
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 16 November 2021
Sec. NK and Innate Lymphoid Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.664218
This article is part of the Research TopicPulmonary Innate Lymphoid Cells - Gatekeepers of Respiratory HealthView all 14 articles
 Barbara C. Mindt1,2
Barbara C. Mindt1,2 Sai Sakktee Krisna1,3
Sai Sakktee Krisna1,3 Claudia U. Duerr4
Claudia U. Duerr4 Mathieu Mancini1,5
Mathieu Mancini1,5 Lara Richer6
Lara Richer6 Silvia M. Vidal1,5Steven Gerondakis7
Silvia M. Vidal1,5Steven Gerondakis7 David Langlais1,5,8
David Langlais1,5,8 Jörg H. Fritz1,2,3,9*
Jörg H. Fritz1,2,3,9*Group 2 innate lymphoid cells (ILC2s) play a key role in the initiation and orchestration of early type 2 immune responses. Upon tissue damage, ILC2s are activated by alarmins such as IL-33 and rapidly secrete large amounts of type 2 signature cytokines. ILC2 activation is governed by a network of transcriptional regulators including nuclear factor (NF)-κB family transcription factors. While it is known that activating IL-33 receptor signaling results in downstream NF-κB activation, the underlying molecular mechanisms remain elusive. Here, we found that the NF-κB subunit c-Rel is required to mount effective innate pulmonary type 2 immune responses. IL-33-mediated activation of ILC2s in vitro as well as in vivo was found to induce c-Rel mRNA and protein expression. In addition, we demonstrate that IL-33-mediated activation of ILC2s leads to nuclear translocation of c-Rel in pulmonary ILC2s. Although c-Rel was found to be a critical mediator of innate pulmonary type 2 immune responses, ILC2-intrinsic deficiency of c-Rel did not have an impact on the developmental capacity of ILC2s nor affected homeostatic numbers of lung-resident ILC2s at steady state. Moreover, we demonstrate that ILC2-intrinsic deficiency of c-Rel alters the capacity of ILC2s to upregulate the expression of ICOSL and OX40L, key stimulatory receptors, and the expression of type 2 signature cytokines IL-5, IL-9, IL-13, and granulocyte-macrophage colony-stimulating factor (GM-CSF). Collectively, our data using Rel−/− mice suggest that c-Rel promotes acute ILC2-driven allergic airway inflammation and suggest that c-Rel may contribute to the pathophysiology of ILC2-mediated allergic airway disease. It thereby represents a promising target for the treatment of allergic asthma, and evaluating the effect of established c-Rel inhibitors in this context would be of great clinical interest.
Group 2 innate lymphoid cells (ILC2s) mediate early type 2 immune responses and thereby exert key roles in the initiation and orchestration of anti-helminth immunity as well as allergic inflammation (1–5). ILC2s exhibit striking functional similarity to adaptive type 2 T helper (Th2) cells. Like Th2 cells, they depend on the transcription factor GATA3 and produce type 2 signature cytokines such as interleukin (IL)-5 and IL-13 upon activation driving eosinophil recruitment as well as goblet cell hyperplasia and mucus production, respectively (6). ILC2s are located at barrier surfaces including the lung and, contrary to Th2 cells, lack the expression of specific antigen receptors (6). They instead become activated in an antigen-independent fashion in response to environmental cues such as alarmins IL-25, IL-33, and/or thymic stromal lymphopoietin (TSLP) that are released upon tissue perturbation (6).
Among these alarmins, IL-33 has been described as the most potent activator of lung ILC2s (7). IL-33 signals through the heterodimeric IL-33 receptor (IL-33R) composed of the ligand-binding chain ST2 and IL-1 receptor accessory protein (IL-1RacP) (8). While it is known that activating IL-33R signaling results in downstream nuclear factor (NF)-κB activation, the underlying molecular mechanisms in ILC2s remain largely elusive (8). NF-κB signaling is mediated by homo- or heterodimers of proteins of the Rel/NF-κB transcription factor superfamily including NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB, and c-Rel (9). Despite sharing a common DNA recognition motif, knockout mice lacking individual Rel/NF-κB family members exhibit non-redundant phenotypes (10, 11). Moreover, differential expression patterns in tissues and responses to receptor signals as well as target gene specificity indicate that distinct NF-κB subunits exert unique physiological roles (10, 11). While stimulation of lung ILC2s with IL-33 results in phosphorylation of RelA and treatment with a pan-NF-κB inhibitor impairs ILC2 effector functions (12–15), RelB was shown to be an intrinsic repressor of ILC2s (16). On the contrary, the function of c-Rel in ILC2s remains undefined. c-Rel has been shown to promote type 2 immune responses upon ovalbumin (OVA)-induced allergic airway inflammation (17). Furthermore, inhibition of c-Rel in a mouse model of house dust mite-mediated allergic inflammation resulted in reduced levels of IL-13 and airway hyper-reactivity as well as lung inflammation (18, 19) and inhibited eosinophil recruitment in an OVA model of chronic asthma (20). Since ILC2s are main drivers of allergic asthma, we aimed to investigate whether c-Rel exhibits similar effects during ILC2-driven allergic airway inflammation.
In the present study, we found that deficiency in c-Rel severely diminished early pulmonary ILC2-driven type 2 responses to intranasal IL-33 administration. We further demonstrate that c-Rel expression and activation in ILC2s are positively regulated by IL-33.
C57BL/6J wild-type (WT) mice were originally purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in house. Rel−/− mice have been previously described and were kindly provided by Dr. Steve Gerondakis (Monash University) (21). All animals were maintained on a C57BL/6J background and were bred and housed under specific pathogen-free conditions with ad libitum access to food and water. Experiments were conducted with adult female age-matched mice (8–12 weeks) in accordance with the guidelines and policies of the Canadian Council on Animal Care and those of McGill University.
Mice were anesthetized with isoflurane followed by intranasal administration of either phosphate-buffered saline (PBS) as a control or 500 ng carrier-free recombinant murine IL-33 (rmIL-33, R&D Systems) per mouse in a total volume of 40 μl. Mice were challenged with PBS or IL-33 for three consecutive days (d0, d1, and d2), and lungs were isolated and analyzed 24 h after the last treatment.
Lungs were inflated with 10% buffered formalin, fixed overnight, and transferred to 70% ethanol. Fixed lungs were embedded in paraffin, sectioned, and stained with hematoxylin and eosin following standard procedures. A histologic disease score from 0 to 4 was attributed based on peribronchial, perivascular, and parenchymal immune cell infiltration.
Lungs were isolated, finely minced, and digested in Roswell Park Memorial Institute (RPMI)-1640 containing 5% fetal bovine serum (FBS), 0.2 mg/ml of Liberase™ TM (Roche), and 0.1 mg/ml of DNase I (Roche) at 37°C. Digested lungs were homogenized with a 5-ml syringe attached to an 18G needle, filtered through a 70 μM cell strainer, and washed with PBS. Red blood cells were lysed using Red Blood Cell Lysing Buffer Hybri-Max™ (Sigma-Aldrich), and cells were washed with fluorescence-activated cell sorting (FACS) buffer (PBS + 2% FBS). Small intestines were isolated, opened longitudinally, cut in small pieces, and vortexed in gut buffer (Hanks’ Balanced Salt Solution (HBSS) + 2% FBS + 15 mM of HEPES) to remove fecal matter. Intestinal epithelial cells were removed by incubation in gut buffer containing 5 mM of EDTA for 20 min. Lamina propria was subsequently digested in RPMI-1640 supplemented with 5% FBS, 15 mM of HEPES, 0.1 mg/ml of Liberase™ TM (Roche), and 0.1 mg/ml of DNase I (Roche) at 37°C for 15 min. Digestion suspensions were filtered through a 70 μM cell strainer to obtain a single-cell suspension, and cells were washed with FACS buffer.
Pelleted single-cell suspensions were resuspended in 2.4G2 hybridoma supernatant dilution and incubated for 15 min on ice to block Fc receptors. Cells were subsequently stained with antibody dilutions prepared in PBS supplemented with 2% FBS for 30 min on ice. Dead cells were excluded by staining with Fixable Viability Dye eFluor™ 780 (eBioscience) following the manufacturer’s instructions. Intracellular staining was performed using the FoxP3/Transcription Factor Staining Buffer Set (eBioscience) according to the manufacturer’s protocol. Stained cell suspensions were acquired on a BD FACSCanto™ II System (BD Biosciences), a BD LSRFortessa™ Cell Analyzer (BD Biosciences) or sorted on a BD FACSAria™ III Cell Sorter (BD Biosciences). Flow cytometry data were analyzed using FlowJo X (BD Biosciences). All antibodies used for flow cytometry analyses are listed in Table 1.

Table 1 Antibodies.
Bone marrow ILC2 progenitors were isolated and expanded as described previously with minor modifications (22). Briefly, bone marrow from the tibias and femurs was pooled, subjected to red blood cell lysis using Red Blood Cell Lysing Buffer Hybri-Max™ (Sigma-Aldrich), and sorted as lineage (CD3ε, CD5, CD11b, CD11c, Gr1, Ly6G, CD45R (B220), NK1.1, TCRβ, TCRγδ, and Ter-119)-negative, Sca-1+c-kit−CD25+ cells. Isolated cells were expanded in complete ILC2 medium supplemented with recombinant murine IL-2, IL-7, IL-25, and IL-33 (all 50 ng/ml; R&D Systems) and TSLP (20 ng/ml; R&D Systems). After 2–3 weeks of expansion, cells were rested for 72 h in IL-2 and IL-7 (both 10 ng/ml), washed, and incubated in complete ILC2 medium without cytokines for 4 h before use in experiments.
Single-cell suspensions from lung and small intestine were stained with respective surface antibodies and viability dye as described above and sorted on a BD FACSAria™ III Cell Sorter (BD Biosciences) as live CD45+Lin−Thy-1+ST2+CD25+ or live CD45+Lin−Thy-1+CD127+KLRG1+ cells, respectively. Isolated cells were cultured for 18 h in complete ILC2 medium containing recombinant murine IL-7 (10 ng/ml; R&D Systems), washed, and rested for 4 h in complete ILC2 medium without cytokines before being used in experiments.
