 Maria-Luisa Schubert1
Maria-Luisa Schubert1 Roman Rohrbach
Roman Rohrbach Michael Schmitt
Michael Schmitt Christoph K. Stein-Thoeringer
Christoph K. Stein-Thoeringer
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 31 May 2021
Sec. Mucosal Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.670286
This article is part of the Research TopicImmunoregulation at Mucosal SurfacesView all 36 articles
Cellular immunotherapy with chimeric antigen receptor (CAR)-T cells (CARTs) represents a breakthrough in the treatment of hematologic malignancies. CARTs are genetically engineered hybrid receptors that combine antigen-specificity of monoclonal antibodies with T cell function to direct patient-derived T cells to kill malignant cells expressing the target (tumor) antigen. CARTs have been introduced into clinical medicine as CD19-targeted CARTs for refractory and relapsed B cell malignancies. Despite high initial response rates, current CART therapies are limited by a long-term loss of antitumor efficacy, the occurrence of toxicities, and the lack of biomarkers for predicting therapy and toxicity outcomes. In the past decade, the gut microbiome of mammals has been extensively studied and evidence is accumulating that human health, apart from our own genome, largely depends on microbes that are living in and on the human body. The microbiome encompasses more than 1000 bacterial species who collectively encode a metagenome that guides multifaceted, bidirectional host-microbiome interactions, primarily through the action of microbial metabolites. Increasing knowledge has been accumulated on the role of the gut microbiome in T cell-driven anticancer immunotherapy. It has been shown that antibiotics, dietary components and gut microbes reciprocally affect the efficacy and toxicity of allogeneic hematopoietic cell transplantation (allo HCT) as the prototype of T cell-based immunotherapy for hematologic malignancies, and that microbiome diversity metrics can predict clinical outcomes of allo HCTs. In this review, we will provide a comprehensive overview of the principles of CD19-CART immunotherapy and major aspects of the gut microbiome and its modulators that impact antitumor T cell transfer therapies. We will outline i) the extrinsic and intrinsic variables that can contribute to the complex interaction of the gut microbiome and host in CART immunotherapy, including ii) antibiotic administration affecting loss of colonization resistance, expansion of pathobionts and disturbed mucosal and immunological homeostasis, and ii) the role of specific gut commensals and their microbial virulence factors in host immunity and inflammation. Although the role of the gut microbiome in CART immunotherapy has only been marginally explored so far, this review may open a new chapter and views on putative connections and mechanisms.
Primary cancer treatment relied for decades almost exclusively on surgery, chemotherapy and radiation therapy. With the development of monoclonal antibodies and the advent of stem cell transplantation, immunotherapy became a clinical option for the treatment of malignant diseases. Agents that induce or enhance antitumor responses, i.e. immune checkpoint inhibition (ICI) and adoptive T cell (ATC) therapy have revolutionized immunotherapeutic approaches. ICI using monoclonal antibodies against the cytotoxic T lymphocyte associated protein 4 (CTLA-4), the programmed cell death protein 1 (PD-1) or the programmed cell death ligand 1 (PD-1 L) (1) are now considered the standard of care in numerous solid and hematologic malignancies including advanced-stage melanoma, non-small-cell lung cancer (NSCLC), head and neck cancer, bladder cancer, or renal cell carcinoma (2). ATC therapy including tumor-infiltrating lymphocytes (TILs), T cell receptor (TCR)-engineered T cells and chimeric antigen receptor (CAR) gene-transduced T cells (CARTs) redirects T cells to tumor antigens. Therapy with TILs has achieved promising therapeutic results in melanoma (3–6) and TCR T cell receptor therapy is under clinical evaluation for different malignancies (NCT03686124, NCT03970382, NCT03691376). TILs and TCR T cell therapy are restricted to Human Leukocyte Antigen (HLA)-expressing antigens. In contrast, CARTs act in an HLA-independent manner and have the potential to effectively recognize target surface antigens, thus restricting immune evasion of malignant cells by HLA-downregulation.
CARTs constitute synthetic receptors composed of 1. an extracellular antigen-specific domain derived from an antibody’s single chain variable fragment (scFv), 2. a hinge and transmembrane segment, and 3. an intracellular domain that mediates activation and co-stimulation of the T cell that has been genetically engineered to express the CAR on the surface. Hence, CARTs combine the antigen-binding properties of antibodies with the effector functions of T cells. The intracellular CAR-domain defines different CART generations. First-generation CARs contain only the tyrosine-based ζ-signal-transducing subunit from the TCR/CD3 receptor complex and have displayed limited in vivo expansion and persistence of transduced T cells (7). Second-generation CARTs carry a costimulatory domain, e.g. CD28, 4-1BB (CD137), DAP-12 (8), OX40 (CD134) (9) or inducible T cell co-stimulator (ICOS) (10), adjacent to the TCR/CD3ζ-domain to mediate superior CART activation, proliferation and in vivo persistence (11). When directed against CD19, second-generation CARTs have demonstrated unprecedented clinical responses in a variety of relapsed and/or refractory (r/r) B cell malignancies including pediatric (12, 13) and adult (14, 15) acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL) (16–19), and other non-Hodgkin’s lymphoma (NHL) (20, 21).
Second-generation CARTs evaluated in the ZUMA-1 (diffuse large B-cell lymphoma (DLBCL) and primary mediastinal B-cell lymphoma (PMBCL)) (22, 23), ELIANA (ALL) (13) and JULIET (DLBCL) (24) as well as the ZUMA-2 trial (mantle cell lymphoma (MCL)) (21), achieved clinical responses in up to 93% of ALL (13, 25–27) and 82% (ZUMA-1) (22), 52% (JULIET) (28) and 92% (21) of NHL patients. Based on this pivotal trials, commercial CARTs axicabtagene ciloleucel (axi-cel) (29), tisagenlecleucel (tisa-cel) (30) and brexucabtagene autoleucel (brexu-cel) (31) were approved. These CARTs have become an integral part of the clinical hematologic practice within the authorized indications and have demonstrated efficacy also in the real-world setting (32–35).
Third-generation CARTs include two co-stimulatory molecules within their CAR constructs and have shown enhanced T cell activation in vitro and in vivo, sustained proliferation and tumor-lytic activity as well as reduced activation-induced cell death (36–39). Nonetheless, clinical evaluation is ongoing to assess if third-generation CARTs are advantageous in the clinical setting with regards to efficacy and safety (40, 41). Fourth-generation CARTs, i.e. engineered T cells redirected for universal cytokine killing (TRUCKs), endow additional modifications such as additional co-stimulatory ligands or cytokines to enhance their efficacy recruiting other effectors of the immune system (42, 43).
Besides CD19 as target, CARTs are under development against other tumor antigens, e. g. CD22 for ALL (44–46), CD30 for Hodgkin lymphoma and anaplastic large cell lymphoma (ALCL) (47, 48) or CD5 for T cell lymphoma (49). For multiple myeloma (MM), CARTs targeting the B cell maturation antigen (BCMA) (50–53) and for acute myeloid leukemia (AML) CARTs targeting CD33 (54), CD123 (55, 56) or CLL-1 (57) are under evaluation. Non-hematologic malignancies addressed by CARTs include glioblastoma (58–60) or neuroblastoma (61–63). CARTs to treat non-malignant indications, i.e. autoimmune (64) or infectious diseases, e. g. human immunodeficiency virus (HIV) infections (NCT0361719; NCT03240328) are also being clinically evaluated. Besides of T cells as sources for CART production, natural killer (NK) cells are broadening the application of CAR cells beyond the autologous T cell setting (65). Currently, 861 CAR trials are ongoing (clinicaltrials.gov; search for CAR cells; February 14th 2021).
Upon encounter and binding of the CAR with the target antigen, CARTs get activated. Activation results in cytotoxicity towards the targeted cell and in immune activation by recruitment of other T cells and bystander immune cells. Depletion of recipient lymphocytes before CART administration enhances engraftment, persistence, and efficacy of CARTs due to the reduction of resident lymphocytes and the reduction of regulatory T cells (66). Furthermore, lymphodepletion has been shown to stimulate stromal cells to produce the cytokines IL-7 and IL-15, both associated with enhanced expansion of CARTs (67–69).
As cellular products, CARTs do not exhibit typical pharmacokinetic properties of traditional drugs and the unique biology of CARTs explains the specific toxicities associated with this therapy including cytokine release syndrome (CRS), neurotoxicity, cytopenia, on-target-off tumor effects (i.e., B cell aplasia and consecutive hypogammaglobulinemia in CD19 CART therapy) and infections.
CRS is frequently observed after CART treatment with CD19-specific CARTs (12, 13, 22) but also with CARTs targeting BCMA (50) and with other T-cell engaging therapies (70, 71). CRS is triggered by inflammatory mediators released directly by the CART and the activated bystander immune cell and results in a supraphysiologic inflammatory state (72–74). CRS manifests with constitutional symptoms such as fever associated with fatigue, myalgia, arthralgia, rigors or anorexia, but can progress to hypotension, tachycardia, tachypnea and hypoxia, arrhythmia, capillary-leak, coagulopathy, respiratory failure, shock and organ dysfunction (22, 75). The treatment of CRS involves symptomatic treatment as well as anti-cytokine treatment with anti-IL-6 antibodies and corticosteroids (28, 76, 77).
Neurotoxicity, referred to as immune effector cell-associated neurotoxicity syndrome (ICANS), is another toxicity commonly observed after CART treatment. ICANS typically presents with impairment of attention and confusion (26, 78–80) and can progress to depressed level of consciousness, coma, seizures, motor weakness, and cerebral edema (34, 81). Trafficking of CARTs, passive diffusion of cytokines into the central nervous system (CNS), endothelial activation with subsequent disruption of the blood-brain barrier and microglial and/or myeloid cell activation in the CNS have been suggested as underlying the pathophysiology of ICANs (14, 25, 80–83). For isolated ICANS, steroids are the first-line of therapy (76).
High-grade and long-term cytopenias are frequently observed after CART therapy and prone patients to infectious complications (13, 22, 23, 25, 28, 84–86). Also, B cell aplasia with secondary hypogammaglobulinemia due to the effects of CD19-directed CARTs on normal B cells can be associated with an increased risk of infections (87, 88).
Besides cytopenia and hypogammaglobulinemia, further risk factors for infections in CART patients include the number of prior chemotherapeutic treatment regimens, impaired performance status at immunotherapy start, ALL as underlying disease, a reduced absolute neutrophil count at baseline, a high dose of administered CARTs and the use of corticosteroid treatment for management of CART toxicities (15, 73, 89). In fact, early and late infectious complications, primarily of bacterial and viral origin, after CART administration are common (32, 85, 90–92). Besides, invasive fungal infections (90, 93) and reactivation of latent DNA viruses are observed after CART treatment (79, 85, 94).
Universal evidence-based guidelines for anti-infective prophylaxis of CART patients are pending. Although for all patients, herpes simplex (HSV) and varicella zoster (VZV) prophylaxis up to one year after CART treatment and/or until sufficient peripheral CD4 cell counts were reconstituted are recommended (95–97), fungal and bacterial prophylaxes are not routinely recommended after CART treatment. Antibiotic prophylaxis standards vary between different institutions (96, 97), but most regimens include the use of fluoroquinolons (96).
However, in neutropenic CART patients, antibiotic treatment is consensually strongly advised (97, 98), especially considering the high number of infections in these patients (92, 97). A recent study in children reported that infections occurs in about half of them within 3 months before the intervention and in about 40% of the patients in the first days after CART infusion. Bacteria accounted for half of the infections causing a high number of severe and life-threatening bacteremia, notably E. coli, Klebsiella spp., Enterococcus spp. and Staphylococcus spp (99). In adult patients, infections were more common within the first 2 months after CART cell therapy, and again bacteria were the most common causative pathogens. Intriguingly, the gut as site of infection and intestinal commensals were found to account for a considerable fraction of infections (89).
Antibiotics are commonly used in patients undergoing antitumor therapies to prevent and/or reduce infectious complications. Nonetheless, antibiotics have been shown to promote development of chronic diseases and to affect the clinical outcome of patients treated with immunotherapies (100), which is suggested to be (at least in part) due to negative effects on the gut commensal microbiome. Despite their essential role in managing infections and, thereby, saving lives, there is a growing body of evidence showing that antibiotics have detrimental impact on the antitumor efficacy of T cell-based immunotherapies, notably ICI therapies (see review of the clinical studies in Table 1). Such adverse influence on the ICI outcomes are hypothesized to occur through modulation of the intestinal microbiome. Therefore, we will focus in the following chapters on the intestinal microbiome, individual commensals and their potential role in anticancer T cell therapies.
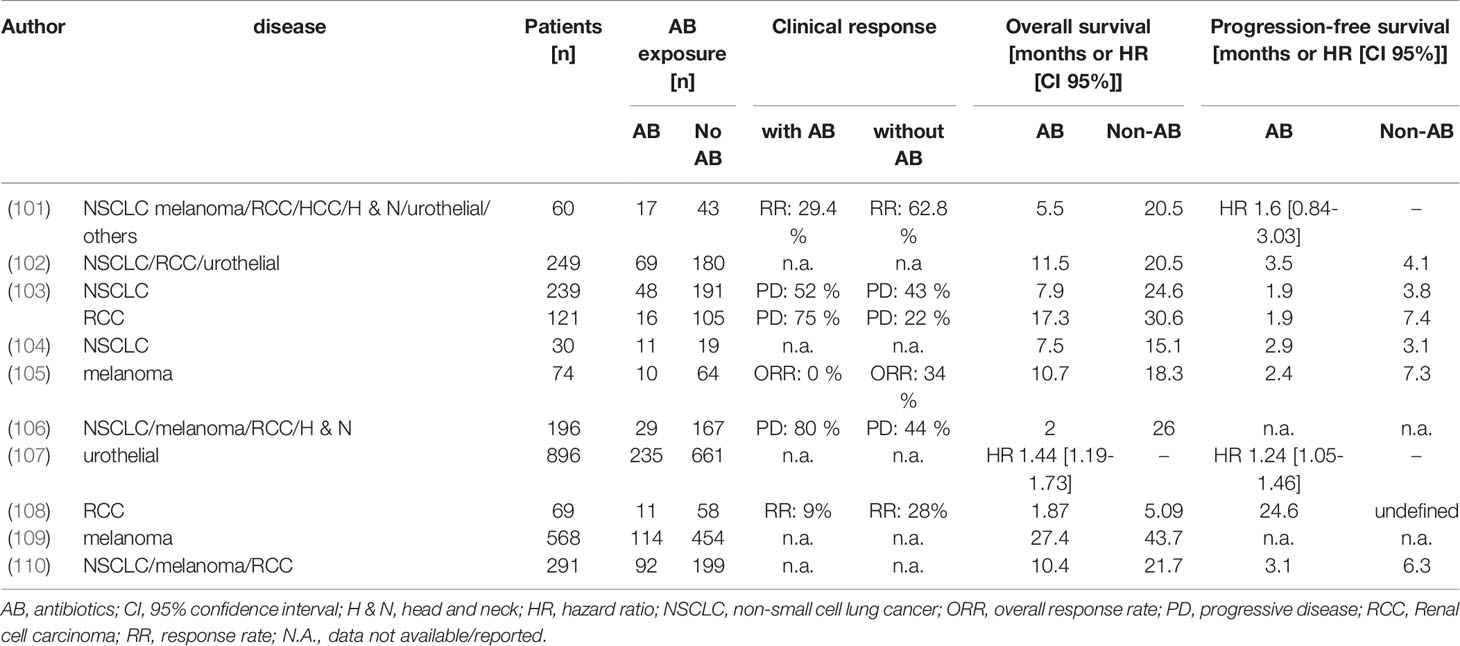
Table 1 Outcome of patients treated with immune checkpoint inhibitors (ICI) with or without receiving antibiotic (AB) treatment.
