 Krista D. DiSano
Krista D. DiSano Francesca Gilli
Francesca Gilli Andrew R. Pachner
Andrew R. Pachner- Department of Neurology, Geisel School of Medicine & Dartmouth-Hitchcock Medical Center, Lebanon, NH, United States
Multiple Sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system. Once thought to be primarily driven by T cells, B cells are emerging as central players in MS immunopathogenesis. Interest in multiple B cell phenotypes in MS expanded following the efficacy of B cell-depleting agents targeting CD20 in relapsing-remitting MS and inflammatory primary progressive MS patients. Interestingly, these therapies primarily target non-antibody secreting cells. Emerging studies seek to explore B cell functions beyond antibody-mediated roles, including cytokine production, antigen presentation, and ectopic follicle-like aggregate formation. Importantly, memory B cells (Bmem) are rising as a key B cell phenotype to investigate in MS due to their antigen-experience, increased lifespan, and rapid response to stimulation. Bmem display diverse effector functions including cytokine production, antigen presentation, and serving as antigen-experienced precursors to antibody-secreting cells. In this review, we explore the cellular and molecular processes involved in Bmem development, Bmem phenotypes, and effector functions. We then examine how these concepts may be applied to the potential role(s) of Bmem in MS pathogenesis. We investigate Bmem both within the periphery and inside the CNS compartment, focusing on Bmem phenotypes and proposed functions in MS and its animal models. Finally, we review how current immunomodulatory therapies, including B cell-directed therapies and other immunomodulatory therapies, modify Bmem and how this knowledge may be harnessed to direct therapeutic strategies in MS.
Introduction
Multiple Sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system (CNS), with a highly variable and unpredictable disease course that can manifest as a variety of physical and cognitive symptoms. Although cellular inflammation in MS has historically focused on one key player in adaptive immunity, T cells, B cells are now recognized as central mediators in MS pathogenesis. B cell antibody-mediated immunity has been implicated in MS pathogenesis since the discovery of elevated CSF IgG in 1942 (1). Subsequently, in 1959 oligoclonal bands (OCBs) in the cerebrospinal fluid (CSF) were identified (2) and, to date, OCBs remain a diagnostic hallmark in MS (3). OCB presence indicates niches of clonally-related antibody-secreting cells (ASC), including plasmablasts and plasma cells, within the CNS. Since the discovery of OCBs in MS, researchers have dedicated intense focus towards identifying the antigenic targets of ASC in the CNS compartment. However, in contrast to CNS neuroinflammatory diseases such as neuromyelitis optica, with clear autoantibody targets (aquaporin-4), probing antibody specificity in MS has not revealed consistent targets (4, 5), with some studies implicating diverse CNS self-antigens (6, 7) and viral antigens (8). The role of ASCs and OCBs in MS still remains elusive, with suggested involvement in pro-inflammatory functions, including autoantibody production, antibody- or complement-dependent cellular cytotoxicity, and opsonization, or anti-inflammatory functions, including production of the anti-inflammatory cytokine IL-10 (9, 10).
Further interest in the role of non-ASC B cells as key players in the MS immunopathogenesis followed the relatively recent success of B cell depletion therapies targeting CD20. These therapies, including rituximab (11, 12), ocrelizumab (13), and ofatumumab (14) reduced new inflammatory lesions and relapses despite the sparing of most ASCs, i.e. CD20- plasma cells and some plasmablasts. These novel findings fueled considerable interest in examining the phenotype and function of non-ASC B cells in MS. Current research seeks to explore B cell function in MS beyond antibody-dependent roles to define antibody-independent mechanisms, including antigen presentation, cytokine production, and ectopic lymphoid follicle-like structures. Among non-ASC B cell subtypes, increased attention has been directed towards the role of memory B cells (Bmem) in regulating immune processes in MS. Bmem have several unique features, including increased longevity, the capacity to rapidly respond to re-exposure to antigen, and the ability to serve as direct antigen-experienced precursors to antibody-secreting cells. Due to the relatively recent interest in Bmem, our knowledge regarding the exact functions of Bmem in MS is expanding. This review aims to explore our current understanding of this key component of immunological memory in MS and its animal models.
In the first part of this review, we summarize the current knowledge regarding Bmem development, trafficking, phenotypes, and function during homeostasis and inflammatory conditions, providing a basis for understanding the mechanisms in which Bmem may contribute to MS and are targeted by immunomodulatory therapies.
In the second part of this review, we describe Bmem in MS and its animal models reviewing phenotypes and putative functions, and finally, we examine the effectiveness of current therapeutic approaches in targeting Bmem.
Bmem Development
A key player in immunological memory, Bmem can be defined as a B cell that has encountered antigen and remains in a quiescent state until re-exposed to antigen, at which point the cell rapidly responds to the second challenge. Upon first pathogen encounter, the majority of Bmem are derived from germinal center (GC) reactions. GCs are specialized structures within secondary lymphoid tissue (SLT) where mature, antigen-experienced B cells undergo cognate interactions with T cells, proliferate, undergo somatic hypermutation to increase B cell receptor (BCR) affinity for antigen, perform immunoglobulin (Ig) isotype switching, and are selected based on affinity for a specific antigenic target. Select GC B cells ultimately differentiate to produce antigen-specific, isotype-switched ASC or Bmem. Though GC B cells serve as the precursor for both ASC and Bmem, the mechanisms regulating Bmem versus ASC differentiation remain poorly understood. Numerous factors have been proposed to contribute to Bmem formation, but no “master regulator” for Bmem differentiation has been identified. Animal models have suggested the transcription factor BACH2 selects GC B cells with intermediate affinity to differentiate into Bmem (15). Additionally, Bmem generation is associated with an increased expression of factors including ZBTB32 (16), KLF2 (17, 18), ABF-1 (19), STAT5, BCL-6 (20, 21), and SKI (21), which, in general, repress differentiation to an ASC phenotype. Cytokines, including IL-24 (22) and IL-9 (17) can enhance Bmem formation. Moreover, in vitro, IL-2, IL-10, and CD40L were demonstrated to be involved in differentiating GC B cells to a Bmem phenotype (23). Outside of GC, a small proportion of antigen-experienced B cells may additionally be selected for based on low affinity to form Bmem in an early wave prior to GC formation (24, 25). GC-independent isotype-unswitched (IgM) or –switched (IgG) Bmem exhibit low affinity due to unmutated Ig variable genes (26). In humans, few Bmem lack somatic mutations for antigen (27), suggesting most Bmem are GC-derived. Following Bmem formation, these cells may reside in survival niches including SLT such as the spleen (28) for years in a resting state independent of antigen; however, these niches are localized near areas of antigen encounter (29). Bmem are also observed in the tonsils and the bone marrow and may enter into circulation to patrol at low levels (28). Bmem express higher levels of the adhesion molecules LFA-1 and VLA-4 compared to naive B cells, with VLA-4 primarily mediating Bmem retention in SLT (30). In vitro, Bmem migrate towards CXCL12 (23, 31), CCL19, and CXCL13 (23, 32) suggesting these chemokines may be involved in movement within the SLT and trafficking to survival niches or sites of inflammation. If the humoral immunity generated from long-lived plasma cells residing in the bone marrow is not sufficient to eliminate pathogens, Bmem become actively involved in the inflammatory response. Upon re-exposure to antigen, Bmem will generate a more rapid and potent antigen-specific response relative to naïve B cells (33).
Bmem Phenotypes
In humans, Bmem are conventionally identified by the expression of tumor necrosis factor superfamily member CD27, a protein regulating entry into plasma cell lineage and properties associated with Bmem including isotype switching and Ig variable gene mutation (34, 35). However, CD27 is not exclusive to Bmem and is likely a marker of GC and post-GC activation as CD27 is also expressed on GC B cells and post-GC B cells, including ASC (Figure 1). Thus, CD27 expression should be coupled with low levels of CD23 (36) and the lack of expression of the ASC marker CD138 (syndecan-1) to identify Bmem in humans. Further inclusion of specific patterns of CD38 (37), CD21 (38), CD24 (39), CD19 (40), B220 (41), FCRL4 (FcRH4) (38, 42) and CD25 (43, 44) can delineate heterogeneous Bmem populations (Figure 1). Thus far, the main populations of CD19+CD27+CD138- Bmem present in peripheral blood and bone marrow include three isotype-unswitched Bmem phenotypes, including IgM+IgD+, IgM- IgD+, IgM+IgD− (IgM-only memory cells), and isotype-switched IgM-IgD- phenotypes, including IgG, IgA, or IgE+ Bmem. Bmem are typically isotype-switched and primarily express IgG subclasses. IgG+ Bmem comprise 15-20% of peripheral blood B cells, including predominately IgG1, IgG2, IgG3 subclasses (45). Among IgG Bmem, it should be noted that a small proportion of “atypical” IgG Bmem may lack CD27 (38, 45, 46). Isotype-switched IgA Bmem comprise around 10% of B cells in peripheral blood and are generally implicated in mucosa-associated tissues (45) while IgE Bmem involved in allergic responses are rarely detectable in humans and mice and their development and lifespan is poorly understood (45). Among isotype-unswitched phenotypes, IgM and IgD-expressing Bmem, including IgM+ IgD+ (15% of B cells), IgM- IgD+ (1%), or IgM+IgD- (5%) may be found within the blood or bone marrow (34, 47, 48).

Figure 1 Bmem phenotyping markers in humans. Overview of B cell differentiation in humans, focusing on naïve B cell to ASC phenotypes, with BCR isotype and surface marker expression. Surface marker levels simplified to highlight relative expression, including low, intermediate, or high levels.
In rodent models, Bmem identification is hampered by the low frequency of Bmem (49) and the lack of CD27 expression among Bmem (50). Further definitive Bmem markers in mice have remained elusive. Exploration of novel Bmem markers in mice have relied on several methods including 1) boosting Bmem frequencies using antigen-based cell enrichment protocols (51, 52), 2) protein immunization in BCR transgenic mice with a fixed BCR specificity (29), 3) adoptive transfer of antigen-specific B cells (53), or 4) genetic tagging of activation-induced cytidine deaminase (AID), an enzyme essential for isotype switching and somatic hypermutation identifying GC-derived B cells including Bmem and ASC (33). Murine studies have proposed at least 10 Bmem subsets utilizing Ig isotyping combined with surface expression of CD80 (49, 54, 55), PDL2 (54, 55), CD73 (55, 56), CD38 (57). However, these markers may be expressed on other murine B cell subtypes, so a diverse panel of surface markers is necessary for identifying Bmem (Figure 2). For isotype-switched Bmem, IgG surface (IgGs) versus intracellular (IgGi) expression (58, 59) in combination with CD138 or Blimp-1 (60, 61) may be used to distinguish ASC (IgGihi/+, igGslow, CD138+, Blimp-1+) (62) and Bmem (IgGilow, IgGshi/+, CD138-, Blimp-1-). Moreover, similar to assaying human Bmem, in vitro stimulation using polyclonal activators (i.e. CpG DNA, R848 TLR7/8 agonist) to convert Bmem into ASC, combined with a conventional Enzyme-linked ImmunoSPOT (ELISPOT) assay, may be used to quantify Bmem and determine antigen specificity and Ig isotype in mice (63–65).
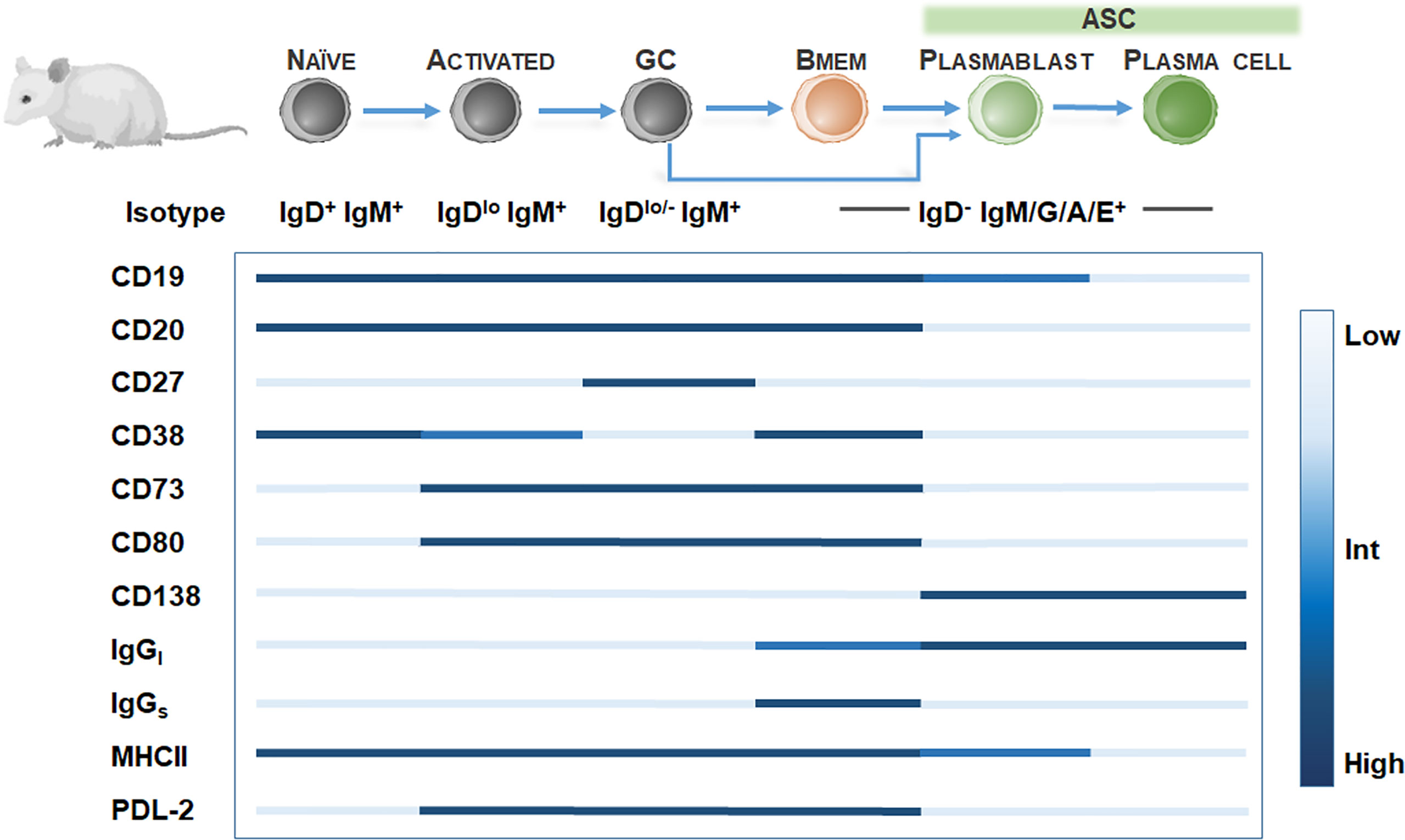
Figure 2 Bmem phenotyping markers in murine models. Overview of B cell differentiation in mice, focusing on naïve B cell to ASC phenotypes, with BCR isotype and surface marker expression. Surface marker levels simplified to highlight relative expression, including low, intermediate, or high levels.