Bone marrow-derived ILC2s were stimulated for the indicated time points with rmIL-33, and sub-cellular fractionation was performed as previously described (23). Briefly, pelleted cells were resuspended in 900 μl of PBS/0.1% NP-40 containing protease inhibitor and triturated with a micropipette to lyse the cell membranes. Three hundred microliters of lysate was collected (WC=whole-cell lysate). The remaining 600 μl were centrifuged at 13,000 g for 10 s, and supernatant was collected (C=cytoplasmic fraction). Finally, the remaining pellet, containing intact nuclei, was resuspended in 300 μl of PBS/0.1% NP-40 with protease inhibitor. All fractionated samples were probe-sonicated for 15 s at 60% amplitude. Protein was quantified using the Bio-Rad Protein Assay (#500-0006), as per manufacturer’s instructions. Prior to loading samples were denaturated in 3× Laemmli buffer containing sodium dodecyl sulfate (SDS) and 15% β-mercaptoethanol at 95°C for 5 min. 15 µg of protein for whole-cell lysate and cytoplasmic fraction samples, or 30 μl/one-tenth of the total nuclear fractionated samples, was separated on 8% polyacrylamide gels, with a 3% polyacrylamide stacking gel. Proteins were wet-transferred onto nitrocellulose membranes. For immunoblotting, membranes were probed with anti-c-Rel; anti-GAPDH or anti-H2A (all Cell Signaling Technology) in 0.1% T-TBS + 5% milk powder followed by incubation with anti-rabbit IgG-HRP (Cell Signaling Technology). The details of the antibodies are listed in Table 1.
Isolated lung ILC2s were left for 4 h in medium without cytokines and left untreated or stimulated with rmIL-33 (10 ng/ml; R&D Systems) for the indicated time points. Cells were fixed and permeabilized using the FoxP3/Transcription factor staining kit (eBioscience) according to the manufacturer’s instructions and stained with anti-c-Rel, anti-CD25, and DAPI (NucBlue™ Fixed Cell ReadyProbes™ Reagent, Life Technologies). Samples were run on an ImageStreamX Mark II imaging flow cytometer (Amnis), and nuclear translocation of c-Rel in CD25+c-Rel+DAPI+ cells was analyzed using the similarity feature of the IDEAS software (Amnis). All antibodies are listed in Table 1.
Total RNA from cultured ILC2s was extracted using the Quick-RNA MicroPrep Kit (Zymo Research) according to the manufacturer’s instructions. For preparation of total lung RNA, tissue was mechanically disrupted in a MagNA Lyser (Roche) followed by RNA extraction with TRIzol™ Reagent (Life Technologies) and clean-up using the RNeasy Mini kit (QIAGEN) according to the manufacturer’s instructions. cDNA was prepared using Oligo(dT)12-18 Primer (Life Technologies) and SuperScript™ III Reverse Transcriptase (Life Technologies). qRT-PCRs were performed with PowerUp™ SYBR™ Green Master Mix (Applied Biosystems) in a StepOnePlus™ Real-Time PCR System (Applied Biosystems). Transcript expression was normalized to Hprt expression levels and quantified using the comparative 2−ΔΔCT method. Data are depicted as either relative expression or relative fold change compared to the mean of the control group. Primers used in this study were designed using the PrimerQuest Tool (Integrated DNA Technologies) and purchased from Integrated DNA Technologies. Sequence details of primers are provided in Table 2.

Table 2 qRT-PCR primers.
IL-5, IL-9, IL-13, and granulocyte-macrophage colony-stimulating factor (GM-CSF) in tissue culture supernatants were determined using the respective mouse DuoSet ELISA kits (R&D Systems) according to the manufacturer’s instructions. Absorbance was measured using an Enspire™ 2300 Multilabel Reader (PerkinElmer).
To determine intracellular cytokine production by isolated murine lung ILC2s, sorted cells in complete ILC2 medium (see above) were stimulated in 96-well round-bottom plates (15,000 cells/well) for 48 h with medium only; recombinant murine IL-2, IL-7, IL-9, and IL-33 (10 ng/ml; R&D Systems); or combinations thereof. GolgiPlug (BD Biosciences) was added for the last 6 h of culture. To analyze cytokine production following in vivo stimulation, lung cell suspensions (1 × 106 cells/well in a 96-well plate) in complete ILC2 medium were stimulated with Cell Stimulation Cocktail (eBioscience) according to the manufacturer’s instructions in the presence of GolgiPlug (BD Biosciences). Cells were stained with respective surface antibodies and viability dye as described above, and intracellular cytokine staining was performed using the FoxP3/Transcription Factor Staining Buffer Set (eBioscience) according to the manufacturer’s protocol. Stained cells were acquired on a BD LSRFortessa™ Cell Analyzer (BD Biosciences) and analyzed using FlowJo X (BD Biosciences). All antibodies used for flow cytometry analyses are listed in Table 1.
All data were analyzed with GraphPad Prism software (GraphPad Software). p-Values below 0.05 were defined as statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001). Unless otherwise indicated, figures display means ± standard deviation (SD). Experiment sample sizes (n), experiment replicate numbers, and statistical tests used are included in the respective figure legends.
To investigate the role of c-Rel during ILC2-driven allergic airway inflammation, we challenged WT and c-Rel-deficient (Rel−/−) mice intranasally with either PBS or IL-33 for three consecutive days and analyzed parameters of airway inflammation (Figure 1A). Pulmonary tissue histology showed that, when exposed to IL-33, lack of c-Rel resulted in a significant reduction in perivascular and peribronchial immune cell infiltration into lung tissue as compared with WT mice (Figures 1B, C). These findings could be recapitulated by flow cytometry, showing markedly decreased numbers of pulmonary CD45+ leukocytes in c-Rel-deficient animals after IL-33 treatment (Figure 1D). Moreover, while challenge of WT mice with IL-33 resulted in a significant increase in total numbers of type 2 immunity-associated cell populations including eosinophils, type 2 dendritic cells (DC2s), and ILC2s, numbers were significantly diminished in Rel−/− mice (Figure 1D and Supplementary Figures 1A–D). Furthermore, c-Rel deficiency led to reduced expression of lung Il5 transcripts with a trend towards lower Il13 levels (Figure 1E). In addition, the type 2-associated chemokines Ccl17 and Ccl22 were greatly reduced in Rel−/− compared with WT mice (Figure 1F). In summary, compared with WT animals, c-Rel-deficient mice mount a reduced innate type 2 immune response with a significant reduction in pulmonary leukocyte infiltration, eosinophilia, DC2s, and ILC2s. Our results thus suggest that c-Rel is critical for the development of IL-33-driven allergic lung inflammation.
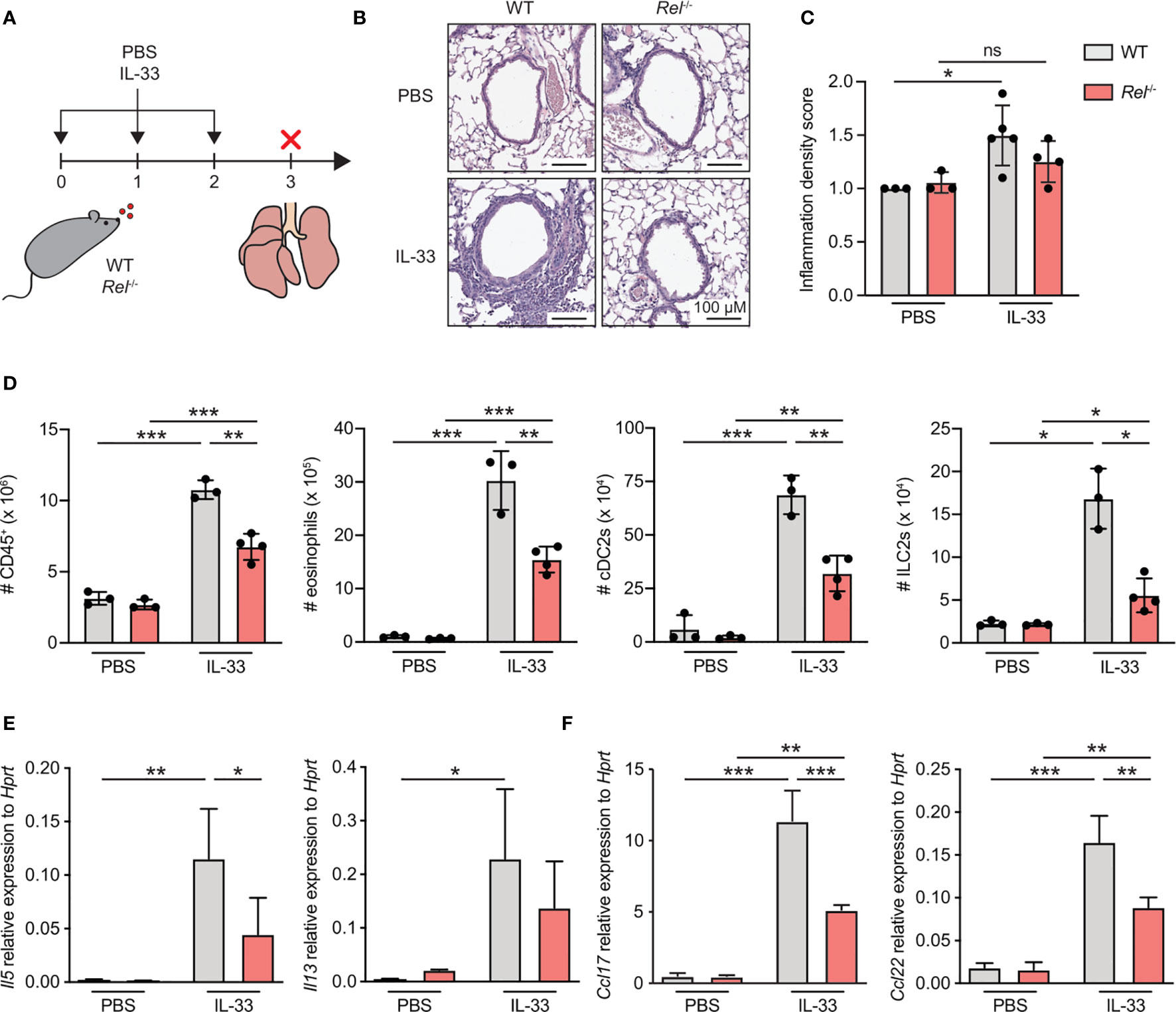
Figure 1 c-Rel is a critical mediator of innate type 2 immune responses. (A) Wild-type (WT) and c-Rel-deficient (Rel−/−) mice were intranasally challenged for three consecutive days (day 0, 1, and 2) with phosphate-buffered saline (PBS) as a control or IL-33 (500 ng/mouse). Lungs were isolated and analyzed 24 h after the last challenge (day 3). (B) Microscopy of lung sections stained with hematoxylin and eosin (H&E) from WT (left panel) and Rel−/− mice (right panel) treated with PBS or IL-33. Scale bars, 100 µM. (C) Pathology score of inflammatory infiltration density assessed microscopically from H&E-stained lung sections. ns, not significant. (D) Total numbers of CD45+ leukocytes, eosinophils, type 2 conventional dendritic cells (cDC2s), and group 2 innate lymphoid cells (ILC2s) in lungs of WT (gray) or Rel−/− (red) mice were determined by flow cytometric analysis. Expression levels of (E) Il5 and Il13 as well as (F) Ccl17 and Ccl22 in whole lung tissue of WT (gray) and Rel−/− (red) animals were assessed by qRT-PCR. Data are representative of two independent experiments with n = 3–4 mice per group. Data are shown as mean ± SD with *p < 0.05, **p < 0.01, ***p < 0.001 as determined by one-way ANOVA followed by Tukey’s multiple comparisons test.
