 Francesco Saettini1*
Francesco Saettini1* Grazia Fazio2Daniele Moratto3Marta Galbiati2Nicola Zucchini4Davide Ippolito5Marco Emilio Dinelli6
Grazia Fazio2Daniele Moratto3Marta Galbiati2Nicola Zucchini4Davide Ippolito5Marco Emilio Dinelli6 Luisa Imberti7
Luisa Imberti7 Mario Mauri8Maria Luisa Melzi9Sonia Bonanomi1
Mario Mauri8Maria Luisa Melzi9Sonia Bonanomi1 Alessio Gerussi10,11
Alessio Gerussi10,11 Marinella Pinelli12Chiara Barisani12Cristina Bugarin2Marco Chiarini3
Marinella Pinelli12Chiara Barisani12Cristina Bugarin2Marco Chiarini3 Mauro Giacomelli12
Mauro Giacomelli12 Rocco Piazza8
Rocco Piazza8 Giovanni Cazzaniga2,8
Giovanni Cazzaniga2,8 Pietro Invernizzi10,11
Pietro Invernizzi10,11 Silvia Clara Giliani12
Silvia Clara Giliani12 Raffaele Badolato13
Raffaele Badolato13 Andrea Biondi1,2
Andrea Biondi1,2- 1Pediatric Hematology Outpatient Clinic, Department of Pediatrics, Fondazione MBBM, Monza, Italy
- 2Centro Ricerca Tettamanti, University of Milano Bicocca, Monza, Italy
- 3Flow Cytometry Laboratory, Diagnostic Department, ASST Spedali Civili, Brescia, Italy
- 4Division of Pathology, San Gerardo Hospital, ASST Monza, Monza, Italy
- 5Department of Diagnostic Radiology, San Gerardo Hospital, Monza, Italy
- 6Endoscopy Unit, San Gerardo Hospital, ASST, Monza, Italy
- 7Centro di Ricerca Emato-oncologica AIL (CREA), ASST Spedali Civili, Brescia, Italy
- 8Department of Medicine and Surgery, University of Milano Bicocca and San Gerardo Hospital, Monza, Italy
- 9Department of Pediatrics, Fondazione MBBM, Monza, Italy
- 10Division of Gastroenterology, Centre for Autoimmune Liver Diseases, Department of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy
- 11European Reference Network on Hepatological Diseases (ERN RARE-LIVER), San Gerardo Hospital, Monza, Italy
- 12Cytogenetic and Medical Genetic Unit, Department of Molecular and Translational medicine, A. Nocivelli Institute for Molecular Medicine, University of Brescia, Spedali Civili, Brescia, Italy
- 13Department of Clinical and Experimental Sciences, Pediatrics Clinic and A. Nocivelli Institute for Molecular Medicine A, University of Brescia, ASST-Spedali Civili, Brescia, Italy
DOCK8 deficiency is a combined immunodeficiency due to biallelic variants in dedicator of cytokinesis 8 (DOCK8) gene. The disease has a wide clinical spectrum encompassing recurrent infections (candidiasis, viral and bacterial infections), virally driven malignancies and immune dysregulatory features, including autoimmune (cytopenia and vasculitis) as well as allergic disorders (eczema, asthma, and food allergy). Hypomorphic function and somatic reversion of DOCK8 has been reported to result in incomplete phenotype without IgE overproduction. Here we describe a case of DOCK8 deficiency in a 8-year-old Caucasian girl. The patient’s disease was initially classified as autoimmune thrombocytopenia, which then evolved toward a combined immunodeficiency phenotype with recurrent infections, persistent EBV infection and lymphoproliferation. Two novel variants (one deletion and one premature stop codon) were characterized, resulting in markedly reduced, but not absent, DOCK8 expression. Somatic reversion of the DOCK8 deletion was identified in T cells. Hypomorphic function and somatic reversion were associated with restricted T cell repertoire, decreased STAT5 phosphorylation and impaired immune synapse functioning in T cells. Although the patient presented with incomplete phenotype (absence of markedly increase IgE and eosinophil count), sclerosing cholangitis was incidentally detected, thus indicating that hypomorphic function and somatic reversion of DOCK8 may delay disease progression but do not necessarily prevent from severe complications.
Introduction
The dedicator of cytokinesis 8 (DOCK8) protein exerts critical roles in both humoral and cellular immune responses. It mediates cell differentiation, survival, adhesion and migration, promoting the immune synapse (IS) by active cytoskeletal response (1–4).
DOCK8 deficiency was described for the first time in 2009 in patients with biallelic loss-of-function variants (1, 2). Patients with DOCK8 deficiency are prone to recurrent infections, including candidiasis, viral and bacterial infections, and virally driven malignancies. DOCK8 deficiency has been associated with a number of immune dysregulatory features, including autoimmune disorders (cytopenia and vasculitis), as well as allergic disorders (eczema, asthma, and food allergy). Blood tests reveal T-cell lymphopenia with hypereosinophilia. Serum immunoglobulin (Ig) levels usually show hyperIgE with low IgM. The current consensus is to perform allogenic hematopoietic stem cell transplantation (HSCT) as early as possible, due to high morbidity and premature mortality (5, 6). Patients with less severe phenotype have been subsequently reported (7, 8). In these individuals, hypomorphic function and somatic reversion of the mutated DOCK8 allele have been identified and associated with a milder phenotype. Yet, the long-term prognosis of this subgroup of DOCK8-deficient patients does not seem to be ameliorated (7, 8) and the only curative treatment remains HSCT (5).