Bmem Function
Compared to naïve mature B cells, Bmem display several distinctive features. Bmem have enhanced longevity and can survive for years and perhaps for the lifetime of the host independent of antigen (66, 67). In comparison, naïve mature B cells have a lifespan of weeks (68). Furthermore, since most Bmem are GC-derived, Bmem are generally clonally expanded, isotype-switched, and have undergone somatic hypermutation of Ig variable genes to increase antigen affinity. Unlike naïve or activated mature B cells, Bmem are able to rapidly proliferate and differentiate into ASC with minimal stimulation requirements, including re-exposure to low levels of antigen (69, 70), T cell help (71–73), or polyclonal stimulation (73, 74). Bmem enter cell cycle, differentiate into ASC, and potentially re-seed GC quicker than mature B cells (75, 76). These advantages are likely due to a combination of factors, including reduced quiescence factors (Kruppel-like factor 4 and 9; PLZF) (77), higher expression of co-stimulatory molecules (CD80, CD86) (78, 79), CD27 (50), IL21R (80), SLAM (signaling lymphocytic activation molecule) (79), TLR7/9 (81), and anti-apoptotic molecules (BCL2) (82). Once activated, Bmem can follow two paths: 1) rapidly differentiating into ASC or 2) re-entering into secondary GC reactions to undergo further affinity maturation and isotype-switching. In murine studies, IgG Bmem show a greater proclivity to differentiate into ASC, while IgM Bmem are often selected for re-entry in GC reactions (33, 51). Bmem differentiating to ASC can contribute to the rapid and copious production of high affinity antibodies to supplement antibody produced by terminally differentiated plasma cells residing in niches, such as the bone marrow. In addition to rapid differentiation to ASC, Bmem are potent antigen-presenting cells (APCs), expressing MHCII (83) that enables not only the efficient recognition of antigen, but the ability to process antigen for presentation to activate other immune cells, including T cells (84). Finally, Bmem produce a wide array of cytokines including TNF (85, 86), GM-CSF (86), IL-6 (86, 87), lymphotoxin (LT) (85), and IL-10 (85).
Bmem in Multiple Sclerosis
In MS, B cells are located within multiple compartments in the CNS, including the CSF, parenchyma, and meninges. However, studies exploring Bmem in MS have primarily focused on the peripheral blood and CSF, with few studies examining Bmem localization in the parenchyma and meninges. Among these studies, there are notable discrepancies in defining Bmem, with the majority of studies defining Bmem based exclusively on CD27 expression. Therefore, for each mentioned study, the surface markers utilized to define Bmem will be noted.
Phenotype, Trafficking, and Localization
In MS, Bmem frequencies are elevated in the CSF compared to peripheral blood (88, 89) and Bmem comprise the majority of B cells populating the CSF (90, 91) (CD27+ IgD- (88, 91); CD19+CD27+ (89); CD27+ CD138- (90); CD19+ CD27+ IgD- and IgD+). In contrast to the peripheral blood, the proportion of CD19+ B cells among total lymphocytes is significantly lower in the CSF (91). However, the proportion of class-switched B cells, including isotype-switched Bmem, among CD19+ B cells is enriched in the CSF (91). Further studies have confirmed the majority of Bmem populating the CSF display an isotype-switched phenotype (71%; CD19+ CD27+ IgD- IgM-) (92). In agreement with these findings, B cells populating the CSF, including Bmem, bear extensive somatic mutations and exhibit clonal expansion (88). Conversely, in a recent pre-print, Bmem in peripheral blood from MS patients displayed an Ig isotype distribution of 50% IgM, 30% IgA, and 20% IgG (93). In MS patients, ASC populating the CSF exhibit a selective enrichment towards the IgG1 allotype G1m1 compared to the peripheral blood (94). In a recent pre-print, Bmem in the intrathecal compartment did not exhibit the same dominance towards the G1m1 allotype constant region polymorphism, suggesting certain B cell-lineages may preferentially differentiate (95). To date, it remains unclear if skewed Ig allotypes influence MS risk and phenotype (96, 97).
Bmem are not restricted to the CSF compartment, and Bmem (CD27+) are found within the brain parenchyma (98, 99). Furthermore, B cells recovered from MS plaques display mutations and clonal expansion (100, 101), suggesting primarily differentiated B cells (Bmem/ASC) occupy the parenchymal space, similar to the CSF. It has been suggested that BCR mutations and clonal expansion may be acquired in the CNS compartment (89), possibly aided by inflammatory aggregates in the brain meninges mimicking some features of ectopic lymphoid follicles (102). In a recent pre-print, extensive clonal connections were found among Bmem and ASC in the CSF compartment (95). Clonal connections between Bmem and ASC were also found to span different isotypes, including IgM/IgG1, IgG1/IgG2, and IgM/IgA1. These findings suggest ASC and Bmem share a common origin, although it remains unclear whether these clonal similarities originate in the periphery or the intrathecal compartment. At least a proportion of B cells appear to undergo an active exchange between the periphery and CNS in MS, with CD27+ IgD- B cells sharing similar repertoires between the peripheral blood and CSF (91, 103). Moreover, Stern et al. demonstrated the B cell clonal families observed in MS brain tissue were frequently derived from founders in the deep cervical lymph nodes (104). Regardless of the mechanism promoting Bmem persistence in the CNS, the exact chemokines initiating and/or sustaining Bmem trafficking to the CNS compartment in MS remain to be determined. Several chemokine receptors including CXCR4 (105), CXCR5 (91), CXCR3 (95), CCR1, CCR2 and CCR4 (88) have been implicated in trafficking and are upregulated on CSF B cells compared to paired-peripheral blood. Adhesion molecules regulating Bmem entry into the CNS meninges and parenchymal compartments are less clearly understood. VLA-4 has been implicated in aiding B cell transmigration in ex vivo culture studies (106) and murine studies (107), though these studies have examined global B cell migration and further studies are required to determine whether VLA-4 is essential for Bmem transmigration.
Function
Antibody Production and Antigen Specificity
Tracking Bmem conversion into ASC to investigate antibody production and specificity in vivo remains challenging and often requires specialized murine models. Alternatively, in vitro, Bmem can be stimulated to convert into ASC utilizing polyclonal activators specifically triggering Bmem differentiation, including the TLR7/8 agonist R848 (108, 109). Bmem may subsequently be quantified and Ig isotype and antibody production may be evaluated. Limited studies exist examining Bmem conversion to ASC and antibody production in MS. Hohmann et al. isolated B cells from the peripheral blood of MS patients and compared IgG antibodies produced by ASCs or Bmem-derived ASCs, i.e. B cells in vitro stimulated using R848 and IL-2 by ELISPOT (110). Bmem-derived ASCs generated larger spot size compared to ASCs, suggesting enhanced IgG secretion from Bmem-derived ASCs.
B cell antigen specificity in MS has remained unclear and is documented as heterogeneous, with antibody targets ranging from self-antigens to viral antigens. With regards to Bmem, there have been few studies on this topic. Hohmann et al. exclusively examined reactivity to normal human brain lysates (110). Among 15 of the 30 relapsing-remitting MS (RRMS) patients tested, brain-reactive Bmem-derived ASC were present in the peripheral blood. In some patients, brain-reactive Bmem were present in relapse and remission, while other patients displayed brain-reactive Bmem in the relapse only. The presence of brain-reactive B cells, including Bmem, predicted relapse. Brain-reactive B cells were not observed in the peripheral blood of healthy donors or other neurological disease controls (111).
Antigen Presentation
Bmem are conventionally regarded as potent APCs. In MS, CSF Bmem (CD27+ IgD-) display upregulated expression of two co-stimulatory molecules key in antigen presenting functions, CD80 and CD86, compared to naïve B cells (88). Although this is a well-known feature of Bmem regardless of disease pathogenesis, this finding suggests Bmem in the CSF of MS patients also display an enhanced ability to engage with immune cells, including T cells. In alignment with these findings, ex vivo Bmem (CD19+ CD27+) isolated from RRMS patients elicited autologous CD4 T cell proliferation in the presence of antigens including, tetanus toxoid, myelin basic protein (MBP), and myelin oligodendrocyte protein (MOG) (112). Moreover, Bmem isolated from some RRMS patients are capable of activating CD4 T helper (Th) cells in the presence of myelin antigens in vitro, inducing T cell proliferation and IFNγ production (112). Furthermore, the in vitro spontaneous proliferation of Th1 cells observed in patients carrying the risk allele HLA-DR15 was found to be mediated by Bmem (CD27+) with high MHCII surface receptor HLA-DR expression (113).
Cytokine Production
B cells, including Bmem, in MS patients may exhibit a propensity towards a dysregulated cytokine network. An increased frequency of Bmem (CD27+) producing GM-CSF was observed in the peripheral blood obtained from MS patients compared to healthy controls (86). Furthermore, in vitro stimulated B cells isolated from the peripheral blood of RRMS and SPMS patients exhibit a decreased production of the anti-inflammatory cytokine IL-10 compared to healthy controls, while LT and TNF levels were comparable (85). Further studies demonstrated stimulated Bmem (CD19+ CD27+) obtained from RRMS patients produce elevated LT and lower IL-10 than naïve B cells (112). However, Bmem isolated from healthy donors produced comparable levels of both cytokines. In vitro stimulated Bmem obtained from healthy donors also exhibited lower levels of IL-10 production compared to naïve B cells (85), thus, low levels of IL-10 production seems to be typical Bmem feature regardless of disease pathogenesis. The reduced IL-10 production by B cells observed in RRMS and SPMS patients may therefore be attributed to another B cell phenotype, including IL-10-producing regulatory B cells or ASC (9, 114).
Associations With Clinical Disease
Recent studies have sought to investigate the association of Bmem with clinical outcomes in MS. In RRMS patients, an increased CD5+ Bmem subpopulation was associated with the remitting stage compared to the relapsing stage (115). Furthermore, Nissimov et al. demonstrated elevated peripheral blood Bmem frequencies were associated with a lower expanded disability status scale score (116). Conversely, Comabella et al. determined that increases in isotype-unswitched and -switched Bmem (CD19+ CD27+ IgD+ or IgD-) in the peripheral blood from RRMS patients were associated with an MRI phenotype with high neurodegeneration, defined by increased contrast-enhancing lesions and non-enhancing black holes on T1-weighted images, and decreased brain parenchymal fraction (117). Bmem populations also differ in peripheral blood obtained from pediatric and adult MS patients (118). In pediatric MS, Bmem (CD20+ CD27+) are elevated in the peripheral blood compared to healthy children and adolescents. In contrast to adult MS patients who display elevated isotype-switched Bmem (CD20+ CD27+ IgD-) and plasma cells in peripheral blood, non-switched Bmem (CD20+ CD27+ IgD+) and plasmablasts were increased in frequency in pediatric MS patients.
Bmem in Animal Models of MS
Murine models of MS generally have been limited in exploring Bmem due to the lack of conventional Bmem markers, the low quantity of Bmem (25), the shifted surface expression of proposed murine markers on Bmem isolated from CNS compartment (63), and the time-consuming methods utilized to isolate Bmem and quantify by in vitro stimulation assays (64, 65). In this section, we will review data on Bmem obtained from pre-clinical models of MS, including two viral models of demyelination, mouse hepatitis virus (MHV) and Theiler’s murine encephalomyelitis virus (TMEV), and the autoimmune model, experimental autoimmune encephalomyelitis (EAE).
Viral Models of Demyelination
Viral immune-mediated demyelination models emulating features of MS, including MHV (coronavirus family) and TMEV (picornavirus family), require B cell and antibody responses for viral control (119, 120) and recruit diverse B cell subtypes CNS (59, 121). There is also evidence for B cell involvement in demyelination and clinical disability (122–125).
