uptake Ca2+, which is released later via the Na+/Ca2+ exchanger. In addition to ER, lysosomes also release a significant amount of Ca2+ following cell activation. Loss of GIMAP5 does not affect TCR- or thapsigargin- induced Ca2+ release from the ER stores but reduces Ca2+entry from extracellular milieu. GIMAP5 resides on lysosomes and the loss of GIMAP5 reduces lysosomal and mitochondrial Ca2+ content, presumably leading to feedback inhibition of the CRAC channels by cytosolic Ca2+. How GIMAP5 integrates TCR signaling to regulate lysosomal and mitochondrial Ca2+ to promote T cell survival and functions remains to be elucidated (28, 30, 31).
We observed that TCR-induced Ca2+ flux is reduced in T cells from BB-DP rats, which lack a functional GIMAP5 protein (30). The IP3-mediated Ca2+ release from the ER stores into the cytosol (88), can be mimicked by blocking the sarco/endoplasmic reticulum Ca2+ ATPases (SERCA) pump that refills the ER Ca2+ reserve, using thapsigargin (89) (Figure 3). We observed that GIMAP5 deficiency does not affect Ca2+ release from the ER in primary rat T cells (30). Similarly, overexpression of rGIMAP5 did not influence the emptying of the ER Ca2+ stores (31). However, Ca2+ influx from the extracellular milieu which occurs mainly via the CRAC channels, was reduced in Gimap5-deficient rat T cells (28, 30).
Following sustained Ca2+ entry via the CRAC channels, the rising concentration of cytosolic Ca2+ ([Ca2+]c) activates the Ca2+ uniporter on the mitochondrial membrane. This induces a slow, membrane potential-driven uptake of Ca2+, which is released later via the Na+/Ca2+ exchanger (90) (Figure 3). This process ensures that Ca2+ entering via the CRAC channel does not cause a feedback inhibition of the CRAC channel activity (91, 92). Thus, mitochondria act like a slow, non-saturable, non-linear buffer for intracellular Ca2+, as they sequester [Ca2+]c during periods of rapid Ca2+ entry and sustain the [Ca2+]c level by releasing it slowly even after the cessation of Ca2+ influx (91) (Figure 3). We observed that the reduced Ca2+ influx in Gimap5-deficient T cells is associated with the inability of their mitochondria to sequester Ca2+ ([Ca2+]m) following capacitative entry (28). This reduced mitochondrial Ca2+ was also observed following stimulation of the ryanodine receptors that are present on ER membrane (28) and are implicated in T cell functions (93–95). As a corollary, overexpression of rGIMAP5 in HEK293 cells resulted in increased Ca2+ accumulation within the mitochondria (28). As a consequence, Ca2+ influx from extracellular milieu is reduced in cells expressing GIMAP5, probably due to early saturation of the mitochondrial Ca2+ store (28).
Even though the ER is the major intracellular Ca2+ store, and mitochondria uptake Ca2+ to prevent feedback inhibition of the CRAC channels, a significant amount of Ca2+ can be released from the Golgi complex, lysosomes, nucleus and secretory granules (96). As GIMAP5 does not directly interact with mitochondria but is present on lysosomes and certain endocytic vesicles (31–33, 40), we postulated that GIMAP5 might regulate the Ca2+ content of lysosomes (Figure 3). Ca2+ was released from lysosomes by Gly-Phe β-naphthylamide (GPN) whose hydrolysis by cathepsin C results in osmotic lysis of the acidic compartment (97). We observed that GPN-mediated Ca2+ release was increased in T cells from Gimap5lyp/lyp rats, and the Ca2+ influx from the extracellular milieu was also higher than that of T cells from control rats (31). These observations suggest that the intracellular partitioning of Ca2+ in Gimap5lyp/lyp rat T cells is different from that of normal rat T cells. These observations were reflected in stable transfectants of full-length GIMAP5 in HEK293T cells, which displayed reduced GPN-induced Ca2+ release. Regulation of Ca2+ homeostasis was dependent on the full-length GIMAP5 protein as C-terminal or N-terminal deletion mutants were unable to do so. Further analyses of the truncated GIMAP5 proteins indicated that the GIMAP5 is anchored to lysosomal membranes and certain vesicles through the C-terminal anchor while the N-terminal regions interacted with the microtubules (31, 33) (Figure 4). Thus, the presence of GIMAP5 appears to decrease Ca2+ release from lysosomes. For the first time, our results suggest that lysosomal Ca2+ homeostasis regulates the survival of T cells and that this process requires GIMAP5. We also observed that that the lysosomal Ca2+ content was altered by signaling through the TCR in murine and human T cells (31). Whether and how GIMAP5 integrates TCR signaling to regulate lysosomal Ca2+ in order to promote T cell survival and functions remain to be elucidated (Figure 3).
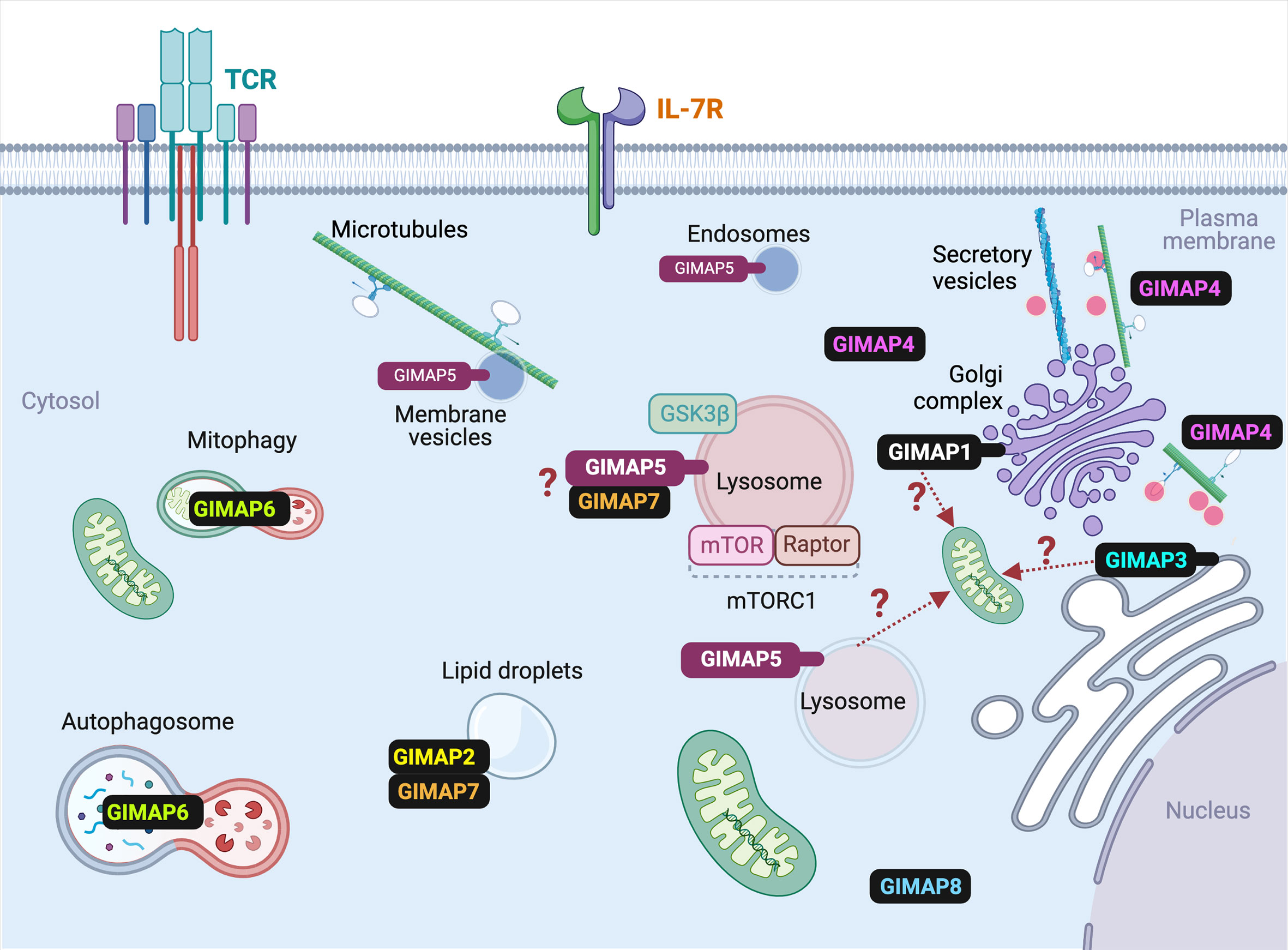
Figure 4 Subcellular localization of various GIMAP proteins in T cells. The known subcellular localization of mammalian GIMAP proteins in T lymphocytes is indicated. For the sake of simplicity, species-specific expression pattern of some GIMAP proteins are omitted. Details are given in the text and Table 1. Events that are predicted but require experimental confirmation are indicated by question marks. Ref- GIMAP5: (31–33); GIMAP1: (33); GIMAP2: (45); GIMAP3: (47); GIMAP4: (49); GIMAP6: (51); GIMAP7: (45); GIMAP8: (55).
