
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 27 April 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.669986
This article is part of the Research Topic Immunopathology of Type 1 Diabetes View all 21 articles
 Brendan K. Reed1,2,3
Brendan K. Reed1,2,3 John W. Kappler1,2,3,4*
John W. Kappler1,2,3,4*The T cell antigens driving autoimmune Type 1 Diabetes (T1D) have been pursued for more than three decades. When diabetogenic CD4 T cell clones and their relevant MHCII antigen presenting alleles were first identified in rodents and humans, the path to discovering the peptide epitopes within pancreatic beta cell proteins seemed straightforward. However, as experimental results accumulated, definitive data were often absent or controversial. Work within the last decade has helped to clear up some of the controversy by demonstrating that a number of the important MHCII presented epitopes are not encoded in the natural beta cell proteins, but in fact are fusions between peptide fragments derived from the same or different proteins. Recently, the mechanism for generating these MHCII diabetogenic chimeric epitopes has been attributed to a form of reverse proteolysis, called transpeptidation, a process that has been well-documented in the production of MHCI presented epitopes. In this mini-review we summarize these data and their implications for T1D and other autoimmune responses.
In the Non-Obese Diabetic (NOD) mouse model of T1D, a variety of CD4 T cell clones or T cell hybridomas were prepared that responded to antigens within the secretory granules of the beta cells of pancreatic islets of Langerhans (1, 2). In some cases, the protein source of the stimulatory activity was identified (1, 3), but in others, no target could be identified. A particularly stringent test for the relative contribution of these T cells to the disease came from introducing the T cell clones into immunodeficient NOD-SCID mouse lacking T cells and observing whether the clone was sufficient to induce T1D (4). A number of T cell clones failed this test, but others, originally isolated by investigators at the Barbara Davis Center (BDC) (1, 2) were very active. It has now taken an effort of more than two decades to identify the functional peptide epitopes recognized by these BDC CD4 T cells. This work has now identified diabetogenic CD4 T cell epitopes derived in part from three beta cell proteins - insulin, chromogranin A (ChgA) and islet amyloid polypeptide (IAPP). In each case a fusion to the N- or C-terminus fragment of these proteins to a peptide from another or the same protein was required to construct a fully stimulatory chimeric epitope. We begin with a short review of how these three altered antigenic epitopes were discovered.
Insulin has become recognized as a major CD4 T cell target in T1D in humans and the NOD mouse model of the disease, reviewed in (5). In the 1990’s at the BDC, a series of CD4 T cells clones were produced from the NOD mouse, including the prototypical BDC-12-4.1 and BDC-12-4.4 clones, that were reactive to a peptide from the insulin B chain, B:9-23, presented by the NOD MHCII allele, IAg7 (2, 3). Many of these clones were diabetogenic when introduced into NOD mice. Subsequently, the BDC (6) and other institutions (7, 8) went on to produce many other T cell clones and T cell hybridomas reactive to this peptide. Later, similar T cells were identified in human T1D reactive to the same peptide presented by human DQ8 (9–11). In IAg7 (12, 13), HLA-DQA1*03:01/DQB1*03:02 (HLA-DQ8) (14) and other MHCII alleles, the core of the peptide binding groove accepts 9 amino acids in the p1 to p9 positions. Therefore the 15 amino acid B:9-23 peptide could theoretically bind in multiple positions or “registers” (Regs) in the MHCII groove, each with different amino side chains interacting within anchoring pockets in the binding groove versus appearing on the surface for T cell receptor (TCR) recognition. Three registers for this peptide have been studied the most. The 9 amino acid cores of these epitopes are: Reg1-B:12-20, VEALYLVCG, Reg2-B:13−21, EALYLVCGE and Reg3-B:14-22, ALYLVCGER. Several studies proposed Reg1 or Reg2 bound epitopes as the relevant peptide register for two groups of B:9-23 reactive T cells (termed Type B and Type A, respectively after the nomenclature of the Unanue laboratory) (7, 8, 15). However, we performed many experiments that have led us to conclude that the relevant register for both types of T cells is actually Reg3.
To study these registers, we made versions of the B chain peptide in which the amino acids predicted at the p1 and p9 positions in the various registers were mutated to optimize binding to IAg7 in that register, but to inhibit T cell recognition if bound in a different register (16). When tested with the BDC-12-4.1 T cell, as well as others reported to respond to the peptide in bound in Reg1 and Reg2 (7), only the peptide forced to bind in Reg3 stimulated these T cells. Additional experiments established that the key modification to the peptide for Reg3 binding was the mutation of B:22R to E at p9, thus changing a very unfavorable amino acid for the IAg7 p9 pocket for an optimal acidic one (12, 13, 17). Similar experiments with human T cells responding to B:9-23 bound to HLA-DQ8 established that the B:22R to E mutation at p9 greatly improved T cell reactivity. Eventually, crystal structures of the peptide bound to IAg7 or DQ8 confirmed the Reg3 binding of the mutated peptide (10, 11). This modified peptide has been used as a tolerogen for in vivo prevention of T1D in NOD mice (18).
We subsequently performed other experiments (19) showing that, while the peptide with p9R to E mutation strongly stimulated Type A T cells, it remained a weak antigen for Type B T cells. This was eventually tracked to interference of the Type B T cell responses by the exposed side chain of B:21E at p8 in Reg3. Combining the p9R to E with a p8E to G mutation to remove the interfering p8 side chain created a strong agonist for Type B T cells, but reduced the Type A T cell responses. Subsequently, crystal structures of these complexes and of Type A and Type B TCRs bound to them explained the Type A vs. Type B discriminating activity of the Reg3 mutations (10, 11). These studies also showed that, for a subset of Type B T cells, changing the p8E to V or L, rather than G, resulted in epitopes that were even stronger stimulators, sometimes even 100-fold better than the p8G modified version (11). Therefore, creating the appropriate CD4 T cell epitopes from the B:9-23 peptide required modifications of the peptide at B:22R (p9) to greatly improve IAg7 binding and sometimes also at B:21E to greatly improve TCR interaction.
