
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 02 June 2021
Sec. Mucosal Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.674313
This article is part of the Research TopicLessons on T-Cells and Immune-Targeting Therapeutics in Coeliac DiseaseView all 10 articles
 Jordan Voisine1,2
Jordan Voisine1,2 Valérie Abadie1,3*
Valérie Abadie1,3*Several environmental, genetic, and immune factors create a “perfect storm” for the development of coeliac disease: the antigen gluten, the strong association of coeliac disease with HLA, the deamidation of gluten peptides by the enzyme transglutaminase 2 (TG2) generating peptides that bind strongly to the predisposing HLA-DQ2 or HLA-DQ8 molecules, and the ensuing unrestrained T cell response. T cell immunity is at the center of the disease contributing to the inflammatory process through the loss of tolerance to gluten and the differentiation of HLA-DQ2 or HLA-DQ8-restricted anti-gluten inflammatory CD4+ T cells secreting pro-inflammatory cytokines and to the killing of intestinal epithelial cells by cytotoxic intraepithelial CD8+ lymphocytes. However, recent studies emphasize that the individual contribution of each of these cell subsets is not sufficient and that interactions between these different populations of T cells and the simultaneous activation of innate and adaptive immune pathways in distinct gut compartments are required to promote disease immunopathology. In this review, we will discuss how tissue destruction in the context of coeliac disease results from the complex interactions between gluten, HLA molecules, TG2, and multiple innate and adaptive immune components.
Coeliac disease (CeD) is a multifactorial intestinal immune-mediated disorder with autoimmune features that leads to inflammatory and destructive lesions in the proximal small intestine. CeD, similar to other organ-specific autoimmune disorders, is marked by its complexity both at the epidemiological and immunological levels, which translates into a spectrum of clinical manifestations (1). CeD is characterized by an infiltration of intraepithelial lymphocytes in the proximal part of the small intestine, crypt hyperplasia and the development of villous atrophy in the latest stages of the disease. In addition, CeD patients produce highly disease-specific antibodies against deamidated gluten peptides and the enzyme tissue transglutaminase 2 (TG2) (2–4). CeD is triggered by gluten consumption in genetically susceptible individuals carrying certain major histocompatibility complex (MHC) class II human leukocyte antigen (HLA) variants (5, 6). 90-95% of CeD patients carry the HLA-DQ2.5 variant (DQA1*05:01, DQB1*02:01) that confers the highest risk of developing CeD while the remaining patients carry HLA-DQ2.2 (DQA1*02:01, DQB1*02:02) or HLA-DQ8 (DQA1*03, DQB1*03:02) (5, 7). However, while up to 40% of the general population in Western countries express one of the predisposing HLA molecules, the global prevalence of CeD is just 1% (8). This finding suggests that these HLA variants contribute to, but are not sufficient for, the development of the disease and that additional genetic and environmental factors are needed to mount a pathogenic immune response against gluten (9). In fact, the HLA locus which is the main inherited genetic susceptibility factor for CeD, only accounts for ~ 40% of the genetic variance of the disease. Hence, non-MHC susceptibility loci explaining ~ 15% of the disease risk (10–13), as well as additional environmental factors other than gluten, are thought to contribute to disease development. Among them are early life gastrointestinal infections, which have been associated with an increased risk of developing CeD in several cohorts of genetically susceptible children (14–16). Of particular interest are enteric viruses such as reovirus, norovirus and rotavirus, which are the most common causes of diarrheal disease in early childhood. Recurrent infections in young individuals with a permissive genetic background could interfere with the maturation of the mucosal immune system and the composition of the microbiome (17), and thus favor the subsequent induction of an inflammatory T cell responses and the loss of oral tolerance to dietary gluten (18, 19). Although much less documented, intestinal infections caused by bacteria such as Campylobacter jejuni or parasites such as Giardia lamblia, could also contribute to the onset or maintenance of the disease (20, 21). In strong support of a role of the microbial environment in promoting the development of CeD is the identification of microbially derived mimics of gliadin epitopes that can activate HLA-DQ2.5-restricted gliadin-specific T cells isolated from CeD patients (22).
The complexity of CeD is also reflected by the contribution of multiple immune pathways for the induction of the disease and intestinal tissue remodeling and destruction (23–25). It has been known for decades that HLA-DQ2 and HLA-DQ8 present TG2-deamidated gluten peptides to CD4+ T cells in the intestinal lamia propria compartment, driving TH1 differentiation (26–31). These gluten-specific TH1 cells contribute to the inflammatory process through the production of the inflammatory cytokines Interferon (IFN)-γ (32) and Interleukin (IL)-21 (33). Yet, this gluten-specific adaptive immunity is not sufficient to promote the licensing of intraepithelial cytotoxic CD8+ T cells (IE-CTLs) that are responsible for the destruction of distressed intestinal epithelial cells. This lack of sufficiency can be seen in potential CeD patients, who carry HLA-DQ2 or HLA-DQ8 and display adaptive immune responses against gluten (proxied by anti-TG2 and anti-endomysium antibodies) but lack villous atrophy (6, 34). In particular, potential CeD patients do not display an accumulation of IE-CTLs with an active killer phenotype (upregulated granzyme B expression, upregulated activating NK receptor expression, downregulated inhibitory NK receptor expression) and also lack upregulation of the pro-inflammatory cytokine IL-15 and the non-classical MHC class I stress molecules MICA/B and HLA-E in intestinal epithelial cells (25, 35, 36), immune features that are both required for the development of villous atrophy. Additionally no tissue destruction was observed in HLA-DQ2 or HLA-DQ8 humanized mice that develop anti-gluten immunity (37–39). Only in recent years has it become clear that the interplay between gluten-specific CD4+ T cells and intraepithelial cytotoxic CD8+ T cells, as well as the simultaneous activation of innate immune pathways in distinct gut compartments, are required to cause villous atrophy observed in the active form of the disease (Figure 1).
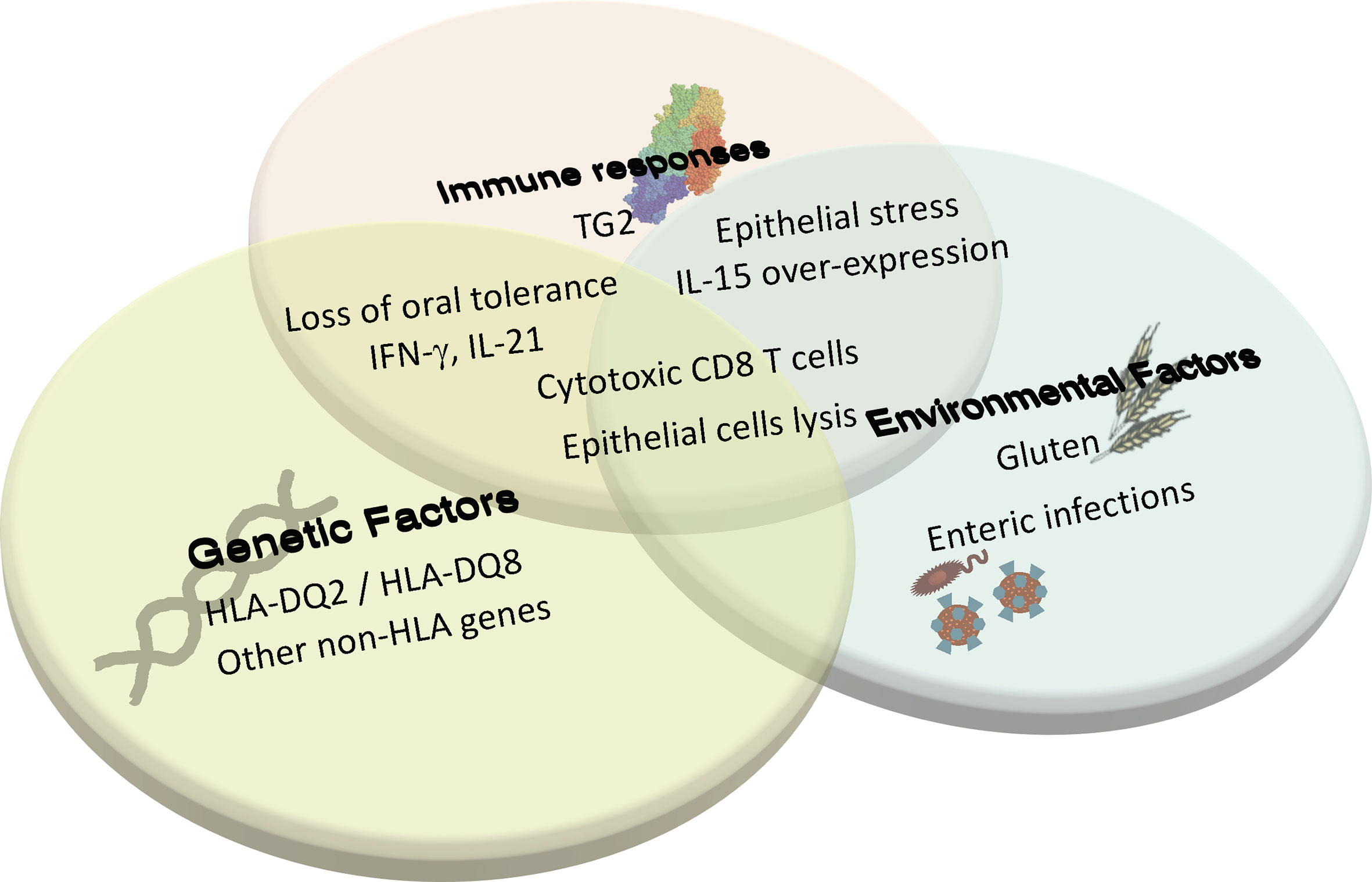
Figure 1 Coeliac disease is a multifactorial complex autoimmune disorder that requires the interplay between genetics, innate and adaptive immunity, and environmental triggers to cause tissue destruction. In individuals with HLA-DQ2/DQ8, induction of adaptive immune response against gluten and the loss of oral tolerance to gluten can occur when there is environmentally triggered epithelial stress and IL-15 overexpression. This adaptive response to gluten and the associated cytokine production promotes further tissue stress, leading to the licensing of cytotoxic CD8 T cells to lyse epithelial cells and cause villous atrophy. Additional environmental triggers and immune factors yet to be determined are also thought to contribute to disease development.
The identification of HLA-DQ2 or HLA-DQ8 restricted CD4+ T cells in the lamina propria of CeD patients (26, 40) that preferentially recognize deamidated gluten peptides over native gluten peptides (41) stressed the connection between gluten, disease-associated HLA molecules and TG2 for the initiation of the pathogenic immune response. The generation of such a gluten-specific T cell response arises from the high affinity binding of gluten peptides post-translationally modified by TG2 to HLA-DQ2 or HLA-DQ8.
Two properties of gluten explain its ability to elicit a mucosal immune response. First, the high content of proline in gluten proteins makes the proteins resistant to degradation by intestinal proteases in the gut lumen (42). Second, the long undigested gluten-derived proteins are good substrates for the enzyme TG2, an ubiquitous and multifunctional enzyme expressed in many organs including the gut (43). It has been acknowledged for a long time that TG2 plays a key role in CeD pathogenesis as the enzyme is the target of autoantibodies that are highly-disease specific and used for the diagnostic work-up (2, 4). In addition, TG2 catalyzes the conversion of glutamine residues present in gluten peptides into glutamate (28–30). This deamidation process is key to initiate a pathogenic response in CeD as it promotes the generation of immunogenic peptides with negatively charged carboxylate residues that anchor with high affinity in the positively charged pockets of HLA-DQ2 or HLA-DQ8 binding grooves (5, 44).
