
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 April 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.668207
 Panagiotis F. Christopoulos1*†
Panagiotis F. Christopoulos1*† Torleif T. Gjølberg2,3,4†
Torleif T. Gjølberg2,3,4† Stig Krüger1
Stig Krüger1 Guttorm Haraldsen1
Guttorm Haraldsen1 Jan Terje Andersen2,4
Jan Terje Andersen2,4 Eirik Sundlisæter1*
Eirik Sundlisæter1*The Notch signaling pathway regulates developmental cell-fate decisions and has recently also been linked to inflammatory diseases. Although therapies targeting Notch signaling in inflammation in theory are attractive, their design and implementation have proven difficult, at least partly due to the broad involvement of Notch signaling in regenerative and homeostatic processes. In this review, we summarize the supporting role of Notch signaling in various inflammation-driven diseases, and highlight efforts to intervene with this pathway by targeting Notch ligands and/or receptors with distinct therapeutic strategies, including antibody designs. We discuss this in light of lessons learned from Notch targeting in cancer treatment. Finally, we elaborate on the impact of individual Notch members in inflammation, which may lay the foundation for development of therapeutic strategies in chronic inflammatory diseases.
The Notch signaling pathway is a major regulator of cell-fate determination during development and cellular differentiation (1, 2). It was identified more than 100 years ago in Drosophila melanogaster, in which haploinsufficiency of the Notch locus and the associated partial loss of Notch function gave rise to a notched wing (3). Unlike soluble ligands, like tumor necrosis factor (TNF) and interleukin (IL) 1 beta, triggering conventional inflammatory receptor signaling, Notch ligands are, like their receptors, transmembrane proteins with large extracellular domains. Therefore, Notch signaling represents a short-range signaling system for direct cell-to-cell communication.
Emerging evidence points to Notch signaling as a modulator of innate and adaptive immune responses (4), and manipulating the pathway has been suggested as an attractive therapeutic strategy to target immunological diseases (5, 6). Numerous reports have provided insights into the outcome of manipulating Notch signaling in experimentally induced inflammation by a range of genetic and pharmacological strategies, including lymphoid- or myeloid-lineage-specific deletion of components of the pathway, systemic pan-Notch inhibition, and selective blockade of individual Notch ligands and receptors (7–10). Although the studies have revealed conflicting roles for Notch in the development, differentiation, and activation of different immune cell subsets, the collective data strongly indicates that Notch activation in most cell types supports a pro-inflammatory phenotype, and that pharmacologic inhibition of this signaling cascade may hold therapeutic potentials in several inflammatory diseases.
In this review, we summarize the emerging roles of Notch signaling in regulating immune responses and highlight strategies that may be used to target the Notch pathway in chronic inflammation. From a disease-oriented standpoint, we focus on classical autoimmune diseases such as rheumatoid arthritis, inflammatory bowel disease and uveitis, and also discuss recent findings from preclinical models of graft-versus-host disease. To date, cancer has been the main focus area of potential therapeutic strategies, for both selective Notch modulation and pan-Notch inhibition, generating an accumulating body of evidence regarding possible disadvantages and side-effects that must be considered to tailor design of future treatment options. Finally, we briefly summarize lessons learned in tumor models, discuss potential therapeutic strategies for Notch modulation in chronic inflammation and suggest improvements in the field.
Notch signaling is mediated by a highly conserved ligand-receptor apparatus and plays a key role in the regulation of cellular proliferation, survival, apoptosis, and differentiation, thereby critically affecting organ development and function (11, 12). However, as with most signaling pathways in human pathophysiology, it is also implicated in the development and progression of various human cancers and autoimmune diseases (Figure 1). Notch genes encode single-pass transmembrane receptors, activated by a dual proteolytic cleavage, which releases the Notch intracellular domain (NICD), allowing its nuclear translocation. Notch ligand binding mediates receptor activation by instigating a conformational change required for such cleavage to take place. In the nucleus, NICD binds to the transcription factor CSL family proteins [recombination signal-binding protein 1 for J-kappa (RBP-Jk) in mammals], and co-activators like the Mastermind-like transcriptional co-activator 1 (MAML-1). The assemble of this transcription activation complex leads to the transcription of various downstream target genes (Figure 2, left panel). To date, four genes encoding Notch receptors (Notch 1-4) have been identified in mammals, including humans. In addition, five genes encoding Notch receptor ligands have been reported. The mammalian ligands are classified on the basis of structural homology to the two Drosophila ligands, Delta and Serrate, and designated as either Delta-like (DLL1, -3, and -4) or as Serrate-like, known as Jagged in mammals (Jagged-1 and Jagged-2).
Figure 1 The “yin and yang” of the Notch signaling. As with most major signaling pathways, the Notch system is implicated in both; (A) pathological processes (yin; indicated in black frame), including, but not limited to, cancer progression and metastasis, as well as, autoimmune diseases, acute and chronic inflammation and (B) favorable homeostatic processes (yang; indicated in white frame), including, but not limited to, organ development and function, as well as, immune cells differentiation and activation.
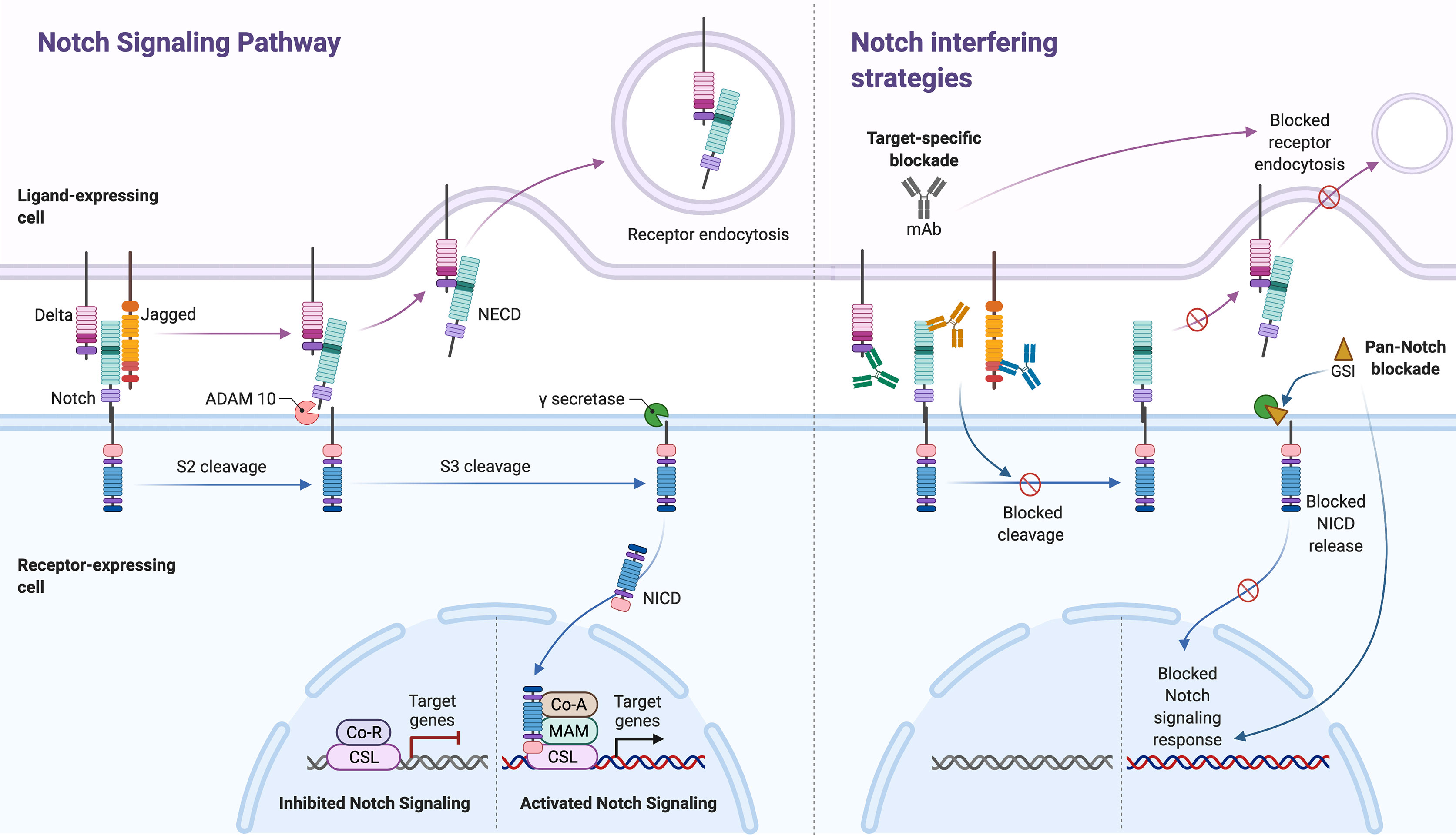
Figure 2 The Notch signaling pathway and its inhibitors. Notch signaling (left panel, separated by stapled line) is activated upon ligand (Delta or Jagged) binding to an adjacent Notch receptor on a neighboring cell. Upon activation, Notch receptors are cleaved, first by ADAM, in the NRR of the extracellular domain (S2 cleavage). The released extracellular domain (NECD) is then trans-endocytosed by the ligand-expressing cell. The second cleavage (S3 cleavage) is mediated by the γ-secretase (GS) complex, which releases the Notch intracellular domain (NICD) into the cytoplasm, thus allowing its activation and nuclear translocation. In the nucleus, NICD binds to the transcription factor CSL family proteins and the Mastermind (MAM)-like transcriptional co-activator 1. The assemble of this transcription activation complex on the recombination signal-binding protein for immunoglobulin kappa J (RBP-J) promoter region, leads to the expression of various downstream target genes. Two main Notch-interfering strategies (right panel) exists; monoclonal antibodies (mAbs) against the ligands and/or receptors (target-specific blockade) and GS inhibitors (GSI, pan-Notch blockade). While mAbs may block specific ligand-induced S2 cleavage and consecutively trans-endocytosis of NECD, GSI strategies will non-specifically block NICD release following all ligand-receptor interactions. The figure was made in BioRender (https://biorender.com).
The four Notch receptors share similarities in their structural architecture, with distinct subtle differences in their extracellular and intracellular domains (13). Following proteolytic cleavage in the secretory pathway, the receptors form a heterodimer of which the extracellular domain is non-covalently bound to the transmembrane-intracellular domain. The extracellular domain of Notch receptors contains up to 36 repeated copies of an epidermal growth factor (EGF)-like motif, which are involved in ligand interaction. These amino-terminal EGF-like repeats are followed by a negative regulatory region (NRR), which contains three cysteine-rich Lin12-Notch repeats (LNR) and a heterodimerization domain (HD), and together, these function to inhibit Notch activation in the absence of ligands. The NICD contains a RBP-Jk-association molecule (RAM) module, ankyrin (ANK) repeats, nuclear localization signals (NLS), a trans-activation domain (TAD), and a negative regulatory peptide sequence, rich in proline, glutamine, serine and threonine residues (PEST). The RAM and ANK domains enable docking of the NICD to other proteins while the PEST sequence is involved in Notch protein turnover (12).
Notch ligands also have multiple EGF-like repeats in their extracellular domain. The Serrate ligands Jagged-1 and Jagged-2 have almost two-fold as many EGF-like repeats compared to Delta, and, in addition, feature a cysteine-rich region (CR) that is absent in Delta.
Notch signaling is activated upon ligand binding to an adjacent Notch receptor on a neighboring cell. In contrast, receptor association with ligands presented by the receptor-bearing cell (cis-presented ligand) inhibit receptor activation (14). Upon activation, Notch receptors are cleaved twice. The first step is mediated by ADAM17 (a disintegrin and metalloprotease) or ADAM10 (15), which cleaves the receptor at the extracellular S2 cleavage site (Site 2), situated deep in the NRR in the posed receptor. Ligand binding in trans causes a pulling force, which opens up the NRR and exposes the S2, thus allowing this cleavage to initiate the activation process. The released extracellular domain is then trans-endocytosed by the ligand-expressing cell. The second cleavage, mediated by the γ-secretase complex (comprised of presenilin-1/2, nicastrin, Pen-2, and Aph-1), releases NICD into the cytoplasm, thus allowing its nuclear translocation (Figure 2, left panel). This sequence has become known as the canonical Notch pathway.
There is also some evidence to support the existence of non-canonical Notch signaling [reviewed in (16, 17)]. Type I non-canonical signaling involves Notch activation and transfer of activation signals independent of RBP-Jk (NICD-dependent but RBP-Jk-independent) (18, 19); Type II involves activation of Notch target genes independent of proteolytic cleavage of Notch and RBP-Jk activation (NICD- and RBP-Jk-independent) (20); Type III involves RBP-Jk-dependent gene activation without receptor cleavage and NICD release (21, 22). Several signaling pathways might be involved, such as Wnt, NfkB, HIF and PI3K/AKT. In conclusion, data on non-canonical Notch signaling is limited, and it is less characterized than the canonical pathway.
In hematopoiesis, the best-studied functions of Notch signaling are its roles during lymphocyte development, in particular during T cell lineage commitment and maturation in the thymus, and during marginal zone B cell development in the spleen [reviewed in (4, 23)]. After common lymphoid progenitors have migrated from the bone marrow to the thymus, thymic epithelial cells expressing DLL4 trigger canonical Notch1 signaling in early thymic progenitors (24–26). DLL4-Notch1 signaling is essential for T cell lineage commitment and is further required during early phases of thymocyte differentiation. Notch signaling also inhibits other lineage potentials, such as B cell and myeloid cell potential (27). Furthermore, after newly formed B cells migrate from bone marrow to the spleen, Notch2 activation by DLL1 is required to generate the marginal zone B cells (28, 29).
However, reports on Notch functions in T helper cell polarization are contradictory, as it surfaced as a potential dictator of mutually exclusive subtypes depending on the experimental context; Notch requirement has been reported for Th1 (30), Th17 (31) and Th2 (32) polarization, as well as in Treg polarization (33). This has led to the proposed theory of Notch as an unbiased amplifier of polarizing signals, like cytokines, whose requirement was not instructive in the T helper polarization process (34). The original instructive theory of Notch was based on the observation that expression of Jagged or Delta-like ligands on antigen-presenting cells (APCs), e.g., dendritic cells, varied depending on the identity of the activating stimulus (7). Dictated by the ligand expressed on APCs, the conveyed Notch signal in approaching T-cells could thus be perceived differently, and subsequently instruct polarization in a ligand-specific manner. The unbiased amplifier model is, however, intuitively a more unifying explanation, suggesting that Notch simultaneously reinforce Th1, Th2 and Th17 specific transcriptional programs irrespective of the cytokine stimuli. In this way, Notch could allow - rather than direct – polarization. Instructive functions of Notch in T helper cell polarization may still be true in some contexts, such as at defined stages of differentiation. Also, there are reports of ligand-independent functions of Notch upon T-cell receptor activation (35). In addition, Notch is also required for cytotoxic (CD8+) T-cell effector differentiation (36), as well as T-cell stimulating processes, such as increased production of the T-cell proliferating factor IL2 (37).
Furthermore, loss-of-function studies have revealed that Notch signaling is playing important roles in innate immunity. One example is Notch 2 signaling in dendritic cell differentiation and homeostasis in the spleen and intestine (38, 39). Notch signaling also seems to be involved in promoting the pro-inflammatory activation of primary human blood monocyte-derived macrophages, where pro-inflammatory stimuli, such as lipopolysaccharide (LPS), induced expression of DLL4 in a Toll-like receptor 4 (TLR4)-NfkB dependent manner (40). Interestingly, exposure of macrophages to DLL4 induced inflammatory nitric oxide synthase (iNOS) (40), a key regulator in the classical (pro-inflammatory) activation of murine macrophages, according to the traditional definitions of macrophages polarization (41, 42). Of note, the “don’t eat me” signal axis CD47-SIRPa, currently under clinical investigation for antibody-based blocking in cancer trials (43), may also be affected by Notch signaling – Notch negatively regulates downstream effectors (Src homology region 2 domain-containing phosphatase-1, SHP-1) of the axis, and pan-Notch inhibition through a γ-secretase inhibitor (GSI) reduced the phagocytic properties of mouse (C57BL/6) bone marrow-derived macrophages (44). Of note, recent evidence points to the tissue-imprinting role of Notch signaling in the establishment and maintenance of liver-resident macrophages (Kupffer cells). Direct interaction of recruited monocytes with DLL4 on sunisoidal endothelial cells was shown to be a critical step in the re-population of depleted Kupffer cells (45, 46), highlighting the role of Notch in monocyte engraftment following liver infection or injury and the acquirement of a tissue-resident macrophage phenotype. Moreover, the development of innate lymphoid cells (ILCs), which may have a key role in regulating immunity, inflammation as well as tissue repair in multiple anatomical compartments (47), has also been reported to be influenced by Notch signaling (48).
Thus, the Notch signaling pathway plays a regulatory role in the differentiation and activation of a range of immune cell types. Although conflicting evidence exists, results support that Notch activation in most of these cell types triggers a pro-inflammatory phenotype. Table 1 gives an overview of the role of Notch in various immune cell subsets.