A similar multi-decade effort led to the identification of the epitope for other T cells identified at the BDC, BDC-2.5 and BDC-10.1 (1, 20). These T cell clones were shown to be extremely diabetogenic in NOD mice (4, 21) and responded to pancreatic islets in vitro, but the source of the antigen and the target epitope of these clones eluded researchers for many years. The first clues to its nature came from the identification of stimulatory epitopes for these T cells in various types of peptide libraries (22–25). These independently discovered “mimotopes” eventually pinpointed ChgA as the likely source of the natural antigen (25), since they bore a C-terminal 5 amino acid (p5-p9) motif that was similar to a sequence in ChgA (WSRMD).
A synthetic 9 amino acid ChgA peptide KDRKWSRMD was synthesized, which placed the WSRMD in p5 to p9 positions to mimic the active library mimotope peptides, but this peptide had no activity with the T cells, which we attributed to inhibitory amino acids for T cell recognition (p3R) (25) and IAg7 binding (p4K) (17)within the KDRK portion of this peptide. However, we noticed that there was a conserved 14 amino peptide (WE14) (26) released from ChgA during prohormone convertase processing leaving the WSRMD sequence at its C-terminus, while removing the inhibitory amino acids. This peptide stimulated BDC-2.5 and BDC-10.1 weakly, presumably because of the missing p1 to p4 amino acids, but we found that pancreatic islets from mice lacking a functional ChgA gene failed to stimulate these T cell leading us to the conclusion that, while the WE14 peptide was in some way involved in the ChgA derived epitope, a post-translational modification was likely required to make up for the loss at the p1 to p4 positions in the epitope (25).
Delong et al. pursued the idea that the modification was due to the action of the tissue transglutaminase enzyme (TG) on the glutamine within WE14 (27, 28). However, reminiscent of our results with the insulin B:9-23 peptide, we postulated that a more likely modification was one that would change the p1 to p4 positions with optimal TCR and IAg7 amino acids. To test this idea, we replaced the natural amino acid extension of WE14 peptide with the N-terminal fragment (RLGL) from our library mimotope peptide (19, 25, 29). This peptide remarkably improved the stimulatory activity of the peptide nearly a million-fold. A crystal structure of this RLGL extended WE14 peptide bound to IAg7 confirmed the positions of these amino acids in the peptide binding groove (29). Therefore, we concluded that, in the reciprocal case to that of the insulin B:9-23 derived epitopes, the major epitope for ChgA specific T cells required replacement of the natural ChgA amino acids at the N-terminus, rather than the C-terminus, of the epitope with optimal ones.
The BDC-6.9 T cell was produced at the BDC at about the same time as the insulin and ChgA specific clones (1). As with the BDC-2.5 and BDC-10.1 clones, it was highly diabetogenic in vivo (30), but the source and nature of the epitope was not known. In this case the clue to the source came from the fact that the stimulatory activity was absent in the islets of BALB/c mice (20). Genetic analyses of the stimulatory activity in backcrossed mice mapped it to a section of NOD chromosome 6 and pointed to the IAPP gene as the likely source (30). Several polymorphisms in the IAPP gene coding region between NOD and BALB/c strengthened this idea (31, 32). Disappointingly in vitro stimulations at the time with overlapping peptides throughout the IAPP protein failed to identify a stimulating epitope, but experiments in which NOD mice bred to carry the BALB/c genomic region were protected from T1D induction by the BDC-6.9 clone (33) leading to the conclusion that the functional epitope was probably a post-translational modified form of an IAPP peptide. It has taken several decades to confirm this idea.
The results of the studies above, led to the idea that the functional epitopes for these diabetogenic CD4 T cells were likely post-translational versions of the natural peptides, derived from these proteins. Post-translational modifications of CD4 T cell epitopes had been well-established in other autoimmune diseases, for example, conversion of arginines to citrullines by peptidylarginine deiminases (PADs) in rheumatoid arthritis (34–37) and of glutamines to glutamic acids by tissue transglutaminase (TG) in celiac disease (38). In fact, the presence of these modified amino acids as well as antibodies to the modification or to the modifying enzyme has become diagnostic markers of the diseases.
In T1D, neither of these two types of post translational modification has been established to be a component of the disease driven by the three CD4 T cell specificities discussed here. While Delong, et al. demonstrated an increase in the stimulatory activity of the WE14 peptide after in vitro TG treatment (27, 28), the active products of the treatment have not been identified nor did the simple conversion of the glutamine to glutamic acid in the peptide account for the increased activity. Furthermore, the increase in activity was orders of magnitude less than that seen with the library mimotopes (22, 24, 25).
An alternate hypothesis has arisen from studies of post-translationally modified MHCI bound epitopes generated in the proteasome. During the 2000’s a series of studies documented the creation of chimeric MHCI epitopes by the fusion of peptides from the same or different proteins (39, 40)through a form of reverse proteolysis often referred to as “transpeptidation” (41–43). Subsequently, new methods developed to look for these chimeric peptides among those eluted directly from MHCI molecules revealed that they are much more frequent than previously appreciated (44, 45), raising the question that mass spectrometry methods that simply match MHCI bound peptides to sequences in naturally encoded proteins may miss many important MHCI epitopes. These results spurred us (11, 29, 46) and others (33, 47, 48) to test whether synthetic versions of chimeric peptides between pieces of beta cell proteins could create MHCII compatible chimeric epitopes for the diabetogenic CD4 T cells discussed here.
