 Anna Gioseffi
Anna Gioseffi Mariola J. Edelmann
Mariola J. Edelmann Peter E. Kima
Peter E. Kima
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 20 April 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.662944
This article is part of the Research TopicPhagocytes in Immunity: Linking Material Internalization to Immune Responses and Therapeutic StrategiesView all 17 articles
Extracellular vesicles (EVs) have garnered significant interest in recent years due to their contributions to cell-to-cell communication and disease processes. EVs are composed of a complex profile of bioactive molecules, which include lipids, nucleic acids, metabolites, and proteins. Although the biogenesis of EVs released by cells under various normal and abnormal conditions has been well-studied, there is incomplete knowledge about how infection influences EV biogenesis. EVs from infected cells contain specific molecules of both host and pathogen origin that may contribute to pathogenesis and the elicitation of the host immune response. Intracellular pathogens exhibit diverse lifestyles that undoubtedly dictate the mechanisms by which their molecules enter the cell’s exosome biogenesis schemes. We will discuss the current understanding of the mechanisms used during infection to traffic molecules from their vacuolar niche to host EVs by selected intravacuolar pathogens. We initially review general exosome biogenesis schemes and then discuss what is known about EV biogenesis in Mycobacterium, Plasmodium, Toxoplasma, and Leishmania infections, which are pathogens that reside within membrane delimited compartments in phagocytes at some time in their life cycle within mammalian hosts. The review includes discussion of the need for further studies into the biogenesis of EVs to better understand the contributions of these vesicles to host-pathogen interactions, and to uncover potential therapeutic targets to control these pathogens.
There are myriad facets of host–pathogen interactions that contribute to infection outcomes. Such is the case with intracellular pathogenesis, which includes organisms that use the host biosynthetic machinery to propagate themselves, organisms that reside in the cell cytoplasm and organisms that reside within vacuolar compartments in host cells. Progress in our understanding of mechanisms deployed by pathogens to exploit or subvert host mechanisms is uneven. It should therefore be enlightening to consider current knowledge of pathogen-host interactions by pathogens that share important characteristics. Our focus will be on pathogens that reside within membrane delimited vacuolar compartments in the cell. Even among this relatively small subset of pathogens, there is great diversity in pathogen strategies to interface with the cell, including differences in the composition and interactions of the pathogen-containing vacuolar compartments. Each pathogen elaborates unique strategies to translocate molecules from their intravacuolar niche into the cell cytosol or to other host cell organelles where they target host cell processes. It is presumed that some of the molecules that are released into the cell cytosol may access the exosome biogenesis machinery of host cells. The intravacuolar pathogens that we will discuss infect phagocytes at some point in their life cycle. Phagocytes are residents of tissues where they play important roles in tissue homeostasis and disease [reviewed in (1)]. Phagocytes are also recruited to sites of tissue damage from infection or other insults, where they proceed to limit the infection by various strategies, including ingesting and destroying any intruders, elaboration of molecular mediators to recruit and activate cells, removal of damaged host cells, or attempts to wall off the site. Paradoxically, phagocytes are unwitting hosts of a wide range of pathogens, including viruses, bacteria, and eukaryotic parasites. Infection of phagocytes and other host cells by the intravacuolar pathogens discussed here results in diseases that cause tremendous human suffering.
Numerous studies have shown that extracellular vesicles (EVs) released by phagocytic cells infected with intracellular pathogens hold infection-specific molecular cargo that may contribute to pathogenesis and host immune responses [reviewed in (2–4)]. Despite the abundance of proteomic and functional data, the process of EV biogenesis during infection and mechanisms by which pathogen-derived molecules are packaged into host exosomes are poorly understood. Insightful studies of EV biogenesis in the context of infection have focused on viral pathogens, revealing that many viruses utilize the endosomal sorting complex required for transport (ESCRT) machinery for viral egress. However, the biogenesis of EVs from infection by intracellular bacteria or intracellular eukaryotic pathogens remains poorly understood. The purpose of this review is to discuss the current understanding of the mechanisms of EV biogenesis in the context of intravacuolar pathogen infection to highlight each pathogen’s strategies to exploit host EV biogenesis schemes. We initially discuss universal characteristics and EV biogenesis schemes, after which the situation in the context of infection is considered.
Extracellular vesicles are secreted by all mammalian cells and can be isolated from various bodily fluids, including blood, urine, breast milk, tears, and cerebrospinal fluid (5). Extracellular vesicles have garnered significant interest in recent years because of their ability to transfer potentially important intercellular communication mediators, including proteins (6). This function is of particular interest in the context of host-pathogen interactions, as EVs may be critical mediators of host-pathogen communication and contribute to pathogenesis. Exosomes may be classified by their size, cell of origin, biogenesis, or proposed function (7–9). Apoptotic bodies are membrane-enveloped vesicles that range in diameter between 50 - 5000nm released via the blebbing of cells undergoing apoptosis (5, 8, 10). The existence of apoptotic bodies has been long known and studied. Healthy cells also shed extracellular vesicles, including microvesicles and exosomes (11). Microvesicles are membrane-bound vesicles released through outward budding and fission of the plasma (11, 12). Microvesicles are a heterogeneous group of vesicles that have been referred to by various terms including, ectosomes, shedding vesicles, microparticles, or platelet dust; that range in size from 100 - >1000nm in diameter (12, 13). Lastly, exosomes are membrane-enclosed vesicles between 50-150 nm in diameter that are secreted when multivesicular bodies (MVBs) in the endosomal pathway fuse with the plasma membrane and release intraluminal vesicles (ILVs) into the extracellular space (13). Together, these three classes of vesicles whose minimal characteristics and nomenclature have been standardized (14), are set apart in significant ways by both their biogenesis and biological functions.
Exosomes were first described in 1987 (15) and have since been found to be secreted by nearly all eukaryotic cells except for mature red blood cells, which do not possess endocytic capacities (16). Exosomes are secreted by healthy cells continuously, and their cargo and pattern of release can be altered by conditions of stress or infection (17). These nanosized vesicles contain thousands of cell-specific molecules, including proteins, nucleic acids, and lipids, enclosed within a single lipid bilayer membrane (18). Some of these molecules are conserved across exosomes from different origins, including tetraspanins (CD9, CD63, and CD81), proteins involved in intracellular trafficking (Rab GTPases, annexins), chaperones (Hsc70), biogenesis factors (ALIX), and proteins associated with signal transduction (14-3-3 proteins) (11). However, the exact composition of exosomes is dynamic and reflective of cellular context and the health of its cell of origin. Because the composition of exosomes is adaptive and reflective of the cell environment and condition, they are intriguing candidates for disease biomarker discovery through the isolation and screening of exosomes from patient bodily fluids (19).
Exosomes are of endosomal origin and are created when intraluminal vesicles (ILVs) form by inward budding of the early endosome (EE) (20). Early endosomes containing ILVs then mature into MVBs and are directed either to the lysosome for degradation or fuse with the plasma membrane to release the ILVs to the extracellular space as exosomes (20). Exosomes contain an assortment of molecular cargo, including membrane proteins with exposed extracellular domains on the exosomal surface and cytosolic proteins enclosed within their lumen (21). The protein composition of exosomes is specific and not reflective of the total cell, indicating that there are specific mechanisms to control the loading of select molecules into exosomes (22). Exosome biogenesis and the process of selective protein loading remain poorly understood phenomena. It is known, however, that the process of exosome biogenesis is driven by at least two general mechanisms: endosomal sorting complexes required for transport (ESCRT)-dependent and ESCRT-independent (tetraspanin or lipid-dependent) processes (22).
The ESCRT machinery was first shown to play a role in sorting proteins into MVBs in yeast in 2001 (23). The ESCRT complex comprises nearly 30 proteins assembled into five coordinating subcomplexes that function in a stepwise fashion (24): ESCRT-0, -I, -II, and -III, and associated AAA ATPase Vps4 complex (25). The primary function of ESCRT in ancestral organisms was to constrict and sever narrow membrane necks by a currently unknown mechanism of membrane scission (26). The ESCRT machinery is involved in many eukaryotic cellular processes, including the sorting of ubiquitinated proteins into ILVs for lysosomal degradation, viral egress, and membrane scission during cytokinesis (27). More recently, ESCRT components such as Tsg101 and Alix have been identified in exosome preparations from different sources, which suggested the involvement of ESCRT in the biogenesis of these vesicles (25).
The ESCRT subcomplexes perform a series of synchronized tasks to drive both cargo loading and physical membrane-remodeling and scission, leading to the production of ILVs: ESCRT-0 sequesters ubiquitinated cargo, ESCRT-I, -II, and -III control ILV budding, and Vps4 regulates membrane scission (28). ESCRT-0 consists of two subunits, hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs) and signal-transducing adaptor molecule 1/2 (STAM1/2), and can bind both phosphatidylinositol 3-phosphate (PtdIns3P) and ubiquitin, providing membrane recruitment, endosomal specificity, and interaction with ubiquitinated target proteins (29). ESCRT-0, ESCRT-I, and ESCRT-II all contain ubiquitin-binding domains, suggesting that ubiquitination is important for the selective loading of cargo proteins into exosomes (30). ESCRT-I consists of tumor susceptibility gene 101 (Tsg101), Vps28, Vps37, and multivesicular body 12 (hMvb12) and interacts with both ESCRT-0 and ESCRT-II using domains located on opposite ends of the complex (31). ESCRT-I then recruits ESCRT-II, a Y-shaped subcomplex consisting of EAP30, EAP45, and two subunits of EAP20, which cooperates with ESCRT-0 to provide further endosomal specificity and recruits ESCRT-III (32). ESCRT-III is composed of four main subunits- charged multivesicular body proteins (CHMPs) CHMP2A, B, CHMP6, CHMP3, and CHMP4A,B,C- and several adaptors and accessory proteins such as ALIX, which can recruit a deubiquitinating enzyme and is essential for cargo loading (33, 34), Deubiquitylation appears to be involved in loading cargo into ILVs, which are destined for degradation, while exosomal proteins remain ubiquitinated (25). The multimeric mechanoenzyme class I AAA (ATPase associated with various cellular activities) ATPase Vps4 is required to remove assembled ESCRT-III from the membrane before vesicle formation (35).