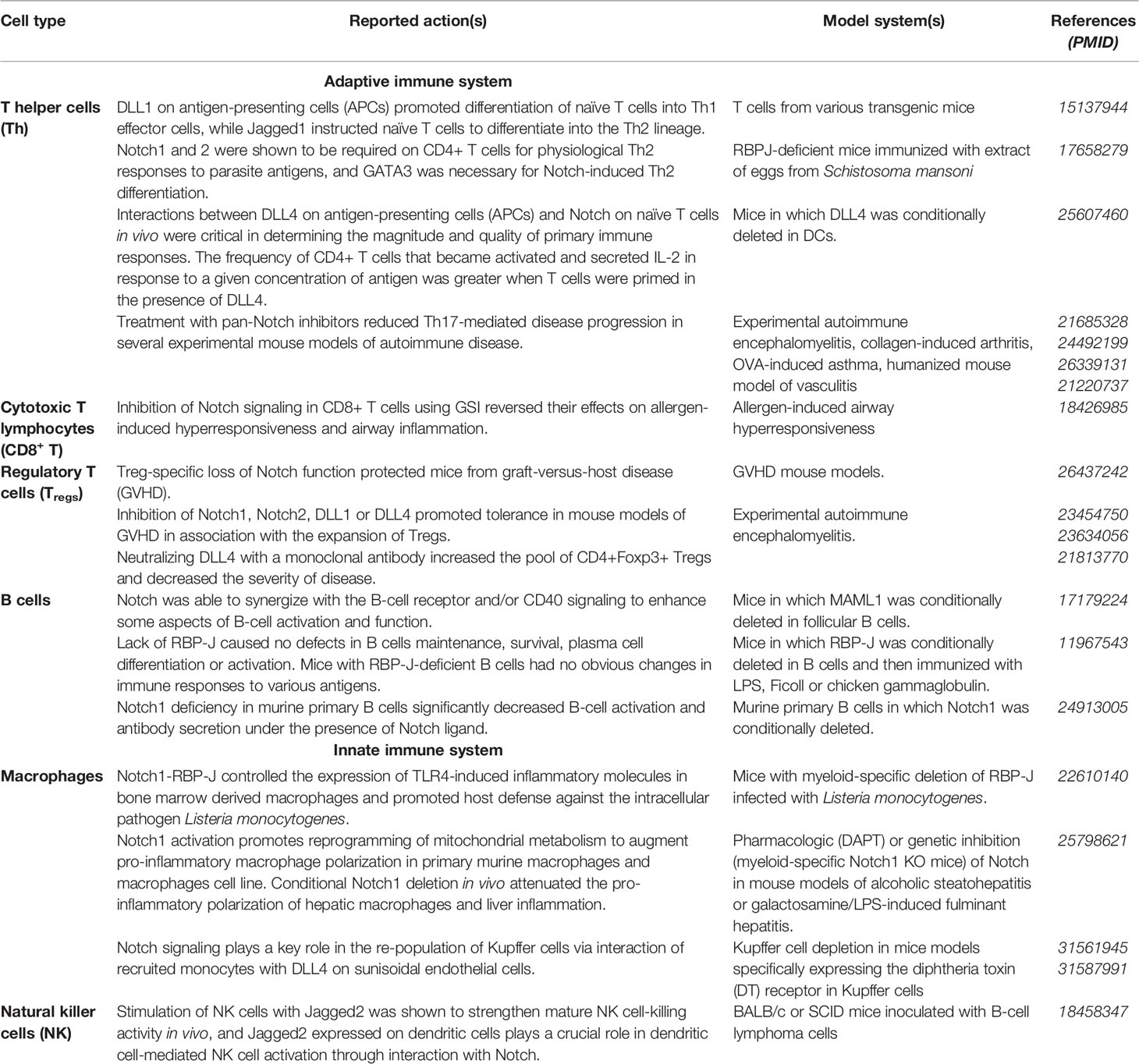
Table 1 The Notch signaling in immune cell differentiation and activation.
Given the distinct features of Notch signaling, targeting this pathway may open new doors for treatment of a range of diseases. While other signaling pathways rely on enzymatic signal amplification, Notch signaling does not, but rather relies on stoichiometric interactions between elements of the pathway (49). Thus, signal intensity can be modulated precisely by cellular regulatory mechanisms. Although there are examples of heterogeneous phenotypic responses to NICD overexpression (50), the downstream effects of Notch activation are in general dose dependent. This implies that complete shutdown of the pathway may not be necessary to achieve a therapeutic effect (51).
Another key feature is that the half-life of the active form of Notch in the nucleus is short, which is important for the dynamic control of Notch signaling (52). As such, Notch signal is a transient, short pulse of gene regulation (53). This implies that sustained inhibition may not always be necessary and that intermittent inhibition can be sufficient to obtain a therapeutic benefit.
Importantly, the effects of Notch may be context dependent. This means that Notch signals trigger distinct responses in different cell types at different time points, and that systemic inhibition of Notch signaling is likely to have a number of effects in a variety of tissues. Thus, for therapeutic utility it will be mandatory to determine whether there is a level and timing of Notch inhibition that is sufficient to attain efficacy in disease control without causing intolerable adverse side-effects. Additional challenges in this context relate to unexplored, but likely roles of different Notch receptors and ligands and their combinations, as specific signaling outcomes might underline particular ligand-receptor pairs. Moreover, all Notch ligands and receptors are transmembrane proteins with extracellular domains that are required for binding and receptor activation, making them reachable for circulating therapeutics. Combined with increasing evidence for aberrant regulation of Notch signaling in several inflammatory disorders, this makes Notch ligands and receptors possibly attractive therapeutic targets. To date, the two main strategies targeting Notch (Figure 2, right panel) are 1) GSIs that block the release of active NCID and 2) monoclonal antibodies (mAbs) that either block ligation upon cell contact or stabilize the NRR-region to prevent proteolytic cleavage.
GSIs were originally developed to block the production of amyloidogenic peptides in Alzheimer’s disease, which also relies on γ-secretase. This multi-subunit enzyme complex targets about 100 transmembrane proteins (54), including the amyloid precursor protein, involved in this neurodegenerative disease. Inhibiting gamma-secretase function prevents the cleavage of the Notch receptors and block Notch signal transduction. Thus, GSIs, such as, DAPT (N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester) or DBZ (dibenzazepine) could pave the way for treatment of diseases where the Notch signaling pathway is central in the pathogenesis and/or progression of disease. Consistent with this rationale, several clinical trials of GSIs in cancer have been conducted (55). The first clinical trial with a GSI Notch inhibitor (MK-0752) was performed for more than 10 years ago on patients with T cell acute lymphoblastic leukemia/lymphoma (T-ALL) (NCT00100152). While deemed optimistic at the time, follow-up studies for T-ALL have not been launched (https://www.clinicaltrials.gov/ct2/results?term=MK-0752&draw=1&rank=9#rowId8). However, MK-0752 has been investigated against refractory central nervous system cancer, pancreatic cancer as well as both metastatic and early-stage breast cancer. Notably, 3 out of these 9 clinical trials have been terminated and follow-up studies of the remaining 6 have yet to be announced. Serious adverse events (SAEs) are common, for instance occurring in 16 out of 30 participants in a 2014 study of metastatic breast cancer (NCT00645333). One of the first and most prominent SAE from Notch inhibition is severe diarrhea. This is most likely due to on-target Notch1 inhibition in intestinal stem cells, resulting in excessive numbers of mucus-secreting goblet cells and inhibition of epithelial cell division (56). Diarrhea has been observed as a dose-limiting toxicity in several clinical trials stratifying GSIs (57, 58), and will likely be the downfall of any systemically delivered pan-Notch inhibitor. Notably, intermittent dosing of a GSI, improved gastrointestinal toxicity to tolerable levels in patients with advanced solid tumors (57).
A phase 3 trial of the GSI semagacestat (NCT00594568), also developed initially for treatment of Alzheimer’s disease, was terminated before completion due to lack of clinical efficacy and safety concerns (59). Adverse events in this case included infections, skin reactions and skin cancers, suggesting on-target Notch inhibition-related events. In line with this, laboratory findings included alterations of immune-cell populations, such as reduced lymphocyte counts, and reduction in Immunoglobulin G (IgG) levels. Despite that the immunosuppressive role of Notch inhibition is well established, its precise roles and effects on the various immune cell subsets are not yet clear. Thus, increased infections in patients treated with semagacestat is not surprising given the significant role of Notch signaling in the activation and differentiation of immune cells serving as the defense line against pathogenic bacteria, viruses and fungi. The increased rate of skin cancer is underpinned by loss-of-function experiments demonstrating a tumor suppressor function of Notch signaling in both mice and human keratinocytes (60–62). Notably, loss of Notch signaling in the murine skin led to atopic dermatitis-like disease and associated myeloproliferative disorder, demonstrating a key role for Notch in homeostatic regulation of the skin and the hematopoietic system (63).
Furthermore, while never reported as an adverse event in humans, administration of a GSI (LY 411575, 5 mg/kg per day for 3 months) in mice resulted in morphologic changes of the liver in the form of minor periportal lymphocytic infiltration (33). The authors hypothesized that Notch inhibition blocked TGFβ-mediated Foxp3 expression, resulting in defective ability to suppress naive T-cell proliferation. These findings are, however, in contrast to a recent study showing that Notch signaling has inhibitory effects on Treg cell function and that Treg cell-specific loss of Notch function actually protects mice from graft-versus-host disease (GVHD) (64). Nevertheless, one cannot rule out that long-term systemic pan-Notch inhibition is the primary causal factor for these morphological changes, since examination of livers from Notch1 antisense mice, which have had reduced Notch1 signaling throughout their entire life span, revealed a more striking infiltration (33). Thus, patients treated long-term with GSIs should most likely be monitored using liver function tests and ultrasound liver imaging.
In addition, hair graying can also be caused by systemic Notch inhibition (65), as conditional deletion of Notch1 and Notch2 in the melanocyte lineage revealed that both are required for proper hair pigmentation in a dose dependent manner (66). Furthermore, conditional ablation of the RBP-J gene in the melanocyte lineage led to defects in hair pigmentation followed by intensive hair graying (67). In line with this, administration of a GSI to wild-type adult mice resulted in gradual hair graying that was irreversible (68). Asides the Notch family members and the amyloid precursor protein, numerous other proteins can also be cleaved by γ-secretase, such as CD44, ErbB4, LRP, syndecan-3, p75 NTR, Apo ER2, DCC, Nectin-1α, E-cadherin and N-cadherin (69). For some of these proteins, such as CD44 binding to hyaluronic acid, the influence of γ-secretase could be to allow cells to become mobile by losing attachment to matrix components (70). For others, like ErbB4, γ-secretase converts it from a membrane-bound tyrosine kinase receptor to an intracellular protein with nuclear localization and the ability to bind and regulate multiple transcription factors (71). Interestingly, ErbB4 is for instance associated with the acquired resistance to ErbB2-inhibitors in HER2+ breast tumors, probably via a shift in the dependency towards ErbB4 signaling following continuous ErbB2 blocking, though additional resistance mechanisms should account (72). Regarding cadherins, γ-secretase activity may affect the epithelial-to-mesenchymal transition or the mesenchymal-to-epithelial transition, depending on the cadherin target [E-cadherin (73), or N-cadherin, respectively (74)]. However, the exact molecular mechanisms involved and the clinical implications remain to be elucidated. In conclusion, the fact that γ-secretase plays a broad biological role and cleaves multiple proteins make the use of inhibitors less attractive for Notch blockade.
A more attractive and specific approach compared with pan-Notch inhibition, is the use of mAbs. The reason for this is that mAbs offer benefits over conventional pharmacotherapy in terms of potency, dosing frequency and specificity for the target. Also, mAbs are generally well tolerated, with off-target effects such as hypersensitivity reactions being relatively rare compared to target-dependent adverse events (75, 76). Thus, development of mAbs with tailored Notch-targeting properties may be a suitable approach. The most preferred molecular scaffold for design of mAbs is IgG1. The reason for this is that it has a long plasma half-life combined with the ability to induce potent effector functions (77). Structurally, it can be divided into a constant fragment crystallizable (Fc) part that mediates effector functions, which is linked to two fragment antigen binding (Fab) arms harboring target specificity (Figure 3, top left panel). This structure has several inherent traits that must be considered for Notch-targeting.
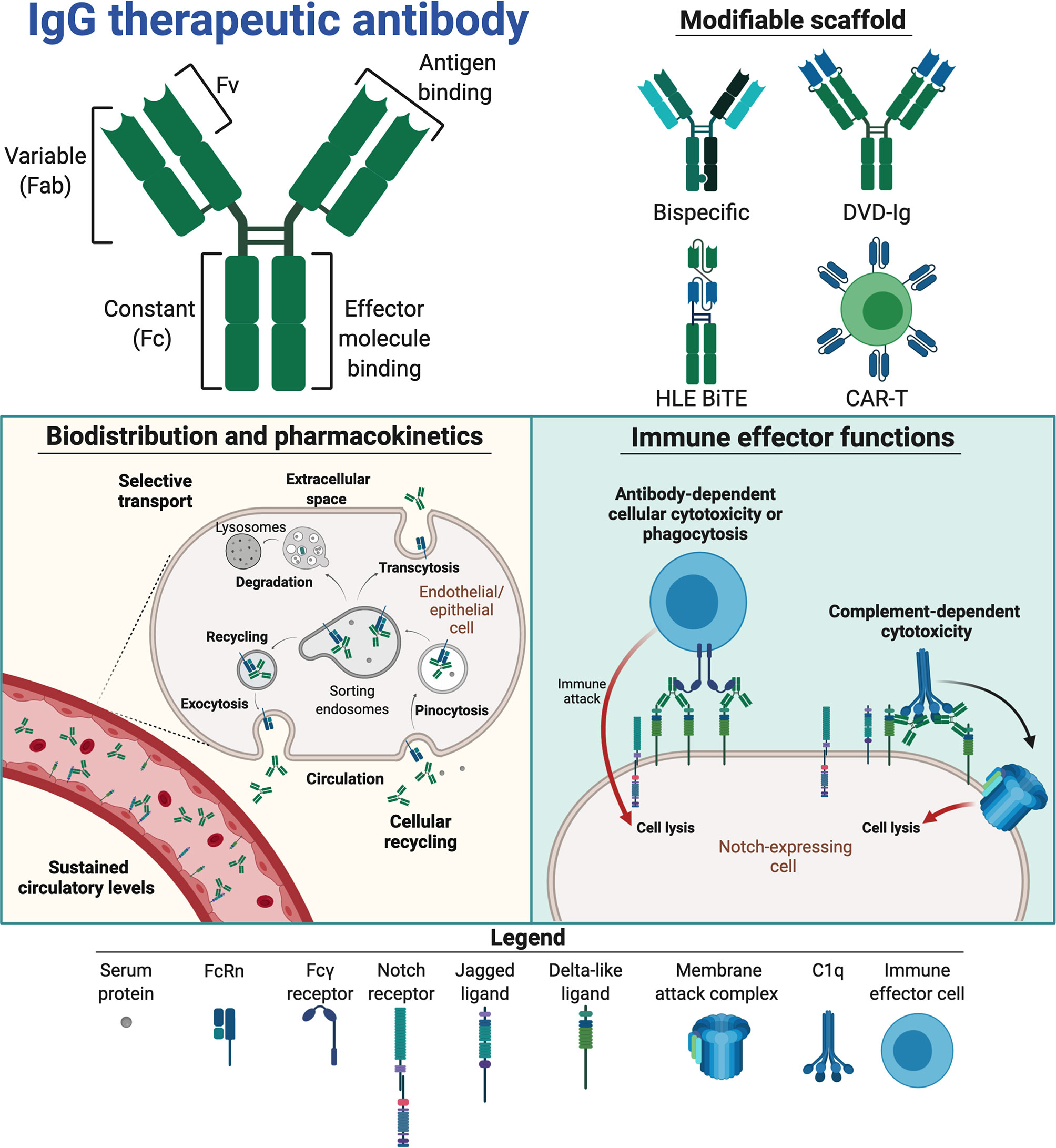
Figure 3 Therapeutic traits of mAbs for Notch interference. Notch interfering by mAbs is based on the therapeutic scaffold of immunoglobulin G (IgG, top left panel). The separate domains of IgG entail different capacities, linking antigen binding in the fragment variable (Fv) to the framework of fragment antigen binding (Fab) arms, which in turn are linked to the constant fragment crystallizable (Fc), containing effector molecule binding sites, via a disulphide-containing hinge. These domains constitute a modifiable scaffold and have been combined in various Notch-targeting moieties (top right panel) to create bispecific and T cell-based therapeutics. If incorporated into therapeutics, the IgG Fc facilitates binding to the neonatal Fc receptor (FcRn, bottom left panel), enabling cellular recycling and specific transcytosis. This may grant therapeutics a long circulatory half-life and affect their biodistribution. IgG Fc fragments may also engage Fcγ receptors and complement proteins (bottom right panel) to mark antigen-expressing cells for destruction via immune cells (antibody-dependent cellular cytotoxicity (ADCC) or phagocytosis (ADCP) or complement-mediated lysis (complement-dependent cytotoxicity, CDC). The figure was made in BioRender (https://biorender.com).
The long plasma half-life of three weeks at average is regulated by binding of the Fc part to the neonatal Fc receptor (FcRn) (78), which is a broadly expressed cellular receptor that rescues IgG from intracellular degradation via a recycling process (Figure 3, bottom left panel). In general, this provides favorably sustained systemic levels of therapeutic mAbs (78). Target availability, and whether the target is soluble or membrane-bound, does however affect the pharmacokinetic properties of mAbs. Regarding targeting of cell-bound Notch ligands and receptors, the pharmacokinetics will be affected through the so-called antigen-sink effect (79). While this will likely affect low-dosage efficacy, it may enable prolonged, specific Notch inhibition with appropriate dosing regimens (80).