In our studies on the B:9-23 peptide, a scan of the sequence of proinsulin C-peptide revealed short sequences that when synthetically added to the C-terminus of fragments the B:9-23 peptide truncated to B:21 or to B:22 would be predicted to create chimeric peptides with the amino acids at p8 and/or p9 required for stimulation of Type A or Type B insulin reactive T cells (11). In vitro testing of synthetic versions of these chimeric epitopes showed strong activation of the appropriate Type A and Type B CD4 NOD T cells and Type A human CD4 T cells. These results are summarized in Table 1. For ChgA, based on the highly stimulatory activity of the RLGL when added to the N-terminus of WE14 (19, 29), we looked in well expressed beta cell granule proteins for similar sequences that could be added to WE14 to make similar complete epitopes predicted to stimulate the BDC-10.1 and/or BCD-2.5 T cell (29). When synthesized, many of these chimeric peptides stimulated BCD-10.1 and or BDC-2.5 T cells, bearing out the predictions (46). These results are summarized in Table 1. One of the predicted epitopes involving a fragment of C-peptide with an C-terminal TLAL added to the N-terminus of WE14 has been shown by Delong and colleagues not only to be active, but also present in pancreatic beta cell tumors and in the islets of Langerhans in mouse pancreata (47). This approach of testing candidate fused peptides also turned up the long-sought IAPP-derived epitope for the BDC-6.9 diabetogenic T cell (33). In this case, the same C-peptide fragment ending in TLAL that was used to complete the ChgA WE14 epitope was fused to N-terminus of a peptide released from proIAPP during its natural processing to mature IAPP. This epitope was a very strong agonist for the BDC-6.9 T cell. Importantly the G (p8) from the donor fragment is an R in the corresponding peptide in the BALB/c proIAPP, accounting for the difference between the strains in creating the epitope. As with ChgA, this chimeric peptide has been identified in NOD beta cell tumors and in pancreatic islets (33).
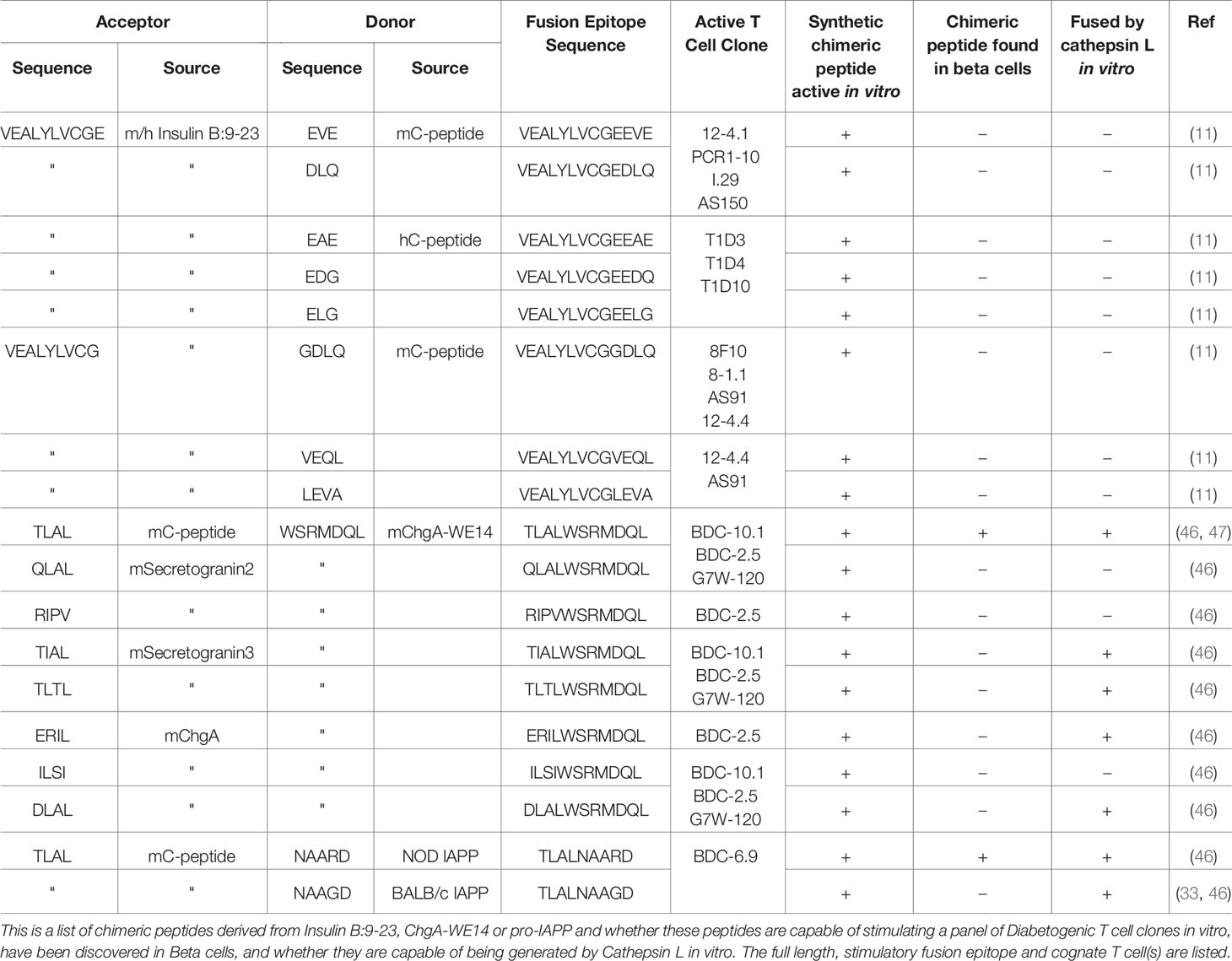
Table 1 Chimeric Peptides Derived from Insulin B:9-23, ChgA-WE14 or pro-IAPP.
Recently, numerous chimeric epitopes have been reported by others for mouse and human CD4 and CD8 T cells in T1D [reviewed in (49, 50)]. The presence of CD4 and CD8 T cells responding to fusion peptides in mouse and human have now all been described and these findings have bridged the gap in our understanding of the T cell mediated pathogenesis in both the mouse and human diseases (51–53). Additionally, the use of these hybrid peptides as therapeutics to tolerize the cognate T cells and prevent the onset of disease has gained a lot of traction (54), but significant limitations still exist in translating these findings to humans.
These accumulating results with chimeric peptides make it highly likely that addition of amino acids to the N- or C-terminus of fragments of insulin B:9-23, WE14 or IAPP derived peptides create the functional epitopes for the corresponding CD4 T cells in T1D. This conclusion begs the question of what mechanism can lead to the generation of these chimeric epitopes in vivo. As mentioned above, the best clues comes from the expanding work on the role of proteasomal transpeptidation in creating many chimeric peptides for MHCI presentation (39, 40, 44, 45, 55–58).
