 Cody D. Moorman
Cody D. Moorman Sue J. Sohn
Sue J. Sohn Hyewon Phee
Hyewon Phee
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 29 March 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.657768
This article is part of the Research TopicEmerging Therapeutics for Immune ToleranceView all 24 articles
Autoimmune diseases affect roughly 5-10% of the total population, with women affected more than men. The standard treatment for autoimmune or autoinflammatory diseases had long been immunosuppressive agents until the advent of immunomodulatory biologic drugs, which aimed at blocking inflammatory mediators, including proinflammatory cytokines. At the frontier of these biologic drugs are TNF-α blockers. These therapies inhibit the proinflammatory action of TNF-α in common autoimmune diseases such as rheumatoid arthritis, psoriasis, ulcerative colitis, and Crohn’s disease. TNF-α blockade quickly became the “standard of care” for these autoimmune diseases due to their effectiveness in controlling disease and decreasing patient’s adverse risk profiles compared to broad-spectrum immunosuppressive agents. However, anti-TNF-α therapies have limitations, including known adverse safety risk, loss of therapeutic efficacy due to drug resistance, and lack of efficacy in numerous autoimmune diseases, including multiple sclerosis. The next wave of truly transformative therapeutics should aspire to provide a cure by selectively suppressing pathogenic autoantigen-specific immune responses while leaving the rest of the immune system intact to control infectious diseases and malignancies. In this review, we will focus on three main areas of active research in immune tolerance. First, tolerogenic vaccines aiming at robust, lasting autoantigen-specific immune tolerance. Second, T cell therapies using Tregs (either polyclonal, antigen-specific, or genetically engineered to express chimeric antigen receptors) to establish active dominant immune tolerance or T cells (engineered to express chimeric antigen receptors) to delete pathogenic immune cells. Third, IL-2 therapies aiming at expanding immunosuppressive regulatory T cells in vivo.
The mammalian immune system evolved to protect our bodies from foreign pathogens and intrinsic aberrant malignancies while concurrently preventing deleterious immune responses toward self (1). Immune tolerance co-evolved as a safety system that maintains a state of immune unresponsiveness to autoantigens and self-tissues (2, 3). There are two mechanisms that maintain immunological tolerance denominated central and peripheral tolerance. Central tolerance occurs during lymphocyte development in the primary lymphoid organs (i.e. thymus and bone marrow), where T or B cell clones that recognize autoantigens with high-affinity are deleted. Peripheral tolerance evolved to counteract autoantigen-recognizing T or B cells that escape central tolerance. Peripheral tolerance occurs in the secondary lymphoid organs (e.g. spleen, lymph nodes, and mucosal/gut associated lymphoid tissues) and peripheral tissues. Mechanisms of peripheral tolerance include inactivation of autoantigen-recognizing T and B cells by the induction of apoptosis, anergy or conversion into immunosuppressive regulatory cells. In addition, suppressor immune cells such as FOXP3+ regulatory T cells (Tregs) exert dominant immune suppression to control autoreactive T and B cells. Evidence suggest that a patient’s genetic predisposition together with environmental factors, such as exposure to pathogens that exhibit molecular mimicry, disturb immune tolerance (4). Loss of immune tolerance to autoantigens associated with a specific organ results in the activation of organ-specific T and B cells that in turn cause organ-specific inflammation and the development of autoimmune diseases such as multiple sclerosis (MS) (5), rheumatoid arthritis (RA) (6), psoriasis (7), and type 1 diabetes (T1D) (8). Thus, therapeutics that induce, restore, and maintain immune tolerance toward these autoantigens represent the “Holy Grail” of treatments for autoimmune diseases.
For the last two decades, our understanding of immunology exploded with the advent of technologies that allowed high throughput screening and generation of large molecule biologics such as monoclonal antibodies (mAb) and small molecule compounds. As an outcome, there was a huge success of anti-TNF-α blockers, which became the “standard of care” for many autoimmune diseases (9). The success of anti-TNF-α blockers stimulated the development of large molecule biologics that block the function of various cytokines (i.e. IL-1, IL-6, IL-12, IL-17, and IL-23) (10, 11) and small molecule compounds that inhibit molecular interactions involved in the generation of inflammation (i.e. JAKs, TYK2, IRAK4, BTK, SYK, RIPs, and TPL2) (12–14).
Despite the success of anti-TNF-α blockers and other immunomodulatory therapies, there are still significant unmet clinical needs. First, currently approved therapies are immunomodulatory and provide a remedy to relieve symptoms but do not provide a cure by directly addressing the loss of immune tolerance. Second, because most of these therapeutics work by reducing systemic inflammation, they have numerous adverse safety risks including increased susceptibility to opportunistic infections or malignancies and have detrimental side effects (15–17). Third, these agents lack efficacy in refractory autoimmune diseases (18) and in patients that are or become unresponsive to treatment (19).
Novel therapeutics are emerging to achieve immune tolerance in autoimmunity. Most of these novel therapeutics harness known immune tolerance mechanisms, such as increasing FOXP3+ Tregs and inducing anergy or deletion of pathologic immune cells. A subset of these therapeutics includes coinhibitory checkpoint agonists and costimulatory checkpoint antagonists to expand Tregs and/or dampening pathogenic effector cells. However, these therapies are beyond the scope of this review, but are discussed in multiple outstanding reviews (20–23). In this review, we will focus on three main categories of therapeutics that drive immune tolerance. First, tolerogenic vaccines designed to elicit autoantigen-specific immune tolerance. Second, T cell therapies using Tregs to establish active dominant immune tolerance or T cells to delete pathogenic immune cells for the treatment of autoimmunity. Third, IL-2 based therapies to expand immunosuppressive Tregs.
Risks associated systemic immune suppression or immunomodulatory therapies for the treatment of autoimmune diseases could be reduced or even halted if antigen-specific tolerance was generated. Unlike general immune suppression, antigen-specific tolerance inhibits the pathogenic autoantigen-specific immune responses that drive autoimmune diseases, while leaving the rest of the immune system intact. Therefore, antigen-specific tolerance is the logical next step in treating autoimmune diseases. Among the emerging therapeutics, tolerogenic vaccines are gaining noticeable traction (24–28). Despite the potentials of antigen-specific tolerance, there are at least two outstanding questions to resolve. First, although knowledge of which autoantigens are associated with specific autoimmune diseases continues to grow, most of them are still not known (29–31). Second, because it is unclear how many autoantigens are involved in individual autoimmune diseases, it is still uncertain whether tolerance toward one or few autoantigens can reverse autoimmunity. Therefore, therapies that not only drive antigen-specific but more extensive organ-specific tolerance would likely have the greatest efficacy in a clinical setting. Here we discuss novel therapeutics that derive antigen-specific and/or organ-specific tolerance.
Immunosuppressive CD4+ FOXP3+ Tregs are required for immune tolerance. Genetic deficiencies of the transcription factor Foxp3, the Treg master regulator, results in the fatal systemic autoimmune diseases, immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) in humans and Scurfy in mice (32–34). CD4+ FOXP3+ Tregs comprise approximately 4-12% of the peripheral CD4+ T cell population and are identified based on the expression of FOXP3, high levels of CD25 and low levels of CD127 (35). FOXP3 controls the transcriptional programing of Tregs and imparts an immunosuppressive phenotype because experimental ectopic expression of FOXP3 in T cells (36–39), B cells (40), and myeloid cells (41) confers immunosuppressive capabilities. Likewise, genetic disruption or downregulation of FOXP3 impairs immunosuppressive capabilities and renders Tregs immunostimulatory (42, 43). CD4+ FOXP3+ Tregs have a T cell receptor (TCR) repertoire that is skewed toward the recognition of self-antigens (44). The self-reactive TCR directs Treg trafficking to self-tissues where cognate autoantigens are presented, resulting in TCR engagement and immunosuppressive effector functions (24). Tregs require TCR activation via their cognate antigen (45, 46) and the cytokine IL-2 (47) to suppress multiple facets of the immune system including T conventional cells (Tcons), B cells, and myeloid cells (48, 49). For example, when activated FOXP3+ Tregs express the immunosuppressive cytokines IL-10, IL-35, and TGF-β that inhibit Tcon and DC activation (50); suppress antigen presenting cells (APCs) expression of antigen presentation molecules MHCI and MHCII, costimulatory molecules CD80, CD86, and CD40 and proinflammatory cytokines IL-12 and IL-6 as well as differentiate dendritic cells (DC) into tolerogenic DCs (tDCs) (51–54); express the ectoenzymes, CD39 and CD72, which catabolize proinflammatory extracellular ATP/ADP into anti-inflammatory AMP (55); express the inhibitory receptors CTLA-4, LAG-3, PD1, TIGIT, GITR, and TIM-3 to block APC maturation and T cell activation (56); produce the cytotoxic molecules Galectin-9, Fas-L, TRAIL, Perforin, and Granzyme-B to kill effector T cells and inflammatory APCs (57); sequester IL-2 to inhibit Tcon access to this critical cytokine required for T cell proliferation, function, and survival (58, 59); and finally, deplete local glucose disrupting the metabolic needs of effector T cells (60). These FOXP3+ Treg effector functions create an immunosuppressive microenvironment at the site of autoantigen recognition preventing autoimmune responses. Because FOXP3+ Tregs play a fundamental role in tolerance, it is crucial that Tregs maintain phenotypical and functional stability in both quiescent and inflammatory environment associated with autoimmune disease. Of the major mechanisms that control Treg stability, demethylation of the Treg-specific demethylated region (TSDR), low-moderate TCR-antigen recognition efficiency, and IL-2 signaling are among the most prominent signals that maintain Treg stability. There are several informative reviews that provide an in-depth review on Treg stability (24, 61–63).
Interestingly, activated antigen-specific Tregs can also maintain tolerance to antigens beyond their cognate antigen specificity via regulatory mechanisms termed, bystander or linked suppression and infectious tolerance (64). Activated Tregs employ numerous effector functions to create an immunosuppressive microenvironment that can suppresses and/or tolerizes local T cells with alternative antigen specificities. This indiscriminate local suppression has been termed “linked/bystander” suppression because both the Treg- and Tcon-cognate antigens must be spatially colocalized and presented on the same APC. Simply, a Treg specific for antigen-X can suppress a Tcon specific for antigen-Y when both antigens X and Y are presented on the same APC (65).
Furthermore, Tregs can induce T cells to differentiate into regulatory T cell subsets. This conversion requires spatial colocalization and coactivation of both the FOXP3+ Treg and T cell. The recruitment of T cells into regulatory T cell subsets has been termed “infectious tolerance” because the new regulatory T cells can maintain tolerance independently of the original stimuli thereby spreading tolerance. Simply, a Treg specific for antigen-X can induce a T cell specific for antigen-Y to become a regulatory T cell when both X and Y are presented on the same APC. The antigen-Y-specific regulatory T cell can then mediate active dominant antigen-specific tolerance for antigen-Y when antigen-X is no longer present (24). For example, FOXP3+ Tregs express TGF-β, IL-10, and IL-35, that in turn differentiate T cells into FOXP3+ Tregs, type 1 regulatory T cells (Tr1), and inducible IL-35 producing regulatory T cells (iTr35), respectively (66–70). Likewise, Tr1 and iTr35 can mediate infectious tolerance (67, 71).
In the context of immune tolerance, therapeutics that elicit Treg responses mediating linked/bystander suppression and infectious tolerance toward organ-specific autoantigens would be ideal to control organ-specific autoimmune disease that involve numerous or unidentified autoantigens. Linked/bystander suppression and infectious tolerance have beneficial roles in autoimmunity, allergy, and organ transplant, while also having a detrimental effect on the clearance of cancers and pathogens (72). It is difficult to experimentally differentiate linked/bystander suppression from infectious tolerance; therefore, we will collectively term them bystander suppression.
DCs are a subset of professional APCs that facilitate T cell responses to generate both protective immune responses toward pathogens/cancer and tolerant immune responses toward self-antigens (73). DCs are the key governing APC of immune tolerance because depletion of CD11c+ DCs resulted in the development of fatal autoimmunity in mice (74). DCs capture, processes, and present antigens while integrating environmental signals, often supplied by T cells (e.g. CD40L or PD1), to modulate the expression of stimulatory and inhibitory molecules to direct T cell responses (73). DCs are a functionally and phenotypically heterologous group of APCs. DCs retain an “immature” phenotype during homeostatic conditions, characterized by minimal expression of co-stimulatory molecules and proinflammatory cytokines. Environmental cues such as pathogen- or danger- associated molecular pattern molecules and co-stimulatory molecules direct immature DCs to differentiate into mature DCs, that upregulate the expression of co-stimulatory molecules and proinflammatory cytokines. These mature DCs, in turn, provide antigen presentation, co-stimulation, and cytokine help, activating effector Tcons to generate protective adaptive immunity (73).
A third subset of “semi-immature” DCs exhibit an intermediate maturation status and may function as tolerogenic DCs (tDCs) (75). Tolerogenic DCs lack a defined transcriptional regulator and instead are classified by their common phenotypical and functional attributes. Typically, tDCs express low levels of MHCI/MHCII, co-stimulatory molecules, and inflammatory cytokines. In addition, tDCs express a unique transcriptional program that results in the expression of immunosuppressive molecules such as nitric oxide, indoleamine 2,3-dioxygenase, anti-inflammatory cytokines IL-10 and TGF-β, and inhibitory co-receptors PD-L1/2, ICOSL, B7-H4, and B7-H3 (73).
How do DCs maintain tolerance? The two pillars of DC mediated tolerance are the induction of apoptosis or anergy of autoreactive T cells and induction, expansion, and maintenance of Tregs. During homeostatic conditions, immature DCs present self-antigens in the absence of extensive co-stimulation or cytokine help, leading to unproductive T cell activation and autoreactive T cell apoptosis and anergy (73, 76). Because tDCs do not express high levels of co-stimulatory receptors or inflammatory mediators, but rather express high levels of inhibitory co-receptors and immunosuppressive molecules, tDC can induce T cell anergy or death under inflammatory conditions. Another way DCs control autoimmune diseases is by expanding and maintain the Treg pool. For example, Treg populations are dependent on DCs because, depletion of CD11c+ DCs reduced Treg populations while increasing DC numbers expanded Treg populations in mice (77, 78). Moreover, DCs support Treg function because Tregs from DC-depleted mice had reduced suppressive capabilities (79). DCs expand Tregs when MHCII-driven autoantigen presentation is paired with immunosuppressive molecules such IL-10, IDO, PGE-2, and TGF-β. Even under inflammatory conditions, DCs expressing autoantigens can expand autoantigen-specific Tregs (79). While there is evidence that both immature and mature DCs can support Treg induction, it is still unclear which subtype of DC confers tolerogenic responses under different circumstances.
Beyond DCs, there are other MHCII expressing APCs that provoke tolerance. These cells include liver sinusoidal endothelial cells (LSECs), macrophages, and T cells. LSECs express MCHII, low levels of costimulatory molecules and do not produce the proinflammatory cytokine IL-12 and thus fail to stimulate pathogenic Th1 responses. Instead, LSEC expand immunosuppressive Tregs via the expression of surface bound TGF-β and Jagged family of Notch ligands (80, 81). Immunosuppressive macrophages such as alternatively activated macrophages (AAMs)/M2-like macrophages, tumor associated macrophages, and marginal zone macrophages produce high levels of IL-10, TGF-β and PD-L1 to promote the expansion and differentiation of Tregs or deletion of autoreactive T cell (82–85). T cells can also express MHCII and favor tolerance via the induction of T cell anergy and apoptosis (86).
Because immune tolerance is orchestrated by APCs, numerous tolerogenic vaccine platforms have been developed to deliver autoantigens to specific APC subtypes. Some of these tolerogenic vaccine platforms include protein/peptide-, nanoparticle-, and DNA/RNA-based vaccines. Furthermore, immunosuppressive cell types such as tDCs, have been manipulated and expanded ex vivo and reintroduced as cell-based tolerogenic vaccines. Together, these agents can be summarized as antigen-specific tolerogenic vaccines.
Due to the breath of tolerogenic vaccines being investigated and broad number of preclinical autoimmune disease models, it is practical to compare different tolerogenic vaccine platforms within the contexts of a single disease model. Thus, for this review, we will focus on tolerogenic vaccines tested in preclinical models of experimental autoimmune encephalomyelitis (EAE), the animal model for MS. EAE models have been extensively utilized as a preclinical animal model for the development of tolerogenic vaccines. EAE is a CD4+ T cell driven CNS-specific autoimmune disease that involves complex immune responses and numerous neuroantigen targets including myelin oligodendrocyte glycoprotein (MOG), myelin basic protein (MBP) and proteolipid protein (PLP), that can mimic some of the complexities of human autoimmune diseases but with limitations. As with any preclinical model, EAE has many differences in comparison to that pathogenesis of MS. Thus, readers should take the results of preclinical EAE studies to understand the mechanism of tolerance, rather than to gauge potential therapeutic efficacy in patients.
In preclinical studies, administration of naked myelin peptides under quiescent homeostatic conditions by various routes including intravenous (IV), subcutaneous (SC), epicutaneous, intraperitoneal (IP), intranasal, intrathymic, or oral induced antigen-specific tolerance and suppressed EAE (87–93). However, when included in the immunogenic complete Freund’s adjuvant (CFA), these same naked myelin peptides provoked encephalitogenic priming and the active induction of EAE (94). Therefore, strategies were devised to induce antigen-specific tolerance in proinflammatory environments during active EAE. These strategies included targeting autoantigens to immunologic niches that favor tolerance such as the skin and oral mucosa (95, 96); administering high doses of peptide IV to induce effector Tcon apoptosis (97); and modifying myelin peptides by altering the amino acid sequence (altered peptide ligands) to decrease TCR-antigen recognition efficiency and induce regulatory T cells and/or effector T cell anergy and apoptosis (98).
