 Guanqun Li1†
Guanqun Li1† Hongze Chen1†Liwei Liu1Peng Xiao1Yu Xie1
Hongze Chen1†Liwei Liu1Peng Xiao1Yu Xie1 Xinglong Geng1Tao Zhang1
Xinglong Geng1Tao Zhang1 Yang Zhang1Tianqi Lu1Hongtao Tan1
Yang Zhang1Tianqi Lu1Hongtao Tan1 Le Li1*
Le Li1* Bei Sun1,2*
Bei Sun1,2*- 1Department of Pancreatic and Biliary Surgery, The First Affiliated Hospital of Harbin Medical University, Harbin, China
- 2Key Laboratory of Hepatosplenic Surgery, Ministry of Education, Harbin, China
Acute pancreatitis (AP) is a leading cause of death and is commonly accompanied by systemic manifestations that are generally associated with a poor prognosis. Many cytokines contribute to pancreatic tissue damage and cause systemic injury. Interleukin-17 (IL-17) is a cytokine that may play a vital role in AP. Specifically, IL-17 has important effects on the immune response and causes interactions between different inflammatory mediators in the AP-related microenvironment. In this literature review, we will discuss the existing academic understanding of IL-17 and the impacts of IL-17 in different cells (especially in acinar cells and immune system cells) in AP pathogenesis. The clinical significance and potential mechanisms of IL-17 on AP deterioration are emphasized. The evidence suggests that inhibiting the IL-17 cytokine family could alleviate the pathogenic process of AP, and we highlight therapeutic strategies that directly or indirectly target IL-17 cytokines in acute pancreatitis.
Background
Acute pancreatitis (AP) is an inflammatory disorder of the pancreas related to tissue damage and includes a cascade of adverse cellular events (1, 2). Starting from the initial premature activation of enzymes with the involvement of the immune system in a potential systemic inflammatory reaction and organ failure, acute pancreatitis has a high lethality and poor prognosis. Currently, there is no effective therapeutic agent that reduces the risks and consequences of AP, which has a mortality rate of up to 30% (3, 4). Over the past decade, immune system activation has been identified as a key trigger and regulator of inflammatory injury in the pancreas, affecting the extent of pancreatic necrosis, organ failure and disease deterioration (5, 6). AP leads to overactivation of leukocytes and increased neutrophil metastasis to inflammation, with a consequent release of proinflammatory factors, including several forms of leukocyte interleukins, procalcitonin, transforming growth factor, and tumor necrosis factor (7, 8). The elevated expression of cytokines seems to provide objective evidence in the progression of AP and is paralleled by a pronounced immune response that amplifies disease severity.
Interleukin-17 (IL-17) is an important proinflammatory cytokine produced by T helper 17 (Th17) cells, γδ T cells and natural killer (NK) cells (9). The primary function of IL-17 is to mediate responses to pathogens and symbioses through various targets, all of which balance the inflammatory response with the immune system (10). In addition to its potential role in regulating the immune response to balance cytotoxic and tolerant immune profiles, it also results in acute injury. As a proinflammatory cytokine, IL-17 essentially incorporates a complicated network of cytokines that is responsible for various inflammatory conditions and pathogenesis (11–14). The inflammatory response and cytokine production are particularly crucial in the progression of AP. To date, few studies have systematically described the role of the IL-17 family in AP. This review outlines the biological properties of IL-17 and its impacts on the pathogenesis of AP. Our review proposes to broaden the treatment of AP by targeting IL-17.
IL-17 Family and Receptors
Commonly, the IL-17 family has six members, in alphabetical order, biologically labeled IL-17A through IL-17F (Figure 1). Among them, IL-17A is a proinflammatory cytokine participating in the primary responses to fungi and bacterial infections, followed by its close homolog IL-17F (15, 16). Novel research demonstrated that intestinal epithelial cell-derived IL-17D, as the least studied member, serves as a critical factor in regulating group 3 innate lymphoid cell (ILC3) functions and intestinal homeostasis by binding the receptor CD93 (17). These results further expand the biological role of the IL-17 family. IL-17 is a characteristic cytokine of T helper 17 (Th17) cells, and it can also be produced by other innate and adaptive immune cells, including CD8+ T cells, γδ T cells, innate lymphoid cells and dendritic cells (18–20). To signal, the biological impacts of IL-17 occur through interactions with its receptor, which is broadly distributed in different tissues and cells of the immune system (21). IL-17RA to IL-17RE are five subtypes of the IL-17 receptor family (IL-17R) (22). Due to the wide expression of IL-17RA and IL-17RC multichain receptors in most cell types, IL-17 has crucial impacts at the local and systemic levels. The activation of IL-17RA and/or IL-17RC signaling upregulates many inflammatory genes, including proinflammatory cytokines and neutrophil-specific chemokines.
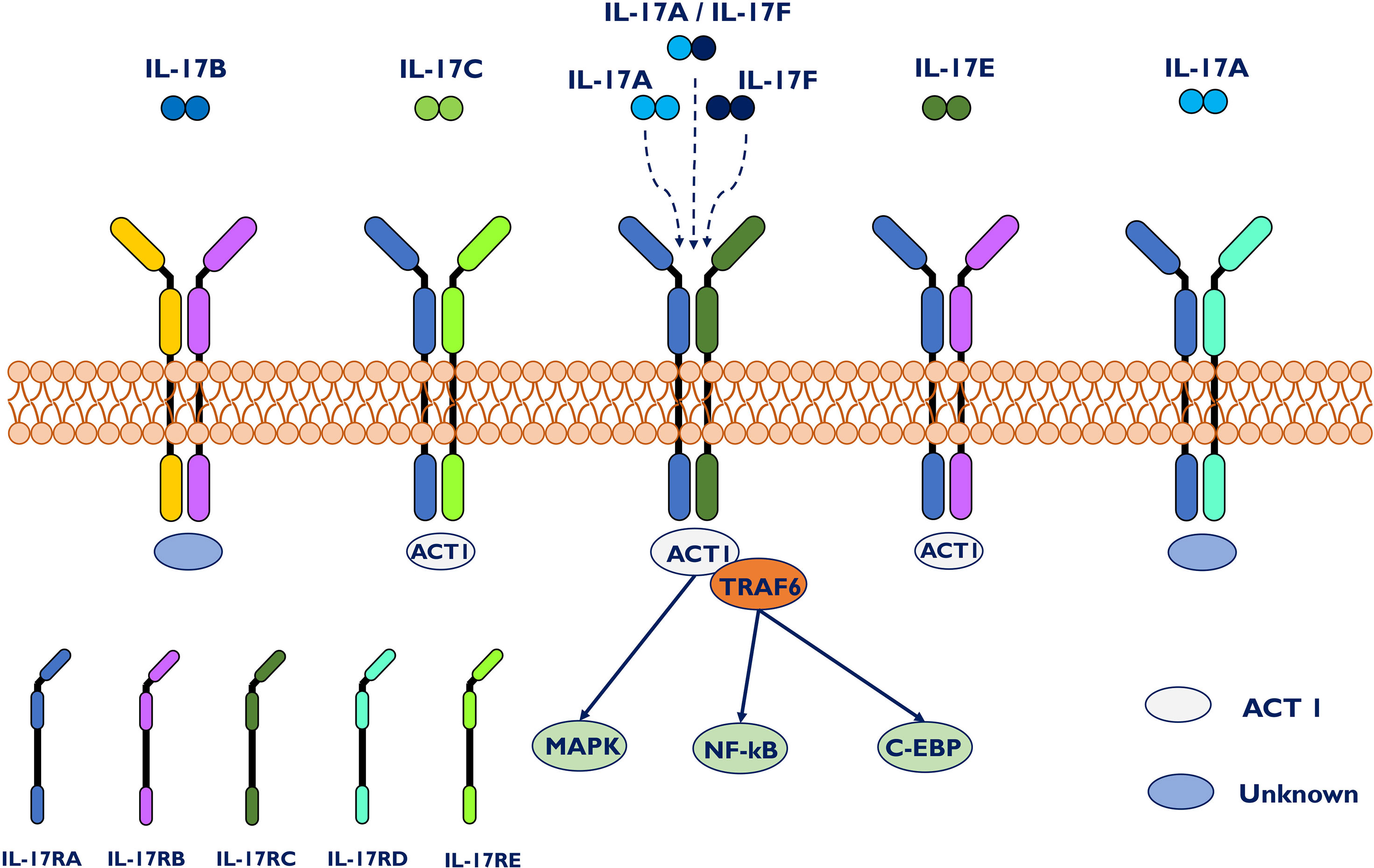
Figure 1 IL-17 family, IL-17 receptors and signal transduction. The IL-17 family is composed of six homologous dimeric proteins (IL-17A to F), while the receptor family consists of five members, IL-17RA to E. IL-17A and IL-17F bind either as individual homodimers or as a heterodimer to a dimeric IL17RA–IL17RC complex. Act1 has been shown to be the key signal-transducing molecule downstream of IL17RA and C, recruiting TRAF6 to further trigger downstream signaling pathways. Act1 has also been identified in IL-17C- and IL-17E-mediated signaling. Specifically, IL-17RA/RD acts only as a receptor for IL-17A/A. IL-17C associates with IL17RA/E, whereas IL-17E binds to IL-17RA/B.
Many studies suggest that upregulation of IL-17 contributes to various acute injury microenvironments (23–26). In pulmonary inflammation, overexpression of IL-17 or induction of exogenous IL-17 through the airway induces severe tissue injury in the lung. However, knockout models lacking critical elements of IL-17 signaling are protected from such damage (27). In acute kidney injury, IL-17 triggers neutrophil recruitment to decrease Th17 activation, verifying the role of IL-17 in Th17 cell activation in inflamed tissue (28). Numerous studies have shown that IL-17 is present at sites of the inflammatory microenvironment and in synergistic interactions, amplifying the inflammation induced by other cytokines, including IL-1, IL-6, IL-8, and TNF-a (14, 29, 30). Thus, current studies have provided a deep understanding of IL-17 and novel strategies for acute injury conditions.
The Role of IL-17 in AP
Novel insights into AP pathophysiology have demonstrated the importance of the immune response on the inflammatory progression of AP (31). Previous studies have shown that the innate immune system is critical in mediating the progression of AP (32, 33). In AP, several types of granulocytes, such as neutrophils, mast cells, macrophages, dendritic cells and platelets, all are the basis for the pathogenesis and development of AP (34, 35). In addition to innate immune cells, T cells are also present in the site of inflamed pancreatic tissue. Immune paralysis occurs, as a result of T cell apoptosis, triggering a breakdown of defense mechanisms during systemic inflammation (36). During AP, cellular damage caused by pancreatic self-digestion can induce the aggregation of CD4+ T helper cells producing IL-17 and stimulate an inflammatory response, which is characteristic of this disease. Thus, recruitment of various immune cells will result in further damage to acinar cells as well as multiple proinflammatory cytokines releasing (37).