Transpeptidation is an inevitable side reaction during digestion of proteins with proteases with a catalytic serine, threonine or cysteine in the protease active site [reviewed in (43, 56)] (Figure 1). During the protein cleavage reaction these amino acids attack the peptide bond at the cleavage site forming a covalent bond between the oxygen or sulfur in the protease active site and the carbonyl carbon in the peptide bond, while releasing the C-terminal fragment of the digestion. This transient covalent bond is usually broken by water to complete the cleavage by releasing the N-terminal fragment of the digestion and restoring the protease active site, but this bond can also be broken by attack with the N-terminus of a nearby donor peptide restoring a peptide bond and replacing the original C-terminal fragment with a new one to create a chimeric peptide. Transpeptidation is generally a predictable, but minor, side reaction in protease digestions, but its efficiency can be greatly improved by adjusting the conditions present during the proteolysis. Especially effective is a high concentration of the donor peptide in close proximity to the cleavage site and a relatively low concentration of the competing water during the reaction. Under ideal conditions, transpeptidation can be efficient enough to be an important mechanism for natural processing of functional proteins in various organisms (43, 59).
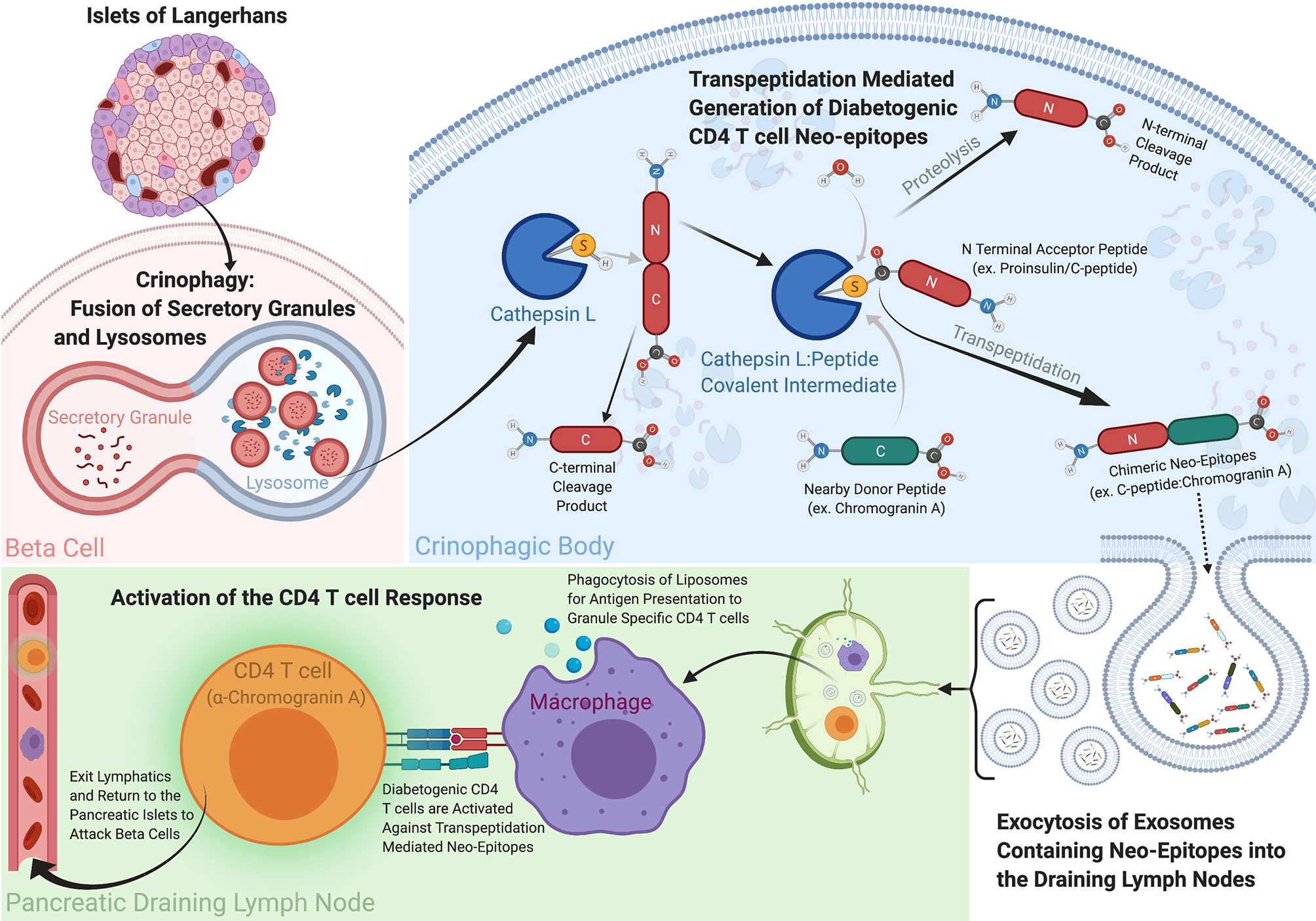
Figure 1 How Transpeptidation in Crinophagic Vesicles Could Create the Chimeric Epitopes Driving T1D. Within the pancreas exist a specialized multicellular network referred to as the Islets of Langerhans. Contained within these islets are the Insulin producing beta cells responsible for maintaining stable blood glucose levels among other neuroendocrine processes. The secretory granules within the beta cells contain prohormones like Proinsulin, Chromogranin A and ProIAPP, and their levels are continually regulated through a catabolic recycling process called crinophagy, whereby secretory granules are fused with lysosomes and their contents are degraded, recycled and secreted. Due to the high concentrations of beta cell hormone donor and acceptor proteins present within these crinophagic bodies, the biochemical conditions are optimal for the reverse proteolysis reaction, transpeptidation to occur. Cathepsin L is a protease capable of cleaving the hormone acceptor peptides (ex. Proinsulin C-peptide) and creating an enzyme linked intermediate complex with the acceptor peptide. Water is generally responsible for breaking this transient bond between the carbonyl carbon of the acceptor peptide and the a sulfur or oxygen in enzyme to complete the digestion, however when high concentrations of a donor peptide with a free N-terminus are present they can outcompete water and generate a new peptide product through transpeptidation. We propose these neo-peptides can be exocytosed and secreted out of the beta cells and to be taken up antigen presenting cells to and presented to diabetogenic CD4 T cells. These peptides are considerably more active than their germline encoded parental counterparts and their presentation can lead to T cell activation and destruction of the beta cells in the islets. This figure was created with Biorender.com.