The PDZ (postsynaptic density protein, disc large, and zonula occludens) domain-containing protein syntenin is important for the biogenesis of a specific subclass of CD63-positive ILVs. Syntenin interacts with ALIX, pieces of the ESCRT machinery, and membrane receptors such as syndecans to produce ILVs using an alternative ESCRT pathway of cargo recruitment and vesicle budding (36), suggesting that ESCRT-dependent exosome production may be a flexible process resulting in a heterogeneous population of vesicles (9).
Depletion of all four ESCRT subcomplexes is insufficient to block exosome secretion entirely in eukaryotic cell lines but does result in drastic changes in cell morphology, such as enlarged, empty MVBs and irregularly shaped ILVs (37). Therefore, these larger, heterogeneous vesicles are produced by ESCRT-independent mechanisms that rely on other lipids and proteins for the loading and budding of exosomes (25).
Lipidomic analysis of EVs reveals that exosomes contain an overall low concentration of lipids, resulting in a high protein/lipid ratio (38). The lipid composition of exosomes is reminiscent of detergent-resistant lipid rafts, and it is enriched in cholesterol, ceramide, and sphingolipids essential for the ESCRT-independent sorting of cargo into ILVs (39). Inhibition of ceramide production using the neutral sphingomyelinase (nSMase) inhibitor GW4869 showed a marked reduction in exosome release, which appeared to be specific and independent from ESCRT-dependent mechanisms rather than a derangement of the entire endosomal system (39). Furthermore, ILVs destined to be secreted as exosomes contain ceramide while ILVs intended for lysosomal degradation contain another related lipid, lysobisphosphatidic acid (LBPA), which is absent in exosomes, suggesting that lipid composition is involved in the determination of vesicle fate (40, 41).
However, depletion of ceramide has no effect on MVB biogenesis or exosome secretion in human melanoma cells (42). In these cells, the tetraspanin CD63 was involved in the ESCRT-independent sorting of cargo into ILVs (42). Other tetraspanins, including CD82 and CD9, have also been shown to participate in the ceramide-dependent biogenesis of exosomes in healthy primary cells (43). Tetraspanin-enriched microdomains (TEMs) may act as sorting platforms for cargo molecules during the ESCRT-independent biogenesis of ILVs (Figure 1C). However, the exact mechanisms by which tetraspanins facilitate exosomal cargo sorting are unknown (44).
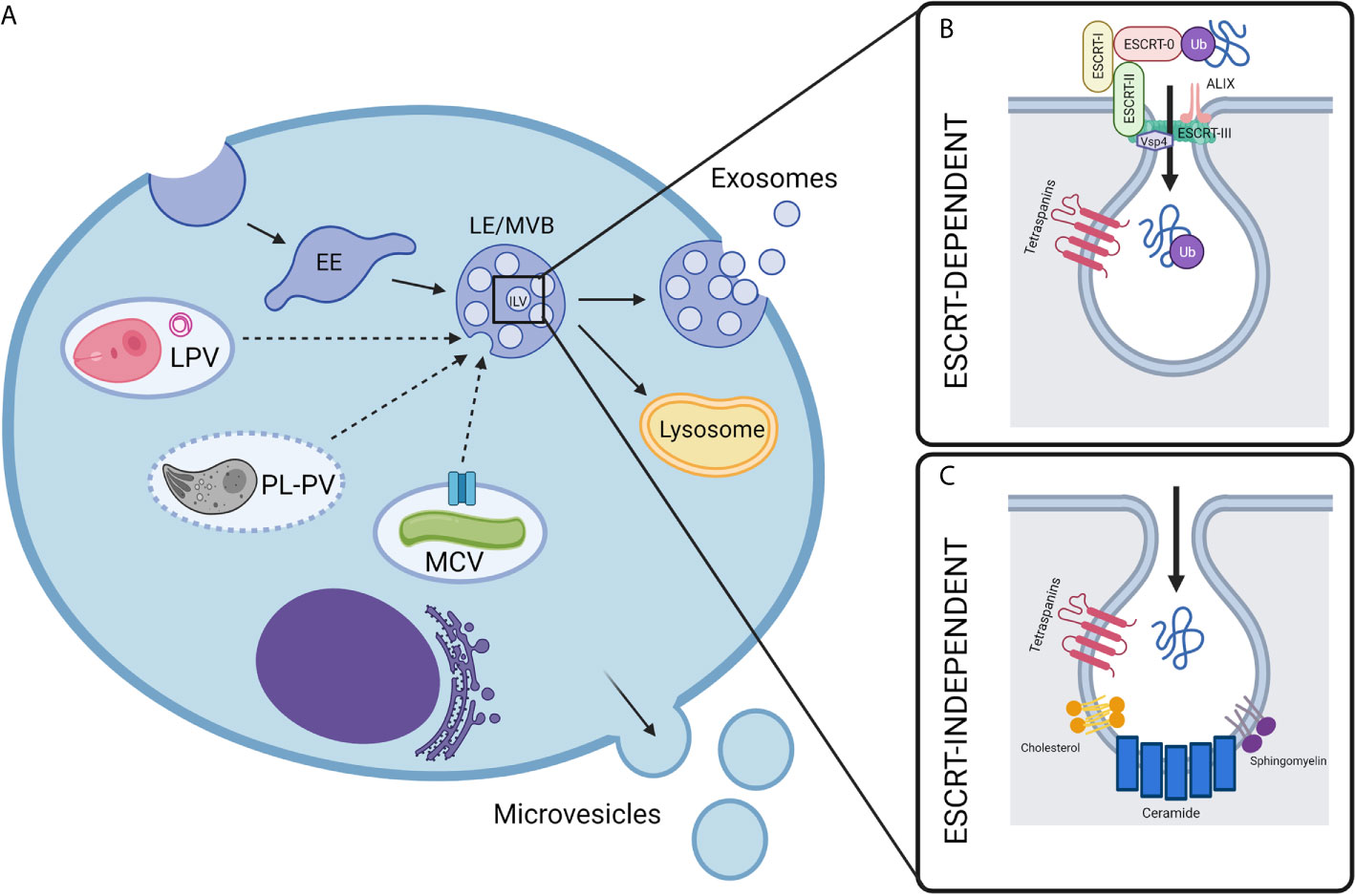
Figure 1 Biogenesis of extracellular vesicles in intravacuolar pathogen infections. (A) Mammalian cell-derived extracellular vesicles include exosomes and microvesicles, which can be characterized by their biogenesis, size, and composition. While microvesicles are generated by the direct outward budding of the cell’s outer membrane, exosomes are derived from an endocytic origin. First, early endosomes (EE) undergo inward budding. This forms intralumenal vesicles (ILVs) inside what is then called the late endosomal vesicle (LE) or multivesicular body (MVB). Depending on molecular signals, the MVB may then be destined for degradation by fusing with lysosomes or will fuse with the plasma membrane, releasing the ILVs to the extracellular space where they are then called exosomes. In the context of intravacuolar pathogens, exosomes are a possible mechanism for the release of pathogen molecules from infected cells. The exact mechanisms for how these pathogen molecules escape their respective vacuoles and are trafficked to host exosomes is not fully understood, but it is likely to be unique for each pathogen as discussed in this review. The composition of pathogen-containing vacuoles is diverse and requires unique methods for the exchange of molecules between host and pathogen. Mycobacteria utilize secA, type VII secretion system (represented by blue channel), and possibly other secretion systems to secrete exosome-bound proteins from the Mycobacterium containing vacuole (MCV). Plasmodium containing vacuoles (PL-PVs) conversely contain pores (represented by dashed vacuole boarder) through which small molecules may pass freely between the PV lumen and host cytosol, presenting easy access of pathogen molecules to host cytosol and exosome processes. Leishmania parasitophorous vacuoles (LPVs) are dynamic compartments that interact with the host’s endocytic and secretory pathways. Multimembranous structures within LPVs whose cellular origin is not known, may transport molecules from LPVs to host cell organelles including MVBs. Dashed arrows represent speculative pathways for trafficking of pathogen molecules into host ILVs. (B, C) Exosome biogenesis can occur by two general mechanisms- either ESCRT-dependent or ESCRT-independent. (B) ESCRT, or endosomal sorting complexes required for transport, is a specialized multi-subunit complex which allows for the recruitment of ubiquitinated proteins and the inward budding and scission of ILVs. ESCRT-0 recruits ubiquitinated proteins while ESCRT-I, ESCRT-II, and ESCRT-III facilitate ILV budding, and finally Vsp4 facilitates membrane scission. Accessory proteins such as ALIX are also involved which perform deubiquitylation for cargo loading. (C) Less is currently known about the mechanisms of ESCRT-independent exosome biogenesis, however it is proposed that ceramide and other lipids, such as sphingomyelin and cholesterol, as well as tetraspanins play a part in the trafficking of proteins and inward budding. Created with BioRender.com.
Exosomal protein composition is selective and dynamic, suggesting that there are regulated mechanisms involved in loading specific target molecules during vesicle biogenesis. Mass spectrometry analysis of exosomes reveals that post-translational modifications (PTMs) such as ubiquitination (mono-ubiquitination and poly-ubiquitination), the addition of small ubiquitin-related modifier (SUMOylation), phosphorylation, and glycosylation are all common modifications of exosome proteins and may be involved in the selective sorting of cargo molecules (45).
Ubiquitin can be bound to a target protein at numerous positions via the isopeptide bond of C-terminal glycine of ubiquitin with the ϵ-amino group of a lysine residue present in the target protein, resulting in a complex network of modifications (45). The relationship between ubiquitination and cargo sorting is unclear as protein sorting can also occur through ESCRT and ubiquitin- independent mechanisms (22). SUMO, another ubiquitin-like modifier, has also been shown to modify exosomal proteins and influence miRNAs packaging into exosomes (46). Also, phosphorylation has been found to work in conjunction with ubiquitination and lipid rafts to regulate the sorting of some specific subsets of proteins into the exosomes (47). Despite these observations, current studies are so far unable to unravel the complicated network of PTMs and their involvement in selective cargo trafficking and vesicle fate.
Extracellular vesicles are known to carry cargo whose composition is unique to the cell of origin and is modulated by the cell’s environment and growth conditions. This implies that the profile of cargo molecules in EVs from cells experiencing stressful growth conditions, including infection, will differ somewhat from the molecular profile of EVs released under non-stressful growth conditions. EVs therefore carry biomarkers that can inform on the staging of a disease process. It is no surprise therefore that exosomes are being touted as a source for promising biomarkers for cancer diagnosis and that they represent new targets for cancer therapy (48). In infectious diseases as well, knowledge of EV composition modulated by infection could similarly be exploited for development of biomarkers for diagnosis of infection and identification of immune targets. Exosomes released by macrophages infected with several intracellular pathogens, including Salmonella enterica and Mycobacterium tuberculosis, have been demonstrated to include pathogen antigens, stimulate a pro-inflammatory response in naïve cells (49) and protect against subsequent infection challenges (50), suggesting that exosomes may also be useful as cell-free vaccines against infectious pathogens.
