 Fatemeh Asgari1,2
Fatemeh Asgari1,2 Domenico Supino1
Domenico Supino1 Raffaella Parente1
Raffaella Parente1 Nadia Polentarutti1
Nadia Polentarutti1 Matteo Stravalaci1,2
Matteo Stravalaci1,2 Remi Porte1Fabio Pasqualini1,2
Remi Porte1Fabio Pasqualini1,2 Marialuisa Barbagallo1Chiara Perucchini1
Marialuisa Barbagallo1Chiara Perucchini1 Camilla Recordati3
Camilla Recordati3 Elena Magrini1
Elena Magrini1 Andrea Mariancini2
Andrea Mariancini2 Federica Riva3Alessia Giordano3
Federica Riva3Alessia Giordano3 Sadaf Davoudian1,2
Sadaf Davoudian1,2 Thierry Roger4Cornelis van’t Veer5
Thierry Roger4Cornelis van’t Veer5 Sebastien Jaillon1,2
Sebastien Jaillon1,2 Alberto Mantovani1,2,6
Alberto Mantovani1,2,6 Andrea Doni7
Andrea Doni7 Cecilia Garlanda1,2*
Cecilia Garlanda1,2*- 1Department of Inflammation and Immunology, IRCCS Humanitas Research Hospital, Rozzano, Italy
- 2Department of Biomedical Sciences, Humanitas University, Pieve Emanuele, Italy
- 3Department of Veterinary Medicine, University of Milano, Lodi, Italy
- 4Infectious Diseases Service, Department of Medicine, Lausanne University Hospital and University of Lausanne, Epalinges, Switzerland
- 5Center of Experimental and Molecular Medicine, Academic Medical Center, Amsterdam, Netherlands
- 6Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London, United Kingdom
- 7Unit of Advanced Optical Microscopy, IRCCS Humanitas Research Hospital, Rozzano, Italy
Klebsiella pneumoniae is a common pathogen in human sepsis. The emergence of multidrug-resistant K. pneumoniae strains represents a major clinical challenge in nosocomial and community acquired infections. The long pentraxin PTX3, a key component of humoral innate immunity, is involved in resistance to selected pathogens by promoting opsonophagocytosis. We investigated the relevance of PTX3 in innate immunity against K. pneumoniae infections using Ptx3-/- mice and mouse models of severe K. pneumoniae infections. Local and systemic PTX3 expression was induced following K. pneumoniae pulmonary infection, in association with the up-regulation of TNF-α and IL-1β. PTX3 deficiency in mice was associated with higher bacterial burden and mortality, release of pro-inflammatory cytokines as well as IL-10 in the lung and systemically. The analysis of the mechanisms responsible of PTX3-dependent control of K. pneumoniae infection revealed that PTX3 did not interact with K. pneumoniae, or promote opsonophagocytosis. The comparison of susceptibility of wild-type, Ptx3-/-, C3-/- and Ptx3-/-/C3-/- mice to the infection showed that PTX3 acted in a complement-independent manner. Lung histopathological analysis showed more severe lesions in Ptx3-/- mice with fibrinosuppurative, necrotizing and haemorrhagic bronchopneumonia, associated with increased fibrin deposition in the lung and circulating fibrinogen consumption. These findings indicate that PTX3 contributes to the control of K. pneumoniae infection by modulating inflammatory responses and tissue damage. Thus, this study emphasizes the relevance of the role of PTX3 as regulator of inflammation and orchestrator of tissue repair in innate responses to infections.
Introduction
Klebsiella pneumoniae is a Gram-negative bacillus belonging to the family of Enterobacteriaceae, normally colonizing the colon and the oropharynx in humans. K. pneumoniae is often involved in pneumonia, urinary tract infections, bacteremia and sepsis, in particular in neonates, elderly and immunocompromised patients (1). Overuse of antibiotics has promoted the emergence of multidrug-resistant K. pneumoniae strains, which are frequently isolated in nosocomial and community-acquired infections (2, 3). Community-acquired pneumonia (CAP) is the most common cause of sepsis and K. pneumoniae is one of the known causative pathogens for CAP with high mortality rates (1, 4, 5).
Sepsis is defined as a life-threatening organ dysfunction caused by a dysregulated host response to an infection with the worldwide mortality rate of almost 11 million individuals per year (6–8). The balance between pro and anti-inflammatory responses contributes to the patient outcome in sepsis (9). The pro-inflammatory responses are associated with cardiovascular disorders, shock, and organ failure, and the anti-inflammatory responses and immune suppression with unpaired control of infections (10–12). In addition to all the complications that sepsis per se brings in clinical setting, the emergence of antibiotic resistant K. pneumoniae strains becomes a notable concern in public health.
The innate immune system is the first line of defense against invading pathogens and tissue damage. It is composed of a cellular and a humoral arm. The humoral arm comprises the complement system and soluble pattern recognition molecules (PRMs), such as pentraxins, collectins and ficolins (13). Pentraxin 3 (PTX3) is the first member and prototype of the long pentraxin subfamily (14, 15). It is produced locally in damaged tissues by a variety of cells, in particular stromal and immune cells, after stimulation by inflammatory cytokines, such as IL-1β and TNF-α, TLR agonists and other microbial components, such as lipopolysaccharide (LPS), outer membrane protein A (OmpA) and lipoarabinomannans, or intact microorganisms (13, 16).
PTX3 is a multifunctional protein involved in a wide range of activities, ranging from microbial recognition to regulation of inflammation and tissue remodeling. In particular, PTX3 is involved in resistance to selected pathogens, acting as an opsonin, promoting phagocytosis in a Fcγ receptor and complement-dependent manner, amplifying the responses to bacteria and neutralizing viruses (17–20). PTX3 has also been shown to regulate inflammation due to infections (16, 21, 22) or sterile tissue damage (23–25), by modulating the complement cascade and neutrophil recruitment (17). Finally, PTX3 is involved in tissue remodeling, by contributing to fibrinolysis, angiogenesis, matrix deposition (20). PTX3 has been reported to play a dual role in K. pneumoniae infections in transgenic mice, depending on the bacterial load and on the amount of overexpressed PTX3 (26). In addition, PTX3 has been shown to contribute to innate immune responses against the outer membrane protein A from K. pneumoniae (KpOmpA) (27). Nevertheless, the role of PTX3 and the mechanism of its action in innate immunity against K. pneumoniae infections are not clearly defined.
In this study, taking advantage of PTX3-deficient mice and well-established in vivo models of K. pneumoniae severe infection, we demonstrated the actual protective role of PTX3 in defense against K. pneumoniae.
Material and Methods
Animals
All mice used were 8-12 weeks old male on a C57BL/6J genetic background. C57BL/6J wild-type mice were purchased from Charles River Laboratories, Calco, Italy. PTX3-deficient mice (Ptx3-/-) were on a C57BL/6J background, generated as described (28) and bred by Charles River Laboratories, Calco, Italy. C3-deficient mice were from The Jackson Laboratories, Bar Harbour, Maine, US. PTX3/C3-double deficient mice were generated by crossing PTX3- and C3- deficient mice. For the generation of bone marrow chimeras, Ptx3-/- and wild-type mice were lethally irradiated with a total dose of 900 cGy. Two h later, mice were injected in the retro-orbital plexus with 5x106 bone marrow cells obtained by flushing of the cavity of femurs from wild-type or Ptx3-/- donor mice. Recipient mice received gentamycin (0.8 mg/ml in drinking water) starting 10 days before and for two weeks after irradiation and used 12 weeks later. For the generation of endothelial conditional PTX3 deficient mice, we used the tamoxifen inducible cre/lox site-specific recombination system: Ptx3fl/flCdh5-cre/ERT2+/- mice were generated crossing mice carrying a loxP-flanked Ptx3 gene generated by Oxgene with Cdh5-Cre/ERT2+/- mice from Taconic. Tamoxifen was injected ip for five days (2 mg/mice) to activate cre recombinase in endothelial cells.
Experiments were performed in the animal facility of Humanitas Research Hospital in individually ventilated cages. Mice were randomized based on sex, age, and weight.
K. Pneumoniae Infection Models
Pneumonia and peritonitis were induced by intranasal inoculation with 1x104 CFUs or intraperitoneal injection with 500 CFUs K. pneumoniae serotype 2 (ATCC 43816), respectively. Mice were sacrificed at different time points (6, 18, 24, 44 hours) after infection for sample collection. In the survival study, mice were observed 2 times a day for clinical sign of distress and mortality, and sacrificed by humanized euthanasia when they reached the human endpoints indicated by clinical signs (body temperature dropping, intermittent respiration, solitude presence, hunched posture, fur erection, unresponsive alertness, and inability to ascend when induced).
In selected experiments, mice were treated intraperitoneally with 10 μg recombinant mouse-PTX3, produced as previously described (29), with one dose at 24 hours or two doses at 0 and 24 hours after infection, and the outcome of infection was evaluated by following mortality (one treatment) and counting lung and spleen CFU (two treatments).
Sample Collection
Lung, liver and spleen were collected and homogenized in 1 ml PBS containing protease inhibitors (Complete tablets EDTA-free, Roche Diagnostic) and 1 mM PMSF (Sigma) and used for CFU count. The homogenates were then centrifuged at 13000 rpm for 20 minutes at 4°C and the supernatants were stored at -80°C. Blood was drawn either from the cava vein or the retro-orbital plexus, collected in an appropriate anti-coagulant (heparin, EDTA or citrate) vacutainer tube.
Lung, liver and spleen homogenates and blood were serially diluted 1:10 in PBS, plated in Tryptic Soy Broth (TSB) agar (BD) and incubated overnight at 37°C for CFU count. EDTA-blood samples from retro-orbital plexus were analyzed by the laser automated counter ADVIA 120 (Siemens).
Citrate-blood from cava vein was used to measure fibrinogen by converting the clotting time under bovine thrombin activation (Futurlab srl, Limena, PD Italy). ELISA assays were used to measure murine cytokines, MPO, PTX3 (R&D DuoSet ELISA Development System, USA) and TAT (Thrombin-Antithrombin Complexes- Abcam, Cambridge, UK) according to manufacturer’s instructions. Magnetic beads multiplex immunoassay (ProcartaPlex- Invitrogen) was used to measure cytokines in the blood. Protein quantitation was performed on the Luminex instrument platform (Bio-plex 200 system- Bio-Rad).
