 Azzeddine Tahiat1*
Azzeddine Tahiat1* Abdelghani Yagoubi2Mohamed Samir Ladj3Reda Belbouab3
Abdelghani Yagoubi2Mohamed Samir Ladj3Reda Belbouab3 Samira Aggoune4Laziz Atek4Djamila Bouziane5Souhila Melzi6
Samira Aggoune4Laziz Atek4Djamila Bouziane5Souhila Melzi6 Chahinez Boubidi7
Chahinez Boubidi7 Warda Drali8Chafa Bendahmane9Hamza Iguerguesdaoune1Sihem Taguemount1
Warda Drali8Chafa Bendahmane9Hamza Iguerguesdaoune1Sihem Taguemount1 Asma Soufane1Asma Oukil1Abdalbasset Ketfi10Hassen Messaoudi11Nadia Boukhenfouf12
Asma Soufane1Asma Oukil1Abdalbasset Ketfi10Hassen Messaoudi11Nadia Boukhenfouf12 Mohamed Amine Ifri13
Mohamed Amine Ifri13 Tahar Bencharif Madani14Hayet Belhadj15Keltoum Nafissa Benhala16Mokhtar Khiari16Nacera Cherif17Leila Smati18Zakia Arada8Zoulikha Zeroual7Zair Bouzerar6Ouardia Ibsaine5Hachemi Maouche4
Tahar Bencharif Madani14Hayet Belhadj15Keltoum Nafissa Benhala16Mokhtar Khiari16Nacera Cherif17Leila Smati18Zakia Arada8Zoulikha Zeroual7Zair Bouzerar6Ouardia Ibsaine5Hachemi Maouche4 Rachida Boukari3
Rachida Boukari3 Kamel Djenouhat1
Kamel Djenouhat1- 1Department of Medical Biology, Rouiba Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
- 2Pediatric Gastroenterology, Centre Algérois de Pédiatrie, Algiers, Algeria
- 3Department of Pediatrics, Mustapha University Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
- 4Department of Pediatrics, El-Harrach Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
- 5Department of Pediatrics, Ain Taya Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
- 6Department of Pediatrics, Bab El-Oued University Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
- 7Department of Pediatrics A, Hussein Dey University Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
- 8Department of Pediatrics B, Hussein Dey University Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
- 9Department of Pediatrics, Meftah Hospital, Blida, Algeria
- 10Department of Pneumology, Rouiba Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
- 11Department of Internal Medicine, Mustapha University Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
- 12Department of Pediatrics, Rouiba Hospital, Algiers, Algeria
- 13Department of Pediatrics, Thenia Hospital, Boumerdes, Algeria
- 14Department of Pediatrics, Mansourah Hospital, Constantine, Algeria
- 15Department of Pediatrics, Central Hospital of the Army, Algiers, Algeria
- 16Department of Pediatrics A, Beni Messous University Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
- 17Department of Pediatrics B, Beni Messous University Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
- 18Department of Pediatrics, Bologhine Hospital, Algiers Faculty of Medicine, University of Algiers 1, Algiers, Algeria
Objectives: To evaluate the diagnostic and predictive contribution of autoantibodies screening in patients with primary immunodeficiencies (PIDs).
Methods: In the present study, PID patients and healthy controls have been screened for 54 different autoantibodies. The results of autoantibodies screening in PID patients were correlated to the presence of autoimmune diseases.
Results: A total of 299 PID patients were included in this study with a predominance of antibody deficiencies (27.8%) followed by immunodeficiencies affecting cellular and humoral immunity (26.1%) and complement deficiencies (22.7%). Autoimmune manifestations were present in 82 (27.4%) patients. Autoimmune cytopenia (10.4%) was the most common autoimmune disease followed by gastrointestinal disorders (10.0%), rheumatologic diseases (3.7%), and endocrine disorders (3.3%). Autoantibodies were found in 32.4% of PID patients and 15.8% of healthy controls (P < 0.0005). Anti-nuclear antibodies (ANA) (10.0%), transglutaminase antibody (TGA) (8.4%), RBC antibodies (6.7%), anti-smooth muscle antibody (ASMA) (5.4%), and ASCA (5.0%) were the most common autoantibodies in our series. Sixty-seven out of the 82 patients with autoimmune manifestations (81.7%) were positive for one or more autoantibodies. Eleven out of the 14 patients (78.6%) with immune thrombocytopenia had positive platelet-bound IgM. The frequencies of ASCA and ANCA among patients with IBD were 47.4% and 21.0% respectively. All patients with celiac disease had TGA-IgA, while six out of the 11 patients with rheumatologic diseases had ANA (54.5%). Almost one third of patients (30/97) with positive autoantibodies had no autoimmune manifestations. ANA, rheumatoid factor, ASMA, anti-phospholipid antibodies and ANCA were often detected while specific AID was absent. Despite the low positive predictive value of TGA-IgA and ASCA for celiac disease and inflammatory bowel disease respectively, screening for these antibodies identified undiagnosed disease in four patients with positive TGA-IgA and two others with positive ASCA.
Conclusion: The present study provides valuable information about the frequency and the diagnostic/predictive value of a large panel of autoantibodies in PIDs. Given the frequent association of some AIDs with certain PIDs, screening for corresponding autoantibodies would be recommended. However, positivity for autoantibodies should be interpreted with caution in patients with PIDs due to their low positive predictive value.
Introduction
Primary immunodeficiencies (PIDs) are a heterogeneous group of genetic disorders that affect distinct components of both humoral and cellular arms of the immune system (1). PIDs are no longer defined by recurrent infections alone. Patients with such disorders are increasingly recognized with features of immune dysregulation, including autoimmunity and inflammation (2–5). In a retrospective study of the French Registry, authors have reported that more than 26% of patients with PIDs developed one or more autoimmune or inflammatory manifestations throughout their lifetime (6). The risk for autoimmune diseases (AID) was at least 10 times higher than in the general population (6). Mechanisms underlying the development of autoimmunity in PIDs include defective T and B cell development and tolerance, defective regulatory T cell (Treg) development or function, increased type I interferon signature and lack of clearance of immune complexes and apoptotic debris (7).
The presence of serum autoantibodies directed to multiple cell surface and intracellular antigens is a serological hallmark of autoimmune diseases and a helpful biomarker for establishing an early and accurate diagnosis. Autoimmunity has been widely studied in PIDs. However, most of published studies were focused on clinical manifestations and only few is known about the clinical relevance of autoantibody testing in such monogenic defects (6, 8). In the present study, a large series of patients with PIDs have been screened for a broad panel of autoantibodies. The study’s main objective was to evaluate the diagnostic and predictive contribution of autoantibodies screening in patients with PIDs. A secondary goal was to report the frequency of autoantibodies in different PID categories.
Material and Methods
Patients and Healthy Controls
In the present study, 299 Algerian patients with PIDs have been enrolled in two-year period (January 2018 to January 2020). All patients met the updated criteria of the European Society for Immunodeficiency (ESID) (www.esid.org). Secondary immunodeficiencies were ruled out for each patient. Patients were categorized according to the International Union of Immunological Societies (IUIS), Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity (2019) (1). In addition, the study included 120 healthy subjects (70 children and 50 adults) as a control group. The study was approved by the local ethics Committee and it conforms to the provisions of the World Medical Association’s Declaration of Helsinki.