Mycobacteria are acid-fast bacteria that prefer an intracellular lifestyle. Mycobacterium tuberculosis is a human pathogen that causes tuberculosis, and M. avium complex bacteria also lead to lung infection, although primarily in immunocompromised individuals. Finally, M. bovis bacillus Calmette-Guérin (BCG) is a vaccine strain of Mycobacterium. Mycobacteria can infect several cell types, such as neutrophils, macrophages, and monocytes, although alveolar macrophages appear to be the preferred host of Mycobacterium tuberculosis in the early phases of infection (51). During the host cell infection, Mycobacterium binds to the macrophage surface, followed by internalization of the bacteria into Mycobacterium containing vacuoles (MCV) that do not fuse with lysosomes, which ensures that the bacterium survives in the vacuole (52). This bacterium recruits several host proteins to the MCV surface, including rab5, to prevent phagolysosomal fusion at the early endosome stage. This phenomenon is reflected by the fact that MCVs have a low abundance of lysosomal markers, such as CD63, LAMP-1 and -2, or rab7 (53, 54). Mycobacterium uses the SecA2 pathway to secrete SapM and PknG effector proteins that affect phagosome and autophagosome maturation (55).
While surviving in the MCV, Mycobacterium components, pathogen-associated molecular patterns (PAMPs) appear to be translocated from the vacuole via secretion systems. It has been shown that cell wall constituents of M. avium, glycopeptidolipids, are released by infected macrophages (56). These glycopeptidolipids are trafficked from the MCV to multivesicular bodies (MVBs), and the trafficking process is enabled by the endocytic network. After the MVBs fuse with the plasma membrane, the released exosomes still contain the glycopeptidolipids, which are transferred from infected to uninfected macrophages where they stimulate proinflammatory mediators via Toll-like receptors (TLRs) -2, -4, that are dependent on the MyD88 pathway (56). Other studies showed that exosomes isolated from the bronchoalveolar lavage fluid (BALF) of M. bovis BCG–infected mice also stimulated proinflammatory responses in macrophages, resulting in TNF-α production (57). These vesicles contained the Mycobacteria components lipoarabinomannan and the 19-kDa lipoprotein. Intranasal vaccination with exosomes isolated from macrophages previously infected with M. bovis BCG and M. tuberculosis stimulated TNF-α or IL-12 production and aided in neutrophil and macrophage recruitment to the lung of mice (57). A similar experiment was performed with exosomes isolated from Mycobacterium-infected macrophages, which were used to treat naïve bone marrow-derived macrophages, revealing that these exosomes also stimulate other cytokines, such as GCSF, sICAM1, IL-1ra, MIP-1a, MIP-1b, MIP-2, RANTES, and MCP-5. In addition to inducing the release of cytokines, exosomes from Mycobacteria-infected cells have been shown to induce migration of macrophages. This phenomenon has been demonstrated in vivo, based on the fact that the intranasally injected exosomes resulted in the recruitment of CD11b+ cells into the lung (58).
Apart from exosomes’ effect on innate immune cells, exosomes derived from Mycobacteria-infected cells also stimulate adaptive immunity in the M. tuberculosis model. Exosomes derived from macrophages exposed to culture filtrate proteins (CFP) of M. tuberculosis induced pathogen‐specific IFN‐γ and IL‐2‐expressing CD4+ and CD8+ T cells. This Th1-biased immune response was specific to mice vaccinated with exosomes obtained from CFP-treated macrophages, while the BCG vaccine boosted the Th2 response. Those exosomes were also shown to prime a protective immune response at a level comparable to BCG and provided a booster to a previous BCG immunization, leading to a decrease in mycobacterial count in lung and spleen of mice challenged with aerosolized M. tuberculosis (50). Exosomes released in vivo during infection with M. tuberculosis also contribute to T cell response, which was shown by using rab27a-deficient mice (59), which have a defect in the exosome generation (60). The deletion of rab27a was correlated with an increased bacterial burden and decreased T cell activation, indicating the importance of exosomes in the T cell function (59).
Mycobacterial proteins are trafficked to exosomes, shown first for the 19-kDa lipoprotein (57). A proteomic study conducted on exosomes derived from M. tuberculosis-infected J774 cells identified 41 mycobacterial proteins, mostly predicted or known to be secreted proteins, including previously known immunogenic proteins ESAT-6, Ag85 complex proteins, MPT64, or MPT63 (61). Interestingly, some of the mycobacterial proteins in exosomes released from macrophages treated with M. tuberculosis CFP were identical to the ones contained in exosomes isolated from infected cells. This observation implies that mycobacterial proteins may have a signal for trafficking into exosomes upon entry into the cell via phagocytosis or endocytosis-based mechanisms (61). Similarly, exosomes obtained from sera of active TB patients also include M. tuberculosis proteins. This observation suggested that exosomes can serve as a source of peptide biomarkers for TB. In comparison to the exosomes isolated from the bronchoalveolar lavage (BAL) fluid from M. tuberculosis-infected BALB/c mice and infected J774 macrophages (61), five of these proteins were in common, including DnaK, PstS2, GlcB, HspX, and AcpM (62). The mycobacterial proteins contained within the host exosomes appear to be secreted via the SecA and type VII secretion system, although at least one of these proteins is not expected to be secreted (GabD1). This might mean that several different mechanisms, including bacterial lysis, are responsible for the release of mycobacterial proteins prior to their incorporation into exosomes (61).
The mechanisms that control the trafficking of these soluble bacterial antigens to the exosomal compartments are not currently known. For M. tuberculosis several pathways have been shown to be important in the antigen trafficking to exosomes. In addition to rab27a discussed above, they also showed reduced trafficking of M. tuberculosis 19 kDa lipoprotein, which were (59). This observation suggested the importance of rab27a pathway in the trafficking of mycobacterial proteins to exosomes.
The pathway that is crucial for the trafficking of proteins into macrophages is ESCRT dependent pathway. Knockdown of Tsg101 and Hrs, which are the ubiquitin-binding domains of ESCRT-1 and ESCRT-0, respectively, resulted in the reduction of exosomes produced in RAW 264.7 macrophages, which indicated that ubiquitin might be one of the mechanisms involved in the trafficking of proteins to MVBs and exosomes (63). Schorey et al. examined whether ubiquitination is a post-translational modification necessary for the trafficking of soluble mycobacterial antigens into the exosomes. Toward this goal, exosomes were purified from Mycobacterium-infected RAW264.7 macrophages, followed by pull-down of mono-ubiquitinated proteins. The western blotting of such mono-ubiquitinated proteins was done using an antibody that recognizes culture filtrate proteins of M. tuberculosis, and the results indicated that several mycobacterial proteins are ubiquitinated. Moreover, specific mycobacterial proteins KatG, HspX, and GroES were shown to be ubiquitinated by using this western blot technique (63). An inhibitor PYR-41 was used to treat cells prior to the exosome purification since this molecule inhibits the thioester bond formation between ubiquitin and E1. This compound treatment leads to a complete depletion of mycobacterial proteins in the collected exosomes. Further, mutation of a specific lysine residue in the mycobacterial protein HspX diminished its trafficking into the exosomes that was dependent on the clathrin-mediated endocytosis. Mono-ubiquitination is required for the trafficking of some proteins into the endosomes. Indeed, the trafficking of HspX was shown to be dependent on clathrin-mediated endocytosis, based on the treatment with Dynasore, which is an inhibitor of this uptake mechanism (63). In addition, expression of a C‐terminal fusion of ubiquitin to EGFP and M. tuberculosis proteins Ag85B and ESAT-6 in HEK 293 cells enhanced the delivery of these proteins into exosomes by ten-fold when they were coupled to ubiquitin (Figure 1). These exosomes were able to elicit a T cell response by stimulating the production of INFɣ‐secreting T lymphocytes in the lung and spleen (64).
These results collectively suggested that mono-ubiquitination could serve as a mechanism for the trafficking of bacterial proteins into the exosomes via clathrin-mediated endocytosis. The ubiquitin E3 ligases responsible for the ubiquitination of these bacterial proteins have not yet been identified, but it is likely that multiple E3 ligases exist that play this function in sorting of proteins to exosomes. Furthermore, apart from a better characterization of ubiquitination in this context, it would be interesting to investigate additional mechanisms that might guide mycobacterial proteins to vesicular compartments. Moreover, the fate of the mycobacterial proteins that are carried in exosomes to target cells is also unknown. It would be important to track the bacterial proteins carried to target cells via exosomes, in order to identify the colocalization of these proteins with intracellular compartments, such as endocytic vesicles and lysosomes. Furthermore, since exosomes can carry these mycobacterial proteins to the antigen-presenting cells, and because exosomes formed during the Mtb infection stimulate protective immunity against Mtb (50, 59), it is possible that the Mtb antigens are either loaded on MHC molecules or the entire exosomal complex containing MHC and Mtb antigenic peptide are exposed on the cell surface. The trafficking of the exosomal content within the target cell clearly deserves further mechanistic studies.
Apart from proteins, exosomes can also transmit other molecules from infected cells. In the case of M. tuberculosis infection, exosomes carry a smaller amount of microRNA (miRNA) than uninfected cells. However, transcripts regulating immune response are more abundant in exosomes derived from M. tuberculosis-infected macrophages. Apart from the host RNA, the vesicles are also capable of carrying mycobacterial RNA. Similar to proteins, the RNA cargo is transferred between cells via exosomes (65). Currently, a mechanism responsible for the trafficking of bacterial transcripts to the exosomes remains uncharacterized, although RNA‐binding protein hnRNPA2B1 has been shown to bind specific miRNAs (46). This and other RNA‐binding proteins are, therefore, likely capable of sorting relevant miRNAs into the vesicles.
