 Marta López-Nevado1,2
Marta López-Nevado1,2 Luis I. González-Granado2,3
Luis I. González-Granado2,3 Raquel Ruiz-García4
Raquel Ruiz-García4 Daniel Pleguezuelo1,2
Daniel Pleguezuelo1,2 Oscar Cabrera-Marante1,2
Oscar Cabrera-Marante1,2 Nerea Salmón2,3
Nerea Salmón2,3 Pilar Blanco-Lobo5
Pilar Blanco-Lobo5 Nerea Domínguez-Pinilla2,6
Nerea Domínguez-Pinilla2,6 Rebeca Rodríguez-Pena7
Rebeca Rodríguez-Pena7 Elena Sebastián8Jaime Cruz-Rojo9
Elena Sebastián8Jaime Cruz-Rojo9 Peter Olbrich5Jesús Ruiz-Contreras2,3,10
Peter Olbrich5Jesús Ruiz-Contreras2,3,10 Estela Paz-Artal1,2,10
Estela Paz-Artal1,2,10 Olaf Neth5
Olaf Neth5 Luis M. Allende1,2,10*
Luis M. Allende1,2,10*- 1Immunology Department, University Hospital 12 de Octubre, Madrid, Spain
- 2Research Institute Hospital 12 Octubre (imas12), Madrid, Spain
- 3Immunodeficiency Unit, Department of Pediatrics, University Hospital 12 de Octubre, Madrid, Spain
- 4Immunology Department, Centre Diagnòstic Biomèdic, Hospital Clínic, Barcelona, Spain
- 5Paediatric Infectious Diseases, Rheumatology and Immunology Unit, University Hospital Virgen del Rocío, Institute of Biomedicine, Biomedicine Institute (IBiS)/University of Seville/Superior Council of Scientific Investigations (CSIC), Seville, Spain
- 6Pediatric Hematology and Oncology Unit, Toledo Hospital Complex, Toledo, Spain and University Hospital 12 de Octubre, Madrid, Spain
- 7Immunology Department, University Hospital La Paz, Madrid, Spain
- 8Hematology and Hemotherapy Unit, University Children’s Hospital Niño Jesús, Madrid, Spain
- 9Endocrine Unit, Department of Pediatrics, University Hospital 12 de Octubre, Madrid, Spain
- 10School of Medicine, Complutense University of Madrid, Madrid, Spain
Primary immune regulatory disorders (PIRD) are associated with autoimmunity, autoinflammation and/or dysregulation of lymphocyte homeostasis. Autoimmune lymphoproliferative syndrome (ALPS) is a PIRD due to an apoptotic defect in Fas-FasL pathway and characterized by benign and chronic lymphoproliferation, autoimmunity and increased risk of lymphoma. Clinical manifestations and typical laboratory biomarkers of ALPS have also been found in patients with a gene defect out of the Fas-FasL pathway (ALPS-like disorders). Following the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA), we identified more than 600 patients suffering from 24 distinct genetic defects described in the literature with an autoimmune lymphoproliferative phenotype (ALPS-like syndromes) corresponding to phenocopies of primary immunodeficiency (PID) (NRAS, KRAS), susceptibility to EBV (MAGT1, PRKCD, XIAP, SH2D1A, RASGRP1, TNFRSF9), antibody deficiency (PIK3CD gain of function (GOF), PIK3R1 loss of function (LOF), CARD11 GOF), regulatory T-cells defects (CTLA4, LRBA, STAT3 GOF, IL2RA, IL2RB, DEF6), combined immunodeficiencies (ITK, STK4), defects in intrinsic and innate immunity and predisposition to infection (STAT1 GOF, IL12RB1) and autoimmunity/autoinflammation (ADA2, TNFAIP3,TPP2, TET2). CTLA4 and LRBA patients correspond around to 50% of total ALPS-like cases. However, only 100% of CTLA4, PRKCD, TET2 and NRAS/KRAS reported patients had an ALPS-like presentation, while the autoimmunity and lymphoproliferation combination resulted rare in other genetic defects. Recurrent infections, skin lesions, enteropathy and malignancy are the most common clinical manifestations. Some approaches available for the immunological study and identification of ALPS-like patients through flow cytometry and ALPS biomarkers are provided in this work. Protein expression assays for NKG2D, XIAP, SAP, CTLA4 and LRBA deficiencies and functional studies of AKT, STAT1 and STAT3 phosphorylation, are showed as useful tests. Patients suspected to suffer from one of these disorders require rapid and correct diagnosis allowing initiation of tailored specific therapeutic strategies and monitoring thereby improving the prognosis and their quality of life.
Introduction
Human Inborn Errors of Immunity (IEI), also referred to as Primary Immunodeficiencies (PID) are a group of heterogeneous genetic diseases that disrupt the function of the immune system. Patients manifest increased susceptibility to develop a broad or narrow constellation of infectious, autoimmune, autoinflammatory, allergic, and/or malignant phenotypes. IEI represent more than 400 distinct disorders with more than 430 different gene defects listed in the classical and phenotypical 2019 International Union of Immunological Societies (IUIS) classification (1, 2). These diseases are mainly monogenic, but increasingly more complex genetic cases such as incomplete penetrance, polygenicity, transcriptional, somatic and epigenetic alterations are being described (3–6).
According to the European Society for Immunodeficiencies (ESID), Primary Immune Regulatory Disorders (PIRD) represent approximately 5.3% of IEI and currently comprise 45 disease-causing genes divided into four categories: hemophagocytic lymphohistiocytosis (HLH), susceptibility to EBV, syndromes with autoimmunity and immune dysregulation with colitis (1, 2). Autoimmune lymphoproliferative syndrome (ALPS), a well-known syndrome with autoimmunity, is a PIRD characterized by chronic and benign lymphoproliferation, autoimmune manifestations and an increased risk of lymphoma due to a defect in lymphocyte apoptosis (7, 8). Expanded CD3+TcRαβ+CD4-CD8- double negative T-cells (DNT) are a hallmark of ALPS patients who can also present elevated plasma biomarkers including interleukin-10 (IL-10), soluble FasL (sFasL) (diminished in patients with defect in FASL) and vitamin B12 (9). Genetic causes comprise the apoptotic Fas-FasL pathway components (FAS, FASL, FADD, CASP10 and CASP8), being germinal and somatic mutations in FAS gene (ALPS-FAS and ALPS-sFAS, respectively) the most common cause of ALPS (60% and 15%, respectively) (7, 8, 10–14). However, approximately 20% of patients with ALPS do not have an identifiable genetic mutation (ALPS-U) (9).
Advances in NGS technologies have enabled an expanding list of monogenic defects in PIRD. Numerous genetic defects outside the Fas-FasL pathway mimicking an ALPS phenotype have been identified (ALPS-like syndromes).
Clinically, autoimmunity and lymphoproliferation is a common finding also in patients with common variable immunodeficiency (CVID) without known genetic defect and therefore cannot be used as a diagnostic marker. Therefore, ALPS immunophenotyping, biomarkers and genetic testing by NGS could be useful for the identification of patients with an overlapping clinical phenotype such as ALPS, ALPS-like, CVID, combined immunodeficiency (CID) and immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) disorders (15–17). To date, thirteen genes have been related with ALPS-like syndrome in the literature: phenocopies of PID (NRAS and KRAS), susceptibility to EBV (MAGT1, PRKCD), regulatory T-cells defects (CTLA4, LRBA, STAT3 GOF), combined immunodeficiencies (ITK, STK4), defects in intrinsic and innate immunity and predisposition to infection (STAT1 GOF, IL12RB1) and autoimmunity/autoinflammation (ADA2, TET2) (18–29).
The approach to the diagnosis and initial management of these immune dysregulation disorders is often similar. However, identification of specific genetic defects and validation by immune evaluation can facilitate the use of specific targeted treatments for the defects that share similar pathophysiology mechanism (30–33).
In the manuscript, we show clinical and immunological characteristics of ALPS-like patients with the aim to identify specific and overlapping phenotypes in the amazing setting of numerous novel disorders, in which a basic immune evaluation can be misleading. Since the broader application of NGS has provided a greater knowledge within this field, rendering the diagnostic approach and the management of these disorders challenging, this study aims to provide important information to identify specificities of each group of disorders and it will hopefully present an important and practical tool to orientate the first steps of the diagnostic process.
Methods
Case Reports Search Strategy and Study Selection
The literature search was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) guidelines (34). Clinical ALPS-like criteria were immune dysregulation defined as autoimmunity and lymphoproliferation (17, 29, 35). The genes in which genetic defects gave rise to an ALPS-like phenotype, excluding those already described in the Fas-FasL pathway, were selected based on the IUIS classification (1, 2) and a literature search in MEDLINE (pubmed), Google scholar databases, Web Of Science (WOS) and congress communications. The following research keywords in English were used: (“ALPS”), (“ALPS-like”), (“Autoimmune lymphoproliferative syndrome”), (“autoimmune lymphoproliferative syndrome-like”), (“autoimmunity” AND “lymphoproliferation”), (“immune dysregulation” AND “autoimmunity”). Once ALPS-like-causing genes were identified, we classified them into the 10 groups of IUIS classification (1, 2). All types of publications reporting patients, in English or in Spanish, published until October 26, 2020, were collected and total of reported patients were counted (Supplementary Tables 1, 2). Those patients described in more than one article or cohort were only counted in the first report. Total cases were filtered according to the clinical criteria for ALPS-like. Those patients who presented lymphoproliferation in the form of lymphadenopathy, splenomegaly, or hepatomegaly together with autoimmune features were selected and clinical, laboratory, and molecular data were collected in a database.
Laboratory Studies
We show diverse functional studies by flow cytometry of not reported ALPS-like patients, diagnosed in the authors’ laboratories. Immunophenotyping of T, B and NK compartments, protein expression (NKG2D, XIAP, SAP, CTLA4 and LRBA) and phosphorylation assays (AKT, STAT3 and STAT1) could be rapid and useful to direct the diagnosis in some patients. Serial monitoring of serum levels of soluble CD25 (sCD25) (R&D system, Abingdon, UK) could be very useful as a measure of disease activity and response to therapy. For further details, see methods in Supplementary Material and Supplementary Table 3.
Results
ALPS-Like Related Genes
The search identified 526 publications describing 2245 unique patients suffering from defects in 24 distinct genes, distributed between the 10 large groups of the IUIS classification: phenocopies of PID (NRAS, KRAS), susceptibility to EBV (MAGT1, PRKCD, XIAP, SH2D1A, RASGRP1, TNFRSF9), antibody deficiency (PIK3CD GOF, PIK3R1 LOF, CARD11 GOF), regulatory T-cells defects (CTLA4, LRBA, STAT3 GOF, IL2RA, IL2RB, DEF6), combined immunodeficiencies (ITK, STK4), defects in intrinsic and innate immunity and predisposition to infection (STAT1 GOF, IL12RB1) and autoimmunity/autoinflammation (ADA2, TNFAIP3, TPP2, TET2). Supplementary Figure 1 summarizes the PRISMA diagram. Of total counted, 645 patients were filtered fulfilling the clinical ALPS-like inclusion criteria (Figure 1 and Supplementary Table 1).