Diagnosis of Autoimmune Diseases
Data on autoimmune manifestations were collected and analyzed for each patient. The diagnosis of AID was based on clinical and complementary paraclinical findings (ex., radiology, endoscopy, colonoscopy and biopsy results) and laboratory tests (ex., Coombs test, antinuclear antibodies, and transglutaminase antibody), according to international criteria for a specific disease. For instance, patients who were diagnosed with autoimmune hemolytic anemia (AIHA) presented with anemia that was associated with a positive Coombs test.
Autoantibody Testing
Patients and healthy controls were systematically screened for a broad panel of autoantibodies (54 distinct autoantibodies) that included : anti-nuclear antibodies (ANA), rheumatoid factor (RF), anti-citrullinated protein antibody (ACPA), anti-neutrophil cytoplasmic antibodies (ANCA), anti-phospholipid antibodies (aPL), anti-tissue antibodies (such as anti-smooth muscle antibody (ASMA)), anti-saccharomyces cerevisiae antibody (ASCA), transglutaminase antibody (TGA), red blood cell (RBC) antibodies, platelet-bound antibodies (PA) and anti-neutrophil antibodies (Table 1).

Table 1 Immunoassays used for antibody testing.
ANA screening was performed at a dilution of 1:80 by indirect immunofluorescence (IIF) using HEp-2 cell line as substrate. For positive sera, nuclear and cytoplasmic staining patterns were read by two experts and ANA specificities, including dsDNA, extractable nuclear antigens (ENA), nucleolar antigens and cytoplasmic antigens were identified by enzyme-like immunosorbent assay (ELISA) and/or Immunodot. IIF and ELISA have been used for ANCA screening and identification, respectively. Detection of RBC antibodies was performed by Coombs test. PA-IgG, PA-IgM and PA-IgA were detected by flow cytometry, using a commercial kit (THROMBOCYTEST, BD Biosciences, CA, USA), according to the manufacturer’s instructions. Briefly, isolated platelets from EDTA whole blood were incubated with PE-labeled goat Ig anti-human IgG, IgM and IgA to detect platelet-bound antibodies. Binding of the antibodies was measured with a FACSCanto (BD Biosciences, CA, USA). Anti-neutrophil antibodies (IgG and IgM) were detected by indirect granulocyte immunofluorescence test (GIFT) as described previously (9). Briefly, a mixture of fresh anticoagulated blood samples obtained from 10 healthy subjects was used as a source of neutrophils. The rationale for using multiple donors was to obtain a good representation of two common human neutrophil antigens (HNA) alloforms 1 and 2. The mixed blood sample was treated with ammonium chloride-based RBC-lysing reagent. The cells were thoroughly washed, resuspended and incubated at room temperature with the patients’ sera for 30 minutes, washed twice and incubated for 15 minutes with FICT-conjugated anti-human IgG and IgM rabbit F(ab’)2 antibody fragment.
Statistical Analysis
The comparison of frequencies was performed by chi-square test or Fisher’s exact test. The threshold for statistical significance was set to a P value of less than 0.05 in all analyses. The statistical analyses were performed using SPSS version 23.0 (software package (IBM), Chicago, IL, USA).
Results
Demographic Data
The demographic data of PID patients and healthy controls are shown in Table 2. Of the 299 patients included in this study, 169 (56.5%) were males and 130 (43.5%) were females. The mean age of patients was 12.8 years (0.1 – 80 years), the mean age at onset of PID symptoms was 4,2 years (0 – 60 years) and the mean disease duration was 5.7 years (0.1 – 53 years). Two hundred three (68%) patients were children and 96 (32%) were adults. Ninety patients (30.1%) were born from consanguineous parents. Diagnosis of PID was confirmed by immunological testing in all patients and genetically in 60 (20%) patients. The distribution of patients according to the classification from the IUIS Expert Committee is shown in Table 2. Predominantly antibody deficiencies (27.8%) were the most common, followed by immunodeficiencies affecting cellular and humoral immunity (26.1%) and complement deficiencies (22.7%).
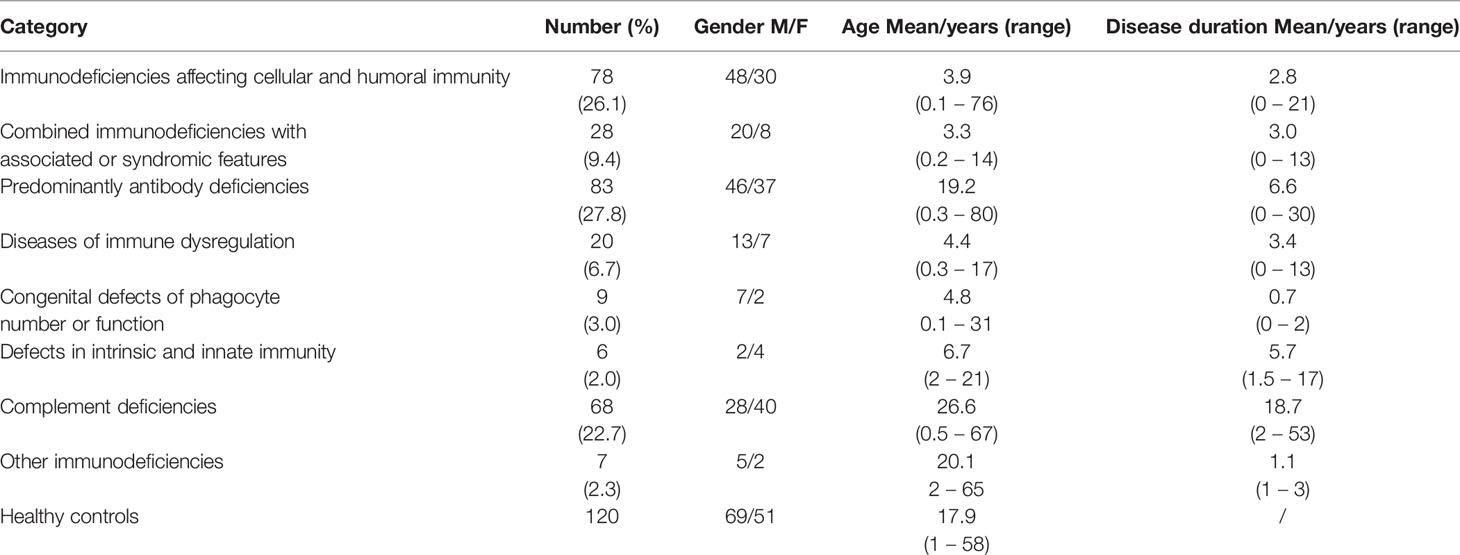
Table 2 Demographic data of PID patients and healthy controls.
Autoimmune Diseases
Autoimmune manifestations were present in 82 (27.4%) patients, including 45 (55%) males and 37 (45%) females. There was no statistically significant association between having AID and gender (P = 0.72). AIDs was more common in patients with immune dysregulation (70.0%), followed by immunodeficiencies affecting cellular and humoral immunity (33.3%) (P < 0.0005). The lowest frequency of AID was seen in patients with complement deficiencies (10.3%) (Table 3). Unlike patients with “classical” severe combined immunodeficiencies (SCIDs), patients with atypical forms of SCID, including Omenn syndrome (OS) (n = 6) and leaky SCID (n = 9) presented a high frequency of AID (14.3% vs. 66.7%; P = 0,006). The frequency of autoimmune manifestations in patients with predominantly antibody deficiencies was 24.1%. Such manifestations were more frequent in patients with common variable immunodeficiencies (CVID) compared to patients with other antibody deficiencies (30.9% vs. 17.1%), but the difference did not reach level of significance (P = 0.14) (Tables 3 and 5).