Flow Cytometry Analysis of Leukocyte Populations
Analysis of leukocyte populations in lung and blood was performed by flow cytometry. Representative gating strategy is shown in Supplementary Figure S1A. The following murine antibodies were used: Anti-mouse CD11b-BV785 (Clone M1/70) and anti-mouse F4/80-PE-Cy7 (Clone BM8) from Biolegend, anti-mouse Ly6G-PE-CF594 (Clone 1A8), anti-mouse CD45-BV605 (Clone 30-F11), anti-mouse CD11b-APC-Cy7 (Clone M1/70), anti-mouse Ly6C-BV421 (Clone AL-21), anti-mouse CD11c-AF700 (Clone HL3), anti-mouse MHCII-FITC (Clone 2G9) and anti-mouse Siglec-F-PE (Clone E50-2440) from BD, and anti-mouse CD103-PerCP (Clone 2E7) from eBioscience. Briefly, lungs were cut into small pieces with razor blades and digested for 20 min at 37 °C in PBS containing 0.1 mg/ml collagenase A (Roche). Then the suspension was smashed and passed through a cell strainer (70 μm pore size). RPMI (Gibco) containing 10% FBS was added to stop the digestion process. Lung and blood total cells were counted on Neubauer chamber. Cell viability was determined by LIVE/DEAD™ Fixable Aqua Stain Kit (Invitrogen) and CD16/32 Fc block (Clone 93- eBioscience) was used to block non-specific Fc receptor binding. Samples were incubated with the mixture of the antibodies and red blood cells were lysed by adding ice-cold ammonium-chloride-potassium (ACK) lysing buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.2). Samples were fixed with 1% paraformaldehyde (PFA) and analyzed on LSR Fortessa (BD Bioscience). Data were analyzed with FlowJo software 10 (Treestar).
Histological Examination
Mouse organs were fixed in 4% neutral buffered formalin. Paraffin embedded mouse tissues were cut at 3 μm and sections were stained by hematoxylin-eosin. Slides were examined blindly under light microscopy by a veterinary pathologist (C.R.).
Confocal Microscopy
Lung tissues were frozen in OCT and cut at 8μm. Sections were fixed with 4% PFA and blocked with 2% BSA in PBS2+. Sections were then incubated with the following primary antibodies for 2 h at room temperature: affinity purified goat IgG anti-PTX3/TSG-14 (5 µg/ml; R&D Systems), rabbit polyclonal anti-fibrin(ogen) (5 µg/ml; Dako), mouse monoclonal anti–α-SMA FITC (4 µg/ml; Sigma-Aldrich); rat monoclonal anti-CD31 APC (0.5 µg/ml; BD); and rat monoclonal anti-PDGFRα (CD140a) BV421 (5 µg/ml; BD). Sections were then incubated for 1 h with Alexa Fluor (488, 568, or 647)–conjugated species-specific cross-adsorbed detection antibodies (2 µg/ml; Invitrogen). 4’,6-diamidino-2-phenylindole-DAPI (1:1000; Invitrogen) was used to visualize the nucleus. Working solution of 5% of donkey serum (Sigma-Aldrich), 2% BSA, 0.1% Triton X-100 (Sigma-Aldrich) in PBS2+, pH 7.4, was used for antibody dilution and sections were washed after each step with washing buffer (PBS2+, pH 7.4, containing 0.01% (vol/vol) Tween 20). Lungs from Ptx3−/− mice were used as negative control for immunostaining of PTX3. The slides were then dried, and coverslips were mounted with fluorsave reagent (Calbiochem, Meudon, France). Samples were analyzed with a laser scanning confocal microscope (FluoView FV1000; Olympus) by excitation at 405, 488, 559, and 635 nm and collection of the emission using at 425–475 nm (for DAPI), 416-458 nm (for BV421), 490–530 nm (for Alexa Fluor 488), 570–615 nm (for Alexa Fluor 568), and 655–700 nm (for Alexa Fluor 647) band-pass filters. Three images/mouse (1,024 x 1,024 pixels) were acquired with 20x objective (20x 0.75 NA Plan-Apochromat; Olympus).
In Vitro Phagocytosis in Murine Whole Blood
K. pneumoniae bacteria were labelled using 10 µM Vybrant™ CFDA, SE Cell Tracer Kit (V12883-Thermo Fisher), incubating for 1 hour at room temperature, followed by 3 times washing with HBSS (Hank’s Balanced Salt Solution) and resuspend in PBS2+. Efficiency of labeling was measured by flow cytometry. Phagocytosis of K. pneumoniae was assessed using whole blood from wild-type and Ptx3-/- mice, collected in heparin vacutainer. Briefly, 4x106 CFSE-labelled K. pneumoniae, either pre-opsonized (85 μg/ml) or not with recombinant human PTX3, were added to 70 μl of whole blood to reach the final concentration of 25 μg/ml and incubated for 20 min at 37°C. Phagocytosis was blocked by placing samples on ice and red blood cells were lysed by adding ice-cold ACK lysing buffer. The samples were then stained by a combination of anti-mouse Ly6G- PECF594, anti-mouse CD45- BV605, anti-mouse CD11b- APC-Cy7, anti-mouse Ly6C- BV421. Samples were fixed with 1% PFA and analyzed by flow cytometry (LSR Fortessa). Representative neutrophil gating strategy is shown in Supplementary Figure S1B.
PTX3 Binding to K. Pneumoniae
Binding of PTX3 to K. pneumoniae was assessed by flow cytometry and wild-field microscopy. Polyclonal rabbit anti-hPTX3 antibody (29) was biotinylated with NHS-LC-Biotin (Thermo Scientific, Waltham, USA), according to the manufacturer’s instructions. 106 CFSE-labelled K. pneumoniae were incubated with PTX3 (ranging from 1 to 20 μg/ml) in PBS2+ containing 0.2% (w/v) BSA for 1h at room temperature. Unbound PTX3 was removed by washing. The samples were then incubated with anti-hPTX3 biotinylated polyclonal antibody (0.5 μg/ml) followed by incubation with Streptavidin-Alexa Fluor 647 (Invitrogen™) (1:500) to detect bound proteins. Samples were fixed with 1% PFA and analyzed either on a FACS Canto II flow cytometry system (BD Biosciences, East Rutherford, NJ) or wild-field microscopy. FITC-labeled Aspergillus fumigatus conidia were used as positive control (28).
Production of PTX3 by Fibroblasts
Mouse embryonic fibroblasts (MEF) were obtained from a wild-type pregnant mouse at 14 day post-coitum as previously described (30). Cells were plated at a density of 105/ml and expanded in 6-well plates in RPMI 1640 medium supplemented with 2 mM L-glutamine, 10% FCS, and B-mercaptoethanol. MEF (5x105 cells/well) were plated in a 24-well plate at passage 5-6 in RPMI 1640 medium supplemented with 2 mM L-glutamine, 1% FCS and stimulated with heat-killed K. pneumoniae (30 min at 70°C) at ratios of 5:1, 1:1 and 1:5 (bacteria:cells). LPS (10ng/ml) and TNFα (10ng/ml) were used as controls of PTX3 induction. Murine PTX3 concentration in supernatants was analyzed by ELISA (R&D DuoSet ELISA Development System, USA).
Statistical Analysis
Values were expressed as mean ± SEM or median of biological replicates, as specified. Two-tailed Mann-Whitney test or ANOVA multiple analysis were used. Survival curves are shown as Kaplan-Meier plots and compared using log-rank test. Linear regression analysis (best-fit curve) was used to analyze correlation between PTX3 and cytokine levels. A ROUT test was applied to exclude outliers. The analyses were done using GraphPad Prism 6 (San Diego, CA). P < 0.05 was considered statistically significant.
Results
PTX3 Expression Is Induced During K. Pneumoniae Infection
Pneumonia was induced in C57BL/6J mice by intranasal inoculation with K. pneumoniae serotype 2 (ATCC 43816). Intranasal administration of K. pneumoniae leads to a gradually localized infection in the lung followed by systemic dissemination of bacteria, along with inflammatory responses, organ injury, and eventually death, mimicking sepsis (31, 32). To investigate the relevance of PTX3 in this model, we first measured the local and systemic levels of PTX3 during the infection and we observed robust induction of PTX3 starting from 24 hours post-infection and reaching a peak at 48h post-infection, following systemic dissemination, as indicated by spleen bacterial load (Figures 1A, B and Supplementary Figure S2A, B).
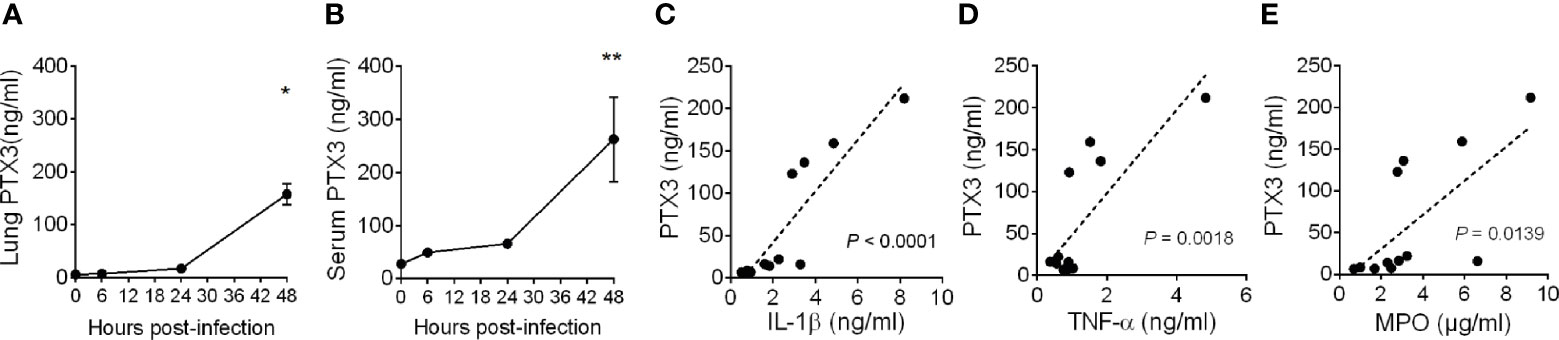
Figure 1 Induction of PTX3 expression during Klebsiella pneumoniae infection. PTX3 concentration in lung homogenate (A) and serum (B) of wild-type mice infected intranasally with 104 CFUs K. pneumoniae serotype 2 (ATCC 43816). Each dot is mean ± SEM (n = 4 mice). PTX3 concentration at 6, 24 and 48 hours after infection were compared with basal levels using ANOVA multiple analysis. *6h vs 48h, **0h vs 48h. (C–E) Correlation between the concentration of PTX3 and IL-1β (C), TNF-α (D) or MPO (E) in the lung during the course of the infection. Linear best-fit curve.
PTX3 expression is induced by both microbial components, including KpOmpA (27), and proinflammatory cytokines, such as TNF-α and IL-1β, and rapidly released by activated neutrophils (16, 33). We thus analyzed the local and systemic concentration of these inflammatory mediators and of MPO, a marker of myeloid cell infiltration and activation. As shown in Supplementary Figures S3A–F, we observed an increase of the concentration of IL-1β, TNF-α and MPO during the progression of the infection, which positively correlated with the concentration of PTX3 (P < 0.0001, P = 0.0018, P = 0.0139, respectively) (Figures 1C–E). These results suggest an association between K. pneumoniae infection-induced inflammation and PTX3 expression.