Intracerebral MHV infection, including the A59 and JHM strains, induces an acute inflammatory demyelinating disease, with prominent B cell CNS infiltration mimicking the acute inflammatory stages of MS. In MHV models, Bmem are present in the CNS parenchyma as evaluated by flow cytometry (59), genetic tagging of AID-expressing B cells (126), and in vitro stimulation and evaluation via ELISPOT assays (63). Among total CNS-infiltrating Bmem (CD19+, CD138-, IgD-, IgG2a/b surface+, IgG2a/b intracellularlow) the majority comprise an IgG2a/2b isotype-switched phenotype. ELISPOT analysis of in vitro stimulated Bmem determined that ASC and Bmem are initially recruited to the CNS (brain/spinal cord) with similar kinetics, but during the chronic phase of infection (day 35 post infection-p.i.), virus-specific IgG ASC persisted at higher frequencies than IgG Bmem in the spinal cord, the predominant site of inflammation and demyelination (63). ELISPOT analyses revealed that antibody production levels were similar between ASC and Bmem-derived ASC in both brain and spinal cord tissues. Gene expression analysis of chemokine receptors on CNS-infiltrating Bmem (CD19+ IgD- CD138-) revealed highly upregulated expression of CXCR3 and CCR7, with moderate expression of CXCR4 and CXCR5 (59). Compared to ASC (CD138+), Bmem expressed higher levels of CCR7 and CXCR5, with similar expression of CXCR4, and lower expression of CXCR3. These results suggest multiple chemokine receptors may be simultaneously regulated on Bmem to direct recruitment. AID-genetically tagged Bmem and ASC were continually recruited from the periphery to the CNS concurrent with GC maturation (126). Moreover, once recruited to the CNS, there was no evidence of AID mRNA expression among Bmem, suggesting these cells were not undergoing somatic hypermutation or isotype switching in the CNS compartment during chronic infection (59). It still remains unclear whether Bmem are required for sustaining the local antibody production responsible for controlling viral recrudescence. Future studies are also required to determine if Bmem contribute to antibody-independent functions, including local cytokine production and antigen presentation.
In the chronic progressive demyelinating disease model, TMEV-induced demyelinating disease (TMEV-IDD), intracranial infection with TMEV mimics several neurodegenerative and clinical features of progressive MS (127). In chronic disease (day 120 p.i.) a phase of accumulating disability, Bmem (IgG+ CD138-) were identified in spinal cord tissue (121). Although the function of Bmem in TMEV-IDD remains to be determined, B cell depletion therapy (anti-CD20) targeting non-ASC B cells, including Bmem, exacerbated microglial activation, increased T cell infiltration, demyelination, and axonal damage (123).
Autoimmune Models
Although a wide array of EAE models exist, the most commonly utilized EAE models emulate the acute or relapsing/remitting stages of MS (128) and are induced independent of B cells (128–130). Due to the limited B cell involvement in these models, including the MOG35-55 peptide model induced in C57BL/6 mice, the role of Bmem in EAE models remains relatively unexplored.
Several therapeutic interventions targeting B cell subtypes including Bmem may provide insights into Bmem function in EAE autoimmune models of MS. In anti-CD20 studies in EAE, clinical disease is suppressed in murine MOG35-55 (131, 132) and marmoset EAE models (133, 134). CD20 depletion was also found to ablate IL-6 producing B cells (131), including Bmem. In a T-independent protein immunization murine model (TNP-LPS) anti-CD20 administration depleted existing and adoptively transferred Bmem (135). Mice deficient in B cell maturation antigen (BCMA), an important receptor for B cell-activating factor (BAFF) and a proliferating inducing ligand (APRIL) regulating ASC differentiation and survival, showed exacerbated EAE disease severity (136). In vitro, BCMA expression directly inhibited Bmem expansion and anti-inflammatory cytokine production, suggesting BCMA deficient mice may show increased proportions of Bmem. Together, these studies suggest Bmem may contribute to EAE pathogenesis. However, other therapeutic interventions have suggested Bmem may play a dispensable or, perhaps, beneficial role in EAE pathogenesis. Atacicept, a TACI fusion protein that inhibits the B cell survival factors B lymphocyte stimulator (BlyS) and APRIL, spares B cell progenitors and Bmem (137). Atacicept’s use has been explored in both the B cell-dependent recombinant human MOG1-125 (rhMOG) and B cell-independent MOG35-55 models. In both models, prophylactic treatment resulted in reduced B cell infiltration into the CNS, delayed disease onset, and attenuated disease severity (138). In addition, a key cytokine promoting Bmem survival, IL-15, was found to be enhanced in a murine lupus model following TACI-IgG treatment (139).
Altogether, further studies are required to determine Bmem function in EAE models of MS as anti-CD20 therapies, atacicept, and BCMA deficiency all affect multiple B cell subsets. Following the success of B cell-depleting therapies in MS, increasingly studies are utilizing B cell-dependent EAE models, including rhMOG EAE and EAE induced in IgHMOG transgenic mice where 30% of B cells are specific for MOG (140). Future studies utilizing these models may pinpoint the exact Bmem phenotypes and Bmem functions involved in autoimmune models of MS.
MS Immunomodulatory Therapies and the Effect on Bmem
B Cell-Directed Immunomodulatory Therapies
B cell depletion therapies targeting CD20, including rituximab, ocrelizumab, and ofatumumab, deplete all B cells except ASC and pro-B cells (141) (Figure 1; Table 1) and have shown significant efficacy in reducing clinical relapse rates and new lesion formation in RRMS patients (11, 196). Additionally, in young, inflammatory primary progressive MS (PPMS) patients, ocrelizumab has been shown to reduce clinical disease progression and brain atrophy (197). Following anti-CD20 therapies, B cells including Bmem are significantly decreased in the peripheral blood of MS patients (142, 146) (Table 1), with dramatic peripheral B cell depletion still evident by 6 months post-treatment. In rituximab-treated patients, a reduction in CSF B cells was also observed in RRMS patients (147, 148), while PPMS patients were only shown to exhibit a moderate reduction (149). In RRMS patients, rituximab treatment was shown to normalize the ratio of GM-CSF to IL-10 producing B cells in the peripheral blood (86). Eight-to-24 months post-treatment, reappearing peripheral blood B cells were strongly diminished in memory B cells (116).
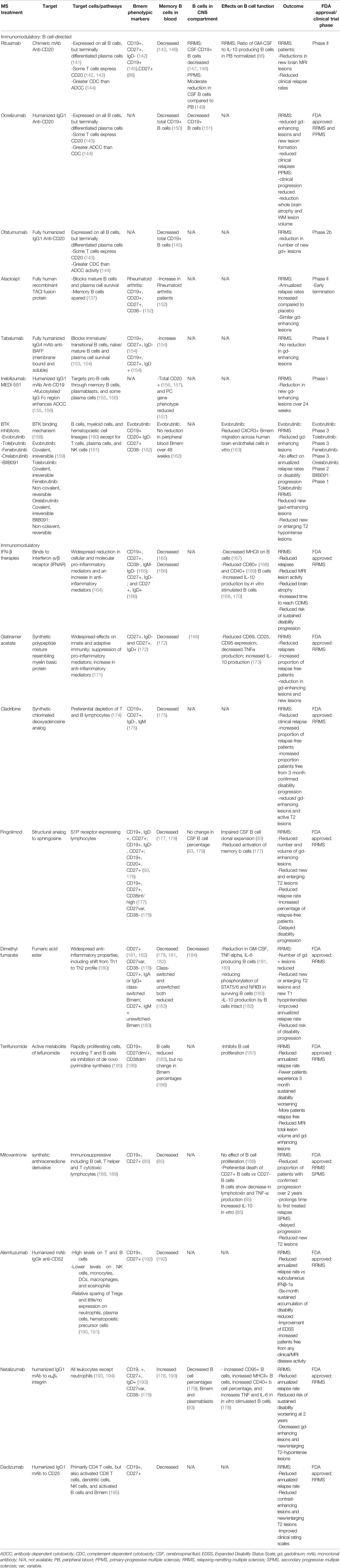
Table 1 Immunomodulatory MS treatment effects on Bmem and B cell function.
Further B cell-directed therapies have sought to target a more diverse range of B cell phenotypes. Inebilizumab (MEDI-551), an anti-CD19 monoclonal antibody targets pro-B cells through memory B cells, plasmablasts, and some plasma cells (155, 198). In contrast to CD20 which is also expressed on a subpopulation of CD4+ T cells, CD19 is exclusively expressed on B cells (198). Similar to anti-CD20 directed therapies, treatment in RRMS patients results in reduced peripheral B cells (156, 157) and decreased gadolinium-enhancing lesions (157). B cell immunomodulatory therapies targeting B cell survival factors have shown contrasting effects on clinical outcomes. Atacicept treatment in RRMS patients resulted in an increased annualized relapse rate and unaltered gadolinium-enhancing lesions leading to the early termination of the phase II clinical trial (199). In rheumatoid arthritis patients, atacicept treatment led to an increase Bmem numbers in the peripheral blood (152), confirming previous studies that Bmem are spared (137). Similarly, tabalumab, an anti-BAFF monoclonal antibody which blocks immature B cells, mature B cells, and ASC survival, also fails to deplete Bmem (153, 154). Bmem were increased in the peripheral blood (154) and no reduction in gadolinium-enhancing lesions was observed in RRMS patients (200). The findings of unchanged or worse clinical outcomes in atacicept and tabalumab may be due to the minimal effect on Bmem (152, 201), although further studies are required.
Recently, the landscape of MS therapies targeting B cells has expanded to include Bruton’s tyrosine kinase (BTK) inhibitors. BTK is a critical enzyme for signaling through the BCR, FcγR, and GM-CSF receptor and is therefore involved in both adaptive and innate immune responses (160, 202). BTK inhibition affects myeloid cells, including microglia (203), and other hematopoietic lineage cells with exception to T cells, plasma cells, and natural killer cells (161). As small molecules, many BTK inhibitors also rapidly penetrate the blood-brain barrier (202, 203). The BTK inhibitors evobrutinib, tolebrutinib, fenebrutinib, orelabrutinib, and B11091 are currently in clinical development for relapsing and progressive forms of MS (Table 1). In clinical trials, BTK inhibitors were shown to reduce gadolinium-enhancing lesions (204) and new or enlarging T2 hypointense lesions (205), but did not reduce annualized relapse rates or disease progression in RRMS patients (204). Preliminary studies monitoring peripheral blood B cells in evobrutinib-treated RRMS and SPMS patients revealed no clinically relevant changes in the number of total B cells or Bmem over the 48 week treatment period (162). However, in vitro assays demonstrated an alteration in Bmem function, with reduced CXCR3+ Bmem migration across human brain endothelial cells (206).
Other Immunomodulatory Therapies
Numerous immunomodulatory therapies utilized in MS have also been observed to affect Bmem. Although not traditionally viewed as modulating the B cell compartment, these therapies can have direct or indirect effects on Bmem survival and function. Interferon (IFN)-β, glatiramer acetate, fingolimod, dimethyl fumarate, and mitoxantrone all reduce Bmem numbers in peripheral blood and alter global B cell function following therapeutic treatment (Table 1). Peripheral blood B cells obtained from IFN-β-treated patients exhibit reductions in MHCII expression (167), reduced co-stimulatory molecules CD80 (168) and CD40 (169), and an increase in IL-10 production (168, 170), suggesting a shift in the overall B cell profile to an anti-inflammatory state. IFN-β treatment was also found to increase Bmem apoptosis (115). Glatiramer acetate-treated MS patients also show alterations in B cell function, resulting in reduced activation markers (CD69, CD95), decreased TNF production, and increased IL-10 production (173). Fingolimod, which targets SIP receptor-expressing lymphocytes such as T cells and B cells results in impaired CSF B cell clonal expansion (93), including Bmem, and reduced Bmem activation in peripheral blood from MS patients (177). Dimethyl fumarate treatment results in similar modulation reducing B cell activation (183) and the production of the pro-inflammatory cytokines GM-CSF, TNF, and IL-6 (181, 183), while IL-10 production is unaltered (182). Mitoxantrone treatment, immunosuppressive to T cells and B cells, does not affect B cell proliferation (188), but results in the preferential death of CD27-expressing B cells and a shift to an anti-inflammatory state, with reduced LT and TNF production, and increased IL-10 production in vitro (85). Conversely, natalizumab, which blocks leukocyte α4β1-mediated entry into the CNS, results in a 2.4-fold increase in Bmem in the peripheral blood (178, 193), but a reduction of Bmem in the CSF (93). In contrast to the aforementioned therapies, B cell activation (CD95, CD40, MHCII expression) and TNF and IL-6 production was increased in the peripheral blood of natalizumab-treated MS patients (178). Multiple other immunomodulatory therapies which have shown to be effective in improving clinical outcomes in RRMS patients, including cladribine, teriflunomide, daclizumab, and alemtuzumab all decrease peripheral Bmem numbers (Table 1), though findings related to the functional changes in B cells following therapeutic treatment remain to be determined.