The human body harbors a massive number of microbial members (likewise the number of human cells) that orchestrate a comprehensive range of physiological processes, diseases and cancer susceptibility. Their 100-fold higher gene diversity encodes outstanding mechanism and metabolic competences that influence their own microbial niche, host tissue specific and immune cells function (111). This microbial ecosystem, collectively termed microbiome, is composed of eukaryotes (fungi and protozoa), virus and prokaryotes (112). The majority of commensal bacteria inhabit the colonic gastrointestinal tract while the minority are colonizing other anatomical regions such as oral-respiratory and urogenital tracts, skin as well as tumors. Overall, under healthy conditions, the host and microbiome exist in a symbiotic equilibrium as a metaorganism by providing a nutrient-rich microenvironment in return for aid in digestion and metabolism, respectively (113–115). As such, the microbiome synthesizes vitamins and breaks down food into absorbable nutrients, e.g., carbohydrates, or host signaling molecules such as short-chain fatty acids (116). Differences in geographic location, ethnicity, and dietary habits cause the human microbiome to be highly variable between and within individuals (117). In the last twenty years, research on the microbiome turned to be a field of enormous interest in a broad scientific community, which was leveraged by the Human Microbiome Project 1 and 2. Numerous diseases including cardiovascular diseases (CVDs), inflammatory bowel disease (IBD), diabetes mellitus, cardiometabolic disease, liver disease, neurodevelopmental illnesses, and cancer have been shown to be associated with and even partially driven by alterations of the intestinal microbiome or of microbe-host interactions, termed dysbiosis (115, 118, 119).
Mucosal surfaces of the intestinal tract are constantly exposed to a huge biomass of commensal bacteria, and the host’s response to gut microbes is compartmentalized to the mucosal surface. As primary gate keepers, the intestinal epithelium and a dense layer of mucus separates the lumen with resident microbes from the underlying host’s tissues. The major building blocks in mucus are mucins which are large, highly glycosylated proteins, secreted by intestinal goblet cells. The mucin domain glycans bind a lot of water and thereby generate the typical gel-like properties of the mucus (120). In addition to forming a mucosal gel, goblet cells have been shown to deliver intestinal luminal material to the lamina propria dendritic cells (DCs) for presentation of oral and intestinal antigens to the immune system (121), and also facilitate colonic translocation of commensal bacteria to host lymphatic organs (122). Antimicrobial peptides (AMP), a diverse group of evolutionary conserved defense proteins and peptides, play another critical role in maintaining mucosal barrier functions. Intestinal AMPs are secreted by Paneth cells and, to a lesser extent, enterocytes in the small intestine (123). AMP families include lysozymes (124), cathelicidins (125), α- and β- defensins (126), and regenerating islet-derived (Reg) proteins (127). AMPs mostly exert barrier function through direct bacterial killing, or indirectly via induction of a diverse array of immunomodulatory mechanisms (128). AMPs such as enteric defensins (129), resistin-like molecule β (130), cathelin-related antimicrobial peptide (131) and lectins of the Reg3 family constitutively shape the indigenous commensal repertoire and microbiome ecology (132). In addition, AMPs protect the host from pathogenic infection, e.g. cathelicidin-related peptides against Salmonella typhimurium infection (133), while the lectin Reg3γ protects mice against Listeria monocytogenes infection (134) and reduces colonization by vancomycin-resistant Enterococcus (135).
Secretion of immunoglobulin A (IgA) represents another feature of the intestinal mucosa to protect the host against intestinal pathogens. IgA, the most abundant antibody isotype produced in our body, primarily induced in the Peyer’s patches of the gut, provides non-inflammatory immune protection against Salmonella typhimurium (136) or Enterobacter cloacae (137). Secretory IgA promotes intestinal immune exclusion by entrapping dietary antigens and microbes in the mucus, down-modulates the expression of proinflammatory bacterial epitopes on commensal bacteria, and, thereby, affects microbial colonization of the gut and maintains a homeostatic ecology of commensal bacteria (138).
These intestinal immunity features are subject to commensal-host mutualisms with extensive bidirectional interaction. For instance, the mucosal expression of Reg3 lectins is regulated by the commensal microbiome through the production of the short-chain fatty acids (SCFAs) propionate and butyrate as major microbial metabolites of Clostridia strains and signaling through G protein-coupled receptor 43 (GPR43) on enterocytes (139). Likewise, the protein and oligosaccharide composition as well as barrier function of the intestinal mucus depends on the colonization with commensal microbes (140). Intestinal secretion of IgA is also determined by the presence of commensal microbes as germ-free animals have ten-fold lower levels of total IgA (141). Specifically, a complex microbiome that contains members of the phylum Proteobacteria promotes a T cell dependent induction of systemic IgA that can further protect against polymicrobial sepsis (142).
Besides these non-immunological features of mucosal microbe – host mutualism, the gut microbiome is essential in the ontogeny, maturation and modulation of the adaptive, T cell-driven immunity both in the intestines, extra-intestinal organs and systemically. This interaction starts very early in life because after birth the colonizing gut commensals induce the development of intestinal lymphoid tissues and maturation of myeloid and lymphoid cells, which imprints the immune system with a reactivity level that persists long into adulthood (143). Germ-free mice, in contrast, show defects in multiple specific immunocyte populations such as reduced Th1, Th17 and regulatory T cells in the intestines and mesenteric lymph nodes, impaired cytotoxicity NK cells or compromised innate lymphoid cell (ILC) function (144). In a seminal mouse study aimed to screen for immunomodulatory human gut symbionts, Geva-Zatorsky et al (Cell 2017) reported the effects of 53 individual bacteria on cellular immune phenotypes after mono-colonization of germ-free laboratory mice. Only a handful of symbionts were found to increase T cell frequencies in the intestines, including segmented filamentous bacteria (SFB) and Th17 cells, Coprobacillus and IL-10+ T cells, or Bifidobacterium longum and IFNγ+ Th1 cells (145). So far, only a few studies have assessed associations between immune cell phenotypes, effector molecules and gut microbiome profiles in healthy humans. For instance, the abundance of the genus Bacteroides has been linked to the Th1 cell numbers in the mucosa of the sigmoid colon (146). Furthermore, pathogen-induced immune cytokine responses are hypothesized to be modulated by the intestinal microbiome in healthy humans. Alistipes, Clostridium or Bilophila spp. were reported to protect against lipopolysaccharide-induced TNFα release from monocytes, whereas Faecalibacterium spp. may protect against IL-17 responses (147).
Immune cells recognize and react to small molecules produced by gut commensal microbes such as the above mentioned SCFAs. Along these lines, propionate and butyrate have been identified as major SCFAs that can stimulate the expansion and immune-suppressive properties of the regulatory T cells in the colon either through GPR43 receptor signaling or inhibition of histone deacetylases (HDACs) on the level of DCs (148, 149). Recently, bacterial transformation of bile acids which creates a complex pool of steroids was also observed to induce peripheral regulatory T cells by acting on DCs to diminish their immunostimulatory properties (150). Bacteroides fragilis and thetaiotaomicron are among the commensals that contribute significantly to bile acid metabolism in the gut (151).
Overall, recent research has greatly enhanced the understandings of the intimate, but complicated crosstalk between the microbiome and the immune system. Nevertheless, many unknowns and challenges remain in disentangling microbiome-immunity interactions in health and disease, notably cancer immunotherapies, which we will cover in the next chapters.
Before widespread use of next-generation sequencing techniques, culture-based methods already provided evidence that chemotherapeutic agents such as 5-fluorouracil (5-FU) perturb the oral and fecal microbiota of laboratory animals with an expansion of gram-negative anaerobes (152). These findings were later expanded by 16S rRNA sequencing results revealing a decrease in Eubacterium and Ruminococcus spp. as beneficial, SCFA producing bacteria (153). The complex interaction of microbes and chemotherapy is also reflected by pre-clinical results showing that the efficacy and toxicity of the drugs (e.g., 5-FU or irinotecan) depend on the intestinal bacterial composition (154, 155).
In humans, there is only a sparse literature on whether chemotherapy affects diversity and composition of the gut microbiome, and the results are often difficult to interpret due to antibiotic treatments, the development of gastrointestinal toxicities with inflammation and diarrhea, and surgical complications (156–159).
Regarding the toxicity of CART therapies, the most notable toxicities are CRS and neurotoxicity. However, gastrointestinal adverse events were reported in up to 28% of patients in a retrospective analysis by Abu-Sbeih et al. (160). All of them developed diarrhea, but also colitis and bloody stools were observed in rare cases, which can confound any microbiome configuration in these patients. Regarding chemotherapy effects without T cell transfer, in patients receiving a myeloablative conditioning therapy for NHL, chemotherapy was associated with an expansion of Enterobacteriaceae and Enterococcaceae, and a loss of Ruminococcaceae, Lachnospiraceae and Bifidobacterium spp. without any additional administration of other drugs such as antibiotics (157). Induction chemotherapy in patients with AML was also observed to reduce the alpha-diversity (i.e., the diversity and species richness within a patient’s biospecimen) of the oral and fecal microbiome during the course of therapy. However, the administration of broad-spectrum antibiotics was found to be the major factor responsible for the loss of diversity in this cohort (156). In patients with advanced colorectal cancer, for instance, adjuvant chemotherapies with irinotecan-, oxaliplatin- and 5-FU-based regimens have also been reported to alter the bacterial and fungal community structure of the gut with outgrowth of Veillonella, Candida, Malassizia spp. and loss of Clostridiales and Faecalibacterium spp. (159). As another anticancer therapy associated with intestinal toxicity, pelvic irradiation for prostate cancer therapy was found to reduce intestinal microbiome diversity, notably in patients developing radiation enteropathy. Radiotherapy also led to a decreased microbial SCFA production capacity and decreased levels of homeostatic rectal mucosa cytokines predisposing to intestinal toxicities and adverse events in these patients (158).
As described above, the intestinal microbiota plays a major role in shaping innate and adaptive immunity (161). Therefore, it is plausible that the efficacy of immunotherapies that rely on peripheral immune cells, such as adoptive cell therapy (ACT) and ICI therapies, may depend on intestinal microbiome configurations and their metabolic outputs as it has been reported repeatedly in recent years (102, 162–164). The role of the microbiome in ICI immunotherapy has already been discussed in extensive reviews (165, 166).
We will rather focus on gut microbiome effects on the outcomes of allogeneic hematopoietic cell transplantation (allo HCT) which is a model and predecessor of modern T cell transfer therapies, such as CART therapy, against hematologic malignancies. During allo HCT, a combination of events such as chemotherapy conditioning, exposure to antibiotics and other drugs such as antacids, or changes in diet greatly affect the structure and function of the gut microbiome leading to injuries and dysbiotic states. As we have shown previously, expansion of Enterococcus spp. within the gut microbiome up to a level of mono-domination early after transplant represents a hallmark of dysbiosis in allo HCT patients (167). This expansion was primarily driven by the administration of broad-spectrum antibiotics, but also diets containing specific nutrients such as lactose that nourish enterococci and related facultative pathogens like streptococci and other members of the Lactobacillales order. Clinically, an Enterococcus mono-domination was associated with reduced overall survival and exacerbated intestinal graft-versus-host disease (GVHD), a major toxicity of allo HCT (167).
Antibiotic treatments and domination events with antibiotic-resistant or pathogenic microbes usually come with a substantial reduction of the diversity of the gut microbiome. Notably, in a multi-centric study with 1362 allo HCT patients, a loss of microbiome diversity early after transplant was associated with a higher risk of transplantation-related death and death attributable to GVHD (168). The risk of relapse, however, was not affected by the diversity. Such a disruption of the intestinal microbial milieu not only leads to the expansion of potential pathogens, but also to a loss of functionality in the host-commensal mutualism. In children undergoing allo HCT and receiving antibiotics, butyrate and propionate, two major microbiome-derived SCFAs, were depleted in the intestinal contents after transplant, and were lower in those children that developed GVHD (169). This is of particular importance as SCFAs are protecting the host, notably gut enterocytes, from deleterious gut GVHD (170).
Besides the development of GVHD, the intestinal microbiome has also been associated with antitumor efficacy and the occurrence of relapse after allo HCT. In a study by Peled et al. (171), a higher peri-transplant abundance of a cluster of intestinal bacteria centered around Eubacterium limosum was associated with a decreased risk of relapse/progression of disease. Eubacterium limosum is a producer of butyrate, propionate, acetate and lactate (172), and these SCFA haven been attributed to the antitumor efficacy of ICI therapies in humans (163).
The development of relapse after allo HCT is determined by the ability of the engrafting immune system to remove residual leukemia cells through a graft-versus-leukemia (GVL) effect. GVL depends on alloreactive, antigen-specific T cells, NK cell alloreactivity and activated DCs (173). In a large patient-centered study, Schluter et al. (174) described associations of the human gut microbiome with the dynamics of the immune system focusing on peripheral immune cells after allo HCT. Abundances of intestinal Ruminococcus gnavus and Staphylococcus spp. were positively associated with blood lymphocyte counts and post-transplant reconstitution, whereas Faecalibacterium was associated with increases in monocyte rates. Although this study lacks details on the subtypes of lymphocytes and other immune cells, it is the first of its kind to demonstrate a clinical relevance of microbiome-immune interactions in humans which has so far only been reported in animal models.
So far, there is only little reporting on the role of the gut microbiome in CART therapy. However, there are several lines of evidence highlighting plausible connections as described above and coming from other preclinical experiments. Intriguingly, the efficacy of ACT against HPV-associated cancers in a mouse model was observed to depend on the microbiome configuration of the host at steady-state by comparing mice obtained from two different vendors (Jackson vs. Harlan laboratories) (175). The microbiome differences were attributed to a diverse range of Bacteroides, Prevotella and Rikenellaceae. Following treatment with vancomycin, ACT efficacy was enhanced in Jackson mice, but not in Harlan mice. The increased ACT efficacy was also phenocopied by fecal microbiota transfers, with the microbiome affecting both tumor infiltration and expansion of reactive T cells (175). Along these lines, Paulos et al. reported that translocation of microbiome-derived components such as lipopolysaccharide (LPS) from a radiation-injured gastrointestinal tract activated the innate immune system of tumor-bearing mice and, thereby, enhanced the ACT efficacy through Toll-like receptor (TLR) 4 signaling (176). Antibiotic treatment, in turn, reduced the activation of the innate immune system in irradiated mice, and impaired the effectiveness of ACT. The broad-spectrum antibiotic-induced reduction of ACT efficacy was also shown for adoptive CD4+ T cell transfers against implanted colorectal tumors in mice (177). In the only published data on antibiotic effects in mouse CARTs so far, it was reported that the administration of broad-spectrum antibiotics did not mitigate the tumor killing and survival of mice carrying A20 B-cell lymphomas and treated with CD19-CARTs (177). However, the depletion of the gut microbiome significantly prolonged the persistence of CARTs and the duration of B-cell aplasia in these mice.
The different sensitivity of T cells for ACT and CD19-CARTs to antibiotics may be explained by a differential dependence of these T cells on the intestinal microbiome for executing their effector functions. As described above, irradiation-induced microbial translocation augments the function of adoptively transferred tumor-specific T cells via an increased activation of DCs. As a side note, chemotherapy is also known to cause translocation of bacteria across the intestinal epithelium, and thereby amplifies the function of effector T cells (178, 179). Kuzma et al. also described that the presence of antibiotics in cell culture did not affect T cell expansion and cytokine production during in vitro antigenic stimulation (177). The loss of sensitivity to antibiotics by CD19-CART may indicate that microbial translocation occurring during the conditioning process does not impact the function of CD19-CART. This phenomenon may be attributable to the unique feature of CARTs as these cells are equipped to exert effector functions instantly upon tumor encounter without the need to be reactivated by DCs as it is the case in ACT.