PTX3 Deficiency Is Associated With Higher Susceptibility to K. Pneumoniae Infection
Ptx3-/- mice were used to address if PTX3 is involved in the defense against K. pneumoniae infection. Early (18 h) after intranasal infection, lung and blood bacterial loads were comparable in Ptx3+/+ and Ptx3-/- mice (Figures 2A, B). In contrast, at the late stage of infection (44 h), PTX3-deficiency was associated with more severe local and systemic infection. In particular, bacterial loads were 15-fold higher in the lungs (P = 0.0005), 55-fold higher in the blood (P = 0.003), 14-fold higher in the liver (P = 0.04) of Ptx3-/- mice (Figures 2C–E). These results indicate that PTX3 is involved in controlling K. pneumoniae burden and dissemination at the late stage of infection.
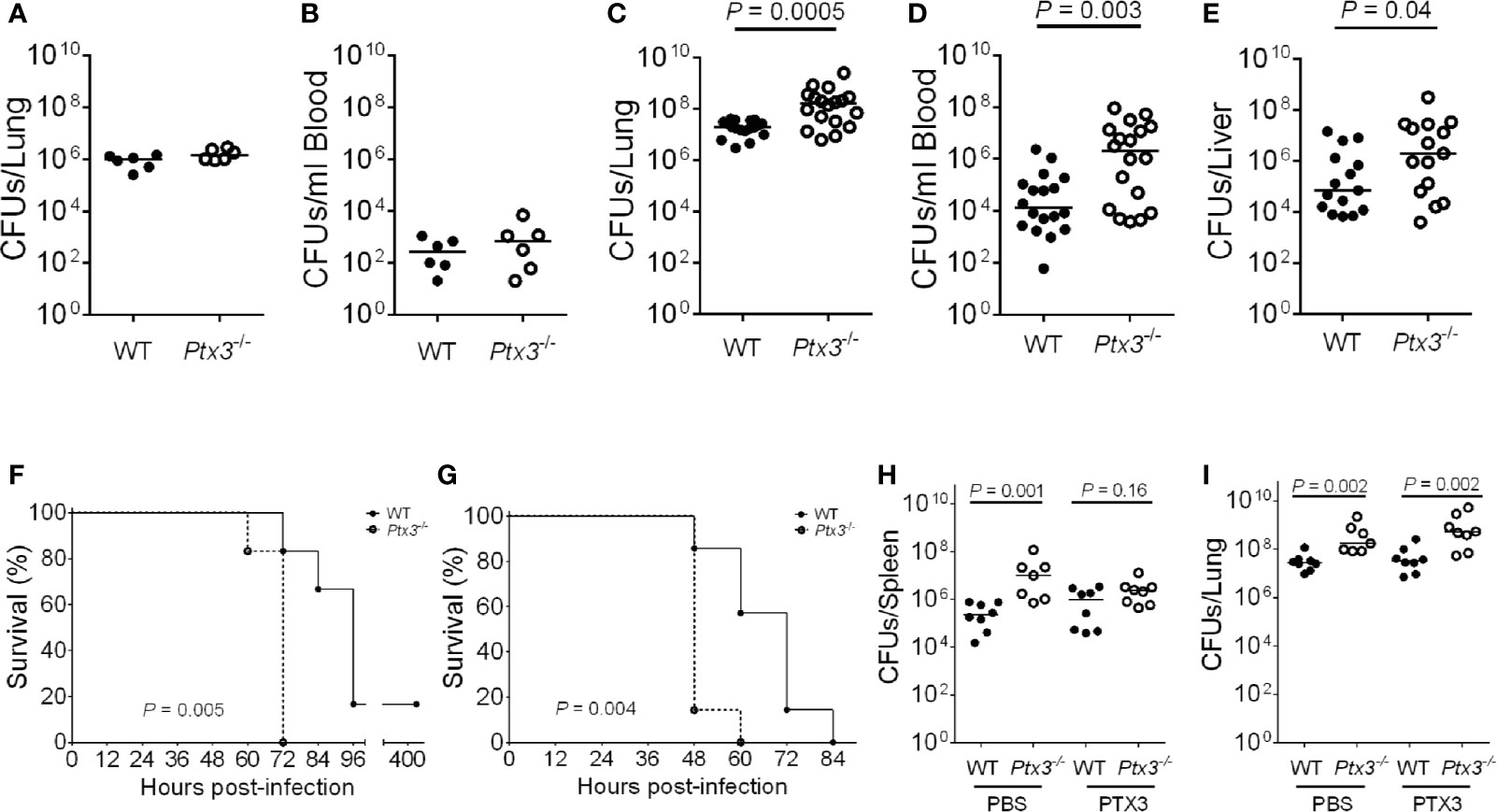
Figure 2 Susceptibility of Ptx3-/- mice to K. pneumoniae infection. (A–E) Bacterial load in the lung (A) and blood (B) at 18 hours after intranasal infection (n = 6 mice) and in the lung (C), blood (D), and liver (E), at 44 hours after intranasal infection (n = 15-18). The median of CFU count is shown. Two-tailed Mann-Whitney test. (F, G) Kaplan-Meier survival curves of wild-type and Ptx3−/− mice inoculated either intranasally with 104 CFUs (n = 6 mice) (F) or intraperitoneally with 500 CFUs (n = 7 mice) K. pneumoniae (G). Survival curves were compared with the Log-Rank test. (H, I) CFU count in the spleen (H) and lung (I) at 44 hours after intranasal infection in mice treated with mPTX3 (10 μg/mouse at 0 and 24 hours after infection) (n = 7-8 mice). The median of CFU count is shown. Two-tailed Mann-Whitney test. (A, F) show one representative experiment out of two (A, F). (C–E) are the pool of two separate experiments. (B, G–I); one experiment performed.
To assess the importance of PTX3 in the outcome of K. pneumoniae infection, the mortality rate was investigated using two different models of infection. Wild-type and Ptx3-/- mice were inoculated either intranasally or intraperitoneally with K. pneumoniae and observed for 5 days. Ptx3-/- mice showed a higher mortality rate compared to wild-type mice in both models of infection, with 100% Ptx3-/- mice dead compared to 17% wild-type mice 72 h post intranasal infection (P = 0.005), and 100% Ptx3-/- mice dead compared to 42% wild-type mice 60 h post intraperitoneal infection (P = 0.004) (Figures 2F, G). Hence, PTX3 deficiency is associated with worst outcome in K. pneumoniae infections.
Finally, we investigated the specificity of the phenotype of Ptx3-/- mice in pneumosepsis induced by K. pneumoniae by treating them with recombinant PTX3. Recombinant mouse PTX3 (mPTX3) was administered at the time of infection and 24 h later in wild-type and Ptx3-/- mice. With this schedule of treatment, we observed a better control of the infection in Ptx3-/- mice, since PTX3 treatment abolished the difference in spleen CFUs between Ptx3-/- and wild-type mice (Figure 2H). In contrast, the treatment did not improve the control of lung infection in either group nor systemic infection in wild-type mice (Figures 2H, I). In addition, in one experiment of survival, the increased mortality of Ptx3-/- mice compared to wild-type mice was abolished in treated mice (P=0.039 in untreated vs P=0.27 in treated mice, n=7, not shown).
PTX3 Promotes Innate Resistance to K. Pneumoniae Infection in an Opsonophagocytosis and Complement-Independent Manner
PTX3 was previously shown to be involved in innate resistance to specific infections by opsonizing microorganisms (e.g. A. fumigatus conidia and Pseudomonas aeruginosa), directly (15, 28, 34, 35) or through ficolins and collectins (13, 36–38), and promoting complement-dependent phagocytosis. Previous studies showed that PTX3 did not interact with K. pneumoniae, strain ATCC-27736 (26), even though it bound to purified KpOmpA, cooperating with TLR2 and scavenger receptors in the activation of innate immune responses to this microbial moiety (27). To assess the mechanisms responsible for the protective role of PTX3 in K. pneumoniae infections, we further investigated a potential interaction between PTX3 and K. pneumoniae. The binding of PTX3 to K. pneumoniae was assessed using wild-field microscopy and flow cytometry. We did not observe any interaction with either approach (Figures 3A, B). In contrast and as expected, PTX3 interacted with A. fumigatus conidia, which were used as positive control in these assays.
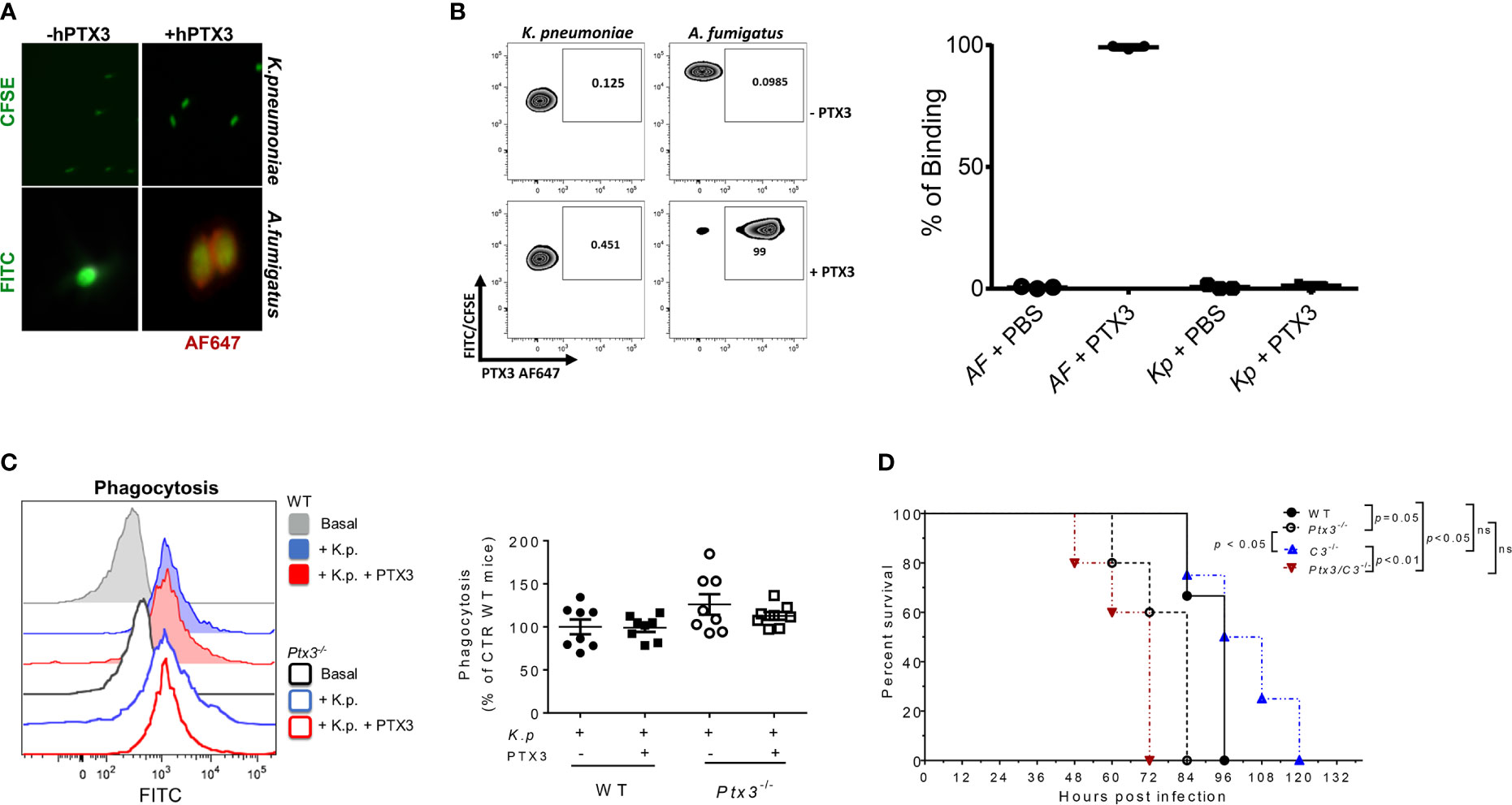
Figure 3 Analysis of opsonophagocytosis and complement in PTX3-dependent resistance to K. pneumoniae. (A) Immunofluorescence analysis by wild-field microscopy and (B) flow cytometry analysis of CFSE-labelled K. pneumoniae (upper panels in (A), left zebra plot panels in (B) and FITC- labeled A. fumigatus (lower panels in (A) and right zebra plot panels in (B) incubated with or without PTX3, followed by anti-hPTX3 biotinylated antibody and streptavidin-Alexa Fluor 647 (AF647). Quantification of percentage of binding of hPTX3 (20 μg/ml) to K. pneumoniae (Kp) and A. fumigatus (AF) of the three experiments performed is shown in the right panel in (B) FITC-labelled A. fumigatus was used as positive control. (C) Left panel: Representative histogram showing phagocytosis (CFSE fluorescent intensity in CD11b+Ly6G+ neutrophils) in wild-type or Ptx3-/- blood. Basal = blood without K. pneumoniae. Right panel: Phagocytosis of CFSE-labelled (K) pneumoniae by wild-type or Ptx3-/- neutrophils with or without PTX3 (25 μg/ml). Results are reported as a percentage of wild-type neutrophil phagocytosis (n = 8 mice, from two separate experiments). Graphs show Mean ± SEM. Two-tailed Mann Withney test and ANOVA. (D) Kaplan-Meier survival curve of wild-type, Ptx3-/-, C3-/- and Ptx3/C3-/- mice inoculated intranasally with K. pneumoniae. N= 3 wild-type, 5 Ptx3-/-, 4 C3-/- and 5 Ptx3/C3-/- mice. Survival curves were compared with the Log-Rank test.