Bmem and Tailoring Therapeutic Treatment
Bmem in peripheral blood may prove useful for monitoring therapeutic effects in MS. In one study, Novi et al. utilized a Bmem-based reinfusion protocol for rituximab administration. Bmem monitoring (CD19+ CD27+ PBMCs) was used to orchestrate rituximab reinfusion, leading to a reduced number of reinfusions while still reducing disease activity (146). This study highlights the potential role for monitoring Bmem to tailor immunomodulatory treatments in MS. Future studies may also investigate the utility of monitoring Bmem in peripheral blood to predict response to therapy, including B cell depletion, in MS. Bmem monitoring in peripheral blood is a currently utilized strategy for predicting response to B cell depletion therapies in several autoimmune diseases implicating B cells including Sjogren’s syndrome, system lupus erythematosus, and rheumatoid arthritis (207–209).
Altogether, future studies are required to determine the exact effects on Bmem function following immunomodulatory treatment, including whether Bmem are central to the efficacy of disease-modifying therapies, and whether Bmem monitoring can be used to “personalize” immunotherapy.
Concluding Remarks and Future Directions
The cause of MS is unknown but growing evidence suggests multiple B cell phenotypes are central players in MS pathogenesis. In MS, Bmem in both the peripheral and CNS compartments are increasingly being explored to define the exact relationship with disease development and progression. Important observations highlighted in the current review include the presence of Bmem alterations in both the peripheral blood and CNS compartments in MS; evidence for potential roles in antibody production, antigen presentation, and cytokine production (Figure 3); and effective targeting of Bmem using currently available immunomodulatory therapies. Future studies should aim to address several key unresolved questions to provide more in-depth insights regarding Bmem in MS (Table 2), including trafficking mechanisms, action within the CNS compartment, functional relevance in MS immunopathogenesis, and defining associations with clinical outcomes. These insights may help to guide therapeutic strategies to develop novel agents specific for Bmem and tailor current therapeutic treatment regimens.
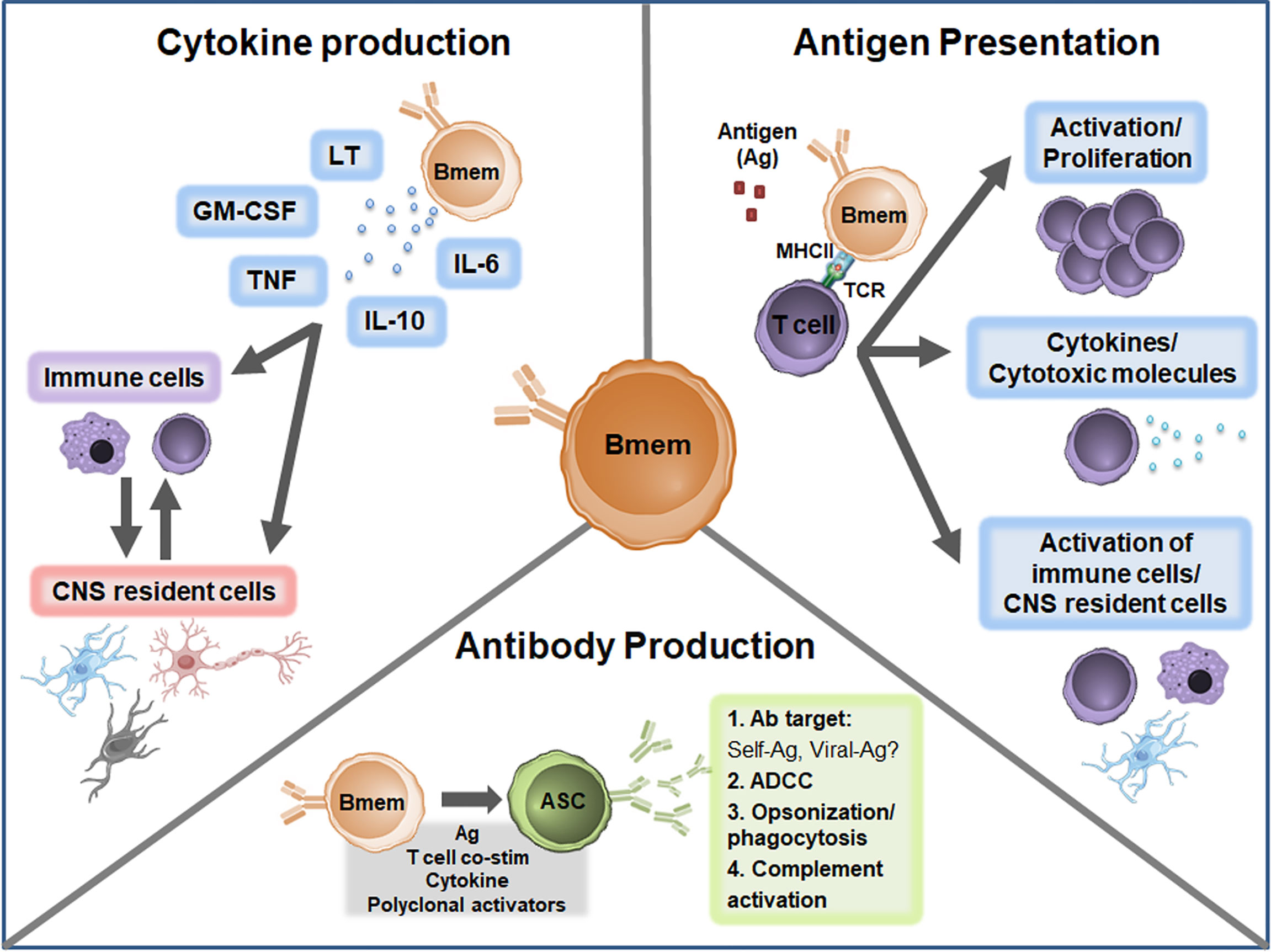
Figure 3 Proposed Bmem functions in MS. Canonical Bmem functions include cytokine production, antigen presentation, and antibody production. Bmem may produce a variety of pro-inflammatory and anti-inflammatory cytokines in MS, including lymphotoxin (LT), GM-CSF, TNF, IL-10, and IL-6. The production of these cytokines may 1) modulate the inflammatory function of immune cells, including monocytes and T cells in the periphery or CNS or 2) may alter the function and survival of CNS resident cells, including neurons, astrocytes, microglia, and oligodendrocytes. Bmem-derived cytokines may also modulate the interactions between CNS-localized immune cells and CNS resident cells. Bmem are potent antigen presenting cells (APCs) and upon uptake and presentation of antigen (Ag) may interact with other immune cells, including T cells, to enhance cell proliferation and effector functions. For example, following T cell-Bmem interaction, activated T cells may engage in cytokine production or cytotoxic molecule secretion. Bmem antigen presentation to T cells may also modify T cell engagement with other immune cells in the periphery or CNS and interaction with CNS resident cells. Bmem can differentiate into ASC following stimulation, including re-exposure to antigen, T cell co-stimulation (co-stim), cytokine stimulation, or T cell-independent polyclonal stimulation. Upon differentiation, Bmem-derived ASC may be involved in sustaining antibody responses in the CNS compartment. Bmem-derived ASC may also contribute to several antibody-dependent functions implicated in MS, including targeting self or viral antigens, antibody-dependent cellular cytotoxicity (ADCC), opsonization/phagocytosis, and complement engagement.

Table 2 Bmem in MS: Unresolved questions.
Author Contributions
KD and AP outlined the subject for the review. KD reviewed the literature, drafted the figures and tables, and wrote the manuscript. AP and FG edited and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Dartmouth College’s Open-Access Publication Equity Fund sponsored by the Dartmouth Library and Office of the Provost.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author’s thank Dartmouth College’s Library and Office of the Provost for sponsoring the open-access fees for the submission of this manuscript.
References
1. Kabat EA, Moore DH, Landow H. An Electrophoretic Study of the Protein Components in Cerebrospinal Fluid and Their Relationship to the Serum Proteins. J Clin Invest (1942) 21:571–7. doi: 10.1172/JCI101335
2. Karcher D, Van Sande M, Lowenthal A. Micro-Electrophoresis in Agar Gel of Proteins of the Cerebrospinal Fluid and Central Nervous System. J Neurochem (1959) 4:135–40. doi: 10.1111/j.1471-4159.1959.tb13182.x
3. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of Multiple Sclerosis: 2017 Revisions of the McDonald Criteria. Lancet Neurol (2018) 17:162–73. doi: 10.1016/S1474-4422(17)30470-2
4. Willis SN, Stathopoulos P, Chastre A, Compton SD, Hafler DA, O’Connor KC. Investigating the Antigen Specificity of Multiple Sclerosis Central Nervous System-Derived Immunoglobulins. Front Immunol (2015) 6:600. doi: 10.3389/fimmu.2015.00600
5. Weber MS, Hemmer B, Cepok S. The Role of Antibodies in Multiple Sclerosis. Biochim Biophys Acta (2011) 1812:239–45. doi: 10.1016/j.bbadis.2010.06.009
6. Olsson T, Baig S, Hojeberg B, Link H. Antimyelin Basic Protein and Antimyelin Antibody-Producing Cells in Multiple Sclerosis. Ann Neurol (1990) 27:132–6. doi: 10.1002/ana.410270207
7. Reindl M, Linington C, Brehm U, Egg R, Dilitz E, Deisenhammer F, et al. Antibodies Against the Myelin Oligodendrocyte Glycoprotein and the Myelin Basic Protein in Multiple Sclerosis and Other Neurological Diseases: A Comparative Study. Brain (1999) 122( Pt 11):2047–56. doi: 10.1093/brain/122.11.2047
8. Gilden DH. Infectious Causes of Multiple Sclerosis. Lancet Neurol (2005) 4:195–202. doi: 10.1016/S1474-4422(05)70023-5
9. Rojas OL, Probstel AK, Porfilio EA, Wang AA, Charabati M, Sun T, et al. Recirculating Intestinal IgA-Producing Cells Regulate Neuroinflammation Via IL-10. Cell (2019) 176:610–24.e18. doi: 10.1016/j.cell.2018.11.035
10. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-Producing Plasmablasts Exert Regulatory Function in Autoimmune Inflammation. Immunity (2014) 41:1040–51. doi: 10.1016/j.immuni.2014.10.016
11. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-Cell Depletion With Rituximab in Relapsing-Remitting Multiple Sclerosis. N Engl J Med (2008) 358:676–88. doi: 10.1056/NEJMoa0706383
12. Hawker K, O’Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, et al. Rituximab in Patients With Primary Progressive Multiple Sclerosis: Results of a Randomized Double-Blind Placebo-Controlled Multicenter Trial. Ann Neurol (2009) 66:460–71. doi: 10.1002/ana.21867
13. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, et al. Ocrelizumab Versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N Engl J Med (2017) 376:221–34. doi: 10.1056/NEJMoa1601277
14. Hauser SL, Bar-Or A, Cohen JA, Comi G, Correale J, Coyle PK, et al. Ofatumumab Versus Teriflunomide in Multiple Sclerosis. N Engl J Med (2020) 383:546–57. doi: 10.1056/NEJMoa1917246
15. Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated Selection of Germinal-Center Cells Into the Memory B Cell Compartment. Nat Immunol (2016) 17:861–9. doi: 10.1038/ni.3460
16. Jash A, Zhou YW, Gerardo DK, Ripperger TJ, Parikh BA, Piersma S, et al. ZBTB32 Restrains Antibody Responses to Murine Cytomegalovirus Infections, But Not Other Repetitive Challenges. Sci Rep (2019) 9:15257. doi: 10.1038/s41598-019-51860-z
17. Wang Y, Shi J, Yan J, Xiao Z, Hou X, Lu P, et al. Germinal-Center Development of Memory B Cells Driven by IL-9 From Follicular Helper T Cells. Nat Immunol (2017) 18:921–30. doi: 10.1038/ni.3788
18. Bhattacharya D, Cheah MT, Franco CB, Hosen N, Pin CL, Sha WC, et al. Transcriptional Profiling of Antigen-Dependent Murine B Cell Differentiation and Memory Formation. J Immunol (2007) 179:6808–19. doi: 10.4049/jimmunol.179.10.6808
19. Chiu YK, Lin IY, Su ST, Wang KH, Yang SY, Tsai DY, et al. Transcription Factor ABF-1 Suppresses Plasma Cell Differentiation But Facilitates Memory B Cell Formation. J Immunol (2014) 193:2207–17. doi: 10.4049/jimmunol.1400411
20. Scheeren FA, Naspetti M, Diehl S, Schotte R, Nagasawa M, Wijnands E, et al. STAT5 Regulates the Self-Renewal Capacity and Differentiation of Human Memory B Cells and Controls Bcl-6 Expression. Nat Immunol (2005) 6:303–13. doi: 10.1038/ni1172
21. Laidlaw BJ, Duan L, Xu Y, Vazquez SE, Cyster JG. The Transcription Factor Hhex Cooperates With the Corepressor Tle3 to Promote Memory B Cell Development. Nat Immunol (2020) 21:1082–93. doi: 10.1038/s41590-020-0713-6
22. Maarof G, Bouchet-Delbos L, Gary-Gouy H, Durand-Gasselin I, Krzysiek R, Dalloul A. Interleukin-24 Inhibits the Plasma Cell Differentiation Program in Human Germinal Center B Cells. Blood (2010) 115:1718–26. doi: 10.1182/blood-2009-05-220251
23. Roy MP, Kim CH, Butcher EC. Cytokine Control of Memory B Cell Homing Machinery. J Immunol (2002) 169:1676–82. doi: 10.4049/jimmunol.169.4.1676