IL-17 as a Novel Biomarker of AP
Studies have found that IL-17A upregulates the transcription of proinflammatory and neutrophil-mobilizing cytokines or chemokines in acute inflammatory diseases (38–40). To date, some researchers have confirmed the correlation between IL-17 and AP and suggest that IL-17 is a predictive marker of AP and correlates with the severity of organ failure (Table 1) (41, 42). The serum level of IL-17 correlates with the severity of AP and is a valuable prognostic factor in assessing disease progression in AP patients (P < 0.01) (43). Compared with healthy controls, AP patients had a significant increase in IL-17 during the first 24 hours, with a positive predictive value of 85.3% (44). In a previous study, researchers reported that higher serum IL-17 is an independent risk factor for adverse outcomes in severe acute pancreatitis (SAP) and is related to excessive bacterial load. Furthermore, IL-17 could be applied as a prognostic factor for length of stay, organ dysfunction, and mortality in SAP patients with continuous blood purification therapy (45). A combination of different proinflammatory factors can provide more comprehensive information to diagnose and treat AP. The serum levels of IL-17 in the progression of AP were correlated with CRP and IL-23 levels. Consistent with current clinical evidence, animal models also confirmed that AP induces the expression of various proinflammatory cytokines, including IL-17, which reach a high level during the first week of AP (46). Although IL-17A is associated with the initiation of systemic inflammatory response syndrome in AP, IL-17A may not be the cause of sepsis in the second stage (pancreatic infection and necrosis) (47). Considering its high prognostic value and rapid availability, IL-17 is considered a promising reference marker among single indicators and enhances the predictive validity of AP.
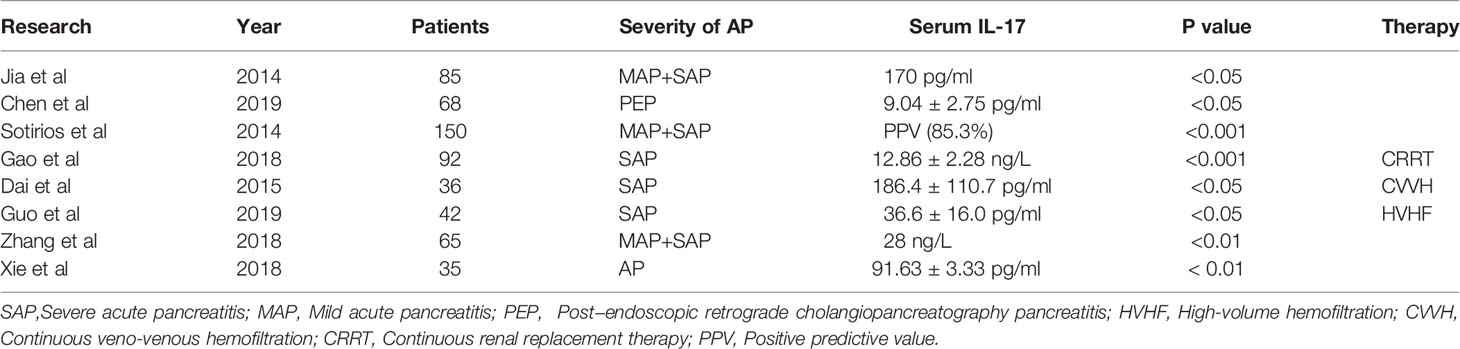
Table 1 IL-17 as a novel biomarker of AP with current clinical results.
IL-17A Induces Pancreatic Acinar Cell Damage
Clinically, inflammation, edema, necrosis of pancreatic tissue and extrapancreatic organs are the main pathobiological courses of AP. Apoptotic death represents the capacity of acinar cells to recombine their genetic program after the initial phase of mild AP, and necrosis is the main process of SAP cell death (48, 49). Indeed, acinar cells synthesize and release cytokines and chemokines that recruit immune cells, such as neutrophils and macrophages (50). Subsequently, an uncontrollable inflammatory event within acinar cells develops into systemic inflammatory response syndrome (SIRS), which eventually contributes to the high incidence rate and poor prognosis.
Pancreatic resident cells, pancreatic acinar cells and pancreatic stellate cells (PSCs) can produce inflammatory mediators (51). In AP, damaged acinar cells and recruited inflammatory monocytes/macrophages release IL-1β and IL-6 to recruit naïve CD4+ T cells into pancreatic tissue, which differentiate into Th17 cells to produce IL-17 acting on IL-17 receptor-positive cells to release various inflammatory mediators exacerbating AP. The IL-17A receptor is a heterodimer complex consisting of IL-17RA and IL-17RC and is broadly expressed in acinar cells, stellate cells, monocytes/macrophages, neutrophils and other immune cells. Mechanistically, IL-17A interacts with acinar cells, infiltrating immune cells, and other cytokines to aggravate the progression of AP and related complications. Acinar cells express DC-SIGN, a phenotype of dendritic cell, promoting the differentiation of naïve CD4+ T cells into CD4+/IFN-γ+ Th1 and CD4+/IL-17A+ Th17 cells in pancreatic tissues during AP (52).
In addition, neutrophils and macrophages directly induce intra-acinar cellular protease activation and acinar necrosis to determine the severity of AP (53). The inflammatory and immune activation of AP is thought to be characterized by neutrophil infiltration and the production of various proinflammatory cytokines (54). IL-17A strengthens neutrophil recruitment to the area of sepsis, thus ameliorating bacterial clearance (55). Neutrophil consumption assays characterized neutrophils as crucial promoters of acinar cell necrosis and inhibitors of acinar cell apoptosis during AP (56). Thus, IL-17A may amplify the inflammatory cascade during AP by recruiting neutrophils and macrophages to the damaged area, contributing to the severity of AP. Neutrophil extracellular traps (NETs), the second bactericidal mechanism of neutrophils, promotes pancreatic tissue injury and plays key role in the pathophysiological mechanisms of AP (57). IL-17 could enhance reactive oxygen species concentrations and NETs production in neutrophils during AP (58). Leppkes et al. also found that IL-17 recruits neutrophils in a PADI4-dependent manner to induce AP (59). Inhibition of PADIs prevents NETs formation (60). Both animal and clinical studies have shown that IL-17A is elevated during the primary phase of AP and induces pancreatic injury with acinar cell necrosis (61). Notably, IL-17A analogs also damage acinar cells directly and stimulate these cells to secrete inflammatory cytokines and chemokines, thereby amplifying the cascade of AP. The accumulation of inflammatory mediators is responsible for pancreatic acinar cell necrosis and multiple organ dysfunction (62).
IL-17 Acts in Synergy With Other Proinflammatory Mediators
IL-17A has the ability to act in synergy with other potent proinflammatory mediators to promote the inflammatory response (63). The impacts of IL-17 originate from its capacity to enroll immune cells by producing chemokines and to induce the expression of receptors, particularly the TNF receptor (64), and its synergistic effects with other cytokines. Different proinflammatory factors have their own characteristics in AP. The combination of different proinflammatory factors can provide more comprehensive information for the diagnosis and treatment of AP. IL-17 mainly interacts with nonleukocytic cells, including epithelial cells, endothelial cells and macrophages (65). IL-17 activates the production of other proinflammatory cytokines, such as IL-1, TNF-α, IL-6 and granulocyte-macrophage colony stimulating factor, collectively resulting in an influx of neutrophils (18).
In mice with acute pancreatitis, dendritic cells (DCs) are essential for pancreatic viability and might protect organs against cell stress (66). A recent study reported that Th17 cells from patients with inflammation potently induce the differentiation and activation of DCs that preferentially promote the IL-17 response in a positive feedback loop (67). As an initiator, IL-17 is also involved in T cell-mediated inflammation. In the primary phase of infection or inflammation, IL-17 accelerates proinflammatory cytokine and chemokine release, amplifying inflammatory reactions (68). IL-17 synergizes with other mediators to activate tissue-infiltrating neutrophils and helps eliminate invading pathogenic bacteria. NF-kB, a central molecule, links initial acinar injury to systemic inflammation and perpetuates inflammation in AP (69). IL-17 directly stimulates inflammatory responses or indirectly induces the production of IL-6 by stabilizing IL-6 mRNA through activation of the NF-κB and ERK1/2 MAP kinase pathways (70–72). As a target of IL-17A, IL-6 is necessary for the RORγt-dependent differentiation of Th17 cells (73), suggesting that IL-17A induces positive feedback. In addition, IL-17A triggers the production and release of various cytokines during AP (74), and these cytokines in turn enhance the synergistic secretion of IL-6 and IL-17A in fibroblasts. Yang demonstrated that IL-17 is increased in the early phase of SAP, confirming that the adaptor protein Card9 coordinates IL-17 to balance immune reactions in SAP pathogenesis (75). By activating the IL-23/IL-17/neutrophil axis, IL-17A exacerbates virus-induced AP (76). Thus, the combination of biomarkers will help clinicians develop and adjust the clinical treatment of AP.
IL-17A Induced Chemokines
Chemokines, a subtype of chemotactic cytokines, are necessary in the recruitment and expression of inflammatory responses (77). Another major target gene of IL-17A is chemokines, particularly C-X-C chemokines, including CXCL1, CXCL2, CXCL4, CXCL5, CXCL8, and others (78). According to a recent study, the mRNA levels of CXCL1, CXCL2, and CXCL5 in acinar cells and CXCL1 in PSCs were increased after stimulation with rIL-17A in vitro. Platelet secretion of CXCL4 is a powerful stimulator of pancreatic neutrophil infiltration and tissue damage, and is mainly formed by CXCL2 (79). These chemokines induced by IL-17A are also involved in the proliferation, maturation, and chemotaxis of neutrophils. These results suggest that IL-17 participates in pancreatic injury by regulating the expression of inflammatory cytokines and chemokines during AP (80).
Interaction Between Gut Microbiota and Immune Cells in AP
Innate myeloid cells are crucial in the activation and differentiation of innate and adaptive immune cells in the intestinal mucosa (81, 82). The inflammatory reaction driven by Th17 cells protects the host from harmful microorganisms, while their overactivation is related to the pathogenesis of intestinal inflammation. A recent study reported that ketogenic diet-related microbiota reduce the levels of intestinal proinflammatory IL-17 (83). Fecal microbiota transplantation (FMT) reduced Th17 cells in mice colonized by donors with Crohn’s disease (84). These studies suggest that the gut microbiome appears to be capable of mediating host immune responses to inflammatory diseases (85, 86). Previous studies demonstrated that dysbiosis of intestinal microbiota was often associated with intestinal barrier dysfunction and metabolic disorders was observed during AP (87). Decreased beneficial bacteria and increased potentially pathogenic bacteria, such as E.coli and Enterococcus, may contribute to dysbiosis of intestinal microbiota and bacterial translocation (88). To confront the microenvironment dysbiosis, the intestine must develop a complex immune protection network including a variety of immune cells. In general, we classified the signals of intestinal microbiota to IL-17 into the following four categories during AP: intestinal barrier function, bacterial components, bacteria-derived metabolites and microbiota-propelled Th17 Cells in AP. Thus, the roles of IL-17 in acute pancreatitis are summarized in Figure 2.