In this report, we describe a 8-year-old girl with two novel DOCK8 variants (one nonsense variant and one deletion). Hypomorphic function and somatic reversion of DOCK8 resulted in an incomplete phenotype and sclerosing cholangitis (SC) was incidentally diagnosed.
Case Description
The index patient was a 8-year-old female patient born from non-consanguineous parents (Figure 1A). Eczema started at the age of 6 months and it was treated with topical steroids and emollients. Recurrent skin bacterial infections occurred, but they responded well to topical antibiotics. At the age of two, she had two episodes of parotitis treated with intravenous antibiotics. The second one was complicated by cellulitis. Vulvovaginal candidiasis occurred once, along with abscess of the outer labia. At the age of three, she had thrombocytopenia in the setting of active EBV infection (9). Platelet count did not increase with high doses of intravenous immunoglobulin (IVIg), requiring oral steroids. Thereafter, EBV DNA remained detectable with viral load ranging from 2 x 102 to 5 x 103 copies/ml. During the follow-up, persistent lymphadenopathies and lymphopenia were detected. Hypereosinophilia was never observed. IgE, initially within normal range, mildly increased by the age of 6 (Table 1). Upper and lower respiratory tract infections were frequent. The most common bacterium in sputum was Haemophilus Influenzae. Bacteria were always sensitive to commonly-used antibiotics. Antimicrobial, antiviral, and antifungal prophylaxis and monthly IVIg infusion were started with good clinical improvement. Lung computed tomography performed at the age of six showed diffuse bronchial wall thickening with tree-in-bud opacities and mediastinal adenopathies. Incidentally, marked enlargement of the biliary tree was noticed, which was absent at a previous abdominal ultrasound. Liver test were normal (including alanine transaminase, aspartate transaminasase, gamma-glutamyltransferase, prothrombin time, and total bilirubin) except for mildly increased bile acids. Testing for infectious