To investigate whether PTX3 improved the phagocytic activity, a phagocytosis assay was performed in whole blood drawn from wild-type and Ptx3-/- mice and in the presence of recombinant PTX3 (25 μg/ml). As shown in Figure 3C, there were no significant differences in phagocytosis between wild-type and Ptx3-/- neutrophils and the addition of PTX3 in the assay did not promote it.
Finally, we assessed the contribution of complement in PTX3-mediated activity in K. pneumoniae infection, by comparing the survival of wild-type and Ptx3-/- mice to C3-/- and Ptx3-/-/C3-/- mice after intranasal infection. As shown in Figure 3D, C3-/- mice showed mortality comparable to that of wild-type mice (P = 0.9), suggesting that complement does not play a major role in this infection. This result is in line with a previous study showing that the strain of K. pneumoniae used in this study (ATCC43816) is resistant to complement killing activity (39). In addition, C3-/- mice showed a lower mortality rate than Ptx3-/-and Ptx3/C3-/- mice (P < 0.05 and P < 0.01, respectively). Finally, no significant difference was observed between Ptx3-/-and Ptx3/C3-/- mice, indicating that the phenotype of PTX3-deficient mice did not depend on complement activation or modulation.
PTX3 Deficiency Is Associated With Exacerbated Sepsis-Induced Inflammation and Tissue Damage
We next evaluated local and systemic inflammation at early and late time points post-infection. As shown in Figures 4A, B, significantly higher IL-6 concentration was observed at a late time point (mean ± SEM: 3.6 ± 0.3 ng/ml in WT and 12 ± 1.7 ng/ml in Ptx3-/-, P < 0.0001), whereas only a trend of increased concentration of IL-1β, TNF-α, IL-6 and CXCL1, a key neutrophil chemotactic factor, was observed at the early time point in lung homogenate of Ptx3-/- mice. We observed an exacerbated systemic inflammatory response in Ptx3-/- mice at the late time point, with significantly higher plasma concentration of TNF-α (36.2 ± 6.2 pg/ml in WT and 102.7 ± 12.3 pg/ml in Ptx3-/-, P = 0.004), IL-6 (0.6 ± 0.1 ng/ml in WT and 3.5 ± 0.8 ng/ml in Ptx3-/-, P = 0.004), and CXCL1 (0.3 ± 0.06 ng/ml in WT and 1.6 ± 0.2 ng/ml in Ptx3-/-, P = 0.004) (Figure 4C). Interestingly, PTX3-deficiency was also associated with significantly higher local and systemic concentrations of IL-10 at the late time point (1.7 ± 0.2 ng/ml in WT and 2.3 ± 0.2 ng/ml in Ptx3-/-, P = 0.04 in lung homogenate, and 15.3 ± 4.4 pg/ml in WT and 82.3 ± 10.1 pg/ml in Ptx3-/-, P = 0.004 in blood) (Figure 4D). These results suggest the coexistence of hyperinflammatory and immunosuppressive responses during pneumosepsis in Ptx3-/- mice.
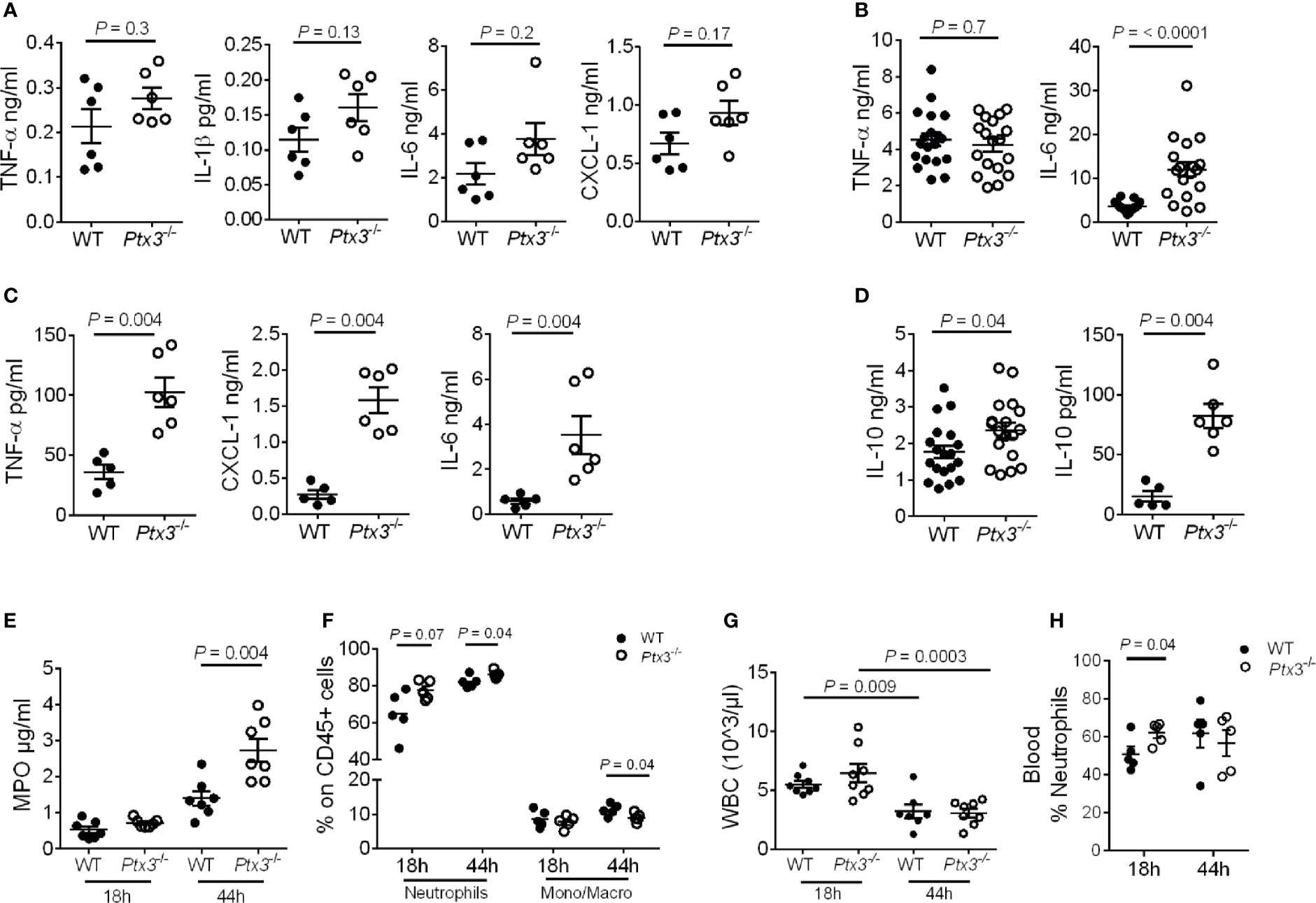
Figure 4 Analysis of inflammation in (K.) pneumoniae infection. (A-C) Cytokine concentration in lung homogenates at 18 hours (n = 6 mice) (A) and 44 hours (n = 18 mice, pool of two separate experiments) post-intranasal infection (B) or in blood 44 hours post-intranasal infection (n = 5-6 mice) (C). (D) IL-10 concentration in lung homogenate (n = 18 mice, pool of two separate experiments) and blood (n = 5-6 mice) 44 hours post-intranasal infection. (E) MPO activity in lung homogenates (n = 7 mice). (F) Flow cytometry analysis of neutrophils and monocytes/macrophages in the lung (n = 5 mice). (G) White blood cell count (n = 7-8 mice). (H) Percentage of neutrophils in blood (n = 5 mice). Mean ± SEM is shown. (A–D, F, H) Mann Whitney test. (G) Multiple t-test. The outliers were removed through ROUT.
In addition, PTX3 deficiency was associated with a higher amount of MPO (P = 0.004 at 44h after infection) (Figure 4E) and a higher percentage of neutrophils in the lung (P = 0.04 at 44 hours after infection) (Figure 4F). We next analyzed total peripheral blood leukocytes and, in the context of comparable sepsis-induced leukopenia (Figure 4G), neutrophilia appeared already at the early time point after infection in Ptx3-/- mice (P = 0.04) (Figure 4H). Since there was not yet a difference in bacterial load in the two groups of mice at this time point, these results suggest that early neutrophilia was not secondary to increased bacterial load, but rather to exacerbated inflammation.
