 Jayashree Srinivasan1†
Jayashree Srinivasan1† Jessica N. Lancaster2†
Jessica N. Lancaster2† Nandini Singarapu3Laura P. Hale4
Nandini Singarapu3Laura P. Hale4 Lauren I. R. Ehrlich1,5‡
Lauren I. R. Ehrlich1,5‡ Ellen R. Richie3*‡
Ellen R. Richie3*‡- 1Department of Molecular Biosciences, Institute of Cellular and Molecular Biology, The University of Texas at Austin, Austin, TX, United States
- 2Department of Immunology, Mayo Clinic, Scottsdale, AZ, United States
- 3Department of Epigenetics and Molecular Carcinogenesis, The University of Texas M.D. Anderson Cancer Center, Smithville, TX, United States
- 4Department of Pathology, Duke University School of Medicine, Durham, NC, United States
- 5Livestrong Cancer Institutes, Dell Medical School, The University of Texas at Austin, Austin, TX, United States
Thymic epithelial cells (TECs) and hematopoietic antigen presenting cells (HAPCs) in the thymus microenvironment provide essential signals to self-reactive thymocytes that induce either negative selection or generation of regulatory T cells (Treg), both of which are required to establish and maintain central tolerance throughout life. HAPCs and TECs are comprised of multiple subsets that play distinct and overlapping roles in central tolerance. Changes that occur in the composition and function of TEC and HAPC subsets across the lifespan have potential consequences for central tolerance. In keeping with this possibility, there are age-associated changes in the cellular composition and function of T cells and Treg. This review summarizes changes in T cell and Treg function during the perinatal to adult transition and in the course of normal aging, and relates these changes to age-associated alterations in thymic HAPC and TEC subsets.
Introduction
Throughout life, the immune system must balance the opposing goals of mounting protective responses against diverse pathogens, while preventing a breakdown in self-tolerance. Maintaining this tenuous balance is complicated by age-related changes in the number and composition of cells that comprise the innate and adaptive immune systems, as well as by changes in hematopoiesis, lymphoid and non-lymphoid tissue microenvironments, and an individual’s history of pathogen exposure. Neonates encounter a barrage of new pathogens, requiring broad and rapid immune protection, at a time when their immune system is skewed towards mounting tolerogenic responses essential for tissue homeostasis (1). In contrast, following a lifetime of pathogen encounters, the T-cell compartment in older individuals contains a higher frequency of memory T cells, which can combat previously encountered pathogens, but often mounts poor responses to newly encountered pathogens and vaccines (2). (Figure 1). Immune responses to self-antigens also exhibit age-associated trends with the onset of many autoimmune disorders peaking in middle age (3) (Figure 1). Notably, there are some similarities between manifestations of immune dysregulation at both ends of the age spectrum, as neonates and elderly individuals have elevated susceptibility to various pathogens relative to adults, but less susceptibility to new-onset autoimmune disorders. For example, neonates are highly susceptible to respiratory syncytial virus (RSV) (4), whereas elderly individuals often mount inadequate immune responses to influenza and West Nile viruses (5, 6). T cells play a central role in modulating the outcome of immune responses by integrating initial signals from the innate immune system with T cell receptor (TCR)-mediated antigen recognition to shift the balance in favor of pathogen-directed protective versus tolerogenic outcomes. Distinct T-cell subset composition, phenotypes, and effector functions have been identified in neonates and in aged individuals compared to younger adults, but the underlying mechanisms responsible for the distinctive age-associated features of T-cell immunity have not been fully established.
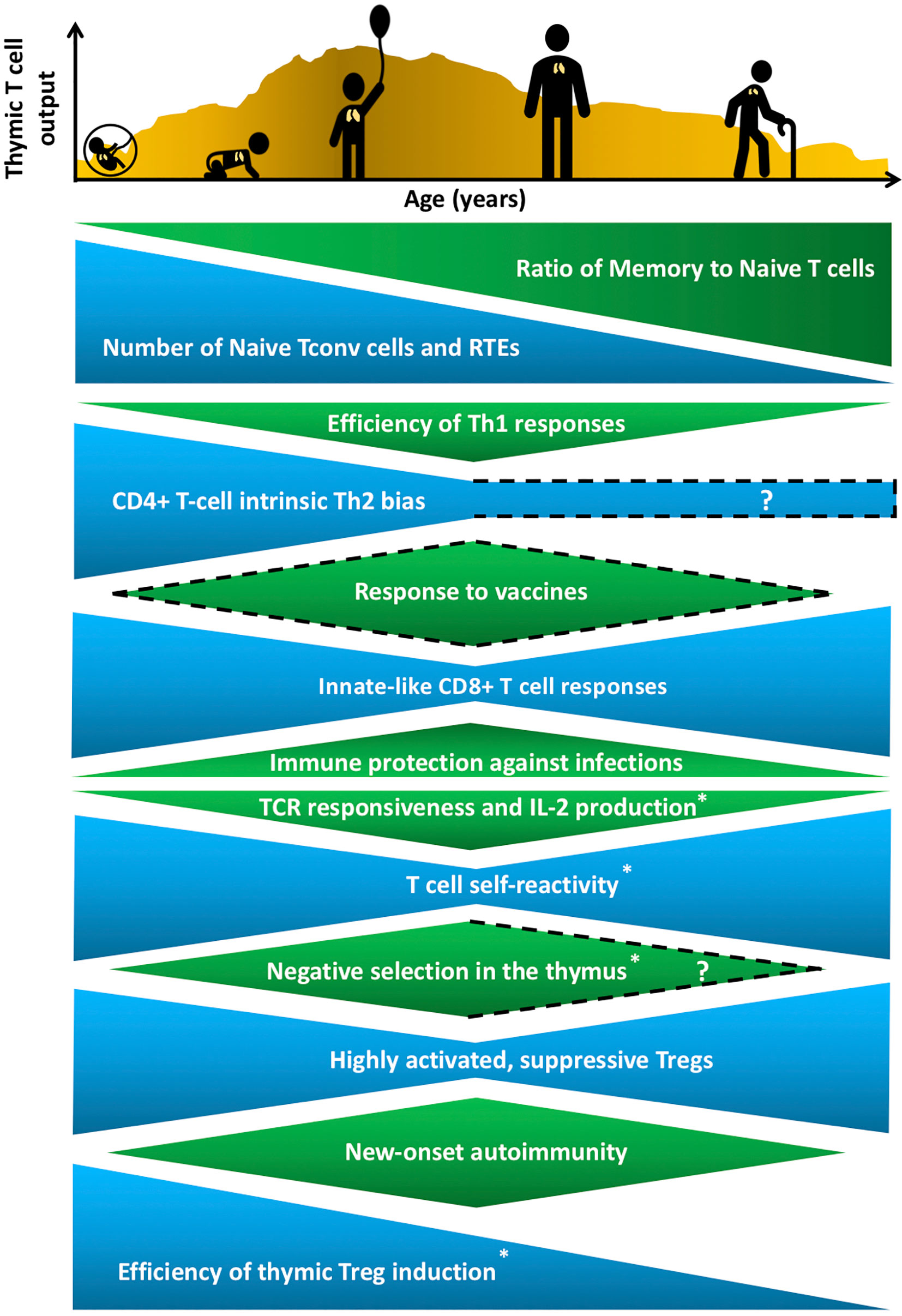
Figure 1 Age-associated changes in T cell generation and function throughout the lifespan. The T cell landscape is in flux throughout life, shaped by age-associated changes in T-cell subset composition and function, which are influenced by cell-intrinsic factors as well as microenvironmental cues that support T cell development and differentiation. While the perinatal T cell pool is dominated by naive conventional T cells (Tconv) and recent thymic emigrants (RTEs), the aged T cell pool contains a higher proportion of memory T cells. Perinatal and aged T cells share several striking similarities in phenotypes and functions. The perinatal and aged CD8+ Tconv cells, including virtual memory T cells (Tvm), are shifted towards short-lived, innate-like, effector responses characterized by increased proliferative potential and rapid cytokine production, at the expense of long-lasting memory generation. Naive CD4+ T cells also display age-associated changes at both ends of the age spectrum, such as reduced T cell receptor (TCR) responsiveness and IL-2 production. In addition, T cells are more self-reactive both early and late in life, which may reflect age-associated changes in thymic selection and/or peripheral maintenance. Regulatory T cells (Treg) generation in the thymus peaks in the perinatal period, but Tregs at both ends of the age spectrum have superior suppressive capacity compared to adult Tregs. These age-associated changes implicate the thymic microenvironment in selecting Tconv cells and Tregs that cater to rapidly changing immune challenges throughout life, while at the same time curbing the risk of triggering autoimmunity. T cell output from the thymus is also lower in both fetal/neonatal periods as well as in the elderly. The uneven pattern of thymic output depicted in the histogram reflects variability throughout life due to numerous extrinsic stressors, such as infections and pregnancy, that alter thymic cellularity and output. In keeping with the above similarities between T cells in the perinatal and elderly stages, immune outcomes, such as overall responsiveness to vaccines and pathogens, as well susceptibility to new onset autoimmunity change in similar directions at both extremes of the lifespan. Phenotypes with question marks are yet to be defined clearly, and dotted lines indicate variable findings in the indicated attributes. All features have been reported in both humans and mice, except those denoted with an asterisk that indicates findings currently reported only in mice in the perinatal to adult and/or adult to aged transitions.
T cells develop in the unique tissue microenvironment of the thymus (Figure 2), in which thymic epithelial cells (TECs) and hematopoietic antigen presenting cells (HAPCs) provide indispensable signals for T-cell maturation and/or the establishment of self-tolerance. Bone-marrow derived hematopoietic progenitors are recruited from circulation into the postnatal thymus. These CD4-CD8- “double negative” (DN) precursors then undergo T-cell lineage specification and differentiation in the thymic cortex. Following productive rearrangement of TCRβ gene segments, DN thymocytes initiate expression of the TCR co-receptors CD4 and CD8 and are referred to as “double positive” (DP) cells. DPs undergo TCRα gene rearrangements resulting in expression of functional αβTCR heterodimers that scan self-peptide/MHC complexes (pMHC) presented by cortical thymic epithelial cells (cTECs). Only thymocytes that express a TCR of sufficient affinity for either MHCI- or MHCII-peptide complexes are signaled to survive and further differentiate to CD8+ or CD4+ single positive (SP) lineages, respectively, through the process of positive selection (7, 8). A range of TCR affinities is compatible with positive selection, and the level of thymocyte self-reactivity has been shown to affect the subsequent threshold of peripheral T cell activation. Positively selected thymocytes migrate into the medulla, a region specialized for the induction of central tolerance. Within the medulla, TECs and HAPCs display a diverse array of self-peptides. Thymocytes expressing TCRs of relatively high affinity for self-pMHC are either triggered to undergo apoptosis, through the process of negative selection, or are diverted to a regulatory T cell (Treg) lineage to establish central tolerance (9). The combined outcomes of positive selection and central tolerance shape the specificity, diversity, and self-reactivity of the TCR repertoire in the peripheral T cell compartment.
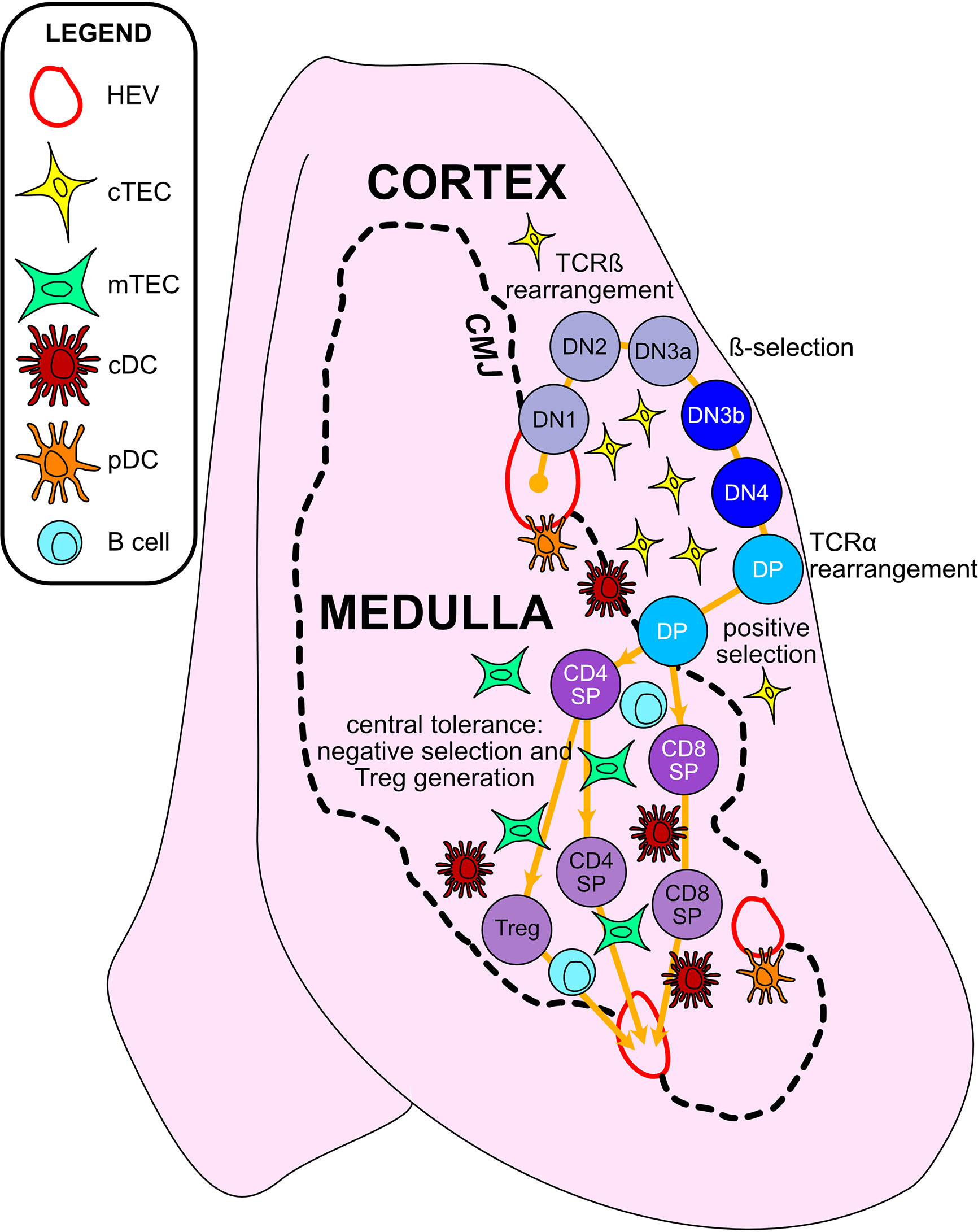
Figure 2 Thymic epithelial cells and hematopoietic antigen presenting cells provide essential signals to guide αβT cell maturation and the induction of central tolerance in the thymus. Cross-sectional view of a thymus lobe reveals cortical and medullary regions, through which thymocytes must travel in an orchestrated manner to encounter heterogeneous stromal cell subsets. Progenitor cells from the bone marrow migrate through the vasculature to seed the thymus at the cortico-medullary junction (CMJ). DN1-DN4 thymocytes require signals from cortical thymic epithelial cells (cTECs) to support their survival, proliferation, and T-lineage commitment. During the DN2-DN3 stages, TCRβ gene segments are recombined, and thymocytes that successfully express TCRβ and signal through the pre-TCR undergo proliferation and further differentiation through the process of β-selection. Subsequently, thymocytes upregulate CD4 and CD8 to become double-positive cells (DPs), which initiate TCRα gene rearrangements. DPs that successfully express a TCRαβ heterodimer are tested for reactivity with self-peptide MHC complexes presented by cTECs. Only those DPs that receive a TCR signal pass positive selection, enabling them to survive and further differentiate. Positively selected DPs transit from the cortex into the medulla. Along the way, some clones may be deleted in an early wave of negative selection in the cortex, driven by strong TCR reactivity to self-peptide MHC complexes displayed by dendritic cells (DCs). In the medulla, DPs downregulate either CD4 or CD8 to become single-positive thymocytes (CD8SP or CD4SP) and interact with medullary APCs to establish central tolerance to a broad array of self-antigens. Strong TCR signals, induced by self-antigens displayed by medullary thymic epithelial cells (mTECs), conventional DCs (cDCs), plasmacytoid DCs (pDCs), or B cells result in either negative selection (apoptosis) or Treg diversion of the autoreactive T cell clones, enforcing central tolerance. SPs that survive these collective thymic selection processes emigrate from the thymus to join the peripheral T cell pool.
Changes in thymus size and thymocyte cellularity are the most apparent age-related changes in the thymus. In both humans and mice, thymus size continues to increase in the neonatal period, then transitions to a homeostatic phase during early life, prior to the initiation of progressive age-associated involution. While the age-associated decline in size and output of T cells is conserved between mice and humans, one notable difference is that only human thymuses accumulate high levels of lipid laden adipocytes, which are interspersed with relatively small functional regions of thymic tissue (Figure 3). Accumulating evidence discussed below indicates that the cellularity and composition of TECs and thymic HAPCs change with age. As TECs and thymic HAPCs play critical roles in establishing central tolerance, age-related changes in the thymic microenvironment likely impact thymocyte selection, tolerance, and thus peripheral T cell responses throughout the lifespan. In this review, we focus on age-associated changes in the thymic microenvironment that can affect the diversity and self-reactivity of T cells that emigrate into the periphery to participate in immune responses. We first review the establishment of central tolerance and the roles of TECs and HAPCs in this process. We then discuss age-associated characteristics of conventional and regulatory T cell responses and how they may be linked to changes in thymic selection. Finally, we explore age-associated changes in the composition of HAPC and TEC subsets that may contribute to altered central tolerance and T cell activity throughout life.
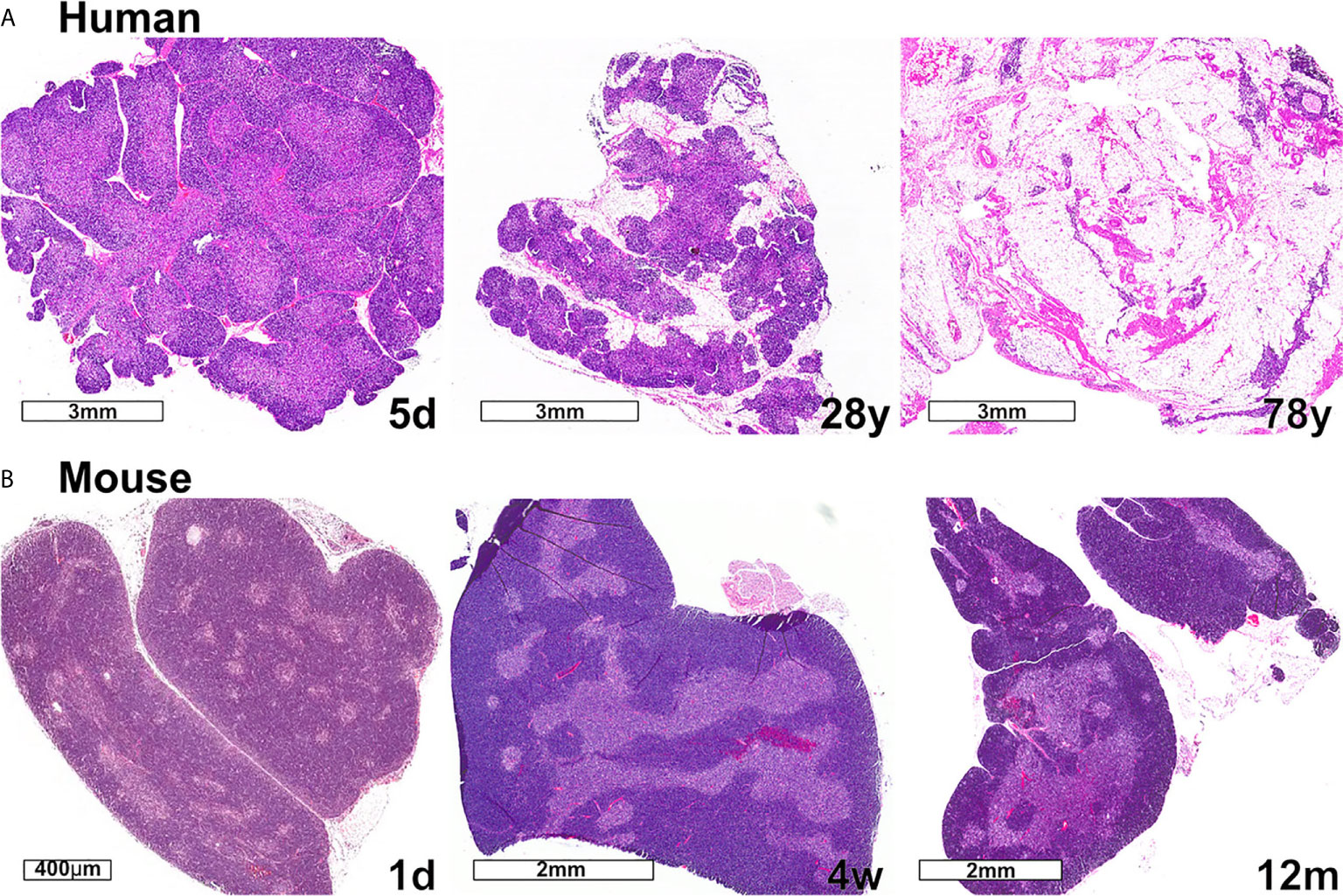
Figure 3 Changes in thymic size, organization, and/or lipid content accompany age-associated thymic involution in humans and mice. (A) In humans, the percentage of the thymus comprised of functional thymic tissue progressively declines with age, and is replaced by adipose tissue, as shown in these hematoxylin and eosin-stained images. The percent of thymus area containing thymic epithelium, representing functional thymic tissue, was calculated via morphometric analysis of cytokeratin immunohistochemical slides. The results for the subjects shown are 91% at 5 days (5d), 55% at 28 years (28y), and 0.5% at 78 years (78y). (B) The mouse thymus grows substantially between postnatal day 1 (1d) (scale bar = 400 µm) and 4 weeks of age (4w) (scale bar = 2 mm), and then declines steadily and is highly involuted by 12 months of age (12m). The small islands of medullary tissue seen at 1d expand and coalesce to form the larger, more organized medullary regions characteristic of adult thymus (4w). Age-associated replacement by adipose tissue is not a prominent characteristic of involution in mice. The corresponding weights (mean ± SD) of murine thymus at the ages shown are 5 ± 0.5 mg at 1d (n = 3), 57 ± 8 mg at 4w (n = 8), and 38 ± 2 mg at 12 m (n = 3).