FcRn also mediates transport of IgG across selective barriers such as the mucosal epithelium and the placenta (78, 81, 82). Transplacental transport may be particularly important to consider – due to the key developmental role of Notch signaling, Notch targeting may be harmful to the fetus. Notably, infant serum levels of the anti-TNFα mAbs infliximab and adalimumab may be equivalent to maternal levels following mAb treatment of rheumatoid arthritis (RA) during pregnancy (83, 84). This should thus be considered in treatment regimens and mAb design. One way to overcome this challenge will be to engineer the IgG:FcRn interaction by introducing amino acid substitutions in the Fc that attenuate receptor engagement, which will reduce both half-life and transcellular transport. Importantly, the FcRn binding site on IgG is distinct and distant from the binding site for classical Fcγ receptors (85). Another option is the use of the IgG2 subclass, which is less well transported to the fetus compared with the other three IgG subclasses. In this way, the transport properties of mAbs are customizable (78).
Furthermore, Fc-mediated effector functions may be initiated upon binding of mAbs to cell surface displayed targets and lead to target cell destruction by cross-binding to Fcγ receptors expressed by immune cells (antibody-dependent cellular cytotoxicity, ADCC and/or antibody-dependent cellular phagocytosis, ADCP) or engagement of the complement system (complement-dependent cytotoxicity, CDC) (Figure 3, bottom right panel). Such effector functions may be either instrumental to therapeutic efficacy, detrimental to Notch-expressing tissue or simply redundant, depending on both target and indication of the mAb. Under normal physiological conditions, these immune processes are tightly regulated by cells to prevent harmful immune activation (86–88). Thus, how novel therapeutics interplays with such regulatory mechanisms must be accounted for. For instance, in tumor targeting, potent effector functions are favorable, as this will enhance the ability of the mAb to induce tumor killing. However, if the aim is to block target-mediated function(s), or if effector functions are not required, they may even lead to adverse side effects. This potential risk may be minimized by the use of less effector potent subclasses, such as IgG2 or IgG4 (89). With the exception of the anti-DLL4 IgG1 mAbs MEDI0639 (90) and enoticumab (91), all anti-Notch mAb candidates under investigation to date are based on IgG2 (52, 92–94). Other options include hybrid mAb scaffolds, engineering of the IgG1 Fc to abolish effector molecule engagement or even excluding the Fc altogether by utilizing only Fab or the fragment variables (Fvs) (95–97). Furthermore, instead of the Fab arms, therapeutic proteins may be fused to the Fc to improve their pharmacokinetic profile and stability (98, 99). DLL4 has for example been fused to IgG1 Fc, creating a protein shown to suppress liver metastasis of small cell lung cancer both in vitro and in vivo (96). A mAb can also be engineered to harbor two Fab arms with different specificities, creating so-called bispecific mAbs. These can grasp onto two different targets, such as CD4/CD70 and HER2/EGFR (100–102). Such an approach may be attractive for Notch targeting to reduce on-target binding in off-target tissues and cells. The first bispecific Notch antibody nearing approval is the anti-vascular endothelial growth factor (VEGF)/DLL4 IgG2 navicixizumab, which in late 2019 was granted fast-track designation for FDA-approval in pretreated ovarian cancer (https://www.targetedonc.com/view/fda-fast-track-designation-granted-to-navicixizumab-for-heavily-pretreated-ovarian-cancer). Alternatively, Fvs from two different mAbs may be coupled together and fused to the Fc, thus creating dual-variable domain immunoglobulins (DVD-Igs). A DVD-Ig binding both DLL4 and VEGF is currently under clinical testing [ABT-165, (103)]. Importantly, this DVD-Ig harbors an IgG1 Fc containing two amino acid substitutions [L234A, L235A (LALA)] that reduce binding to effector molecules. Fv(s) can further be engineered to single-chain (Sc) Fvs capable of independent monovalent binding (104). ScFv as a building block has been utilized in design of anti-DLL3 chimeric antigen receptor T cells, creating an investigational anti-Notch cancer cell therapeutic [AMG 119, (105, 106)]. Similarly, anti-DLL3 has been used to direct CD3 expressing cytotoxic T cells to DLL3-expressing small cell lung cancer as a half-life extended bispecific T cell engager (HLE BiTE; AMG 757) that results in antitumor activity (107). The structure of AMG 757 consists of a bispecific ScFv fused in tandem to one arm of an IgG1 LALA Fc for half-life extension (107, 108). As such, a range of different antibody scaffolds with Notch targeting properties have reached human studies, resulting in a body of knowledge that may guide the design of future Notch targeting modalities.
In animal studies, targeting of individual Notch receptors and/or ligands with mAbs exhibit reduced on-target intestinal toxicity compared to that seen following GSI treatment (109, 110). For example, use of a murine JAG1-targeting IgG1 mAb in a rat breast cancer xenograft model did not cause toxicity or any effects on body weight (111). Moreover, transient blockade of DLL1 and DLL4 with IgG1 mAbs in the peritransplant period provided durable protection against GVHD after allogeneic bone marrow transplantation without inducing limiting toxicity (109). However, mAb-based DLL-blockade in mice, rats or monkeys induced histopathological changes in the liver that were associated with development of subcutaneous neoplasms with uncertain malignant potential, which alludes to the fundamental role of the Notch system in mammalian physiology (112). On the other hand, blocking of DLL4 by the DVD-Ig ABT-165 was well tolerated in cynomolgous monkeys (103), leading to one completed and one currently recruiting clinical trial for metastatic colorectal cancer (NCT03368859) and various advanced solid tumors (NCT01946074), respectively. Furthermore, completed clinical trials of demcizumab have demonstrated DLL4 blockade to be associated with diarrhea, vomiting, fatigue peripheral edema and hypertension (113–115). These studies have enrolled patients with pancreatic cancer, ovarian-associated cancers and lung cancer, where the mAb has been combined with chemotherapy. One study of lung cancer also tackled demcizumab in combination with pembrolizumab, targeting the programmed cell death protein 1 (PD-1) (NCT02722954). These studies conclude on a tolerable toxicity profile at truncated dosing regimens (114), but further clinical testing of demcizumab is halted due to lack of sufficient therapeutic improvement (92). As reviewed elsewhere, other DLL4-specific mAbs (MEDI0639, enoticumab) have undergone similar trials and pause (92). Navicixizumab is however (as ABT-165) moving forward, having demonstrated notable antitumor activity in its first-in-human phase 1a trial of pretreated solid tumors (93).
Of note, targeting individual Notch receptors rather than activating ligands elicit side-effects similar to that of blockade of Notch activation through DLL4, with dose-limiting diarrhea being the most prominent adverse event during clinical testing of both the anti-Notch1 IgG2 brontictuzumab (NCT03031691) (116, 117), and the anti-Notch2/3 IgG2 tarextumab (118, 119). Importantly, both brontictuzumab and tarextumab were tolerable at adjusted dosages. The ability of mAbs to distinguish between Notch1 and -2/3 is likely essential in this context, as simultaneous inhibition of Notch1 and -2 results in intestinal toxicity (56, 110, 120). Still, neither brontictuzumab nor tarextumab is currently moving forward after failure to meet end goals in phase 1b and 2, respectively (https://www.globenewswire.com/news-release/2017/04/17/961251/0/en/OncoMed-s-Phase-2-Trial-of-Tarextumab-in-Small-Cell-Lung-Cancer-Does-Not-Meet-Endpoints.html).
Looking at alternative Notch ligands, DLL3 stands out as a target having reached clinical studies. In contrast to the previously discussed cell-surface displayed activating ligands, DLL3 resides predominantly in the Golgi apparatus and has fewer EGF-like repeats (121–123). DLL3 acts as a Notch antagonist and attenuates the effects of other DSL ligands (124), distinguishing it from the other Notch ligands. Therapeutic attempts aim to kill DLL3-expressing cells in neuroendocrine cancers, where DLL3 acts as an oncogenic driver, through either lymphocyte activation or antibody-drug conjugates (ADCs) (105, 121, 125). One ADC – rovalpituzumab tesirine – reached clinical trials, where side-effects included fatigue and peripheral edema before it failed to meet its end goal (https://news.abbvie.com/news/press-releases/abbvie-discontinues-rovalpituzumab-tesirine-rova-t-research-and-development-program.htm).
Taken together, evidence suggests that 1) blockade of individual Notch ligands and/or receptors is attainable without intolerable adverse effects and 2) efficacy of targeting Notch ligands, in particular DLL4, is dependent on both indication and combinatory treatment strategy. Importantly, in-human testing of anti-Notch mAbs have yet to reveal any disastrous side-effects, such as those seen in the 2006 study of the anti-CD28 mAb TGN1412 (126). While the clinical efforts on anti-Notch mAbs so far have centered on various, often difficult to treat cancers, the growing body of knowledge surrounding specific Notch blockade may pave the way for development against other immuno-dysregulations, such as inflammatory diseases. Such attempts must be tailored to gain specific therapeutic benefits with limited adverse and toxic effects. In addition, the failure to meet the end goals of the cancer trials, might also be related to the supporting role of Notch in maintaining a pro-inflammatory phenotype of various immune cell subsets (Table 1), which is also required for eliciting an anti-tumor immune response. As such, targeting Notch in other pathophysiologies, where inflammation is the driving force for the disease manifestations, may prove a more valid approach.
In view of targeting Notch signaling in inflammation and the increasing emphasis on combination therapy for chronic inflammatory diseases and cancer, a few recent reports are noteworthy. A significantly improved outcome in a mouse xenograft model of glucocorticoid-resistant T cell acute lymphoblastic leukemia (T-ALL) was reported (127), using a combination of the GSI, dibenzazepine, with the glucocorticoid, dexamethasone. Dexamethasone was shown to counteract the lethal gut toxicity induced by the GSI, and the combination therapy induced apoptosis in xenografts of T-ALL cell lines in mice, to a greater extent than either dexamethasone or dibenzazepine alone. Corroborating with these findings, Samon and colleagues (128), reported a synergistic antileukemic response between the clinically relevant GSI, PF-03084014, and dexamethasone in human T-ALL cell lines, primary patient samples, and in a xenograft model of glucocorticoid-resistant T-ALL. Mechanistically, the GSI increased the transcriptional up-regulation of the glucocorticoid receptor and its target genes, suggesting increased cytotoxicity due to synergistic increase in the glucocorticoid activity. Moreover, gastro-intestinal toxicity induced by PF-03084014 was reversed by dexamethasone, although the precise mechanism is not clear. Notably, the therapeutic efficacy of brontictuzumab against Notch1 was also significantly enhanced in combination with dexamethasone in T-ALL xenografts (129), implying that the effects are not dependent on pan-Notch inhibition or the pharmacodynamics of the GSI but rather result from synergy between the Notch1 inhibition and the increased glucocorticoid receptor activity. This approach, if it can be translated to human patients, might have benefits not only over current treatment of T-ALL, but also for the treatment of chronic inflammatory diseases, where Notch signaling is involved in disease pathogenesis. Glucocorticoids are often used in the clinic to treat or prevent inflammation either alone or in combinational therapies. However, a wide range of dose-related and duration of treatment-related adverse effects of glucocorticoids including osteopenia, skin atrophy, diabetes mellitus, hypertension, muscle atrophy, and cataract formation, have been reported (130).
Moreover, almost 1/3 of patients with ulcerative colitis, asthma, systemic lupus erythematosus, RA, and uveitis, have a disease that is refractory to glucocorticoid therapy (131). Since a synergistic benefit is most likely to occur if therapeutic agents affect different, yet complementary pathways in disease pathogenesis, combinational treatment of Notch inhibitors along with anti-inflammatory therapeutics could be an alternative in chronic inflammatory diseases, refractory to other treatment regimens.
Regarding synergy, it must be noted that the closest to clinical success of mAbs in a Notch context to date is when combining DLL4 and VEGF blockade. This is underlined by observations in preclinical studies of enoticumab combined with the anti-VEGF IgG1 Fc-fusion (aflibercept) in different cancer models (132, 133), ABT-165 in mouse xenografts (103) and in clinical studies of navicixizumab (93). Two additional anti-VEGF/DLL4 bispecific IgG1 antibodies exist and have shown promising results in preclinical cancer models – HB105 and HB-32 (134, 135). Of note, chemotherapy is standard of care for most indications mAbs are investigated for in current clinical trials. Thus, the mAbs are administrated in combination with various chemotherapeutic agents, such as paclitaxel for navicixizumab in ovarian, peritoneal and fallopian tube cancer (NCT03030287). The rationale for this is not new; DLL4, like VEGF, is targeted to disrupt tumor angiogenesis, with an intent to prevent further tumor growth and/or improve delivery of alternative therapeutics through normalized tumor vasculature (136–139). Notably, DLL4 blockade may both enhance chemotherapeutic efficacy and elicit tumoricidal effects in anti-VEGF-resistant tumors (137, 138). Thus, Notch-targeting therapeutics may function as both replacement options and as catalysts for enhancing efficacy of existing therapies in difficult-to-treat cases.
There are several reports of dysregulated Notch signaling in clinical samples from patients with chronic inflammatory diseases. For example, transcription levels of NOTCH1 and HES1 were significantly elevated in colonic mucosal biopsies from patients with ulcerative colitis (140). By analyzing gene expression in tissue from arteries of patients with giant cell arteritis and healthy individuals, Wey and colleagues found abundant expression of JAG1 and NOTCH1 in vasculitic arteries (141). Jagged1 protein was expressed on endothelial cells from patients with giant cell arteritis, but not on endothelial cells from healthy individuals. Moreover, Notch1 protein was up-regulated on circulating CD4+ T cells in patients with giant cell arteritis compared to healthy controls. Likewise, circulating CD4+ T cells and Th2 cells in patients with asthma have been found to have higher levels of NOTCH1 and NOTCH2 (142). Higher levels of active NOTCH1 have also been observed in endothelial cells of inflamed human appendix (143). Such clinical observations, but also experimental studies involving different animal models of inflammation, support a pro-inflammatory role of Notch. In the following sections, we discuss some chronic inflammatory diseases where Notch signaling seem to play a role in pathogenesis, and thus, may represent a potential novel therapeutic target.
Giant cell arteritis is a chronic vasculitis of large- and medium-sized vessels. Although it is a systemic illness and vascular involvement may be widespread, symptomatic blood vessel inflammation most frequently involves the cranial branches of arteries originating from the aortic arch. Visual loss is the most feared complication, resulting from vascular stenosis and occlusion of the central retinal artery (144). A critical step in the disease process is the activation of pro-inflammatory T cells in the vessel wall. It has been demonstrated that treatment with a GSI or soluble Jagged1 to inhibit Notch-dependent signaling, effectively suppressed vascular inflammation in a humanized mouse model, in which human T cells form inflammatory lesions in human vessel were used as xenografts (145). Notch blockade suppressed activity of IFN-γ-producing Th1 cells and made IL-17-producing Th17 cells almost undetectable. This effect may have resulted from a disrupted communication between Notch1-expressing, circulating CD4+ T cells and vascular smooth muscle cells or endothelial cells expressing Jagged1 (141). The latter was shown to up-regulate expression of Jagged1 in response to excess circulating VEGF, thereby educating circulating CD4+ T cells and inducing T cell activation. Not only that, aberrant Notch4 signaling was recently identified as a cause of defective CD8+ Treg cell function, promoting vascular inflammation via accumulation of endosomes and sequestering of NADPH oxidase 2 intracellularly (146). Together, these studies point to the immunostimulatory functions of endothelial Jagged1 and the importance of normal Notch4 signaling in anti-inflammatory CD8+ Treg cells, emphasizing the therapeutic potential of targeting Notch in vasculitis.
RA is an autoimmune disease that affects approximately 1% of the worldwide population (147). The etiology is not fully understood, but involves both genetic and environmental factors. The main autoimmune target in RA is the synovium, facilitating the development of a new synovium with aberrant qualities. This transformed, thickened synovium is called pannus, and harbors invading properties capable of eroding cartilage and bone, thus eventually destroying the functional joint. A leading pannus front involving pathologically activated osteoclasts is fueled by microenvironmental cytokine production, angiogenic vessel reorganization and endothelial activation responsible for leukocyte recruitment, including Th1, Th17, monocytes and neutrophils (148).
Several studies in murine models of arthritis have focused on the in vivo anti-inflammatory potential of Notch inhibition. Most of these have employed either GSIs or Notch1-inhibition, though a growing body of selective modulations involving other members of the pathway is reported (149–151).
The impressive anti-inflammatory effect of GSIs has been demonstrated also in these arthritis models; in both collagen induced arthritis (CIA) (152–154) and collagen antibody-induced arthritis (CAIA) (153), as well as in transgenic TNF-overexpressing (TNF-Tg) mice (155). As a strategy to circumvent the intestinal on-target side effects of global Notch-inhibition, nano-vesicle delivery systems have been employed (156); and a similar strategy showed promise with Notch1-siRNA either alone (157) (in CIA mice) or in combination with the disease modifying anti-rheumatic drug methotrexate (158) (in rats). Genetically engineered Notch1 Antisense-expressing (NAS) mice, where global expression of Notch1 is reduced, are less arthritogenic than controls (152, 153), while conditional myeloid overexpression of NICD1 trended towards worsened arthritis in one study (159). Antibody-mediated inhibition of Notch1 signaling by an IgG1 anti-human and anti-mouse Notch1 led to a modest attenuation in arthritis severity and paw swelling (151).
Other Notch member-specific modulations have successfully attenuated experimental arthritis using HMD1-5 hamster anti-mouse Dll1 IgG (150, 160) and anti-human and anti-mouse Notch3 IgG2a (151) mAbs. On the other side, anti-Jagged1 mAb-treated mice worsened CIA in one study (149), further corroborating the importance of specific targeting selective members of the Notch axis.