The proteasome has a milieu very favorable for transpeptidation. It contains threonine proteases (60) and has a steady high concentration of cytoplasmic proteins directed into the organelle for degradation [Reviewed in (61)]. It has an encapsulated interior containing low water content. It is the main source of protein digestion products destined to the endoplasmic reticulum for further processing and MHCI loading. These ideal conditions perhaps explain the high proportion of chimeric peptides found in those eluted from surface expressed MHCI molecules (44, 58). While the conditions in the proteasome may be ideal to catalyze these reactions, the spatial constraints of the proteasome have been shown to prefer Cis-splicing events, where internal deletions are made within the same protein, instead of fusion events between two different proteins (Trans-splicing) (62).
A parallel pathway involving lysosomes exists in pancreatic islet beta cells and can be predicted to favor the generation of chimeric peptides. In beta cells, secretory granules have a high concentration of insulin and other proteins, including ChgA and IAPP [Reviewed in (63)]. Convertase proteases in the granules convert the precursor forms of these proteins into their mature, active forms, by releasing protective prohormone fragments, as well as additional active hormone fragments by internal cleavages (64). The number of granules in a beta cell is strictly regulated (63). Therefore, since new granules are constantly being formed, excess granules need to be eliminated to maintain the optimal number. This is accomplished by a form of autophagy called crinophagy (65), in which granules are fused with lysosomes and their proteins denatured and degraded by a variety of enzymes including cathepsins and other cysteine or serine proteases. Thus, ideal conditions for transpeptidation are set up – a high concentration of actively degrading proteins encapsulated in a vesicle with multiple proteases that are capable of the transpeptidation reaction. Exosomes from these crinophagic vesicles carrying antigenic fragments of insulin and other granule proteins can be released from beta cells and into circulation (66), providing a pathway for chimeric peptides to reach the pancreatic draining lymph nodes for activation of diabetogenic CD4 T cells (67) (Figure 1). While these findings are considered circumstantial by some, these extracellular vesicles have been shown to carry cargo relevant to T1D in the form of prohormone proteins for both CD4 and CD8 T cells, and miRNAs, all of which have been implicated in multiple facets of the disease [reviewed in (68)]. Since the lysosomal/endosomal pathway in MHCII bearing antigen presenting cells (APCs) is the primary site for proteolytic generation of peptides for MHCII presentation [reviewed (69)], a number of laboratories have studied this antigen processing reaction in vitro, by exposing proteins to various lysosomal proteases under lysosomal conditions and testing the products of the digestion for antigenic activity (46, 70–72). We have used this system to see if any of the active chimeric epitopes identified in the beta cells mentioned above could be generated in vitro during lysosomal protease digestion of a suitable acceptor protein fragment in the presence of a donor peptide that when fused to a site within the acceptor would form the active diabetogenic epitope (46).
We tested a number of cathepsin proteases, we settled on cathepsin L for these experiments, due to its ability to generate the necessary complimentary Proinsulin acceptor peptide. In looking for an active WE14 containing chimeric epitope, we used a fragment of C-peptide containing the previously documented TLAL sequence discussed above (47), as well as fragments of other granule proteins with embedded sequences that also were active when fused to the N-terminus of WE14 (46). In each case an internal cleavage at the C-terminus of the embedded fragment was required to create a site for transpeptidation fusion to an N-terminal fragment of WE14. Cathepsin L digestions were performed at lysosomal pH with a molar excess of a WE14 donor fragment to favor the transpeptidation reaction.
When the digests were used to stimulate the prototypical NOD WE14-specific CD4 T cells, BDC-10.1 and/or BCD-2.5, five (Table 1) were active with one or both T cells (46). Tandem mass spectrometric analysis (MS-MS) of the digests revealed the presence of the predicted chimeric peptide in the digests. Synthetic versions of the identified epitope had the same stimulating specificity as the digests. In each case the identification was further confirmed by showing that the MS-MS fractionation pattern of the synthetic peptide was virtually identical to that seen in the corresponding peptide found in the digest.
The MS-MS analyses also showed that these functional peptides were by no means the only chimeric peptides detected in the digests. Hundreds of additional chimeric peptides were identified involving many combinations of the input acceptor and donor peptides. In the case of joining of the WE14 fragment to sites within the input acceptor peptide precursor, fusions were detected at nearly every position (46), but strikingly, the positions that contained a preferred cathepsin L cleavage sequence were highly favored. A similar digestion with the C-peptide fragment containing TLAL using a donor peptide from NOD proIAPP (NAARD) generated the previously reported functional chimeric peptide for the BDC-6.9 T cell (46). Substituting the equivalent donor peptide from BALB/c IAPP (NAAGD) also generated the predicted chimeric peptide, but as expected this peptide was 10x less active than the NOD derived one.
Cysteine proteases have been implicated in disease resistance in Type 1 Diabetes (73), however further investigation by other groups determined that the effect was indirect, due in part to T cell repertoire changes resulting from Cathepsin L being absent during thymic selection. They observed a 2 fold higher incidence of regulatory T cells in the knockout mice compared to their CatL sufficient counterparts, which they attributed to the disease protection (74). Although splenocytes from NOD mice are capable of mounting a response against the CatlL-/- islets, it is not clear whether the absence of CatL has allowed for a compensatory mechanism whereby alternative proteases are utilized to generate these fusion peptides, or if another protease is responsible for it altogether. To date, we have not discovered another protease capable of generating these fusions other than Cathepsin L.
After a decades-long struggle to understand the structures of the diabetic CD4 T cells epitopes in T1D, the door has cracked open with the discovery of the functional chimeric epitopes and their formation by transpeptidation. As more acceptor-donor pairs are tested with multiple lysosomal proteases, it seems likely that this form of post-translational modification will play an important role in epitope formation in other CD4 T cell driven autoimmune diseases, especially those of other neuro-endocrine tissues containing secretory granules. This phenomenon may also contribute to epitopes for CD4 T cells derived from foreign (viral/bacterial) and tumor antigens. As with MHCI, these MHCII results point out that existing peptides databases for MHCII bound to peptides directly encoded in the genome may be incomplete and need to be updated to include chimeric peptides found directly bound to MHCII molecules. One can hope that the computational methods for identifying chimeric epitopes bound to MHCI molecules can be adapted to those bound to MHCII. The variable lengths of MHCII bound epitopes presents a challenge in approaching this task, but the longer MHCII bound peptides may also be an advantage. They could make it easier to identify independently the N- and C-terminal components of a chimeric peptide among the MS-MS generated b-ion versus y-ion fragments. The similarities between these recent findings within the MHCI and MHCII epitope fields might also provide reason to reexamine old data sets for MHC peptide elutions and reprobe them for their presence of transpeptidation mediated fusions.