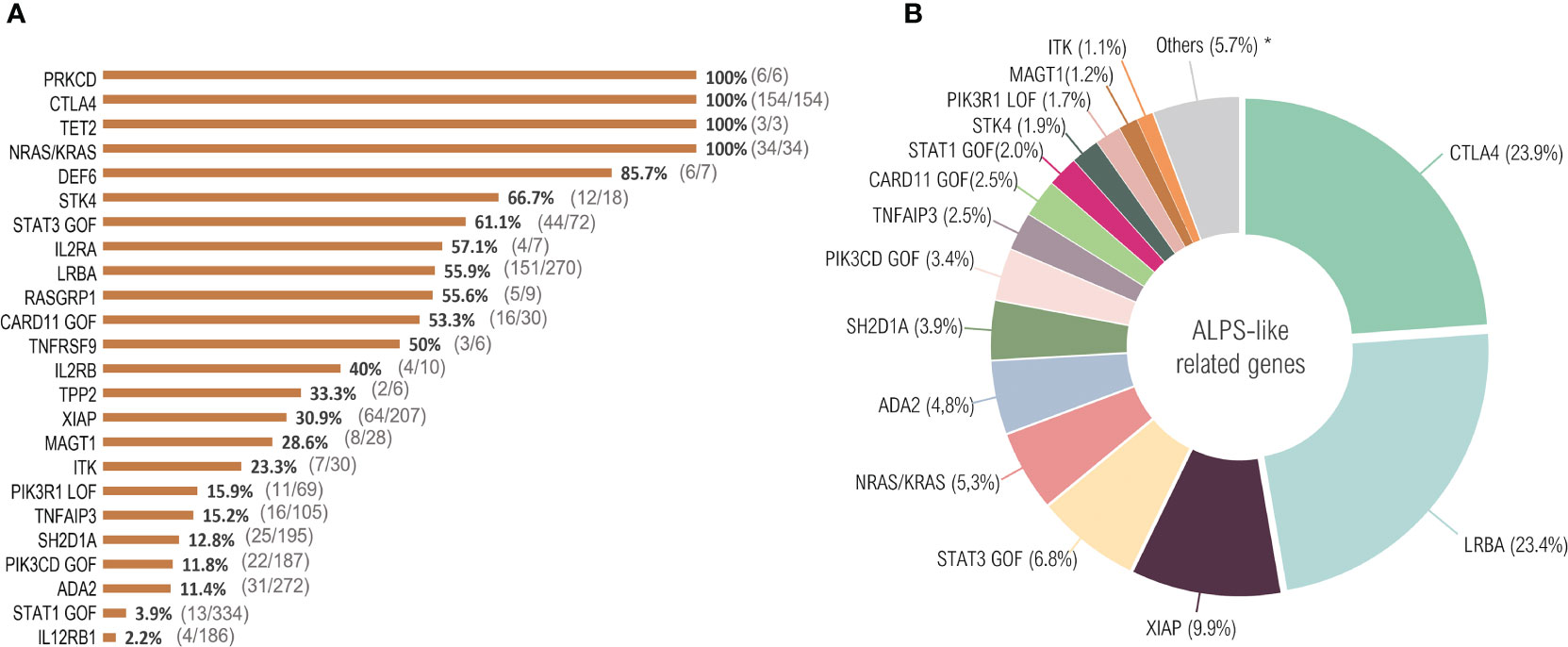
Figure 1 ALPS-like related cases. Twenty-four distinct genetic defects with immune dysregulation, lymphoproliferation and autoimmunity were identified in the literature until October 2020. More than 2000 total cases were reported. After filtering according to the ALPS-like inclusion criteria, 645 patients were selected. (A) percentage of patients that fulfill clinical ALPS-like phenotype criteria. The number of ALPS-like patients with respect to the total number of patients counted in each genetic defect is indicated in parentheses. (B) ALPS-like related genes prevalence. *ALPS-like related genes with a prevalence less than 1%: PRKCD (0.9%), DEF6 (0.9%), RASGRP1 (0.8%), IL2RA (0.6%), IL2RB (0.6%), TNFRSF9 (0.5%), TET2 (0.5%), TPP2 (0.3%).
All PRKCD, CTLA4, TET2 and NRAS/KRAS patients presented clinically with lymphoproliferation and autoimmunity, considering all those reported cases (n=197) as ALPS-like patients (Figure 1A). ALPS-like phenotype was seen in a significant percentage of patients suffering, among others, from mutations in STK4, STAT3 (GOF), LRBA or CARD11 (GOF) (66.7%, 61.1%, 55.9% and 53.3%, respectively, corresponding to 223 out of 390 patients), while the combination of autoimmunity and lymphoproliferation is quite rare in others. Very few cases of STAT1 GOF or IL12RB1 deficiency were suspected of ALPS-like (3.9% and 2.2%, respectively, corresponding to 17 out of 520 patients) (Figure 1A).
CTLA4, together with LRBA, is the most common cause of ALPS-like. Both disorders represent around the 50% of total filtered cases, while none of the rest of disorders exceed the 10% of total representation (Figure 1B).
To reduce the ALPS-like subgroups and due to similar immunopathology pathways, the patients have been grouped into Epstein Barr Virus (EBV) susceptibility, regulatory T-cells defect and others. Clinical and laboratory features as well as treatment are summarized in Table 1.
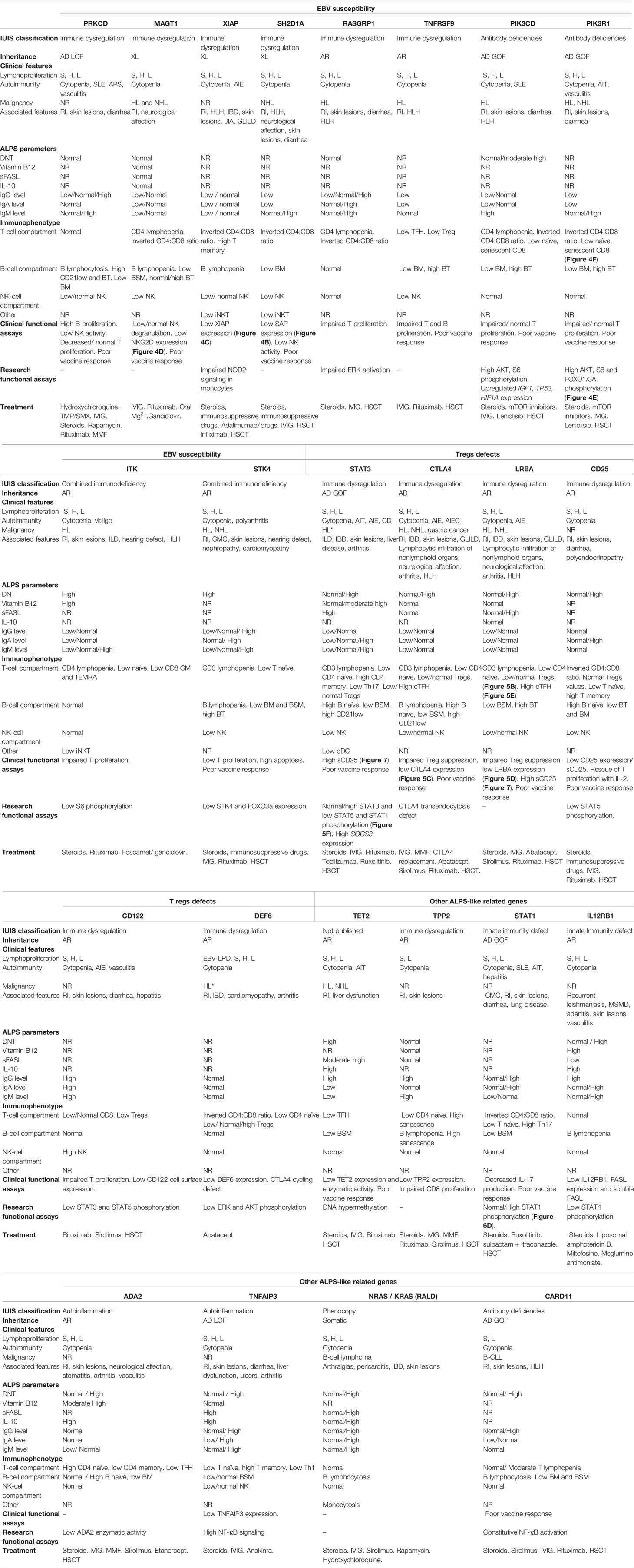
Table 1 Clinical and laboratory features of ALPS-like patients.
ALPS-Like Clinical Phenotype: Beyond Autoimmunity and Lymphoproliferation
According to the working definitions for clinical diagnosis of ALPS from the ESID (35), all ALPS-like cases included in this manuscript presented with lymphadenopathies and splenomegaly or hepatomegaly or all three. Immune-mediated cytopenia were also present in all patients, being the autoimmune hemolytic anemia (AIHA), immune thrombocytopenic purpura (ITP), autoimmune neutropenia (AN) or multilineage cytopenia the most common manifestations of autoimmunity (Table 1), as previously described in other patients with human IEI (33, 36–38). Unlike ALPS, organ-specific autoimmunity was commonly observed in ALPS-like patients, such as autoimmune thyroiditis in PIK3R1 or STAT1 GOF patients (39, 40), autoimmune polyarthritis in STK4 patients (22) autoimmune enteropathy in XIAP, STAT3 GOF, CTLA4, LRBA and IL2RB patients (16, 41–46) or autoimmune hepatitis in STAT1 GOF or LRBA patients (40, 47) (Table 1).
The lymphoproliferation and the immune dysregulation predispose both ALPS and ALPS-like patients to malignancies, especially lymphoma or leukemia (48, 49). In the ALPS-like group with EBV-susceptibility, several patients developed an EBV-driven B-cell lymphoma from uncontrolled EBV infection (23, 50–54). No ALPS-like patient with a defect in the PRKCD, XIAP, IL2RA, IL2RB, TPP2, STAT1 GOF, IL12RB1, ADA2 and TNFAIP3 genes presented malignancy in the reviewed literature (Table 1).
Apart from the clinical conditions shared with ALPS, phenotypic presentation of ALPS-like disorders is highly heterogeneous (Figure 2). Recurrent infections are the most common condition associated to ALPS-like patients, in contrast to ALPS, where very few cases with immunodeficiency have been reported (13, 14, 29). Lower and upper respiratory tract infections, both viral and bacterial, were presented in most ALPS-like patients with Tregs defect, ADA2 and CARD11 GOF patients (46, 47, 55–59). Less frequently, candida or recurrent herpes zoster infections were also found in STAT3 GOF, CTLA4 and LRBA patients (46, 60). Susceptibility to predominantly viral infections was observed in TET2, TPP2 and TNFAIP3 patients (27, 61, 62) while chronic mucocutaneous candidiasis (CMC) and mendelian susceptibility to mycobacterial disease (MSMD) were characteristic of STAT1 GOF and IL12RB1 patients, respectively (40, 63).