24. Takahashi Y, Ohta H, Takemori T. Fas is Required for Clonal Selection in Germinal Centers and the Subsequent Establishment of the Memory B Cell Repertoire. Immunity (2001) 14:181–92. doi: 10.1016/S1074-7613(01)00100-5
25. Weisel F, Shlomchik M. Memory B Cells of Mice and Humans. Annu Rev Immunol (2017) 35:255–84. doi: 10.1146/annurev-immunol-041015-055531
26. Inamine A, Takahashi Y, Baba N, Miyake K, Tokuhisa T, Takemori T, et al. Two Waves of Memory B-Cell Generation in the Primary Immune Response. Int Immunol (2005) 17:581–9. doi: 10.1093/intimm/dxh241
27. Ahmed R, Gray D. Immunological Memory and Protective Immunity: Understanding Their Relation. Science (1996) 272:54–60. doi: 10.1126/science.272.5258.54
28. Giesecke C, Frolich D, Reiter K, Mei HE, Wirries I, Kuhly R, et al. Tissue Distribution and Dependence of Responsiveness of Human Antigen-Specific Memory B Cells. J Immunol (2014) 192:3091–100. doi: 10.4049/jimmunol.1302783
29. Anderson SM, Hannum LG, Shlomchik MJ. Memory B Cell Survival and Function in the Absence of Secreted Antibody and Immune Complexes on Follicular Dendritic Cells. J Immunol (2006) 176:4515–9. doi: 10.4049/jimmunol.176.8.4515
30. Camponeschi A, Gerasimcik N, Wang Y, Fredriksson T, Chen D, Farroni C, et al. Dissecting Integrin Expression and Function on Memory B Cells in Mice and Humans in Autoimmunity. Front Immunol (2019) 10:534. doi: 10.3389/fimmu.2019.00534
31. Bleul CC, Schultze JL, Springer TA. B Lymphocyte Chemotaxis Regulated in Association With Microanatomic Localization, Differentiation State, and B Cell Receptor Engagement. J Exp Med (1998) 187:753–62. doi: 10.1084/jem.187.5.753
32. Badr G, Borhis G, Lefevre EA, Chaoul N, Deshayes F, Dessirier V, et al. BAFF Enhances Chemotaxis of Primary Human B Cells: A Particular Synergy Between BAFF and CXCL13 on Memory B Cells. Blood (2008) 111:2744–54. doi: 10.1182/blood-2007-03-081232
33. Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, et al. Multiple Layers of B Cell Memory With Different Effector Functions. Nat Immunol (2009) 10:1292–9. doi: 10.1038/ni.1814
34. Klein U, Rajewsky K, Küppers R. Human Immunoglobulin (Ig)M+IgD+ Peripheral Blood B Cells Expressing the CD27 Cell Surface Antigen Carry Somatically Mutated Variable Region Genes: CD27 as a General Marker for Somatically Mutated (Memory) B Cells. J Exp Med (1998) 188:1679–89. doi: 10.1084/jem.188.9.1679
35. Tangye SG, Liu YJ, Aversa G, Phillips JH, de Vries JE. Identification of Functional Human Splenic Memory B Cells by Expression of CD148 and CD27. J Exp Med (1998) 188:1691–703. doi: 10.1084/jem.188.9.1691
36. Ellyard JI, Avery DT, Phan TG, Hare NJ, Hodgkin PD, Tangye SG. Antigen-Selected, Immunoglobulin-Secreting Cells Persist in Human Spleen and Bone Marrow. Blood (2004) 103:3805–12. doi: 10.1182/blood-2003-09-3109
37. Sanz I, Wei C, Lee FE, Anolik J. Phenotypic and Functional Heterogeneity of Human Memory B Cells. Semin Immunol (2008) 20:67–82. doi: 10.1016/j.smim.2007.12.006
38. Ehrhardt GR, Hsu JT, Gartland L, Leu CM, Zhang S, Davis RS, et al. Expression of the Immunoregulatory Molecule FcRH4 Defines a Distinctive Tissue-Based Population of Memory B Cells. J Exp Med (2005) 202:783–91. doi: 10.1084/jem.20050879
39. Oleinika K, Mauri C, Salama AD. Effector and Regulatory B Cells in Immune-Mediated Kidney Disease. Nat Rev Nephrol (2019) 15:11–26. doi: 10.1038/s41581-018-0074-7
40. Culton DA, Nicholas MW, Bunch DO, Zhen QL, Kepler TB, Dooley MA, et al. Similar CD19 Dysregulation in Two Autoantibody-Associated Autoimmune Diseases Suggests a Shared Mechanism of B-Cell Tolerance Loss. J Clin Immunol (2007) 27:53–68. doi: 10.1007/s10875-006-9051-1
41. Cappione AJ, Pugh-Bernard AE, Anolik JH, Sanz I. Lupus IgG Vh4.34 Antibodies Bind to a 220-kDa Glycoform of CD45/B220 on the Surface of Human B Lymphocytes. J Immunol (2004) 172:4298–307. doi: 10.4049/jimmunol.172.7.4298
42. Ehrhardt GR, Davis RS, Hsu JT, Leu CM, Ehrhardt A, Cooper MD. The Inhibitory Potential of Fc Receptor Homolog 4 on Memory B Cells. Proc Natl Acad Sci USA (2003) 100:13489–94. doi: 10.1073/pnas.1935944100
43. Brisslert M, Bokarewa M, Larsson P, Wing K, Collins LV, Tarkowski A. Phenotypic and Functional Characterization of Human CD25+ B Cells. Immunology (2006) 117:548–57. doi: 10.1111/j.1365-2567.2006.02331.x
44. Amu S, Tarkowski A, Dorner T, Bokarewa M, Brisslert M. The Human Immunomodulatory CD25+ B Cell Population Belongs to the Memory B Cell Pool. Scand J Immunol (2007) 66:77–86. doi: 10.1111/j.1365-3083.2007.01946.x
45. Shah HB, Smith K, Wren JD, Webb CF, Ballard JD, Bourn RL, et al. Insights From Analysis of Human Antigen-Specific Memory B Cell Repertoires. Front Immunol (2018) 9:3064. doi: 10.3389/fimmu.2018.03064
46. Fecteau JF, Cote G, Neron S. A New Memory CD27-IgG+ B Cell Population in Peripheral Blood Expressing VH Genes With Low Frequency of Somatic Mutation. J Immunol (2006) 177:3728–36. doi: 10.4049/jimmunol.177.6.3728
47. Paramithiotis E, Cooper MD. Memory B Lymphocytes Migrate to Bone Marrow in Humans. Proc Natl Acad Sci USA (1997) 94:208–12. doi: 10.1073/pnas.94.1.208
48. Klein U, Kuppers R, Rajewsky K. Evidence for a Large Compartment of IgM-Expressing Memory B Cells in Humans. Blood (1997) 89:1288–98. doi: 10.1182/blood.V89.4.1288
49. Anderson SM, Tomayko MM, Ahuja A, Haberman AM, Shlomchik MJ. New Markers for Murine Memory B Cells That Define Mutated and Unmutated Subsets. J Exp Med (2007) 204:2103–14. doi: 10.1084/jem.20062571
50. Xiao Y, Hendriks J, Langerak P, Jacobs H, Borst J. CD27 Is Acquired by Primed B Cells at the Centroblast Stage and Promotes Germinal Center Formation. J Immunol (2004) 172:7432–41. doi: 10.4049/jimmunol.172.12.7432
51. Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B Cell Populations Mediate Early and Late Memory During an Endogenous Immune Response. Science (2011) 331:1203–7. doi: 10.1126/science.1201730
52. Taylor JJ, Pape KA, Jenkins MK. A Germinal Center-Independent Pathway Generates Unswitched Memory B Cells Early in the Primary Response. J Vis Exp (2012) 209:597–606. doi: 10.1084/jem.20111696
53. Tomayko MM, Anderson SM, Brayton CE, Sadanand S, Steinel NC, Behrens TW, et al. Systematic Comparison of Gene Expression Between Murine Memory and Naive B Cells Demonstrates That Memory B Cells Have Unique Signaling Capabilities. J Immunol (2008) 181:27–38. doi: 10.4049/jimmunol.181.1.27
54. Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 Define Functionally Distinct Memory B Cell Subsets That are Independent of Antibody Isotype. Nat Immunol (2014) 15:631–7. doi: 10.1038/ni.2914
55. Tomayko MM, Steinel NC, Anderson SM, Shlomchik MJ. Cutting Edge: Hierarchy of Maturity of Murine Memory B Cell Subsets. J Immunol (2010) 185:7146–50. doi: 10.4049/jimmunol.1002163
56. Conter LJ, Song E, Shlomchik MJ, Tomayko MM. CD73 Expression Is Dynamically Regulated in the Germinal Center and Bone Marrow Plasma Cells are Diminished in its Absence. PloS One (2014) 9:e92009. doi: 10.1371/journal.pone.0092009
57. Ridderstad A, Tarlinton DM. Kinetics of Establishing the Memory B Cell Population as Revealed by CD38 Expression. J Immunol (1998) 160:4688–95.
58. Metcalf TU, Griffin DE. Alphavirus-Induced Encephalomyelitis: Antibody-Secreting Cells and Viral Clearance From the Nervous System. J Virol (2011) 85:11490–501. doi: 10.1128/JVI.05379-11
59. Phares TW, DiSano KD, Stohlman SA, Bergmann CC. Progression From IgD+ IgM+ to Isotype-Switched B Cells Is Site Specific During Coronavirus-Induced Encephalomyelitis. J Virol (2014) 88:8853–67. doi: 10.1128/JVI.00861-14
60. Angelin-Duclos C, Cattoretti G, Lin KI, Calame K. Commitment of B Lymphocytes to a Plasma Cell Fate Is Associated With Blimp-1 Expression In Vivo. J Immunol (2000) 165:5462–71. doi: 10.4049/jimmunol.165.10.5462
61. Shapiro-Shelef M, Lin KI, Savitsky D, Liao J, Calame K. Blimp-1 Is Required for Maintenance of Long-Lived Plasma Cells in the Bone Marrow. J Exp Med (2005) 202:1471–6. doi: 10.1084/jem.20051611
62. Kalia V, Sarkar S, Gourley TS, Rouse BT, Ahmed R. Differentiation of Memory B and T Cells. Curr Opin Immunol (2006) 18:255–64. doi: 10.1016/j.coi.2006.03.020
63. DiSano KD, Stohlman SA, Bergmann CC. An Optimized Method for Enumerating CNS Derived Memory B Cells During Viral-Induced Inflammation. J Neurosci Methods (2017) 285:58–68. doi: 10.1016/j.jneumeth.2017.05.011
64. Slifka MK, Ahmed R. Limiting Dilution Analysis of Virus-Specific Memory B Cells by an ELISPOT Assay. J Immunol Methods (1996) 199:37–46. doi: 10.1016/S0022-1759(96)00146-9
65. Amanna IJ, Slifka MK. Quantitation of Rare Memory B Cell Populations by Two Independent and Complementary Approaches. J Immunol Methods (2006) 317:175–85. doi: 10.1016/j.jim.2006.09.005
66. Maruyama M, Lam KP, Rajewsky K. Memory B-Cell Persistence Is Independent of Persisting Immunizing Antigen. Nature (2000) 407:636–42. doi: 10.1038/35036600
67. Hannum LG, Haberman AM, Anderson SM, Shlomchik MJ. Germinal Center Initiation, Variable Gene Region Hypermutation, and Mutant B Cell Selection Without Detectable Immune Complexes on Follicular Dendritic Cells. J Exp Med (2000) 192:931–42. doi: 10.1084/jem.192.7.931
68. Sprent J, Basten A. Circulating T and B Lymphocytes of the Mouse. II. Lifespan. Cell Immunol (1973) 7:40–59. doi: 10.1016/0008-8749(73)90181-0
69. Hebeis BJ, Klenovsek K, Rohwer P, Ritter U, Schneider A, Mach M, et al. Activation of Virus-Specific Memory B Cells in the Absence of T Cell Help. J Exp Med (2004) 199:593–602. doi: 10.1084/jem.20030091
70. Yefenof E, Sanders VM, Uhr JW, Vitetta ES. In Vitro Activation of Murine Antigen-Specific Memory B Cells by a T-Dependent Antigen. J Immunol (1986) 137:85–90.