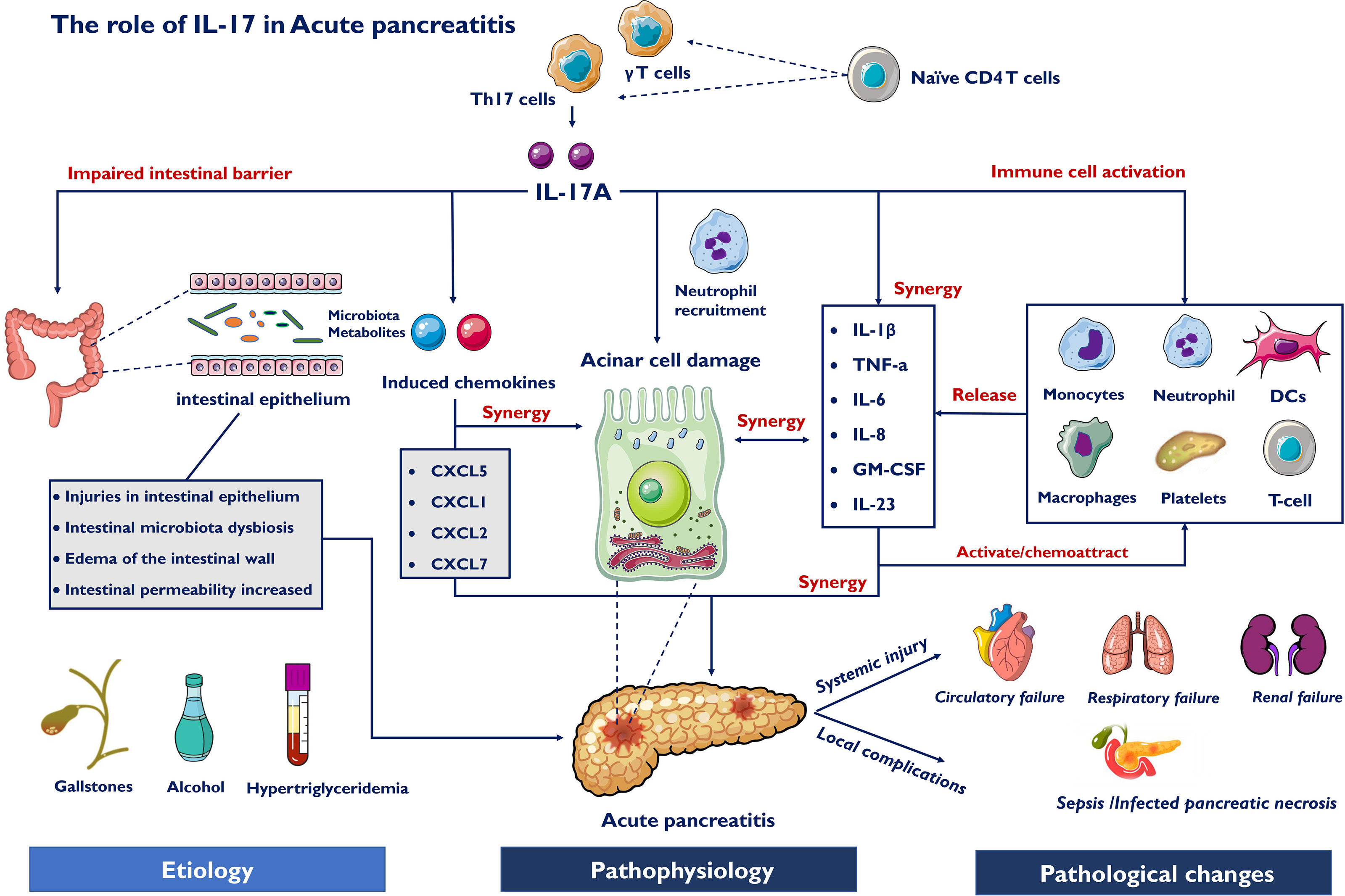
Figure 2 Roles of IL-17 in acute pancreatitis. Etiology for AP include gallstones, alcohol and hypertriglyceridemia, which cause pathophysiological changes of AP. IL-17A induce pancreatic acinar cell damage through synthesizing and releasing cytokines and chemokines which recruit immune cells, such as neutrophils and macrophages. IL-17 also directly induce chemokines and activate innate immune cells, releasing pro-inflammatory mediators. Different inflammatory factors act in synergy with each other to aggravate AP. Furthermore, the intestinal microbiota-derived signals to IL-17 include microbiota alteration, bacteria-derived metabolites and impaired intestinal barrier function. Systemic injury develops early due to IL-17 involving inflammatory response and may precede necrosis, and late secondary organ failure due to infected pancreatic necrosis (IPN) induce sepsis.
IL-17 Induced Impaired Intestinal Barrier Function
Impairment of intestinal barrier function is an adverse event in AP, which leads to the translocation of gut microbes and endotoxin. The excessive release of inflammatory cytokines during AP is a primary reason for intestinal barrier injury (89). Under AP conditions, the expression of ZO-1, claudin-1 and occludin is decreased, changing the tight connections between cells and promoting the translocation of gut microbes. Damage to the intestinal barrier is related to intestinal microcirculation disturbance, excessive release of inflammatory cytokines, intestinal epithelial cell damage and intestinal microorganism disturbance. As a magnifying agent of the inflammatory reaction, recent research has indicated that increased IL-17 in AP is harmful to maintaining intestinal barrier function and epithelial cells. IL-17 functions at mucosal interfaces, such as the gut epithelial lining, maintaining intact physical barrier and inducing antimicrobial peptides to promote invasion by intestinal bacteria (90). Furthermore, IL-17 results in edema of the intestinal wall, and destruction of the mucosal epithelial barrier via expression of CCR6 in enterocytes and mobilization of Th17 cells in the intestines (91). A recent study revealed that IL-17 induces tight junction expression through the ERK-MAPK signaling pathway to alter intestinal permeability (92). Additionally, the level of serum IL-17 is also closely related to bacterial overload, illustrating that the overproduction of IL-17 damages intestinal barrier function, contributing to organ failure in AP.
IL-17 and Host Gut Microbiota in AP
IL-17A is a key mediator of mucosal monitoring and barrier integrity by promoting the production of antimicrobial factors necessary to contain pathogens (93). IL-17A is involved in inducing an appropriate immune response against resident bacteria in the gastrointestinal tract through a variety of mechanisms, including promoting intestinal IgA responses and the expression of epithelial cell-related innate immune receptors, such as Toll-like receptors (TLRs) and antimicrobial peptides (94). The accumulation of proinflammatory and anti-inflammatory immune cells is regulated by the various commensal microbiota populations within the gut. However, gut dysbiosis can lead to disruptions in immune cell activity. Dysbiosis of the intestinal microbiota contributes to excessive release of inflammatory cytokines, common in both the immune system and intestine in the progression of AP (95). Pathogenic bacteria, such as Escherichia coli and Enterococcus, enriched in AP showed a positive correlation, while beneficial bacteria, such as Bifidobacterium, Lactobacillus and Bacteroides, showed a downward trend (96). Intestinal Th17 cells can be induced by specific microbes, including Escherichia coli and Bifidobacterium. Escherichia coli was positively correlated with the proportion of RORγt+ and IL-17A+ Th17 cells in the colon, while the proportion of IL-17A+ Th17 cells in the colon of mice lacking only Escherichia coli was significantly reduced (97). Bifidobacterium and Lactobacillus inhibited the expression of IL-6 and IL-17 while facilitating the protein synthesis of major tight junction proteins (98). In addition to strengthening the intestinal epithelial barrier, the microbiome also modulates the immune system and transmits advantages to the host (99). FMT and probiotics, which are both designed to reverse microbial disorders and renew a broader structural status, may provide promising insights into inflammatory and immune response networks, facilitating the recovery of AP.
IL-17 and Host Gut Microbiota-Derived Metabolites in AP
Metabolites from the microbiota are involved in maintaining intestinal homeostasis or the pathogenesis of inflammation by regulating IL-17 during AP. Short-chain fatty acid (SCFA) attenuated AP, as evidenced by reduced serum amylase and lipase levels and improved pancreatic morphology (100). The regulation of T cells by SCFAs depends on the cytokine environment and immunological microenvironment. In fact, acetate boosts IL-10-producing T cells in a stable state and affects Th1 and Th17 cells in active immune responses. Butyrate depletion appears to play a central role in disease progression towards necrotizing pancreatitis (101). The decrease in butyrate production was associated with the phylum Proteobacteria and the genera Escherichia/Shigella and Streptococcus increasing in patients with AP. Butyrate prophylaxis could mediate the differentiation of CD4+ T cells towards Treg cells, resulting in production of the anti-inflammatory cytokine IL-10 (102). Bile acid metabolites might also regulate the intestinal immune landscape through the balance of Th17 cells and Treg cells (103, 104). Wan found that supplementation with bile acids could relieve pancreatic and intestinal damage, which may be related to the gut microbiome (105). Moreover, the administration of bile acid to an animal model led to a decrease in Th17 cells and an increase in Treg cells in the small intestine lamina propria (103). Tissue-specific anti-inflammatory mechanisms during AP suggest that modulation of gut microbiota-derived metabolite levels could become a potential strategy to improve AP.
Microbiota-Propelled T Helper 17 Cells in AP
T helper 17 cells are a subset of T lymphocytes with many functions in immune adaptation and have a double-edge sword in pathological and physiological processes. The microbiota is a potent promoter of proinflammatory Th17 cells expressing the lineage-specific transcription factor RORγt (106). Increasing evidence indicates that gut microbiota dysbiosis often occurs in the “second hit” induced by AP gut-derived infection (87, 107, 108). Upon microbiota antigen stimulation, Th17 cells produce several proinflammatory effector molecules which mediate inflammatory cells infiltration and tissue destruction, including proinflammatory cytokines (IL-17A, IL-17F and IFN-γ), chemokines, and matrix metalloproteases (109, 110). Proinflammatory cytokines further stimulate innate immune cells and epithelial cells to produce IL-1, IL-6, and G-CSF, inducing neutrophil recruitment and impairing intestinal barrier function (111, 112). The imbalance between Th17 cells and Tregs is common and relevant to the severity and prognosis of AP. Recent study confirmed that the proportion of Th17/Treg was significantly higher in SAP patients and more pronounced in the multiple organ failure group (P<0.05) (113). The homeostasis of the pancreatic microenvironment and rapid response to invaders are tightly regulated by the continuous adjustment between harmful Th17 cells and immunosuppressive Tregs, which establishes an ecological niche for bacterial intestinal border-dwelling bacteria (114). Thus, inhibiting pathogenic Th17 cells and balancing the differentiation of T-cell toward Treg are critical for host defense (115–117).
In conclusion, gut microbiota and metabolic disorders in AP often lead to the imbalance of Th17 and Treg, resulting in the increase of IL-17A. Subsequently, IL-17A recruit neutrophils and act in synergy with other proinflammatory mediators to promote the inflammatory response, which destruct intestinal microcirculation and mucosal intestinal barrier. Based on intestinal barrier damage, IL-17A further leads to a pathological bacterial and metabolic translocation, and eventually promote a positive feedback loop to aggravate pancreatic injury (118–120) (Figure 3).