Several of these strategies were advanced into human clinical trials (Table 1). The first of these was a phase I clinical trial testing daily oral administration of encapsulated bovine myelin in patients with relapsing-remitting MS (99). It was determined that oral bovine myelin was safe and reduced the frequency of MBP-specific T cells; however, in a larger phase III clinical trial, oral bovine myelin did not significantly improve MS (113). Subsequently, phase I and II clinical trials tested the safety and efficacy of “ATX-MS-1467” which was comprised of MBP30-44, MBP131-145, MBP140-154, and MBP83-99 and was administered ID or SC to patients with relapsing-remitting or secondary progressive MS (100). The therapy was safe and resulted in a significant decrease of new/persisting CNS lesions. In addition, a phase I clinical trial examined the safety of a peptide vaccine comprised of MBP85-99, MOG35-55, and PLP139-155 that was administered via skin patch to patients with relapsing-remitting MS (102). The therapy was safe and resulted in a significant reduction of CNS lesions and reduced clinical disease. The vaccine activated skin Langerhans cells, induced Tr1 cells, and decreased myelin‐specific T cell responses. Moreover, a phase III clinical trial tested the efficacy of IV administered MBP83-99 in patients with secondary progressive MS. The therapy was safe but lacked therapeutic efficacy (NCT00468611) (104). Unfortunately, several peptide-based vaccines tested in MS had adverse effects. A phase II clinical trial, testing SC administration of altered peptide ligands of MBP83-99, resulted in systemic hypersensitivity reactions in 9% of patients (105). Additionally, in a separate phase II clinical trial, the altered peptide ligand vaccine, exacerbated MS and resulted in a 3-fold increase of CNS lesions, expansion of Th1 pathogenic MBP83-99-specific Tcons, and increased intramolecular epitope spreading within MBP (106). Both clinical trials were terminated due to safety concerns. Therefore, as peptide-based tolerogenic vaccines are advanced, every effort should be made to select vaccine platforms that support tolerogenic responses and minimize the risk of sensitization.
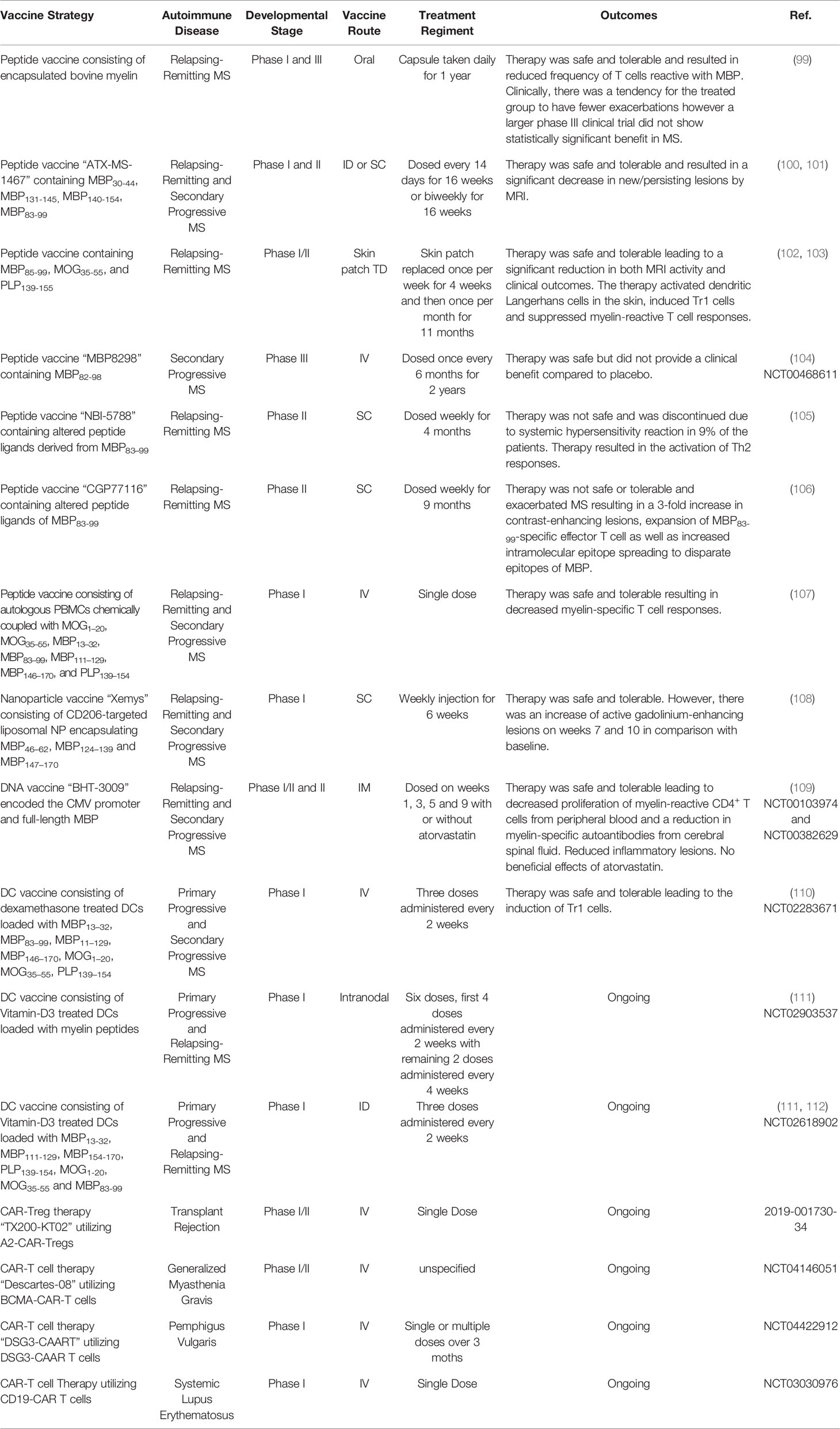
Table 1 Clinical trials.
A common problem with naked peptides is they are rapidly cleared and thus exert transient effects (114). Consequently, peptide vaccines are typically administered repeatedly and often require high doses (90, 115, 116). Therefore, protein carriers are being investigated to increase the stability, half-life, and bioavailability of autoantigen peptides to increase the efficacy of peptide-based tolerogenic vaccines. Protein carriers such as mAb, cytokines, cells, and pathogen derived immunosuppressive or adhesion proteins have served as targeting moieties to introduce tethered peptides into specific immunological niches or as tolerogenic adjuvants to favor tolerance. Here we discuss tolerogenic peptide-carrier vaccine targeting and tolerogenic adjuvant strategies that generated tolerance in EAE (outlined in Table 2).
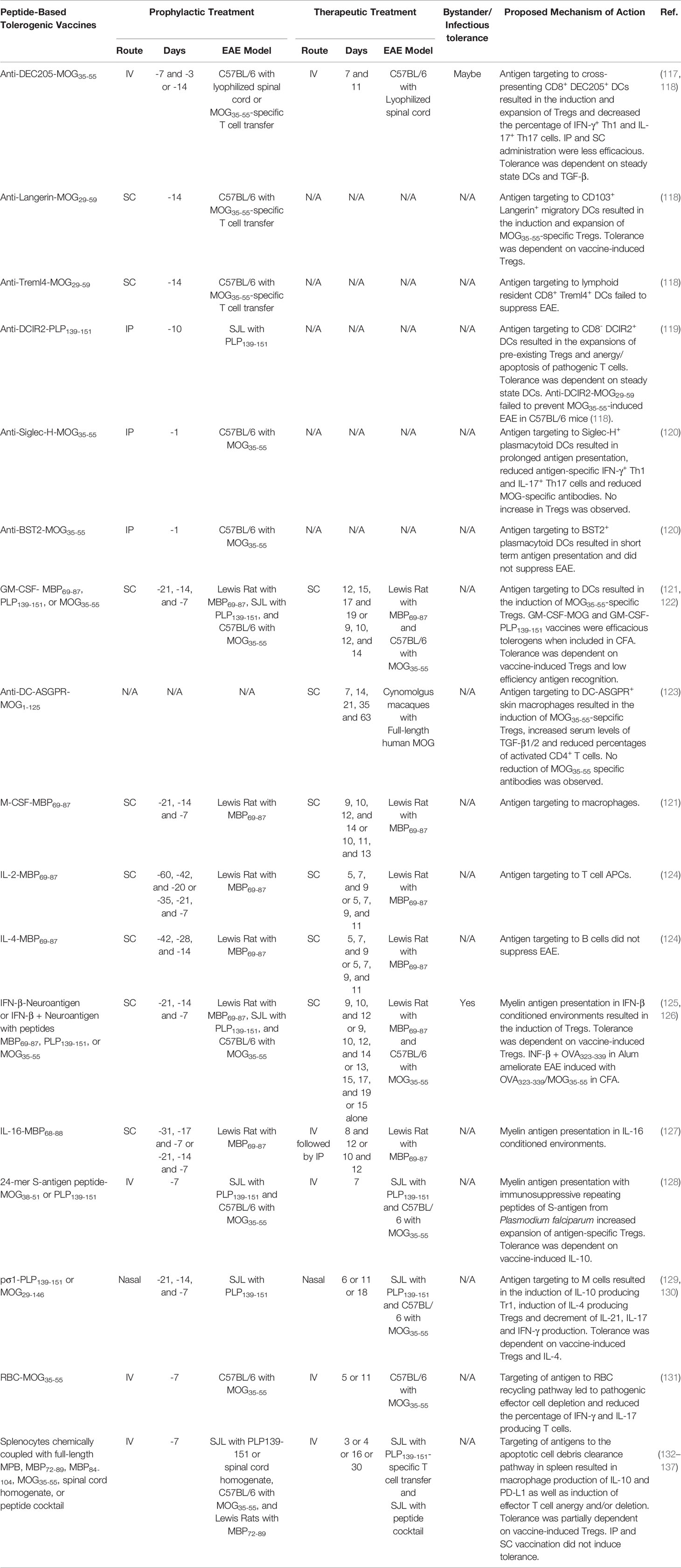
Table 2 Protein- and peptide-based tolerogenic vaccines.
DC-targeting carrier proteins have been extensively explored in preclinical models of EAE. Myelin peptides fused to mAbs specific for DC receptors such as DEC205, DCIR2, Langerin, and Siglec-H targeted tethered myelin peptides to the corresponding receptor expressing DCs populations and prevented EAE (117–120). These antibodies were selected based on their ability to target unique DC subsets including CD8+ DCs, CD8─ DCs, CD103+ migratory DCs, or plasmacytoid DCs. For example, disease-specific antigen peptides fused to anti-DEC205 mAbs or single chain variable fragments (scFv) are targeted to steady state cross-presenting CD8+ conventional DCs and CD103+ migratory DCs (118), and suppressed murine models of CD4+ T cell-driven cartilage proteoglycan induced arthritis, DTH, T1D, and EAE as well as CD8+ T cell-driven contact hypersensitivity and T1D (138–142). Anti-DEC205 vaccines mediated dominant tolerance via the induction of antigen-specific Tregs and passive tolerance via the induction of autoreactive T cell anergy and apoptosis (117, 118). IV vaccination was more effective than both SC and IP. Tolerance was dependent on TGF-β (117) and immature steady state DCs (141).
Other examples of DC-targeting peptide vaccines that suppressed EAE include anti-Langerin, DCIR2 and Siglec-H fusion proteins that targeted migratory CD103+ DCs, CD8- DCs, or plasmacytoid DCs, respectively (Table 2).
Carrier proteins serving as both tolerogenic adjuvants and targeting moieties have demonstrated efficacy in preclinical models of EAE. For example, the cytokine GM-CSF has been used as a tolerogenic adjuvant and a DC targeting moiety to suppress EAE. Peptides tethered to GM-CSF were specifically targeted to myeloid DCs in vitro (121). GM-CSF fused with either MBP69-87, PLP139-151, or MOG35-55 prevented EAE induced with MBP69-87 in Lewis rats, PLP139-151 in SJL mice, and MOG35-55 in C57BL/6 mice, respectively. These GM-CSF-neuroantigen fusion protein vaccines were effective when administered as prophylactic or therapeutic treatments in multiple rodent models of EAE (121, 143–145). GM-CSF-PLP139-151 or -MOG35-55 fusion proteins mediated tolerance in proinflammatory environments and prevented EAE when mixed directly with the encephalitogenic emulsion (144). The GM-CSF-MOG35-55 fusion protein increased Tregs in the blood, spleen, and lymph nodes and decreased circulating Tcon in the blood of MOG35-55-specific 2D2 TCR transgenic mice. Tolerance was dependent on GM-CSF-MOG35-55-induced Tregs (143). The mechanism by which GM-CSF-neuroantigen vaccines elicited tolerance was dependent on low efficiency TCR-antigen recognition that excluded the recruitment of the CD40-CD40L costimulatory pathway (122, 143).
Targeting myelin peptides to macrophages and DCs had tolerogenic efficacy in cynomolgus macaque and Lewis rat models of EAE. For example, anti-DC-ASGPR mAb fused with MOG1-125 selectively targeted MOG1-125 to CD163+ CD40─ resident macrophages and monocyte derived DCs (123). The anti-DC-ASGPR-MOG1-125 fusion protein completely protected cynomolgus macaques from MOG induced EAE when administered after sensitization but before disease onset. This vaccine decreased the percentage of activated CD4+ T cells, increased the percentage of MOG35-55-specific FOXP3+ Tregs, and increased levels of TGF-β1/2. Peptides can also be targeted to macrophages and DCs with the cytokine M-CSF. Recombinant fusion proteins comprised of M-CSF covalently linked with MBP69-87 inhibited MBP69-87-induced EAE in Lewis rats when administered prophylactically or therapeutically (121).
A fusion protein comprised of IL-2 and MBP69-87 targeted MBP69-87 to MHCII+ T cell for enhanced antigen presentation (121). The IL-2-MBP69-87 fusion protein prevented MBP69-87-induced EAE in Lewis rats when injected SC prophylactically or therapeutically (124). The tolerogenic activity was not due to the immunomodulatory properties of IL-2 because IL-2 alone did not suppress EAE. It was proposed that autoantigen presentation by MHCII+ CD4+ T cells induces T cell anergy and apoptosis (146–148). Interestingly, B cell targeting with IL-4-neurotantigen fusion protein did not elicit tolerance in EAE (121) (124).
IFN-β was used as a tolerogenic adjuvant and carrier protein because of its ability to suppresses T cell priming, inhibit Tcon proliferation, induce Tregs, and prompt tDC differentiation (149). IFN-β-neuroantigen fusion proteins were potent therapeutic or prophylactic interventions that inhibited EAE in Lewis rats and mice (125, 126, 150). The tolerogenic activity of IFN-β-neuroantigen was not due to the immunomodulatory activity of IFN-β because Lewis rats treated with an equimolar dose of IFN-β alone were not protected from EAE (150). IFN-β could also be non-covalently linked with peptides via hydrostatic binding in Alum to mediate tolerance. For example, a tolerogenic vaccine comprised of IFN-β + MOG35-55 in Alum ameliorated MOG35-55-induced EAE in C57BL/6 mice when administered at peak disease (125). The IFN-β + MOG35-55 in Alum vaccine induced Tregs in MOG35-55-specific 2D2 TCR transgenic mice. In addition, vaccine-induced tolerance was dependent on vaccine-induced Tregs. Interestingly, the vaccine mediated bystander suppression because IFN-β + ovalbumin (OVA323-339) in Alum protected mice from EAE when mice were challenged with OVA323-339 + MOG35-55 in CFA but not MOG35-55 in CFA. These results suggested that the IFN-β + OVA323-339 in Alum induced OVA-specific Treg and blocked the priming of encephalitogenic MOG35-55-specific T cells. Likewise, fusion proteins comprised of the immunomodulatory cytokine IL-16 and MBP69-88 ameliorated MPB69-88-induced EAE in Lewis rats when administered prophylactically or therapeutically (127). Other tolerogenic vaccine strategies included fusing myelin peptides to the immunosuppressive S-antigen from Plasmodium falciparum (128) or targeting myelin peptides with the viral adhesion protein pσ1, to M cells, which mediate mucosal antigen sampling and mucosal tolerance, to suppress EAE (Table 2) (129, 130).
Cell have been used as peptide carriers and targeting moieties. Red blood cells (RBCs) have been employed to introduce autoantigens into the RBC recycling pathway to mediate tolerance. Autoantigen loaded RBCs suppressed mouse models of EAE and T1D (131, 151). For example, RBCs carrying MOG35-55 protected C57BL/6 mice from MOG35-55-induced EAE when administered IV before or during disease onset (131). The RBC-antigen vaccine depleted antigen-specific immunocytes. Likewise, apoptotic leukocytes have been exploited to introduce autoantigens into the apoptotic cell debris clearance pathway and induced tolerance in rodent models of DTH, Theiler’s murine encephalomyelitis, allergy, experimental autoimmune thyroiditis, uveitis, neuritis, T1D, and EAE (132, 133, 152–161). For example, prophylactic IV vaccination of SJL mice with splenocytes coupled to PLP139-151 suppressed EAE induced with mouse spinal cord homogenate. The route of vaccine administration was critical since IV, but neither SC or IP vaccination, prevented EAE (134). The vaccine decreased antigen-specific T cell proliferation and increased the production of IL-10 and TGF-β (133). Tregs transferred tolerance from vaccinated donor mice to recipient mice and prevented the subsequent induction of EAE. Interestingly, Tregs were not required for tolerance but instead maintained long-term tolerance since Treg depletion only abrogated tolerance during late stage disease (day 63) but not during early disease (day 35). Mechanistically, it was determined that the vaccine was targeted to marginal zone macrophages in the spleen, that upregulated IL-10 and PD-L1 resulting in the expansion of Tregs and depletion of antigen-specific effectors cells (135, 162).