71. Ise W, Inoue T, McLachlan JB, Kometani K, Kubo M, Okada T, et al. Memory B Cells Contribute to Rapid Bcl6 Expression by Memory Follicular Helper T Cells. Proc Natl Acad Sci USA (2014) 111:11792–7. doi: 10.1073/pnas.1404671111
72. Aiba Y, Kometani K, Hamadate M, Moriyama S, Sakaue-Sawano A, Tomura M, et al. Preferential Localization of IgG Memory B Cells Adjacent to Contracted Germinal Centers. Proc Natl Acad Sci USA (2010) 107:12192–7. doi: 10.1073/pnas.1005443107
73. Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of Serological Memory by Polyclonal Activation of Human Memory B Cells. Science (2002) 298:2199–202. doi: 10.1126/science.1076071
74. Obukhanych TV, Nussenzweig MC. T-Independent Type II Immune Responses Generate Memory B Cells. J Exp Med (2006) 203:305–10. doi: 10.1084/jem.20052036
75. Palm AE, Henry C. Remembrance of Things Past: Long-Term B Cell Memory After Infection and Vaccination. Front Immunol (2019) 10:1787. doi: 10.3389/fimmu.2019.01787
76. Kurosaki T, Kometani K, Ise W. Memory B Cells. Nat Rev Immunol (2015) 15:149–59. doi: 10.1038/nri3802
77. Good KL, Tangye SG. Decreased Expression of Kruppel-Like Factors in Memory B Cells Induces the Rapid Response Typical of Secondary Antibody Responses. Proc Natl Acad Sci USA (2007) 104:13420–5. doi: 10.1073/pnas.0703872104
78. Liu YJ, Barthelemy C, de Bouteiller O, Arpin C, Durand I, Banchereau J. Memory B Cells From Human Tonsils Colonize Mucosal Epithelium and Directly Present Antigen to T Cells by Rapid Up-Regulation of B7-1 and B7-2. Immunity (1995) 2:239–48. doi: 10.1016/1074-7613(95)90048-9
79. Good KL, Avery DT, Tangye SG. Resting Human Memory B Cells are Intrinsically Programmed for Enhanced Survival and Responsiveness to Diverse Stimuli Compared to Naive B Cells. J Immunol (2009) 182:890–901. doi: 10.4049/jimmunol.182.2.890
80. Good KL, Bryant VL, Tangye SG. Kinetics of Human B Cell Behavior and Amplification of Proliferative Responses Following Stimulation With IL-21. J Immunol (2006) 177:5236–47. doi: 10.4049/jimmunol.177.8.5236
81. Bernasconi NL, Onai N, Lanzavecchia A. A Role for Toll-Like Receptors in Acquired Immunity: Up-Regulation of TLR9 by BCR Triggering in Naive B Cells and Constitutive Expression in Memory B Cells. Blood (2003) 101:4500–4. doi: 10.1182/blood-2002-11-3569
82. Laidlaw BJ, Cyster JG. Transcriptional Regulation of Memory B Cell Differentiation. Nat Rev Immunol (2020) 21:209–20. doi: 10.1038/s41577-020-00446-2
83. Shimoda M, Li T, Pihkala JP, Koni PA. Role of MHC Class II on Memory B Cells in Post-Germinal Center B Cell Homeostasis and Memory Response. J Immunol (2006) 176:2122–33. doi: 10.4049/jimmunol.176.4.2122
84. McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. Molecular Programming of B Cell Memory. Nat Rev Immunol (2011) 12:24–34. doi: 10.1038/nri3128
85. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, et al. Distinct Effector Cytokine Profiles of Memory and Naive Human B Cell Subsets and Implication in Multiple Sclerosis. J Immunol (2007) 178:6092–9. doi: 10.4049/jimmunol.178.10.6092
86. Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, et al. Proinflammatory GM-CSF-Producing B Cells in Multiple Sclerosis and B Cell Depletion Therapy. Sci Transl Med (2015) 7:310ra166. doi: 10.1126/scitranslmed.aab4176
87. Cognasse F, Hamzeh-Cognasse H, Lafarge S, Chavarin P, Pozzetto B, Richard Y, et al. Identification of Two Subpopulations of Purified Human Blood B Cells, CD27- CD23+ and CD27high CD80+, That Strongly Express Cell Surface Toll-Like Receptor 9 and Secrete High Levels of Interleukin-6. Immunology (2008) 125:430–7. doi: 10.1111/j.1365-2567.2008.02844.x
88. Corcione A, Casazza S, Ferretti E, Giunti D, Zappia E, Pistorio A, et al. Recapitulation of B Cell Differentiation in the Central Nervous System of Patients With Multiple Sclerosis. Proc Natl Acad Sci USA (2004) 101:11064–9. doi: 10.1073/pnas.0402455101
89. Harp C, Lee J, Lambracht-Washington D, Cameron E, Olsen G, Frohman E, et al. Cerebrospinal Fluid B Cells From Multiple Sclerosis Patients Are Subject to Normal Germinal Center Selection. J Neuroimmunol (2007) 183:189–99. doi: 10.1016/j.jneuroim.2006.10.020
90. Cepok S, Rosche B, Grummel V, Vogel F, Zhou D, Sayn J, et al. Short-Lived Plasma Blasts Are the Main B Cell Effector Subset During the Course of Multiple Sclerosis. Brain (2005) 128:1667–76. doi: 10.1093/brain/awh486
91. Eggers EL, Michel BA, Wu H, Wang SZ, Bevan CJ, Abounasr A, et al. Clonal Relationships of CSF B Cells in Treatment-Naive Multiple Sclerosis Patients. JCI Insight (2017) 2(22):e92724. doi: 10.1172/jci.insight.92724
92. Cepok S, von Geldern G, Grummel V, Hochgesand S, Celik H, Hartung H, et al. Accumulation of Class Switched IgD-IgM- Memory B Cells in the Cerebrospinal Fluid During Neuroinflammation. J Neuroimmunol (2006) 180:33–9. doi: 10.1016/j.jneuroim.2006.06.031
93. Kowarik MC AD, Lepennetier G, Ritchie A, Hemmer B, Owens GP, Bennett J. Fingolimod Alters Intrathecal B Cell Maturation in Multiple Sclerosis Patients. Res Square (2020). doi: 10.21203/rs.3.rs-23388/v1
94. Lossius A, Tomescu-Baciu A, Holmoy T, Vedeler CA, Rosjo E, Lorentzen AR, et al. Selective Intrathecal Enrichment of G1m1-Positive B Cells in Multiple Sclerosis. Ann Clin Transl Neurol (2017) 4:756–61. doi: 10.1002/acn3.451
95. Lindeman I, Polak J, Qiao SW, Holmoy T, Hoglund RA, Vartdal F, et al. Stereotyped B-Cell Responses Are Linked to IgG Constant Region Polymorphisms in Multiple Sclerosis. bioRxiv (2021). doi: 10.1101/2021.04.23.441098
96. Raknes G, Fernandes Filho JA, Pandey JP, Myhr KM, Ulvestad E, Nyland H, et al. IgG Allotypes and Subclasses in Norwegian Patients With Multiple Sclerosis. J Neurol Sci (2000) 175:111–5. doi: 10.1016/S0022-510X(00)00292-6
97. Blanc M, Clanet M, Berr C, Dugoujon JM, Ruydavet B, Ducos SJ, et al. Immunoglobulin Allotypes and Susceptibility to Multiple Sclerosis. An Epidemiological and Genetic Study in the Hautes-Pyrenees County of France. J Neurol Sci (1986) 75:1–5. doi: 10.1016/0022-510X(86)90045-6
98. Machado-Santos J, Saji E, Troscher AR, Paunovic M, Liblau R, Gabriely G, et al. The Compartmentalized Inflammatory Response in the Multiple Sclerosis Brain Is Composed of Tissue-Resident CD8+ T Lymphocytes and B Cells. Brain (2018) 141:2066–82. doi: 10.1093/brain/awy151
99. Serafini B, Severa M, Columba-Cabezas S, Rosicarelli B, Veroni C, Chiappetta G, et al. Epstein-Barr Virus Latent Infection and BAFF Expression in B Cells in the Multiple Sclerosis Brain: Implications for Viral Persistence and Intrathecal B-Cell Activation. J Neuropathol Exp Neurol (2010) 69:677–93. doi: 10.1097/NEN.0b013e3181e332ec
100. Owens GP, Kraus H, Burgoon MP, Smith-Jensen T, Devlin ME, Gilden DH. Restricted Use of VH4 Germline Segments in an Acute Multiple Sclerosis Brain. Ann Neurol (1998) 43:236–43. doi: 10.1002/ana.410430214
101. Baranzini SE, Jeong MC, Butunoi C, Murray RS, Bernard CC, Oksenberg JR. B Cell Repertoire Diversity and Clonal Expansion in Multiple Sclerosis Brain Lesions. J Immunol (1999) 163:5133–44.
102. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of Ectopic B-Cell Follicles With Germinal Centers in the Meninges of Patients With Secondary Progressive Multiple Sclerosis. Brain Pathol (2004) 14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x
103. Palanichamy A, Apeltsin L, Kuo TC, Sirota M, Wang S, Pitts SJ, et al. Immunoglobulin Class-Switched B Cells Form an Active Immune Axis Between CNS and Periphery in Multiple Sclerosis. Sci Transl Med (2014) 6:248ra106. doi: 10.1126/scitranslmed.3008930
104. Stern JN, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, et al. B Cells Populating the Multiple Sclerosis Brain Mature in the Draining Cervical Lymph Nodes. Sci Transl Med (2014) 6:248ra107. doi: 10.1126/scitranslmed.3008879
105. Ramesh A, Schubert RD, Greenfield AL, Dandekar R, Loudermilk R, Sabatino JJ Jr., et al. A Pathogenic and Clonally Expanded B Cell Transcriptome in Active Multiple Sclerosis. Proc Natl Acad Sci USA (2020) 117:22932–43. doi: 10.1073/pnas.2008523117
106. Alter A, Duddy M, Hebert S, Biernacki K, Prat A, Antel JP, et al. Determinants of Human B Cell Migration Across Brain Endothelial Cells. J Immunol (2003) 170:4497–505. doi: 10.4049/jimmunol.170.9.4497
107. Parker Harp CR, Archambault AS, Cheung M, Williams JW, Czepielewski RS, Duncker PC, et al. Neutrophils Promote VLA-4-Dependent B Cell Antigen Presentation and Accumulation Within the Meninges During Neuroinflammation. Proc Natl Acad Sci USA (2019) 116:24221–30. doi: 10.1073/pnas.1909098116
108. Walsh PN, Friedrich DP, Williams JA, Smith RJ, Stewart TL, Carter DK, et al. Optimization and Qualification of a Memory B-Cell Elispot for the Detection of Vaccine-Induced Memory Responses in HIV Vaccine Trials. J Immunol Methods (2013) 394:84–93. doi: 10.1016/j.jim.2013.05.007
109. Jahnmatz M, Kesa G, Netterlid E, Buisman AM, Thorstensson R, Ahlborg N. Optimization of a Human IgG B-Cell ELISpot Assay for the Analysis of Vaccine-Induced B-Cell Responses. J Immunol Methods (2013) 391:50–9. doi: 10.1016/j.jim.2013.02.009
110. Hohmann C, Milles B, Schinke M, Schroeter M, Ulzheimer J, Kraft P, et al. Categorization of Multiple Sclerosis Relapse Subtypes by B Cell Profiling in the Blood. Acta Neuropathol Commun (2014) 2:138. doi: 10.1186/s40478-014-0138-2
111. Kuerten S, Pommerschein G, Barth SK, Hohmann C, Milles B, Sammer FW, et al. Identification of a B Cell-Dependent Subpopulation of Multiple Sclerosis by Measurements of Brain-Reactive B Cells in the Blood. Clin Immunol (2014) 152:20–4. doi: 10.1016/j.clim.2014.02.014
112. Harp CT, Ireland S, Davis LS, Remington G, Cassidy B, Cravens PD, et al. Memory B Cells From a Subset of Treatment-Naive Relapsing-Remitting Multiple Sclerosis Patients Elicit CD4(+) T-Cell Proliferation and IFN-gamma Production in Response to Myelin Basic Protein and Myelin Oligodendrocyte Glycoprotein. Eur J Immunol (2010) 40:2942–56. doi: 10.1002/eji.201040516
113. Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory B Cells Activate Brain-Homing, Autoreactive Cd4(+) T Cells in Multiple Sclerosis. Cell (2018) 175:85–100.e23. doi: 10.1016/j.cell.2018.08.011
114. Maseda D, Smith SH, DiLillo DJ, Bryant JM, Candando KM, Weaver CT, et al. Regulatory B10 Cells Differentiate Into Antibody-Secreting Cells After Transient IL-10 Production In Vivo. J Immunol (2012) 188:1036–48. doi: 10.4049/jimmunol.1102500
115. Niino M, Hirotani M, Miyazaki Y, Sasaki H. Memory and Naive B-Cell Subsets in Patients With Multiple Sclerosis. Neurosci Lett (2009) 464:74–8. doi: 10.1016/j.neulet.2009.08.010
116. Nissimov N, Hajiyeva Z, Torke S, Grondey K, Bruck W, Hausser-Kinzel S, et al. B Cells Reappear Less Mature and More Activated After Their anti-CD20-Mediated Depletion in Multiple Sclerosis. Proc Natl Acad Sci USA (2020) 117:25690–9. doi: 10.1073/pnas.2012249117
117. Comabella M, Canto E, Nurtdinov R, Rio J, Villar LM, Picon C, et al. MRI Phenotypes With High Neurodegeneration Are Associated With Peripheral Blood B-Cell Changes. Hum Mol Genet (2016) 25:308–16. doi: 10.1093/hmg/ddv473
118. Schwarz A, Balint B, Korporal-Kuhnke M, Jarius S, von Engelhardt K, Furwentsches A, et al. B-Cell Populations Discriminate Between Pediatric- and Adult-Onset Multiple Sclerosis. Neurol Neuroimmunol Neuroinflamm (2017) 4:e309. doi: 10.1212/NXI.0000000000000309
119. Lin MT, Hinton DR, Marten NW, Bergmann CC, Stohlman SA. Antibody Prevents Virus Reactivation Within the Central Nervous System. J Immunol (1999) 162:7358–68.