agents, including Rotavirus, Adenovirus, Clostridium Difficile, Giardia, and Cryptosporidium was negative. With respect to Cryptosporidium, fecal samples were repeatedly tested by means of microscopic analysis and chromatographic immunoassay for the detection of Cryptosporidium antigens. Marked dilation of intrahepatic bile ducts (more pronounced at the right system) and dilation of the common bile duct (up to 7 mm and without any mass forming lesion in the lumen) were then confirmed by means of abdominal ultrasound and magnetic resonance imaging (Figure 2A). Liver biopsy showed one dilated duct with “onion-skin” type of periductal fibrosis and portal and acinar inflammation consistent with SC (Figure 2B). Fecal calprotectin was mildly increased (185 μg/mg; normal up to 50 μg/mg). Upper gastroenterology endoscopy showed enlarged duodenal papilla. Histology revealed moderate chronic gastritis and mild chronic inflammation of the duodenal mucosa. Colonoscopy showed mild mucosal inflammation with mixed cell infiltrate, including macrophages, eosinophils, and plasma cells. Considering the good clinical condition, endoscopic retrograde cholangiopancreatography with endoscopic sphincterotomy of the major duodenal papilla was successfully performed with disposable duodenoscope (10) and no immunosuppressive treatment was initiated. Patient remained well, without any abdominal symptom.
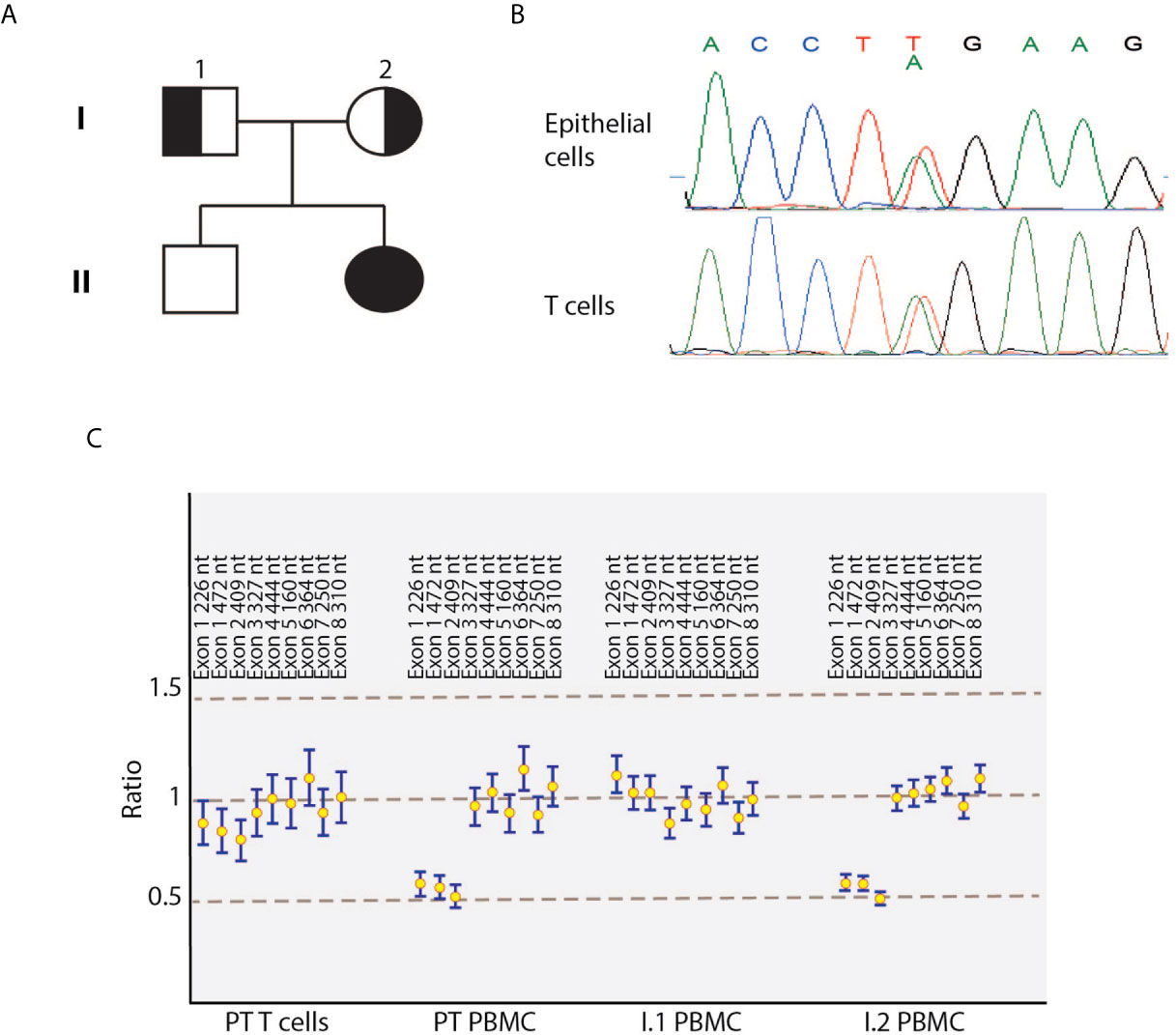
Figure 1 Identification of compound heterozygous DOCK8 variants in the index patient. (A) Pedigree of the family. Solid symbols indicate affected patient; half solid symbols, heterozygous persons; circles, female family members; square, male family members. (B) Electropherograms showing the novel c.T824A mutation in DOCK8 patient’s epithelial cells and T-cell blasts. (C) Reversion of the deletion in patient’s T cells blasts detected by multiplex ligation-dependent probe amplification. PBMC, peripheral blood mononuclear cells; PT, patient.
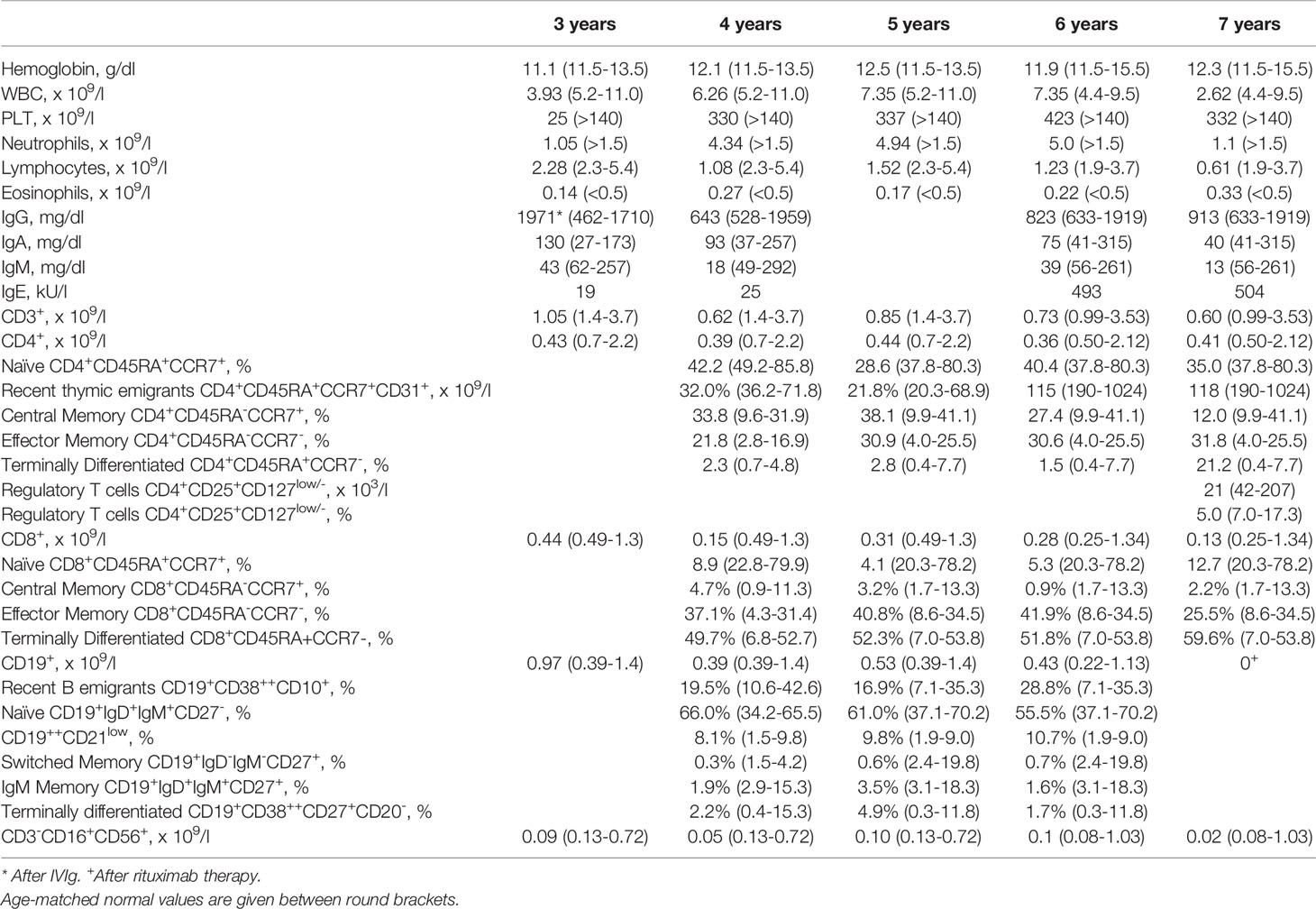
Table 1 Immunological characteristics of the patient.