Defective Ca2+ signaling in BB-DP rat T cells is evident within 30 minutes of TCR signaling induced by CD3 cross-linking (30). However, T cells from Gimap5Sph/Sph mice do not display any defect in Ca2+ influx induced by TCR signaling (40). Nonetheless, the TCR proximal signaling events showed similar impairments in Gimap5 mutant T cells in both the species (29, 34, 40, 41) (Figure 2). Rat T cells were activated by cross-linking CD3/CD28, and T cells from OT-II TCR transgenic control and Gimap5sph/sph mice were activated using anti-CD3/CD28 antibodies or dendritic cells pulsed with the cognate OVA-peptide. Gimap5 mutant rat and mouse T cells displayed reduced phosphorylation of LCK and the scaffold protein LAT following anti-CD3 stimulation (29). Notably, T cells from the Gimap5 mutant mice and rats exhibited constitutive phosphorylation of ATK at the basal level (34). This constitutive phosphorylation of AKT was inhibited by PI3K inhibitors. While aberrant activation of the PI3K/AKT pathway results in malignant growth in most cell types (98), it causes cell death in non-transformed T lymphocytes, leading to immunodeficiency (99–101). Spontaneous activating mutations of PI3K that results in constitutively active PI3K/AKT pathway in T cells has been observed in a new class of patients with primary immunodeficiency (99–101). T cells from these patients do not proliferate in response to mitogenic signals. These published reports and our observation indicate that aberrant activation of the PI3K/AKT pathway in the absence of TCR engagement may result in T cell death. The constitutive phosphorylation of AKT can also explain the reduced phosphorylation of STAT5 that we have observed following IL-7 stimulation (29), as pAKT can suppress the phosphorylation of STAT5 (102).
Spontaneous AKT activation observed in Gimap5 deficient T cells is reflected in the phosphorylation of downstream substrates including mTORC1 and FOXO1 (34). In fact, FOXO1 proteins are depleted progressively with age in Gimap5Sph/Sph mice (41). It is possible that the persistent phosphorylation of FOXO1 can lead to their degradation by proteasomes (103, 104). The Hoebe group recently showed that GIMAP5, which is localized on lysosomes and in certain vesicles, is required for+ the inactivation of GSK3β (40). GSK3β is a serine threonine kinase that is constitutively active in all cell types (105). Phosphorylation on Ser-9 and Ser-389 is required to inactivate GSK3β and promote T cell proliferation (106–108). Absence of GIMAP5 in T cells prevented CD3/CD28-induced inactivation of GSK3β as its inhibition by lithium chloride permitted the survival of T cells in Gimap5Sph/Sph mice (40). As GIMAP5 is present in certain vesicles, GIMAP5-mediated sequestration of GSK3β may play a role inhibiting its activity and promote cell proliferation (40).
GIMAP5 is essential to maintain the hematopoietic stem cell (HSC) niche as HSC numbers diminish with age in mice carrying mutant GIMAP5 (38), although this is not the case in rats. The absolute numbers of HSCs as defined by Lin-IL-7R-cKit+Sca+ (LSK) cells, was comparable between wildtype and Gimap5-/- mice (38, 41). However, Gimap5-deficient HSCs have intrinsic defects in long-term engrafting capacity (38). The exhaustion of HSC in Gimap5-/- mice may be the consequence of their inability to stay in the quiescent state. In line with these observations, Gimap5 expression is observed in murine HSC (38). Interaction of GIMAP5 with MCL1 and HSC70 appears to contribute to quiescence, however the underlying mechanisms are not yet known (38, 40). Simultaneous deletion of the two pro-apoptotic proteins, BAX and BAK, which rescues survival in the absence of MCL1, did not alter the survival of T cells in Gimap5Sph/Sph mice (40). It is possible that GSK3β, activated in the absence of GIMAP5 (40), might accelerate the degradation of MCL1 (109). These in vivo observations stand in contrast to over-expression studies in vitro where GIMAP5 was shown to interact with BCL-2 and BCL-xL (110). The requirement of GIMAP5 to maintain quiescence in HSCs is further supported by the observation that extramedullary hematopoiesis and reduced lifespan are still observed Gimap5-/-Rag2-/- mice (36).
Polymorphisms in human GIMAP5 gene locus has been shown to be associated with autoimmune T1D and lupus in independent studies (111–114). Recently, mutations in GIMAP5 have been described in human primary immunodeficiency. The Hoebe group (40) described one patient who had mild lymphopenia with impaired T cell proliferation in vitro that was rescued by GSK3β inhibition. Lenardo’s group (42) identified 4 different family clusters with mutations in GIMAP5 gene. Similar to what was reported in rats (31), GIMAP5 exists as 2 distinct isoforms in humans (42). GIMAP5 expression was observed in NK and T cells but not in B cells or monocytes. Nonetheless, frequency of T lymphocytes was reduced in all the patients studied while some of them also showed reduction in B lymphocytes and neutrophils. These patients exhibit splenomegaly and lymphadenopathy with abnormal liver and were susceptible to recurrent infections. T cells from the patients recapitulate the spontaneous mTORC1 activation observed in GIMAP5 mutant T cells from rats and mice (34). Similar to the in vitro findings (34), Gimap5Sph/Sph mice that received rapamycin, showed reduced activation of the mTORC1 pathway (42). Treatment of one of the patients with rapamycin for over 6 years diminished splenomegaly and lymphadenopathy, suggesting aberrant activation of the AKT/mTORC1 pathway in vivo in humans in the absence of functional GIMAP5. The abnormal activation of T cells in these patients is accompanied by replicative senescence in T cells as seen from reduced telomere length. These observations indicate that mutations in Gimap5 profoundly compromises the survival of T cells. In rats only one of the two isoforms was shown to regulate calcium homeostasis (31). While the abundantly expressed GIMAP5v2 regulates lysosomal calcium, GIMAP5v1 that is expressed at a lower level did not. It appears that the GIMAP5v1 homolog is functional in mice and in humans but not in rats.
GIMAP1 was initially identified as Imap38 that was induced in the spleen following Plasmodium chaboudi infection (115). However later studies carried out with anti-GIMAP1 antibody in infected tissues was unable to confirm these findings (116). GIMAP1, which is located in the Golgi complex (33), is expressed during all the stages of thymocyte development and in mature T, B cells and NK cells but minimally in macrophages (116). In humans, GIMAP1 is upregulated following Th1, but not Th2 differentiation in vitro (117). Constitutive and induced deletion of Gimap1 in T cells compromised their survival in the periphery (43, 118). Following T cell activation via the TCR, Gimap1-/- CD4+ T cells upregulate markers of activation such as CD25 but fail to proliferate and expand (43). In vitro, Gimap1-/- CD4+ T cells showed accelerated loss of mitochondrial potential with a concomitant reduction in oxygen consumption. GIMAP1 is also essential for the survival and functioning of mature B cells (119). Transgenic expression of BCL-2 did not rescue the loss of Gimap1-/- B cells. Deletion of GIMAP1 in germinal center B cells prevented the generation of efficient T-dependent antibody responses. GIMAP1 expression is increased in diffuse large B-cell lymphoma (DLBCL) due to hypomethylation of the GIMAP locus (120). Additional studies are needed to understand the role of GIMAP1 in cell survival and leukemia.
Functional GIMAP2 is present in humans but is absent in mice and rats (3, 4). The functions of GIMAP2 have not been characterized. The C-terminal double hydrophobic domain, which is unique to GIMAP2, localizes it with the lipid droplets marker BIODIPY following overexpression in JURKAT cells (45). Structural studies of GTP-bound and unbound GIMAP2 indicate that the nucleotide binding domains of GIMAPs are related to those found in dynamin, chloroplast proteins Toc and septin-GTPases (45, 46). GTP binding induces the formation of dimers of GIMAP2 and presumably, of GIMAP5. It has been proposed that the membrane bound GIMAPs such as GIMAP5 and GIMAP2 may form scaffolds in the GTP associated forms (121). GTP hydrolysis may be initiated by dimerization-dependent mechanisms involving GIMAP7 and GIMAP4 that do not have a membrane anchor, to promote GTP catalysis (44, 63). If this hypothesis is supported by experimental evidence, it is possible that GTP-bound GIMAP5 scaffolds may inhibit apoptosis, whereas heterodimerization with GIMAP4 and consequent GTP hydrolysis may antagonize the pro-survival functions of GIMAP5 (44, 49). These studies may also help in identifying other associated proteins and delineating their functional contributions.
GIMAP3 is a pseudogene in humans and in rats (4). Mouse Gimap3 is present in the ER (47) and influences the segregation of mitochondrial DNA in hematopoietic tissues (48). Mammalian mitochondrial DNA (mtDNA) is inherited from the mother. When all the cells in an organism carry the same haplotype of mtDNA it is referred to as homoplasmy (122, 123). In the case of mtDNA mutations, the cell carries two different haplotypes of mtDNA, referred to as heteroplasmy. Many of these mutations are associated with maternally inherited disorders in humans (122, 123). The two mitochondrial haplotypes are not inherited by daughter somatic cells in a stochastic manner (124). Additionally, mtDNA segregation in tissues and cell types are regulated by distinct mechanisms in an age-dependent manner (125). In inbred strains of Mus musculus domesticus mtDNA exists in two distinct haplotypes namely, BALB and NZB, which are transmitted in a random manner in the germline, but show segregation in the post-natal stage (126). For example, the BALB haplotype is enriched in hematopoietic cells while NZB is enriched in the liver and kidneys (127). While nuclear factors are implicated in tissue-specific segregation of mtDNA, the identity of proteins/RNA that could influence mtDNA segregation and the underlying mechanisms are not yet fully characterized (128, 129).