To assess whether lack of c-Rel results in ILC2-intrinsic alterations that may explain why Rel−/−animals mount a strongly reduced innate type 2 immune response, we analyzed ILC2 progenitor numbers in the bone marrow of c-Rel sufficient (Rel+/+), heterozygous (Rel+/−), and c-Rel-deficient (Rel−/−) littermate control animals. We observed that mice of all three genotypes harbored comparable numbers of common lymphoid progenitors (CLPs), as well as the more downstream common helper ILC progenitors (CHILPs), and ILC2 progenitors (ILC2Ps) (Figure 2A and Supplementary Figure 2A). In addition, there was no difference in total numbers of mature lung ILC2s (Figure 2B). Together, these results indicate that c-Rel is dispensable for the development of ILC2s in the bone marrow and that c-Rel deficiency does not affect homeostatic numbers of lung-resident ILC2s.
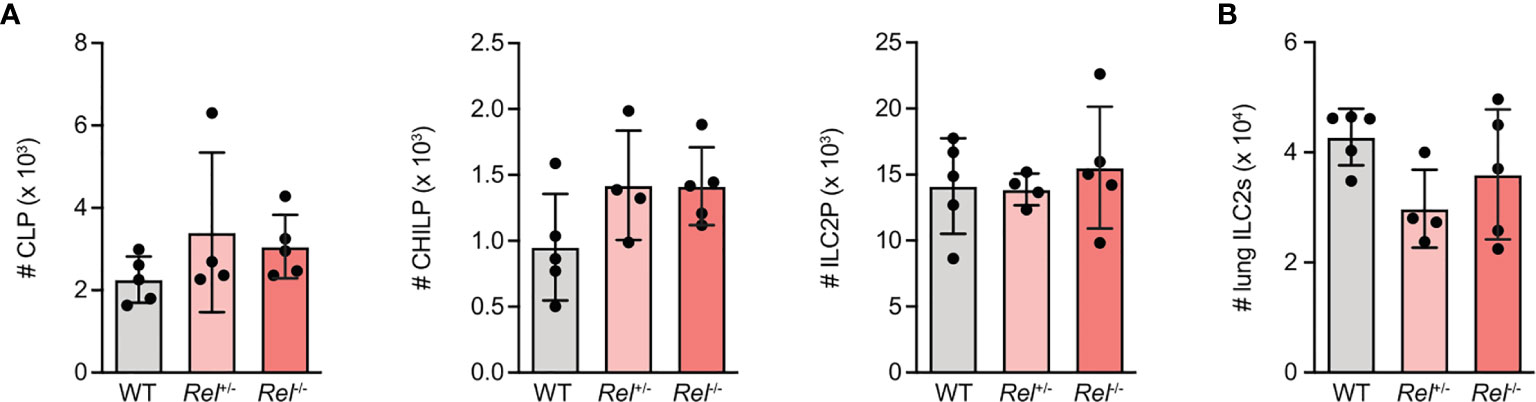
Figure 2 c-Rel-deficient group 2 innate lymphoid cells (ILC2s) exhibit no intrinsic defects in their developmental capacity. Flow cytometric analysis of total numbers of (A) ILC2 precursor populations CLP (left), CHILP, (middle) and ILC2 progenitor (ILC2P) (right) as well as (B) pulmonary ILC2s in wild-type (WT), heterozygous Rel+/−, and homozygous Rel−/− littermate control mice. Data points are representative of two independent experiments with n = 4–5 mice per treatment group (A, B). Data are shown as mean ± SD as determined by one-way ANOVA followed by Tukey’s multiple comparisons test. CLP, common lymphoid progenitor; CHILP, common helper ILC2 progenitor; ILC2P, ILC2 progenitor.
Although it is well established that IL-33 signaling via ST2 triggers downstream activation of NF-κB pathways (8), the specific roles of the NF-κB subunit c-Rel in ILC2s remain unknown. We therefore analyzed the direct effect of IL-33 on c-Rel expression and activation in ILC2s. Ex vivo culture of bone marrow-derived (Figure 3A and Supplementary Figure 3A) as well as lung ILC2s (Figure 3B and Supplementary Figure 2B) with IL-33 resulted in a rapid increase in c-Rel transcripts levels compared with unstimulated cells. Consistently, lung c-Rel protein levels increased in a dose-dependent manner and remained elevated for at least 72 h of culture with IL-33 (Figures 3C, D). Furthermore, c-Rel protein expression in lung, small intestine, and bone marrow-derived ILC2s was significantly elevated over levels found in unstimulated cells as early as 4 h following stimulation with IL-33 and was further induced 24 h post stimulation (Figures 3E, F and Supplementary Figure 3B). Taken together, these findings show that IL-33 potently induces c-Rel expression in ILC2s derived from different anatomical locations, indicating a tissue-spanning role of the IL-33–c-Rel axis.
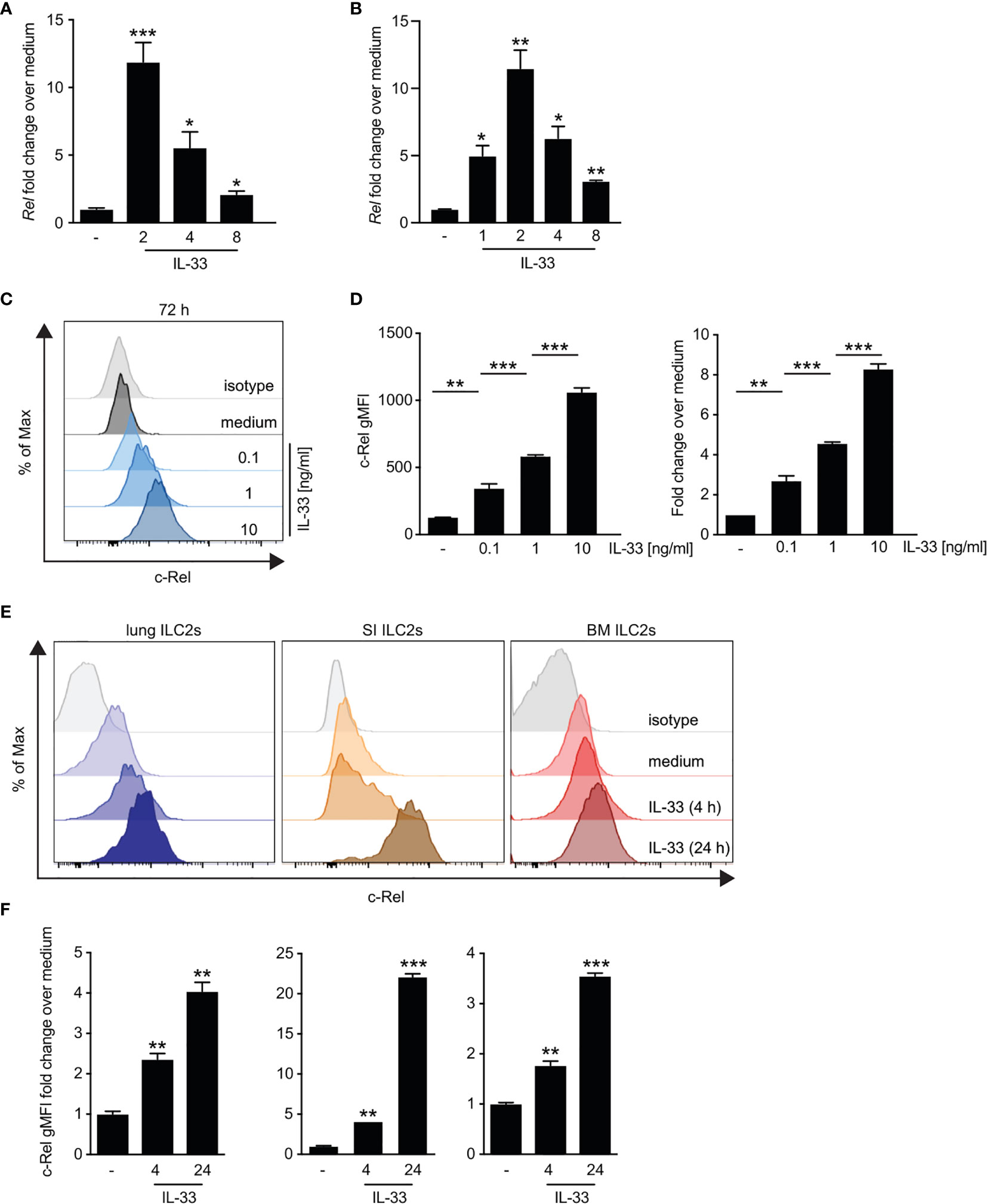
Figure 3 IL-33-driven ex vivo activation of group 2 innate lymphoid cells (ILC2s) induces c-Rel expression. (A–D) Murine lung, small intestine, or bone marrow-derived ILC2s were isolated by flow cytometry and left unstimulated (−) or cultured in the presence of recombinant murine IL-33 for the indicated time points. Kinetics of c-Rel (Rel) transcript expression in (A) ex vivo expanded bone marrow-derived ILC2s stimulated with IL-33 (10 ng/ml) or (B) primary murine lung ILC2s. Asterisks indicate significance over untreated control. Transcript levels were assessed by qRT-PCR and are depicted as fold change over unstimulated (−) ILC2s. Asterisks indicate significance over untreated control. (C, D) Flow cytometric analysis of intracellular expression of c-Rel of isolated lung ILC2s after 72 h of ex vivo stimulation with medium (−) or IL-33 (0.1, 1, and 10 ng/ml). c-Rel staining is shown as (C) histogram plots in comparison with an isotype control antibody (gray), (D) gMFI (left panel), or fold change over unstimulated ILC2s (right panel). Asterisks indicate significance over unstimulated control. (E) Representative histogram plots of intracellular flow cytometric staining of c-Rel in lung (left), small intestine (middle), or bone marrow-derived (right) ILC2s left untreated (medium) or cultured with IL-33 (10 ng/ml) for 4 or 24 h. c-Rel staining is shown in comparison with staining with an isotype control antibody (gray). (F) c-Rel staining intensity in lung (left), small intestinal (middle), or bone-marrow-derived (right) ILC2s quantified as gMFI fold change over unstimulated cells. Asterisks indicate significance over respective untreated controls. Data are representative of at least two independent experiments including two or more biological replicates. Data are shown as mean ± SD with *p < 0.05, **p < 0.01, and ***p < 0.001 as determined by two-tailed t-test (unpaired). gMFI, geometric mean fluorescence intensity.
To determine whether c-Rel is activated upon IL-33 stimulation, we assessed nuclear translocation in ILC2s upon ex vivo IL-33 stimulation by Western blotting using cytoplasmic and nuclear ILC2 lysates (Figure 4A) as well as by ImageStream analysis (Figures 4B, C). With both methods, we observed that c-Rel remained confined to the cytoplasm in unstimulated ILC2s, while activation with IL-33 resulted in translocation of c-Rel from the cytoplasm to the nucleus, indicating a potential role of c-Rel in transcriptional regulation of ILC2 effector functions.