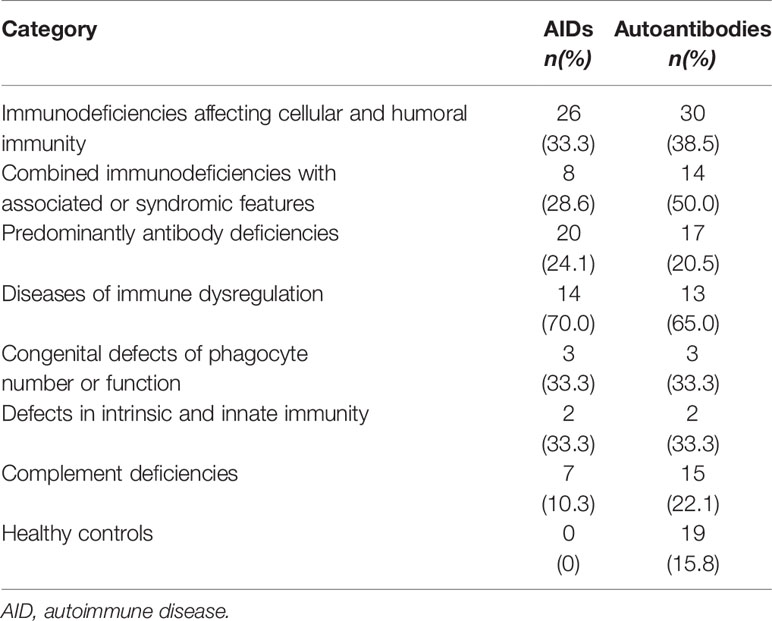
Table 3 AID and autoantibodies in different PID categories and healthy controls.
A wide range of autoimmune manifestations was observed in our series; autoimmune cytopenia (10.4%) was the most common AID followed by gastrointestinal disorders (10.0%), rheumatologic diseases (3.7%), skin manifestations (3.3%) and endocrine disorders (3.3%) (Table 4). The distribution of different AIDs with respect to PID categories is shown in Table 5. The most striking findings are that autoimmune cytopenia and skin diseases were common in patients with combined immunodeficiencies (CID), especially atypical SCID, while IBD and endocrine disorders predominated in patients with diseases of immune dysregulation. IBD was also common in patients with chronic granulomatous disease (CGD).
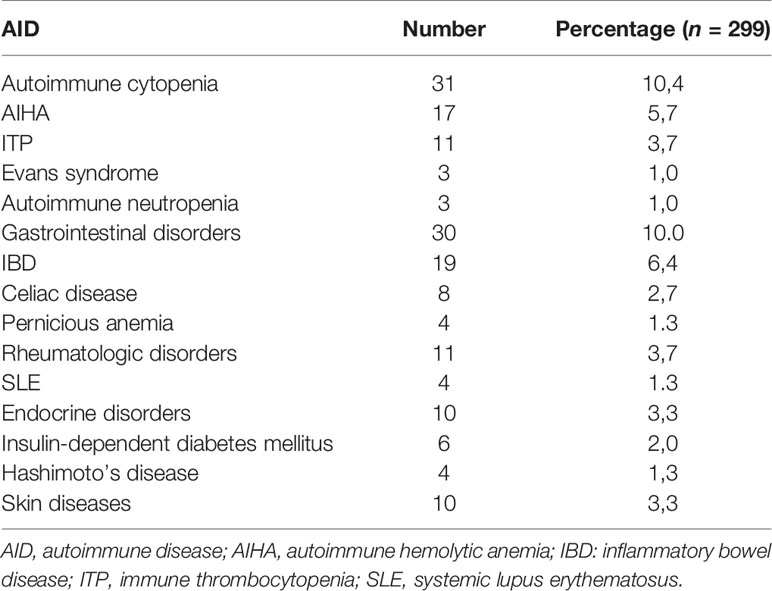
Table 4 Main autoimmune manifestations in our series.
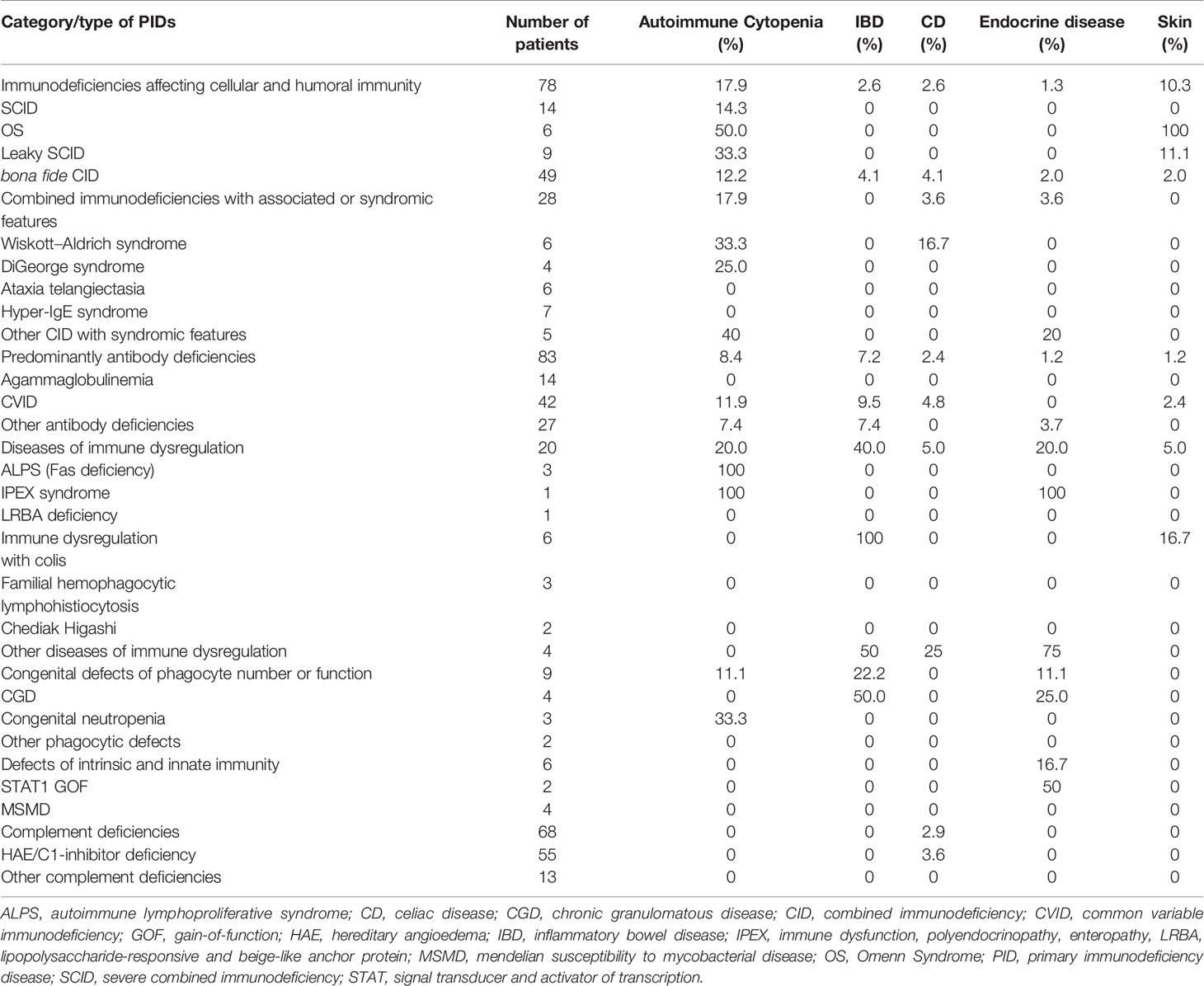
Table 5 Distribution of major autoimmune diseases among different categories/types of PIDs.