The group of Battersby showed that GIMAP3 is involved in regulating mtDNA segregation in hematopoietic cells (48). A mutation in the splice acceptor site of Gimap3 in Mus musculus castaneus (CAST/Ei), results in Gimap3 mRNA that codes for an additional 58 amino acids at the N-terminus (47). However, the longer N-terminus interferes with efficient translation, rendering CAST Gimap3 a functional null mutant (47). Absence of functional Gimap3 (CAST/Ei allele) resulted in equivalent representation of NZB mtDNA haplotype in hematopoietic tissues and in neutral tissues (where there is no haplotype selection), whereas mice expressing the wildtype Giamp3 allele showed enrichment of BALB mtDNA haplotype in hematopoietic tissues (48). Analysis of segregation of BALB mtDNA in Gimap5 heterozygous mice generated by the Weiler’s group (36) indicated that the abundance of GIMAP5 protein influenced that of GIMAP3 protein and was accompanied by differential segregation of BALB mtDNA in hematopoietic cells (47). In this context, it is interesting to note that Yano et al. (37), observed that T cell survival was significantly affected in Gimap5/Gimap3 double knock out mice indicating as yet unknown interactions between these GIMAP members in hematopoietic cells.
Gimap4 is expressed in developing T lymphocytes at the DN4 stage in response to pre-TCR signaling, is transiently downregulated in the DP stage and re-expressed in SP thymocytes, peripheral T cells as well as in B cells, NK cells and to a lesser extent in macrophages (49). The absence of GIMAP4 expression in RAG1-deficient thymocytes indicates that it is not expressed in DN 1-3 stages (49). Of the GIMAP proteins, interacting partners have been characterized in detail for GIMAP4 (49). GIMAP4 lacks a membrane anchor and is expressed in the cytosol (Figure 4). The C-terminal IQ domain, that is unique to the GIMAP4 (Figure 1) binds calmodulin. GIMAP4 harbours four PKC phosphorylation sites that are phosphorylated following T cell activation. Absence of GIMAP4 did not affect the generation or survival of T cells in the periphery. However, the frequency of apoptotic cells was increased in Gimap4-deficient T cells following exposure to gamma-irradiation, etoposide or dexamethasone, suggesting that GIMAP4 may be involved in promoting cell death following induction of apoptosis. In support of this notion, wildtype T cells undergoing apoptotic death display increased phosphorylation of GIMAP4 (49). The mitochondrial membrane potential and cytochrome c levels were comparable between the wildtype and Gimap4-deficient T cells indicating that GIMAP4 modulates apoptosis downstream of mitochondria. Furthermore, the apoptotic phenotype was inhibited by effector-caspase inhibitor in Gimap4-deficient T cells (49). Similar to other GIMAP proteins, GIMAP4 shows association with cytoskeletal elements and is implicated in membrane trafficking, movement of vesicles and cytokine secretion in T cells (31, 33, 47, 50, 130). Polymorphisms in GIMAP4 was shown to be associated with asthma and allergy, although the underlying mechanisms remain to be elucidated (111).
Human GIMAP6 has been shown to be involved in autophagy (51). Mass spectrometry analysis identified GABARAPL2, the mammalian homolog of yeast ATG8, as the binding partner for GIMAP6. GIMAP6 is localized to the punctate structures along with GABARAPL2 and MAP1LC3B, an autophagosome marker. Knockdown of GIMAP6 in JURKAT T cells resulted in reduced levels of GABARAPL2, suggesting that GIMAP6 may regulate the expression of the latter (51). Knockdown of GIMAP6 in JURKAT cells also increased their sensitivity to apoptosis inducing agents (52). Similar to human GIMAP6, mouse GIMAP6 is also implicated in autophagy. CD2-Cre mediated deletion of Gimap6 in mice caused in a significant reduction in T and B cell numbers in the periphery even though antigen-specific responses of Gimap6-/- T and B cells remained unaffected (53). The half-life of T cells lacking GIMAP6 was estimated to be around 4-5 days based on 4-hydroxytamoxifen mediated deletion in Gimap6fl/flERT2Cre mice. Recently, genetic loss of GIMAP6 protein was reported in humans, but with different degrees of clinical manifestations, presumably influenced by additional genetic and environmental factors, as one sibling exhibited lymphopenia while the other was asymptomatic even though both of them were homozygous for the mutant allele (54). Lymphocytes from these patients exhibited accelerated apoptosis, while maintaining normal activation, proliferation and cytokine secretion in vitro. Given the lymphocyte-specific expression of GIMAP proteins, it is possible that GIMAP6 may confer additional level of control over apoptotic and autophagic pathways in T cells.
In contrast to other GIMAP proteins, GIMAP8 possesses three GTPase domains and it is expressed in DN1, DN2 and mature thymocytes and T cells similar to GIMAP5 (23, 55). Gimap8-deficient mice show normal T cell development but show a reduction of recirculating B cells in the bone marrow (55). Nonetheless, the responses of B cells to a T-dependent antigen appear to be normal in these mice.
The importance of GIMAP proteins in cell survival and their regulation during the ontogeny of different HSC-derived cell populations suggest that their deregulation may contribute to oncogenesis. The GIMAP locus has been implicated in T acute lymphoblastic leukemia (T-ALL) as a target of NOTCH signaling. During T cell development in the thymus, transition through DN1 to DN4 stages is accompanied by a progressive increase in NOTCH1 signaling (131). The DN3 to DN4 transition is a critical checkpoint that selects for productive rearrangement of the TCRB locus. Subsequent pre-TCR signaling induces NOTCH1 activation, which is required to expand the pool of cells that rearrange the TCRA locus, become CD4+CD8+ DP thymocytes and undergo positive and negative selection processes to generate SP CD4+ or CD8+ naïve T cells. A majority of thymocytes die due to the lack of survival signals in the absence of productively rearranged TCR genes (131, 132). Aberrant oncogenic signaling arising from faulty TCR rearrangement is often implicated in the pathogenesis of T-ALL. Aberrant NOTCH1 activation plays a key oncogenic role in T-ALL (133). In addition to NOTCH1, T-ALL development is associated with oncogenic activation of transcription factors such as TAL1, LYL1, LMO2, TLX1, TLX3 etc., which interfere with progression through T cell developmental stages (134–141).
GIMAP5 was identified as one of the NOTCH1 targets that contributed to the survival of leukemic T cell lines (142, 143). Many GIMAP genes are expressed in T-ALL cell lines (144, 145). The expression of GIMAP5 occurs in many T cell leukemic cell lines and in anaplastic large cell lymphoma (ALCL) cell lines while the expression of GIMAP1, GIMAP2, GIMAP6 and GIMAP7 were down-regulated in ALCLs (44, 146). KMT2A/GIMAP8 rearrangements were detected in a patient with acute undifferentiated leukemia (147). Gimap1 is upregulated in a murine leukemic cell line during p53-induced apoptosis (148). Most of the Gimap genes are expressed in HSC and/or in mature T cells but not in DN thymocytes in zebrafish (145). The GIMAP super-enhancer region was shown to be activated by the oncogenic transcription factor TAL1 and is repressed by E- proteins. Knocking out the TAL1 binding domain in JURKAT cells abrogated the expression of GIMAP genes. Ectopic expression of human GIMAP5 and GIMAP7 under the Rag2 promoter did not induce leukemia but was capable of accelerating T-ALL induced by MYC in the zebrafish (145). As knockdown of GIMAP in T-ALL cells reduced their survival, it is possible that GIMAP proteins play an important role in maintaining the survival of transformed cells.
GTPases with similarities to mammalian GIMAPs appear to have evolved independently in different species (56, 58, 59, 149–152). Proteins containing the AIG domain are present in angiosperms (flowering plants) and are induced by infections and stress (57, 61). Gimap genes are upregulated in certain invertebrates and zebrafish in response to infections (58, 59, 149, 151). Future studies aimed at understanding the functions of the related genes in the context of stress and infections in different species will further our knowledge on GIMAP proteins.