Despite the lack of effect of CD19-CART efficacy in mice as shown in this very first study, there is preliminary evidence from human studies pointing towards the role of the gut microbiome and dysbiosis in outcomes of CART immunotherapies. In a single-center study published as meeting abstract, Smith et al. observed differences in the gut microbiome composition assessed by 16S rRNA gene sequencing before CD19-CART infusion associated with therapy outcomes. Lachnospiraceae and Ruminococcaceae were found to be more prevalent in patients who achieved complete remission, whereas Peptostreptococcaceae and Clostridiales were more abundant in non-responders; microbiome diversity was not observed to be different in responder vs. non-responders (180). Another small study, again published as meeting abstract, reported that the administration of broad-spectrum antibiotics up to 14 days before CART infusion was associated with reduced 3-months response rates. The administration of antibiotics was also found to reduce gut microbiome diversity, and, in turn, facilitated the expansion of enterococci in the patients’ stools (181). These preliminary studies corroborate our hypothesis of associations between microbial diversity and taxonomic composition around the days of CART administration and clinical outcomes of this immunotherapy.
In the following chapters we will focus on mechanistic details of microbe - immune interactions that are of particular relevance for patients receiving CD19-CART immunotherapies, notably the effects of broad-spectrum antibiotic treatments and the role of Enterococcus spp. and Klebsiella spp. as facultative pathogenic commensals frequently observed in cohorts of blood cancer patients.
Antibiotic treatment is one of the major causes of perturbation of the human gut microbiome. Alterations in the microbial composition is dependent on dosage, duration of treatment, form of application, and class of antibiotics (182). Various studies investigated the short- and long-term effects on the gut microbiome during and after antibiotic treatment. The most common observation was a decrease of alpha-diversity of the microbiome. For example, a 6-day cefuroxime administration led to a 5% loss of alpha-diversity, whereas 6-days of ciprofloxacin administration resulted in a 40% loss of microbial diversity (183). The loss of diversity was reported to come at the expense of bacteria belonging to Actinobacteria and Firmicutes whereas, the relative abundance of Proteobacteria was found increased (182, 184–186).
Assessing the effects of individual antibiotic classes on the composition of the microbiome, macrolide antibiotics were associated with a decrease of Actinobacteria, especially bifidobacteria, and Firmicutes, especially lactobacilli, and with an increase of the relative abundance of Bacteroidetes and Proteobacteria (182, 184). Beta-lactam targeting antibiotics were reported to influence the abundance of Firmicutes and Actinobacteria negatively while leading to an increase of Proteobacteria (184). In addition, cephalosporine administration can also lead to an increase of Bacteroidetes (182–184). Glycopeptides do not undergo reabsorption in the gut, and are, thus, considered to perturb the gut microbiome. Vancomycin, for instance, was found to reduce the abundance of Firmicutes while increasing Proteobacteria abundances, especially Enterobacteriaceae, both in human and mice (182–184, 186, 187). Fluoroquinolones, like ciprofloxacin, decreased the abundances of Firmicutes and Actinobacteria, especially bifidobacteria, but increased the relative abundance of Bacteroidetes (182, 184). Clindamycin, a lincosamide, reduced the abundance of Actinobacteria, mainly bifidobacteria, and of Firmicutes, notably lactobacilli (182). Interestingly, the frequently used antibiotic amoxicillin was found to exert only minor effects on microbial diversity (184, 187).
As presented above on some examples, antibiotic treatments can lead to a loss of potentially beneficial bacteria, resulting in alterations in bacterial metabolites, like SCFA, and colonization with potentially pathogenic bacteria, like Enterococcus or Klebsiella spp. (188–190).
The relationship between SCFA, including acetic acid, propionic acid and butyric acid, and mucosal homeostasis is well studied (189, 191). SCFAs are involved in several homeostatic processes, including inhibition of histone deacetylases and regulation of hematopoietic cell and non-hematopoietic cell differentiation, resulting in an anti-inflammatory and tolerant condition for the mucosal homeostasis. Furthermore, SCFAs are capable to suppress nuclear factor-κB (NF-κB) in immune cells, resulting in inhibition of the production of proinflammatory cytokines (191–194). SCFAs are also involved in maintaining the physical mucosal barrier as they have been shown to increase transcription of mucin genes and production of mucus in intestinal goblet cells. In addition, cell-cell contacts are influenced by SCFAs, improving tight-junction integrity (191, 195).
Infection of pathogenic bacteria is a major cause of death in cancer patients. To elucidate the mechanisms underlying infections, it is important to take a closer look into the healthy microbiome and its interplay with potentially harmful microbes. There are direct and indirect microbiome-related mechanisms to provide colonization resistance. Direct mechanisms include production of antimicrobial molecules, called bacteriocins, with the ability of killing other bacteria, mostly active against closely related species providing selection advantages. Also known is the ability of microbial metabolites, such as SCFAs, to inhibit growth of pathogenic bacteria, like Salmonella typhimurium (189, 196). Indirect mechanisms involve the host’s immune system, which is shaped by the commensal microbiome and involves antimicrobial peptides as described above. The disruption of the gut microbiome by antibiotic treatment has been shown to reduce colonization resistance against pathogenic bacteria with an increased susceptibility to infection (196–198). In laboratory rats, a 14-day ceftriaxone treatment led to increased abundance of E. coli, Staphylococcus spp., and hemolytic bacteria that was associated with reduction in fecal propionic acid and higher colonic epithelial permeability and a disturbance of oxidant-antioxidant balance. This mucosal injury was accompanied by increased bacterial translocation, suggesting antibiotic-driven susceptibility to blood-stream invasion of potential pathogenic bacteria (196). In a murine model, a single dose of either streptomycin, clindamycin or a cocktail of metronidazole, neomycin, vancomycin and clindamycin, led to susceptibility to Listeria monocytogenes infections. In ex vivo experiments, contents of the small intestine of untreated mice, co-cultured under anaerobic conditions, were efficient in eliminating Listeria monocytogenes, suggesting a microbiota dependent but immune independent role of colonization resistance. Especially Clostridia strains were associated with protection against Listeria monocytogenes infection (197). It was also demonstrated that Bacteroides spp. effectively inhibited Salmonella typhimurium growth by production of propionic acid leading to an intracellular acidification of the pathogen (199). Secondary bile-acids, produced by commensals through 7-α-dihydroxylation of primary bile-acids, as for instance by Clostridium scindens, promoted resistance to colonization with Clostridioides difficile in mice (200). The role of the gut microbiome in providing resistance against colonization and infection was repeatedly demonstrated in patients with recurrent infection with Clostridioides difficile, and restoring a functional microbiome through fecal-microbiota-transfer (FMT) is known to treat recurrent infection more efficiently than antibiotic therapy (201).
Part of the human gut microbiome is the mycobiome, which is barely investigated yet. A recent study showed the influence of antibiotic treatment on fungi in the gut environment. Especially Candida albicans, an opportunistic pathogen was found to expand during antibiotic treatment in patients, suggesting a regulatory function of commensal bacteria for Candida albicans in vivo growth. Recovery of the microbial community, in turn, led to an effective suppression of the initial outgrowth of the fungus (183). Thus, continuous antibiotic administration may lead to increased susceptibility to intestinal and/or blood-stream infections with Candida and other fungi (202).
Colonization and infection with potentially pathogenic bacteria after antibiotic treatment is a widely observed phenomenon, especially in hospitalized patients (186, 189, 190, 197, 199, 201, 203, 204). Underlying mechanisms include the development of antibiotic resistances or the loss of colonization resistance against potential pathogenic bacteria. In addition, intrinsic antibiotic resistances can facilitate colonization and expansion, like resistances of Enterococcus faecalis and Enterococcus faecium to cephalosporines and aminoglycosides (205).
The development of antibiotic resistances is a major subject of research, even more after it was declared by the WHO as one of the top 10 global public health threats (95). Antibiotic resistance genes (ARGs) are phylogenetically conserved genes and their existence was dated back before the age of antibiotics (206). Due to the excessive use of antibiotics all over the world, the amount of ARGs in commensal and environmental bacteria was presumably never higher as today. It is well documented that the increased ARG-copy number in bacteria correlates with the use of antibiotics (207, 208). As reviewed recently (207), antibiotic pressure drives horizontal gene transfer (HGT) of ARGs with a specific selection of more differentiated ARGs. This selection occurs on the single nucleotide level indicating the high efficiency in gene selection conferring survival advantage. A process that facilitates the development of antibiotic resistance is antibiotic treatment under sub-inhibitory concentrations. It has been shown that antibiotic administration under sub-inhibitory conditions augment gene transfer and gene recombination (208, 209). This effective antibiotic resistance development of potential pathogens is associated with the expansion of individual microbes during antibiotic administration with potentially fatal consequences for the host.
Several microbes are known for their pathologic colonization properties, causing disturbance in the microbial community and contributing to severe infections. Among them are bacteria belonging to the genus Enterococcus, Klebsiella, Salmonella and Streptococcus (186, 189, 199, 203–205, 210, 211). Various mechanisms of colonization advantages and virulence development were described in recent studies, but little is known about immune regulation during colonization or the infection with these pathobionts.
Clinical and experimental research has focused on the cause and consequence of expansion of Enterococcus spp. in hospitalized patient. We have shown previously that antibiotic therapy, but also diet, contribute to mono-domination of the gut with enterococci in allo HCT patients (167), and preliminary data found similar pattern in CD19-CART treated patients (181). As observed in several other clinical conditions, antibiotic treatments facilitate intestinal outgrowth of commensal Enterococcus spp., mostly Enterococcus faecium and Enterococcus faecalis. Recent studies enabled a more comprehensive view on mechanisms forcing a commensal to become a pathogenic bacterium.
Enterococcus spp. are extremely flexible in adapting to their environment, as reviewed recently (205), especially in response to environmental stress, e.g., the exposure to antibiotics. A recent study investigated the impact of Enterococcus faecalis on the host under various conditions, like mono-colonization or co-colonization with a colitogenic bacterial consortium. Mono-colonization of germ-free mice with two different strains of Enterococcus faecalis has been shown to increase the number of DCs and regulatory T cells in the colon at steady-state (145). However, using IL-10 deficient germ-free mice as a background mouse strain that is more susceptible to inflammation, mono-colonization with Enterococcus faecalis resulted in a severe inflammation of the colon accompanied by an upregulation of genes involved in stress responses towards unfavorable conditions in enterococci, including protease and chaperone genes and oxidative stress resistance (212). Co-colonization of germ-free, IL-10 deficient mice with Enterococcus faecalis together with a colitogenic bacterial consortium, showed an oppositional gene expression pattern involved in growth and replication and only moderate intestinal inflammation. Thus, gene expression and behavior of Enterococcus faecalis is dependent on the microbial environment (213). These results indicate a crucial role of the microbial environment in maintaining virulence of enterococci and could explain the ambivalent role of Enterococcus spp. as harmless commensal versus being a pathogen. Interestingly, environmental stress in bacteria could also be induced by catecholamines like norepinephrine (214). Norepinephrine led to differently expressed protein patterns, associated with higher bile acid tolerance, aggregation capability and biofilm forming abilities, indicating enhanced environmental resistance with potential role in pathogenesis of colonization and infection during increased stress response of the host (215).
Mechanisms of virulence are, among others, dependent on virulence factors expressed in microbes. Several virulence factors of Enterococcus spp. have been shown to interfere with the intestinal environment. Gelatinase E (GelE), a matrix metalloproteinase found in enterococci, is one of the major virulence factors shown to impair the integrity of gut mucosal barriers. In vitro experiments showed that the media of GelE-producing enterococci co-incubated with macrophages lead to an altered morphology of intestinal epithelial cells (IECs) (216). In vivo mono-associations of germ-free, IL-10 deficient mice with GelE producing Enterococcus strains resulted in colitis, whereas colonization with GelE deficient enterococci attenuated the inflammation (212). Other virulence factors associated with aggregation, adhesion or β-hemolysis, contributing to environmental resistance or cell destruction are described in detail elsewhere (217, 218).
Regarding their immune system interactions, Klebsiella pneumoniae isolated from various infection sides of hospitalized patients revealed different immune stimulatory patterns in in vitro and in vivo-experimental studies. Intraperitoneal injection with Klebsiella strains, for instance, was associated with lower survival of mice when challenged with strains that induced a low TNFα response in peripheral blood mononuclear cells (PBMCs) in vitro, suggesting a potential correlation of immune evasion ability and severity of infection (211).
Amoxicillin treatment in mice led to drastically increased abundance of Klebsiella variicola with a significantly increased antibiotic resistance profile and elevated virulence compared to control. Inoculation of antibiotic pre-treated mice with these virulent strains resulted in increased pro-inflammatory cytokine production with more severe colon damage, whereas isolated strains of untreated mice led to almost no inflammation, confirming the hypothesis of antibiotic-enhanced virulence. Inflammation was accompanied by modulated Th1 and regulatory T cell differentiation in peripheral lymphoid tissues. Especially Th1 cells were increased and regulatory T cells were decreased in cervical and mesenterial lymph nodes compared to mice inoculated with Klebsiella variicola isolated from non-antibiotic treated mice (219). Besides the effect of antibiotics to induce expansion of individual microbes of the intestinal microbiota, it is hypothesized that antibiotics can favor the de novo intestinal colonization with microbes of the oral cavity. Ectopic colonization of germ-free mice with saliva samples of patients with inflammatory bowel disease (IBD) showed a significant Klebsiella spp. dependent induction of Th1 cells in the intestinal lamina propria. Other commensals inhabiting the oral cavity failed to induce Th1 cells in these experiments. Knock-out models revealed TLR signaling and IL-18 to contribute to DC mediated Th1 cell induction (220).
Taken together, the expansion of individual microbes can have various impact on the mucosal and systemic immunity. However, there are still several open questions regarding the interplay of individual microbes, their virulence and the host immune system, notably T cell-driven anticancer immunity.
Effector function of T cells defined by the magnitude of cytokine production depends on triggering of the TCR by antigen recognition, engagement of costimulatory molecules and availability of proinflammatory cytokines (221, 222). Increasing evidence indicates that microbial metabolites and cell wall components can regulate the T cell function via host receptors and other target molecules (Figure 1).