The Players in Thymic Central Tolerance
TCR gene rearrangements can generate >1015 distinct TCRs, enabling recognition of an extensive array of diverse antigens (10, 11). Given the random nature of the TCR gene rearrangement process, it is inevitable that some TCRs will recognize self-antigens. To achieve self-tolerance, thymocytes must be screened for autoreactivity and either purged or directed into the Treg lineage to prevent autoimmunity. Multiple factors influence whether a self-reactive thymocyte will undergo negative selection or Treg lineage diversion. One critical determinant is the avidity of TCR binding to pMHC complexes presented by thymic APCs, which is a combined function of both individual TCR-pMHC binding affinities and the abundance of pMHC on APC surfaces. High-avidity binding results in thymocyte negative selection, eliminating autoreactive clones from the TCR repertoire (7). Selection into the Treg lineage is generally induced by somewhat lower avidity TCR-pMHC interactions (9). However, the broad and partially overlapping TCR repertoires of conventional T cells (Tconv) and Tregs (12) demonstrate that this cell fate decision is not dictated solely by TCR avidity. Another factor influencing fate choice is intraclonal competition for limited Treg niches. Thymocytes expressing a Treg-derived TCR transgene efficiently divert to the Treg lineage only when present at low clonal frequencies (9, 13, 14). Thus, the fate of a self-reactive thymocyte is determined by cell-intrinsic and -extrinsic factors. Cell-extrinsic factors include the abundance and local availability of self-pMHC (15–17), CD80 and CD86 co-stimulatory molecules (18, 19), and IL-2, with some contribution from IL-15 and IL-7 (20, 21). Altogether, multiple factors in the thymic environment shape the self-reactivity and diversity of emerging T cells, regulating their responsiveness to self- and foreign antigens.
A variety of thymic APC types present self-peptides to induce central tolerance. The importance of the thymic medulla in negative selection is well-established. Nevertheless, two studies reported that most antigen-induced clonal deletion occurs in DP thymocytes, suggesting that cortical APCs can induce negative selection (22, 23). Although antigen presentation by cTECs is required for positive selection, cTECs have not been associated with negative selection (7). Instead, several studies indicate that HAPCs, such as DCs, in the cortex and near the cortico-medullary junction (CMJ) present ubiquitous self-antigens to induce cortical negative selection (24–26). Thymocytes clearly undergo negative selection at the DP stage, but further studies are needed to determine if DPs undergo negative selection in the cortex or medulla. Prior research predominantly relied on CCR7 expression as a proxy for cortical versus medullary localization of thymocytes undergoing negative section. However, live imaging studies indicate that positively-selected DPs can enter the medulla before upregulating CCR7 (27, 28), raising the possibility that medullary APCs may also contribute to this early wave of DP negative selection.
Thymocytes are screened for reactivity against non-ubiquitous self-antigens primarily in the medulla. The requirement for medullary localization was demonstrated in mice deficient for the chemokine receptor CCR7 for its ligand CCL21, which together promote the medullary accumulation of post-positive selection SP thymocytes (29–33). In the absence of CCR7 signaling, thymocyte migration into the medulla is compromised, resulting in diminished central tolerance and subsequent autoimmune exocrinopathy (34). mTECs play a key role in negative selection due to their unique ability to collectively express >85% of the proteome, allowing them to induce central tolerance against a wide array of self-antigens (35–37). Importantly, mTECs express tissue-restricted antigens (TRAs), encoded by 2,000-3,000 genes that are otherwise expressed only in a small number of terminally-differentiated tissues (38–40). TRA expression is largely under control of the transcriptional regulator AIRE (35–37), which is expressed predominantly by MHCIIhi CD80hi mature mTECs (41, 42). AIRE deficiency impairs mTEC maturation and prevents expression of Aire-dependent TRAs, resulting in failed central tolerance and export of autoreactive T cells that induce multi-organ autoimmunity (43, 44). Analogous to Aire-deficient mice, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) patients have mutations in the AIRE gene, resulting in autoimmunity affecting multiple endocrine glands (45, 46). TCR repertoire analysis of Treg and Tconv cells from Aire-deficient and -sufficient mice demonstrated that AIRE is also required to select autoreactive clones into the Treg lineage (47, 48).
In addition to expressing diverse self-antigens, mTECs play a critical role in central tolerance by directly presenting self-antigens to thymocytes. MHCIIhi mTECs express the costimulatory molecules CD80 and CD86, which are required for negative selection and Treg induction (49, 50). Reducing MHCII expression selectively in mTECs revealed that efficient negative selection requires antigen presentation by mTECs (51). Moreover, tolerance to the Aire-dependent RIP-mOVA neoantigen remained intact when MHC was expressed only by TECs, and not by thymic HAPCs (52, 53). Furthermore, our imaging studies using thymic slices from RIP-mOVA transgenic mice, in which MHC is physiologically expressed by both mTECs and HAPCs, revealed that AIRE+ mTECs contribute to roughly half of the negatively-selecting interactions with both MHCI and MHCII-restricted thymocytes (54). mTECs also have the capacity to induce Tregs independently of HAPCs (55). Interestingly, while AIRE+MHCIIhi mTECs clearly express and present diverse TRAs that are essential for central tolerance, recent findings demonstrate that MHCIIlo mTECs also contribute to selection of the T cell repertoire (56). Thus, mTECs play a pivotal role in central tolerance, not only as sources of TRAs, but also as APCs that directly present self-antigens to thymocytes to induce central tolerance.
The importance of HAPCs, in particular thymic dendritic cells (DCs), in negative selection is well-established, as genetic ablation of DCs leads to defective central tolerance and autoimmunity (57). DCs efficiently mediate negative selection and Treg induction in reaggregate thymic organ cultures (RTOCs) (58). In vivo, DCs can acquire antigens in peripheral tissues and traffic them into the thymus to induce tolerance (59), and those positioned near thymus vasculature can acquire and present blood-borne antigens (60, 61). Thymic DCs also acquire and present mTEC-derived TRAs (53, 62, 63). Thymic DCs with an activated signature, including elevated expression of MHCII and CD86, function as highly efficient APCs (64). Thymocyte-DC crosstalk is important for DC maturation and function. CD40L on SP thymocytes induces CD40 signaling in DCs, which is required for DCs to induce Tregs in vitro (65). Interestingly, TCR repertoire sequencing demonstrated that mTECs and DCs select distinct clones into the Tconv and Treg repertoires (48, 66). Our imaging studies also indicated that DCs present mTEC-derived TRAs to MHCI- and MHCII-restricted thymocytes (54). For both monoclonal TCR transgenic and polyclonal thymocytes, DCs were engaged in slightly more than half of the interactions between thymocytes and APCs that induced TCR signaling (54), further supporting the fundamental contribution of DCs to central tolerance. Collectively, these studies demonstrate that both mTECs and DCs are required to establish self-tolerance.
DCs are a heterogenous group of HAPCs that include conventional DCs (cDC) and plasmacytoid DCs (pDCs) (67). The mouse cDC1 subset expresses CD8α and XCR1, and the cDC2 subset expresses CD11b and Sirpα/CD172a (68). While both cDC subsets contribute to tolerance induction, thymic cDC2s have greater CD4+ T cell stimulatory capacity (69) and are especially proficient at Treg induction (70, 71). The cDC1 subset plays a role in clonal deletion, but was reported to be dispensable for Treg induction and to have a negligible impact on the Treg TCR repertoire (72, 73). In contrast, other studies concluded that cDC1s are essential for inducing Tregs in response to mTEC-derived antigens (48, 66). Thus, further studies are needed to resolve the contributions of distinct DC subsets to central tolerance. When compared to cDCs, pDCs in the thymus have a reduced capacity to stimulate T cells and to acquire antigens from TECs (74). However, CCR9+ pDCs can transport peripheral antigens to the thymus to induce negative selection (75). In humans, comparable cDC1 and cDC2 subsets have been identified and defined by expression of CD141 and CD1c, respectively (68). It has been technically challenging to dissect the roles of human thymic APC subsets in establishing central tolerance, although in vitro studies have confirmed that DCs have tolerogenic capacity (76), including the ability to induce Tregs (77). Human thymic cDCs are activated by thymic stromal lymphopoietin (TSLP), which is expressed by Hassall’s corpuscles that consist of terminally differentiated mTECs, and the activated CD80hi CD86hi cDCs can induce Tregs in vitro (78). Like cDCs, human thymic CD123+ pDCs support Treg induction in vitro (79, 80). Thus, multiple thymic DC subsets have been shown to promote central tolerance, though the distinct contributions of DC subsets are not entirely resolved.
B cells have also been shown to contribute to central tolerance. Thymic B cells are localized in the medulla and express high levels of MHCII, CD80, and CD86, distinguishing them from splenic B cells (81, 82). Thymic B cells with specificity for self-antigens can present self-peptides to CD4SPs, driving activation-induced cytidine deaminase (AID)-dependent B cell class switching. Class-switched thymic B cells promote negative selection (81, 82). CD40 activation in thymic B cells, driven by CD40L on SP thymocytes, is required to support B cell proliferation, differentiation, and class switching, as well as upregulation of Aire, and these licensed B cells present self-antigens to induce negative selection (83, 84). Thus, B cells may play a significant role in central tolerance, but whether licensed B cells are autoreactive and the nature of the self-antigens they present remain to be resolved. Collectively, these studies demonstrate the cooperative roles of multiple TEC and HAPC subsets in enforcing central tolerance against a broad range of self-antigens.
Changes in Tconv Function and Thymic Selection Throughout the Lifespan
Function of Tconv Cells in the Perinatal Period
Tconv cells generated during the perinatal period face the daunting task of mounting a rapid, protective immune response against a sudden surge of pathogen encounters, while at the same time ensuring they do not trigger autoimmunity. Neonatal T cells differ substantially from adult T cells in composition and function, helping them to achieve this balance (85). Generally, T cell responses in neonates are diminished relative to those of adults. This may partly be attributed to a shift in the ratio of naive to memory subsets, as naive T cells are predominant in perinatal tissues, whereas memory T cells become more abundant in adults (86). In keeping with this concept, T cells from pediatric lymph nodes (LNs) produce relatively lower levels of cytokines, including IFN-γ, IL-2, and IL-4 relative to adult T cells (86). Moreover, in the context of infections such as malaria (87) and congenital Cytomegalovirus (CMV) (88) human neonatal T cells express lower levels of Th1 and Th2-associated cytokines compared to adult T cells. Cell-intrinsic properties of perinatal T cells such as high PD-1 expression (88), low NFAT expression (89) and diminished Ca2+ influx after TCR stimulation (90) may contribute to the diminished responsiveness of neonatal T cells. Overall, the relative paucity of memory T cells and reduced functionality of T cells in the perinate are consistent with increased susceptibility to infection in early life.
Multiple studies in mice and humans have demonstrated that neonatal T cell responses are strongly skewed towards Th2 versus Th1 differentiation (91–95). Mouse neonatal T cells and human cord blood T cells readily produce the Th2 cytokines IL-4 and IL-13 following in vitro stimulation (92, 94–96). Also, CD4+ T cells isolated from neonatal mice immunized with bacille Calmette-Guerin (BCG), Tetanus toxoid and other vaccines expressed higher levels of IL-5 and lower IFN-γ upon antigen re-stimulation in vitro, compared to adults (97). The neonatal Th2 bias is due at least in part to permissive epigenetic regulation of Th2-associated cytokine genes (95, 98). Also, the IFN-γ promoter region is hypermethylated in cord blood CD4+ T cells consistent with their deficient production of IFN-γ after in vitro stimulation (99). Moreover, stimulated CD4+ cord blood T cells express higher levels of GATA3, a key transcriptional regulator of Th2 fate (94, 100). The strong Th2 bias could be beneficial both in suppressing development of damaging inflammatory Th1 responses, as well as in promoting tolerance towards allogeneic maternal antigens in utero. Consistent with the latter idea, cord blood from preterm infants contains higher levels of proinflammatory cytokines and alloreactive Th1-like central memory CD4+ T cells, which were absent in term infants, suggesting their potential role in promoting premature uterine contractions (101). However, Th2-skewing could leave the newborn vulnerable to infections and unable to respond to some vaccines, which require Th1 responses (102–104).
Interestingly, studies have demonstrated that with appropriate stimuli, such as exogenous IFN-γ and IL-12 (105, 106), exposure to helminth and mycobacterial antigens (107), low viral doses (108), various adjuvants (93), or DNA vaccines (109), neonates can mount Th1-like responses in addition to Th2 responses (110–113). In contrast to findings in mice (97), BCG vaccination of infants induces a strong Th1 response, comparable to adults, supported by high IFN-γ and low IL-4/IL-5 expression after antigen re-stimulation in vitro (112, 113). Moreover, Th1 responses are elicited by CMV in the fetus and B. pertussis in infants (88, 114). The capacity of neonatal T cells to mount a Th1 response under some conditions may reflect the extent of DC maturation, as mycobacterial and pertussis toxin antigens are particularly effective at activating DCs (115, 116). Nonetheless, studies with neonatally immunized mice suggest that while Th1 responses can be induced in adults following antigen re-challenge, Th2 memory responses still predominated (93, 117). In addition, while a balanced Th1 and Th2 primary response could be induced in neonates early after exposure to a foreign antigen, a Th2 secondary response was dominant in mice re-challenged as adults (111).
Cell-intrinsic properties of neonatal T cells, as well as extrinsic microenvironmental cues have been implicated in driving the reduced responsiveness and Th2 bias of neonatal T cell responses. Adoptive transfer experiments in mice revealed that Th2 skewing was observed only when fetal, but not adult CD4+ T cells were primed regardless of whether the host microenvironment was fetal or adult (110, 118, 119). These results suggest a cell-intrinsic difference in the fate potential of neonatal versus adult CD4+ T cells. Interestingly, when both Th1 and Th2 responses were elicited by primary antigen challenge in neonates, Th1 cells upregulated IL-13Rα1 which associated with IL-4Rα (119). Upon antigen re-challenge, the activated Th2 cells secreted IL-4 which bound the IL-4Rα/IL-13Rα1 heterodimer, triggering Th1 apoptosis, tipping the balance towards Th2-mediated immunity. Moreover, upregulation of IL-13Rα expression during initial activation of Th1 cells is developmentally regulated; antigen exposure after postnatal day 6 does not induce IL-13Rα expression. These results are due to the delayed maturation of a subset of splenic CD8α+ cDC1s, which secrete IL-12 that inhibits IL-13Rα expression on Th1 cells (120). These findings demonstrate that cell extrinsic factors can regulate the Th2 bias in neonates.
Neonatal CD8+ T cell responses also differ from their adult counterparts (reviewed in (85)). Co-transfer of neonatal and adult CD8+ T cells into adult recipients revealed a cell-intrinsic bias of neonatal cells towards a short-lived effector fate, whereas adult T cells differentiated into both effector and memory subsets (121). Thus, upon pathogen re-challenge, the immune response was dominated by adult CD8+ T cells. Further studies demonstrated that neonatal and adult CD8+ T cells are derived from distinct hematopoietic progenitors (122). Notably, neonatally-derived CD8+ T cells persist into adulthood, where they continue to play an important role in responding to pathogens due to their preferential differentiation into effectors that proliferate rapidly and produce cytokines (123, 124). In contrast, adult-derived CD8+ T cells in the same environment have a greater propensity to generate memory T cells (124). In uninfected mice, CD8+ T cells generated during the neonatal period tend to differentiate into “virtual memory” T cells (Tvm), expressing high levels of CD44, Eomes, and CD122, and they proliferate more rapidly and differentiate into short-lived effector cells following pathogen challenge, mirroring the neonatal CD8+ T cell pool (122, 124, 125). Consistent with findings in mice, human cord blood CD8+ T cells are also highly proliferative upon TCR stimulation (123), and undergo bystander activation, producing IFN-γ, TNFα, or IL-4, depending on the cytokine receptor (126, 127). Collectively, these findings suggest that the functional potential of neonatal naive CD8+ T cells is biased towards an innate-like effector phenotype.
Thus, perinatal CD4+ and CD8+ Tconv cells have distinct functional properties compared to their adult counterparts. Both cell-intrinsic changes in differentiation potential and priming by different microenvironmental cues result in CD4+ T cell responses biased towards a Th2 or Treg (see below) fate, which may protect the neonate from damaging inflammatory Th1 responses at a time when tissue homeostasis, including responses to commensal colonization, is being established. During this period, CD8+ T cells are biased to differentiate into short-lived effector cells, which can rapidly combat pathogenic threats at the expense of generating memory responses.
Selection of Tconv Cells in the Perinatal Period
Previous studies suggest that negative selection is impaired in the perinatal compared to the adult thymus, resulting in decreased deletion of self-reactive thymocytes (69, 128, 129). In mice, susceptibility to experimental autoimmune encephalomyelitis (EAE) decreases between the perinatal and adult period, which correlates with increasing age-dependent negative selection of MBP (myelin basic protein) specific T cells (129). Also, in mice and humans, Tconv cells that mature in the perinatal thymus express higher levels of CD5 and Nur77 compared to those generated in adults (130, 131). CD5 and Nur77 levels correlate with TCR affinity for peptide-MHC (132, 133), suggesting that perinatal Tconv cells are more self-reactive compared to those generated in the adult thymus. While heightened self-reactivity could reflect impaired central tolerance, as discussed above, it is also possible that the threshold for positive selection is higher in the perinatal thymus, such that thymocytes with low-affinity TCRs are not efficiently positively selected, resulting in elevated CD5 levels on perinatal T cells (130). Regardless, higher TCR self-reactivity could enable T cells to respond quickly and effectively against multiple foreign antigens, despite the limited perinatal TCR repertoire (131, 134). Studies in mice have suggested another potential advantage of increased TCR self-reactivity: neonatal recent thymic emigrants enter a lymphopenic periphery, where CD5hi T cells outcompete their CD5lo counterparts for CD28 co-stimulation due to their increased affinity for self-pMHC. Their resultant increased sensitivity to the homeostatic cytokines IL-7 and IL-15 promote lymphopenia-induced proliferation to fill empty niches (135–139). Moreover, CD5hi T cells have skewed effector potential in the periphery. CD5hi CD4+ T cells are more prone to differentiate into Tregs (140), while CD5hi CD8+ T cells express effector molecules such as Eomes, T-bet and Helios that promote T cell differentiation to an effector or virtual memory fate (134, 139). Thus, the increased self-reactivity of T cells selected in a neonatal thymus likely contributes to the characteristic rapid proliferation of neonatal CD4+ and CD8+ T cells in response to cytokine or antigen stimulation, as well as the altered differentiation potential biasing neonatal CD8+ T cells to become short-lived effector cells or Tvm, and CD4+ T cells to adopt a Treg fate. These studies emphasize that the strength of TCR signaling during thymic selection not only determines lineage fate decisions in the thymus but also influences peripheral effector T cell function.
Declining Function of Tconv Cells During Aging
It is well established that T cell function declines with age, correlating with increased morbidity and mortality to infectious diseases and reduced responses to vaccination (2, 141, 142). While following a general pattern of age-associated decline, there is increased variability in immune responses between individuals with age, due in part to their lifetime histories of acute and persistent pathogen encounters (143, 144). As in the perinatal period, both cell-intrinsic and microenvironmental changes contribute to the age-associated decline in T cell function; however, the complex mechanisms that drive waning T cell immunity are not yet fully resolved (2).
CD4 T cells exhibit multiple functional changes with age. Reduced expression and production of IL-2 has been demonstrated following stimulation of mouse CD4+ T cells (145, 146). There is evidence for reduced IL-2 production in CD4+ T cells from elderly humans, but this decline has not been universally observed (2). CD4+ T cells from old mice were found to be functionally deficient in B cell activation, indicating reduced T follicular helper cell (Tfh) activity with age (147). Consistent with this notion, an age-associated decline in Tfh responses, along with diminished class-switched antibody levels were reported following viral infections in mice, non-human primates, and humans (143, 148). In addition, aging is associated with reduced IFN-γ+ CD4 T cell responses to viral pathogens (143, 149). Age associated defects in CD4 activity could result from impaired T cell priming as aged CD4+ T cells exhibit cytoskeletal defects and poor immunologic synapse formation (150), reduced calcium flux upon TCR cross-linking (151), and defective metabolic reprogramming upon activation (152). These findings suggest cell-intrinsic defects impair CD4+ T cell responses in aged individuals. Cell-extrinsic defects also contribute to the decline in CD4 T cell function with age. For example, aged CD4+ T cells showed reduced homing to LNs following viral infections, despite the finding that expression levels of LN homing molecules (CCR7, CXCR4, PSGL1, and LFA1) were not decreased (148). However, levels of CCL21, which recruits naive T cells to LNs, were lower in draining LNs from old mice early after infection (148). Further support for cell-extrinsic defects was demonstrated by studies showing that the LN microenvironment deteriorates with age due, in part, to reduced IL-7 presentation and increased fibrosis (2, 153, 154),
Because T cells consist of multiple functionally distinct subsets, the defects in T cell function with age described above could reflect a change in subset composition and/or alterations in activity on a per-cell basis. Indeed, phenotypic analyses revealed an age-associated reduction in the proportion and numbers of naive T cells in humans, non-human primates, and mice (154–158). Recent comprehensive single-cell transcriptional profiling studies confirm the shift towards a higher frequency of effector-memory T cell subsets with age (159, 160). Notably, in mice, aging was associated with a stark increase in representation of cytotoxic CD4+ T cells, exhausted CD4+ T cells, and activated Treg (159). The shift to an increased frequency of these CD4+ T cell subsets correlated with elevated levels of cytokines associated with inflammaging, such as IL-27, IFN-β, and IL-6. Thus, the altered distribution of CD4+ T cell subsets likely has a profound impact on immune responses with age. However, such alterations do not fully account for age-related changes in T cell function. For example, antigen-inexperienced CD4+ recent thymic emigrants (RTEs) from old mice produce less IL-2 and proliferate poorly after in vitro stimulation compared to young RTEs (161). In addition, naive CD4+ T cells from older mice have a longer lifespan, reflecting increased Bim expression, but proliferate poorly after in vitro and in vivo stimulation (162, 163). Although profound functional deficiencies in naive human CD4+ T cells have not been reported, naïve CD4+ T cells from elderly humans exhibit reduced TCR signaling and expansion following in vitro stimulation due, at least in part, to the age-associated decline in miR-181a expression (164–166). Interestingly, naive polyclonal CD4+ T cells in aged mice are more self-reactive, as indicated by increased CD5 expression, display higher TCR affinity for foreign antigens, and are more promiscuous in antigen recognition. The increase in self-reactivity and promiscuity of the aged CD4+ T cell compartment implicate altered thresholds of CD4+ T cell selection in the thymus with age (155).