The data available to date overwhelmingly indicate that the GIMAP proteins have important and non-redundant functions in the survival of lymphocytes and in the maintenance of quiescence (GIMAP5) in HSCs. While some GIMAP proteins may interact with proteins involved in classical pathways of cell survival such as BCL2 family members and caspases, they also appear to promote survival by distinct mechanisms. GIMAP proteins are distinctly related to septins and share structural similarities with dynamins, Toc and other TRAnslation FACtor (TRAFAC) proteins (153). Structural analyses of GIMAP2 and GIAMP7 suggest that their GTPase activity is regulated by dimerization (44). GIMAP1, GIMAP2, GIMAP3 and GIMAP5 are membrane-associated while the rest of the GIMAP proteins do not have membrane anchor domains (Figures 1, 4). Overexpressed GIMAP2 increases the formation of lipid droplets in JURKAT cells and can dimerize with GIMAP7 (44–46, 121). It is possible that the homo- and hetero- oligomers can form membrane scaffolds that recruit additional interacting proteins (121). Many of these interactions can be presumed to be dynamic and temporal, making it difficult to identify the interacting partners by classical methods (53). GIMAP5 and GIMAP4 have been observed to be associated with microtubules and actin (31, 50), implicating them in the transport of cellular cargo. Given the implication of GIMAP proteins in the survival of T lymphocytes, it is not surprising that GIMAPs have been associated with different types of leukemias. Unravelling the functions of GIMAP proteins will predicate a better understanding of their role in T cell survival and their contribution to the development of leukemias.
SR and M-AL designed the content and wrote the manuscript. SI, MC, and MN contributed to the discussions and specific sub-titles in the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by SR’s NSERC Discovery grant RGPIN-2016-04349, MN’s FRQS post-doctoral fellowship #288811.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Illustrations were generated using the platform BioRender.com
BB-DP, Biobreeding diabetes prone; CRAC, Ca2+ release activated Ca2+ channel; GIMAP, GTPase of the immune associated nucleotide binding protein; GPN, Gly-Phe β-naphthylamide; HSC, hematopoietic stem cell; MHC:p, MHC peptide complex; RTE, recent thymic emigrants; T1D, Type 1 diabetes.
1. Ramanathan S, Poussier P. BB Rat Lyp Mutation and Type 1 Diabetes. Immunol Rev (2001) 184:161–71. doi: 10.1034/j.1600-065x.2001.1840115.x
2. Hornum L, Romer J, Markholst H. The Diabetes-Prone BB Rat Carries a Frameshift Mutation in Ian4, a Positional Candidate of Iddm1. Diabetes (2002) 51(6):1972–9. doi: 10.2337/diabetes.51.6.1972
3. MacMurray AJ, Moralejo DH, Kwitek AE, Rutledge EA, Van Yserloo B, Gohlke P, et al. Lymphopenia in the BB Rat Model of Type 1 Diabetes is Due to a Mutation in a Novel Immune-Associated Nucleotide (Ian)-Related Gene. Genome Res (2002) 12(7):1029–39. doi: 10.1101/gr.412702
4. Krucken J, Schroetel RM, Muller IU, Saidani N, Marinovski P, Benten WP, et al. Comparative Analysis of the Human Gimap Gene Cluster Encoding a Novel GTPase Family. Gene (2004) 341:291–304. doi: 10.1016/j.gene.2004.07.005
5. Colle E, Guttmann RD, Seemayer T. Spontaneous Diabetes Mellitus Syndrome in the Rat. I. Association With the Major Histocompatibility Complex. J Exp Med (1981) 154(4):1237–42. doi: 10.1084/jem.154.4.1237
6. Jackson R, Rassi N, Crump T, Haynes B, Eisenbarth GS. The BB Diabetic Rat. Profound T-Cell Lymphocytopenia. Diabetes (1981) 30(10):887–9. doi: 10.2337/diabetes.30.10.887
7. Marliss EB, Nakhooda AF, Poussier P, Sima AA. The Diabetic Syndrome of the ‘BB’ Wistar Rat: Possible Relevance to Type 1 (Insulin-Dependent) Diabetes in Man. Diabetologia (1982) 22(4):225–32. doi: 10.1007/BF00281296
8. Elder ME, Maclaren NK. Identification of Profound Peripheral T Lymphocyte Immunodeficiencies in the Spontaneously Diabetic BB Rat. J Immunol (1983) 130(4):1723–31.
9. Jacob HJ, Pettersson A, Wilson D, Mao Y, Lernmark A, Lander ES. Genetic Dissection of Autoimmune Type I Diabetes in the BB Rat. Nat Genet (1992) 2(1):56–60. doi: 10.1038/ng0992-56
10. Poussier P, Nakhooda AF, Falk JA, Lee C, Marliss EB. Lymphopenia and Abnormal Lymphocyte Subsets in the “BB” Rat: Relationship to the Diabetic Syndrome. Endocrinology (1982) 110(5):1825–7. doi: 10.1210/endo-110-5-1825
11. Woda BA, Padden C. BioBreeding/Worcester (BB/Wor) Rats are Deficient in the Generation of Functional Cytotoxic T Cells. J Immunol (1987) 139(5):1514–7.
12. Prud’homme GJ, Lapchak PH, Parfrey NA, Colle E, Guttmann RD. Autoimmunity-Prone BB Rats Lack Functional Cytotoxic T Cells. Cell Immunol (1988) 114(1):198–208. doi: 10.1016/0008-8749(88)90266-3
13. Colle E, Fuks A, Poussier P, Edouard P, Guttmann RD. Polygenic Nature of Spontaneous Diabetes in the Rat. Permissive MHC Haplotype and Presence of the Lymphopenic Trait of the BB Rat are Not Sufficient to Produce Susceptibility. Diabetes (1992) 41(12):1617–23. doi: 10.2337/diabetes.41.12.1617
14. Sarkar P, Crisa L, McKeever U, Bortell R, Handler E, Mordes JP, et al. Loss of RT6 Message and Most Circulating T Cells After Thymectomy of Diabetes Prone BB Rats. Autoimmunity (1994) 18(1):15–22. doi: 10.3109/08916939409014675
15. Lau A, Ramanathan S, Poussier P. Excessive Production of Nitric Oxide by Macrophages From DP-BB Rats is Secondary to the T-Lymphopenic State of These Animals. Diabetes (1998) 47(2):197–205. doi: 10.2337/diabetes.47.2.197
16. Lee KU. Nitric Oxide Produced by Macrophages Mediates Suppression of ConA-Induced Proliferative Responses of Splenic Leukocytes in the Diabetes-Prone BB Rat. Diabetes (1994) 43(10):1218–20. doi: 10.2337/diabetes.43.10.1218
17. Ramanathan S, Marandi L, Poussier P. Evidence for the Extrathymic Origin of Intestinal TCRgammadelta(+) T Cells in Normal Rats and for an Impairment of This Differentiation Pathway in BB Rats. J Immunol (2002) 168(5):2182–7. doi: 10.4049/jimmunol.168.5.2182
18. Poussier P, Ning T, Murphy T, Dabrowski D, Ramanathan S. Impaired Post-Thymic Development of Regulatory CD4+25+ T Cells Contributes to Diabetes Pathogenesis in BB Rats. J Immunol (2005) 174(7):4081–9. doi: 10.4049/jimmunol.174.7.4081
19. Whalen BJ, Greiner DL, Mordes JP, Rossini AA. Adoptive Transfer of Autoimmune Diabetes Mellitus to Athymic Rats: Synergy of CD4+ and CD8+ T Cells and Prevention by RT6+ T Cells. J Autoimmun (1994) 7(6):819–31. doi: 10.1006/jaut.1994.1065
20. Edouard P, Hiserodt JC, Plamondon C, Poussier P. CD8+ T-cells are Required for Adoptive Transfer of the BB Rat Diabetic Syndrome. Diabetes (1993) 42(3):390–7. doi: 10.2337/diabetes.42.3.390
21. Zadeh HH, Greiner DL, Wu DY, Tausche F, Goldschneider I. Abnormalities in the Export and Fate of Recent Thymic Emigrants in Diabetes-Prone BB/W Rats. Autoimmunity (1996) 24(1):35–46. doi: 10.3109/08916939608995355
22. Groen H, Klatter FA, Brons NH, Mesander G, Nieuwenhuis P, Kampinga J. Abnormal Thymocyte Subset Distribution and Differential Reduction of CD4+ and CD8+ T Cell Subsets During Peripheral Maturation in Diabetes-Prone BioBreeding Rats. J Immunol (1996) 156(3):1269–75.
23. Dion C, Carter C, Hepburn L, Coadwell WJ, Morgan G, Graham M, et al. Expression of the Ian Family of Putative GTPases During T Cell Development and Description of an Ian With Three Sets of GTP/GDP-Binding Motifs. Int Immunol (2005) 17(9):1257–68. doi: 10.1093/intimm/dxh302
24. Plamondon C, Kottis V, Brideau C, Metroz-Dayer MD, Poussier P. Abnormal Thymocyte Maturation in Spontaneously Diabetic BB Rats Involves the Deletion of CD4-8+ Cells. J Immunol (1990) 144(3):923–8.
25. Hernandez-Hoyos G, Joseph S, Miller NG, Butcher GW. The Lymphopenia Mutation of the BB Rat Causes Inappropriate Apoptosis of Mature Thymocytes. Eur J Immunol (1999) 29(6):1832–41. doi: 10.1002/(SICI)1521-4141(199906)29:06<1832::AID-IMMU1832>3.0.CO;2-F
26. Lang JA, Kominski D, Bellgrau D, Scheinman RI. Partial Activation Precedes Apoptotic Death in T Cells Harboring an IAN Gene Mutation. Eur J Immunol (2004) 34(9):2396–406. doi: 10.1002/eji.200324751
27. Ramanathan S, Norwich K, Poussier P. Antigen Activation Rescues Recent Thymic Emigrants From Programmed Cell Death in the BB Rat. J Immunol (1998) 160(12):5757–64.