How Notch drives the arthritogenic inflammation is thus multifaceted, but involve inflammatory events like endothelial activation and leukocyte recruitment [e.g (143).,], leukocyte activation and function (discussed above), and pathologic angiogenesis (see AMD-section below). Notch1-signaling is involved in hypoxia-induced angiogenesis and potentially also in VEGF/Ang2-induced expression of IL6, IL8, MMP-2 and MMP-9 (161). On the other hand, Notch1 seems to inhibit osteoclastogenesis, while DLL1/Notch2-signaling promotes osteoclast formation and bone resorption (150). Endothelial DLL1 is also involved in Notch2-mediated differentiation of the Ly6Clow monocyte subtype (162), a subtype involved in both initiation and progression of experimental arthritis (163). Notch also has important functions in fibroblast-like synoviocyte, because DLL1, expressed on synovial macrophages, has the potential to activate Notch2 (upregulated on synoviocytes by TNFα), to secrete pro-inflammatory cytokines and matrix metalloproteinases (164). In addition, Notch3 is involved in the differentiation and arthrogenic expansion of a special subset of CD90-positive fibroblasts in the inflamed synovium (151).
A more detailed understanding of the various Notch members’ contributions to RA pathogenesis may aid future treatment strategies, but requires enhanced focus on possible on-target side effects to enable identification of the truly beneficial targets and/or strategies.
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease affecting different organ systems due to the deposition of immune complexes that activate complement (165). Disturbed apoptosis, following UV radiation and impaired removal of apoptotic material, may lead to an increased burden of nuclear antigens, and development of anti-nuclear auto-antibodies (ANA), such as, anti-double stranded DNA (anti-dsDNA), anti-histone, and anti-Sm [reviewed in (166)].
Notch signaling may affect SLE in several ways. GSI treatment reduced elevated numbers of double negative (CD4-/CD8-) T cells and several other disease parameters in the well-studied murine MRL/lpr model of SLE (167). Focusing on the putative role of the different macrophage activation subtypes in SLE pathogenesis [reviewed in (168)], GSI treatment may also ameliorate lupus by impeding the polarization of a particular macrophage subtype (characterized among others by the markers MHCII, CD86, TNFα, IL-10high, IL-12low) in a murine SLE model, generated by immunization with activated lymphocyte-derived DNA (169).
Moreover, cleaved Notch1, cleaved Notch2, and Jagged1 have been shown to be expressed on podocytes in proteinuric nephropathies including lupus nephritis, one of the most serious manifestations of SLE, and their level of expression correlated with the amount of proteinuria, across all disease groups (170). Stronger mechanistic data, obtained by genetic deletion or knockdown technology, revealed that Notch3 is involved in the progression of nephritis, by promoting migratory and pro-inflammatory pathways (171). In line with these findings, persistent Notch activation induced podocyte death, and it has been suggested that Notch acts as a regulator of the balance between podocyte death and regeneration during glomerular disorders (172).
Although the above-mentioned studies demonstrate efficacy of Notch inhibition in experimental SLE or nephritis models, and suggest that Notch inhibition could be an attractive new therapeutic approach, it should be mentioned that clinical observations appear to point in other directions. Comparing T cells from SLE patients with those from healthy controls, the former failed to up-regulate Notch1 expression upon in vitro stimulation. They also exhibited significantly less up-regulation of Notch 1 expression compared with SLE patients with remission (173), and therefore, a reversed correlation with the disease severity. Reduced Notch1 levels in human T cells from SLE patients are also associated with increased IL-17A production (174). However, while GSI treatment inhibits Notch signaling in all receptors and all cell types, signaling via Notch1 in one cell type could possibly elicit responses that counteract the global Notch effect.
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) characterized by an auto-reactive, presumably Th1/Th17-initiated, immune response targeting myelin. The disease most frequently follows a relapsing-remitting course, eventually leading to chronic progression and disability as irreversible damage to axons become prominent (175). Axonal damage is preceded by a loss of oligodendrocyte maturation and their remyelinating capacity.
Notch signaling prevents the neural progenitor stem-cell pool from developing into neurons, thereby ensuring oligodendroglial precursor cell (OPC) potential (176). However, up-regulation of Jagged1 on activated astrocytes inhibits oligodendroglial maturation through a canonical Notch1-Hes5 dependent response in OPCs (177, 178). Thus, since OPCs are readily present but fail to mature in MS patients, it has been suggested that therapeutic inhibition of Notch signaling could favor remyelination in MS lesions (179). However, other studies propose that Notch signaling do not inhibit remyelination (180, 181), but rather promotes it (182, 183). It is possible that Notch signaling serves to maintain the OPC pool (their proliferation and migratory potential), while also providing local, ligand and/or receptor specific environmental cues from demyelinated axons to modulate late stages of oligodendrocyte maturation.
Regardless of the underlying mechanisms, the overall effect of Notch inhibition in neuro-inflammation appears beneficial in several studies, where various approaches have been used. In the experimental autoimmune encephalomyelitis (EAE) model in mice, GSIs reduce clinical and histopathological signs when given either systemically or intraventricularly (30, 184).
While GSIs are capable of penetrating the blood-brain barrier and mount a direct effect on remyelination, mAb therapies given systemically probably exert their effects through modulating the peripheral immune response, and only indirectly affect the CNS. Nevertheless, anti-DLL1 (HMD1-5 hamster anti-mouse Dll1 IgG) (185, 186) or anti-DLL4 (HMD4-2 hamster anti-mouse Dll4 IgG) (10, 187) given systemically reduced the disease severity in mouse models. By contrast, anti-Jagged1 treatment worsened the disease, apparently through inhibiting a Jagged1-dependent up-regulation of splenic IL10-producing cells (185). Altogether, there seem to be a relative consensus that inhibitions of the delta-like ligands cause a reduction in the Th1- and Th17- responses and their CNS infiltration and/or accumulation, perhaps by activating Notch3 (184). The release of Treg-inhibition is suggested as an additional means of action during DLL4 inhibition (10). Some of the positive preclinical findings observed in MS animal models with GSIs might also be the result of Notch inhibition in CNS-resident microglia (188), or tissue infiltrating monocytes/macrophages (189). Reduced Notch2-expression on ‘classical’ (CD14+) monocytes has proved its potential as biomarker to predict drug-neutralizing antibody development in IFNβ-naive MS patients, and a functional link between Notch2-activity and the differentiation into ‘non-classical’ (CD14lowCD16+), pro-inflammatory monocytes was indicated and certainly advocate future studies (190).
Given the possibility that Notch signaling affects both the inflammation and the failed remyelination occurring during MS pathogenesis, future strategies could focus on enhancing CNS delivery of specific Notch modulators administered systemically (191). Targeting specific ligands and receptors could ensure the beneficial effects of Notch inhibition in inflammation and, putatively, in remyelinisation.
Systemic sclerosis (SSc) is a chronic fibrotic disease of unknown etiology that affects the skin, and several internal organs (192). Histopathological hallmarks of early stages are perivascular inflammatory infiltrates and a reduced capillary density (193). Later stages are characterized by an excessive accumulation of extracellular matrix (ECM), caused by persistently activated fibroblasts (192). The resulting fibrosis disrupts the physiological structure of the affected tissues, interferes with proper organ function and is the major cause of death in SSc patients (192, 193). Some advances have been made to treat vascular complications, but no treatment has shown convincing effectiveness to reduce skin and visceral fibrosis. Thus, SSc is still considered incurable.
While research has long been focusing on pro-fibrotic cytokines such as TGFβ, PDGF, MCP-1, IL-4, IL-13 and endothelin, accumulating evidence highlights key roles of morphogen pathways like Wnt, Hedgehog and Notch in the pathogenesis of SSc and other fibrotic diseases (193).
An increasing amount of evidence suggests that the Notch pathway is implicated in the fibrosis development that characterizes SSc. Indeed, Notch1 was activated in the lesional skin of SSc patients and in their fibroblasts (194, 195). Mice with ROS-induced SSc, bleomycin-induced SSc and Tsk1-mice also displayed elevated levels of NICD, overexpression of the ligand Jagged-1, and increased transcription of the target gene HES-1 in the skin and lungs. This accumulation of NICD was associated with the over activation of ADAM17 (also known as tumor necrosis factor-α-converting enzyme, TACE) (194). Moreover, treating mice with a GSI could reduce the collagen content in both skin and lungs and the production of autoantibodies, thus preventing the development of SSc in various in vivo models (192, 194, 195). Similarly, treating SSc-mice with Notch siRNA prevented dermal thickening and fibrosis (192). In the same study, inhibition of Notch signaling prevented the development, of dermal bleomycin-induced fibrosis and inflammation-independent fibrosis in TSK-1 mice. These data suggest that targeting of Notch directly reduces collagen synthesis in fibroblasts and might be effective in the early inflammatory stages of SSc, as well as, in later stages when the inflammatory infiltrates have been resolved. These results are also consistent with those of a study in which the GSI DAPT prevented hypochlorite-induced fibrosis (196).
In summary, the Notch pathway is activated in SSc and inhibition of Notch signaling seems to exert potent anti-fibrotic effects in preclinical models. Thus, the Notch signaling pathway might be a promising molecular target for anti-fibrotic therapeutic approaches, however, the relevant evidence from clinical trials is yet lacking.
Ulcerative colitis (UC) and Crohn’s disease (CD) are the two major phenotypes of inflammatory bowel disease (IBD). Like many other chronic inflammatory diseases, IBD is thought to occur in genetically susceptible individuals exposed to environmental risk factors. Notch signaling plays an indispensable role in gut formation, from the earliest stages of primitive gastrointestinal tube development, to the differentiation and proliferation of both epithelial stem cells and mature cells (197). Beyond its critical roles in normal development and homeostasis of the gut, Notch signaling may also be crucial to the maintenance of the intestinal architecture and barrier function (198).
Functional studies of the role of Notch in the gut have for the most part used either GSIs or genetic modulation of RBP-J. GSIs cause a dose-dependent conversion of enterocyte progenitors to the secretory cell fate, accompanied by activation of the basic helix-loop-helix transcription factor Atoh1/Math1, and down regulation of Hes1 in intestinal crypt progenitors (56). A similar phenotype is obtained by knocking out RBP-J in the intestinal epithelium. Most of the effects observed are probably the result of Notch1 inhibition, since blocking Notch2 with a mAb (anti-human and anti-mouse IgG1, YW169.60.79) did not induce any noticeable effect in the intestinal morphology (110).
It has been demonstrated that Notch signaling increases IL-6 production through an NFκB and RBP-Jκ-mediated pathway in colon epithelial cells, and also that, transient blockage of Notch signaling reduces intestinal inflammation during experimental colitis (199). On the contrary, mice harboring intestinal epithelial cell-specific deletion of RBP-J spontaneously developed chronic colitis characterized by the accumulation of Th17 cells in colonic lamina propria (200). Moreover, absence of colonic Notch1 increased disease severity in acute dextran sulfate sodium (DSS)-colitis (197). Although supporting evidence of a pro-inflammatory role of Notch in experimental IBD exists (199), it’s probably unlikely that blocking the Notch signaling, at least with pan-Notch inhibitors or Notch1-selective inhibitors, is a promising future strategy for treatment of intestinal inflammation. Convincing evidence now indicates that Notch signaling ensures integrity and homeostasis of the intestinal epithelium, and thus any possible positive effects of Notch inhibition in colitis may be overshadowed by the unwanted effects. It should however be emphasized again that individual Notch ligands and receptors can have divergent functions, and future functional studies in IBD models should examine the effect of individual ligand and/or receptor blockade.
Asthma is characterized by persistent airway inflammation, airway hyper responsiveness and mucin hypersecretion. The pathogenesis is complex and involves both genetic and environmental factors (201). Allergen-specific IgE is elevated and the Th2 cytokines IL-4, IL-5, IL-9, and IL-13 are secreted by T helper cells followed by subsequent activation of mast cells, infiltration of eosinophils and airway smooth muscle constriction (202). After antigenic stimulation, naive CD4+ T cells differentiate into two distinct helper T cell subsets; Th1 and Th2 cells (203). It has been generally accepted that disturbed T helper cell balance plays an important role in the pathogenesis of asthma (204).
In a mouse model of asthma, siRNA-mediated Notch1 blockade decreased IL-4 and increased IFN-γ in activated lung T cells (205), suggesting that Notch1 could play a role in regulating the Th1/Th2 imbalance (205, 206). Other studies have also reported that pharmacologic inhibitors of Notch signaling can reduce allergic pulmonary inflammation by modulating Th1 and/or Th2 responses (203, 207, 208). These observations may be mechanistically explained by a critical requirement for Notch to license the Th2 response via promoting lymph node egress of effector Th2 cells (209). Moreover IL-17-producing T helper cells (Th17) are important players in asthma pathogenesis (203). Numerous studies have shown an increase of Th17 cells in the inflammatory airway (205). Therefore, suppressing Th17 response may serve as a promising therapeutic strategy (210). An important role for Notch signaling in Th17 cell differentiation was recently emphasized since, GSI treatment of mice with ovalbumin-induced asthma, ameliorated disease development (211). This treatment also suppressed the Th17-cell responses in spleen and reduced the IL-17 levels in serum. It is worth noting that circulating T cells from asthma patients exhibit increased Notch expression (142). Thus, targeting Notch signaling may represent a promising therapeutic strategy to treat allergic asthma.
Uveitis is inflammation of the uvea, which is made up of the iris, the ciliary body and the choroid. It may be caused by infections, eye traumas, toxins that penetrate the eye, or autoimmunity. It is usually an isolated illness but can also be part of a systemic condition, such as ankylosing spondylitis, juvenile rheumatoid arthritis or Behcet’s disease. Autoimmune uveitis is thought to be related to an aberrant T cell-mediated immune response. Notably, increased activation of the Notch pathway has been demonstrated in CD4+ T cells from patients with active uveitis (212). Moreover, it has been shown that DLL4-induced signaling is critical for the development of experimental autoimmune uveitis (EAU) in mice (213), and that global inhibition of Notch signaling with DAPT, reduced the levels of various proinflammatory cytokines (214). In this model, auto-aggressive CD4+ T cells are the major pathogenic population. Anti-DLL4 mAb (HMD4-2 hamster anti-mouse Dll4 IgG) given systemically during the induction and effector phases reduced the severity of EAU (213). GSI treatment in mice has also been shown to suppress EAU progression, presumably via decreased IL-22 production in CD4+ T cells. This effect was even observed when treatment was administered 13 days after immunization (215). Furthermore, the role of Notch might be more nuanced than captured by these studies, because Notch1 knockdown in infiltrating Tregs contributed to the suppression of EAU (216). Together, these findings indicate that Dll4-mediated Notch signaling is critical for EAU development during the induction phase, but also that inhibition of Notch1 is important for the Treg-mediated suppression of EAU. Perhaps selective Notch blockade could hold promising therapeutic potentials in autoimmune uveitis. Future studies are warranted to explore this possibility.
A concrete body of evidence suggests that low-grade chronic inflammation contributes to the progression of human diseases that were previously not considered as inflammatory disorders, including age-related macular degeneration (AMD) [reviewed in (217)]. In early “dry” AMD, extracellular deposits, known as drusen, accumulate between the retinal pigment epithelium (RPE) and Bruch’s membrane. Most likely, chronic inflammation and complement activation play some roles in the formation of these, and drusen material itself is pro-inflammatory (218).
Although the relationship between drusen formation and AMD is only correlative, increasing accumulation of the former enhances the risk of developing advanced disease; geographic atrophy or exudative (wet) AMD. Wet AMD is characterized by invasion of abnormal choroidal blood vessels (choroidal neovascularization, CNV), and fluid leakage into the retina. At least for wet AMD, be it in AMD patients or experimental CNV, evidence supports the involvement of immune cells (219), such as T cells, macrophages and monocytes that have been identified in eyes from AMD patients (220, 221).
To date, the role of Notch signaling in AMD pathogenesis remains elusive and warrants further research. As previously mentioned, the complement pathway is involved in drusen formation and emerging evidence suggests its causative role in the development of AMD (222). Interestingly, newly identified functions of complement include a role as a nexus for interactions with other effector systems, including the Notch signaling (223). The complement regulator CD46 is a potent co-stimulator for the induction of IFN-γ-secreting Th1 cells and their subsequent switch into regulatory T cells. Jagged1 was recently identified as a new physiological ligand for CD46, and CD46 activation on T cells was shown to regulate the expression of Notch and Notch ligands (224). It is thus legitimate to hypothesize that dysregulation of Jagged1 and/or CD46, leading to Notch hyperactivity, would increase Th1 responses that in turn may contribute to low-grade inflammation in AMD (225). Consequently, interference with Notch signaling could have beneficial effects in AMD progression.
Undoubtedly, Notch plays an important role during pathological angiogenesis, and pan-Notch inhibition was shown to increase the CNV volume, in a study of laser-induced CNV in rats (226). Many of the changes observed were presumably due to the modulation of the endothelial Notch signaling, as mice with conditionally deleted RBP-J in endothelial cells, display patterns similar to those observed after pan-notch inhibition, such as increased leakage shown on angiography and larger CNVs after laser photocoagulation compared to control mice (227). In contrast to treatment with a pan-Notch inhibitor, intravitreal injections of a Jagged1 peptide reduced the CNV volume (226). Nevertheless, Jagged1 is a potent proangiogenic regulator (228), and thus, it is unclear whether the effects observed were due to inhibition of Jagged1-Notch, cell-to-cell interactions or activation of Notch. Administration of Jagged1 peptide reduced sprouting in the postnatal mouse retina (228), suggesting an inhibitory effect. It is tempting to speculate that some of the effects on laser-induced CNV lesions after Jagged1 modulation could be the result of immune cell suppression. Interestingly, a recent genome-wide association study (GWAS), showed an association between AMD and single-nucleotide polymorphisms of the NOTCH4 gene (229). Most of the association signal came from CNV, but the subgroup analysis also suggested an association with geographic atrophy. Thus, Notch signaling, such as its role in the function of immune cells, may be of particular importance.