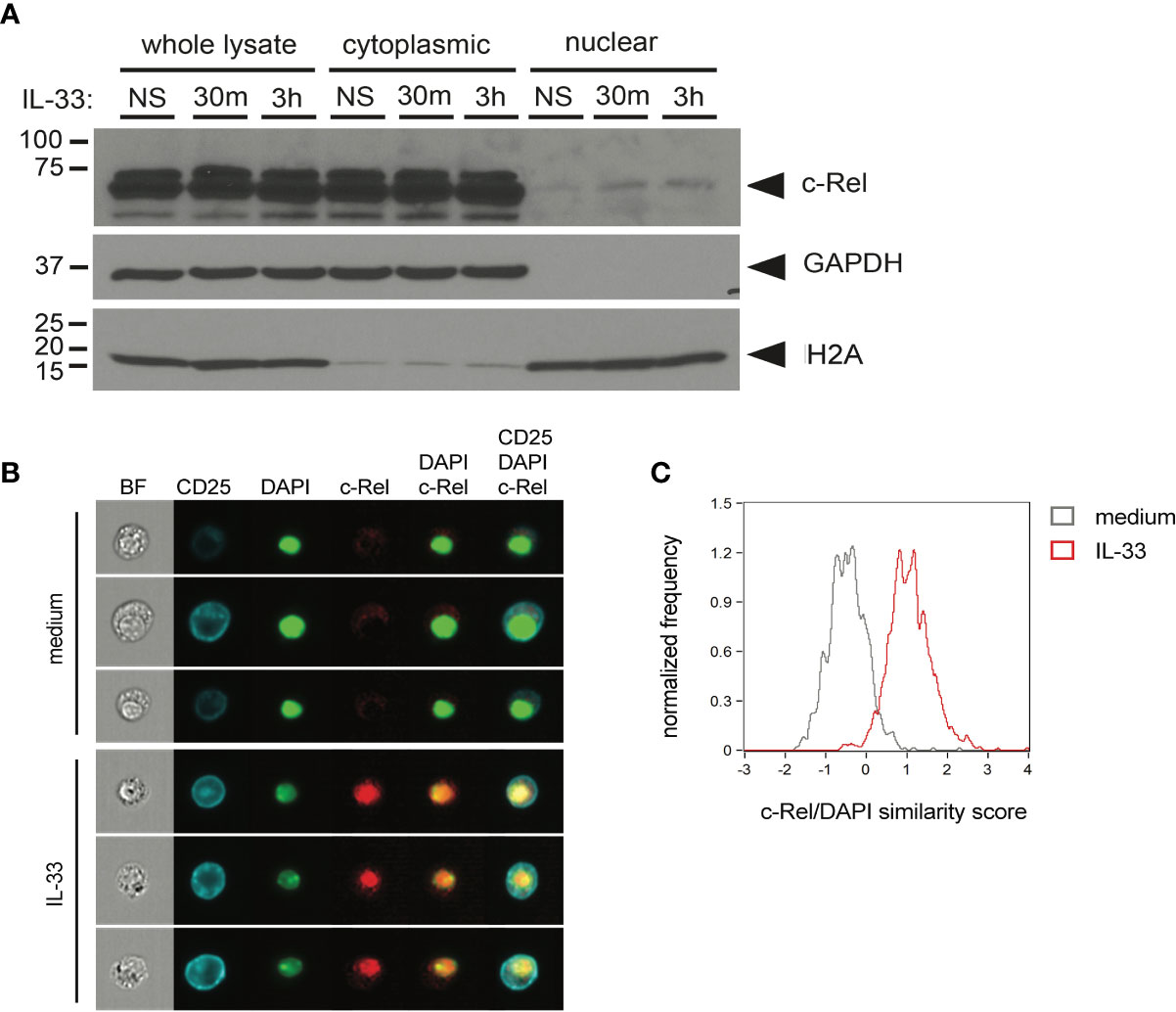
Figure 4 c-Rel translocates to the nucleus upon ex vivo activation of group 2 innate lymphoid cells (ILC2s) by IL-33. (A) Ex vivo expanded bone-marrow derived ILC2s were left unstimulated (NS) or cultured with IL-33 (100 ng/ml) for 30 min or 3 h. Whole-cell, cytoplasmic, and nuclear lysates were probed for c-Rel by Western blotting. Expression of GAPDH and H2A served as fractionation and loading controls. (B, C) ImageStream analysis of isolated lung ILC2s left unstimulated (medium) or cultured with IL-33 (10 ng/ml) for 3 h. (B) Representative images (from left to right) of bright-field (BF), CD25, DAPI, and c-Rel as well as merged images of DAPI + c-Rel and CD25 + DAPI + c-Rel. (C) Quantification of nuclear translocation of c-Rel by overlay of c-Rel/DAPI similarity scores of untreated (gray histogram) and IL-33-activated ILC2s (red histogram). Data are representative of at least two independent experiments.
To assess if c-Rel expression is also increased upon in vivo activation of ILC2s, we induced allergic airway inflammation by treating mice intranasally with IL-33 for 3 days. c-Rel-deficient mice that underwent the same treatment were used as negative controls. c-Rel transcript levels were significantly higher in lung tissues of IL-33-challenged mice compared with mice that received PBS, while no transcripts were detected in the lungs of control Rel−/− mice (Figure 5A). Consistent with ILC2s stimulated ex vivo, intranasal administration of IL-33 in WT mice resulted in a 10-fold induction of c-Rel expression in lung ILC2s over PBS-treated mice (Figures 5B, C). Taken together, these findings show that c-Rel expression is induced upon ex vivo stimulation of ILC2s as well as during IL-33- and allergen-induced allergic airway inflammation.
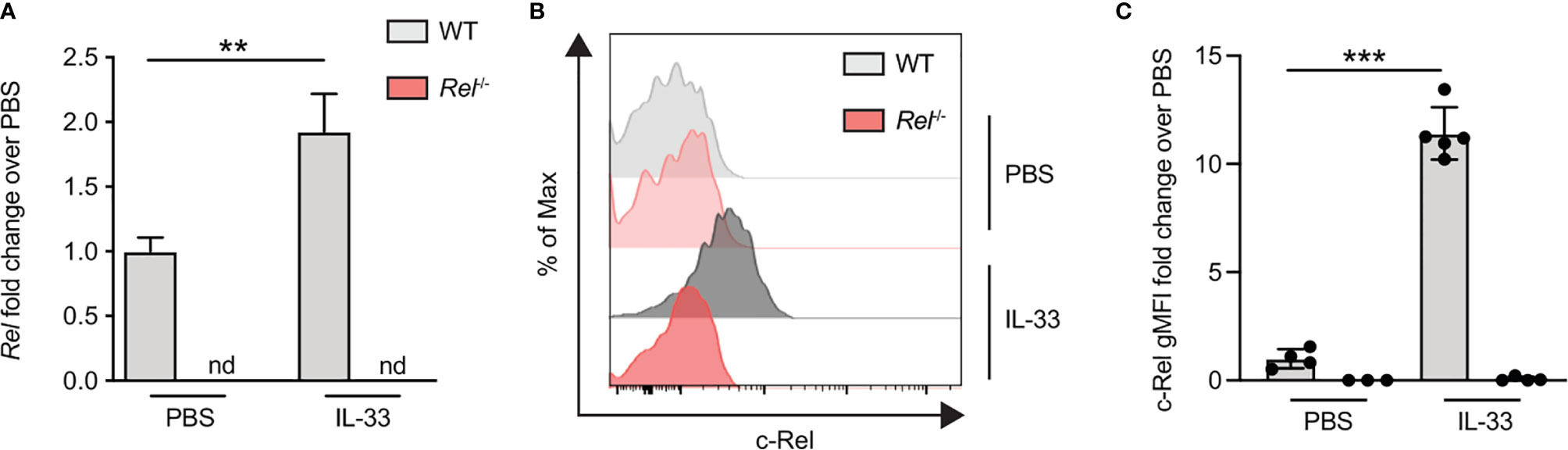
Figure 5 c-Rel expression is induced in lung group 2 innate lymphoid cells (ILC2s) following intranasal challenge with IL-33. Wild-type (WT) (gray) and Rel−/− (red) mice were intranasally challenged for three consecutive days (days 0, 1, and 2) with either phosphate-buffered saline (PBS) or IL-33 (500 ng/mouse), and lungs were analyzed 24 h after the last administration. (A) Lung c-Rel transcript levels in WT and Rel−/− mice following intranasal challenge were determined by qRT-PCR, and (B, C) intracellular expression of c-Rel protein in lung ILC2s was analyzed by flow cytometry. (B) Representative histogram plots of intracellular c-Rel staining in pulmonary WT and Rel−/− ILC2s following intranasal challenge with PBS or IL-33 quantified as (C) gMFI fold change over the PBS-treated control group. Data points are representative of at least two independent experiments with n = 3–5 mice per treatment group. Data are shown as mean ± SD with **p < 0.01, and ***p < 0.001 as determined by one-way ANOVA followed by Tukey’s multiple comparisons test or by two-tailed t-test (unpaired). gMFI, geometric mean fluorescence intensity; nd, not detectable.
Besides activating signals provided by alarmins, ILC2s require cues from STAT5-activating cytokines such as IL-2, IL-7, or TSLP, as well as costimulatory signals for optimal activation and exertion of effector functions (6, 26). We therefore assessed whether observed reduction in Rel−/− ILC2 numbers during allergic airway inflammation may stem from lowered sensitivity to activating signals due to diminished expression of the respective surface receptors. To this end, we assessed surface expression levels of CD25 (IL-2Rα), CD127 (IL-7R), and other key activating ILC2 receptors as well as the ILC2 master transcription factor GATA3 in isolated pulmonary WT and Rel−/− ILC2s at steady state and after ex vivo stimulation with IL-33 in the presence or absence of IL-2 or IL-7. We found that c-Rel deficiency did not result in diminished expression of surface CD25, CD127, or GATA3 by ILC2s at steady state or upon IL-33-mediated activation under the tested conditions (Figures 6A and Supplementary Figure 4A). Moreover, the IL-33R chain ST2 and the costimulatory receptor ICOS (Figure 6B) were slightly enhanced upon ILC2 activation in c-Rel-deficient ILC2s when compared with WT cells.