Defects in CD8+ T cell responses with age are well established. Early studies reported defective CD8+ T cell responses following primary and secondary influenza challenges (167). Additionally, CD8+ T cells have an age-associated decrease in their capacity to proliferate and produce effector molecules, such as IFN-γ, following in vitro stimulation or infection with viral or bacterial pathogens (168–175). In humans, the frequency of activated CD8+ T cells induced by yellow fever vaccination was significantly diminished with age (176), underscoring the potential impact of a declining CD8+ T cell compartment on vaccine-induced as well as on natural protection against pathogens (143).
Similar to CD4+ T cells, the overall decline in CD8+ T cell function with age could reflect changes in the proportions of functionally distinct CD8+ T cell subsets. Indeed, one of the most notable hallmarks of the aged immune system in humans and mice is a substantial decline in both the number and frequency of naive CD8+ T cells (143, 173, 177). At the same time, the CD8+ T cell pool becomes progressively enriched in clonally expanded, antigen-inexperienced CD8+ Tvm cells in mice and in humans (139, 174, 177–180). The homeostatic cytokine IL-15 is required for differentiation and function of Tvm cells (139), which in turn respond to IL-12 and IL-18 stimulation in a TCR-independent manner, resulting in secretion of IFN-γ (139, 174, 177–179). Notably, Tvm can provide antigen-independent bystander protection in bacterial infections, proliferating more rapidly than naive T cells, but differentiating preferentially into short-lived effector cells (181), strikingly reminiscent of perinatal Tconv cells. While Tvm cells can provide effective protection against pathogens in a bystander or TCR-dependent manner (139, 181), and increase in frequency with age, there is a seemingly incongruous age-associated decline in the overall response of CD8+ T cells to pathogen challenge. Previous studies partially resolved this conundrum by showing that aged Tvm in mice and humans have a reduced capacity to proliferate in response to TCR stimulation relative to young Tvm. The mouse Tvm response to homeostatic cytokines is sustained with age, but whether human Tvm have a similarly sustained response has not been tested (173, 174). Regardless of age, Tvm mount a monofunctional cytokine response to TCR stimulation, while naive CD8+ T cells produce multiple cytokines in response to mouse influenza infection (173, 174). These studies indicate that with age Tvm cells accumulate in the CD8+ compartment, respond poorly to TCR stimulation, and produce a less diverse cytokine response. In contrast, while naive CD8+ T cells retain a robust capacity to proliferate to TCR stimulation with age, they do not survive or proliferate well in response to the homeostatic cytokines IL-2 and IL-15, explaining the decreased proportion of naive CD8+ T cells relative to Tvm with age (173, 174). Transcriptional profiling revealed that Tvm cells that accumulate with age have a senescent signature, consistent with the lower frequency of cells that respond to TCR stimulation as well as the reduced burst size of individual responding cells (173). Together, these results partially explain why the composition and function of the CD8+ T cell compartment changes with age. Additional insights into the declining function of aged CD8+ T cells were revealed in a recent single cell transcriptional profiling study that identified a subset of CD8+ T cells that expresses and secretes granzyme K (GZMK) and accumulates with age in mice and humans (160). In contrast to Tvm, age-associated GZMK+ CD8+ T cells have a transcriptional profile and surface marker phenotype (PD-1+ Tox+) consistent with a state of terminal exhaustion (160). Strikingly, upon TCR stimulation, these cells secrete GZMK, which alone or in combination with IFN-γ, induces fibroblasts to secrete pro-inflammatory factors, such as IL-6 and CCL5. Thus, GZMK+ CD8 T cells may contribute to inflammaging. GZMK+ CD8+ T cells also express the integrin CD49d, reminiscent of a previously described clonally expanded CD49d+ CD8+ T cell subset in aged mice (182). These cells home to multiple tissues and fail to secrete granzyme B (GZMB) or IFN-γ upon TCR stimulation, further distinguishing them from Tvm (160). Notably, single-cell TCR repertoire analysis of human PBMCs revealed that the well-documented clonal restriction of the CD8+ T cell pool with age (177, 183–185) was due in part to clonal expansion of this novel GZMK+ CD8+ T cell subset, which was distinct from the clonally expanded GZMB-producing cells that are enriched for recognition of CMV or Epstein-Barr virus (EBV) (158, 186). Clonal expansion of Tvm with age has also been reported (174, 177, 185). Collectively, these studies reveal that aging is associated with a profound shift in the composition of CD8+ T cell subsets, resulting in reduced responses to newly encountered antigens and a shift towards a pro-inflammatory phenotype.
Age-associated changes in the composition of the CD8+ T cell compartment could reflect cell-intrinsic and/or extrinsic influences. Several lines of evidence indicate that the aged environment is a causative factor in the decline in CD8+ T cell functionality. When young naive CD8+ T or Tvm cells are transferred into an aged host, their proliferative potential declines (173). Similarly, an aged host environment induces young CD8+ T cells to adopt an exhausted phenotype, including upregulation of GZMK (160). Additionally, in heterochronic parabiosis experiments fewer young CD8+ T cells were recovered in old compared to young partners (187). Conversely, in each of these studies, the young environment did not restore function, cellularity or phenotype to old CD8+ T cells. Strikingly, the number of T cells declined in the lymph node of a young mouse when parabiosed to an old partner (187). Together, these data indicate that the old environment contains soluble factors that negatively impact CD8+ T cell cellularity and function. Additional cell-extrinsic influences that can diminish CD8+ T cell responses with age include ineffective antigen presentation by aged DCs (188, 189) and disrupted architecture of secondary lymphoid organs that could impair recruitment, maintenance or priming of CD8+ T cells (153, 154, 190, 191). Despite clear evidence that cell-extrinsic factors in the aged environment modulate CD8+ T cell responses, there is evidence that age-associated cell-intrinsic changes also contribute to diminished T cell responses with age. In addition to the declining responsiveness of aged Tvm to TCR stimulation (173, 174), another characteristic of Tvm cells that accumulate with age is their increased self-reactivity, as reflected by elevated expression of CD5 (139, 185). Furthermore, there is an apparent enrichment in naive T cells with higher CD5 levels in the CD8+ T cell repertoire with age (177, 192), and naive CD8+ T cells expressing higher levels of CD5 have an increased propensity to differentiate into Tvm cells. Together, these data indicate that the naive CD8+ T cell pool is more self-reactive with age. Further studies are needed to determine whether the increased self-reactivity of naive T cells is driven by age-associated cell-extrinsic changes in the thymic microenvironment that affect selection thresholds, peripheral maintenance of self-reactive T cells, and/or intrinsic transcriptional profiles of T cells that alter their capacity to respond to TCR signals.
Changes in Negative Selection of Tconv Cells During Aging
Aging induces profound changes in the thymic microenvironment (see section Changes In Thymic Apcs And Implications For Selection Throughout The Lifespan), which could negatively affect central tolerance. For example, TRA expression decreases with age (193, 194), reflecting both a decline in the frequency of Aire+ mTECs and reduced Aire expression per mTEC (195, 196). Thus, thymocytes may not encounter the full spectrum of self-antigens responsible for central tolerance in an aged thymus, potentially contributing to the increased incidence of autoimmunity with age. Consistent with this possibility, Aire haploinsufficiency results in decreased negative selection and an increased incidence of diabetes (197). Also, in an inducible Foxn1-deletion model of accelerated thymic atrophy, TRA expression was reported to decline, and negative selection was impaired (198). In addition to age-associated changes in TECs, changes in thymic B cells could impact central tolerance during aging. The number and frequency of thymic B cells increase in old mice; however, their expression of Aire and TRAs diminishes with age (199–201). A decline in AIRE-dependent TRA expression is also observed in human thymic B cells (199). Despite the clear association between aging and thymic involution, and recognition that the thymic microenvironment is critical for establishing self-tolerance, surprisingly little is known about the impact of aging on central tolerance. Further investigations are needed to determine if central tolerance is altered during aging, to elucidate the underlying mechanisms, and to determine the impact on autoimmunity.
Changes in Treg Function and Thymic Selection Throughout the Lifespan
Function of Tregs in the Perinatal Period
The critical role of Tregs in suppressing damaging inflammatory immune responses in a broad range of tissues has been well documented [reviewed in (202)]. Immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) patients, in whom Treg lineage differentiation is impaired, develop severe gastrointestinal pathology, type-1 diabetes mellitus and severe skin inflammation, in addition to other autoimmune manifestations within the first few weeks to months after birth (203–206). Studies in mice have demonstrated that organ-specific Tregs play a crucial role in promoting peripheral tolerance in both lymphoid and non-lymphoid organs (207–209). Tregs control inflammatory T cell responses towards food antigens (210) and commensal microbiota in the gut (211), and intestinal Tregs have been shown to expand in response to microbial cues (211–214). Tregs also migrate to the hair follicles in the skin, where they are critical for tolerance to skin commensals (215, 216). Retinal antigen-specific Tregs in the eye control inflammation in experimental autoimmune uveitis and help resolve disease pathology (217, 218). Other experimental models of organ-specific diseases such as diabetes (219) and EAE (220) have reinforced the crucial role played by Tregs in suppressing autoimmune pathology.
Tregs control T cell responses through multiple mechanisms (221, 222). For example, expression of CTLA-4 on Tregs reduces the ability of DCs to stimulate T cell responses by masking the costimulatory molecules CD80 and CD86 (223). Tregs also express CD39 and CD73, which catalyze the release of adenosine into the extracellular milieu, thus inhibiting effector T cell proliferation (224). In addition, Tregs outcompete effector T cells for IL-2, inhibiting their proliferation (225), and Tregs produce suppressive cytokines like IL-10 and TGFβ (226, 227). Tregs can also suppress effector T-cell differentiation and induce apoptosis of Tconv cells (228, 229).
The importance of intrathymic Treg generation in the neonatal period is illustrated by an experiment performed nearly 50 years ago in which neonatal thymectomy in mice was shown to cause autoimmune pathology in the ovaries (230). Later studies showed that transfer of adult T cells, in particular CD25+ Tregs, prevented autoimmune destruction of ovaries in these mice, implying that a defect in neonatal thymic Treg generation failed to curb activation of autoreactive T cells (231, 232). Differentiation of Foxp3+ CD25+ Tregs in the mouse thymus lags behind that of Tconv cell development (233). In newborn mice, Tregs comprise only 0.09% of CD4SP thymocytes and do not reach adult levels (~4% of CD4SP thymocytes) until 21 days after birth (233). In contrast, CD25+ Tregs constitute ~6-8% of CD4SP thymocytes in humans by 14-17 gestational weeks (GW), and this frequency remains relatively constant after birth (234, 235). Perinatal expansion of the human Treg compartment is observed in the periphery, with a striking surge in the frequency and number of peripheral blood CD25+ Foxp3+ Tregs during the early neonatal period (7-8 days post birth) compared to those in cord blood or present at a later neonatal period (2-4 weeks after birth) (236). Additionally, compared to adult tissues, a higher frequency of Foxp3+ CD25+ Tregs is observed in human fetal as well as in several pediatric lymphoid and mucosal tissues, indicating their importance in early life (86, 237). Tregs in neonatal circulation display an activated phenotype, with a predominantly Foxp3hi CTLA-4hi CCR7lo CD25+ phenotype (236). Similarly, an activated (CD69hi GITRhi CCR7lo CTLA-4hi), memory (CD62Llo CD45RO+) Treg phenotype was documented in fetal LN and cord blood (235, 237).
The high frequency of Tregs in the fetal and perinatal periods may be due to a higher propensity of fetal hematopoietic progenitors to differentiate into the Treg lineage. HSCs transplanted from human fetal liver or bone marrow into humanized mice give rise to a higher frequency of CD25+ Foxp3+ Tregs compared to HSCs from adult bone marrow (238). Additionally, studies in mice have demonstrated that perinatal CD4SP thymocytes are more prone to differentiate into Tregs upon TCR stimulation when compared to adult CD4SP thymocytes, both in vitro and in vivo (239, 240). Gene expression profiles of adult Tregs are more similar to fetal naive CD4+ T cells than to adult naive CD4+ T cells, indicating that fetal T-cells may be transcriptionally primed to be suppressive. Consistent with this finding, naive CD4+ T cells from human fetuses give rise to more Tregs than adult CD4+ T cells in vitro (236, 238). The increased Treg induction efficiency of perinatal progenitors could be a protective mechanism required to establish initial immune tolerance in multiple peripheral tissues, particularly in light of elevated Tconv self-reactivity in the perinatal period (see above). Supporting this theory, Aire expression in the perinatal period is necessary and sufficient to prevent autoimmunity in mice (241), and Treg ablation in perinates induces profound multiorgan autoimmunity characteristic of Aire deficiency (242). Together, these findings suggest that Aire expression in the perinatal thymus is essential for selecting perinatal Tregs that suppress multiorgan autoimmunity. Tregs are required for self-tolerance throughout life, as demonstrated by the autoimmunity that ensues following Foxp3 elimination in adult mice (207). Notably perinatally-derived Tregs persist into adulthood, and relative to adult-derived Tregs, are uniquely capable of protecting against autoimmunity when transplanted into Aire-deficient mice. Perinatally-derived Tregs also express an activated gene signature and have an increased capacity to suppress Tconv cell proliferation in vitro relative to adult-derived Tregs (242). These mouse studies are consistent with human studies showing distinct gene expression patterns in fetal versus adult Tregs (238), as well as increased protein expression and suppressive activity of pediatric compared to adult Tregs (86). Taken together, these findings suggest that perinatally-derived Tregs persist into adulthood, where they suppress damaging autoreactive T-cell responses in multiple organs.
Many studies of Treg-mediated protection in tissues have been performed in adults, raising questions of whether neonatally derived Tregs play a critical role in these processes, and if so, what mechanisms underlie their suppressive activity. Some progress has been made towards answering these questions. Tregs generated in the neonatal thymus migrate to the skin in a CCR6-CCL20 dependent manner, where they are essential for establishing tolerance to newly colonizing commensal bacteria (216). Recent studies have also reported that a wave of neonatal thymus-derived Tregs migrates to the liver (243, 244). Interestingly, perinatal liver-resident Tregs are more suppressive than their splenic counterparts, and they are activated in a TCR-dependent manner in the liver microenvironment (243). Ablating these perinatal Tregs resulted in Th1-type inflammation and breakdown of lipid metabolism, highlighting their role in establishing liver homeostasis (243). Another study demonstrated that perinatal Tregs promote and maintain anergy of self-reactive PD-1+ CD44+ Tconv cells in the liver; notably development of these perinatal Tregs was Aire independent (244). These results contrast with the Aire-dependence of perinatal Tregs that confer protection against autoimmune infiltrates in Aire-deficient mice (242). Thus, the contribution of Aire to selection of perinatal thymic Tregs that suppress tissue-specific autoreactivity requires further investigation. Tolerance to commensals at mucosal barriers is established in the neonatal period and is mediated by peripherally-induced Tregs. In neonatal mice, the lung microbiota induce differentiation of a Helios negative Treg subset that suppresses Th2-like hyper-responsiveness to aeroallergens (245). Additionally, neonatal T cells encounter a wide variety of antigens derived from gut microbiota which induce Treg differentiation required for tolerance to gut commensals throughout life (246). Thus, thymus-derived and peripherally-induced Tregs are generated early in life and are critical for tissue-specific immune homeostasis in multiple organs.
Selection of Tregs in the Perinatal Period
In mice and humans, Foxp3, the master transcriptional regulator of Treg lineage commitment and maintenance, is predominantly induced in self-reactive CD4SP thymocytes, although it can be detected as early as the DP stage (233–235). Perinatal Tregs express higher CD5 levels compared to adult Tregs, suggesting increased self-reactivity (130).Two distinct Treg populations that differ in their affinity towards self-antigens have been identified in adults (247). Triplehi (PD-1hi GITRhi CD25hi) Tregs are more self-reactive, as indicated by higher Nur77 and CD5 levels, and are efficient at suppressing Tconv cell proliferation in lymphoid organs. In contrast, Triplelo (PD-1lo GITRlo CD25lo) Tregs express less Nur77 and CD5, indicative of lower self-reactivity, and more effectively limit the induction of colitis by inducing peripheral Tregs in the gut (247). However, both Triplehi and Triplelo Tregs in the perinatal thymus express elevated CD5 levels relative to their adult counterparts (130). Taken together with the evidence that Tregs selected in the perinatal thymus are critical for suppressing autoimmunity at multiple tissue sites, higher CD5 expression by thymic perinatal Tregs suggests that the perinatal thymic environment may be specialized for selecting tissue-protective Tregs.
Recent studies support the possibility that self-antigen presentation differs in the perinatal versus adult thymus microenvironment, resulting in efficient Treg selection. A self-peptide derived from peptidyl arginine deaminase type IV (Padi4) was found to efficiently induce selection Treg only in the perinatal thymus (248). Interestingly, in adults, Padi4-specific thymocytes were subject to negative selection as early as the post-positive selection DP stage, whereas in perinates, negative selection was delayed until the CD4SP stage. Thus, in the adult thymus, Padi4-specific DP precursors were deleted before they could give rise to CD4SP cells or Tregs, likely underlying the switch from perinatal Treg induction to adult negative selection (248). The age-associated shift towards clonal deletion could reflect cell-intrinsic changes in signaling downstream of TCR stimulation in perinatal versus adult DP thymocytes and/or changes in the perinatal versus adult thymic microenvironment. In this regard, bone marrow chimera experiments revealed that expression of Padi4 by HAPCs induced negative selection in the adult thymus, but when Padi4 expression was restricted to radioresistant thymic stromal cells, Treg induction was restored in adults. These findings suggest that antigen presentation by adult HAPCs preferentially drives negative selection, as opposed to Treg induction. Conversely, unique properties of the thymic APC compartment in neonates may selectively promote thymic Treg induction over negative selection. The concept that age-associated changes in the thymic microenvironment play a role in the outcome of self-antigen recognition is supported by the lower expression of H2-DO relative to H2-DM in perinatal versus adult mTECs, which would increase the diversity of peptides presented in the perinatal thymus (242), thus altering the TCR repertoire during thymic selection. Collectively, these studies demonstrate that both negative selection and Treg induction differ in the perinatal versus adult thymus, yielding more autoreactive Treg and Tconv cells in the perinatal period. However, the mechanisms driving these age-dependent changes in selection thresholds and TCR specificities, including whether these differences are due to cell-intrinsic changes in T cell progenitors and/or cell-extrinsic factors in the thymic microenvironment, remain to be resolved.
Changes in Treg Function During Aging
The prevalence of Tregs in the blood of adult mice and humans ranges from 5-10% of the CD4+ T cell compartment (249). The frequency of Tregs does not increase in mouse blood with age (250). In contrast, elevated frequencies of circulating Tregs have been reported in aging humans (251, 252). Furthermore, aging is associated with an increase in both the frequency and number of Tregs in mouse spleen and lymph nodes, but not in the lung, liver or peritoneum (160, 250, 251, 253, 254) In fact, a recent single-cell transcriptional profiling study confirmed that the frequency of Tregs increases in aging mouse spleens, but revealed that this increase was driven almost entirely by an emerging subset of activated Tregs (159). Taken together, these studies indicate that the abundance, distribution, and function of Tregs shift with age towards increasing immunosuppression.
Two single-cell transcriptomics reports show that with age, Tregs express elevated levels of genes associated with Treg activation and suppressive activity, including Foxp3, S100a11, IL1r2, Pdcd1, Tigit, Lag3, and Batf (159, 160). Moreover, expression of proteins that promote Treg suppressive activity, such as FOXP3, CD25, CTLA-4, and GITR, is maintained, and in some cases increased in aged Tregs (251, 252, 254, 255). A recent study reported that old activated Tregs are more suppressive than young Tregs (159), consistent with previous findings showing increased functional activity of Tregs with age (251). In contrast, other studies report that the in vitro suppressive capacity of Tregs does not differ between young and aged mice (256, 257) or humans (255). Nevertheless, whether due to increased frequency or increased suppressive capacity, it is likely that aged Tregs may impair T-cell mediated control of infection with age, thus contributing to pathology. In keeping with this concept, young mice are able to resolve primary Leishmania major infection, whereas aged mice experience increasing reactivation of lesions. However, Treg depletion in the aged mice increased cytokine production by effector T cells and decreased disease severity (251). In addition, CD4+ CD25hi Tregs recovered from Alzheimer’s disease and Parkinson’s disease patients displayed increased suppressive activity in vitro when compared to young and control elderly donors, suggesting that Treg suppressive capacity is also associated with age-related neurodegeneration (252). Increasing Treg activity may also contribute to diminished anti-tumor responses with age. Whereas young mice were able to reject transplanted BM-185 tumor cells, aged mice succumbed, and their ability to reject tumors was restored by Treg depletion (254). Because there are multiple subsets of functionally distinct Tregs (258), some of the discrepancies above regarding alterations of Treg functionality with age may reflect changes in the composition of Treg subsets, which could be impacted by organ sites and the assays chosen to measure Treg suppressive activity. Consistent with this possibility, Tregs were found to be more abundant in the oral mucosa of aged mice and humans, although, counterintuitively, inflammation associated with Candida albicans infection was exacerbated despite pathogen control (259). Notably, an age-related shift in favor of IFN-γ-producing relative to IL-17-producing Tregs and Tconv cells was associated with decreased IL-1β and increased IL-6 levels in the mucosa. IL-1R1 deficiency decreased induction of IL-17-producing Tregs after Candida albicans infection, whereas there was a relative increase in IFN-γ-producing Tregs, which required IL-6 for their expansion (259). In a mouse model of autoimmune colitis, aged Tregs could suppress IFN-γ+ Th1 cells, but not IL-17+ Th17 cells (260). Restraint of Th17 cells requires STAT3 activation in Tregs (261), and aged Tregs do not activate STAT3 in response to inflammatory IL-6 to the same extent as young Tregs (260). Collectively, these studies demonstrate that age-associated changes in the relative abundance of different cytokines, as well as the responsiveness of aged Tregs to cytokine stimulation, can alter Treg subset differentiation and thus, the ability to suppress inflammatory T cell responses to self-antigens, pathogens, and commensals in a tissue-specific manner. While changes in cytokine levels would contribute to extrinsic alterations in Treg differentiation and function with age, changes in the ability of aged Treg to respond to cytokines suggest that age-associated cell-intrinsic changes affect Treg function.