We next investigated lung histopathology 44 hours after intranasal infection. First, gross examination of lung tissue showed more severe hemorrhaging pattern and congestion in Ptx3-/- mice compared to wild type mice (Figure 5A). Histopathology showed multiple foci of suppurative bronchopneumonia with intrabronchial and interalveolar bacteria, consistent with K. pneumoniae infection, in wild-type mice (Figures 5B–E). In contrast, Ptx3-/- mice developed multifocal areas of fibrinosuppurative and necrotizing and/or hemorrhagic bronchopneumonia with bacteria mainly localized in the perivascular interstitium (Figures 5F, G). In wild-type mice, bacteria were not readily visible and were occasionally found in the bronchiolar and alveolar spaces or within foamy macrophages (Figure 5E), while in Ptx3-/- mice perivascular aggregates of bacteria with a mucoid aspect associated with edema were observed (Figures 5H, I). Microscopic hemorrhagic lesions were scored, and a higher grade was attributed to Ptx3-/- mice, but it did not reach the statistical significance (Figure 5J). These results suggest that the infection was confined to the bronchial and alveolar spaces in wild-type mice, whereas it spread to the perivascular interstitium and was associated with edema, fibrinosuppurative, and necrotizing and/or hemorrhagic lesions in Ptx3-/- mice.
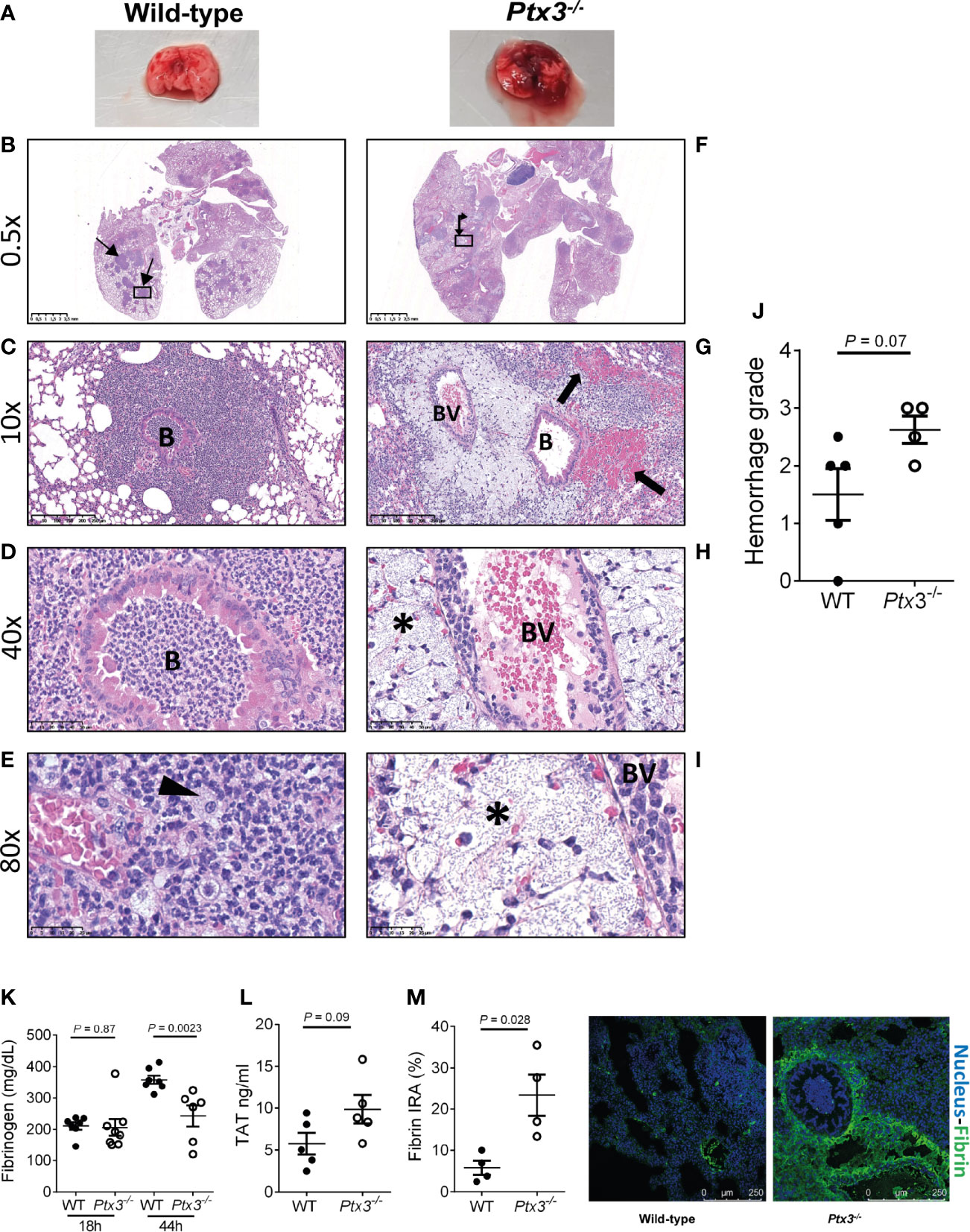
Figure 5 Analysis of lung damage and fibrin deposition of K. pneumoniae intranasally infected mice. (A) Macroscopic aspect of lungs of wild-type and Ptx3-/- mice infected with K. pneumoniae. A representative picture out of five is shown. (B–E): Histological analysis of wild-type mice. (B) Multiple foci of suppurative bronchopneumonia (arrow line). (C, D) Lesions centered on bronchiolar structures. (E) Bacteria in the bronchiolar and alveolar spaces or within foamy macrophages (arrowhead). (F–I): Histological analysis of Ptx3-/- mice. (F) Multifocal areas of fibrinosuppurative and necrotizing and/or hemorrhagic bronchopneumonia (arrow left-up). (G) Intra alveolar and intrabronchial hemorrhages (arrow). (H, I) Perivascular aggregates of bacteria associated with edema (*). B, Bronchiole; BV, blood vessel. (J) Quantification of the hemorrhage grade in the lung (n = 4 mice). (K) Blood fibrinogen concentration (n = 7-8 mice) at 18 and 44 hours after infection and (L) TAT levels (n = 5 mice) at 44 hours after infection. (M) Fibrin deposition in the lung by confocal microscopy. Left panels: representative images of wild-type and Ptx3-/- mice. Right panel: Quantitation of fibrin/ogen deposition. Each dot represents the mean percentage of the immunoreactive area (IRA) of four fields per mice (n = 4 mice). (J–M) Mean ± SEM is shown. Groups were compared using two-tailed Mann-Whitney test.
Fibrinogen plasma concentration increases in inflammatory conditions as part of the acute phase response, but can decrease in sepsis conditions as a consequence of increased consumption due to disseminated intravascular coagulation (DIC) (40, 41). Fibrinogen plasma levels increased during the infection in wild-type mice. At the early time point, fibrinogen levels were comparable in the two groups, but were significantly lower in Ptx3-/- mice at the late time point compared to wild-type mice (Figure 5K). Further, a trend of increase of TAT levels was observed (Figure 5L). These results show that despite increased inflammation (Figure 4), PTX3-deficiency was associated with lower fibrinogen levels, suggesting DIC and fibrinogen consumption. In addition, immunofluorescence analysis revealed higher fibrin deposition in the lung tissue of Ptx3-/- mice compared to wild-type mice (4-fold increase of the immunoreactive area, P = 0.028) (Figure 5M). Finally, the analysis of different organs at 24 hours after intraperitoneal infection indicated an increase in the number of thrombotic lesions in Ptx3-/- mice when compared to wild-type mice, although it did not reach the statistical significance (Supplementary Figure S4A–F).
Collectively, these results suggest that PTX3-deficiency is associated with more severe local and systemic inflammation, tissue damage, as well as signs compatible with DIC and coagulopathy, including increased fibrin deposition in the lungs, which may contribute to increased susceptibility and mortality in this model of pneumosepsis.
PTX3 Is Produced by Non-Hematopoietic Cells During Klebsiella Pneumoniae Infections
Having observed that PTX3-mediated opsonophagocytosis was not relevant, we further dissected the role of PTX3 in this model by investigated its cell source during K. pneumoniae infection. Bone marrow chimeric mice were used to address the involvement of hematopoietic and stromal cells. The evaluation of PTX3 expression during K. pneumoniae infection in chimeric mice showed that only the genotype of recipient mice impacted on PTX3 plasma levels of infected mice, indicating that non-hematopoietic cells were a major source of PTX3 expression, whereas hematopoietic cells were irrelevant (Figure 6A).
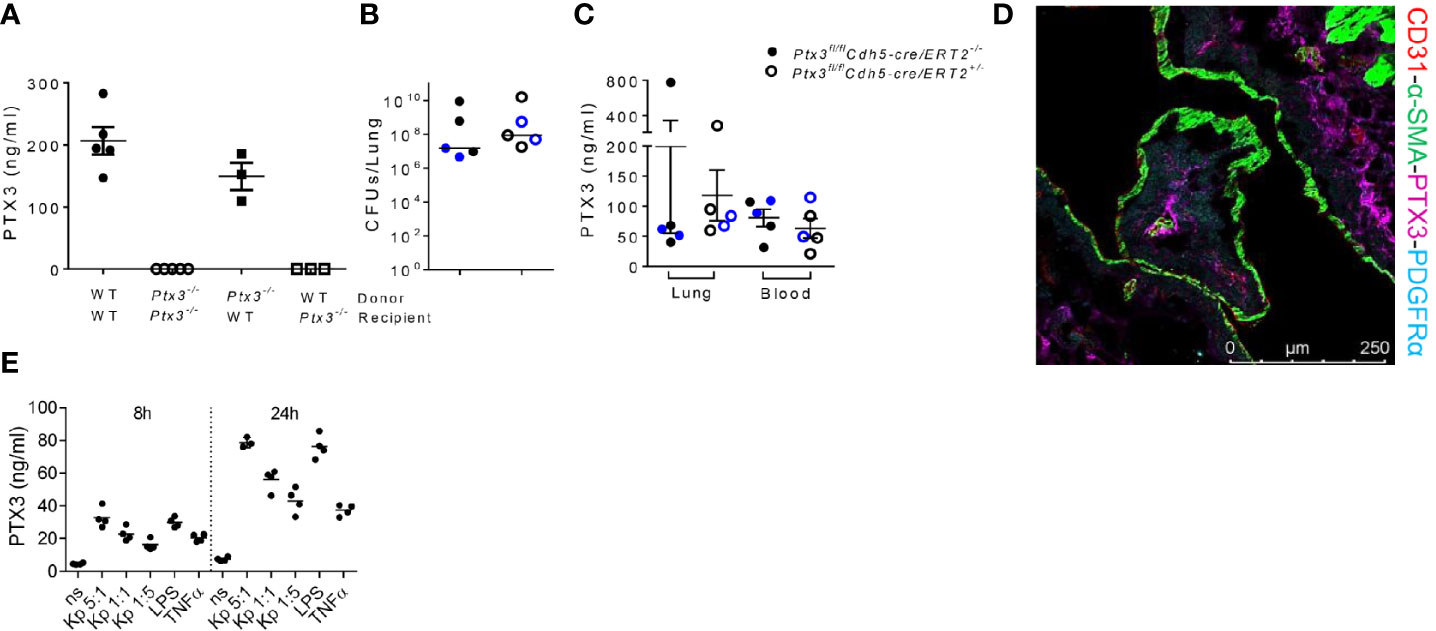
Figure 6 Analysis of the cellular source of PTX3 (A) PTX3 protein concentration in serum of chimeric mice 44 hours after infection. Mean ± SEM is shown (n = 3-5 mice). (B) Bacterial load in the lung of Ptx3fl/flCdh5-cre/ERT2+/- and Ptx3fl/flCdh5-cre/ERT2-/- mice 44 hours after infection. The median of CFU count is shown. (C) PTX3 concentration in lung homogenate and blood of Ptx3fl/flCdh5-cre/ERT2+/- and Ptx3fl/flCdh5-cre/ERT2-/- mice. Mean ± SEM is shown. n = 5 male and female mice (shown in black and blue, respectively). Groups were compared using two-tailed Mann-Whitney test. (D) Immunofluorescence confocal microscopy images of PTX3 (magenta), PDGFRα (blue), CD31 (red) and α-SMA (green) staining in the lung of wild-type mice 44 hours after infection. Representative merged image is shown (n = 3 mice; 3 fields were observed for each mouse). Scale bar = 250μm. (E) PTX3 production by embryonic fibroblasts after stimulation with heat killed K. pneumoniae at different bacteria:cells ratio for 8 and 24 h. TNF and LPS were used as positive control.