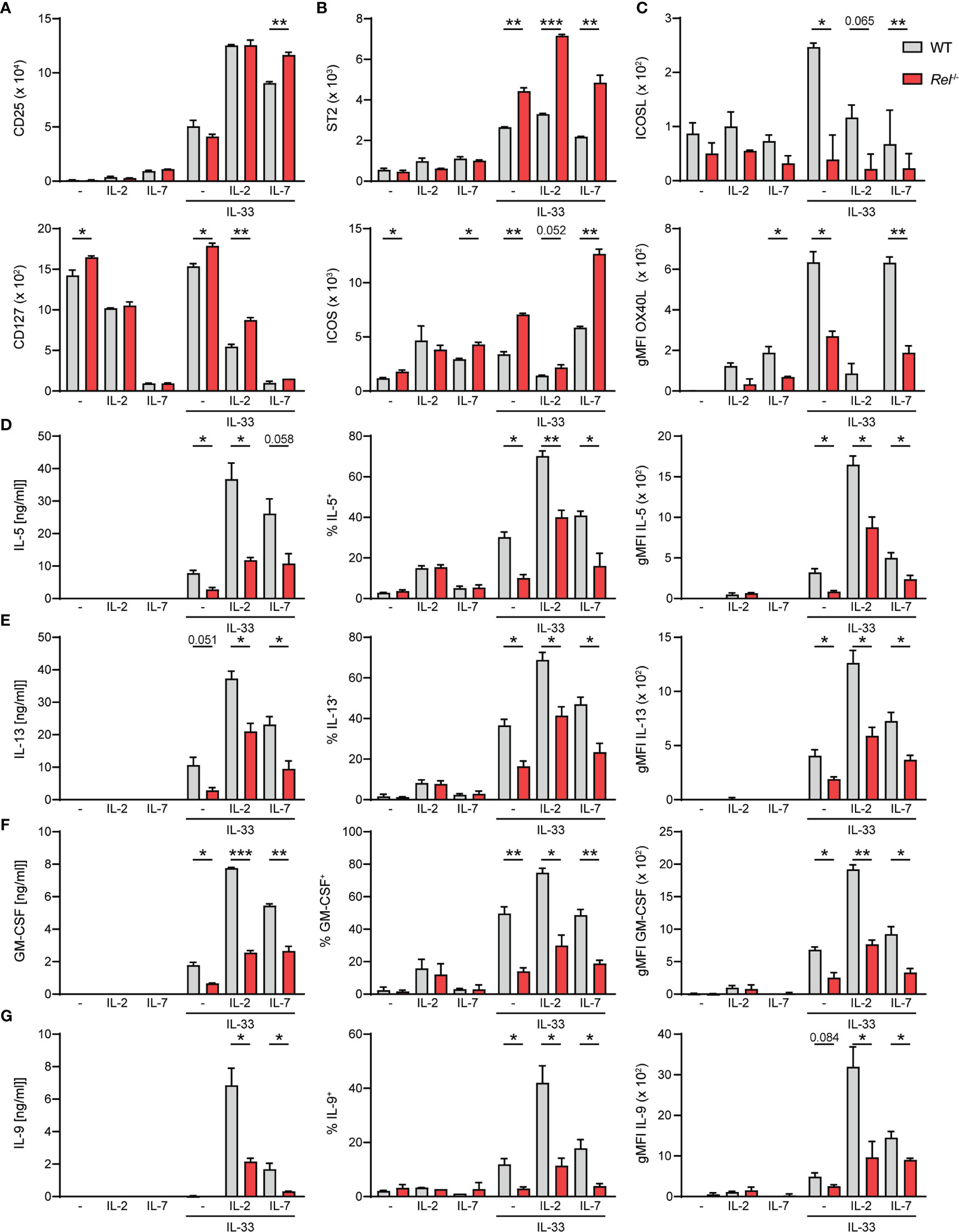
Figure 6 c-Rel deficiency limits group 2 innate lymphoid cell (ILC2) effector functions following ex vivo activation. Lung ILC2s from wild-type (WT) and Rel−/− mice were isolated by flow cytometry and cultured ex vivo in the absence (−) or presence of IL-2, IL-7, or IL-33 and combinations thereof (all 10 ng/ml). Expression levels of surface (A) CD25 and CD127, (B) ST2 and ICOS, and (C) ICOSL and OX40L on isolated WT (gray) and Rel−/− (red) lung ILC2s after 24-h stimulation under indicated conditions. Analysis of (D) IL-5, (E) IL-13, (F) granulocyte-macrophage colony-stimulating factor (GM-CSF), and (G) IL-9 expression by WT (gray) and Rel−/− (red) lung ILC2s left untreated or cultured with IL-33 for 48 h. Cytokine concentration in culture supernatants is depicted in the left panel, frequencies of cytokine-producing cells are depicted in the middle panel, and cytokine expression levels are depicted as gMFI in the right panel. Data points are representative of two independent experiments with two biological replicates for each stimulation condition. Data are shown as mean ± SD with *p < 0.05, **p < 0.01, and ***p < 0.001 as determined by two-tailed t-test (unpaired). gMFI, geometric mean fluorescence intensity.
Besides ICOS, murine ILC2s also express ICOS ligand (ICOSL), whose interaction with ICOS promotes ILC2 survival and effector cytokine production upon IL-33-mediated activation (27, 28). Furthermore, constitutive as well as activation-induced expression of other costimulatory ligands and receptors including DR3, GITR, and OX40L on ILC2s have been reported (13, 29–32). We therefore assessed whether c-Rel regulates the expression of costimulatory ligands and/or receptors that promote ILC2 functions directly as well as are expressed by ILC2s to aid in instructing adaptive immune responses. While no changes in DR3 or GITR expression were observed (data not shown), deficiency in c-Rel resulted in significantly reduced expression of surface ICOSL as well as OX40L (Figure 6C), indicating that c-Rel might further enhance ILC2 functions by driving ICOSL expression and also potentially drive adaptive type 2 responses by positively regulating surface OX40L levels.
Earlier studies demonstrated that c-Rel promotes cell cycle progression and survival in activated lymphocytes (21, 33–36). We therefore analyzed the proliferative capacity of lung ILC2s at steady state or upon IL-33-driven activation by Ki-67 staining after 48 h of activation. Ki-67 expression levels in lung ILC2s isolated from WT and Rel−/− mice were comparable upon IL-33 stimulation, indicating that c-Rel does not impact early expansion of IL-33-activated ILC2s (Supplementary Figure 4B).
ILC2s mediate their effector mechanisms by rapidly secreting large amounts of type 2 signature cytokines upon activation. To determine whether c-Rel directly regulates lung ILC2 type 2 signature cytokine production, we determined the concentrations of IL-5, IL-13, GM-CSF, and IL-9 in culture supernatants of ILC2s that were left unstimulated or were cultured with IL-2, IL-7, or IL-33 alone, or with indicated combinations (Figures 6D–G). We additionally assessed frequencies of cytokine-producing ILC2 and intracellular cytokine expression levels. All measured type 2 cytokines were induced upon activation of ILC2s with IL-33, and expression was further elevated when ILC2s were stimulated with IL-33 together with IL-2 or IL-7. Importantly, effector cytokine levels in the culture supernatant were significantly reduced in the absence of c-Rel (Figures 6D–G, left panel), which may be a direct result of significantly lower frequencies of cytokine-producing ILC2s (Figures 6D–G, middle panel) as well as significantly diminished production of cytokines within those cells (Figures 6D–G, right panel). Collectively, our data show that c-Rel positively regulates the expression of costimulatory ligands as well as ILC2 effector cytokines, thereby driving innate and potentially also adaptive type 2 immune responses.
To further decipher the in vivo impact of c-Rel on ILC2 function, WT and Rel−/− mice were challenged intranasally for three consecutive days with PBS as a control or IL-33 (Figure 7A). Mice were sacrificed and lung ILC2 surface expression levels of CD25, CD127, and ST2 (Figure 7B), ICOS as well as ICOSL and OX40L were determined. Additionally, we assessed intracellular expression levels of GATA3 (Figure 7F) and frequencies of Ki-67+ proliferating lung ILC2s following in vivo challenge. In accordance with our ex vivo stimulation results, no significant differences were observed in the expression of the key stimulatory ILC2 receptors CD25, CD127, and ST2 and ICOS (Figures 7B, C). Importantly, both ICOSL and OX40L were significantly downregulated in the absence of c-Rel, suggesting an in vivo role of c-Rel in the regulation of ILC2 costimulation (Figures 7D, E). While GATA3 expression was comparable in ILC2s of WT and c-Rel-deficient mice (Figure 7F), frequencies of proliferating ILC2s were significantly reduced in the absence of c-Rel (Figure 7G).