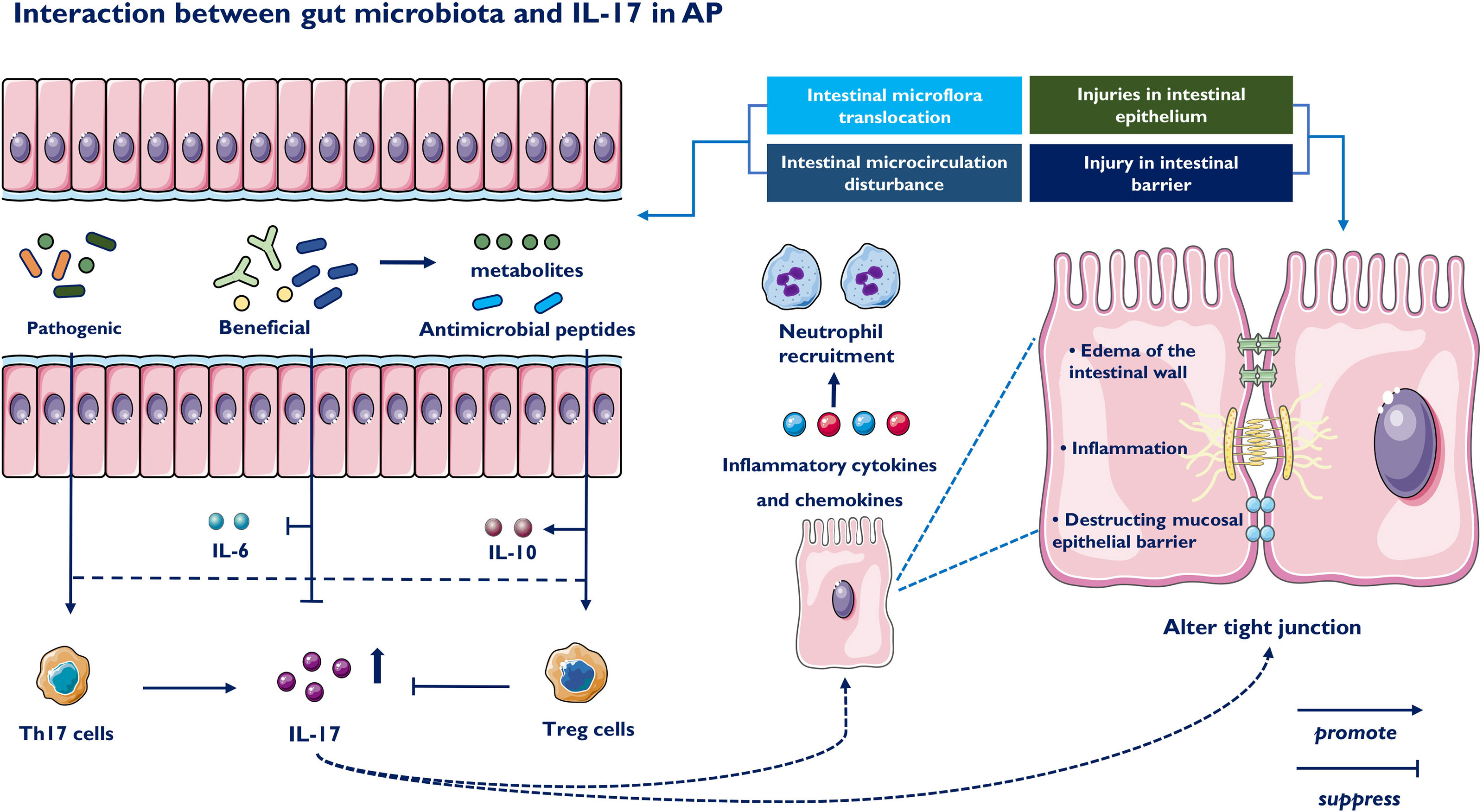
Figure 3 Impact of the gut microbiota on IL-17 and Th17 immune responses. The intestinal microbiota-derived signals to immune cells are classified into the following three parts: bacterial components, bacteria-derived metabolites and intestinal barrier function. Beneficial bacteria suppressed the expression of proinflammatory cytokine IL-6 and IL-17 and promoted the expression of major tight junction proteins. On the contrary, colonization of pathogenic commensal bacteria induces generation of Th17 and downregulates Tregs immune responses. Beneficial bacteria produce short-chain fatty acids, which participate in the generation of Tregs and IL-10 by suppressing proinflammatory cytokines. IL-17 injuries intestinal epithelium, releases inflammatory cytokines and cause intestinal microcirculation disturbance. IL-17 also induces impaired intestinal barrier function, including edema of the intestinal wall and destruction of the mucosal epithelial barrier.
Therapeutic Implications of AP
In AP, a cascade of responses starts with the discharge of endogenous molecules as a result of tissue damage and leukocyte activation. In addition to conservative supportive treatment, drug therapy targeting immunomodulation has achieved promising results. The accumulated discoveries from early clinical research with anti-IL-17 agents yielded strong evidence for the role of IL-17 signaling within the pathophysiology of inflammatory diseases and the potential utilization of these operators in AP therapy (121, 122) Based on the above discussion, this approach represents a strategic therapeutic concept to balance this cascade response.
Targeting the Th17 Pathway in AP
Moderate blockade of the inflammatory pathway and regulation of the cascade response through different mechanisms may be an effective approach in developing pharmacological drugs for AP. Inhibition of Th17 cell differentiation could reduce IL-17 secretion and delay the progression of AP. Currently, most drugs that block Th17 differentiation are inhibitors targeting IL-23 and RORγt. IL-23 inhibitors are specific antibodies against the p19 subunit of IL-23, including tidrakizumab, guselkumab and AMG139 (123, 124). These antibodies are in clinical trials in patients with autoimmune diseases and have shown remarkable effectiveness for targeting psoriasis. In addition to targeting downstream effectors of Th17 pathways, the primary transcriptional regulator RORγt could be a promising therapeutic target. Several RORγt antagonists are used in ongoing clinical trials, such as GSK805 and TAK-828F, which both have shown prospective efficacy in preclinical models and animal models of inflammatory diseases (125, 126). However, several questions remain about RORγt antagonism, including potential mechanisms of action, key target cells and effects on the homeostatic balance within Th17 cells and Treg cells (127). Furthermore, miR-155 is closely related to the differentiation of CD4+ T cells to Th17 cells. Pretreatment of human CD4+ T cells with a miR-155 inhibitor might reduce the transition from CD4+ T cells to Th17 cells, thereby reducing the secretion of IL-17 and relevant pathological damage to the pancreas (128).
Targeting IL-17 and Its Receptor in AP
IL-17A released by Th17 and γδ T cells stimulates acinar cells, stellate cells, and various immune cells to augment the release of inflammatory cytokines, recruiting more immune cells, and ultimately causing inflammatory cascades in AP. The suppression of IL-17A secretion or specific blockade of IL-17A receptors may offer a potential therapy for AP. Currently, an anti-IL-17A antibody reduces the impact of IL-17A on pancreatic stellate cells and attenuates pancreatic fibrosis in chronic pancreatitis (129). Several molecules inhibiting IL-17 signaling, including antibodies targeting IL-17A (secukinumab and ixekizumab), have been prepared with the target of IL-17 activity normalization to improve anti-inflammatory reactions. Of note, no obvious safety issue was found with either secukinumab or ixekizumab. IL-17RA inhibitors, such as brodalumab, also block the anti-inflammatory reactions regulated by IL-25 (IL-17E) by inhibiting the proinflammatory impacts mediated by IL-17A, IL-17F and IL-17A/F (130). The combination of IL-17R and different subtypes of IL-17 may upset the anti-inflammatory or proinflammatory balance mediated by IL-17 and produce certain side effects while controlling disease progression. It is possible that anti-IL-17 and IL-17R treatment may be a prospective therapeutic target for AP treatment in the near future.
Inhibition of IL-17 Synergistic Cytokines
IL-17 and other inflammatory factors, including IL-22, IFNγ, TNF-α, and GM-CSF as well as other synergistic actions, constitute a positive feedback circuit and promote recruitment of inflammatory mediators, aggravating the inflammatory response. TNF-α has been defined as a crucial cytokine that mediates the systemic inflammatory response. Pentoxifyllin, an oral TNF-α antagonist, reduced ICU admissions and shortened hospital stays (131). IL-17 and TNF-α cooperate to induce the expression of receptor activator of membrane protein nuclear factor-κ-ligand, which is involved in tissue remodeling and dendritic cell maturation (132). The combination of IL-17 and TNF-α inhibitors has shown better efficacy in inflammatory diseases. In SAP, ellipticine attenuated NF-κB and MAPK activation in response to IL-17A and TNF-α treatment, inhibited Act1- and TRAF6-mediated NF-κB activation, and blocked the interaction of Act1 with TRAF6 (133). The IL-6/STAT3 signaling pathway is a vital regulator of Th17 and Treg cells by promoting Th17 cells and inhibiting the differentiation of Tregs. Recent studies have shown that three novel small molecule IL-6 inhibitors, madindoline-5 (MDL-5), MDL-16 and MDL-101, significantly suppress IL-17 production (134). Targeting the IL-23–IL-17 axis has shown encouraging results for psoriasis and Crohn’s disease. Pancreas-specific deletion of IκBα leads to nuclear translocation of RelA and diminishes AP induction and trypsin activity (135, 136). (IκB)-ζ, an inhibitor of NF-κB, promotes IL-17 downstream gene expression by inhibiting miR-23b, which acts as a central regulator of the IL-17 signaling pathway. RelA was constitutively activated in mice with pancreatic-specific IκB deficiency, and the gene expression profiling was consistent with NF-κB activation. Targeted knockout of the (IκB)-ζ target gene may effectively inhibit the IL-17-mediated inflammatory response. Thus, the combination of other cytokines will help inhibit the progression of AP.
Interdicting the AP Cascade Reaction Through Gut Microbiota
As IL-17 exacerbates AP through different mechanisms, pharmacological blockade of the gut microbiota may exert a partial effect on treating AP. Recent studies found that oxymatrine plays an anti-inflammatory role in AP intestinal injury by suppressing Th1/Th17 secretion of cytokines (137). Probiotics have protective functions against intestinal mucosal damage diseases. Oral administration of Enterococcus durans, as a probiotic, to an animal model of colitis confirmed that the GM-related bacterium suppresses the expression of IL-17A and alleviate disease development (138). The Th1/Th2 balance and Treg activity are key elements in the immunomodulatory impacts of probiotics. Recent research has confirmed that certain commensal bacterial species (139), such as segmented filamentous bacteria (SFB), initiate the accumulation of Th17 cells within the digestive tract in numerous species. Furthermore, probiotics changed the intestinal microbial community towards certain advantageous microbes, such as Streptococcus, Lactobacilli and Bifidobacteria, which generated anti-inflammatory metabolites, suppressed Th17 polarization and promoted anti-inflammatory Treg/Tr1 cell differentiation (140). Probiotic strains can potentially inhibit Th17/IL-17 activity by enhancing Treg and/or Th1 subsets (139). However, probiotics have been shown to prevent intestinal infection while unintentionally increasing mortality, leading to early termination of this trial. Fecal microbiota transplantation means transplanting functional bacteria in the feces of healthy controls to the gastrointestinal tracts of patients, and the new microbiota appropriate for the receptors can be modified (141). A few trials have confirmed the ability to improve intestinal function in AP utilizing this strategy (142, 143), but advanced RCTs are still needed.
The Efficacy of Continuous Renal Replacement of SAP With IL-17
Continuous renal replacement therapy (CRRT) is a new model of renal replacement therapy and was first used in the treatment of renal failure. In the comparison of conventional therapy and CRRT, Ning et al. found that IL-17 showed a continuous decrease at 6, 12, and 24 hours after CRRT and was significantly lower than conventional treatment at different time points (P<0.001) (144). Dai reported that earlier and higher levels of IL-17 evaluated the extended length of hospital stay, organ failure and death, possibly due to a disruption of intestinal barrier function (45). Researchers found that continuous veno-venous hemofiltration (CVVH) removed IL-17 and other proinflammatory cytokines from serum, improved intestinal barrier function, and relieved systemic reactions. In addition, high-volume hemofiltration (HVHF) effectively attenuated the Th17/Treg imbalance during SAP (113). The Th17/Treg ratio and IL-17 were significantly reduced (P<0.05) after HVHF, while no significant change was found in the non-HVHF group. These alterations illustrate that IL-17 plays a key role in the occurrence and development of removing excess inflammatory mediators in SAP patients (Figure 4). Therefore, these results suggest the clinical significance of IL-17 and may improve the outcomes of SAP.