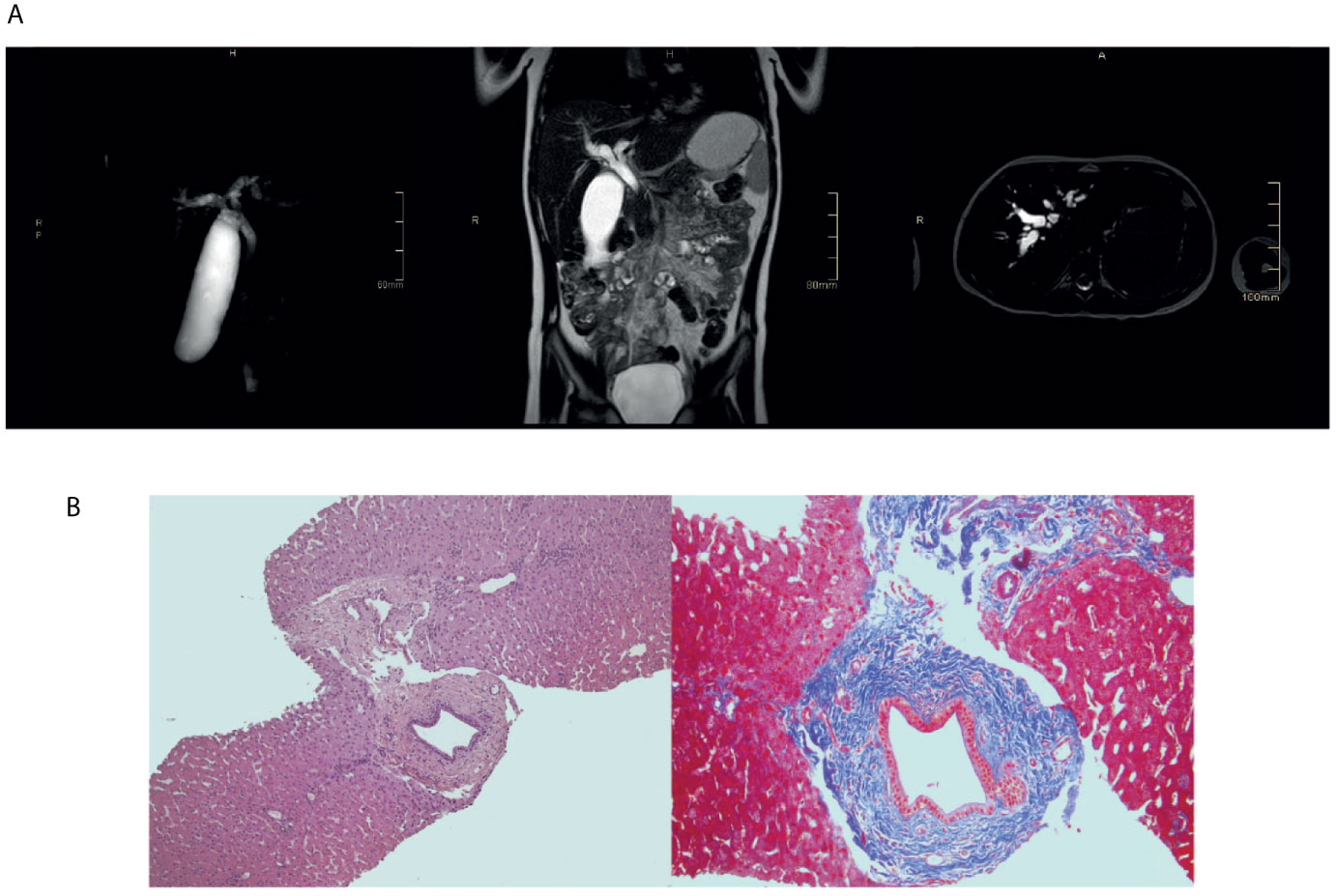
Figure 2 Sclerosing cholangitis in the index patient. (A) Upper abdomen magnetic resonance showing diffuse and marked enlargement of intra- and extrahepatic bile ducts. (B) Liver biopsy showing one dilated duct with “onion-skin” type of periductal fibrosis.