In support of the role of TG2 in orchestrating mucosal immune responses to dietary gluten, TG2 is mostly found catalytically inactive in the intestine under physiological conditions but its expression and activity are increased in inflamed tissues and in cells with inflammatory stress (45). Interestingly, the administration of poly(I:C) (Polyinosinic:polycytidylic acid), which results in the rapid induction of villous atrophy that is the typical intestinal tissue injury observed in CeD patients, promotes TG2 activation (46). TG2 can be released into the gut lumen by small intestine enterocyte shedding, allowing TG2 to become catalytically active in the extracellular environment (47). This feature that allows the TG2 to be in close vicinity of gluten peptides could explain the formation of enzyme-substrate complexes between the two proteins that can bind to the B cell receptor of TG2-specific B cells, hence contributing to the generation of TG2-specific autoantibodies (24, 48, 49). In addition, using T84 enterocytic cell line as a model, it was shown that extracellular TG2 can be activated in a phosphatidylinositol-3-kinase-dependent mechanism by IFN-γ (50), the pro-inflammatory cytokine abundantly produced by gluten-specific T cells and released in the inflamed intestinal mucosa of CeD patients (31, 32). In accordance with a role for IFN-γ in TG2 activation, TG2 enzymatic activity can be triggered by the protein cofactor thioredoxin-1 (TRX) whose release from monocytic cells is also elicited by IFN-γ (51).
The requirement of gluten, predisposing HLA variant, TG2, and CD4 T cells to elicit the disease was formally demonstrated using a newly engineered DQ8-Dd-villin-IL-15tg mouse model of CeD that develops villous atrophy upon gluten exposure (23). Using this model, we showed that villous atrophy, anti-deamidated gluten peptide antibodies and TH1 immunity recede on a gluten free diet and reoccur after gluten introduction. In addition, intestinal tissue destruction only occurred in mice carrying HLA-DQ8 and depletion of CD4+ T cells or administration of TG2 inhibitors in gluten-fed animals prevented the development of villous atrophy (23). Hence, the priming of a gluten-specific immune response depends on the coordinated interaction between gluten, the coeliac predisposing HLA-DQ8 molecule, activated TG2, and CD4+ T cells.
Interestingly, there is a gene dosage effect of the HLA-DQ2 or HLA-DQ8 allotypes in CeD whereby disease susceptibility depends on the HLA-DQ genotype and HLA homozygous individuals are at higher risk of developing the disease (52, 53). The haplotype HLA-DQ2.5 that binds and presents the full repertoire of gluten peptides, with many proline-rich α- and ω-gliadin-derived peptides that are protected from the degradation by gastrointestinal enzymes, confers a higher risk of developing CeD as compared to HLA-DQ8 that presents a smaller repertoire of immunogenic peptides more prone to proteolytic degradation and HLA-DQ2.2 that can only bind and present a few peptides (42, 54, 55). In addition, DQ2.5 has an increased ability to retain its peptide cargo as compared to DQ2.2. thanks to the presence of a polymorphism in the DQα chains allowing DQ2.5 to establish a hydrogen bond to the peptide main chain that stabilizes peptide-MHC complexes at the surface of antigen-presenting cells leading to sustained gluten peptides presentation to T cells (56). Interestingly, differences in the nature of HLA-DQ2.2 or HLA-DQ2.5-bound epitopes translates into a more diverse TCR repertoire generated in the context of HLA-DQ2.2, as compared to HLA-DQ2.5-mediated CeD and with a lower disease penetrance (57). Homozygosity for HLA-DQ2.5 that is linked to a heightened expression of HLA-DQ2.5 on the surface of antigen-presenting cells and increased antigen presentation is also more strongly associated with CeD as compared to heterozygosity (52, 55). Hence, the amounts of HLA-DQ-gluten peptides complexes correlates with the magnitude and breadth of the gluten-specific T cell response (52, 55). This suggests that CeD development and the ensuing intestinal tissue destruction will only occur when the T cell response has reached a certain magnitude to become pathogenic (35).
Under homeostatic conditions, ingested dietary antigens induce oral tolerance, a state of local and systemic immune ignorance against orally ingested innocuous antigens (58, 59). However, in patients harboring the CeD-predisposing HLA-DQ2 or HLA-DQ8 molecules, orally ingested gluten can initiate a gluten-specific pro-inflammatory TH1 response rather than a tolerogenic response (35). In both adults and children, these gluten-specific CD4 T cells produce high levels of the pro-inflammatory cytokines IFN-γ and IL-21, hallmarks of TH1 cells (32, 33, 60). As loss of oral tolerance to gluten is a preceding event for the development of villous atrophy in CeD patients (25, 32), much work has been done to uncover the mechanisms behind this loss of tolerance against gluten and how this pro-inflammatory TH1 CD4 T cell response leads to villous atrophy.
One of the early culprits implicated in promoting an inflammatory vs tolerogenic T cell response to gluten is the cytokine interferon-α (IFN-α). IFN-α is a Type-1 interferon that is produced by almost all cells as an innate response to viral infection (61). Among its many immune effector-promoting roles, it has been shown to drive pro-inflammatory dendritic cells activation as well as to promote the differentiation of CD4 T cells to the TH1 lineage (61, 62). The connection between IFN-α and CeD was made by Monteleone and colleagues, who identified a CeD-like enteropathy with villous atrophy and high intraepithelial lymphocytes infiltration in a chronic myeloid leukemia patient receiving an IFN-α treatment (63). The association between high expression of IFN-α and high levels of IFN-γ in CeD patients compared to controls suggested that IFN-α in CeD patients may be one factor leading to induction of a TH1 response against gluten. It remains unclear what directly is driving the increase in IFN-α production, but recent studies have also implicated viral infection as a driver for loss of oral tolerance.
While viral infections, such as with adenovirus or hepatitis C, have long been known to be associated with a higher risk of developing CeD (64), only recently have viral infections been mechanistically shown to induce loss of oral tolerance to gluten and dietary antigens. Using the Type-I Lang (T1L) reovirus strain, and murine norovirus (MNV) that both infect the gut, Bouziat and colleagues demonstrated that both viruses were capable of mediating TH1 responses to dietary antigens (18, 19). Type-1 IFN signaling was required for the blockade of peripheral regulatory T cells conversion while Interferon Regulatory Factor (IRF)1 expression was required for the induction of a TH1 immunity characterized by the differentiation of IL-12p40-producing dendritic cells, the production of gluten-specific IgG2c antibodies in the serum, TG2 activation in the proximal small intestine and a delayed type hypersensitivity reaction to gluten, all hallmarks of loss of oral tolerance to gluten in virus-infected HLA-DQ8 transgenic mice (18, 19). Taken together, these studies demonstrated that viral infections can be triggers for loss of oral tolerance towards dietary antigens and TH1-skewed responses to gluten.
Another major player implicated in the loss of oral tolerance to gluten is IL-15. The first signs that IL-15 may have been involved in the proinflammatory TH1 response to gluten came with the finding that IL-15 is heavily upregulated in the lamina propria of active CeD patients, the effector site where dendritic cells will encounter gluten peptides (65, 66). Using HLA-DQ8 transgenic mice that overexpressed IL-15 in the lamina propria and mesenteric lymph nodes (DQ8-Dd-IL15tg mice) but not in the intestinal epithelium (38), we demonstrated that IL-15 overexpression in combination with retinoic acid altered the tolerogenic phenotype of intestinal dendritic cells and endowed them with a pro-inflammatory phenotype, hindering the development of Foxp3+ regulatory T cells and instead promoting the differentiation of IFNγ-producing TH1 cells. Additionally, these gluten-fed DQ8-Dd-IL15tg mice displayed elevated levels of anti-gliadin and anti-TG2 antibodies, mimicking potential CeD patients who display a loss of oral tolerance and the development of a TH1 response to gluten in the absence of villous atrophy (38). In addition, IL-15 can block the immunosuppressive effects of TGF-β on CD4 and CD8 T cells by inhibiting Smad3-signalling and additionally render effector CD4 and CD8 T cells resistant to regulatory T cells-mediated suppression by activating PI3K-signaling (67, 68). Whether Foxp3+ regulatory T cells play an active role in dampening harmful immune responses to gluten in the small intestine remains poorly understood. Although it was shown that Foxp3+ regulatory T cells expand in the celiac lesion (69–71), it remains controversial whether regulatory T cells retain or loss their suppressive function (71–73). Moreover, regulatory CD4+ T cells specific for immunodominant gluten peptides haven’t been identified so far in the small intestine of genetically predisposed healthy individuals (74). Therefore, additional investigations are warranted to determine whether a regulatory response to gluten exists and whether an altered mucosal suppressive CD4+ T cell response to gluten contributes to CeD pathogenesis.
In the context of CeD, TH1 immunity is accompanied by the production of IFN-γ and IL-21 by mucosal gluten-specific CD4+ T cells (32, 33, 60). The idea of crosstalk between lamina propria and epithelium mediated by cytokines was put forward several years ago based on in vitro observations (75). Indeed, it had been shown that IFN-γ released by stimulated mucosal T cells was required for the optimal killing of human colonic epithelial cell lines in ex-vivo cytotoxic assays (76). In addition, the incubation of intestinal tissue specimens with the supernatants from gluten-stimulated T cell clones or with IFN-γ lead to epithelial cell damage, and the cytotoxic effect of the supernatants could be counteracted by the addition of neutralizing IFN-γ (77). We recently confirmed the requirement of IFN-γ for the activation of cytotoxic intraepithelial lymphocytes and the ensuing development of villous atrophy in vivo using a relevant mouse model of CeD (23). Although the exact mechanism underlying this effect remains to be uncovered, it has been shown that local production of IFN-γ can promote the upregulation of the non-classical MHC class Ib molecule HLA-E on epithelial cells (78, 79) hence potentializing the expression of the ligand for the activating NK receptor CD94/NKG2C present on cytotoxic intraepithelial lymphocytes during disease development. Although it has been shown that IL-21 can increase the cytotoxicity of human intraepithelial lymphocytes (80), the administration of an IL-21R blocking antibody in our mouse model didn’t reveal any significant direct role of IL-21 in promoting cytotoxic properties on intraepithelial lymphocytes but instead demonstrated that IL-21 and IFN-γ both play a role in the development of anti-deamidated gluten peptides antibodies (23). Interestingly, IL-15 can promote IL-21 production in lamina propria cells (81) reinforcing the idea of the involvement of a cytokine network in CeD.
While IFN-α, viral infections and IL-15 overexpression in the lamina propria can induce a loss of oral tolerance and a pro-inflammatory TH1 response to gluten, several studies in mice have demonstrated that the CD4 T cell response to gluten alone is nevertheless not sufficient to license intraepithelial lymphocytes and induce villous atrophy (23, 37, 38, 82). This is in accordance with observations in potential CeD patients who not only lack IL-15 and stress molecules expression on intestinal tissue cells, but also do not display an accumulation of intraepithelial cytotoxic T lymphocytes with an active killer phenotype, as seen in active CeD patients, despite the development of an inflammatory CD4 T cell response (25).
Interestingly, a distinct cytokine signature has been identified in the peripheral blood of treated CeD patients after oral gluten challenge (83, 84) or subcutaneous administration of T-cell stimulatory gluten peptides (85). Indeed, secretion of IL-2, IL-17A, TNF-α, IL-6 and IL-10 was detected as soon as 2h after gluten-re-exposure reflecting the rapid mobilization of gluten-specific memory CD4 T cells. Serum cytokine elevations, particularly IL-2 levels, correlated with the severity of acute digestive symptoms (83) and were specific to CeD (84, 86, 87), demonstrating a direct impact of gluten on the adaptive immune system in genetically susceptible individuals. Not only could serum cytokine release contribute to some extra-intestinal manifestations of CeD driven by inflammation, but it could be used as immune marker to diagnose and monitor the development of CeD.