Selection of Treg With Age
The absolute number of Tregs in the thymus decreases with age, reflecting the reduction in cellularity that accompanies age-associated thymic involution (250, 253). Although the frequency of FOXP3+ cells does not change with age (250, 253), initial studies did not distinguish between thymic Tregs generated in the aged thymus versus those that had recirculated into the thymus from the periphery. Subsequent studies using RAG2p-GFP mice revealed that the frequency of newly generated Tregs declines rapidly with age, while the proportion of recirculating Tregs increases (262, 263). Moreover, mature Tregs inhibit de novo generation of Tregs in fetal thymic organ cultures, suggesting that recirculating Tregs reduce selection of new Tregs in the aged thymus, perhaps by sequestering IL-2, a limiting cytokine required for Treg induction (264). In this regard, Treg generation was increased in the presence of exogenously administered IL-2 (262). These studies suggest that thymic Treg induction is reduced with age. In contrast, Treg selection was favored over clonal deletion in an inducible Foxn1-deletion model of accelerated thymic involution, in which TECs are precipitously depleted (198). Thus, it remains to be resolved whether Treg generation is generally reduced in an aged thymus, or is actually increased under some conditions, such as limited self-antigen availability.
Given that the number of Tregs in the periphery does not decline with age, and in fact increases in some organs (see above), the decline in thymic output of newly generated Tregs during age-associated thymic involution must be compensated for in the periphery either by increased proliferation/survival of extant Tregs or increased Treg induction. Naïve CD4+ T cells from old mice have a diminished ability to differentiate into Tregs in vitro and in vivo (188, 265). However, aged Tregs have a survival advantage relative to young Tregs due to lower expression of the pro-apoptotic factor Bim (253, 266). It is important to note that there are multiple subsets of peripheral Tregs (267–271), such that age-associated increased Treg survival could reflect an increased proportion of a long-lived subset. In keeping with this possibility, CD25lo Tregs accumulate with age in the periphery (256, 266). CD25lo Tregs express lower levels of Bim than CD25hi Tregs, even though Bim levels decline in CD25hi Treg with age (266). Notably, IL-2 is critical for homeostasis of CD25hi Tregs and IL-2 levels decline with age, whereas the CD25lo subset requires IL-15 for survival (266). Thus, altered access to homeostatic cytokines could impact the relative proportions of different Treg subsets with age, which would be in keeping with both the observed decline in circulating IL-2 and the age-associated deterioration of a supportive T-cell microenvironment in secondary lymphoid organs (2), especially given that autoreactive CD4+ T cells in secondary lymphoid organs are an important source of IL-2 for Tregs (272). Thus, there are age-related consequences for Treg selection, induction, and maintenance in the thymus and in the periphery.
Changes in Thymic APCs and Implications for Selection Throughout the Lifespan
Changes in TECs Across the Lifespan
The composition and function of TEC subsets undergo major changes throughout the lifespan, and there is mounting evidence that the dynamic nature of the TEC compartment is a critical determinant of age-associated alterations in the immune response. As previously discussed, mTECs play a critical role in establishing and maintaining central tolerance. Not only are mTECs uniquely capable of expressing and presenting Aire-dependent and Aire-independent TRAs (38, 43), but they also transfer TRAs to DCs for subsequent cross-presentation to thymocytes (53, 62, 63). In addition, mTECs produce chemokines such as XCL1, CCL19, and CCL21 that promote DC medullary recruitment and localization (70, 273, 274). Moreover, in response to Toll-like receptor (TLR) signaling, mTECs secrete chemokines that recruit CD14+ monocyte-derived DCs into the medulla to promote Treg generation (275). Thus, mTECs play multifunctional and essential roles in negative selection and Treg generation.
The mTEC compartment in both humans and mice is phenotypically and functionally heterogeneous. Initially, mTECs were classified into two major subsets, namely an immature MHCIIlo CD80lo AIRE- subset (mTEClo) and a functionally mature MHCIIhi CD80hi AIRE+ subset (mTEChi). There is long-standing evidence that the mTEClo compartment contains progenitor cells that generate mTEChi progeny (276–278). However, it is now evident that the mTEClo population is highly diverse and contains multiple functionally and developmentally distinct subsets that have been identified in investigations using flow cytometric as well as lineage tracing and transcriptomic analyses of mouse and human mTECs. For example, a subset of mTEClo cells expresses CCL21, indicating their functional importance in recruiting positively selected thymocytes into the medulla (279–283). Interestingly, despite the initial association of an mTEClo phenotype with an immature stage of differentiation, the mTEClo subset also contains mature cells that have downregulated Aire and MHCII expression (44, 279, 280, 282). Studies employing single-cell RNA sequencing (scRNAseq) analyses have shown that post-Aire mTECs include a unique population of thymic tuft cells, which are sensory epithelial cells similar to those present in the intestine and other mucosal sites (279, 280, 282). It has been suggested that tuft cells play a role in central tolerance, as the abundance of Foxp3lo Treg precursors decreases in tuft cell-deficient mice (284). Hassall’s corpuscles (HCs) are another cell type in the heterogeneous post-Aire mTEClo subset. HCs form distinctive concentric structures of flattened epithelial cells that are prominent in the human thymus medulla, and small clusters of TECs that may be analogous are found in mouse medullary regions. Transcriptional profiling studies have identified genes that are highly expressed by both HCs and terminally differentiated keratinocytes (279, 282). Moreover, HCs resemble keratinocytes in that both cell types produce proteins found in terminally differentiated epithelial cells such as keratin 10, involucrin, filaggrin and TSLP (44, 285–287). It has been suggested that HCs play a role in regulating central tolerance as TSLP produced by human HCs activates DCs to express co-stimulatory molecules that enhance Treg induction (78). A recent study in which scRNAseq analysis was performed on index-sorted TECs identified a novel TEC subtype, referred to as intertypical, which has both mTEC and cTEC characteristics (193). Thus, studies to date have shown that the mTEC compartment is highly diverse, consisting of multiple subsets whose phenotypic and functional characteristics, as well as lineage relationships have not yet been fully deciphered. Nevertheless, there is mounting evidence that various mTEC subsets significantly impact the establishment and/or maintenance of central tolerance.
The TEC compartment is highly dynamic during the perinatal to adult transition. TEC numbers expand exponentially during mouse fetal thymus development, and TEC cellularity continues to increase in the perinatal period prior to temporarily leveling off in young adults (288, 289). In parallel, there is a higher frequency of proliferating TECs in the perinatal compared to adult thymus (279, 288, 290). Remodeling of the TEC compartment during the perinatal period in mice is reflected by an increase in the percentage of mTECs and a corresponding decline in the percentage of cTECs (289, 291). Interestingly, functional blockade of vascular endothelial growth factor (VEGF) receptors in neonatal mice inhibits perinatal thymus expansion and accelerates the shifted mTEC to cTEC ratio despite the lack of VEGF receptors on thymocytes and TECs (292). These effects were independent of changes in the vasculature; however, VEGF inhibition altered expression of genes regulating cellular adhesion, migration, adipogenesis and inflammation in CD140a+ mesenchymal cells suggesting that VEGF-mediated effects on mesenchymal stromal cells influences changes in the TEC compartment during the perinatal period (293). The relative increase in mTECs during the perinatal to adult transition was found not only by flow cytometric analysis, but also by microscopic analysis of histological sections (289) showing that this change is not merely an artifact of the enzymatic digestion procedure required to obtain single thymus cell suspensions. This is a matter of concern because enzymatic disaggregation results in suboptimal recovery of cTECs, particularly those present in cage-like structures, referred to as thymic nurse cells, which encompass DP thymocytes (288, 289, 294). An increase in the frequency of mTECs relative to cTECs was also demonstrated by single cell transcriptional profiling of neonatal versus adult human thymuses (295). Furthermore, a recent scRNAseq analysis revealed the presence of a unique cTEC subset in the perinatal mouse thymus that rapidly declined and was replaced by mature cTECs in the adult thymus (193). The composition of the mTEC compartment also changes as perinates transition into adulthood. For example, few tuft cells are present in the neonatal mouse thymus, but their numbers increase substantially in adults (279, 281). Similarly, HCs become more abundant after the perinatal to adult transition (296). Taken together, these studies show that the network of TEC subsets undergoes extensive remodeling during the perinatal to adult transition.
TECs also undergo dynamic changes at the opposite end of the age spectrum as the thymus undergoes involution, a general feature of vertebrate aging. Thymus involution is characterized by progressive organ atrophy, reduced T cell output, disruption of thymus architecture and collapse of the TEC compartment (193, 297–300). Although both thymocyte and TEC cellularity decline as the thymus undergoes involution (161, 194, 288, 289, 291, 297), TEC depletion is a primary factor driving thymus involution. FOXN1, a transcription factor required for TEC development and maintenance, declines with age in mice and humans (291, 301–303). Genetic models in which Foxn1 expression is upregulated in TECs prior to or after thymus involution can attenuate or reverse this process (195, 304), whereas downregulation of Foxn1 results in early degeneration of the TEC compartment and premature involution (291). Furthermore, thymus involution can be prevented by expressing either a Cyclin D1 or c-myc transgene in TECs, or by deleting Retinoblastoma family genes, all of which result in a continuous thymus growth phenotype despite the fact that thymocytes are not genetically manipulated (290, 305, 306). Furthermore, heterochronic parabiosis experiments showed that migration of thymus-seeding hematopoietic cells from a young partner into the thymus of an aged partner failed to restore cellularity of the old, involuted thymus (307). Collectively, these investigations indicate that degradation of the TEC compartment is a major factor contributing to thymus involution, a finding that is not surprising given that TEC-derived signals are indispensable for T cell differentiation and selection.
Although thymus involution is generally thought to result in a progressive decline in the number of both cTEC and mTEC compartments, this view was challenged by a recent investigation showing that the extensive cytoplasmic projections characteristic of cTECs contract during involution (308). Based on these findings, it was suggested that changes in cTEC morphology, rather than cellular depletion, are responsible for the apparent reduction in cTEC cellularity and associated cortical thinning (308). In contrast, morphological changes in mTECs were not observed during thymus involution consistent with an age-associated decline in the number of mTECs. In addition, changes in mTEC gene expression patterns, including increased expression of inflammatory pathway genes (193, 309) occur during thymus involution. The mechanisms responsible for transcriptional changes may reflect altered mTEC subset composition and/or intrinsic alterations in transcriptional regulation (193, 195, 196, 309). With regard to the former possibility, a recent study combining scRNAseq and lineage tracing approaches demonstrated marked changes in TEC subset composition with age (193). Taken together, these studies show that remodeling of the TEC compartment is a characteristic and progressive feature of age-related thymus involution.
Depletion of the mTEC compartment during aging, particularly the decline in mature mTECs that express Aire-dependent TRAs (195, 196, 288, 291), is likely to compromise central tolerance and result in increased export of self-reactive T cells. Indeed, a decline in expression of Aire-regulated as well as Aire-independent TRAs has been associated with age-related thymus involution (193, 194). Interestingly, however, neither the expression of Aire nor Fezf2 (required for expression of Aire-independent TRAs) was altered in TECs obtained from aged, involuted thymuses suggesting that TRA expression depends on additional, as yet undefined, factors (193, 194). Collectively, these data suggest that the decline in mTEC cellularity, changes in mTEC subset composition and altered transcriptional signatures of mTEC subsets are features of thymus involution that may affect central tolerance and contribute to the age-associated increase in autoimmunity (3, 310).
Changes in HAPCs Across the Lifespan
While mTECs present self-antigens directly to thymocytes to mediate central tolerance, DCs also cross-present mTEC-derived TRAs to induce negative selection and Treg generation (48, 53, 54, 62, 66, 71, 73, 74). Changes have been documented in the composition of thymic DCs during the perinatal to adult transition. Some studies indicate that CD8α+ cDC1s increase in the thymus of adult relative to fetal and neonatal mice (242, 311). However, another study showed that CD8α+ cDC1s decrease during the transition from neonate to adult, whereas Sirpα+ cDC2s and pDCs increase with age in the thymus (69). Adult cDC2s express higher levels of genes associated with antigen processing and presentation and are more efficient at MHCII-dependent antigen processing and presentation to T cells compared to newborn cDC2s. Interestingly, this study indicates that the efficiency of negative selection is diminished in perinatal mice, compared to adults, correlating with a lower frequency of cDC2 cells (69). In the human thymus, XCR1+ cDC1s increase in the second trimester of pregnancy and decline postnatally with increasing age (295), consistent with the trend reported in mice (69). Thus, while multiple studies have established that the DC compartment changes with age, the cellular composition and molecular alterations remain to be elucidated.
Since mTECs influence thymic DC localization and composition, mTEC-DC crosstalk likely plays an important role in mediating central tolerance. For instance, in the adult thymus, mTECs express CCL2, XCL1, JAG1, CCL19, and CCL21, which could promote recruitment, localization and/or maturation of thymic DCs (30, 33, 34, 61, 274, 295, 312–314). CCR7 is expressed not only by thymocytes, but also by some thymic DCs, indicating that expression of CCL19 and CCL21 by mTECs could recruit not only post-positive selection thymocytes to the medulla, but also CCR7+ DCs. CCR7 is upregulated on thymic DCs by interactions with autoreactive thymocytes in a CD40-CD40L dependent manner (65). CCR7+ DCs in human and mouse fetal and postnatal thymuses express high levels of MHCII and costimulatory molecules, suggesting they may have an increased capacity to present self-antigens to medullary thymocytes (64, 65, 295). Indeed, compared to CCR7- cDC1s, CCR7+ cDC1s are more efficient at acquiring and presenting Aire-dependent TRAs to CD8SP cells (64) and CCR7+ cDC1s have also been implicated in playing a central role in presenting mTEC-derived antigens to promote Treg selection (48, 66). We found that CCR7 deficiency results in increased apoptosis of MHCIIhi cDC1s and reduced antigen transfer from mTECs, with a concomitant increase in cDC2 and, surprisingly, Treg induction (70). Thus, production of CCR7 ligands by mTECs alters the composition of the thymic DC compartment, with downstream consequences for central tolerance. Taken together with the age-associated changes in DCs and mTECs, these findings suggest that altered mTEC-DC crosstalk in the perinatal period likely impacts central tolerance induction.
While the perinatal thymic HAPC compartment has slowly garnered interest over the last two decades, there is relatively less information regarding the impact of age-associated thymic involution on thymic HAPCs. The numbers and proportions of CD8α+ cDC1s and pDCs decrease gradually with age in the mouse thymus, while migratory Sirpα+ cDC2s remain constant in number, thus comprising an increased proportion of the thymic DC compartment (315). Though reduced in number, DCs in aged mice express similar levels of the activation markers CD40, CD80, CD86, and MHCII compared to young DCs, suggesting they are functionally intact (315). In humans, the number of total thymic DCs declines in proportion to the overall decrease in cellularity of the thymus with age (316). Similar to the mouse thymus, the proportion of DCs expressing MHCII, CD80, and CD86 was not altered with age, though expression of CD40 was diminished with age (316). Our transcriptional profiling analysis showed that murine thymic cDC1s and cDC2s express an increasingly proinflammatory gene signature with age, including expression of Il1a, Il6, Tnf and Il18 (309). These aging DCs could potentially contribute to the age-associated inflammation observed in the thymus (317) and alter central tolerance. As previously mentioned, B cells mediate negative selection (81–83) and Treg induction in the thymus (318), underscoring their crucial contribution to central tolerance. Although the frequency of thymic B cells increases with age, there is a dramatic age-associated decline in Aire and Aire-dependent TRA expression in mouse and human thymic B cells (199), which could impair negative selection and Treg induction. These studies suggest that age-associated changes in HAPC subset composition and/or gene expression may impact central tolerance, highlighting the need for more comprehensive studies to determine how age-associated changes in thymic HAPCs influence negative selection, Treg induction, and the incidence of autoimmunity.
Conclusions
The cellular composition and transcriptional profiles of TECs and HAPCs undergo profound changes throughout life, suggesting that a concomitant change in central tolerance likely occurs. Indeed, multiple lines of evidence presented above suggest that the perinatal thymic microenvironment is inefficient at inducing negative selection of Tconv cells, but is biased towards selection of auto reactive Tregs that are critical for suppressing autoimmunity in various tissues throughout life. On the other end of the aging spectrum, evidence suggests that the involuting thymus becomes inefficient at supporting both negative selection and Treg induction. The impact of age on the ability of the thymus to support both arms of central tolerance warrants further investigation. Studies to date raise several important issues. For example, while negative selection may be inefficient in both the perinatal and aged thymus, only the aged thymus appears to be impaired in supporting Treg generation, invoking a possible link with the age-associated increase in incidence of autoimmune disorders. Moreover, given that age-related changes in peripheral T cell differentiation and maintenance can impact the outcome of T cell responses to self and foreign antigens, it will be important to distinguish thymic from peripheral contributions with regard to changes in Tconv and Treg cells as a function of age.
Given that expression levels of CD5 on T cells are set during positive selection in the thymus, and that the level of CD5 on Tconv and Treg cells correlates with altered activity in the periphery, elevated CD5 levels on Tconv and/or Treg cells in the neonatal and aged periods indicate that age-associated changes in thymic selection impact peripheral T cell responses. However, changes in thymic selection could reflect either an altered capacity of thymic APCs to induce selection or cell-intrinsic changes in the differentiation potential of hematopoietic progenitors that seed and differentiate within the thymus. For example, the bias of neonatal CD8 T cells towards short-lived effectors reflects altered functional potential of neonatal versus adult hematopoietic cells (122). Whether preferential differentiation of CD4SP cells into Th2 effectors and Tregs in perinates reflects changes in T cell differentiation in the periphery and/or altered thymic selection, due to either an altered microenvironment or changes in hematopoeitic progenitors, requires further investigation. Furthermore, the mechanisms underlying thymic selection of T cells with altered self-reactivity have not been firmly established. Elucidating altered functions of Tconv and Treg cells with age will enhance understanding of how the immune system responds to pathogens throughout life without invoking autoimmunity. Furthermore, determining the mechanisms underlying altered thymic selection and peripheral maintenance of functionally distinct T cell subests will inform future strategies for enhancing T cell mediated immune protection and suppressing autoimmunity.
While studies to date have identified unique aspects of immune responses at both extremes of the age spectrum, and suggested that central tolerance is subject to age-related restrictions, a number of questions remain unanswered. Some of these unresolved questions include:
1. What are the precise roles of diverse mTEC and HAPC subsets in selection across the lifespan?
2. Is the bias of perinatal Tconv towards Th2 and short-lived effector cells due to altered thymic selection? Although this was found not to be the case for short lived CD8 effector cells (85), this question remains open for CD4+ T cells.
3. Do Aire-dependent versus Aire-independent Treg subsets induce anergy in Tconv cells in a tissue-specific manner?
4. What cellular subsets and molecular mechanisms account for the propensity of the perinatal thymic microenvironment to select CD5hi Tregs?
5. Are the activated Tregs that accumulate with age and have heightened suppressive ability (159) essential for maintaining tolerance with age? A related issue is whether Tregs generated in adulthood contribute to tolerance.
Author Contributions
JS and JL reviewed the literature. JS, JL, NS and LH prepared figures and legends, and commented on manuscript drafts. LE and ER revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Work in the laboratories of LH, LE and ER is supported by National Institutes of Health grants P01-AG052359 and P01- AI139449.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Rebecca Deen for her administrative assistance in preparing the manuscript.