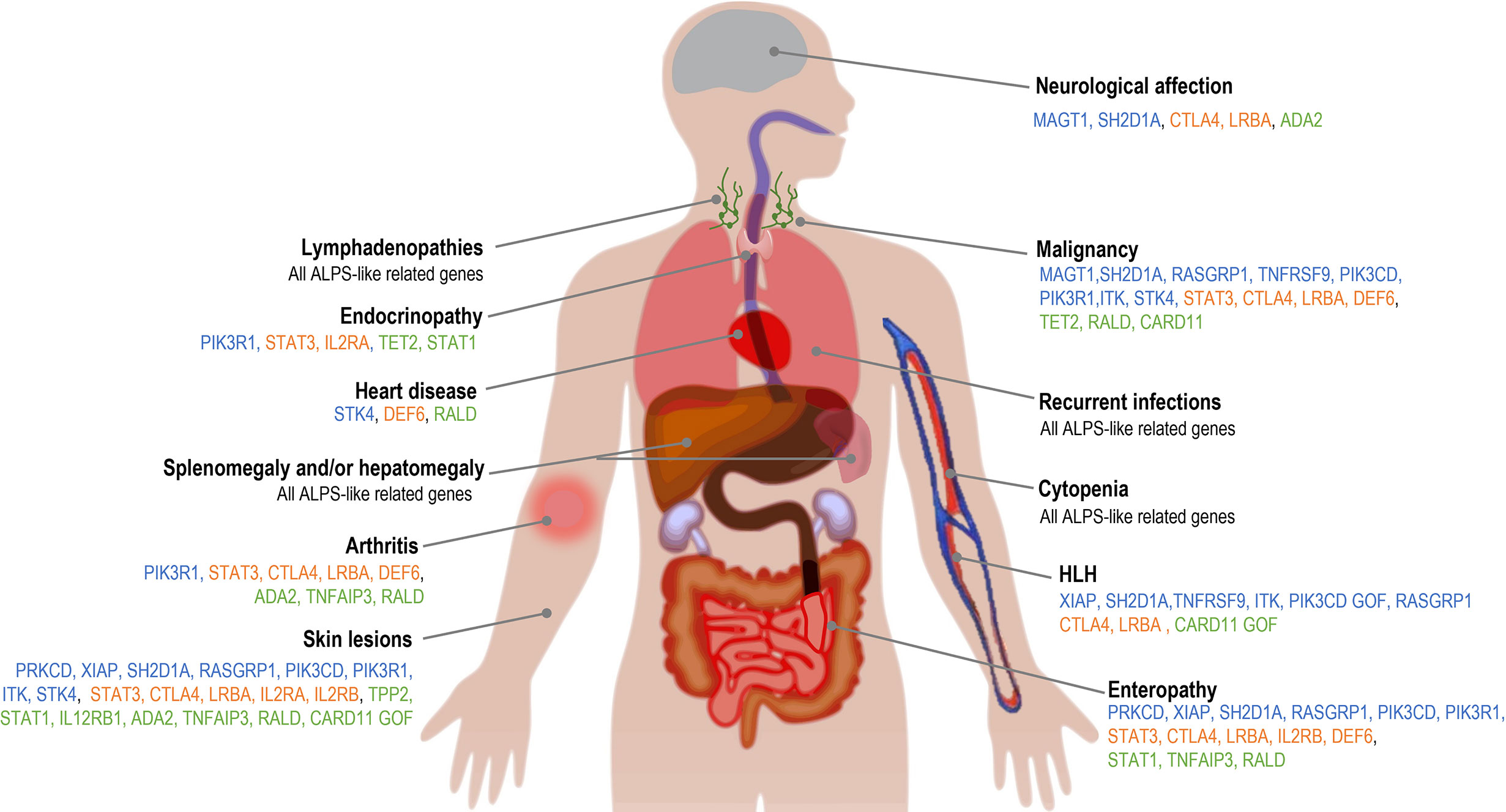
Figure 2 Clinical features associated with ALPS-like disorders. Blue: ALPS-like related genes with EBV-susceptibility; orange: ALPS-like related genes with regulatory T-cells defect; green: other ALPS-like related genes.
The gastrointestinal involvement, both autoimmune or inflammatory, was described in many of these patients with immune dysregulation and ALPS-like phenotype (Figure 2 and Table 1). Inflammatory bowel disease (IBD) was associated to a greater extent with ALPS-like patients with regulatory T-cells defect (Table 1) and specifically the early-onset IBD turned out to be characteristic of LRBA patients compared to others (47, 64–66).
Hemophagocytic lymphohistiocytosis (HLH) is a criterion of severity associated with significant mortality. ALPS-like patients who have developed HLH should undergo HSCT. HLH turned out to be characteristic of patients with X-linked lymphoproliferative syndrome (XIAP and SH2D1A patients) and was also found in ten patients with TNFRSF9 (51), LRBA (67), ITK (53, 68), RASGRP1 (69) deficiencies, CTLA4 haploinsufficiency (55, 70), CARD11 GOF (71) and PIK3CD GOF (52) (Table 1).
Skin involvement was remarkable in TNFAIP3 and ADA2 deficiencies, but it was one of the most common manifestations among ALPS-like disorders (Figure 2), mainly in form of non-infectious skin rashes, psoriatic-like lesions, skin abscess, severe eczema and skin warts (usually due to papilloma virus infection). Candida infection of the skin was characteristic of STAT1 GOF patients. Less frequently, arthritis, neurological affection (with both central and peripheral nervous system implication), cardiomyopathy and endocrinopathy were other clinical complications (Figure 2 and Table 1).
Thus, in a suspected ALPS patient presenting with clinical complications beyond lymphoproliferation and immune-mediated cytopenia, an ALPS-like syndrome should be considered.
Laboratory Features of ALPS-Suspected Patients: Immunophenotyping, ALPS Biomarkers and Functional Assays
There are no clinical or laboratory guidelines for the diagnosis of ALPS-like patients. Immunophenotyping of DNT by flow cytometry and typical ALPS biomarkers should be testing in any suspected ALPS patient (29, 72). A recent study established that 51.5% of probably ALPS-like patients presented abnormally high DNT, meaning that is not pathognomonic in this type of syndromes (73).
Although ALPS parameters are not provided in most of reviewed literature, increased DNT values have been reported in some ALPS-like patients with a defect in PIK3CD GOF (74), ITK (19), STK4 (22), STAT3 GOF (24, 43, 46, 75, 76), CTLA4 (55, 60), LRBA (16, 20, 47, 67, 77–81), IL2RA (82), TET2 (27), IL12RB1 (26, 63), ADA2 (28, 58, 83, 84), TNFAIP3 (62, 85), NRAS/KRAS (86–89) and CARD11 GOF (59, 71, 90, 91) genes, while DNT values within the normal range were observed in PRKCD (92, 93), MAGT1 (23, 94), RASGRP1 (69) and TPP2 (61) ALPS-like patients (Table 1 and Figure 3A). We show that elevated DNT values in ALPS-like patients were found with an average of 3.0% (2.4-3.4%) respect to CD3+ lymphocytes and 7.5% (3.5-27%) respect to CD3+TCRαβ+ lymphocytes (Figure 3B). Serum levels of vitamin B12, sFASL or IL-10 were only reported in seventeen patients. Different combinations of altered parameters were found. The isolated elevation of DNT was the most common alteration among patients (Supplementary Figure 2).
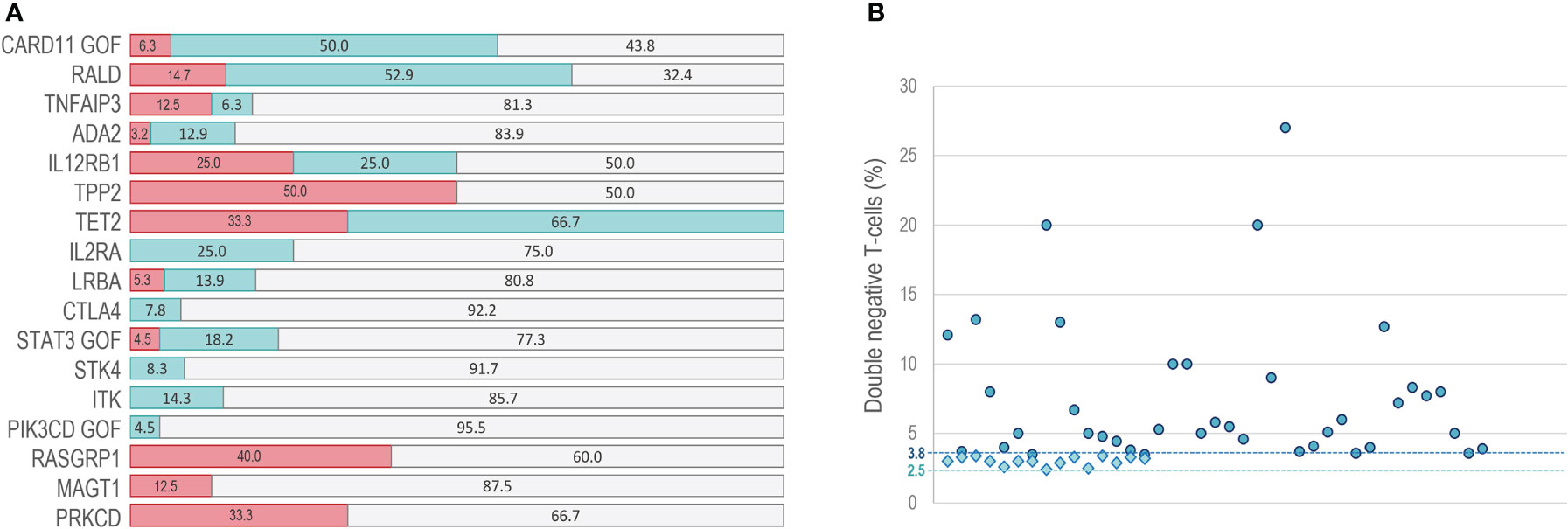
Figure 3 DNT in ALPS-like patients. (A) percentage of patients with normal (red bars) increased (blue bars) or not reported (gray bars) values of DNT. (B) range of DNT percentage presented in patients with high DNT. Circles: percentage of DNT in CD3+TCRαβ+ lymphocytes. Rhombus: percentage of DNT in CD3+ lymphocytes. Dashed line at point 3.8: DNT cutoff in CD3+TCRαβ+ lymphocytes. Dashed line at point 2.5: DNT cutoff in CD3+lymphocytes.
Alteration in the B-cell compartment is a common factor in most ALPS-like disorders in the term of decreased memory B-cells and increased transitional B-cells (Table 1). Inverted CD4/CD8 ratio, decreased naïve and increased memory and effector memory T-cells populations are also an extended phenotype in those ALPS-like patients with a compromised T-cells compartment (Table 1).
In those cases where the disease-causing mutation is not known or the variant is of uncertain significance (VUS), flow cytometry can be a powerful tool for the diagnosis approach through the study of extracellular and intracellular protein expression as well as the functional assessment of specific cell populations. Figures 4–6 show signaling pathways and its role in the pathogenesis of ALPS-like. It is shown some helpful functional assays through flow cytometry in ALPS-like patients diagnosed in the authors’ laboratories that were not part of the PRISMA. Other functional assays reported in the literature are represented in Table 1.