The contribution of innate immunity to CeD pathogenesis was suggested by the observation that non-HLA genomic regions associated with CeD harbor genes involved in stress pathways and innate immunity (5). The activation of innate immune pathways in different gut compartments, in particular at the level of the lamina propria or in the intestinal epithelium, has a substantial impact on the adaptive immune responses taking place in those same compartments. These responses include the loss of oral tolerance to gluten and the associated induction of TH1 immunity, as well as the acquisition of lymphokine killer activity by intraepithelial lymphocytes, all of which contribute to disease development.
In vitro studies where gluten was used to stimulate duodenal biopsy samples, intestinal epithelial cells, monocytes, macrophages, and dendritic cells have shown that gluten may have innate immune stimulatory properties (88–94). Additionally, other molecules contained in wheat and related proteins could also drive immune cells activation, such as wheat amylase trypsin inhibitors (95, 96).
The gluten-derived α-gliadin peptide P31-43, which unlike the 33-mer (P55-87) and the 25-mer (P31-55) does not induce specific T cell responses in the celiac lesion, has been shown to activate innate immune pathways (90, 92, 97–99). However, the observed innate properties have been different across studies and we still need cross-validation. P31-43 peptide could induce enterocyte proliferation and actin rearrangements in an IL-15 and epithelial growth factor (EGF)-dependent manner, leading in particular to crypt hyperplasia, one of the characteristics of tissue remodeling seen in the celiac lesion (100–104). Other reported effects of gliadin on the innate immune system encompass cell structural changes, alterations in epithelial cells signaling, and induction of inflammatory and stress signals [reviewed in (100, 105)]. The finding that enterocytes from CeD patients have a stressed/inflamed phenotype and present a constitutive alteration in the intracellular vesicular trafficking provides an explanation as to why those cells are more sensitive to the effects of the P31-43 peptide (106–108). Indeed, due to sequence similarity between the P31-43 peptide and a region of hepatocyte growth factor regulated substrate (HRS) kinase - an essential protein involved in endocytic maturation-, P31-43 localizes in early endosomes and alters HRS-mediated maturation of early endosomes and the recycling pathway. The ensuing delayed vesicular trafficking leads to a reduction in the degradation of receptor tyrosine kinases including the receptor for EGF and promotes a sustained trans-presentation of IL-15 at the epithelial cell level [reviewed in (100, 105)].
Interestingly, a peptic-tryptic digest of gliadin or the P31-49-derived peptide can induce the upregulation of the expression of the stress-inducible MHC class I polypeptide-related molecules (MIC) via a pathway involving IL-15 (90). This is in accordance with the observation that intestinal epithelial cells in CeD patients express high levels of the MIC molecules (90, 109) and the non-classical MHC class I molecule HLA-E (79, 94). The expression of the inflammatory cytokine IL-15 can also be upregulated in whole biopsies, intestinal epithelial cells, or antigen-presenting cells from CeD patients upon gluten challenge or P31-49 derived peptide stimulation (65, 66, 78, 92, 110). The physiological consequences of the activation of immune pathways by gluten remain unclear given the fact that family members of CeD patients that lack an adaptive response to gluten, yet harbor the pre-disposing HLA-DQ2 and HLA-DQ8 and also show IL-15 upregulation in their intestinal compartment (25), retain normal intestinal morphology. However, the observation that, unlike active CeD patients, potential CeD patients -who display a gluten-specific adaptive immune response in the absence of tissue destruction- lack the innate epithelial stress response suggests that the alteration of the epithelial compartment is required for the development of villous atrophy (25). This observation is in accordance with the mechanism of epithelial cell destruction whereby activated intraepithelial TCRαβ lymphocytes mediate the killing of intestinal epithelial cells based on the recognition of stress signals such as non-classical MHC molecules and IL-15. As discussed in more detail below, the acquisition of innate-like properties by intraepithelial lymphocytes is also driven by IL-15 (111).
In addition to the epithelial upregulation of IL-15, most CeD patients display a chronic upregulation of IL-15 in the lamina propria (66). IL-15 plays a critical role in the lamina propria as it can impart dendritic cells to initiate the polarization of inflammatory TH1 responses and the loss of oral tolerance to gluten (38). This loss of oral tolerance to dietary gluten could also be triggered by type 1 interferons in individuals over-expressing IFN-α in lieu of IL-15 (63). Interestingly, the P31-43 peptide can trigger the expression of inflammatory mediators and increase cell death in a MyD88- and type 1 IFNs-dependent manner and this innate immune activation is enhanced by the TLR3 agonist poly(I:C) (112). This synergistic action of gluten peptides with poly(I:C) and the finding that poly(I:C) (46) or reovirus infection (19) promote the activation of TG2 suggests that multiple environmental hits can trigger and drive disease development. Because high levels of IL-15 can persist in a subset of patients on a gluten-free diet, it remains unclear what drives the excessive chronic upregulation of this innate cytokine. Similarly, both viral and bacterial infections could be the main source of type 1 interferons, yet how this expression is sustained remains to be determined (113).
The association between CeD susceptibility and single nucleotides polymorphisms in genes involved in microbial sensing has also pointed towards a role for bacterial microbes in triggering immune activation (5, 114). Many studies have noted differences in the microbiota composition between CeD patients, treated CeD patients on a gluten-free diet and healthy individuals [reviewed in (17)]. Gluten-degrading proteases produced by some opportunistic pathogens found in the duodenum of CeD patients such as Pseudomonas aeruginosa, can activate PAR-2 initiated inflammatory signaling pathways resulting in the expansion of intraepithelial lymphocytes (115). In addition, thanks to its elastase activity, Pseudomonas aeruginosa could contribute to the initiation of the disease by favoring the generation of immunogenic gluten peptides that can efficiently translocate through the intestinal barrier (116). Neisseria flavescens, abundantly present in the duodenal microbiome of CeD patients, could also contribute to the inflammatory response through its ability to endow a pro-inflammatory phenotype in dendritic cells (117). Dysbiosis is usually associated with a decrease in bacterial diversity and in the production of short-chain fatty acids such as butyrate, propionate and acetate that result from carbohydrate fermentation (118–121) and contribute to the maintenance of the gut homeostasis (122). Interestingly, a decrease in butyrate-producing bacteria such as Bifidobacterium or Faecalibacterium prausnitzii (123, 124) as well as alteration of the fecal metabolites patterns has been observed in children with CeD (125–127), yet these changes have not been observed in adult cohorts (128–130). These observations, together with the findings that genetically predisposed children carrying the HLA-DQ2 molecule present an altered gut microbiota composition, suggest that commensal bacteria could contribute early on to determining disease risk (131). In addition, because alterations in gut microbiota and fecal short-chain fatty acid composition can persist even when gluten is withdrawn, it is unclear whether dysbiosis could contribute to the initiation and enhancement of the disease or if changes in the microbiota reflect the ongoing local inflammation (15).
Overall, innate factors induced by gluten exposure and additional unknown triggers, perhaps of microbial origin, play a critical role in promoting the loss of oral tolerance to gluten and in altering intestinal epithelial cells that become the target of activated intraepithelial lymphocytes. However, studies comparing potential and active CeD patients as well as comparing mice expressing IL-15 in different gut compartments have shown that IL-15 and stress molecules overexpression in the epithelium need to be associated with adaptive immunity for villous atrophy to develop (23, 25).
In addition to the TH1-skewed CD4 T cell response, another hallmark of CeD is the large accumulation of oligoclonal cytotoxic intraepithelial TCRαβ+ CD8+ lymphocytes (IE-CTLs) and TCRγδ+ intraepithelial lymphocytes (78, 109, 132). These cells, and not the gluten-specific CD4+ T cells, are the particular immune cell type thought to mediate the destruction of intestinal epithelial cells and directly lead to villous atrophy (23, 36). Their critical role in tissue destruction was not appreciated until the discovery that these cells are reprogrammed to express high levels of activating NK receptors and associated adaptor molecules. In healthy individuals, intraepithelial lymphocytes predominantly express the dimeric inhibitory CD94/NKG2A receptor with only low levels of the activating CD94/NKG2C and NKG2D receptors (78, 79, 133, 134). However, intraepithelial lymphocytes from CeD patients were found to undergo extensive NK cell-like reprograming, downregulating expression of the inhibitory CD94/NKG2A receptor and upregulating expression of the activating CD94/NKG2C and NKG2D receptors (78, 79, 133, 134). Furthermore, the CD94/NKG2C receptors in CeD patients are associated with the ITAM-bearing adaptor molecule DAP12, enabling cytokine secretion, proliferation and cytolytic activity in response to NK receptor ligands, even independently of TCR activation (79). The stress-inducible, non-classical MHC-like molecule HLA-E is the ligand for CD94/NKG2C and it is selectively upregulated on intestinal epithelial cells in CeD patients, allowing enterocytes to be targeted for killing by IE-CTLs (79). Upregulation of NKG2D on IE-CTLs in CeD patients, as well as upregulation of its adaptor molecule DAP10, was found to be directly caused by high levels of IL-15 on intestinal epithelial cells (109). This IL-15 mediated signaling not only upregulated NKG2D, but also acted in a co-stimulatory manner, synergizing with NKG2D signaling to enable TCR-independent cytolysis of targets expressing both IL-15 and the stress-induced NKG2D ligands MICA/B (109, 134, 135). MICA/B, which are highly expressed in the intestinal mucosa of CeD, enable enterocytes to be excellent targets for IE-CTLs-mediated destruction (90). Taken together, the reprograming of CD8+ intraepithelial lymphocytes into NK-like IE-CTLs with the ability to lyse target cells independently of TCR activation positions this cell type to be the direct mediator of tissue destruction in CeD.
It is important to note however that while no gluten-specific IE-CTLs have been identified in the intestinal epithelium of CeD patients, and IE-CTLs can kill intestinal epithelial cells in a TCR-independent manner, TCR specificity may still be playing a role in tissue destruction. Support for this idea comes from studies showing that inhibitory and activating NK receptor expression was associated with particular TCR specificities (133, 136). Additionally, signaling through CD94/NKG2C, NKG2D, and IL-15 receptors lowers the threshold for TCR activation (79, 109, 134). This co-stimulatory signaling could allow for low-affinity TCR-ligand interactions to activate TCR signaling, while under normal conditions the interaction would be too low affinity for activation. Evidence for this was shown in a study in which IL-15 expressing tumors were selectively controlled and killed by SIY-specific CTLs cells in a non-cognate but TCR-dependent fashion (137). Therefore, there still may be a potential role for TCR specificity among IE-CTLs as low affinity TCR interactions may be playing a part in tissue destruction along with TCR-independent cytolysis. Overall, although TCRαβ+ IE-CTLs are not gluten specific, they destroy specifically intestinal epithelial cells expressing IL-15 and ligands for activating NK receptors.
While the frequencies of TCRαβ+ CD8+ IE-CTLs decrease when gluten is excluded from the diet (35), the expansion of TCRγδ+ intraepithelial lymphocytes persists (132). Intriguingly, the composition of the tissue-resident TCRγδ+ compartment is irreversibly altered by inflammation with the depletion of innate-like Vγ4+/Vδ1+ intraepithelial lymphocytes and the expansion of gluten-sensitive IFN-γ-producing Vδ1+ intraepithelial lymphocytes (138). Although the exact role of the naturally occurring tissue resident TCRγδ+ intraepithelial lymphocytes that have both cytotoxic and tissue repair potential remains to be determined, their loss may lead to a defect in tissue healing and the protection against infections and tumors. Furthermore, the role of gluten-dependent production of IFN-γ by active CeD Vδ1+ intraepithelial lymphocytes remains elusive.