References
1. Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, Levy O. Protecting the Newborn and Young Infant From Infectious Diseases: Lessons From Immune Ontogeny. Immunity (2017) 46(3):350–63. doi: 10.1016/j.immuni.2017.03.009
2. Nikolich-Zugich J. The Twilight of Immunity: Emerging Concepts in Aging of the Immune System. Nat Immunol (2018) 19(1):10–9. doi: 10.1038/s41590-017-0006-x
3. Watad A, Bragazzi NL, Adawi M, Amital H, Toubi E, Porat BS, et al. Autoimmunity in the Elderly: Insights From Basic Science and Clinics - a Mini-Review. Gerontology (2017) 63(6):515–23. doi: 10.1159/000478012
4. Kollmann TR, Marchant A, Way SS. Vaccination Strategies to Enhance Immunity in Neonates. Science (2020) 368(6491):612–5. doi: 10.1126/science.aaz9447
5. McElhaney JE. Influenza Vaccine Responses in Older Adults. Ageing Res Rev (2011) 10(3):379–88. doi: 10.1016/j.arr.2010.10.008
6. Murray K, Baraniuk S, Resnick M, Arafat R, Kilborn C, Cain K, et al. Risk Factors for Encephalitis and Death From West Nile Virus Infection. Epidemiol Infect (2006) 134(6):1325–32. doi: 10.1017/S0950268806006339
7. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and Negative Selection of the T Cell Repertoire: What Thymocytes See (and Don’t See). Nat Rev Immunol (2014) 14(6):377–91. doi: 10.1038/nri3667
8. Lancaster JN, Li Y, Ehrlich LIR. Chemokine-Mediated Choreography of Thymocyte Development and Selection. Trends Immunol (2018) 39(2):86–98. doi: 10.1016/j.it.2017.10.007
9. Klein L, Robey EA, Hsieh CS. Central CD4(+) T Cell Tolerance: Deletion Versus Regulatory T Cell Differentiation. Nat Rev Immunol (2019) 19(1):7–18. doi: 10.1038/s41577-018-0083-6
10. Nikolich-Zugich J, Slifka MK, Messaoudi I. The Many Important Facets of T-Cell Repertoire Diversity. Nat Rev Immunol (2004) 4(2):123–32. doi: 10.1038/nri1292
11. Zarnitsyna VI, Evavold BD, Schoettle LN, Blattman JN, Antia R. Estimating the Diversity, Completeness, and Cross-Reactivity of the T Cell Repertoire. Front Immunol (2013) 4:485. doi: 10.3389/fimmu.2013.00485
12. Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the Peripheral Self by Naturally Arising CD25+ CD4+ T Cell Receptors. Immunity (2004) 21(2):267–77. doi: 10.1016/j.immuni.2004.07.009
13. Bautista JL, Lio CW, Lathrop SK, Forbush K, Liang Y, Luo J, et al. Intraclonal Competition Limits the Fate Determination of Regulatory T Cells in the Thymus. Nat Immunol (2009) 10(6):610–7. doi: 10.1038/ni.1739
14. Leung MW, Shen S, Lafaille JJ. TCR-Dependent Differentiation of Thymic Foxp3+ Cells is Limited to Small Clonal Sizes. J Exp Med (2009) 206(10):2121–30. doi: 10.1084/jem.20091033
15. Khailaie S, Robert PA, Toker A, Huehn J, Meyer-Hermann M. A Signal Integration Model of Thymic Selection and Natural Regulatory T Cell Commitment. J Immunol (2014) 193(12):5983–96. doi: 10.4049/jimmunol.1400889
16. Malhotra D, Linehan JL, Dileepan T, Lee YJ, Purtha WE, Lu JV, et al. Tolerance is Established in Polyclonal CD4(+) T Cells by Distinct Mechanisms, According to Self-Peptide Expression Patterns. Nat Immunol (2016) 17(2):187–95. doi: 10.1038/ni.3327
17. Picca CC, Oh S, Panarey L, Aitken M, Basehoar A, Caton AJ. Thymocyte Deletion Can Bias Treg Formation Toward Low-Abundance Self-Peptide. Eur J Immunol (2009) 39(12):3301–6. doi: 10.1002/eji.200939709
18. Lio CW, Dodson LF, Deppong CM, Hsieh CS, Green JM. CD28 Facilitates the Generation of Foxp3(-) Cytokine Responsive Regulatory T Cell Precursors. J Immunol (2010) 184(11):6007–13. doi: 10.4049/jimmunol.1000019
19. Tai X, Cowan M, Feigenbaum L, Singer A. CD28 Costimulation of Developing Thymocytes Induces Foxp3 Expression and Regulatory T Cell Differentiation Independently of Interleukin 2. Nat Immunol (2005) 6(2):152–62. doi: 10.1038/ni1160
20. Burchill MA, Yang J, Vang KB, Moon JJ, Chu HH, Lio CW, et al. Linked T Cell Receptor and Cytokine Signaling Govern the Development of the Regulatory T Cell Repertoire. Immunity (2008) 28(1):112–21. doi: 10.1016/j.immuni.2007.11.022
21. Lio CW, Hsieh CS. A Two-Step Process for Thymic Regulatory T Cell Development. Immunity (2008) 28(1):100–11. doi: 10.1016/j.immuni.2007.11.021
22. Daley SR, Hu DY, Goodnow CC. Helios Marks Strongly Autoreactive CD4+ T Cells in Two Major Waves of Thymic Deletion Distinguished by Induction of PD-1 or NF-Kappab. J Exp Med (2013) 210(2):269–85. doi: 10.1084/jem.20121458
23. Stritesky GL, Xing Y, Erickson JR, Kalekar LA, Wang X, Mueller DL, et al. Murine Thymic Selection Quantified Using a Unique Method to Capture Deleted T Cells. Proc Natl Acad Sci U S A (2013) 110(12):4679–84. doi: 10.1073/pnas.1217532110
24. Ladi E, Schwickert TA, Chtanova T, Chen Y, Herzmark P, Yin X, et al. Thymocyte-Dendritic Cell Interactions Near Sources of CCR7 Ligands in the Thymic Cortex. J Immunol (2008) 181(10):7014–23. doi: 10.4049/jimmunol.181.10.7014
25. McCaughtry TM, Baldwin TA, Wilken MS, Hogquist KA. Clonal Deletion of Thymocytes Can Occur in the Cortex With No Involvement of the Medulla. J Exp Med (2008) 205(11):2575–84. doi: 10.1084/jem.20080866
26. Melichar HJ, Ross JO, Herzmark P, Hogquist KA, Robey EA. Distinct Temporal Patterns of T Cell Receptor Signaling During Positive Versus Negative Selection in Situ. Sci Signal (2013) 6(297):ra92. doi: 10.1126/scisignal.2004400
27. Hu Z, Lancaster JN, Sasiponganan C, Ehrlich LI. CCR4 Promotes Medullary Entry and Thymocyte-Dendritic Cell Interactions Required for Central Tolerance. J Exp Med (2015) 212(11):1947–65. doi: 10.1084/jem.20150178
28. Ross JO, Melichar HJ, Au-Yeung BB, Herzmark P, Weiss A, Robey EA. Distinct Phases in the Positive Selection of CD8+ T Cells Distinguished by Intrathymic Migration and T-Cell Receptor Signaling Patterns. Proc Natl Acad Sci U S A (2014) 111(25):E2550–8. doi: 10.1073/pnas.1408482111
29. Ehrlich LI, Oh DY, Weissman IL, Lewis RS. Differential Contribution of Chemotaxis and Substrate Restriction to Segregation of Immature and Mature Thymocytes. Immunity (2009) 31(6):986–98. doi: 10.1016/j.immuni.2009.09.020
30. Kozai M, Kubo Y, Katakai T, Kondo H, Kiyonari H, Schaeuble K, et al. Essential Role of CCL21 in Establishment of Central Self-Tolerance in T Cells. J Exp Med (2017) 214(7):1925–35. doi: 10.1084/jem.20161864
31. Nitta T, Nitta S, Lei Y, Lipp M, Takahama Y. CCR7-Mediated Migration of Developing Thymocytes to the Medulla is Essential for Negative Selection to Tissue-Restricted Antigens. Proc Natl Acad Sci U S A (2009) 106(40):17129–33. doi: 10.1073/pnas.0906956106
32. Kwan J, Killeen N. CCR7 Directs the Migration of Thymocytes Into the Thymic Medulla. J Immunol (2004) 172(7):3999–4007. doi: 10.4049/jimmunol.172.7.3999
33. Ueno T, Saito F, Gray DH, Kuse S, Hieshima K, Nakano H, et al. CCR7 Signals are Essential for Cortex-Medulla Migration of Developing Thymocytes. J Exp Med (2004) 200(4):493–505. doi: 10.1084/jem.20040643
34. Kurobe H, Liu C, Ueno T, Saito F, Ohigashi I, Seach N, et al. CCR7-Dependent Cortex-to-Medulla Migration of Positively Selected Thymocytes is Essential for Establishing Central Tolerance. Immunity (2006) 24(2):165–77. doi: 10.1016/j.immuni.2005.12.011
35. Brennecke P, Reyes A, Pinto S, Rattay K, Nguyen M, Kuchler R, et al. Single-Cell Transcriptome Analysis Reveals Coordinated Ectopic Gene-Expression Patterns in Medullary Thymic Epithelial Cells. Nat Immunol (2015) 16(9):933–41. doi: 10.1038/ni.3246
36. Meredith M, Zemmour D, Mathis D, Benoist C. Aire Controls Gene Expression in the Thymic Epithelium With Ordered Stochasticity. Nat Immunol (2015) 16(9):942–9. doi: 10.1038/ni.3247
37. Sansom SN, Shikama-Dorn N, Zhanybekova S, Nusspaumer G, Macaulay IC, Deadman ME, et al. Population and Single-Cell Genomics Reveal the Aire Dependency, Relief From Polycomb Silencing, and Distribution of Self-Antigen Expression in Thymic Epithelia. Genome Res (2014) 24(12):1918–31. doi: 10.1101/gr.171645.113
38. Derbinski J, Gabler J, Brors B, Tierling S, Jonnakuty S, Hergenhahn M, et al. Promiscuous Gene Expression in Thymic Epithelial Cells is Regulated At Multiple Levels. J Exp Med (2005) 202(1):33–45. doi: 10.1084/jem.20050471
39. Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous Gene Expression in Medullary Thymic Epithelial Cells Mirrors the Peripheral Self. Nat Immunol (2001) 2(11):1032–9. doi: 10.1038/ni723
40. Gotter J, Brors B, Hergenhahn M, Kyewski B. Medullary Epithelial Cells of the Human Thymus Express a Highly Diverse Selection of Tissue-Specific Genes Colocalized in Chromosomal Clusters. J Exp Med (2004) 199(2):155–66. doi: 10.1084/jem.20031677
41. Anderson MS, Su MA. AIRE Expands: New Roles in Immune Tolerance and Beyond. Nat Rev Immunol (2016) 16(4):247–58. doi: 10.1038/nri.2016.9
42. Mathis D, Benoist C. Aire. Annu Rev Immunol (2009) 27:287–312. doi: 10.1146/annurev.immunol.25.022106.141532
43. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an Immunological Self Shadow Within the Thymus by the Aire Protein. Science (2002) 298(5597):1395–401. doi: 10.1126/science.1075958
44. Yano M, Kuroda N, Han H, Meguro-Horike M, Nishikawa Y, Kiyonari H, et al. Aire Controls the Differentiation Program of Thymic Epithelial Cells in the Medulla for the Establishment of Self-Tolerance. J Exp Med (2008) 205(12):2827–38. doi: 10.1084/jem.20080046
45. Aaltonen J, Bjorses P, Sandkuijl L, Perheentupa J, Peltonen L. An Autosomal Locus Causing Autoimmune Disease: Autoimmune Polyglandular Disease Type I Assigned to Chromosome 21. Nat Genet (1994) 8(1):83–7. doi: 10.1038/ng0994-83
46. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional Cloning of the APECED Gene. Nat Genet (1997) 17(4):393–8. doi: 10.1038/ng1297-393
47. Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA. Aire Enforces Immune Tolerance by Directing Autoreactive T Cells Into the Regulatory T Cell Lineage. Immunity (2016) 44(5):1102–13. doi: 10.1016/j.immuni.2016.02.009
48. Perry JSA, Lio CJ, Kau AL, Nutsch K, Yang Z, Gordon JI, et al. Distinct Contributions of Aire and Antigen-Presenting-Cell Subsets to the Generation of Self-Tolerance in the Thymus. Immunity (2014) 41(3):414–26. doi: 10.1016/j.immuni.2014.08.007
49. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 Costimulation is Essential for the Homeostasis of the CD4+CD25+ Immunoregulatory T Cells That Control Autoimmune Diabetes. Immunity (2000) 12(4):431–40. doi: 10.1016/S1074-7613(00)80195-8
50. Watanabe M, Lu Y, Breen M, Hodes RJ. B7-CD28 Co-Stimulation Modulates Central Tolerance Via Thymic Clonal Deletion and Treg Generation Through Distinct Mechanisms. Nat Commun (2020) 11(1):6264. doi: 10.1038/s41467-020-20070-x
51. Hinterberger M, Aichinger M, da Costa OP, Voehringer D, Hoffmann R, Klein L. Autonomous Role of Medullary Thymic Epithelial Cells in Central CD4(+) T Cell Tolerance. Nat Immunol (2010) 11(6):512–9. doi: 10.1038/ni.1874
52. Gallegos AM, Bevan MJ. Central Tolerance to Tissue-Specific Antigens Mediated by Direct and Indirect Antigen Presentation. J Exp Med (2004) 200(8):1039–49. doi: 10.1084/jem.20041457
53. Hubert FX, Kinkel SA, Davey GM, Phipson B, Mueller SN, Liston A, et al. Aire Regulates the Transfer of Antigen From Mtecs to Dendritic Cells for Induction of Thymic Tolerance. Blood (2011) 118(9):2462–72. doi: 10.1182/blood-2010-06-286393
54. Lancaster JN, Thyagarajan HM, Srinivasan J, Li Y, Hu Z, Ehrlich LIR. Live-Cell Imaging Reveals the Relative Contributions of Antigen-Presenting Cell Subsets to Thymic Central Tolerance. Nat Commun (2019) 10(1):2220. doi: 10.1038/s41467-019-09727-4
55. Aschenbrenner K, D’Cruz LM, Vollmann EH, Hinterberger M, Emmerich J, Swee LK, et al. Selection of Foxp3+ Regulatory T Cells Specific for Self Antigen Expressed and Presented by Aire+ Medullary Thymic Epithelial Cells. Nat Immunol (2007) 8(4):351–8. doi: 10.1038/ni1444
56. Lebel ME, Coutelier M, Galipeau M, Kleinman CL, Moon JJ, Melichar HJ. Differential Expression of Tissue-Restricted Antigens Among Mtec is Associated With Distinct Autoreactive T Cell Fates. Nat Commun (2020) 11(1):3734. doi: 10.1038/s41467-020-17544-3
57. Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, et al. Constitutive Ablation of Dendritic Cells Breaks Self-Tolerance of CD4 T Cells and Results in Spontaneous Fatal Autoimmunity. J Exp Med (2009) 206(3):549–59. doi: 10.1084/jem.20082394 doi: jem.20082394 [pii].
58. Guerri L, Peguillet I, Geraldo Y, Nabti S, Premel V, Lantz O. Analysis of APC Types Involved in CD4 Tolerance and Regulatory T Cell Generation Using Reaggregated Thymic Organ Cultures. J Immunol (2013) 190(5):2102–10. doi: 10.4049/jimmunol.1202883
59. Bonasio R, Scimone ML, Schaerli P, Grabie N, Lichtman AH, von Andrian UH. Clonal Deletion of Thymocytes by Circulating Dendritic Cells Homing to the Thymus. Nat Immunol (2006) 7(10):1092–100. doi: 10.1038/ni1385
60. Atibalentja DF, Murphy KM, Unanue ER. Functional Redundancy Between Thymic CD8alpha+ and Sirpalpha+ Conventional Dendritic Cells in Presentation of Blood-Derived Lysozyme by MHC Class II Proteins. J Immunol (2011) 186(3):1421–31. doi: 10.4049/jimmunol.1002587
61. Baba T, Nakamoto Y, Mukaida N. Crucial Contribution of Thymic Sirp Alpha+ Conventional Dendritic Cells to Central Tolerance Against Blood-Borne Antigens in a CCR2-Dependent Manner. J Immunol (2009) 183(5):3053–63. doi: 10.4049/jimmunol.0900438
62. Koble C, Kyewski B. The Thymic Medulla: A Unique Microenvironment for Intercellular Self-Antigen Transfer. J Exp Med (2009) 206(7):1505–13. doi: 10.1084/jem.20082449 doi: jem.20082449 [pii].
63. Millet V, Naquet P, Guinamard RR. Intercellular MHC Transfer Between Thymic Epithelial and Dendritic Cells. Eur J Immunol (2008) 38(5):1257–63. doi: 10.1002/eji.200737982
64. Ardouin L, Luche H, Chelbi R, Carpentier S, Shawket A, Montanana Sanchis F, et al. Broad and Largely Concordant Molecular Changes Characterize Tolerogenic and Immunogenic Dendritic Cell Maturation in Thymus and Periphery. Immunity (2016) 45(2):305–18. doi: 10.1016/j.immuni.2016.07.019
65. Oh J, Wu N, Barczak AJ, Barbeau R, Erle DJ, Shin JS. CD40 Mediates Maturation of Thymic Dendritic Cells Driven by Self-Reactive CD4(+) Thymocytes and Supports Development of Natural Regulatory T Cells. J Immunol (2018) 200(4):1399–412. doi: 10.4049/jimmunol.1700768
66. Perry JSA, Russler-Germain EV, Zhou YW, Purtha W, Cooper ML, Choi J, et al. Transfer of Cell-Surface Antigens by Scavenger Receptor CD36 Promotes Thymic Regulatory T Cell Receptor Repertoire Development and Allo-Tolerance. Immunity (2018) 48(6):1271. doi: 10.1016/j.immuni.2018.05.011
67. Wu L, Shortman K. Heterogeneity of Thymic Dendritic Cells. Semin Immunol (2005) 17(4):304–12. doi: 10.1016/j.smim.2005.05.001
68. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic Cells, Monocytes and Macrophages: A Unified Nomenclature Based on Ontogeny. Nat Rev Immunol (2014) 14(8):571–8. doi: 10.1038/nri3712
69. Kroger CJ, Wang B, Tisch R. Temporal Increase in Thymocyte Negative Selection Parallels Enhanced Thymic Sirpalpha(+) DC Function. Eur J Immunol (2016) 46(10):2352–62. doi: 10.1002/eji.201646354
70. Hu Z, Li Y, Van Nieuwenhuijze A, Selden HJ, Jarrett AM, Sorace AG, et al. CCR7 Modulates the Generation of Thymic Regulatory T Cells by Altering the Composition of the Thymic Dendritic Cell Compartment. Cell Rep (2017) 21(1):168–80. doi: 10.1016/j.celrep.2017.09.016
71. Proietto AI, van Dommelen S, Zhou P, Rizzitelli A, D’Amico A, Steptoe RJ, et al. Dendritic Cells in the Thymus Contribute to T-Regulatory Cell Induction. Proc Natl Acad Sci U S A (2008) 105(50):19869–74. doi: 10.1073/pnas.0810268105
72. Herbin O, Bonito AJ, Jeong S, Weinstein EG, Rahman AH, Xiong H, et al. Medullary Thymic Epithelial Cells and CD8alpha(+) Dendritic Cells Coordinately Regulate Central Tolerance But CD8alpha(+) Cells are Dispensable for Thymic Regulatory T Cell Production. J Autoimmun (2016) 75:141–9. doi: 10.1016/j.jaut.2016.08.002
73. Leventhal DS, Gilmore DC, Berger JM, Nishi S, Lee V, Malchow S, et al. Dendritic Cells Coordinate the Development and Homeostasis of Organ-Specific Regulatory T Cells. Immunity (2016) 44(4):847–59. doi: 10.1016/j.immuni.2016.01.025
74. Kroger CJ, Spidale NA, Wang B, Tisch R. Thymic Dendritic Cell Subsets Display Distinct Efficiencies and Mechanisms of Intercellular MHC Transfer. J Immunol (2017) 198(1):249–56. doi: 10.4049/jimmunol.1601516
75. Hadeiba H, Lahl K, Edalati A, Oderup C, Habtezion A, Pachynski R, et al. Plasmacytoid Dendritic Cells Transport Peripheral Antigens to the Thymus to Promote Central Tolerance. Immunity (2012) 36(3):438–50. doi: 10.1016/j.immuni.2012.01.017
76. Matsumoto T, Hasegawa H, Onishi S, Ishizaki J, Suemori K, Yasukawa M. Protein Kinase C Inhibitor Generates Stable Human Tolerogenic Dendritic Cells. J Immunol (2013) 191(5):2247–57. doi: 10.4049/jimmunol.1203053
77. Adnan E, Matsumoto T, Ishizaki J, Onishi S, Suemori K, Yasukawa M, et al. Human Tolerogenic Dendritic Cells Generated With Protein Kinase C Inhibitor are Optimal for Functional Regulatory T Cell Induction - a Comparative Study. Clin Immunol (2016) 173:96–108. doi: 10.1016/j.clim.2016.09.007
78. Watanabe N, Wang YH, Lee HK, Ito T, Wang YH, Cao W, et al. Hassall’s Corpuscles Instruct Dendritic Cells to Induce CD4+CD25+ Regulatory T Cells in Human Thymus. Nature (2005) 436(7054):1181–5. doi: 10.1038/nature03886
79. Hanabuchi S, Ito T, Park WR, Watanabe N, Shaw JL, Roman E, et al. Thymic Stromal Lymphopoietin-Activated Plasmacytoid Dendritic Cells Induce the Generation of FOXP3+ Regulatory T Cells in Human Thymus. J Immunol (2010) 184(6):2999–3007. doi: 10.4049/jimmunol.0804106
80. Martin-Gayo E, Sierra-Filardi E, Corbi AL, Toribio ML. Plasmacytoid Dendritic Cells Resident in Human Thymus Drive Natural Treg Cell Development. Blood (2010) 115(26):5366–75. doi: 10.1182/blood-2009-10-248260
81. Perera J, Meng L, Meng F, Huang H. Autoreactive Thymic B Cells are Efficient Antigen-Presenting Cells of Cognate Self-Antigens for T Cell Negative Selection. Proc Natl Acad Sci U S A (2013) 110(42):17011–6. doi: 10.1073/pnas.1313001110
82. Perera J, Zheng Z, Li S, Gudjonson H, Kalinina O, Benichou JIC, et al. Self-Antigen-Driven Thymic B Cell Class Switching Promotes T Cell Central Tolerance. Cell Rep (2016) 17(2):387–98. doi: 10.1016/j.celrep.2016.09.011
83. Yamano T, Nedjic J, Hinterberger M, Steinert M, Koser S, Pinto S, et al. Thymic B Cells are Licensed to Present Self Antigens for Central T Cell Tolerance Induction. Immunity (2015) 42(6):1048–61. doi: 10.1016/j.immuni.2015.05.013
84. Fujihara C, Williams JA, Watanabe M, Jeon H, Sharrow SO, Hodes RJ. T Cell-B Cell Thymic Cross-Talk: Maintenance and Function of Thymic B Cells Requires Cognate CD40-CD40 Ligand Interaction. J Immunol (2014) 193(11):5534–44. doi: 10.4049/jimmunol.1401655
85. Rudd BD. Neonatal T Cells: A Reinterpretation. Annu Rev Immunol (2020) 38:229–47. doi: 10.1146/annurev-immunol-091319-083608
86. Thome JJ, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, Gordon C, et al. Early-Life Compartmentalization of Human T Cell Differentiation and Regulatory Function in Mucosal and Lymphoid Tissues. Nat Med (2016) 22(1):72–7. doi: 10.1038/nm.4008
87. Winkler S, Willheim M, Baier K, Schmid D, Aichelburg A, Graninger W, et al. Frequency of Cytokine-Producing T Cells in Patients of Different Age Groups With Plasmodium Falciparum Malaria. J Infect Dis (1999) 179(1):209–16. doi: 10.1086/314571
88. Huygens A, Lecomte S, Tackoen M, Olislagers V, Delmarcelle Y, Burny W, et al. Functional Exhaustion Limits CD4+ and CD8+ T-Cell Responses to Congenital Cytomegalovirus Infection. J Infect Dis (2015) 212(3):484–94. doi: 10.1093/infdis/jiv071
89. Kadereit S, Mohammad SF, Miller RE, Woods KD, Listrom CD, McKinnon K, et al. Reduced NFAT1 Protein Expression in Human Umbilical Cord Blood T Lymphocytes. Blood (1999) 94(9):3101–7. doi: 10.1182/blood.V94.9.3101.421k04_3101_3107
90. Schmiedeberg K, Krause H, Rohl FW, Hartig R, Jorch G. Brunner-Weinzierl MC. T Cells of Infants are Mature, But Hyporeactive Due to Limited Ca2+ Influx. PloS One (2016) 11(11):e0166633. doi: 10.1371/journal.pone.0166633
91. Adkins B, Bu Y, Guevara P. Murine Neonatal CD4+ Lymph Node Cells are Highly Deficient in the Development of Antigen-Specific Th1 Function in Adoptive Adult Hosts. J Immunol (2002) 169(9):4998–5004. doi: 10.4049/jimmunol.169.9.4998
92. Adkins B, Hamilton K. Freshly Isolated, Murine Neonatal T Cells Produce IL-4 in Response to Anti-CD3 Stimulation. J Immunol (1992) 149(11):3448–55.