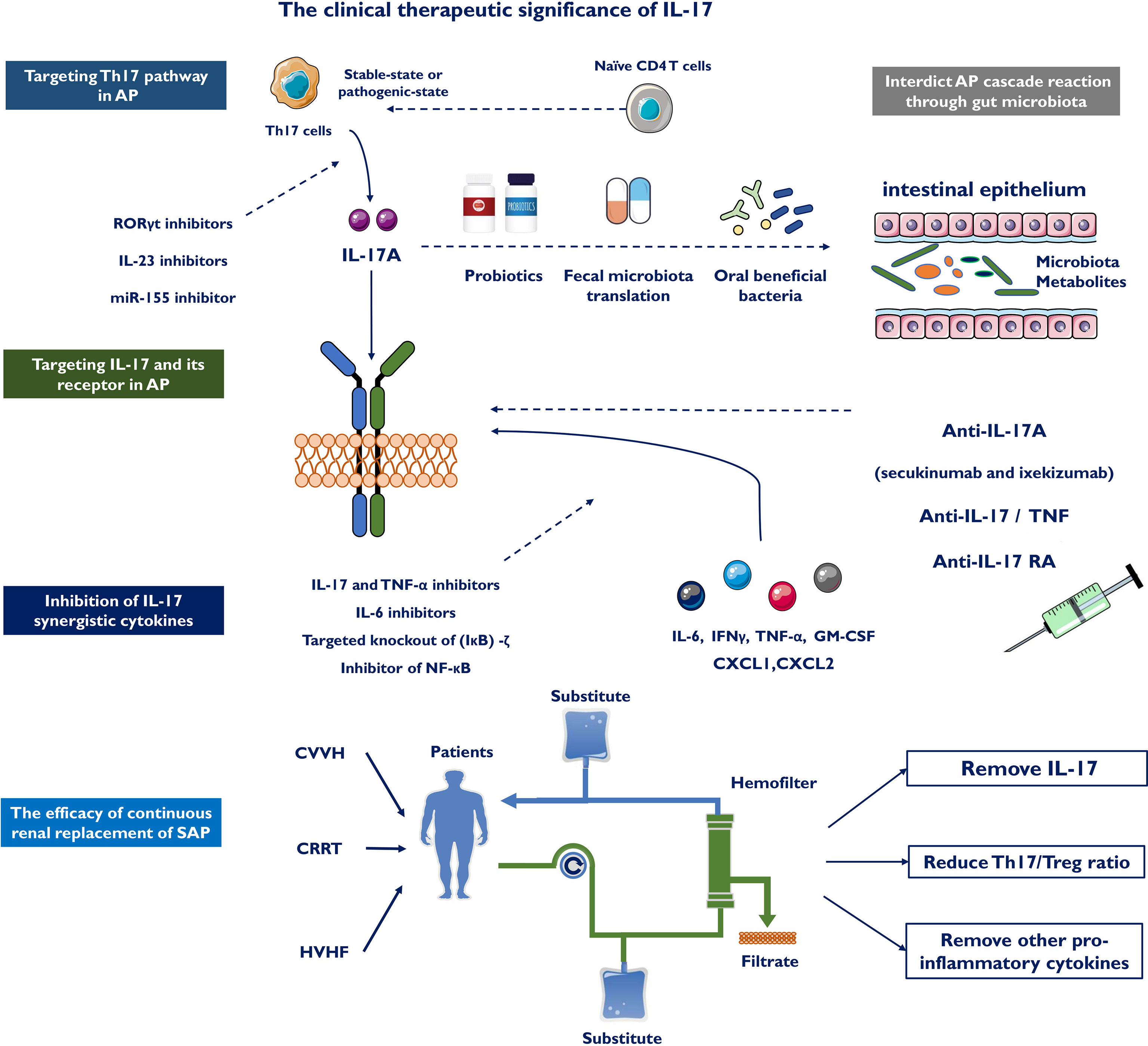
Figure 4 The clinical therapeutic significance of IL-17. IL-17 could boost deeper comprehend of AP and represents a novel immunotherapy for AP by targeting the Th 17 cell/IL-17 immune axis, including targeting Th17 pathway in AP, targeting IL-17 and its receptor in AP, inhibition of IL-17 synergistic cytokines and interdicting AP cascade reaction through gut microbiota. Continuous renal replacement therapy (CRRT), continuous veno-venous hemofiltration (CVVH) and high-volume hemofiltration (HVHF) effectively attenuates the Th17/Treg imbalance and clear serum IL-17 in SAP patients.
Conclusions
Since the discovery of Th17 cells, the cytokine IL-17 has undergone an increase in considerations and revelations. According to the previous literature, IL-17 causes many acute inflammatory diseases, but the precise mechanisms of its contribution are not fully understood. IL-17 is correlated with numerous cell types, acts on a number of cellular targets in tissue and immune cells, and plays vital roles in innate and adaptive immunity. Dysregulated cytokine systems are generally included in AP, and targeted therapy with IL-17 is of great value. Inhibition of IL-17A and its receptor or simultaneous inhibition of IL-17A and IL-17F contributes to interruption of signaling pathways important for AP development and maintenance. Accordingly, biologics targeting IL-17 contribute to quick and dramatic prompts of systemic symptoms during AP. We believe that IL-17 could lead to a deeper understanding of AP and represents a novel immunotherapy for AP by targeting the Th17 cell/IL-17 immune axis.
Author Contributions
LeL, GL, and BS conceived of the study and participated in its design. LiL and TL acquired and analyzed study data. TZ, YX, PX, YZ, HC, HT, and XG participated substantially in the organization and coordination of study. GL and HC wrote the manuscript and prepared the figures. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by The National Natural Science Foundation of China (81871974, 82070658, 81800572) and The Natural Science Foundation of Heilongjiang Province (LH2021H048).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Boxhoorn L, Voermans RP, Bouwense SA, Bruno MJ, Verdonk RC, Boermeester MA, et al. Acute Pancreatitis. Lancet (2020) 396(10252):726–34. doi: 10.1016/S0140-6736(20)31310-6
2. Vege SS, DiMagno MJ, Forsmark CE, Martel M, Barkun AN. Initial Medical Treatment of Acute Pancreatitis: American Gastroenterological Association Institute Technical Review. Gastroenterology (2018) 154(4):1103–39. doi: 10.1053/j.gastro.2018.01.031
3. Habtezion A, Gukovskaya AS, Pandol SJ. Acute Pancreatitis: A Multifaceted Set of Organelle and Cellular Interactions. Gastroenterology (2019) 156(7):1941–50. doi: 10.1053/j.gastro.2018.11.082
4. Trikudanathan G, Wolbrink DRJ, van Santvoort HC, Mallery S, Freeman M, Besselink MG. Current Concepts in Severe Acute and Necrotizing Pancreatitis: An Evidence-Based Approach. Gastroenterology (2019) 156(7):1994–2007.e3. doi: 10.1053/j.gastro.2019.01.269
5. Sendler M, Dummer A, Weiss FU, Krüger B, Wartmann T, Scharffetter-Kochanek K, et al. Tumour Necrosis Factor α Secretion Induces Protease Activation and Acinar Cell Necrosis in Acute Experimental Pancreatitis in Mice. Gut (2013) 62(3):430–9. doi: 10.1136/gutjnl-2011-300771
6. Jiang X, Shi JY, Wang XY, Hu Y, Cui YF. The Impacts of Infectious Complications on Outcomes in Acute Pancreatitis: A Retrospective Study. Mil Med Res (2020) 7(1):38. doi: 10.1186/s40779-020-00265-5
7. Mofidi R, Patil PV, Suttie SA, Parks RW. Risk Assessment in Acute Pancreatitis. Br J Surg (2009) 96(2):137–50. doi: 10.1002/bjs.6431
8. Staubli SM, Oertli D, Nebiker CA. Laboratory Markers Predicting Severity of Acute Pancreatitis. Crit Rev Clin Lab Sci (2015) 52(6):273–83. doi: 10.3109/10408363.2015.1051659
9. Gouda MM, Bhandary YP. Acute Lung Injury: IL-17a-Mediated Inflammatory Pathway and Its Regulation by Curcumin. Inflammation (2019) 42:1160–9. doi: 10.1007/s10753-019-01010-4
10. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional Specialization of Interleukin-17 Family Members. Immunity (2011) 34:149–62. doi: 10.1016/j.immuni.2011.02.012
11. Sun L, Han R, Guo F, Chen H, Wang W, Chen Z, et al. Antagonistic Effects of IL-17 and Astragaloside IV on Cortical Neurogenesis and Cognitive Behavior After Stroke in Adult Mice Through Akt/GSK-3β Pathway. Cell Death Discovery (2020) 10(6):74. doi: 10.1038/s41420-020-00298-8
12. Shabgah AG, Fattahi E, Shahneh FZ. Interleukin-17 in Human Inflammatory Diseases. Postepy Dermatol Alergol (2014) 31(4):256–61. doi: 10.5114/pdia.2014.40954
13. Hohenberger M, Cardwell LA, Oussedik E, Feldman SR. Interleukin-17 Inhibition: Role in Psoriasis and Inflammatory Bowel Disease. J Dermatolog Treat (2018) 29(1):13–8. doi: 10.1080/09546634.2017.1329511
14. Beringer A, Miossec P. Systemic Effects of IL-17 in Inflammatory Arthritis. Nat Rev Rheumatol (2019) 15(8):491–501. doi: 10.1038/s41584-019-0243-5
15. Veldhoen M. Interleukin 17 is a Chief Orchestrator of Immunity. Nat Immunol (2017) 18(6):612–21. doi: 10.1038/ni.3742
16. Lavocat F, Ndongo-Thiam N, Miossec P. Interleukin-25 Produced by Synoviocytes Has Anti-Inflammatory Effects by Acting As a Receptor Antagonist for Interleukin-17a Function. Front Immunol (2017) 31:647(8). doi: 10.3389/fimmu.2017.00647
17. Huang J, Lee HY, Zhao X, Han J, Su Y, Sun Q, et al. Interleukin-17D Regulates Group 3 Innate Lymphoid Cell Function Through Its Receptor CD93. Immunity (2021) 54(4):673–86.e4. doi: 10.1016/j.immuni.2021.03.018
18. Cua DJ, Tato CM. Innate IL-17-Producing Cells: The Sentinels of the Immune System. Nat Rev Immunol (2010) 10(7):479–89. doi: 10.1038/nri2800
19. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 Immune Axis: From Mechanisms to Therapeutic Testing. Nat Rev Immunol (2014) 14(9):585–600. doi: 10.1038/nri3707
20. Kenna TJ, Davidson SI, Duan R, Bradbury LA, McFarlane J, Smith M, et al. Enrichment of Circulating Interleukin-17-Secreting Interleukin-23 Receptor-Positive γ/δ T Cells in Patients With Active Ankylosing Spondylitis. Arthritis Rheum (2012) 64(5):1420–9. doi: 10.1002/art.33507
21. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 Family of Cytokines in Health and Disease. Immunity (2019) 50(4):892–906. doi: 10.1016/j.immuni.2019.03.021
22. Gu C, Wu L, Li X. IL-17 Family: Cytokines, Receptors and Signaling. Cytokine (2013) 64(2):477–85. doi: 10.1016/j.cyto.2013.07.022
23. Lee JH, Shim YR, Seo W, Kim MH, Choi WM, Kim HH, et al. Mitochondrial Double-Stranded RNA in Exosome Promotes Interleukin-17 Production Through Toll-Like Receptor 3 in Alcohol-Associated Liver Injury. Hepatology (2020) 72(2):609–25. doi: 10.1002/hep.31041
24. Liu Y, Zhao Q, Yin Y, McNutt MA, Zhang T, Cao Y. Serum Levels of IL-17 Are Elevated in Patients With Acute Gouty Arthritis. Biochem Biophys Res Commun (2018) 497(3):897–902. doi: 10.1016/j.bbrc.2018.02.166
25. Beringer A, Miossec P. IL-17 and IL-17-Producing Cells and Liver Diseases, With Focus on Autoimmune Liver Diseases. Autoimmun Rev (2018) 17(12):1176–85. doi: 10.1016/j.autrev.2018.06.008
26. Rahimzadeh M, Montazerghaem H, Chegeni SA, Naderi N. Interleukin -17 Serum Levels and Polymorphisms in Acute Kidney Injury Patients. Endocr Metab Immune Disord Drug Targets (2020) 20(3):400–8. doi: 10.2174/1871530319666191009152048
27. Bulek K, Liu C, Swaidani S, Wang L, Page RC, Gulen MF, et al. The Inducible Kinase IKKi Is Required for IL-17-Dependent Signaling Associated With Neutrophilia and Pulmonary Inflammation. Nat Immunol (2011) 12(9):844–52. doi: 10.1038/ni.2080
28. Wang F, Yin J, Lin Y, Zhang F, Liu X, Zhang G, et al. IL-17C has a Pathogenic Role in Kidney Ischemia/Reperfusion Injury. Kidney Int (2020) 97(6):1219–29. doi: 10.1016/j.kint.2020.01.015
29. Zhang J, Qiao Q, Liu M, He T, Shi J, Bai X, et al. IL-17 Promotes Scar Formation by Inducing Macrophage Infiltration. Am J Pathol (2018) 188(7):1693–702. doi: 10.1016/j.ajpath.2018.04.005
30. Mora-Ruíz MD, Blanco-Favela F, Chávez Rueda AK, Legorreta-Haquet MV, Chávez-Sánchez L. Role of Interleukin-17 in Acute Myocardial Infarction. Mol Immunol (2019) 107:71–8. doi: 10.1016/j.molimm.2019.01.008
31. Dawra R, Sah RP, Dudeja V, Rishi L, Talukdar R, Garg P, et al. Intra-Acinar Trypsinogen Activation Mediates Early Stages of Pancreatic Injury But Not Inflammation in Mice With Acute Pancreatitis. Gastroenterology (2011) 141(6):2210–7.e2. doi: 10.1053/j.gastro.2011.08.033