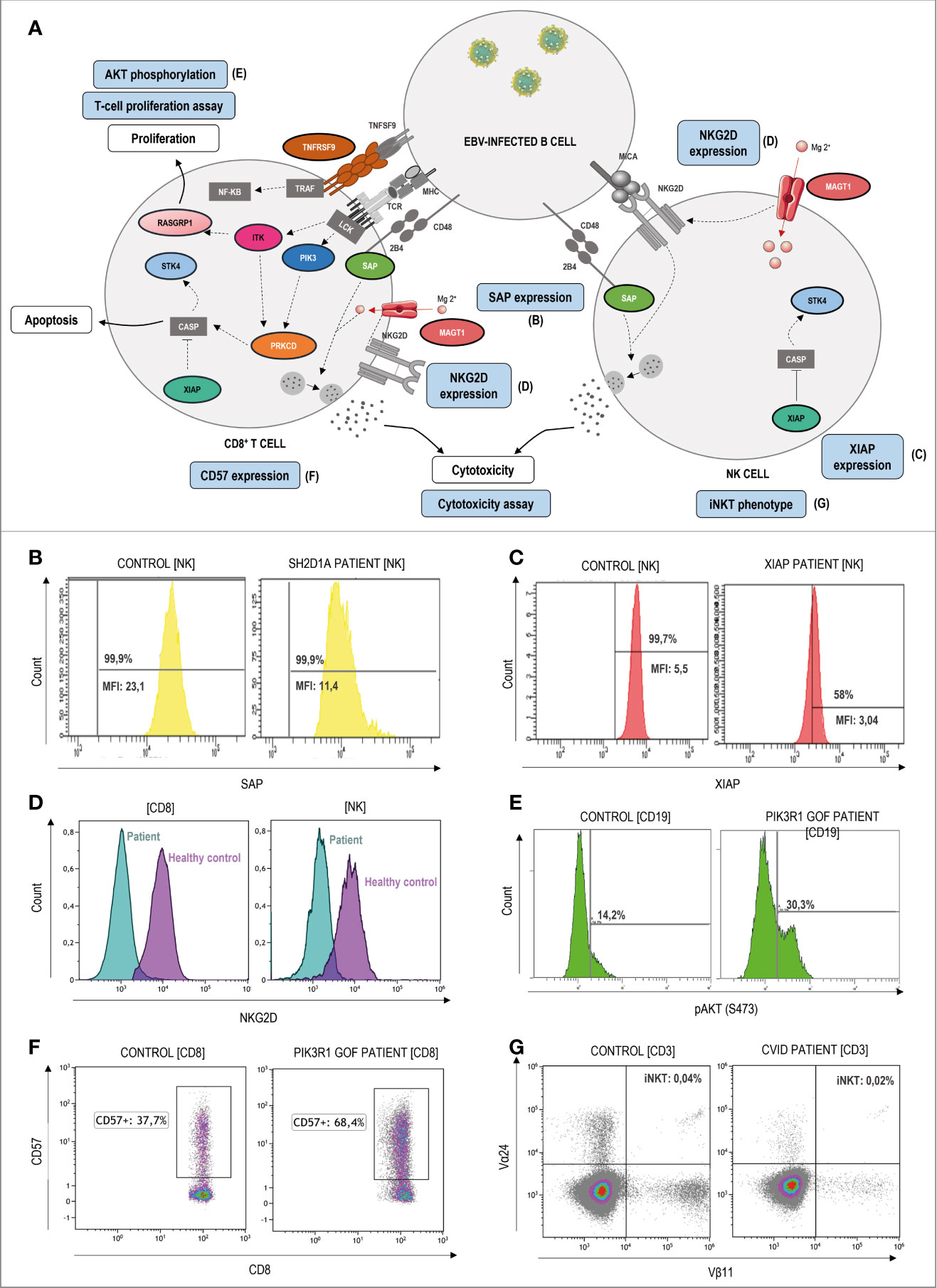
Figure 4 Pathophysiology and functional assays through flow cytometry in ALPS-like patients with EBV-susceptibility. (A) Schematic representation of the genetic defects (in color) and pathways identified in ALPS-like patients predisposing to high susceptibility to Epstein-Barr Virus (EBV)-driven lymphoproliferative disease. Immunologic studies are outlined in blue. The letters in parentheses refer to the functional studies included in figure 4. Defects in the control of EBV infection is mainly due to impairment of CD8, NK and NKT cell cytotoxicity (due to SAP, MAGT1 and TNFRSF9 deficiencies) and/or EBV-specific T cell proliferation (due to ITK, RASGRP1, TNFRSF9 deficiencies, PIK3CD gain of function and PIK3R1 loss of function) or survival (due to XIAP and STK4 deficiencies). (B) Decreased but not absent SAP expression in NK-cells of a patient with SAP deficiency. MFI: median fluorescence Intensity. (C) Decreased but not absent XIAP expression in NK-cells of a patient with XIAP deficiency. (D) Decreased NKG2D expression both in CD8 T and NK-cells of a MAGT1 patient (blue) in comparison with a healthy control (purple). (E) Hyperactivation of the PI3K-AKT signaling pathway is shown as high levels of AKT phosphorylation in a patient with PIK3R1 defect. (F) Senescence phenotype of CD8+CD57+ T-cells in a PIK3R1 patient. (G) Decreased CD3+Vα24+Vβ11+ invariant NKT-cells in a patient with common variable immunodeficiency and ALPS-like features.
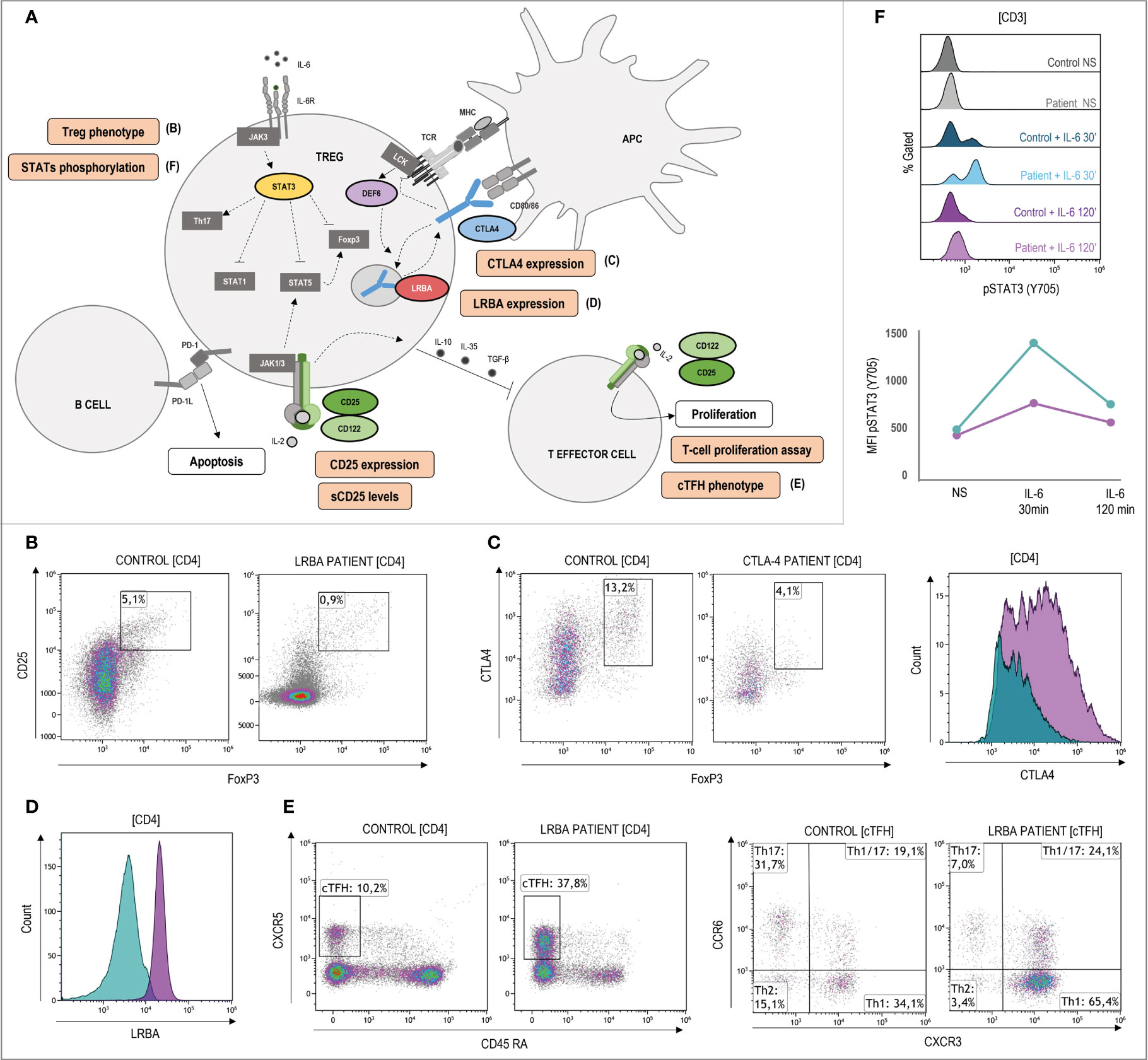
Figure 5 Pathophysiology and functional assays through flow cytometry in ALPS-like patients with regulatory T-cells defect. (A) Schematic representation of the genetic defects (in color) and pathways identified in ALPS-like patients with Treg defect. Immunologic studies are outlined in orange. The letters in parentheses refer to the functional studies included in Figure 5. Altered Tregs levels and/or function is shown in patients with CTLA4, LRBA, DEF6, IL2RA and IL2RB deficiencies and gain of function of STAT3. (B) Decreased CD4+CD25+FoxP3+ Treg cells in a LRBA patient. (C) Lower CTLA4 levels in a patient with CTLA4 haploinsufficiency (blue) in comparison with the healthy control analyzed (purple). (D) Decreased LRBA expression in CD4 T-cells of a patient with LRBA deficiency (blue) in comparison with the healthy control analyzed (purple). (E) Expansion of CD4+CXCR5+CD45RA- circulant follicular helper T cells (cTFH) and polarization to CCR6-CXCR3+ Th1 phenotype in a LRBA patient. (F) Hyperphosphorylation and delayed dephosphorylation of STAT3 (in tyrosine 705) induced by IL-6 in a patient with gain of function of STAT3 in comparison of the healthy control included. Line graph shows the median intensity fluorescence of STAT3 phosphorylated in the STAT3 GOF patient (blue) and the healthy control (purple). NS, no stimulation.
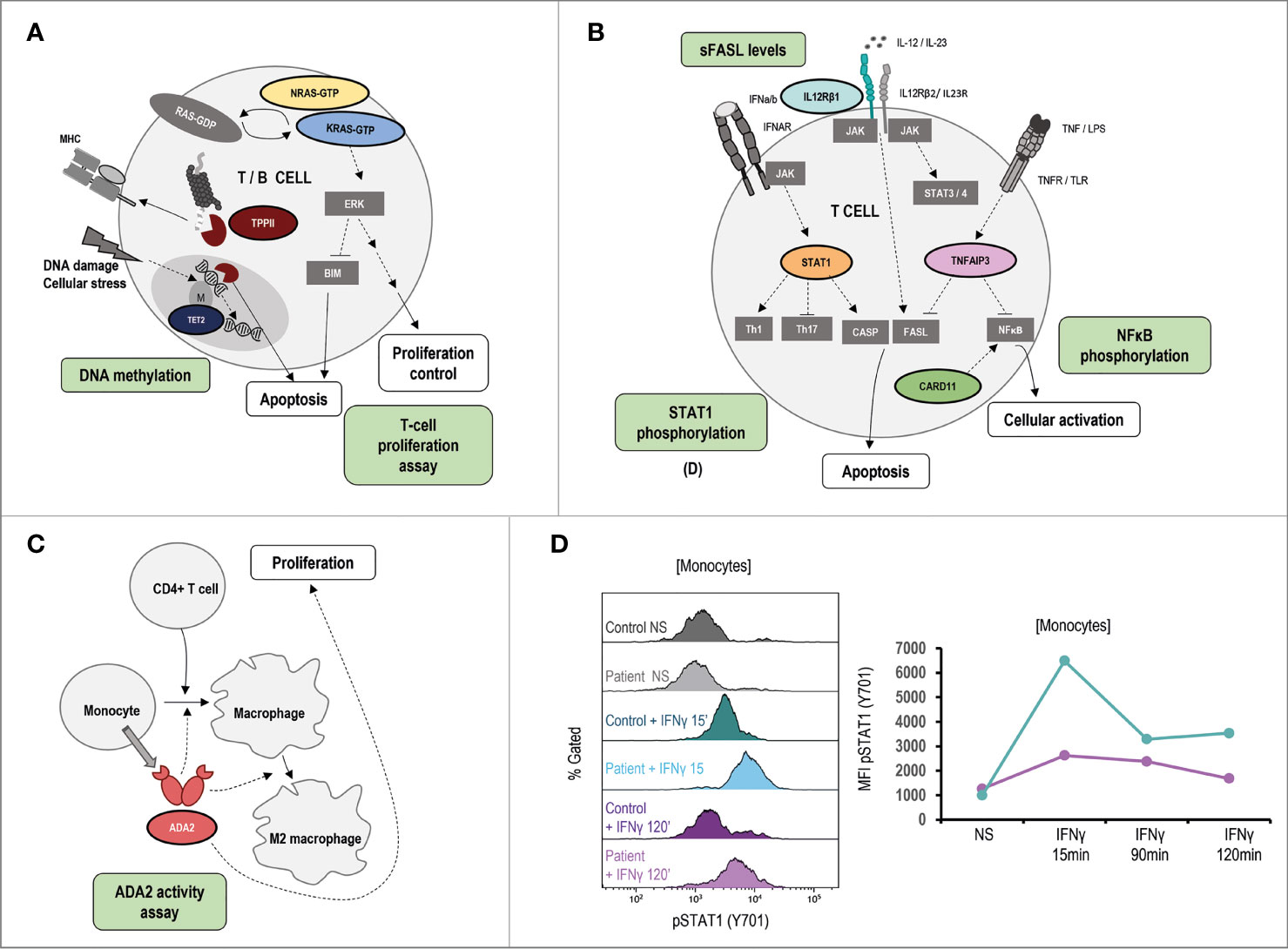
Figure 6 Pathophysiology and functional assays in other ALPS-like related genes. Immunologic studies are outlined in green. It is shown schematic representation of the genetic defects and pathways identified in ALPS-like patients with RALD, TTP2 and TET2 disorders (A), IL12RB1, STAT1 GOF, TNFAIP3, CARD11 GOF (B), ADA2 (C). (D) Hyperphosphorylation and delayed dephosphorylation of STAT1 (in tyrosine 701) induced by IFNγ in patient´s monocytes with gain of function of STAT1 in comparison of the healthy control included. Line graph shows the MFI of STAT1 phosphorylated in the STAT1 GOF patient (blue) and the healthy control (purple). NS, no stimulation.