28. Chen XL, Serrano D, Mayhue M, Wieden HJ, Stankova J, Boulay G, et al. GTPase of the Immune-Associated Nucleotide-Binding Protein 5 (GIMAP5) Regulates Calcium Influx in T-lymphocytes by Promoting Mitochondrial Calcium Accumulation. Biochem J (2013) 449(2):353–64. doi: 10.1042/BJ20120516
29. Chen XL, Serrano D, Ghobadi F, Mayhue M, Hoebe K, Ilangumaran S, et al. TCR and IL-7 Signaling Are Altered in the Absence of Functional Gtpase of the Immune Associated Nucleotide Binding Protein 5 (GIMAP5). PloS One (2016) 11(3):e0151837. doi: 10.1371/journal.pone.0151837
30. Ilangumaran S, Forand-Boulerice M, Bousquet SM, Savard A, Rocheleau P, Chen XL, et al. Loss of GIMAP5 (GTPase of Immunity-Associated Nucleotide Binding Protein 5) Impairs Calcium Signaling in Rat T Lymphocytes. Mol Immunol (2009) 46(6):1256–9. doi: 10.1016/j.molimm.2008.09.031
31. Serrano D, Ghobadi F, Boulay G, Ilangumaran S, Lavoie C, Ramanathan S. GTPase of the Immune-Associated Nucleotide Protein 5 Regulates the Lysosomal Calcium Compartment in T Lymphocytes. Front Immunol (2017) 8:94. doi: 10.3389/fimmu.2017.00094
32. Keita M, Leblanc C, Andrews D, Ramanathan S. GIMAP5 Regulates Mitochondrial Integrity From a Distinct Subcellular Compartment. Biochem Biophys Res Commun (2007) 361(2):481–6. doi: 10.1016/j.bbrc.2007.07.048
33. Wong VW, Saunders AE, Hutchings A, Pascall JC, Carter C, Bright NA, et al. The Autoimmunity-Related GIMAP5 Gtpase is a Lysosome-Associated Protein. Self Nonself (2010) 1(3):259–68. doi: 10.4161/self.1.3.12819
34. Chen XL, Serrano D, Mayhue M, Hoebe K, Ilangumaran S, Ramanathan S. GIMAP5 Deficiency is Associated With Increased AKT Activity in T Lymphocytes. PloS One (2015) 10(10):e0139019. doi: 10.1371/journal.pone.0139019
35. Barnes MJ, Aksoylar H, Krebs P, Bourdeau T, Arnold CN, Xia Y, et al. Loss of T Cell and B Cell Quiescence Precedes the Onset of Microbial Flora-Dependent Wasting Disease and Intestinal Inflammation in Gimap5-Deficient Mice. J Immunol (2010) 184(7):3743–54. doi: 10.4049/jimmunol.0903164
36. Schulteis RD, Chu H, Dai X, Chen Y, Edwards B, Haribhai D, et al. Impaired Survival of Peripheral T Cells, Disrupted NK/NKT Cell Development, and Liver Failure in Mice Lacking Gimap5. Blood (2008) 112(13):4905–14. doi: 10.1182/blood-2008-03-146555
37. Yano K, Carter C, Yoshida N, Abe T, Yamada A, Nitta T, et al. Gimap3 and Gimap5 Cooperate to Maintain T-Cell Numbers in the Mouse. Eur J Immunol (2014) 44(2):561–72. doi: 10.1002/eji.201343750
38. Chen Y, Yu M, Dai X, Zogg M, Wen R, Weiler H, et al. Critical Role for Gimap5 in the Survival of Mouse Hematopoietic Stem and Progenitor Cells. J Exp Med (2011) 208(5):923–35. doi: 10.1084/jem.20101192
39. Endale M, Aksoylar HI, Hoebe K. Central Role of Gimap5 in Maintaining Peripheral Tolerance and T Cell Homeostasis in the Gut. Mediators Inflamm (2015) 2015:436017. doi: 10.1155/2015/436017
40. Patterson AR, Endale M, Lampe K, Aksoylar HI, Flagg A, Woodgett JR, et al. Gimap5-Dependent Inactivation of GSK3beta is Required for CD4(+) T Cell Homeostasis and Prevention of Immune Pathology. Nat Commun (2018) 9(1):430. doi: 10.1038/s41467-018-02897-7
41. Aksoylar HI, Lampe K, Barnes MJ, Plas DR, Hoebe K. Loss of Immunological Tolerance in Gimap5-Deficient Mice is Associated With Loss of Foxo in CD4+ T Cells. J Immunol (2012) 188(1):146–54. doi: 10.4049/jimmunol.1101206
42. Park AY, Leney-Greene M, Lynberg M, Xu X, Zheng L, Zhang Y, et al. Human Immunodeficiency Reveals GIMAP5 as Lymphocyte-Specific Regulator of Senescence. bioRxiv (2021) 2021.02.22.432146. doi: 10.1101/2021.02.22.432146
43. Datta P, Webb LM, Avdo I, Pascall J, Butcher GW. Survival of Mature T Cells in the Periphery is Intrinsically Dependent on GIMAP1 in Mice. Eur J Immunol (2017) 47(1):84–93. doi: 10.1002/eji.201646599
44. Schwefel D, Arasu BS, Marino SF, Lamprecht B, Kochert K, Rosenbaum E, et al. Structural Insights Into the Mechanism of GTPase Activation in the GIMAP Family. Structure (2013) 21(4):550–9. doi: 10.1016/j.str.2013.01.014
45. Schwefel D, Frohlich C, Eichhorst J, Wiesner B, Behlke J, Aravind L, et al. Structural Basis of Oligomerization in Septin-Like GTPase of Immunity-Associated Protein 2 (GIMAP2). Proc Natl Acad Sci U S A (2010) 107(47):20299–304. doi: 10.1073/pnas.1010322107
46. Schwefel D, Frohlich C, Daumke O. Purification, Crystallization and Preliminary X-ray Analysis of Human GIMAP2. Acta Crystallogr Sect F Struct Biol Cryst Commun (2010) 66(Pt 6):725–9. doi: 10.1107/S174430911001537X
47. Jokinen R, Lahtinen T, Marttinen P, Myohanen M, Ruotsalainen P, Yeung N, et al. Quantitative Changes in Gimap3 and Gimap5 Expression Modify Mitochondrial DNA Segregation in Mice. Genetics (2015) 200(1):221–35. doi: 10.1534/genetics.115.175596
48. Jokinen R, Marttinen P, Sandell HK, Manninen T, Teerenhovi H, Wai T, et al. Gimap3 Regulates Tissue-Specific Mitochondrial DNA Segregation. PloS Genet (2010) 6(10):e1001161. doi: 10.1371/journal.pgen.1001161
49. Schnell S, Demolliere C, van den Berk P, Jacobs H. Gimap4 Accelerates T-cell Death. Blood (2006) 108(2):591–9. doi: 10.1182/blood-2005-11-4616
50. Heinonen MT, Kanduri K, Lahdesmaki HJ, Lahesmaa R, Henttinen TA. Tubulin- and Actin-Associating GIMAP4 is Required for IFN-gamma Secretion During Th Cell Differentiation. Immunol Cell Biol (2015) 93(2):158–66. doi: 10.1038/icb.2014.86
51. Pascall JC, Rotondo S, Mukadam AS, Oxley D, Webster J, Walker SA, et al. The Immune System GTPase GIMAP6 Interacts With the Atg8 Homologue GABARAPL2 and is Recruited to Autophagosomes. PloS One (2013) 8(10):e77782. doi: 10.1371/journal.pone.0077782
52. Ho CH, Tsai SF. Functional and Biochemical Characterization of a T Cell-Associated Anti-Apoptotic Protein, GIMAP6. J Biol Chem (2017) 292(22):9305–19. doi: 10.1074/jbc.M116.768689
53. Pascall JC, Webb LMC, Eskelinen EL, Innocentin S, Attaf-Bouabdallah N, Butcher GW. GIMAP6 is Required for T Cell Maintenance and Efficient Autophagy in Mice. PloS One (2018) 13(5):e0196504. doi: 10.1371/journal.pone.0196504
54. Shadur B, Asherie N, Kfir-Erenfeld S, Dubnikov T, NaserEddin A, Schejter YD, et al. A Human Case of GIMAP6 Deficiency: A Novel Primary Immune Deficiency. Eur J Hum Genet (2020) 29(4):657–62. doi: 10.1038/s41431-020-00773-x
55. Webb LM, Pascall JC, Hepburn L, Carter C, Turner M, Butcher GW. Generation and Characterisation of Mice Deficient in the Multi-GTPase Domain Containing Protein, GIMAP8. PloS One (2014) 9(10):e110294. doi: 10.1371/journal.pone.0110294
56. Weinstein DJ, Allen SE, Lau MCY, Erasmus M, Asalone KC, Walters-Conte K, et al. The Genome of a Subterrestrial Nematode Reveals Adaptations to Heat. Nat Commun (2019) 10(1):5268. doi: 10.1038/s41467-019-13245-8
57. Wang Z, Li X. IAN/GIMAPs are Conserved and Novel Regulators in Vertebrates and Angiosperm Plants. Plant Signal Behav (2009) 4(3):165–7. doi: 10.4161/psb.4.3.7722