BR and JK both reviewed relevant literature and drafted the initial manuscript. BR and JK edited and approved the final manuscript.
This work was supported by National Institutes of Health grants T32 AI 074491 (BR), P01 AI-118688 (JK). BR was also supported by the Intersect Fellowship Program for Computational Scientists and Immunologists from the American Association of Immunologists.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work would not have been possible without the enormous efforts of Frances Crawford and all of our other collaborators and scientific friends from over the years. We thank the Howard Hughes Medical Institute, National Jewish Health, the University of Colorado School of Medicine, and the Claire Friedlander Family Foundation for financial support during the course of our studies.
1. Haskins K, Portas M, Bergman B, Lafferty K, Bradley B. Pancreatic islet-specific T-cell clones from nonobese diabetic mice. Proc Natl Acad Sci USA (1989) 86:8000–4. doi: 10.1073/pnas.86.20.8000
2. Wegmann DR, Gill RG, Norbury-Glaser M, Schloot N, Daniel D. Analysis of the spontaneous T cell response to insulin in NOD mice. J Autoimmun (1994) 7:833–43. doi: 10.1006/jaut.1994.1066
3. Daniel D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur J Immunol (1995) 25:1056–62. doi: 10.1002/eji.1830250430
4. Burton AR, Vincent E, Arnold PY, Lennon GP, Smeltzer M, Li CS, et al. On the pathogenicity of autoantigen-specific T-cell receptors. Diabetes (2008) 57:1321–30. doi: 10.2337/db07-1129
5. Zhang L, Nakayama M, Eisenbarth GS. Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol (2008) 20:111–8. doi: 10.1016/j.coi.2007.11.005
6. Zhang L, Jasinski JM, Kobayashi M, Davenport B, Johnson K, Davidson H, et al. Analysis of T cell receptor beta chains that combine with dominant conserved TRAV5D-4*04 anti-insulin B:9-23 alpha chains. J Autoimmun (2009) 33:42–9. doi: 10.1016/j.jaut.2009.02.003
7. Levisetti MG, Suri A, Petzold SJ, Unanue ER. The insulin-specific T cells of nonobese diabetic mice recognize a weak MHC-binding segment in more than one form. J Immunol (2007) 178:6051–7. doi: 10.4049/jimmunol.178.10.6051
8. Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med (2011) 208:2375–83. doi: 10.1084/jem.20111502
9. Yang J, Chow IT, Sosinowski T, Torres-Chinn N, Greenbaum CJ, James EA, et al. Autoreactive T cells specific for insulin B:11-23 recognize a low-affinity peptide register in human subjects with autoimmune diabetes. Proc Natl Acad Sci USA (2014) 111:14840–5. doi: 10.1073/pnas.1416864111
10. Wang Y, Sosinowski T, Novikov A, Crawford F, Neau DB, Yang J, et al. C-terminal modification of the insulin B:11–23 peptide creates superagonists in mouse and human type 1 diabetes. Proc Natl Acad Sci (2018) 115:162–7. doi: 10.1073/pnas.1716527115
11. Wang Y, Sosinowski T, Novikov A, Crawford F, White J, Jin N, et al. How C-terminal additions to insulin B-chain fragments create superagonists for T cells in mouse and human type 1 diabetes. Sci Immunol (2019) 4:1–12. doi: 10.1126/sciimmunol.aav7517
12. Latek RR, Suri A, Petzold SJ, Nelson CA, Kanagawa O, Unanue ER, et al. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity (2000) 12:699–710. doi: 10.1016/S1074-7613(00)80220-4
13. Corper AL, Stratmann T, Apostolopoulos V, Scott CA, Garcia KC, Kang AS, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science (2000) 288:505–11. doi: 10.1126/science.288.5465.505
14. Lee KH, Wucherpfennig KW, Wiley DC. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat Immunol (2001) 2:501–7. doi: 10.1038/88694
15. Gioia L, Holt M, Costanzo A, Sharma S, Abe B, Kain L, et al. Position beta57 of I-A(g7) controls early anti-insulin responses in NOD mice, linking an MHC susceptibility allele to type 1 diabetes onset. Sci Immunol (2019) 4:1–12. doi: 10.1126/sciimmunol.aaw6329
16. Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci USA (2010) 107:10978–83. doi: 10.1073/pnas.1006545107
17. Suri A, Walters JJ, Gross ML, Unanue ER. Natural peptides selected by diabetogenic DQ8 and murine I-A(g7) molecules show common sequence specificity. J Clin Invest (2005) 115:2268–76. doi: 10.1172/JCI25350
18. Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med (2011) 208:1501–10. doi: 10.1084/jem.20110574
19. Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, et al. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci USA (2011) 108:16729–34. doi: 10.1073/pnas.1113954108
20. Haskins K. Pathogenic T-cell clones in autoimmune diabetes: more lessons from the NOD mouse. Adv Immunol (2005) 87:123–62. doi: 10.1016/S0065-2776(05)87004-X
21. Peterson JD, Haskins K. Transfer of diabetes in the NOD-scid mouse by CD4 T-cell clones. Different Requirement CD8 T-cells Diabetes (1996) 45:328–36. doi: 10.2337/diab.45.3.328
22. Judkowski V, Pinilla C, Schroder K, Tucker L, Sarvetnick N, Wilson DB. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J Immunol (2001) 166:908–17. doi: 10.4049/jimmunol.166.2.908
23. You S, Chen C, Lee WH, Wu CH, Judkowski V, Pinilla C, et al. Detection and characterization of T cells specific for BDC2.5 T cell-stimulating peptides. J Immunol (2003) 170:4011–20. doi: 10.4049/jimmunol.170.8.4011