120. Kang BS, Palma JP, Lyman MA, Dal Canto M, Kim BS. Antibody Response is Required for Protection From Theiler’s Virus-Induced Encephalitis in C57BL/6 Mice in the Absence of CD8+ T Cells. Virology (2005) 340:84–94. doi: 10.1016/j.virol.2005.06.028
121. DiSano KD, Royce DB, Gilli F, Pachner AR. Central Nervous System Inflammatory Aggregates in the Theiler’s Virus Model of Progressive Multiple Sclerosis. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.01821
122. Jin YH, Kim CX, Huang J, Kim BS. Infection and Activation of B Cells by Theiler’s Murine Encephalomyelitis Virus (TMEV) Leads to Autoantibody Production in an Infectious Model of Multiple Sclerosis. Cells (2020) 9(8)1787. doi: 10.3390/cells9081787
123. Gilli F, Li L, Campbell SJ, Anthony DC, Pachner AR. The Effect of B-Cell Depletion in the Theiler’s Model of Multiple Sclerosis. J Neurol Sci (2015) 359:40–7. doi: 10.1016/j.jns.2015.10.012
124. Houtman JJ, Fleming JO. Dissociation of Demyelination and Viral Clearance in Congenitally Immunodeficient Mice Infected With Murine Coronavirus JHM. J Neurovirol (1996) 2:101–10. doi: 10.3109/13550289609146543
125. Sutherland RM, Chua MM, Lavi E, Weiss SR, Paterson Y. CD4+ and CD8+ T Cells are Not Major Effectors of Mouse Hepatitis Virus A59-Induced Demyelinating Disease. J Neurovirol (1997) 3:225–8. doi: 10.3109/13550289709018297
126. Atkinson JR, Hwang M, Reyes-Rodriguez A, Bergmann CC. Dynamics of Virus-Specific Memory B Cells and Plasmablasts Following Viral Infection of the Central Nervous System. J Virol (2019) 93(2):e00875–18. doi: 10.1128/JVI.00875-18
127. Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. Theiler’s Virus Infection: A Model for Multiple Sclerosis. Clin Microbiol Rev (2004) 17:174–207. doi: 10.1128/CMR.17.1.174-207.2004
128. Lassmann H, Bradl M. Multiple Sclerosis: Experimental Models and Reality. Acta Neuropathol (2017) 133:223–44. doi: 10.1007/s00401-016-1631-4
129. Kuerten S, Javeri S, Tary-Lehmann M, Lehmann PV, Angelov DN. Fundamental Differences in the Dynamics of CNS Lesion Development and Composition in MP4- and MOG Peptide 35-55-Induced Experimental Autoimmune Encephalomyelitis. Clin Immunol (2008) 129:256–67. doi: 10.1016/j.clim.2008.07.016
130. DiSano KD, Linzey MR, Royce DB, Pachner AR, Gilli F. Differential Neuro-Immune Patterns in Two Clinically Relevant Murine Models of Multiple Sclerosis. J Neuroinflamm (2019) 16:109. doi: 10.1186/s12974-019-1501-9
131. Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, et al. B Cell Depletion Therapy Ameliorates Autoimmune Disease Through Ablation of IL-6-Producing B Cells. J Exp Med (2012) 209:1001–10. doi: 10.1084/jem.20111675
132. Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B Cells Inhibit EAE Initiation in Mice While Other B Cells Promote Disease Progression. J Clin Invest (2008) 118:3420–30. doi: 10.1172/JCI36030
133. Kap YS, van Driel N, Blezer E, Parren PW, Bleeker WK, Laman JD, et al. Late B Cell Depletion With a Human Anti-Human CD20 IgG1kappa Monoclonal Antibody Halts the Development of Experimental Autoimmune Encephalomyelitis in Marmosets. J Immunol (2010) 185:3990–4003. doi: 10.4049/jimmunol.1001393
134. Jagessar SA, Heijmans N, Bauer J, Blezer EL, Laman JD, Hellings N, et al. B-Cell Depletion Abrogates T Cell-Mediated Demyelination in an Antibody-Nondependent Common Marmoset Experimental Autoimmune Encephalomyelitis Model. J Neuropathol Exp Neurol (2012) 71:716–28. doi: 10.1097/NEN.0b013e3182622691
135. DiLillo DJ, Hamaguchi Y, Ueda Y, Yang K, Uchida J, Haas KM, et al. Maintenance of Long-Lived Plasma Cells and Serological Memory Despite Mature and Memory B Cell Depletion During CD20 Immunotherapy in Mice. J Immunol (2008) 180:361–71. doi: 10.4049/jimmunol.180.1.361
136. Gaurav Kumar RMK, Robert C. Axtell, B Cell Maturation Antigen Deficiency Increases Inflammatory B-Cell Functions in Multiple Sclerosis. J Immunol (2020) 204.
137. Gross JA, Dillon SR, Mudri S, Johnston J, Littau A, Roque R, et al. TACI-Ig Neutralizes Molecules Critical for B Cell Development and Autoimmune Disease. Impaired B Cell Maturation in Mice Lacking BLyS. Immunity (2001) 15:289–302. doi: 10.1016/S1074-7613(01)00183-2
138. Hartung HP, Kieseier BC. Atacicept: Targeting B Cells in Multiple Sclerosis. Ther Adv Neurol Disord (2010) 3:205–16. doi: 10.1177/1756285610371146
139. Ma N, Xiao H, Marrero B, Xing C, Wang X, Zheng M, et al. Combination of TACI-IgG and Anti-IL-15 Treats Murine Lupus by Reducing Mature and Memory B Cells. Cell Immunol (2014) 289:140–4. doi: 10.1016/j.cellimm.2014.03.017
140. Litzenburger T, Fassler R, Bauer J, Lassmann H, Linington C, Wekerle H, et al. B Lymphocytes Producing Demyelinating Autoantibodies: Development and Function in Gene-Targeted Transgenic Mice. J Exp Med (1998) 188:169–80. doi: 10.1084/jem.188.1.169
141. Roll P, Palanichamy A, Kneitz C, Dorner T, Tony HP. Regeneration of B Cell Subsets After Transient B Cell Depletion Using Anti-CD20 Antibodies in Rheumatoid Arthritis. Arthritis Rheum (2006) 54:2377–86. doi: 10.1002/art.22019
142. Palanichamy A, Jahn S, Nickles D, Derstine M, Abounasr A, Hauser SL, et al. Rituximab Efficiently Depletes Increased CD20-Expressing T Cells in Multiple Sclerosis Patients. J Immunol (2014) 193:580–6. doi: 10.4049/jimmunol.1400118
143. Gingele S, Skripuletz T, Jacobs R. Role of CD20(+) T Cells in Multiple Sclerosis: Implications for Treatment With Ocrelizumab. Neural Regener Res (2020) 15:663–4. doi: 10.4103/1673-5374.266913
144. Nguyen AL, Gresle M, Marshall T, Butzkueven H, Field J. Monoclonal Antibodies in the Treatment of Multiple Sclerosis: Emergence of B-Cell-Targeted Therapies. Br J Pharmacol (2017) 174:1895–907. doi: 10.1111/bph.13780
145. Bar-Or A, Grove RA, Austin DJ, Tolson JM, VanMeter SA, Lewis EW, et al. Subcutaneous Ofatumumab in Patients With Relapsing-Remitting Multiple Sclerosis: The MIRROR Study. Neurology (2018) 90:e1805–14. doi: 10.1212/WNL.0000000000005516
146. Novi G, Bovis F, Fabbri S, Tazza F, Gazzola P, Maietta I, et al. Tailoring B Cell Depletion Therapy in MS According to Memory B Cell Monitoring. Neurol Neuroimmunol Neuroinflamm (2020) 7(5):e845. doi: 10.1212/NXI.0000000000000845
147. Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab Reduces B Cells and T Cells in Cerebrospinal Fluid of Multiple Sclerosis Patients. J Neuroimmunol (2006) 180:63–70. doi: 10.1016/j.jneuroim.2006.06.029
148. Petereit HF, Moeller-Hartmann W, Reske D, Rubbert A. Rituximab in a Patient With Multiple Sclerosis–Effect on B Cells, Plasma Cells and Intrathecal IgG Synthesis. Acta Neurol Scand (2008) 117:399–403. doi: 10.1111/j.1600-0404.2007.00958.x
149. Monson NL, Cravens PD, Frohman EM, Hawker K, Racke MK. Effect of Rituximab on the Peripheral Blood and Cerebrospinal Fluid B Cells in Patients With Primary Progressive Multiple Sclerosis. Arch Neurol (2005) 62:258–64. doi: 10.1001/archneur.62.2.258
150. Baker D, Pryce G, James LK, Marta M, Schmierer K. The Ocrelizumab Phase II Extension Trial Suggests the Potential to Improve the Risk: Benefit Balance in Multiple Sclerosis. Mult Scler Relat Disord (2020) 44:102279. doi: 10.1016/j.msard.2020.102279
151. Amit Bar-Or JB, Von Budingen H, Carruthers R, Edwards K, Fallis R, Fiore D, et al. B Cells, T Cells and Inflammatory CSF Biomarkers in Primary Progressive MS and Relapsing MS in the OBOE (Ocrelizumab Biomarker Outcome Evaluation) Trial. Neurology (2020) 94.
152. Tak PP, Thurlings RM, Rossier C, Nestorov I, Dimic A, Mircetic V, et al. Atacicept in Patients With Rheumatoid Arthritis: Results of a Multicenter, Phase Ib, Double-Blind, Placebo-Controlled, Dose-Escalating, Single- and Repeated-Dose Study. Arthritis Rheum (2008) 58:61–72. doi: 10.1002/art.23178
153. Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, et al. Cutting Edge: The Dependence of Plasma Cells and Independence of Memory B Cells on BAFF and APRIL. J Immunol (2008) 180:3655–9. doi: 10.4049/jimmunol.180.6.3655
154. Baker D, Pryce G, James LK, Schmierer K, Giovannoni G. Failed B Cell Survival Factor Trials Support the Importance of Memory B Cells in Multiple Sclerosis. Eur J Neurol (2020) 27:221–8. doi: 10.1111/ene.14105
155. Nadler LM, Anderson KC, Marti G, Bates M, Park E, Daley JF, et al. B4, a Human B Lymphocyte-Associated Antigen Expressed on Normal, Mitogen-Activated, and Malignant B Lymphocytes. J Immunol (1983) 131:244–50.
156. Herbst R, Wang Y, Gallagher S, Mittereder N, Kuta E, Damschroder M, et al. B-Cell Depletion In Vitro and In Vivo With an Afucosylated Anti-CD19 Antibody. J Pharmacol Exp Ther (2010) 335:213–22. doi: 10.1124/jpet.110.168062
157. Agius MA, Klodowska-Duda G, Maciejowski M, Potemkowski A, Li J, Patra K, et al. Safety and Tolerability of Inebilizumab (MEDI-551), An Anti-CD19 Monoclonal Antibody, in Patients With Relapsing Forms of Multiple Sclerosis: Results From a Phase 1 Randomised, Placebo-Controlled, Escalating Intravenous and Subcutaneous Dose Study. Mult Scler (2019) 25:235–45. doi: 10.1177/1352458517740641
158. Dolgin E. BTK Blockers Make Headway in Multiple Sclerosis. Nat Biotechnol (2021) 39:3–5. doi: 10.1038/s41587-020-00790-7
159. Caldwell RD, Qiu H, Askew BC, Bender AT, Brugger N, Camps M, et al. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J Med Chem (2019) 62:7643–55. doi: 10.1021/acs.jmedchem.9b00794
160. Mangla A, Khare A, Vineeth V, Panday NN, Mukhopadhyay A, Ravindran B, et al. Pleiotropic Consequences of Bruton Tyrosine Kinase Deficiency in Myeloid Lineages Lead to Poor Inflammatory Responses. Blood (2004) 104:1191–7. doi: 10.1182/blood-2004-01-0207
161. Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al. Deficient Expression of a B Cell Cytoplasmic Tyrosine Kinase in Human X-Linked Agammaglobulinemia. Cell (1993) 72:279–90. doi: 10.1016/0092-8674(93)90667-F
162. Montalban X, Shaw J, Syed S, Dangond F, Martin EC, Grenningloh R, et al. Effect of Evobrutinib, a Bruton’s Tyrosine Kinase Inhibitor, on Immune Cell and Immunoglobulin Levels Over 48 Weeks in a Phase 2 Study in Relapsing Multiple Sclerosis. Stockholm, Sweden: European Committee For Treatment And Research In Multiple Sclerosis (2019).
163. Rijvers L, Melief M-J, van Langelaar J, Hendriks R, Boschert U, Grenningloh R, et al. Brain-Homing B Cells in Multiple Sclerosis: Association With Bruton’s Tyrosine Kinase and Targeting by Evobrutinib. Neurology (2021).