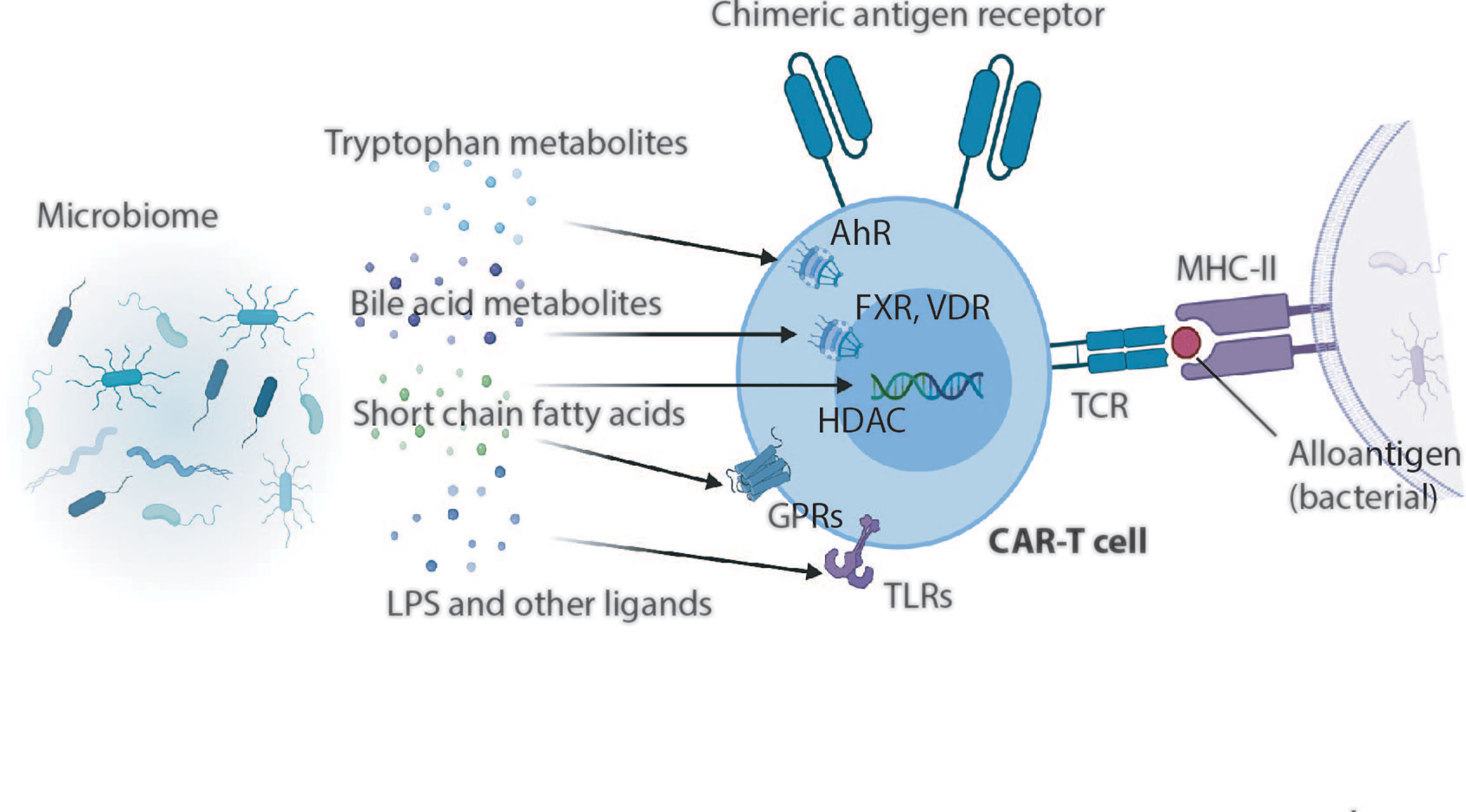
Figure 1 Gut microbial metabolites and microbial ligands can exert far reaching influences on T cells and presumably also CAR-T cells. Whereas tryptophan metabolites can act on T cells through the cytoplasmatic aryl hydrocarbon receptor (AhR), bile acid metabolites induce T cell differentiation and change effector functions through actions on vitamin D receptors (VDR), Farnesoid X receptor (FXR), and also through TGR5, PXR or LXR (no shown in this illustration). Short chain fatty acids act on T cells through G-protein coupled receptors 41, 43 or 109 (GPRs), or regulate immune cell differentiation and function through histone deacetylases (HDACs). Bacterial derived membrane fractions or secreted proteins modulate T cells via Toll-like receptors (TLRs) expressed on activated T cells.
TLRs are widely expressed in the innate immune system, but certain TLRs such as TLR2, TLR3, TLR5 and TLR9 are also expressed in T cells (223). They can act as costimulatory molecules to enhance proliferation and/or cytokine production of TCR-stimulated T cells. For instance, it has been demonstrated that TLR2 provides co-stimulatory signals to amplify the magnitude of IL-2, TNFα and IFNγ production in murine and human CD8+ T cells, and to increase the percentage of polyfunctional T cells against tumor cells (224). Intriguingly, overexpression of TLR2 in CD19- or mesothelin-targeted CARTs was associated with the expansion, persistency and antitumor function of respective CARTs in mouse models (225). This is of particular interest as commensal enterococci specifically stimulate TLR2 on immune cells via lipoteichoic acid (226); therefore, and based on preliminary data evaluating the impact of the microbiome on toxicity and efficacy of CARTs (181) we hypothesize that an intestinal expansion of enterococci and the co-occurrence of severe CRS in CART treated patients may be due to a direct effect of Enterococcus spp. on CARTs via TLR2.
Microbiome derived SCFAs can also act on T cells either through G-protein coupled receptors GPR43 or GPR109 receptor signaling or inhibition of histone deacetylases (HDACs). We have shown recently in an allo HCT mouse model that GPR109 is expressed on activated T cells and significantly contributes to the metabolic fitness of T cells. T cells lacking this receptor are able to proliferate upon antigen stimulation, but undergo activation-induced cell death (227). Notably, antibiotic-induced loss of gut commensal anaerobes leads to a depletion of SCFAs in the host; this, in turn, could explain reduced efficacies of T cell-driven cancer immunotherapies through a reduced metabolic fitness of T cells in an antibiotic-treated host.
By-products of microbial tryptophan metabolism in the gut such as indoles and 5-hydroxytryptophan are further important metabolites that can affect T cell function through the aryl hydrocarbon receptor (AhR). AhR is expressed in different body compartments, but also on T cells. Mounting evidence indicates that AhR plays multiple roles in modulating CD4+ T differentiation and function (228). Recently, it has been shown that 5-hydroxytryptophan induced AhR activation of tumor-infiltrating CD8+ T cells induces a downregulation of cytokines and effector molecules rendering T cells exhausted and dysfunctional in the tumor microenvironment (229). Bile acid metabolites represent another major class of microbiome-derived metabolites that can control T cell differentiation and effector function through the nuclear receptors Farnesoid X receptor (FXR) or vitamin D receptor (150, 230–232).
Besides regulatory and co-stimulatory actions of microbial metabolites on T cell function and antitumor immunity, the concept of “holoimmunity”, i.e., T cell-receptor mediated tolerance against the host and the microbial community residing within the host (233), has emerged as an interesting new area of research in cancer immunity. In this context, cross-reactivity between antigens expressed in commensal bacteria and neoepitopes in melanomas has been demonstrated in a mouse model. In detail, colonization of mice with commensal Bifidobacterium breve shape the TCR repertoire to target a bacterial epitope SVY. These T cells cross-react with the model neoantigen SIY on melanomas leading to decreased tumor growth and extended survival in Bifidobacterium colonized mice (234). In a recent seminal study, this concept of microbe-associated neoantigens in antitumor immunity was developed even further by demonstrating that bacteria residing within melanoma cells can stimulate an HLA presentation of novel peptides that elicit immune response of tumor-infiltrating T cells (235). Although not extensively studied, a considerable number of bacterial reads has also been recently found in DLBCL tissue (236), but whether bacteria-derived HLA-bound neoantigens also stimulate antitumor T cells or even CARTs remains speculative.
The problematic role of antibiotics on immunotherapy outcomes has been discussed in previous chapters. If antibiotics, however, are administered to cancer patients because of medical needs such as infections, strategies to protect a healthy microbiome are currently discussed and evaluated. For instance, a colon-targeted antibiotic adsorbent drug has been shown to protect the gut microbiome from moxifloxacin-induced loss of diversity in healthy volunteers (237). This drug is currently investigated in a phase III study with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) patients to investigate beneficial effects on the occurrence of life-threatening complications and increased survival.
Another strategy to restore microbial homeostasis after antibiotic-induced microbiome injuries or other dysbiotic states relies on the transfer of a healthy microbiome from a healthy donor to a patient. Such a fecal microbiota transfer (FMT) has been very successfully implemented in clinical medicine as a rescue treatment for Clostridioides difficile infections (201). In cancer immunotherapy medicine, two pilot studies recently reported on the induction of de novo responses to ICI by FMT in melanoma patients (238, 239).
Apart from transferring whole microbial ecologies, administering individual bacteria as exogenous probiotics has been shown to reward benefits in immunotherapy. For instance, Bifidobacterium spp. treatment of tumor-bearing mice improved cancer-specific immunity and response to ICI therapy (240). In other studies, administration of Bifidobacterium or Lactobacillus spp. was observed to abrogate ICI-associated colitis in mice (241, 242).
As probiotics may be ineffective as exogenous bacteria colonize poorly and live only for a short time in host intestines, prebiotic strategies to enhance endogenous or exogenous microbes in the gut have been developed. Smectite, a type of mineral clay has been shown to promote the expansion of Lactobacillus and Bifidobacterium by intestinal biofilm formation in mice, and, thereby, enhances the antitumor efficacies of ICI or chemotherapy in tumor mouse models (243).
Diet is considered one of the major modulators of the gut microbiome, and among several nutrients, fibers are essential for microbial homeostasis as they provide essential substrates for microbial growth (244). Low intake of fibers reduces the production of SCFAs and mediates long-term, irreversible shifts in the composition of the microbiome (245). In cancer immunotherapy, a small study (published as meeting abstract) examined the effects of diet and supplement use amongst 46 patients receiving ICIs, and found that patients reporting high-fiber diets were approximately five times more likely to respond to therapy compared to patients with low-fiber intake (246).
To the best of our knowledge, none of the above-mentioned strategies has been studied in CART animal models or within clinical trials. However, these studies would provide enough evidence to initiate trials focusing on dietary or prebiotics approaches to modulate the microbiome and, subsequently, clinical outcomes in CART immunotherapy.
Multiple clinical and preclinical studies add to the growing evidence that the intestinal microbiome acts in concert with the host in determining antitumor immunity and the outcomes of cancer immunotherapy. For this reason, it is becoming increasingly clear that environmental or external injuries to the microbiome such as administration of broad-spectrum antibiotics can attenuate the efficacy of antitumor immunotherapies that even affect long-term survival. This emerging concept has already led to adjust antibiotic prophylaxis in clinical practice for allo HCT by switching to anaerobe sparing antibiotics (247), or by applying FMT to increase the anticancer efficacy of ICI (239).
Due to the short time from their approval, there are only preliminary data suggesting again an important role of the gut microbiome in CD19 CART immunotherapies. In Figure 2, we are summarizing plausible perturbations of the microbiome in CART treated patients and their potential impact on the course of the therapy and outcomes. Finally, several modalities are highlighted including dietary interventions through prebiotics, probiotic therapies, FMT and adjustments in antibiotic regimens or phage-based antimicrobial therapies that can help restoring an injured microbiome. Whether these strategies improve response and prognosis of blood cancer patients treated with CART immunotherapies is subject of current studies.
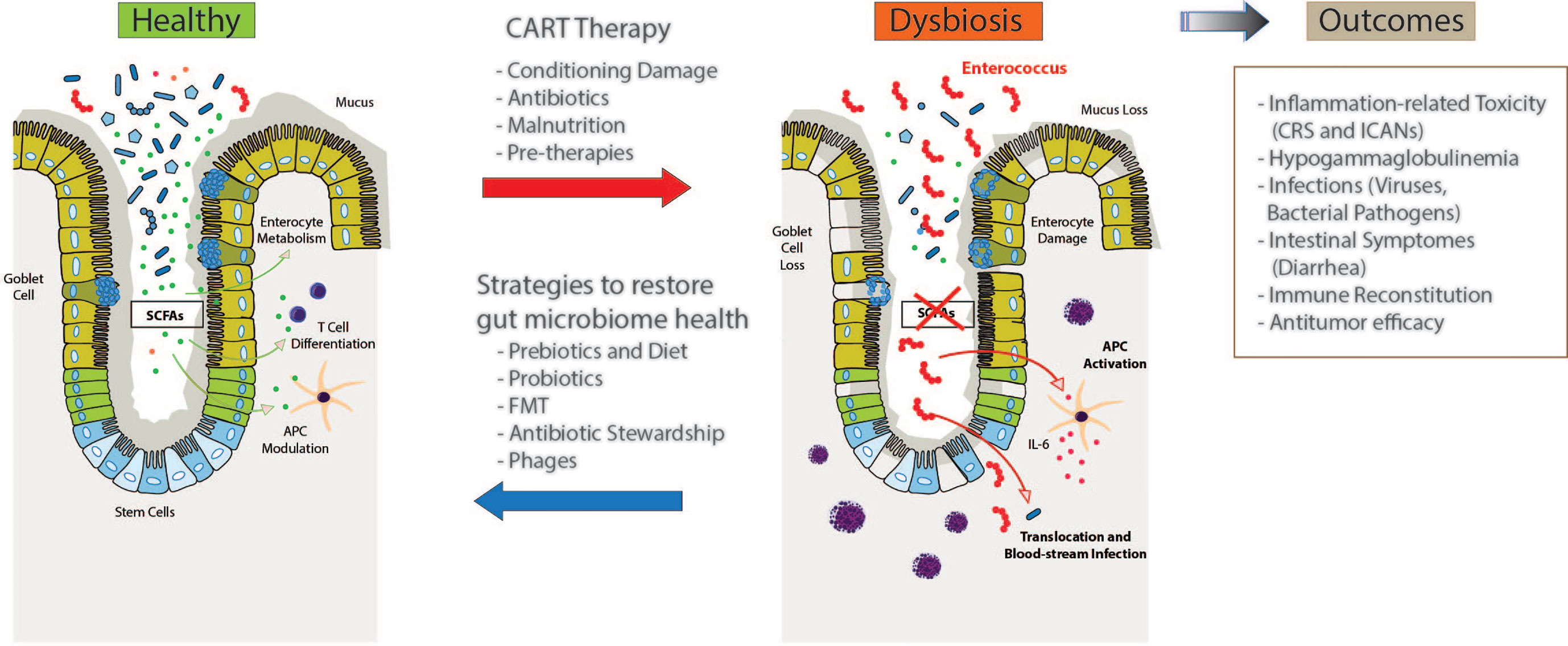
Figure 2 Intestinal microbiome dysbiosis and potential associations with patient outcomes following therapy with chimeric antigen receptor (CAR)-T cells (CART). Patients receiving CARTs are exposed to various environmental conditions including cytotoxic conditioning regimens, antibiotics, and dietary changes or malnutrition that might contribute to changes in the intestinal microbiome. In addition, previous therapies including chemotherapy, stem cell transplantation or even surgeries can affect microbiome homeostasis. These injuries to the intestinal microbiome, in turn, are hypothesized to affect clinical outcome and toxicity to CART treatment, including cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANs), infections, gastrointestinal adverse events, immune reconstitution, and relapse through various different immunological mechanisms involving different hematopoietic and non-hematopoietic cell populations. Several strategies have been proposed, although not specifically for CAR-T cell immunotherapy, to restore the intestinal microbiome health which comprise pre- and probiotics, fecal microbiome transfer (FMT), de-escalated antibiotic exposures or even phage-based therapies to mitigate expansion of potential pathobionts.
M-LS, RR and CS-T wrote the manuscript and performed literature search. MS contributed to concept and interpretation. All authors contributed to the article and approved the submitted version.
This work was supported by funding of the Deutsche José Carreras Leukämie-Stiftung to CS-T. M-LS is supported by the Olympia Morata Program of the Medical Faculty of Heidelberg.
M-LS: consultancy for Kite/Gilead, Takeda. Advisory board Kite/Gilead, Janssen. None of these sources were involved in the writing of this review. MS: research grants from Apogenix, Hexal and Novartis. Travel grants from Hexal and Kite. Financial support for educational activities and conferences from bluebird bio, Kite and Novartis. Advisory board member of MSD. (Co-)PI of clinical trials of MSD, GSK, Kite and BMS. Co-Founder and shareholder of TolerogenixX Ltd. None of these sources were involved in the writing of this review.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank M. Hinkelbein for help editing references.
1. Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol (2015) 33(17):1974–82. doi: 10.1200/JCO.2014.59.4358
2. Xin Yu J, Hubbard-Lucey VM, Tang J. Immuno-Oncology Drug Development Goes Global. Nat Rev Drug Discovery (2019) 18(12):899–900. doi: 10.1038/d41573-019-00167-9
3. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable Complete Responses in Heavily Pretreated Patients With Metastatic Melanoma Using T-cell Transfer Immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. doi: 10.1158/1078-0432.CCR-11-0116
4. Buder-Bakhaya K, Hassel JC. Biomarkers for Clinical Benefit of Immune Checkpoint Inhibitor Treatment-a Review From the Melanoma Perspective and Beyond. Front Immunol (2018) 9:1474. doi: 10.3389/fimmu.2018.01474
5. Rosenberg SA, Dudley ME. Adoptive Cell Therapy for the Treatment of Patients With Metastatic Melanoma. Curr Opin Immunol (2009) 21(2):233–40. doi: 10.1016/j.coi.2009.03.002
6. Dudley M. Adoptive Cell Therapy for Patients With Melanoma. J Cancer (2011) 2:360–2. doi: 10.7150/jca.2.360
7. Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene Rejection Responses Contribute to Attenuated Persistence of Adoptively Transferred CD20/CD19-specific Chimeric Antigen Receptor Redirected T Cells in Humans. Biol Blood Marrow Transplant (2010) 16(9):1245–56. doi: 10.1016/j.bbmt.2010.03.014
8. Wang E, Wang L-C, Tsai C-Y, Bhoj V, Gershenson Z, Moon E, et al. Generation of Potent T-Cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. J Cancer Immunol Res (2015) 3(7):815–26. doi: 10.1158/2326-6066.CIR-15-0054
9. Hombach AA, Chmielewski M, Rappl G, Abken H. Adoptive Immunotherapy With Redirected T Cells Produces CCR7- Cells That are Trapped in the Periphery and Benefit From Combined CD28-OX40 Costimulation. Hum Gene Ther (2013) 24(3):259–69. doi: 10.1089/hum.2012.247
10. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-Based Chimeric Antigen Receptors Program Bipolar TH17/TH1 Cells. Blood (2014) 124(7):1070–80. doi: 10.1182/blood-2013-10-535245
11. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 Costimulation Improves Expansion and Persistence of Chimeric Antigen Receptor-Modified T Cells in Lymphoma Patients. J Clin Invest (2011) 121(5):1822–6. doi: 10.1172/JCI46110
12. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-Treat Leukemia Remission by CD19 Car T Cells of Defined Formulation and Dose in Children and Young Adults. Blood (2017) 129(25):3322–31. doi: 10.1182/blood-2017-02-769208
13. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
14. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and Toxicity Management of 19-28z Car T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Transl Med (2014) 6(224):224ra25. doi: 10.1126/scitranslmed.3008226
15. Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-Term Follow-up of CD19 Car Therapy in Acute Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):449–59. doi: 10.1056/NEJMoa1709919
16. Frey NV, Gill S, Hexner EO, Schuster S, Nasta S, Loren A, et al. Long-Term Outcomes From a Randomized Dose Optimization Study of Chimeric Antigen Receptor Modified T Cells in Relapsed Chronic Lymphocytic Leukemia. J Clin Oncol (2020) 38(25):2862–71. doi: 10.1200/JCO.19.03237
17. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric Antigen Receptor T Cells Persist and Induce Sustained Remissions in Relapsed Refractory Chronic Lymphocytic Leukemia. Sci Transl Med (2015) 7(303):303ra139. doi: 10.1126/scitranslmed.aac5415
18. Siddiqi T, Soumerai JD, Dorritie KA, Stephens DM, Riedell PA, Arnason JE, et al. Rapid Undetectable MRD (Umrd) Responses in Patients With Relapsed/Refractory (R/R) Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (Cll/Sll) Treated With Lisocabtagene Maraleucel (Liso-Cel), a CD19-Directed Car T Cell Product: Updated Results From Transcend CLL 004, a Phase 1/2 Study Including Patients With High-Risk Disease Previously Treated With Ibrutinib. Blood (2019) 134(Supplement_1):503. doi: 10.1182/blood-2019-127603
19. Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With Cd19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J Clin Oncol (2017) 35(26):3010–20. doi: 10.1200/JCO.2017.72.8519
20. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang ML, Arnason JE, et al. Pivotal Safety and Efficacy Results From Transcend NHL 001, a Multicenter Phase 1 Study of Lisocabtagene Maraleucel (Liso-Cel) in Relapsed/Refractory (R/R) Large B Cell Lymphomas. Blood (2019) 134(Supplement_1):241. doi: 10.1182/blood-2019-127508
21. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. Kte-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med (2020) 382(14):1331–42. doi: 10.1056/NEJMoa1914347
22. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel Car T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
23. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1-2 Trial. Lancet Oncol (2019) 20(1):31–42. doi: 10.1016/S1470-2045(18)30864-7
24. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med (2017) 377(26):2545–54. doi: 10.1056/NEJMoa1708566
25. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T Cells Expressing CD19 Chimeric Antigen Receptors for Acute Lymphoblastic Leukaemia in Children and Young Adults: A Phase 1 Dose-Escalation Trial. Lancet (London England) (2015) 385(9967):517–28. doi: 10.1016/S0140-6736(14)61403-3
26. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med (2014) 371(16):1507–17. doi: 10.1056/NEJMoa1407222
27. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. Cd19 CAR-T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J Clin Invest (2016) 126(6):2123–38. doi: 10.1172/JCI85309
28. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med (2019) 380(1):45–56. doi: 10.1056/NEJMoa1804980
29. European Medicines Agency. Yescarta (Axicabtagene Ciloleucel) - An Overview of Yescarta and Why it is Authorised in the EU (2018). Available at: https://www.ema.europa.eu/en/documents/overview/yescarta-epar-medicine-overview_en.pdf.
30. European Medicines Agency. Kymriah (Tisagenlecleucel) - An Overview of Kymriah and Why it is Authorised in the EU (2018). Available at: https://www.ema.europa.eu/en/documents/overview/kymriah-epar-medicine-overview_en.pdf (Accessed Feb. 19, 2021).
31. European Medicines Agency. Tecartus. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/tecartus.
32. Grupp S, Hu Z-H, Zhang Y, Keating A, Pulsipher MA, Philips C, et al. Tisagenlecleucel Chimeric Antigen Receptor (Car) T-Cell Therapy for Relapsed/Refractory Children and Young Adults With Acute Lymphoblastic Leukemia (All): Real World Experience From the Center for International Blood and Marrow Transplant Research (CIBMTR) and Cellular Therapy (Ct) Registry. Blood (2019) 134(Supplement_1):2619. doi: 10.1182/blood-2019-129279
33. Jaglowski S, Hu Z-H, Zhang Y, Kamdar M, Ghosh M, Lulla P, et al. Tisagenlecleucel Chimeric Antigen Receptor (Car) T-Cell Therapy for Adults With Diffuse Large B-Cell Lymphoma (Dlbcl): Real World Experience From the Center for International Blood & Marrow Transplant Research (Cibmtr) Cellular Therapy (Ct) Registry. Blood (2019) 134(Supplement_1):766. doi: 10.1182/blood-2019-130983
34. Nastoupil LJ, Jain MD, Feng L, Spiegel JY, Ghobadi A, Lin Y, et al. Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma Car T Consortium. J Clin Oncol Off J Am Soc Clin Oncol (2020) 38(27):3119–28. doi: 10.1200/jco.19.02104
35. Pasquini MC, Locke FL, Herrera AF, Siddiqi T, Ghobadi A, Komanduri KV, et al. Post-Marketing Use Outcomes of an Anti-CD19 Chimeric Antigen Receptor (Car) T Cell Therapy, Axicabtagene Ciloleucel (Axi-Cel), for the Treatment of Large B Cell Lymphoma (LBCL) in the United States (Us). Blood (2019) 134(Supplement_1):764. doi: 10.1182/blood-2019-124750
36. Han EQ, X-l Li, Wang C-r, Li T-f, Han S-y. Chimeric Antigen Receptor-Engineered T Cells for Cancer Immunotherapy: Progress and Challenges. J Hematol Oncol. 6:47 doi: 10.1186/1756-8722-6-47
37. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat Med (2015) 21(6):581–90. doi: 10.1038/nm.3838
38. Tammana S, Huang X, Wong M, Milone MC, Ma L, Levine BL, et al. 4-1BB and CD28 Signaling Plays a Synergistic Role in Redirecting Umbilical Cord Blood T Cells Against B-cell Malignancies. Hum Gene Ther (2010) 21(1):75–86. doi: 10.1089/hum.2009.122
39. Zhong X-S, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell-Mediated Tumor Eradication. Mol Ther 18: 413-420. Mol Ther J Am Soc Gene Ther (2009) 18:413–20. doi: 10.1038/mt.2009.210
40. Schubert M-L, Schmitt A, Neuber B, Hückelhoven-Krauss A, Kunz A, Wang L, et al. Third-Generation Car T Cells Targeting Cd19 Are Associated With an Excellent Safety Profile and Might Improve Persistence of CAR T Cells in Treated Patients. Blood (2019) 134(Supplement_1):51. doi: 10.1182/blood-2019-125423
41. Schubert M-L, Schmitt A, Sellner L, Neuber B, Kunz J, Wuchter P, et al. Treatment of Patients With Relapsed or Refractory CD19+ Lymphoid Disease With T Lymphocytes Transduced by RV-SFG.CD19.CD28.4-1BBzeta Retroviral Vector: A Unicentre Phase I/II Clinical Trial Protocol. BMJ Open (2019) 9(5):e026644. doi: 10.1136/bmjopen-2018-026644
42. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: Chimeric Antigen Receptor (CAR) T Cells Engineered With an Inducible Cytokine to Modulate the Tumor Stroma. Immunol Rev (2014) 257(1):83–90. doi: 10.1111/imr.12125
43. Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, et al. T Cell–Encoded CD80 and 4-1BBL Induce Auto- and Transcostimulation, Resulting in Potent Tumor Rejection. Nat Med (2007) 13(12):1440–9. doi: 10.1038/nm1676
44. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-Targeted CAR T Cells Induce Remission in B-ALL That is Naive or Resistant to CD19-targeted CAR Immunotherapy. Nat Med (2018) 24(1):20–8. doi: 10.1038/nm.4441
45. Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z, et al. Cd22 CAR T-Cell Therapy in Refractory or Relapsed B Acute Lymphoblastic Leukemia. Leukemia (2019) 33(12):2854–66. doi: 10.1038/s41375-019-0488-7
46. Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. Cd4/Cd8 T-Cell Selection Affects Chimeric Antigen Receptor (Car) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-Cd22 CAR T-Cell Trial. J Clin Oncol (2020) 38(17):1938–50. doi: 10.1200/jco.19.03279
47. Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and Immunological Responses After CD30-specific Chimeric Antigen Receptor-Redirected Lymphocytes. J Clin Invest (2017) 127(9):3462–71. doi: 10.1172/JCI94306
48. Wang CM, Wu ZQ, Wang Y, Guo YL, Dai HR, Wang XH, et al. Autologous T Cells Expressing Cd30 Chimeric Antigen Receptors for Relapsed or Refractory Hodgkin Lymphoma: An Open-Label Phase I Trial. Clin Cancer Res (2017) 23(5):1156–66. doi: 10.1158/1078-0432.CCR-16-1365
49. Hill LC, Rouce RH, Smith TS, Yang L, Srinivasan M, Zhang H, et al. Safety and Anti-Tumor Activity of CD5 Car T-Cells in Patients With Relapsed/Refractory T-Cell Malignancies. Blood (2019) 134(Supplement_1):199. doi: 10.1182/blood-2019-129559
50. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T Cells Expressing an anti-B-cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Multiple Myeloma. Blood (2016) 128(13):1688–700. doi: 10.1182/blood-2016-04-711903
51. Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J Clin Oncol (2018) 36(22):2267–80. doi: 10.1200/JCO.2018.77.8084
52. Zhao WH, Liu J, Wang BY, Chen YX, Cao XM, Yang Y, et al. A Phase 1, Open-Label Study of LCAR-B38M, a Chimeric Antigen Receptor T Cell Therapy Directed Against B Cell Maturation Antigen, in Patients With Relapsed or Refractory Multiple Myeloma. J Hematol Oncol (2018) 11(1):141. doi: 10.1186/s13045-018-0681-6
53. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med (2019) 380(18):1726–37. doi: 10.1056/NEJMoa1817226
54. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJ, et al. CD33-Specific Chimeric Antigen Receptor T Cells Exhibit Potent Preclinical Activity Against Human Acute Myeloid Leukemia. Leukemia (2015) 29(8):1637–47. doi: 10.1038/leu.2015.52
55. Budde L, Song JY, Kim Y, Blanchard S, Wagner J, Stein AS, et al. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment With CD123-Specific Car T Cells: A First-in-Human Clinical Trial. Blood (2017) 130(Supplement 1):811. doi: 10.1182/blood.V130.Suppl_1.811.811
56. Cummins KD, Frey N, Nelson AM, Schmidt A, Luger S, Isaacs RE, et al. Treating Relapsed / Refractory (Rr) AML With Biodegradable Anti-Cd123 CAR Modified T Cells. Blood (2017) 130(Supplement 1):1359. doi: 10.1182/blood.V130.Suppl_1.1359.1359
57. Wang J, Chen S, Xiao W, Li W, Wang L, Yang S, et al. CAR-T Cells Targeting CLL-1 as an Approach to Treat Acute Myeloid Leukemia. J Hematol Oncol (2018) 11(1):7. doi: 10.1186/s13045-017-0553-5
58. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. Her2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol (2017) 3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184
59. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma After Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375(26):2561–9. doi: 10.1056/NEJMoa1610497
60. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A Single Dose of Peripherally Infused EGFRvIII-directed Car T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients With Recurrent Glioblastoma. Sci Trans Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
61. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. Car T Cells Administered in Combination With Lymphodepletion and PD-1 Inhibition to Patients With Neuroblastoma. Mol Ther (2017) 25(9):2214–24. doi: 10.1016/j.ymthe.2017.05.012
62. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor Activity and Long-Term Fate of Chimeric Antigen Receptor–Positive T Cells in Patients With Neuroblastoma. Blood (2011) 118(23):6050–6. doi: 10.1182/blood-2011-05-354449
63. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-Specific T Cells Engineered to Coexpress Tumor-Specific Receptors: Persistence and Antitumor Activity in Individuals With Neuroblastoma. Nat Med (2008) 14(11):1264–70. doi: 10.1038/nm.1882
64. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering Chimeric Antigen Receptor T Cells for Targeted Therapy of Autoimmune Disease. Science (2016) 353(6295):179–84. doi: 10.1126/science.aaf6756
65. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
66. Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s Lymphoma With a Defined Ratio of CD8+ and CD4+ CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Sci Transl Med (2016) 8(355):355ra116. doi: 10.1126/scitranslmed.aaf8621
67. Dudley M, Wunderlich J, Yang J, Sherry R, Topalian S, Restifo N, et al. Adoptive Cell Transfer Therapy Following Non-Myeloablative But Lymphodepleting Chemotherapy for the Treatment of Patients With Refractory Metastatic Melanoma. J Clin Oncol (2005) 23:2346–57. doi: 10.1200/JCO.2005.00.240
68. Guimond M, Veenstra RG, Grindler DJ, Zhang H, Cui Y, Murphy RD, et al. Interleukin 7 Signaling in Dendritic Cells Regulates the Homeostatic Proliferation and Niche Size of CD4+ T Cells. Nat Immunol (2009) 10(2):149–57. doi: 10.1038/ni.1695
69. Wallen H, Thompson JA, Reilly JZ, Rodmyre RM, Cao J, Yee C. Fludarabine Modulates Immune Response and Extends In Vivo Survival of Adoptively Transferred CD8 T Cells in Patients With Metastatic Melanoma. PloS One (2009) 4(3):e4749. doi: 10.1371/journal.pone.0004749
70. Gokbuget N, Dombret H, Bonifacio M, Reichle A, Graux C, Faul C, et al. Blinatumomab for Minimal Residual Disease in Adults With B-cell Precursor Acute Lymphoblastic Leukemia. Blood (2018) 131(14):1522–31. doi: 10.1182/blood-2017-08-798322
71. Kantarjian H, Stein A, Gökbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab Versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N Engl J Med (2017) 376(9):836–47. doi: 10.1056/NEJMoa1609783
72. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and Biomarkers of Severe Cytokine Release Syndrome After CD19 Chimeric Antigen Receptor-Modified T-Cell Therapy. Blood (2017) 130(21):2295–306. doi: 10.1182/blood-2017-06-793141
73. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome After Chimeric Antigen Receptor T-Cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discovery (2016) 6(6):664–79. doi: 10.1158/2159-8290.CD-16-0040
74. Titov A, Petukhov A, Staliarova A, Motorin D, Bulatov E, Shuvalov O, et al. The Biological Basis and Clinical Symptoms of CAR-T Therapy-Associated Toxicites. Cell Death Dis (2018) 9(9):897. doi: 10.1038/s41419-018-0918-x
75. Brudno JN, Kochenderfer JN. Toxicities of Chimeric Antigen Receptor T Cells: Recognition and Management. Blood (2016) 127(26):3321–30. doi: 10.1182/blood-2016-04-703751
76. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. Astct Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated With Immune Effector Cells. Biol Blood Marrow Transplant (2019) 25(4):625–38. doi: 10.1016/j.bbmt.2018.12.758
77. Schubert ML, Schmitt M, Wang L, Ramos CA, Jordan K, Muller-Tidow C, et al. Side-Effect Management of Chimeric Antigen Receptor (CAR) T-cell Therapy. Ann Oncol (2021) 32(1):34–48. doi: 10.1016/j.annonc.2020.10.478
78. Hunter BD, Jacobson CA. Car T-cell Associated Neurotoxicity: Mechanisms, Clinicopathologic Correlates, and Future Directions. J Natl Cancer Institute (2019) 111(7):646–54. doi: 10.1093/jnci/djz017
79. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current Concepts in the Diagnosis and Management of Cytokine Release Syndrome. Blood (2014) 124(2):188–95. doi: 10.1182/blood-2014-05-552729
80. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric Antigen Receptor T-cell Therapy - Assessment and Management of Toxicities. Nat Rev Clin Oncol (2018) 15(1):47–62. doi: 10.1038/nrclinonc.2017.148
81. Gust J, Hay KA, Hanafi L-A, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity After Adoptive Immunotherapy With CD19 Car-T Cells. Cancer Discovery (2017) 7(12):1404–19. doi: 10.1158/2159-8290.CD-17-0698
82. Gust J, Finney OC, Li D, Brakke HM, Hicks RM, Futrell RB, et al. Glial Injury in Neurotoxicity After Pediatric CD19-Directed Chimeric Antigen Receptor T Cell Therapy. Ann Neurol (2019) 86(1):42–54. doi: 10.1002/ana.25502
83. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and Biological Correlates of Neurotoxicity Associated With CAR T-Cell Therapy in Patients With B-cell Acute Lymphoblastic Leukemia. Cancer Discovery (2018) 8(8):958–71. doi: 10.1158/2159-8290.CD-17-1319
84. Fried S, Avigdor A, Bielorai B, Meir A, Besser MJ, Schachter J, et al. Early and Late Hematologic Toxicity Following CD19 CAR-T Cells. Bone Marrow Transplant (2019) 54(10):1643–50. doi: 10.1038/s41409-019-0487-3
85. Logue JM, Zucchetti E, Bachmeier CA, Krivenko GS, Larson V, Ninh D, et al. Immune Reconstitution and Associated Infections Following Axicabtagene Ciloleucel in Relapsed or Refractory Large B-cell Lymphoma. Haematologica (2020) 106(4)>:978–86. doi: 10.3324/haematol.2019.238634 haematol.2019.238634.