Plasmodium parasites are apicomplexan parasites that cause malaria. It is estimated that over 100 million people are infected worldwide, and just under 1 million people succumb to these infections each year (66). Infections in humans are initiated by sporozoites deposited into a host by female Anopheles mosquitoes when they take a blood meal. The sporozoites establish a productive infection when they infect liver cells. Although hepatocytes are the principal hosts of parasites in the liver, it has been shown that sporozoites take up transient residence in Kupfer cells that mediate their access to hepatocytes (67, 68). It was also shown that after infection of hepatocytes, a small subset of liver parasites is acquired by monocyte-derived CD11c+ cells (69). Infection of CD11c+ cells and hepatocytes were shown to be dependent on their expression of the tetraspanin CD81, which coincidentally is a vital component of exosomes (70). The parasitophorous vacuoles (PVs) in which the parasites reside in hepatocytes exhibit unique characteristics compared to PVs that harbor other Apicomplexan parasites. For example, Plasmodium containing PVs rest close to the cell nucleus and establish interactions with the Endoplasmic Reticulum (ER), which is in contrast to Toxoplasma whose PVs establish a tight association with the Golgi apparatus, Mitochondria, and Endoplasmic Reticulum (ER) (71) of the host cell, Furthermore, Plasmodium-containing PVs have been shown to have pores through which nutrients smaller than 3000 Da can be exchanged between the PV lumen and cell cytosol (71). Parasites in PVs within liver cells undergo rapid division into merozoites. The PVs enlarge into giant syncytium-like compartments (71). Upon their release from the liver, merozoites enter the blood circulation, where they infect red blood cells. Of the five species of Plasmodium parasites that infect humans, Plasmodium falciparum and Plasmodium knowlesi infect all red blood cells. In contrast, Plasmodium vivax and Plasmodium ovale infect only reticulocytes. Plasmodium malariae infect older red blood cells. Many mechanistic studies on Plasmodium infections have been performed in experimental models with parasites of rodents, including P. berghei, P. yoelii and P. chabaudi that exhibit similarities and differences with Plasmodium falciparum and Plasmodium vivax, depending on the stage of infection.
As discussed above, during their life cycle, Plasmodium parasites infect distinct cell types that should be expected to employ mechanisms for exosome biogenesis. Several studies have explored the role of exosomes and microvesicles (previously called microparticles) in Plasmodium infections. Mature red blood cells are devoid of a nucleus and the endocytic cell machinery in their cytoplasm, including multivesicular bodies (MVBs) that play a critical role in exosome biogenesis in nucleated cells. Plasmodium within mature red blood cells have been shown to translocate over 300 parasite-derived proteins from their PVs into the red blood cell cytosol. These molecules that are distributed throughout the red blood cell, including on its cell surface, have been implicated in several functions, including the formation of Maurer’s clefts that appear to play some role in exosome formation in Plasmodium-infected cells (72). It is striking that only a subset of the molecules that are translocated into the red cell has been identified in EVs that are recovered from the supernatant fluid of cultured infected erythrocytes (73, 74). While EVs included proteins from the PV membrane and some well-studied surface molecules such as PfEBA and knob-associated molecules, they however, lacked other well studied molecules, including PfEMP1(a knob associated protein) and AMA-1. This observation suggests that there is a machinery outside of the parasite in the red blood cell that plays a pivotal role in selecting molecules for inclusion in EVs. Abdi et al. (74) analyzed exosomes released from red blood cells infected with a relatively earlier parasite passage. They identified over 50 more Plasmodium molecules in those EVs as compared to exosomes produced from a parasite line that had long been adapted to the in vitro culture conditions (74). Not surprisingly, many of the molecules that were identified in the study with the low passage parasites were involved in virulence. That study underscored the need to evaluate recently obtained field isolates and suggested that molecules in exosomes may play a role in the parasites’ virulence.
A couple of elegant studies have shown that exosomes can mediate intracellular communication between parasites within infected red blood cells [reviewed in (75)]. Studies using transgenic parasites expressing a drug resistance marker showed that DNA packaged in EVs could be exchanged by parasites in an infected cell, which results in the spread of a resistance marker (72). Another study showed that infected red blood cells selectively took up EVs produced by other infected cells, which then stimulated gametocyte production in the recipient infected cells (73). Exosomes have also been shown to stimulate the immune response by activating macrophages and neutrophils [reviewed in (75)]. They also play significant roles in cerebral malaria where among other activities, they promote vascular changes, including endothelial cell activation [reviewed in (76)]. Together, those studies demonstrated the critical role of EVs in Plasmodium infections and suggested that a greater understanding of their biogenesis and functions could be exploited to modify the course of malaria.
Studies on the ATP binding cassette transporter A1 have suggested a role for this molecule in EV biogenesis in Plasmodium infections. It had been shown that ATP binding cassette transporter A-1 (ABCA-1) plays a role in phosphatidylserine distribution at the plasma membrane (77). ABCA-1 knock-out mice were subsequently shown to be defective in EV release. Red blood cells from these mice produced reduced levels of EVs, which implicated this molecule in EV biogenesis in red blood cells [reviewed in (76)]. As mentioned briefly above, the molecules localized to Maurer’s clefts were identified in EVs from Plasmodium-infected RBCs. Studies by Regev-Rudzki et al. (72) identified and tracked the PfEMP1 trafficking protein (PfPTP) that associates with Maurer’s cleft (72). They proceeded to show that parasites that were genetically altered to lack expression of PTP-2 were defective in EV release when used to infect red blood cells. This provided compelling evidence of the role of Maurer’s clefts in EV biogenesis in infected cells.
In contrast to mature red blood cells, reticulocytes are nucleated and possess the biosynthetic machinery of mammalian nucleated cells. The composition and functions of exosomes released from Plasmodium-infected reticulocytes and liver cells have been described. The studies of Martin-Jaular et al. (78) that characterized exosomes released by BALB/c mice infected with non-lethal Plasmodium yoelii 17X described the presence of parasite-derived molecules in exosomes from infected reticulocytes (78). In that experimental model, where Plasmodium yoelii 17X infects reticulocytes, infections were initiated by intraperitoneal injection of infected cell blood cells. Up to 31 parasite-derived proteins were found to be included among the molecules in the reticulocyte-derive exosomes. Some of the parasite proteins that were identified included the serine-rich antigen (SERA) that is expressed by a multigene family and has been implicated in virulence [reviewed in (79)], merozoite surface antigens (MSP1 and MSP9), and heat shock protein 70. The mechanisms that led to their inclusion in reticulocyte derived exosomes have not been described. A more recent study by Gualdrón-López et al. (80) described the characterization of exosomes that were secreted from the liver stages of Plasmodium vivax infections (80). The research team took advantage of the human liver-chimeric (FRG huHep) mouse (81) in which an immunocompromised mouse with several genetic mutations was engrafted with human hepatocytes. FRG huHep mice support the complete development of the human parasites, P. falciparum (81) and P. vivax (82) that would not otherwise infect mice. The analysis of exosomes isolated from the blood plasma of the P. vivax infected FRG huHep mice (ExEF) identified 290 and 234 proteins from mouse and human origin, which included liver proteins that had previously been described from liver exosomes (80). This analysis also identified 17 parasite-derived molecules that were included in exosomes from liver-infected cells. When the authors compared human ExEF exosomes from infected animals versus uninfected control mice, they stated that several proteins were differentially associated with P. vivax infections (P-value < 0.05). This finding is consistent with studies from other infections that have shown that the infection influences host-derived molecules’ composition in exosomes. The list of parasite proteins in ExEF included heat shock protein 70 (HSP70), which was also seen in the study on infected reticulocyte-derived exosomes described above. A different variant of merozoite surface antigen (MSP3) was found in the ExEF, which differed from the variants found in the infected reticulocyte derived exosomes discussed above. Whether it is 17 or 31 parasite-derived proteins identified in exosomes from infected liver cells or from infected reticulocytes, it is currently unknown which characteristics of these proteins direct them to the host cellular exosome biogenesis machinery. It is also not known whether ubiquitination plays a role in exosome loading within Plasmodium-infected cells.
Toxoplasma are Apicomplexan parasites that can infect all warm-blooded animals, including mammals and birds. In humans, Toxoplasma have been implicated in a range of clinical presentations whose severity is determined by the individual’s immune status. Toxoplasma is acquired by ingestion of raw or inadequately cooked meat. It can also be acquired upon ingestion of oocyst dispersed in the environment in cat feces. This later mode of acquiring the infection by pregnant women is the subject of public health campaigns that dissuade women from changing cat litter, as infection during pregnancy can lead to congenital transmission resulting in stillbirths or hydrocephalus or retinal infections of the newborn. Toxoplasma can infect all nucleated cells in mammals. Parasite entry into cells involves the sequential deployment of molecules from Apicomplexan-specific organelles. The release of proteins from Rhoptries follows the discharge of micronemal proteins. Dense granule proteins are then released, which contribute to the formation of the parasitophorous vacuole and the intravacuolar network. Rastogi et al. (83), Håkansson (84), and Nadipuram (85) have described the export machinery of proteins from the Toxoplasma PVM and provided examples of molecules that are transported to the host cell (83–85). Whether displayed on the PVM or translocated to other host cell organelles or the cytosol, most of these molecules are potentially accessible to the exosome biogenesis machinery.
Studies of the potential role of exosomes in toxoplasmosis have evaluated exosomes that are released from axenic cultures of parasites (86) or exosomes that are released from dendritic cells that are pulse with Toxoplasma antigens (lysate) (87, 88) or mammalian cells that are infected with Toxoplasma (56, 89). Despite the obvious differences in the sources of Toxoplasma molecules, exosomes containing Toxoplasma molecules were shown to be able to stimulate naïve recipient cells to secrete cytokines. Injection into hosts in experimental studies led to the elaboration of a variety of Toxoplasma specific responses. Some studies of exosomes secreted from Toxoplasma, have shown that infection with Toxoplasma induces the release of a unique profile of protein and nucleic acids that is different from those released by uninfected cells. In infections of human foreskin fibroblast cells, Wowk analyzed exosomes from cells infected with Toxoplasma gondii and compared their protein content to exosomes from axenic cultured tachyzoites. They found 69 unique parasite derived proteins in infection derived EVs, however the number of parasite derived proteins was not stated (90). In another study in which dendritic cells were infected with Toxoplasma gondii, 12 differentially expressed miRNAs compared to exosomes from uninfected cells were identified (91). Further analysis predicted that in recipient cells, these miRNAs could be associated with a variety of biological processes, including signaling pathways involved in host ubiquitin system, innate immunity, biosynthesis, and transferase activity. Future studies will no doubt provide greater insight on the trafficking of these parasites derived molecules in the infected host cells.