A subset of adults with CeD go on to develop refractory coeliac disease (RCD), a rare CeD complication in which patients have persistent severe villous atrophy despite being on a strict gluten-free diet (139, 140). One of the hallmarks of RCD is the expansion of aberrant intraepithelial cytotoxic lymphocytes that lack surface CD3 expression (sCD3-), express intracellular CD3 (iCD3+), and display a highly activated NK cell-like phenotype (140). These aberrant IE-CTLs develop from hematopoietic stem cell-derived CD103+sCD3- IELs that encounter high levels of IL-15 and Notch signals in the gut epithelium and develop gain-of-function JAK1 or STAT3 mutations (141). Since both JAK1 and STAT3 are involved in IL-15 signaling, these gain of function mutations lead to heightened IL-15 signaling in aberrant iCD3+ IE-CTLs resulting in their expansion and survival through activation of anti-apoptotic signaling pathways (e.g. upregulation of anti-apoptotic factors Bcl-2 and Bcl-xl) (66, 142, 143). With IL-15 playing such a major role in the development of CeD and RCD, recent phase 2a clinical trials tested the impact of blocking IL-15 in CeD and RCD patients (144, 145). The trials did not show a significant difference in the primary clinical endpoints (improvement of the mucosal architecture in CeD and reduction in the proportion of aberrant intraepithelial lymphocytes in RCD). However, there were differences in some secondary endpoints, with treated RCD patients having fewer gastrointestinal symptoms and displaying less T cell receptor clonality than the placebo group (144), suggesting that blocking IL-15 may still be an option for treating RCD. There is a need to perform long-term follow-up studies in which the treatment duration will be significantly increased (duration of the anti-IL5 treatment in the published study was only ten weeks).
As discussed throughout this review, both innate and adaptive immune responses possibly connected by a cytokine network contribute to the immunopathogenesis of CeD. Each of the described cell types and their known mechanism of action are required to promote CeD but none of them individually is sufficient to culminate in the full-blown disease characterized by intestinal tissue destruction and remodeling. Observations in mice and humans have suggested or demonstrated the requirement for the simultaneous activation of distinct pathways in different gut locations, some cell-cell interactions, and the existence of a crosstalk between the lamina propria and the epithelial compartments (Figure 2).
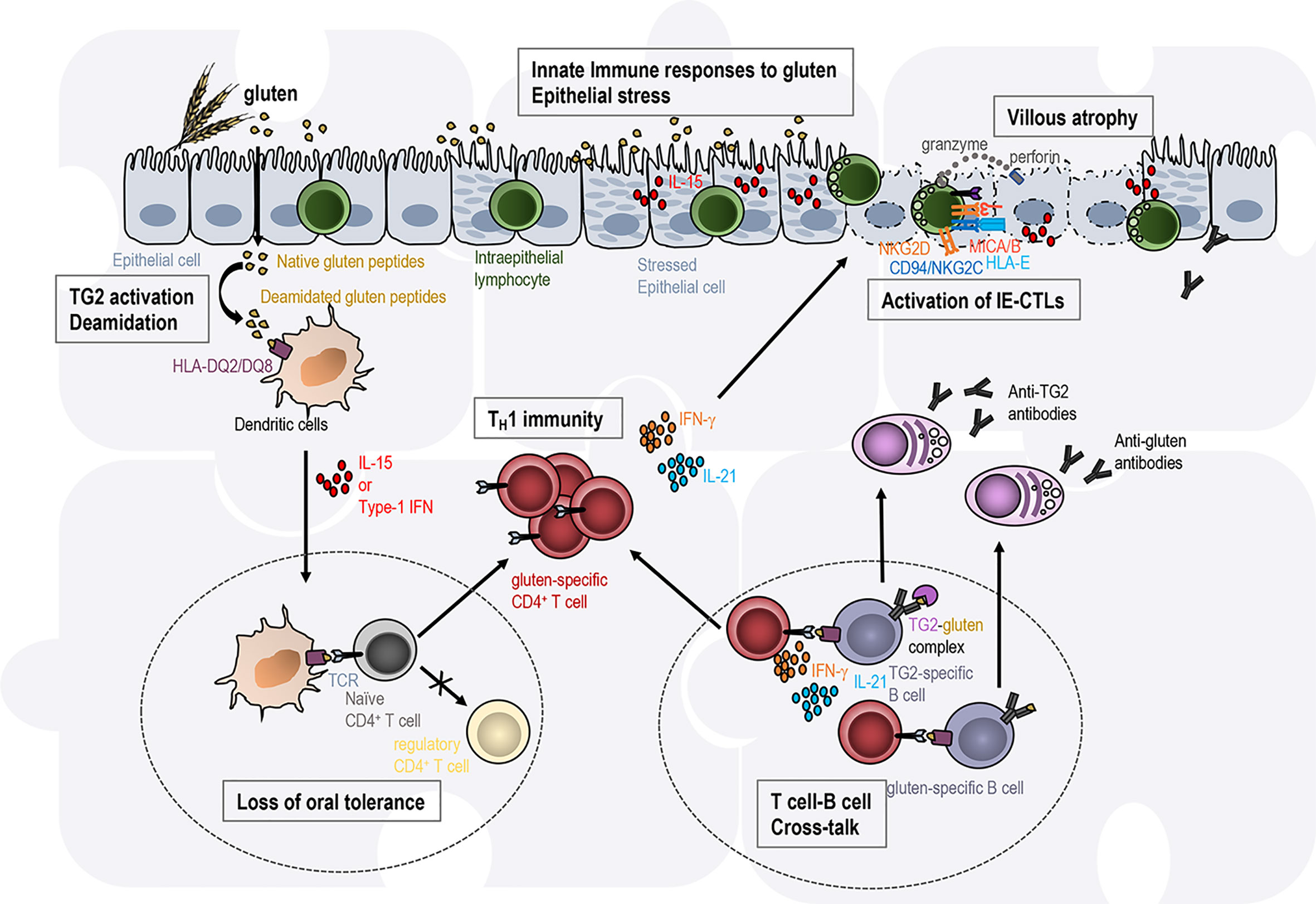
Figure 2 Intestinal tissue destruction in coeliac disease results from the interplay between several immune pathways in distinct gut locations. Transglutaminase 2 (TG2)-deamidated gluten peptides bind with high affinity to the disease-associated HLA-DQ2 or HLA-DQ8 molecules on antigen-presenting cells. In an inflammatory context (presence of IL-15, type 1 IFN), dendritic cells acquire a pro-inflammatory phenotype and migrate to the mesenteric lymph nodes (left circle with dashed line). They present gluten peptides to naïve CD4 T cells and promote T cell differentiation into TH1 effector T cells, while the induction of regulatory T cells involved in oral tolerance is abrogated. Anti-gluten CD4+ T cells directly secrete IFN-γ or IL-21. Anti-gluten CD4+ T cells are thought to provide help to gluten- and TG2-specific B cells in gut-associated secondary lymphoid organs (right circle with dashed line) leading to the production of IgA and IgG anti-gluten and anti-TG2 antibodies. In the presence of high IL-15 expression in the epithelium, intraepithelial lymphocytes acquire cytotoxic properties (activating NK receptors, release of the cytotoxic molecules granzyme B and perforin) and the ability to kill stressed epithelial cells expressing the ligands (HLA-E, MICA/B) for the NK receptors. Each of these immune events are required to promote coeliac disease but none of them individually is sufficient to promote intestinal tissue destruction. Hence the immunopathogenesis of coeliac disease is often presented as a jigsaw where each piece associated with one immune event needs to be connected to promote the disease.
First, the simultaneous analysis of IE-CTLs and intestinal epithelial cells features in patients encompassing the spectrum of the disease - i.e. family members, potential CeD patients and active CeD patients- has led to the hypothesis that the combination of epithelial stress associated with high IL-15 expression in enterocytes and an anti-gluten adaptive immune response induced in the lamina propria in the presence of inflammatory mediators such as IL-15 is needed for the development of villous atrophy (25). The cooperation between epithelial IL-15 and CD4+ T cells to promote tissue destruction was also suggested by a study performed in mice overexpressing IL-15 in the gut epithelium and fed with the dietary antigen ovalbumin (146). The analysis of our DQ8-Dd-villin-IL-15g mice modeling CeD patients upon gluten oral challenge unequivocally confirmed that the development of villous atrophy requires the concomitant presence of epithelial stress and anti-gluten adaptive immunity (23).
Next, in agreement with the findings that CD4+ T cells are required for the development of CeD, the prevention of villous atrophy in DQ8-Dd-villin-IL-15tg mice treated with an anti-CD4 depleting antibody, anti-IFNγ depleting antibody or TG2 inhibitors concomitantly to gluten administration also underlined the existence of a cross-talk between TH1 immunity in the lamina propria and the activation of IE-CTLs (23).
In addition, a recent study using a mouse model of CeD lacking B cells has demonstrated that B lymphocytes are required for the development of villous atrophy and the induction of a killer phenotype in IE-CTLs (147). The mechanisms underlying the exact contribution of B-cell mediated immune responses to the immunopathogenesis of CeD remains to be determined. Most of our current understanding of the role of antibodies is based upon in vitro experiments. Although anti-TG2 antibodies can exert several effects, no consensus has been reached regarding their potential pathogenic role [reviewed in (148)]. While cytokines produced by B cells, that need yet to be identified, could contribute to the inflammatory process in the CeD lesion, B lymphocytes and/or plasma cells themselves could contribute to disease pathogenesis through their role as antigen-presenting cells. Indeed, plasma cells were found to be the most abundant gluten peptide MHC-expressing cells in the lamina propria of CD patients (149). Human studies have also suggested that gluten CD4+ T cells and B cells having internalized TG2-gluten complexes interact to promote the generation of anti-TG2 antibodies, whose formation rely on the presence of gluten (24, 48, 150, 151). This T cell-B cell crosstalk is supported by in vitro assays showing that transduced lymphoma B cells expressing HLA-DQ2.5 and binding catalytically active TG2 can activate gluten-specific hybridoma T cells in the presence of non-deamidated gluten peptides (151). However, whether this interaction also benefits CD4+ T cells by promoting their activation and expansion helping to reach the threshold needed to reach a pathogenic T cell response remains to be determined.
T cell clones have proven to be an invaluable research tool to gain insights into the immune mechanisms underlying CeD pathogenesis. Early analysis of CD4 T cell clones in celiac patients uncovered TH1-skewed gluten-specific CD4 T cells restricted to the disease-associated HLA-DQ2 and HLA-DQ8 molecules (26, 31, 40). Further interrogation of gut derived gluten-specific T cell clones helped to demonstrate that these clones respond to TG2-modified gluten peptides and that deamidation of these peptides leads to the strongest T cell response (29, 30). The usage of animal models of the disease combined with our ability to track gluten-specific T cells should help identify the intestinal location of the pathogenic cellular interactions described in this review, such as the B cell-T cell crosstalk (37, 152). Leveraging single-cell approaches technologies to study human samples could also help identify additional understudied cell subsets such as innate lymphoid cells [reviewed in (138, 153)] that could participate in the establishment and maintenance of the disease.
Altogether, CeD represents a perfect example of a multifactorial complex autoimmune disorder. The priming of a gluten-specific inflammatory immune response depends on the coordinated interaction between gluten, the celiac predisposing HLA-DQ molecule, activated TG2, and CD4+ T cells, while the alteration of epithelial cells expressing stress molecules, and the subsequent activation of IE-CTLs are all required to promote intestinal tissue destruction. How T lymphocytes in the lamina propria and cells present in the epithelial compartment communicate and where the cross-talk between distinct cell types occur is not yet understood and will certainly lead to the identification of signaling pathways that could potentially represent novel therapeutic targets.
All authors contributed to the article and approved the submitted version.