The success of antigen-coupled cells in preclinical models of EAE led to a phase I clinical trial testing the safety of myelin peptide-coupled PBMCs in patients with relapsing-remitting or secondary progressive MS (Table 1) (107). The study concluded that myelin peptide-coupled PBMC vaccination was safe and associated with decreased myelin reactive T cell recall responses in treated patients.
Particulate vaccines, such as nanoparticles (NP) and microparticles (MP) are being explored as protein/peptide carriers to extend autoantigen half-life and target autoantigens to distinct immunological niches. Particle-based vaccines can coordinate the delivery of tolerogenic adjuvants and autoantigens to different cell types or anatomical locations by modulating the particles size, charge, composition, and route of administration. For example, SC administered particles that are 1-6 nm, 9-100 nm, and 100 nm or greater drain to the blood, lymphatics, or form a local depot, respectively. When administered IV, particles that are 20–100 nm, 100-200 nm, and 200 nm or greater accumulate in the liver, liver/spleen or spleen, respectively (163). Ideally, particles should be biodegradable to prevent bioaccumulation and cytotoxicity (163). Here we discuss particle-based tolerogenic vaccine-targeting strategies that generate immune tolerance in rodent models of EAE (outlined in Table 3).
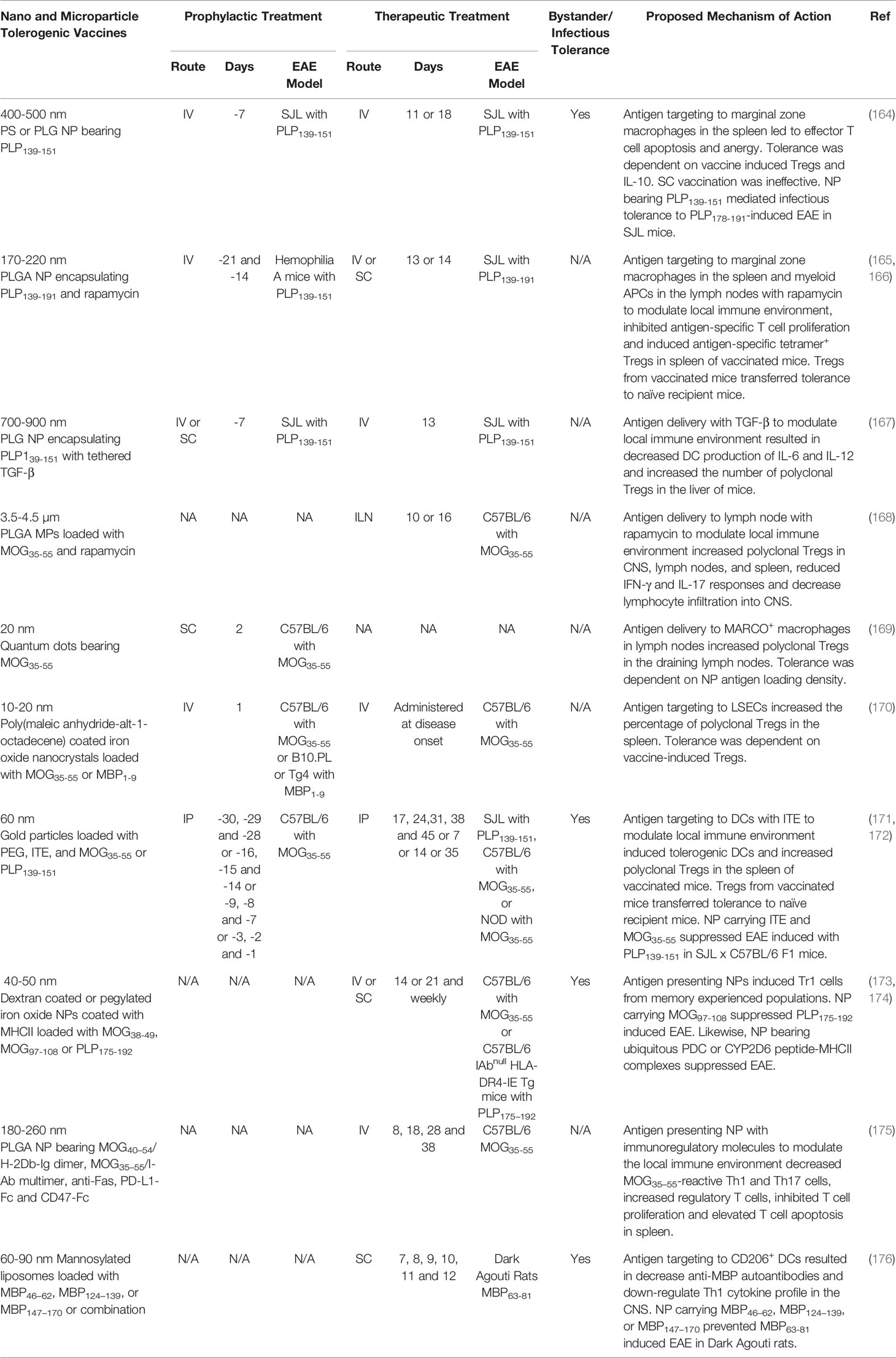
Table 3 Particle-based tolerogenic vaccines.
One strategy is to target autoantigens to the spleen using microparticle-based (MP) tolerogenic vaccines. MP vaccines composed of either non-biodegradable polystyrene and biodegradable PLGA (400-500 nm) mimicked apoptotic cell debris and target tethered autoantigens to MARCO+ marginal zone macrophages in the spleen when injected IV. The MP coupled with disease-specific antigens suppressed preclinical rodent models of EAE, T1D, celiac, and allergic airway disease (164, 177–180). For example, PLP139-151-MP suppressed PLP139-151-induced EAE in SJL mice when administered prophylactically or therapeutically. The particle size was crucial because smaller (100 nm) and larger (1.75 and 4.5 µm) particles were less effective (164). Likewise, the administration route was critical because IV vaccination demonstrated robust tolerogenic activity, while IP and SC vaccination had moderate to no tolerogenic activity, respectively (178). Collectively, these data suggested that MP must be a size that is conducive for spleen targeting and marginal zone macrophage phagocytosis to elicit robust tolerance. Mechanistically, the MP induced tolerance through the induction of T cell anergy and deletion. The PLP139-151-MP reduced the PLP139-151-specific T cell recall response and decreased the percentage of PLP139-151-specific IFN-γ and IL-17 producing T cells in vivo. In addition, MP-induced tolerance was partially dependent on Tregs and IL-10 (164). Moreover, tolerogenic adjuvants, including TGF-β and rapamycin, have been added to reinforce anti-inflammatory pathways and enhance the efficacy of spleen-targeted NP/MP vaccines (165–167).
A second strategy is to target particles to the lymph nodes, a site of T cell priming. Lymph node targeting has been achieved via SC injection of small NP (10-100 nm) that drain to local lymph nodes and are subsequently retained (181). Additionally, intra-lymph node injection of large MPs that are too large to subsequently migrate to other anatomical sites can be used to influence the lymph node environment. For example, intra-lymph node injection of MP (3.5-4.5 µm) vaccines comprised of biodegradable PLGA bearing MOG35-55 and the tolerogenic adjuvant rapamycin suppressed MOG35-55-induced EAE in C57BL/6 mice when administered prophylactically or therapeutically. The MP were not inherently immunosuppressive, because MP bearing an irrelevant antigen and rapamycin did not suppress EAE. Tolerance was dependent on lymph node targeting because IM vaccination did not restrain EAE. The MP increased the number of Tregs in the lymph nodes and spleen of vaccinated mice (168). Moreover, a tolerogenic NP vaccine comprised of 20 nm quantum dots fused with MOG35-55 was targeted to MARCO+ macrophages in the draining lymph nodes following SC vaccination. The quantum dot-MOG35-55 NP vaccine prevented MOG35-55-induced EAE in C57BL/6 mice when administered after the encephalitogenic challenge but before disease onset. Tolerance was dependent on antigen density because NP loaded with increasing quantities of MOG35-55 were less efficacious. Tolerance was associated with increased numbers of Tregs in the draining lymph nodes of vaccinated mice (169).
A third strategy includes targeting tolerogenic NP to the liver. Tolerogenic NP comprised of autoantigens tethered to Poly (maleic anhydride-alt-1-octadecene) coated superparamagnetic iron oxide nanocrystals (10-20 nm) were specifically enriched in the liver and internalized by LSECs following IV administration (170). When loaded with MOG35-55, the LSEC targeted NP suppressed MOG35-55-induced EAE in C57BL/6 mice when administered IV before or during active disease (170). The LSEC-targeted NP increased the percentage of polyclonal Tregs in the spleens of vaccinated mice and Treg depletion abrogated vaccine-induced tolerance.
A fourth strategy is to target NP to DCs with tolerogenic adjuvants that promote the differentiation of tDC. AHR ligand 2-(1’H-indole-3’-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) is an adjuvant that activates the aryl hydrocarbon receptor and promoted the differentiation of tDCs. DC targeted gold nanoparticles (60 nm) coated with disease-specific autoantigen, ITE, and PEG suppressed rodent models of EAE and T1D (171, 172, 182). The ITE-antigen-NP induced the differentiation of tDCs in vitro, that expressed low levels of the co-stimulatory molecules (MHCII, CD40, and CD86) and proinflammatory cytokines (IL-6 and IL-12) and increased the production of anti-inflammatory cytokines (TGF-β and IL-10) (171, 182). When administered SC or IV, the NP were selectively targeted to CD11c+ DCs. SC administration of the NP increased the percentage of MOG35-55-specific (tetramer+) FOXP3+ Tregs and IL-10 producing Tr1 cells in addition to reducing the percentage of MOG35-55-specific (tetramer+) IL-17 and IFN-γ producing Tcons in the spleen (182). The NP loaded with ITE and MOG35-55 suppressed MOG-induced EAE when administered IP, IV, or SC and were effective when administered prophylactically or therapeutically (171, 182). Tregs transferred tolerance from vaccinated donor mice to recipient mice and prevented the subsequent induction of EAE (171). The NP vaccine elicited bystander suppression because NP containing ITE and MOG35-55 suppressed PLP139-151-induced EAE in C57BL/6 x SJL F1 mice (182).
A fifth strategy is to directly target autoreactive T cells with artificial APC NP decorated with autoantigen-MHCII complexes. This strategy is based on the premise that chronic autoantigen stimulation in the absence of co-stimulation results in T cell anergy and expansion of suppressive Tr1 cells. NP (40-50 nm) loaded with diseases-specific autoantigen-MHCII complexes suppressed autoimmune disease in rodent models of collagen induced arthritis, autoimmune hepatitis, T1D, and EAE (173, 174, 183, 184). For example, NP bearing MOG38-49-MHCII complexes inhibited MOG35-55 induced EAE in C57BL/6 mice when administered at the peak of the disease (174). The disease resolution was associated with the conversion of antigen-experienced autoreactive T cell into suppressive Tr1 cells that expressed ICOS, LAP, CD49b, and LAG-3 as well as the anti-inflammatory cytokines IL-10 and IL-21. Effects of this vaccine were independent of FOXP3+ Tregs. Impressively, the autoantigen-MHCII nanoparticles elicited bystander suppression. Specifically, NP loaded with CNS-specific MOG97-108-MHCII but not joint-specific collagen type II (CII259–273)-MHCII ameliorated PLP-induced CNS autoimmunity in MHCII humanized C57BL/6 mice. Particulate size was critical because vaccines comprised of larger MP loaded with MHCII-autoantigen or smaller monomeric MHCII-autoantigen were unable to restrain EAE (174).
NP displaying peptide-MHC complexes, in which peptides are from ubiquitous autoantigens such as mitochondrial pyruvate dehydrogenase (PDC) or cytochrome P450 (CYP2D6), could induce autoantigen-specific Tr1 cells that ameliorate hepatic autoimmunity, T1D, and EAE in mice (173, 184). The consensus was that the ubiquitous self-proteins PDC and CYP2D6 are released during inflammation leading to antigen-specific CD4+ T cell responses. Following vaccination, the PDC- or CYP2D6-specific effector CD4+ T cell were converted to Tr1 cells that trafficked to sites of inflammation where PDC and CYP2D6 were released during tissue damage to mediate tolerance. To our knowledge this is the first account in which tolerance to ubiquitous autoantigens elicited organ-specific tolerance. Impressively, immunity toward vaccinia virus, influenzas and Listeria Monocytogenes or allogeneic colon carcinoma (CT26) and melanoma (B16/F10) liver metastases was maintained, thus the NP were not overtly immunosuppressive (173).
Tolerogenic adjuvants have been added to NP-based artificial APC platforms to enhance vaccine efficacy. For example, biodegradable PLGA nanoparticles (180-260 nm) containing surface tethered peptide-MHC complexes (MOG40–54/H-2Db-Ig dimer, MOG35–55/I-Ab multimer), regulatory molecules (anti-Fas, PD-L1-Fc), self-marker (CD47-Fc), and encapsulated TGF-β1 modulated T cell responses to induce tolerance. These NP suppressed MOG35-55-induced EAE in C57BL/6 mice when administered during disease onset. Tolerogenic NP without neuroantigen but with regulatory molecules lacked therapeutic activity in EAE suggesting the NP were not inherently immunosuppressive. Mechanistically, the MOG-MHC loaded NP decreased the percentage of MOG35–55-reactive Th1 and Th17 cells and increased the percentage of Tregs in the spleen (175).
One tolerogenic NP vaccine has been advanced into a clinical trial for MS (108). The NP was composed of mannosylated liposome unilamellar vesicles that were 60-90 nm in diameter and encapsulated MBP peptides. Preclinical studies determined that mannosylated liposome NP were selectively phagocytosed by DCs in vitro, via the mannose receptor CD206. The mannosylated liposome NP loaded with either MBP46–62, MBP124–139, or MBP147–170 as well as a mixture of the three peptides together prevented MBP63-81-induced EAE in Dark Agouti rats when administered SC at disease onset (176). These NP decreased serum anti-MBP autoantibodies and down-regulated Th1 cytokine profile. Following the preclinical success of mannosylated liposome NP, a phase I clinical trial was initiated in patients with relapsing-remitting or secondary progressive MS. Patients were treated SC with ascending doses of “Xemys” a mannosylated liposome NP loaded with MBP46–62, MBP124–139, and MBP147–170 (Table 1). The study determined that Xemys was safe and well tolerated. However, there was an increase of active CNS lesions on weeks 7 and 10 in comparison with baseline.
DNA-based vaccines are made of DNA vectors that include nucleic acid sequences encoding target antigens. When injected, the DNA-based vaccine is internalized by local or target cells and translated to generate in situ protein products that are subjected to traditional antigen presentation on MCHII. Presentation of DNA vaccine protein products (e.g. peptide) on MCHII then elicits immunogenic or tolerogenic immune responses. These DNA-based vaccines have been administered as naked plasmid DNA or as DNA constructs packaged in cationic lipids, liposomes, microparticles, and viruses to mediate DNA uptake and ectopic expression of the encoded autoantigens (185). Numerous DNA-based tolerogenic vaccines have been devised that induce antigen-specific tolerance in animal models of autoinflammatory disease (186), including murine models of EAE (185). However, several DNA-based vaccines encoding myelin antigens failed to induce tolerance and instead resulted in sensitization and exacerbated EAE (187–189). Therefore, several strategies have been conceived to specifically tailor DNA-vaccines to promote immune tolerance and prevent sensitization. These strategies include: (1) targeting DNA vaccines to anatomical sites that favor immune regulation such as the skin, muscle, and liver; (2) limiting expression of DNA vaccine to DC subsets; (3) co-expressing or administering tolerogenic adjuvants alongside DNA vaccines; and (4) modifying DNA vaccines to reduce the number of immunogenic CpG motifs to inhibit proinflammatory TLR-9 activation. Here we discuss DNA-based tolerogenic vaccine strategies that generate immune tolerance in rodent models of EAE (outlined in Table 4).