24. Yoshida K, Martin T, Yamamoto K, Dobbs C, Munz C, Kamikawaji N, et al. Evidence for shared recognition of a peptide ligand by a diverse panel of non-obese diabetic mice-derived, islet-specific, diabetogenic T cell clones. Int Immunol (2002) 14:1439–47. doi: 10.1093/intimm/dxf106
25. Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol (2010) 11:225–31. doi: 10.1038/ni.1844
26. Curry WJ, Norlen P, Barkatullah SC, Johnston CF, Hakanson R, Hutton JC, et al. and its derived peptides in the rat and porcine gastro-entero-pancreatic system. Expression Localization Characterization Adv Exp Med Biol (2000) 482:205–13. doi: 10.1007/0-306-46837-9_16
27. Delong T, Baker RL, He J, Haskins K. Novel autoantigens for diabetogenic CD4 T cells in autoimmune diabetes. Immunol Res (2013) 55:167–72. doi: 10.1007/s12026-012-8375-6
28. Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes (2012) 61:3239–46. doi: 10.2337/db12-0112
29. Jin N, Wang Y, Crawford F, White J, Marrack P, Dai S, et al. N-terminal additions to the WE14 peptide of chromogranin A create strong autoantigen agonists in type 1 diabetes. Proc Natl Acad Sci USA (2015) 112:13318–23. doi: 10.1073/pnas.1517862112
30. Dallas-Pedretti A, McDuffie M, Haskins K. A diabetes-associated T-cell autoantigen maps to a telomeric locus on mouse chromosome 6. Proc Natl Acad Sci USA (1995) 92:1386–90. doi: 10.1073/pnas.92.5.1386
31. Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature (2011) 477:289–94. doi: 10.10381/nature10413
32. Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev (2011) 91:795–826. doi: 10.1152/physrev.00042.2009
33. Wiles TA, Delong T, Baker RL, Bradley B, Barbour G, Powell RL, et al. An insulin-IAPP hybrid peptide is an endogenous antigen for CD4 T cells in the non-obese diabetic mouse. J Autoimmun (2017) 78:11–8. doi: 10.1016/j.jaut.2016.10.007
34. Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest (1998) 101:273–81. doi: 10.1172/JCI1316
35. Goldbach-Mansky R, Lee J, McCoy A, Hoxworth J, Yarboro C, Smolen JS, et al. Rheumatoid arthritis associated autoantibodies in patients with synovitis of recent onset. Arthritis Res (2000) 2:236–43. doi: 10.1186/ar93
36. Bas S, Genevay S, Meyer O, Gabay C. Anti-cyclic citrullinated peptide antibodies, IgM and IgA rheumatoid factors in the diagnosis and prognosis of rheumatoid arthritis. Rheumatol (Oxford) (2003) 42:677–80. doi: 10.1093/rheumatology/keg184
37. Doyle HA, Mamula MJ. Posttranslational modifications of self-antigens. Ann N Y Acad Sci (2005) 1050:1–9. doi: 10.1196/annals.1313.001
38. Sollid LM. Molecular basis of celiac disease. Annu Rev Immunol (2000) 18:53–81. doi: 10.1146/annurev.immunol.18.1.53
39. Hanada K, Yewdell JW, Yang JC. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature (2004) 427:252–6. doi: 10.1038/nature02240
40. Vigneron N, Stroobant V, Chapiro J, Ooms A, Degiovanni G, Morel S, et al. An antigenic peptide produced by peptide splicing in the proteasome. Science (2004) 304:587–90. doi: 10.1126/science.1095522
41. Lorand L, Konishi K, Jacobsen A. Transpeptidation mechanism in blood clotting. Nature (1962) 194:1148–9. doi: 10.1038/1941148a0
42. Cresswell P. Cell biology. Cutting and pasting antigenic peptides. Science (2004) 304:525–7. doi: 10.1126/science.1097508
43. Berkers CR, de Jong A, Ovaa H, Rodenko B. Transpeptidation and reverse proteolysis and their consequences for immunity. Int J Biochem Cell Biol (2009) 41:66–71. doi: 10.1016/j.biocel.2008.08.036
44. Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science (2016) 354:354–8. doi: 10.1126/science.aaf4384
45. Faridi P, Li C, Ramarathinam SH, Vivian JP, Illing PT, Mifsud NA, et al. A subset of HLA-I peptides are not genomically templated: Evidence for cis- and trans-spliced peptide ligands. Sci Immunol (2018) 3:1–11. doi: 10.1126/sciimmunol.aar3947
46. Reed B, Crawford F, Hill RC, Jin N, White J, Krovi SH, et al. Lysosomal cathepsin creates chimeric epitopes for diabetogenic CD4 T cells via transpeptidation. J Exp Med (2021) 218:1–17. doi: 10.1084/jem.20192135
47. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science (2016) 351:711–4. doi: 10.1126/science.aad2791
48. Williams T, Krovi HS, Landry LG, Crawford F, Jin N, Hohenstein A, et al. Development of T cell lines sensitive to antigen stimulation. J Immunol Methods (2018) 462:65–73. doi: 10.1016/j.jim.2018.08.011
49. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med (2016) 22:1482–7. doi: 10.1038/nm.4203
50. James EA, Mallone R, Kent SC, DiLorenzo TP. T-Cell Epitopes and Neo-epitopes in Type 1 Diabetes: A Comprehensive Update and Reappraisal. Diabetes (2020) 69:1311–35. doi: 10.2337/dbi19-0022
51. Arribas-Layton D, Guyer P, Delong T, Dang M, Chow IT, Speake C, et al. Hybrid Insulin Peptides Are Recognized by Human T Cells in the Context of DRB1*04:01. Diabetes (2020) 69:1492–502. doi: 10.2337/db19-0620
52. Baker RL, Rihanek M, Hohenstein AC, Nakayama M, Michels A, Gottlieb PA, et al. Hybrid Insulin Peptides Are Autoantigens in Type 1 Diabetes. Diabetes (2019) 68:1830–40. doi: 10.2337/db19-0128