This work was supported by a Young Investigator Award from the Celiac Disease Foundation to VA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Tack GJ, Verbeek WH, Schreurs MW, Mulder CJ. The Spectrum of Celiac Disease: Epidemiology, Clinical Aspects and Treatment. Nat Rev Gastroenterol Hepatol (2010) 7:204–13. doi: 10.1038/nrgastro.2010.23
2. Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, et al. Identification of Tissue Transglutaminase as the Autoantigen of Celiac Disease. Nat Med (1997) 3:797–801. doi: 10.1038/nm0797-797
3. Dieterich W, Laag E, Schopper H, Volta U, Ferguson A, Gillett H, et al. Autoantibodies to Tissue Transglutaminase as Predictors of Celiac Disease. Gastroenterology (1998) 115:1317–21. doi: 10.1016/S0016-5085(98)70007-1
4. Husby S, Koletzko S, Korponay-Szabo I, Kurppa K, Mearin ML, Ribes-Koninckx C, et al. European Society Paediatric Gastroenterology, Hepatology and Nutrition Guidelines for Diagnosing Coeliac Disease 2020. J Pediatr Gastroenterol Nutr (2020) 70:141–56. doi: 10.1097/MPG.0000000000002497
5. Abadie V, Sollid LM, Barreiro LB, Jabri B. Integration of Genetic and Immunological Insights Into a Model of Celiac Disease Pathogenesis. Annu Rev Immunol (2011) 29:493–525. doi: 10.1146/annurev-immunol-040210-092915
6. Ludvigsson JF, Leffler DA, Bai JC, Biagi F, Fasano A, Green PH, et al. The Oslo Definitions for Coeliac Disease and Related Terms. Gut (2013) 62:43–52. doi: 10.1136/gutjnl-2011-301346
7. Sollid LM, Markussen G, Ek J, Gjerde H, Vartdal F, Thorsby E. Evidence for a Primary Association of Celiac Disease to a Particular HLA-DQ Alpha/Beta Heterodimer. J Exp Med (1989) 169:345–50.
8. Singh P, Arora A, Strand TA, Leffler DA, Catassi C, Green PH, et al. Global Prevalence of Celiac Disease: Systematic Review and Meta-Analysis. Clin Gastroenterol Hepatol (2018) 16:823–36.e2. doi: 10.1016/j.cgh.2017.06.037
9. Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple Common Variants for Celiac Disease Influencing Immune Gene Expression. Nat Genet (2010) 42:295–302. doi: 10.1038/ng.543
10. Gutierrez-Achury J, Zhernakova A, Pulit SL, Trynka G, Hunt KA, Romanos J, et al. Fine Mapping in the MHC Region Accounts for 18% Additional Genetic Risk for Celiac Disease. Nat Genet (2015) 47:577–8. doi: 10.1038/ng.3268
11. Kumar V, Gutierrez-Achury J, Kanduri K, Almeida R, Hrdlickova B, Zhernakova DV, et al. Systematic Annotation of Celiac Disease Loci Refines Pathological Pathways and Suggests a Genetic Explanation for Increased Interferon-Gamma Levels. Hum Mol Genet (2015) 24:397–409. doi: 10.1093/hmg/ddu453
12. Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, Szperl A, et al. Dense Genotyping Identifies and Localizes Multiple Common and Rare Variant Association Signals in Celiac Disease. Nat Genet (2011) 43:1193–201. doi: 10.1038/ng.998
13. van Heel DA, Franke L, Hunt KA, Gwilliam R, Zhernakova A, Inouye M, et al. A Genome-Wide Association Study for Celiac Disease Identifies Risk Variants in the Region Harboring IL2 and IL21. Nat Genet (2007) 39:827–9. doi: 10.1038/ng2058
14. Kemppainen KM, Lynch KF, Liu E, Lonnrot M, Simell V, Briese T, et al. Factors That Increase Risk of Celiac Disease Autoimmunity After a Gastrointestinal Infection in Early Life. Clin Gastroenterol Hepatol (2017) 15:694–702.e5. doi: 10.1016/j.cgh.2016.10.033
15. Marild K, Kahrs CR, Tapia G, Stene LC, Stordal K. Infections and Risk of Celiac Disease in Childhood: A Prospective Nationwide Cohort Study. Am J Gastroenterol (2015) 110:1475–84. doi: 10.1038/ajg.2015.287
16. Stene LC, Honeyman MC, Hoffenberg EJ, Haas JE, Sokol RJ, Emery L, et al. Rotavirus Infection Frequency and Risk of Celiac Disease Autoimmunity in Early Childhood: A Longitudinal Study. Am J Gastroenterol (2006) 101:2333–40. doi: 10.1111/j.1572-0241.2006.00741.x
17. Caminero A, Verdu EF. Celiac Disease: Should We Care About Microbes? Am J Physiol Gastrointest Liver Physiol (2019) 317:G161–70. doi: 10.1152/ajpgi.00099.2019
18. Bouziat R, Biering SB, Kouame E, Sangani KA, Kang S, Ernest JD, et al. Murine Norovirus Infection Induces Th1 Inflammatory Responses to Dietary Antigens. Cell Host Microbe (2018) 24:677–88.e5. doi: 10.1016/j.chom.2018.10.004
19. Bouziat R, Hinterleitner R, Brown JJ, Stencel-Baerenwald JE, Ikizler M, Mayassi T, et al. Reovirus Infection Triggers Inflammatory Responses to Dietary Antigens and Development of Celiac Disease. Science (2017) 356:44–50. doi: 10.1126/science.aah5298
20. Carroccio A, Cavataio F, Montalto G, Paparo F, Troncone R, Iacono G. Treatment of Giardiasis Reverses “Active” Coeliac Disease to “Latent” Coeliac Disease. Eur J Gastroenterol Hepatol (2001) 13:1101–5. doi: 10.1097/00042737-200109000-00018
21. Verdu EF, Mauro M, Bourgeois J, Armstrong D. Clinical Onset of Celiac Disease After an Episode of Campylobacter Jejuni Enteritis. Can J Gastroenterol (2007) 21:453–5. doi: 10.1155/2007/169591
22. Petersen J, Ciacchi L, Tran MT, Loh KL, Kooy-Winkelaar Y, Croft NP, et al. T Cell Receptor Cross-Reactivity Between Gliadin and Bacterial Peptides in Celiac Disease. Nat Struct Mol Biol (2020) 27:49–61. doi: 10.1038/s41594-019-0353-4
23. Abadie V, Kim SM, Lejeune T, Palanski BA, Ernest JD, Tastet O, et al. Il-15, Gluten and HLA-DQ8 Drive Tissue Destruction in Coeliac Disease. Nature (2020) 578:600–4. doi: 10.1038/s41586-020-2003-8
24. Iversen R, Sollid LM. Autoimmunity Provoked by Foreign Antigens. Science (2020) 368:132–3. doi: 10.1126/science.aay3037
25. Setty M, Discepolo V, Abadie V, Kamhawi S, Mayassi T, Kent A, et al. Distinct and Synergistic Contributions of Epithelial Stress and Adaptive Immunity to Functions of Intraepithelial Killer Cells and Active Celiac Disease. Gastroenterology (2015) 149:681–91.e10. doi: 10.1053/j.gastro.2015.05.013
26. Lundin KE, Scott H, Hansen T, Paulsen G, Halstensen TS, Fausa O, et al. Gliadin-Specific, HLA-DQ(alpha 1*0501,Beta 1*0201) Restricted T Cells Isolated From the Small Intestinal Mucosa of Celiac Disease Patients. J Exp Med (1993) 178:187–96. doi: 10.1084/jem.178.1.187
27. Molberg O, Kett K, Scott H, Thorsby E, Sollid LM, Lundin KE, et al. DQ2-Restricted T Cells are Commonly Found in Small Intestinal Biopsies From Coeliac Disease Patients, But Not From Controls. Scand J Immunol (1997) 46:103–9. doi: 10.1046/j.1365-3083.1996.d01-17.x
28. Arentz-Hansen H, Korner R, Molberg O, Quarsten H, Vader W, Kooy YM, et al. The Intestinal T Cell Response to Alpha-Gliadin in Adult Celiac Disease is Focused on a Single Deamidated Glutamine Targeted by Tissue Transglutaminase. J Exp Med (2000) 191:603–12. doi: 10.1084/jem.191.4.603
29. Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, et al. Tissue Transglutaminase Selectively Modifies Gliadin Peptides That are Recognized by Gut-Derived T Cells in Celiac Disease. Nat Med (1998) 4:713–7. doi: 10.1038/nm0698-713
30. van de Wal Y, Kooy Y, van Veelen P, Pena S, Mearin L, Papadopoulos G, et al. Selective Deamidation by Tissue Transglutaminase Strongly Enhances Gliadin-Specific T Cell Reactivity. J Immunol (1998) 161:1585–8.
31. Nilsen EM, Lundin KE, Krajci P, Scott H, Sollid LM, Brandtzaeg P, et al. Restricted T Cells From Coeliac Mucosa Produce Cytokines With Th1 or Th0 Profile Dominated by Interferon Gamma. Gut (1995) 37:766–76. doi: 10.1136/gut.37.6.766
32. Nilsen EM, Jahnsen FL, Lundin KE, Johansen FE, Fausa O, Sollid LM, et al. Gluten Induces an Intestinal Cytokine Response Strongly Dominated by Interferon Gamma in Patients With Celiac Disease. Gastroenterology (1998) 115:551–63. doi: 10.1016/S0016-5085(98)70134-9
33. Bodd M, Raki M, Tollefsen S, Fallang LE, Bergseng E, Lundin KE, et al. Hla-DQ2-restricted Gluten-Reactive T Cells Produce IL-21 But Not IL-17 or IL-22. Mucosal Immunol (2012) 3:594–601. doi: 10.1038/mi.2010.36
34. Tosco A, Salvati VM, Auricchio R, Maglio M, Borrelli M, Coruzzo A, et al. Natural History of Potential Celiac Disease in Children. Clin Gastroenterol Hepatol (2011) 9:320–5. quiz e36. doi: 10.1016/j.cgh.2010.09.006
35. Jabri B, Sollid LM. Tissue-Mediated Control of Immunopathology in Coeliac Disease. Nat Rev Immunol (2009) 9:858–70. doi: 10.1038/nri2670
36. Jabri B, Sollid LM. T Cells in Celiac Disease. J Immunol (2017) 198:3005–14. doi: 10.4049/jimmunol.1601693
37. de Kauwe AL, Chen Z, Anderson RP, Keech CL, Price JD, Wijburg O, et al. Resistance to Celiac Disease in Humanized HLA-DR3-DQ2-transgenic Mice Expressing Specific Anti-Gliadin CD4+ T Cells. J Immunol (2009) 182:7440–50. doi: 10.4049/jimmunol.0900233
38. DePaolo RW, Abadie V, Tang F, Fehlner-Peach H, Hall JA, Wang W, et al. Co-Adjuvant Effects of Retinoic Acid and IL-15 Induce Inflammatory Immunity to Dietary Antigens. Nature (2011) 471:220–4. doi: 10.1038/nature09849
39. Marietta E, Black K, Camilleri M, Krause P, Rogers RS 3rd, David C, et al. A New Model for Dermatitis Herpetiformis That Uses HLA-DQ8 Transgenic NOD Mice. J Clin Invest (2004) 114:1090–7. doi: 10.1172/JCI200421055
40. Lundin KE, Scott H, Fausa O, Thorsby E, Sollid LM. T Cells From the Small Intestinal Mucosa of a DR4, Dq7/Dr4, DQ8 Celiac Disease Patient Preferentially Recognize Gliadin When Presented by DQ8. Hum Immunol (1994) 41:285–91. doi: 10.1016/0198-8859(94)90047-7
41. Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F. Nomenclature and Listing of Celiac Disease Relevant Gluten T-cell Epitopes Restricted by HLA-DQ Molecules. Immunogenetics (2012) 64:455–60. doi: 10.1007/s00251-012-0599-z
42. Shan L, Molberg O, Parrot I, Hausch F, Filiz F, Gray GM, et al. Structural Basis for Gluten Intolerance in Celiac Sprue. Science (2002) 297:2275–9. doi: 10.1126/science.1074129
43. Fesus L, Piacentini M. Transglutaminase 2: An Enigmatic Enzyme With Diverse Functions. Trends Biochem Sci (2002) 27:534–9. doi: 10.1016/S0968-0004(02)02182-5
44. Tollefsen S, Arentz-Hansen H, Fleckenstein B, Molberg O, Raki M, Kwok WW, et al. Hla-DQ2 and -DQ8 Signatures of Gluten T Cell Epitopes in Celiac Disease. J Clin Invest (2006) 116:2226–36. doi: 10.1172/JCI27620
45. Ientile R, Caccamo D, Griffin M. Tissue Transglutaminase and the Stress Response. Amino Acids (2007) 33:385–94. doi: 10.1007/s00726-007-0517-0
46. Siegel M, Strnad P, Watts RE, Choi K, Jabri B, Omary MB, et al. Extracellular Transglutaminase 2 is Catalytically Inactive, But is Transiently Activated Upon Tissue Injury. PLoS One (2008) 3:e1861. doi: 10.1371/journal.pone.0001861
47. Iversen R, Amundsen SF, Kleppa L, du Pre MF, Stamnaes J, Sollid LM. Evidence That Pathogenic Transglutaminase 2 in Celiac Disease Derives From Enterocytes. Gastroenterology (2020) 159:788–90. doi: 10.1053/j.gastro.2020.04.018
48. Sollid LM, Molberg O, McAdam S, Lundin KE. Autoantibodies in Coeliac Disease: Tissue Transglutaminase–Guilt by Association? Gut (1997) 41:851–2. doi: 10.1136/gut.41.6.851
49. Maki M. “C.O.F”. In: Feighery C, editor. Seventh INternational Symposium on Coeliac Disease. Oak Tree Press (1992). p. 246–52.