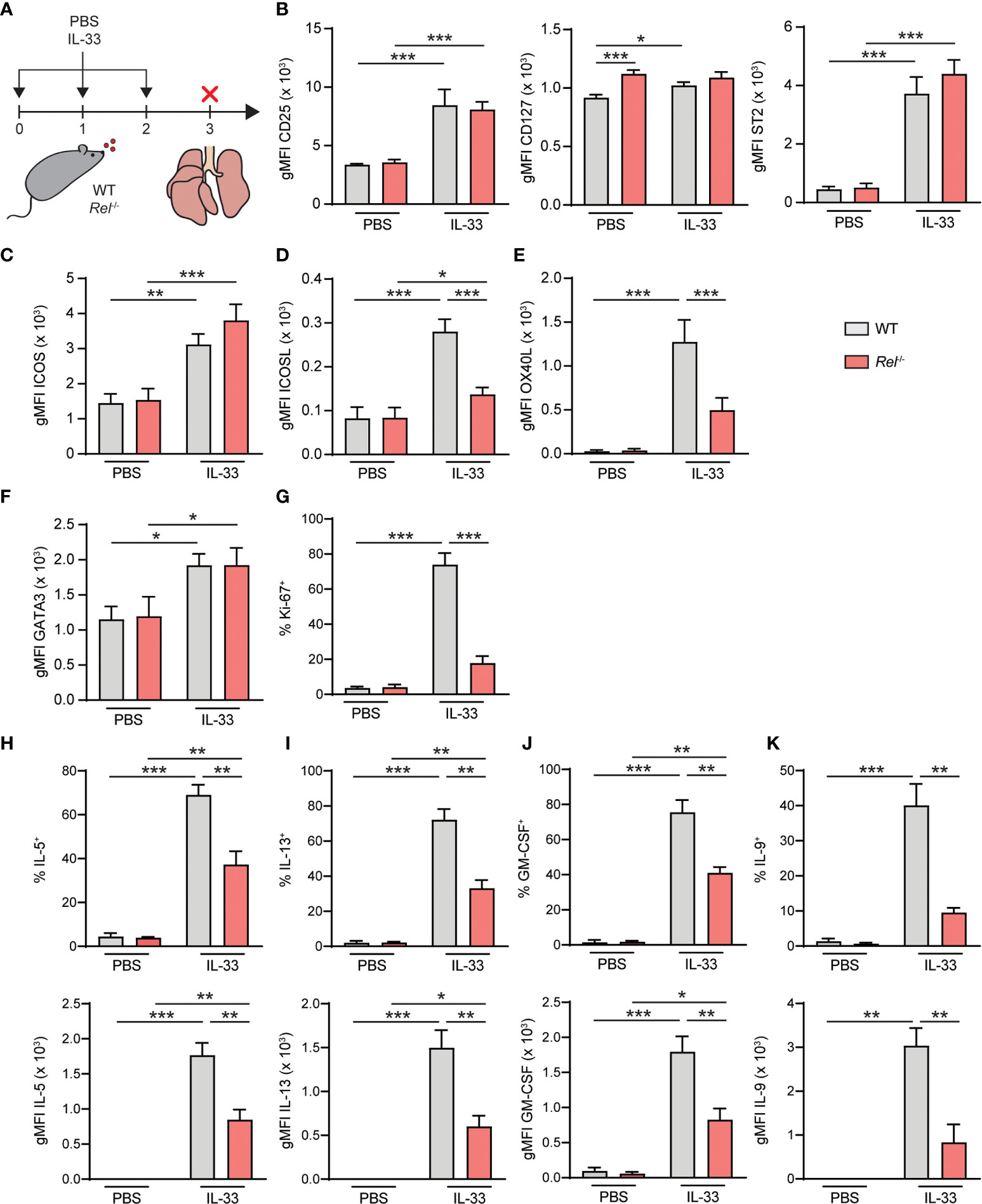
Figure 7 c-Rel drives group 2 innate lymphoid cell (ILC2) effector responses during IL-33-induced allergic airway inflammation. (A) Wild-type (WT) and Rel−/− mice were challenged intranasally for 3 days (days 0, 1, and 2) with phosphate-buffered saline (PBS) or IL-33 (500 ng/mouse). Mice were sacrificed, and lungs were analyzed 24 h after the last administration. Flow cytometry analysis of surface expression levels of (B) CD25, CD127, and ST2, (C) ICOS, (D) ICOSL, and (E) OX40L, and intracellular expression levels of (F) GATA3 in lung ILC2s following intranasal challenge of WT (gray bars) and Rel−/− (red bars) mice. (G) Frequencies of Ki-67+ WT (gray bars) and Rel−/− (red bars) lung ILC2s following in vivo challenge. Analysis of intracellular expression of (H) IL-5, (I) IL-13, (J) granulocyte-macrophage colony-stimulating factor (GM-CSF), and (K) IL-9 in WT (gray bars) and Rel−/− (red bars) in lung ILC2s following intranasal PBS or IL-33 challenge and ex vivo restimulation with phorbol myristate acetate (PMA)/ionomycin. Frequencies of cytokine-expressing cells are depicted in the top panel; and expression levels, measured as gMFI, are shown in the bottom panel. Data points are representative of two independent experiments with two biological replicates for each stimulation condition. Data are shown as mean ± SD with *p < 0.05, **p < 0.01, and ***p < 0.001 as determined by two-tailed t-test (unpaired). gMFI, geometric mean fluorescence intensity.
To determine the effect of c-Rel on ILC2 effector cytokine production, lung cells from challenged animals were activated with phorbol myristate acetate (PMA)/ionomycin; and intracellular levels of IL-5 (Figure 7H), IL-13 (Figure 7I), GM-CSF (Figure 7J), and IL-9 (Figure 7K) in ILC2s were analyzed. While production of all cytokines was induced, frequencies of cytokine-expressing ILC2s were significantly reduced in Rel−/− mice following in vivo challenge (Figures 7H–K, top panel). Furthermore, intracellular cytokine expression levels, measured as gMFI, were markedly lower in the absence of c-Rel. Together, these data show that c-Rel is critical for ILC2 proliferation as well as type 2 effector cytokine production during allergic airway inflammation and might potentially drive subsequent adaptive immune responses by promoting the expression of OX40L.
ILC2s are critical mediators of early type 2 immune responses and allergic airway disease. They depend on an intricate network of transcriptional regulators to ensure efficient activation and execution of effector functions. Rel/NF-κB transcription factors modulate immune responses by regulating the expression of hundreds of genes involved in lymphoid cell development, proliferation, survival, and immune cell effector functions (37).
We here observed that the NF-κB transcription factor c-Rel promotes type 2 immunity in an ILC2-driven mouse model of allergic airway inflammation. Challenge of c-Rel-deficient mice resulted in reduced lung immune cell infiltration and diminished numbers of key type 2 immune cell populations including ILC2s, cDC2s, and eosinophils. Consistent with reduced ILC2 numbers, markedly lower transcript and protein levels of the type 2 signature cytokine IL-5 were observed, which is critical for the recruitment of eosinophils to the lung during allergic airway inflammation (38). Moreover, c-Rel deficiency led to lower pulmonary IL-13 transcript and protein levels, a hallmark cytokine of type 2 immunity. Attenuated induction of the type 2 chemokine ligands CCL17 and CCL22 in the lungs of Rel−/− mice following exposure to IL-33 was observed as well, which may be a direct result of reduced numbers of CCL17- and CCL22-producing DC2s (39). Moreover, CCL17 and CCL22 have been described previously to promote ILC2 chemotaxis, and reduced levels might affect positioning of ILC2s within the airways, which could contribute to the observed reduction of type 2 responses (40). In addition, we demonstrate that c-Rel deficiency in ILC2s leads to markedly reduced IL-9 and GM-CSF production in vitro and in vivo. IL-9 has been shown to increase ILC2 fitness (41) and reinforces innate and adaptive type 2 immune responses (42, 43). In addition, GM-CSF has been shown to drive type 2 immunity (44) and allergic sensitization (45). This suggests that the attenuated response of c-Rel-deficient mice to IL-33 may be the result of defective activation and cytokine production by ILC2s.
Consistent with earlier reports showing that c-Rel is dispensable for normal hemopoiesis and lymphocyte development, the absence of c-Rel did not result in altered numbers of ILC2 precursors (21). In addition, numbers of lung ILC2s were comparable in naive WT and Rel−/− mice, indicating no homeostatic defects due to c-Rel deficiency that might contribute to the inability of c-Rel-deficient mice to mount an efficient type 2 response upon IL-33 challenge.
Our data show that c-Rel expression is potently induced in a dose-dependent manner in pulmonary ILC2s at both transcript and protein levels following activation with IL-33. Upregulation of c-Rel protein levels was also observed in IL-33-stimulated ILC2s isolated from other tissues, indicating a general role of c-Rel in ILC2 function. Importantly, c-Rel expression was induced in lung ILC2s following intranasal challenge with IL-33, suggesting a physiological function during allergic airway inflammation. We furthermore demonstrated nuclear translocation of c-Rel in lung ILC2s upon IL-33 stimulation, and since c-Rel harbors a transcription transactivation domain, it can thereby act as a direct transcriptional activator once in the nucleus (37).
Our data suggest that the attenuated response of c-Rel-deficient mice to IL-33 may be the result of defective activation and expansion of ILC2s. IL-33 acts in concert with signals provided by costimulatory STAT5-activating cytokines IL-2 and IL-7, and costimulatory surface molecules to promote ILC2 survival, expansion, and exertion of effector functions (6, 26). Previous work demonstrated that c-Rel upregulates the expression of IL-2Rα (CD25) in T cells (21, 33, 46, 47), and it is known that IL-2 enhances ILC2 survival as well as activation-mediated expansion and type 2 cytokine production (48–50). Moreover, earlier studies demonstrated that c-Rel positively modulates cell cycle progression and the expression of anti-apoptotic proteins in activated T and B cells (21, 33–36). In addition, genes encoding the type 2 signature cytokines IL-4 and IL-13 were proposed as putative c-Rel target genes based on differential transcript expression observed in gain-of-function and loss-of-function experiments in T cells (51). While our work showed no effect of c-Rel on ILC2-intrinsic expression of the cytokine receptors CD25 and CD127, we observed a strong reduction of IL-5, IL-9, IL-13, and GM-CSF production upon IL-33 stimulation in vitro and in vivo. Similar to T cells, ILC2s express activating costimulatory molecules, including DR3, GITR, and ICOS, that promote ILC2 function in disease settings as well as during homeostasis (13, 29–31, 52, 53). While no reduction in ICOS expression was observed, ICOSL and OX40L expression was found to be significantly lower in c-Rel deficient ILC2 after activation. This could explain the markedly reduced ILC2 response in the absence of c-Rel during experimental acute allergic airway inflammation.
Interestingly, WT and c-Rel deficient ILC2s proliferated equally well in vitro. In contrast, the proliferative potential was found to be altered in vivo. Since c-Rel is expressed in a variety of cells within the hematopoietic compartment (10, 54), it cannot be excluded that c-Rel deficiency impacts effector functions of other innate as well as adaptive pulmonary immune cell populations such as IL-33-reactive macrophages, DCs, or CD4+ T helper cells, which could potentially promote ILC2 activation by providing costimulation and/or activating cytokines. IL-33 was shown to induce IL-2 expression in DCs (55) and IL-2 is known to strongly enhance ILC2 survival as well as activation-mediated expansion and type 2 cytokine production (48–50). Moreover, interaction of ILC2 with CD4+ T cells has been suggested to enhance the ability of CD4+ T cells to produce IL-2, thereby reinforcing ILC2 activation (49). Interestingly, c-Rel deficiency was shown to alter the ability of CD4+ T cells to produce IL-2 (46, 56). It is therefore possible that c-Rel deficiency also alters the capacity of CD4+ T cells to produce IL-2 in response to IL-33, which could contribute to the altered proliferative capacity of ILC2 observed in vivo.
Our data using Rel−/− mice suggest that c-Rel promotes acute ILC2-driven allergic airway inflammation and suggests that c-Rel may contribute to the pathophysiology of ILC2-mediated allergic airway disease. It thereby represents a promising target for the treatment of allergic asthma, and evaluating the effect of established c-Rel inhibitors in this context would be of great clinical interest. In addition, while RelA deficiency results in embryonic lethality and RelA−/− cells exhibit severe defects regarding survival, proliferation, and effector functions (57), mice lacking c-Rel are viable and only show limited immunological defects (21). Thus, targeting c-Rel specifically may avoid the adverse side effects that have halted advancement of pan-NF-κB inhibitors for therapeutic applications (19, 58). IL-33-dependent ILC2 activation has also been reported during cancer (59, 60), viral infections (22, 61, 62), and murine models of autoimmune diseases (63–66); and given the observed induction of c-Rel expression in ILC2s from different tissues, modulation of c-Rel function in these conditions may be worth evaluating.
The datasets presented in this article are not readily available because no datasets were generated as listed in 2. Requests to access the datasets should be directed to am9yZy5mcml0ekBtY2dpbGwuY2E=.
The animal study was reviewed and approved by the Canadian Council on Animal Care, McGill University, ethics committee.
JF and BM designed the intellectual framework of the study and the layout of the manuscript. BM, SK, and CD performed in vivo allergic airway inflammation models, and BM and SK conducted ex vivo cell culture experiments. BM, SK, CD, and DL analyzed data. MM carried out cellular fractionation and Western blotting analysis. LR performed histological scoring. SV and SG provided critical reagents and mouse strains. BM and JF designed experiments and wrote the manuscript. All authors contributed to the article and approved the submitted version.