53. Laban S, Suwandi JS, van Unen V, Pool J, Wesselius J, Hollt T, et al. Heterogeneity of circulating CD8 T-cells specific to islet, neo-antigen and virus in patients with type 1 diabetes mellitus. PloS One (2018) 13:e0200818. doi: 10.1371/journal.pone.0200818
54. Jamison BL, Neef T, Goodspeed A, Bradley B, Baker RL, Miller SD, et al. Nanoparticles Containing an Insulin-ChgA Hybrid Peptide Protect from Transfer of Autoimmune Diabetes by Shifting the Balance between Effector T Cells and Regulatory T Cells. J Immunol (2019) 203:48–57. doi: 10.4049/jimmunol.1900127
55. Ebstein F, Textoris-Taube K, Keller C, Golnik R, Vigneron N, Van den Eynde BJ, et al. Proteasomes generate spliced epitopes by two different mechanisms and as efficiently as non-spliced epitopes. Sci Rep (2016) 6:24032. doi: 10.1038/srep24032
56. Mishto M, Liepe J. Post-Translational Peptide Splicing and T Cell Responses. Trends Immunol (2017) 38:904–15. doi: 10.1016/j.it.2017.07.011
57. Specht G, Roetschke HP, Mansurkhodzhaev A, Henklein P, Textoris-Taube K, Urlaub H, et al. Large database for the analysis and prediction of spliced and non-spliced peptide generation by proteasomes. Sci Data (2020) 7:146. doi: 10.1038/s41597-020-0487-6
58. Mishto M. What We See, What We Do Not See, and What We Do Not Want to See in HLA Class I Immunopeptidomes. Proteomics (2020) e2000112. doi: 10.1002/pmic.202000112
59. Conlan BF, Gillon AD, Craik DJ, Anderson MA. Circular proteins and mechanisms of cyclization. Biopolymers (2010) 94:573–83. doi: 10.1002/bip.21422
60. Kisselev AF, Songyang Z, Goldberg AL. Why does threonine, and not serine, function as the active site nucleophile in proteasomes? J Biol Chem (2000) 275:14831–7. doi: 10.1074/jbc.275.20.14831
61. Vigneron N, Van den Eynde BJ. Proteasome subtypes and regulators in the processing of antigenic peptides presented by class I molecules of the major histocompatibility complex. Biomolecules (2014) 4:994–1025. doi: 10.3390/biom4040994
62. Paes W, Leonov G, Partridge T, Nicastri A, Ternette N, Borrow P. Elucidation of the Signatures of Proteasome-Catalyzed Peptide Splicing. Front Immunol (2020) 11:563800. doi: 10.3389/fimmu.2020.563800
63. Hou JC, Min L, Pessin JE. Insulin granule biogenesis, trafficking and exocytosis. Vitam Horm (2009) 80:473–506. doi: 10.1016/S0083-6729(08)00616-X
64. Marzban L, Trigo-Gonzalez G, Zhu X, Rhodes CJ, Halban PA, Steiner DF, et al. Role of beta-cell prohormone convertase (PC)1/3 in processing of pro-islet amyloid polypeptide. Diabetes (2004) 53:141–8. doi: 10.2337/diabetes.53.1.141
65. Sandberg M, Borg LA. Intracellular degradation of insulin and crinophagy are maintained by nitric oxide and cyclo-oxygenase 2 activity in isolated pancreatic islets. Biol Cell (2006) 98:307–15. doi: 10.1042/BC20050085
66. Wan X, Zinselmeyer BH, Zakharov PN, Vomund AN, Taniguchi R, Santambrogio L, et al. Pancreatic islets communicate with lymphoid tissues via exocytosis of insulin peptides. Nature (2018) 560:107–11. doi: 10.1038/s41586-018-0341-6
67. Zinselmeyer BH, Vomund AN, Saunders BT, Johnson MW, Carrero JA, Unanue ER. The resident macrophages in murine pancreatic islets are constantly probing their local environment, capturing beta cell granules and blood particles. Diabetologia (2018) 61:1374–83. doi: 10.1007/s00125-018-4592-4
68. Pang H, Luo S, Xiao Y, Xia Y, Li X, Huang G, et al. Emerging Roles of Exosomes in T1DM. Front Immunol (2020) 11:593348. doi: 10.3389/fimmu.2020.593348
69. Marrack P, Kappler JW. Do MHCII-presented neoantigens drive type 1 diabetes and other autoimmune diseases? Cold Spring Harb Perspect Med (2012) 2:a007765. doi: 10.1101/cshperspect.a007765
70. Kim A, Hartman IZ, Poore B, Boronina T, Cole RN, Song N, et al. Divergent paths for the selection of immunodominant epitopes from distinct antigenic sources. Nat Commun (2014) 5:5369. doi: 10.1038/ncomms6369
71. Sadegh-Nasseri S, Kim A. Exogenous antigens bind MHC class II first, and are processed by cathepsins later. Mol Immunol (2015) 68:81–4. doi: 10.1016/j.molimm.2015.07.018
72. Sadegh-Nasseri S, Kim A. Selection of immunodominant epitopes during antigen processing is hierarchical. Mol Immunol (2019) 113:115–9. doi: 10.1016/j.molimm.2018.08.011
73. Maehr R, Mintern JD, Herman AE, Lennon-Dumenil AM, Mathis D, Benoist C, et al. Cathepsin L is essential for onset of autoimmune diabetes in NOD mice. J Clin Invest (2005) 115:2934–43. doi: 10.1172/JCI25485
Keywords: antigen presenting cell, transpeptidation, immune tolerance, type 1 diabetes mellitus, chimeric peptide, CD4 T cell, beta cell, antigen
Citation: Reed BK and Kappler JW (2021) Hidden in Plain View: Discovery of Chimeric Diabetogenic CD4 T Cell Neo-Epitopes. Front. Immunol. 12:669986. doi: 10.3389/fimmu.2021.669986
Received: 19 February 2021; Accepted: 01 April 2021;
Published: 27 April 2021.
Edited by:
Jon D. Piganelli, University of Pittsburgh, United StatesReviewed by:
Sally C. Kent, University of Massachusetts Medical School, United StatesCopyright © 2021 Reed and Kappler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John W. Kappler, a2FwcGxlcmpAbmpoZWF0bGgub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.