In respect to the ALPS-like group with EBV-susceptibility (Figure 4A), an abolished or diminished SAP and XIAP expression may be indicative of mutations in SH2D1A and XIAP genes, respectively (Figures 4B, C). Alteration of MAGT1 gene abolishes TCR-induced transient Mg2+ influx and, consequently, the expression of NKG2D receptor is decreased (Figure 4D). The hyperactivation of the PI3K-AKT pathway is translated in an increased AKT phosphorylation in B-cells by flow cytometry. This assay can be used to screen patients with GOF and LOF mutations in PIK3CD and PI3KR1, respectively (Figure 4E), which are also characterized by a senescence phenotype of CD8 T-cells with high CD57 expression (Figure 4F). XIAP, SAP and ITK-deficient patients have low or absent numbers of iNKT cells (see iNKT in a CVID patient with an ALPS-like phenotype in Figure 4G) suggesting that these genes are required for the maturation, survival and/or differentiation of NKT cells.
CTLA4, LRBA, DEF6, CD25, CD122 and STAT3 are involved in Treg function and regulation (Figure 5A). The low percentage of Treg detected by flow cytometry (Figure 5B) should alert of these disorders, although some patients show normal or high levels, and it is questioned if the Treg deficiency could be secondary to the immune dysregulation presented in these patients. Determination of LRBA (and sometimes CTLA4) expression levels upon T-cell stimulation can be decisive for diagnostic approaches using flow cytometry (Figures 5C, D). In case of CTLA4, transendocytosis assay is recommendable to elucidate the result obtained by flow cytometry. CTLA4 and LRBA deficiencies show increased frequencies of circulating follicular helper T-cells (cTFH) with a polarized TH1-like phenotype (Figure 5E). Although it is not a fact exclusively of these genetic defects, the phenotypic characterization of TFH can be useful to rule out other ALPS-like related genes that also lead to decreased levels and/or function of Tregs. STAT3 hyperphosphorylation (Figure 5F) can explain the gain of function only in few patients whereas a normal result cannot exclude a STAT3 GOF patient, due to the high heterogeneity shown in these patients. Therefore, it is considered as a research functional assay with low diagnostic yield. In the case of CD25 deficiency, very low levels of soluble CD25 and/or CD25 expression could also be used for the diagnosis.
TET2, TPP2, STAT1 (GOF), IL12RB1, ADA2, TNFAIP3, CARD11 (GOF) and KRAS/NRAS (Ras-associated autoimmune leukoproliferative disorder, RALD) are other ALPS-like related genes (Figures 6A–C). As in the case of STAT3 GOF, some STAT1 GOF patients show STAT1 hyperphosphorylation (Figure 6D).
Treatment, Management, and Monitoring of ALPS-Like Patients
It is recommended that patients with a probable diagnosis of ALPS be managed in the same way as patients with a definitive diagnosis (9). The first-line management of ALPS-like patients is based on treatment of the disease manifestations and complications, including lymphoproliferation, autoimmunity and infections.
Short-term corticosteroid in combination with immunosuppressive drugs as Mycophenolate (MMF) was the first-line treatment for autoimmune cytopenia and specifically AIHA in most of revised patients (Table 1). High-dose intravenous immunoglobulin G (IVIG) therapy, together with corticosteroids was also a first-line option in case of immune thrombocytopenia. As second-line option, rituximab was used when first-line treatments failed (Table 1).
Sirolimus is a mTOR inhibitor used for management of ALPS patients (95). Several ALPS-like patients with elevated DNT suffering from CTLA4 haploinsufficiency, LRBA, TPP2, ADA2 and IL2RB deficiencies, RALD syndrome or CARD11 GOF were treated and responded to this drug (31, 44, 55, 61, 71, 83, 96–98) (Table 1).
Targeted therapies for certain molecular defects allowed patients to benefit from treatment for their underlying disease. For example, abatacept in CTLA4 haploinsufficiency, LRBA and DEF6 deficiencies; tocilizumab in STAT3 GOF; JAK inhibitors in STAT3 and STAT1 GOF patients; alemtuzumab for CD25 deficiency; PIK3δ inhibitors for APDS or anti-TNFα therapy for vasculitis in ADA2 deficiency (Table 1). In ALPS patients, splenectomy and rituximab should be discouraged unless they are the only remaining measures to control chronic refractory life-threatening cytopenia. In case of ALPS-like patients, these decisions should be made on a case-by-case basis (29, 98).
Hematopoietic stem cell transplantation (HSCT) was carried out when the patient progressed to severe disease, as is the case of severe immunodeficiency, refractory cytopenia, HLH and high risk of malignancy. In this study we show HSCT as a possible therapeutic option in 17 of the 24 ALPS-like disorders (70.8% of genetic defects) (Table 1). Considering the cohort of 645 ALPS-like patients, HSCT was a therapeutic option in 108 of them (16.7%). The outcome had positive effect in 82 patients (75.9%) vs. negative effect in 26 (24.1%) (Supplementary Table 4). HSCT in ALPS-like patients is recommended in cases of SH2D1A, APDS, ADA2, and probably increasingly for LRBA. There is sufficient evidence of the potential benefit in XIAP, CTLA4, but less in STAT3, STAT1, CARD11 GOF, CD25 or CD122 deficiencies (99).
Finding the therapeutic target is as important as monitoring the prognosis of the patient and the effectiveness of the treatment. In the context of immune dysregulation, elevated serum sCD25 is indicative of chronic immune activation and it can be helpful as a clinical biomarker of the disease and treatment monitoring. Here we show two cases of LRBA deficiency and STAT3 GOF, where measuring sCD25 allowed to evaluate the course of the disease and the efficacy of the treatment (Figure 7). In the case of LRBA patients, sCD25 tightly correlated with other markers of immune dysregulation such as the study of cTFH (100). Thus, monitoring sCD25 and cTFH cell frequencies in LRBA and CTLA4 deficiencies maybe particularly useful in tracking disease activity and response to different therapies. Accordingly, increased frequencies of cTFH cells have been involved in the pathogenesis of several autoimmune diseases and their frequency positively correlated with serum autoantibody titers (101).
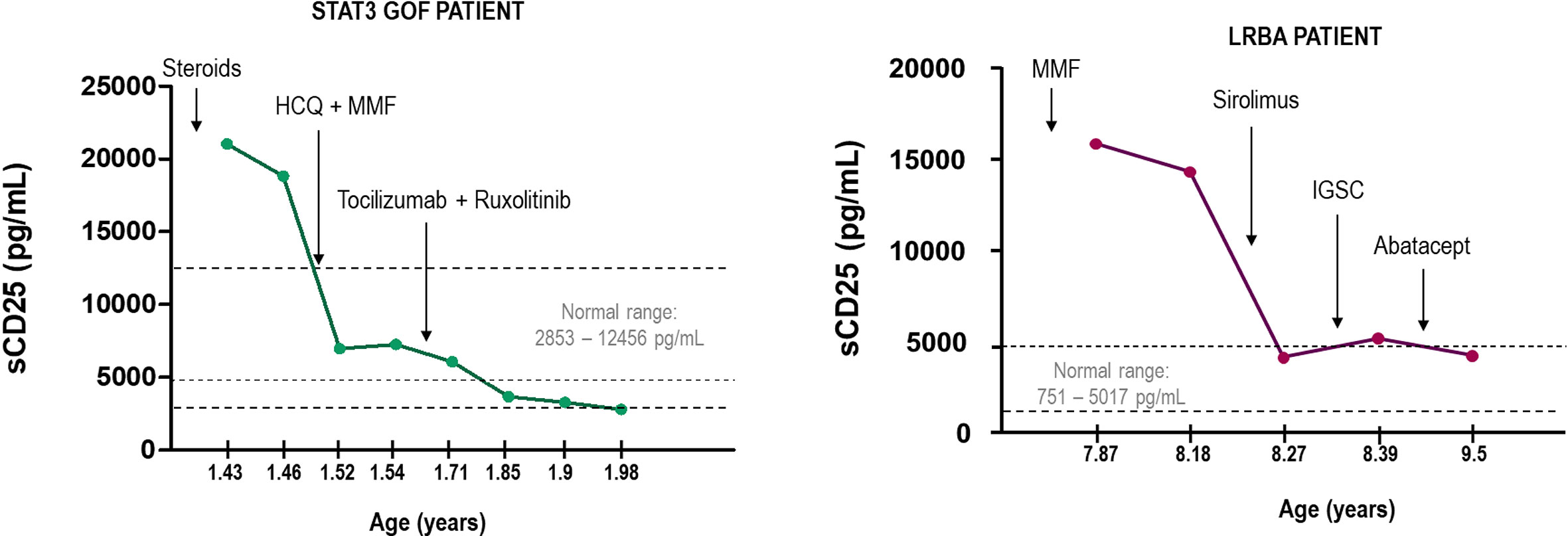
Figure 7 Monitorization of two cases of STAT3 GOF and LRBA deficiency measuring sCD25. Arrows indicate the changes in the treatment. HCQ, hydroxychloroquine; MMF, mycophenolate mofetil; SCIg, subcutaneous immunoglobulin therapy.
Concluding Remarks
In recent years, multiple monogenic causes of PIRD have been identified, some of them overlap clinical and immunologic features with the well-known ALPS syndrome. The prevalence and true incidence of ALPS-like disorders is unknown, probably because many cases remain undiagnosed or misdiagnosed.
More and more genes responsible for lymphoproliferation and autoimmunity are being described. In this study, twenty-four genetic disorders with ALPS-like characteristics have been identified in the literature: PRKCD, MAGT1, XIAP, SH2D1A, RASGRP1, TNFRSF9, ITK, STK4, CTLA4, LRBA, CD25, CD122, DEF6, TET2, TPP2, IL12RB1, ADA2 and TNFAIP3 deficiencies, PIK3CD, STAT3, STAT1 and CARD11 gain of function, PIK3R1 loss of function and RALD (caused by somatic gain of function mutations in NRAS and KRAS genes). Furthermore, other genetic defects as IKZF1 (75), NFKB1 (102), RAG1 (103) or NFKB2 (104) deficiencies have been associated with autoimmune and lymphoproliferative features in several patients cohorts.
This genetic heterogeneity makes the diagnostic approach through immunological and functional studies can be misleading. Currently, the gold standard for the diagnosis of these diseases is the performance of molecular studies by NGS, either by a targeted gene panel or by whole exome sequencing. However, flow cytometry is a quick and effective diagnostic approach tool in the case of protein expression studies and functional evaluation of different cell subpopulations, especially in those cases of VUS.
In the management of these patients, the balance between the long-term disease control and the risk of a HSCT must be considered. Targeted and individualized therapies existing for selected PIRD can solve the fine tuning of immunosuppressive treatments in patients with increased risk of infections, mitigating the possible adverse effects and saving time to offer HSCT in better circumstances.