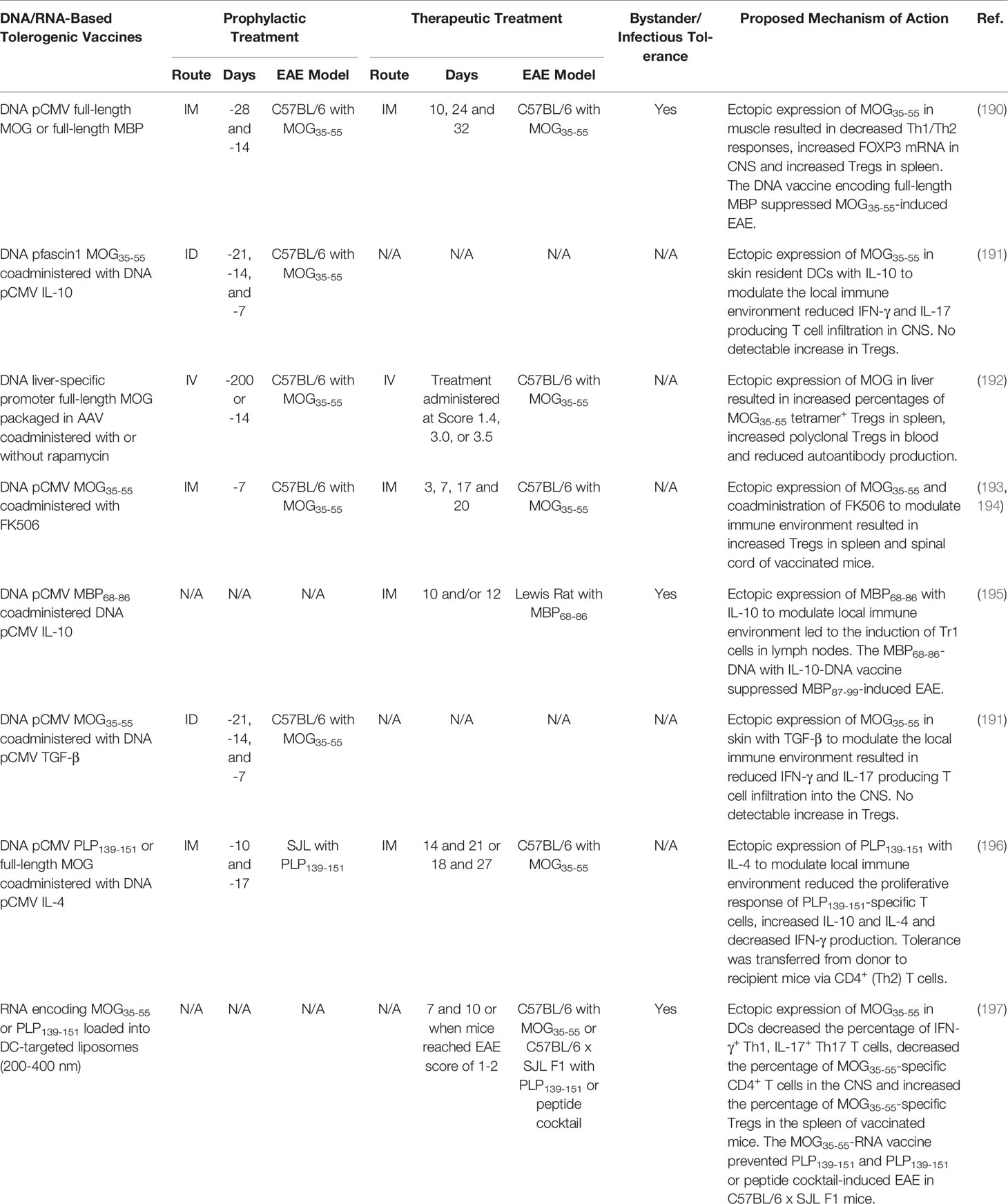
Table 4 DNA-based tolerogenic vaccines.
The anatomical site of autoantigen recognition can influence tolerance. Therefore, DNA-based vaccines are often introduced into anatomical sites that are immunologically quiescent (e.g. muscle) or promote Treg responses (e.g. skin and liver). For example, intramuscular (IM) vaccination of C57BL/6 mice with a DNA-vaccine encoding full-length MOG under the ubiquitous CMV promoter suppressed MOG35-55-induced EAE when administered prophylactically or therapeutically (197). The vaccine increased the percentage of polyclonal Tregs in the spleen and increased the expression of the Treg-specific transcription factor FOXP3 in the CNS. Splenocytes from MOG-DNA vaccinated mice produced less INF-γ, IL-17, and IL-4 following re-stimulation with MOG35-55. Of note, a full-length MBP-DNA vaccine elicited bystander suppression because the MBP-DNA vaccine suppressed MOG35-55-induced EAE when administered before disease induction. However, a full-length PLP-DNA vaccine failed to suppress MOG35-55-induced EAE as a therapeutic or prophylactic vaccine (197). In addition, DNA-based vaccines targeted to skin resident DCs suppressed EAE (198) (Table 4).
DNA-based tolerogenic vaccines encoding autoantigens have also been targeted to the liver. For example, an adeno-associated virus (AAV) packaged DNA-vaccine encoding full-length MOG under the control of a liver specific promoter resulted in the stable and restricted expression of MOG in the liver of C57BL/6 mice (199). The AAV liver targeted MOG-DNA vaccine elicited robust and lasting (>335 days) tolerance to MOG35-55-induced EAE and was effective when administered as prophylactic or therapeutic vaccine. The vaccine increased the percentage of MOG35-55-specific tetramer+ Tregs in the spleen and increased the abundance of polyclonal Tregs in the blood of vaccinated mice. Additionally, DNA-based tolerogenic vaccines have been co-administered with the tolerogenic adjuvants FK506 and rapamycin to suppress EAE (199–201) (Table 4).
Furthermore, DNA vaccines encoding autoantigens in conjunction with the immunoregulatory cytokines IL-10, TGF-β, or IL-4 were tested. For example, co-administration of separate plasmids encoding MBP68-86 or IL-10 under the CMV promoter suppressed MBP68-89-induced EAE in Lewis rats when administered prophylactically or therapeutically (202). The vaccine was associated with the induction of Tr1 cells. Interestingly, the tolerogenic vaccine demonstrated bystander suppression because the IL-10 + MBP68-86-DNA vaccine blocked EAE induced with MBP87-99 (202). DNA-based vaccines encoding autoantigens in conjunction with DNA vaccines encoding TGF-β (198) or IL-4 (203) suppressed EAE in rodents (Table 4).
The success of DNA-based tolerogenic vaccines in rodent models of EAE led to the development of a phase I/II and II clinical trials testing “BHT-3009”, a DNA-based tolerogenic vaccine encoding the CMV promoter and full-length MBP (Table 1). The DNA backbone contained reduced numbers of immunostimulatory CpG motifs to limit TLR-9 activation. The BHT-3009 vaccine was injected IM into patients with relapsing-remitting or secondary progressive MS (NCT00103974) (109). The DNA vaccine was found to be safe and decreased the number of CNS lesions in patients, however the differences did not reach statistical significance. Interestingly, the vaccine decreased the antigen-specific T and B cell response not only to MBP, but also PLP and MOG, suggesting that DNA-based vaccination may promote bystander suppression in humans.
RNA-based tolerogenic vaccines have been explored (outline in Table 4) (204). Extracellular and double stranded RNA molecules are inherently proinflammatory and trigger TLR activation, DC maturation, IFN-α production, and Th1 responses. Therefore, efforts were made to reduce the proinflammatory nature of RNA-vaccines by replacing uridine with 1-methylpseudouridine (m1Ψ) to abrogate TLR-3, TLR-7, and TLR-8 activation (205) and by removing double-stranded RNA contaminants to abrogate TLR-7 activation (206). In addition, RNA-based vaccines have been loaded into DC targeting liposomes to specifically introduce the RNA-based vaccine into DCs for translation and MHCII presentation (207). The nanoparticle-formulated m1Ψ-modified single stranded mRNA vaccine that encoded MOG35-55 inhibited MOG35-55-induced EAE in C57BL/6 mice when administered before or after disease development (204). The vaccine decreased the percentage of IFN-γ+ Th1, IL-17+ Th17 and MOG35-55-specific (tetramer+) CD4+ T cells in the CNS and increased the percentage of MOG35-55-specific (tetramer+) FOXP3+ Tregs in the spleen. The vaccine mediated bystander suppression as the MOG35-55-RNA vaccine also prevented PLP139-151-induced EAE in C57BL/6 x SJL F1 mice when administered before disease onset. Likewise, the MOG35-55-RNA tolerogenic vaccine ameliorate EAE induced with a cocktail of MOG35-55, PLP139-151, PLP178-191, MBP84-104, and Myelin-associated oligodendrocyte basic protein (MOBP15-36) in C57BL/6 x SJL F1 mice when administered as a prophylactic vaccine. These results suggest that RNA-based tolerogenic vaccines encoding a single myelin epitope can induced antigen specific-Tregs that mediate bystander suppression toward multiple noncognate myelin epitopes to control CNS autoimmunity in mice.
Transplant of autologous cells including stem cells, Tregs, Tr1, iTr35, Bregs, DC, myeloid suppressor cells, microglia, and macrophages among others, suppressed EAE (66, 208–214). Autologous autoantigen loaded DCs have garnered interest as cell-based tolerogenic vaccine strategy due to the unique capacity of DCs to favor Treg response under inflammatory conditions. DCs can be loaded with autoantigens to confers antigen-specificity and treated with pharmacological agents and biologics to derive stable immunosuppressive tDCs. Determining the best methods for generating robust tDCs are currently being investigated. In Table 5, we summarized DC-based tolerogenic vaccine strategies that generate immune tolerance in rodent models of EAE. Pharmacological agents that inhibit the NF-kB pathway (190), a selective JAK1/JAK3 inhibitor Tofacitinib (191), a selective JAK3/STAT5 inhibitor BD750 (192), cytotoxic agents dexamethasone and/or minocycline (193), VitD3 (215), and biologics IL-27 (195) and IL-10 (196) were used to differentiate tDCs. When loaded with autoantigens these tDCs suppressed EAE.
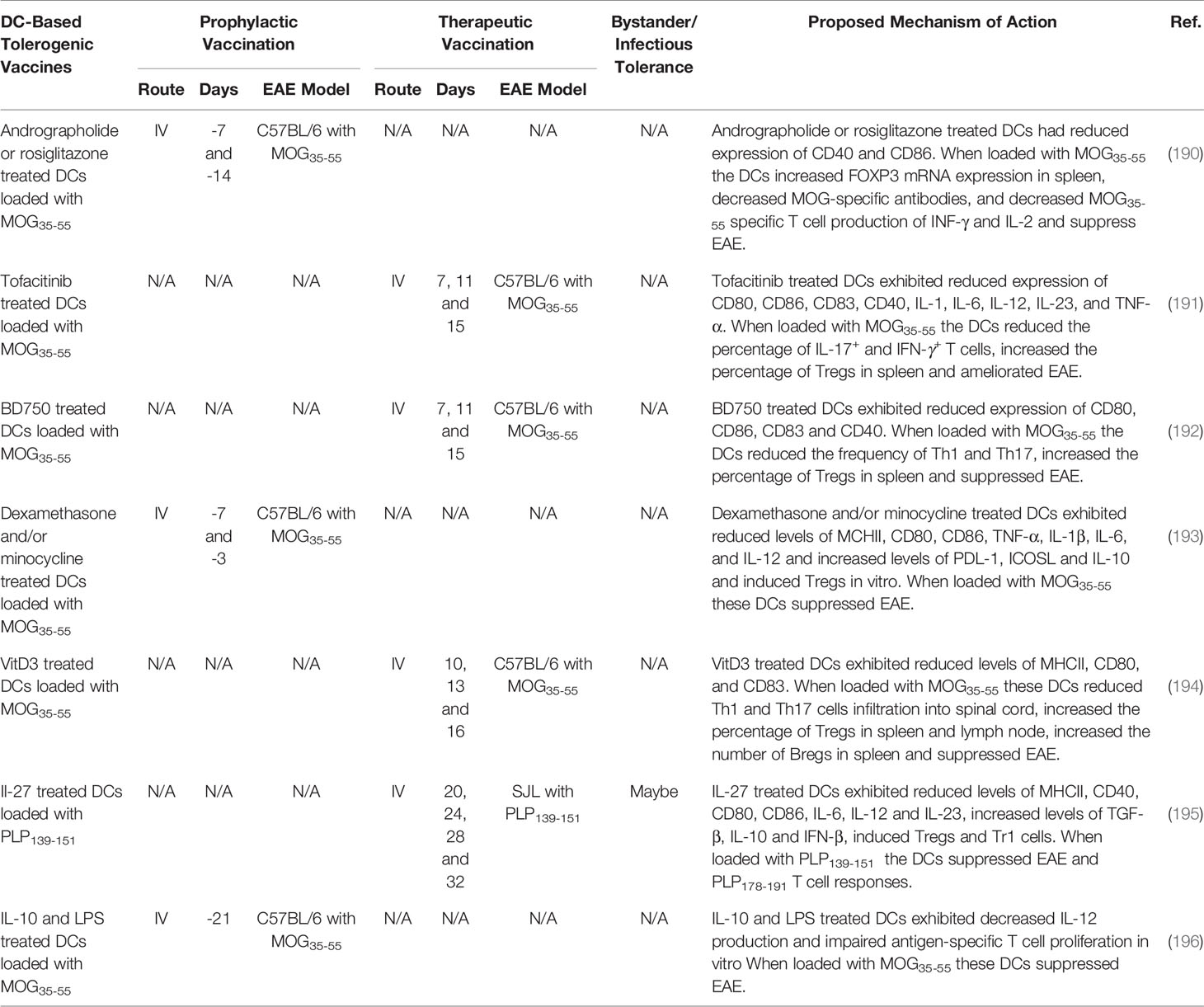
Table 5 DC-based tolerogenic vaccines.
Several DC-based tolerogenic vaccines have been advanced into clinical trials for the treatment of MS (Table 1). First, a phase I clinical trial tested the safety of IV administered myelin peptide loaded DCs in patients with primary or secondary progressive MS (NCT02283671). To generate DCs, PBMC monocytes were cultured with IL-4, and GM-CSF and treated with dexamethasone to induce a tDC phenotype. The tDCs were further treated with IL-1β, IL-6, TNF-α, and prostaglandin E2, and loaded with a mixture of the myelin peptides, MBP13–32, MBP83–99, MBP11–129, MBP146–170, MOG1–20, MOG35–55, and PLP139–154. The myelin-loaded tDC vaccine was safe because patients remained stable in terms of relapse and disability. The DC vaccination increased IL-10 production and increased Tr1 cells (110). In addition, two more phase I clinical trials are underway testing the feasibility and safety of intranodal (IN) and intradermal (ID) administration of VitD3 treated DCs loaded with a mix of myelin peptides (NCT02618902 and NCT02903537) (27, 213).
Recent progress in single cell sorting, cell monoculture techniques, and genetic engineering paired with the clinical success of chimeric antigen receptor (CAR)-T cell therapy in treating hematologic cancer has renewed enthusiasm for the use of T cell-based therapies to treat autoimmune diseases. Treg adoptive transfer therapy has become a major focus of cell-based therapy as these cells suppress antigen-specific autoimmunity. Although there are multiple substantial barriers to overcome, T cell therapy is an elegant concept: as these therapies would directly provide antigen-specific Tregs to suppress immune response or cytotoxic T cells to remove autoreactive immune cells, a common goal among tolerogenic vaccine platforms.
Mounting evidence suggest that Treg adoptive transfer may be an efficacious treatment for inflammatory autoimmune diseases. Indeed, the adoptive transfer of polyclonal Tregs suppresses numerous animal models of autoimmunity, allergic disease, and transplant rejection (216). These realizations initiated multiple clinical trials investigating autologous polyclonal Treg transfer as a treatment for autoimmune disease and transplant rejection. In these clinical trials, polyclonal Tregs were purified from the patient’s blood, expanded ex-vivo, and then subsequently reinfused into patient as an autologous Treg cell therapy. Polyclonal Tregs have been or currently are being tested in patients with T1D, pemphigus vulgaris (PV), autoimmune hepatitis, systemic lupus erythematosus (SLE), Crohn’s disease, and organ transplant [reviewed here (217)]. Completed clinical trials in T1D (218) and transplant rejection (219) have revealed that polyclonal Treg therapy is safe and, in some cases, demonstrated modest but limited efficacy.
Antigen-specific Tregs, suppress antigen-specific immune responses more potently compared to polyclonal Tregs, and are being investigated as cell-based therapy for autoimmunity (220, 221). However, multiple technical hurdles must be overcome for widespread use in the clinic. First, generating large quantities of antigen-specific Tregs is difficult due to their low precursor frequency in vivo and their limited proliferative potential. Moreover, Treg cell surface markers are limited, thus Tcon contamination poses a concern. Nevertheless, preclinical studies utilizing TCR transgenic mice as a source of antigen-specific Tregs demonstrated the potential use of antigen-specific Tregs to treat autoimmunity. For example, TCR transgenic Tregs that recognized MOG35-55, PLP139-151, and MBP1-9 were able to suppress EAE driven with each respective neuroantigen (58, 222, 223). These antigen-specific Tregs suppressed disease not only as a prophylactic, as did polyclonal Tregs, but also as a therapeutic treatment during active disease. For example, MBP1-9-specific Tregs completely inhibited MBP1-9-induced EAE in B10.PL while non-specific polyclonal Tregs only transiently inhibited EAE (222). Furthermore, antigen-specific Tregs exhibited bystander suppression to suppress CNS autoimmunity elicited against nonrelated myelin antigens. For example, MBP1-9-specific Tregs partially inhibit EAE induced with PLP139-151 (222). Likewise, PLP139-151-specific Tregs were able to restrain EAE induced with a disparate epitope of PLP178-191 (223). Together these studies support the development of antigen-specific Treg therapy for autoimmunity.
Expression of chimeric antigen receptors (CAR) or synthetic TCR confers antigen-specificity to polyclonal Tregs. CAR- and TCR-Tregs are being developed as a strategy to suppress pathogenic immune responses (outlined in Table 6).