93. Forsthuber T, Yip HC, Lehmann PV. Induction of TH1 and TH2 Immunity in Neonatal Mice. Science (1996) 271(5256):1728–30. doi: 10.1126/science.271.5256.1728
94. Hebel K, Weinert S, Kuropka B, Knolle J, Kosak B, Jorch G, et al. CD4+ T Cells From Human Neonates and Infants are Poised Spontaneously to Run a Nonclassical IL-4 Program. J Immunol (2014) 192(11):5160–70. doi: 10.4049/jimmunol.1302539
95. Rose S, Lichtenheld M, Foote MR, Adkins B. Murine Neonatal CD4+ Cells are Poised for Rapid Th2 Effector-Like Function. J Immunol (2007) 178(5):2667–78. doi: 10.4049/jimmunol.178.5.2667
96. Ribeiro-do-Couto LM, Boeije LC, Kroon JS, Hooibrink B, Breur-Vriesendorp BS, Aarden LA, et al. High IL-13 Production by Human Neonatal T Cells: Neonate Immune System Regulator? Eur J Immunol (2001) 31(11):3394–402. doi: 10.1002/1521-4141(200111)31:11<3394::aid-immu3394>3.0.co;2-b
97. Barrios C, Brawand P, Berney M, Brandt C, Lambert PH, Siegrist CA. Neonatal and Early Life Immune Responses to Various Forms of Vaccine Antigens Qualitatively Differ From Adult Responses: Predominance of a Th2-Biased Pattern Which Persists After Adult Boosting. Eur J Immunol (1996) 26(7):1489–96. doi: 10.1002/eji.1830260713
98. Webster RB, Rodriguez Y, Klimecki WT, Vercelli D. The Human IL-13 Locus in Neonatal CD4+ T Cells is Refractory to the Acquisition of a Repressive Chromatin Architecture. J Biol Chem (2007) 282(1):700–9. doi: 10.1074/jbc.M609501200
99. White GP, Watt PM, Holt BJ, Holt PG. Differential Patterns of Methylation of the IFN-Gamma Promoter At Cpg and Non-Cpg Sites Underlie Differences in IFN-Gamma Gene Expression Between Human Neonatal and Adult CD45RO- T Cells. J Immunol (2002) 168(6):2820–7. doi: 10.4049/jimmunol.168.6.2820
100. Zheng W, Flavell RA. The Transcription Factor GATA-3 is Necessary and Sufficient for Th2 Cytokine Gene Expression in CD4 T Cells. Cell (1997) 89(4):587–96. doi: 10.1016/s0092-8674(00)80240-8
101. Frascoli M, Coniglio L, Witt R, Jeanty C, Fleck-Derderian S, Myers DE, et al. Alloreactive Fetal T Cells Promote Uterine Contractility in Preterm Labor Via IFN-Gamma and TNF-Alpha. Sci Transl Med (2018) 10(438):eaan2263. doi: 10.1126/scitranslmed.aan2263
102. Gans H, Yasukawa L, Rinki M, DeHovitz R, Forghani B, Beeler J, et al. Immune Responses to Measles and Mumps Vaccination of Infants At 6, 9, and 12 Months. J Infect Dis (2001) 184(7):817–26. doi: 10.1086/323346
103. Ota MO, Vekemans J, Schlegel-Haueter SE, Fielding K, Whittle H, Lambert PH, et al. Hepatitis B Immunisation Induces Higher Antibody and Memory Th2 Responses in New-Borns Than in Adults. Vaccine (2004) 22(3-4):511–9. doi: 10.1016/j.vaccine.2003.07.020
104. Vekemans J, Ota MO, Wang EC, Kidd M, Borysiewicz LK, Whittle H, et al. T Cell Responses to Vaccines in Infants: Defective Ifngamma Production After Oral Polio Vaccination. Clin Exp Immunol (2002) 127(3):495–8. doi: 10.1046/j.1365-2249.2002.01788.x
105. Donckier V, Abramowicz D, Bruyns C, Florquin S, Vanderhaeghen ML, Amraoui Z, et al. IFN-Gamma Prevents Th2 Cell-Mediated Pathology After Neonatal Injection of Semiallogenic Spleen Cells in Mice. J Immunol (1994) 153(6):2361–8.
106. Mingari MC, Maggi E, Cambiaggi A, Annunziato F, Schiavetti F, Manetti R, et al. Development in Vitro of Human CD4+ Thymocytes Into Functionally Mature Th2 Cells. Exogenous Interleukin-12 is Required for Priming Thymocytes to Produce Both Th1 Cytokines and Interleukin-10. Eur J Immunol (1996) 26(5):1083–7. doi: 10.1002/eji.1830260519
107. Malhotra I, Ouma J, Wamachi A, Kioko J, Mungai P, Omollo A, et al. In Utero Exposure to Helminth and Mycobacterial Antigens Generates Cytokine Responses Similar to That Observed in Adults. J Clin Invest (1997) 99(7):1759–66. doi: 10.1172/JCI119340
108. Sarzotti M, Robbins DS, Hoffman PM. Induction of Protective CTL Responses in Newborn Mice by a Murine Retrovirus. Science (1996) 271(5256):1726–8. doi: 10.1126/science.271.5256.1726
109. Martinez X, Brandt C, Saddallah F, Tougne C, Barrios C, Wild F, et al. DNA Immunization Circumvents Deficient Induction of T Helper Type 1 and Cytotoxic T Lymphocyte Responses in Neonates and During Early Life. Proc Natl Acad Sci U S A (1997) 94(16):8726–31. doi: 10.1073/pnas.94.16.8726
110. Adkins B. Peripheral CD4+ Lymphocytes Derived From Fetal Versus Adult Thymic Precursors Differ Phenotypically and Functionally. J Immunol (2003) 171(10):5157–64. doi: 10.4049/jimmunol.171.10.5157
111. Adkins B, Du RQ. Newborn Mice Develop Balanced Th1/Th2 Primary Effector Responses in Vivo But are Biased to Th2 Secondary Responses. J Immunol (1998) 160(9):4217–24.
112. Marchant A, Goetghebuer T, Ota MO, Wolfe I, Ceesay SJ, De Groote D, et al. Newborns Develop a Th1-Type Immune Response to Mycobacterium Bovis Bacillus Calmette-Guerin Vaccination. J Immunol (1999) 163(4):2249–55.
113. Vekemans J, Amedei A, Ota MO, D’Elios MM, Goetghebuer T, Ismaili J, et al. Neonatal Bacillus Calmette-Guerin Vaccination Induces Adult-Like IFN-Gamma Production by CD4+ T Lymphocytes. Eur J Immunol (2001) 31(5):1531–5. doi: 10.1002/1521-4141(200105)31:5<1531::AID-IMMU1531>3.0.CO;2-1
114. Mascart F, Verscheure V, Malfroot A, Hainaut M, Pierard D, Temerman S, et al. Bordetella Pertussis Infection in 2-Month-Old Infants Promotes Type 1 T Cell Responses. J Immunol (2003) 170(3):1504–9. doi: 10.4049/jimmunol.170.3.1504
115. Ausiello CM, Fedele G, Urbani F, Lande R, Di Carlo B, Cassone A. Native and Genetically Inactivated Pertussis Toxins Induce Human Dendritic Cell Maturation and Synergize With Lipopolysaccharide in Promoting T Helper Type 1 Responses. J Infect Dis (2002) 186(3):351–60. doi: 10.1086/341510
116. Henderson RA, Watkins SC, Flynn JL. Activation of Human Dendritic Cells Following Infection With Mycobacterium Tuberculosis. J Immunol (1997) 159(2):635–43.
117. Garza KM, Griggs ND, Tung KS. Neonatal Injection of an Ovarian Peptide Induces Autoimmune Ovarian Disease in Female Mice: Requirement of Endogenous Neonatal Ovaries. Immunity (1997) 6(1):89–96. doi: 10.1016/s1074-7613(00)80245-9
118. Ki S, Thyagarajan HM, Hu Z, Lancaster JN, Ehrlich LIR. EBI2 Contributes to the Induction of Thymic Central Tolerance in Mice by Promoting Rapid Motility of Medullary Thymocytes. Eur J Immunol (2017) 47(11):1906–17. doi: 10.1002/eji.201747020
119. Li L, Lee HH, Bell JJ, Gregg RK, Ellis JS, Gessner A, et al. IL-4 Utilizes an Alternative Receptor to Drive Apoptosis of Th1 Cells and Skews Neonatal Immunity Toward Th2. Immunity (2004) 20(4):429–40. doi: 10.1016/s1074-7613(04)00072-x
120. Lee HH, Hoeman CM, Hardaway JC, Guloglu FB, Ellis JS, Jain R, et al. Delayed Maturation of an IL-12-Producing Dendritic Cell Subset Explains the Early Th2 Bias in Neonatal Immunity. J Exp Med (2008) 205(10):2269–80. doi: 10.1084/jem.20071371
121. Smith NL, Wissink E, Wang J, Pinello JF, Davenport MP, Grimson A, et al. Rapid Proliferation and Differentiation Impairs the Development of Memory CD8+ T Cells in Early Life. J Immunol (2014) 193(1):177–84. doi: 10.4049/jimmunol.1400553
122. Wang J, Wissink EM, Watson NB, Smith NL, Grimson A, Rudd BD. Fetal and Adult Progenitors Give Rise to Unique Populations of CD8+ T Cells. Blood (2016) 128(26):3073–82. doi: 10.1182/blood-2016-06-725366
123. Reynaldi A, Smith NL, Schlub TE, Tabilas C, Venturi V, Rudd BD, et al. Fate Mapping Reveals the Age Structure of the Peripheral T Cell Compartment. Proc Natl Acad Sci U S A (2019) 116(10):3974–81. doi: 10.1073/pnas.1811634116
124. Smith NL, Patel RK, Reynaldi A, Grenier JK, Wang J, Watson NB, et al. Developmental Origin Governs CD8(+) T Cell Fate Decisions During Infection. Cell (2018) 174(1):117–30 e14. doi: 10.1016/j.cell.2018.05.029
125. Akue AD, Lee JY, Jameson SC. Derivation and Maintenance of Virtual Memory CD8 T Cells. J Immunol (2012) 188(6):2516–23. doi: 10.4049/jimmunol.1102213
126. Jacomet F, Cayssials E, Basbous S, Levescot A, Piccirilli N, Desmier D, et al. Evidence for Eomesodermin-Expressing Innate-Like CD8(+) KIR/NKG2A(+) T Cells in Human Adults and Cord Blood Samples. Eur J Immunol (2015) 45(7):1926–33. doi: 10.1002/eji.201545539
127. Zhang Y, Maksimovic J, Huang B, De Souza DP, Naselli G, Chen H, et al. Cord Blood CD8(+) T Cells Have a Natural Propensity to Express IL-4 in a Fatty Acid Metabolism and Caspase Activation-Dependent Manner. Front Immunol (2018) 9:879. doi: 10.3389/fimmu.2018.00879
128. He Q, Morillon YM,2, Spidale NA, Kroger CJ, Liu B, Sartor RB, et al. Thymic Development of Autoreactive T Cells in NOD Mice is Regulated in an Age-Dependent Manner. J Immunol (2013) 191(12):5858–66. doi: 10.4049/jimmunol.1302273
129. Huseby ES, Sather B, Huseby PG, Goverman J. Age-Dependent T Cell Tolerance and Autoimmunity to Myelin Basic Protein. Immunity (2001) 14(4):471–81. doi: 10.1016/s1074-7613(01)00127-3
130. Dong M, Artusa P, Kelly SA, Fournier M, Baldwin TA, Mandl JN, et al. Alterations in the Thymic Selection Threshold Skew the Self-Reactivity of the TCR Repertoire in Neonates. J Immunol (2017) 199(3):965–73. doi: 10.4049/jimmunol.1602137
131. Mandl JN, Monteiro JP, Vrisekoop N, Germain RN. T Cell-Positive Selection Uses Self-Ligand Binding Strength to Optimize Repertoire Recognition of Foreign Antigens. Immunity (2013) 38(2):263–74. doi: 10.1016/j.immuni.2012.09.011
132. Azzam HS, DeJarnette JB, Huang K, Emmons R, Park CS, Sommers CL, et al. Fine Tuning of TCR Signaling by CD5. J Immunol (2001) 166(9):5464–72. doi: 10.4049/jimmunol.166.9.5464
133. Osborne BA, Smith SW, Liu ZG, McLaughlin KA, Grimm L, Schwartz LM. Identification of Genes Induced During Apoptosis in T Lymphocytes. Immunol Rev (1994) 142:301–20. doi: 10.1111/j.1600-065x.1994.tb00894.x
134. Fulton RB, Hamilton SE, Xing Y, Best JA, Goldrath AW, Hogquist KA, et al. The TCR’s Sensitivity to Self Peptide-MHC Dictates the Ability of Naive CD8(+) T Cells to Respond to Foreign Antigens. Nat Immunol (2015) 16(1):107–17. doi: 10.1038/ni.3043
135. Kieper WC, Burghardt JT, Surh CD. A Role for TCR Affinity in Regulating Naive T Cell Homeostasis. J Immunol (2004) 172(1):40–4. doi: 10.4049/jimmunol.172.1.40
136. Min B, McHugh R, Sempowski GD, Mackall C, Foucras G, Paul WE. Neonates Support Lymphopenia-Induced Proliferation. Immunity (2003) 18(1):131–40. doi: 10.1016/s1074-7613(02)00508-3
137. Stoklasek TA, Colpitts SL, Smilowitz HM, Lefrancois L, MHC class I. And TCR Avidity Control the CD8 T Cell Response to IL-15/IL-15Ralpha Complex. J Immunol (2010) 185(11):6857–65. doi: 10.4049/jimmunol.1001601
138. Vrisekoop N, Artusa P, Monteiro JP, Mandl JN. Weakly Self-Reactive T-Cell Clones Can Homeostatically Expand When Present At Low Numbers. Eur J Immunol (2017) 47(1):68–73. doi: 10.1002/eji.201646540
139. White JT, Cross EW, Burchill MA, Danhorn T, McCarter MD, Rosen HR, et al. Virtual Memory T Cells Develop and Mediate Bystander Protective Immunity in an IL-15-Dependent Manner. Nat Commun (2016) 7:11291. doi: 10.1038/ncomms11291
140. Henderson JG, Opejin A, Jones A, Gross C, Hawiger D. CD5 Instructs Extrathymic Regulatory T Cell Development in Response to Self and Tolerizing Antigens. Immunity (2015) 42(3):471–83. doi: 10.1016/j.immuni.2015.02.010
141. Ciabattini A, Nardini C, Santoro F, Garagnani P, Franceschi C, Medaglini D. Vaccination in the Elderly: The Challenge of Immune Changes With Aging. Semin Immunol (2018) 40:83–94. doi: 10.1016/j.smim.2018.10.010
142. Simon AK, Hollander GA, McMichael A. Evolution of the Immune System in Humans From Infancy to Old Age. Proc Biol Sci (2015) 282(1821):20143085. doi: 10.1098/rspb.2014.3085
143. Nikolich-Zugich J, Bradshaw CM, Uhrlaub JL, Watanabe M. Immunity to Acute Virus Infections With Advanced Age. Curr Opin Virol (2020) 46:45–58. doi: 10.1016/j.coviro.2020.09.007
144. Shen-Orr SS, Furman D. Variability in the Immune System: of Vaccine Responses and Immune States. Curr Opin Immunol (2013) 25(4):542–7. doi: 10.1016/j.coi.2013.07.009
145. Fong TC, Makinodan T. In Situ Hybridization Analysis of the Age-Associated Decline in IL-2 Mrna Expressing Murine T Cells. Cell Immunol (1989) 118(1):199–207. doi: 10.1016/0008-8749(89)90369-9
146. Haynes L, Linton PJ, Eaton SM, Tonkonogy SL, Swain SL. Interleukin 2, But Not Other Common Gamma Chain-Binding Cytokines, Can Reverse the Defect in Generation of CD4 Effector T Cells From Naive T Cells of Aged Mice. J Exp Med (1999) 190(7):1013–24. doi: 10.1084/jem.190.7.1013
147. Eaton SM, Burns EM, Kusser K, Randall TD, Haynes L. Age-Related Defects in CD4 T Cell Cognate Helper Function Lead to Reductions in Humoral Responses. J Exp Med (2004) 200(12):1613–22. doi: 10.1084/jem.20041395
148. Richner JM, Gmyrek GB, Govero J, Tu Y, van der Windt GJ, Metcalf TU, et al. Age-Dependent Cell Trafficking Defects in Draining Lymph Nodes Impair Adaptive Immunity and Control of West Nile Virus Infection. PloS Pathog (2015) 11(7):e1005027. doi: 10.1371/journal.ppat.1005027
149. Uhrlaub JL, Pulko V, DeFilippis VR, Broeckel R, Streblow DN, Coleman GD, et al. Dysregulated TGF-Beta Production Underlies the Age-Related Vulnerability to Chikungunya Virus. PloS Pathog (2016) 12(10):e1005891. doi: 10.1371/journal.ppat.1005891
150. Garcia GG, Miller RA. Age-Related Defects in the Cytoskeleton Signaling Pathways of CD4 T Cells. Ageing Res Rev (2011) 10(1):26–34. doi: 10.1016/j.arr.2009.11.003
151. Clise-Dwyer K, Huston GE, Buck AL, Duso DK, Swain SL. Environmental and Intrinsic Factors Lead to Antigen Unresponsiveness in CD4(+) Recent Thymic Emigrants From Aged Mice. J Immunol (2007) 178(3):1321–31. doi: 10.4049/jimmunol.178.3.1321
152. Ron-Harel N, Notarangelo G, Ghergurovich JM, Paulo JA, Sage PT, Santos D, et al. Defective Respiration and One-Carbon Metabolism Contribute to Impaired Naive T Cell Activation in Aged Mice. Proc Natl Acad Sci U S A (2018) 115(52):13347–52. doi: 10.1073/pnas.1804149115
153. Becklund BR, Purton JF, Ramsey C, Favre S, Vogt TK, Martin CE, et al. The Aged Lymphoid Tissue Environment Fails to Support Naive T Cell Homeostasis. Sci Rep (2016) 6:30842. doi: 10.1038/srep30842
154. Thompson HL, Smithey MJ, Uhrlaub JL, Jeftic I, Jergovic M, White SE, et al. Lymph Nodes as Barriers to T-Cell Rejuvenation in Aging Mice and Nonhuman Primates. Aging Cell (2019) 18(1):e12865. doi: 10.1111/acel.12865
155. Deshpande NR, Parrish HL, Kuhns MS. Self-Recognition Drives the Preferential Accumulation of Promiscuous CD4(+) T-Cells in Aged Mice. Elife (2015) 4:e05949. doi: 10.7554/eLife.05949
156. Maue AC, Yager EJ, Swain SL, Woodland DL, Blackman MA, Haynes L. T-Cell Immunosenescence: Lessons Learned From Mouse Models of Aging. Trends Immunol (2009) 30(7):301–5. doi: 10.1016/j.it.2009.04.007
157. Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, et al. Spatial Map of Human T Cell Compartmentalization and Maintenance Over Decades of Life. Cell (2014) 159(4):814–28. doi: 10.1016/j.cell.2014.10.026
158. Wertheimer AM, Bennett MS, Park B, Uhrlaub JL, Martinez C, Pulko V, et al. Aging and Cytomegalovirus Infection Differentially and Jointly Affect Distinct Circulating T Cell Subsets in Humans. J Immunol (2014) 192(5):2143–55. doi: 10.4049/jimmunol.1301721
159. Elyahu Y, Hekselman I, Eizenberg-Magar I, Berner O, Strominger I, Schiller M, et al. Aging Promotes Reorganization of the CD4 T Cell Landscape Toward Extreme Regulatory and Effector Phenotypes. Sci Adv (2019) 5(8):eaaw8330. doi: 10.1126/sciadv.aaw8330
160. Mogilenko DA, Shpynov O, Andhey PS, Arthur L, Swain A, Esaulova E, et al. Comprehensive Profiling of an Aging Immune System Reveals Clonal GZMK(+) CD8(+) T Cells as Conserved Hallmark of Inflammaging. Immunity (2021) 54(1):99–115 e12. doi: 10.1016/j.immuni.2020.11.005
161. Hale JS, Boursalian TE, Turk GL, Fink PJ. Thymic Output in Aged Mice. Proc Natl Acad Sci U S A (2006) 103(22):8447–52. doi: 10.1073/pnas.0601040103
162. Tsukamoto H, Clise-Dwyer K, Huston GE, Duso DK, Buck AL, Johnson LL, et al. Age-Associated Increase in Lifespan of Naive CD4 T Cells Contributes to T-Cell Homeostasis But Facilitates Development of Functional Defects. Proc Natl Acad Sci U S A (2009) 106(43):18333–8. doi: 10.1073/pnas.0910139106
163. Tsukamoto H, Huston GE, Dibble J, Duso DK, Swain SL. Bim Dictates Naive CD4 T Cell Lifespan and the Development of Age-Associated Functional Defects. J Immunol (2010) 185(8):4535–44. doi: 10.4049/jimmunol.1001668
164. Kim C, Jadhav RR, Gustafson CE, Smithey MJ, Hirsch AJ, Uhrlaub JL, et al. Defects in Antiviral T Cell Responses Inflicted by Aging-Associated Mir-181a Deficiency. Cell Rep (2019) 29(8):2202–16 e5. doi: 10.1016/j.celrep.2019.10.044
165. Li G, Yu M, Lee WW, Tsang M, Krishnan E, Weyand CM, et al. Decline in Mir-181a Expression With Age Impairs T Cell Receptor Sensitivity by Increasing DUSP6 Activity. Nat Med (2012) 18(10):1518–24. doi: 10.1038/nm.2963
166. Schaffert SA, Loh C, Wang S, Arnold CP, Axtell RC, Newell EW, et al. Mir-181a-1/B-1 Modulates Tolerance Through Opposing Activities in Selection and Peripheral T Cell Function. J Immunol (2015) 195(4):1470–9. doi: 10.4049/jimmunol.1401587