Leishmania are members of the family Trypanomastidae in the order Kinetoplastida. Leishmania parasites are grouped into two subgenera: Leishmania (Leishmania) and Leishmania (Viannia) that are further classified into species and subspecies. Infection of humans results in a disease presentation that is mostly dependent on the parasite species. Parasites of the Leishmania (Leishmania) subgenus including L.(L) donovani, L.(L) infantum and L.(L) chagasi (with a few exceptions including L.(L) amazonensis and L.(L) major, L.(L) tropica that cause cutaneous infections) are the primary causative agents of visceral disease. In contrast, parasites of the Leishmania (Viannia) subgenus, including L.(V) braziliensis, L.(V) panamensis, cause cutaneous infections that can manifest as self-limiting lesions or disseminated lesions or mucocutaneous infections [reviewed in (92, 93)]. However, there are reports of visceral disease caused by parasites that ordinarily lead to cutaneous lesions, while other reports of cutaneous lesions have implicated parasites species that typically lead to visceral disease [reviewed in (94)]. These ‘unexpected’ disease presentations underscore the complexity of these infections and may be due to host genetics contributions and poorly defined environmental factors (94, 95). Leishmania parasites are transmitted by sandflies. Once inside the mammalian host, Leishmania infects phagocytic cells wherein they reside in Leishmania-containing parasitophorous vacuoles (LPVs). The Leishmania species determine LPV morphology. At the extremes of morphological differences, parasites of the L. mexicana complex (L. mexicana, L. amazonensis) reside within large communal LPVs that continuously distend. At the other extreme, L. donovani, L. chagasi/L. Infantum reside in tight LPVs that harbor a single parasite. After parasite replication and fission, daughter parasites segregate into secondary LPVs that also house individual parasites. All other Leishmania species reside in LPVs that may house one to four parasites. It is presently not known how LPV morphology differences affect Leishmania pathogenesis. LPVs are dynamic compartments composed of molecules from the host secretory pathway, including the endoplasmic reticulum (ER) and the endocytic pathway, including late endosomes and lysosomes (96). Although much has been learned about the molecular composition of LPVs, there are still unanswered questions about the biology of LPVs, including how Leishmania derived molecules are translocated across the LPV membrane and which signals mediate the trafficking of parasite-derived molecules to the intracellular sites where they express their functions. On this topic, a recent publication provided evidence of traffic of Leishmania proteins from LPVs to the ER in vesicles that otherwise transmit cargo between the Golgi and the ER (97). The authors proposed that this could be one route through which parasite molecules are retrieved from LPVs for distribution in the infected cell and beyond.
In natural infections, the parasite’s promastigote form is deposited in the skin of the host by the sandfly. The parasites commence a skin phase of the infection. After a transient residence in neutrophils, parasites are transferred to macrophages wherein they undergo replication as amastigotes (98, 99). In cutaneous infections, inflammatory cells are recruited to the bite site, which results in a cutaneous lesion over time. In infections by parasites that cause visceral infections, there is also an initial cutaneous phase (can last for several weeks) after which infected cells migrate to visceral organs where they replicate and form inflammatory lesions; this was first demonstrated in infections of hamsters (100). Parasites such as L. amazonensis and L. braziliensis that disseminate to secondary sites, proliferate for much longer periods at the primary site (months to years) before dissemination. Recent studies that have explored the phenotype of cells at primary and secondary sites in experimental infections with L. major, highlighted the expression of chemokine receptors CCR2+ and CX3CR1+ on the monocyte-derived cells that are the primary host cells of these parasites (99, 101). Infections in knockout mice that lack these receptors were limited, which suggested a role for chemokines in Leishmania-infected cell dissemination [Also reviewed in (102)]. Nonetheless, neither the triggers for disseminating the infection nor the factors that determine the selection of secondary sites to which parasites spread are not known. Could some of the mechanisms that promote metastatic tumor dissemination, such as the roles played by exosomes in tissue homing or organotropism (reviewed in (103, 104), be also important in the dissemination of Leishmania infections?
After parasites take up residence at a tissue site, remodeling of the site ensues. Analyses of skin lesions in humans revealed the noticeable presence of blood vessels of varying morphologies at these sites (105, 106). Experimental infections of cutaneous and visceral leishmaniasis also undergo vascular changes at the lesion site. Horst et al. (107) reported that cutaneous infection by L. major parasites in the hindfoot of C57BL/6 wild-type mice resulted in extensively vascularized lesions because the lymphatic and blood vessels were readily evident as the infection established (107). The authors implicated the expression of carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) on mononuclear cells (CD11bhi cells) as essential mediators of angiogenesis in L. major infected lesions. Also performing studies with L. major, Weinkopff et al. (108), showed that there was increased expression of vascular endothelial growth factor-A (VEGF-A) and vascular endothelial growth factor receptor (VEGFR-2) at the site of infection that mirrored the increase in lesion size and parasite numbers (108). In infections with L. donovani, where lesions form in visceral tissue, Yurdakul et al. (109), described vascularization and neovascularization of the red pulp and white pulp regions of the spleen, respectively (109). They attributed splenic vascularization to Ly6C+ inflammatory monocytes. In a more recent study, Dalton et al. (110), showed that neurotrophic tyrosine kinase receptor type 2 (Ntrk2, also known as TrkB) was aberrantly expressed on splenic endothelial cells following Leishmania infections (110). The study then showed that macrophages expressed the ligand(s) for Ntrk2 in the infected spleen and that inhibition of signaling through Ntrk2 blocked white pulp neovascularization.
Natural Leishmania infections are initiated with promastigotes, which are a transient stage within the mammalian host. Within 24 hours of internalization into macrophages, they transform into amastigotes. The characteristics of this transformation that is triggered by temperature and pH changes were described by Zilberstein and colleagues (111, 112). The present challenge is to determine which molecules are released from long-term infections. Many Leishmania virulence factors have been described; however, as Kaye et al. (113) noted in their recent review, no Leishmania parasite-derived factors that cause tissue damage are known (113). It is likely, though, that exosomes released from infected cells carry molecules that could play a role in lesion development and immune response activation. Leishmania parasites themselves produce a variety of extracellular vesicles, including exosomes, which enable them to interact with and respond to their environment. Leishmania promastigotes were found to release vesicles with an average diameter of 30-70nm, consistent with exosomes released by other cell types (114). The molecular composition of these parasite-derived exosomes has been evaluated and shown to contain homologs of some mammalian exosome markers, as well as molecules that may enhance infectivity (114, 115). Up to 329 molecules have been identified in exosomes released from axenic promastigotes, accounting for greater than 52% of the parasite secretome (115). Atayde et al. (116) showed that L. major parasites secrete exosomes within the sandfly midgut, which, when injected with parasites during the initial insect bite of a mammalian host, enhance infection and lesion development in mice (116). Additionally, the exosomes produced by axenic Leishmania promastigotes modulate the chemotactic activity and cytokine secretion of macrophages in vitro to suppress the immune response and enhance permissiveness to subsequent infection (115). The vesicles also have the capacity to activate the immune system of the host. Exosomes released from L. major parasites were shown to induce Th2 polarization and enhanced disease progression in mice, indicating that parasite-derived vesicles are immunosuppressive and proparasitic in nature.
The Leishmania major surface protease (MSP), also referred to as GP63, is an important virulence factor, which contributes to enhanced phagocytosis by macrophages and promoting the survival of the parasite during both promastigote and amastigote life stages. There are three pools of GP63: surface-localized, internal, or released, that are believed to traffic separately through the cell and then released into the extracellular environment (117). Upon infection of macrophages, released GP63 is captured in vesicles and may also access the macrophage cytoplasm, though the exact mechanism of GP63 delivery to the host cytoplasm remains unclear; Leishmania parasites lack secretion systems comparable to those found in pathogenic bacteria (118). Exosomes released by L. mexicana-infected J774 macrophages for 24 hours were found to contain GP63, suggesting that parasite molecules from this intracellular pathogen can access host exosomes and then be released widely (119).
Recent research also indicates that macrophages infected with L. donovani amastigotes release exosomes containing a mixture of unique host and parasite proteins contributing to pathogenic processes (120). In that study, infections of RAW264.7 macrophages were initiated with L. donovani promastigotes, and infections were allowed to continue for 72 hours (about 3 days) to evaluate mature infections. After 72 hours of infection, extracellular vesicles were purified from media supernatants using differential centrifugation and then subjected to liquid chromatography combined with tandem mass spectrometry. In-solution and in-gel protocols for tryptic digestion were used prior to the mass spectrometry-based analysis, and data combined to yield protein composition profiles for uninfected and infected macrophage-derived EVs. Consequently, this approach led to a confident identification of 59 parasite proteins in EVs released by infected macrophages. These proteins included a putative Vasohibin, nucleoside transporter 1, kinesin, DNA directed RNA polymerase II subunit 2, dynein heavy chain, and putative protein kinases. Interestingly, some of the same protein families were also identified in a study of circulating immune complexes in the peripheral blood of 115 human patients with active L. donovani infections (121). In that clinical study, circulating immune complexes (CICs) were purified from patient serum using PEG-assisted precipitation and centrifugation, CIC antigens and antibodies were then dissociated using an acidic buffer, and the antibody was removed using protein A agarose to yield purified antigens. The researchers then used 2D gel electrophoresis and mass spectrometry of these purified CIC antigens to identify parasite proteins, revealing 31 proteins present during active infection before drug treatment. While there appears to be an elevated level of congruence between the parasite-derived protein profiles of these CICs and EVs isolated from infected macrophages, it is difficult to resolve this relationship past the family level. Finding similarities when comparing data between projects can be complicated due to differences in mass spectrometry approach, poor protein annotations, and the overall redundancy of Leishmania protein databases currently in use. In addition, it can be challenging to match proteins exactly across different experiments by reported accession numbers. However, the parity between these profiles at the protein family level remains intriguing. Along with what is known about exosomes and their ability to circulate through the body and deliver their contents to tissues distant from their origin, these findings suggest that L. donovani infected macrophages release exosomes containing parasite factors that may aid in their circulation throughout the host.