Definitely, a negative diagnostic workup for patients with suspected ALPS should alert clinical immunologists to consider the possibility of an ALPS-like disorder. Although the heterogeneity of the phenotype makes a clinical definition difficult, these patients require a fast and precise diagnosis to offer them the best prognostic, therapeutic considerations and monitoring, including biomarkers and organ function.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité Ético de Investigación Clínica, University Hospital 12 de Octubre. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
ML-N and LA contributed to conception, design of the study and drafted the manuscript. ML-N developed the database, analysis, tables and figures. LG-G, DP, OC-M, NS, ND-P, RR-P, ES, JC-R, PO, JR-C and ON conducted the clinical and immunological follow-up of the patients. LG-G, JR-C, RR-P, and ON informed the patients about the study and collected the informed consents approved by the ethics committee. ML-N, RR-G, EP-A and PB-L contributed to the performance of functional tests. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from Fondo de Investigación Sanitaria (FIS-PI16/2053) to LA and LG-G. The project has been co-financed with FEDER funds. ML-N was co-financed by Fondo Social Europeo, Programa Operativo de empleo juvenil (YEI).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.671755/full#supplementary-material
Abbreviations
ADA2, Adenosine Deaminase 2; ALPS, Autoimmune Lymphoproliferative Syndrome; CARD11, Caspase Recruitment Domain Family Member 11; CTLA4, Cytotoxic T-Lymphocyte Associated Protein 4; DNT, Double Negative T-cells; FASL, FAS ligand; GOF, Gain of Function; IEI, Inborn Errors of Immunity; IL12RB1, Interleukin 12 Receptor Subunit Beta 1; ITK, IL2 Inducible T Cell Kinase; KRAS, KRAS Proto-Oncogene; GTPase; LRBA, LPS Responsive Beige-Like Anchor Protein; LOF, Loss of Function; MAGT1, Magnesium Transporter 1; NRAS, NRAS Proto-Oncogene; GTPase; PID, Primary Immunodeficiency; PIK3CD, Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Delta; PIK3R1, Phosphoinositide-3-Kinase Regulatory Subunit 1; PIRD, Primary Immune Regulatory Disorders; PRKCD, Protein Kinase C Delta; RASGRP1, RAS Guanyl Releasing Protein 1; SH2D1A, SH2 Domain Containing 1A; STAT1, Signal Transducer and Activator of Transcription 1; STAT3, Signal Transducer and Activator of Transcription 3; STK4, Serine/Threonine Kinase 4; TNFAIP3, TNF Alpha Induced Protein 3; UNC13D, Unc-13 Homolog D; XIAP, X-Linked Inhibitor of Apoptosis.
References
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
2. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
3. de Valles-Ibáñez G, Esteve-Solé A, Piquer M, Azucena González-Navarro E, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the Genetics of Common Variable Immunodeficiency: Monogenetic Model and Beyond. Front Immunol (2018) 9:636. doi: 10.3389/fimmu.2018.00636
4. Casanova JL, Conley ME, Seligman SJ, Abel L, Notarangelo LD. Guidelines for Genetic Studies in Single Patients: Lessons From Primary Immunodeficiencies. J Exp Med (2014) 211(11):2137–49. doi: 10.1084/jem.20140520
5. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal Dominant Immune Dysregulation Syndrome in Humans With CTLA4 Mutations. Nat Med (2014) 20(12):1410–6. doi: 10.1038/nm.3746
6. Rieux-Laucat F, Casamova JL. Autoimmunity by Haploinsufficiency. In: Science, vol. 345. American Association for the Advancement of Science (2014). p. 1560–1.
7. Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middelton LA, Lin AY, et al. Dominant Interfering Fas Gene Mutations Impair Apoptosis in a Human Autoimmune Lymphoproliferative Syndrome. Cell (1995) 81(6):935–46. doi: 10.1016/0092-8674(95)90013-6
8. Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IAG, Debatin KM, Fischer A, et al. Mutations in Fas Associated With Human Lymphoproliferative Syndrome and Autoimmunity. Sci (80-) (1995) 268(5215):1347–9. doi: 10.1126/science.7539157
9. Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, et al. Revised Diagnostic Criteria and Classification for the Autoimmune Lymphoproliferative Syndrome (ALPS): Report From the 2009 NIH International Workshop. In: Blood. Blood (2010) 116(14):e35–40. doi: 10.1182/blood-2010-04-280347
10. Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, et al. Autoimmune Lymphoproliferative Syndrome With Somatic Fas Mutations. N Engl J Med (2004) 351(14):1409–18. doi: 10.1056/NEJMoa040036
11. Del-Rey M, Ruiz-Contreras J, Bosque A, Calleja S, Gomez-Rial J, Roldan E, et al. A Homozygous Fas Ligand Gene Mutation in a Patient Causes a New Type of Autoimmune Lymphoproliferative Syndrome. Blood (2006) 108(4):1306–12. doi: 10.1182/blood-2006-04-015776
12. Jackson CE, Fischer RE, Hsu AP, Anderson SM, Choi Y, Wang J, et al. Autoimmune Lymphoproliferative Syndrome With Defective Fas: Genotype Influences Penetrance. Am J Hum Genet (1999) 64(4):1002–14. doi: 10.1086/302333
13. Bolze A, Byun M, McDonald D, Morgan NV, Abhyankar A, Premkumar L, et al. Whole-Exome-Sequencing-Based Discovery of Human FADD Deficiency. Am J Hum Genet (2010) 87(6):873–81. doi: 10.1016/j.ajhg.2010.10.028
14. Chun HJ, Zheng L, Ahmad M, Wang J, Speirs CK, Siegel RM, et al. Pleiotropic Defects in Lymphocyte Activation Caused by Caspase-8 Mutations Lead to Human Immunodeficiency. Nature (2002) 419(6905):395–9. doi: 10.1038/nature01063
15. Bussone G, Mouthon L. Autoimmune Manifestations in Primary Immune Deficiencies. Autoimmun Rev Autoimmun Rev (2009) 8:332–6. doi: 10.1016/j.autrev.2008.11.004
16. Cagdas D, Halaçlı SO, Tan Ç, Lo B, Çetinkaya PG, Esenboğa S, et al. A Spectrum of Clinical Findings From ALPS to CVID: Several Novel LRBA Defects. J Clin Immunol (2019) 39(7):726–38. doi: 10.1007/s10875-019-00677-6
17. Chandrakasan S, Chandra S, Davila Saldana BJ, Torgerson TR, Buchbinder D. Primary Immune Regulatory Disorders for the Pediatric Hematologist and Oncologist: A Case-Based Review. Pediatr Blood Cancer (2019) 66(5):e27619. doi: 10.1002/pbc.27619
18. Takagi M, Shinoda K, Piao J, Mitsuiki N, Takagi M, Matsuda K, et al. Autoimmune Lymphoproliferative Syndrome-Like Disease With Somatic KRAS Mutation. Blood (2011) 117(10):2887–90. doi: 10.1182/blood-2010-08-301515
19. Wallace JG, Alosaimi MF, Khayat CD, Jaber F, Almutairi A, Beaussant-Cohen S, et al. ITK Deficiency Presenting as Autoimmune Lymphoproliferative Syndrome. J Allergy Clin Immunol (2020) 147(2):743–5.e1.
20. Revel-Vilk S, Fischer U, Keller B, Nabhani S, Gámez-Díaz L, Rensing-Ehl A, et al. Autoimmune Lymphoproliferative Syndrome-Like Disease in Patients With LRBA Mutation. Clin Immunol (2015) 159(1):84–92. doi: 10.1016/j.clim.2015.04.007
21. Kucuk ZY, Charbonnier LM, McMasters RL, Chatila T, Bleesing JJ. CTLA-4 Haploinsufficiency in a Patient With an Autoimmune Lymphoproliferative Disorder. J Allergy Clin Immunol (2017) 140(3):862–4.e4. doi: 10.1016/j.jaci.2017.02.032
22. Schipp C, Schlütermann D, Hönscheid A, Nabhani S, Höll J, Oommen PT, et al. EBV Negative Lymphoma and Autoimmune Lymphoproliferative Syndrome Like Phenotype Extend the Clinical Spectrum of Primary Immunodeficiency Caused by STK4 Deficiency. Front Immunol (2018) 9:2400. doi: 10.3389/fimmu.2018.02400
23. Patiroglu T, Haluk Akar H, Gilmour K, Unal E, Akif Ozdemir M, Bibi S, et al. A Case of XMEN Syndrome Presented With Severe Auto-Immune Disorders Mimicking Autoimmune Lymphoproliferative Disease. Clin Immunol (2015) 159(1):58–62. doi: 10.1016/j.clim.2015.04.015
24. Nabhani S, Schipp C, Miskin H, Levin C, Postovsky S, Dujovny T, et al. STAT3 Gain-of-Function Mutations Associated With Autoimmune Lymphoproliferative Syndrome Like Disease Deregulate Lymphocyte Apoptosis and can be Targeted by BH3 Mimetic Compounds. Clin Immunol (2017) 181:32–42. doi: 10.1016/j.clim.2017.05.021
25. Fernández KS, Antony R, Kumar A. Patients With “ALPS-Like Phenotype” Diagnosed With Immune Dysregulation Due to LRBA Deficiency. Pediatr Blood Cancer (2019) 66(3):e27558. doi: 10.1002/pbc.27558
26. Nabhani S, Ginzel S, Miskin H, Revel-Vilk S, Harlev D, Fleckenstein B, et al. Deregulation of Fas Ligand Expression as a Novel Cause of Autoimmune Lymphoproliferative Syndrome-Like Disease. Haematologica (2015) 100(9):1189–98. doi: 10.3324/haematol.2014.114967
27. Stremenova Spegarova J, Lawless D, Mohamad SMB, Engelhardt KR, Doody G, Shrimpton J, et al. Germline TET2 Loss of Function Causes Childhood Immunodeficiency and Lymphoma. Blood (2020) Aug 27136(9):1055–66. doi: 10.1182/blood.2020005844
28. Alsultan A, Basher E, Alqanatish J, Mohammed R, Alfadhel M. Deficiency of ADA2 Mimicking Autoimmune Lymphoproliferative Syndrome in the Absence of Livedo Reticularis and Vasculitis. Pediatr Blood Cancer (2018) 65(4):10–3. doi: 10.1002/pbc.26912
29. Casamayor-Polo L, López-Nevado M, Paz-Artal E, Anel A, Rieux-Laucat F, Allende LM. Immunologic Evaluation and Genetic Defects of Apoptosis in Patients With Autoimmune Lymphoproliferative Syndrome (ALPS). Crit Rev Clin Lab Sci(2020) 58(4):253–74. doi: 10.1080/10408363.2020.1855623
30. Cabral-Marques O, Schimke LF, de Oliveira EB, El Khawanky N, Ramos RN, Al-Ramadi BK, et al. Flow Cytometry Contributions for the Diagnosis and Immunopathological Characterization of Primary Immunodeficiency Diseases With Immune Dysregulation. Front Immunol (2019) 10:2742. Frontiers Media S.A. doi: 10.3389/fimmu.2019.02742
31. Delmonte OM, Castagnoli R, Calzoni E, Notarangelo LD. Inborn Errors of Immunity With Immune Dysregulation: From Bench to Bedside. Front Pediatr (2019) 7:353. doi: 10.3389/fped.2019.00353
32. Teachey D, Bride K. Autoimmune Lymphoproliferative Syndrome: More Than a Fascinating Disease. F1000Res (2017) 6:1928. doi: 10.12688/f1000research.11545.1
33. Palmisani E, Miano M, Micalizzi C, Calvillo M, Pierri F, Terranova P, et al. Clinical Features and Therapeutic Challenges of Cytopenias Belonging to Alps and Alps-Related (ARS) Phenotype. Br J Haematol (2019) 184(5):861–4. doi: 10.1111/bjh.15178
34. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. J Clin Epidemiol (2009) 62(10):1006–12. doi: 10.1016/j.jclinepi.2009.06.005
35. ESID - European Society for Immunodeficiencies. Available at: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria.