Notch also has a role in mediating the cellular response to hypoxia through crosstalk with hypoxia-inducible factors 1- and 2-α (HIF1α, HIF2α) (230, 231). Inhibiting Notch signaling has been suggested as a therapeutic approach to prevent hypoxia-induced tumor invasion in uveal melanoma (232). In the same study, hypoxia was found to increase the levels of Notch ligands, including Jagged1. Blocking Notch signaling could thus be a promising approach to reduce hypoxia-mediated stress and thereby pro-angiogenic signaling, both suggested as driving factors in wet AMD (233). A recent study of combined targeting of HIF1α and VEGF in a hypoxia-driven model of retinal neovascularization concluded on beneficial effects of combinatory treatment (234). This raises the hypothesis that the interplay with the hypoxic pathways may also enable Notch blockade to increase the efficacy of the current VEGF-based treatment regimens in neovascular eye diseases. Hypoxia has also been found to up-regulate Notch3 in lung tissue, alluding to the hypothesis of Notch3 serving as a potential therapeutic target to reduce pulmonary arterial hypertension (235). Interestingly, Notch3 has also been shown to be crucial in pathological neovascularization in an oxygen-induced retinopathic model, by stimulating production of angiopoietin-2 and thereby driving angiogenesis (236). Angiopoietin-2 is a known disease driver in AMD and is being targeted by the bispecific mAb IgG1 faricimab, currently in clinical trials for both diabetic macular edema (NCT04432831, phase 3) (237) and wet AMD (NCT03823287, phase 3) (238). Targeting Notch3, to inhibit angiopietin-2 production, could thus be a potential therapeutic avenue for mAb-based anti-Notch approaches that could act upstream of strategies currently in use and/or development.
In addition to their potential effects in eye diseases, Notch-interfering compounds may have an important niche for ocular delivery as the molecular format can be engineered to minimize systemic exposure. This is an active field in mAb design, which has not yet arrived at a clear conclusion. Important perspectives are tissue penetration into the retina, active and passive transport out of the ocular space, interaction with effector molecules and systemic clearance rate (239). Following injection into the eye, therapeutics eventually end up in the circulation, where the aforementioned inherent traits of mAbs may allow them to persist for prolonged periods of time. Therefore, it is not given that anti-Notch mAbs previously tested in clinical trials, would be optimal in an ophthalmological setting. Thus, both the optimal disease-specific Notch target(s) and the molecular format(s) remain to be determined.
Atherosclerosis is an inflammatory disease and not merely the passive accumulation of lipids within artery walls (240). The chronic inflammatory process involving the arterial endothelium that results in the complications of atherosclerosis may be caused by a response to the oxidative components of the modified low-density lipoprotein (LDL), chronic infection, free radicals or other factors (241). Recent evidence has shown that Notch signaling was activated in luminal endothelial cells at atherosclerotic lesions from human aortas (242). Enforced Notch activation also resulted in endothelial cell senescence and upregulated expression of several molecules implicated in the inflammatory response. Additional experimental data, point to the potential of targeting the Notch signaling in atherosclerosis (243). Blockade of DLL4-Notch signaling using an anti-DLL4 mAb (HMD4-2 hamster anti-mouse Dll4 IgG), attenuated the development of atherosclerosis, diminished plaque calcification, improved insulin resistance, and decreased fat accumulation in LDL-receptor-deficient mice fed with a high-fat, high-cholesterol, diet (244). In the same study, DLL4 inhibition appeared to reverse the NF-κB/MCP-1-mediated macrophage accumulation in arteries and adipose tissues. Furthermore, systemic administration of a GSI, reduced plaque areas and fatty streak content in the aortic sinus of apolipoprotein E-deficient mice, fed with a Western diet (245). Similarly, this treatment also suppressed the migratory activity of primary murine peritoneal macrophages and of a macrophage cell line (RAW 264.7) and reduced the expression of ICAM-1, resulting in significantly decreased macrophage infiltration in the atherosclerotic plaques in mice (245). As previously discussed, activation of Notch probably promotes atherosclerosis by inducing a pro-inflammatory activation phenotype in macrophages at the expense of the anti-inflammatory subtype. Even though the Notch signaling has attractive potentials both as a biomarker and as a therapeutic target candidate in atherosclerosis (246), more studies are needed to dissect the precise roles of the various Notch members in the atheromatous plague formation and to explore their individual potentials as novel treatment strategies.
The GVHD induced by donor-derived T cells remains the major limitation of allogeneic bone marrow transplantation (allo-BMT). Current strategies to control GVHD rely on global immunosuppression, however, these strategies do not work in all cases and also decrease the anticancer activity of the allogeneic graft. A critical role for the Notch signaling was discovered in pathogenic host-reactive T cells after allo-BMT (247). A dominant negative Mastermind-like (DNMAML) pan-Notch inhibitor was conditionally expressed in mature CD4+ and CD8+ T cells, leading to reduced GVHD severity. Furthermore, Treg cell-specific deletion of Notch signaling components, protected against full MHC-mismatched GVHD (64). A recent report has provided additional evidence underpinning a clinical relevance (109); transient DLL1 [anti-human and anti-mouse Dll1 IgG1 (109),] and DLL4 [anti-human and anti-mouse Dll4 IgG1, YW152F (138),] inhibition using specific humanized mAbs was sufficient to provide long-lasting protection against GVHD in mice models. Protection was associated with persistent Treg expansion. Notch1 inhibition also controlled GVHD, but led to treatment-limiting toxicity. These findings highlight the therapeutic potential of targeting individual Notch receptors or ligands as a new strategy for controlling GVHD after allo-BMT.
To date, no compounds interfering with the Notch signaling are approved for patients with inflammatory diseases, neither have any reached clinical trials investigating the potential thereof. Clinical studies inhibiting the Notch system have so far focused on cancer, although an increasing interest in defining the roles of Notch in inflammation is clearly evident. Several pharmaceutical and biotech companies have designed pan-Notch inhibitors, Notch receptor or ligand subtype-specific mAbs, and an extensive overview of ongoing and completed clinical studies stratifying Notch targeting strategies in patients with cancer have been reported (52). Based on the available preclinical data from mouse models of inflammation, we propose that modulating the Notch signaling could be an attractive new therapeutic option in several diseases where inflammation plays a fundamental role. Nevertheless, translating the preclinical findings from these models into the clinic remains a challenge. Table 2 summarizes the antibodies used in animal experimentation, up to date. The ubiquitous expression of Notch receptors and ligands on many cell types makes intolerable side effects a distinct possibility, and poses challenges on both therapeutic mode of action and delivery. In these ways, Notch can indeed be said to be a “high hanging fruit” among therapeutic targets (77). Importantly, lessons learned regarding the outcome of targeting individual Notch receptors and ligands in cancer-centered clinical trials may guide future therapeutic efforts. Furthermore, developing drug delivery systems for specific cell-targeted inhibition, intermittent administration schedules and dosage optimization have shown promising results and may pave the way from bench to bedside.
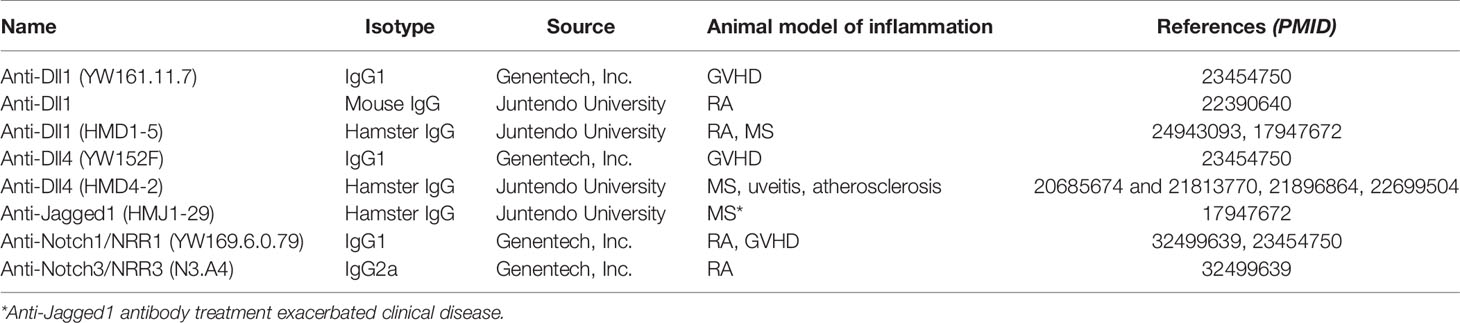
Table 2 Antibodies against the Notch system used in animal studies.
PC and TG performed the literature research, wrote, reviewed and formatted the ms. SK contributed to the literature research, wrote and reviewed the ms. GH and JA wrote and reviewed the ms and provided critical scientific input. ES designed the lay out, wrote and reviewed the ms and supervised the study. All authors contributed to the article and approved the submitted version.
This research was supported by funds provided by the Research Council of Norway (Grant no. 240814) to ES. Supported in part by the South-Eastern Norway Regional Health Authority (Grant no. 2019059) to PC, by internal funding from Division of Head, Neck and Reconstructive Surgery, Oslo University Hospital and Dr. Jon S Larsens Foundation to TG, and by the Research Council of Norway (Grant no. 287927) and South-Eastern Norway Regional Health Authority (Grant no. 2021069 and 2018052) to JA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ALL, acute lymphoblastic leukemia; AMD, age-related macular degeneration; BMT, bone marrow transplantation; CD, Crohn’s disease; CNS, central nervous system; CNV, choroidal neovascularization; DLL-1, Delta-like 1; DSS, dextran sulfate sodium; EAE, autoimmune encephalomyelitis; EAU, experimental autoimmune uveitis; ECM, extracellular matrix; EGF, epidermal growth factor; GSI, γ-secretase inhibitor; GVHD, graft-versus-host disease; HIF1α, hypoxia-inducible factor 1 alpha; IBD, inflammatory bowel disease; IgG, immunoglobulin G; iNOS, inducible Nitric Oxide Synthase; LDL, low-density lipoprotein; LPS, Lipopolysaccharide; mAbs, monoclonal antibodies; MAML-1, Mastermind-like transcriptional co-activator 1; MCP-1, monocyte chemoattractant protein-1; MS, Multiple sclerosis; NICD, Notch intracellular domain; NRR, negative regulatory region; OPC, oligodendroglial precursor cell; PDGF, platelet-derived growth factor; RA, rheumatoid arthritis; RBP-Jk, Recombination Signal-Binding Protein 1 for J-kappa; SHP-1, Src homology region 2 domain-containing phosphatase-1; SLE, Systemic lupus erythematosus; Ssc, Systemic sclerosis; TACE, tumor necrosis factor-α-converting enzyme; TGFb, transforming growth factor beta; TNF, tumor necrosis factor; TLR, Toll-like receptor; UC, Ulcerative colitis; VEGF, vascular endothelial growth factor; ADC, antibody-drug conjugates; DVD-Ig, dual-variable domain immunoglobulin; HLE BiTE, half-life extended bispecific T cell engager; CAR-T, chimeric antigen receptor T cell; ScFv, single-chain fragment variable; Fab, fragment antigen binding; Fc, fragment crystallizable; FcRn, neonatal Fc receptor.
1. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science (1999) 284:770–6. doi: 10.1126/science.284.5415.770
2. Lai EC. Notch signaling: control of cell communication and cell fate. Dev Camb Engl (2004) 131:965–73. doi: 10.1242/dev.01074
4. Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol (2013) 13:427–37. doi: 10.1038/nri3445
5. Hoyne GF, Dallman MJ, Champion BR, Lamb JR. Notch signalling in the regulation of peripheral immunity. Immunol Rev (2001) 182:215–27. doi: 10.1034/j.1600-065x.2001.1820118.x
6. McKenzie GJ, Khan M, Briend E, Stallwood Y, Champion BR. Notch: a unique therapeutic target for immunomodulation. Expert Opin Ther Targ (2005) 9:395–410. doi: 10.1517/14728222.9.2.395
7. Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell (2004) 117:515–26. doi: 10.1016/s0092-8674(04)00451-9
8. Xu J, Chi F, Guo T, Punj V, Lee WNP, French SW, et al. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest (2015) 125:1579–90. doi: 10.1172/JCI76468
9. Okamoto M, Takeda K, Joetham A, Ohnishi H, Matsuda H, Swasey CH, et al. Essential role of Notch signaling in effector memory CD8+ T cell-mediated airway hyperresponsiveness and inflammation. J Exp Med (2008) 205:1087–97. doi: 10.1084/jem.20072200
10. Bassil R, Zhu B, Lahoud Y, Riella LV, Yagita H, Elyaman W, et al. Notch ligand delta-like 4 blockade alleviates experimental autoimmune encephalomyelitis by promoting regulatory T cell development. J Immunol Baltim Md 1950 (2011) 187:2322–8. doi: 10.4049/jimmunol.1100725
11. Hori K, Sen A, Artavanis-Tsakonas S. Notch signaling at a glance. J Cell Sci (2013) 126:2135–40. doi: 10.1242/jcs.127308
12. Kopan R, Ilagan MXG. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell (2009) 137:216–33. doi: 10.1016/j.cell.2009.03.045
13. Gordon WR, Arnett KL, Blacklow SC. The molecular logic of Notch signaling–a structural and biochemical perspective. J Cell Sci (2008) 121:3109–19. doi: 10.1242/jcs.035683
14. del Álamo D, Rouault H, Schweisguth F. Mechanism and significance of cis-inhibition in Notch signalling. Curr Biol CB (2011) 21:R40–47. doi: 10.1016/j.cub.2010.10.034
15. van Tetering G, van Diest P, Verlaan I, van der Wall E, Kopan R, Vooijs M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J Biol Chem (2009) 284:31018–27. doi: 10.1074/jbc.M109.006775
16. Ayaz F, Osborne BA. Non-canonical notch signaling in cancer and immunity. Front Oncol (2014) 4:345:345. doi: 10.3389/fonc.2014.00345
17. Sanalkumar R, Dhanesh SB, James J. Non-canonical activation of Notch signaling/target genes in vertebrates. Cell Mol Life Sci CMLS (2010) 67:2957–68. doi: 10.1007/s00018-010-0391-x
18. Turkoz M, Townsend RR, Kopan R. The Notch Intracellular Domain Has an RBPj-Independent Role during Mouse Hair Follicular Development. J Invest Dermatol (2016) 136:1106–15. doi: 10.1016/j.jid.2016.02.018
19. Dongre A, Surampudi L, Lawlor RG, Fauq AH, Miele L, Golde TE, et al. Non-Canonical Notch Signaling Drives Activation and Differentiation of Peripheral CD4(+) T Cells. Front Immunol (2014) 5:54. doi: 10.3389/fimmu.2014.00054
20. Hayashi Y, Nishimune H, Hozumi K, Saga Y, Harada A, Yuzaki M, et al. A novel non-canonical Notch signaling regulates expression of synaptic vesicle proteins in excitatory neurons. Sci Rep (2016) 6:23969. doi: 10.1038/srep23969
21. Hori K, Cholewa-Waclaw J, Nakada Y, Glasgow SM, Masui T, Henke RM, et al. A nonclassical bHLH Rbpj transcription factor complex is required for specification of GABAergic neurons independent of Notch signaling. Genes Dev (2008) 22:166–78. doi: 10.1101/gad.1628008
22. Díaz-Trelles R, Scimia MC, Bushway P, Tran D, Monosov A, Monosov E, et al. Notch-independent RBPJ controls angiogenesis in the adult heart. Nat Commun (2016) 7:12088. doi: 10.1038/ncomms12088
23. Cruickshank MN, Ulgiati D. The role of notch signaling in the development of a normal B-cell repertoire. Immunol Cell Biol (2010) 88:117–24. doi: 10.1038/icb.2009.73
24. Hozumi K, Mailhos C, Negishi N, Hirano K, Yahata T, Ando K, et al. Delta-like 4 is indispensable in thymic environment specific for T cell development. J Exp Med (2008) 205:2507–13. doi: 10.1084/jem.20080134
25. Koch U, Fiorini E, Benedito R, Besseyrias V, Schuster-Gossler K, Pierres M, et al. Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J Exp Med (2008) 205:2515–23. doi: 10.1084/jem.20080829
26. Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity (1999) 10:547–58. doi: 10.1016/s1074-7613(00)80054-0
27. Kawamata S, Du C, Li K, Lavau C. Overexpression of the Notch target genes Hes in vivo induces lymphoid and myeloid alterations. Oncogene (2002) 21:3855–63. doi: 10.1038/sj.onc.1205487
28. Saito T, Chiba S, Ichikawa M, Kunisato A, Asai T, Shimizu K, et al. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity (2003) 18:675–85. doi: 10.1016/s1074-7613(03)00111-0
29. Hozumi K, Negishi N, Suzuki D, Abe N, Sotomaru Y, Tamaoki N, et al. Delta-like 1 is necessary for the generation of marginal zone B cells but not T cells in vivo. Nat Immunol (2004) 5:638–44. doi: 10.1038/ni1075
30. Minter LM, Turley DM, Das P, Shin HM, Joshi I, Lawlor RG, et al. Inhibitors of gamma-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nat Immunol (2005) 6:680–8. doi: 10.1038/ni1209x
31. Mukherjee S, Schaller MA, Neupane R, Kunkel SL, Lukacs NW. Regulation of T cell activation by Notch ligand, DLL4, promotes IL-17 production and Rorc activation. J Immunol Baltim Md 1950 (2009) 182:7381–8. doi: 10.4049/jimmunol.0804322
32. Fang TC, Yashiro-Ohtani Y, Del Bianco C, Knoblock DM, Blacklow SC, Pear WS. Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity (2007) 27:100–10. doi: 10.1016/j.immuni.2007.04.018
33. Samon JB, Champhekar A, Minter LM, Telfer JC, Miele L, Fauq A, et al. Notch1 and TGFbeta1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood (2008) 112:1813–21. doi: 10.1182/blood-2008-03-144980
34. Bailis W, Yashiro-Ohtani Y, Fang TC, Hatton RD, Weaver CT, Artis D, et al. Notch simultaneously orchestrates multiple helper T cell programs independently of cytokine signals. Immunity (2013) 39:148–59. doi: 10.1016/j.immuni.2013.07.006
35. Steinbuck MP, Arakcheeva K, Winandy S. Novel TCR-Mediated Mechanisms of Notch Activation and Signaling. J Immunol Baltim Md 1950 (2018) 200:997–1007. doi: 10.4049/jimmunol.1700070
36. Backer RA, Helbig C, Gentek R, Kent A, Laidlaw BJ, Dominguez CX, et al. A central role for Notch in effector CD8(+) T cell differentiation. Nat Immunol (2014) 15:1143–51. doi: 10.1038/ni.3027
37. Adler SH, Chiffoleau E, Xu L, Dalton NM, Burg JM, Wells AD, et al. Notch signaling augments T cell responsiveness by enhancing CD25 expression. J Immunol Baltim Md 1950 (2003) 171:2896–903. doi: 10.4049/jimmunol.171.6.2896
38. Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med (2007) 204:1653–64. doi: 10.1084/jem.20062648
39. Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity (2011) 35:780–91. doi: 10.1016/j.immuni.2011.08.013
40. Fung E, Tang S-MT, Canner JP, Morishige K, Arboleda-Velasquez JF, Cardoso AA, et al. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation (2007) 115:2948–56. doi: 10.1161/CIRCULATIONAHA.106.675462
41. Mills CD, Kincaid K, Alt JM, Heilman MJ. Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol Baltim Md 1950 (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
42. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23:549–55. doi: 10.1016/s1471-4906(02)02302-5
43. Andrechak JC, Dooling LJ, Discher DE. The macrophage checkpoint CD47 : SIRPα for recognition of “self” cells: from clinical trials of blocking antibodies to mechanobiological fundamentals. Philos Trans R Soc Lond B Biol Sci (2019) 374:20180217. doi: 10.1098/rstb.2018.0217
44. Lin Y, Zhao J-L, Zheng Q-J, Jiang X, Tian J, Liang S-Q, et al. Notch Signaling Modulates Macrophage Polarization and Phagocytosis Through Direct Suppression of Signal Regulatory Protein α Expression. Front Immunol (2018) 9:1744. doi: 10.3389/fimmu.2018.01744
45. Bonnardel J, T’Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity (2019) 51:638–654.e9. doi: 10.1016/j.immuni.2019.08.017
46. Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, et al. Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity (2019) 51:655–670.e8. doi: 10.1016/j.immuni.2019.09.002
47. Mazzurana L, Rao A, Van Acker A, Mjösberg J. The roles for innate lymphoid cells in the human immune system. Semin Immunopathol (2018) 40:407–19. doi: 10.1007/s00281-018-0688-7
48. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol (2011) 13:144–51. doi: 10.1038/ni.2187
49. Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet (2012) 13:654–66. doi: 10.1038/nrg3272
50. Mazzone M, Selfors LM, Albeck J, Overholtzer M, Sale S, Carroll DL, et al. Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc Natl Acad Sci U S A (2010) 107:5012–7. doi: 10.1073/pnas.1000896107
51. Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene (2008) 27:5124–31. doi: 10.1038/onc.2008.226
52. Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discovery (2014) 13:357–78. doi: 10.1038/nrd4252
53. Miele L, Golde T, Osborne B. Notch signaling in cancer. Curr Mol Med (2006) 6:905–18. doi: 10.2174/156652406779010830
54. Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Proteomic profiling of gamma-secretase substrates and mapping of substrate requirements. PloS Biol (2008) 6:e257. doi: 10.1371/journal.pbio.0060257
55. Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S, et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett (2015) 369:20–7. doi: 10.1016/j.canlet.2015.07.048
56. van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature (2005) 435:959–63. doi: 10.1038/nature03659
57. Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol Off J Am Soc Clin Oncol (2012) 30:2307–13. doi: 10.1200/JCO.2011.39.1540
58. Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, et al. dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin Cancer Res Off J Am Assoc Cancer Res (2015) 21:60–7. doi: 10.1158/1078-0432.CCR-14-0607
59. Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med (2013) 369:341–50. doi: 10.1056/NEJMoa1210951
60. Demarest RM, Ratti F, Capobianco AJ. It’s T-ALL about Notch. Oncogene (2008) 27:5082–91. doi: 10.1038/onc.2008.222
61. Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet (2003) 33:416–21. doi: 10.1038/ng1099
62. Lefort K, Mandinova A, Ostano P, Kolev V, Calpini V, Kolfschoten I, et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev (2007) 21:562–77. doi: 10.1101/gad.1484707
63. Dumortier A, Durham A-D, Di Piazza M, Vauclair S, Koch U, Ferrand G, et al. Atopic dermatitis-like disease and associated lethal myeloproliferative disorder arise from loss of Notch signaling in the murine skin. PloS One (2010) 5:e9258. doi: 10.1371/journal.pone.0009258
64. Charbonnier L-M, Wang S, Georgiev P, Sefik E, Chatila TA. Control of peripheral tolerance by regulatory T cell-intrinsic Notch signaling. Nat Immunol (2015) 16:1162–73. doi: 10.1038/ni.3288
65. Schouwey K, Beermann F. The Notch pathway: hair graying and pigment cell homeostasis. Histol Histopathol (2008) 23:609–19. doi: 10.14670/HH-23.609
66. Schouwey K, Delmas V, Larue L, Zimber-Strobl U, Strobl LJ, Radtke F, et al. Notch1 and Notch2 receptors influence progressive hair graying in a dose-dependent manner. Dev Dyn Off Publ Am Assoc Anat (2007) 236:282–9. doi: 10.1002/dvdy.21000
67. Moriyama M, Osawa M, Mak S-S, Ohtsuka T, Yamamoto N, Han H, et al. Notch signaling via Hes1 transcription factor maintains survival of melanoblasts and melanocyte stem cells. J Cell Biol (2006) 173:333–9. doi: 10.1083/jcb.200509084
68. Kumano K, Masuda S, Sata M, Saito T, Lee S-Y, Sakata-Yanagimoto M, et al. Both Notch1 and Notch2 contribute to the regulation of melanocyte homeostasis. Pigment Cell Melanoma Res (2008) 21:70–8. doi: 10.1111/j.1755-148X.2007.00423.x
69. Purow B. Notch inhibitors as a new tool in the war on cancer: a pathway to watch. Curr Pharm Biotechnol (2009) 10:154–60. doi: 10.2174/138920109787315060
70. Pelletier L, Guillaumot P, Frêche B, Luquain C, Christiansen D, Brugière S, et al. Gamma-secretase-dependent proteolysis of CD44 promotes neoplastic transformation of rat fibroblastic cells. Cancer Res (2006) 66:3681–7. doi: 10.1158/0008-5472.CAN-05-3870
71. Lee H-J, Jung K-M, Huang YZ, Bennett LB, Lee JS, Mei L, et al. Presenilin-dependent gamma-secretase-like intramembrane cleavage of ErbB4. J Biol Chem (2002) 277:6318–23. doi: 10.1074/jbc.M110371200
72. Canfield K, Li J, Wilkins OM, Morrison MM, Ung M, Wells W, et al. Receptor tyrosine kinase ERBB4 mediates acquired resistance to ERBB2 inhibitors in breast cancer cells. Cell Cycle Georget Tex (2015) 14:648–55. doi: 10.4161/15384101.2014.994966
73. Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, et al. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J (2002) 21:1948–56. doi: 10.1093/emboj/21.8.1948
74. Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, et al. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell (2003) 114:635–45. doi: 10.1016/j.cell.2003.08.008
75. Isabwe GAC, Garcia Neuer M, de Las Vecillas Sanchez L, Lynch D-M, Marquis K, Castells M. Hypersensitivity reactions to therapeutic monoclonal antibodies: Phenotypes and endotypes. J Allergy Clin Immunol (2018) 142:159–70. doi: 10.1016/j.jaci.2018.02.018
76. Santos RB, Galvão VR. Monoclonal Antibodies Hypersensitivity: Prevalence and Management. Immunol Allergy Clin North Am (2017) 37:695–711. doi: 10.1016/j.iac.2017.07.003
77. Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the “high-hanging fruit.” Nat Rev Drug Discovery (2018) 17:197–223. doi: 10.1038/nrd.2017.227
78. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol (2007) 7:715–25. doi: 10.1038/nri2155
79. Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet (2010) 49:493–507. doi: 10.2165/11531280-000000000-00000
80. Wang B, Lau YY, Liang M, Vainshtein I, Zusmanovich M, Lu H, et al. Mechanistic modeling of antigen sink effect for mavrilimumab following intravenous administration in patients with rheumatoid arthritis. J Clin Pharmacol (2012) 52:1150–61. doi: 10.1177/0091270011412964
81. Eliesen GAM, van Drongelen J, van Hove H, Kooijman NI, van den Broek P, de Vries A, et al. Assessment of Placental Disposition of Infliximab and Etanercept in Women With Autoimmune Diseases and in the Ex Vivo Perfused Placenta. Clin Pharmacol Ther (2020) 108:99–106. doi: 10.1002/cpt.1827
82. Pyzik M, Sand KMK, Hubbard JJ, Andersen JT, Sandlie I, Blumberg RS. The Neonatal Fc Receptor (FcRn): A Misnomer? Front Immunol (2019) 10:1540:1540. doi: 10.3389/fimmu.2019.01540
83. Mahadevan U, Wolf DC, Dubinsky M, Cortot A, Lee SD, Siegel CA, et al. Placental Transfer of Anti-Tumor Necrosis Factor Agents in Pregnant Patients with Inflammatory Bowel Disease. Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc (2013) 11:286–e24. doi: 10.1016/j.cgh.2012.11.011
84. Julsgaard M, Christensen LA, Gibson PR, Gearry RB, Fallingborg J, Hvas CL, et al. Concentrations of Adalimumab and Infliximab in Mothers and Newborns, and Effects on Infection. Gastroenterology (2016) 151:110–9. doi: 10.1053/j.gastro.2016.04.002
85. Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol Baltim Md 1950 (1989) 143:2595–601.
86. Davies A, Lachmann PJ. Membrane defence against complement lysis: the structure and biological properties of CD59. Immunol Res (1993) 12:258–75. doi: 10.1007/BF02918257
87. Tay MZ, Wiehe K, Pollara J. Antibody-Dependent Cellular Phagocytosis in Antiviral Immune Responses. Front Immunol (2019) 10:332. doi: 10.3389/fimmu.2019.00332
88. Tang Y, Lou J, Alpaugh RK, Robinson MK, Marks JD, Weiner LM. Regulation of antibody-dependent cellular cytotoxicity by IgG intrinsic and apparent affinity for target antigen. J Immunol Baltim Md 1950 (2007) 179:2815–23. doi: 10.4049/jimmunol.179.5.2815
89. Beers SA, Glennie MJ, White AL. Influence of immunoglobulin isotype on therapeutic antibody function. Blood (2016) 127:1097–101. doi: 10.1182/blood-2015-09-625343
90. Jenkins DW, Ross S, Veldman-Jones M, Foltz IN, Clavette BC, Manchulenko K, et al. MEDI0639: a novel therapeutic antibody targeting Dll4 modulates endothelial cell function and angiogenesis in vivo. Mol Cancer Ther (2012) 11:1650–60. doi: 10.1158/1535-7163.MCT-11-1027
91. Vallee BL, Auld DS. Short and long spacer sequences and other structural features of zinc binding sites in zinc enzymes. FEBS Lett (1989) 257:138–40. doi: 10.1016/0014-5793(89)81805-8
92. Moore G, Annett S, McClements L, Robson T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells (2020) 9:1503. doi: 10.3390/cells9061503
93. Jimeno A, Moore KN, Gordon M, Chugh R, Diamond JR, Aljumaily R, et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Invest New Drugs (2019) 37:461–72. doi: 10.1007/s10637-018-0665-y
94. Smith DC, Eisenberg PD, Manikhas G, Chugh R, Gubens MA, Stagg RJ, et al. A phase I dose escalation and expansion study of the anticancer stem cell agent demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin Cancer Res Off J Am Assoc Cancer Res (2014) 20:6295–303. doi: 10.1158/1078-0432.CCR-14-1373
95. An Z, Forrest G, Moore R, Cukan M, Haytko P, Huang L, et al. IgG2m4, an engineered antibody isotype with reduced Fc function. mAbs (2009) 1:572–9. doi: 10.4161/mabs.1.6.10185
96. Kuramoto T, Goto H, Mitsuhashi A, Tabata S, Ogawa H, Uehara H, et al. Dll4-Fc, an inhibitor of Dll4-notch signaling, suppresses liver metastasis of small cell lung cancer cells through the downregulation of the NF-κB activity. Mol Cancer Ther (2012) 11:2578–87. doi: 10.1158/1535-7163.MCT-12-0640
97. Kang TH, Jung ST. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp Mol Med (2019) 51:1–9. doi: 10.1038/s12276-019-0345-9
98. Jafari R, Zolbanin NM, Rafatpanah H, Majidi J, Kazemi T. Fc-fusion Proteins in Therapy: An Updated View. Curr Med Chem (2017) 24:1228–37. doi: 10.2174/0929867324666170113112759
99. Rath T, Baker K, Dumont JA, Peters RT, Jiang H, Qiao S-W, et al. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol (2015) 35:235–54. doi: 10.3109/07388551.2013.834293
100. Mazor Y, Sachsenmeier KF, Yang C, Hansen A, Filderman J, Mulgrew K, et al. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci Rep (2017) 7:40098. doi: 10.1038/srep40098
101. Mazor Y, Hansen A, Yang C, Chowdhury PS, Wang J, Stephens G, et al. Insights into the molecular basis of a bispecific antibody’s target selectivity. mAbs (2015) 7:461–9. doi: 10.1080/19420862.2015.1022695
102. Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, et al. Improving target cell specificity using a novel monovalent bispecific IgG design. mAbs (2015) 7:377–89. doi: 10.1080/19420862.2015.1007816
103. Li Y, Hickson JA, Ambrosi DJ, Haasch DL, Foster-Duke KD, Eaton LJ, et al. ABT-165, a Dual Variable Domain Immunoglobulin (DVD-Ig) Targeting DLL4 and VEGF, Demonstrates Superior Efficacy and Favorable Safety Profiles in Preclinical Models. Mol Cancer Ther (2018) 17:1039–50. doi: 10.1158/1535-7163.MCT-17-0800
104. Nie S, Wang Z, Moscoso-Castro M, D’Souza P, Lei C, Xu J, et al. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antib Ther (2020) 3:18–62. doi: 10.1093/abt/tbaa003
105. Owen DH, Giffin MJ, Bailis JM, Smit M-AD, Carbone DP, He K. DLL3: an emerging target in small cell lung cancer. J Hematol OncolJ Hematol Oncol (2019) 12:61. doi: 10.1186/s13045-019-0745-2
106. Giffin M, Cooke K, Lobenhofer E, Friedrich M, Raum T, Coxon A. P3.12-03 Targeting DLL3 with AMG 757, a BiTE® Antibody Construct, and AMG 119, a CAR-T, for the Treatment of SCLC. J Thorac Oncol (2018) 13:S971. doi: 10.1016/j.jtho.2018.08.1826
107. Hipp S, Voynov V, Drobits-Handl B, Giragossian C, Trapani F, Nixon AE, et al. A Bispecific DLL3/CD3 IgG-Like T-Cell Engaging Antibody Induces Antitumor Responses in Small Cell Lung Cancer. Clin Cancer Res Off J Am Assoc Cancer Res (2020) 26:5258–68. doi: 10.1158/1078-0432.CCR-20-0926
108. Venkataramani S, Low S, Weigle B, Dutcher D, Jerath K, Menzenski M, et al. Design and characterization of Zweimab and Doppelmab, high affinity dual antagonistic anti-TSLP/IL13 bispecific antibodies. Biochem Biophys Res Commun (2018) 504:19–24. doi: 10.1016/j.bbrc.2018.08.064
109. Tran IT, Sandy AR, Carulli AJ, Ebens C, Chung J, Shan GT, et al. Blockade of individual Notch ligands and receptors controls graft-versus-host disease. J Clin Invest (2013) 123:1590–604. doi: 10.1172/JCI65477
110. Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature (2010) 464:1052–7. doi: 10.1038/nature08878
111. Masiero M, Li D, Whiteman P, Bentley C, Greig J, Hassanali T, et al. Development of Therapeutic Anti-JAGGED1 Antibodies for Cancer Therapy. Mol Cancer Ther (2019) 18:2030–42. doi: 10.1158/1535-7163.MCT-18-1176
112. Yan M, Callahan CA, Beyer JC, Allamneni KP, Zhang G, Ridgway JB, et al. Chronic DLL4 blockade induces vascular neoplasms. Nature (2010) 463:E6–7. doi: 10.1038/nature08751
113. McKeage MJ, Kotasek D, Markman B, Hidalgo M, Millward MJ, Jameson MB, et al. Phase IB Trial of the Anti-Cancer Stem Cell DLL4-Binding Agent Demcizumab with Pemetrexed and Carboplatin as First-Line Treatment of Metastatic Non-Squamous NSCLC. Target Oncol (2018) 13:89–98. doi: 10.1007/s11523-017-0543-0
114. Coleman RL, Handley KF, Burger R, Molin GZD, Stagg R, Sood AK, et al. Demcizumab combined with paclitaxel for platinum-resistant ovarian, primary peritoneal, and fallopian tube cancer: The SIERRA open-label phase Ib trial. Gynecol Oncol (2020) 157:386–91. doi: 10.1016/j.ygyno.2020.01.042
115. Hidalgo M, Cooray P, Carrato A, Jameson MB, Parnis F, Jeffery M, et al. A phase 1b study of the anti-cancer stem cell agent demcizumab (DEM) and gemcitabine (GEM) +/- nab-paclitaxel in patients with pancreatic cancer. J Clin Oncol (2016) 34:341–1. doi: 10.1200/jco.2016.34.4_suppl.341
116. Ferrarotto R, Eckhardt G, Patnaik A, LoRusso P, Faoro L, Heymach JV, et al. A phase I dose-escalation and dose-expansion study of brontictuzumab in subjects with selected solid tumors. Ann Oncol Off J Eur Soc Med Oncol (2018) 29:1561–8. doi: 10.1093/annonc/mdy171
117. Casulo C, Ruan J, Dang NH, Gore L, Diefenbach C, Beaven AW, et al. Safety and Preliminary Efficacy Results of a Phase I First-in-Human Study of the Novel Notch-1 Targeting Antibody Brontictuzumab (OMP-52M51) Administered Intravenously to Patients with Hematologic Malignancies. Blood (2016) 128:5108–8. doi: 10.1182/blood.V128.22.5108.5108
118. Yen W-C, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B, et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin Cancer Res Off J Am Assoc Cancer Res (2015) 21:2084–95. doi: 10.1158/1078-0432.CCR-14-2808
119. Smith DC, Chugh R, Patnaik A, Papadopoulos KP, Wang M, Kapoun AM, et al. A phase 1 dose escalation and expansion study of Tarextumab (OMP-59R5) in patients with solid tumors. Invest New Drugs (2019) 37:722–30. doi: 10.1007/s10637-018-0714-6
120. Riccio O, van Gijn ME, Bezdek AC, Pellegrinet L, van Es JH, Zimber-Strobl U, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep (2008) 9:377–83. doi: 10.1038/embor.2008.7
121. Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med (2015) 7:302ra136. doi: 10.1126/scitranslmed.aac9459
122. Geffers I, Serth K, Chapman G, Jaekel R, Schuster-Gossler K, Cordes R, et al. Divergent functions and distinct localization of the Notch ligands DLL1 and DLL3 in vivo. J Cell Biol (2007) 178:465–76. doi: 10.1083/jcb.200702009
123. Fehon RG, Kooh PJ, Rebay I, Regan CL, Xu T, Muskavitch MA, et al. Molecular interactions between the protein products of the neurogenic loci Notch and Delta, two EGF-homologous genes in Drosophila. Cell (1990) 61:523–34. doi: 10.1016/0092-8674(90)90534-l
124. Ladi E, Nichols JT, Ge W, Miyamoto A, Yao C, Yang L-T, et al. The divergent DSL ligand Dll3 does not activate Notch signaling but cell autonomously attenuates signaling induced by other DSL ligands. J Cell Biol (2005) 170:983–92. doi: 10.1083/jcb.200503113
125. Lashari BH, Vallatharasu Y, Kolandra L, Hamid M, Uprety D. Rovalpituzumab Tesirine: A Novel DLL3-Targeting Antibody-Drug Conjugate. Drugs RD (2018) 18:255–8. doi: 10.1007/s40268-018-0247-7
126. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med (2006) 355:1018–28. doi: 10.1056/NEJMoa063842
127. Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med (2009) 15:50–8. doi: 10.1038/nm.1900
128. Samon JB, Castillo-Martin M, Hadler M, Ambesi-Impiobato A, Paietta E, Racevskis J, et al. Preclinical analysis of the γ-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther (2012) 11:1565–75. doi: 10.1158/1535-7163.MCT-11-0938
129. Agnusdei V, Minuzzo S, Frasson C, Grassi A, Axelrod F, Satyal S, et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia (2014) 28:278–88. doi: 10.1038/leu.2013.183
130. Yasir M, Goyal A, Bansal P, Sonthalia S. “Corticosteroid Adverse Effects,”, in: StatPearls (Treasure Island (FL): StatPearls Publishing). Available at: http://www.ncbi.nlm.nih.gov/books/NBK531462/ (Accessed February 11, 2021).
131. Lee RWJ, Creed TJ, Schewitz LP, Newcomb PV, Nicholson LB, Dick AD, et al. CD4+CD25(int) T cells in inflammatory diseases refractory to treatment with glucocorticoids. J Immunol Baltim Md 1950 (2007) 179:7941–8. doi: 10.4049/jimmunol.179.11.7941
132. Huang J, Hu W, Hu L, Previs RA, Dalton HJ, Yang X-Y, et al. Dll4 Inhibition plus Aflibercept Markedly Reduces Ovarian Tumor Growth. Mol Cancer Ther (2016) 15:1344–52. doi: 10.1158/1535-7163.MCT-15-0144
133. Miles KM, Seshadri M, Ciamporcero E, Adelaiye R, Gillard B, Sotomayor P, et al. Dll4 blockade potentiates the anti-tumor effects of VEGF inhibition in renal cell carcinoma patient-derived xenografts. PloS One (2014) 9:e112371. doi: 10.1371/journal.pone.0112371
134. Lee D, Kim D, Choi YB, Kang K, Sung E-S, Ahn J-H, et al. Simultaneous blockade of VEGF and Dll4 by HD105, a bispecific antibody, inhibits tumor progression and angiogenesis. mAbs (2016) 8:892–904. doi: 10.1080/19420862.2016.1171432
135. Zhou R, Wang S, Wen H, Wang M, Wu M. The bispecific antibody HB-32, blockade of both VEGF and DLL4 shows potent anti-angiogenic activity in vitro and anti-tumor activity in breast cancer xenograft models. Exp Cell Res (2019) 380:141–8. doi: 10.1016/j.yexcr.2019.04.025
136. Sitohy B, Nagy JA, Dvorak HF. Anti-VEGF/VEGFR therapy for cancer: reassessing the target. Cancer Res (2012) 72:1909–14. doi: 10.1158/0008-5472.CAN-11-3406
137. Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature (2006) 444:1032–7. doi: 10.1038/nature05355
138. Ridgway J, Zhang G, Wu Y, Stawicki S, Liang W-C, Chanthery Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature (2006) 444:1083–7. doi: 10.1038/nature05313
139. Thurston G, Noguera-Troise I, Yancopoulos GD. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer (2007) 7:327–31. doi: 10.1038/nrc2130
140. Ghorbaninejad M, Heydari R, Mohammadi P, Shahrokh S, Haghazali M, Khanabadi B, et al. Contribution of NOTCH signaling pathway along with TNF-α in the intestinal inflammation of ulcerative colitis. Gastroenterol Hepatol Bed Bench (2019) 12:S80–6. doi: 10.22037/GHFBB.V12I0.1821
141. Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med (2017) 9:aal3322. doi: 10.1126/scitranslmed.aal3322
142. Tindemans I, Vroman H, Lukkes M, van Nimwegen M, de Bruijn MJW, Li BWS, et al. Increased surface expression of NOTCH on memory T cells in peripheral blood from patients with asthma. J Allergy Clin Immunol (2019) 143:769–771.e3. doi: 10.1016/j.jaci.2018.09.012
143. Poulsen L la C, Edelmann RJ, Krüger S, Diéguez-Hurtado R, Shah A, Stav-Noraas TE, et al. Inhibition of Endothelial NOTCH1 Signaling Attenuates Inflammation by Reducing Cytokine-Mediated Histone Acetylation at Inflammatory Enhancers. Arterioscler Thromb Vasc Biol (2018) 38:854–69. doi: 10.1161/ATVBAHA.117.310388
144. Vodopivec I, Rizzo JF. Ophthalmic manifestations of giant cell arteritis. Rheumatol Oxf Engl (2018) 57:ii63–72. doi: 10.1093/rheumatology/kex428
145. Piggott K, Deng J, Warrington K, Younge B, Kubo JT, Desai M, et al. Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis. Circulation (2011) 123:309–18. doi: 10.1161/CIRCULATIONAHA.110.936203
146. Jin K, Wen Z, Wu B, Zhang H, Qiu J, Wang Y, et al. NOTCH-induced rerouting of endosomal trafficking disables regulatory T cells in vasculitis. J Clin Invest (2021) 131 (1) e136042. doi: 10.1172/JCI136042
147. Scott DL, Wolfe F, Huizinga TWJ. Rheumatoid arthritis. Lancet Lond Engl (2010) 376:1094–108. doi: 10.1016/S0140-6736(10)60826-4
148. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Primer (2018) 4:18001. doi: 10.1038/nrdp.2018.1
149. Kijima M, Iwata A, Maekawa Y, Uehara H, Izumi K, Kitamura A, et al. Jagged1 suppresses collagen-induced arthritis by indirectly providing a negative signal in CD8+ T cells. J Immunol Baltim Md 1950 (2009) 182:3566–72. doi: 10.4049/jimmunol.0803765
150. Sekine C, Koyanagi A, Koyama N, Hozumi K, Chiba S, Yagita H. Differential regulation of osteoclastogenesis by Notch2/Delta-like 1 and Notch1/Jagged1 axes. Arthritis Res Ther (2012) 14:R45. doi: 10.1186/ar3758
151. Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature (2020) 582:259–64. doi: 10.1038/s41586-020-2222-z
152. Park J-S, Kim S-H, Kim K, Jin C-H, Choi KY, Jang J, et al. Inhibition of notch signalling ameliorates experimental inflammatory arthritis. Ann Rheum Dis (2015) 74:267–74. doi: 10.1136/annrheumdis-2013-203467
153. Choi BY, Choi Y, Park J-S, Kang L-J, Baek SH, Park JS, et al. Inhibition of Notch1 induces population and suppressive activity of regulatory T cell in inflammatory arthritis. Theranostics (2018) 8:4795–804. doi: 10.7150/thno.26093
154. Jiao Z, Wang W, Hua S, Liu M, Wang H, Wang X, et al. Blockade of Notch signaling ameliorates murine collagen-induced arthritis via suppressing Th1 and Th17 cell responses. Am J Pathol (2014) 184:1085–93. doi: 10.1016/j.ajpath.2013.12.010
155. Sun W, Zhang H, Wang H, Chiu YG, Wang M, Ritchlin CT, et al. Targeting Notch-Activated M1 Macrophages Attenuates Joint Tissue Damage in a Mouse Model of Inflammatory Arthritis. J Bone Miner Res Off J Am Soc Bone Miner Res (2017) 32:1469–80. doi: 10.1002/jbmr.3117
156. Heo R, Park J-S, Jang HJ, Kim S-H, Shin JM, Suh YD, et al. Hyaluronan nanoparticles bearing γ-secretase inhibitor: in vivo therapeutic effects on rheumatoid arthritis. J Control Release Off J Control Release Soc (2014) 192:295–300. doi: 10.1016/j.jconrel.2014.07.057
157. Kim MJ, Park J-S, Lee SJ, Jang J, Park JS, Back SH, et al. Notch1 targeting siRNA delivery nanoparticles for rheumatoid arthritis therapy. J Control Release Off J Control Release Soc (2015) 216:140–8. doi: 10.1016/j.jconrel.2015.08.025
158. Zhao G, Zhang H. Notch-1 siRNA and Methotrexate towards a Multifunctional Approach in Rhematoid Arthritis Management: a Nanomedicine Approach. Pharm Res (2018) 35:123. doi: 10.1007/s11095-018-2401-x
159. Zhao B, Grimes SN, Li S, Hu X, Ivashkiv LB. TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. J Exp Med (2012) 209:319–34. doi: 10.1084/jem.20111566
160. Sekine C, Nanki T, Yagita H. Macrophage-derived delta-like protein 1 enhances interleukin-6 and matrix metalloproteinase 3 production by fibroblast-like synoviocytes in mice with collagen-induced arthritis. Arthritis Rheumatol Hoboken NJ (2014) 66:2751–61. doi: 10.1002/art.38743
161. Gao W, Sweeney C, Walsh C, Rooney P, McCormick J, Veale DJ, et al. Notch signalling pathways mediate synovial angiogenesis in response to vascular endothelial growth factor and angiopoietin 2. Ann Rheum Dis (2013) 72:1080–8. doi: 10.1136/annrheumdis-2012-201978
162. Gamrekelashvili J, Giagnorio R, Jussofie J, Soehnlein O, Duchene J, Briseño CG, et al. Regulation of monocyte cell fate by blood vessels mediated by Notch signalling. Nat Commun (2016) 7:12597. doi: 10.1038/ncomms12597
163. Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK, et al. Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep (2014) 9:591–604. doi: 10.1016/j.celrep.2014.09.032
164. Jiao Z, Wang W, Ma J, Wang S, Su Z, Xu H. Notch signaling mediates TNF-α-induced IL-6 production in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Clin Dev Immunol (2012) 2012:350209. doi: 10.1155/2012/350209
165. Paley MA, Strand V, Kim AHJ. From mechanism to therapies in systemic lupus erythematosus. Curr Opin Rheumatol (2017) 29:178–86. doi: 10.1097/BOR.0000000000000369
166. Rekvig OP, Van der Vlag J. The pathogenesis and diagnosis of systemic lupus erythematosus: still not resolved. Semin Immunopathol (2014) 36:301–11. doi: 10.1007/s00281-014-0428-6
167. Teachey DT, Seif AE, Brown VI, Bruno M, Bunte RM, Chang YJ, et al. Targeting Notch signaling in autoimmune and lymphoproliferative disease. Blood (2008) 111:705–14. doi: 10.1182/blood-2007-05-087353
168. Orme J, Mohan C. Macrophage subpopulations in systemic lupus erythematosus. Discovery Med (2012) 131(1):e136042.
169. Zhang W, Xu W, Xiong S. Blockade of Notch1 signaling alleviates murine lupus via blunting macrophage activation and M2b polarization. J Immunol Baltim Md 1950 (2010) 184:6465–78. doi: 10.4049/jimmunol.0904016
170. Murea M, Park J-K, Sharma S, Kato H, Gruenwald A, Niranjan T, et al. Expression of Notch pathway proteins correlates with albuminuria, glomerulosclerosis, and renal function. Kidney Int (2010) 78:514–22. doi: 10.1038/ki.2010.172
171. El Machhour F, Keuylian Z, Kavvadas P, Dussaule J-C, Chatziantoniou C. Activation of Notch3 in Glomeruli Promotes the Development of Rapidly Progressive Renal Disease. J Am Soc Nephrol JASN (2015) 26:1561–75. doi: 10.1681/ASN.2013090968
172. Lasagni L, Ballerini L, Angelotti ML, Parente E, Sagrinati C, Mazzinghi B, et al. Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem Cells Dayt Ohio (2010) 28:1674–85. doi: 10.1002/stem.492
173. Sodsai P, Hirankarn N, Avihingsanon Y, Palaga T. Defects in Notch1 upregulation upon activation of T Cells from patients with systemic lupus erythematosus are related to lupus disease activity. Lupus (2008) 17:645–53. doi: 10.1177/0961203308089406
174. Rauen T, Grammatikos AP, Hedrich CM, Floege J, Tenbrock K, Ohl K, et al. cAMP-responsive element modulator α (CREMα) contributes to decreased Notch-1 expression in T cells from patients with active systemic lupus erythematosus (SLE). J Biol Chem (2012) 287:42525–32. doi: 10.1074/jbc.M112.425371
175. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol (2015) 15:545–58. doi: 10.1038/nri3871
176. Imayoshi I, Sakamoto M, Yamaguchi M, Mori K, Kageyama R. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J Neurosci Off J Soc Neurosci (2010) 30:3489–98. doi: 10.1523/JNEUROSCI.4987-09.2010
177. Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, et al. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron (1998) 21:63–75. doi: 10.1016/s0896-6273(00)80515-2
178. Genoud S, Lappe-Siefke C, Goebbels S, Radtke F, Aguet M, Scherer SS, et al. Notch1 control of oligodendrocyte differentiation in the spinal cord. J Cell Biol (2002) 158:709–18. doi: 10.1083/jcb.200202002
179. John GR, Shankar SL, Shafit-Zagardo B, Massimi A, Lee SC, Raine CS, et al. Multiple sclerosis: re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat Med (2002) 8:1115–21. doi: 10.1038/nm781
180. Stidworthy MF, Genoud S, Li W-W, Leone DP, Mantei N, Suter U, et al. Notch1 and Jagged1 are expressed after CNS demyelination, but are not a major rate-determining factor during remyelination. Brain J Neurol (2004) 127:1928–41. doi: 10.1093/brain/awh217
181. Seifert T, Bauer J, Weissert R, Fazekas F, Storch MK. Notch1 and its ligand Jagged1 are present in remyelination in a T-cell- and antibody-mediated model of inflammatory demyelination. Acta Neuropathol (Berl) (2007) 113:195–203. doi: 10.1007/s00401-006-0170-9
182. Hu Q-D, Ang B-T, Karsak M, Hu W-P, Cui X-Y, Duka T, et al. F3/contactin acts as a functional ligand for Notch during oligodendrocyte maturation. Cell (2003) 115:163–75. doi: 10.1016/s0092-8674(03)00810-9
183. Aparicio E, Mathieu P, Pereira Luppi M, Almeira Gubiani MF, Adamo AM. The Notch signaling pathway: its role in focal CNS demyelination and apotransferrin-induced remyelination. J Neurochem (2013) 127:819–36. doi: 10.1111/jnc.12440
184. Jurynczyk M, Jurewicz A, Raine CS, Selmaj K. Notch3 inhibition in myelin-reactive T cells down-regulates protein kinase C theta and attenuates experimental autoimmune encephalomyelitis. J Immunol Baltim Md 1950 (2008) 180:2634–40. doi: 10.4049/jimmunol.180.4.2634
185. Elyaman W, Bradshaw EM, Wang Y, Oukka M, Kivisäkk P, Chiba S, et al. JAGGED1 and delta1 differentially regulate the outcome of experimental autoimmune encephalomyelitis. J Immunol Baltim Md 1950 (2007) 179:5990–8. doi: 10.4049/jimmunol.179.9.5990
186. Tsugane S, Takizawa S, Kaneyama T, Ichikawa M, Yagita H, Kim BS, et al. Therapeutic effects of anti-Delta1 mAb on Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J Neuroimmunol (2012) 252:66–74. doi: 10.1016/j.jneuroim.2012.08.003
187. Takeichi N, Yanagisawa S, Kaneyama T, Yagita H, Jin Y-H, Kim BS, et al. Ameliorating effects of anti-Dll4 mAb on Theiler’s murine encephalomyelitis virus-induced demyelinating disease. Int Immunol (2010) 22:729–38. doi: 10.1093/intimm/dxq059
188. Wei Z, Chigurupati S, Arumugam TV, Jo D-G, Li H, Chan SL. Notch activation enhances the microglia-mediated inflammatory response associated with focal cerebral ischemia. Stroke (2011) 42:2589–94. doi: 10.1161/STROKEAHA.111.614834
189. Wongchana W, Lawlor RG, Osborne BA, Palaga T. Impact of Notch1 Deletion in Macrophages on Proinflammatory Cytokine Production and the Outcome of Experimental Autoimmune Encephalomyelitis. J Immunol Baltim Md 1950 (2015) 195:5337–46. doi: 10.4049/jimmunol.1401770
190. Adriani M, Nytrova P, Mbogning C, Hässler S, Medek K, Jensen PEH, et al. Monocyte NOTCH2 expression predicts IFN-β immunogenicity in multiple sclerosis patients. JCI Insight (2018) 3(11):e99274. doi: 10.1172/jci.insight.99274
191. Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med (2011) 3:84ra44. doi: 10.1126/scitranslmed.3002230
192. Dees C, Zerr P, Tomcik M, Beyer C, Horn A, Akhmetshina A, et al. Inhibition of Notch signaling prevents experimental fibrosis and induces regression of established fibrosis. Arthritis Rheumatol (2011) 63:1396–404. doi: 10.1002/art.30254
193. Beyer C, Dees C, Distler JHW. Morphogen pathways as molecular targets for the treatment of fibrosis in systemic sclerosis. Arch Dermatol Res (2013) 305:1–8. doi: 10.1007/s00403-012-1304-7
194. Kavian N, Servettaz A, Mongaret C, Wang A, Nicco C, Chéreau C, et al. Targeting ADAM-17/notch signaling abrogates the development of systemic sclerosis in a murine model. Arthritis Rheumatol (2010) 62:3477–87. doi: 10.1002/art.27626
195. Dees C, Tomcik M, Zerr P, Akhmetshina A, Horn A, Palumbo K, et al. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Ann Rheum Dis (2011) 70:1304–10. doi: 10.1136/ard.2010.134742
196. Kavian N, Servettaz A, Weill B, Batteux F. New insights into the mechanism of notch signalling in fibrosis. Open Rheumatol J (2012) 6:96–102. doi: 10.2174/1874312901206010096
197. Mathern DR, Laitman LE, Hovhannisyan Z, Dunkin D, Farsio S, Malik TJ, et al. Mouse and human Notch-1 regulate mucosal immune responses. Mucosal Immunol (2014) 7:995–1005. doi: 10.1038/mi.2013.118
198. Dahan S, Rabinowitz KM, Martin AP, Berin MC, Unkeless JC, Mayer L. Notch-1 signaling regulates intestinal epithelial barrier function, through interaction with CD4+ T cells, in mice and humans. Gastroenterology (2011) 140:550–9. doi: 10.1053/j.gastro.2010.10.057
199. Aziz M, Ishihara S, Ansary MU, Sonoyama H, Tada Y, Oka A, et al. Crosstalk between TLR5 and Notch1 signaling in epithelial cells during intestinal inflammation. Int J Mol Med (2013) 32:1051–62. doi: 10.3892/ijmm.2013.1501
200. Obata Y, Takahashi D, Ebisawa M, Kakiguchi K, Yonemura S, Jinnohara T, et al. Epithelial cell-intrinsic Notch signaling plays an essential role in the maintenance of gut immune homeostasis. J Immunol Baltim Md 1950 (2012) 188:2427–36. doi: 10.4049/jimmunol.1101128
201. Okamoto M, Takeda K, Lucas JJ, Joetham A, Yasutomo K, Gelfand EW. Low-dose lipopolysaccharide affects lung allergic responses by regulating Jagged1 expression on antigen-pulsed dendritic cells. Int Arch Allergy Immunol (2012) 157:65–72. doi: 10.1159/000324836
202. Corry DB. Emerging immune targets for the therapy of allergic asthma. Nat Rev Drug Discovery (2002) 1:55–64. doi: 10.1038/nrd702
203. Gu W, Xu W, Ding T, Guo X. Fringe controls naïve CD4(+)T cells differentiation through modulating notch signaling in asthmatic rat models. PloS One (2012) 7:e47288. doi: 10.1371/journal.pone.0047288
204. Robinson DS. The role of the T cell in asthma. J Allergy Clin Immunol (2010) 126:1081–91; quiz 1092–1093. doi: 10.1016/j.jaci.2010.06.025
205. Zhang W, Nie Y, Chong L, Cai X, Zhang H, Lin B, et al. PI3K and Notch signal pathways coordinately regulate the activation and proliferation of T lymphocytes in asthma. Life Sci (2013) 92:890–5. doi: 10.1016/j.lfs.2013.03.005
206. Guo X, Zhou M, Ren L, Yang M, Huang S, Xu W. Small interfering RNA-mediated knockdown of Notch1 in lung. Chin Med J (Engl) (2009) 122:2647–51. doi: 10.3760/cma.j.issn.0366-6999.2009.21.023
207. Kang JH, Kim BS, Uhm TG, Lee S-H, Lee GR, Park C-S, et al. Gamma-secretase inhibitor reduces allergic pulmonary inflammation by modulating Th1 and Th2 responses. Am J Respir Crit Care Med (2009) 179:875–82. doi: 10.1164/rccm.200806-893OC
208. KleinJan A, Tindemans I, Montgomery JE, Lukkes M, de Bruijn MJW, van Nimwegen M, et al. The Notch pathway inhibitor stapled α-helical peptide derived from mastermind-like 1 (SAHM1) abrogates the hallmarks of allergic asthma. J Allergy Clin Immunol (2018) 142:76–85.e8. doi: 10.1016/j.jaci.2017.08.042
209. Tindemans I, van Schoonhoven A, KleinJan A, de Bruijn MJ, Lukkes M, van Nimwegen M, et al. Notch signaling licenses allergic airway inflammation by promoting Th2 cell lymph node egress. J Clin Invest (2020) 130:3576–91. doi: 10.1172/JCI128310
210. Dos Santos TM, Righetti RF, Camargo L do N, Saraiva-Romanholo BM, Aristoteles LRCRB, de Souza FCR, et al. Effect of Anti-IL17 Antibody Treatment Alone and in Combination With Rho-Kinase Inhibitor in a Murine Model of Asthma. Front Physiol (2018) 9:1183:1183. doi: 10.3389/fphys.2018.01183
211. Zhang W, Zhang X, Sheng A, Weng C, Zhu T, Zhao W, et al. γ-Secretase Inhibitor Alleviates Acute Airway Inflammation of Allergic Asthma in Mice by Downregulating Th17 Cell Differentiation. Mediators Inflamm (2015) 2015:258168. doi: 10.1155/2015/258168
212. Qi J, Yang Y, Hou S, Qiao Y, Wang Q, Yu H, et al. Increased Notch pathway activation in Behçet’s disease. Rheumatol Oxf Engl (2014) 53:810–20. doi: 10.1093/rheumatology/ket438
213. Ishida W, Fukuda K, Sakamoto S, Koyama N, Koyanagi A, Yagita H, et al. Regulation of experimental autoimmune uveoretinitis by anti-delta-like ligand 4 monoclonal antibody. Invest Ophthalmol Vis Sci (2011) 52:8224–30. doi: 10.1167/iovs.11-7756
214. Yang H, Zheng S, Mao Y, Chen Z, Zheng C, Li H, et al. Modulating of ocular inflammation with macrophage migration inhibitory factor is associated with notch signalling in experimental autoimmune uveitis. Clin Exp Immunol (2016) 183:280–93. doi: 10.1111/cei.12710
215. Bhuyan ZA, Asanoma M, Iwata A, Ishifune C, Maekawa Y, Shimada M, et al. Abrogation of Rbpj attenuates experimental autoimmune uveoretinitis by inhibiting IL-22-producing CD4+ T cells. PloS One (2014) 9:e89266. doi: 10.1371/journal.pone.0089266
216. Rong H, Shen H, Xu Y, Yang H. Notch signalling suppresses regulatory T-cell function in murine experimental autoimmune uveitis. Immunology (2016) 149:447–59. doi: 10.1111/imm.12663
217. Chen M, Xu H. Parainflammation, chronic inflammation, and age-related macular degeneration. J Leukoc Biol (2015) 98:713–25. doi: 10.1189/jlb.3RI0615-239R
218. Kauppinen A, Paterno JJ, Blasiak J, Salminen A, Kaarniranta K. Inflammation and its role in age-related macular degeneration. Cell Mol Life Sci CMLS (2016) 73:1765–86. doi: 10.1007/s00018-016-2147-8
219. Ambati J, Atkinson JP, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol (2013) 13:438–51. doi: 10.1038/nri3459
220. Lopez PF, Grossniklaus HE, Lambert HM, Aaberg TM, Capone A, Sternberg P, et al. Pathologic features of surgically excised subretinal neovascular membranes in age-related macular degeneration. Am J Ophthalmol (1991) 112:647–56. doi: 10.1016/s0002-9394(14)77270-8
221. Oh H, Takagi H, Takagi C, Suzuma K, Otani A, Ishida K, et al. The potential angiogenic role of macrophages in the formation of choroidal neovascular membranes. Invest Ophthalmol Vis Sci (1999) 40:1891–8.
222. Wu J, Sun X. Complement system and age-related macular degeneration: drugs and challenges. Drug Des Devel Ther (2019) 13:2413–25. doi: 10.2147/DDDT.S206355
223. Hess C, Kemper C. Complement-Mediated Regulation of Metabolism and Basic Cellular Processes. Immunity (2016) 45:240–54. doi: 10.1016/j.immuni.2016.08.003
224. Le Friec G, Sheppard D, Whiteman P, Karsten CM, Shamoun SA-T, Laing A, et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat Immunol (2012) 13:1213–21. doi: 10.1038/ni.2454
225. Chen J, Wang W, Li Q. Increased Th1/Th17 Responses Contribute to Low-Grade Inflammation in Age-Related Macular Degeneration. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2017) 44:357–67. doi: 10.1159/000484907
226. Ahmad I, Balasubramanian S, Del Debbio CB, Parameswaran S, Katz AR, Toris C, et al. Regulation of ocular angiogenesis by Notch signaling: implications in neovascular age-related macular degeneration. Invest Ophthalmol Vis Sci (2011) 52:2868–78. doi: 10.1167/iovs.10-6608
227. Dou G-R, Wang Y-C, Hu X-B, Hou L-H, Wang C-M, Xu J-F, et al. RBP-J, the transcription factor downstream of Notch receptors, is essential for the maintenance of vascular homeostasis in adult mice. FASEB J Off Publ Fed Am Soc Exp Biol (2008) 22:1606–17. doi: 10.1096/fj.07-9998com
228. Benedito R, Roca C, Sörensen I, Adams S, Gossler A, Fruttiger M, et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell (2009) 137:1124–35. doi: 10.1016/j.cell.2009.03.025
229. Cipriani V, Leung H-T, Plagnol V, Bunce C, Khan JC, Shahid H, et al. Genome-wide association study of age-related macular degeneration identifies associated variants in the TNXB-FKBPL-NOTCH4 region of chromosome 6p21.3. Hum Mol Genet (2012) 21:4138–50. doi: 10.1093/hmg/dds225
230. Zheng X, Linke S, Dias JM, Zheng X, Gradin K, Wallis TP, et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci U S A (2008) 105:3368–73. doi: 10.1073/pnas.0711591105
231. Mutvei AP, Landor SK-J, Fox R, Braune E-B, Tsoi YL, Phoon YP, et al. Notch signaling promotes a HIF2α-driven hypoxic response in multiple tumor cell types. Oncogene (2018) 37:6083–95. doi: 10.1038/s41388-018-0400-3
232. Asnaghi L, Lin MH, Lim KS, Lim KJ, Tripathy A, Wendeborn M, et al. Hypoxia promotes uveal melanoma invasion through enhanced Notch and MAPK activation. PloS One (2014) 9:e105372. doi: 10.1371/journal.pone.0105372
233. Zhang L, Cui X, Han Y, Park KS, Gao X, Zhang X, et al. Hypoxic drive caused type 3 neovascularization in a preclinical model of exudative age-related macular degeneration. Hum Mol Genet (2019) 28:3475–85. doi: 10.1093/hmg/ddz159
234. Usui-Ouchi A, Aguilar E, Murinello S, Prins M, Gantner ML, Wright PE, et al. An allosteric peptide inhibitor of HIF-1α regulates hypoxia-induced retinal neovascularization. Proc Natl Acad Sci U S A (2020) 117:28297–306. doi: 10.1073/pnas.2017234117
235. Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, et al. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med (2009) 15:1289–97. doi: 10.1038/nm.2021
236. Liu H, Zhang W, Kennard S, Caldwell RB, Lilly B. Notch3 is critical for proper angiogenesis and mural cell investment. Circ Res (2010) 107:860–70. doi: 10.1161/CIRCRESAHA.110.218271
237. Sahni J, Patel SS, Dugel PU, Khanani AM, Jhaveri CD, Wykoff CC, et al. Simultaneous Inhibition of Angiopoietin-2 and Vascular Endothelial Growth Factor-A with Faricimab in Diabetic Macular Edema: BOULEVARD Phase 2 Randomized Trial. Ophthalmology (2019) 126:1155–70. doi: 10.1016/j.ophtha.2019.03.023
238. Khanani AM, Patel SS, Ferrone PJ, Osborne A, Sahni J, Grzeschik S, et al. Efficacy of Every Four Monthly and Quarterly Dosing of Faricimab vs Ranibizumab in Neovascular Age-Related Macular Degeneration: The STAIRWAY Phase 2 Randomized Clinical Trial. JAMA Ophthalmol (2020) 138:964–72. doi: 10.1001/jamaophthalmol.2020.2699
239. Gadkar K, Pastuskovas CV, Le Couter JE, Elliott JM, Zhang J, Lee CV, et al. Design and Pharmacokinetic Characterization of Novel Antibody Formats for Ocular Therapeutics. Invest Ophthalmol Vis Sci (2015) 56:5390–400. doi: 10.1167/iovs.15-17108
240. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
241. Paoletti R, Gotto AM, Hajjar DP. Inflammation in atherosclerosis and implications for therapy. Circulation (2004) 109:III20–26. doi: 10.1161/01.CIR.0000131514.71167.2e
242. Liu Z-J, Tan Y, Beecham GW, Seo DM, Tian R, Li Y, et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: implication of Notch signaling in atherosclerosis. Atherosclerosis (2012) 225:296–303. doi: 10.1016/j.atherosclerosis.2012.04.010
243. Aquila G, Pannella M, Morelli MB, Caliceti C, Fortini C, Rizzo P, et al. The role of Notch pathway in cardiovascular diseases. Glob Cardiol Sci Pract (2013) 2013:364–71. doi: 10.5339/gscp.2013.44
244. Fukuda D, Aikawa E, Swirski FK, Novobrantseva TI, Kotelianski V, Gorgun CZ, et al. Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc Natl Acad Sci U S A (2012) 109:E1868–1877. doi: 10.1073/pnas.1116889109
245. Aoyama T, Takeshita K, Kikuchi R, Yamamoto K, Cheng XW, Liao JK, et al. gamma-Secretase inhibitor reduces diet-induced atherosclerosis in apolipoprotein E-deficient mice. Biochem Biophys Res Commun (2009) 383:216–21. doi: 10.1016/j.bbrc.2009.03.154
246. Vieceli Dalla Sega F, Fortini F, Aquila G, Campo G, Vaccarezza M, Rizzo P. Notch Signaling Regulates Immune Responses in Atherosclerosis. Front Immunol (2019) 10:1130:1130. doi: 10.3389/fimmu.2019.01130
Keywords: notch, inflammation, autoimmunity, gamma-secretase inhibitors (GSIs), neutralizing antibodies, biological therapeutics
Citation: Christopoulos PF, Gjølberg TT, Krüger S, Haraldsen G, Andersen JT and Sundlisæter E (2021) Targeting the Notch Signaling Pathway in Chronic Inflammatory Diseases. Front. Immunol. 12:668207. doi: 10.3389/fimmu.2021.668207
Received: 15 February 2021; Accepted: 24 March 2021;
Published: 12 April 2021.
Edited by:
Markus H. Hoffmann, University of Erlangen Nuremberg, GermanyReviewed by:
Jaba Gamrekelashvili, Hannover Medical School, GermanyCopyright © 2021 Christopoulos, Gjølberg, Krüger, Haraldsen, Andersen and Sundlisæter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panagiotis F. Christopoulos, Y2hyaXN0b3BvdWxvcy5wYW5hZ2lvdGlzQHJyLXJlc2VhcmNoLm5v; Eirik Sundlisæter, ZWlyaWtzdW5kbGlzYXRlckBnbWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.