It is also worth noting that exosomes released by infected macrophages contain only 59 parasite-derived proteins, a significantly smaller number than the 329 parasite proteins identified in Leishmania promastigote secreted exosomes (122). Comparing the parasite-derived profiles of exosomes from promastigotes versus exosomes from amastigote infected macrophages, we again see several overlapping protein families, including elongation factor 1-alpha, serine/threonine-protein kinase, kinesin, and calpain-like cysteine peptidase. Strikingly, many of the molecules present only in promastigote-derived exosomes are homologs of mammalian exosome structural components (5). The absence of these parasite proteins in exosomes released by infected macrophages may be the result of two biological phenomena. First, the expression of parasite proteins may be differentially regulated between the promastigote and amastigote life stages. For example, a sizable portion of promastigote proteins are downregulated during macrophage infection as the parasites adapt to life in the intracellular environment. Alternatively, it may be unnecessary for the structural proteins of EVs to be contributed by the parasites during infection, as the parasites may be able to hijack the existing host machinery for protein secretion and exosome biogenesis. To be sure, the mammalian counterparts of these structural proteins were identified in EVs from infected macrophages, suggesting that Leishmania amastigotes may translocate their molecules across the LPVM into the host cytosol to utilize normal host processes to their advantage for the secretion of specific parasite molecules. EVs released by Leishmania-infected macrophages were also found to enhance several measures of endothelial cell activation in vitro, including tube formation, cell migration, and enhanced production of VEGF and IL8, which suggests that infection-induced EVs may play a role in neovascularization and pathogenesis (120).
All eukaryotic cells release EVs, including exosomes. The biogenesis of EVs released by cells under various normal and abnormal conditions has been well-studied. There is ample evidence that intrinsic cell characteristics and environmental queues determine the composition of EVs. This results in unique exosome compositions that could be monitored for diagnostic value. Ongoing studies have shown that intracellular pathogens, including viruses, bacteria, and parasites, can take advantage of the host exosome machinery to release virulence factors. Pathogens may smuggle pathogen-encoded virulence factors and other molecules that collectively contribute to their pathogenesis. The existence of these mechanisms suggests that pathogens have evolved adaptations to take advantage of host protein trafficking mechanisms for exosomal packaging. For its part, the host cells also modulate exosome composition as an anti-pathogen strategy since exosomes induce immune responses directed against specific pathogens. More research is needed to evaluate specific mechanisms by which pathogen-derived molecules are targeted to multivesicular bodies within infected cells and further packaged into host-derived exosomes. This information will elucidate a large gap in our understanding of intracellular host-pathogen interactions and identify novel drug targets for infection control.
AG wrote and edited the manuscript. ME wrote and edited the manuscript. PEK developed the manuscript focus, wrote and edited manuscript. All authors contributed to the article and approved the submitted version.
PEK was supported by an R56 AI143293 grant from the NIH. ME was supported by the R03 AI-135610 grant from NIH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity (2014) 41(1):21–35. doi: 10.1016/j.immuni.2014.06.013
2. Schorey JS, Cheng Y, Singh PP, Smith VL. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep (2015) 16(1):24–43. doi: 10.15252/embr.201439363
3. Atayde VD, Hassani K, da Silva Lira Filho A, Borges AR, Adhikari A, Martel C, et al. Leishmania exosomes and other virulence factors: Impact on innate immune response and macrophage functions. Cell Immunol (2016) 309:7–18. doi: 10.1016/j.cellimm.2016.07.013
4. Alenquer M, Amorim MJ. Exosome Biogenesis, Regulation, and Function in Viral Infection. Viruses (2015) 7(9):5066–83. doi: 10.3390/v7092862
5. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol (2013) 200(4):373–83. doi: 10.1083/jcb.201211138
6. Yáñez-Mó M, Siljander PR, Andreu Z, Zavec AB, Borràs FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles (2015) 4:27066. doi: 10.3402/jev.v4.27066
7. Gould SJ, Raposo G. As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracell Vesicles (2013) 2. doi: 10.3402/jev.v2i0.20389
8. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer (1972) 26(4):239–57. doi: 10.1038/bjc.1972.33
9. Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
10. Merchant ML, Rood IM, Deegens JKJ, Klein JB. Isolation and characterization of urinary extracellular vesicles: implications for biomarker discovery. Nat Rev Nephrol (2017) 13(12):731–49. doi: 10.1038/nrneph.2017.148
11. van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol (2018) 19(4):213–28. doi: 10.1038/nrm.2017.125
12. Tricarico C, Clancy J, D’Souza-Schorey C. Biology and biogenesis of shed microvesicles. Small GTPases (2017) 8(4):220–32. doi: 10.1080/21541248.2016.1215283
13. Maas SLN, Breakefield XO, Weaver AM. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol (2017) 27(3):172–88. doi: 10.1016/j.tcb.2016.11.003
14. Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles (2014) 3:26913. doi: 10.3402/jev.v3.26913
15. Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem (1987) 262(19):9412–20. doi: 10.1016/S0021-9258(18)48095-7
16. Vidal M. Exosomes: Revisiting their role as “garbage bags”. Traffic (2019) 20(11):815–28. doi: 10.1111/tra.12687
17. O’Neill CP, Gilligan KE, Dwyer RM. Role of Extracellular Vesicles (EVs) in Cell Stress Response and Resistance to Cancer Therapy. Cancers (Basel) (2019) 11(2). doi: 10.3390/cancers11020136
18. Morshed A, Karawdeniya BI, Bandara YMND, Kim MJ, Dutta P. Mechanical characterization of vesicles and cells: A review. Electrophoresis (2020) 41(7-8):449–70. doi: 10.1002/elps.201900362
19. Pant S, Hilton H, Burczynski ME. The multifaceted exosome: biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem Pharmacol (2012) 83(11):1484–94. doi: 10.1016/j.bcp.2011.12.037
20. Doyle LM, Wang MZ. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells (2019) 8(7). doi: 10.3390/cells8070727
21. Christianson HC, Svensson KJ, van Kuppevelt TH, Li JP, Belting M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc Natl Acad Sci U S A (2013) 110(43):17380–5. doi: 10.1073/pnas.1304266110
22. Villarroya-Beltri C, Baixauli F, Gutiérrez-Vázquez C, Sánchez-Madrid F, Mittelbrunn M. Sorting it out: regulation of exosome loading. Semin Cancer Biol (2014) 28:3–13. doi: 10.1016/j.semcancer.2014.04.009
23. Katzmann DJ, Babst M, Emr SD. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell (2001) 106(2):145–55. doi: 10.1016/s0092-8674(01)00434-2
24. Catalano M, O’Driscoll L. Inhibiting extracellular vesicles formation and release: a review of EV inhibitors. J Extracell Vesicles (2020) 9(1):1703244. doi: 10.1080/20013078.2019.1703244
25. Juan T, Fürthauer M. Biogenesis and function of ESCRT-dependent extracellular vesicles. Semin Cell Dev Biol (2018) 74:66–77. doi: 10.1016/j.semcdb.2017.08.022
27. Hurley JH. ESCRT complexes and the biogenesis of multivesicular bodies. Curr Opin Cell Biol (2008) 20(1):4–11. doi: 10.1016/j.ceb.2007.12.002
28. Henne WM, Stenmark H, Emr SD. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol (2013) 5(9). doi: 10.1101/cshperspect.a016766
29. Raiborg C, Bache KG, Mehlum A, Stang E, Stenmark H. Hrs recruits clathrin to early endosomes. EMBO J (2001) 20(17):5008–21. doi: 10.1093/emboj/20.17.5008
30. Henne WM, Buchkovich NJ, Emr SD. The ESCRT pathway. Dev Cell (2011) 21(1):77–91. doi: 10.1016/j.devcel.2011.05.015
31. Kostelansky MS, Schluter C, Tam YY, Lee S, Ghirlando R, Beach B, et al. Molecular architecture and functional model of the complete yeast ESCRT-I heterotetramer. Cell (2007) 129(3):485–98. doi: 10.1016/j.cell.2007.03.016
32. Langelier C, von Schwedler UK, Fisher RD, De Domenico I, White PL, Hill CP, et al. Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J Virol (2006) 80(19):9465–80. doi: 10.1128/JVI.01049-06
33. Babst M, Katzmann DJ, Snyder WB, Wendland B, Emr SD. Endosome-associated complex, ESCRT-II, recruits transport machinery for protein sorting at the multivesicular body. Dev Cell (2002) 3(2):283–9. doi: 10.1016/s1534-5807(02)00219-8
34. Odorizzi G, Katzmann DJ, Babst M, Audhya A, Emr SD. Bro1 is an endosome-associated protein that functions in the MVB pathway in Saccharomyces cerevisiae. J Cell Sci (2003) 116(Pt 10):1893–903. doi: 10.1242/jcs.00395
35. Babst M, Wendland B, Estepa EJ, Emr SD. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J (1998) 17(11):2982–93. doi: 10.1093/emboj/17.11.2982
36. Ghossoub R, Lembo F, Rubio A, Gaillard CB, Bouchet J, Vitale N, et al. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat Commun (2014) 5:3477. doi: 10.1038/ncomms4477
37. Stuffers S, Sem Wegner C, Stenmark H, Brech A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic (2009) 10(7):925–37. doi: 10.1111/j.1600-0854.2009.00920.x
38. Chen S, Datta-Chaudhuri A, Deme P, Dickens A, Dastgheyb R, Bhargava P, et al. Lipidomic characterization of extracellular vesicles in human serum. J Circ Biomark (2019) 8:1849454419879848. doi: 10.1177/1849454419879848
39. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science (2008) 319(5867):1244–7. doi: 10.1126/science.1153124
40. Wubbolts R, Leckie RS, Veenhuizen PT, Schwarzmann G, Möbius W, Hoernschemeyer J, et al. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J Biol Chem (2003) 278(13):10963–72. doi: 10.1074/jbc.M207550200
41. Matsuo H, Chevallier J, Mayran N, Le Blanc I, Ferguson C, Fauré J, et al. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science (2004) 303(5657):531–4. doi: 10.1126/science.1092425
42. van Niel G, Charrin S, Simoes S, Romao M, Rochin L, Saftig P, et al. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev Cell (2011) 21(4):708–21. doi: 10.1016/j.devcel.2011.08.019
43. Chairoungdua A, Smith DL, Pochard P, Hull M, Caplan MJ. Exosome release of β-catenin: a novel mechanism that antagonizes Wnt signaling. J Cell Biol (2010) 190(6):1079–91. doi: 10.1083/jcb.201002049
44. Perez-Hernandez D, Gutiérrez-Vázquez C, Jorge I, López-Martín S, Ursa A, Sánchez-Madrid F, et al. The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes. J Biol Chem (2013) 288(17):11649–61. doi: 10.1074/jbc.M112.445304
45. Moreno-Gonzalo O, Villarroya-Beltri C, Sánchez-Madrid F. Post-translational modifications of exosomal proteins. Front Immunol (2014) 5:383. doi: 10.3389/fimmu.2014.00383
46. Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, Pérez-Hernández D, Vázquez J, Martin-Cofreces N, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun (2013) 4:2980. doi: 10.1038/ncomms3980
47. Zuccato E, Blott EJ, Holt O, Sigismund S, Shaw M, Bossi G, et al. Sorting of Fas ligand to secretory lysosomes is regulated by mono-ubiquitylation and phosphorylation. J Cell Sci (2007) 120(Pt 1):191–9. doi: 10.1242/jcs.03315
48. Zhang X, Yuan X, Shi H, Wu L, Qian H, Xu W. Exosomes in cancer: small particle, big player. J Hematol Oncol (2015) 8:83. doi: 10.1186/s13045-015-0181-x
49. Hui WW, Hercik K, Belsare S, Alugubelly N, Clapp B, Rinaldi C, et al. Salmonella enterica Serovar Typhimurium Alters the Extracellular Proteome of Macrophages and Leads to the Production of Proinflammatory Exosomes. Infect Immun (2018) 86(2). doi: 10.1128/IAI.00386-17
50. Cheng Y, Schorey JS. Exosomes carrying mycobacterial antigens can protect mice against Mycobacterium tuberculosis infection. Eur J Immunol (2013) 43(12):3279–90. doi: 10.1002/eji.201343727
51. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar Macrophages Provide an Early Mycobacterium tuberculosis Niche and Initiate Dissemination. Cell Host Microbe (2018) 24(3):439–46.e4. doi: 10.1016/j.chom.2018.08.001
52. Armstrong JA, Hart PD. Phagosome-lysosome interactions in cultured macrophages infected with virulent tubercle bacilli. Reversal of the usual nonfusion pattern and observations on bacterial survival. J Exp Med (1975) 142(1):1–16. doi: 10.1084/jem.142.1.1
53. Via LE, Deretic D, Ulmer RJ, Hibler NS, Huber LA, Deretic V. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J Biol Chem (1997) 272(20):13326–31. doi: 10.1074/jbc.272.20.13326
54. Xu S, Cooper A, Sturgill-Koszycki S, van Heyningen T, Chatterjee D, Orme I, et al. Intracellular trafficking in Mycobacterium tuberculosis and Mycobacterium avium-infected macrophages. J Immunol (1994) 153(6):2568–78.