86. Schubert ML, Dietrich S, Stilgenbauer S, Schmitt A, Pavel P, Kunz A, et al. Feasibility and Safety of CD19 Car T Cell Treatment for B-cell Lymphoma Relapse After Allogeneic Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant (2020) 26(9):1575–80. doi: 10.1016/j.bbmt.2020.04.025
87. Arnold DE, Maude SL, Callahan CA, DiNofia AM, Grupp SA, Heimall JR. Subcutaneous Immunoglobulin Replacement Following CD19-specific Chimeric Antigen Receptor T-cell Therapy for B-cell Acute Lymphoblastic Leukemia in Pediatric Patients. Pediatr Blood Cancer (2020) 67(3):e28092. doi: 10.1002/pbc.28092
88. Doan A, Pulsipher MA. Hypogammaglobulinemia Due to CAR T-Cell Therapy. Pediatr Blood Cancer (2018) 65(4):e26914. doi: 10.1002/pbc.26914
89. Wudhikarn K, Palomba ML, Pennisi M, Garcia-Recio M, Flynn JR, Devlin SM, et al. Infection During the First Year in Patients Treated With CD19 Car T Cells for Diffuse Large B Cell Lymphoma. Blood Cancer J (2020) 10(8):79. doi: 10.1038/s41408-020-00346-7
90. Cordeiro A, Bezerra ED, Hirayama AV, Hill JA, Wu QV, Voutsinas J, et al. Late Events After Treatment With CD19-Targeted Chimeric Antigen Receptor Modified T Cells. Biol Blood Marrow Transplant (2020) 26(1):26–33. doi: 10.1016/j.bbmt.2019.08.003
91. Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious Complications of CD19-targeted Chimeric Antigen Receptor-Modified T-cell Immunotherapy. Blood (2018) 131(1):121–30. doi: 10.1182/blood-2017-07-793760
92. Park JH, Romero FA, Taur Y, Sadelain M, Brentjens RJ, Hohl TM, et al. Cytokine Release Syndrome Grade as a Predictive Marker for Infections in Patients With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia Treated With Chimeric Antigen Receptor T Cells. Clin Infect Dis (2018) 67(4):533–40. doi: 10.1093/cid/ciy152
93. Haidar G, Dorritie K, Farah R, Bogdanovich T, Nguyen MH, Samanta P. Invasive Mold Infections After Chimeric Antigen Receptor-Modified T-cell Therapy: A Case Series, Review of the Literature, and Implications for Prophylaxis. Clin Infect Dis (2019) 71(3):672–6. doi: 10.1093/cid/ciz1127
94. Wei J, Zhu X, Mao X, Huang L, Meng F, Zhou J. Severe Early Hepatitis B Reactivation in a Patient Receiving Anti-CD19 and Anti-CD22 Car T Cells for the Treatment of Diffuse Large B-cell Lymphoma. J Immunotherapy Cancer (2019) 7:315. doi: 10.1186/s40425-019-0790-y
95. Kansagra AJ, Frey NV, Bar M, Laetsch TW, Carpenter PA, Savani BN, et al. Clinical Utilization of Chimeric Antigen Receptor T Cells in B Cell Acute Lymphoblastic Leukemia: An Expert Opinion From the European Society for Blood and Marrow Transplantation and the American Society for Transplantation and Cellular Therapy. Biol Blood Marrow Transplant (2019) 25(3):e76–85. doi: 10.1016/j.bbmt.2018.12.068
96. Mahmoudjafari Z, Hawks KG, Hsieh AA, Plesca D, Gatwood KS, Culos KA. American Society for Blood and Marrow Transplantation Pharmacy Special Interest Group Survey on Chimeric Antigen Receptor T Cell Therapy Administrative, Logistic, and Toxicity Management Practices in the United States. Biol Blood Marrow Transplant (2019) 25(1):26–33. doi: 10.1016/j.bbmt.2018.09.024
97. Yakoub-Agha I, Chabannon C, Bader P, Basak GW, Bonig H, Ciceri F, et al. Management of Adults and Children Undergoing Chimeric Antigen Receptor T-Cell Therapy: Best Practice Recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (Jacie). Haematologica (2020) 105(2):297–316. doi: 10.3324/haematol.2019.229781
98. Neelapu SS. Managing the Toxicities of CAR T-Cell Therapy. Hematological Oncol (2019) 37(S1):48–52. doi: 10.1002/hon.2595
99. Vora SB, Waghmare A, Englund JA, Qu P, Gardner RA, Hill JA. Infectious Complications Following CD19 Chimeric Antigen Receptor T-cell Therapy for Children, Adolescents, and Young Adults. Open Forum Infect Dis (2020) 7(5):ofaa121. doi: 10.1093/ofid/ofaa121
100. Kamada N, Seo SU, Chen GY, Núñez G. Role of the Gut Microbiota in Immunity and Inflammatory Disease. Nat Rev Immunol (2013) 13(5):321–35. doi: 10.1038/nri3430
101. Ahmed J, Kumar A, Parikh K, Anwar A, Knoll BM, Puccio C, et al. Use of Broad-Spectrum Antibiotics Impacts Outcome in Patients Treated With Immune Checkpoint Inhibitors. Oncoimmunology (2018) 7(11):e1507670. doi: 10.1080/2162402x.2018.1507670
102. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut Microbiome Influences Efficacy of PD-1-based Immunotherapy Against Epithelial Tumors. Science (2018) 359(6371):91–7. doi: 10.1126/science.aan3706
103. Derosa L, Hellmann MD, Spaziano M, Halpenny D, Fidelle M, Rizvi H, et al. Negative Association of Antibiotics on Clinical Activity of Immune Checkpoint Inhibitors in Patients With Advanced Renal Cell and Non-Small-Cell Lung Cancer. Ann Oncol Off J Eur Soc Med Oncol (2018) 29(6):1437–44. doi: 10.1093/annonc/mdy103
104. Huemer F, Rinnerthaler G, Westphal T, Hackl H, Hutarew G, Gampenrieder SP, et al. Impact of Antibiotic Treatment on Immune-Checkpoint Blockade Efficacy in Advanced Non-Squamous non-Small Cell Lung Cancer. Oncotarget (2018) 9(23):16512–20. doi: 10.18632/oncotarget.24751
105. Elkrief A, El Raichani L, Richard C, Messaoudene M, Belkaid W, Malo J, et al. Antibiotics are Associated With Decreased Progression-Free Survival of Advanced Melanoma Patients Treated With Immune Checkpoint Inhibitors. Oncoimmunology (2019) 8(4):e1568812. doi: 10.1080/2162402x.2019.1568812
106. Pinato DJ, Howlett S, Ottaviani D, Urus H, Patel A, Mineo T, et al. Association of Prior Antibiotic Treatment With Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA Oncol (2019) 5(12):1774–8. doi: 10.1001/jamaoncol.2019.2785
107. Hopkins AM, Kichenadasse G, Karapetis CS, Rowland A, Sorich MJ. Concomitant Antibiotic Use and Survival in Urothelial Carcinoma Treated With Atezolizumab. Eur Urol (2020) 78(4):540–3. doi: 10.1016/j.eururo.2020.06.061
108. Derosa L, Routy B, Fidelle M, Iebba V, Alla L, Pasolli E, et al. Gut Bacteria Composition Drives Primary Resistance to Cancer Immunotherapy in Renal Cell Carcinoma Patients. Eur Urol (2020) 78(2):195–206. doi: 10.1016/j.eururo.2020.04.044
109. Mohiuddin JJ, Chu B, Facciabene A, Poirier K, Wang X, Doucette A, et al. Association of Antibiotic Exposure With Survival and Toxicity in Patients With Melanoma Receiving Immunotherapy. J Natl Cancer Institute (2021) 113(2):162–70. doi: 10.1093/jnci/djaa057
110. Tinsley N, Zhou C, Tan G, Rack S, Lorigan P, Blackhall F, et al. Cumulative Antibiotic Use Significantly Decreases Efficacy of Checkpoint Inhibitors in Patients With Advanced Cancer. Oncologist (2020) 25(1):55–63. doi: 10.1634/theoncologist.2019-0160
111. Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current Understanding of the Human Microbiome. Nat Med (2018) 24(4):392–400. doi: 10.1038/nm.4517
112. Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of Mammals and Their Gut Microbes. Science (2008) 320(5883):1647–51. doi: 10.1126/science.1155725
113. Zitvogel L, Galluzzi L, Viaud S, Vetizou M, Daillere R, Merad M, et al. Cancer and the Gut Microbiota: An Unexpected Link. Sci Transl Med (2015) 7(271):271ps1. doi: 10.1126/scitranslmed.3010473
114. Schwabe RF, Jobin C. The Microbiome and Cancer. Nat Rev Cancer (2013) 13(11):800–12. doi: 10.1038/nrc3610
115. Xavier JB, Young VB, Skufca J, Ginty F, Testerman T, Pearson AT, et al. The Cancer Microbiome: Distinguishing Direct and Indirect Effects Requires a Systemic View. Trends Cancer (2020) 6(3):192–204. doi: 10.1016/j.trecan.2020.01.004
116. Gill PA, van Zelm MC, Muir JG, Gibson PR. Review Article: Short Chain Fatty Acids as Potential Therapeutic Agents in Human Gastrointestinal and Inflammatory Disorders. Aliment Pharmacol Ther (2018) 48(1):15–34. doi: 10.1111/apt.14689
117. González A, Vázquez-Baeza Y, Knight R. SnapShot: The Human Microbiome. Cell (2014) 158(3):690–.e1. doi: 10.1016/j.cell.2014.07.019
118. Durack J, Lynch SV. The Gut Microbiome: Relationships With Disease and Opportunities for Therapy. J Exp Med (2019) 216(1):20–40. doi: 10.1084/jem.20180448
119. Fan Y, Pedersen O. Gut Microbiota in Human Metabolic Health and Disease. Nat Rev Microbiol (2021) 19(1):55–71. doi: 10.1038/s41579-020-0433-9
120. Johansson ME, Sjövall H, Hansson GC. The Gastrointestinal Mucus System in Health and Disease. Nat Rev Gastroenterol Hepatol (2013) 10(6):352–61. doi: 10.1038/nrgastro.2013.35
121. Pelaseyed T, Bergström JH, Gustafsson JK, Ermund A, Birchenough GM, Schütte A, et al. The Mucus and Mucins of the Goblet Cells and Enterocytes Provide the First Defense Line of the Gastrointestinal Tract and Interact With the Immune System. Immunol Rev (2014) 260(1):8–20. doi: 10.1111/imr.12182
122. Knoop KA, Gustafsson JK, McDonald KG, Kulkarni DH, Kassel R, Newberry RD. Antibiotics Promote the Sampling of Luminal Antigens and Bacteria Via Colonic Goblet Cell Associated Antigen Passages. Gut Microbes (2017) 8(4):400–11. doi: 10.1080/19490976.2017.1299846
123. Zheng D, Liwinski T, Elinav E. Interaction Between Microbiota and Immunity in Health and Disease. Cell Res (2020) 30(6):492–506. doi: 10.1038/s41422-020-0332-7
124. Repaske R. Lysis of Gram-Negative Bacteria by Lysozyme. Biochim Biophys Acta (1956) 22(1):189–91. doi: 10.1016/0006-3002(56)90240-2
125. Cowland JB, Johnsen AH, Borregaard N. hCAP-18, a Cathelin/Pro-Bactenecin-Like Protein of Human Neutrophil Specific Granules. FEBS Lett (1995) 368(1):173–6. doi: 10.1016/0014-5793(95)00634-l
126. Ganz T. Defensins: Antimicrobial Peptides of Innate Immunity. Nat Rev Immunol (2003) 3(9):710–20. doi: 10.1038/nri1180
127. Mukherjee S, Zheng H, Derebe MG, Callenberg KM, Partch CL, Rollins D, et al. Antibacterial Membrane Attack by a Pore-Forming Intestinal C-type Lectin. Nature (2014) 505(7481):103–7. doi: 10.1038/nature12729
128. Mookherjee N, Anderson MA, Haagsman HP, Davidson DJ. Antimicrobial Host Defence Peptides: Functions and Clinical Potential. Nat Rev Drug Discovery (2020) 19(5):311–32. doi: 10.1038/s41573-019-0058-8
129. Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjöberg J, Amir E, et al. Enteric Defensins are Essential Regulators of Intestinal Microbial Ecology. Nat Immunol (2010) 11(1):76–83. doi: 10.1038/ni.1825
130. Propheter DC, Chara AL, Harris TA, Ruhn KA, Hooper LV. Resistin-Like Molecule β is a Bactericidal Protein That Promotes Spatial Segregation of the Microbiota and the Colonic Epithelium. Proc Natl Acad Sci USA (2017) 114(42):11027–33. doi: 10.1073/pnas.1711395114
131. Yoshimura T, McLean MH, Dzutsev AK, Yao X, Chen K, Huang J, et al. The Antimicrobial Peptide CRAMP is Essential for Colon Homeostasis by Maintaining Microbiota Balance. J Immunol (2018) 200(6):2174–85. doi: 10.4049/jimmunol.1602073
132. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating Nlrp6 Inflammasome Signaling. Cell (2015) 163(6):1428–43. doi: 10.1016/j.cell.2015.10.048
133. Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. Protection Against Enteric Salmonellosis in Transgenic Mice Expressing a Human Intestinal Defensin. Nature (2003) 422(6931):522–6. doi: 10.1038/nature01520
134. Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated Signals Induce the Bactericidal Lectin RegIII Gamma and Protect Mice Against Intestinal Listeria Monocytogenes Infection. J Exp Med (2007) 204(8):1891–900. doi: 10.1084/jem.20070563
135. Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, et al. Vancomycin-Resistant Enterococci Exploit Antibiotic-Induced Innate Immune Deficits. Nature (2008) 455(7214):804–7. doi: 10.1038/nature07250
136. Martinoli C, Chiavelli A, Rescigno M. Entry Route of Salmonella Typhimurium Directs the Type of Induced Immune Response. Immunity (2007) 27(6):975–84. doi: 10.1016/j.immuni.2007.10.011
137. Macpherson AJ, Uhr T. Induction of Protective IgA by Intestinal Dendritic Cells Carrying Commensal Bacteria. Science (2004) 303(5664):1662–5. doi: 10.1126/science.1091334
138. Cerutti A, Rescigno M. The Biology of Intestinal Immunoglobulin A Responses. Immunity (2008) 28(6):740–50. doi: 10.1016/j.immuni.2008.05.001
139. Bajic D, Niemann A, Hillmer AK, Mejias-Luque R, Bluemel S, Docampo M, et al. Gut Microbiota-Derived Propionate Regulates the Expression of Reg3 Mucosal Lectins and Ameliorates Experimental Colitis in Mice. J Crohns Colitis (2020) 14(10):1462–72. doi: 10.1093/ecco-jcc/jjaa065
140. Johansson ME, Jakobsson HE, Holmén-Larsson J, Schütte A, Ermund A, Rodríguez-Piñeiro AM, et al. Normalization of Host Intestinal Mucus Layers Requires Long-Term Microbial Colonization. Cell Host Microbe (2015) 18(5):582–92. doi: 10.1016/j.chom.2015.10.007