58. Weiss Y, Foret S, Hayward DC, Ainsworth T, King R, Ball EE, et al. The Acute Transcriptional Response of the Coral Acropora Millepora to Immune Challenge: Expression of GiMAP/IAN Genes Links the Innate Immune Responses of Corals With Those of Mammals and Plants. BMC Genomics (2013) 14:400. doi: 10.1186/1471-2164-14-400
59. Lu L, Loker ES, Zhang SM, Buddenborg SK, Bu L. Genome-Wide Discovery, and Computational and Transcriptional Characterization of an AIG Gene Family in the Freshwater Snail Biomphalaria Glabrata, a Vector for Schistosoma Mansoni. BMC Genomics (2020) 21(1):190. doi: 10.1186/s12864-020-6534-z
60. Liu C, Wang T, Zhang W, Li X. Computational Identification and Analysis of Immune-Associated Nucleotide Gene Family in Arabidopsis Thaliana. J Plant Physiol (2008) 165(7):777–87. doi: 10.1016/j.jplph.2007.06.002
61. Reuber TL, Ausubel FM. Isolation of Arabidopsis Genes That Differentiate Between Resistance Responses Mediated by the RPS2 and RPM1 Disease Resistance Genes. Plant Cell (1996) 8(2):241–9. doi: 10.1105/tpc.8.2.241
62. Zenz T, Roessner A, Thomas A, Frohling S, Dohner H, Calabretta B, et al. hIan5: The Human Ortholog to the Rat Ian4/Iddm1/lyp is a New Member of the Ian Family That is Overexpressed in B-Cell Lymphoid Malignancies. Genes Immun (2004) 5(2):109–16. doi: 10.1038/sj.gene.6364044
63. Cambot M, Aresta S, Kahn-Perles B, de Gunzburg J, Romeo PH. Human Immune Associated Nucleotide 1: A Member of a New Guanosine Triphosphatase Family Expressed in Resting T and B Cells. Blood (2002) 99(9):3293–301. doi: 10.1182/blood.V99.9.3293
64. Filen S, Lahesmaa R. Gimap Proteins in T-Lymphocytes. J Signal Transduct (2010) 2010:268589. doi: 10.1155/2010/268589
65. Yui MA, Rothenberg EV. Developmental Gene Networks: A Triathlon on the Course to T Cell Identity. Nat Rev Immunol (2014) 14(8):529–45. doi: 10.1038/nri3702
66. Ashton-Rickardt PG, Van Kaer L, Schumacher TN, Ploegh HL, Tonegawa S. Peptide Contributes to the Specificity of Positive Selection of CD8+ T Cells in the Thymus. Cell (1993) 73(5):1041–9. doi: 10.1016/0092-8674(93)90281-t
67. Klein L, Hinterberger M, Wirnsberger G, Kyewski B. Antigen Presentation in the Thymus for Positive Selection and Central Tolerance Induction. Nat Rev Immunol (2009) 9(12):833–44. doi: 10.1038/nri2669
68. Kirberg J, Berns A, von Boehmer H. Peripheral T Cell Survival Requires Continual Ligation of the T Cell Receptor to Major Histocompatibility Complex-Encoded Molecules. J Exp Med (1997) 186(8):1269–75. doi: 10.1084/jem.186.8.1269
69. Modigliani Y, Coutinho G, Burlen-Defranoux O, Coutinho A, Bandeira A. Differential Contribution of Thymic Outputs and Peripheral Expansion in the Development of Peripheral T Cell Pools. Eur J Immunol (1994) 24(5):1223–7. doi: 10.1002/eji.1830240533
70. Cousins L, Graham M, Tooze R, Carter C, Miller JR, Powrie FM, et al. Eosinophilic Bowel Disease Controlled by the BB Rat-Derived Lymphopenia/Gimap5 Gene. Gastroenterology (2006) 131(5):1475–85. doi: 10.1053/j.gastro.2006.09.023
71. Fischer HJ, Witte AK, Walter L, Grone HJ, van den Brandt J, Reichardt HM. Distinct Roles of T-Cell Lymphopenia and the Microbial Flora for Gastrointestinal and CNS Autoimmunity. FASEB J (2016) 30(5):1724–32. doi: 10.1096/fj.15-277384
72. Sandal T, Ahlgren R, Lillehaug J, Doskeland SO. Establishment of Okadaic Acid Resistant Cell Clones Using a cDNA Expression Library. Cell Death Differ (2001) 8(7):754–66. doi: 10.1038/sj.cdd.4400873
73. Sandal T, Aumo L, Hedin L, Gjertsen BT, Doskeland SO. Irod/Ian5: An Inhibitor of Gamma-Radiation- and Okadaic Acid-Induced Apoptosis. Mol Biol Cell (2003) 14(8):3292–304. doi: 10.1091/mbc.E02-10-0700
74. Takada K, Jameson SC. Self-Class I MHC Molecules Support Survival of Naive CD8 T Cells, But Depress Their Functional Sensitivity Through Regulation of CD8 Expression Levels. J Exp Med (2009) 206(10):2253–69. doi: 10.1084/jem.20082553
75. MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The Liver Kinase B1 is a Central Regulator of T Cell Development, Activation, and Metabolism. J Immunol (2011) 187(8):4187–98. doi: 10.4049/jimmunol.1100367
76. Blagih J, Krawczyk CM, Jones RG. LKB1 and AMPK: Central Regulators of Lymphocyte Metabolism and Function. Immunol Rev (2012) 249(1):59–71. doi: 10.1111/j.1600-065X.2012.01157.x
77. Wu Q, Liu Y, Chen C, Ikenoue T, Qiao Y, Li CS, et al. The Tuberous Sclerosis Complex-Mammalian Target of Rapamycin Pathway Maintains the Quiescence and Survival of Naive T Cells. J Immunol (2011) 187(3):1106–12. doi: 10.4049/jimmunol.1003968
78. Yang K, Neale G, Green DR, He W, Chi H. The Tumor Suppressor Tsc1 Enforces Quiescence of Naive T Cells to Promote Immune Homeostasis and Function. Nat Immunol (2011) 12(9):888–97. doi: 10.1038/ni.2068
79. Malissen B, Gregoire C, Malissen M, Roncagalli R. Integrative Biology of T Cell Activation. Nat Immunol (2014) 15(9):790–7. doi: 10.1038/ni.2959
80. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of Immune Responses by mTOR. Annu Rev Immunol (2012) 30:39–68. doi: 10.1146/annurev-immunol-020711-075024
81. Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr., et al. STIM is a Ca2+ Sensor Essential for Ca2+-Store-Depletion-Triggered Ca2+ Influx. Curr Biol (2005) 15(13):1235–41. doi: 10.1016/j.cub.2005.05.055
82. Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. STIM1, an Essential and Conserved Component of Store-Operated Ca2+ Channel Function. J Cell Biol (2005) 169(3):435–45. doi: 10.1083/jcb.200502019
83. Berridge MJ. Inositol Trisphosphate and Calcium Signalling. Nature (1993) 361(6410):315–25. doi: 10.1038/361315a0
84. Hogan PG, Lewis RS, Rao A. Molecular Basis of Calcium Signaling in Lymphocytes: STIM and ORAI. Annu Rev Immunol (2010) 28:491–533. doi: 10.1146/annurev.immunol.021908.132550
85. Feske S. Calcium Signalling in Lymphocyte Activation and Disease. Nat Rev Immunol (2007) 7(9):690–702. doi: 10.1038/nri2152
86. Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, et al. A Mutation in Orai1 Causes Immune Deficiency by Abrogating CRAC Channel Function. Nature (2006) 441(7090):179–85. doi: 10.1038/nature04702
87. Valitutti S, Dessing M, Aktories K, Gallati H, Lanzavecchia A. Sustained Signaling Leading to T Cell Activation Results From Prolonged T Cell Receptor Occupancy. Role of T Cell Actin Cytoskeleton. J Exp Med (1995) 181(2):577–84. doi: 10.1084/jem.181.2.577
88. Lewis RS. Calcium Signaling Mechanisms in T Lymphocytes. Annu Rev Immunol (2001) 19:497–521. doi: 10.1146/annurev.immunol.19.1.497
89. Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a Tumor Promoter, Discharges Intracellular Ca2+ Stores by Specific Inhibition of the Endoplasmic Reticulum Ca2(+)-Atpase. Proc Natl Acad Sci U S A (1990) 87(7):2466–70. doi: 10.1073/pnas.87.7.2466
90. Pfeiffer DR, Gunter TE, Eliseev R, Broekemeier KM, Gunter KK. Release of Ca2+ From Mitochondria Via the Saturable Mechanisms and the Permeability Transition. IUBMB Life (2001) 52(3-5):205–12. doi: 10.1080/15216540152846019
91. Hoth M, Fanger CM, Lewis RS. Mitochondrial Regulation of Store-Operated Calcium Signaling in T Lymphocytes. J Cell Biol (1997) 137(3):633–48. doi: 10.1083/jcb.137.3.633