The work in the laboratory of JF is supported by a foundation (FDN-148405) followed by a project grant (PJT-175173) from the Canadian Institutes of Health Research (CIHR), and a Leaders Opportunity Fund infrastructure grant from the Canadian Foundation of Innovation (CFI; #38958). JF is supported by a CIHR New Investigator Award and by a Junior 1 & 2 Investigator Award by the (FRQS). DL’s work is supported by a Canada Foundation for Innovation John R. Evans Leaders Fund award (CFI-JELF #38033), Startup Funds from the McGill University Faculty of Medicine, and a CIHR Project Grant (#168959). DL is also recipient of a FRQS, Chercheur-Boursier Junior 1 Award (#284497). CD acknowledges support by a Rahel Hirsch stipend.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors want to thank Julien Leconte and Camille Stegen from the Flow Cytometry and Cell Sorting Facility at the Life Sciences Complex at McGill University. The facility acknowledges support by the Canada Foundation for Innovation (CFI) and McGill University’s Faculty of Medicine. The authors further thank Marie-Hélène Lacombe from the Research Institute of the McGill University Health Centre (RI-MUHC) immunophenotyping platform for excellent technical support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.664218/full#supplementary-material
Supplementary Figure 1 | Gating strategies for the identification of pulmonary type 2 cell populations. (A) Multicolor flow cytometry gating strategies to identify murine pulmonary (B) eosinophils (single, live CD45+SSChiSiglec-F+CD11c-), (C) cDC2s (single, live CD45+CD64- Lin-MHCII+CD26+CD11c+XCR1-CD172a+) as well as (D) ILC2s (single, live CD45+ Lin-Thy-1+CD127+KLRG1+). cDC2, conventional type 2 dendritic cell.
Supplementary Figure 2 | Gating strategies for the identification of bone marrow ILC2 progenitor populations and isolation of murine lung ILC2s. Flow cytometry gating strategies to (A) identify bone marrow CLP (single live Lin-CD127+Flt3+a4b7-), CHILP (single live Lin-CD127+Flt3+a4b7+CD25-) and ILC2P (single live Lin-CD127+Flt3+a4b7+ CD25+) populations and (B) isolate murine lung ILC2s (single live Lin-CD45+Thy-1+ST2+CD25+). CLP, common lymphoid progenitor; CHILP, common helper ILC2 progenitor; ILC2P, ILC2 progenitor.
Supplementary Figure 3 | Gating strategies for the isolation of murine bone marrow-derived ILC2 progenitors and small intestinal ILC2s. Flow cytometry gating strategies to isolate (A) bone marrow-derived ILC2 progenitors (single Lin- Sca-1+c-Kit- CD25+) and (B) murine small intestinal ILC2s (single live CD45+Lin- KLRG1+CD127+).
Supplementary Figure 4 | Absence of c-Rel does not affect GATA3 expression or early ILC2 proliferation upon ex vivo IL-33 challenge. Isolated WT (grey bars) and Rel-/- (red bars) lung ILC2s were left untreated (-) or cultured with IL-2, IL-7, IL-33 (all 10 ng/ml) or indicated combinations for 48 hours and analyzed by flow cytometry. (A) GATA3 expression, shown as gMFI. (B) Frequencies of Ki-67-expressing lung ILC2s. Data points are representative of two independent experiments with two biological replicates for each stimulation condition. Data are shown as mean ± SD with **p < 0.01 as determined by two-tailed t test (unpaired). gMFI, geometric mean fluorescence intensity.
1. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate Production of T(H)2 Cytokines by Adipose Tissue-Associated C-Kit(+)Sca-1(+) Lymphoid Cells. Nature (2010) 463:540–4. doi: 10.1038/nature08636
2. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes Represent a New Innate Effector Leukocyte That Mediates Type-2 Immunity. Nature (2010) 464:1367–70. doi: 10.1038/nature08900
3. Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically Dispersed Innate IL-13-Expressing Cells in Type 2 Immunity. Proc Natl Acad Sci USA (2010) 107:11489–94. doi: 10.1073/pnas.1003988107
4. Halim TY, Krauss RH, Sun AC, Takei F. Lung Natural Helper Cells Are a Critical Source of Th2 Cell-Type Cytokines in Protease Allergen-Induced Airway Inflammation. Immunity (2012) 36:451–63. doi: 10.1016/j.immuni.2011.12.020
5. Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13-Producing Nuocytes Arise During Allergic Lung Inflammation and Contribute to Airways Hyperreactivity. J Allergy Clin Immunol (2012) 129:191–8.e191–4. doi: 10.1016/j.jaci.2011.09.041
6. Mindt BC, Fritz JH, Duerr CU. Group 2 Innate Lymphoid Cells in Pulmonary Immunity and Tissue Homeostasis. Front Immunol (2018) 9:840. doi: 10.3389/fimmu.2018.00840
7. Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, et al. IL-33 Is More Potent Than IL-25 in Provoking IL-13-Producing Nuocytes (Type 2 Innate Lymphoid Cells) and Airway Contraction. J Allergy Clin Immunol (2013) 132:933–41. doi: 10.1016/j.jaci.2013.05.012
8. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity (2015) 42:1005–19. doi: 10.1016/j.immuni.2015.06.006
9. Hayden MS, Ghosh S. Shared Principles in NF-KappaB Signaling. Cell (2008) 132:344–62. doi: 10.1016/j.cell.2008.01.020
10. Liou HC, Hsia CY. Distinctions Between C-Rel and Other NF-kappaB Proteins in Immunity and Disease. Bioessays (2003) 25:767–80. doi: 10.1002/bies.10306
11. Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, et al. An NF-kappaB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity (2017) 47:450–65.e455. doi: 10.1016/j.immuni.2017.08.010
12. Nagashima H, Mahlakoiv T, Shih HY, Davis FP, Meylan F, Huang Y, et al. Neuropeptide CGRP Limits Group 2 Innate Lymphoid Cell Responses and Constrains Type 2 Inflammation. Immunity (2019) 51:682–95.e686. doi: 10.1016/j.immuni.2019.06.009
13. Nagashima H, Okuyama Y, Fujita T, Takeda T, Motomura Y, Moro K, et al. GITR Cosignal in ILC2s Controls Allergic Lung Inflammation. J Allergy Clin Immunol (2018) 141:1939–43.e1938. doi: 10.1016/j.jaci.2018.01.028
14. Galle-Treger L, Suzuki Y, Patel N, Sankaranarayanan I, Aron JL, Maazi H, et al. Nicotinic Acetylcholine Receptor Agonist Attenuates ILC2-Dependent Airway Hyperreactivity. Nat Commun (2016) 7:13202. doi: 10.1038/ncomms13202
15. Li Y, Chen S, Chi Y, Yang Y, Chen X, Wang H, et al. Kinetics of the Accumulation of Group 2 Innate Lymphoid Cells in IL-33-Induced and IL-25-Induced Murine Models of Asthma: A Potential Role for the Chemokine CXCL16. Cell Mol Immunol (2019) 16:75–86. doi: 10.1038/s41423-018-0182-0
16. Zhang L, Ying Y, Chen S, Arnold PR, Tian F, Minze LJ, et al. The Transcription Factor RelB Restrains Group 2 Innate Lymphoid Cells and Type 2 Immune Pathology In Vivo. Cell Mol Immunol (2020) 18(1):230–42. doi: 10.1038/s41423-020-0404-0
17. Donovan CE, Mark DA, He HZ, Liou HC, Kobzik L, Wang Y, et al. NF-Kappa B/Rel Transcription Factors: C-Rel Promotes Airway Hyperresponsiveness and Allergic Pulmonary Inflammation. J Immunol (1999) 163:6827–33.
18. Kim SJ, Kim JW, Kim YH, Lee SH, Yoon HK, Kim CH, et al. Effects of Tranilast and Pentoxifylline in a Mouse Model of Chronic Asthma Using House Dust Mite Antigen. J Asthma (2009) 46:884–94. doi: 10.3109/02770900903089998
19. Grinberg-Bleyer Y, Oh H, Desrichard, Bhatt DM, Caron R, Chan TA, et al. NF-KappaB C-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell (2017) 170:1096–108.e1013. doi: 10.1016/j.cell.2017.08.004
20. Kumar RK, Herbert C, Thomas PS, Wollin L, Beume R, Yang M, et al. Inhibition of Inflammation and Remodeling by Roflumilast and Dexamethasone in Murine Chronic Asthma. J Pharmacol Exp Ther (2003) 307:349–55. doi: 10.1124/jpet.103.053819
21. Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, et al. Mice Lacking the C-Rel Proto-Oncogene Exhibit Defects in Lymphocyte Proliferation, Humoral Immunity, and Interleukin-2 Expression. Genes Dev (1995) 9:1965–77. doi: 10.1101/gad.9.16.1965
22. Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I Interferon Restricts Type 2 Immunopathology Through the Regulation of Group 2 Innate Lymphoid Cells. Nat Immunol (2016) 17:65–75. doi: 10.1038/ni.3308
23. Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, Riabowol K. REAP: A Two Minute Cell Fractionation Method. BMC Res Notes (2010) 3:294. doi: 10.1186/1756-0500-3-294
24. Hernandez PP, Mahlakoiv T, Yang I, Schwierzeck V, Nguyen N, Guendel F, et al. Interferon-Lambda and Interleukin 22 Act Synergistically for the Induction of Interferon-Stimulated Genes and Control of Rotavirus Infection. Nat Immunol (2015) 16:698–707.