141. Pabst O, Slack E. Iga and the Intestinal Microbiota: The Importance of Being Specific. Mucosal Immunol (2020) 13(1):12–21. doi: 10.1038/s41385-019-0227-4
142. Wilmore JR, Gaudette BT, Gomez Atria D, Hashemi T, Jones DD, Gardner CA, et al. Commensal Microbes Induce Serum Iga Responses That Protect Against Polymicrobial Sepsis. Cell Host Microbe (2018) 23(3):302–11.e3. doi: 10.1016/j.chom.2018.01.005
143. Al Nabhani Z, Eberl G. Imprinting of the Immune System by the Microbiota Early in Life. Mucosal Immunol (2020) 13(2):183–9. doi: 10.1038/s41385-020-0257-y
144. Kennedy EA, King KY, Baldridge MT. Mouse Microbiota Models: Comparing Germ-Free Mice and Antibiotics Treatment as Tools for Modifying Gut Bacteria. Front Physiol (2018) 9:1534. doi: 10.3389/fphys.2018.01534
145. Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, et al. Mining the Human Gut Microbiota for Immunomodulatory Organisms. Cell (2017) 168(5):928–43.e11. doi: 10.1016/j.cell.2017.01.022
146. James KR, Gomes T, Elmentaite R, Kumar N, Gulliver EL, King HW, et al. Distinct Microbial and Immune Niches of the Human Colon. Nat Immunol (2020) 21(3):343–53. doi: 10.1038/s41590-020-0602-z
147. Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, et al. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell (2016) 167(4):1125–36.e8. doi: 10.1016/j.cell.2016.10.020
148. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-cell Generation. Nature (2013) 504(7480):451–5. doi: 10.1038/nature12726
149. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science (2013) 341(6145):569–73. doi: 10.1126/science.1241165
150. Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, et al. Bacterial Metabolism of Bile Acids Promotes Generation of Peripheral Regulatory T Cells. Nature (2020) 581(7809):475–9. doi: 10.1038/s41586-020-2193-0
151. Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, et al. Microbial Bile Acid Metabolites Modulate Gut RORgamma(+) Regulatory T Cell Homeostasis. Nature (2020) 577(7790):410–5. doi: 10.1038/s41586-019-1865-0
152. Von Bültzingslöwen I, Adlerberth I, Wold AE, Dahlén G, Jontell M. Oral and Intestinal Microflora in 5-Fluorouracil Treated Rats, Translocation to Cervical and Mesenteric Lymph Nodes and Effects of Probiotic Bacteria. Oral Microbiol Immunol (2003) 18(5):278–84. doi: 10.1034/j.1399-302x.2003.00075.x
153. Le Bastard Q, Ward T, Sidiropoulos D, Hillmann BM, Chun CL, Sadowsky MJ, et al. Fecal Microbiota Transplantation Reverses Antibiotic and Chemotherapy-Induced Gut Dysbiosis in Mice. Sci Rep (2018) 8(1):6219. doi: 10.1038/s41598-018-24342-x
154. García-González AP, Ritter AD, Shrestha S, Andersen EC, Yilmaz LS, Walhout AJM. Bacterial Metabolism Affects the C. Elegans Response to Cancer Chemotherapeutics. Cell (2017) 169(3):431–41.e8. doi: 10.1016/j.cell.2017.03.046
155. Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, et al. Alleviating Cancer Drug Toxicity by Inhibiting a Bacterial Enzyme. Science (2010) 330(6005):831–5. doi: 10.1126/science.1191175
156. Galloway-Peña JR, Shi Y, Peterson CB, Sahasrabhojane P, Gopalakrishnan V, Brumlow CE, et al. Gut Microbiome Signatures Are Predictive of Infectious Risk Following Induction Therapy for Acute Myeloid Leukemia. Clin Infect Dis (2020) 71(1):63–71. doi: 10.1093/cid/ciz777
157. Montassier E, Gastinne T, Vangay P, Al-Ghalith GA, Bruley des Varannes S, Massart S, et al. Chemotherapy-Driven Dysbiosis in the Intestinal Microbiome. Aliment Pharmacol Ther (2015) 42(5):515–28. doi: 10.1111/apt.13302
158. Reis Ferreira M, Andreyev HJN, Mohammed K, Truelove L, Gowan SM, Li J, et al. Microbiota- and Radiotherapy-Induced Gastrointestinal Side-Effects (Mars) Study: A Large Pilot Study of the Microbiome in Acute and Late-Radiation Enteropathy. Clin Cancer Res (2019) 25(21):6487–500. doi: 10.1158/1078-0432.Ccr-19-0960
159. Shuwen H, Xi Y, Yuefen P, Jiamin X, Quan Q, Haihong L, et al. Effects of Postoperative Adjuvant Chemotherapy and Palliative Chemotherapy on the Gut Microbiome in Colorectal Cancer. Microb Pathog (2020) 149:104343. doi: 10.1016/j.micpath.2020.104343
160. Abu-Sbeih H, Ali FS, Naqash AR, Owen DH, Patel S, Otterson GA, et al. Resumption of Immune Checkpoint Inhibitor Therapy After Immune-Mediated Colitis. J Clin Oncol (2019) 37(30):2738–45. doi: 10.1200/jco.19.00320
161. Belkaid Y, Hand TW. Role of the Microbiota in Immunity and Inflammation. Cell (2014) 157(1):121–41. doi: 10.1016/j.cell.2014.03.011
162. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-Derived Inosine Modulates Response to Checkpoint Inhibitor Immunotherapy. Science (2020) 369(6510):1481–9. doi: 10.1126/science.abc3421
163. Nomura M, Nagatomo R, Doi K, Shimizu J, Baba K, Saito T, et al. Association of Short-Chain Fatty Acids in the Gut Microbiome With Clinical Response to Treatment With Nivolumab or Pembrolizumab in Patients With Solid Cancer Tumors. JAMA Netw Open (2020) 3(4):e202895. doi: 10.1001/jamanetworkopen.2020.2895
164. Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, et al. A Defined Commensal Consortium Elicits CD8 T Cells and Anti-Cancer Immunity. Nature (2019) 565(7741):600–5. doi: 10.1038/s41586-019-0878-z
165. Elkrief A, Derosa L, Kroemer G, Zitvogel L, Routy B. The Negative Impact of Antibiotics on Outcomes in Cancer Patients Treated With Immunotherapy: A New Independent Prognostic Factor? Ann Oncol (2019) 30(10):1572–9. doi: 10.1093/annonc/mdz206
166. Halsey T, Ologun G, Wargo J, Jenq RR. Uncovering the Role of the Gut Microbiota in Immune Checkpoint Blockade Therapy: A Mini-Review. Semin Hematol (2020) 57(1):13–8. doi: 10.1053/j.seminhematol.2020.05.002
167. Stein-Thoeringer CK, Nichols KB, Lazrak A, Docampo MD, Slingerland AE, Slingerland JB, et al. Lactose Drives Enterococcus Expansion to Promote Graft-Versus-Host Disease. Science (2019) 366(6469):1143–9. doi: 10.1126/science.aax3760
168. Peled JU, Gomes ALC, Devlin SM, Littmann ER, Taur Y, Sung AD, et al. Microbiota as Predictor of Mortality in Allogeneic Hematopoietic-Cell Transplantation. N Engl J Med (2020) 382(9):822–34. doi: 10.1056/NEJMoa1900623
169. Romick-Rosendale LE, Haslam DB, Lane A, Denson L, Lake K, Wilkey A, et al. Antibiotic Exposure and Reduced Short Chain Fatty Acid Production After Hematopoietic Stem Cell Transplant. Biol Blood Marrow Transplant (2018) 24(12):2418–24. doi: 10.1016/j.bbmt.2018.07.030
170. Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, Toubai T, et al. Gut Microbiome-Derived Metabolites Modulate Intestinal Epithelial Cell Damage and Mitigate Graft-Versus-Host Disease. Nat Immunol (2016) 17(5):505–13. doi: 10.1038/ni.3400
171. Peled JU, Devlin SM, Staffas A, Lumish M, Khanin R, Littmann ER, et al. Intestinal Microbiota and Relapse After Hematopoietic-Cell Transplantation. J Clin Oncol (2017) 35(15):1650–9. doi: 10.1200/jco.2016.70.3348
172. Kanauchi O, Fukuda M, Matsumoto Y, Ishii S, Ozawa T, Shimizu M, et al. Eubacterium Limosum Ameliorates Experimental Colitis and Metabolite of Microbe Attenuates Colonic Inflammatory Action With Increase of Mucosal Integrity. World J Gastroenterol (2006) 12(7):1071–7. doi: 10.3748/wjg.v12.i7.1071
173. Dickinson AM, Norden J, Li S, Hromadnikova I, Schmid C, Schmetzer H, et al. Graft-Versus-Leukemia Effect Following Hematopoietic Stem Cell Transplantation for Leukemia. Front Immunol (2017) 8:496. doi: 10.3389/fimmu.2017.00496
174. Schluter J, Peled JU, Taylor BP, Markey KA, Smith M, Taur Y, et al. The Gut Microbiota is Associated With Immune Cell Dynamics in Humans. Nature (2020) 588(7837):303–7. doi: 10.1038/s41586-020-2971-8
175. Uribe-Herranz M, Bittinger K, Rafail S, Guedan S, Pierini S, Tanes C, et al. Gut Microbiota Modulates Adoptive Cell Therapy Via CD8alpha Dendritic Cells and IL-12. JCI Insight (2018) 3(4):e94952. doi: 10.1172/jci.insight.94952
176. Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, et al. Microbial Translocation Augments the Function of Adoptively Transferred Self/Tumor-Specific CD8+ T Cells Via TLR4 Signaling. J Clin Invest (2007) 117(8):2197–204. doi: 10.1172/jci32205
177. Kuczma MP, Ding ZC, Li T, Habtetsion T, Chen T, Hao Z, et al. The Impact of Antibiotic Usage on the Efficacy of Chemoimmunotherapy is Contingent on the Source of Tumor-Reactive T Cells. Oncotarget (2017) 8(67):111931–42. doi: 10.18632/oncotarget.22953
178. Roberti MP, Yonekura S, Duong CPM, Picard M, Ferrere G, Tidjani Alou M, et al. Chemotherapy-Induced Ileal Crypt Apoptosis and the Ileal Microbiome Shape Immunosurveillance and Prognosis of Proximal Colon Cancer. Nat Med (2020) 26(6):919–31. doi: 10.1038/s41591-020-0882-8
179. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The Intestinal Microbiota Modulates the Anticancer Immune Effects of Cyclophosphamide. Science (2013) 342(6161):971–6. doi: 10.1126/science.1240537
180. Smith M, Littmann ER, Slingerland JB, Clurman A, Slingerland AE, Taur Y, et al. Intestinal Microbiota Composition Prior to CAR T Cell Infusion Correlates With Efficacy and Toxicity. Blood (2018) 132(Supplement 1):3492. doi: 10.1182/blood-2018-99-118628
181. Blumenberg V, Schubert ML, Zamir E, Schmidt S, Rohrbach R, Waldhoff P, et al. Antibiotic Therapy and Low Gut Microbiome Diversity Is Associated With Decreased Response and High Toxicity in BCP-ALL and DLBCL Patients After Treatment With CD19. Car T-Cells. Blood (2020) 136:33–34. doi: 10.1182/blood-2020-141210
182. Iizumi T, Battaglia T, Ruiz V, Perez Perez GI. Gut Microbiome and Antibiotics. Arch Med Res (2017) 48(8):727–34. doi: 10.1016/j.arcmed.2017.11.004
183. Seelbinder B, Chen J, Brunke S, Vazquez-Uribe R, Santhaman R, Meyer AC, et al. Antibiotics Create a Shift From Mutualism to Competition in Human Gut Communities With a Longer-Lasting Impact on Fungi Than Bacteria. Microbiome (2020) 8(1):133. doi: 10.1186/s40168-020-00899-6
184. Ianiro G, Tilg H, Gasbarrini A. Antibiotics as Deep Modulators of Gut Microbiota: Between Good and Evil. Gut (2016) 65(11):1906–15. doi: 10.1136/gutjnl-2016-312297
185. Shono Y, Docampo MD, Peled JU, Perobelli SM, Velardi E, Tsai JJ, et al. Increased GVHD-related Mortality With Broad-Spectrum Antibiotic Use After Allogeneic Hematopoietic Stem Cell Transplantation in Human Patients and Mice. Sci Transl Med (2016) 8(339):339ra71. doi: 10.1126/scitranslmed.aaf2311
186. Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, et al. Vancomycin-Resistant Enterococcus Domination of Intestinal Microbiota is Enabled by Antibiotic Treatment in Mice and Precedes Bloodstream Invasion in Humans. J Clin Invest (2010) 120(12):4332–41. doi: 10.1172/JCI43918
187. Vrieze A, Out C, Fuentes S, Jonker L, Reuling I, Kootte RS, et al. Impact of Oral Vancomycin on Gut Microbiota, Bile Acid Metabolism, and Insulin Sensitivity. J Hepatol (2014) 60(4):824–31. doi: 10.1016/j.jhep.2013.11.034
188. Geirnaert A, Calatayud M, Grootaert C, Laukens D, Devriese S, Smagghe G, et al. Butyrate-Producing Bacteria Supplemented In Vitro to Crohn’s Disease Patient Microbiota Increased Butyrate Production and Enhanced Intestinal Epithelial Barrier Integrity. Sci Rep (2017) 7(1):11450. doi: 10.1038/s41598-017-11734-8
189. Rivera-Chavez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, et al. Depletion of Butyrate-Producing Clostridia From the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe (2016) 19(4):443–54. doi: 10.1016/j.chom.2016.03.004
190. Palleja A, Mikkelsen KH, Forslund SK, Kashani A, Allin KH, Nielsen T, et al. Recovery of Gut Microbiota of Healthy Adults Following Antibiotic Exposure. Nat Microbiol (2018) 3(11):1255–65. doi: 10.1038/s41564-018-0257-9
191. Rooks MG, Garrett WS. Gut Microbiota, Metabolites and Host Immunity. Nat Rev Immunol (2016) 16(6):341–52. doi: 10.1038/nri.2016.42
192. Usami M, Kishimoto K, Ohata A, Miyoshi M, Aoyama M, Fueda Y, et al. Butyrate and Trichostatin A Attenuate Nuclear Factor Kappab Activation and Tumor Necrosis Factor Alpha Secretion and Increase Prostaglandin E2 Secretion in Human Peripheral Blood Mononuclear Cells. Nutr Res (2008) 28(5):321–8. doi: 10.1016/j.nutres.2008.02.012
193. Chang PV, Hao L, Offermanns S, Medzhitov R. The Microbial Metabolite Butyrate Regulates Intestinal Macrophage Function Via Histone Deacetylase Inhibition. Proc Natl Acad Sci USA (2014) 111(6):2247–52. doi: 10.1073/pnas.1322269111