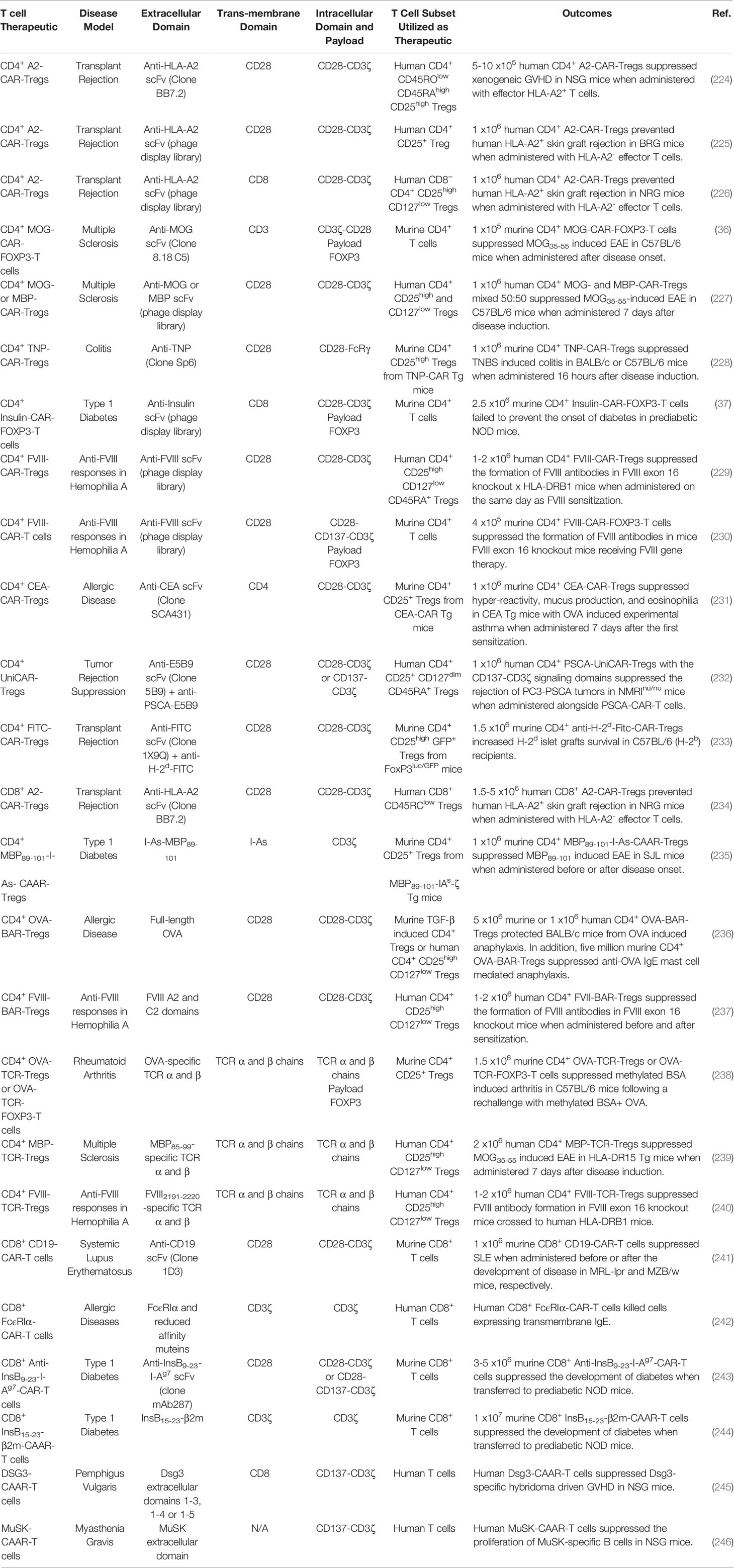
Table 6 T cell therapies for the treatment of inflammatory disease.
CD4+ CAR-Tregs: CD4+ CAR-Tregs are being investigated to treat GVHD and transplant rejection. The hypothesis is that Tregs expressing a CAR that recognizes a graft-specific MHCI haplotype can suppress allogenic graft transplant rejection. Several MHCI-recognizing CAR constructs have been devised. One such CAR construct encoded an extracellular HLA-A2-specific scFv, transmembrane CD28 domain, and the intracellular signaling domains of CD28 and CD3ζ (224). Human Tregs transduced with this receptor (A2-CAR-Tregs) were activated, proliferated, and upregulated the Treg activation markers when co-cultured with HLA-A2 expressing cells. The A2-CAR-Tregs selectively interacted with HLA-A2+ PBMCs and suppressed allogenic Tcon responses against HLA-A2 better than polyclonal Tregs in vitro. The human HLA-A2-CAR-Tregs were also superior to polyclonal Tregs at preventing xenogeneic GVHD, when transferred together with HLA-A2- Tcons to NSG mice (224). In two independent systems, human A2-CAR-Tregs were also able to inhibit allogenic rejection of human HLA-A2+ skin grafts more effectively than polyclonal Tregs in BRG or NSG mice (225, 226). In addition, an extensive panel of intracellular co-signaling domains were tested in combination with the intracellular CD3ζ signaling domain in human A2-CAR-Tregs (247) and determined that the CD28 co-signaling domain was the most potent at suppressing HLA-A2+ PBMC-mediated GVHD in NSG mice. These murine studies led to the initiation of a phase I/II clinical trial investigating the safety and efficacy of A2-CAR-Tregs in MHCI-mismatched HLA-A2+ kidney transplant patients in end-stage renal failure (Table 1) (EudraCT number 2019-001730-34).
CD4+ CAR-Tregs are also being investigated as an intervention for organ-specific autoimmune diseases in mice. The hypothesis is that CAR-Tregs can be targeted to specific-organs and suppress organ-specific autoimmunity using CARs that recognize organ-restricted autoantigens. For example, CAR-Tregs that recognized the CNS restricted antigen MOG, have been tested in EAE. The CAR construct encoded an extracellular anti-MOG scFv, transmembrane CD3 domain, intracellular signaling domains of CD3ζ and CD28, internal ribosomal entry site (IRES), and the transcription factor Foxp3 (36) to confer myelin-specificity and redirect T cells into the Treg lineage (36). Murine CD4+ T cells transfected with the MOG-CAR-FOXP3 construct were immunosuppressive and suppressed Tcons in coculture as well as ameliorated MOG35-55-induced EAE in C57BL/6 mice when administered therapeutically at peak of disease (36). Likewise, human Tregs transduced with a CAR encoding anti-MBP or anti-MOG scFv, transmembrane CD28 and the intracellular signaling domains of CD28 and CD3ζ, suppressed MOG35-55-induced EAE in C57BL/6 mice (227). Similarly, CD4+ CAR-Tregs recognizing 2,4,6 Trinitrophenol (TNP) suppressed TNP-induced colitis in mice, decreased colonoscopy colitis scores, and increased survival (Table 6) (228).
CD4+ CAR-Tregs have also been tested in a murine model of T1D. The CAR construct encoded an extracellular anti-insulin scFv, transmembrane CD8 domain, the intracellular signaling domains of CD28 and CD3ζ, T2A self-cleaving peptide, and the transcription factor Foxp3 (37). The insulin-CAR-FOXP3-T cells were recruited into the Treg lineage and were activated and proliferated in response to aggregated insulin, but not monomeric insulin. Therefore, it was hypothesized that insulin-CAR-FOXP3-T cells would only be activated in the pancreas where aggregated insulin is secreted. Nonetheless, the insulin-CAR-FOXP3-T cells were unable to suppress the development of T1D when adoptively transferred into prediabetic NOD mice. Although the insulin-CAR-FOXP3-T cells lacked efficacy, these CAR-Tregs were long lived and could identified 4 months after adoptive transfer.
CD4+ CAR-Tregs are also being investigated as a strategy to suppress undesirable immune responses against recombinant protein-based therapeutics that result in the formation of neutralizing antibodies which inhibit therapeutic efficacy. For example, hemophilia A patients treated with Factor VIII (FVIII) replacement therapy often develop FVIII-neutralizing antibodies that abrogate therapeutic efficacy (248). Therefore, FVIII-CAR-Tregs were tested to prevent FVIII-specific immune responses in mice. The CAR construct encoded an extracellular anti-FVIII scFv, transmembrane CD28 domain, and the intracellular signaling domains of CD28 and CD3ζ (229). Human FVIII-CAR-Tregs exhibited antigen-specific suppression and prevented the proliferation of a FVIII-CAR-T cells, better than natural polyclonal Tregs, when cocultured with FVIII and PBMCs in vitro. Human FVIII-CAR-Tregs were tested in FVIII-knockout mice (E16) x humanized DR1 mice immunized with FVIII in incomplete Freund’s adjuvant and suppressed the development of FVIII-specific antibodies.
Furthermore, CD4+ CAR-Tregs have been investigated as an intervention for allergic disease. CEA-CAR-Tregs were tested in a murine model of OVA induced allergic airway inflammation in carcinoembryonic antigen (CEA) Tg mice that express CEA on the luminal surface of the pulmonary and the gastrointestinal tract epithelia (231). The CAR construct encoded an extracellular anti-CEA scFv, transmembrane CD4 domain, and the intracellular signaling domains of CD28 and CD3ζ. The CEA-CAR-Tregs homed to the lungs which expresses high levels of CEA in CEA-Tg mice. Following adoptive transfer to CEA-Tg mice, the CEA-CAR-Tregs reduced airways hyper-reactivity, inflammation, mucus production, and eosinophilia in experimental OVA induced asthma. Furthermore, CEA-CAR-Tregs reduced the production of proinflammatory cytokines IL-5 and IL-15 as well as prevented the accumulation of pathogenic IgE antibodies.
CD8+ CAR-Tregs: Although the specific function of CD8+ Tregs remains to be determined, it was reported that CD8+ Tregs are immunosuppressive and contribute to immune tolerance in mice and human (249, 250). Human CD8+ A2-CAR-Tregs were activated, as seen with CD4+ A2-CAR-Tregs, in the presence of HLA-A2+ cells (234). The CD8+ A2-CAR-Tregs were not inherently cytotoxic as they did not kill HLA-A2+ cells or induce weight loss when adoptively transferred into in HLA-A2 transgenic NSG mice. Additionally, human CD8+ A2-CAR-Tregs prevented human HLA-A2- T cell rejection of human allogenic HLA-A2+ skin grafts and xenograft GVHD induced with HLA-A2+ PBMCs in NSG mice.
CAAR- and BAR-Tregs: Antigen-specificity can also be conferred to Tregs using a specific type of CAR receptor known as chimeric autoantigen receptor (CAAR) or B cell-targeting antibody receptor (BAR). These receptors express antigens that are recognized by autoreactive T or B cells, instead of the conventional extracellular antigen recognition domains such as scFv. CAAR- and BAR-Tregs are activated when antigen-specific B and T cell recognize their cognate antigens included in the CAAR/BAR construct and drive CAAR/BAR crosslinking. These receptors act as bait to trap and suppress/kill antigen-specific lymphocytes.
Antigen-specificity has been conferred to Tregs using CAAR constructs that encode peptide-MHCII complexes. For example, a CAAR construct encoded an extracellular MBP89-101-I-As chains fused to the intracellular signaling domain of CD3ζ (235). The I-As-MBP89-101 CAAR domain was designed to lure pathogenic MBP89-101-specific CD4+ T cells to I-As-MBP89-101 CAAR-Tregs for suppression. The CD4+ MBP89-101-I-As-CAAR-Tregs suppressed MBP89-101-induced EAE in SJL mice, when administered at the time of disease induction or at peak disease. Lymphocytes from MBP89-101-I-As-CAAR-Treg treated mice exhibited decreased antigen-specific T cell responses and increased IL-4 and IL-10 production. Monoclonal antibody mediated neutralization of IL-10 and IL-4 abrogated MBP89-101-I-As-CAAR-Tregs mediated suppression. Interestingly, T cells from MBP89-101-I-As-CAAR-Treg treated donor mice suppress MBP89-101-induced EAE in recipient mice even after depletion of MBP89-101-I-As-CAAR-Treg. Therefore MBP89-101-I-As-CAAR-Treg induced MBP89-101-specific suppressor cells that could prevent EAE.
BAR-Tregs are being investigated as a therapeutic for allergic disease and were tested in a murine model of anaphylaxis. The BAR construct encoded an OVA extracellular antigen domain, transmembrane CD28 domain, and intracellular CD28 and CD3ζ signaling domains (236). Upon adoptive transfer into OVA sensitized BLAB/c mice the CD4+ OVA-BAR-Tregs did not result in anaphylaxis, in response to the OVA contained in the BAR construct. Instead, murine or human OVA-BAR-Tregs suppressed anaphylaxis in OVA sensitized BALB/c mice when mice were re-challenged with OVA. The OVA-BAR-Tregs also suppressed anti-OVA IgE mediated mast cell induced anaphylaxis.
BAR-Tregs are also being investigated to prevent undesirable immune responses to recombinant therapeutic drugs. Human FVIII-BAR-Tregs blocked the formation of FVIII-specific antibodies when administered before FVIII sensitization and prevented the further development of FVIII-specific antibodies when administered between FVIII sensitizations in FVIII knockout x DR1 mice (237).
The antigen specificity of polyclonal Tregs can be redirected using genetically engineered TCR. TCR-Tregs can recognize both extracellular and intracellular antigens in the contexts of MHCII while CAR-Tregs are limited to extracellular antigens. However, TCR-Tregs are restricted by MHCII/antigen availability as a well as MCHII haplotype.
TCR-Tregs are being tested to suppress organ-specific autoimmunity. For example, TCR-Tregs have been tested in a murine model of arthritis. The TCR constructs encoded OVA-reactive TCRα and β chains and in some cases included a Foxp3 transcript (238). Murine CD4+ Tregs were transduced with the OVA-reactive TCR construct that lacked Foxp3 while CD4+ T cells were transduced with the OVA-reactive TCR construct containing Foxp3. Expression of FOXP3 redirected T cells into the Treg lineage. Both OVA-TCR-Tregs and OVA-TCR-FOXP3-T cells elicited bystander suppression and suppressed a nucleoprotein-reactive CD8+ T cell line when cocultured with DCs, OVA, and nucleoprotein. The OVA-TCR Tregs and OVA-TCR-FOXP3-T cells suppressed methylated bovine serum albumin (mBSA)-induced arthritis in C57BL/6 mice and reduced knee swelling when mice were re-challenged with mBSA and OVA but not mBSA alone. However, OVA-TCR-FOXP3-T cells were less effective at suppressing disease compared to OVA-TCR-Tregs.
Additionally, myelin-specificity has been conferred to human Tregs via the expression of an MBP85-99-specific HLA-DR15-restircted TCR (239). The engineered MBP85-99-TCR-Tregs displayed antigen-specific suppression and prevented the proliferation of MBP85-99-specific effector T cells in vitro. Furthermore, the MBP85-99-TCR-Tregs were superior compared to OVA323-339-TCR Tregs at protecting HLA-DR15 transgenic mice from MOG35-55-induced EAE. Therefore, TCR-Tregs exhibit organ-specific bystander suppression. In addition, human polyclonal Tregs have been redirected to pancreatic antigens using islet-specific TCR (Table 6) (251).
In addition, human TCR-Tregs expressing a TCR-specific for FVIII were tested for their ability to block FVIII generated immune responses in FVIII knockout x DR1 humanized mice (240). The FVIII-TCR-Tregs were immunosuppressive and blocked FVIII-TCR-Tcon responses as well as the formation of FVIII-specific antibodies more efficiently than polyclonal Tregs in vitro.
CAR-T cell therapy: Cytotoxic CD8+ CAR-T cells are being explored as a therapeutic strategy to deplete pathogenic lymphocytes involved in the etiology of autoimmune and allergic disease. CAR-T cells may have several advantages over mAb-based cell depletion strategies currently used to treat autoimmune disease, including anti-CD20 and anti-CD52 which deplete B cells and B/T cells, respectively (252, 253). First, CAR-T cells are long lived cells that can multiply, while antibodies are constrained by a pharmacological half-life and require repeated administrations to achieve and maintain therapeutic efficacy. Furthermore, CAR-T cells can traffic to the lymphoid tissues or target organs and develop into memory populations that can prevent the reemergence of pathogenic lymphocytes. Therefore, CAR-T cells therapy may only require a single dose to achieve lasting therapeutic efficacy. Second, CAR-T cell may have a higher potency than mAb therapies and may more efficiently delete pathogenic immune cells. These attributes suggest that CAR-T cell therapies may be an efficacious means to deplete pathogenic immune cells to alleviate autoimmunity. However, caution should be taken as CAR-T could produce massive amounts of inflammatory cytokines following activation, which could exacerbate autoimmunity.
CD8+ CAR-T cells are being designed to kill B cells that express the B cell restricted surface molecule CD19, as a potential therapeutic for SLE (241). CD8+ CD19-CAR-T cells eliminated B cells, reduced circulating IgM, IgG, and pathogenic anti-DNA antibodies as well as reduced the ratio of CD4/CD8+ T cells in murine models of SLE. A single dose of the CD19-CAR-T cells suppressed murine SLE when administered before or after the development of clinical disease in MRL-lpr and MZB/w mice, respectively. The CD19-CAR-T cells depleted B cells throughout the experiment which lasted approximately one year. Moreover, CD8+ T cells transferred from donor mice treated with CD19-CAR-T cell 7 months prior, depleted B cells in recipient mice. These results suggest that B cell depletion with CAR-T cells may provide long term protection in autoimmune diseases. In addition to CD19, the transmembrane IgE receptor expressed on B cells was explored as a B cell target for the CD8+ CAR-T cells (Table 6) (242).