50. Diraimondo TR, Klock C, Khosla C. Interferon-Gamma Activates Transglutaminase 2 Via a phosphatidylinositol-3-kinase-dependent Pathway: Implications for Celiac Sprue Therapy. J Pharmacol Exp Ther (2012) 341:104–14. doi: 10.1124/jpet.111.187385
51. Jin X, Stamnaes J, Klock C, DiRaimondo TR, Sollid LM, Khosla C. Activation of Extracellular Transglutaminase 2 by Thioredoxin. J Biol Chem (2011) 286:37866–73. doi: 10.1074/jbc.M111.287490
52. Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L, et al. HLA Types in Celiac Disease Patients Not Carrying the DQA1*05-DQB1*02 (DQ2) Heterodimer: Results From the European Genetics Cluster on Celiac Disease. Hum Immunol (2003) 64:469–77. doi: 10.1016/S0198-8859(03)00027-2
53. PLoSki R, Ek J, Thorsby E, Sollid LM. On the HLA-DQ(alpha 1*0501, Beta 1*0201)-Associated Susceptibility in Celiac Disease: A Possible Gene Dosage Effect of DQB1*0201. Tissue Antigens (1993) 41:173–7. doi: 10.1111/j.1399-0039.1993.tb01998.x
54. Koning F. Celiac Disease: Quantity Matters. Semin Immunopathol (2012) 34:541–9. doi: 10.1007/s00281-012-0321-0
55. Vader W, Stepniak D, Kooy Y, Mearin L, Thompson A, van Rood JJ, et al. The HLA-DQ2 Gene Dose Effect in Celiac Disease is Directly Related to the Magnitude and Breadth of Gluten-Specific T Cell Responses. Proc Natl Acad Sci U S A (2003) 100:12390–5. doi: 10.1073/pnas.2135229100
56. Fallang LE, Bergseng E, Hotta K, Berg-Larsen A, Kim CY, Sollid LM. Differences in the Risk of Celiac Disease Associated With HLA-DQ2.5 or HLA-DQ2.2 are Related to Sustained Gluten Antigen Presentation. Nat Immunol (2009) 10:1096–101. doi: 10.1038/ni.1780
57. Ting YT, Dahal-Koirala S, Kim HSK, Qiao SW, Neumann RS, Lundin KEA, et al. A Molecular Basis for the T Cell Response in HLA-DQ2.2 Mediated Celiac Disease. Proc Natl Acad Sci U S A (2020) 117:3063–73. doi: 10.1073/pnas.1914308117
58. Faria AM, Weiner HL. Oral Tolerance. Immunol Rev (2005) 206:232–59. doi: 10.1111/j.0105-2896.2005.00280.x
59. Pabst O, Mowat AM. Oral Tolerance to Food Protein. Mucosal Immunol (2012) 5:232–9. doi: 10.1038/mi.2012.4
60. van Leeuwen MA, Lindenbergh-Kortleve DJ, Raatgeep HC, de Ruiter LF, de Krijger RR, Groeneweg M, et al. Increased Production of interleukin-21, But Not interleukin-17A, in the Small Intestine Characterizes Pediatric Celiac Disease. Mucosal Immunol (2013) 6:1202–13. doi: 10.1038/mi.2013.19
61. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I Interferons in Infectious Disease. Nat Rev Immunol (2015) 15:87–103. doi: 10.1038/nri3787
62. Brinkmann V, Geiger T, Alkan S, Heusser CH. Interferon Alpha Increases the Frequency of Interferon Gamma-Producing Human CD4+ T Cells. J Exp Med (1993) 178:1655–63. doi: 10.1084/jem.178.5.1655
63. Monteleone G, Pender SL, Alstead E, Hauer AC, Lionetti P, McKenzie C, et al. Role of Interferon Alpha in Promoting T Helper Cell Type 1 Responses in the Small Intestine in Coeliac Disease. Gut (2001) 48:425–9. doi: 10.1136/gut.48.3.425
64. Plot L, Amital H. Infectious Associations of Celiac Disease. Autoimmun Rev (2009) 8:316–9. doi: 10.1016/j.autrev.2008.10.001
65. Maiuri L, Ciacci C, Auricchio S, Brown V, Quaratino S, Londei M. Interleukin 15 Mediates Epithelial Changes in Celiac Disease. Gastroenterology (2000) 119:996–1006. doi: 10.1053/gast.2000.18149
66. Mention JJ, Ben Ahmed M, Begue B, Barbe U, Verkarre V, Asnafi V, et al. Interleukin 15: A Key to Disrupted Intraepithelial Lymphocyte Homeostasis and Lymphomagenesis in Celiac Disease. Gastroenterology (2003) 125:730–45. doi: 10.1016/S0016-5085(03)01047-3
67. Ben Ahmed M, Belhadj Hmida N, Moes N, Buyse S, Abdeladhim M, Louzir H, et al. Il-15 Renders Conventional Lymphocytes Resistant to Suppressive Functions of Regulatory T Cells Through Activation of the Phosphatidylinositol 3-Kinase Pathway. J Immunol (2009) 182:6763–70. doi: 10.4049/jimmunol.0801792
68. Benahmed M, Meresse B, Arnulf B, Barbe U, Mention JJ, Verkarre V, et al. Inhibition of TGF-beta Signaling by IL-15: A New Role for IL-15 in the Loss of Immune Homeostasis in Celiac Disease. Gastroenterology (2007) 132:994–1008. doi: 10.1053/j.gastro.2006.12.025
69. Sudhir Kumar Chauhan RBC, Landsverk OJB, Jahnsen J, Horneland R, Yaqub S, Aandahl EM, et al. The Human Small Intestine Comprises Two Subsets of Regulatory Foxp3+ Cd4 T Cells With Very Different Life Span and Functional Properties. bioRxiv [Preprint] (2020). doi: 10.1101/2020.02.12.941369
70. Vorobjova T, Uibo O, Heilman K, Rago T, Honkanen J, Vaarala O, et al. Increased FOXP3 Expression in Small-Bowel Mucosa of Children With Coeliac Disease and Type I Diabetes Mellitus. Scand J Gastroenterol (2009) 44:422–30. doi: 10.1080/00365520802624177
71. Zanzi D, Stefanile R, Santagata S, Iaffaldano L, Iaquinto G, Giardullo N, et al. Il-15 Interferes With Suppressive Activity of Intestinal Regulatory T Cells Expanded in Celiac Disease. Am J Gastroenterol (2011) 106:1308–17. doi: 10.1038/ajg.2011.80
72. Gianfrani C, Levings MK, Sartirana C, Mazzarella G, Barba G, Zanzi D, et al. Gliadin-Specific Type 1 Regulatory T Cells From the Intestinal Mucosa of Treated Celiac Patients Inhibit Pathogenic T Cells. J Immunol (2006) 177:4178–86. doi: 10.4049/jimmunol.177.6.4178
73. Hmida NB, Ben Ahmed M, Moussa A, Rejeb MB, Said Y, Kourda N, et al. Impaired Control of Effector T Cells by Regulatory T Cells: A Clue to Loss of Oral Tolerance and Autoimmunity in Celiac Disease? Am J Gastroenterol (2012) 107:604–11. doi: 10.1038/ajg.2011.397
74. Christophersen A, Risnes LF, Bergseng E, Lundin KE, Sollid LM, Qiao SW. Healthy HLA-DQ2.5+ Subjects Lack Regulatory and Memory T Cells Specific for Immunodominant Gluten Epitopes of Celiac Disease. J Immunol (2016) 196:2819–26. doi: 10.4049/jimmunol.1501152
75. van Bergen J, Mulder CJ, Mearin ML, Koning F. Local Communication Among Mucosal Immune Cells in Patients With Celiac Disease. Gastroenterology (2015) 148:1187–94. doi: 10.1053/j.gastro.2015.01.030
76. Deem RL, Shanahan F, Targan SR. Triggered Human Mucosal T Cells Release Tumour Necrosis Factor-Alpha and Interferon-Gamma Which Kill Human Colonic Epithelial Cells. Clin Exp Immunol (1991) 83:79–84. doi: 10.1111/j.1365-2249.1991.tb05592.x
77. Przemioslo RT, Lundin KE, Sollid LM, Nelufer J, Ciclitira PJ. Histological Changes in Small Bowel Mucosa Induced by Gliadin Sensitive T Lymphocytes can be Blocked by Anti-Interferon Gamma Antibody. Gut (1995) 36:874–9. doi: 10.1136/gut.36.6.874
78. Jabri B, de Serre NP, Cellier C, Evans K, Gache C, Carvalho C, et al. Selective Expansion of Intraepithelial Lymphocytes Expressing the HLA-E-specific Natural Killer Receptor CD94 in Celiac Disease. Gastroenterology (2000) 118:867–79. doi: 10.1016/S0016-5085(00)70173-9
79. Meresse B, Curran SA, Ciszewski C, Orbelyan G, Setty M, Bhagat G, et al. Reprogramming of CTLs Into Natural Killer-Like Cells in Celiac Disease. J Exp Med (2006) 203:1343–55. doi: 10.1084/jem.20060028
80. Ebert EC. Interleukin 21 Up-Regulates Perforin-Mediated Cytotoxic Activity of Human Intra-Epithelial Lymphocytes. Immunology (2009) 127:206–15. doi: 10.1111/j.1365-2567.2008.02941.x
81. Sarra M, Cupi ML, Monteleone I, Franze E, Ronchetti G, Di Sabatino A, et al. Il-15 Positively Regulates IL-21 Production in Celiac Disease Mucosa. Mucosal Immunol (2013) 6:244–55. doi: 10.1038/mi.2012.65
82. Black KE, Murray JA, David CS. Hla-DQ Determines the Response to Exogenous Wheat Proteins: A Model of Gluten Sensitivity in Transgenic Knockout Mice. J Immunol (2002) 169:5595–600. doi: 10.4049/jimmunol.169.10.5595
83. Goel G, Daveson AJM, Hooi CE, Tye-Din JA, Wang S, Szymczak E, et al. Serum Cytokines Elevated During Gluten-Mediated Cytokine Release in Coeliac Disease. Clin Exp Immunol (2020) 199:68–78. doi: 10.1111/cei.13369
84. Goel G, Tye-Din JA, Qiao SW, Russell AK, Mayassi T, Ciszewski C, et al. Cytokine Release and Gastrointestinal Symptoms After Gluten Challenge in Celiac Disease. Sci Adv (2019) 5:eaaw7756. doi: 10.1126/sciadv.aaw7756
85. Goel G, King T, Daveson AJ, Andrews JM, Krishnarajah J, Krause R, et al. Epitope-Specific Immunotherapy Targeting CD4-Positive T Cells in Coeliac Disease: Two Randomised, Double-Blind, Placebo-Controlled Phase 1 Studies. Lancet Gastroenterol Hepatol (2017) 2:479–93. doi: 10.1016/S2468-1253(17)30110-3
86. Tye-Din JA, Daveson AJM, Ee HC, Goel G, MacDougall J, Acaster S, et al. Elevated Serum Interleukin-2 After Gluten Correlates With Symptoms and is a Potential Diagnostic Biomarker for Coeliac Disease. Aliment Pharmacol Ther (2019) 50:901–10. doi: 10.1111/apt.15477