194. Vinolo MA, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R. Suppressive Effect of Short-Chain Fatty Acids on Production of Proinflammatory Mediators by Neutrophils. J Nutr Biochem (2011) 22(9):849–55. doi: 10.1016/j.jnutbio.2010.07.009
195. Willemsen LE, Koetsier MA, van Deventer SJ, van Tol EA. Short Chain Fatty Acids Stimulate Epithelial Mucin 2 Expression Through Differential Effects on Prostaglandin E(1) and E(2) Production by Intestinal Myofibroblasts. Gut (2003) 52(10):1442–7. doi: 10.1136/gut.52.10.1442
196. Holota Y, Dovbynchuk T, Kaji I, Vareniuk I, Dzyubenko N, Chervinska T, et al. The Long-Term Consequences of Antibiotic Therapy: Role of Colonic Short-Chain Fatty Acids (SCFA) System and Intestinal Barrier Integrity. PloS One (2019) 14(8):e0220642. doi: 10.1371/journal.pone.0220642
197. Becattini S, Littmann ER, Carter RA, Kim SG, Morjaria SM, Ling L, et al. Commensal Microbes Provide First Line Defense Against Listeria Monocytogenes Infection. J Exp Med (2017) 214(7):1973–89. doi: 10.1084/jem.20170495
198. Baumler AJ, Sperandio V. Interactions Between the Microbiota and Pathogenic Bacteria in the Gut. Nature (2016) 535(7610):85–93. doi: 10.1038/nature18849
199. Jacobson A, Lam L, Rajendram M, Tamburini F, Honeycutt J, Pham T, et al. A Gut Commensal-Produced Metabolite Mediates Colonization Resistance to Salmonella Infection. Cell Host Microbe (2018) 24(2):296–307 e7. doi: 10.1016/j.chom.2018.07.002
200. Studer N, Desharnais L, Beutler M, Brugiroux S, Terrazos MA, Menin L, et al. Functional Intestinal Bile Acid 7alpha-Dehydroxylation by Clostridium Scindens Associated With Protection From Clostridium Difficile Infection in a Gnotobiotic Mouse Model. Front Cell Infect Microbiol (2016) 6:191. doi: 10.3389/fcimb.2016.00191
201. van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, et al. Duodenal Infusion of Donor Feces for Recurrent Clostridium Difficile. N Engl J Med (2013) 368(5):407–15. doi: 10.1056/NEJMoa1205037
202. Zhai B, Ola M, Rolling T, Tosini NL, Joshowitz S, Littmann ER, et al. High-Resolution Mycobiota Analysis Reveals Dynamic Intestinal Translocation Preceding Invasive Candidiasis. Nat Med (2020) 26(1):59–64. doi: 10.1038/s41591-019-0709-7
203. Al-Nassir WN, Sethi AK, Li Y, Pultz MJ, Riggs MM, Donskey CJ. Both Oral Metronidazole and Oral Vancomycin Promote Persistent Overgrowth of Vancomycin-Resistant Enterococci During Treatment of Clostridium Difficile-Associated Disease. Antimicrob Agents Chemother (2008) 52(7):2403–6. doi: 10.1128/AAC.00090-08
204. Martin RM, Bachman MA. Colonization, Infection, and the Accessory Genome of Klebsiella Pneumoniae. Front Cell Infect Microbiol (2018) 8:4. doi: 10.3389/fcimb.2018.00004
205. Garcia-Solache M, Rice LB. The Enterococcus: A Model of Adaptability to Its Environment. Clin Microbiol Rev (2019) 32(2):e00058–18. doi: 10.1128/CMR.00058-18
206. Datta N, Hughes VM. Plasmids of the Same Inc Groups in Enterobacteria Before and After the Medical Use of Antibiotics. Nature (1983) 306(5943):616–7. doi: 10.1038/306616a0
207. Li J, Rettedal EA, van der Helm E, Ellabaan M, Panagiotou G, Sommer MOA. Antibiotic Treatment Drives the Diversification of the Human Gut Resistome. Genomics Proteomics Bioinf (2019) 17(1):39–51. doi: 10.1016/j.gpb.2018.12.003
208. Couce A, Blazquez J. Side Effects of Antibiotics on Genetic Variability. FEMS Microbiol Rev (2009) 33(3):531–8. doi: 10.1111/j.1574-6976.2009.00165.x
209. Davies J, Spiegelman GB, Yim G. The World of Subinhibitory Antibiotic Concentrations. Curr Opin Microbiol (2006) 9(5):445–53. doi: 10.1016/j.mib.2006.08.006
210. Rendueles O. Deciphering the Role of the Capsule of Klebsiella Pneumoniae During Pathogenesis: A Cautionary Tale. Mol Microbiol (2020) 113(5):883–8. doi: 10.1111/mmi.14474
211. Pantelidou IM, Galani I, Georgitsi M, Daikos GL, Giamarellos-Bourboulis EJ. Interactions of Klebsiella Pneumoniae With the Innate Immune System Vary in Relation to Clone and Resistance Phenotype. Antimicrob Agents Chemother (2015) 59(11):7036–43. doi: 10.1128/AAC.01405-15
212. Steck N, Hoffmann M, Sava IG, Kim SC, Hahne H, Tonkonogy SL, et al. Enterococcus Faecalis Metalloprotease Compromises Epithelial Barrier and Contributes to Intestinal Inflammation. Gastroenterology (2011) 141(3):959–71. doi: 10.1053/j.gastro.2011.05.035
213. Lengfelder I, Sava IG, Hansen JJ, Kleigrewe K, Herzog J, Neuhaus K, et al. Complex Bacterial Consortia Reprogram the Colitogenic Activity of Enterococcus Faecalis in a Gnotobiotic Mouse Model of Chronic, Immune-Mediated Colitis. Front Immunol (2019) 10:1420. doi: 10.3389/fimmu.2019.01420
214. Belay T, Sonnenfeld G. Differential Effects of Catecholamines on In Vitro Growth of Pathogenic Bacteria. Life Sci (2002) 71(4):447–56. doi: 10.1016/s0024-3205(02)01683-1
215. Scardaci R, Varese F, Manfredi M, Marengo E, Mazzoli R, Pessione E. Enterococcus Faecium NCIMB10415 Responds to Norepinephrine by Altering Protein Profiles and Phenotypic Characters. J Proteomics (2021) 231:104003. doi: 10.1016/j.jprot.2020.104003
216. Belogortseva N, Krezalek M, Guyton K, Labno C, Poroyko V, Zaborina O, et al. Media From Macrophages Co-Incubated With Enterococcus Faecalis Induces Epithelial Cell Monolayer Reassembly and Altered Cell Morphology. PloS One (2017) 12(8):e0182825. doi: 10.1371/journal.pone.0182825
217. Fisher K, Phillips C. The Ecology, Epidemiology and Virulence of Enterococcus. Microbiol (Reading) (2009) 155(Pt 6):1749–57. doi: 10.1099/mic.0.026385-0
218. Ben Braiek O, Smaoui S. Enterococci: Between Emerging Pathogens and Potential Probiotics. BioMed Res Int (2019) 2019:5938210. doi: 10.1155/2019/5938210
219. Lin H, Wang Q, Liu L, Chen Z, Das R, Zhao Y, et al. Colonization of Mice With Amoxicillin-Associated Klebsiella Variicola Drives Inflammation Via Th1 Induction and Treg Inhibition. Front Microbiol (2020) 11:1256. doi: 10.3389/fmicb.2020.01256
220. Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, et al. Ectopic Colonization of Oral Bacteria in the Intestine Drives TH1 Cell Induction and Inflammation. Science (2017) 358(6361):359–65. doi: 10.1126/science.aan4526
221. Dong C. Cytokine Regulation and Function in T Cells. Annu Rev Immunol (2021) 39:51–76. doi: 10.1146/annurev-immunol-061020-053702
222. Hwang JR, Byeon Y, Kim D, Park SG. Recent Insights of T Cell Receptor-Mediated Signaling Pathways for T Cell Activation and Development. Exp Mol Med (2020) 52(5):750–61. doi: 10.1038/s12276-020-0435-8
223. Kabelitz D. Expression and Function of Toll-Like Receptors in T Lymphocytes. Curr Opin Immunol (2007) 19(1):39–45. doi: 10.1016/j.coi.2006.11.007
224. Salerno F, Freen-van Heeren JJ, Guislain A, Nicolet BP, Wolkers MC. Costimulation Through TLR2 Drives Polyfunctional Cd8(+) T Cell Responses. J Immunol (2019) 202(3):714–23. doi: 10.4049/jimmunol.1801026
225. Lai Y, Weng J, Wei X, Qin L, Lai P, Zhao R, et al. Toll-Like Receptor 2 Costimulation Potentiates the Antitumor Efficacy of CAR T Cells. Leukemia (2018) 32(3):801–8. doi: 10.1038/leu.2017.249
226. Park OJ, Han JY, Baik JE, Jeon JH, Kang SS, Yun CH, et al. Lipoteichoic Acid of Enterococcus Faecalis Induces the Expression of Chemokines Via TLR2 and PAFR Signaling Pathways. J Leukoc Biol (2013) 94(6):1275–84. doi: 10.1189/jlb.1012522
227. Docampo MD, Stein-Thoeringer CK, Lazrak A, da Silva MDB, Cross J, van den Brink MRM. Expression of the Butyrate/Niacin Receptor, GPR109a on T Cells Plays an Important Role in a Mouse Model of Graft Versus Host Disease. Blood (2018) 132(Supplement 1):61. doi: 10.1182/blood-2018-99-118783
228. Rothhammer V, Quintana FJ. The Aryl Hydrocarbon Receptor: An Environmental Sensor Integrating Immune Responses in Health and Disease. Nat Rev Immunol (2019) 19(3):184–97. doi: 10.1038/s41577-019-0125-8
229. Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, et al. IL-2 Regulates Tumor-Reactive CD8(+) T Cell Exhaustion by Activating the Aryl Hydrocarbon Receptor. Nat Immunol (2021) 22(3):358–69. doi: 10.1038/s41590-020-00850-9
230. Pols TWH, Puchner T, Korkmaz HI, Vos M, Soeters MR, de Vries CJM. Lithocholic Acid Controls Adaptive Immune Responses by Inhibition of Th1 Activation Through the Vitamin D Receptor. PloS One (2017) 12(5):e0176715. doi: 10.1371/journal.pone.0176715
231. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile Acid Metabolites Control TH17 and Treg Cell Differentiation. Nature (2019) 576(7785):143–8. doi: 10.1038/s41586-019-1785-z
232. Campbell C, Marchildon F, Michaels AJ, Takemoto N, van der Veeken J, Schizas M, et al. FXR Mediates T Cell-Intrinsic Responses to Reduced Feeding During Infection. Proc Natl Acad Sci USA (2020) 117(52):33446–54. doi: 10.1073/pnas.2020619117
233. Root-Bernstein R. Autoimmunity and the Microbiome: T-cell Receptor Mimicry of “Self” and Microbial Antigens Mediates Self Tolerance in Holobionts: The Concepts of “Holoimmunity” (TcR-mediated Tolerance for the Holobiont) and “Holoautoimmunity” (Loss of Tolerance for the Holobiont) are Introduced. Bioessays (2016) 38(11):1068–83. doi: 10.1002/bies.201600083
234. Bessell CA, Isser A, Havel JJ, Lee S, Bell DR, Hickey JW, et al. Commensal Bacteria Stimulate Antitumor Responses Via T Cell Cross-Reactivity. JCI Insight (2020) 5(8):e135597. doi: 10.1172/jci.insight.135597
235. Kalaora S, Nagler A, Nejman D, Alon M, Barbolin C, Barnea E, et al. Identification of Bacteria-Derived HLA-bound Peptides in Melanoma. Nature (2021) 592(7852):138–43. doi: 10.1038/s41586-021-03368-8
236. Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, et al. Microbiome Analyses of Blood and Tissues Suggest Cancer Diagnostic Approach. Nature (2020) 579(7800):567–74. doi: 10.1038/s41586-020-2095-1
237. de Gunzburg J, Ghozlane A, Ducher A, Le Chatelier E, Duval X, Ruppe E, et al. Protection of the Human Gut Microbiome From Antibiotics. J Infect Dis (2018) 217(4):628–36. doi: 10.1093/infdis/jix604
238. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal Microbiota Transplant Overcomes Resistance to anti-PD-1 Therapy in Melanoma Patients. Science (2021) 371(6529):595–602. doi: 10.1126/science.abf3363
239. Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal Microbiota Transplant Promotes Response in Immunotherapy-Refractory Melanoma Patients. Science (2021) 371(6529):602–9. doi: 10.1126/science.abb5920
240. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium Promotes Antitumor Immunity and Facilitates anti-PD-L1 Efficacy. Science (2015) 350(6264):1084–9. doi: 10.1126/science.aac4255
241. Wang F, Yin Q, Chen L, Davis MM. Bifidobacterium can Mitigate Intestinal Immunopathology in the Context of CTLA-4 Blockade. Proc Natl Acad Sci USA (2018) 115(1):157–61. doi: 10.1073/pnas.1712901115
242. Wang T, Zheng N, Luo Q, Jiang L, He B, Yuan X, et al. Probiotics Lactobacillus Reuteri Abrogates Immune Checkpoint Blockade-Associated Colitis by Inhibiting Group 3 Innate Lymphoid Cells. Front Immunol (2019) 10:1235. doi: 10.3389/fimmu.2019.01235
243. Han C, Song J, Hu J, Fu H, Feng Y, Mu R, et al. Smectite Promotes Probiotic Biofilm Formation in the Gut for Cancer Immunotherapy. Cell Rep (2021) 34(6):108706. doi: 10.1016/j.celrep.2021.108706
244. Makki K, Deehan EC, Walter J, Backhed F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe (2018) 23(6):705–15. doi: 10.1016/j.chom.2018.05.012
245. Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL. Diet-Induced Extinctions in the Gut Microbiota Compound Over Generations. Nature (2016) 529(7585):212–5. doi: 10.1038/nature16504
246. Lee KA, Shaw HM, Bataille V, Nathan P, Spector TD. Role of the Gut Microbiome for Cancer Patients Receiving Immunotherapy: Dietary and Treatment Implications. Eur J Cancer (2020) 138:149–55. doi: 10.1016/j.ejca.2020.07.026
Keywords: microbiome, immunotherapy, antibiotic, CAR (chimeric antigen receptor) T cells, microbe-host association
Citation: Schubert M-L, Rohrbach R, Schmitt M and Stein-Thoeringer CK (2021) The Potential Role of the Intestinal Micromilieu and Individual Microbes in the Immunobiology of Chimeric Antigen Receptor T-Cell Therapy. Front. Immunol. 12:670286. doi: 10.3389/fimmu.2021.670286
Received: 20 February 2021; Accepted: 04 May 2021;
Published: 31 May 2021.
Edited by:
Christoph Siegfried Niki Klose, Charité – Universitätsmedizin Berlin, GermanyReviewed by:
Francesca Ronchi, University of Bern, SwitzerlandCopyright © 2021 Schubert, Rohrbach, Schmitt and Stein-Thoeringer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph K. Stein-Thoeringer, Yy5zdGVpbi10aG9lcmluZ2VyQGRrZnouZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.