APCs presenting autoantigens including the insulin beta chain peptide 9-23 (InsB9-23) which binds to the MHCII (I-Ag7) in NOD mice can activate pathogenic insulin-specific T cells that drive T1D (254). Consistent with these findings, a mAb specific for InsB9-23-I-Ag7 complex prevented diabetes in NOD mice by a mechanism that was hypothesized to be dependent on APC depletion (243, 254). Therefore, a CAR-T cell construct was designed to kill InsB9-8 presenting APCs. The CAR encoded anti-InsB9-23-I-Ag7 scFv, transmembrane CD28 domain, and intracellular CD28 and CD3ζ signaling domains (243). A second-generation construct also contained the intracellular signaling domain of CD137 which increased CAR-T cell persistence in vivo. The CD8+ anti-InsB9-23-I-Ag7-CAR-T cells were activated and killed cells that expressed InsB9-23-I-Ag7 but not control I-Ag7construct. Adoptively transferred anti-InsB9-23-I-Ag7-CAR-T cell homed to the pancreatic lymph nodes and delay the development of T1D by approximately 5 weeks.
CAAR- and BAR-T cell therapy: Cytotoxic T cells have been engineered to express a CAAR construct encoding the peptide-MCHI complex that autoreactive CD8 T cell recognizes. For example, a mRNA based CAAR construct encoding InsB peptide 15-23 (InsB15-23) fused to the extracellular β2m and intracellular CD3ζ signaling domain redirected CD8+ T cells against pathogenic insulin-specific CD8+ T cells that recognize the MHCI/InsB15-23 complexes (244). The InsB15-23-β2m-CAAR construct killed InsB15-23-specific CD8+ T cell hybridoma line. When adoptively transferred into prediabetic 5-6-week-old NOD mice, the CD8+ InsB15-23-β2m-CAAR-T cells reduced CD4+ and CD8+ T cell infiltration into pancreatic islets and protected NOD mice from the development of T1D.
Likewise, antigen-specificity has also been conferred to T cells using CAAR constructs that encode antigen domains. For example, CAAR-T cells that express extracellular Desmoglein 3 (DSG3) domains were designed to deplete DSG3-specific B cells, which drive the pathogenesis of PV. A CAAR-T cell construct was designed that encoded an extracellular DSG3 domain, transmembrane CD8α domain, and the intracellular signaling domains of CD137 and CD3ζ (245). Human DSG3-CAAR-T cells produced IFN-γ and specifically lysed a DSG3-specific hybridoma cells even in the presence of soluble DSG3-specific antibodies that could potentially block the CAAR extracellular DSG3 domain. Likewise, human DSG3-CAAR-T cell eliminated DSG3-specific B cells in humanized NSG mice. The DSG3-CAAR-T cells did not exhibit off target cytotoxicity and prevented DSG3-specific hybridoma driven GVHD in NSG mice. In addition, CAAR-T cells that express extracellular muscle-specific receptor kinase (MuSK) domains were designed to delete pathogenic MuSK-specific B cells, that drive MG (Table 6) (246).
These preclinical studies led to the initiation of 3 clinical trials using CAR- or CAAR-T cells to deplete pathogenic B cells in MG, PV, and SLE (Table 1). For MG, a phase I/II clinical trial has been devised to test the safety and efficacy of anti-B-cell maturation antigen (BCMA)-CAR-T cells designed to deplete activated B cells (NCT04146051). For PV, a phase I clinical trial has been devised to test the safety of a DSG3-CAAR-T cells designed to deplete DSG3-specific autoreactive B cells (NCT04422912). Finally, for SLE, a phase I clinical trial will test the safety of CD19-CAR-T cells designed to deplete B cells (NCT03030976). In addition, CD19-CAR-T cells will be tested with or without an intracellular signaling CD137 domain (255).
Differentiation, function, and maintenance of Tregs are dependent on IL-2. In mouse models, deficiency of IL-2 or its receptor components leads to spontaneous autoimmune phenotype due to defects in the generation of functional Tregs (256–259). In humans, allelic variants associated with reduced expression of IL-2 or its receptor chains, or genes that impact the IL-2 receptor signaling pathway, have been associated with increased risks of multiple autoimmune and inflammatory diseases, including T1D, SLE, RA, and MS (260–266). IL-2 drives proliferation of Tregs and enhances expression of FOXP3 and additional genes that define Treg cell phenotype and function [review by (267, 268)]. These effects of IL-2 can be recapitulated by expression of a constitutively activated form of STAT5 in mouse Tregs (269), further highlighting the role of the IL-2 receptor-JAK/STAT signaling pathway in these cells. Additionally, although not required, IL-2 can induce proliferation and activation of other cell types including FOXP3─ T cells (Teff/Tcon), NK cells, and ILC2 (type 2 innate lymphoid cells) (268).
Recombinant IL-2 at high doses is an approved therapy for the treatment of advanced and metastatic cancer [review by (270)]. In high dose IL-2 therapy, efficacy is driven by the ability of IL-2 to enhance conventional and effector T and NK cell responses against tumor cells, but the therapeutic window is limited by significant and often severe toxicity. In contrast, evidence from over a decade of research has indicated that Tregs not only require IL-2 for function and stability, but that they are exquisitely sensitive to IL-2 (271, 272). Tregs constitutively express high levels of CD25 (IL2Rα) (273), which captures IL-2 and is assembled with CD122 (IL2Rβ) and the common γ chain CD132 (IL2Rγ), into a high-affinity heterotrimeric receptor complex. As a result, Tregs exhibit increased affinity to IL-2 compared to other cell types that express lower levels of or lack CD25. Increased affinity to IL-2 allows Tregs to compete and “soak up” free IL-2 (274). Moreover, Tregs can proliferate in response to limiting amounts of IL-2 compared to non-Treg cells in vivo (275). These findings have formed the basis of the low-dose (LD) IL-2 therapy for treatment of autoimmune and inflammatory diseases, where limiting amounts of recombinant IL-2 preferentially expand and enhance function of Tregs which in turn suppress pathogenic inflammatory responses.
Data emerging from multiple clinical studies support the therapeutic rationale for LD IL-2 therapy. For example, LD IL-2 therapy ranging from 1.0x106 IU/day (276), 1.5x106 IU/day (277), or 1.0x106 IU every other day (278) led to 2-5 fold expansion of Tregs over baseline in HCV-induced vasculitis (277), chronic GVHD (275, 276), and SLE (278, 279) patients, which appeared to correlate with disease improvement in a subset of patients. Nonetheless, dose-limiting toxicity (DLT) was observed, which confined the therapeutic dose range of IL-2 to a narrow 2-3-fold window. Specifically, significant toxicity was observed at 3.0x106 IU/day which required dose reduction to 1.5x106 IU/day (280) and similarly 3.0x106 IU/m2/day was reduced to 1x106 IU/m2/day (281). As a potential explanation for the observed toxicity, significant Tcon/Teff and NK cell expansion was observed, in some cases greater than that observed for Tregs (276, 281). With better understanding of safe dose range that induces Treg expansion in disease patients, efficacy of low LD IL-2 either alone or in combination therapies continues to be evaluated in a broader range of indications in multiple ongoing phase II clinical trials (Table 7) (291, 292).

Table 7 Summary of IL-2 therapies in development.
Results from the LD IL-2 therapy have encouraged subsequent development of approaches to increase the Treg-selective effects of IL-2 while reducing potentially disease-exacerbating effects of non-Treg cells, to achieve a wider therapeutic dose range. Engineered versions of IL-2 to modify the activity and binding specificity to Treg versus non-Tregs and IL-2 complexed with anti-IL-2 antibody that indirectly impacts activity and selectivity to the high affinity IL2 receptor are among these approaches. In most cases, these approaches also include a modification to extend the in vivo half-life of IL-2.
Several examples of engineered human and murine IL-2 molecules harboring mutations that attenuate interactions with the IL-2R complex have been described as IL-2 muteins. Among these, IL-2 muteins that decrease interactions with the signaling components of the IL2R complex, namely CD122 and CD132, but retain affinity to CD25 have been reported and are currently being tested in clinical trials for autoimmune and inflammatory diseases (Table 7). Due to the mutations that inhibit interaction between IL-2 and CD122/CD132, these IL-2 muteins induced weaker STAT5 activation compared to wildtype IL-2 (WT). However, because Tregs express high affinity trimeric IL2R, IL-2 muteins had activity that was less diminished in Tregs than non-Tregs, resulting in enhanced Treg selectivity. Fused to full IgG (293) or the Fc portion of IgG (294, 295). human IL-2 muteins showed prolonged in vivo half-life compared to recombinant human IL-2 (Proleukin) and induced preferential expansion of Tregs over CD4+ Teff or NK cells in vivo in non-human primates (293) and in humanized mouse models (296). Data from the first-in-human trial for IL-2 mutein (AMG 592 “Efavaleukin alpha”) supports the rationale for this approach in human subjects, where Treg expansion was observed to a similar degree as the LD IL-2 therapy, but the expansion of non-Tregs (particularly Tcon and NK cells) (281, 297) was significantly reduced (295). In parallel, mouse IL-2 muteins with enhanced Treg selectivity and in vivo half-life, expanded highly suppressive Tregs in tissues (298) and controlled autoimmunity in the mouse NOD T1D model (299). Moreover, the IL-2 mutein prevented antibody response to FVIII in a mouse model of hemophilia A (300), further supporting that Tregs responding to an attenuated IL-2 mutein exert immune suppressive effects. Another class of IL-2 muteins with enhanced affinity to CD25 has also been described, although the activity and selectivity profile of such muteins need to be further evaluated (294).
Since endogenous WT IL-2 also activates non-Tregs, the in vivo efficacy of IL-2 muteins in inflammatory settings is likely to be influenced by the combined effects on Tregs and IL-2-sensitive non-Tregs that might be pathogenic, rather than strictly by its effects on Tregs alone. In fact, an antagonist IL-2 mutein with enhanced binding to IL2Rβ chain but reduced signaling capacity reduced inflammatory response in a mouse GVHD model (282), presumably by displacing and antagonizing the effects of endogenous WT IL-2 on pathogenic cells such as Teff and NK cells. The mechanism of action of this class of “engineered receptor signaling clamp” IL-2 muteins is distinct from the above-mentioned IL-2 muteins that preferentially act on Tregs by retaining affinity to CD25 and delivering agonist activity.
In addition to Teff and NK cells, ILC2 and T follicular helper (TFH) cells are particularly relevant targets of IL-2 and may impact disease activity. ILC2s express high levels of CD25, produce IL-5 and induce eosinophil expansion in response to the LD IL-2 therapy (301). Consistently, multiple LD IL-2 clinical studies reported transient treatment-related increases in serum IL-5 levels and eosinophil numbers (276, 280, 292). Additionally, IL-2 is thought to inhibit TFH cell differentiation during T cell priming (302) which may reduce autoantibody production. Interestingly, LD IL-2 suppressed the number of TFH cells in SLE patients (278), suggesting that limiting amounts of IL-2 was sufficient to inhibit the TFH cell differentiation.
Characterization of a panel of human IL-2 muteins that had activity spanning a broad spectrum of STAT5 activation signaling demonstrated that attenuation of IL-2 signaling asymmetrically impacted Treg and non-Treg cell responses (296). As the affinity of IL-2 to its receptor declined, STAT5 signal in non-Tregs decreased first, while it was maintained over a wider range of attenuation in Tregs. Different sensitivity of Treg versus non-Tregs toward IL-2 was partly but not entirely due to the differences in CD25 levels, and additional cell-intrinsic mechanisms are likely to contribute (272). If attenuated IL-2 is inherently better at maintaining Treg responses, it also explains why engineering or modifications that weakens the IL-2 and its receptor interaction via steric hindrance enhances Treg selectivity. These include PEGylated IL-2 (NKTR-358 “LY3471851”) (303), which induced clinical response and dose-dependent changes in Treg functional markers in a phase I trial in mild-to-moderate SLE, various IL-2:IL-2 Ab complexes described as CD25-directed IL-2cx (283, 284) and CD25-IL2 fusion proteins (287, 288). Anti-IL-2 antibodies that form complex with IL-2 can attenuate the IL2R signal by sterically inhibiting the IL-2:IL2R interactions but they can also modify the IL-2 structure to influence the bias with which it interacts with the receptor subunits (285, 286). Likewise, CD25-IL-2 fusion proteins formed multimers via intermolecular binding, effectively limiting the amount of free IL-2 that could bind to CD25 on cell surfaces. While the concept behind attenuated IL-2 muteins is similar to that of the LD IL-2 therapy, the intended advantage of the engineered attenuated IL-2 is the increased dose range where Treg-to-non-Treg selectivity is safely enhanced.
The next generation of IL-2-based modality is likely to include tissue- and/or Treg-directed and combination cytokine approaches. It may be possible to target IL-2 to inflamed tissue sites via tissue-specific or inflammation-specific markers. The most challenging task to this approach is identification of suitable markers for targeting, and to maintain Treg-to-non-Treg selectivity when delivering IL-2 to sites where both cell types reside in close contact. Recently, a hybrid cytokine that can simultaneously engage IL-2 and IL-33 receptors (IL233) expanded Treg and ILC2 and conferred protective effects in an animal model of kidney injury (289). Likewise, a dimeric dual-acting cytokine containing both IL-2 and TNF receptor 2 (TNFR2) agonist enhanced Treg expansion and phenotype better than either IL-2 or TNFR2 agonist alone in vitro (290). Although the in vivo activity and selectivity of this molecule was not evaluated, it presents an interesting possibility of simultaneously engaging synergistic pathways for more robust or selective induction of Treg responses.
Therapeutics are emerging with the aspiration to achieve lasting immune tolerance and provide a cure by addressing the root cause of disease. In this review, we covered three areas of emerging therapeutics which induce immune tolerance. How knowledge gained from the bench will be translated into bedside remains to be seen. It is critical to note that there is no tolerogenic vaccine nor T cell therapy approved for the treatment of autoimmune disease as of today. The advancement of tolerogenic vaccines has been difficult due to a lack of understanding of which mechanism of action is the most effective to achieve immune tolerance, shortage of biomarkers, and complexities within the pathogenesis of individual autoimmune diseases. Equipped with new technology platforms and growing knowledge of the autoantigen repertoire that drives specific autoimmune diseases, the next wave of emerging tolerogenic vaccines hold promises to overcome these obstacles. When considering a tolerogenic vaccine for the treatment of autoimmune disease, researchers should investigate the dose, administration route, and epitope components for tolerogenic vaccines as these components affect the immunological outcomes. Tolerogenic adjuvants can be used to augment the local environment and favor tolerance under proinflammatory conditions. However, special care must be taken to address whether these tolerogenic adjuvants induce systemic immune suppression or engender immune tolerance. Current evidence suggests that DC, LSEC, and marginal zone macrophage encompass a group of specialized APCs that favor tolerance via the induction of Tregs or deletion of autoreactive T cells. An ideal tolerogenic vaccine should induce organ-specific tolerance which can restrain autoantigen responses beyond those included within the tolerogenic vaccine. Finally, while most tolerogenic vaccines tested in the clinic have been safe, caution is required, as these vaccine platforms have the potential to activate autoantigen-specific T effector and B cell responses thereby exacerbating disease or even resulting in IgG-induced anaphylactic shock. Thus, it is critical to determine the best therapeutic window at which a therapeutic achieves the greatest benefit without resulting in unacceptable side-effects or toxicity. For example, recombinant WT IL-2 therapy is restrained to a narrow “low dose” therapeutic window due to dose-limiting toxicities. Likewise, one must consider the timing at which to stage a therapeutic intervention. For example, tolerogenic vaccines may be more effective when administered during early disease when epitope spreading is minimal or during disease remissions when clinical disease is suppressed. Careful design of clinical trials, especially for the phase I studies along with the development of clinical biomarkers associated with therapeutic benefit will likely determine the success of future clinical trials.
Although promising, there are still many barriers to overcome for the practical use of T cell therapies. For therapies using Tregs as a drug product, considerable efforts will be needed to establish a process to manufacture engineered Tregs within reasonable costs to benefit broad patient populations. The production of suitable quantities of functional, stable, and pure Tregs would be the most crucial step. Unlike CD8 or even CD4 T cell counterparts used in CAR-T cell therapies with greater proliferative potentials, it is difficult to expand Tregs. Moreover, there is no Treg-exclusive markers to confirm the purity of the Treg product. For example, CD25 and FOXP3 are used as markers for Tregs, however these markers are also transiently expressed by proinflammatory pathogenic T cells. Thus, standard protocols that ensure the quality of Treg cell therapy drug needs to be established. Furthermore, the transfer of high numbers of Tregs can lead to systematic immunosuppression similar to that of immunosuppressive agents. For T cell therapies using engineered cytotoxic T cells as a drug product, how transferred T cells behave under inflammatory environments associated with autoimmune disease is unclear. CAR/CAAR/BAR T cells produce multiple arrays of inflammatory cytokines which may exacerbate autoimmune disease. Finally, it remains to be determined which therapies discussed can establish long-lasting tolerance. To establish long-lasting tolerance, the treatment will likely need to induce long-lived memory Tregs or suppressor cells specific to disease-relevant antigens or induce differentiation of memory T cells that continue to kill pathogenic autoreactive cells. Additionally, it is unclear if therapeutics which are antigen independent such as IL-2 therapies which expand polyclonal Tregs or broad immune cell depletion (e.g. CD19+ B cell depletion) that rebalance or reset the immune system can lead to lasting immune tolerance.
In this review, we explored three areas of therapeutics that aim to achieve immune tolerance. The breadth of the therapeutic modalities designed to mediate immune tolerance continues to grow. We are excited for the future of immune tolerance therapies and the potential benefits they bring to alleviate patient suffering.
All authors contributed to the article and approved the submitted version. CM, SS, and HP wrote the manuscript and approved it for publication. HP edited and finalized the manuscript.
CM, SS, and HP are employees of Amgen Inc.
We thank Ashutosh Chaudry for reading the manuscript and providing input.