32. Chand SK, Singh RG, Pendharkar SA, Bharmal SH, Petrov MS. Interplay Between Innate Immunity and Iron Metabolism After Acute Pancreatitis. Cytokine (2018) 103:90–8. doi: 10.1016/j.cyto.2017.09.014
33. Zhao Q, Wei Y, Pandol SJ, Li L, Habtezion A. STING Signaling Promotes Inflammation in Experimental Acute Pancreatitis. Gastroenterology (2018) 154(6):1822–35.e2. doi: 10.1053/j.gastro.2018.01.065
34. Shamoon M, Deng Y, Chen YQ, Bhatia M, Sun J. Therapeutic Implications of Innate Immune System in Acute Pancreatitis. Expert Opin Ther Targets (2016) 20(1):73–87. doi: 10.1517/14728222.2015.1077227
35. Hoque R. Update on Innate Immunity and Perspectives on Metabolite Regulation in Acute Pancreatitis. Curr Opin Gastroenterol (2016) 32(6):507–12. doi: 10.1097/MOG.0000000000000311
36. Schmidt MV, Paulus P, Kuhn AM, Weigert A, Morbitzer V, Zacharowski K, et al. Peroxisome Proliferator-Activated Receptor γ-Induced T Cell Apoptosis Reduces Survival During Polymicrobial Sepsis. Am J Respir Crit Care Med (2011) 184(1):64–74. doi: 10.1164/rccm.201010-1585OC
37. Jakkampudi A, Jangala R, Reddy R, Mitnala S, Rao GV, Pradeep R, et al. Acinar Injury and Early Cytokine Response in Human Acute Biliary Pancreatitis. Sci Rep (2017) 7(1):15276. doi: 10.1038/s41598-017-15479-2
38. Syed SN, Raue R, Weigert A, von Knethen A, Brüne B. Macrophage S1PR1 Signaling Alters Angiogenesis and Lymphangiogenesis During Skin Inflammation. Cells (2019) 8(8):785. doi: 10.3390/cells8080785
39. Akitsu A, Iwakura Y. Interleukin-17-Producing γδ T (γδ17) Cells in Inflammatory Diseases. Immunology (2018) 155(4):418–26. doi: 10.1111/imm.12993
40. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and Effector Functions of T(H)17 Cells. Nature (2008) 453(7198):1051–7. doi: 10.1038/nature07036
41. Xie R, Chen Y, Qi M, Zhao Z, Zhang J, Xu Z, et al. Increased Frequency of Circulating Tfh Cells in Patients With Acute Pancreatitis. Int J Clin Exp Pathol (2018) 11(11):5300–8.
42. Chen Z, Fu H, Fang J, Yang J, Zhu X, Cheng B, et al. Preventive and Therapeutic Significance of Octreotide Combined With Lansoprazole on Post-ERCP Pancreatitis and Its Effect on Serum Amylase, Inflammatory Factors and Immune Function. Exp Ther Med (2021) 21(3):251. doi: 10.3892/etm.2021.9682
43. Jia R, Tang M, Qiu L, Sun R, Cheng L, Ma X, et al. Increased Interleukin-23/17 Axis and C-Reactive Protein Are Associated With Severity of Acute Pancreatitis in Patients. Pancreas (2015) 44(2):321–5. doi: 10.1097/MPA.0000000000000284
44. Vlachos S, Tsaroucha AK, Konstantoudakis G, Papachristou F, Trypsianis G, Schizas D, et al. Serum Profiles of M30, M65 and Interleukin-17 Compared With C-Reactive Protein in Patients With Mild and Severe Acute Pancreatitis. J Hepatobiliary Pancreat Sci (2014) 21(12):911–8. doi: 10.1002/jhbp.162
45. Dai SR, Li Z, Zhang JB. Serum Interleukin 17 as an Early Prognostic Biomarker of Severe Acute Pancreatitis Receiving Continuous Blood Purification. Int J Artif Organs (2015) 38(4):192–8. doi: 10.5301/ijao.5000406
46. Ni J, Hu G, Xiong J, Shen J, Shen J, Yang L, et al. Involvement of Interleukin-17A in Pancreatic Damage in Rat Experimental Acute Necrotizing Pancreatitis. Inflammation (2013) 36(1):53–65. doi: 10.1007/s10753-012-9519-5
47. Thomson JE, Brand M, Fonteh P. The Immune Imbalance in the Second Hit of Pancreatitis Is Independent of IL-17a. Pancreatology (2018) 18(3):246–52. doi: 10.1016/j.pan.2018.01.007
48. Sung KF, Odinokova IV, Mareninova OA, Rakonczay Z Jr, Hegyi P, Pandol SJ, et al. Prosurvival Bcl-2 Proteins Stabilize Pancreatic Mitochondria and Protect Against Necrosis in Experimental Pancreatitis. Exp Cell Res (2009) 315(11):1975–89. doi: 10.1016/j.yexcr.2009.01.009
49. Liu L, Guo Y, Zheng J, Lu Y, Shen Y, Huang C, et al. Paneth Cell Ablation Increases the Small Intestinal Injury During Acute Necrotizing Pancreatitis in Rats. Mol Med Rep (2019) 20(1):473–84. doi: 10.3892/mmr.2019.10274
50. Tokoro T, Makino I, Harada S, Okamoto K, Nakanuma S, Sakai S, et al. Interactions Between Neutrophils and Platelets in the Progression of Acute Pancreatitis. Pancreas (2020) 49(6):830–6. doi: 10.1097/MPA.0000000000001585
51. Omary MB, Lugea A, Lowe AW, Pandol SJ. The Pancreatic Stellate Cell: A Star on the Rise in Pancreatic Diseases. J Clin Invest (2007) 117(1):50–9. doi: 10.1172/JCI30082
52. Xu D, Xie R, Xu Z, Zhao Z, Ding M, Chen W, et al. mTOR-Myc Axis Drives Acinar-to-Dendritic Cell Transition and the CD4+ T Cell Immune Response in Acute Pancreatitis. Cell Death Dis (2020) 11(6):416. doi: 10.1038/s41419-020-2517-x
53. Mayerle J. A Novel Role for Leucocytes in Determining the Severity of Acute Pancreatitis. Gut (2009) 58(11):1440–1. doi: 10.1136/gut.2009.186692
54. Chi DZ, Chen J, Huang DP. Influence of Interleukin-1β and Interleukin-6 Gene Polymorphisms on the Development of Acute Pancreatitis. Genet Mol Res (2015) 14(1):975–80. doi: 10.4238/2015
55. Chousterman BG, Swirski FK, Weber GF. Cytokine Storm and Sepsis Disease Pathogenesis. Semin Immunopathol (2017) 5):517–28. doi: 10.1007/s00281-017-0639-8
56. Nakamura Y, Do JH, Yuan J, Odinokova IV, Mareninova O, Gukovskaya AS, et al. Inflammatory Cells Regulate P53 and Caspases in Acute Pancreatitis. Am J Physiol Gastrointest Liver Physiol (2010) 298(1):G92–100. doi: 10.1152/ajpgi.00324.2009
57. Wan J, Ren Y, Yang X, Li X, Xia L, Lu N. The Role of Neutrophils and Neutrophil Extracellular Traps in Acute Pancreatitis. Front Cell Dev Biol (2021) 21(8):565758. doi: 10.3389/fcell.2020.565758
58. Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, et al. Interleukin-17-Induced Neutrophil Extracellular Traps Mediate Resistance to Checkpoint Blockade in Pancreatic Cancer. J Exp Med (2020) 217(12):e20190354. doi: 10.1084/jem.20190354
59. Leppkes M, Maueröder C, Hirth S, Nowecki S, Günther C, Billmeier U, et al. Externalized Decondensed Neutrophil Chromatin Occludes Pancreatic Ducts and Drives Pancreatitis. Nat Commun (2016) 7:10973. doi: 10.1038/ncomms10973
60. Knight JS, Luo W, O'Dell AA, Yalavarthi S, Zhao W, Subramanian V, et al. Peptidylarginine Deiminase Inhibition Reduces Vascular Damage and Modulates Innate Immune Responses in Murine Models of Atherosclerosis. Circ Res (2014) 114(6):947–56. doi: 10.1161/CIRCRESAHA.114.303312
61. Klöppel G. Chronic Pancreatitis, Pseudotumors and Other Tumor-Like Lesions. Mod Pathol (2007) 20 Suppl 1:S113–31. doi: 10.1038/modpathol.3800690
62. Hoque R, Malik AF, Gorelick F, Mehal WZ. Sterile Inflammatory Response in Acute Pancreatitis. Pancreas (2012) 41(3):353–7. doi: 10.1097/MPA.0b013e3182321500
63. Gaffen SL. Structure and Signalling in the IL-17 Receptor Family. Nat Rev Immunol (2009) 9(8):556–67. doi: 10.1038/nri2586
64. Zrioual S, Ecochard R, Tournadre A, Lenief V, Cazalis MA, Miossec P. Genome-Wide Comparison Between IL-17A- and IL-17F-Induced Effects in Human Rheumatoid Arthritis Synoviocytes. J Immunol (2009) 182(5):3112–20. doi: 10.4049/jimmunol.0801967
65. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T Cell Interleukin-17 Induces Stromal Cells to Produce Proinflammatory and Hematopoietic Cytokines. J Exp Med (1996) 183(6):2593–603. doi: 10.1084/jem.183.6.2593
66. Bedrosian AS, Nguyen AH, Hackman M, Connolly MK, Malhotra A, Ibrahim J, et al. Dendritic Cells Promote Pancreatic Viability in Mice With Acute Pancreatitis. Gastroenterology (2011) 141(5):1915–26.e1-14. doi: 10.1053/j.gastro.2011.07.033
67. Wang J, Sun W, Bond A, Xu C, Li K, Ren D, et al. A Positive Feedback Loop Between Th17 Cells and Dendritic Cells in Patients With Endplate Inflammation. Immunol Invest (2019) 48(1):39–51. doi: 10.1080/08820139.2018.1496097
68. Flierl MA, Rittirsch D, Gao H, Hoesel LM, Nadeau BA, Day DE, et al. Adverse Functions of IL-17A in Experimental Sepsis. FASEB J (2008) 22(7):2198–205. doi: 10.1096/fj.07-105221
69. Jakkampudi A, Jangala R, Reddy BR, Mitnala S, Nageshwar Reddy D, Talukdar R. NF-κb in Acute Pancreatitis: Mechanisms and Therapeutic Potential. Pancreatology (2016) 16(4):477–88. doi: 10.1016/j.pan.2016.05.001
70. Hata K, Andoh A, Shimada M, Fujino S, Bamba S, Araki Y, et al. IL-17 Stimulates Inflammatory Responses via NF-kappaB and MAP Kinase Pathways in Human Colonic Myofibroblasts. Am J Physiol Gastrointest Liver Physiol (2002) 282(6):G1035–44. doi: 10.1152/ajpgi.00494.2001