92. Hajnoczky G, Csordas G, Yi M. Old Players in a New Role: Mitochondria-Associated Membranes, VDAC, and Ryanodine Receptors as Contributors to Calcium Signal Propagation From Endoplasmic Reticulum to the Mitochondria. Cell Calcium (2002) 32(5-6):363–77. doi: 10.1016/s0143416002001872
93. Conrad DM, Hanniman EA, Watson CL, Mader JS, Hoskin DW. Ryanodine Receptor Signaling is Required for Anti-CD3-Induced T Cell Proliferation, Interleukin-2 Synthesis, and Interleukin-2 Receptor Signaling. J Cell Biochem (2004) 92(2):387–99. doi: 10.1002/jcb.20064
94. Hakamata Y, Nishimura S, Nakai J, Nakashima Y, Kita T, Imoto K. Involvement of the Brain Type of Ryanodine Receptor in T-Cell Proliferation. FEBS Lett (1994) 352(2):206–10. doi: 10.1016/0014-5793(94)00955-4
95. Hosoi E, Nishizaki C, Gallagher KL, Wyre HW, Matsuo Y, Sei Y. Expression of the Ryanodine Receptor Isoforms in Immune Cells. J Immunol (2001) 167(9):4887–94. doi: 10.4049/jimmunol.167.9.4887
96. Rizzuto R, Pozzan T. Microdomains of Intracellular Ca2+: Molecular Determinants and Functional Consequences. Physiol Rev (2006) 86(1):369–408. doi: 10.1152/physrev.00004.2005
97. Haller T, Volkl H, Deetjen P, Dietl P. The Lysosomal Ca2+ Pool in MDCK Cells can be Released by Ins(1,4,5)P3-Dependent Hormones or Thapsigargin But Does Not Activate Store-Operated Ca2+ Entry. Biochem J (1996) 319( Pt 3):909–12. doi: 10.1042/bj3190909
98. Curran E, Smith SM. Phosphoinositide 3-Kinase Inhibitors in Lymphoma. Curr Opin Oncol (2014) 26(5):469–75. doi: 10.1097/CCO.0000000000000113
99. Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J, et al. PIK3R1 Mutations Cause Syndromic Insulin Resistance With Lipoatrophy. Am J Hum Genet (2013) 93(1):141–9. doi: 10.1016/j.ajhg.2013.05.019
100. Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A Human Immunodeficiency Caused by Mutations in the PIK3R1 Gene. J Clin Invest (2014) 124(9):3923–8. doi: 10.1172/JCI75746
101. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-Activating Germline Mutations in the Gene Encoding the PI(3)K Catalytic Subunit p110delta Result in T Cell Senescence and Human Immunodeficiency. Nat Immunol (2014) 15(1):88–97. doi: 10.1038/ni.2771
102. Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, et al. Differential Effects of STAT5 and PI3K/AKT Signaling on Effector and Memory CD8 T-Cell Survival. Proc Natl Acad Sci U S A (2010) 107(38):16601–6. doi: 10.1073/pnas.1003457107
103. Aoki M, Jiang H, Vogt PK. Proteasomal Degradation of the FoxO1 Transcriptional Regulator in Cells Transformed by the P3k and Akt Oncoproteins. Proc Natl Acad Sci U S A (2004) 101(37):13613–7. doi: 10.1073/pnas.0405454101
104. Huang H, Tindall DJ. Regulation of FOXO Protein Stability Via Ubiquitination and Proteasome Degradation. Biochim Biophys Acta (2011) 1813(11):1961–4. doi: 10.1016/j.bbamcr.2011.01.007
105. Woodgett JR. Molecular Cloning and Expression of Glycogen Synthase Kinase-3/factor A. EMBO J (1990) 9(8):2431–8. doi: 10.1002/j.1460-2075.1990.tb07419.x
106. Stambolic V, Woodgett JR. Mitogen Inactivation of Glycogen Synthase Kinase-3 Beta in Intact Cells Via Serine 9 Phosphorylation. Biochem J (1994) 303( Pt 3):701–4. doi: 10.1042/bj3030701
107. Thornton TM, Pedraza-Alva G, Deng B, Wood CD, Aronshtam A, Clements JL, et al. Phosphorylation by P38 MAPK as an Alternative Pathway for GSK3beta Inactivation. Science (2008) 320(5876):667–70. doi: 10.1126/science.1156037
108. Thornton TM, Delgado P, Chen L, Salas B, Krementsov D, Fernandez M, et al. Inactivation of Nuclear GSK3beta by Ser(389) Phosphorylation Promotes Lymphocyte Fitness During DNA Double-Strand Break Response. Nat Commun (2016) 7:10553. doi: 10.1038/ncomms10553
109. Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen Synthase Kinase-3 Regulates Mitochondrial Outer Membrane Permeabilization and Apoptosis by Destabilization of MCL-1. Mol Cell (2006) 21(6):749–60. doi: 10.1016/j.molcel.2006.02.009
110. Nitta T, Nasreen M, Seike T, Goji A, Ohigashi I, Miyazaki T, et al. IAN Family Critically Regulates Survival and Development of T Lymphocytes. PloS Biol (2006) 4(4):e103. doi: 10.1371/journal.pbio.0040103
111. Heinonen MT, Laine AP, Soderhall C, Gruzieva O, Rautio S, Melen E, et al. GIMAP GTPase Family Genes: Potential Modifiers in Autoimmune Diabetes, Asthma, and Allergy. J Immunol (2015) 194(12):5885–94. doi: 10.4049/jimmunol.1500016
112. Hellquist A, Zucchelli M, Kivinen K, Saarialho-Kere U, Koskenmies S, Widen E, et al. The Human GIMAP5 Gene has a Common Polyadenylation Polymorphism Increasing Risk to Systemic Lupus Erythematosus. J Med Genet (2007) 44(5):314–21. doi: 10.1136/jmg.2006.046185
113. Lim MK, Sheen DH, Kim SA, Won SK, Lee SS, Chae SC, et al. IAN5 Polymorphisms are Associated With Systemic Lupus Erythematosus. Lupus (2009) 18(12):1045–52. doi: 10.1177/0961203309106830
114. Shin JH, Janer M, McNeney B, Blay S, Deutsch K, Sanjeevi CB, et al. IA-2 Autoantibodies in Incident Type I Diabetes Patients are Associated With a Polyadenylation Signal Polymorphism in GIMAP5. Genes Immun (2007) 8(6):503–12. doi: 10.1038/sj.gene.6364413
115. Krucken J, Schmitt-Wrede HP, Markmann-Mulisch U, Wunderlich F. Novel Gene Expressed in Spleen Cells Mediating Acquired Testosterone-Resistant Immunity to Plasmodium Chabaudi Malaria. Biochem Biophys Res Commun (1997) 230(1):167–70. doi: 10.1006/bbrc.1996.5876
116. Saunders A, Lamb T, Pascall J, Hutchings A, Dion C, Carter C, et al. Expression of GIMAP1, a GTPase of the Immunity-Associated Protein Family, is Not Up-Regulated in Malaria. Malar J (2009) 8:53. doi: 10.1186/1475-2875-8-53
117. Filen JJ, Filen S, Moulder R, Tuomela S, Ahlfors H, West A, et al. Quantitative Proteomics Reveals GIMAP Family Proteins 1 and 4 to be Differentially Regulated During Human T Helper Cell Differentiation. Mol Cell Proteomics (2009) 8(1):32–44. doi: 10.1074/mcp.M800139-MCP200
118. Saunders A, Webb LM, Janas ML, Hutchings A, Pascall J, Carter C, et al. Putative GTPase GIMAP1 is Critical for the Development of Mature B and T Lymphocytes. Blood (2010) 115(16):3249–57. doi: 10.1182/blood-2009-08-237586
119. Webb LM, Datta P, Bell SE, Kitamura D, Turner M, Butcher GW. GIMAP1 Is Essential for the Survival of Naive and Activated B Cells In Vivo. J Immunol (2016) 196(1):207–16. doi: 10.4049/jimmunol.1501582
120. Chambwe N, Kormaksson M, Geng H, De S, Michor F, Johnson NA, et al. Variability in DNA Methylation Defines Novel Epigenetic Subgroups of DLBCL Associated With Different Clinical Outcomes. Blood (2014) 123(11):1699–708. doi: 10.1182/blood-2013-07-509885
121. Schwefel D, Daumke O. GTP-Dependent Scaffold Formation in the GTPase of Immunity Associated Protein Family. Small GTPases (2011) 2(1):27–30. doi: 10.4161/sgtp.2.1.14938
122. Wai T, Teoli D, Shoubridge EA. The Mitochondrial DNA Genetic Bottleneck Results From Replication of a Subpopulation of Genomes. Nat Genet (2008) 40(12):1484–8. doi: 10.1038/ng.258
123. Taylor RW, Turnbull DM. Mitochondrial DNA Mutations in Human Disease. Nat Rev Genet (2005) 6(5):389–402. doi: 10.1038/nrg1606
124. Chinnery PF, Thorburn DR, Samuels DC, White SL, Dahl HM, Turnbull DM, et al. The Inheritance of Mitochondrial DNA Heteroplasmy: Random Drift, Selection or Both? Trends Genet (2000) 16(11):500–5. doi: 10.1016/s0168-9525(00)02120-x
125. Jokinen R, Junnila H, Battersby BJ. Gimap3: A Foot-in-the-Door to Tissue-Specific Regulation of Mitochondrial DNA Genetics. Small GTPases (2011) 2(1):31–5. doi: 10.4161/sgtp.2.1.14937
126. Jenuth JP, Peterson AC, Fu K, Shoubridge EA. Random Genetic Drift in the Female Germline Explains the Rapid Segregation of Mammalian Mitochondrial DNA. Nat Genet (1996) 14(2):146–51. doi: 10.1038/ng1096-146
127. Battersby BJ, Shoubridge EA. Selection of a mtDNA Sequence Variant in Hepatocytes of Heteroplasmic Mice is Not Due to Differences in Respiratory Chain Function or Efficiency of Replication. Hum Mol Genet (2001) 10(22):2469–79. doi: 10.1093/hmg/10.22.2469
128. Battersby BJ, Loredo-Osti JC, Shoubridge EA. Nuclear Genetic Control of Mitochondrial DNA Segregation. Nat Genet (2003) 33(2):183–6. doi: 10.1038/ng1073
129. Battersby BJ, Redpath ME, Shoubridge EA. Mitochondrial DNA Segregation in Hematopoietic Lineages Does Not Depend on MHC Presentation of Mitochondrially Encoded Peptides. Hum Mol Genet (2005) 14(17):2587–94. doi: 10.1093/hmg/ddi293
130. Simpson JC, Joggerst B, Laketa V, Verissimo F, Cetin C, Erfle H, et al. Genome-Wide RNAi Screening Identifies Human Proteins With a Regulatory Function in the Early Secretory Pathway. Nat Cell Biol (2012) 14(7):764–74. doi: 10.1038/ncb2510
131. Shah DK, Zuniga-Pflucker JC. An Overview of the Intrathymic Intricacies of T Cell Development. J Immunol (2014) 192(9):4017–23. doi: 10.4049/jimmunol.1302259
132. von Boehmer H. Unique Features of the Pre-T-Cell Receptor Alpha-Chain: Not Just a Surrogate. Nat Rev Immunol (2005) 5(7):571–7. doi: 10.1038/nri1636
133. Belver L, Ferrando A. The Genetics and Mechanisms of T Cell Acute Lymphoblastic Leukaemia. Nat Rev Cancer (2016) 16(8):494–507. doi: 10.1038/nrc.2016.63
134. Girardi T, Vicente C, Cools J, De Keersmaecker K. The Genetics and Molecular Biology of T-ALL. Blood (2017) 129(9):1113–23. doi: 10.1182/blood-2016-10-706465
135. Aifantis I, Raetz E, Buonamici S. Molecular Pathogenesis of T-Cell Leukaemia and Lymphoma. Nat Rev Immunol (2008) 8(5):380–90. doi: 10.1038/nri2304
136. Teitell MA, Pandolfi PP. Molecular Genetics of Acute Lymphoblastic Leukemia. Annu Rev Pathol (2009) 4:175–98. doi: 10.1146/annurev.pathol.4.110807.092227
137. Zhong Y, Jiang L, Hiai H, Toyokuni S, Yamada Y. Overexpression of a Transcription Factor LYL1 Induces T- and B-Cell Lymphoma in Mice. Oncogene (2007) 26(48):6937–47. doi: 10.1038/sj.onc.1210494
138. Hsu HL, Wadman I, Baer R. Formation of In Vivo Complexes Between the TAL1 and E2A Polypeptides of Leukemic T Cells. Proc Natl Acad Sci U S A (1994) 91(8):3181–5. doi: 10.1073/pnas.91.8.3181
139. Curtis DJ, Robb L, Strasser A, Begley CG. The CD2-scl Transgene Alters the Phenotype and Frequency of T-lymphomas in N-ras Transgenic or p53 Deficient Mice. Oncogene (1997) 15(24):2975–83. doi: 10.1038/sj.onc.1201467
140. Condorelli GL, Facchiano F, Valtieri M, Proietti E, Vitelli L, Lulli V, et al. T-Cell-Directed TAL-1 Expression Induces T-Cell Malignancies in Transgenic Mice. Cancer Res (1996) 56(22):5113–9.
141. Dadi S, Le Noir S, Payet-Bornet D, Lhermitte L, Zacarias-Cabeza J, Bergeron J, et al. TLX Homeodomain Oncogenes Mediate T Cell Maturation Arrest in T-ALL Via Interaction With ETS1 and Suppression of TCRalpha Gene Expression. Cancer Cell (2012) 21(4):563–76. doi: 10.1016/j.ccr.2012.02.013
142. Chadwick N, Zeef L, Portillo V, Fennessy C, Warrander F, Hoyle S, et al. Identification of Novel Notch Target Genes in T Cell Leukaemia. Mol Cancer (2009) 8:35. doi: 10.1186/1476-4598-8-35
143. Chadwick N, Zeef L, Portillo V, Boros J, Hoyle S, van Doesburg JC, et al. Notch Protection Against Apoptosis in T-ALL Cells Mediated by GIMAP5. Blood Cells Mol Dis (2010) 45(3):201–9. doi: 10.1016/j.bcmd.2010.07.006
144. Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, et al. Genome-Wide Analysis Reveals Conserved and Divergent Features of Notch1/RBPJ Binding in Human and Murine T-Lymphoblastic Leukemia Cells. Proc Natl Acad Sci U S A (2011) 108(36):14908–13. doi: 10.1073/pnas.1109023108
145. Liau WS, Tan SH, Ngoc PCT, Wang CQ, Tergaonkar V, Feng H, et al. Aberrant Activation of the GIMAP Enhancer by Oncogenic Transcription Factors in T-Cell Acute Lymphoblastic Leukemia. Leukemia (2017) 31(8):1798–807. doi: 10.1038/leu.2016.392
146. Eckerle S, Brune V, Doring C, Tiacci E, Bohle V, Sundstrom C, et al. Gene Expression Profiling of Isolated Tumour Cells From Anaplastic Large Cell Lymphomas: Insights Into its Cellular Origin, Pathogenesis and Relation to Hodgkin Lymphoma. Leukemia (2009) 23(11):2129–38. doi: 10.1038/leu.2009.161
147. Berg HE, Blackburn PR, Baughn LB, Ketterling RP, Xu X, Greipp PT, et al. Identification of a Novel KMT2A/GIMAP8 Gene Fusion in a Pediatric Patient With Acute Undifferentiated Leukemia. Genes Chromosomes Cancer (2021) 60(2):108–11. doi: 10.1002/gcc.22902
148. Kannan K, Kaminski N, Rechavi G, Jakob-Hirsch J, Amariglio N, Givol D. DNA Microarray Analysis of Genes Involved in p53 Mediated Apoptosis: Activation of Apaf-1. Oncogene (2001) 20(26):3449–55. doi: 10.1038/sj.onc.1204446
149. Milan M, Dalla Rovere G, Smits M, Ferraresso S, Pastore P, Marin MG, et al. Ecotoxicological Effects of the Herbicide Glyphosate in Non-Target Aquatic Species: Transcriptional Responses in the Mussel Mytilus Galloprovincialis. Environ Pollut (2018) 237:442–51. doi: 10.1016/j.envpol.2018.02.049
150. McDowell IC, Modak TH, Lane CE, Gomez-Chiarri M. Multi-Species Protein Similarity Clustering Reveals Novel Expanded Immune Gene Families in the Eastern Oyster Crassostrea Virginica. Fish Shellfish Immunol (2016) 53:13–23. doi: 10.1016/j.fsi.2016.03.157
151. Balla KM, Rice MC, Gagnon JA, Elde NC. Linking Virus Discovery to Immune Responses Visualized During Zebrafish Infections. Curr Biol (2020) 30(11):2092–103.e5. doi: 10.1016/j.cub.2020.04.031
152. Ye X, Zhou L, Jia J, Wei L, Wen Y, Yan X, et al. ITRAQ Proteomic Analysis of Yellow and Black Skin in Jinbian Carp (Cyprinus Carpio). Life (Basel) (2020) 10(10):1–19. doi: 10.3390/life10100226
Keywords: GIMAP5, gimap, lymphopenia, AIG domain, T lymphocyte, B cells
Citation: Limoges M-A, Cloutier M, Nandi M, Ilangumaran S and Ramanathan S (2021) The GIMAP Family Proteins: An Incomplete Puzzle. Front. Immunol. 12:679739. doi: 10.3389/fimmu.2021.679739
Received: 16 March 2021; Accepted: 12 May 2021;
Published: 31 May 2021.
Edited by:
Thomas Herrmann, Julius Maximilian University of Würzburg, GermanyReviewed by:
Oliver Daumke, Max Delbrueck Center for Molecular Medicine, GermanyCopyright © 2021 Limoges, Cloutier, Nandi, Ilangumaran and Ramanathan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheela Ramanathan, c2hlZWxhLnJhbWFuYXRoYW5AdXNoZXJicm9va2UuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.