36. Teachey DT, Manno CS, Axsom KM, Andrews T, Choi JK, Greenbaum BH, et al. Unmasking Evans Syndrome: T-Cell Phenotype and Apoptotic Response Reveal Autoimmune Lymphoproliferative Syndrome (ALPS). Blood (2005) 105(6):2443–8. doi: 10.1182/blood-2004-09-3542
37. Aladjidi N, Leverger G, Leblanc T, Picat MQ, Michel G, Bertrand Y, et al. New Insights Into Childhood Autoimmune Hemolytic Anemia: A French National Observational Study of 265 Children. Haematologica (2011) 96(5):655–63. doi: 10.3324/haematol.2010.036053
38. Al Ghaithi I, Wright NAM, Breakey VR, Cox K, Warias A, Wong T, et al. Combined Autoimmune Cytopenias Presenting in Childhood. Pediatr Blood Cancer (2016) 63(2):292–8. doi: 10.1002/pbc.25769
39. Bravo García-Morato M, García-Miñaúr S, Molina Garicano J, Santos Simarro F, Del Pino Molina L, López-Granados E, et al. Mutations in PIK3R1 Can Lead to APDS2, SHORT Syndrome or a Combination of the Two. Clin Immunol (2017) 179:77–80. doi: 10.1016/j.clim.2017.03.004
40. Chen X, Xu Q, Li X, Wang L, Yang L, Chen Z, et al. Molecular and Phenotypic Characterization of Nine Patients With STAT1 GOF Mutations in China. J Clin Immunol (2020) 40(1):82–95. doi: 10.1007/s10875-019-00688-3
41. Girardelli M, Arrigo S, Barabino A, Loganes C, Morreale G, Crovella S, et al. The Diagnostic Challenge of Very Early-Onset Enterocolitis in an Infant With XIAP Deficiency. BMC Pediatr (2015) 15(1):208. doi: 10.1186/s12887-015-0522-5
42. Fabre A, Marchal S, Forbes LR, Vogel TP, Barlogis V, Triolo V, et al. STAT3 Gain of Function: A New Kid on the Block in Interstitial Lung Diseases. In: American Journal of Respiratory and Critical Care Medicine, vol. 197. American Thoracic Society (2018). p. e22–3.
43. Haapaniemi EM, Kaustio M, Rajala HLM, Van Adrichem AJ, Kainulainen L, Glumoff V, et al. Autoimmunity, Hypogammaglobulinemia, Lymphoproliferation, and Mycobacterial Disease in Patients With Activating Mutations in STAT3. Blood (2015) 125(4):639–48. doi: 10.1182/blood-2014-04-570101
44. Zhang Z, Gothe F, Pennamen P, James JR, McDonald D, Mata CP, et al. Human Interleukin-2 Receptor B Mutations Associated With Defects in Immunity and Peripheral Tolerance. J Exp Med (2019) 216(6):1311–27. doi: 10.1084/jem.20182304
45. Navarini AA, Hruz P, Berger CT, Hou TZ, Schwab C, Gabrysch A, et al. Vedolizumab as a Successful Treatment of CTLA-4–Associated Autoimmune Enterocolitis. J Allergy Clin Immunol (2017) 139(3):1043–1046.e5. doi: 10.1016/j.jaci.2016.08.042
46. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-Onset Lymphoproliferation and Autoimmunity Caused by Germline STAT3 Gain-of-Function Mutations. Blood (2015) 125(4):591–9.
47. Gámez-Díaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The Extended Phenotype of LPS-Responsive Beige-Like Anchor Protein (LRBA) Deficiency. J Allergy Clin Immunol (2016) 137(1):223–30. doi: 10.1016/j.jaci.2015.09.025
48. Mayor PC, Eng KH, Singel KL, Abrams SI, Odunsi K, Moysich KB, et al. Cancer in Primary Immunodeficiency Diseases: Cancer Incidence in the United States Immune Deficiency Network Registry. J Allergy Clin Immunol (2018) 141(3):1028–35. doi: 10.1016/j.jaci.2017.05.024
49. Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rösen-Wolff A, et al. The Development of Lymphomas in Families With Autoimmune Lymphoproliferative Syndrome With Germline Fas Mutations and Defective Lymphocyte Apoptosis. Blood (2001) 98(1):194–200. doi: 10.1182/blood.V98.1.194
50. Platt CD, Fried AJ, Hoyos-Bachiloglu R, Usmani GN, Schmidt B, Whangbo J, et al. Combined Immunodeficiency With EBV Positive B Cell Lymphoma and Epidermodysplasia Verruciformis Due to a Novel Homozygous Mutation in RASGRP1. Clin Immunol (2017) 183:142–4. doi: 10.1016/j.clim.2017.08.007
51. Alosaimi MF, Hoenig M, Jaber F, Platt CD, Jones J, Wallace J, et al. Immunodeficiency and EBV-Induced Lymphoproliferation Caused by 4-1BB Deficiency. J Allergy Clin Immunol (2019) 144(2):574–583.e5. doi: 10.1016/j.jaci.2019.03.002
52. Cansever M, Zietara N, Chiang SCC, Ozcan A, Yilmaz E, Karakukcu M, et al. A Rare Case of Activated Phosphoinositide 3-Kinase Delta Syndrome (APDS) Presenting With Hemophagocytosis Complicated With Hodgkin Lymphoma. J Pediatr Hematol Oncol (2020) 42(2):156–9. doi: 10.1097/MPH.0000000000001487
53. Huck K, Feyen O, Niehues T, Rüschendorf F, Hübner N, Laws HJ, et al. Girls Homozygous for an IL-2-Inducible T Cell Kinase Mutation That Leads to Protein Deficiency Develop Fatal EBV-Associated Lymphoproliferation. J Clin Invest (2009) 119(5):1350–8. doi: 10.1172/JCI37901
54. Linka RM, Risse SL, Bienemann K, Werner M, Linka Y, Krux F, et al. Loss-of-Function Mutations Within the IL-2 Inducible Kinase ITK in Patients With EBV-Associated Lymphoproliferative Diseases. Leukemia (2012) 26(5):963–71. doi: 10.1038/leu.2011.371
55. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, Penetrance, and Treatment of 133 Cytotoxic T-Lymphocyte Antigen 4–Insufficient Subjects. J Allergy Clin Immunol (2018) 142(6):1932–46.
56. Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Mannurita SC, et al. Human IL2RA Null Mutation Mediates Immunodeficiency With Lymphoproliferation and Autoimmunity. Clin Immunol (2013) 146(3):248–61. doi: 10.1016/j.clim.2013.01.004
57. Serwas NK, Hoeger B, Ardy RC, Stulz SV, Sui Z, Memaran N, et al. Publisher Correction: Human DEF6 Deficiency Underlies an Immunodeficiency Syndrome With Systemic Autoimmunity and Aberrant CTLA-4 Homeostasis (Nature Communications, (2019), 10, 1, (3106), 10.1038/s41467-019-10812-x) [Internet]. Nat Commun (2019) 10(1):4555. doi: 10.1038/s41467-019-12454-5
58. Trotta L, Martelius T, Siitonen T, Hautala T, Hämäläinen S, Juntti H, et al. ADA2 Deficiency: Clonal Lymphoproliferation in a Subset of Patients. J Allergy Clin Immunol (2018) 141(4):1534–1537.e8. doi: 10.1016/j.jaci.2018.01.012
59. Brohl AS, Stinson JR, Su HC, Badgett T, Jennings CD, Sukumar G, et al. Germline CARD11 Mutation in a Patient With Severe Congenital B Cell Lymphocytosis. J Clin Immunol (2015) 35(1):32–46. doi: 10.1007/s10875-014-0106-4
60. Besnard C, Levy E, Aladjidi N, Stolzenberg MC, Magerus-Chatinet A, Alibeu O, et al. Pediatric-Onset Evans Syndrome: Heterogeneous Presentation and High Frequency of Monogenic Disorders Including LRBA and CTLA4 Mutations. Clin Immunol (2018) 188:52–7. doi: 10.1016/j.clim.2017.12.009
61. Stepensky P, Rensing-Ehl A, Gather R, Revel-Vilk S, Fischer U, Nabhani S, et al. Early-Onset Evans Syndrome, Immunodeficiency, and Premature Immunosenescence Associated With Tripeptidyl-Peptidase II Deficiency. in: Blood. Am Soc Hematol (2015) 125(5):753–61. doi: 10.1182/blood-2014-08-593202
62. Takagi M, Ogata S, Ueno H, Yoshida K, Yeh T, Hoshino A, et al. Haploinsufficiency of TNFAIP3 (A20) by Germline Mutation is Involved in Autoimmune Lymphoproliferative Syndrome. J Allergy Clin Immunol (2017) 139(6):1914–22. doi: 10.1016/j.jaci.2016.09.038
63. Göktürk B, Reisli İ, Çalışkan Ü, Oleaga-Quintas C, Deswarte C, Turul-Özgür T, et al. Infectious Diseases, Autoimmunity and Midline Defect in a Patient With a Novel Bi-Allelic Mutation in IL12RB1 Gene. Turk J Pediatr (2016) 58(3):331–6. doi: 10.24953/turkjped.2016.03.019
64. Alangari A, Alsultan A, Adly N, Massaad MJ, Kiani IS, Aljebreen A, et al. LPS-Responsive Beige-Like Anchor (LRBA) Gene Mutation in a Family With Inflammatory Bowel Disease and Combined Immunodeficiency. J Allergy Clin Immunol (2012) 130(2):481. doi: 10.1016/j.jaci.2012.05.043
65. Serwas NK, Kansu A, Santos-Valente E, Kuloǧlu Z, Demir A, Yaman A, et al. Atypical Manifestation of LRBA Deficiency With Predominant IBD-Like Phenotype. Inflammation Bowel Dis (2015) 21(1):40–7. doi: 10.1097/MIB.0000000000000266
66. Eren Akarcan S, Edeer Karaca N, Aksu G, Aykut A, Yilmaz Karapinar D, Cetin F, et al. Two Male Siblings With a Novel LRBA Mutation Presenting With Different Findings of IPEX Syndrome. JMM Case Rep (2018) 5(10). doi: 10.1099/jmmcr.0.005167
67. Kiykim A, Ogulur I, Dursun E, Charbonnier LM, Nain E, Cekic S, et al. Abatacept as a Long-Term Targeted Therapy for LRBA Deficiency. J Allergy Clin Immunol Pract (2019) 7(8):2790–2800.e15.
68. Stepensky P, Weintraub M, Yanir A, Revel-Vilk S, Krux F, Huck K, et al. IL-2-Inducible T-Cell Kinase Deficiency: Clinical Presentation and Therapeutic Approach. Haematologica (2011) 96(3):472–6. doi: 10.3324/haematol.2010.033910
69. Mao H, Yang W, Latour S, Yang J, Winter S, Zheng J, et al. RASGRP1 Mutation in Autoimmune Lymphoproliferative Syndrome-Like Disease. J Allergy Clin Immunol (2018) 142(2):595–604.e16.