Autoantibodies
The results of autoantibody testing in PID patients and healthy controls are shown in Table 3 and 6. One or more autoantibodies were found in 32.4% of PID patients and 15.8% of healthy individuals (P < 0.0005). Among the 97 patients with positive autoantibodies, 58 (60%) were males and 39 (40%) were females. There was no statistically significant association between having autoantibodies and gender (P = 0.54). The distribution of patients according to the number of positive autoantibodies showed that 46 patients (47.4%) were positive for 1 autoantibody, 22 patients (22.7%) were positive for 2 autoantibodies, 15 patients (15.5%) had 3 autoantibodies, while 14 patients (14.4%) developed 4 or more autoantibodies. There was a statistically significant association between having autoantibodies and PID category (P = 0.001). Serum autoantibodies were more frequent in patients with diseases of immune dysregulation followed by T cell immunodeficiencies, including immunodeficiencies affecting cellular and humoral immunity (38.5%) and CID with associated or syndromic features (50%) (Table 3).
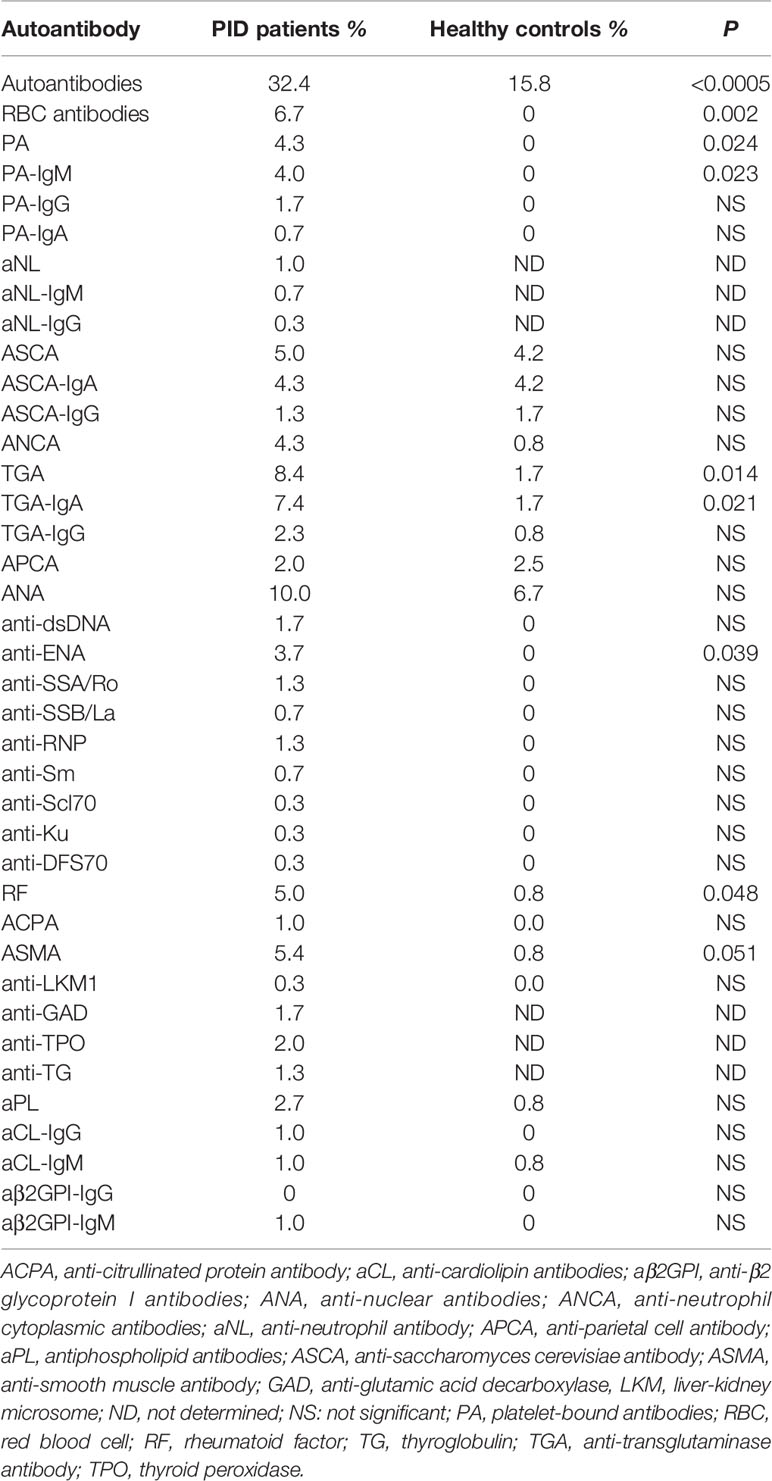
Table 6 Frequencies of different autoantibodies in PID patients and healthy controls.
The most frequent autoantibodies in our series were: ANA (10.0%), TGA (8.4%), RBC antibodies (6.7%), ASMA (5.4%), RF (5.0%), ASCA (5.0%), ANCA (4.3%), and PA (4.3%). RBC antibodies (P = 0.002), PA-IgM (P = 0.023), TGA-IgA (P = 0.021), anti-ENA antibodies (P = 0.039), and RF (P = 0.048) were significantly more frequent in PID patients when compared to healthy controls (Table 6). The distribution of different autoantibodies with respect to PID categories is shown in Table 7. The frequencies of most tested autoantibodies were higher in patients with diseases of immune dysregulation (Table 7). However, when considering PID patients with increased susceptibility to infections, and after excluding patients who have defects of phagocyte number or function, and defects in intrinsic and innate immunity due to low numbers, the frequency of RBC antibodies was significantly higher in patients with T cell immunodeficiency (P = 0.003). The frequency of PA was also higher in patients with T cell immunodeficiency but the difference did not reach level of significance (P = 0.26). There was no statistically significant association between having ANA, ASCA, ANCA and TGA-IgA and PID category. However, it is interesting to note that TGA-IgA (8.8%) was common in patients with complement deficiencies, especially in patients with hereditary angioedema due to C1-inhibitor deficiency (HAE/C1-inhibitor deficiency) (9.1%) (Table 7).