During the follow-up, neutropenia was detected with occasionally normal neutrophil count (Table 1). Analyses of main lymphocyte population consistently displayed reduced T cell counts compared to age-matched healthy controls (HC), while evaluation of lymphocyte subsets revealed a moderate reduction of naïve T cells and memory B cells. At 7 years of age, regulatory T cells (Treg), defined as CD4+CD25+CD127low/- cells, were analyzed, resulting in a percentage that was below the detection limit of age-matched HC and in a significant reduction of their absolute count (below 2SD from mean; Table 1). The release of new lymphocytes by the thymus, either quantified by means of T-cell receptor (TR) excision circles (TRECs) or recent thymic emigrants’ counts, was reduced in comparison to that of age-matched HC. Flow cytometric analysis of T lymphocyte proliferation by incorporation of carboxyfluorescein diacetate succinimidyl ester was assessed, displaying a markedly reduced proliferation ability at the level of CD8+ cells either upon stimulation with phytoemoagglutinin (PHA) or with anti-CD3 monoclonal antibody. Spectratyping analysis of T-cell receptor variable beta (TRBV) chain subgroups revealed a restricted T-cell repertoire. The production of new B-lymphocytes from the bone marrow, measured by means of K-deleting recombination excision circles (KRECs), was comparable to that HC. Evaluation of serum immunoglobulins indicated reduced IgM and moderately elevated IgE (Table 1).
Considering the clinical manifestations and the immunology evaluation, combined immunodeficiency was suspected, and molecular analysis performed. At the age of seven, using targeted next generation sequencing (NGS) and multiplex ligation-dependent probe amplification (MLPA), we identified distinct biallelic variants in DOCK8, which were not present in public databases (gnomAD, ESP, and 1000 Genomes) and were exclusive to this family in our internal databases. NGS revealed a heterozygous NM_203447:c.T824A variant in exon 7 of the DOCK8 gene, resulting in a premature stop codon (p.L275*), which was confirmed by Sanger sequencing (Figure 1B). The father was heterozygous carrier of the variant. Single nucleotide polymorphism (SNP) array and MLPA revealed the maternally inherited deletion of exons 1 to 3 (Figure 1C). Both variants are supposed for their nature to lead to a null DOCK8 phenotype being pathogenetic (class 5). Flow cytometric and western blot analysis showed markedly reduced, but not absent, DOCK8 expression in patient’s interleukin-2 (IL2)-PHA expanded T-cells, CD3+CD4+ and CD3+CD8+ while DOCK8 expression was absent in monocytes. These results collectively suggested hypomorphic residual function in T-cells (Figures 3A, B). Sanger sequencing analysis performed on epithelial cells as well as on peripheral T-cells confirmed the presence of the heterozygous status for the T824A variant in all the lineages, excluding the possibility of somatic reversion for this variant, while a somatic reversion of the deletion was detected by MLPA analysis in patient’s T-cells blasts (Figures 1B, C). STAT5 phosphorylation in T-cells (both CD3+CD4+ and CD3+CD8+) was reduced either after anti-CD3/CD28 or IL-2 stimulation (Figure 3C). To examine the role of the novel variants in the IS, CD8+ were stimulated with anti-CD3 and anti-CD28 monoclonal antibodies to mimic the interaction between CD8+ cells and dendritic cells. Patient’s cells exhibited a significantly reduced actin polymerization in response to stimuli, whose distribution pattern was similar to the one displayed by LFA-1 (Figure 3D).
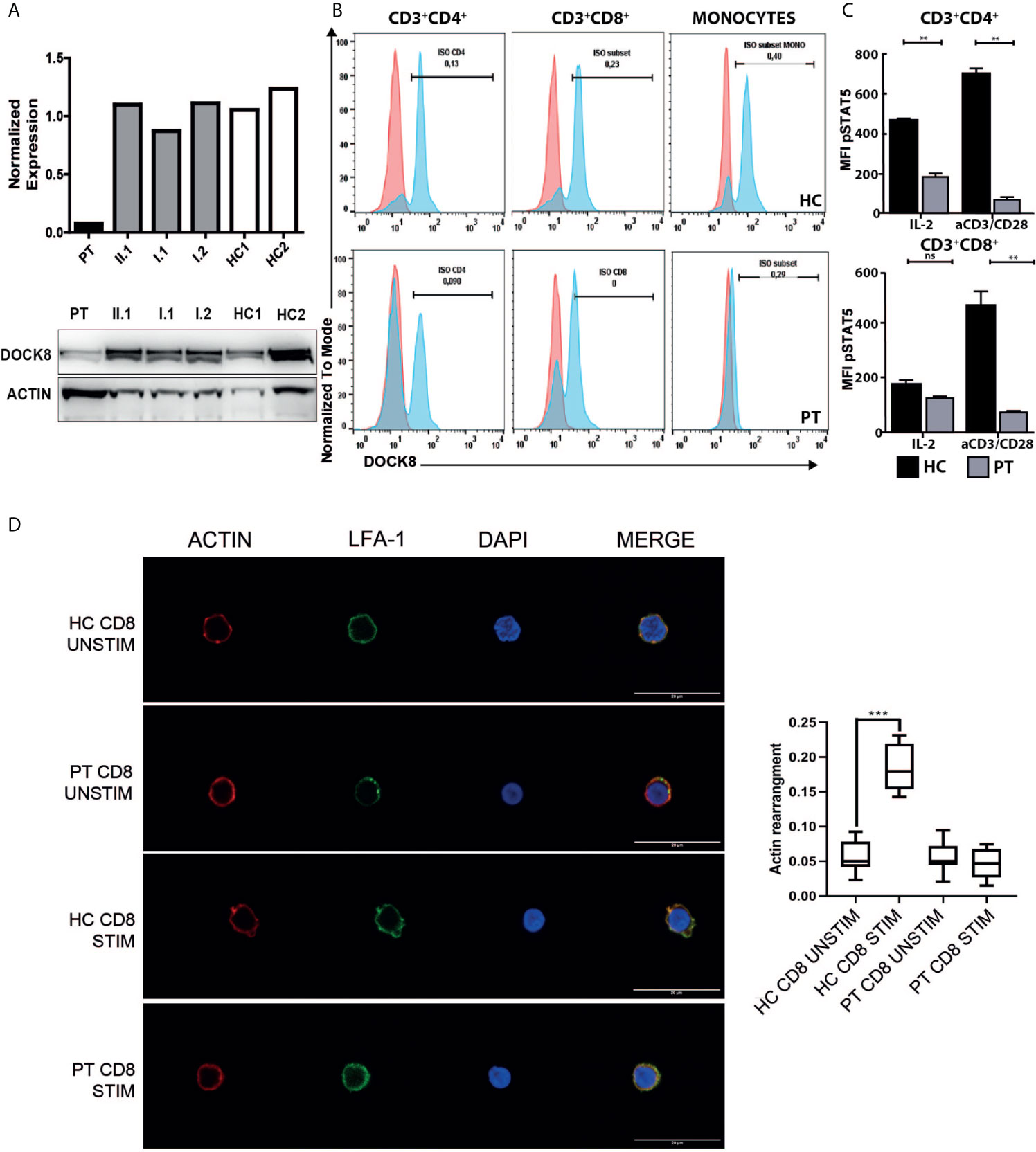