167. Effros RB, Walford RL. The Immune Response of Aged Mice to Influenza: Diminished T-Cell Proliferation, Interleukin 2 Production and Cytotoxicity. Cell Immunol (1983) 81(2):298–305. doi: 10.1016/0008-8749(83)90237-x
168. Briceno O, Lissina A, Wanke K, Afonso G, von Braun A, Ragon K, et al. Reduced Naive CD8(+) T-Cell Priming Efficacy in Elderly Adults. Aging Cell (2016) 15(1):14–21. doi: 10.1111/acel.12384
169. Decman V, Laidlaw BJ, Dimenna LJ, Abdulla S, Mozdzanowska K, Erikson J, et al. Cell-Intrinsic Defects in the Proliferative Response of Antiviral Memory CD8 T Cells in Aged Mice Upon Secondary Infection. J Immunol (2010) 184(9):5151–9. doi: 10.4049/jimmunol.0902063
170. Decman V, Laidlaw BJ, Doering TA, Leng J, Ertl HC, Goldstein DR, et al. Defective CD8 T Cell Responses in Aged Mice are Due to Quantitative and Qualitative Changes in Virus-Specific Precursors. J Immunol (2012) 188(4):1933–41. doi: 10.4049/jimmunol.1101098
171. Heng TS, Reiseger JJ, Fletcher AL, Leggatt GR, White OJ, Vlahos K, et al. Impact of Sex Steroid Ablation on Viral, Tumour and Vaccine Responses in Aged Mice. PloS One (2012) 7(8):e42677. doi: 10.1371/journal.pone.0042677
172. Jiang J, Fisher EM, Murasko DM. Intrinsic Defects in CD8 T Cells With Aging Contribute to Impaired Primary Antiviral Responses. Exp Gerontol (2013) 48(6):579–86. doi: 10.1016/j.exger.2013.02.027
173. Quinn KM, Fox A, Harland KL, Russ BE, Li J, Nguyen THO, et al. Age-Related Decline in Primary CD8(+) T Cell Responses is Associated With the Development of Senescence in Virtual Memory CD8(+) T Cells. Cell Rep (2018) 23(12):3512–24. doi: 10.1016/j.celrep.2018.05.057
174. Renkema KR, Li G, Wu A, Smithey MJ, Nikolich-Zugich J. Two Separate Defects Affecting True Naive or Virtual Memory T Cell Precursors Combine to Reduce Naive T Cell Responses With Aging. J Immunol (2014) 192(1):151–9. doi: 10.4049/jimmunol.1301453
175. Smithey MJ, Renkema KR, Rudd BD, Nikolich-Zugich J. Increased Apoptosis, Curtailed Expansion and Incomplete Differentiation of CD8+ T Cells Combine to Decrease Clearance of L. Monocytogenes Old Mice Eur J Immunol (2011) 41(5):1352–64. doi: 10.1002/eji.201041141
176. Schulz AR, Malzer JN, Domingo C, Jurchott K, Grutzkau A, Babel N, et al. Low Thymic Activity and Dendritic Cell Numbers are Associated With the Immune Response to Primary Viral Infection in Elderly Humans. J Immunol (2015) 195(10):4699–711. doi: 10.4049/jimmunol.1500598
177. Rudd BD, Venturi V, Li G, Samadder P, Ertelt JM, Way SS, et al. Nonrandom Attrition of the Naive CD8+ T-Cell Pool With Aging Governed by T-Cell Receptor:Pmhc Interactions. Proc Natl Acad Sci U S A (2011) 108(33):13694–9. doi: 10.1073/pnas.1107594108
178. Haluszczak C, Akue AD, Hamilton SE, Johnson LD, Pujanauski L, Teodorovic L, et al. The Antigen-Specific CD8+ T Cell Repertoire in Unimmunized Mice Includes Memory Phenotype Cells Bearing Markers of Homeostatic Expansion. J Exp Med (2009) 206(2):435–48. doi: 10.1084/jem.20081829
179. Jacomet F, Cayssials E, Barbarin A, Desmier D, Basbous S, Lefevre L, et al. The Hypothesis of the Human Inkt/Innate CD8(+) T-Cell Axis Applied to Cancer: Evidence for a Deficiency in Chronic Myeloid Leukemia. Front Immunol (2016) 7:688. doi: 10.3389/fimmu.2016.00688
180. Callahan JE, Kappler JW, Marrack P. Unexpected Expansions of CD8-Bearing Cells in Old Mice. J Immunol (1993) 151(12):6657–69.
181. Lee JY, Hamilton SE, Akue AD, Hogquist KA, Jameson SC. Virtual Memory CD8 T Cells Display Unique Functional Properties. Proc Natl Acad Sci U S A (2013) 110(33):13498–503. doi: 10.1073/pnas.1307572110
182. Clambey ET, White J, Kappler JW, Marrack P. Identification of Two Major Types of Age-Associated CD8 Clonal Expansions With Highly Divergent Properties. Proc Natl Acad Sci U S A (2008) 105(35):12997–3002. doi: 10.1073/pnas.0805465105
183. Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T Cell Maintenance and Function in Human Aging. J Immunol (2015) 194(9):4073–80. doi: 10.4049/jimmunol.1500046
184. Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY, et al. Diversity and Clonal Selection in the Human T-Cell Repertoire. Proc Natl Acad Sci U S A (2014) 111(36):13139–44. doi: 10.1073/pnas.1409155111
185. Rudd BD, Venturi V, Davenport MP, Nikolich-Zugich J. Evolution of the Antigen-Specific CD8+ TCR Repertoire Across the Life Span: Evidence for Clonal Homogenization of the Old TCR Repertoire. J Immunol (2011) 186(4):2056–64. doi: 10.4049/jimmunol.1003013
186. Goronzy JJ, Weyand CM. Successful and Maladaptive T Cell Aging. Immunity (2017) 46(3):364–78. doi: 10.1016/j.immuni.2017.03.010
187. Davies JS, Thompson HL, Pulko V, Padilla Torres J, Nikolich-Zugich J. Role of Cell-Intrinsic and Environmental Age-Related Changes in Altered Maintenance of Murine T Cells in Lymphoid Organs. J Gerontol A Biol Sci Med Sci (2018) 73(8):1018–26. doi: 10.1093/gerona/glx102
188. Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of Phagocytic and Antigen Cross-Presenting Capacity in Aging Dendritic Cells is Associated With Mitochondrial Dysfunction. J Immunol (2015) 195(6):2624–32. doi: 10.4049/jimmunol.1501006
189. Li G, Smithey MJ, Rudd BD, Nikolich-Zugich J. Age-Associated Alterations in CD8alpha+ Dendritic Cells Impair CD8 T-Cell Expansion in Response to an Intracellular Bacterium. Aging Cell (2012) 11(6):968–77. doi: 10.1111/j.1474-9726.2012.00867.x
190. LeLievre CS, Le Douarin N. Mesenchymal Derivatives of the Neural Crest: Analysis of Chimaeric Quail and Chick Embryos. J Embryol Exp Morph (1975) 34:125–54.
191. Turner VM, Mabbott NA. Structural and Functional Changes to Lymph Nodes in Ageing Mice. Immunology (2017) 151(2):239–47. doi: 10.1111/imm.12727
192. Quinn KM, Zaloumis SG, Cukalac T, Kan WT, Sng XY, Mirams M, et al. Heightened Self-Reactivity Associated With Selective Survival, But Not Expansion, of Naive Virus-Specific CD8+ T Cells in Aged Mice. Proc Natl Acad Sci U S A (2016) 113(5):1333–8. doi: 10.1073/pnas.1525167113
193. Baran-Gale J, Morgan MD, Maio S, Dhalla F, Calvo-Asensio I, Deadman ME, et al. Ageing Compromises Mouse Thymus Function and Remodels Epithelial Cell Differentiation. Elife (2020) 9:1–27. doi: 10.7554/eLife.56221
194. Griffith AV, Fallahi M, Venables T, Petrie HT. Persistent Degenerative Changes in Thymic Organ Function Revealed by an Inducible Model of Organ Regrowth. Aging Cell (2012) 11(1):169–77. doi: 10.1111/j.1474-9726.2011.00773.x
195. Bredenkamp N, Nowell CS, Blackburn CC. Regeneration of the Aged Thymus by a Single Transcription Factor. Development (2014) 141(8):1627–37. doi: 10.1242/dev.103614
196. Lepletier A, Hun ML, Hammett MV, Wong K, Naeem H, Hedger M, et al. Interplay Between Follistatin, Activin a, and BMP4 Signaling Regulates Postnatal Thymic Epithelial Progenitor Cell Differentiation During Aging. Cell Rep (2019) 27(13):3887–901. doi: 10.1016/j.celrep.2019.05.045
197. Liston A, Gray DH, Lesage S, Fletcher AL, Wilson J, Webster KE, et al. Gene Dosage–Limiting Role of Aire in Thymic Expression, Clonal Deletion, and Organ-Specific Autoimmunity. J Exp Med (2004) 200(8):1015–26. doi: 10.1084/jem.20040581
198. Oh J, Wang W, Thomas R, Su DM. Capacity of Ttreg Generation is Not Impaired in the Atrophied Thymus. PloS Biol (2017) 15(11):e2003352. doi: 10.1371/journal.pbio.2003352
199. Cepeda S, Cantu C, Orozco S, Xiao Y, Brown Z, Semwal MK, et al. Age-Associated Decline in Thymic B Cell Expression of Aire and Aire-Dependent Self-Antigens. Cell Rep (2018) 22(5):1276–87. doi: 10.1016/j.celrep.2018.01.015
200. Flores KG, Li J, Hale LP. B Cells in Epithelial and Perivascular Compartments of Human Adult Thymus. Hum Pathol (2001) 32(9):926–34. doi: 10.1053/hupa.2001.27106
201. Flores KG, Li J, Sempowski GD, Haynes BF, Hale LP. Analysis of the Human Thymic Perivascular Space During Aging. J Clin Invest (1999) 104(8):1031–9. doi: 10.1172/JCI7558
202. Campbell C, Rudensky A. Roles of Regulatory T Cells in Tissue Pathophysiology and Metabolism. Cell Metab (2020) 31(1):18–25. doi: 10.1016/j.cmet.2019.09.010
203. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) is Caused by Mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
204. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a New Forkhead/Winged-Helix Protein, Scurfin, Results in the Fatal Lymphoproliferative Disorder of the Scurfy Mouse. Nat Genet (2001) 27(1):68–73. doi: 10.1038/83784
205. Godfrey VL, Wilkinson JE, Russell LB. X-Linked Lymphoreticular Disease in the Scurfy (Sf) Mutant Mouse. Am J Pathol (1991) 138(6):1379–87.
206. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-Linked Neonatal Diabetes Mellitus, Enteropathy and Endocrinopathy Syndrome is the Human Equivalent of Mouse Scurfy. Nat Genet (2001) 27(1):18–20. doi: 10.1038/83707
207. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T Cells Prevent Catastrophic Autoimmunity Throughout the Lifespan of Mice. Nat Immunol (2007) 8(2):191–7. doi: 10.1038/ni1428
208. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T Cell-Derived Interleukin-10 Limits Inflammation At Environmental Interfaces. Immunity (2008) 28(4):546–58. doi: 10.1016/j.immuni.2008.02.017
209. Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, et al. Altering the Distribution of Foxp3(+) Regulatory T Cells Results in Tissue-Specific Inflammatory Disease. J Exp Med (2007) 204(6):1335–47. doi: 10.1084/jem.20070081
210. Kim KS, Hong SW, Han D, Yi J, Jung J, Yang BG, et al. Dietary Antigens Limit Mucosal Immunity by Inducing Regulatory T Cells in the Small Intestine. Science (2016) 351(6275):858–63. doi: 10.1126/science.aac5560
211. Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL, et al. Thymus-Derived Regulatory T Cells Contribute to Tolerance to Commensal Microbiota. Nature (2013) 497(7448):258–62. doi: 10.1038/nature12079
212. Round JL, Mazmanian SK. Inducible Foxp3+ Regulatory T-Cell Development by a Commensal Bacterium of the Intestinal Microbiota. Proc Natl Acad Sci U S A (2010) 107(27):12204–9. doi: 10.1073/pnas.0909122107
213. Russler-Germain EV, Rengarajan S, Hsieh CS. Antigen-Specific Regulatory T-Cell Responses to Intestinal Microbiota. Mucosal Immunol (2017) 10(6):1375–86. doi: 10.1038/mi.2017.65
214. Yissachar N, Zhou Y, Ung L, Lai NY, Mohan JF, Ehrlicher A, et al. An Intestinal Organ Culture System Uncovers a Role for the Nervous System in Microbe-Immune Crosstalk. Cell (2017) 168(6):1135–48 e12. doi: 10.1016/j.cell.2017.02.009
215. Rodriguez RS, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW, et al. Memory Regulatory T Cells Reside in Human Skin. J Clin Invest (2014) 124(3):1027–36. doi: 10.1172/JCI72932
216. Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, et al. A Wave of Regulatory T Cells Into Neonatal Skin Mediates Tolerance to Commensal Microbes. Immunity (2015) 43(5):1011–21. doi: 10.1016/j.immuni.2015.10.016
217. McPherson SW, Heuss ND, Gregerson DS. Local “on-Demand” Generation and Function of Antigen-Specific Foxp3+ Regulatory T Cells. J Immunol (2013) 190(10):4971–81. doi: 10.4049/jimmunol.1202625
218. Silver PB, Horai R, Chen J, Jittayasothorn Y, Chan CC, Villasmil R, et al. Retina-Specific T Regulatory Cells Bring About Resolution and Maintain Remission of Autoimmune Uveitis. J Immunol (2015) 194(7):3011–9. doi: 10.4049/jimmunol.1402650
219. Szanya V, Ermann J, Taylor C, Holness C, Fathman CG. The Subpopulation of CD4+CD25+ Splenocytes That Delays Adoptive Transfer of Diabetes Expresses L-Selectin and High Levels of CCR7. J Immunol (2002) 169(5):2461–5. doi: 10.4049/jimmunol.169.5.2461
220. Hori S, Haury M, Coutinho A, Demengeot J. Specificity Requirements for Selection and Effector Functions of CD25+4+ Regulatory T Cells in Anti-Myelin Basic Protein T Cell Receptor Transgenic Mice. Proc Natl Acad Sci U S A (2002) 99(12):8213–8. doi: 10.1073/pnas.122224799
221. Plitas G, Rudensky AY. Regulatory T Cells: Differentiation and Function. Cancer Immunol Res (2016) 4(9):721–5. doi: 10.1158/2326-6066.CIR-16-0193
222. Wing JB, Tanaka A, Sakaguchi S. Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity (2019) 50(2):302–16. doi: 10.1016/j.immuni.2019.01.020
223. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 Control Over Foxp3+ Regulatory T Cell Function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
224. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine Generation Catalyzed by CD39 and CD73 Expressed on Regulatory T Cells Mediates Immune Suppression. J Exp Med (2007) 204(6):1257–65. doi: 10.1084/jem.20062512
225. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ Regulatory T Cells Induce Cytokine Deprivation-Mediated Apoptosis of Effector CD4+ T Cells. Nat Immunol (2007) 8(12):1353–62. doi: 10.1038/ni1536
226. Sundstedt A, O’Neill EJ, Nicolson KS, Wraith DC. Role for IL-10 in Suppression Mediated by Peptide-Induced Regulatory T Cells in Vivo. J Immunol (2003) 170(3):1240–8. doi: 10.4049/jimmunol.170.3.1240
227. Li MO, Sanjabi S, Flavell RA. Transforming Growth Factor-β Controls Development, Homeostasis, and Tolerance of T Cells by Regulatory T Cell-Dependent and -Independent Mechanisms. Immunity (2006) 25(3):455–71. doi: 10.1016/j.immuni.2006.07.011
228. Dowling MR, Kan A, Heinzel S, Marchingo JM, Hodgkin PD, Hawkins ED. Regulatory T Cells Suppress Effector T Cell Proliferation by Limiting Division Destiny. Front Immunol (2018) 9:2461. doi: 10.3389/fimmu.2018.02461
229. Shen S, Ding Y, Tadokoro CE, Olivares-Villagomez D, Camps-Ramirez M, Curotto de Lafaille MA, et al. Control of Homeostatic Proliferation by Regulatory T Cells. J Clin Invest (2005) 115(12):3517–26. doi: 10.1172/JCI25463
230. Nishizuka Y, Sakakura T. Thymus and Reproduction: Sex-Linked Dysgenesia of the Gonad After Neonatal Thymectomy in Mice. Science (1969) 166(3906):753–5. doi: 10.1126/science.166.3906.753
231. Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune Disease as a Consequence of Developmental Abnormality of a T Cell Subpopulation. J Exp Med (1996) 184(2):387–96. doi: 10.1084/jem.184.2.387
232. Sakaguchi S, Takahashi T, Nishizuka Y. Study on Cellular Events in Post-Thymectomy Autoimmune Oophoritis in Mice. II. Requirement of Lyt-1 Cells in Normal Female Mice for the Prevention of Oophoritis. J Exp Med (1982) 156(6):1577–86. doi: 10.1084/jem.156.6.1577
233. Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental Regulation of Foxp3 Expression During Ontogeny. J Exp Med (2005) 202(7):901–6. doi: 10.1084/jem.20050784
234. Cupedo T, Nagasawa M, Weijer K, Blom B, Spits H. Development and Activation of Regulatory T Cells in the Human Fetus. Eur J Immunol (2005) 35(2):383–90. doi: 10.1002/eji.200425763
235. Darrasse-Jeze G, Marodon G, Salomon BL, Catala M, Klatzmann D. Ontogeny of CD4+CD25+ Regulatory/Suppressor T Cells in Human Fetuses. Blood (2005) 105(12):4715–21. doi: 10.1182/blood-2004-10-4051
236. Hayakawa S, Ohno N, Okada S, Kobayashi M. Significant Augmentation of Regulatory T Cell Numbers Occurs During the Early Neonatal Period. Clin Exp Immunol (2017) 190(2):268–79. doi: 10.1111/cei.13008
237. Michaelsson J, Mold JE, McCune JM, Nixon DF. Regulation of T Cell Responses in the Developing Human Fetus. J Immunol (2006) 176(10):5741–8. doi: 10.4049/jimmunol.176.10.5741
238. Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina SA, et al. Fetal and Adult Hematopoietic Stem Cells Give Rise to Distinct T Cell Lineages in Humans. Science (2010) 330(6011):1695–9. doi: 10.1126/science.1196509
239. Cheng C, Wang S, Ye P, Huang X, Liu Z, Wu J, et al. ‘Default’ Generated Neonatal Regulatory T Cells are Hypomethylated At Conserved Non-Coding Sequence 2 and Promote Long-Term Cardiac Allograft Survival. Immunology (2014) 143(4):618–30. doi: 10.1111/imm.12343
240. Wang G, Miyahara Y, Guo Z, Khattar M, Stepkowski SM, Chen W. “Default” Generation of Neonatal Regulatory T Cells. J Immunol (2010) 185(1):71–8. doi: 10.4049/jimmunol.0903806
241. Guerau-de-Arellano M, Martinic M, Benoist C, Mathis D. Neonatal Tolerance Revisited: A Perinatal Window for Aire Control of Autoimmunity. J Exp Med (2009) 206(6):1245–52. doi: 10.1084/jem.20090300
242. Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Immune Tolerance. Regulatory T Cells Generated Early in Life Play a Distinct Role in Maintaining Self-Tolerance. Science (2015) 348(6234):589–94. doi: 10.1126/science.aaa7017
243. Li M, Zhao W, Wang Y, Jin L, Jin G, Sun X. A Wave of Foxp3+ Regulatory T Cell Accumulation in the Neonatal Liver Plays Unique Roles in Maintaining Self-Tolerance. Cell Mol Immunol (2019). doi: 10.1038/s41423-019-0246-9
244. Tuncel J, Benoist C, Mathis D. T Cell Anergy in Perinatal Mice is Promoted by T Reg Cells and Prevented by IL-33. J Exp Med (2019) 216(6):1328–44. doi: 10.1084/jem.20182002
245. Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, McCoy KD, et al. Lung Microbiota Promotes Tolerance to Allergens in Neonates Via PD-L1. Nat Med (2014) 20(6):642–7. doi: 10.1038/nm.3568
246. Knoop KA, Gustafsson JK, McDonald KG, Kulkarni DH, Coughlin PE, McCrate S, et al. Microbial Antigen Encounter During a Preweaning Interval is Critical for Tolerance to Gut Bacteria. Sci Immunol (2017) 2(18):1–11. doi: 10.1126/sciimmunol.aao1314