Endothelial cells (ECs) have been known as one of the non-hematopoietic sources of PTX3 (42). To determine whether endothelial cells were the source of PTX3 during K. pneumoniae infection, we investigated the expression of PTX3 using tamoxifen-responsive cdh5-Cre/lox-dependent conditional Ptx3 knockout mice (Ptx3fl/flCdh5-cre/ERT2). As shown in Figures 6B, C, we did not observe differences in lung bacterial load (Figure 6B) and in local or systemic expression of PTX3 (Figure 6C) in Ptx3fl/flCdh5-cre/ERT2+/- mice compared to Ptx3fl/flCdh5-cre/ERT2-/-. These results indicate that endothelial cells are not the main non-hematopoietic source of PTX3.
We next investigated the localization of PTX3 in the lung by confocal microscopy. In particular, we investigated the association of PTX3 with CD31 (a marker of endothelial cells), α-smooth muscle actin (α-SMA) (a marker of smooth muscle cells), and with platelet-derived growth factor receptor-α (PDGFRα) (a marker of mesenchymal cells). As shown in Figure 6D, PTX3 expression was mostly localized in the extracellular space in close proximity of PDGFRα+ mesenchymal cells, whereas minimal or no association was observed with endothelial cells or smooth muscle cells, respectively. We finally investigated PTX3 expression by stromal cells after in vitro stimulation with K. pneumoniae, using embryonic fibroblasts as cellular model for mesenchymal cells. As shown in Figure 6E, PTX3 was induced in this cell type in a dose- and time-dependent manner after incubation with heat killed K. pneumoniae, as well as by TNF and LPS. These results are in line with previous observations in mouse tissue damage models, such as skin wound healing or liver and lung damage, where PDGFRα+ cells were found to be a major source of PTX3 expression (24). Bioinformatic analysis of single cell RNA-seq publicly available datasets confirmed that in addition to vascular endothelial cells, PDGFRα+ and PDGFRβ+ stromal cells are the major source of PTX3 in mouse and human lung in healthy conditions (43, 44).
Discussion
The present study was designed to address the role of the long pentraxin PTX3 in innate resistance to K. pneumoniae infection, taking advantage of PTX3-deficient mice and well-established in vivo models of K. pneumoniae severe infection. The results obtained demonstrate a clear involvement of PTX3 in the control of the infection, in terms of bacterial load, mortality and inflammation. PTX3 was shown to act through mechanisms independent of opsonophagocytosis or complement regulation. PTX3-dependent regulation of inflammation and tissue damage were found the major mechanisms responsible of the outcome of the infection.
Previous studies showed that PTX3 has a dual role in K. pneumoniae infection in PTX3-overexpressing mice, depending on the severity of the inflammatory response induced by the given bacterial load and the amount of protein expressed (26). Overexpression of PTX3 in mice receiving low dose of bacteria promoted the resistance against K. pneumoniae respiratory infection, while in mice receiving a high inoculum, overexpression of PTX3 was associated with faster lethality, suggesting the relevance of PTX3-dependent inflammation tuning in this infection (26). In addition, PTX3 was shown to interact with KpOmpA, contributing to the innate response to this microbial moiety (27). However, the effect of PTX3 in the response to K. pneumoniae infection could not be inferred by this result. Our study was performed to elucidate the actual role of PTX3 in this infection and its mechanisms of action.
Previous studies showed that PTX3 deficiency is associated with higher bacterial load, more severe outcome and mortality after infection with A. fumigatus, P. aeruginosa, Escherichia coli and Shigella flexneri (22, 28, 45). We obtained similar results upon intranasal or intraperitoneal infection with K. pneumoniae, where PTX3 deficiency was associated with higher local and systemic bacterial load and accelerated mortality. These data confirm the role of PTX3 in innate immune responses and resistance to selected pathogens and widen its spectrum of activity in infectious diseases, in particular toward a pathogen frequently involved in human sepsis and associated with multi-drug resistance (2, 4).
The protective role of PTX3 during A. fumigatus and P. aeruginosa infection was attributed to an opsonic activity followed by enhanced opsonophagocytosis in a complement and Fcγ receptors-dependent manner (15, 28, 34). In this study, we did not observe direct interaction of PTX3 with the K2 strain of K. pneumoniae. This suggests that even if PTX3 binds to purified KpOmpA (27), the binding to the bacteria is impaired, possibly by the presence of the capsule. Furthermore, exogenous recombinant PTX3 did not enhance bacterial phagocytosis. These results indicate that the protective role of PTX3 in this model of infection was not mediated by opsophagocytosis, in line with the capacity of K. pneumoniae to evade this first line mechanism of defence through the expression of a polysaccharide capsule (2). Resistance to phagocytosis of K1 and K2 K. pneumoniae strains by alveolar macrophages and neutrophils has been attributed to various factors, for instance, sialic acid residues which mimic endogenous molecules, and lack of specific mannose residue repeats which prevents efficient lectin-dependent phagocytosis through macrophage mannose receptor and lung-secreted SP-A (2). Therefore, the marginal phagocytosis observed in our experiments could be due to these escaping mechanisms of K. pneumoniae, which were not counteracted by PTX3.
Treatment with recombinant PTX3 reduced P. aeruginosa colonization in cystic fibrosis mice and synergized with antifungals in the therapy of A. fumigatus infections (15, 18, 28, 46), paving the way for the development of PTX3 as potential new therapeutic molecule in these opportunistic infections. In our study, treatment with recombinant PTX3 decreased the systemic spread of the infection and mortality in Ptx3-/- mice, but did not ameliorate the control of the infection in wild-type mice. These results demonstrate that the phenotypes observed were specifically due to PTX3-deficiency and confirm the role of PTX3 in this infection, but further experiments with different treatment schedules would be necessary to demonstrate its therapeutic potential in PTX3-competent individuals.
The crosstalk between PTX3 and the complement system has a dual nature. By interacting with complement recognition molecules, e.g. C1q, ficolins and MBL, PTX3 increases the recognition potential of the humoral innate immunity, leading to increased complement-dependent opsonophagocytosis (13). On the other hand, by binding to factor H or recruiting C4BP, PTX3 tunes activation of complement, preventing excessive activation of complement and inflammation (13, 47). The latter effect has been observed in myocardial infarction and demonstrated in cancer (20, 25, 48–50). By using mice deficient of C3 and mice deficient of both C3 and PTX3, we showed that the complement system does not play a major role during K. pneumoniae infection. This is in line with studies showing that either the capsule or LPS can prevent killing by complement of various strains of K. pneumoniae. In particular, it has been shown that the K2 strain (ATCC43816) used in our study is protected against killing by human serum (39). In addition, Ptx3/C3 double deficient mice were not protected compared to Ptx3-/- mice, indicating that PTX3-mediated regulation of complement-dependent inflammation was not involved in the phenotype. Thus, neither complement-dependent opsonophagocytosis nor complement-dependent inflammation were involved in the phenotype of PTX3-deficient mice.
Balanced inflammatory responses contribute to the elimination of infections, whereas exaggerated inflammation can contribute to tissue damage and immunopathology, promoting sepsis (12, 51, 52). In particular, neutrophil-dependent inflammation may contribute to lung immunopathology in pneumonia (35, 53). Through exacerbated release of toxic agents such as proteinases (neutrophil elastase, cathepsin G and proteinase-3), cationic polypeptides, cytokines, ROS, and finally, neutrophil extracellular traps (NETs), neutrophils can contribute to increased epithelial and vascular permeability leading to pulmonary damage, which allow for accelerated transmigration of neutrophils into the alveolar space and bacterial systemic spread (53). During systemic infection, NETs and their content also contribute to endothelial damage promoting diffuse thrombosis, DIC and acute organ injury (54). We observed higher inflammatory responses as well as neutrophil recruitment and activation in Ptx3-/- mice. In particular, higher neutrophilia was observed in an early time point of infection, when there was still no difference in bacterial load between the two groups of mice, which emphasizes that the higher inflammatory responses in Ptx3-/- mice is not secondary to higher bacterial load. PTX3 was shown to tune neutrophil recruitment by binding to endothelial P-selectin and following inflammation-induced tissue damage (19). This mechanism has been observed in acute lung injury, pleurisy and mesenteric inflammation, as well as in acute kidney injury (13, 19, 23). Increased MPO levels and neutrophil recruitment in the lung could suggest that P-selectin-dependent deregulated mechanisms account for the phenotypes observed in our model. However, P-selectin deficiency has been associated with higher bacterial burdens and more severe late-stage K. pneumoniae pneumosepsis, thus indicating that P-selectin dependent neutrophil recruitment is essential to establish innate immune responses against K. pneumoniae (55).