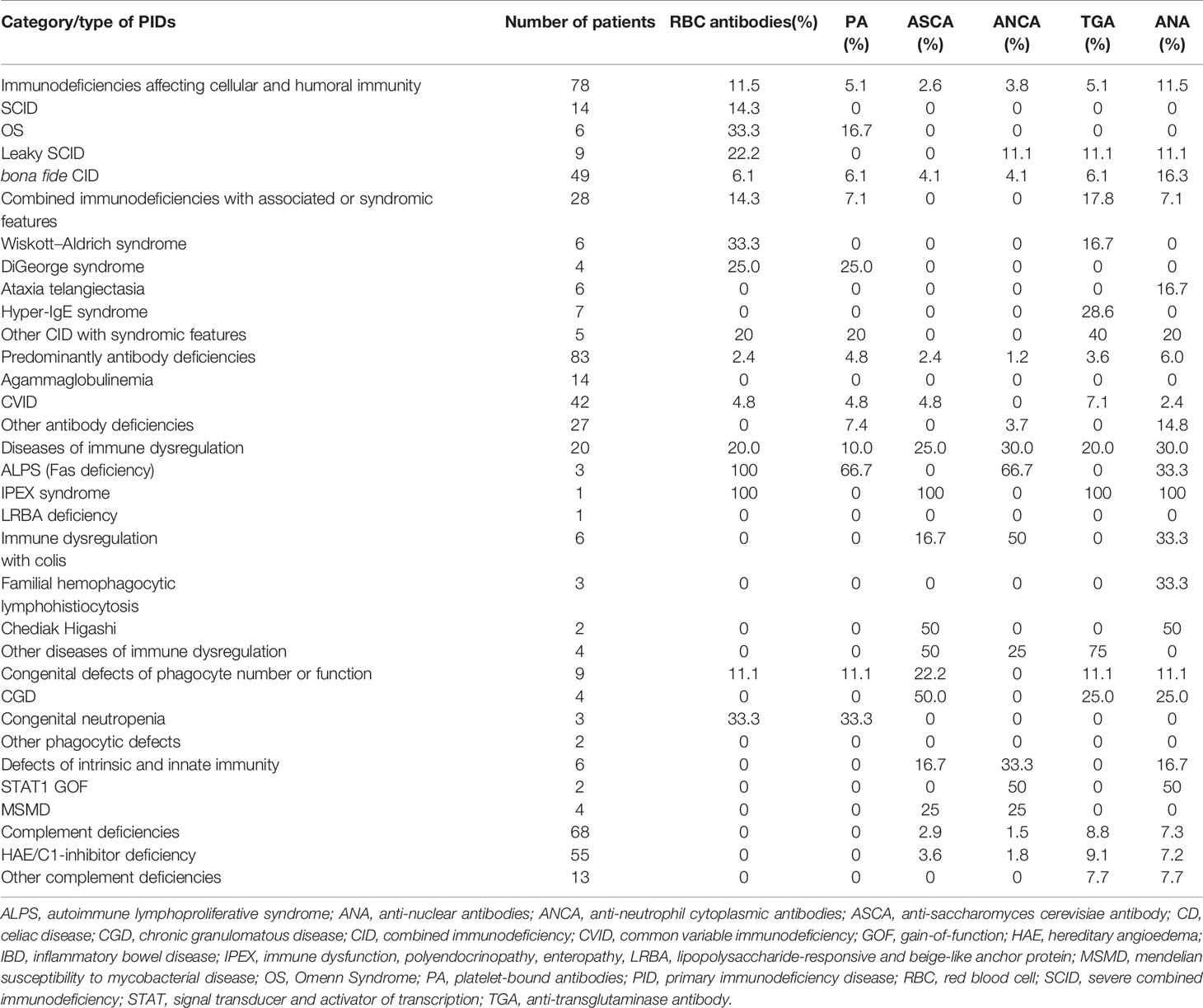
Table 7 Distribution of autoantibodies among different categories/types of PIDs.
Sixty-seven out of the 82 patients (81.7%) with autoimmune manifestations were positive for one or more autoantibodies. All patients with AIHA had positive Coombs test. Twelve out of the 14 patients (85.7%) with immune thrombocytopenia (ITP) had positive PA. Among these patients, six (50%) were positive for PA-IgM, three (25%) for PA-IgM and PA-IgG, two (16.7%) for PA-IgM and PA-IgA and one (8.3%) for PA-IgG. Interestingly, PA-IgM and PA-IgG were present in a patient with autoimmune lymphoproliferative syndrome (ALPS) (due to homozygous mutation in FAS) and normal platelet count (188000 per uL). IgM (n = 2) and IgG (n = 1) anti-neutrophil have been detected by indirect GIFT in three patients with autoimmune neutropenia. The prevalence of ASCA and ANCA among patients with IBD (n = 19) were 47.4% and 21.0% respectively. All patients with celiac disease (CD) (n = 8) had TGA-IgA, while none of them had TGA-IgG. Among patients with rheumatologic diseases (n = 11), six had ANA (54.5%), two were positive for RF (18.2%) while none had ACPA.
Almost one third of patients (31%) with positivity for one or more autoantibodies had no autoimmune manifestations. ANA, RF, ASMA, aPL and ANCA were often detected while specific AID was absent (Figure 1). Thus, only 20% of patients with positivity for ANA had rheumatologic disease. Such AID was found in only 13.3% of patients with RF. Similarly, 18.7% of patients with positive ASMA had autoimmune hepatitis (AIH) and none of the eight patients who were positive for aPL had anti-phospholipid syndrome (APS).
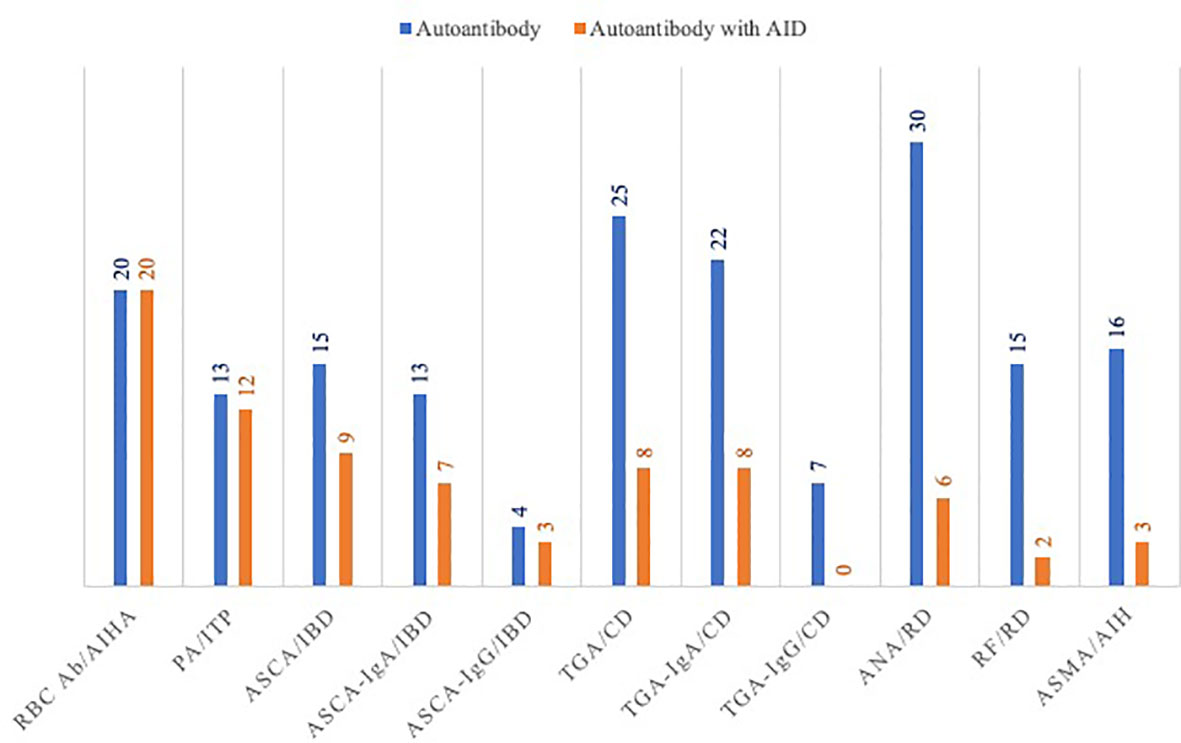
Figure 1 Autoimmune diseases in patients with positive autoantibodies. AIHA, autoimmune hemolytic anemia; ANA, anti-nuclear antibodies; ASCA, anti-saccharomyces cerevisiae antibody; ASMA, anti-smooth muscle antibody; CD, celiac disease; IBD: inflammatory bowel disease; PA, platelet-bound antibodies; RBC Ab, red blood cell antibodies; RD, rheumatoid disease; TGA, anti-transglutaminase antibody.