247. Wyss L, Stadinski BD, King CG, Schallenberg S, McCarthy NI, Lee JY, et al. Affinity for Self Antigen Selects Treg Cells With Distinct Functional Properties. Nat Immunol (2016) 17(9):1093–101. doi: 10.1038/ni.3522
248. Stadinski BD, Blevins SJ, Spidale NA, Duke BR, Huseby PG, Stern LJ, et al. A Temporal Thymic Selection Switch and Ligand Binding Kinetics Constrain Neonatal Foxp3(+) Treg Cell Development. Nat Immunol (2019) 20(8):1046–58. doi: 10.1038/s41590-019-0414-1
249. Rodriguez-Perea AL, Arcia ED, Rueda CM, Velilla PA. Phenotypical Characterization of Regulatory T Cells in Humans and Rodents. Clin Exp Immunol (2016) 185(3):281–91. doi: 10.1111/cei.12804
250. Chiu BC, Stolberg VR, Zhang H, Chensue SW. Increased Foxp3(+) Treg Cell Activity Reduces Dendritic Cell Co-Stimulatory Molecule Expression in Aged Mice. Mech Ageing Dev (2007) 128(11-12):618–27. doi: 10.1016/j.mad.2007.09.002
251. Lages CS, Suffia I, Velilla PA, Huang B, Warshaw G, Hildeman DA, et al. Functional Regulatory T Cells Accumulate in Aged Hosts and Promote Chronic Infectious Disease Reactivation. J Immunol (2008) 181(3):1835–48. doi: 10.4049/jimmunol.181.3.1835
252. Rosenkranz D, Weyer S, Tolosa E, Gaenslen A, Berg D, Leyhe T, et al. Higher Frequency of Regulatory T Cells in the Elderly and Increased Suppressive Activity in Neurodegeneration. J Neuroimmunol (2007) 188(1-2):117–27. doi: 10.1016/j.jneuroim.2007.05.011
253. Chougnet CA, Tripathi P, Lages CS, Raynor J, Sholl A, Fink P, et al. A Major Role for Bim in Regulatory T Cell Homeostasis. J Immunol (2011) 186(1):156–63. doi: 10.4049/jimmunol.1001505
254. Sharma S, Dominguez AL, Lustgarten J. High Accumulation of T Regulatory Cells Prevents the Activation of Immune Responses in Aged Animals. J Immunol (2006) 177(12):8348–55. doi: 10.4049/jimmunol.177.12.8348
255. Gregg R, Smith CM, Clark FJ, Dunnion D, Khan N, Chakraverty R, et al. The Number of Human Peripheral Blood CD4+ CD25high Regulatory T Cells Increases With Age. Clin Exp Immunol (2005) 140(3):540–6. doi: 10.1111/j.1365-2249.2005.02798.x
256. Nishioka T, Shimizu J, Iida R, Yamazaki S, Sakaguchi S. CD4+CD25+Foxp3+ T Cells and CD4+CD25-Foxp3+ T Cells in Aged Mice. J Immunol (2006) 176(11):6586–93. doi: 10.4049/jimmunol.176.11.6586
257. Williams-Bey Y, Jiang J, Murasko DM. Expansion of Regulatory T Cells in Aged Mice Following Influenza Infection. Mech Ageing Dev (2011) 132(4):163–70. doi: 10.1016/j.mad.2011.03.001
258. Liston A, Gray DH. Homeostatic Control of Regulatory T Cell Diversity. Nat Rev Immunol (2014) 14(3):154–65. doi: 10.1038/nri3605
259. Bhaskaran N, Faddoul F, Paes da Silva A, Jayaraman S, Schneider E, Mamileti P, et al. IL-1beta-Myd88-Mtor Axis Promotes Immune-Protective IL-17A(+)Foxp3(+) Cells During Mucosal Infection and is Dysregulated With Aging. Front Immunol (2020) 11:595936. doi: 10.3389/fimmu.2020.595936
260. Sun L, Hurez VJ, Thibodeaux SR, Kious MJ, Liu A, Lin P, et al. Aged Regulatory T Cells Protect From Autoimmune Inflammation Despite Reduced STAT3 Activation and Decreased Constraint of IL-17 Producing T Cells. Aging Cell (2012) 11(3):509–19. doi: 10.1111/j.1474-9726.2012.00812.x
261. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ Regulatory T Cells Control TH17 Responses in a Stat3-Dependent Manner. Science (2009) 326(5955):986–91. doi: 10.1126/science.1172702
262. Thiault N, Darrigues J, Adoue V, Gros M, Binet B, Perals C, et al. Peripheral Regulatory T Lymphocytes Recirculating to the Thymus Suppress the Development of Their Precursors. Nat Immunol (2015) 16(6):628–34. doi: 10.1038/ni.3150
263. Yang E, Zou T, Leichner TM, Zhang SL, Kambayashi T. Both Retention and Recirculation Contribute to Long-Lived Regulatory T-Cell Accumulation in the Thymus. Eur J Immunol (2014) 44(9):2712–20. doi: 10.1002/eji.201444529
264. Weist BM, Kurd N, Boussier J, Chan SW, Robey EA. Thymic Regulatory T Cell Niche Size is Dictated by Limiting IL-2 From Antigen-Bearing Dendritic Cells and Feedback Competition. Nat Immunol (2015) 16(6):635–41. doi: 10.1038/ni.3171
265. Carpentier M, Chappert P, Kuhn C, Lalfer M, Flament H, Burlen-Defranoux O, et al. Extrathymic Induction of Foxp3(+) Regulatory T Cells Declines With Age in a T-Cell Intrinsic Manner. Eur J Immunol (2013) 43(10):2598–604. doi: 10.1002/eji.201343532
266. Raynor J, Sholl A, Plas DR, Bouillet P, Chougnet CA, Hildeman DA. IL-15 Fosters Age-Driven Regulatory T Cell Accrual in the Face of Declining IL-2 Levels. Front Immunol (2013) 4:161. doi: 10.3389/fimmu.2013.00161
267. Darrigues J, van Meerwijk JPM, Romagnoli P. Age-Dependent Changes in Regulatory T Lymphocyte Development and Function: A Mini-Review. Gerontology (2018) 64(1):28–35. doi: 10.1159/000478044
268. Gratz IK, Campbell DJ. Organ-Specific and Memory Treg Cells: Specificity, Development, Function, and Maintenance. Front Immunol (2014) 5:333. doi: 10.3389/fimmu.2014.00333
269. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional Delineation and Differentiation Dynamics of Human CD4+ T Cells Expressing the Foxp3 Transcription Factor. Immunity (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
270. Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol (2016) 34:609–33. doi: 10.1146/annurev-immunol-032712-095948
271. Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 Provides Localized Access to IL-2 and Defines Homeostatically Distinct Regulatory T Cell Subsets. J Exp Med (2014) 211(1):121–36. doi: 10.1084/jem.20131142
272. Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, Germain RN. Immune Homeostasis Enforced by Co-Localized Effector and Regulatory T Cells. Nature (2015) 528(7581):225–30. doi: 10.1038/nature16169
273. Cosway EJ, Lucas B, James KD, Parnell SM, Carvalho-Gaspar M, White AJ, et al. Redefining Thymus Medulla Specialization for Central Tolerance. J Exp Med (2017) 214(11):3183–95. doi: 10.1084/jem.20171000
274. Lei Y, Ripen AM, Ishimaru N, Ohigashi I, Nagasawa T, Jeker LT, et al. Aire-Dependent Production of XCL1 Mediates Medullary Accumulation of Thymic Dendritic Cells and Contributes to Regulatory T Cell Development. J Exp Med (2011) 208(2):383–94. doi: 10.1084/jem.20102327 doi: jem.20102327 [pii].
275. Voboril M, Brabec T, Dobes J, Splichalova I, Brezina J, Cepkova A, et al. Toll-Like Receptor Signaling in Thymic Epithelium Controls Monocyte-Derived Dendritic Cell Recruitment and Treg Generation. Nat Commun (2020) 11(1):2361. doi: 10.1038/s41467-020-16081-3
276. Gray D, Abramson J, Benoist C, Mathis D. Proliferative Arrest and Rapid Turnover of Thymic Epithelial Cells Expressing Aire. J Exp Med (2007) 204(11):2521–8. doi: 10.1084/jem.20070795
277. Hamazaki Y, Sekai M, Minato N. Medullary Thymic Epithelial Stem Cells: Role in Thymic Epithelial Cell Maintenance and Thymic Involution. Immunol Rev (2016) 271(1):38–55. doi: 10.1111/imr.12412
278. Rossi SW, Chidgey AP, Parnell SM, Jenkinson WE, Scott HS, Boyd RL, et al. Redefining Epithelial Progenitor Potential in the Developing Thymus. Eur J Immunol (2007) 37(9):2411–8. doi: 10.1002/eji.200737275
279. Bornstein C, Nevo S, Giladi A, Kadouri N, Pouzolles M, Gerbe F, et al. Single-Cell Mapping of the Thymic Stroma Identifies IL-25-Producing Tuft Epithelial Cells. Nature (2018) 559(7715):622–6. doi: 10.1038/s41586-018-0346-1
280. Dhalla F, Baran-Gale J, Maio S, Chappell L, Hollander GA, Ponting CP. Biologically Indeterminate Yet Ordered Promiscuous Gene Expression in Single Medullary Thymic Epithelial Cells. EMBO J (2020) 39(1):e101828. doi: 10.15252/embj.2019101828
281. Lucas B, White AJ, Cosway EJ, Parnell SM, James KD, Jones ND, et al. Diversity in Medullary Thymic Epithelial Cells Controls the Activity and Availability of Inkt Cells. Nat Commun (2020) 11(1):2198. doi: 10.1038/s41467-020-16041-x
282. Miller CN, Proekt I, von Moltke J, Wells KL, Rajpurkar AR, Wang H, et al. Thymic Tuft Cells Promote an IL-4-Enriched Medulla and Shape Thymocyte Development. Nature (2018) 559(7715):627–31. doi: 10.1038/s41586-018-0345-2
283. Lkhagvasuren E, Sakata M, Ohigashi I, Takahama Y. Lymphotoxin Beta Receptor Regulates the Development of CCL21-Expressing Subset of Postnatal Medullary Thymic Epithelial Cells. J Immunol (2013) 190(10):5110–7. doi: 10.4049/jimmunol.1203203
284. Owen DL, Mahmud SA, Sjaastad LE, Williams JB, Spanier JA, Simeonov DR, et al. Thymic Regulatory T Cells Arise Via Two Distinct Developmental Programs. Nat Immunol (2019) 20(2):195–205. doi: 10.1038/s41590-018-0289-6
285. Hale LP, Markert ML. Corticosteroids Regulate Epithelial Cell Differentiation and Hassall Body Formation in the Human Thymus. J Immunol (2004) 172(1):617–24. doi: 10.4049/jimmunol.172.1.617
286. Nishikawa Y, Hirota F, Yano M, Kitajima H, Miyazaki J, Kawamoto H, et al. Biphasic Aire Expression in Early Embryos and in Medullary Thymic Epithelial Cells Before End-Stage Terminal Differentiation. J Exp Med (2010) 207(5):963–71. doi: 10.1084/jem.20092144 doi: jem.20092144 [pii].
287. Watanabe N, Hanabuchi S, Soumelis V, Yuan W, Ho S, de Waal Malefyt R, et al. Human Thymic Stromal Lymphopoietin Promotes Dendritic Cell-Mediated CD4+ T Cell Homeostatic Expansion. Nat Immunol (2004) 5(4):426–34. doi: 10.1038/ni1048
288. Gray DH, Seach N, Ueno T, Milton MK, Liston A, Lew AM, et al. Developmental Kinetics, Turnover, and Stimulatory Capacity of Thymic Epithelial Cells. Blood (2006) 108(12):3777–85. doi: 10.1182/blood-2006-02-004531
289. Hirakawa M, Nagakubo D, Kanzler B, Avilov S, Krauth B, Happe C, et al. Fundamental Parameters of the Developing Thymic Epithelium in the Mouse. Sci Rep (2018) 8(1):11095. doi: 10.1038/s41598-018-29460-0
290. Cowan JE, Malin J, Zhao Y, Seedhom MO, Harly C, Ohigashi I, et al. Myc Controls a Distinct Transcriptional Program in Fetal Thymic Epithelial Cells That Determines Thymus Growth. Nat Commun (2019) 10(1):5498. doi: 10.1038/s41467-019-13465-y
291. Chen L, Xiao S, Manley NR. Foxn1 is Required to Maintain the Postnatal Thymic Microenvironment in a Dosage-Sensitive Manner. Blood (2009) 113(3):567–74. doi: 10.1182/blood-2008-05-156265
292. Cuddihy AR, Ge S, Zhu J, Jang J, Chidgey A, Thurston G, et al. VEGF-Mediated Cross-Talk Within the Neonatal Murine Thymus. Blood (2009) 113(12):2723–31. doi: 10.1182/blood-2008-06-162040
293. de Barros SC, Suterwala BT, He C, Ge S, Chick B, Blumberg GK, et al. Pleiotropic Roles of VEGF in the Microenvironment of the Developing Thymus. J Immunol (2020) 205(9):2423–36. doi: 10.4049/jimmunol.1901519
294. Sakata M, Ohigashi I, Takahama Y. Cellularity of Thymic Epithelial Cells in the Postnatal Mouse. J Immunol (2018) 200(4):1382–8. doi: 10.4049/jimmunol.1701235
295. Park JE, Botting RA, Dominguez Conde C, Popescu DM, Lavaert M, Kunz DJ, et al. A Cell Atlas of Human Thymic Development Defines T Cell Repertoire Formation. Science (2020) 367(6480):1–11. doi: 10.1126/science.aay3224
296. White AJ, Nakamura K, Jenkinson WE, Saini M, Sinclair C, Seddon B, et al. Lymphotoxin Signals From Positively Selected Thymocytes Regulate the Terminal Differentiation of Medullary Thymic Epithelial Cells. J Immunol (2010) 185(8):4769–76. doi: 10.4049/jimmunol.1002151 doi: jimmunol.1002151 [pii].
297. Chinn IK, Blackburn CC, Manley NR, Sempowski GD. Changes in Primary Lymphoid Organs With Aging. Semin Immunol (2012) 24(5):309–20. doi: 10.1016/j.smim.2012.04.005
298. Lynch HE, Goldberg GL, Chidgey A, Van den Brink MR, Boyd R, Sempowski GD. Thymic Involution and Immune Reconstitution. Trends Immunol (2009) 30(7):366–73. doi: 10.1016/j.it.2009.04.003
299. Aw D, Silva AB, Maddick M, von Zglinicki T, Palmer DB. Architectural Changes in the Thymus of Aging Mice. Aging Cell (2008) 7(2):158–67. doi: 10.1111/j.1474-9726.2007.00365.x
300. Hun ML, Wong K, Gunawan JR, Alsharif A, Quinn K, Chidgey AP. Gender Disparity Impacts on Thymus Aging and LHRH Receptor Antagonist-Induced Thymic Reconstitution Following Chemotherapeutic Damage. Front Immunol (2020) 11:302. doi: 10.3389/fimmu.2020.00302
301. Nowell CS, Bredenkamp N, Tetelin S, Jin X, Tischner C, Vaidya H, et al. Foxn1 Regulates Lineage Progression in Cortical and Medullary Thymic Epithelial Cells But is Dispensable for Medullary Sublineage Divergence. PloS Genet (2011) 7(11):e1002348. doi: 10.1371/journal.pgen.1002348
302. Rode I, Martins VC, Kublbeck G, Maltry N, Tessmer C, Rodewald HR. Foxn1 Protein Expression in the Developing, Aging, and Regenerating Thymus. J Immunol (2015) 195(12):5678–87. doi: 10.4049/jimmunol.1502010
303. Reis MD, Csomos K, Dias LP, Prodan Z, Szerafin T, Savino W, et al. Decline of FOXN1 Gene Expression in Human Thymus Correlates With Age: Possible Epigenetic Regulation. Immun Ageing (2015) 12:18. doi: 10.1186/s12979-015-0045-9
304. Zook EC, Krishack PA, Zhang S, Zeleznik-Le NJ, Firulli AB, Witte PL, et al. Overexpression of Foxn1 Attenuates Age-Associated Thymic Involution and Prevents the Expansion of Peripheral CD4 Memory T Cells. Blood (2011) 118(22):5723–31. doi: 10.1182/blood-2011-03-342097 doi: blood-2011-03-342097 [pii].
305. Garfin PM, Min D, Bryson JL, Serwold T, Edris B, Blackburn CC, et al. Inactivation of the RB Family Prevents Thymus Involution and Promotes Thymic Function by Direct Control of Foxn1 Expression. J Exp Med (2013) 210(6):1087–97. doi: 10.1084/jem.20121716
306. Klug DB, Crouch E, Carter C, Coghlan L, Conti CJ, Richie ER. Transgenic Expression of Cyclin D1 in Thymic Epithelial Precursors Promotes Epithelial and T Cell Development. J Immunol (2000) 164:1881–8. doi: 10.4049/jimmunol.164.4.1881
307. Kim MJ, Miller CM, Shadrach JL, Wagers AJ, Serwold T. Young, Proliferative Thymic Epithelial Cells Engraft and Function in Aging Thymuses. J Immunol (2015) 194(10):4784–95. doi: 10.4049/jimmunol.1403158
308. Venables T, Griffith AV, DeAraujo A, Petrie HT. Dynamic Changes in Epithelial Cell Morphology Control Thymic Organ Size During Atrophy and Regeneration. Nat Commun (2019) 10(1):4402. doi: 10.1038/s41467-019-11879-2
309. Ki S, Park D, Selden HJ, Seita J, Chung H, Kim J, et al. Global Transcriptional Profiling Reveals Distinct Functions of Thymic Stromal Subsets and Age-Related Changes During Thymic Involution. Cell Rep (2014) 9(1):402–15. doi: 10.1016/j.celrep.2014.08.070
310. Muller L, Pawelec G. As We Age: Does Slippage of Quality Control in the Immune System Lead to Collateral Damage? Ageing Res Rev (2015) 23(Pt A):116–23. doi: 10.1016/j.arr.2015.01.005
311. Dakic A, Shao QX, D’Amico A, O’Keeffe M, Chen WF, Shortman K, et al. Development of the Dendritic Cell System During Mouse Ontogeny. J Immunol (2004) 172(2):1018–27. doi: 10.4049/jimmunol.172.2.1018
312. Cosway EJ, Ohigashi I, Schauble K, Parnell SM, Jenkinson WE, Luther S, et al. Formation of the Intrathymic Dendritic Cell Pool Requires CCL21-Mediated Recruitment of CCR7(+) Progenitors to the Thymus. J Immunol (2018) 201(2):516–23. doi: 10.4049/jimmunol.1800348
313. Martin-Gayo E, Gonzalez-Garcia S, Garcia-Leon MJ, Murcia-Ceballos A, Alcain J, Garcia-Peydro M, et al. Spatially Restricted JAG1-Notch Signaling in Human Thymus Provides Suitable DC Developmental Niches. J Exp Med (2017) 214(11):3361–79. doi: 10.1084/jem.20161564
314. Ueno T, Hara K, Willis MS, Malin MA, Hopken UE, Gray DH, et al. Role for CCR7 Ligands in the Emigration of Newly Generated T Lymphocytes From the Neonatal Thymus. Immunity (2002) 16(2):205–18. doi: 10.1016/S1074-7613(02)00267-4
315. van Dommelen SL, Rizzitelli A, Chidgey A, Boyd R, Shortman K, Wu L. Regeneration of Dendritic Cells in Aged Mice. Cell Mol Immunol (2010) 7(2):108–15. doi: 10.1038/cmi.2009.114
316. Varas A, Sacedon R, Hernandez-Lopez C, Jimenez E, Garcia-Ceca J, Arias-Diaz J, et al. Age-Dependent Changes in Thymic Macrophages and Dendritic Cells. Microsc Res Tech (2003) 62(6):501–7. doi: 10.1002/jemt.10411
317. Sempowski GD, Hale LP, Sundy JS, Massey JM, Koup RA, Douek DC, et al. Leukemia Inhibitory Factor, Oncostatin M, IL-6, and Stem Cell Factor Mrna Expression in Human Thymus Increases With Age and is Associated With Thymic Atrophy. J Immunol (2000) 164(4):2180–7. doi: 10.4049/jimmunol.164.4.2180
Keywords: central tolerance, life span, thymus, thymic epithelial cells, dendritic cells
Citation: Srinivasan J, Lancaster JN, Singarapu N, Hale LP, Ehrlich LIR and Richie ER (2021) Age-Related Changes in Thymic Central Tolerance. Front. Immunol. 12:676236. doi: 10.3389/fimmu.2021.676236
Received: 04 March 2021; Accepted: 06 April 2021;
Published: 22 April 2021.
Edited by:
Jennifer Elizabeth Cowan, National Institutes of Health (NIH), United StatesReviewed by:
Avinash Bhandoola, National Institutes of Health (NIH), United StatesYousuke Takahama, National Institute of Health (NIH), United States
Copyright © 2021 Srinivasan, Lancaster, Singarapu, Hale, Ehrlich and Richie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ellen R. Richie, RVJpY2hpZUBtZGFuZGVyc29uLm9yZw==
†These authors share first authorship
‡These authors share last authorship