Figure 3 Functional characterization of the DOCK8 variants. (A) Evaluation of DOCK8 levels in IL2-PHA expanded T-cell of the index patient, family members and two healthy controls (HC1 and HC2) by means of flow cytometry (upper panel; one representative experiment of 2 independent experiments) and western blot (lower panel; one representative experiment of 2 independent experiments). (B) Flow cytometric analysis of DOCK8 expression in peripheral T-cells and monocytes of the patient and a healthy control, showing hypomorphic DOCK8 expression in CD3+CD4+ and CD3+CD8+. (representative experiment) (C) Reduction of STAT5 phosphorylation in patient’s peripheral CD4+ and CD8+ T-cells at baseline and following stimulation with IL2 or CD3/CD28 monoclonal antibodies. (one representative experiment performed in duplicates) (D) Representative images of Actin (Red), LFA-1 (green) and DAPI (Blue) staining on unstimulated or CD3/CD28 stimulated CD8+ cells from the index patient and a healthy donor (representative experiment). Scale bar: 20um. In the right panel, box plots show the quantification of actin signal in analyzed samples. (**p < 0.01; ***p < 0.001). HC, healthy control; PT, patient; Stim, stimulated; Unstim, unstimulated. NS, not significant.
Haploidentical related donor HSCT was performed at 7 years and 6 months of age. Due to donor specific anti-human leukocyte antigen antibodies, desensitization protocol included a single dose of 375 mg/m2 rituximab and plasma exchange. Conditioning regimen included 2 days of low dose cyclofosmamide, 5 days of fludarabin, 3 days of busulfan and 200 cGy total body irradiation. Graf-versus-host disease (GVHD) prophylaxis consisted of cyclosporine (from day -1, ongoing), cyclofosfamide (on day +3 and +4) and mycophenolate (from day +1 to day +26). Neutrophil engraftment (0.5 x 109/l) occurred at day +17 (1.0 x 109/l at day +19) and platelet engraftment (20 x 109/l) occurred on day +18 (50 x 109/l at day +19). Complete donor chimerism was detected on day +25 and thereafter checked every 14 days. Main complication after HSCT was hyperacute stage 2 grade 1 skin GVHD on day +16, which required additional therapy with methylprednisolone at 1 mg/kg/day and resolved on day +18. CMV reactivation on day +34 was successfully treated with valganciclovir. The patient is now alive and well 122 days after HSCT, with no signs of GVHD. Chimerism will be checked every 14 days until day +180 and afterward once a month until 1 year after HSCT.
Discussion
The variant spectrum of inborn errors of immunity (IEI)/primary immunodeficiencies (PID) comprises unconventional genetic etiologies. Large deletions have been frequently reported in DOCK8-deficient patients (11), thus requiring including copy number variants (CNV) analysis in patients with complex phenotypes (12). If whole exome/whole genome sequencing with CNV analysis cannot be performed, a combination of NGS, SNP array, or MLPA should be used.
The low-level DOCK8 protein expression in patient’s T cells suggests either residual protein synthesis by means of alternative splicing (13) or somatic reversion (7, 8). Somatic reversions have been observed in several immunodeficiencies (i.e. Wiskott Aldrich syndrome) (14). In DOCK8 deficiency this phenomenon is enriched in T cells, particularly antigen-experienced T cells. Chronic antigenic stimulation, especially viral, contributes to the expansion of such subgroup of T cells. It has been proposed that reversion may confer a selective advantage and may correct Th2 skewing due to abnormal cytokine production thereby modulating disease’s phenotype (7). This phenomenon, nonetheless, does not ameliorate the defective formation of the IS in CD8+ T lymphocytes (4). Although hypormorphic DOCK8-deficient patients may present with an incomplete phenotype (i.e. lack of markedly elevated IgE levels, hypereosinophilia, and/or severe viral skin infections), patients still experienced disease progression and severe or fatal complications (7, 8). Interestingly, while preparing this manuscript, clinical improvement over time has been described in 3 DOCK8-deficient patients (15). Authors demonstrated that somatic reversion of one of the mutated alleles restored lymphocyte survival, differentiation and function. We did not assess revertant T and B subsets, but we show that the qualitative defect in CD4+ and CD8+ was not reverted. In our patient, in vitro stimulation with anti-CD3 or PHA, T-cell repertoire, STAT5 phosphorylation and IS polarization were impaired (4, 16). In keeping with this, the frequency of infectious episodes decreased only when prophylaxes and IVIg had been started. Therefore, despite our patient showed spontaneous repair, we decided to perform HSCT.