71. Shimada M, Andoh A, Hata K, Tasaki K, Araki Y, Fujiyama Y, et al. IL-6 Secretion by Human Pancreatic Periacinar Myofibroblasts in Response to Inflammatory Mediators. J Immunol (2002) 168(2):861–8. doi: 10.4049/jimmunol.168.2.861
72. Andoh A, Shimada M, Bamba S, Okuno T, Araki Y, Fujiyama Y, et al. Extracellular Signal-Regulated Kinases 1 and 2 Participate in Interleukin-17 Plus Tumor Necrosis Factor-Alpha-Induced Stabilization of Interleukin-6 mRNA in Human Pancreatic Myofibroblasts. Biochim Biophys Acta (2002) 1591(1-3):69–74. doi: 10.1016/s0167-4889(02)00250-1
73. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 Programs T(H)-17 Cell Differentiation by Promoting Sequential Engagement of the IL-21 and IL-23 Pathways. Nat Immunol (2007) 8(9):967–74. doi: 10.1038/ni1488
74. Vonlaufen A, Apte MV, Imhof BA, Frossard JL. The Role of Inflammatory and Parenchymal Cells in Acute Pancreatitis. J Pathol (2007) 213(3):239–48. doi: 10.1002/path.2231
75. Yang ZW, Weng CZ, Wang J, Xu P. The Role of Card9 Overexpression in Peripheral Blood Mononuclear Cells From Patients With Aseptic Acute Pancreatitis. J Cell Mol Med (2016) 20(3):441–9. doi: 10.1111/jcmm.12738
76. Yan K, Yang J, Qian Q, Xu D, Liu H, Wei L, et al. Pathogenic Role of an IL-23/γδt17/Neutrophil Axis in Coxsackievirus B3-Induced Pancreatitis. J Immunol (2019) 203(12):3301–12. doi: 10.4049/jimmunol.1900787
77. Lau HY, Wong FL, Bhatia M. A Key Role of Neurokinin 1 Receptors in Acute Pancreatitis and Associated Lung Injury. Biochem Biophys Res Commun (2005) 327(2):509–15. doi: 10.1016/j.bbrc.2004.12.030
78. Onishi RM, Gaffen SL. Interleukin-17 and its Target Genes: Mechanisms of Interleukin-17 Function in Disease. Immunology (2010) 129(3):311–21. doi: 10.1111/j.1365-2567.2009.03240.x
79. Wetterholm E, Linders J, Merza M, Regner S, Thorlacius H. Platelet-Derived CXCL4 Regulates Neutrophil Infiltration and Tissue Damage in Severe Acute Pancreatitis. Transl Res (2016) 176:105–18. doi: 10.1016/j.trsl.2016.04.006
80. Xu S, Cao X. Interleukin-17 and its Expanding Biological Functions. Cell Mol Immunol (2010) 7(3):164–74. doi: 10.1038/cmi.2010.21
81. Zhang XM, Zhang ZY, Zhang CH, Wu J, Wang YX, Zhang GX. Intestinal Microbial Community Differs Between Acute Pancreatitis Patients and Healthy Volunteers. BioMed Environ Sci (2018) 31(1):81–6. doi: 10.3967/bes2018.010
82. Klose CS, Artis D. Innate Lymphoid Cells as Regulators of Immunity, Inflammation and Tissue Homeostasis. Nat Immunol (amp]]#xFF08;2016) 17(7):765–74. doi: 10.1038/ni.3489
83. Ang QY, Alexander M, Newman JC, Tian Y, Cai J, Upadhyay V, et al. Ketogenic Diets Alter the Gut Microbiome Resulting in Decreased Intestinal Th17 Cells. Cell (2020) 181(6):1263–75.e16. doi: 10.1016/j.cell.2020.04.027
84. Ueno A, Jeffery L, Kobayashi T, Hibi T, Ghosh S, Jijon H. Th17 Plasticity and Its Relevance to Inflammatory Bowel Disease. J Autoimmun (2018) 87:38–49. doi: 10.1016/j.jaut.2017.12.004
85. Brevi A, Cogrossi LL, Grazia G, Masciovecchio D, Impellizzieri D, Lacanfora L, et al. Much More Than IL-17a: Cytokines of the IL-17 Family Between Microbiota and Cancer. Front Immunol (2020) 11:565470. doi: 10.3389/fimmu.2020.565470
86. Rooks MG, Garrett WS. Gut Microbiota, Metabolites and Host Immunity. Nat Rev Immunol (2016) 16(6):341–52. doi: 10.1038/nri.2016.42
87. Shen Y, Cui NQ. Clinical Observation of Immunity in Patients With Secondary Infection From Severe Acute Pancreatitis. Inflammation Res (2012) 61(7):743–8. doi: 10.1007/s00011-012-0467-1
88. Brubaker L, Luu S, Hoffman K, Wood A, Navarro Cagigas M, Yao Q, et al. Microbiome Changes Associated With Acute and Chronic Pancreatitis: A Systematic Review. Pancreatology (2021) 21(1):1–14. doi: 10.1016/j.pan.2020.12.013
89. Li XY, He C, Zhu Y, Lu NH. Role of Gut Microbiota on Intestinal Barrier Function in Acute Pancreatitis. World J Gastroenterol (2020) 26(18):2187–93. doi: 10.3748/wjg.v26.i18.2187
90. Martínez-López M, Iborra S, Conde-Garrosa R, Mastrangelo A, Danne C, Mann ER, et al. Microbiota Sensing by Mincle-Syk Axis in Dendritic Cells Regulates Interleukin-17 and -22 Production and Promotes Intestinal Barrier Integrity. Immunity (2019) 50(2):446–61.e9. doi: 10.1016/j.immuni.2018.12.020
91. Beurel E, Lowell JA. Th17 Cells in Depression. Brain Behav Immun (2018) 69:28–34. doi: 10.1016/j.bbi.2017.08.001
92. Kinugasa T, Sakaguchi T, Gu X, Reinecker HC. Claudins Regulate the Intestinal Barrier in Response to Immune Mediators. Gastroenterology (2000) 118(6):1001–11. doi: 10.1016/s0016-5085(00)70351-9
93. Abusleme L, Moutsopoulos NM. IL-17: Overview and Role in Oral Immunity and Microbiome. Oral Dis (2017) 23(7):854–65. doi: 10.1111/odi.12598
94. Douzandeh-Mobarrez B, Kariminik A. Gut Microbiota and IL-17a: Physiological and Pathological Responses. Probiotics Antimicrob Proteins (2019) 11(1):1–10. doi: 10.1007/s12602-017-9329-z
95. Adolph TE, Mayr L, Grabherr F, Schwärzler J, Tilg H. Pancreas-Microbiota Cross Talk in Health and Disease. Annu Rev Nutr (2019) 39:249–66. doi: 10.1146/annurev-nutr-082018-124306
96. Zhu Y, He C, Li X, Cai Y, Hu J, Liao Y, et al. Gut Microbiota Dysbiosis Worsens the Severity of Acute Pancreatitis in Patients and Mice. J Gastroenterol (2019) 54(4):347–58. doi: 10.1007/s00535-018-1529-0
97. Britton GJ, Contijoch EJ, Spindler MP, Aggarwala V, Dogan B, Bongers G, et al. Defined Microbiota Transplant Restores Th17/Rorγt+ Regulatory T Cell Balance in Mice Colonized With Inflammatory Bowel Disease Microbiotas. Proc Natl Acad Sci USA (2020) 117(35):21536–45. doi: 10.1073/pnas.1922189117
98. Wang H, Gong J, Wang W, Long Y, Fu X, Fu Y, et al. Are There Any Different Effects of Bifidobacterium, Lactobacillus and Streptococcus on Intestinal Sensation, Barrier Function and Intestinal Immunity in PI-IBS Mouse Model? PloS One (2014) 9(3):e90153. doi: 10.1371/journal.pone.0090153
99. Shi N, Li N, Duan X, Niu H. Interaction Between the Gut Microbiome and Mucosal Immune System. Mil Med Res (2017) 4:14. doi: 10.1186/s40779-017-0122-9
100. Pan X, Fang X, Wang F, Li H, Niu W, Liang W, et al. Butyrate Ameliorates Caerulein-Induced Acute Pancreatitis and Associated Intestinal Injury by Tissue-Specific Mechanisms. Br J Pharmacol (2019) 176(23):4446–61. doi: 10.1111/bph.14806
101. van den Berg FF, van Dalen D, Hyoju SK, van Santvoort HC, Besselink MG, Wiersinga WJ, et al. Western-Type Diet Influences Mortality From Necrotising Pancreatitis and Demonstrates a Central Role for Butyrate. Gut (2021) 70(5):915–27. doi: 10.1136/gutjnl-2019-320430
102. Omenetti S, Pizarro TT. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front Immunol (2015) 6:639. doi: 10.3389/fimmu.2015.00639
103. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile Acid Metabolites Control TH17 and Treg Cell Differentiation. Nature (2019) 576(7785):143–8. doi: 10.1038/s41586-019-1785-z
104. Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, et al. Microbial Bile Acid Metabolites Modulate Gut Rorγ+ Regulatory T Cell Homeostasis. Nature (2020) 577(7790):410–5. doi: 10.1038/s41586-019-1865-0
105. Wan YD, Zhu RX, Pan XT, Sun TW. Bile Acid Supplementation Improves Murine Pancreatitis in Association With the Gut Microbiota. Front Physiol (2020) 11:650. doi: 10.3389/fphys.2020.00650
106. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell (2009) 139(3):485–98. doi: 10.1016/j.cell.2009.09.033
107. Cen ME, Wang F, Su Y, Zhang WJ, Sun B, Wang G. Gastrointestinal Microecology: A Crucial and Potential Target in Acute Pancreatitis. Apoptosis (2018) 23(7-8):377–87. doi: 10.1007/s10495-018-1464-9
108. Wang C, Li Q, Ren J. Microbiota-Immune Interaction in the Pathogenesis of Gut-Derived Infection. Front Immunol (2019) 7:1873(10). doi: 10.3389/fimmu.2019.01873
109. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A Distinct Lineage of CD4 T Cells Regulates Tissue Inflammation by Producing Interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi: 10.1038/ni1261
110. Cosmi L, Santarlasci V, Maggi L, Liotta F, Annunziato F. Th17 Plasticity: Pathophysiology and Treatment of Chronic Inflammatory Disorders. Curr Opin Pharmacol (2014) 17:12–6. doi: 10.1016/j.coph.2014.06.004
111. Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 Cells Transdifferentiate Into Regulatory T Cells During Resolution of Inflammation. Nature (2015) 523(7559):221–5. doi: 10.1038/nature14452
112. Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, Tolaini M, et al. The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity (2019) 51(1):77–89.e6. doi: 10.1016/j.immuni.2019.05.004