AAV, Adeno-Associated Virus; AHR, Aryl Hydrocarbon Receptor; APC, Antigen Presenting Cell; BAR, B Cell-Targeting Antibody Receptor; BTK, Bruton’s Tyrosine Kinase; CAAR, Chimeric Autoantigen Receptor; CAR, Chimeric Antigen Receptor; CD, Cluster of Differentiation; CEA, Carcinoembryonic Antigen; CNS, Central Nervous System; CTLA4, Cytotoxic T-Lymphocyte Associated Protein-4; CYP2D6, Cytochrome P450; DC, Dendritic Cell; DCIR2, DC Immunoreceptor 2; DC-ASGR, DC-Asialoglycoprotein Receptor; DLT, Dose-Limiting Toxicity; DNA, Deoxyribonucleic Acid; DTH, Delayed-Type Hypersensitivity; DSG3, Desmoglein 3; EAE, Experimental Autoimmune Encephalomyelitis; FOXP3, Forkhead Box Protein P3; FVIII, Factor VIII; GARP, Glycoprotein A Repetitions Predominant; GITR, Glucocorticoid-Induced TNFR-Related Protein; GM-CSF, Granulocyte Macrophage-Colony Stimulating Factor; GVHD, Graft Versus Host Disease; HLA, Human Leukocyte Antigen; ICOSL, Inducible T Cell Costimulator Ligand; IDO, Indoleamine 2,3-dioxygenase; ILC2, Type 2 Innate Lymphoid Cells; IL, Interleukin; IP, Intraperitoneal; IRAK4, Interleukin 1 Receptor Associated Kinase 4; IRES, Internal Ribosomal Entry Site; iTr35, inducible IL-35 producing regulatory T cells; ITE, 2-(1’ H-indole-3’-carbonyl)-thiazole-4-carboxylic acid methyl ester; IV, Intravenous; JAK, Janus Kinase; LAG3, Lymphocyte Activating Protein 3; LAP, Latency-associated peptide; LD, Low Dose; LSEC, Liver Sinusoidal Endothelial Cells; mAb, Monoclonal Antibody; MBP, Myelin Basic Protein; Mbsa, Methylated Bovine Serum Albumin; M-CSF, Macrophage Colony-Stimulating Factor; MG, Myasthenia Gravis; MHC, Major Histocompatibility Complex; MOG, Myelin Oligodendrocyte Glycoprotein; MS, Multiple Sclerosis; MuSK, Muscle-Specific Receptor Kinase; MP, Microparticle; NF-KB, Nuclear Factor Kappa B; NK, Natural Killer Cell; NO, Nitric Oxide; NP, Nanoparticle; NSG, NOD Scid Gamma mice; OVA, Ovalbumin; PBMC, Peripheral Blood Mononuclear Cell; PDC, Pyruvate Dehydrogenase Complex; PDL1, Programmed Cell Death 1 Ligand; PD1, Programmed Cell Death 1; PEG, Polyethylene Glycol; PGE-2, Prostaglandin E2; PLGA, Poly(lactic-co-glycolic acid); PLP, Proteolipid Protein; PV, Pemphigus Vulgaris; RA, Rheumatoid Arthritis; RBC, Red Blood Cell; RIP, Receptor-Interacting Protein; RNA, Ribonucleic Acid; SC, Subcutaneous; scFv, Single Chain Variable Fragment; SLE, Systemic Lupus Erythematosus; SYK, Spleen Associated Tyrosine Kinase; T1D, Type 1 Diabetes; Tcon, T Conventional Cell; TCR, T Cell Receptor; tDC, Tolerogenic Dendric Cell; Teff, T Effector Cell; TGF-β, Transforming Growth Factor-beta; Th cell, T Helper Cell; TIGIT, T Cell Immunoreceptor With Ig And ITIM Domains; TIM-3, T Cell Immunoglobulin Mucin 3; TLR, Toll Like Receptor; TNF-α, Tumor Necrosis Factor-alpha; TNFR2, TNF Receptor 2; TNP, 2,4,6 Trinitrophenol; TPL2, Tumor Progression Locus 2; TRAIL, TNF-Related Apoptosis-Inducing Ligand; Treg, T Regulatory Cell; Tr1, Type 1 Regulatory T Cell; TYK2, Tyrosine Kinase 2.
1. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S3–S23. doi: 10.1016/j.jaci.2009.12.980
2. Schwartz RH. Historical overview of immunological tolerance. Cold Spring Harb Perspect Biol (2012) 4(4):a006908–a. doi: 10.1101/cshperspect.a006908
3. Bluestone JA. Mechanisms of tolerance. Immunol Rev (2011) 241(1):5–19. doi: 10.1111/j.1600-065X.2011.01019.x
4. Wang L, Wang FS, Gershwin ME. Human autoimmune diseases: a comprehensive update. J Intern Med (2015) 278(4):369–95. doi: 10.1111/joim.12395
5. Goverman JM. Immune tolerance in multiple sclerosis. Immunol Rev (2011) 241(1):228–40. doi: 10.1111/j.1600-065X.2011.01016.x
6. Firestein GS, McInnes IB. Immunopathogenesis of Rheumatoid Arthritis. Immunity (2017) 46(2):183–96. doi: 10.1016/j.immuni.2017.02.006
7. Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol (2014) 32:227–55. doi: 10.1146/annurev-immunol-032713-120225
8. Jeker LT, Bour-Jordan H, Bluestone JA. Breakdown in peripheral tolerance in type 1 diabetes in mice and humans. Cold Spring Harb Perspect Med (2012) 2(3):a007807–a. doi: 10.1101/cshperspect.a007807
9. Feldmann M, Maini RN. Anti-TNF therapy, from rationale to standard of care: what lessons has it taught us? J Immunol (2010) 185(2):791–4. doi: 10.4049/jimmunol.1090051
10. Lai Y, Dong C. Therapeutic antibodies that target inflammatory cytokines in autoimmune diseases. Int Immunol (2015) 28(4):181–8. doi: 10.1093/intimm/dxv063
11. Li P, Zheng Y, Chen X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front Pharmacol (2017) 8:460. doi: 10.3389/fphar.2017.00460
12. Ghoreschi K, Gadina M. Jakpot! New small molecules in autoimmune and inflammatory diseases. Exp Dermatol (2014) 23(1):7–11. doi: 10.1111/exd.12265
13. Peña D, Dasgupta B. Biologic agents and small-molecule inhibitors in systemic autoimmune conditions: an update. Pol Arch Intern Med (2020) 131(2):171–81. doi: 10.20452/pamw.15438
14. Zarrin AA, Bao K, Lupardus P, Vucic D. Kinase inhibition in autoimmunity and inflammation. Nat Rev Drug Discovery (2021) 20(1):39–63. doi: 10.1038/s41573-020-0082-8
15. Kim SY, Solomon DH. Tumor necrosis factor blockade and the risk of viral infection. Nat Rev Rheumatol (2010) 6(3):165–74. doi: 10.1038/nrrheum.2009.279
16. Murdaca G, Spanò F, Contatore M, Guastalla A, Penza E, Magnani O, et al. Infection risk associated with anti-TNF-α agents: A review. Expert Opin Drug Saf (2015) 14:1–12. doi: 10.1517/14740338.2015.1009036
17. Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF Antibody Therapy in Rheumatoid Arthritis and the Risk of Serious Infections and MalignanciesSystematic Review and Meta-analysis of Rare Harmful Effects in Randomized Controlled Trials. JAMA (2006) 295(19):2275–85. doi: 10.1001/jama.295.19.2275
18. Kemanetzoglou E, Andreadou E. CNS Demyelination with TNF-α Blockers. Curr Neurol Neurosci Rep (2017) 17(4):36–. doi: 10.1007/s11910-017-0742-1
19. Finckh A, Simard JF, Gabay C, Guerne PA, Physicians S. Evidence for differential acquired drug resistance to anti-tumour necrosis factor agents in rheumatoid arthritis. Ann Rheum Dis (2006) 65(6):746–52. doi: 10.1136/ard.2005.045062
20. Murakami N, Riella LV. Co-Inhibitory Pathways and Their Importance in Immune Regulation. Transplantation (2014) 98(1):3–14. doi: 10.1097/TP.0000000000000169
21. Hosseini A, Gharibi T, Marofi F, Babaloo Z, Baradaran B. CTLA-4: From mechanism to autoimmune therapy. Int Immunopharmacol (2020) 80:106221. doi: 10.1016/j.intimp.2020.106221
22. Paluch C, Santos AM, Anzilotti C, Cornall RJ, Davis SJ. Immune Checkpoints as Therapeutic Targets in Autoimmunity. Front Immunol (2018) 9:2306–. doi: 10.3389/fimmu.2018.02306
23. Carballido JM, Regairaz C, Rauld C, Raad L, Picard D, Kammüller M. The Emerging Jamboree of Transformative Therapies for Autoimmune Diseases. Front Immunol (2020) 11:472. doi: 10.3389/fimmu.2020.00472
24. Mannie MD, DeOca KB, Bastian AG, Moorman CD. Tolerogenic vaccines: Targeting the antigenic and cytokine niches of FOXP3+ regulatory T cells. Cell Immunol (2020) 355:104173. doi: 10.1016/j.cellimm.2020.104173
25. Mannie MD, Curtis AD,2. Tolerogenic vaccines for Multiple sclerosis. Hum Vaccin Immunother (2013) 9(5):1032–8. doi: 10.4161/hv.23685
26. Serra P, Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nat Biotechnol (2019) 37(3):238–51. doi: 10.1038/s41587-019-0015-4
27. Willekens B, Cools N. Beyond the Magic Bullet: Current Progress of Therapeutic Vaccination in Multiple Sclerosis. CNS Drugs (2018) 32(5):401–10. doi: 10.1007/s40263-018-0518-4
28. Kammona O, Kiparissides C. Recent Advances in Antigen-Specific Immunotherapies for the Treatment of Multiple Sclerosis. Brain Sci (2020) 10(6):333. doi: 10.3390/brainsci10060333
29. Ganesan V, Ascherman DP, Minden JS. Immunoproteomics technologies in the discovery of autoantigens in autoimmune diseases. Biomol Concepts (2016) 7(2):133–43. doi: 10.1515/bmc-2016-0007
30. Moritz CP, Paul S, Stoevesandt O, Tholance Y, Camdessanché J-P, Antoine J-C. Autoantigenomics: Holistic characterization of autoantigen repertoires for a better understanding of autoimmune diseases. Autoimmun Rev (2020) 19(2):102450. doi: 10.1016/j.autrev.2019.102450
31. Purcell AW, Sechi S, DiLorenzo TP. The Evolving Landscape of Autoantigen Discovery and Characterization in Type 1 Diabetes. Diabetes (2019) 68(5):879–86. doi: 10.2337/dbi18-0066
32. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
33. van der Vliet HJJ, Nieuwenhuis EE. IPEX as a result of mutations in FOXP3. Clin Dev Immunol (2007) 2007:89017–. doi: 10.1155/2007/89017
34. Ramsdell F, Ziegler SF. FOXP3 and scurfy: how it all began. Nat Rev Immunol (2014) 14(5):343–9. doi: 10.1038/nri3650
35. Churlaud G, Pitoiset F, Jebbawi F, Lorenzon R, Bellier B, Rosenzwajg M, et al. Human and Mouse CD8(+)CD25(+)FOXP3(+) Regulatory T Cells at Steady State and during Interleukin-2 Therapy. Front Immunol (2015) 6:171–. doi: 10.3389/fimmu.2015.00171
36. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation (2012) 9:112–. doi: 10.1186/1742-2094-9-112
37. Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J Autoimmun (2019) 103:102289. doi: 10.1016/j.jaut.2019.05.017
38. Allan SE, Alstad AN, Merindol N, Crellin NK, Amendola M, Bacchetta R, et al. Generation of Potent and Stable Human CD4+ T Regulatory Cells by Activation-independent Expression of FOXP3. Mol Ther (2008) 16(1):194–202. doi: 10.1038/sj.mt.6300341
39. Honaker Y, Hubbard N, Xiang Y, Fisher L, Hagin D, Sommer K, et al. Gene editing to induce FOXP3 expression in human CD4(+) T cells leads to a stable regulatory phenotype and function. Sci Transl Med (2020) 12(546):eaay6422. doi: 10.1126/scitranslmed.aay6422
40. Park MK, Jung YO, Lee S-Y, Lee SH, Heo YJ, Kim EK, et al. Amelioration of autoimmune arthritis by adoptive transfer of Foxp3-expressing regulatory B cells is associated with the Treg/Th17 cell balance. J Transl Med (2016) 14(1):191. doi: 10.1186/s12967-016-0980-z
41. Lipscomb MW, Taylor JL, Goldbach CJ, Watkins SC, Wesa AK, Storkus WJ. DC expressing transgene Foxp3 are regulatory APC. Eur J Immunol (2010) 40(2):480–93. doi: 10.1002/eji.200939667
42. Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol (2007) 8(3):277–84. doi: 10.1038/ni1437
43. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol (2009) 10(9):1000–7. doi: 10.1038/ni.1774
44. Pacholczyk R, Kern J. The T-cell receptor repertoire of regulatory T cells. Immunology (2008) 125(4):450–8. doi: 10.1111/j.1365-2567.2008.02992.x
45. Schmidt AM, Lu W, Sindhava VJ, Huang Y, Burkhardt JK, Yang E, et al. Regulatory T cells require TCR signaling for their suppressive function. J Immunol (2015) 194(9):4362–70. doi: 10.4049/jimmunol.1402384
46. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous T Cell Receptor Signals Maintain a Functional Regulatory T Cell Pool. Immunity (2014) 41(5):722–36. doi: 10.1016/j.immuni.2014.10.012
47. Fan MY, Low JS, Tanimine N, Finn KK, Priyadharshini B, Germana SK, et al. Differential Roles of IL-2 Signaling in Developing versus Mature Tregs. Cell Rep (2018) 25(5):1204–13.e4. doi: 10.1016/j.celrep.2018.10.002
48. Iikuni N, Lourenço EV, Hahn BH, La Cava A. Cutting edge: Regulatory T cells directly suppress B cells in systemic lupus erythematosus. J Immunol (2009) 183(3):1518–22. doi: 10.4049/jimmunol.0901163
49. Schmidt A, Oberle N, Krammer P. Molecular Mechanisms of Treg-Mediated T Cell Suppression. Front Immunol (2012) 3:51. doi: 10.3389/fimmu.2012.00051
50. Arce-Sillas A, Álvarez-Luquín DD, Tamaya-Domínguez B, Gomez-Fuentes S, Trejo-García A, Melo-Salas M, et al. Regulatory T Cells: Molecular Actions on Effector Cells in Immune Regulation. J Immunol Res (2016) 2016:1720827. doi: 10.1155/2016/1720827
51. Lan Q, Zhou X, Fan H, Chen M, Wang J, Ryffel B, et al. Polyclonal CD4+Foxp3+ Treg cells induce TGFβ-dependent tolerogenic dendritic cells that suppress the murine lupus-like syndrome. J Mol Cell Biol (2012) 4(6):409–19. doi: 10.1093/jmcb/mjs040
52. Yang J, Yang Y, Fan H, Zou H. Tolerogenic splenic IDO (+) dendritic cells from the mice treated with induced-Treg cells suppress collagen-induced arthritis. J Immunol Res (2014) 2014:831054. doi: 10.1155/2014/831054
53. Mavin E, Nicholson L, Rafez Ahmed S, Gao F, Dickinson A, Wang X-N. Human Regulatory T Cells Mediate Transcriptional Modulation of Dendritic Cell Function. J Immunol (2017) 198(1):138. doi: 10.4049/jimmunol.1502487
54. Mahnke K, Ring S, Johnson TS, Schallenberg S, Schönfeld K, Storn V, et al. Induction of immunosuppressive functions of dendritic cells in vivo by CD4+CD25+ regulatory T cells: role of B7-H3 expression and antigen presentation. Eur J Immunol (2007) 37(8):2117–26. doi: 10.1002/eji.200636841
55. Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med (2013) 19(6):355–67. doi: 10.1016/j.molmed.2013.03.005
56. Zhao H, Liao X, Kang Y. Tregs: Where We Are and What Comes Next? Front Immunol (2017) 8:1578. doi: 10.3389/fimmu.2017.01578
57. Safinia N, Scotta C, Vaikunthanathan T, Lechler RI, Lombardi G. Regulatory T Cells: Serious Contenders in the Promise for Immunological Tolerance in Transplantation. Front Immunol (2015) 6:438. doi: 10.3389/fimmu.2015.00438
58. Wilkinson DS, Ghosh D, Nickle RA, Moorman CD. Mannie MD. Partial CD25 Antagonism Enables Dominance of Antigen-Inducible CD25(high) FOXP3(+) Regulatory T Cells As a Basis for a Regulatory T Cell-Based Adoptive Immunotherapy. Front Immunol (2017) 8:1782. doi: 10.3389/fimmu.2017.01782
59. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation–mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8(12):1353–62. doi: 10.1038/ni1536
60. Li L, Liu X, Sanders KL, Edwards JL, Ye J, Si F, et al. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab (2019) 29(1):103–23.e5. doi: 10.1016/j.cmet.2018.09.020
61. Korn T, Muschaweckh A. Stability and Maintenance of Foxp3(+) Treg Cells in Non-lymphoid Microenvironments. Front Immunol (2019) 10:2634. doi: 10.3389/fimmu.2019.02634
62. Barbi J, Pardoll D, Pan F. Treg functional stability and its responsiveness to the microenvironment. Immunol Rev (2014) 259(1):115–39. doi: 10.1111/imr.12172
63. Colamatteo A, Carbone F, Bruzzaniti S, Galgani M, Fusco C, Maniscalco GT, et al. Molecular Mechanisms Controlling Foxp3 Expression in Health and Autoimmunity: From Epigenetic to Post-translational Regulation. Front Immunol (2020) 10:3136. doi: 10.3389/fimmu.2019.03136
64. Walsh PT, Taylor DK, Turka LA. Tregs and transplantation tolerance. J Clin Invest (2004) 114(10):1398–403. doi: 10.1172/JCI200423238