Ptx3-/- mice developed multifocal areas of fibrinosuppurative and necrotizing and/or haemorrhagic bronchopneumonia, and the infection diffused to the perivascular interstitium, compared with milder tissue damage and confinement of bacteria in the bronchial and alveolar spaces of wild-type mice. Increased fibrin/fibrinogen deposition in the inflamed lung of Ptx3-/- mice associated with decreased blood fibrinogen levels were also observed. These last phenotypes are in line with previous findings obtained in Ptx3-/- mice in models of tissue damage, including acute lung injury, where altered thrombotic responses to injury and augmented fibrin deposition were observed (21, 24, 56–59). Furthermore, pharmacologic inhibitors of fibrin deposition rescued the phenotype of PTX3-deficient mice in specific tissue damage models (24). In these contexts, in the acidic environment due to metabolic adaptation during tissue repair, PTX3 interacted with fibrinogen and plasminogen and promoted plasmin-mediated fibrinolysis (24). It was shown by in vivo and in vitro studies that thrombin improved the host defense against K. pneumoniae (ATCC 43816) via thrombin-induced fibrin generation and polymerization (60), a finding which is in contrast with ours. On the opposite, our results remind the condition described in human sepsis patients, where activation of coagulation and fibrin deposition are associated with microvascular and multiple organ failure (61), thus suggesting the complexity of the consequences of coagulation and fibrin formation in this infection. Finally, the major cell source of PTX3 during K. pneumoniae was represented by stromal cells and not hematopoietic cells. In infected lungs, PTX3 localized in proximity of PDGFRα+ mesenchymal cells, further supporting its role in this infection is associated with tissue remodeling and not in pro-phagocytic activity. Further studies will be necessary to assess which of the phenotypes observed in PTX3-deficient mice depend on the lack of the profibrinolytic activity of PTX3.
PTX3 genetic variants have been associated to innate resistance to infections in humans (62, 63) and PTX3 is a potential therapeutic target in opportunistic infections. Our study clearly demonstrates that PTX3 contributes to the innate resistance to K. pneumoniae lung and systemic infections and suggests that a combination of PTX3-mediated mechanisms, which are not related to opsonophagocytosis, concur to the protection. These include regulation of local and systemic inflammation, as well as tuning of immunopathology. Thus, this study emphasizes the relevance of the role of PTX3 as orchestrator of inflammation and tissue repair in innate responses to infections. The identification of endogenous mechanisms of resistance may lead to the early identification of susceptible individuals based on genetic variability and to the development of new therapeutic targets.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
Procedures involving animals and their care were conformed with protocols approved by the Humanitas Research Hospital (Milan, Italy) in compliance with national (D.L. N.116, G.U., suppl. 40, 18-2-1992 and N. 26, G.U. March 4, 2014) and international law and policies (EEC Council Directive 2010/63/EU, OJ L 276/33, 22-09-2010; National Institutes of Health Guide for the Care and Use of Laboratory Animals, US National Research Council, 2011). The study was approved by the Italian Ministry of Health (approvals n. 742/2016-PR, issued on 26/07/2016). All efforts were made to minimize the number of animals used and their suffering.
Author Contributions
CG and AlM contributed to the conception of the study. CG, FA, ReP, SJ, and AD contributed to the design of the study. FA, DS, RaP, NP, MS, ReP, EM, AnM, CR, MB, FR, FP, and CP performed the experiments. FA contributed to the data collection. FA, AD, TR, CV, and CG contributed to the data analysis. FA, DS, SD, and MS performed the statistical analysis. CG and FA wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The project was funded by the European Commission (European Sepsis Academy Horizon 2020 Marie Skłodowska-Curie Action: Innovative Training Network (MSCA-ESA-ITN, grant number 676129; and European Research Council PHII – 669415 to AM).
Conflict of Interest
AIM and CG obtain royalties on pentraxin-3 related reagents.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Marina Sironi and Tilo Schorn for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.666198/full#supplementary-material
References
1. Bengoechea JA, Sa Pessoa J. Klebsiella Pneumoniae Infection Biology: Living to Counteract Host Defences. FEMS Microbiol Rev (2018) 43:123–44. doi: 10.1093/femsre/fuy043
2. Paczosa MK, Mecsas J. Klebsiella Pneumoniae: Going on the Offense With a Strong Defense. Microbiol Mol Biol Rev (2016) 80:629–61. doi: 10.1128/MMBR.00078-15
3. Fodah RA, Scott JB, Tam H-H, Yan P, Pfeffer TL, Bundschuh R, et al. Correlation of Klebsiella Pneumoniae Comparative Genetic Analyses With Virulence Profiles in a Murine Respiratory Disease Model. PloS One (2014) 9:e107394. doi: 10.1371/journal.pone.0107394
4. Cilloniz C, Dominedo C, Torres A. Multidrug Resistant Gram-Negative Bacteria in Community-Acquired Pneumonia. Crit Care (2019) 23:79. doi: 10.1186/s13054-019-2371-3
5. Chung DR, Song J-H, Kim SH, Thamlikitkul V, Huang S-G, Wang H, et al. High Prevalence of Multidrug-Resistant Nonfermenters in Hospital-Acquired Pneumonia in Asia. Am J Resp Crit Care Med (2011) 184:1409–17. doi: 10.1164/rccm.201102-0349OC
6. Schrijver IT, Théroude C, Roger T. Myeloid-Derived Suppressor Cells in Sepsis. Front Immunol (2019) 10:327. doi: 10.3389/fimmu.2019.00327
7. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA (2016) 315:801–10. doi: 10.1001/jama.2016.0287
8. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, Regional, and National Sepsis Incidence and Mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet (2020) 395:200–11. doi: 10.1016/S0140-6736(19)32989-7
9. Cavaillon J-M, Giamarellos-Bourboulis EJ. Immunosuppression is Inappropriately Qualifying the Immune Status of Septic and SIRS Patients. Shock (2019) 52:307–17. doi: 10.1097/SHK.0000000000001266
10. Karnatovskaia LV, Festic E. Sepsis: A Review for the Neurohospitalist. Neurohospitalist (2012) 2:144–53. doi: 10.1177/1941874412453338
11. Savva A, Roger T. Targeting Toll-Like Receptors: Promising Therapeutic Strategies for the Management of Sepsis-Associated Pathology and Infectious Diseases. Front Immunol (2013) 4:387. doi: 10.3389/fimmu.2013.00387
12. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The Immunopathology of Sepsis and Potential Therapeutic Targets. Nat Revs Immunol (2017) 17:407. doi: 10.1038/nri.2017.36
13. Ma YJ, Garred P. Pentraxins in Complement Activation and Regulation. Front Immunol (2018) 9:3046. doi: 10.3389/fimmu.2018.03046
14. Porte R, Davoudian S, Asgari F, Parente R, Mantovani A, Garlanda C, et al. The Long Pentraxin PTX3 as a Humoral Innate Immunity Functional Player and Biomarker of Infections and Sepsis. Front Immunol (2019) 10:794. doi: 10.3389/fimmu.2019.00794
15. Moalli F, Paroni M, Rodriguez TV, Riva F, Polentarutti N, Bottazzi B, et al. The Therapeutic Potential of the Humoral Pattern Recognition Molecule PTX3 in Chronic Lung Infection Caused by Pseudomonas Aeruginosa. J Immunol (2011) 186:5425–34. doi: 10.4049/jimmunol.1002035
16. Bottazzi B, Doni A, Garlanda C, Mantovani A. An Integrated View of Humoral Innate Immunity: Pentraxins as a Paradigm. Ann Rev Immunol (2009) 28:157–83. doi: 10.1146/annurev-immunol-030409-101305
17. Garlanda C, Jaillon S, Doni A, Bottazzi B, Mantovani A. PTX3, a Humoral Pattern Recognition Molecule At the Interface Between Microbe and Matrix Recognition. Curr Opin Immunol (2016) 38:39–44. doi: 10.1016/j.coi.2015.11.002
18. Paroni M, Moalli F, Nebuloni M, Pasqualini F, Bonfield T, Nonis A, et al. Response of CFTR-deficient Mice to Long-Term Chronic Pseudomonas Aeruginosa Infection and PTX3 Therapy. J Infect Dis (2013) 208:130–8. doi: 10.1093/infdis/jis636
19. Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, et al. Regulation of Leukocyte Recruitment by the Long Pentraxin PTX3. Nat Immunol (2010) 11:328–34. doi: 10.1038/ni.1854
20. Garlanda C, Bottazzi B, Magrini E, Inforzato A, Mantovani A. PTX3, a Humoral Pattern Recognition Molecule, in Innate Immunity, Tissue Repair, and Cancer. Physiol Rev (2018) 98:623–39. doi: 10.1152/physrev.00016.2017
21. Han B, Ma X, Zhang J, Zhang Y, Bai X, Hwang DM, et al. Protective Effects of Long Pentraxin PTX3 on Lung Injury in a Severe Acute Respiratory Syndrome Model in Mice. Lab Invest (2012) 92:1285–96. doi: 10.1038/labinvest.2012.92
22. Jaillon S, Moalli F, Ragnarsdottir B, Bonavita E, Puthia M, Riva F, et al. The Humoral Pattern Recognition Molecule PTX3 is a Key Component of Innate Immunity Against Urinary Tract Infection. Immunity (2014) 40:621–32. doi: 10.1016/j.immuni.2014.02.015
23. Lech M, Rommele C, Grobmayr R, Eka Susanti H, Kulkarni OP, Wang S, et al. Endogenous and Exogenous Pentraxin-3 Limits Postischemic Acute and Chronic Kidney Injury. Kidney Int (2013) 83:647–61. doi: 10.1038/ki.2012.463
24. Doni A, Musso T, Morone D, Bastone A, Zambelli V, Sironi M, et al. An Acidic Microenvironment Sets the Humoral Pattern Recognition Molecule PTX3 in a Tissue Repair Mode. J Exp Med (2015) 212:905–25. doi: 10.1084/jem.20141268