113. Guo J, Li Z, Tang D, Zhang J. Th17/Treg Imbalance in Patients With Severe Acute Pancreatitis: Attenuated by High-Volume Hemofiltration Treatment. Med (Baltimore) (2020) 99(31):e21491. doi: 10.1097/MD.0000000000021491
114. Campbell C, Dikiy S, Bhattarai SK, Chinen T, Matheis F, Calafiore M, et al. Extrathymically Generated Regulatory T Cells Establish a Niche for Intestinal Border-Dwelling Bacteria and Affect Physiologic Metabolite Balance. Immunity (2018) 48(6):1245–57.e9. doi: 10.1016/j.immuni.2018.04.013
115. Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined Immunodeficiency Associated With DOCK8 Mutations. N Engl J Med (2009) 361(21):2046–55. doi: 10.1056/NEJMoa0905506
116. Curtis MM, Way SS. Interleukin-17 in Host Defence Against Bacterial, Mycobacterial and Fungal Pathogens. Immunology (2009) 126(2):177–85. doi: 10.1111/j.1365-2567.2008.03017.x
117. Zhang S, Takaku M, Zou L, Gu AD, Chou WC, Zhang G, et al. Reversing SKI-SMAD4-Mediated Suppression Is Essential for TH17 Cell Differentiation. Nature (2017) 551(7678):105–9. doi: 10.1038/nature24283
118. Dolff S, Witzke O, Wilde B. Th17 Cells in Renal Inflammation and Autoimmunity. Autoimmun Rev (2019) 18(2):129–36. doi: 10.1016/j.autrev.2018.08.006
119. Tanoue T, Atarashi K, Honda K. Development and Maintenance of Intestinal Regulatory T Cells. Nat Rev Immunol (2016) 16(5):295–309. doi: 10.1038/nri.2016.36
120. Evans-Marin H, Rogier R, Koralov SB, Manasson J, Roeleveld D, van der Kraan PM, et al. Microbiota-Dependent Involvement of Th17 Cells in Murine Models of Inflammatory Arthritis. Arthritis Rheumatol (2018) 70(12):1971–83. doi: 10.1002/art.40657
121. Nirula A, Nilsen J, Klekotka P, Kricorian G, Erondu N, Towne JE, et al. Effect of IL-17 Receptor A Blockade With Brodalumab in Inflammatory Diseases. Rheumatol (Oxford) (2016) 55(suppl 2):ii43–55. doi: 10.1093/rheumatology/kew346
122. Beringer A, Noack M, Miossec P. IL-17 in Chronic Inflammation: From Discovery to Targeting. Trends Mol Med (2016) 22(3):230–41. doi: 10.1016/j.molmed.2016.01.001
123. Rich P, Sigurgeirsson B, Thaci D, Ortonne JP, Paul C, Schopf RE, et al. Secukinumab Induction and Maintenance Therapy in Moderate-to-Severe Plaque Psoriasis: A Randomized, Double-Blind, Placebo-Controlled, Phase II Regimen-Finding Study. Br J Dermatol (2013) 168(2):402–11. doi: 10.1111/bjd.12112
124. Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson-Heredia E, et al. Anti-Interleukin-17 Monoclonal Antibody Ixekizumab in Chronic Plaque Psoriasis. N Engl J Med (2012) 366(13):1190–9. doi: 10.1056/NEJMoa1109997
125. Shibata A, Uga K, Sato T, Sagara M, Igaki K, Nakamura Y, et al. Pharmacological Inhibitory Profile of TAK-828F, a Potent and Selective Orally Available Rorγt Inverse Agonist. Biochem Pharmacol (2018) 150:35–45. doi: 10.1016/j.bcp.2018.01.023
126. Withers DR, Hepworth MR, Wang X, Mackley EC, Halford EE, Dutton EE, et al. Transient Inhibition of ROR-γt Therapeutically Limits Intestinal Inflammation by Reducing TH17 Cells and Preserving Group 3 Innate Lymphoid Cells. Nat Med (2016) 22(3):319–23. doi: 10.1038/nm.4046
127. Tan J, Liu H, Huang M, Li N, Tang S, Meng J, et al. Small Molecules Targeting Rorγt Inhibit Autoimmune Disease by Suppressing Th17 Cell Differentiation. Cell Death Dis (2020) 11(8):697. doi: 10.1038/s41419-020-02891-2
128. Wang D, Tang M, Zong P, Liu H, Zhang T, Liu Y, et al. MiRNA-155 Regulates the Th17/Treg Ratio by Targeting SOCS1 in Severe Acute Pancreatitis. Front Physiol (2018) 9:686. doi: 10.3389/fphys.2018.00686
129. Zhao Q, Manohar M, Wei Y, Pandol SJ, Habtezion A. STING Signalling Protects Against Chronic Pancreatitis by Modulating Th17 Response. Gut (2019) 68(10):1827–37. doi: 10.1136/gutjnl-2018-317098
130. Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL. IL-17 Receptor-Based Signaling and Implications for Disease. Nat Immunol (2019) 20(12):1594–602. doi: 10.1038/s41590-019-0514-y
131. Vege SS, Atwal T, Bi Y, Chari ST, Clemens MA, Enders FT. Pentoxifylline Treatment in Severe Acute Pancreatitis: A Pilot, Double-Blind, Placebo-Controlled, Randomized Trial. Gastroenterology (2015) 149(2):318–20.e3. doi: 10.1053/j.gastro.2015.04.019
132. Awane M, Andres PG, Li DJ, Reinecker HC. NF-Kappa B-Inducing Kinase is a Common Mediator of IL-17-, TNF-Alpha-, and IL-1 Beta-Induced Chemokine Promoter Activation in Intestinal Epithelial Cells. J Immunol (1999) 162(9):5337–44. doi: 10.0000/PMID10228009
133. Li X, Ye C, Mulati M, Sun L, Qian F. Ellipticine Blocks Synergistic Effects of IL-17A and TNF-α in Epithelial Cells and Alleviates Severe Acute Pancreatitis-Associated Acute Lung Injury. Biochem Pharmacol (2020) 177:113992. doi: 10.1016/j.bcp.2020.113992
134. Aqel SI, Kraus EE, Jena N, Kumari V, Granitto MC, Mao L, et al. Novel Small Molecule IL-6 Inhibitor Suppresses Autoreactive Th17 Development and Promotes Treg Development. Clin Exp Immunol (2019) 196(2):215–25. doi: 10.1111/cei.13258
135. Vecellio M, Hake VX, Davidson C, Carena MC, Wordsworth BP, Selmi C. The IL-17/IL-23 Axis and Its Genetic Contribution to Psoriatic Arthritis. Front Immunol (2021) 11:596086. doi: 10.3389/fimmu.2020.596086
136. Neuhöfer P, Liang S, Einwächter H, Schwerdtfeger C, Wartmann T, Treiber M, et al. Deletion of Iκbα Activates RelA to Reduce Acute Pancreatitis in Mice Through Up-Regulation of Spi2A. Gastroenterology (2013) 144(1):192–201. doi: 10.1053/j.gastro.2012.09.058
137. Zhang Z, Liu Q, Zang H, Shao Q, Sun T. Oxymatrine Protects Against L-Arginine-Induced Acute Pancreatitis and Intestine Injury Involving Th1/Th17 Cytokines and MAPK/NF-κb Signalling. Pharm Biol (2019) 57(1):595–603. doi: 10.1080/13880209.2019.1657906
138. Kanda T, Nishida A, Ohno M, Imaeda H, Shimada T, Inatomi O, et al. Enterococcus Durans TN-3 Induces Regulatory T Cells and Suppresses the Development of Dextran Sulfate Sodium (DSS)-Induced Experimental Colitis. PloS One (2016) 11(7):e0159705. doi: 10.1371/journal.pone.0159705
139. Tanabe S. The Effect of Probiotics and Gut Microbiota on Th17 Cells. Int Rev Immunol (2013) 32(5-6):511–25. doi: 10.3109/08830185.2013.839665
140. Westfall S, Caracci F, Zhao D, Wu QL, Frolinger T, Simon J, et al. Microbiota Metabolites Modulate the T Helper 17 to Regulatory T Cell (Th17/Treg) Imbalance Promoting Resilience to Stress-Induced Anxiety- and Depressive-Like Behaviors. Brain Behav Immun (2021) 91:350–68. doi: 10.1016/j.bbi.2020.10.013
141. El-Salhy M, Hatlebakk JG, Gilja OH, Bråthen Kristoffersen A, Hausken T. Efficacy of Faecal Microbiota Transplantation for Patients With Irritable Bowel Syndrome in a Randomised, Double-Blind, Placebo-Controlled Study. Gut (2020) 69(5):859–67. doi: 10.1136/gutjnl-2019-319630
142. Chen H, Chen Z, Shen L, Wu X, Ma X, Lin D, et al. Fecal Microbiota Transplantation From Patients With Autoimmune Encephalitis Modulates Th17 Response and Relevant Behaviors in Mice. Cell Death Discovery (2020) 6:75. doi: 10.1038/s41420-020-00309-8
143. Li X, He C, Li N, Ding L, Chen H, Wan J, et al. The Interplay Between the Gut Microbiota and NLRP3 Activation Affects the Severity of Acute Pancreatitis in Mice. Gut Microbes (2020) 11(6):1774–89. doi: 10.1080/19490976.2020.1770042
144. Gao N, Yan C, Zhang G. Changes of Serum Procalcitonin (PCT), C-Reactive Protein (CRP), Interleukin-17 (IL-17), Interleukin-6 (IL-6), High Mobility Group Protein-B1 (HMGB1) and D-Dimer in Patients With Severe Acute Pancreatitis Treated With Continuous Renal Replacement Therapy (CRRT) and Its Clinical Significance. Med Sci Monit (2018) 23(24):5881–6. doi: 10.12659/MSM.910099
Keywords: interleukin-17, acute pancreatitis, T helper 17 cells, immune response, gut microbiome
Citation: Li G, Chen H, Liu L, Xiao P, Xie Y, Geng X, Zhang T, Zhang Y, Lu T, Tan H, Li L and Sun B (2021) Role of Interleukin-17 in Acute Pancreatitis. Front. Immunol. 12:674803. doi: 10.3389/fimmu.2021.674803
Received: 02 March 2021; Accepted: 31 August 2021;
Published: 14 September 2021.
Edited by:
Andreas Von Knethen, Goethe University Frankfurt, GermanyReviewed by:
Dragana Odobasic, Monash University, AustraliaSonia Andrea Leon-Cabrera, National Autonomous University of Mexico, Mexico
Copyright © 2021 Li, Chen, Liu, Xiao, Xie, Geng, Zhang, Zhang, Lu, Tan, Li and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bei Sun, c3VuYmVpNzBAb3V0bG9vay5jb20=; Le Li, bGlsZUBocmJtdS5lZHUuY24=
†These authors have contributed equally to this work