87. Tye-Din JA, Skodje GI, Sarna VK, Dzuris JL, Russell AK, Goel G, et al. Cytokine Release After Gluten Ingestion Differentiates Coeliac Disease From Self-Reported Gluten Sensitivity. United Eur Gastroenterol J (2020) 8:108–18. doi: 10.1177/2050640619874173
88. Barone MV, Gimigliano A, Castoria G, Paolella G, Maurano F, Paparo F, et al. Growth Factor-Like Activity of Gliadin, an Alimentary Protein: Implications for Coeliac Disease. Gut (2007) 56:480–8. doi: 10.1136/gut.2005.086637
89. Cinova J, Palova-Jelinkova L, Smythies LE, Cerna M, Pecharova B, Dvorak M, et al. Gliadin Peptides Activate Blood Monocytes From Patients With Celiac Disease. J Clin Immunol (2007) 27:201–9. doi: 10.1007/s10875-006-9061-z
90. Hue S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, et al. A Direct Role for NKG2D/MICA Interaction in Villous Atrophy During Celiac Disease. Immunity (2004) 21:367–77. doi: 10.1016/j.immuni.2004.06.018
91. Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, et al. Gliadin Induces an Increase in Intestinal Permeability and Zonulin Release by Binding to the Chemokine Receptor CXCR3. Gastroenterology (2008) 135:194–204.e3. doi: 10.1053/j.gastro.2008.03.023
92. Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, et al. Association Between Innate Response to Gliadin and Activation of Pathogenic T Cells in Coeliac Disease. Lancet (2003) 362:30–7. doi: 10.1016/S0140-6736(03)13803-2
93. Nikulina M, Habich C, Flohe SB, Scott FW, Kolb H. Wheat Gluten Causes Dendritic Cell Maturation and Chemokine Secretion. J Immunol (2004) 173:1925–33. doi: 10.4049/jimmunol.173.3.1925
94. Terrazzano G, Sica M, Gianfrani C, Mazzarella G, Maurano F, De Giulio B, et al. Gliadin Regulates the NK-dendritic Cell Cross-Talk by HLA-E Surface Stabilization. J Immunol (2007) 179:372–81. doi: 10.4049/jimmunol.179.1.372
95. Caminero A, McCarville JL, Zevallos VF, Pigrau M, Yu XB, Jury J, et al. Lactobacilli Degrade Wheat Amylase Trypsin Inhibitors to Reduce Intestinal Dysfunction Induced by Immunogenic Wheat Proteins. Gastroenterology (2019) 156:2266–80. doi: 10.1053/j.gastro.2019.02.028
96. Junker Y, Zeissig S, Kim SJ, Barisani D, Wieser H, Leffler DA, et al. Wheat Amylase Trypsin Inhibitors Drive Intestinal Inflammation Via Activation of Toll-Like Receptor 4. J Exp Med (2012) 209:2395–408. doi: 10.1084/jem.20102660
97. de Ritis G, Auricchio S, Jones HW, Lew EJ, Bernardin JE, Kasarda DD. In Vitro (Organ Culture) Studies of the Toxicity of Specific A-gliadin Peptides in Celiac Disease. Gastroenterology (1988) 94:41–9. doi: 10.1016/0016-5085(88)90607-5
98. Maiuri L, Troncone R, Mayer M, Coletta S, Picarelli A, De Vincenzi M, et al. In Vitro Activities of A-gliadin-related Synthetic Peptides: Damaging Effect on the Atrophic Coeliac Mucosa and Activation of Mucosal Immune Response in the Treated Coeliac Mucosa. Scand J Gastroenterol (1996) 31:247–53. doi: 10.3109/00365529609004874
99. Sturgess R, Day P, Ellis HJ, Lundin KE, Gjertsen HA, Kontakou M, et al. Wheat Peptide Challenge in Coeliac Disease. Lancet (1994) 343:758–61. doi: 10.1016/S0140-6736(94)91837-6
100. Barone MV, Troncone R, Auricchio S. Gliadin Peptides as Triggers of the Proliferative and Stress/Innate Immune Response of the Celiac Small Intestinal Mucosa. Int J Mol Sci (2014) 15:20518–37. doi: 10.3390/ijms151120518
101. Barone MV, Zanzi D, Maglio M, Nanayakkara M, Santagata S, Lania G, et al. Gliadin-Mediated Proliferation and Innate Immune Activation in Celiac Disease are Due to Alterations in Vesicular Trafficking. PLoS One (2011) 6:e17039. doi: 10.1371/journal.pone.0017039
102. Clemente MG, De Virgiliis S, Kang JS, Macatagney R, Musu MP, Di Pierro MR, et al. Early Effects of Gliadin on Enterocyte Intracellular Signalling Involved in Intestinal Barrier Function. Gut (2003) 52:218–23. doi: 10.1136/gut.52.2.218
103. Juuti-Uusitalo K, Maki M, Kainulainen H, Isola J, Kaukinen K. Gluten Affects Epithelial Differentiation-Associated Genes in Small Intestinal Mucosa of Coeliac Patients. Clin Exp Immunol (2007) 150:294–305. doi: 10.1111/j.1365-2249.2007.03500.x
104. Nanayakkara M, Lania G, Maglio M, Discepolo V, Sarno M, Gaito A, et al. An Undigested Gliadin Peptide Activates Innate Immunity and Proliferative Signaling in Enterocytes: The Role in Celiac Disease. Am J Clin Nutr (2013) 98:1123–35. doi: 10.3945/ajcn.112.054544
105. Chirdo FG, Auricchio S, Troncone R, Barone MV. The Gliadin p31-43 Peptide: Inducer of Multiple Proinflammatory Effects. Int Rev Cell Mol Biol (2021) 358:165–205. doi: 10.1016/bs.ircmb.2020.10.003
106. Dieterich W, Neurath MF, Zopf Y. Intestinal Ex Vivo Organoid Culture Reveals Altered Programmed Crypt Stem Cells in Patients With Celiac Disease. Sci Rep (2020) 10:3535. doi: 10.1038/s41598-020-60521-5
107. Fernandez-Jimenez N, Castellanos-Rubio A, Plaza-Izurieta L, Irastorza I, Elcoroaristizabal X, Jauregi-Miguel A, et al. Coregulation and Modulation of NFkappaB-related Genes in Celiac Disease: Uncovered Aspects of Gut Mucosal Inflammation. Hum Mol Genet (2014) 23:1298–310. doi: 10.1093/hmg/ddt520
108. Lania G, Nanayakkara M, Maglio M, Auricchio R, Porpora M, Conte M, et al. Constitutive Alterations in Vesicular Trafficking Increase the Sensitivity of Cells From Celiac Disease Patients to Gliadin. Commun Biol (2019) 2:190. doi: 10.1038/s42003-019-0443-1
109. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated Induction by IL15 of a TCR-independent NKG2D Signaling Pathway Converts CTL Into Lymphokine-Activated Killer Cells in Celiac Disease. Immunity (2004) 21:357–66. doi: 10.1016/j.immuni.2004.06.020
110. Bernardo D, Garrote JA, Fernandez-Salazar L, Riestra S, Arranz E. Is Gliadin Really Safe for non-Coeliac Individuals? Production of Interleukin 15 in Biopsy Culture From non-Coeliac Individuals Challenged With Gliadin Peptides. Gut (2007) 56:889–90. doi: 10.1136/gut.2006.118265
111. Abadie V, Jabri B. Il-15: A Central Regulator of Celiac Disease Immunopathology. Immunol Rev (2014) 260:221–34. doi: 10.1111/imr.12191
112. Araya RE, Gomez Castro MF, Carasi P, McCarville JL, Jury J, Mowat AM, et al. Mechanisms of Innate Immune Activation by Gluten Peptide p31-43 in Mice. Am J Physiol Gastrointest Liver Physiol (2016) 311:G40–9. doi: 10.1152/ajpgi.00435.2015
113. Sollid LM, Jabri B. Triggers and Drivers of Autoimmunity: Lessons From Coeliac Disease. Nat Rev Immunol (2013) 13:294–302. doi: 10.1038/nri3407
114. Zhernakova A, Elbers CC, Ferwerda B, Romanos J, Trynka G, Dubois PC, et al. Evolutionary and Functional Analysis of Celiac Risk Loci Reveals SH2B3 as a Protective Factor Against Bacterial Infection. Am J Hum Genet (2010) 86:970–7. doi: 10.1016/j.ajhg.2010.05.004
115. Caminero A, McCarville JL, Galipeau HJ, Deraison C, Bernier SP, Constante M, et al. Duodenal Bacterial Proteolytic Activity Determines Sensitivity to Dietary Antigen Through Protease-Activated Receptor-2. Nat Commun (2019) 10:1198. doi: 10.1038/s41467-019-09037-9
116. Caminero A, Galipeau HJ, McCarville JL, Johnston CW, Bernier SP, Russell AK, et al. Duodenal Bacteria From Patients With Celiac Disease and Healthy Subjects Distinctly Affect Gluten Breakdown and Immunogenicity. Gastroenterology (2016) 151:670–83. doi: 10.1053/j.gastro.2016.06.041
117. D’Argenio V, Casaburi G, Precone V, Pagliuca C, Colicchio R, Sarnataro D, et al. Metagenomics Reveals Dysbiosis and a Potentially Pathogenic N. Flavescens Strain in Duodenum of Adult Celiac Patients. Am J Gastroenterol (2016) 111:879–90. doi: 10.1038/ajg.2016.95
118. Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-Omics of the Gut Microbial Ecosystem in Inflammatory Bowel Diseases. Nature (2019) 569:655–62. doi: 10.1038/s41586-019-1237-9
119. Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A Decrease of the Butyrate-Producing Species Roseburia Hominis and Faecalibacterium Prausnitzii Defines Dysbiosis in Patients With Ulcerative Colitis. Gut (2014) 63:1275–83. doi: 10.1136/gutjnl-2013-304833
120. Scher JU, Ubeda C, Artacho A, Attur M, Isaac S, Reddy SM, et al. Decreased Bacterial Diversity Characterizes the Altered Gut Microbiota in Patients With Psoriatic Arthritis, Resembling Dysbiosis in Inflammatory Bowel Disease. Arthritis Rheumatol (2015) 67:128–39. doi: 10.1002/art.38892
121. Wang W, Chen L, Zhou R, Wang X, Song L, Huang S, et al. Increased Proportions of Bifidobacterium and the Lactobacillus Group and Loss of Butyrate-Producing Bacteria in Inflammatory Bowel Disease. J Clin Microbiol (2014) 52:398–406. doi: 10.1128/JCM.01500-13
122. Rooks MG, Garrett WS. Gut Microbiota, Metabolites and Host Immunity. Nat Rev Immunol (2016) 16:341–52. doi: 10.1038/nri.2016.42
123. Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Specific Duodenal and Faecal Bacterial Groups Associated With Paediatric Coeliac Disease. J Clin Pathol (2009) 62:264–9. doi: 10.1136/jcp.2008.061366
124. De Palma G, Nadal I, Medina M, Donat E, Ribes-Koninckx C, Calabuig M, et al. Intestinal Dysbiosis and Reduced Immunoglobulin-Coated Bacteria Associated With Coeliac Disease in Children. BMC Microbiol (2010) 10:63. doi: 10.1186/1471-2180-10-63
125. Di Cagno R, Rizzello CG, Gagliardi F, Ricciuti P, Ndagijimana M, Francavilla R, et al. Different Fecal Microbiotas and Volatile Organic Compounds in Treated and Untreated Children With Celiac Disease. Appl Environ Microbiol (2009) 75:3963–71. doi: 10.1128/AEM.02793-08
126. Tjellstrom B, Hogberg L, Stenhammar L, Falth-Magnusson K, Magnusson KE, Norin E, et al. Faecal Short-Chain Fatty Acid Pattern in Childhood Coeliac Disease is Normalised After More Than One Year’s Gluten-Free Diet. Microb Ecol Health Dis (2013) 24. doi: 10.3402/mehd.v24i0.20905
127. Tjellstrom B, Stenhammar L, Hogberg L, Falth-Magnusson K, Magnusson KE, Midtvedt T, et al. Gut Microflora Associated Characteristics in Children With Celiac Disease. Am J Gastroenterol (2005) 100:2784–8. doi: 10.1111/j.1572-0241.2005.00313.x
128. Caminero A, Nistal E, Herran AR, Perez-Andres J, Ferrero MA, Vaquero Ayala L, et al. Differences in Gluten Metabolism Among Healthy Volunteers, Coeliac Disease Patients and First-Degree Relatives. Br J Nutr (2015) 114:1157–67. doi: 10.1017/S0007114515002767
129. Niccolai E, Baldi S, Ricci F, Russo E, Nannini G, Menicatti M, et al. Evaluation and Comparison of Short Chain Fatty Acids Composition in Gut Diseases. World J Gastroenterol (2019) 25:5543–58. doi: 10.3748/wjg.v25.i36.5543
130. Nistal E, Caminero A, Herran AR, Arias L, Vivas S, de Morales JM, et al. Differences of Small Intestinal Bacteria Populations in Adults and Children With/Without Celiac Disease: Effect of Age, Gluten Diet, and Disease. Inflammation Bowel Dis (2012) 18:649–56. doi: 10.1002/ibd.21830
131. Olivares M, Neef A, Castillejo G, Palma GD, Varea V, Capilla A, et al. The HLA-DQ2 Genotype Selects for Early Intestinal Microbiota Composition in Infants at High Risk of Developing Coeliac Disease. Gut (2015) 64:406–17. doi: 10.1136/gutjnl-2014-306931
132. Kutlu T, Brousse N, Rambaud C, Le Deist F, Schmitz J, Cerf-Bensussan N. Numbers of T Cell Receptor (TCR) Alpha Beta+ But Not of TcR Gamma Delta+ Intraepithelial Lymphocytes Correlate With the Grade of Villous Atrophy in Coeliac Patients on a Long Term Normal Diet. Gut (1993) 34:208–14. doi: 10.1136/gut.34.2.208
133. Jabri B, Selby JM, Negulescu H, Lee L, Roberts AI, Beavis A, et al. TCR Specificity Dictates CD94/NKG2A Expression by Human CTL. Immunity (2002) 17:487–99. doi: 10.1016/S1074-7613(02)00427-2
134. Roberts AI, Lee L, Schwarz E, Groh V, Spies T, Ebert EC, et al. NKG2D Receptors Induced by IL-15 Costimulate CD28-negative Effector CTL in the Tissue Microenvironment. J Immunol (2001) 167:5527–30. doi: 10.4049/jimmunol.167.10.5527
135. Jabri B, Abadie V. Il-15 Functions as a Danger Signal to Regulate Tissue-Resident T Cells and Tissue Destruction. Nat Rev Immunol (2015) 15:771–83. doi: 10.1038/nri3919
136. Uhrberg M, Valiante NM, Young NT, Lanier LL, Phillips JH, Parham P. The Repertoire of Killer Cell Ig-like Receptor and CD94:NKG2A Receptors in T Cells: Clones Sharing Identical Alpha Beta TCR Rearrangement Express Highly Diverse Killer Cell Ig-like Receptor Patterns. J Immunol (2001) 166:3923–32. doi: 10.4049/jimmunol.166.6.3923
137. Liu RB, Engels B, Schreiber K, Ciszewski C, Schietinger A, Schreiber H, et al. Il-15 in Tumor Microenvironment Causes Rejection of Large Established Tumors by T Cells in a Noncognate T Cell Receptor-Dependent Manner. Proc Natl Acad Sci USA (2013) 110:8158–63. doi: 10.1073/pnas.1301022110
138. Mayassi T, Ladell K, Gudjonson H, McLaren JE, Shaw DG, Tran MT, et al. Chronic Inflammation Permanently Reshapes Tissue-Resident Immunity in Celiac Disease. Cell (2019) 176:967–81.e19. doi: 10.1016/j.cell.2018.12.039
139. Cellier C, Delabesse E, Helmer C, Patey N, Matuchansky C, Jabri B, et al. Refractory Sprue, Coeliac Disease, and Enteropathy-Associated T-cell Lymphoma. French Coeliac Disease Study Group. Lancet (2000) 356:203–8. doi: 10.1016/S0140-6736(00)02481-8
140. Cellier C, Patey N, Mauvieux L, Jabri B, Delabesse E, Cervoni JP, et al. Abnormal Intestinal Intraepithelial Lymphocytes in Refractory Sprue. Gastroenterology (1998) 114:471–81. doi: 10.1016/S0016-5085(98)70530-X
141. Ettersperger J, Montcuquet N, Malamut G, Guegan N, Lopez-Lastra S, Gayraud S, et al. Interleukin-15-Dependent T-Cell-like Innate Intraepithelial Lymphocytes Develop in the Intestine and Transform Into Lymphomas in Celiac Disease. Immunity (2016) 45:610–25. doi: 10.1016/j.immuni.2016.07.018
142. Malamut G, El Machhour R, Montcuquet N, Martin-Lanneree S, Dusanter-Fourt I, Verkarre V, et al. Il-15 Triggers an Antiapoptotic Pathway in Human Intraepithelial Lymphocytes That is a Potential New Target in Celiac Disease-Associated Inflammation and Lymphomagenesis. J Clin Invest (2010) 120:2131–43. doi: 10.1172/JCI41344
143. Yokoyama S, Watanabe N, Sato N, Perera PY, Filkoski L, Tanaka T, et al. Antibody-Mediated Blockade of IL-15 Reverses the Autoimmune Intestinal Damage in Transgenic Mice That Overexpress IL-15 in Enterocytes. Proc Natl Acad Sci USA (2009) 106:15849–54. doi: 10.1073/pnas.0908834106
144. Cellier C, Bouma G, van Gils T, Khater S, Malamut G, Crespo L, et al. Safety and Efficacy of AMG 714 in Patients With Type 2 Refractory Coeliac Disease: A Phase 2a, Randomised, Double-Blind, Placebo-Controlled, Parallel-Group Study. Lancet Gastroenterol Hepatol (2019) 4:960–70. doi: 10.1016/S2468-1253(19)30265-1
145. Lahdeaho ML, Scheinin M, Vuotikka P, Taavela J, Popp A, Laukkarinen J, et al. Safety and Efficacy of AMG 714 in Adults With Coeliac Disease Exposed to Gluten Challenge: A Phase 2a, Randomised, Double-Blind, Placebo-Controlled Study. Lancet Gastroenterol Hepatol (2019) 4:948–59. doi: 10.1016/S2468-1253(19)30264-X
146. Korneychuk N, Ramiro-Puig E, Ettersperger J, Schulthess J, Montcuquet N, Kiyono H, et al. Interleukin 15 and CD4(+) T Cells Cooperate to Promote Small Intestinal Enteropathy in Response to Dietary Antigen. Gastroenterology (2014) 146:1017–27. doi: 10.1053/j.gastro.2013.12.023
147. Lejeune T, Meyer C, Abadie V. B Lymphocytes Contribute to Celiac Disease Pathogenesis. Gastroenterology (2021) 160(7):2608–10.e4. doi: 10.1053/j.gastro.2021.02.063
148. Maglio M, Troncone R. Intestinal Anti-tissue Transglutaminase2 Autoantibodies: Pathogenic and Clinical Implications for Celiac Disease. Front Nutr (2020) 7:73. doi: 10.3389/fnut.2020.00073
149. Hoydahl LS, Richter L, Frick R, Snir O, Gunnarsen KS, Landsverk OJB, et al. Plasma Cells are the Most Abundant Gluten Peptide MHC-expressing Cells in Inflamed Intestinal Tissues From Patients With Celiac Disease. Gastroenterology (2019) 156:1428–39.e10. doi: 10.1053/j.gastro.2018.12.013
150. Di Niro R, Mesin L, Zheng NY, Stamnaes J, Morrissey M, Lee JH, et al. High Abundance of Plasma Cells Secreting Transglutaminase 2-Specific IgA Autoantibodies With Limited Somatic Hypermutation in Celiac Disease Intestinal Lesions. Nat Med (2012) 18:441–5. doi: 10.1038/nm.2656
151. Iversen R, Roy B, Stamnaes J, Hoydahl LS, Hnida K, Neumann RS, et al. Efficient T Cell-B Cell Collaboration Guides Autoantibody Epitope Bias and Onset of Celiac Disease. Proc Natl Acad Sci USA (2019) 116:15134–9. doi: 10.1073/pnas.1901561116
152. Du Pre MF, Kozijn AE, van Berkel LA, ter Borg MN, Lindenbergh-Kortleve D, Jensen LT, et al. Tolerance to Ingested Deamidated Gliadin in Mice is Maintained by Splenic, Type 1 Regulatory T Cells. Gastroenterology (2011) 141:610–20. 620 e1-2. doi: 10.1053/j.gastro.2011.04.048
Keywords: coeliac disease, villous atrophy, gluten, transglutaminase 2, HLA-DQ2/8, T lymphocytes
Citation: Voisine J and Abadie V (2021) Interplay Between Gluten, HLA, Innate and Adaptive Immunity Orchestrates the Development of Coeliac Disease. Front. Immunol. 12:674313. doi: 10.3389/fimmu.2021.674313
Received: 01 March 2021; Accepted: 18 May 2021;
Published: 02 June 2021.
Edited by:
Robert Paul Anderson, The Wesley-St Andrew’s Research Institute, AustraliaReviewed by:
Jason Allan Tye-Din, Walter and Eliza Hall Institute of Medical Research, AustraliaCopyright © 2021 Voisine and Abadie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valérie Abadie, dmFiYWRpZUBtZWRpY2luZS5ic2QudWNoaWNhZ28uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.