Eight out of the 22 patients (36.4%) with positive TGA-IgA had CD. The screening of TGA-IgA in our series identified four earlier undiagnosed CD. The diagnostic was confirmed by a biopsy in two patients with CID and CVID and by a “no-biopsy approach” (i.e., TGA-IgA values ≥10 times the upper limit of normal and positive endomysial antibodies (EMA-IgA) in a second serum sample) in two patients with HAE/C1-inhibitor deficiency. Nine out of the 15 patients (60%) with positive ASCA (IgA and or IgG) had IBD. The screening for ASCA identified two undiagnosed IBD in two patients with CGD and CVID.
Discussion
In the present study, a large series of Algerian patients with PIDs were systematically tested for a broad panel of autoantibodies. The results of autoantibodies screening were correlated to the presence of autoimmune manifestations. Although the occurrence of autoimmunity in patients with PIDs has been previously reported, to the best of our knowledge, this is the first report to evaluate the diagnostic and predictive values of autoantibodies for AIDs in the context of PIDs. Moreover this is the first systematic report of autoimmunity and autoantibody profile in patients with PIDs from Africa.
A total of 299 Algerian patients with 60 different PIDs, belonging to 7 different categories of IUIS classification, were included in this study. Autoimmune manifestations were present in 27.4% of patients. A similar prevalence (26.2%) has been reported in a previous study of the French National Primary Immunodeficiencies Registry (6). However, the frequency of AIDs in the Slovenian (22%) and Kuwaiti (20%) studies were lower (8, 10). Many factors, such as ethnicity, environment, cohort size, age of patients, disease duration, and access to hematopoietic stem cell transplantation could have contributed to such differences between series. In our study, AIDs were more common in patients with immune dysregulation (70.0%) followed by immunodeficiencies affecting cellular and humoral immunity (33.3%). Although autoimmunity was less prevalent in patients with complement deficiencies, it is worth to be mentioned that six out of the 55 patients (10.9%) with HAE/C1-inhibitor deficiency (which is the most common complement deficiency in our series) had autoimmune diseases, including two patients (3.6%) with systemic lupus erythematosus and two others (3.6%) with CD.
Autoimmune cytopenia (10.4%) was the most common autoimmune manifestation in our series. This finding is in accordance with the French and the Kuwaiti studies and emphasize the importance of recognizing autoimmune cytopenia as ‘warning sign’ of immunodeficiency (especially in children) that should trigger an immune evaluation. Furthermore, gastrointestinal disorders (10.0%), rheumatologic diseases (3.7%) and endocrine disorders (3.3%) were also common in our series. This is comparable to the percentages found in the French study (9.5%, 5.0% and 3.2%, respectively) (6, 8). However, skin manifestations, which have been observed in 3.3% of our patients were more frequent in the French registry (5.5%) (6).
In our series, autoantibodies were present in 97 patients (32.4%), they were significantly more frequent than in the control group (P < 0.0005). Among patients with positive autoantibodies, 67 (69%) had AIDs while 30 (31%) did not exhibit any autoimmune manifestation. The presence of autoantibodies was a common feature of all types of PIDs. However, their prevalence was significantly higher in patients with defects of immune dysregulation. In our study, all patients with ALPS, lipopolysaccharide-responsive and beige-like anchor protein (LRBA) deficiency and immune dysfunction, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome developed autoantibodies, highlighting the role of B-cell homeostasis (Fas deficiency), Treg (IPEX syndrome) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) surface expression (LRBA deficiency) in the control of autoreactive B cells and autoantibody production. Furthermore, 38.5% of patients with immunodeficiencies affecting cellular and humoral immunity were positive for at least one autoantibody. Although this category is mainly characterized by defects in T-cell development and function (1), aberrant autoantibody production is likely to be related to defects in B-cell tolerance and function. In recombination-activating gene (RAG) deficiency, the presence of a wide spectrum of autoantibodies, both in patients (11) and in animal models (12, 13) has been attributed to defects in central and peripheral B-cell tolerance. Re-expression of RAG proteins in mice allows receptor editing in bone marrow immature B cells and reduce the frequency of self-reactive B-cells (14).
Red blood cell and platelet-bound antibodies were detected in 6.7% and 4.3% of patients respectively. All patients with AIHA had positive Coombs test, while 85.7% of patients with ITP had positive PA. The detection of PA by flow cytometry is an excellent diagnostic tool for ITP, but it is worth noting that PA could be detected in the absence of ITP. Unlike RBC antibodies, most of detected platelet-bound antibodies were IgM. PA-IgM was present in 78.6% of patients with ITP (mostly children), while PA-IgG was detected in only 28.6% of patients. This finding is in accordance with a recent Danish study, in which PA-IgM were significantly more frequent than PA-IgG in children with ITP (63% vs. 44%, P = 0.03) (15). Schmidt et al have also reported that anti-platelet antibodies were predominantly of the IgM class in children with newly diagnosed ITP (16). In the same study, the authors have noted that IgM responses were present short term without evidence of class-switching to IgG (16). Such responses are likely to be triggered by infections, and molecular mimicry may play a central role in the development of self-directed anti-platelet immunity (17, 18). Furthermore, a pathophysiological role of IgM anti-platelet antibodies in ITP has been demonstrated (16, 19). Unlike IgM anti-erythrocyte autoantibody that promotes anemia through a massive agglutination of RBCs in spleen and liver, PA-IgM induces thrombocytopenia through uptake of opsonized platelets (19, 20). In the mouse, it has been reported that IgM autoantibody-mediated thrombocytopenia was macrophage dependent and the Fcα/μR was required for macrophage uptake of opsonized thrombocytes (21). Complement fixation on IgM might also potentially activate phagocytosis involving complement receptors expressed on macrophages (16). Taken all findings together, childhood ITP unlike chronic adult ITP is mainly IgM-mediated. Patients should be tested for both PA-IgM and PA-IgG in order to improve the sensitivity of the flow cytometric test and to determine the prognosis of the ITP (16).