25. Salio M CS, De Angelis N, Molla F, Maina V, Nebuloni M, Pasqualini F, et al. Cardioprotective Function of the Long Pentraxin PTX3 in Acute Myocardial Infarction. Circulation (2008) 117:10551064. doi: 10.1161/CIRCULATIONAHA.107.749234
26. Soares AC, Souza DG, Pinho V, Vieira AT, Nicoli JR, Cunha FQ, et al. Dual Function of the Long Pentraxin PTX3 in Resistance Against Pulmonary Infection With Klebsiella Pneumoniae in Transgenic Mice. Microbes Infect (2006) 8:1321–29. doi: 10.1016/j.micinf.2005.12.017
27. Jeannin P, Bottazzi B, Sironi M, Doni A, Rusnati M, Presta M, et al. Complexity and Complementarity of Outer Membrane Protein A Recognition by Cellular and Humoral Innate Immunity Receptors. Immunity (2005) 22:551–60. doi: 10.1016/j.immuni.2005.03.008
28. Garlanda C, Hirsch E, Bozza S, Salustri A, De Acetis M, Nota R, et al. Non-Redundant Role of the Long Pentraxin PTX3 in Anti-Fungal Innate Immune Response. Nature (2002) 420:182. doi: 10.1038/nature01195
29. Bottazzi B, Vouret-Craviari V, Bastone A, De Gioia L, Matteucci C, Peri G, et al. Multimer Formation and Ligand Recognition by the Long Pentraxin PTX3. Similarities and Differences With the Short Pentraxins C-reactive Protein and Serum Amyloid P Component. J Biol Chem (1997) 272:32817–23. doi: 10.1074/jbc.272.52.32817
30. Doni A, Mantovani G, Porta C, Tuckermann J, Reichardt HM, Kleiman A, et al. Cell-Specific Regulation of PTX3 by Glucocorticoid Hormones in Hematopoietic and Nonhematopoietic Cells. J Biol Chem (2008) 283:29983–92. doi: 10.1074/jbc.M805631200
31. Perlee D, de Vos AF, Scicluna BP, Mancheño P, De la Rosa O, Dalemans W, et al. Human Adipose-Derived Mesenchymal Stem Cells Modify Lung Immunity and Improve Antibacterial Defense in Pneumosepsis Caused by Klebsiella Pneumoniae. Stem Cells Transl Med (2019) 8:785–96. doi: 10.1002/sctm.18-0260
32. Ding C, Yang J, van’t Veer C, van der Poll T. Bradykinin Receptor Deficiency or Antagonism do Not Impact the Host Response During Gram-Negative Pneumonia-Derived Sepsis. Intensive Care Med Exp (2019) 7:14. doi: 10.1186/s40635-019-0228-3
33. Jaillon S, Peri G, Delneste Y, Frémaux I, Doni A, Moalli F, et al. The Humoral Pattern Recognition Receptor PTX3 is Stored in Neutrophil Granules and Localizes in Extracellular Traps. J Exp Med (2007) 204:793–804. doi: 10.1084/jem.20061301
34. Moalli F, Doni A, Deban L, Zelante T, Zagarella S, Bottazzi B, et al. Role of Complement and Fcγ Receptors in the Protective Activity of the Long Pentraxin PTX3 Against Aspergillus Fumigatus. Blood (2010) 116:5170–80. doi: 10.1182/blood-2009-12-258376
35. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the Activation and Regulation of Innate and Adaptive Immunity. Nat Rev Immunol (2011) 11:519–31. doi: 10.1038/nri3024
36. Foo SS, Reading PC, Jaillon S, Mantovani A, Mahalingam S. Pentraxins and Collectins: Friend or Foe During Pathogen Invasion? Trends Microbiol (2015) 23:799–811. doi: 10.1016/j.tim.2015.09.006
37. Ma YJ, Doni A, Hummelshøj T, Honoré C, Bastone A, Mantovani A, et al. Synergy Between Ficolin-2 and Pentraxin 3 Boosts Innate Immune Recognition and Complement Deposition. J Biol Chem (2009) 284:28263–75. doi: 10.1074/jbc.M109.009225
38. Gout E, Moriscot C, Doni A, Dumestre-Pérard C, Lacroix M, Pérard J, et al. M-Ficolin Interacts With the Long Pentraxin PTX3: A Novel Case of Cross-Talk Between Soluble Pattern-Recognition Molecules. J Immunol (2011) 186:5815–22. doi: 10.4049/jimmunol.1100180
39. Tzouvelekis L, Miriagou V, Kotsakis S, Spyridopoulou K, Athanasiou E, Karagouni E, et al. KPC-Producing, Multidrug-Resistant Klebsiella Pneumoniae Sequence Type 258 as a Typical Opportunistic Pathogen. Antimicrob Agents Chemother (2013) 57:5144–46. doi: 10.1128/AAC.01052-13
40. Simmons J, Pittet J-F. The Coagulopathy of Acute Sepsis. Curr Op Anaesthesiol (2015) 28:227. doi: 10.1097/ACO.0000000000000163
41. Levi M, de Jonge E, van der Poll T. Sepsis and Disseminated Intravascular Coagulation. J Thromb Thrombolysis (2003) 16:43–7. doi: 10.1023/B:THRO.0000014592.27892.11
42. Doni A, Stravalaci M, Inforzato A, Magrini E, Mantovani A, Garlanda C, et al. The Long Pentraxin PTX3 as a Link Between Innate Immunity, Tissue Remodeling, and Cancer. Front Immunol (2019) 10:1–13. doi: 10.3389/fimmu.2019.00712
43. Raredon MSB, Adams TS, Suhail Y, Schupp JC, Poli S, Neumark N, et al. Single-Cell Connectomic Analysis of Adult Mammalian Lungs. Sci Adv (2019) 5:eaaw3851. doi: 10.1126/sciadv.aaw3851
44. Tabula Muris Consortium, Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis, et al. Single-cell Transcriptomics of 20 Mouse Organs Creates a Tabula Muris. Nature (2018) 562:367–72. doi: 10.1038/s41586-018-0590-4
45. Ciancarella V, Lembo-Fazio L, Paciello I, Bruno A-K, Jaillon S, Berardi S, et al. Role of a Fluid-Phase PRR in Fighting an Intracellular Pathogen: PTX3 in Shigella Infection. PloS Pathog (2018) 14:e1007469. doi: 10.1371/journal.ppat.1007469
46. Salvatori G, Campo S. Current Understanding of PTX3 Protective Activity on Aspergillus Fumigatus Infection. Med Mycol (2012) 50:225–33. doi: 10.3109/13693786.2011.648215
47. Deban L, Jarva H, Lehtinen MJ, Bottazzi B, Bastone A, Doni A, et al. Binding of the Long Pentraxin PTX3 to Factor H: Interacting Domains and Function in the Regulation of Complement Activation. J Immunol (2008) 181:8433–40. doi: 10.4049/jimmunol.181.12.8433
48. Ristagno G, Fumagalli F, Bottazzi B, Mantovani A, Olivari D, Novelli D, et al. Pentraxin 3 in Cardiovascular Disease. Front Immunol (2019) 10:823. doi: 10.3389/fimmu.2019.00823
49. Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P, et al. PTX3 is an Extrinsic Oncosuppressor Regulating Complement-Dependent Inflammation in Cancer. Cell (2015) 160:700–14. doi: 10.1016/j.cell.2015.01.004
50. Magrini E, Mantovani A, Garlanda C. The Dual Complexity of PTX3 in Health and Disease: A Balancing Act? Trends Mol Med (2016) 22:497–510. doi: 10.1016/j.molmed.2016.04.007
51. Hotchkiss RS, Karl IE. The Pathophysiology and Treatment of Sepsis. N Engl J Med (2003) 348:138–50. doi: 10.1056/NEJMra021333
52. Tidswell R, Singer M. Sepsis–Thoughtful Management for the Non-Expert. Clin Med (2018) 18:62. doi: 10.7861/clinmedicine.18-1-62
53. Pechous RD. With Friends Like These: The Complex Role of Neutrophils in the Progression of Severe Pneumonia. Front Cell Infect Microbiol (2017) 7:160. doi: 10.3389/fcimb.2017.00160
54. Camicia G, Pozner R, de Larranaga G. Neutrophil Extracellular Traps in Sepsis. Shock (2014) 42:286–94. doi: 10.1097/SHK.0000000000000221
55. De Stoppelaar S, Van’t Veer C, Roelofs J, Claushuis T, De Boer O, Tanck M, et al. Platelet and Endothelial Cell P-selectin are Required for Host Defense Against Klebsiella Pneumoniae-Induced Pneumosepsis. J Thromb Haemost (2015) 13:1128–38. doi: 10.1111/jth.12893
56. Cappuzzello C, Doni A, Dander E, Pasqualini F, Nebuloni M, Bottazzi B, et al. Mesenchymal Stromal Cell-Derived PTX3 Promotes Wound Healing Via Fibrin Remodeling. J Invest Dermatol (2016) 136:293–300. doi: 10.1038/JID.2015.346
57. Bonacina F, Barbieri SS, Cutuli L, Amadio P, Doni A, Sironi M, et al. Vascular Pentraxin 3 Controls Arterial Thrombosis by Targeting Collagen and Fibrinogen Induced Platelets Aggregation. Biochim Biophys Acta (2016) 1862:1182–90. doi: 10.1016/j.bbadis.2016.03.007
58. Doni A, Garlanda C, Mantovani A. Innate Immunity, Hemostasis and Matrix Remodeling: PTX3 as a Link. Sem Immunol (2016) 28:570–77. doi: 10.1016/j.smim.2016.10.012
59. Mauri T, Zambelli V, Cappuzzello C, Bellani G, Dander E, Sironi M, et al. Intraperitoneal Adoptive Transfer of Mesenchymal Stem Cells Enhances Recovery From Acid Aspiration Acute Lung Injury in Mice. Intensive Care Med Exp (2017) 5:13. doi: 10.1186/s40635-017-0126-5
60. Claushuis T, De Stoppelaar S, Stroo I, Roelofs J, Ottenhoff R, Van der Poll T, et al. Thrombin Contributes to Protective Immunity in Pneumonia-Derived Sepsis Via Fibrin Polymerization and Platelet–Neutrophil Interactions. J Thromb Haemostasis (2017) 15:744–57. doi: 10.1111/jth.13625
61. Winer LK, Salyer C, Beckmann N, Caldwell CC, Nomellini V. Enigmatic Role of Coagulopathy Among Sepsis Survivors: A Review of Coagulation Abnormalities and Their Possible Link to Chronic Critical Illness. Trauma Surg Acute Care Open (2020) 5:e000462. doi: 10.1136/tsaco-2020-000462
62. Cunha C, Aversa F, Lacerda JF, Busca A, Kurzai O, Grube M, et al. Genetic PTX3 Deficiency and Aspergillosis in Stem-Cell Transplantation. N Engl J Med (2014) 370:421–32. doi: 10.1056/NEJMoa1211161
Keywords: Pentraxin 3 (PTX3), Klebsiella pneumoniae, sepsis, innate immunity, inflammation, complement – immunological term
Citation: Asgari F, Supino D, Parente R, Polentarutti N, Stravalaci M, Porte R, Pasqualini F, Barbagallo M, Perucchini C, Recordati C, Magrini E, Mariancini A, Riva F, Giordano A, Davoudian S, Roger T, Veer C, Jaillon S, Mantovani A, Doni A and Garlanda C (2021) The Long Pentraxin PTX3 Controls Klebsiella Pneumoniae Severe Infection. Front. Immunol. 12:666198. doi: 10.3389/fimmu.2021.666198
Received: 09 February 2021; Accepted: 04 May 2021;
Published: 20 May 2021.
Edited by:
Ivan Zanoni, Harvard Medical School, United StatesReviewed by:
Laura Marongiu, University of Milano-Bicocca, ItalyAchille Broggi, Boston Children’s Hospital and Harvard Medical School, United States
Copyright © 2021 Asgari, Supino, Parente, Polentarutti, Stravalaci, Porte, Pasqualini, Barbagallo, Perucchini, Recordati, Magrini, Mariancini, Riva, Giordano, Davoudian, Roger, Veer, Jaillon, Mantovani, Doni and Garlanda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecilia Garlanda, Y2VjaWxpYS5nYXJsYW5kYUBodW1hbml0YXNyZXNlYXJjaC5pdA==