As noted in the larger DOCK8 cohorts so-far described (3, 4), some of our patient’s manifestations could overlap with other IEI presenting with hypogammaglobulinemia and lymphoproliferation (17), thrombocytopenia (9), lymphopenia with low but detectably persistent EBV copies (18), and occasionally normal neutrophils count (19). Some forms of PID/IEI have a high-incidence of liver disease and SC, i.e. common variable immunodeficiency and hyperIgM syndrome. Up to 70% of them are colonized with Cryptosporidium parvum, and this has been associated with significant chronic liver disease (20, 21). In case of abnormal liver function tests results, Cryptosporidium should be always ruled out in DOCK8-deficient patients, as it is a under-recognized cause of liver disease. Ideally, Cryptosporidium should be investigated by means of PCR, as it has been proven to be more sensitive than microscopic analysis and chromatographic immunoassay (22). In a large cohort of DOCK8-deficient patients, SC has been reported in 5% of patients but authors described no autoimmune gastrointestinal manifestations (6). Subsequently, IPEX-like phenotype and active colitis have been associated with DOCK8 deficiency (13, 23). Autoimmune phenomena, in our case SC, have been related to defective number and function of Treg, depending on DOCK8 deficiency (15, 23).
With this report, we suggest that SC can be incidentally detected even in asymptomatic patients with apparently mild phenotype due to hypormorphic DOCK8 function. Regular biochemical and radiological follow up should be scheduled even in patients with no history of liver disease. These findings can lead to personalized follow-up and to early intervention in case of evolution of the manifestations. Larger studies focusing on hypomorphic DOCK8-deficient patients should investigate the immunological differences in individuals with clinical improvement over time compared to patients who still experience severe complications.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
Written informed consent was obtained from the legal guardian for the publication of any potentially identifiable images or data included in this article.
Author Contributions
FS and RB contributed to conception and design of the study. FS wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Acknowledgments
AG and PI are members of the European Reference Network on Hepatological Diseases (ERN RARE LIVER). These authors thank AMAF Monza ONLUS and AIRCS for the unrestricted research funding. We also acknowledge that this research was partially supported by the Italian Ministry of University and Research (MIUR) - Department of Excellence project PREMIA (PREcision MedIcine Approach: bringing biomarker research to clinic).
Supplementary Material
The Supplementary Material for this article can be found online at:https://www.frontiersin.org/articles/10.3389/fimmu.2021.673487/full#supplementary-material
References
1. Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med (2009) 361:2046–55. doi: 10.1056/NEJMoa0905506
2. Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol (2009) 124:1289–302. doi: 10.1016/j.jaci.2009.10.038
3. Janssen E, Morbach H, Ullas S, Bannock JM, Massad C, Menard L, et al. Dedicator of cytokinesis 8-deficient patients have a breakdown in peripheral B-cell tolerance and defective regulatory T cells. J Allergy Clin Immunol (2014) 134(6):1365–74. doi: 10.1016/j.jaci.2014.07.042
4. Randall KL, Chan SS, Ma CS, Fung I, Mei Y, Yabas M, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. J Exp Med (2011) 208(11):2305–20. doi: 10.1084/jem.20110345
5. Engelhardt KR, Gertz ME, Keles S, Schäffer AA, Sigmund EC, Glocker C, et al. The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol (2015) 136(2):402–12. doi: 10.1016/j.jaci.2014.12.1945
6. Aydin SE, Sebnem Kilic S, Aytekin C, Kumar A, Porras O, Kainulainen L, et al. DOCK8 Deficiency: Clinical and Immunological Phenotype and Treatment Options - a Review of 136 Patients. J Clin Immunol (2015) 35:189–98. doi: 10.1007/s10875-014-0126-0
7. Jing H, Zhang Q, Zhang Y, Hill BJ, Dove CG, Gelfand EW, et al. Somatic Reversion in Dedicator of Cytokinesis 8 Immunodeficiency Modulates Disease Phenotype. J Allergy Clin Immunol (2014) 133(6):1667–75. doi: 10.1016/j.jaci.2014.03.025
8. Kienzler AK, van Schouwenburg PA, Taylor J, Marwah I, Sharma RU, Noakes C, et al. Hypomorphic function and somatic reversion of DOCK8 cause combined immunodeficiency without hyper-IgE. Clin Immunol (2016) 163:17–21. doi: 10.1016/j.clim.2015.12.003