When considering PID patients with increased susceptibility to infections (i.e., after excluding patients with diseases of immune regulation), RBC antibodies and PA were more common in patients with T-cell immunodeficiency. Of 8 RAG-deficient patients, 3 (37.5%) were positive for RBC antibodies or PA and exhibited autoimmune cytopenia. In previous studies, biallelic RAG1 and RAG2 mutations have been associated with a wide range of clinical and immunological phenotypes (22). While null mutations result in SCID with the absence of T and B lymphocytes (T− B− NK+ SCID) (23), hypomorphic mutations allowing for residual protein function are associated with atypical forms including OS (24–26), and leaky SCID (23, 27). Autoimmune manifestations are rare in RAG-deficient patients presenting with typical SCID. However, cytopenias, in particular AIHA, have been reported in more than half of patients with hypomorphic mutations in RAG1 and RAG2 (28). In our series, none of the two RAG-deficient patients with SCID had autoimmune cytopenia while three out of the 6 patients with hypomorphic mutations (manifesting as OS in 4 patients and leaky SCID in 2 others) presented with autoimmune cytopenias, including AIHA in two patients and ITP in another one.
Inflammatory bowel disease was the most common gastrointestinal disease in our cohort. IBD was more frequent in patients with defects of immune dysregulation (40%), CGD (50%), and CVID (9.5%). The frequencies of ASCA and ANCA among patients with IBD were 47.4% and 21.0%, respectively. ASCA was associated with IBD in 60% of patients. Interestingly, the systematic testing for ASCA identified two undiagnosed IBD, representing 10.5% of total IBD cases. Hence, patients presenting PIDs with a high risk for IBD, such as CGD and CVID, should be tested for ASCA even in the absence of digestive symptoms. In case of positive ASCA, additional investigations (ex., radiology, colonoscopy and biopsy) are necessary to confirm the diagnosis.
One of the most striking results of our study is the high frequency of TGA. We were somewhat surprised to observe that 25 patients (8.4%) were positive for TGA-IgA and/or TGA-IgG (compared to 1.7% in the control group). Among patients with TGA-IgA, only 8 (36.4%) had CD. Despite their low positive predictive value (PPV), TGA-IgA testing identified four earlier undiagnosed CD, representing 50% of total CD cases in our series. Interestingly enough, the screening of HAE patients for TGA-IgA revealed a frequent association of CD with C1-inhibitor deficiency (3.6%). Such association was also reported by a Hungarian study in which 3.1% of patients with HAE had CD (29). Considering the high prevalence of CD among patients with HAE, screening for the former is warranted. In addition, it may help in differential diagnosis as well as in the selection of the most appropriate therapy, which is very important known the similarities between the symptoms of HAE and CD.
In our study, ANA and RF were present in 10% and 5% of patients respectively. However, their PPVs for rheumatologic diseases were very low (20% and 13.3% for ANA and FR, respectively). Similarly, only 18.7% of patients with positive ASMA had AIH and none of the positive patients for aPL had APS. The aberrant autoantibody production in patients with PIDs may be related to several intrinsic and environmental factors (7, 30). Infections would play a central role in triggering autoimmunity and autoantibody production in patients with PIDs. In fact, the presence of autoantibodies such as ANA, RF, ACPA, aPL, ANCA and ASMA has been described as an epiphenomenon in several bacterial and viral infections (ex., Staphylococcus aureus, Mycobacterium tuberculosis and HCV) (30–34).
In conclusion, the present study provides valuable information about the frequency and the diagnostic/predictive value of a large panel of autoantibodies in PIDs. Considering the frequent association of some AIDs with certain PIDs, such as autoimmune cytopenia/Fas and RAG deficiencies, CD/C1-inhibitor deficiency and IBD/CGD, systematic screening for corresponding autoantibodies would be recommended. However, positivity for autoantibodies, especially ANA, RF, ANCA, aPL and ASMA, should be interpreted with caution due to their low positive predictive value.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Local Ethics Committee of Rouiba Hospital, Algiers, Algeria. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
AT: designed the study, conducted flow cytometry assays and autoantibody testing, collected and analyzed data, performed statistical analysis, and wrote the manuscript. AY: contributed to data analysis and interpretation, and edited the manuscript. HI, ST, AS and AO: contributed to autoantibody testing. SL, RBe, SA, LA, DB, SM, CBo, WD, CBe, AK, HMe, NB, AI, TBM, HB, KB, MK, NC, LS, ZA, ZZ, ZB, OI, HMa, and RBo: provided clinical diagnosis of patients. KD: designed and supervised the study, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Raif Geha for his contribution to the identification of genetic defects in our patients and for the critical review of the manuscript. We express our deepest gratitude for the patients and their families to whom our work is dedicated.
References
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40:24–64. doi: 10.1007/s10875-019-00737-x
2. Goyal R, Bulua A, Nikolov N, Schwartzberg P, Siegel R. Rheumatologic and autoimmune manifestations of primary immunodeficiency disorders. Curr Opin Rheumatol (2009) 21:78–84. doi: 10.1097/BOR.0b013e32831cb939
3. Kolhatkar NS, Brahmandam A, Thouvenel CD, Becker-Herman S, Jacobs HM, Schwartz MA, et al. Altered BCR and TLR signals promote enhanced positive selection of autoreactive transitional B cells in Wiskott-Aldrich syndrome. J Exp Med (2015) 212:1663–77. doi: 10.1084/jem.20150585
4. Magnani A, Brosselin P, Beaute J, de Vergnes N, Mouy R, Debre M, et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol (2014) 134:655–62. doi: 10.1016/j.jaci.2014.04.014
5. Hoyt K, Chatila T, Notarangelo L, Hazen M, Janssen E, Henderson L. The immunologic features of patients with early-onset and polyautoimmunity. Clin Immunol (2020) 211:108326. doi: 10.1016/j.clim.2019.108326
6. Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N. members of the CEREDIH French PID study group. Autoimmune inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol (2017) 140:1388–93. doi: 10.1016/j.jaci.2016.12.978
7. Grimbacher B, Warnatz K, Yong P, Korganow A, Peter H. The crossroads of autoimmunity and immunodeficiency: lessons from polygenic traits and monogenic defects. J Allergy Clin Immunol (2016) 37:3–17. doi: 10.1016/j.jaci.2015.11.004
8. Massaad MJ, Zainal M, Al-Herz W. Frequency and Manifestations of autoimmunity among children registered in the Kuwait National Primary Immunodeficiency Registry. Front Immunol (2020) 11:1119. doi: 10.3389/fimmu.2020.01119
9. Sella R, Flomenblit L, Goldstein I, Kaplinsky C. Detection of anti-neutrophil antibodies in autoimmune neutropenia of infancy: a multicenter study. Isr Med Assoc J (2010) 12(2):91–6.