164. Dhib-Jalbut S, Marks S. Interferon-Beta Mechanisms of Action in Multiple Sclerosis. Neurology (2010) 74 Suppl 1:S17–24. doi: 10.1212/WNL.0b013e3181c97d99
165. Rizzo F, Giacomini E, Mechelli R, Buscarinu MC, Salvetti M, Severa M, et al. Interferon-Beta Therapy Specifically Reduces Pathogenic Memory B Cells in Multiple Sclerosis Patients by Inducing a FAS-Mediated Apoptosis. Immunol Cell Biol (2016) 94:886–94. doi: 10.1038/icb.2016.55
166. Maimaitijiang G, Watanabe M, Shinoda K, Isobe N, Nakamura Y, Masaki K, et al. Long-Term Use of Interferon-Beta in Multiple Sclerosis Increases Vdelta1(-)Vdelta2(-)Vgamma9(-) Gammadelta T Cells That Are Associated With a Better Outcome. J Neuroinflamm (2019) 16:179. doi: 10.1186/s12974-019-1574-5
167. Jiang H, Milo R, Swoveland P, Johnson KP, Panitch H, Dhib-Jalbut S. Interferon Beta-1b Reduces Interferon Gamma-Induced Antigen-Presenting Capacity of Human Glial and B Cells. J Neuroimmunol (1995) 61:17–25. doi: 10.1016/0165-5728(95)00072-A
168. Huang H, Ito K, Dangond F, Dhib-Jalbut S. Effect of Interferon Beta-1a on B7.1 and B7.2 B-Cell Expression and its Impact on T-Cell Proliferation. J Neuroimmunol (2013) 258:27–31. doi: 10.1016/j.jneuroim.2013.02.010
169. Liu Z, Pelfrey CM, Cotleur A, Lee JC, Rudick RA. Immunomodulatory Effects of Interferon Beta-1a in Multiple Sclerosis. J Neuroimmunol (2001) 112:153–62. doi: 10.1016/S0165-5728(00)00403-3
170. Schubert RD, Hu Y, Kumar G, Szeto S, Abraham P, Winderl J, et al. IFN-Beta Treatment Requires B Cells for Efficacy in Neuroautoimmunity. J Immunol (2015) 194:2110–6. doi: 10.4049/jimmunol.1402029
171. Lalive PH, Neuhaus O, Benkhoucha M, Burger D, Hohlfeld R, Zamvil SS, et al. Glatiramer Acetate in the Treatment of Multiple Sclerosis: Emerging Concepts Regarding its Mechanism of Action. CNS Drugs (2011) 25:401–14. doi: 10.2165/11588120-000000000-00000
172. Ireland SJ, Guzman AA, O’Brien DE, Hughes S, Greenberg B, Flores A, et al. The Effect of Glatiramer Acetate Therapy on Functional Properties of B Cells From Patients With Relapsing-Remitting Multiple Sclerosis. JAMA Neurol (2014) 71:1421–8. doi: 10.1001/jamaneurol.2014.1472
173. Hausler D, Hajiyeva Z, Traub JW, Zamvil SS, Lalive PH, Bruck W, et al. Glatiramer Acetate Immune Modulates B-Cell Antigen Presentation in Treatment of MS. Neurol Neuroimmunol Neuroinflamm (2020) 7(3):e698. doi: 10.1212/NXI.0000000000000698
174. Deeks ED. Cladribine Tablets: A Review in Relapsing MS. CNS Drugs (2018) 32:785–96. doi: 10.1007/s40263-018-0562-0
175. Ceronie B, Jacobs BM, Baker D, Dubuisson N, Mao Z, Ammoscato F, et al. Cladribine Treatment of Multiple Sclerosis is Associated With Depletion of Memory B Cells. J Neurol (2018) 265:1199–209. doi: 10.1007/s00415-018-8830-y
176. Claes N DT, Fraussen J, Broux B, Van Wijmeersch B, Stinissen P, Hupperts R, et al. Compositional Changes of B and T Cell Subtypes During Fingolimod Treatment in Multiple Sclerosis Patients: A 12-Month Follow-Up Study. PloS One (2014) 31(10):e111115. doi: 10.1371/journal.pone.0111115
177. Nakamura M, Matsuoka T, Chihara N, Miyake S, Sato W, Araki M, et al. Differential Effects of Fingolimod on B-Cell Populations in Multiple Sclerosis. Mult Scler (2014) 20:1371–80. doi: 10.1177/1352458514523496
178. Traub JW, Pellkofer HL, Grondey K, Seeger I, Rowold C, Bruck W, et al. Natalizumab Promotes Activation and Pro-Inflammatory Differentiation of Peripheral B Cells in Multiple Sclerosis Patients. J Neuroinflamm (2019) 16:228. doi: 10.1186/s12974-019-1593-2
179. Kowarik MC, Pellkofer HL, Cepok S, Korn T, Kumpfel T, Buck D, et al. Differential Effects of Fingolimod (FTY720) on Immune Cells in the CSF and Blood of Patients With MS. Neurology (2011) 76:1214–21. doi: 10.1212/WNL.0b013e3182143564
180. Bomprezzi R. Dimethyl Fumarate in the Treatment of Relapsing-Remitting Multiple Sclerosis: An Overview. Ther Adv Neurol Disord (2015) 8:20–30. doi: 10.1177/1756285614564152
181. Smith MD, Martin KA, Calabresi PA, Bhargava P. Dimethyl Fumarate Alters B-Cell Memory and Cytokine Production in MS Patients. Ann Clin Transl Neurol (2017) 4:351–5. doi: 10.1002/acn3.411
182. Lundy SK, Wu Q, Wang Q, Dowling CA, Taitano SH, Mao G, et al. Dimethyl Fumarate Treatment of Relapsing-Remitting Multiple Sclerosis Influences B-Cell Subsets. Neurol Neuroimmunol Neuroinflamm (2016) 3:e211. doi: 10.1212/NXI.0000000000000211
183. Li R, Rezk A, Ghadiri M, Luessi F, Zipp F, Li H, et al. Dimethyl Fumarate Treatment Mediates an Anti-Inflammatory Shift in B Cell Subsets of Patients With Multiple Sclerosis. J Immunol (2017) 198:691–8. doi: 10.4049/jimmunol.1601649
184. Hoglund RA, Polak J, Vartdal F, Holmoy T, Lossius A. B-Cell Composition in the Blood and Cerebrospinal Fluid of Multiple Sclerosis Patients Treated With Dimethyl Fumarate. Mult Scler Relat Disord (2018) 26:90–5. doi: 10.1016/j.msard.2018.08.032
185. Gandoglia I, Ivaldi F, Laroni A, Benvenuto F, Solaro C, Mancardi G, et al. Teriflunomide Treatment Reduces B Cells in Patients With MS. Neurol Neuroimmunol Neuroinflamm (2017) 4:e403. doi: 10.1212/NXI.0000000000000403
186. Medina S, Sainz de la Maza S, Villarrubia N, Alvarez-Lafuente R, Costa-Frossard L, Arroyo R, et al. Teriflunomide Induces a Tolerogenic Bias in Blood Immune Cells of MS Patients. Ann Clin Transl Neurol (2019) 6:355–63. doi: 10.1002/acn3.711
187. Li L, Liu J, Delohery T, Zhang D, Arendt C, Jones C. The Effects of Teriflunomide on Lymphocyte Subpopulations in Human Peripheral Blood Mononuclear Cells In Vitro. J Neuroimmunol (2013) 265:82–90. doi: 10.1016/j.jneuroim.2013.10.003
188. Chan A, Weilbach FX, Toyka KV, Gold R. Mitoxantrone Induces Cell Death in Peripheral Blood Leucocytes of Multiple Sclerosis Patients. Clin Exp Immunol (2005) 139:152–8. doi: 10.1111/j.1365-2249.2005.02653.x
189. Neuhaus O, Wiendl H, Kieseier BC, Archelos JJ, Hemmer B, Stuve O, et al. Multiple Sclerosis: Mitoxantrone Promotes Differential Effects on Immunocompetent Cells In Vitro. J Neuroimmunol (2005) 168:128–37. doi: 10.1016/j.jneuroim.2005.01.024
190. Hu Y, Turner MJ, Shields J, Gale MS, Hutto E, Roberts BL, et al. Investigation of the Mechanism of Action of Alemtuzumab in a Human CD52 Transgenic Mouse Model. Immunology (2009) 128:260–70. doi: 10.1111/j.1365-2567.2009.03115.x
191. Gilleece MH, Dexter TM. Effect of Campath-1H Antibody on Human Hematopoietic Progenitors In Vitro. Blood (1993) 82:807–12. doi: 10.1182/blood.V82.3.807.bloodjournal823807
192. Thompson SA, Jones JL, Cox AL, Compston DA, Coles AJ. B-Cell Reconstitution and BAFF After Alemtuzumab (Campath-1H) Treatment of Multiple Sclerosis. J Clin Immunol (2010) 30:99–105. doi: 10.1007/s10875-009-9327-3
193. Planas R, Jelcic I, Schippling S, Martin R, Sospedra M. Natalizumab Treatment Perturbs Memory- and Marginal Zone-Like B-Cell Homing in Secondary Lymphoid Organs in Multiple Sclerosis. Eur J Immunol (2012) 42:790–8. doi: 10.1002/eji.201142108
194. Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A Randomized, Placebo-Controlled Trial of Natalizumab for Relapsing Multiple Sclerosis. N Engl J Med (2006) 354:899–910. doi: 10.1056/NEJMoa044397
195. Baldassari LE, Rose JW. Daclizumab: Development, Clinical Trials, and Practical Aspects of Use in Multiple Sclerosis. Neurotherapeutics (2017) 14:842–58. doi: 10.1007/s13311-017-0553-8
196. Kappos L, Li D, Calabresi PA, O’Connor P, Bar-Or A, Barkhof F, et al. Ocrelizumab in Relapsing-Remitting Multiple Sclerosis: A Phase 2, Randomised, Placebo-Controlled, Multicentre Trial. Lancet (2011) 378:1779–87. doi: 10.1016/S0140-6736(11)61649-8
197. Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab Versus Placebo in Primary Progressive Multiple Sclerosis. N Engl J Med (2017) 376:209–20. doi: 10.1056/NEJMoa1606468
198. Chen D, Gallagher S, Monson NL, Herbst R, Wang Y. Inebilizumab, a B Cell-Depleting Anti-CD19 Antibody for the Treatment of Autoimmune Neurological Diseases: Insights From Preclinical Studies. J Clin Med (2016) 5(12):107. doi: 10.3390/jcm5120107
199. Kappos L, Hartung HP, Freedman MS, Boyko A, Radu EW, Mikol DD, et al. Atacicept in Multiple Sclerosis (ATAMS): A Randomised, Placebo-Controlled, Double-Blind, Phase 2 Trial. Lancet Neurol (2014) 13:353–63. doi: 10.1016/S1474-4422(14)70028-6
200. Maria Silk EN. Efficacy and Safety of Tabalumab in Patients With Relapsing-Remitting Multiple Sclerosis: A Randomized, Double-Blind, Placebo-Controlled Study. Neurology (2018) 90.
201. Genovese MC, Kinnman N, de La Bourdonnaye G, Pena Rossi C, Tak PP. Atacicept in Patients With Rheumatoid Arthritis and an Inadequate Response to Tumor Necrosis Factor Antagonist Therapy: Results of a Phase II, Randomized, Placebo-Controlled, Dose-Finding Trial. Arthritis Rheum (2011) 63:1793–803. doi: 10.1002/art.30373
202. Francesco M, Wong M, LaStant J, Finkle D, Lowenstein N, Macsata R, et al. PRN2246, A Potent and Selective Blood Brain Barrier Penetrating BTK Inhibitor, Exhibits Efficacy in Central Nervous System Immunity European Committee For Treatment And Research In Multiple Sclerosis. (2017).
203. Keaney J, Gasser J, Gillet G, Scholz D, Kadiu I. Inhibition of Bruton’s Tyrosine Kinase Modulates Microglial Phagocytosis: Therapeutic Implications for Alzheimer’s Disease. J Neuroimmune Pharmacol (2019) 14:448–61. doi: 10.1007/s11481-019-09839-0
204. Montalban X, Arnold DL, Weber MS, Staikov I, Piasecka-Stryczynska K, Willmer J, et al. Placebo-Controlled Trial of an Oral BTK Inhibitor in Multiple Sclerosis. N Engl J Med (2019) 380:2406–17. doi: 10.1056/NEJMoa1901981
205. Syed S, Yonkers N, LaGanke C, Honeycutt WD, Traboulsee A, Wynn D, et al. Efficacy and Safety of Tolebrutinib in Patients With Highly Active Relapsing MS: Subgroup Analysis of the Phase 2b Study. (2021). American Academy of Neurology.
206. Rijvers L, Melief M-J, van Langelaar J, Hendriks R, Boschert U, Grenningloh R, et al. Brain-Homing B Cells in Multiple Sclerosis: Association With Bruton’s Tyrosine Kinase and Targeting by Evobrutinib (4095). Neurology (2021) 96.
207. Roll P, Dorner T, Tony HP. Anti-CD20 Therapy in Patients With Rheumatoid Arthritis: Predictors of Response and B Cell Subset Regeneration After Repeated Treatment. Arthritis Rheum (2008) 58:1566–75. doi: 10.1002/art.23473
208. Leandro MJ, Cambridge G, Ehrenstein MR, Edwards JC. Reconstitution of Peripheral Blood B Cells After Depletion With Rituximab in Patients With Rheumatoid Arthritis. Arthritis Rheum (2006) 54:613–20. doi: 10.1002/art.21617
Keywords: memory B cells, multiple sclerosis, neuroinflammation, B cells, multiple sclerosis-drug therapy
Citation: DiSano KD, Gilli F and Pachner AR (2021) Memory B Cells in Multiple Sclerosis: Emerging Players in Disease Pathogenesis. Front. Immunol. 12:676686. doi: 10.3389/fimmu.2021.676686
Received: 05 March 2021; Accepted: 11 May 2021;
Published: 08 June 2021.
Edited by:
Valentina Tomassini, University of Studies G. d’Annunzio Chieti and Pescara, ItalyReviewed by:
Trygve Holmøy, Akershus University Hospital, NorwayMarco Capobianco, University Hospital S. Luigi, Orbassano, Italy
Copyright © 2021 DiSano, Gilli and Pachner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Krista D. DiSano, S3Jpc3RhLkQuRGlTYW5vQGRhcnRtb3V0aC5lZHU=