9. Saettini F, Cattoni A, Redaelli M, Silvestri D, Ferrari GM, Biondi A, et al. Primary immunodeficiencies, autoimmune hyperthyroidism, coeliac disease and systemic lupus erythematosus in childhood immune thrombocytopenia. Acta Paediatr (2021) 110(2):643–51. doi: 10.1111/apa.15593
10. Costamagna G, Bove V, Ruggiero A, Rovelli A, Tringali A. Pediatric ERCP with a single-use duodenoscope in animmunocompromised child. VideoGIE (2021). doi: 10.1016/j.vgie.2020.12.004
11. Su HC. DOCK8 (Dedicator of cytokinesis 8) deficiency. Curr Opin Allergy Clin Immunol (2010) 10(6):515–20. doi: 10.1097/ACI.0b013e32833fd718
12. Saettini F, Poli C, Vengoechea J, Bonanomi S, Orellana JC, Fazio G, et al. Absent B cells, agammaglobulinemia, and hypertrophic cardiomyopathy in folliculin-interacting protein 1 deficiency. Blood (2021) 137(4):493–9. doi: 10.1182/blood.2020006441
13. Dinwiddie DL, Kingsmore SF, Caracciolo S, Rossi G, Moratto D, Mazza C, et al. Combined DOCK8 and CLEC7A Mutations Causing Immunodeficiency in 3 Brothers With Diarrhea, Eczema, and Infections. J Allergy Clin Immunol (2013) 131(2):594–7.e1-3. doi: 10.1016/j.jaci.2012.10.062
14. Davis BR, Candotti F. Revertant somatic mosaicism in the Wiskott-Aldrich syndrome. Immunol Res (2009) 44(1-3):127–31. doi: 10.1007/s12026-008-8091-4
15. Pillay BA, Fusaro M, Gray PE, Statham AL, Burnett L, Bezrodnik L, et al. Somatic reversion of pathogenic DOCK8 variants alters lymphocyte differentiation and function to effectively cure DOCK8 deficiency. J Clin Invest (2021) 131(3):e142434. doi: 10.1172/JCI142434
16. Janssen E, Kumari S, Tohme M, Ullas S, Barrera V, Tas JM, et al. DOCK8 enforces immunological tolerance by promoting IL-2 signaling and immune synapse formation in Tregs. JCI Insight (2017) 2(19):e94298. doi: 10.1172/jci.insight.94298
17. Saettini F, Fazio G, Corti P, Quadri M, Bugarin C, Gaipa G, et al. Two siblings presenting with novel ADA2 variants, lymphoproliferation, persistence of large granular lymphocytes, and T-cell perturbations. Clin Immunol (2020) 218:108525. doi: 10.1016/j.clim.2020.108525
18. Tessarin G, Rossi S, Baronio M, Gazzurelli L, Colpani M, Benvenuto A, et al. Activated Phosphoinositide 3-Kinase Delta Syndrome 1: Clinical and Immunological Data from an Italian Cohort of Patients. J Clin Med (2020) 9(10):3335. doi: 10.3390/jcm9103335
19. Saettini F, Cattoni A, D’Angio’ M, Corti P, Maitz S, Pagni F, et al. Intermittent granulocyte maturation arrest, hypocellular bone marrow, and episodic normal neutrophil count can be associated with SRP54 mutations causing Shwachman-Diamond-like syndrome. Br J Haematol (2020) 189(4):e171–4. doi: 10.1111/bjh.16585
20. Rodrigues F, Davies EG, Harrison P, McLauchlin J, Karani J, Portmann B, et al. Liver disease in children with primary immunodeficiencies. J Pediatr (2004) 145(3):333–9. doi: 10.1016/j.jpeds.2004.05.037
21. Barbouche MR, Chen Q, Carbone M, Ben-Mustapha I, Shums Z, Trifa M, et al. Comprehensive review of autoantibodies in patients with hyper-IgM syndrome. Cell Mol Immunol (2018) 15(6):610–7. doi: 10.1038/cmi.2017.140
22. Ben Yakov G, Sharma D, Cho MH, Shah NN, Hickstein D, Urban A, et al. Cryptosporidium infection in Dedicator of Cytokinesis 8 (DOCK 8) Deficiency. J Allergy Clin Immunol Pract (2020) 8(10):3663–6.e1. doi: 10.1016/j.jaip.2020.06.021
Keywords: primary immumunodeficiencies, DOCK 8, EBV - Epstein-Barr Virus, sclerosing cholangitis, thrombocytopenia, lymphopenia, somatic reversion
Citation: Saettini F, Fazio G, Moratto D, Galbiati M, Zucchini N, Ippolito D, Dinelli ME, Imberti L, Mauri M, Melzi ML, Bonanomi S, Gerussi A, Pinelli M, Barisani C, Bugarin C, Chiarini M, Giacomelli M, Piazza R, Cazzaniga G, Invernizzi P, Giliani SC, Badolato R and Biondi A (2021) Case Report: Hypomorphic Function and Somatic Reversion in DOCK8 Deficiency in One Patient With Two Novel Variants and Sclerosing Cholangitis. Front. Immunol. 12:673487. doi: 10.3389/fimmu.2021.673487
Received: 27 February 2021; Accepted: 30 March 2021;
Published: 16 April 2021.
Edited by:
Stuart G. Tangye, Garvan Institute of Medical Research, AustraliaReviewed by:
Capucine Picard, Hôpital Necker-Enfants Malades, Assistance Publique Hopitaux De Paris, FranceAlexandra Freeman, National Institutes of Health (NIH), United States
Copyright © 2021 Saettini, Fazio, Moratto, Galbiati, Zucchini, Ippolito, Dinelli, Imberti, Mauri, Melzi, Bonanomi, Gerussi, Pinelli, Barisani, Bugarin, Chiarini, Giacomelli, Piazza, Cazzaniga, Invernizzi, Giliani, Badolato and Biondi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Saettini, Zi5zYWV0dGluaUBnbWFpbC5jb20=