10. Blazina Š, Markelj G, Jeverica AK, Toplak N, Bratanič N, Jazbec J, et al. Autoimmune and Inflammatory Manifestations in 247 Patients with Primary Immunodeficiency-a Report from the Slovenian National Registry. J Clin Immunol (2016) 36(8):764–73. doi: 10.1007/s10875-016-0330-1
11. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest (2015) 125(11):4135–48. doi: 10.1172/JCI91162
12. Cassani B, Poliani PL, Marrella V, Schena F, Sauer AV, Ravanini M, et al. Homeostatic expansion of autoreactive immunoglobulinsecreting cells in the Rag2 mouse model of Omenn syndrome. J Exp Med (2010) 207(7):1525–40. doi: 10.1084/jem.20091928
13. Walter JE, Rucci F, Patrizi L, Recher M, Regenass S, Paganini T, et al. Expansion of immunoglobulin-secreting cells and defects in B cell tolerance in Rag-dependent immunodeficiency. J Exp Med (2010) 207(7):1541–54. doi: 10.1084/jem.20091927
14. Jankovic M, Casellas R, Yannoutsos N, Wardemann H, Nussenzweig MC. RAGs and regulation of autoantibodies. Annu Rev Immunol (2004) 22:485–501. doi: 10.1146/annurev.immunol.22.012703.104707
15. Nielsen OH, Tuckuviene R, Nielsen KR, Rosthøj S. Flow cytometric measurement of platelet-associated immunoglobulin in children with newly diagnosed Immune Thrombocytopenia. Eur J Haematol (2016) 96(4):397–403. doi: 10.1111/ejh.12605
16. Schmidt DE, Heitink-Polle KMJ, Porcelijn L, van der Schoot CE, Vidarsson G, Bruin MCA, et al. Anti-platelet antibodies in childhood immune thrombocytopenia: Prevalence and prognostic implications. M J Thromb Haemost (2020) 18(5):1210–20. doi: 10.1111/jth.14762
17. Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood (2009) 113(26):6511–21. doi: 10.1182/blood-2009-01-129155
18. Heitink-Polle KMJ, Nijsten J, Boonacker CWB, de Haas M, Bruin MCA. Clinical and laboratory predictors of chronic immune thrombocytopenia in children: a systematic review and meta-analysis. Blood (2014) 124(22):3295–307. doi: 10.1182/blood-2014-04-570127
19. Hoemberg M, Stahl D, Schlenke P, Sibrowski W, Pachmann U, Cassens U. The isotype of autoantibodies influences the phagocytosis of antibody-coated platelets in autoimmune thrombocytopenic purpura. Scand J Immunol (2011) 74(5):489–95. doi: 10.1111/j.1365-3083.2011.02600.x
20. Detalle ,L, . Su D, van Rooijen N, Coutelier J-P. Immunoglobulin M antiplatelet autoantibodies from mice immunized with rat platelets induce thrombocytopenia and platelet function impairment. Exp Biol Med (Maywood) (2010) 235:1464–71. doi: 10.1258/ebm.2010.010018
21. Legrain S, Su D, Breukel C, Detalle L, Claassens JW, van der Kaa J, et al. Involvement of Fcα/μ Receptor in IgM Anti-Platelet, but Not Anti-Red Blood Cell Autoantibody Pathogenicity in Mice. J Immunol (2015) 195(9):4171–5. doi: 10.4049/jimmunol.1500798
22. Niehues T, Perez-Becker R, Schuetz C. More than just SCID–the phenotypic range of combined immunodeficiencies associated with mutations in the recombinase activating genes (RAG) 1 and 2. Clin Immunol (2010) 135(2):183–92. doi: 10.1016/j.clim.2010.01.013
23. Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human B cell-negative SCID. Science (1996) 274(5284):97–9. doi: 10.1126/science.274.5284.97
24. Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, et al. Partial V(D)J recombination activity leads to Omenn syndrome. Cell (1998) 93(5):885–96. doi: 10.1016/s0092-8674(00)81448-8
25. de Saint-Basile G, Le Deist F, de Villartay JP, Cerf-Bensussan N, Journet O, Brousse N, et al. Restricted heterogeneity of T lymphocytes in combined immunodeficiency with hypereosinophilia (Omenn’s syndrome). J Clin Invest (1991) 87(4):1352–9. doi: 10.1172/JCI115139
26. Rieux-Laucat F, Bahadoran P, Brousse N, Selz F, Fischer A, Le Deist F, et al. Highly restricted human Tcell repertoire in peripheral blood and tissue-infiltrating lymphocytes in Omenn’s syndrome. J Clin Invest (1998) 102(2):312–21. doi: 10.1172/JCI332
27. Villa A, Sobacchi C, Notarangelo LD, Bozzi F, Abinun M, Abrahamsen TG, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood (2001) 97(1):81–8. doi: 10.1182/blood.v97.1.81
28. Delmonte OM, Schuetz C, Notarangelo LD. RAG Deficiency: Two Genes, Many Diseases. J Clin Immunol (2018) 38(6):646–55. doi: 10.1007/s10875-018-0537-4
29. Csuka D, Kelemen Z, Czaller I, Molnár K, Füst G, Varga L, et al. Association of celiac disease and hereditary angioedema due to C1- inhibitor deficiency. Screening patients with hereditary angioedema for celiac disease: is it worth the effort? Eur J Gastroenterol Hepatol (2011) 23(3):238–44. doi: 10.1097/MEG.0b013e328343d3b2
30. Jara LJ, Medina G, Saavedra MA. Autoimmune manifestations of infections. Curr Opin Rheumatol (2018) 30(4):373–79. doi: 10.1097/BOR.0000000000000505
31. Alam J, Yong CK, Choi Y. Potential role of bacterial infection in autoimmune diseases: a new aspect of molecular mimicry. Immune Netw (2014) 14:7–13. doi: 10.4110/in.2014.14.1.7
32. Chen M, Kallenberg CG. The environment, geoepidemiology and ANCA-associated vasculitides. Autoimmun Rev (2010) 9:A293–8. doi: 10.1016/j.autrev.2009.10.008
33. Sasaki H, Inagaki M, Shioda M, Nagasaka K. Poncet’s disease with high titers of rheumatoid factor and anticitrullinated peptide antibodies mimicking rheumatoid arthritis. J Infect Chemother (2015) 21:65–9. doi: 10.1016/j.jiac.2014.07.015
Keywords: primary immunodeficiencies, autoantibody, screening, autoimmune cytopenia, celiac disease, platelet-bound IgM, transglutaminase antibody
Citation: Tahiat A, Yagoubi A, Ladj MS, Belbouab R, Aggoune S, Atek L, Bouziane D, Melzi S, Boubidi C, Drali W, Bendahmane C, Iguerguesdaoune H, Taguemount S, Soufane A, Oukil A, Ketfi A, Messaoudi H, Boukhenfouf N, Ifri MA, Bencharif Madani T, Belhadj H, Benhala KN, Khiari M, Cherif N, Smati L, Arada Z, Zeroual Z, Bouzerar Z, Ibsaine O, Maouche H, Boukari R and Djenouhat K (2021) Diagnostic and Predictive Contribution of Autoantibodies Screening in a Large Series of Patients With Primary Immunodeficiencies . Front. Immunol. 12:665322. doi: 10.3389/fimmu.2021.665322
Received: 07 February 2021; Accepted: 22 March 2021;
Published: 01 April 2021.
Edited by:
Mohamed-Ridha Barbouche, Pasteur Institute of Tunis, TunisiaReviewed by:
Craig Platt, Boston Children’s Hospital and Harvard Medical School, United StatesMichel J. Massaad, American University of Beirut, Lebanon
Copyright © 2021 Tahiat, Yagoubi, Ladj, Belbouab, Aggoune, Atek, Bouziane, Melzi, Boubidi, Drali, Bendahmane, Iguerguesdaoune, Taguemount, Soufane, Oukil, Ketfi, Messaoudi, Boukhenfouf, Ifri, Bencharif Madani, Belhadj, Benhala, Khiari, Cherif, Smati, Arada, Zeroual, Bouzerar, Ibsaine, Maouche, Boukari and Djenouhat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Azzeddine Tahiat, YXp6ZWRpbmV0YWhpYXRAZ21haWwuY29t