70. Greil C, Roether F, La Rosée P, Grimbacher B, Duerschmied D, Warnatz K. Rescue of Cytokine Storm Due to HLH by Hemoadsorption in a CTLA4-Deficient Patient. J Clin Immunol (2017) 37:273–6. Springer New York LLC. doi: 10.1007/s10875-017-0377-7
71. Gupta M, Aluri J, Desai M, Lokeshwar M, Taur P, Lenardo M, et al. Clinical, Immunological, and Molecular Findings in Four Cases of B Cell Expansion With NF-κb and T Cell Anergy Disease for the First Time From India. Front Immunol (2018) 9:1049. doi: 10.3389/fimmu.2018.01049
72. Magerus-Chatinet A, Stolzenberg MC, Loffredo MS, Neven B, Schaffner C, Ducrot N, et al. FAS-L, IL-10, and Double-Negative CD4 -CD8 - TCR α/β + T Cells are Reliable Markers of Autoimmune Lymphoproliferative Syndrome (ALPS) Associated With FAS Loss of Function. Blood (2009) 113(13):3027–30. doi: 10.1182/blood-2008-09-179630
73. Molnár E, Radwan N, Kovács G, Andrikovics H, Henriquez F, Zarafov A, et al. Key Diagnostic Markers for Autoimmune Lymphoproliferative Syndrome With Molecular Genetic Diagnosis. Blood (2020) 136(17):1933–45. doi: 10.1182/blood.2020005486
74. Chiriaco M, Brigida I, Ariganello P, Di Cesare S, Di Matteo G, Taus F, et al. The Case of an APDS Patient: Defects in Maturation and Function and Decreased in Vitro Anti-Mycobacterial Activity in the Myeloid Compartment. Clin Immunol (2017) 178:20–8. doi: 10.1016/j.clim.2015.12.008
75. Takagi M, Hoshino A, Yoshida K, Ueno H, Imai K, Piao J, et al. Genetic Heterogeneity of Uncharacterized Childhood Autoimmune Diseases With Lymphoproliferation. Pediatr Blood Cancer (2018) 65(2):e26831. doi: 10.1002/pbc.26831
76. Weinreich MA, Vogel TP, Rao VK, Milner JD. Up, Down, and All Around: Diagnosis and Treatment of Novel STAT3 Variant. Front Pediatr (2017) 5:49. doi: 10.3389/fped.2017.00049
77. Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of Phenotypes Associated With Mutations in LRBA. J Clin Immunol (2016) 36:33–45. Springer New York LLC. doi: 10.1007/s10875-015-0224-7
78. Azizi G, Abolhassani H, Habibi S, Rahamooz T, Mohammadi H, Jafarnezhad-Ansariha F, et al. Two Faces of LRBA Deficiency in Siblings: Hypogammaglobulinemia and Normal Immunoglobulin Levels. J Investigational Allergol Clin Immunol (2018) 28:48–50. ESMON Publicidad S.A. doi: 10.18176/jiaci.0205
79. Kostel Bal S, Haskologlu S, Serwas NK, Islamoglu C, Aytekin C, Kendirli T, et al. Multiple Presentations of LRBA Deficiency: A Single-Center Experience. J Clin Immunol (2017) 37(8):790–800. doi: 10.1007/s10875-017-0446-y
80. Kutluǧ Ş, Boztuǧ K, Yildiran A. Development of Multiple Gallstones in a Child With Lipopolysaccharide-Responsive Beige-Like Anchor Protein Mutation. Cent Eur J Immunol (2019) 44(3):332–5. doi: 10.5114/ceji.2019.89613
81. Maggiore R, Grossi A, Fioredda F, Palmisani E, Terranova P, Cappelli E, et al. Unusual Late-Onset Enteropathy in a Patient With Lipopolysaccharide-Responsive Beige-Like Anchor Protein Deficiency. J Pediatr Hematol Oncol (2020) 42(8):e768–71. doi: 10.1097/MPH.0000000000001708
82. Lai N, Liu L, Lin L, Cui C, Wang Y, Min Q, et al. Effective and Safe Treatment of a Novel IL2RA Deficiency With Rapamycin. J Allergy Clin Immunol Pract (2020) 8(3):1132–1135.e4. doi: 10.1016/j.jaip.2019.09.027
83. Barzaghi F, Minniti F, Mauro M, Bortoli M, Balter R, Bonetti E, et al. ALPS-Like Phenotype Caused by ADA2 Deficiency Rescued by Allogeneic Hematopoietic Stem Cell Transplantation. Front Immunol (2019) 9:2767/full. doi: 10.3389/fimmu.2018.02767/full
84. Sundin M, Marits P, Nierkens S, Kolios AGA, Nilsson J. Immune Thrombocytopenia as Key Feature of a Novel ADA2 Deficiency Variant: Implication on Differential Diagnostics of ITP in Children. J Pediatr Hematol Oncol (2019) 41(2):155–7. doi: 10.1097/MPH.0000000000001132
85. Dong X, Liu L, Wang Y, Yang X, Wang W, Lin L, et al. Novel Heterogeneous Mutation of TNFAIP3 in a Chinese Patient With Behçet-Like Phenotype and Persistent EBV Viremia. J Clin Immunol (2019) 39(2):188–94. doi: 10.1007/s10875-019-00604-9
86. Calvo KR, Price S, Braylan RC, Oliveira JB, Lenardo M, Fleisher TA, et al. JMML and RALD (Ras-Associated Autoimmune Leukoproliferative Disorder): Common Genetic Etiology Yet Clinically Distinct Entities. Blood (2015) 125(18):2753–8. doi: 10.1182/blood-2014-11-567917
87. Levy-Mendelovich S, Lev A, Rechavi E, Barel O, Golan H, Bielorai B, et al. T and B Cell Clonal Expansion in Ras-Associated Lymphoproliferative Disease (RALD) as Revealed by Next-Generation Sequencing. Clin Exp Immunol (2017) 189(3):310–7. doi: 10.1111/cei.12986
88. Shiota M, Yang X, Kubokawa M, Morishima T, Tanaka K, Mikami M, et al. Somatic Mosaicism for a NRAS Mutation Associates With Disparate Clinical Features in RAS-Associated Leukoproliferative Disease: A Report of Two Cases. J Clin Immunol (2015) 35(5):454–8. doi: 10.1007/s10875-015-0163-3
89. Niemela JE, Lu L, Fleisher TA, Davis J, Caminha I, Natter M, et al. Somatic KRAS Mutations Associated With a Human Nonmalignant Syndrome of Autoimmunity and Abnormal Leukocyte Homeostasis. Blood (2011) 117(10):2883–6. doi: 10.1182/blood-2010-07-295501
90. Outinen T, Syrjänen J, Rounioja S, Saarela J, Kaustio M, Helminen M. Constant B Cell Lymphocytosis Since Early Age in a Patient With CARD11 Mutation: A 20-Year Follow-Up. Clin Immunol (2016) 165:19–20. doi: 10.1016/j.clim.2016.02.002
91. Snow AL, Xiao W, Stinson JR, Lu W, Benjamin CD, Zheng L, et al. Congenital B Cell Lymphocytosis Explained by Novel Germline CARD11 Mutations. J Exp Med (2012) 209(12):2247–61. doi: 10.1084/jem.20120831
92. Kiykim A, Ogulur I, Baris S, Salzer E, Karakoc-Aydiner E, Ozen AO, et al. Potentially Beneficial Effect of Hydroxychloroquine in a Patient With a Novel Mutation in Protein Kinase Cδ Deficiency. J Clin Immunol (2015) 35(6):523–6. doi: 10.1007/s10875-015-0178-9
93. Kuehn HS, Niemela JE, Rangel-Santos A, Zhang M, Pittaluga S, Stoddard JL, et al. Loss-of-Function of the Protein Kinase C Δ (PKCδ) Causes a B-Cell Lymphoproliferative Syndrome in Humans. Blood (2013) 121(16):3117–25. doi: 10.1182/blood-2012-12-469544
94. Hoyos-Bachiloglu R, Concha S, Sepúlveda P, Campos R, Perez-Mateluna G, King A, et al. The Many Faces of XMEN Disease, Report of Two Patients With Novel Mutations. J Clin Immunol (2020) 40:415–7. doi: 10.1007/s10875-020-00746-1
95. Teachey DT, Greiner R, Seif A, Attiyeh E, Bleesing J, Choi J, et al. Treatment With Sirolimus Results in Complete Responses in Patients With Autoimmune Lymphoproliferative Syndrome. Br J Haematol (2009) 145(1):101–6. doi: 10.1111/j.1365-2141.2009.07595.x
96. Ben-Ami T, Revel-Vilk S, Brooks R, Shaag A, Hershfield MS, Kelly SJ, et al. Extending the Clinical Phenotype of Adenosine Deaminase 2 Deficiency. J Pediatr (2016) 177:316–20. doi: 10.1016/j.jpeds.2016.06.058
97. Azizi G, Abolhassani H, Zaki-Dizaji M, Habibi S, Mohammadi H, Shaghaghi M, et al. Polyautoimmunity in Patients With LPS-Responsive Beige-Like Anchor (LRBA) Deficiency. Immunol Invest (2018) 47(5):457–67. doi: 10.1080/08820139.2018.1446978
98. Seidel MG. Treatment of Immune-Mediated Cytopenias in Patients With Primary Immunodeficiencies and Immune Regulatory Disorders (PIRDs). Hematol (United States) (2020) 20(1):673–9. doi: 10.1182/hematology.2020000153
99. Castagnoli R, Delmonte OM, Calzoni E, Notarangelo LD. Hematopoietic Stem Cell Transplantation in Primary Immunodeficiency Diseases: Current Status and Future Perspectives. Front Pediatr (2019) 7:295. Frontiers Media S.A. doi: 10.3389/fped.2019.00295
100. Alroqi FJ, Charbonnier LM, Baris S, Kiykim A, Chou J, Platt CD, et al. Exaggerated Follicular Helper T-Cell Responses in Patients With LRBA Deficiency Caused by Failure of CTLA4-Mediated Regulation. J Allergy Clin Immunol (2018) 141(3):1050–1059.e10. doi: 10.1016/j.jaci.2017.05.022
101. Gensous N, Charrier M, Duluc D, Contin-Bordes C, Truchetet ME, Lazaro E, et al. T Follicular Helper Cells in Autoimmune Disorders. Front Immunol (2018) 9:1637. Frontiers Media S.A. doi: 10.3389/fimmu.2018.01637
102. Schröder C, Sogkas G, Fliegauf M, Dörk T, Liu D, Hanitsch LG, et al. Late-Onset Antibody Deficiency Due to Monoallelic Alterations in NFKB1. Front Immunol (2019) 10:2618. doi: 10.3389/fimmu.2019.02618
103. Westermann-Clark E, Grossi A, Fioredda F, Giardino S, Cappelli E, Terranova P, et al. Rag Deficiency With Alps Features Successfully Treated With TCRαβ/CD19 Cell Depleted Haploidentical Stem Cell Transplant. Clin Immunol (2018) 187:102–3. doi: 10.1016/j.clim.2017.10.012
Keywords: ALPS, ALPS-like, autoimmunity, lymphoproliferation, malignancy, immune dysregulation
Citation: López-Nevado M, González-Granado LI, Ruiz-García R, Pleguezuelo D, Cabrera-Marante O, Salmón N, Blanco-Lobo P, Domínguez-Pinilla N, Rodríguez-Pena R, Sebastián E, Cruz-Rojo J, Olbrich P, Ruiz-Contreras J, Paz-Artal E, Neth O and Allende LM (2021) Primary Immune Regulatory Disorders With an Autoimmune Lymphoproliferative Syndrome-Like Phenotype: Immunologic Evaluation, Early Diagnosis and Management. Front. Immunol. 12:671755. doi: 10.3389/fimmu.2021.671755
Received: 24 February 2021; Accepted: 16 July 2021;
Published: 10 August 2021.
Edited by:
Isabelle Meyts, KU Leuven, BelgiumReviewed by:
Maurizio Miano, Giannina Gaslini Institute (IRCCS), ItalyKlaus Warnatz, University of Freiburg, Germany
Copyright © 2021 López-Nevado, González-Granado, Ruiz-García, Pleguezuelo, Cabrera-Marante, Salmón, Blanco-Lobo, Domínguez-Pinilla, Rodríguez-Pena, Sebastián, Cruz-Rojo, Olbrich, Ruiz-Contreras, Paz-Artal, Neth and Allende. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis M. Allende, bHVpcy5hbGxlbmRlQHNhbHVkLm1hZHJpZC5vcmc=