25. Mohapatra A, Van Dyken SJ, Schneider C, Nussbaum JC, Liang HE, Locksley RM. Group 2 Innate Lymphoid Cells Utilize the IRF4-IL-9 Module to Coordinate Epithelial Cell Maintenance of Lung Homeostasis. Mucosal Immunol (2016) 9:275–86. doi: 10.1016/j.celrep.2018.06.005
26. Kabata H, Moro K, Koyasu S. The Group 2 Innate Lymphoid Cell (ILC2) Regulatory Network and Its Underlying Mechanisms. Immunol Rev (2018) 286:37–52. doi: 10.1111/imr.12706
27. Paclik D, Stehle C, Lahmann A, Hutloff A, Romagnani C. ICOS Regulates the Pool of Group 2 Innate Lymphoid Cells Under Homeostatic and Inflammatory Conditions in Mice. Eur J Immunol (2015) 45:2766–72. doi: 10.1002/eji.201545635
28. Maazi H, Patel N, Sankaranarayanan I, Suzuki Y, Rigas D, Soroosh P, et al. ICOS:ICOS-Ligand Interaction Is Required for Type 2 Innate Lymphoid Cell Function, Homeostasis, and Induction of Airway Hyperreactivity. Immunity (2015) 42:538–51. doi: 10.1016/j.immuni.2015.02.007
29. Meylan F, Hawley ET, Barron L, Barlow JL, Penumetcha P, Pelletier M, et al. The TNF-Family Cytokine TL1A Promotes Allergic Immunopathology Through Group 2 Innate Lymphoid Cells. Mucosal Immunol (2014) 7:958–68. doi: 10.1038/mi.2013.114
30. Yu X, Pappu R, Ramirez-Carrozzi V, Ota N, Caplazi P, Zhang J, et al. TNF Superfamily Member TL1A Elicits Type 2 Innate Lymphoid Cells at Mucosal Barriers. Mucosal Immunol (2014) 7:730–40. doi: 10.1038/mi.2013.92
31. Galle-Treger L, Sankaranarayanan I, Hurrell BP, Howard E, Lo R, Maazi H, et al. Costimulation of Type-2 Innate Lymphoid Cells by GITR Promotes Effector Function and Ameliorates Type 2 Diabetes. Nat Commun (2019) 10:713. doi: 10.1038/s41467-019-08449-x
32. Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, et al. Tissue-Restricted Adaptive Type 2 Immunity Is Orchestrated by Expression of the Costimulatory Molecule OX40L on Group 2 Innate Lymphoid Cells. Immunity (2018) 48:1195–207.e1196. doi: 10.1016/j.immuni.2018.05.003
33. Liou HC, Jin Z, Tumang J, Andjelic S, Smith KA, Liou ML, et al. C-Rel Is Crucial for Lymphocyte Proliferation But Dispensable for T Cell Effector Function. Int Immunol (1999) 11:361–71. doi: 10.1093/intimm/11.3.361
34. Tumang JR, Owyang A, Andjelic S, Jin Z, Hardy RR, Liou ML, et al. Et al. C-Rel Is Essential for B Lymphocyte Survival and Cell Cycle Progression. Eur J Immunol (1998) 28:4299–312. doi: 10.1002/(SICI)1521-4141(199812)28:12<4299::AID-IMMU4299>3.0.CO;2-Y
35. Grossmann M, O'Reilly LA, Gugasyan R, Strasser A, Adams JM, Gerondakis S, et al. The Anti-Apoptotic Activities of Rel and RelA Required During B-Cell Maturation Involve the Regulation of Bcl-2 Expression. EMBO J (2000) 19:6351–60. doi: 10.1093/emboj/19.23.6351
36. Gilmore TD, Kalaitzidis D, Liang MC, Starczynowski DT. The C-Rel Transcription Factor and B-Cell Proliferation: A Deal With the Devil. Oncogene (2004) 23:2275–86. doi: 10.1038/sj.onc.1207410
37. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-Kappab: A Blossoming of Relevance to Human Pathobiology. Cell (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
38. Collins PD, Marleau S, Griffiths-Johnson DA, Jose PJ, Williams TJ. Cooperation Between Interleukin-5 and the Chemokine Eotaxin to Induce Eosinophil Accumulation In Vivo. J Exp Med (1995) 182:1169–74. doi: 10.1084/jem.182.4.1169
39. Perros F, Hoogsteden HC, Coyle AJ, Lambrecht BN, Hammad H. Blockade of CCR4 in a Humanized Model of Asthma Reveals a Critical Role for DC-Derived CCL17 and CCL22 in Attracting Th2 Cells and Inducing Airway Inflammation. Allergy (2009) 64:995–1002. doi: 10.1111/j.1398-9995.2009.02095.x
40. Knipfer L, Schulz-Kuhnt A, Kindermann M, Greif V, Symowski C, Voehringer D, et al. A CCL1/CCR8-Dependent Feed-Forward Mechanism Drives ILC2 Functions in Type 2-Mediated Inflammation. J Exp Med (2019) 216:2763–77. doi: 10.1084/jem.20182111
41. Turner JE, Morrison JP, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, et al. IL-9-Mediated Survival of Type 2 Innate Lymphoid Cells Promotes Damage Control in Helminth-Induced Lung Inflammation. J Exp Med (2013) 210:2951–65. doi: 10.1084/jem.20130071
42. Koch S, Sopel N, Finotto S. Th9 and Other IL-9-Producing Cells in Allergic Asthma. Semin Immunopathol (2017) 39:55–68. doi: 10.1007/s00281-016-0601-1
43. Licona-Limon P, Arias-Rojas A, Olguin-Martinez E. IL-9 and Th9 in Parasite Immunity. Semin Immunopathol (2017) 39:29–38. doi: 10.1007/s00281-016-0606-9
44. Willart MA, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, et al. Interleukin-1alpha Controls Allergic Sensitization to Inhaled House Dust Mite via the Epithelial Release of GM-CSF and IL-33. J Exp Med (2012) 209:1505–17. doi: 10.1084/jem.20112691
45. Sheih A, Parks WC, Ziegler SF. GM-CSF Produced by the Airway Epithelium Is Required for Sensitization to Cockroach Allergen. Mucosal Immunol (2017) 10:705–15. doi: 10.1038/mi.2016.90
46. Rao S, Gerondakis S, Woltring D, Shannon MF. C-Rel Is Required for Chromatin Remodeling Across the IL-2 Gene Promoter. J Immunol (2003) 170:3724–31. doi: 10.4049/jimmunol.170.7.3724
47. Tan TH, Huang GP, Sica A, Ghosh P, Young HA, Longo DL, et al. Kappa B Site-Dependent Activation of the Interleukin-2 Receptor Alpha-Chain Gene Promoter by Human C-Rel. Mol Cell Biol (1992) 12:4067–75. doi: 10.1128/MCB.12.9.4067
48. Roediger B, Kyle R, Tay SS, Mitchell AJ, Bolton HA, Guy TV, et al. IL-2 Is a Critical Regulator of Group 2 Innate Lymphoid Cell Function During Pulmonary Inflammation. J Allergy Clin Immunol (2015) 136:1653–.e1657. doi: 10.1016/j.jaci.2015.03.043
49. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-Mediated Dialog Between Group 2 Innate Lymphoid Cells and CD4(+) T Cells Potentiates Type 2 Immunity and Promotes Parasitic Helminth Expulsion. Immunity (2014) 41:283–95. doi: 10.1016/j.immuni.2014.06.016
50. Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 Innate Lymphoid Cells Drive CD4+ Th2 Cell Responses. J Immunol (2014) 192:2442–8. doi: 10.4049/jimmunol.1300974
51. Bunting K, Rao S, Hardy K, Woltring D, Denyer GS, Wang J, et al. Genome-Wide Analysis of Gene Expression in T Cells to Identify Targets of the NF-Kappa B Transcription Factor C-Rel. J Immunol (2007) 178:7097–109. doi: 10.4049/jimmunol.178.11.7097
52. Chen L, Flies DB. Molecular Mechanisms of T Cell Co-Stimulation and Co-Inhibition. Nat Rev Immunol (2013) 13:227–42. doi: 10.1038/nri3405
53. Hurrell BP, Shafiei Jahani P, Akbari O. Social Networking of Group Two Innate Lymphoid Cells in Allergy and Asthma. Front Immunol (2018) 9:2694. doi: 10.3389/fimmu.2018.02694
54. Carrasco D, Weih F, Bravo R. Developmental Expression of the Mouse C-Rel Proto-Oncogene in Hematopoietic Organs. Development (1994) 120:2991–3004. doi: 10.1242/dev.120.10.2991
55. Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, et al. IL-33 Is an Unconventional Alarmin That Stimulates IL-2 Secretion by Dendritic Cells to Selectively Expand IL-33r/ST2+ Regulatory T Cells. J Immunol (2014) 193:4010–20. doi: 10.4049/jimmunol.1400481
56. Iwashima M, Takamatsu M, Yamagishi H, Hatanaka Y, Huang YY, McGinty C, et al. Genetic Evidence for Shc Requirement in TCR-Induced C-Rel Nuclear Translocation and IL-2 Expression. Proc Natl Acad Sci USA (2002) 99:4544–9. doi: 10.1073/pnas.082647499
57. Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic Lethality and Liver Degeneration in Mice Lacking the RelA Component of NF-Kappa B. Nature (1995) 376:167–70. doi: 10.1038/376167a0
58. DiDonato JA, Mercurio F, Karin M. NF-kappaB and the Link Between Inflammation and Cancer. Immunol Rev (2012) 246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x
59. Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, et al. ILC2s Amplify PD-1 Blockade by Activating Tissue-Specific Cancer Immunity. Nature (2020) 579:130–5. doi: 10.1038/s41586-020-2015-4
60. Lu B, Yang M, Wang Q. Interleukin-33 in Tumorigenesis, Tumor Immune Evasion, and Cancer Immunotherapy. J Mol Med (Berl) (2016) 94:535–43. doi: 10.1007/s00109-016-1397-0
61. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate Lymphoid Cells Promote Lung-Tissue Homeostasis After Infection With Influenza Virus. Nat Immunol (2011) 12:1045–54. doi: 10.1038/ni.2131
62. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate Lymphoid Cells Mediate Influenza-Induced Airway Hyper-Reactivity Independently of Adaptive Immunity. Nat Immunol (2011) 12:631–8. doi: 10.1038/ni.2045
63. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D, et al. IL-33 Promotes an Innate Immune Pathway of Intestinal Tissue Protection Dependent on Amphiregulin-EGFR Interactions. Proc Natl Acad Sci USA (2015) 112:10762–7. doi: 10.1073/pnas.1509070112
64. Jiang HR, Milovanovic M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 Attenuates EAE by Suppressing IL-17 and IFN-Gamma Production and Inducing Alternatively Activated Macrophages. Eur J Immunol (2012) 42:1804–14. doi: 10.1002/eji.201141947
65. Milovanovic M, Volarevic V, Ljujic B, Radosavljevic G, Jovanovic I, Arsenijevic N, et al. Deletion of IL-33r (ST2) Abrogates Resistance to EAE in BALB/C Mice by Enhancing Polarization of APC to Inflammatory Phenotype. PloS One (2012) 7:e45225. doi: 10.1371/journal.pone.0045225
Keywords: group 2 innate lymphoid cell (ILC2), IL-33, c-Rel, type 2 immunity, airway inflammation
Citation: Mindt BC, Krisna SS, Duerr CU, Mancini M, Richer L, Vidal SM, Gerondakis S, Langlais D and Fritz JH (2021) The NF-κB Transcription Factor c-Rel Modulates Group 2 Innate Lymphoid Cell Effector Functions and Drives Allergic Airway Inflammation. Front. Immunol. 12:664218. doi: 10.3389/fimmu.2021.664218
Received: 04 February 2021; Accepted: 27 August 2021;
Published: 16 November 2021.
Edited by:
Malcolm Ronald Starkey, Monash University, AustraliaReviewed by:
Fumio Takei, University of British Columbia, CanadaCopyright © 2021 Mindt, Krisna, Duerr, Mancini, Richer, Vidal, Gerondakis, Langlais and Fritz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jörg H. Fritz, am9yZy5mcml0ekBtY2dpbGwuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.