55. Zulauf KE, Sullivan JT, Braunstein M. The SecA2 pathway of Mycobacterium tuberculosis exports effectors that work in concert to arrest phagosome and autophagosome maturation. PloS Pathog (2018) 14(4):e1007011. doi: 10.1371/journal.ppat.1007011
56. Bhatnagar S, Schorey JS. Exosomes released from infected macrophages contain Mycobacterium avium glycopeptidolipids and are proinflammatory. J Biol Chem (2007) 282(35):25779–89. doi: 10.1074/jbc.M702277200
57. Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JS. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood (2007) 110(9):3234–44. doi: 10.1182/blood-2007-03-079152
58. Singh PP, Smith VL, Karakousis PC, Schorey JS. Exosomes isolated from mycobacteria-infected mice or cultured macrophages can recruit and activate immune cells in vitro and in vivo. J Immunol (2012) 189(2):777–85. doi: 10.4049/jimmunol.1103638
59. Smith VL, Cheng Y, Bryant BR, Schorey JS. Exosomes function in antigen presentation during an in vivo Mycobacterium tuberculosis infection. Sci Rep (2017) 7:43578. doi: 10.1038/srep43578
60. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol (2010) 12(1):19–30; sup pp 1-13. doi: 10.1038/ncb2000
61. Giri PK, Kruh NA, Dobos KM, Schorey JS. Proteomic analysis identifies highly antigenic proteins in exosomes from M. tuberculosis-infected and culture filtrate protein-treated macrophages. Proteomics (2010) 10(17):3190–202. doi: 10.1002/pmic.200900840
62. Kruh-Garcia NA, Wolfe LM, Chaisson LH, Worodria WO, Nahid P, Schorey JS, et al. Detection of Mycobacterium tuberculosis peptides in the exosomes of patients with active and latent M. tuberculosis infection using MRM-MS. PloS One (2014) 9(7):e103811. doi: 10.1371/journal.pone.0103811
63. Smith VL, Jackson L, Schorey JS. Ubiquitination as a Mechanism To Transport Soluble Mycobacterial and Eukaryotic Proteins to Exosomes. J Immunol (2015) 195(6):2722–30. doi: 10.4049/jimmunol.1403186
64. Cheng Y, Schorey JS. Targeting soluble proteins to exosomes using a ubiquitin tag. Biotechnol Bioeng (2016) 113(6):1315–24. doi: 10.1002/bit.25884
65. Singh PP, Li L, Schorey JS. Exosomal RNA from Mycobacterium tuberculosis-Infected Cells Is Functional in Recipient Macrophages. Traffic (2015) 16(6):555–71. doi: 10.1111/tra.12278
66. World Health Organization. Malaria (2020). Available at: https://www.who.int/news-room/fact-sheets/detail/malaria.
67. Sibley LD. Intracellular parasite invasion strategies. Science (2004) 304(5668):248–53. doi: 10.1126/science.1094717
68. Pradel G, Frevert U. Malaria sporozoites actively enter and pass through rat Kupffer cells prior to hepatocyte invasion. Hepatology (2001) 33(5):1154–65. doi: 10.1053/jhep.2001.24237
69. Kurup SP, Anthony SM, Hancox LS, Vijay R, Pewe LL, Moioffer SJ, et al. Monocyte-Derived CD11c. Cell Host Microbe (2019) 25(4):565–77.e6. doi: 10.1016/j.chom.2019.02.014
70. Silvie O, Rubinstein E, Franetich JF, Prenant M, Belnoue E, Rénia L, et al. Hepatocyte CD81 is required for Plasmodium falciparum and Plasmodium yoelii sporozoite infectivity. Nat Med (2003) 9(1):93–6. doi: 10.1038/nm808
71. Bano N, Romano JD, Jayabalasingham B, Coppens I. Cellular interactions of Plasmodium liver stage with its host mammalian cell. Int J Parasitol (2007) 37(12):1329–41. doi: 10.1016/j.ijpara.2007.04.005
72. Regev-Rudzki N, Wilson DW, Carvalho TG, Sisquella X, Coleman BM, Rug M, et al. Cell-cell communication between malaria-infected red blood cells via exosome-like vesicles. Cell (2013) 153(5):1120–33. doi: 10.1016/j.cell.2013.04.029
73. Mantel PY, Hoang AN, Goldowitz I, Potashnikova D, Hamza B, Vorobjev I, et al. Malaria-infected erythrocyte-derived microvesicles mediate cellular communication within the parasite population and with the host immune system. Cell Host Microbe (2013) 13(5):521–34. doi: 10.1016/j.chom.2013.04.009
74. Abdi A, Yu L, Goulding D, Rono MK, Bejon P, Choudhary J, et al. Proteomic analysis of extracellular vesicles from a. Wellcome Open Res (2017) 2:50. doi: 10.12688/wellcomeopenres.11910.2
75. Mantel PY, Marti M. The role of extracellular vesicles in Plasmodium and other protozoan parasites. Cell Microbiol (2014) 16(3):344–54. doi: 10.1111/cmi.12259
76. Debs S, Cohen A, Hosseini-Beheshti E, Chimini G, Hunt NH, Grau GER. Interplay of extracellular vesicles and other players in cerebral malaria pathogenesis. Biochim Biophys Acta Gen Subj (2019) 1863(2):325–31. doi: 10.1016/j.bbagen.2018.10.012
77. Combes V, Coltel N, Faille D, Wassmer SC, Grau GE. Cerebral malaria: role of microparticles and platelets in alterations of the blood-brain barrier. Int J Parasitol (2006) 36(5):541–6. doi: 10.1016/j.ijpara.2006.02.005
78. Martin-Jaular L, Nakayasu ES, Ferrer M, Almeida IC, Del Portillo HA. Exosomes from Plasmodium yoelii-infected reticulocytes protect mice from lethal infections. PloS One (2011) 6(10):e26588. doi: 10.1371/journal.pone.0026588
79. Arisue N, Palacpac NMQ, Tougan T, Horii T. Characteristic features of the SERA multigene family in the malaria parasite. Parasit Vectors (2020) 13(1):170. doi: 10.1186/s13071-020-04044-y
80. Gualdrón-López M, Flannery EL, Kangwanrangsan N, Chuenchob V, Fernandez-Orth D, Segui-Barber J, et al. Characterization of Plasmodium vivax Proteins in Plasma-Derived Exosomes from Malaria-Infected Liver-Chimeric Humanized Mice. Characterization of Front Microbiol (2018) 9:1271. doi: 10.3389/fmicb.2018.01271
81. Vaughan AM, Mikolajczak SA, Wilson EM, Grompe M, Kaushansky A, Camargo N, et al. Complete Plasmodium falciparum liver-stage development in liver-chimeric mice. J Clin Invest (2012) 122(10):3618–28. doi: 10.1172/JCI62684
82. Mikolajczak SA, Vaughan AM, Kangwanrangsan N, Roobsoong W, Fishbaugher M, Yimamnuaychok N, et al. Plasmodium vivax liver stage development and hypnozoite persistence in human liver-chimeric mice. Cell Host Microbe (2015) 17(4):526–35. doi: 10.1016/j.chom.2015.02.011
83. Rastogi S, Cygan AM, Boothroyd JC. Translocation of effector proteins into host cells by Toxoplasma gondii. Curr Opin Microbiol (2019) 52:130–8. doi: 10.1016/j.mib.2019.07.002
84. Håkansson S, Charron AJ, Sibley LD. Toxoplasma evacuoles: a two-step process of secretion and fusion forms the parasitophorous vacuole. EMBO J (2001) 20(12):3132–44. doi: 10.1093/emboj/20.12.3132
85. Nadipuram SM, Kim EW, Vashisht AA, Lin AH, Bell HN, Coppens I, et al. In Vivo Biotinylation of the Toxoplasma Parasitophorous Vacuole Reveals Novel Dense Granule Proteins Important for Parasite Growth and Pathogenesis. mBio (2016) 7(4). doi: 10.1128/mBio.00808-16
86. Li Y, Liu Y, Xiu F, Wang J, Cong H, He S, et al. Characterization of exosomes derived from. Int J Nanomed (2018) 13:467–77. doi: 10.2147/IJN.S151110
87. Aline F, Bout D, Amigorena S, Roingeard P, Dimier-Poisson I. Toxoplasma gondii antigen-pulsed-dendritic cell-derived exosomes induce a protective immune response against T. gondii infection. Infect Immun (2004) 72(7):4127–37. doi: 10.1128/IAI.72.7.4127-4137.2004
88. Beauvillain C, Ruiz S, Guiton R, Bout D, Dimier-Poisson I. A vaccine based on exosomes secreted by a dendritic cell line confers protection against T. gondii infection in syngeneic and allogeneic mice. Microbes Infect (2007) 9(14-15):1614–22. doi: 10.1016/j.micinf.2007.07.002
89. Pope SM, Lässer C. Toxoplasma gondii infection of fibroblasts causes the production of exosome-like vesicles containing a unique array of mRNA and miRNA transcripts compared to serum starvation. J Extracell Vesicles (2013) 2. doi: 10.3402/jev.v2i0.22484
90. Wowk PF, Zardo ML, Miot HT, Goldenberg S, Carvalho PC, Mörking PA. Proteomic profiling of extracellular vesicles secreted from Toxoplasma gondii. Proteomics (2017) 17(15-16). doi: 10.1002/pmic.201600477