65. Kendal AR, Waldmann H. Infectious tolerance: therapeutic potential. Curr Opin Immunol (2010) 22(5):560–5. doi: 10.1016/j.coi.2010.08.002
66. Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol (2010) 11(12):1093–101. doi: 10.1038/ni.1952
67. Sullivan JA, Tomita Y, Jankowska-Gan E, Lema DA, Arvedson MP, Nair A, et al. Treg-Cell-Derived IL-35-Coated Extracellular Vesicles Promote Infectious Tolerance. Cell Rep (2020) 30(4):1039–51.e5. doi: 10.1016/j.celrep.2019.12.081
68. Jonuleit H, Schmitt E, Kakirman H, Stassen M, Knop J, Enk AH. Infectious tolerance: human CD25(+) regulatory T cells convey suppressor activity to conventional CD4(+) T helper cells. J Exp Med (2002) 196(2):255–60. doi: 10.1084/jem.20020394
69. Stassen M, Fondel S, Bopp T, Richter C, Müller C, Kubach J, et al. Human CD25+ regulatory T cells: two subsets defined by the integrins α4β7 or α4β1 confer distinct suppressive properties upon CD4+ T helper cells. Eur J Immunol (2004) 34(5):1303–11. doi: 10.1002/eji.200324656
70. Gravano DM, Vignali DAA. The battle against immunopathology: infectious tolerance mediated by regulatory T cells. Cell Mol Life Sci CMLS (2012) 69(12):1997–2008. doi: 10.1007/s00018-011-0907-z
71. Zeng H, Zhang R, Jin B, Chen L. Type 1 regulatory T cells: a new mechanism of peripheral immune tolerance. Cell Mol Immunol (2015) 12(5):566–71. doi: 10.1038/cmi.2015.44
72. Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Rev Immunol (2007) 7(11):875–88. doi: 10.1038/nri2189
73. Fucikova J, Palova-Jelinkova L, Bartunkova J, Spisek R. Induction of Tolerance and Immunity by Dendritic Cells: Mechanisms and Clinical Applications. Front Immunol (2019) 10:2393. doi: 10.3389/fimmu.2019.02393
74. Ohnmacht C, Pullner A, King SBS, Drexler I, Meier S, Brocker T, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med (2009) 206(3):549–59. doi: 10.1084/jem.20082394
75. Hasegawa H, Matsumoto T. Mechanisms of Tolerance Induction by Dendritic Cells In Vivo. Front Immunol (2018) 9:350–. doi: 10.3389/fimmu.2018.00350
76. Domogalla MP, Rostan PV, Raker VK, Steinbrink K. Tolerance through Education: How Tolerogenic Dendritic Cells Shape Immunity. Front Immunol (2017) 8:1764. doi: 10.3389/fimmu.2017.01764
77. Darrasse-Jèze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao K-H, et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med (2009) 206(9):1853–62. doi: 10.1084/jem.20090746
78. Suffner J, Hochweller K, Kühnle M-C, Li X, Kroczek RA, Garbi N, et al. Dendritic Cells Support Homeostatic Expansion of Foxp3+ Regulatory T Cells in Foxp3.LuciDTR Mice. J Immunol (2010) 184(4):1810–20. doi: 10.4049/jimmunol.0902420
79. Yogev N, Frommer F, Lukas D, Kautz-Neu K, Karram K, Ielo D, et al. Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor(+) regulatory T cells. Immunity (2012) 37(2):264–75. doi: 10.1016/j.immuni.2012.05.025
80. Neumann K, Rudolph C, Neumann C, Janke M, Amsen D, Scheffold A. Liver sinusoidal endothelial cells induce immunosuppressive IL-10-producing Th1 cells via the Notch pathway. Eur J Immunol (2015) 45(7):2008–16. doi: 10.1002/eji.201445346
81. Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol (2016) 13(3):347–53. doi: 10.1038/cmi.2016.5
82. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J BioMed Sci (2019) 26(1):78. doi: 10.1186/s12929-019-0568-z
83. Ravishankar B, Shinde R, Liu H, Chaudhary K, Bradley J, Lemos HP, et al. Marginal zone CD169+ macrophages coordinate apoptotic cell-driven cellular recruitment and tolerance. Proc Natl Acad Sci USA (2014) 111(11):4215–20. doi: 10.1073/pnas.1320924111
84. Getts DR, McCarthy DP, Miller SD. Exploiting apoptosis for therapeutic tolerance induction. J Immunol (2013) 191(11):5341–6. doi: 10.4049/jimmunol.1302070
85. Sun S-W, Chen L, Zhou M, Wu J-H, Meng Z-J, Han H-L, et al. BAMBI regulates macrophages inducing the differentiation of Treg through the TGF-β pathway in chronic obstructive pulmonary disease. Respir Res (2019) 20(1):26. doi: 10.1186/s12931-019-0988-z
86. Tsang JYS, Chai JG, Lechler R. Antigen presentation by mouse CD4+ T cells involving acquired MHC class II:peptide complexes: another mechanism to limit clonal expansion? Blood (2003) 101(7):2704–10. doi: 10.1182/blood-2002-04-1230
87. Khoury SJ, Sayegh MH, Hancock WW, Gallon L, Carpenter CB, Weiner HL. Acquired tolerance to experimental autoimmune encephalomyelitis by intrathymic injection of myelin basic protein or its major encephalitogenic peptide. J Exp Med (1993) 178(2):559–66. doi: 10.1084/jem.178.2.559
88. Higgins PJ, Weiner HL. Suppression of experimental autoimmune encephalomyelitis by oral administration of myelin basic protein and its fragments. J Immunol (1988) 140(2):440–5.
89. Al-Sabbagh A, Nelson PA, Akselband Y, Sobel RA, Weiner HL. Antigen-driven peripheral immune tolerance: suppression of experimental autoimmmune encephalomyelitis and collagen-induced arthritis by aerosol administration of myelin basic protein or type II collagen. Cell Immunol (1996) 171(1):111–9. doi: 10.1006/cimm.1996.0180
90. Staykova MA, Simmons RD, Willenborg DO. Infusion of soluble myelin basic protein protects long term against induction of experimental autoimmune encephalomyelitis. Immunol Cell Biol (1997) 75(1):54–64. doi: 10.1038/icb.1997.9
91. Chou FC-H, Chou C-HJ, Fritz RB, Kibler RF. Prevention of experimental allergic encephalomyelitis in lewis rats with peptide 68-88 of guinea pig myelin basic protein. Ann Neurol (1980) 7(4):336–9. doi: 10.1002/ana.410070409
92. Tutaj M, Szczepanik M. Epicutaneous (EC) immunization with myelin basic protein (MBP) induces TCRalphabeta+ CD4+ CD8+ double positive suppressor cells that protect from experimental autoimmune encephalomyelitis (EAE). J Autoimmun (2007) 28(4):208–15. doi: 10.1016/j.jaut.2007.02.017
93. Jiang Z, Li H, Fitzgerald DC, Zhang G-X, Rostami A. MOG(35-55) i.v suppresses experimental autoimmune encephalomyelitis partially through modulation of Th17 and JAK/STAT pathways. Eur J Immunol (2009) 39(3):789–99. doi: 10.1002/eji.200838427
94. Glatigny S, Bettelli E. Experimental Autoimmune Encephalomyelitis (EAE) as Animal Models of Multiple Sclerosis (MS). Cold Spring Harb Perspect Med (2018) 8(11):a028977. doi: 10.1101/cshperspect.a028977
95. Rezende RM, Cox LM, Weiner HL. Mucosal tolerance therapy in humans: Past and future. Clin Exp Neuroimmunol (2019) 10(S1):20–31. doi: 10.1111/cen3.12500
96. Wildner P, Selmaj KW. Multiple sclerosis: Skin-induced antigen-specific immune tolerance. J Neuroimmunol (2017) 311:49–58. doi: 10.1016/j.jneuroim.2017.08.001
97. Racke MK, Ratts RB, Arredondo L, Perrin PJ, Lovett-Racke A. The role of costimulation in autoimmune demyelination. J Neuroimmunol (2000) 107(2):205–15. doi: 10.1016/S0165-5728(00)00230-7
98. Katsara M, Minigo G, Plebanski M, Apostolopoulos V. The good, the bad and the ugly: how altered peptide ligands modulate immunity. Expert Opin Biol Ther (2008) 8(12):1873–84. doi: 10.1517/14712590802494501
99. Weiner H, Mackin G, Matsui M, Orav E, Khoury S, Dawson D, et al. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science (1993) 259(5099):1321–4. doi: 10.1126/science.7680493
100. Chataway J, Martin K, Barrell K, Sharrack B, Stolt P, Wraith DC. Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology (2018) 90(11):e955–e62. doi: 10.1212/WNL.0000000000005118
101. Streeter HB, Rigden R, Martin KF, Scolding NJ, Wraith DC. Preclinical development and first-in-human study of ATX-MS-1467 for immunotherapy of MS. Neurol(R) Neuroimmunol Neuroinflammation (2015) 2(3):e93–e. doi: 10.1212/NXI.0000000000000093
102. Walczak A, Siger M, Ciach A, Szczepanik M, Selmaj K. Transdermal Application of Myelin Peptides in Multiple Sclerosis Treatment. JAMA Neurol (2013) 70(9):1105–9. doi: 10.1001/jamaneurol.2013.3022
103. Juryńczyk M, Walczak A, Jurewicz A, Jesionek-Kupnicka D, Szczepanik M, Selmaj K. Immune regulation of multiple sclerosis by transdermally applied myelin peptides. Ann Neurol (2010) 68(5):593–601. doi: 10.1002/ana.22219
104. Freedman MS, Bar-Or A, Oger J, Traboulsee A, Patry D, Young C, et al. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology (2011) 77(16):1551–60. doi: 10.1212/WNL.0b013e318233b240
105. Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. Nat Med (2000) 6(10):1176–82. doi: 10.1038/80525
106. Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med (2000) 6(10):1167–75. doi: 10.1038/80516
107. Lutterotti A, Yousef S, Sputtek A, Stürner KH, Stellmann J-P, Breiden P, et al. Antigen-Specific Tolerance by Autologous Myelin Peptide–Coupled Cells: A Phase 1 Trial in Multiple Sclerosis. Sci Transl Med (2013) 5(188):188ra75–ra75. doi: 10.1126/scitranslmed.3006168
108. Belogurov A, Zakharov K, Lomakin Y, Surkov K, Avtushenko S, Kruglyakov P, et al. CD206-Targeted Liposomal Myelin Basic Protein Peptides in Patients with Multiple Sclerosis Resistant to First-Line Disease-Modifying Therapies: A First-in-Human, Proof-of-Concept Dose-Escalation Study. Neurotherapeutics (2016) 13(4):895–904. doi: 10.1007/s13311-016-0448-0
109. Bar-Or A, Vollmer T, Antel J, Arnold DL, Bodner CA, Campagnolo D, et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch Neurol (2007) 64(10):1407–15. doi: 10.1001/archneur.64.10.nct70002
110. Zubizarreta I, Flórez-Grau G, Vila G, Cabezón R, España C, Andorra M, et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc Natl Acad Sci USA (2019) 116(17):8463–70. doi: 10.1073/pnas.1820039116
111. Willekens B, Presas-Rodriguez S, Mansilla MJ, Derdelinckx J, Lee WP, Nijs G, et al. Tolerogenic dendritic cell-based treatment for multiple sclerosis (MS): a harmonised study protocol for two phase I clinical trials comparing intradermal and intranodal cell administration. BMJ Open (2019) 9(9):e030309. doi: 10.1136/bmjopen-2019-030309
112. Cools N, Presas-Rodríguez S, Mansilla M, Derdelinckx J, Lee W, Nijs G, et al. Towards a dendritic cell-based vaccine for the treatment of multiple sclerosis (MS): interim safety data of the first dose cohort of the MS-tolDC phase I clinical trial. Front Neurosci (2018) 12. doi: 10.3389/conf.fnins.2018.95.00060
113. Benson JM, Stuckman SS, Cox KL, Wardrop RM, Gienapp IE, Cross AH, et al. Oral Administration of Myelin Basic Protein Is Superior to Myelin in Suppressing Established Relapsing Experimental Autoimmune Encephalomyelitis. J Immunol (1999) 162(10):6247–54.
114. Strohl WR. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. Biodrugs (2015) 29(4):215–39. doi: 10.1007/s40259-015-0133-6
115. Devaux B, Enderlin F, Wallner B, Smilek DE. Induction of EAE in mice with recombinant human MOG, and treatment of EAE with a MOG peptide. J Neuroimmunol (1997) 75(1):169–73. doi: 10.1016/S0165-5728(97)00019-2
116. Critchfield JM, Racke MK, Zúñiga-Pflücker JC, Cannella B, Raine CS, Goverman J, et al. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science (1994) 263(5150):1139–43. doi: 10.1126/science.7509084
117. Ring S, Maas M, Nettelbeck DM, Enk AH, Mahnke K. Targeting of autoantigens to DEC205(+) dendritic cells in vivo suppresses experimental allergic encephalomyelitis in mice. J Immunol (2013) 191(6):2938–47. doi: 10.4049/jimmunol.1202592
118. Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, et al. Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest (2013) 123(2):844–54. doi: 10.1172/JCI65260
119. Tabansky I, Keskin DB, Watts D, Petzold C, Funaro M, Sands W, et al. Targeting DEC-205–DCIR2+ dendritic cells promotes immunological tolerance in proteolipid protein-induced experimental autoimmune encephalomyelitis. Mol Med (2018) 24(1):17. doi: 10.1186/s10020-018-0017-6
120. Loschko J, Heink S, Hackl D, Dudziak D, Reindl W, Korn T, et al. Antigen Targeting to Plasmacytoid Dendritic Cells via Siglec-H Inhibits Th Cell-Dependent Autoimmunity. J Immunol (2011) 187(12):6346. doi: 10.4049/jimmunol.1102307
121. Blanchfield JL, Mannie MD. A GMCSF-neuroantigen fusion protein is a potent tolerogen in experimental autoimmune encephalomyelitis (EAE) that is associated with efficient targeting of neuroantigen to APC. J Leukoc Biol (2010) 87(3):509–21. doi: 10.1189/jlb.0709520
122. Moorman CD, Bastian AG, DeOca KB, Mannie MD. A GM-CSF-neuroantigen tolerogenic vaccine elicits inefficient antigen recognition events below the CD40L triggering threshold to expand CD4+ CD25+ FOXP3+ Tregs that inhibit experimental autoimmune encephalomyelitis (EAE). J Neuroinflamm (2020) 17(1):180. doi: 10.1186/s12974-020-01856-8
123. Fovet CM, Stimmer L, Contreras V, Horellou P, Hubert A, Seddiki N, et al. Intradermal vaccination prevents anti-MOG autoimmune encephalomyelitis in macaques. EBioMedicine (2019) 47:492–505. doi: 10.1016/j.ebiom.2019.08.052
124. Mannie MD, Clayson BA, Buskirk EJ, DeVine JL, Hernandez JJ, Abbott DJ. IL-2/Neuroantigen Fusion Proteins as Antigen-Specific Tolerogens in Experimental Autoimmune Encephalomyelitis (EAE): Correlation of T Cell-Mediated Antigen Presentation and Tolerance Induction. J Immunol (2007) 178(5):2835–43. doi: 10.4049/jimmunol.178.5.2835
125. Wang D, Ghosh D, Islam SM, Moorman CD, Thomason AE, Wilkinson DS, et al. IFN-beta Facilitates Neuroantigen-Dependent Induction of CD25+ FOXP3+ Regulatory T Cells That Suppress Experimental Autoimmune Encephalomyelitis. J Immunol (2016) 197(8):2992–3007. doi: 10.4049/jimmunol.1500411
126. Mannie MD, Blanchfield JL, Islam SMT, Abbott DJ. Cytokine-neuroantigen fusion proteins as a new class of tolerogenic, therapeutic vaccines for treatment of inflammatory demyelinating disease in rodent models of multiple sclerosis. Front Immunol (2012) 3:255–. doi: 10.3389/fimmu.2012.00255
127. Mannie MD, Abbott DJ. A Fusion Protein Consisting of IL-16 and the Encephalitogenic Peptide of Myelin Basic Protein Constitutes an Antigen-Specific Tolerogenic Vaccine That Inhibits Experimental Autoimmune Encephalomyelitis. J Immunol (2007) 179(3):1458–65. doi: 10.4049/jimmunol.179.3.1458
128. Puentes F, Dickhaut K, Hofstätter M, Pfeil J, Lauer U, Hamann A, et al. Immune Modulation and Prevention of Autoimmune Disease by Repeated Sequences from Parasites Linked to Self Antigens. J Neuroimmune Pharmacol (2016) 11(4):749–62. doi: 10.1007/s11481-016-9701-x
129. Rynda A, Maddaloni M, Ochoa-Repáraz J, Callis G, Pascual DW. IL-28 Supplants Requirement for Treg Cells in Protein σ1-Mediated Protection against Murine Experimental Autoimmune Encephalomyelitis (EAE). PloS One (2010) 5(1):e8720. doi: 10.1371/journal.pone.0008720
130. Rynda-Apple A, Huarte E, Maddaloni M, Callis G, Skyberg JA, