 Amber Papillion
Amber Papillion André Ballesteros-Tato
André Ballesteros-Tato- Division of Clinical Immunology and Rheumatology, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
Immunosuppressive drugs can partially control Antibody (Ab)-dependent pathology. However, these therapeutic regimens must be maintained for the patient’s lifetime, which is often associated with severe side effects. As research advances, our understanding of the cellular and molecular mechanisms underlying the development and maintenance of auto-reactive B cell responses has significantly advanced. As a result, novel immunotherapies aimed to restore immune tolerance and prevent disease progression in autoimmune patients are underway. In this regard, encouraging results from clinical and preclinical studies demonstrate that subcutaneous administration of low-doses of recombinant Interleukin-2 (r-IL2) has potent immunosuppressive effects in patients with autoimmune pathologies. Although the exact mechanism by which IL-2 induces immunosuppression remains unclear, the clinical benefits of the current IL-2-based immunotherapies are attributed to its effect on bolstering T regulatory (Treg) cells, which are known to suppress overactive immune responses. In addition to Tregs, however, rIL-2 also directly prevent the T follicular helper cells (Tfh), T helper 17 cells (Th17), and Double Negative (DN) T cell responses, which play critical roles in the development of autoimmune disorders and have the ability to help pathogenic B cells. Here we discuss the broader effects of rIL-2 immunotherapy and the potential of combining rIL-2 with other cytokine-based therapies to more efficiently target Tfh cells, Th17, and DN T cells and subsequently inhibit auto-antibody (ab) production in autoimmune patients.
Introduction
Self-reactive auto-antibodies (auto-Abs) against nuclear and cytoplasmic antigens play critical roles in autoimmune disease development and severity (1, 2). Auto-Abs contribute to disease pathogenesis by direct and indirect mechanisms. On the one hand, immune complexes (IC) formed by Auto-Abs and self-antigens activate antigens presenting cells and innate cells through the activation of Fc receptors (FCRs), thereby initiating a feedback loop of immune activation that ultimately leads to unwarranted inflammation and tolerance breakdown (3–5). Auto-Abs also engage the complement system, which mediates tissue damage and further contributes to triggering systemic inflammation. Deposition of IC in the blood vessels, kidney, joints, and lungs amplifies the local inflammatory response and enhance tissue damage. In agreement with their pathogenic roles, the serum levels of auto-Abs strongly correlate with disease activity and severity in multiple forms of autoimmune disease, including systemic lupus erythematosus (SLE), type 1 diabetes (T1D), or rheumatoid arthritis (RA), among others. Furthermore, Auto-Abs can be detected years before the onset of clinical manifestations (6), thus suggesting that loss of B cell tolerance and production of Auto-Abs is a critical step that precedes the development of the autoimmune disease. While Ab-dependent pathology can be partially controlled by immunosuppression, there is currently no cure for systemic autoimmune disorders.
Auto-Abs are produced by autoreactive-plasma cells (PCs), a subset of terminally differentiated B cells that secrete large amounts of Abs (7). PCs can be originated in the germinal centers (GCs), the site of B cell maturation, where B cells undergo rapid rounds of proliferation, somatic hypermutation, and affinity maturation leading to the generation of high-affinity antibodies (8–10). Autoreactive PCs can also be generated outside the GCs via the extrafollicular pathway (11–13). Recent studies demonstrate a critical role for the extrafollicular PCs in the development of pathogenic Ab responses (11, 13, 14).
In the last decade, B cell depleting therapies were designed based on the rationale that depletion of self-reactive B cells would reduce the production of auto-Ab and subsequent Auto-Ab-mediated immunopathology (15, 16). However, the clinical efficacy of these therapies is lower than initially anticipated (17–19). The inability of B cell depleting agents to eliminate self-reactive PCs efficiently has been suggested as a plausible explanation for the relatively low effectiveness of these approaches (17). The life-threatening side effects of sustained immunosuppression and the failure of new therapies, such as B cell depletion, vindicate looking for new therapeutic alternatives to treat Ab-mediated pathologies. In this manuscript, we review the potential of low-dose IL-2-based immunotherapies to target T cell populations with B cell helper activity, mainly T follicular helper cells (Tfh), T helper 17 (Th17) cells, and Double-negative (DN) CD3+CD4-CD8- T cells (Figure 1). While IL-2 also induces immunosuppression by Treg-dependent mechanisms, more extensive reviews on this topic are available elsewhere. Hence, the role of IL-2 in promoting Treg-mediated immunosuppression will be only briefly discussed in this review.
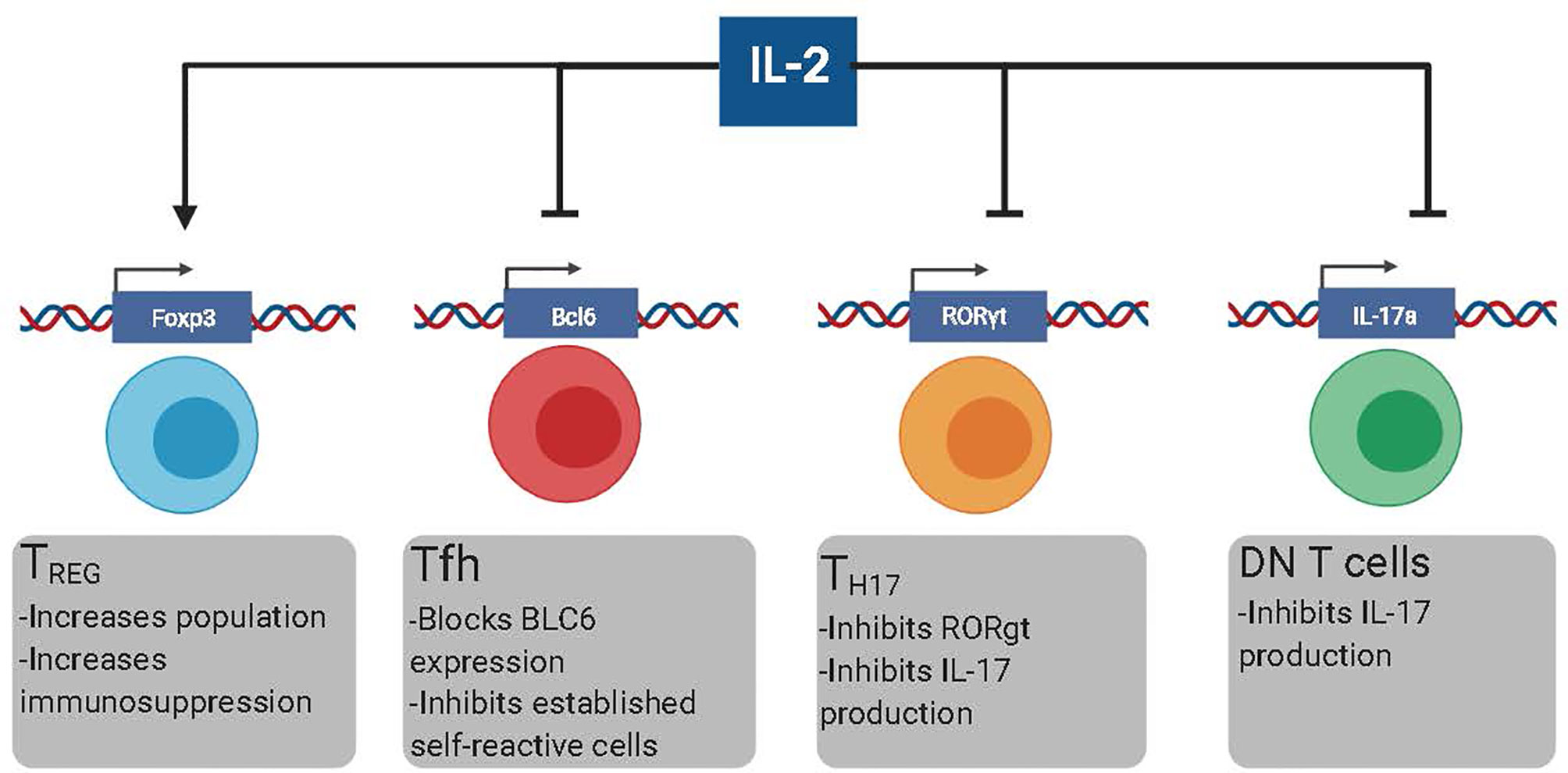
Figure 1 The different effects of low dose rIL-2 therapy in autoimmunity. Low dose rIL-2 stabilizes FoxP3 program in Treg cells which increases both the size of the population and enhances immunosuppression. Low dose rIL-2 therapy can both inhibit the generation of new self-reactive Tfh cells and decrease already present self-reactive Tfh cells by blocking Bcl6. IL-6 blockade will make Tfh cells more suspectable to IL-2 signaling. Low dose rIL-2 can inhibit Th17 cells by diminishing expression of RORgt and inhibiting IL17a expression. Low dose rIL2 can also inhibit IL-17 production by DN T cells by directly inhibiting IL17a.
Pathogenic B Cell Helper T Cell Subsets
Tfh Cells
T follicular helper (Tfh) cells are a subset of CD4+ T cells that provide co-stimulatory signals and cytokines that are required for the development and maintenance of GCs (20–22) and extrafollicular PC differentiation (23, 24) (Figure 2). In the absence of pathogen-specific Tfh cells, GCs do not develop, and pathogen-specific PC responses are impaired. Phenotypically, Tfh cells are characterized by the expression of CXCR5, PD1, ICOS, and Bcl6, among other markers (25). CXCR5 is a chemokine receptor that allows Tfh cells to localize in the proximity of the B/T cell border in response to CXCL13. The inhibitory receptor PD-1 and the co-stimulatory receptor ICOS ultimately direct Tfh cells into the B cell follicles (26–28), where they provide CD40L (22, 29) and IL-21 (30–32) to the responding B cells. Bcl6 is a transcription factor that promotes the expression of genes required for Tfh cell development and function while preventing the up-regulation of transcription factors implicated in T effector (Teff) cell differentiation (33–35). Bcl6 is critical for the differentiation of Tfh cells (33–35). Thus, it is considered the master regulator of Tfh differentiation. While Bcl6 promotes Tfh formation, the transcription factor Blimp-1 represses it (33–35). Importantly, Blimp-1 and Bcl6 are mutually antagonistic transcription factors that directly repress one another in CD4+ T cells. Thus, the balance between the relative expression of Bcl6 and Blimp-1, rather than the expression of Bcl6 alone, fine-tunes the commitment into the Tfh cell pathway (20).
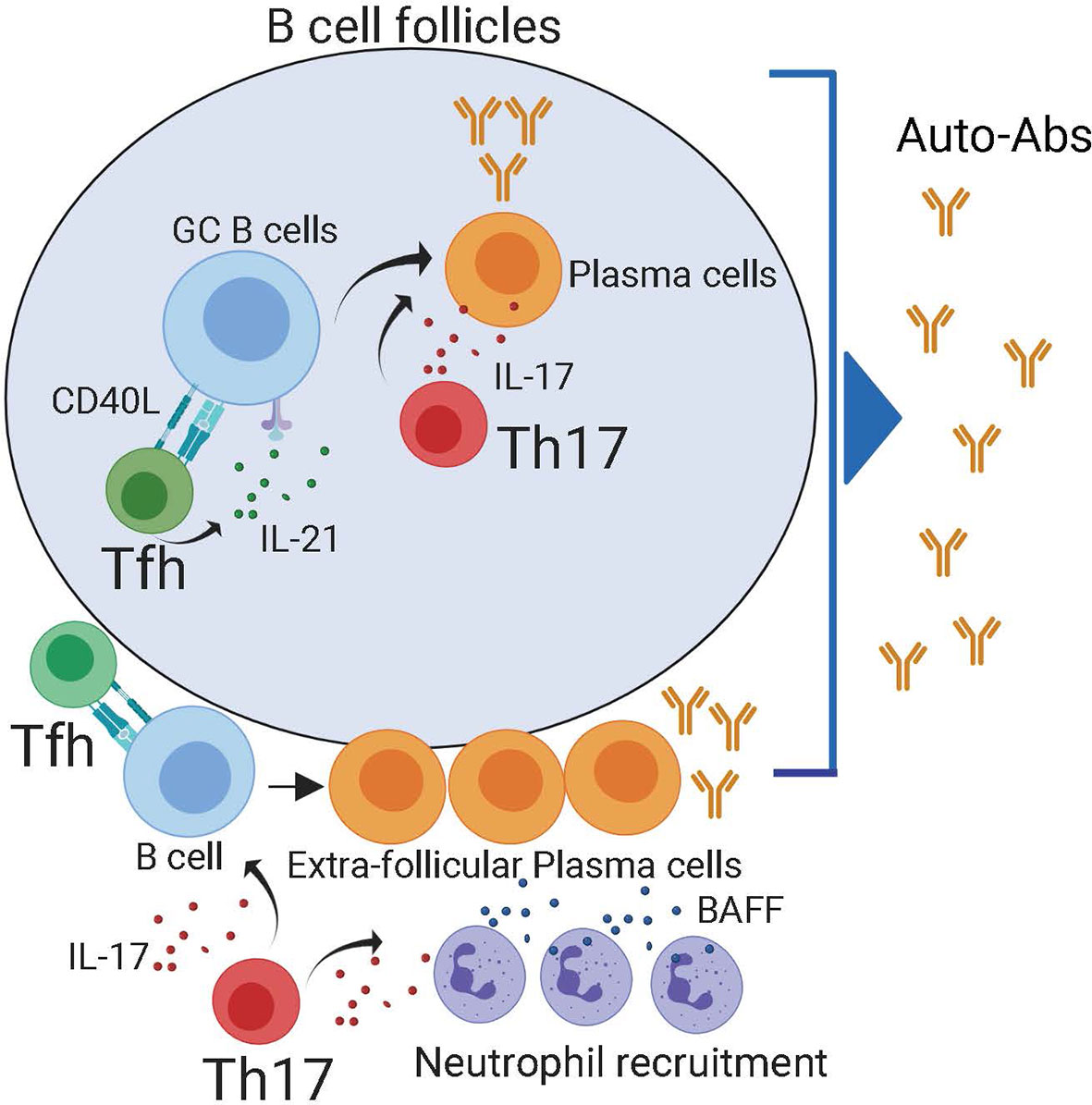
Figure 2 Both follicular and extra-follicular pathways contribute to auto-antibody production. Within the B cell follicles, Tfh cells maintain auto-reactive germinal center reactions which generate self-reactive plasma cells. Tfh cells can also provide help in to extra-follicular PCs in the B cell border. Reports have demonstrated that IL-21 produced by Th17 cells can also contribute to the production of self-reactive plasma cells in the follicle. Besides, in the extra-follicular space, IL-17 produced by Th17 recruits BAFF producing neutrophils. The combination of IL-17, BAFF, and Tfh cells promotes the generation of self-reactive plasma cells.
Under homeostatic conditions, Tfh cells help germinal center B cells to facilitate somatic hypermutation and class switch to generate long-term Ab protection to pathogens (25). However, when there is a break in intolerance, self-reactive Tfh cells are not depleted from the repertoire and provide co-stimulatory signals and cytokines to self-reactive B cells, leading to pathogenic auto-Ab responses (36, 37). In agreement, the presence of Tfh cells correlates with elevated levels of Auto-Ab and disease activity in preclinical animal models and autoimmune patients (37–41). When Tfh cells are depleted or decreased, autoimmune disease pathogenesis and auto-Ab responses are reduced (40, 42, 43). Based on these findings, Tfh cells are considered a potential target for autoimmune disorders (44). However, to date, there are no therapeutic agents approved to selectively deplete Tfh cells in vivo.
Th17 Cells
Th17 cells are a specialized subset of CD4+ T cells that play an essential role in aiding host defense by recruiting neutrophils and macrophages. Th17 cells differentiate in response to TGFβ and IL-6, and their development is driven by the transcription factor RORγt (45). Excessive Th17 cell responses are implicated in the pathogenesis of multiple forms of autoimmune diseases. As such, the expansion of self-reactive Th17 cells correlates with disease activity in common autoimmune diseases, including RA, psoriasis, asthma, and lupus (46–48). Due to its characteristic pro-inflammatory properties, it is generally believed that Th17 cells contribute to autoimmune disease pathogenesis by inducing tissue inflammation. However, recent studies demonstrate that Th17 cells can also help the development of auto-reactive GC B cells (48–51) and extrafollicular PCs (52). In agreement with this idea, IL-17 deficiency prevents auto-Ab production and disease progression in lupus-prone mice (53–55). In addition, the self-reactive GC responses were reduced by IL-17 deficiency in autoimmune Roquinsan/san mice, thereby suggesting a cause-effect relationship between IL-17 and pathogenic GC B cell responses. Collectively, these studies suggest an important role for IL-17 producing cells in promoting pathogenic-B cell responses in the context of autoimmune disorders.
The capacity of Th17 cells to help B cell responses is not entirely surprising since, like Tfh cells, Th17 characteristically produce large amounts of IL-21 (56), a cytokine that promotes GC and PC differentiation. Furthermore, IL-17 synergizes with the B cell-activating factor belonging to the TNF family, BAFF, to protect responding cells from BCR-induced apoptosis (57), demonstrating an intrinsic effect of IL-17 in promoting B cell survival. Besides, IL-17 promotes the recruitment of neutrophils (58, 59), which secrete both BAFF and APRIL and can facilitate the survival and devolvement of extrafollicular PCs (60, 61). Collectively, these studies suggest that IL-17-producing cells can, directly and indirectly, help auto-reactive B cells responses, thereby contributing to Ab-mediated pathology in autoimmune patients (Figure 2). However, whether IL-17-producing cells promote rather than merely correlate with self-reactive B cell responses in autoimmune patients has not yet been formally demonstrated.
Hybrid IL-17+Tfh Cells?
An additional important question remaining is how the putative “IL-17+ helper” cells gain access to the B cell follicles to provide B cell help. One exciting possibility is that pathogenic “IL-17+ helper” cells are indeed Tfh cells that secrete IL-17. In agreement with this possibility, studies suggest the presence of hybrid IL-17-producing cells with “Tfh-like” characteristics in autoimmune prone BXD2 mice (48) and human tonsils (45). Furthermore, the culture of human CD4+ T cells with a combination of TGFβ and IL-23, which is frequently used for the in vitro differentiation of Th17 cells (45), triggers the acquisition of a Tfh-like transcriptional signature characterized by the up-regulation of Bcl6, c-Maf, and CXCR5, and the down-regulation of Blimp-1, thereby resulting in the acquisition of a hybrid Bcl6+RORγt+ Tfh/Th17 signature (62). Whether hybrid Tfh/Th17 cells are Tfh cells that secondary acquire the capacity of secrete IL-17 or represent a separate lineage of Tfh cells is still unclear. Further investigations are needed in order to clarify the potential relationship between these two lineages.
Interestingly, the ‘pro-Tfh” effect of TGF-β is restricted to humans, as TGF-β does not significantly affect Tfh cell differentiation in mice (62, 63). Nevertheless, the commonality between the Tfh and Th17 differentiation requirements extends beyond TGF-β. For example, ICOS, which is required for the survival and the migration of Tfh cells into the B cell follicles (26, 28, 64), is also critical for the differentiation and maintenance of Th17 cells (65, 66). Besides, similar to Tfh cells, the IL-6/STAT3 pathway is also a key positive regulator of Th17 differentiation (67). Thus, critical signaling pathways implicated in Tfh cell differentiation also critically regulate the Th17 program. Therefore, it is reasonable to speculate that the same inflammatory conditions that promote Tfh differentiation in autoimmune patients also favor the development of Th17 cells and/or the generation of hybrid IL-17-producing Tfh cells.
The concept of a highly pathogenic hybrid Tfh/IL-17 population with superior helper activity is, however, at odds with early studies suggesting that Bcl6 functions as a direct transcriptional repressor that prevents the acquisition of Teff programs, including the Th17 program (33–35). Indeed, Bcl6hiCXCR5hiPD-1hi Tfh cells present in the B cell follicles (we will refer to these cells as GC-Tfh cells) do not normally produce IL-17, which is consistent with studies showing that high-expression of Bcl6 in GC-Tfh cells directly represses RORγt and Th17 differentiation (33, 34). Nevertheless, the role of Bcl6 in controlling alternative differentiation programs in Tfh cells is puzzling. As such, while studies suggest that Bcl6 binds to the Rorc promoter and inhibits its expression (33), other studies show no evidence of Bcl6 binding (34, 68). Furthermore, while Bcl6-expressing cells do not normally express Teff cytokines, some studies indicate that Tfh cells can produce effector cytokines in the context of high inflammatory conditions, such as viral infections (69–72) or autoimmune diseases (48). In addition, extrafollicular-Tfh cells (which express medium levels of Bcl6) have a more heterogeneous transcriptional signature than Bcl6hi GC-Tfh cells (which express high levels of Bcl6) (73–75). These results suggest that the ability of Bcl6 to inhibit the initiation of secondary Teff differentiation programs in developing Tfh cells is dose-dependent and can be partially overcome in highly reactive environments, such as in autoimmune diseases, thereby leading to the acquisition of hybrid Tfh/Teff phenotypes, such as IL-17+ Tfh cells, with enhanced pathogenic functions.
Double-Negative T Cells
Double-negative (DN) CD3+CD4-CD8- T cells are a rare population of TCR-αβ+ T cells that lack CD4 and CD8 expression and express high levels of B220 (76, 77). While DN T cells are relatively scarce in healthy individuals, they abnormally expand in lupus patients and children with autoimmune diseases, such as mixed connective tissue disease or juvenile idiopathic arthritis (78). Aberrant accumulation of DN T cells is also a clinical hallmark of the Autoimmune Lymphoproliferative Syndrome (ALPS, also known as Canale-Smith syndrome), a genetic disorder caused by defective FAS-mediated apoptosis that is characterized by the development of autoimmune disease, splenomegaly, lymphadenopathy, and an increased risk of secondary lymphomas during childhood (79, 80). In aged MRL/lpr mice, DN T cells represent nearly 70% of the total cells in the enlarged lymph nodes, accounting for the characteristic lymphadenopathy observed in these mice.
The exact origin of DN T cells remains controversial (76, 77). Early studies suggest they derive from activated CD4 T cells that fail to undergo apoptosis (81). However, a more detailed examination of the DN T cell origin in vivo indicates that DN T cells derive from CD8+ T cells that down-regulate their co-receptor after continuous stimulation by self-antigens derived from apoptotic cells (82, 83). Thus, it is generally believed that DN T cells derive from CD8+ T cells.
Expansion of DN T cells correlates with disease activity in lupus-prone mice (83, 84) and systemic SLE patients (85), leading to the idea that these cells play an important pathogenic role in autoimmune disease development. Despite the evidence supporting a pathogenic role for DN T cells, their exact function remains largely elusive (76, 77). Interestingly, DN T cells express high levels of CXCR5, localize in the B cells follicles (86), and stimulate Ab production in vitro (85, 87). Moreover, similar to Tfh cells, the presence of DN T cells correlates with disease activity and autoantibody production in SLE patients and MRL.lpr mice (76, 77, 83–85, 88). Moreover, new studies show that DN T cells produce large amounts of IL-17 (54, 83, 88). Given the putative role of IL-17 in helping B cell responses (48–52, 56, 89), it is tempting to speculate that CXCR5+DN T cells contribute to autoimmune pathology by promoting auto-reactive B cell responses in an IL-17-dependent manner. Corresponding with this idea, a recent study demonstrated that DN T cells are sufficient to promote autoantibody production and renal immune complex deposition after adoptive transfer into B6 Rag1−/− mice that also received B cells from 12-month-old B6.lpr mice (83). These findings provide evidence that DN T cells can contribute to pathogenic Ab responses in vivo. Further investigations will be required to compare the capacity of DN T cells, Tfh cells, and bona fide Th17 to help self-reactive B cell responses and determine how each of these subsets relatively contribute to sustaining pathogenic-Ab responses.
IL-2 and Immunosuppression
IL-2 Signaling
IL-2 is a member of the common γ-chain family of cytokines that was initially characterized and as a growth factor for T and NK T cells (90–92). IL-2 signaling is transmitted through the IL-2 receptor (IL-2R), which can exist in two conformations (93). The high-affinity receptor is a heterotrimeric receptor that consists of the α chain (CD25), the β chain (CD122), and the common γ chain (CD132) (94, 95). The high-affinity receptor is constitutively expressed by FoxP3-expressing CD4+ regulatory T-cells (Tregs), which require IL-2 signaling for their differentiation and function (96–99). In contrast, NK T cells, naïve and memory T cells express the intermediate-affinity IL-2R, a heterodimer composed of the β and γ chain. Following TCR activation, however, they transiently up-regulate CD25 and temporarily express the high-affinity IL-2R. The differential expression of CD25 by regulatory T cells and conventional T cells has important therapeutic consequences. When administered at high doses, IL-2 can help conventional T cells and NK T cells, hence favoring effector responses. In contrast, because Tregs express high levels of CD25 and better compete for IL-2 than other cells, low IL-2 regimes preferentially target IL-2 to Tregs, thus promoting immunosuppression (100).
The binding of IL-2 to the IL-2R triggers the phosphorylation of the Janus-Activated Kinase 1 (JAK1) and 3 (JAK3), leading to the activation of the transcription factor STAT5 (101). In addition, phosphorylation of the adaptor Shc in response to IL-2 activates the Ras-Raf MAP Kinase and PI-3K pathways. The combined effects of STAT5, Ras-Raf MAP Kinase, and PI-3K signaling pathways results in the regulation of the transcription of a broad range of IL-2-target genes, including the forkhead box P3 (FOXP3) (102–104), eomesodermin (Eomes) (105), the B Lymphocyte Induced Maturation Protein 1 (BLIMP1) (105), the T-box transcription factor TBX21 (T-bet) (106, 107), Retinoic acid-related Orphan Receptor (ROR)γ (108–110) and B cell lymphoma (Bcl6) (107, 111–114). Due to its pleiotropic transcriptional effects, IL-2 has been implicated in regulating multiple, and often contradictory, critical immunoregulatory pathways. For example, IL-2–STAT5 signaling positively regulates IL-4R and GATA-3 expression and subsequent Th2 differentiation (106). On the other hand, IL-2 induces IL-12Rβ2, Blimp-1, and IFN-γ up-regulation, which are required for Th1 cell polarization (106).
Importantly, while IL-2 signaling can help effector responses, the development of a lethal multiorgan autoimmune syndrome in the IL-2 and IL-2R deficient mice revealed that the critical non-redundant function of IL-2 is to promote immunosuppression (115–118). Rather than immunodeficiency, diminished IL-2 production is associated with autoimmune disease development in mice and humans (119–123), highlighting the critical role of this cytokine in maintaining immunological tolerance. Given that Treg cells fail to normally develop in the absence of IL-2 signaling (98, 124–126) and that they are essential for maintaining immune tolerance (127–130), it is generally accepted that the principal mechanism by which IL-2 contributes to preserving immune tolerance is by supporting the development and function of Tregs. Supporting this view, early transfer of Tregs into neonatal CD122 deficient mice prevents autoimmune pathology (96).
Low-Dose IL-2 Therapy
Work done over the last fifteen years demonstrate the potential of leveraging the immunosuppressive properties of IL-2 to treat autoimmune disorders. Early studies show that exogenous IL-2 supplementation prevents disease progression and contributes to inducing immunosuppression in mice with established autoimmune diseases, including Type I diabetes, EA, experimental myasthenia, and lupus (131–138). More recently, a novel immunotherapy based on subcutaneous administration of low-dose recombinant human IL-2 (r-IL2, (Aldesleukin/Proleukin) has shown potent immunosuppressive effects in patients with autoimmune pathologies (139), including Type I diabetes (140), hepatitis C-associated vasculitis (141), SLE (142–145), and chronic graft-versus-host disease (146–148). The recent TRANSREG clinical trial further demonstrated that the same dose of rIL-2 selectively expands Tregs and clinical benefits across eleven selected autoimmune diseases (149). Collectively these studies demonstrate that low-dose rIL-2 regimes have therapeutic effects across a broad range of heterogonous autoimmune disorders.
Current low-dose rIL-2 treatment schemes consist of 3-4 cycles of 7-10 million IU of rIL-2 per cycle administered over 1-2 weeks separated by resting periods of 9-16 days. Importantly, low-dose rIL-2 can be safely administered to humans. Thus numerous clinical trials to further explore the potential benefits of low-dose IL-2 in SLE are now underway.
Based on the critical functional relationship between IL-2 and Treg-mediated immunosuppression, the current paradigm suggests that low-dose rIL-2 regimes contribute to restoring immune homeostasis in autoimmune patients by a Treg-dependent mechanism (139, 150). In agreement with this view, low-dose rIL-2 supplementation induces Treg cell expansion in vivo (139). Hence considerable effort has been invested in developing new therapeutic approaches to selectively target IL-2 to Tregs.
Intriguingly, though the frequency of Tregs increases after low-dose rIL-2 administration, the changes in Treg cell numbers are transient and drop to placebo control levels quickly after the last rIL-2 cycle (142, 143). Nevertheless, despite a nearly normal frequency of Tregs, the improved clinical outcomes persist for weeks after the last cycle of rIL-2 (142, 143). Thus, while it is clear that Treg-mediated immunosuppression is critical for achieving the clinical benefits observed after rIL-2 treatment, additional underlying mechanisms might synergize with Treg-mediated effects to provide long-lasting immunosuppression after low-dose rIL-2 immunotherapy. In this regard, recent studies demonstrate that prolonged IL-2 signaling prevents the expression of (RORγt) (110) and Bcl6 (107, 111–114), thereby repressing Th17 and Tfh cell development, respectively. Correspondingly, low-dose rIL-2 treatment significantly reduced the frequency of Tfh and Th17 in humans and preclinical animal models (71, 114, 138, 143, 151, 152). Similarly, CD3+CD4-CD8- DN T cells are depleted after rIL-2 administration (138). Based on the inhibitory role of IL-2 in Tfh, Th17, and DN T cells and their putative roles in promoting self-reactive B cell responses, targeting IL-2 to these T cell populations could represent a good therapeutic strategy to prevented Ab-mediated pathology in autoimmune patients without inducing profound immunosuppression.
Targeting B Cell Helpers with IL-2
IL-2 and Tfh Cells
Studies by us and others demonstrate that IL-2 signaling inhibits Tfh cell differentiation (107, 111, 113, 114, 152). Mechanistically, IL-2 indirectly inhibits Tfh cells by inducing BLIMP, which in turn represses Bcl6 expression and Tfh cell differentiation (111, 113). Besides, STAT5 in response to IL-2 binds to the Bcl6 promoter and directly prevents Bcl6 transcription (107, 112), thereby inhibiting the initiation of the Tfh cell program. In support of these findings, the lack of IL-2/STAT5 signaling during T cell differentiation skews the CD4+ T cell response towards the Tfh cell differentiation pathway (111, 113, 114). Data from the Weinmann’s laboratory also suggest that, in addition to directly repressing Bcl6 expression, IL-2 signaling favors the formation of T-bet/Bcl6 complexes that block Bcl6 activity (107).
Corresponding with the inhibitory role of IL-2 in Tfh cell development, Tfh cell differentiation can be fine-tuned in vivo by altering the environmental levels of IL-2. As such, limiting IL-2 signaling in vivo results in enhanced Tfh cell responses (111, 113, 114, 151–153). Contrariwise, treatment with rIL-2 prevents Tfh cell differentiation and ensuing GC responses in mice infected with influenza virus (71, 114, 151). Importantly, in these studies, the ability of IL-2 to suppress Tfh cell responses is independent of the presence of Tregs (114, 152). Hence, these studies demonstrate that lL-2 intrinsically inhibits Tfh cell development by repressing Bcl6 expression and activity in a Treg-independent manner.
Notably, there are significant differences between human and mouse Tfh cell developmental requirements (154, 155). However, recent studies demonstrate that IL-2 is also a potent inhibitor of human Tfh cell responses. Corresponding with this, while IL-2 blockade increases human Tfh cell differentiation in vitro (156), treatment with low-dose rIL-2 reduces the frequency of Tfh cells in SLE patients (143). These results provide evidence to support the potential of IL-2-based therapies to deplete Tfh cells in vivo. Furthermore, these studies offer a new interpretation for how impaired IL-2 production by T cells (120–123) and single nucleotide polymorphisms in the IL-2 and IL-2 receptor genes (122, 157, 158), which are associated with various autoimmune diseases, affects autoimmune disease development. In this regard, one would predict that a low IL-2 environment favors self-reactive Tfh cell differentiation and subsequent Auto-Ab production in autoimmune patients.
Synergistic Low-Dose rIL-2 Therapies
Recent studies suggest that in addition to secreting a low amount of IL-2, T cells from SLE patients poorly respond to exogenous IL-2 (159). Thus, the lack of IL-2 responsiveness could be a potential limitation when designing low-IL-2-based therapies to efficiently deplete Tfh cells in vivo. Importantly, data from our laboratory demonstrate that the IL-6/STAT3 pathway is an important regulator of the IL-2 responsiveness of Tfh cells. Briefly, using a combination of in vivo and genetic studies, we found that STAT3 in response to IL-6 binds to the Il2rβ locus and prevents CD122 up-regulation in Tfh cells, thereby limiting the capacity of these cells to respond to IL-2 (151). Hence, blockade of IL-6 signaling renders Tfh cells hyperresponsive to IL-2, thus lowering the threshold of IL-2 required to deplete Tfh cells (Figure 3). As a consequence, the frequency of Tfh cells was dramatically reduced in influenza-infected mice treated with an anti-IL-6 blockade in combination with rIL-2 compared to mice treated with rIL-2 alone, even when rIL-2 was administered at ultra-low doses.
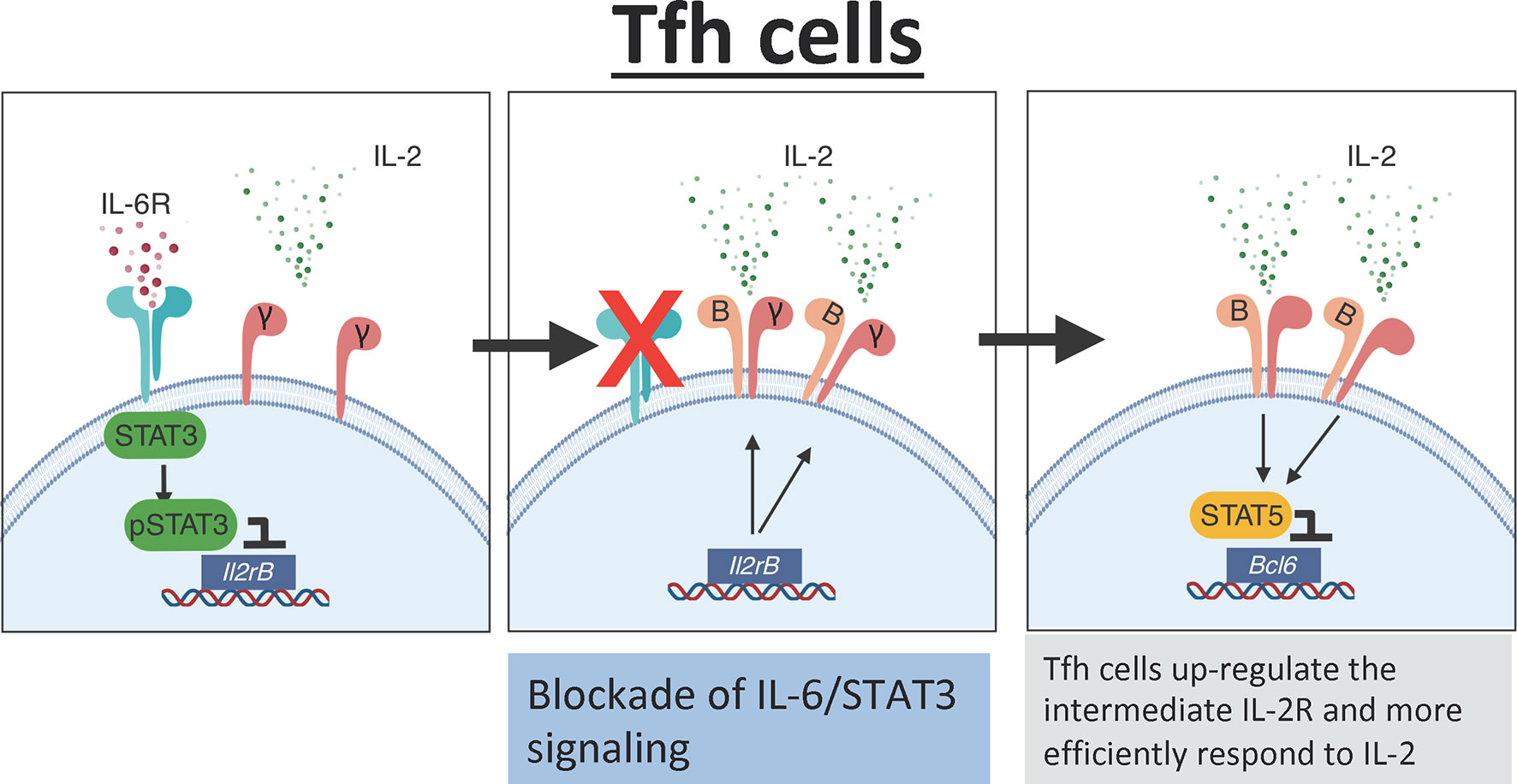
Figure 3 IL-6 mediates the IL-2 responsiveness of Tfh cells. In the presence of IL-6 signaling, Tfh cells activate STAT3 which directly binds to the Il2rb promoter preventing expression of CD122. Low expression of CD122 limits IL-2 signaling in Tfh cells. In the absence of IL-6 signaling, there is increased expression of CD122 on the surface of Tfh cells leading to increased responsiveness to IL-2. The increased IL-2 signaling activates STAT5 which binds to the promoter region of Bcl6 and suppresses the Tfh cell program.
These findings have important therapeutic and conceptual implications. In this regard, one would predict that Tocilizumab, a humanized anti-IL-6 receptor monoclonal antibody, administered together with rIL-2 will synergize to target the Tfh cell population more efficiently than rIL-2 administered alone. In this scenario, an IL-6 blockade would increase the expression of CD122 on the surface of Tfh cells, making them more susceptible to rIL-2 signaling and subsequent depletion. This combination therapy will likely achieve the same biological effect using a lower dose of rIL-2. In addition, IL-2 consumption by CD25+Tregs limits the amount of available IL-2 (152, 160). Thus, it is likely that, in the presence of high numbers of CD25+Tregs, the environmental levels of IL-2 are scarce, and only cells with a relatively low response threshold will be able to respond to IL-2. Hence, another significant advantage of this approach is that, by increasing IL-2 responsiveness, this synergistic therapy will allow Tfh cells to react to rIL-2 even in the presence of IL-2-consuming Tregs, thereby simultaneously targeting Tregs and Tfh cells.
Importantly, similar to IL-6 (161, 162), the serum levels of the STAT3-activating cytokines IL-23 and IL-27 are increased in autoimmune patients (163–167). Thus, IL-6 signaling blockade alone might not be sufficient to enhance IL-2 responsiveness of Tfh cells due to the capacity of additional STAT3-activating cytokines to compensate for the lack of IL-6. Furthermore, IL-6, IL-23, and IL-27 all induce STAT3 activation via JAK2. In contrast, JAK2 is not required for IL-2 signaling. Notably, a recent study shows that the specific JAK2 inhibitor CEP-33779 can be safely administered to MRL.lpr mice, which show significant improvement in disease pathogenesis and reduced pSTAT3 levels after treatment (168). Given that JAK2 is required for STAT3 but not STAT5 activation, it is tempting to speculate that “non-cytokine specific” STAT3 inhibition after treatment with a JAK2 inhibitor will lower the threshold of IL-2 required for suppressing Tfh cells regardless of the presence of redundant STAT3-activating cytokines. In any case, altogether, these studies suggest a model in which STAT3 activation in response to STAT3-activating cytokines counterbalances IL-2-mediated suppression of Tfh cells by limiting IL-2 responsiveness of Tfh cells. A better knowledge of how the crosstalk between different cytokine pathways regulates Tfh cell development will allow us to design more efficient therapeutic strategies to prevent self-reactive Tfh cell responses in autoimmune patients, thereby precluding ensuing pathogenic B cells responses and Ab-mediated pathology.
IL-2 and Th17 Cells
The clinical benefits of targeting Th17 cells to prevent autoimmune manifestations have been explored in preclinical and clinical settings, and additional clinical trials are being conducted. The results, however, are conflicting (169–171). Independent randomized clinical trials demonstrate the clinical efficacy of targeting IL-17 to treat moderate to severe psoriasis. As a result, two monoclonal anti-IL-17A antibodies (secukinumab and ixekizumab) and one antibody targeting the IL-17 receptor (brodalumab) are now FDA approved for the treatment of this disease. However, the studies assessing the clinical benefits of anti-IL-17 biologics for the treatment of systemic rheumatologic disorders, such as RA or SLE, have yielded mixed results. While some preclinical studies and clinical trials show promising results after IL-17 blockade (169), the therapeutic effect of anti-IL-17 anti-IL-17A Abs is lower than anticipated.
The relatively low efficacy of these treatments is, to some extent, surprising, given the abundance of publications showing reduced pathology and severity after IL-17 blockade in preclinical animal models (172). One potential explanation for this discrepancy is patient sample heterogeneity. In this regard, while most of the studies show that elevated levels of IL-17 and high frequency of Th17 cells correlate with disease activity in SLE patients, some found no significant differences between patients and healthy controls (172). Since anti-IL-17 biological-based treatments will likely only be effective in patients with a “high IL-17” profile, the lack of proper patient stratification based on their IL-17 profile could explain the lack of consistency in the results. An alternative, but mutually complementary, explanation is the inability of the current anti-IL-17 biologics to sufficiently block the aberrantly increased IL-17 pathway in these patients. Besides, IL-17/IL-17A blockade alone might not be sufficient to effectively disrupt the inflammatory cycle leading to disease pathogenesis once this has already been initiated.
As aforementioned, IL-2 signaling inhibits Th17 differentiation (106, 108, 110). As a consequence of this inhibitory effect, Th17 cells fail to differentiate in relatively high IL-2 environments. Contrariwise, IL-2 quenching facilitates Th17 cell development (109). Mechanistically, IL-2 antagonizes IL-17 differentiation via STAT5, which outcompetes STAT3 binding at the IL-17 locus, hence preventing binding of STAT3 and its enhancer elements in response to IL-6 (108). IL-2 signaling also represses IL-6R expression and recruits the histone deacetylator adaptor protein NCoR2 to the Il17 locus, thereby contributing to further inhibiting IL-17 production (108). In agreement with the suppressor role of IL-2 in Th17 cell differentiation, the regulatory mechanisms that control IL-2 production also indirectly control IL-17 production. For example, the cAMP-responsive element modulator alpha (CREMα) negatively regulates IL-2 transcription by binding to the Il2 locus (121, 173–175). IL-2 shortage after CREMα overexpression in T cells contributes to enhancing IL-17 differentiation, a phenomenon that can be reserved after IL-2 supplementation (175). Similarly, by suppressing IL-2 production, the phosphatase and tensin homologue (PTEN) indirectly favors Th17 differentiation (176). At a cellular level, IL-2 consumption by Treg cells favors Th17 development by creating a low IL-2 environment permissive for Th17 differentiation (160). Collectively, these studies demonstrate that Th17 cells preferably differentiate in “low-IL-2” environments.
Importantly, works from multiple laboratories demonstrate a causative relationship between IL-2 deficiency, subsequent excessive IL-17 responses, and autoimmune pathology development. For example, elegant work has shown that a lack of STAT3 activation prevents the accumulation of Th17 cells in IL-2-deficient mice, resulting in prolonged lifespan and reduced autoimmunity associated with IL-2 deficiency (108). Additional studies have shown that MRL/Fas(lpr/lpr) mice treated with rIL-2 have reduced frequency of IL-17 producing cells, which correlated with diminished disease manifestations (138). Moreover, in SLE patients, low-dose rIL-2 treatment resulted in reduced frequencies of Th17 cells, which correlated with the induction of remission in a recent open-labeled trial (143). Collectively, these results provide strong evidence for the therapeutic potential of rIL-2 to prevent unwanted Th17 responses in vivo. Importantly, current anti-IL-17 biologics target the product of Th17 cells (i.e., IL-17). In contrast, low-dose rIL-2 precludes the development of these cells, which has the potential to more effectively prevent IL-17-dependent immunopathology by preventing the continuous replenishment of Th17 cells from their precursors, thereby inducing long-lasting effects. Besides, anti-IL-17 biologics are limited in that their effect is restricted to limiting IL-17 responses. In contrast, rIL-2 therapy has broader effects beyond dampening IL-17, such as bolstering the Treg–mediated immunosuppression and/or decreasing autoreactive Tfh cells, which are likely to synergize with Th17 suppression to further prevent immunopathology. In this regard, given that Tfh and Th17 cells are similarly regulated by the IL-2/STA5 and IL-6/STAT3 pathways, the aforementioned combinational therapy with STAT3 blocking agents and rIL-2 is likely to simultaneously target Tfh and Th17 cells efficiently. In summary, the inhibitory effects of IL-2 in Th17 cells, and its subsequent effects on Auto-Ab responses and systemic inflammation, need to be evaluated when considering IL-2-based therapies for the treatment of autoimmune disorders.
IL-2 and DN T Cells
Despite the accumulating evidence supporting a pathogenic role for DN T cells, the exact mechanisms that regulate DN T cell homeostasis are unknown, and there are currently no therapies to selectively deplete DN T cells in vivo. Importantly, however, work from George Tsokos’s group demonstrates that treatment with an inducible recombinant adeno-associated virus vector encoding IL-2 significantly reduced the frequency of IL-17+ DN T cells in MRL/lpr mice, which was accompanied by reduced pathology and kidney infiltration (138). The effect of IL-2 on DN T cells is likely independent of the role of IL-2 in Treg cells, as treatment with IL-2 is complexed with the anti-IL-2 monoclonal JES6-1, which selectively target CD25-expressing Tregs did not affect DN T cells. Nevertheless, because DN T cells express neglectable levels of CD25 and CD122 and poorly phosphorylate STAT5 in response to IL-2, the authors suggest that the effect of IL-2 on DN T cells is indirect. In any case, whereas the exact mechanism by which IL-2 prevents DN T cell accumulation remains elusive, these studies demonstrate a critical role for IL-2 in preventing DN T cell expansion in vivo. Given the potential pathological role of these cells and their contribution to sustaining pathogenic Ab responses, the effects of low-dose rIL-2 immunotherapies on DN T cells should be carefully examined in future low-dose rIL-2 clinical trials.
Concluding Remarks
In conclusion, we present here the rationale for using new therapeutic regimens based on the combination of low-dose rIL-2 with other biologics to achieve ideal immunosuppression and improved disease scores. Since the original observation that in the absence of IL-2 signaling mice develop catastrophic autoimmune disease, our knowledge of the complex intersection of multiple underlining conditions contributing to autoimmunity has grown to include multiple T cell populations in addition to Treg cells. Armed with the understanding that Tfh, Th17, and DN T cells play critical roles in autoimmune disease progression and that they are efficiently depleted after rIL-2 treatment, it is time to consider how to leverage the broad-ranging effects of rIL-2 therapy to synergistically induce Treg cell immunosuppression along with the Tfh/Th17/DN T cells axis to efficiently prevent inflammation and auto-Ab-mediated pathology in autoimmune patients without the undesired side effects associated to systemic immunosuppression.
While some studies suggest that B cells do not express CD25 and STAT5 signaling is dispensable for B cell maturation and function (177), IL-2 favors B cell survival and PC differentiation in vitro (178–180). These potential “positive” effects of IL-2 in B cells could, to some extent, compensate for the absence of T cell help and be detrimental in the context of B-cell mediated pathologies, particularly when administered for short periods of time. Therefore, though it is clear that low-dose rIL-2 therapies promote immunosuppression, the potential intrinsic effects of low-dose rIL-2 treatment in B cells should be carefully examined. Similarly, IL-2 inhibits the development of T follicular regulatory (TFR) cells (181–183), a particular subset of Tregs that express Bcl6 and CXCR5 and localize into the B cell follicles where they suppress Tfh and GC B cell responses (184–186). Mechanistically, IL-2 signaling induces Blimp-1 expression in conventional Tregs cells, thereby preventing them from up-regulating Bcl6 and becoming TFR cells. Given that TFR cells have a suppressive function in Tfh and GCs, the lack of these cells after low-dose rIL-2 could enhance pathogenic B cell responses. Corresponding with this idea, the absence of TFR cells favors the outgrowth of self-reactive B cell clones in some models (181, 185). Nevertheless, the role of TFR cells is more complex than initially expected, as, rather than inhibit, they promote GC and Ab responses in some models (187, 188). Besides, TFR cells express low levels of CD25 (181, 182). Thus, it is unlikely that rIL2 therapy will have a preferential impact on TFR cells. In any case, despite the putative adverse effects of IL-2, treatment with low-dose rIL-2 and IL-2/anti-IL-2 Ab complexes efficiently decreases anti-DNA Ab titers in NZB/W F1 mice (189) and hinders influenza-specific B cell responses in influenza-infected mice (114). These data support the view that, when used in vivo, the dominant effect of IL-2 in the B cell response is immunosuppression.
Author Contributions
Both authors contributed equally. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the University of Alabama at Birmingham (UAB), the National Institutes of Health grant R01AI150664 to A. Ballesteros-Tato, and the Lupus Research Alliance Novel Research Award to A. Ballesteros-Tato.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Figures were created with BioRender.com.
References
1. Xiao ZX, Miller JS, Zheng SG. An Updated Advance of Autoantibodies in Autoimmune Diseases. Autoimmun Rev (2021) 20:102743. doi: 10.1016/j.autrev.2020.102743
2. Naparstek Y, Plotz PH. The Role of Autoantibodies in Autoimmune Disease. Annu Rev Immunol (1993) 11:79–104. doi: 10.1146/annurev.iy.11.040193.000455
3. Woodroffe AJ, Border WA, Theofilopoulos AN, Gotze O, Glassock RJ, Dixon FJ, et al. Detection of Circulating Immune Complexes in Patients With Glomerulonephritis. Kidney Int (1977) 12:268–78. doi: 10.1038/ki.1977.111
4. Greisman SG, Redecha PB, Kimberly RP, Christian CL. Differences Among Immune Complexes: Association of C1q in SLE Immune Complexes With Renal Disease. J Immunol (1987) 138:739–45.
5. Das A, Heesters BA, Bialas A, O’Flynn J, Rifkin IR, Ochando J, et al. Follicular Dendritic Cell Activation by TLR Ligands Promotes Autoreactive B Cell Responses. Immunity (2017) 46:106–19. doi: 10.1016/j.immuni.2016.12.014
6. Leslie D, Lipsky P, Notkins AL. Autoantibodies as Predictors of Disease. J Clin Invest (2001) 108:1417–22. doi: 10.1172/JCI14452
7. Nguyen DC, Joyner CJ, Sanz I, Lee FE. Factors Affecting Early Antibody Secreting Cell Maturation Into Long-Lived Plasma Cells. Front Immunol (2019) 10:2138. doi: 10.3389/fimmu.2019.02138
8. Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dorner T, et al. Competence and Competition: The Challenge of Becoming a Long-Lived Plasma Cell. Nat Rev Immunol (2006) 6:741–50. doi: 10.1038/nri1886
9. Bannard O, Cyster JG. Germinal Centers: Programmed for Affinity Maturation and Antibody Diversification. Curr Opin Immunol (2017) 45:21–30. doi: 10.1016/j.coi.2016.12.004
10. Malkiel S, Barlev AN, Atisha-Fregoso Y, Suurmond J, Diamond B. Plasma Cell Differentiation Pathways in Systemic Lupus Erythematosus. Front Immunol (2018) 9:427. doi: 10.3389/fimmu.2018.00427
11. William J, Euler C, Christensen S, Shlomchik MJ. Evolution of Autoantibody Responses Via Somatic Hypermutation Outside of Germinal Centers. Science (2002) 297:2066–70. doi: 10.1126/science.1073924
12. Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, et al. ICOS-Dependent Extrafollicular Helper T Cells Elicit IgG Production Via IL-21 in Systemic Autoimmunity. J Exp Med (2008) 205:2873–86. doi: 10.1084/jem.20080840
13. Jenks SA, Cashman KS, Woodruff MC, Lee FE, Sanz I. Extrafollicular Responses in Humans and SLE. Immunol Rev (2019) 288:136–48. doi: 10.1111/imr.12741
14. Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, et al. Diversity, Cellular Origin and Autoreactivity of Antibody-Secreting Cell Population Expansions in Acute Systemic Lupus Erythematosus. Nat Immunol (2015) 16:755–65. doi: 10.1038/ni.3175
15. Ramos-Casals M, Sanz I, Bosch X, Stone JH, Khamashta MA. B-Cell-Depleting Therapy in Systemic Lupus Erythematosus. Am J Med (2012) 125:327–36. doi: 10.1016/j.amjmed.2011.09.010
16. Lund FE, Randall TD. Effector and Regulatory B Cells: Modulators of CD4(+) T Cell Immunity. Nat Rev Immunol (2010) 10:236–47. doi: 10.1038/nri2729
17. Calero I, Sanz I. Targeting B Cells for the Treatment of SLE: The Beginning of the End or the End of the Beginning? Discov Med (2010) 10:416–24.
18. Cambridge G, Leandro MJ, Teodorescu M, Manson J, Rahman A, Isenberg DA, et al. B Cell Depletion Therapy in Systemic Lupus Erythematosus: Effect on Autoantibody and Antimicrobial Antibody Profiles. Arthritis Rheum (2006) 54:3612–22. doi: 10.1002/art.22211
19. Sanz I, Lee FE. B Cells as Therapeutic Targets in SLE. Nat Rev Rheumatol (2010) 6:326–37. doi: 10.1038/nrrheum.2010.68
20. Crotty S. Follicular Helper CD4 T Cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
21. Yu D, Vinuesa CG. The Elusive Identity of T Follicular Helper Cells. Trends Immunol (2010) 31:377–83. doi: 10.1016/j.it.2010.07.001
22. Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control Systems and Decision Making for Antibody Production. Nat Immunol (2010) 11:681–8. doi: 10.1038/ni.1900
23. Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, et al. B Cell Priming for Extrafollicular Antibody Responses Requires Bcl-6 Expression by T Cells. J Exp Med (2011) 208:1377–88. doi: 10.1084/jem.20102065
24. Rankin AL, Guay H, Herber D, Bertino SA, Duzanski TA, Carrier Y, et al. IL-21 Receptor is Required for the Systemic Accumulation of Activated B and T Lymphocytes in MRL/MpJ-Fas(lpr/lpr)/J Mice. J Immunol (2012) 188:1656–67. doi: 10.4049/jimmunol.1003871
25. Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
26. Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-Helper Cell Recruitment Governed by Bystander B Cells and ICOS-Driven Motility. Nature (2013) 496:523–7. doi: 10.1038/nature12058
27. Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. PD-1 Controls Follicular T Helper Cell Positioning and Function. Immunity (2018) 49:264–74.e4. doi: 10.1016/j.immuni.2018.06.012
28. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS Receptor Instructs T Follicular Helper Cell Versus Effector Cell Differentiation Via Induction of the Transcriptional Repressor Bcl6. Immunity (2011) 34:932–46. doi: 10.1016/j.immuni.2011.03.023
29. Han S, Hathcock K, Zheng B, Kepler TB, Hodes R, Kelsoe G. Cellular Interaction in Germinal Centers. Roles of CD40 Ligand and B7-2 in Established Germinal Centers. J Immunol (1995) 155:556–67.
30. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 Acts Directly on B Cells to Regulate Bcl-6 Expression and Germinal Center Responses. J Exp Med (2010) 207:353–63. doi: 10.1084/jem.20091738
31. Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A Fundamental Role for Interleukin-21 in the Generation of T Follicular Helper Cells. Immunity (2008) 29:127–37. doi: 10.1016/j.immuni.2008.06.001
32. Bryant VL, Ma CS, Avery DT, Li Y, Good KL, Corcoran LM, et al. Cytokine-Mediated Regulation of Human B Cell Differentiation Into Ig-Secreting Cells: Predominant Role of IL-21 Produced by CXCR5+ T Follicular Helper Cells. J Immunol (2007) 179:8180–90. doi: 10.4049/jimmunol.179.12.8180
33. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The Transcriptional Repressor Bcl-6 Directs T Follicular Helper Cell Lineage Commitment. Immunity (2009) 31:457–68. doi: 10.1016/j.immuni.2009.07.002
34. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 Mediates the Development of T Follicular Helper Cells. Science (2009) 325:1001–5. doi: 10.1126/science.1176676
35. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are Reciprocal and Antagonistic Regulators of T Follicular Helper Cell Differentiation. Science (2009) 325:1006–10. doi: 10.1126/science.1175870
36. Mountz JD, Hsu HC, Ballesteros-Tato A. Dysregulation of T Follicular Helper Cells in Lupus. J Immunol (2019) 202:1649–58. doi: 10.4049/jimmunol.1801150
37. Gensous N, Charrier M, Duluc D, Contin-Bordes C, Truchetet ME, Lazaro E, et al. T Follicular Helper Cells in Autoimmune Disorders. Front Immunol (2018) 9:1637. doi: 10.3389/fimmu.2018.01637
38. Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, et al. Expansion of Circulating T Cells Resembling Follicular Helper T Cells is a Fixed Phenotype That Identifies a Subset of Severe Systemic Lupus Erythematosus. Arthritis Rheum (2010) 62:234–44. doi: 10.1002/art.25032
39. Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S, et al. Circulating Follicular Helper-Like T Cells in Systemic Lupus Erythematosus: Association With Disease Activity. Arthritis Rheumatol (2015) 67:988–99. doi: 10.1002/art.39020
40. Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, et al. Follicular Helper T Cells are Required for Systemic Autoimmunity. J Exp Med (2009) 206:561–76. doi: 10.1084/jem.20081886
41. Zhang X, Lindwall E, Gauthier C, Lyman J, Spencer N, Alarakhia A, et al. Circulating CXCR5+CD4+Helper T Cells in Systemic Lupus Erythematosus Patients Share Phenotypic Properties With Germinal Center Follicular Helper T Cells and Promote Antibody Production. Lupus (2015) 24:909–17. doi: 10.1177/0961203314567750
42. Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC3, et al. A Critical Role for IL-21 Receptor Signaling in the Pathogenesis of Systemic Lupus Erythematosus in BXSB-Yaa Mice. Proc Natl Acad Sci USA (2009) 106:1518–23. doi: 10.1073/pnas.0807309106
43. Hu YL, Metz DP, Chung J, Siu G, Zhang M. B7RP-1 Blockade Ameliorates Autoimmunity Through Regulation of Follicular Helper T Cells. J Immunol (2009) 182:1421–8. doi: 10.4049/jimmunol.182.3.1421
44. Sawaf M, Dumortier H, Monneaux F. Follicular Helper T Cells in Systemic Lupus Erythematosus: Why Should They Be Considered as Interesting Therapeutic Targets? J Immunol Res (2016) 2016:5767106. doi: 10.1155/2016/5767106
45. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 Family Cytokines and the Expanding Diversity of Effector T Cell Lineages. Annu Rev Immunol (2007) 25:821–52. doi: 10.1146/annurev.immunol.25.022106.141557
46. Raymond W, Ostli-Eilertsen G, Griffiths S, Nossent J. IL-17A Levels in Systemic Lupus Erythematosus Associated With Inflammatory Markers and Lower Rates of Malignancy and Heart Damage: Evidence for a Dual Role. Eur J Rheumatol (2017) 4:29–35. doi: 10.5152/eurjrheum.2017.16059
47. Nalbandian A, Crispin JC, Tsokos GC. Interleukin-17 and Systemic Lupus Erythematosus: Current Concepts. Clin Exp Immunol (2009) 157:209–15. doi: 10.1111/j.1365-2249.2009.03944.x
48. Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, et al. Interleukin 17-Producing T Helper Cells and Interleukin 17 Orchestrate Autoreactive Germinal Center Development in Autoimmune BXD2 Mice. Nat Immunol (2008) 9:166–75. doi: 10.1038/ni1552
49. Ding Y, Li J, Wu Q, Yang P, Luo B, Xie S, et al. IL-17RA is Essential for Optimal Localization of Follicular Th Cells in the Germinal Center Light Zone to Promote Autoantibody-Producing B Cells. J Immunol (2013) 191:1614–24. doi: 10.4049/jimmunol.1300479
50. Tarlinton D. IL-17 Drives Germinal Center B Cells? Nat Immunol (2008) 9:124–6. doi: 10.1038/ni0208-124
51. Mitsdoerffer M, Lee Y, Jager A, Kim HJ, Korn T, Kolls JK, et al. Proinflammatory T Helper Type 17 Cells are Effective B-Cell Helpers. Proc Natl Acad Sci USA (2010) 107:14292–7. doi: 10.1073/pnas.1009234107
52. Patakas A, Benson RA, Withers DR, Conigliaro P, McInnes IB, Brewer JM, et al. Th17 Effector Cells Support B Cell Responses Outside of Germinal Centres. PloS One (2012) 7:e49715. doi: 10.1371/journal.pone.0049715
53. Amarilyo G, Lourenco EV, Shi FD, La Cava A. IL-17 Promotes Murine Lupus. J Immunol (2014) 193:540–3. doi: 10.4049/jimmunol.1400931
54. Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting Edge: IL-23 Receptor Deficiency Prevents the Development of Lupus Nephritis in C57BL/6-lpr/lpr Mice. J Immunol (2010) 184:4605–9. doi: 10.4049/jimmunol.0903595
55. Dai H, He F, Tsokos GC, Kyttaris VC. IL-23 Limits the Production of IL-2 and Promotes Autoimmunity in Lupus. J Immunol (2017) 199:903–10. doi: 10.4049/jimmunol.1700418
56. Wei L, Laurence A, Elias KM, O’Shea JJ. IL-21 is Produced by Th17 Cells and Drives IL-17 Production in a STAT3-Dependent Manner. J Biol Chem (2007) 282:34605–10. doi: 10.1074/jbc.M705100200
57. Doreau A, Belot A, Bastid J, Riche B, Trescol-Biemont MC, Ranchin B, et al. Interleukin 17 Acts in Synergy With B Cell-Activating Factor to Influence B Cell Biology and the Pathophysiology of Systemic Lupus Erythematosus. Nat Immunol (2009) 10:778–85. doi: 10.1038/ni.1741
58. Flannigan KL, Ngo VL, Geem D, Harusato A, Hirota SA, Parkos CA, et al. IL-17A-Mediated Neutrophil Recruitment Limits Expansion of Segmented Filamentous Bacteria. Mucosal Immunol (2017) 10:673–84. doi: 10.1038/mi.2016.80
59. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of Interleukin 17 Receptor Signaling for Lung CXC Chemokine and Granulocyte Colony-Stimulating Factor Expression, Neutrophil Recruitment, and Host Defense. J Exp Med (2001) 194:519–27. doi: 10.1084/jem.194.4.519
60. Parsa R, Lund H, Georgoudaki AM, Zhang XM, Ortlieb Guerreiro-Cacais A, Grommisch D, et al. BAFF-Secreting Neutrophils Drive Plasma Cell Responses During Emergency Granulopoiesis. J Exp Med (2016) 213:1537–53. doi: 10.1084/jem.20150577
61. Cerutti A, Puga I, Magri G. The B Cell Helper Side of Neutrophils. J Leukoc Biol (2013) 94:677–82. doi: 10.1189/jlb.1112596
62. Schmitt N, Liu Y, Bentebibel SE, Munagala I, Bourdery L, Venuprasad K, et al. The Cytokine TGF-Beta Co-Opts Signaling Via STAT3-STAT4 to Promote the Differentiation of Human TFH Cells. Nat Immunol (2014) 15:856–65. doi: 10.1038/ni.2947
63. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T Follicular Helper Cells is Mediated by Interleukin-21 But Independent of T Helper 1, 2, or 17 Cell Lineages. Immunity (2008) 29:138–49. doi: 10.1016/j.immuni.2008.05.009
64. Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai CY, Emslie E, et al. ICOS Coreceptor Signaling Inactivates the Transcription Factor FOXO1 to Promote Tfh Cell Differentiation. Immunity (2015) 42:239–51. doi: 10.1016/j.immuni.2015.01.017
65. Bauquet AT, Jin H, Paterson AM, Mitsdoerffer M, Ho IC, Sharpe AH, et al. The Costimulatory Molecule ICOS Regulates the Expression of c-Maf and IL-21 in the Development of Follicular T Helper Cells and TH-17 Cells. Nat Immunol (2009) 10:167–75. doi: 10.1038/ni.1690
66. Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, Golovina TN, et al. The Inducible Costimulator (ICOS) is Critical for the Development of Human T(H)17 Cells. Sci Transl Med (2010) 2:55ra78. doi: 10.1126/scitranslmed.3000448
67. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 Controls Th17 Immunity in Vivo by Inhibiting the Conversion of Conventional T Cells Into Foxp3+ Regulatory T Cells. Proc Natl Acad Sci USA (2008) 105:18460–5. doi: 10.1073/pnas.0809850105
68. Mondal A, Sawant D, Dent AL. Transcriptional Repressor BCL6 Controls Th17 Responses by Controlling Gene Expression in Both T Cells and Macrophages. J Immunol (2010) 184:4123–32. doi: 10.4049/jimmunol.0901242
69. Weinstein JS, Herman EI, Lainez B, Licona-Limon P, Esplugues E, Flavell R, et al. TFH Cells Progressively Differentiate to Regulate the Germinal Center Response. Nat Immunol (2016) 17:1197–205. doi: 10.1038/ni.3554
70. Luthje K, Kallies A, Shimohakamada Y, Belz GT, Light A, Tarlinton DM, et al. The Development and Fate of Follicular Helper T Cells Defined by an IL-21 Reporter Mouse. Nat Immunol (2012) 13:491–8. doi: 10.1038/ni.2261
71. Ballesteros-Tato A, Randall TD, Lund FE, Spolski R, Leonard WJ, Leon B. T Follicular Helper Cell Plasticity Shapes Pathogenic T Helper 2 Cell-Mediated Immunity to Inhaled House Dust Mite. Immunity (2016) 44:259–73. doi: 10.1016/j.immuni.2015.11.017
72. Gowthaman U, Chen JS, Zhang B, Flynn WF, Lu Y, Song W, et al. Identification of a T Follicular Helper Cell Subset That Drives Anaphylactic Ige. Science (2019) 365:eaaw6433. doi: 10.1126/science.aaw6433
73. Liu X, Yan X, Zhong B, Nurieva RI, Wang A, Wang X, et al. Bcl6 Expression Specifies the T Follicular Helper Cell Program in Vivo. J Exp Med (2012) 209:1841–52, S1-24. doi: 10.1084/jem.20120219
74. Trub M, Barr TA, Morrison VL, Brown S, Caserta S, Rixon J, et al. Heterogeneity of Phenotype and Function Reflects the Multistage Development of T Follicular Helper Cells. Front Immunol (2017) 8:489. doi: 10.3389/fimmu.2017.00489
75. Crotty S. T Follicular Helper Cell Differentiation, Function, and Roles in Disease. Immunity (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
76. D’Acquisto F, Crompton T. CD3+CD4-CD8- (Double Negative) T Cells: Saviours or Villains of the Immune Response? Biochem Pharmacol (2011) 82:333–40. doi: 10.1016/j.bcp.2011.05.019
77. Martina MN, Noel S, Saxena A, Rabb H, Hamad AR. Double Negative (DN) Alphabeta T Cells: Misperception and Overdue Recognition. Immunol Cell Biol (2015) 93:305–10. doi: 10.1038/icb.2014.99
78. Tarbox JA, Keppel MP, Topcagic N, Mackin C, Ben Abdallah M, Baszis KW, et al. Elevated Double Negative T Cells in Pediatric Autoimmunity. J Clin Immunol (2014) 34:594–9. doi: 10.1007/s10875-014-0038-z
79. Lim MS, Straus SE, Dale JK, Fleisher TA, Stetler-Stevenson M, Strober W, et al. Pathological Findings in Human Autoimmune Lymphoproliferative Syndrome. Am J Pathol (1998) 153:1541–50. doi: 10.1016/S0002-9440(10)65742-2
80. Price S, Shaw PA, Seitz A, Joshi G, Davis J, Niemela JE, et al. Natural History of Autoimmune Lymphoproliferative Syndrome Associated With FAS Gene Mutations. Blood (2014) 123:1989–99. doi: 10.1182/blood-2013-10-535393
81. Laouar Y, Ezine S. In Vivo CD4+ Lymph Node T Cells From Lpr Mice Generate CD4-CD8-B220+TCR-Beta Low Cells. J Immunol (1994) 153:3948–55.
82. Crispin JC, Tsokos GC. Human TCR-Alpha Beta+ CD4- CD8- T Cells Can Derive From CD8+ T Cells and Display an Inflammatory Effector Phenotype. J Immunol (2009) 183:4675–81. doi: 10.4049/jimmunol.0901533
83. Li H, Adamopoulos IE, Moulton VR, Stillman IE, Herbert Z, Moon JJ, et al. Systemic Lupus Erythematosus Favors the Generation of IL-17 Producing Double Negative T Cells. Nat Commun (2020) 11:2859. doi: 10.1038/s41467-020-16636-4
84. Masuda T, Ohteki T, Abo T, Seki S, Nose S, Nagura H, et al. Expansion of the Population of Double Negative CD4-8- T Alpha Beta-Cells in the Liver is a Common Feature of Autoimmune Mice. J Immunol (1991) 9:2907–12, 147.
85. Shivakumar S, Tsokos GC, Datta SK. T Cell Receptor Alpha/Beta Expressing Double-Negative (CD4-/CD8-) and CD4+ T Helper Cells in Humans Augment the Production of Pathogenic Anti-DNA Autoantibodies Associated With Lupus Nephritis. J Immunol (1989) 143:103–12.
86. Ansel KM, McHeyzer-Williams LJ, Ngo VN, McHeyzer-Williams MG, Cyster JG. In Vivo-Activated CD4 T Cells Upregulate CXC Chemokine Receptor 5 and Reprogram Their Response to Lymphoid Chemokines. J Exp Med (1999) 190:1123–34. doi: 10.1084/jem.190.8.1123
87. Sieling PA, Porcelli SA, Duong BT, Spada F, Bloom BR, Diamond B, et al. Human Double-Negative T Cells in Systemic Lupus Erythematosus Provide Help for IgG and are Restricted by CD1c. J Immunol (2000) 165:5338–44. doi: 10.4049/jimmunol.165.9.5338
88. Crispin JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded Double Negative T Cells in Patients With Systemic Lupus Erythematosus Produce IL-17 and Infiltrate the Kidneys. J Immunol (2008) 181:8761–6. doi: 10.4049/jimmunol.181.12.8761
89. Liu SM, King C. IL-21-Producing Th Cells in Immunity and Autoimmunity. J Immunol (2013) 191:3501–6. doi: 10.4049/jimmunol.1301454
90. Morgan DA, Ruscetti FW, Gallo R. Selective in Vitro Growth of T Lymphocytes From Normal Human Bone Marrows. Science (1976) 193:1007–8. doi: 10.1126/science.181845
91. Ruscetti FW, Morgan DA, Gallo RC. Functional and Morphologic Characterization of Human T Cells Continuously Grown in Vitro. J Immunol (1977) 119:131–8.
92. Mier JW, Gallo RC. Purification and Some Characteristics of Human T-Cell Growth Factor From Phytohemagglutinin-Stimulated Lymphocyte-Conditioned Media. Proc Natl Acad Sci USA (1980) 77:6134–8. doi: 10.1073/pnas.77.10.6134
93. Bayer AL, Pugliese A, Malek TR. The IL-2/IL-2R System: From Basic Science to Therapeutic Applications to Enhance Immune Regulation. Immunol Res (2013) 57:197–209. doi: 10.1007/s12026-013-8452-5
94. Wang X, Rickert M, Garcia KC. Structure of the Quaternary Complex of Interleukin-2 With Its Alpha, Beta, and Gammac Receptors. Science (2005) 310:1159–63. doi: 10.1126/science.1117893
95. Rickert M, Wang X, Boulanger MJ, Goriatcheva N, Garcia KC. The Structure of Interleukin-2 Complexed With Its Alpha Receptor. Science (2005) 308:1477–80. doi: 10.1126/science.1109745
96. Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 Regulatory T Cells Prevent Lethal Autoimmunity in IL-2Rbeta-Deficient Mice. Implications for the Nonredundant Function of IL-2. Immunity (2002) 17:167–78. doi: 10.1016/S1074-7613(02)00367-9
97. Rudensky AY. Regulatory T Cells and Foxp3. Immunol Rev (2011) 241:260–8. doi: 10.1111/j.1600-065X.2011.01018.x
98. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A Function for Interleukin 2 in Foxp3-Expressing Regulatory T Cells. Nat Immunol (2005) 6:1142–51. doi: 10.1038/ni1263
99. Yuan X, Cheng G, Malek TR. The Importance of Regulatory T-Cell Heterogeneity in Maintaining Self-Tolerance. Immunol Rev (2014) 259:103–14. doi: 10.1111/imr.12163
100. Ballesteros-Tato A. Beyond regulatory T cells: the potential role for IL-2 to deplete T-follicular helper cells and treat autoimmune diseases. Immunotherapy (2014) 6(11):1207–20. doi: 10.2217/imt.14.83
101. Malek TR. The Biology of Interleukin-2. Annu Rev Immunol (2008) 26:453–79. doi: 10.1146/annurev.immunol.26.021607.090357
102. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 Regulates FOXP3 Expression in Human CD4+CD25+ Regulatory T Cells Through a STAT-Dependent Mechanism and Induces the Expansion of These Cells in Vivo. Blood (2006) 108:1571–9. doi: 10.1182/blood-2006-02-004747
103. Murawski MR, Litherland SA, Clare-Salzler MJ, Davoodi-Semiromi A. Upregulation of Foxp3 Expression in Mouse and Human Treg is IL-2/STAT5 Dependent: Implications for the NOD STAT5B Mutation in Diabetes Pathogenesis. Ann N Y Acad Sci (2006) 1079:198–204. doi: 10.1196/annals.1375.031
104. Passerini L, Allan SE, Battaglia M, Di Nunzio S, Alstad AN, Levings MK, et al. STAT5-Signaling Cytokines Regulate the Expression of FOXP3 in CD4+CD25+ Regulatory T Cells and CD4+CD25- Effector T Cells. Int Immunol (2008) 20:421–31. doi: 10.1093/intimm/dxn002
105. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and Inflammation Induce Distinct Transcriptional Programs That Promote the Differentiation of Effector Cytolytic T Cells. Immunity (2010) 32:79–90. doi: 10.1016/j.immuni.2009.11.012
106. Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of Cytokine Receptors by IL-2 Broadly Regulates Differentiation Into Helper T Cell Lineages. Nat Immunol (2011) 12:551–9. doi: 10.1038/ni.2030
107. Oestreich KJ, Mohn SE, Weinmann AS. Molecular Mechanisms That Control the Expression and Activity of Bcl-6 in TH1 Cells to Regulate Flexibility With a TFH-Like Gene Profile. Nat Immunol (2012) 13:405–11. doi: 10.1038/ni.2242
108. Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing Regulation of the Locus Encoding IL-17 Through Direct, Reciprocal Actions of STAT3 and STAT5. Nat Immunol (2011) 12:247–54. doi: 10.1038/ni.1995
109. Chen Y, Haines CJ, Gutcher I, Hochweller K, Blumenschein WM, McClanahan T, et al. Foxp3(+) Regulatory T Cells Promote T Helper 17 Cell Development in Vivo Through Regulation of Interleukin-2. Immunity (2011) 34:409–21. doi: 10.1016/j.immuni.2011.02.011
110. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 Signaling Via STAT5 Constrains T Helper 17 Cell Generation. Immunity (2007) 26:371–81. doi: 10.1016/j.immuni.2007.02.009
111. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a Potent Negative Regulator of TFH Cell Differentiation. J Exp Med (2012) 209:243–50. doi: 10.1084/jem.20111174
112. Walker SR, Nelson EA, Frank DA. STAT5 Represses BCL6 Expression by Binding to a Regulatory Region Frequently Mutated in Lymphomas. Oncogene (2007) 26:224–33. doi: 10.1038/sj.onc.1209775
113. Nurieva RI, Podd A, Chen Y, Alekseev AM, Yu M, Qi X, et al. STAT5 Protein Negatively Regulates T Follicular Helper (Tfh) Cell Generation and Function. J Biol Chem (2012) 287:11234–9. doi: 10.1074/jbc.M111.324046
114. Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 Inhibits Germinal Center Formation by Limiting T Follicular Helper Cell Differentiation. Immunity (2012) 36:847–56. doi: 10.1016/j.immuni.2012.02.012
115. Suzuki H, Duncan GS, Takimoto H, Mak TW. Abnormal Development of Intestinal Intraepithelial Lymphocytes and Peripheral Natural Killer Cells in Mice Lacking the IL-2 Receptor Beta Chain. J Exp Med (1997) 185:499–505. doi: 10.1084/jem.185.3.499
116. Sadlack B, Lohler J, Schorle H, Klebb G, Haber H, Sickel E, et al. Generalized Autoimmune Disease in Interleukin-2-Deficient Mice is Triggered by an Uncontrolled Activation and Proliferation of CD4+ T Cells. Eur J Immunol (1995) 25:3053–9. doi: 10.1002/eji.1830251111
117. Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, et al. Deregulated T Cell Activation and Autoimmunity in Mice Lacking Interleukin-2 Receptor Beta. Science (1995) 268:1472–6. doi: 10.1126/science.7770771
118. Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative Colitis-Like Disease in Mice With a Disrupted Interleukin-2 Gene. Cell (1993) 75:253–61. doi: 10.1016/0092-8674(93)80067-O
119. Ballesteros-Tato A, Papillion A. Mechanisms of Action of Low-Dose IL-2 Restoration Therapies in SLE. Curr Opin Immunol (2019) 61:39–45. doi: 10.1016/j.coi.2019.07.003
120. Lieberman LA, Tsokos GC. The IL-2 Defect in Systemic Lupus Erythematosus Disease Has an Expansive Effect on Host Immunity. J BioMed Biotechnol (2010) 2010:740619. doi: 10.1155/2010/740619
121. Solomou EE, Juang YT, Gourley MF, Kammer GM, Tsokos GC. Molecular Basis of Deficient IL-2 Production in T Cells From Patients With Systemic Lupus Erythematosus. J Immunol (2001) 166:4216–22. doi: 10.4049/jimmunol.166.6.4216
122. Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, et al. Interleukin-2 Gene Variation Impairs Regulatory T Cell Function and Causes Autoimmunity. Nat Genet (2007) 39:329–37. doi: 10.1038/ng1958
123. Linker-Israeli M, Bakke AC, Kitridou RC, Gendler S, Gillis S, Horwitz DA. Defective Production of Interleukin 1 and Interleukin 2 in Patients With Systemic Lupus Erythematosus (SLE). J Immunol (1983) 130:2651–5.
124. Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic Maintenance of Natural Foxp3(+) CD25(+) CD4(+) Regulatory T Cells by Interleukin (IL)-2 and Induction of Autoimmune Disease by IL-2 Neutralization. J Exp Med (2005) 201:723–35. doi: 10.1084/jem.20041982
125. Brandenburg S, Takahashi T, de la Rosa M, Janke M, Karsten G, Muzzulini T, et al. IL-2 Induces in Vivo Suppression by CD4(+)CD25(+)Foxp3(+) Regulatory T Cells. Eur J Immunol (2008) 38:1643–53. doi: 10.1002/eji.200737791
126. Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ. Interleukin 2 Signaling is Required for CD4(+) Regulatory T Cell Function. J Exp Med (2002) 196:851–7. doi: 10.1084/jem.20020190
127. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T Cells Prevent Catastrophic Autoimmunity Throughout the Lifespan of Mice. Nat Immunol (2007) 8:191–7. doi: 10.1038/ni1428
128. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a New Forkhead/Winged-Helix Protein, Scurfin, Results in the Fatal Lymphoproliferative Disorder of the Scurfy Mouse. Nat Genet (2001) 27:68–73. doi: 10.1038/83784
129. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) is Caused by Mutations of FOXP3. Nat Genet (2001) 27:20–1. doi: 10.1038/83713
130. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-Linked Neonatal Diabetes Mellitus, Enteropathy and Endocrinopathy Syndrome is the Human Equivalent of Mouse Scurfy. Nat Genet (2001) 27:18–20. doi: 10.1038/83707
131. Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central Role of Defective Interleukin-2 Production in the Triggering of Islet Autoimmune Destruction. Immunity (2008) 28:687–97. doi: 10.1016/j.immuni.2008.03.016
132. Diaz-de-Durana Y, Lau J, Knee D, Filippi C, Londei M, McNamara P, et al. IL-2 Immunotherapy Reveals Potential for Innate Beta Cell Regeneration in the Non-Obese Diabetic Mouse Model of Autoimmune Diabetes. PloS One (2013) 8:e78483. doi: 10.1371/journal.pone.0078483
133. Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, et al. IL-2 Reverses Established Type 1 Diabetes in NOD Mice by a Local Effect on Pancreatic Regulatory T Cells. J Exp Med (2010) 207:1871–8. doi: 10.1084/jem.20100209
134. Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, et al. In Vivo Expansion of T Reg Cells With IL-2-mAb Complexes: Induction of Resistance to EAE and Long-Term Acceptance of Islet Allografts Without Immunosuppression. J Exp Med (2009) 206:751–60. doi: 10.1084/jem.20082824
135. Liu R, Zhou Q, La Cava A, Campagnolo DI, Van Kaer L, Shi FD. Expansion of Regulatory T Cells Via IL-2/Anti-IL-2 mAb Complexes Suppresses Experimental Myasthenia. Eur J Immunol (2010) 40:1577–89. doi: 10.1002/eji.200939792
136. Shapiro AM, Suarez-Pinzon WL, Power R, Rabinovitch A. Combination Therapy With Low Dose Sirolimus and Tacrolimus is Synergistic in Preventing Spontaneous and Recurrent Autoimmune Diabetes in Non-Obese Diabetic Mice. Diabetologia (2002) 45:224–30. doi: 10.1007/s00125-001-0745-x
137. Baeyens A, Perol L, Fourcade G, Cagnard N, Carpentier W, Woytschak J, et al. Limitations of IL-2 and Rapamycin in Immunotherapy of Type 1 Diabetes. Diabetes (2013) 62:3120–31. doi: 10.2337/db13-0214
138. Mizui M, Koga T, Lieberman LA, Beltran J, Yoshida N, Johnson MC, et al. IL-2 Protects Lupus-Prone Mice From Multiple End-Organ Damage by Limiting CD4-CD8- IL-17-Producing T Cells. J Immunol (2014) 193:2168–77. doi: 10.4049/jimmunol.1400977
139. Klatzmann D, Abbas AK. The Promise of Low-Dose Interleukin-2 Therapy for Autoimmune and Inflammatory Diseases. Nat Rev Immunol (2015) 15:283–94. doi: 10.1038/nri3823
140. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-Dose Interleukin 2 in Patients With Type 1 Diabetes: A Phase 1/2 Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Diabetes Endocrinol (2013) 1:295–305. doi: 10.1016/S2213-8587(13)70113-X
141. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-Cell Responses to Low-Dose Interleukin-2 in HCV-Induced Vasculitis. N Engl J Med (2011) 365:2067–77. doi: 10.1056/NEJMoa1105143
142. He J, Zhang R, Shao M, Zhao X, Miao M, Chen J, et al. Efficacy and Safety of Low-Dose IL-2 in the Treatment of Systemic Lupus Erythematosus: A Randomised, Double-Blind, Placebo-Controlled Trial. Ann Rheum Dis (2020) 79:141–9. doi: 10.1136/annrheumdis-2019-215396
143. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-Dose Interleukin-2 Treatment Selectively Modulates CD4(+) T Cell Subsets in Patients With Systemic Lupus Erythematosus. Nat Med (2016) 22:991–3. doi: 10.1038/nm.4148
144. Humrich JY, Riemekasten G. Low-Dose Interleukin-2 Therapy for the Treatment of Systemic Lupus Erythematosus. Curr Opin Rheumatol (2019) 31:208–12. doi: 10.1097/BOR.0000000000000575
145. Humrich JY, von Spee-Mayer C, Siegert E, Alexander T, Hiepe F, Radbruch A, et al. Rapid Induction of Clinical Remission by Low-Dose Interleukin-2 in a Patient With Refractory SLE. Ann Rheum Dis (2015) 74:791–2. doi: 10.1136/annrheumdis-2014-206506
146. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-Dose Interleukin-2 Therapy Restores Regulatory T Cell Homeostasis in Patients With Chronic Graft-Versus-Host Disease. Sci Transl Med (2013) 5:179ra43. doi: 10.1126/scitranslmed.3005265
147. Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu MF, Liu H, et al. Ultra Low-Dose IL-2 for GVHD Prophylaxis After Allogeneic Hematopoietic Stem Cell Transplantation Mediates Expansion of Regulatory T Cells Without Diminishing Antiviral and Antileukemic Activity. Clin Cancer Res (2014) 20:2215–25. doi: 10.1158/1078-0432.CCR-13-3205
148. von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-Dose Interleukin-2 Selectively Corrects Regulatory T Cell Defects in Patients With Systemic Lupus Erythematosus. Ann Rheum Dis (2016) 75:1407–15. doi: 10.1136/annrheumdis-2015-207776
149. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and Clinical Effects of Low-Dose Interleukin-2 Across 11 Autoimmune Diseases in a Single, Open Clinical Trial. Ann Rheum Dis (2019) 78:209–17. doi: 10.1136/annrheumdis-2018-214229
150. Shevach EM. Application of IL-2 Therapy to Target T Regulatory Cell Function. Trends Immunol (2012) 33:626–32. doi: 10.1016/j.it.2012.07.007
151. Papillion A, Powell MD, Chisolm DA, Bachus H, Fuller MJ, Weinmann AS, et al. Inhibition of IL-2 Responsiveness by IL-6 is Required for the Generation of GC-TFH Cells. Sci Immunol (2019) 4:eaaw7636. doi: 10.1126/sciimmunol.aaw7636
152. Leon B, Bradley JE, Lund FE, Randall TD, Ballesteros-Tato A. FoxP3+ Regulatory T Cells Promote Influenza-Specific Tfh Responses by Controlling IL-2 Availability. Nat Commun (2014) 5:3495. doi: 10.1038/ncomms4495
153. Leon B, Ballesteros-Tato A, Browning JL, Dunn R, Randall TD, Lund FE. Regulation of T(H)2 Development by CXCR5+ Dendritic Cells and Lymphotoxin-Expressing B Cells. Nat Immunol (2012) 13:681–90. doi: 10.1038/ni.2309
154. Schmitt N, Liu Y, Bentebibel SE, Ueno H. Molecular Mechanisms Regulating T Helper 1 Versus T Follicular Helper Cell Differentiation in Humans. Cell Rep (2016) 16:1082–95. doi: 10.1016/j.celrep.2016.06.063
155. Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T Follicular Helper Cells in Humans and Mice. Nat Immunol (2015) 16:142–52. doi: 10.1038/ni.3054
156. Locci M, Wu JE, Arumemi F, Mikulski Z, Dahlberg C, Miller AT, et al. Activin a Programs the Differentiation of Human TFH Cells. Nat Immunol (2016) 17:976–84. doi: 10.1038/ni.3494
157. Brand OJ, Lowe CE, Heward JM, Franklyn JA, Cooper JD, Todd JA, et al. Association of the Interleukin-2 Receptor Alpha (IL-2Ralpha)/CD25 Gene Region With Graves’ Disease Using a Multilocus Test and Tag SNPs. Clin Endocrinol (Oxf) (2007) 66:508–12. doi: 10.1111/j.1365-2265.2007.02762.x
158. Matesanz F, Fedetz M, Collado-Romero M, Fernandez O, Guerrero M, Delgado C, et al. Allelic Expression and Interleukin-2 Polymorphisms in Multiple Sclerosis. J Neuroimmunol (2001) 119:101–5. doi: 10.1016/S0165-5728(01)00354-X
159. Comte D, Karampetsou MP, Kis-Toth K, Yoshida N, Bradley SJ, Kyttaris VC, et al. Brief Report: CD4+ T Cells From Patients With Systemic Lupus Erythematosus Respond Poorly to Exogenous Interleukin-2. Arthritis Rheumatol (2017) 69:808–13. doi: 10.1002/art.40014
160. Pandiyan P, Conti HR, Zheng L, Peterson AC, Mathern DR, Hernandez-Santos N, et al. CD4(+)CD25(+)Foxp3(+) Regulatory T Cells Promote Th17 Cells in Vitro and Enhance Host Resistance in Mouse Candida Albicans Th17 Cell Infection Model. Immunity (2011) 34:422–34. doi: 10.1016/j.immuni.2011.03.002
161. Tackey E, Lipsky PE, Illei GG. Rationale for Interleukin-6 Blockade in Systemic Lupus Erythematosus. Lupus (2004) 13:339–43. doi: 10.1191/0961203304lu1023oa
162. Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR. Elevated Levels of Endogenous IL-6 in Systemic Lupus Erythematosus. a Putative Role in Pathogenesis. J Immunol (1991) 147:117–23.
163. Wong CK, Lit LC, Tam LS, Li EK, Wong PT, Lam CW. Hyperproduction of IL-23 and IL-17 in Patients With Systemic Lupus Erythematosus: Implications for Th17-Mediated Inflammation in Auto-Immunity. Clin Immunol (2008) 127:385–93. doi: 10.1016/j.clim.2008.01.019
164. Du J, Li Z, Shi J, Bi L. Associations Between Serum Interleukin-23 Levels and Clinical Characteristics in Patients With Systemic Lupus Erythematosus. J Int Med Res (2014) 42:1123–30. doi: 10.1177/0300060513509130
165. Leng RX, Pan HF, Chen GM, Wang C, Qin WZ, Chen LL, et al. IL-23: A Promising Therapeutic Target for Systemic Lupus Erythematosus. Arch Med Res (2010) 41:221–5. doi: 10.1016/j.arcmed.2010.02.011
166. Qiu F, Song L, Yang N, Li X. Glucocorticoid Downregulates Expression of IL-12 Family Cytokines in Systemic Lupus Erythematosus Patients. Lupus (2013) 22:1011–6. doi: 10.1177/0961203313498799
167. Xia LP, Li BF, Shen H, Lu J. Interleukin-27 and Interleukin-23 in Patients With Systemic Lupus Erythematosus: Possible Role in Lupus Nephritis. Scand J Rheumatol (2015) 44:200–5. doi: 10.3109/03009742.2014.962080
168. Lu LD, Stump KL, Wallace NH, Dobrzanski P, Serdikoff C, Gingrich DE, et al. Depletion of Autoreactive Plasma Cells and Treatment of Lupus Nephritis in Mice Using CEP-33779, a Novel, Orally Active, Selective Inhibitor of JAK2. J Immunol (2011) 187:3840–53. doi: 10.4049/jimmunol.1101228
169. Taams LS. Interleukin-17 in Rheumatoid Arthritis: Trials and Tribulations. J Exp Med (2020) 217:3840–53. doi: 10.1084/jem.20192048
170. Yang J, Sundrud MS, Skepner J, Yamagata T. Targeting Th17 Cells in Autoimmune Diseases. Trends Pharmacol Sci (2014) 35:493–500. doi: 10.1016/j.tips.2014.07.006
171. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 Family of Cytokines in Health and Disease. Immunity (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
172. Robert M, Miossec P. Interleukin-17 and Lupus: Enough to Be a Target? for Which Patients? Lupus (2020) 29:6–14. doi: 10.1177/0961203319891243
173. Juang YT, Wang Y, Solomou EE, Li Y, Mawrin C, Tenbrock K, et al. Systemic Lupus Erythematosus Serum IgG Increases CREM Binding to the IL-2 Promoter and Suppresses IL-2 Production Through CaMKIV. J Clin Invest (2005) 115:996–1005. doi: 10.1172/JCI22854
174. Tenbrock K, Juang YT, Gourley MF, Nambiar MP, Tsokos GC. Antisense Cyclic Adenosine 5’-Monophosphate Response Element Modulator Up-Regulates IL-2 in T Cells From Patients With Systemic Lupus Erythematosus. J Immunol (2002) 169:4147–52. doi: 10.4049/jimmunol.169.8.4147
175. Lippe R, Ohl K, Varga G, Rauen T, Crispin JC, Juang YT, et al. Cremalpha Overexpression Decreases IL-2 Production, Induces a T(H)17 Phenotype and Accelerates Autoimmunity. J Mol Cell Biol (2012) 4:121–3. doi: 10.1093/jmcb/mjs004
176. Kim HS, Jang SW, Lee W, Kim K, Sohn H, Hwang SS, et al. PTEN Drives Th17 Cell Differentiation by Preventing IL-2 Production. J Exp Med (2017) 214:3381–98. doi: 10.1084/jem.20170523
177. Dai X, Chen Y, Di L, Podd A, Li G, Bunting KD, et al. Stat5 is Essential for Early B Cell Development But Not for B Cell Maturation and Function. J Immunol (2007) 179:1068–79. doi: 10.4049/jimmunol.179.2.1068
178. Mingari MC, Gerosa F, Carra G, Accolla RS, Moretta A, Zubler RH, et al. Human Interleukin-2 Promotes Proliferation of Activated B Cells Via Surface Receptors Similar to Those of Activated T Cells. Nature (1984) 312:641–3. doi: 10.1038/312641a0
179. Hipp N, Symington H, Pastoret C, Caron G, Monvoisin C, Tarte K, et al. IL-2 Imprints Human Naive B Cell Fate Towards Plasma Cell Through ERK/ELK1-Mediated BACH2 Repression. Nat Commun (2017) 8:1443. doi: 10.1038/s41467-017-01475-7
180. Le Gallou S, Caron G, Delaloy C, Rossille D, Tarte K, Fest T. IL-2 Requirement for Human Plasma Cell Generation: Coupling Differentiation and Proliferation by Enhancing MAPK-ERK Signaling. J Immunol (2012) 189:161–73. doi: 10.4049/jimmunol.1200301
181. Botta D, Fuller MJ, Marquez-Lago TT, Bachus H, Bradley JE, Weinmann AS, et al. Dynamic Regulation of T Follicular Regulatory Cell Responses by Interleukin 2 During Influenza Infection. Nat Immunol (2017) 18:1249–60. doi: 10.1038/ni.3837
182. Wing JB, Kitagawa Y, Locci M, Hume H, Tay C, Morita T, et al. A Distinct Subpopulation of CD25(-) T-Follicular Regulatory Cells Localizes in the Germinal Centers. Proc Natl Acad Sci USA (2017) 114:E6400–9. doi: 10.1073/pnas.1705551114
183. Fonseca VR, Ribeiro F, Graca L. T Follicular Regulatory (Tfr) Cells: Dissecting the Complexity of Tfr-Cell Compartments. Immunol Rev (2019) 288:112–27. doi: 10.1111/imr.12739
184. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular Regulatory T Cells Expressing Foxp3 and Bcl-6 Suppress Germinal Center Reactions. Nat Med (2011) 17:983–8. doi: 10.1038/nm.2426
185. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ Follicular Regulatory T Cells Control the Germinal Center Response. Nat Med (2011) 17:975–82. doi: 10.1038/nm.2425
186. Wollenberg I, Agua-Doce A, Hernandez A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the Germinal Center Reaction by Foxp3+ Follicular Regulatory T Cells. J Immunol (2011) 187:4553–60. doi: 10.4049/jimmunol.1101328
187. Laidlaw BJ, Lu Y, Amezquita RA, Weinstein JS, Vander Heiden JA, Gupta NT, et al. Interleukin-10 From CD4(+) Follicular Regulatory T Cells Promotes the Germinal Center Response. Sci Immunol (2017) 2:eaan4767. doi: 10.1126/sciimmunol.aan4767
188. Wu H, Chen Y, Liu H, Xu LL, Teuscher P, Wang S, et al. Follicular Regulatory T Cells Repress Cytokine Production by Follicular Helper T Cells and Optimize IgG Responses in Mice. Eur J Immunol (2016) 46:1152–61. doi: 10.1002/eji.201546094
Keywords: IL-2 (interleukin-2), Tfh and immunity, autoimmune disease, Th17 & Tregs cells, auto-antibodies
Citation: Papillion A and Ballesteros-Tato A (2021) The Potential of Harnessing IL-2-Mediated Immunosuppression to Prevent Pathogenic B Cell Responses. Front. Immunol. 12:667342. doi: 10.3389/fimmu.2021.667342
Received: 12 February 2021; Accepted: 06 April 2021;
Published: 27 April 2021.
Edited by:
David A. Horwitz, University of Southern California, United StatesReviewed by:
Bergithe Eikeland Oftedal, University of Bergen, NorwayMatthew Cook, Australian National University, Australia
Copyright © 2021 Papillion and Ballesteros-Tato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: André Ballesteros-Tato, YW5kcmViYWxsZXN0ZXJvc3RhdG9AdWFibWMuZWR1