91. Li DL, Zou WH, Deng SQ, Peng HJ. Analysis of the Differential Exosomal miRNAs of DC2.4 Dendritic Cells Induced by. Int J Mol Sci (2019) 20(21). doi: 10.3390/ijms20215506
92. Pearson RD, Sousa AQ. Clinical spectrum of Leishmaniasis. Clin Infect Dis (1996) 22(1):1–13. doi: 10.1093/clinids/22.1.1
93. McMahon-Pratt D, Alexander J. Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunol Rev (2004) 201:206–24. doi: 10.1111/j.0105-2896.2004.00190.x
94. Wilson ME, Jeronimo SM, Pearson RD. Immunopathogenesis of infection with the visceralizing Leishmania species. Microb Pathog (2005) 38(4):147–60. doi: 10.1016/j.micpath.2004.11.002
95. Blackwell JM, Fakiola M, Ibrahim ME, Jamieson SE, Jeronimo SB, Miller EN, et al. Genetics and visceral leishmaniasis: of mice and man. Parasite Immunol (2009) 31(5):254–66. doi: 10.1111/j.1365-3024.2009.01102.x
96. Ndjamen B, Kang BH, Hatsuzawa K, Kima PE. Leishmania parasitophorous vacuoles interact continuously with the host cell’s endoplasmic reticulum; parasitophorous vacuoles are hybrid compartments. Cell Microbiol (2010) 12(10):1480–94. doi: 10.1111/j.1462-5822.2010.01483.x
97. Arango Duque G, Jardim A, Gagnon É, Fukuda M, Descoteaux A. The host cell secretory pathway mediates the export of Leishmania virulence factors out of the parasitophorous vacuole. PloS Pathog (2019) 15(7):e1007982. doi: 10.1371/journal.ppat.1007982
98. Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, et al. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science (2008) 321(5891):970–4. doi: 10.1126/science.1159194
99. Romano A, Carneiro MBH, Doria NA, Roma EH, Ribeiro-Gomes FL, Inbar E, et al. Divergent roles for Ly6C+CCR2+CX3CR1+ inflammatory monocytes during primary or secondary infection of the skin with the intra-phagosomal pathogen Leishmania major. PloS Pathog (2017) 13(6):e1006479. doi: 10.1371/journal.ppat.1006479
100. Wilson ME, Innes DJ, Sousa AD, Pearson RD. Early histopathology of experimental infection with Leishmania donovani in hamsters. J Parasitol (1987) 73(1):55–63. doi: 10.2307/3282344
101. Costa DL, Lima-Júnior DS, Nascimento MS, Sacramento LA, Almeida RP, Carregaro V, et al. CCR2 signaling contributes to the differentiation of protective inflammatory dendritic cells in Leishmania braziliensis infection. J Leukoc Biol (2016) 100(2):423–32. doi: 10.1189/jlb.4A0715-288R
102. Kaye PM, Beattie L. Lessons from other diseases: granulomatous inflammation in leishmaniasis. Semin Immunopathol (2016) 38(2):249–60. doi: 10.1007/s00281-015-0548-7
103. Steinbichler TB, Dudás J, Riechelmann H, Skvortsova II. The role of exosomes in cancer metastasis. Semin Cancer Biol (2017) 44:170–81. doi: 10.1016/j.semcancer.2017.02.006
104. Wortzel I, Dror S, Kenific CM, Lyden D. Exosome-Mediated Metastasis: Communication from a Distance. Dev Cell (2019) 49(3):347–60. doi: 10.1016/j.devcel.2019.04.011
105. Caltagirone F, Pistone G, Arico M, Vitale F, Sorbello D, Rizzo D, et al. Vascular patterns in cutaneous leishmaniasis: A videodermatoscopic study. Indian J Dermatol Venereol Leprol (2015) 81(4):394–8. doi: 10.4103/0378-6323.159939
106. Taheri AR, Pishgooei N, Maleki M, Goyonlo VM, Kiafar B, Banihashemi M, et al. Dermoscopic features of cutaneous leishmaniasis. Int J Dermatol (2013) 52(11):1361–6. doi: 10.1111/ijd.12114
107. Horst AK, Bickert T, Brewig N, Ludewig P, van Rooijen N, Schumacher U, et al. CEACAM1+ myeloid cells control angiogenesis in inflammation. Blood (2009) 113(26):6726–36. doi: 10.1182/blood-2008-10-184556
108. Weinkopff T, Konradt C, Christian DA, Discher DE, Hunter CA, Scott P. Leishmania major Infection-Induced VEGF-A/VEGFR-2 Signaling Promotes Lymphangiogenesis That Controls Disease. J Immunol (2016) 197(5):1823–31. doi: 10.4049/jimmunol.1600717
109. Yurdakul P, Dalton J, Beattie L, Brown N, Erguven S, Maroof A, et al. Compartment-specific remodeling of splenic micro-architecture during experimental visceral leishmaniasis. Am J Pathol (2011) 179(1):23–9. doi: 10.1016/j.ajpath.2011.03.009
110. Dalton JE, Glover AC, Hoodless L, Lim EK, Beattie L, Kirby A, et al. The neurotrophic receptor Ntrk2 directs lymphoid tissue neovascularization during Leishmania donovani infection. PloS Pathog (2015) 11(2):e1004681. doi: 10.1371/journal.ppat.1004681
111. Barak E, Amin-Spector S, Gerliak E, Goyard S, Holland N, Zilberstein D. Differentiation of Leishmania donovani in host-free system: analysis of signal perception and response. Mol Biochem Parasitol (2005) 141(1):99–108. doi: 10.1016/j.molbiopara.2005.02.004
112. Tsigankov P, Gherardini PF, Helmer-Citterich M, Späth GF, Myler PJ, Zilberstein D. Regulation dynamics of Leishmania differentiation: deconvoluting signals and identifying phosphorylation trends. Mol Cell Proteomics (2014) 13(7):1787–99. doi: 10.1074/mcp.M114.037705
113. Kaye PM, Cruz I, Picado A, Van Bocxlaer K, Croft SL. Leishmaniasis immunopathology-impact on design and use of vaccines, diagnostics and drugs. Semin Immunopathol (2020) 42(3):247–64. doi: 10.1007/s00281-020-00788-y
114. Silverman JM, Clos J, de’Oliveira CC, Shirvani O, Fang Y, Wang C, et al. An exosome-based secretion pathway is responsible for protein export from Leishmania and communication with macrophages. J Cell Sci (2010) 123(Pt 6):842–52. doi: 10.1242/jcs.056465
115. Silverman JM, Clos J, Horakova E, Wang AY, Wiesgigl M, Kelly I, et al. Leishmania exosomes modulate innate and adaptive immune responses through effects on monocytes and dendritic cells. J Immunol (2010) 185(9):5011–22. doi: 10.4049/jimmunol.1000541
116. Atayde VD, Aslan H, Townsend S, Hassani K, Kamhawi S, Olivier M. Exosome Secretion by the Parasitic Protozoan Leishmania within the Sand Fly Midgut. Cell Rep (2015) 13(5):957–67. doi: 10.1016/j.celrep.2015.09.058
117. Yao C, Donelson JE, Wilson ME. Internal and surface-localized major surface proteases of Leishmania spp. and their differential release from promastigotes. Eukaryot Cell (2007) 6(10):1905–12. doi: 10.1128/EC.00073-07
118. Isnard A, Christian JG, Kodiha M, Stochaj U, McMaster WR, Olivier M. Impact of Leishmania infection on host macrophage nuclear physiology and nucleopore complex integrity. PloS Pathog (2015) 11(3):e1004776. doi: 10.1371/journal.ppat.1004776
119. Hassani K, Olivier M. Immunomodulatory impact of leishmania-induced macrophage exosomes: a comparative proteomic and functional analysis. PloS Negl Trop Dis (2013) 7(5):e2185. doi: 10.1371/journal.pntd.0002185
120. Gioseffi A, Hamerly T, Van K, Zhang N, Dinglasan RR, Yates PA, et al. -infected macrophages release extracellular vesicles that can promote lesion development. Life Sci Alliance (2020) 3(12). doi: 10.26508/lsa.202000742
121. Jamal F, Shivam P, Kumari S, Singh MK, Sardar AH, Pushpanjali, et al. Identification of Leishmania donovani antigen in circulating immune complexes of visceral leishmaniasis subjects for diagnosis. PloS One (2017) 12(8):e0182474. doi: 10.1371/journal.pone.0182474
Keywords: extracellular vesicles, exosomes, infections, Leishmania, Mycobacterium, Plasmodium, Toxoplasma
Citation: Gioseffi A, Edelmann MJ and Kima PE (2021) Intravacuolar Pathogens Hijack Host Extracellular Vesicle Biogenesis to Secrete Virulence Factors. Front. Immunol. 12:662944. doi: 10.3389/fimmu.2021.662944
Received: 01 February 2021; Accepted: 30 March 2021;
Published: 20 April 2021.
Edited by:
Johnathan Canton, University of Calgary, CanadaReviewed by:
Fernando Real, Centre National de la Recherche Scientifique (CNRS), FranceCopyright © 2021 Gioseffi, Edelmann and Kima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter E. Kima, cGtpbWFAdWZsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.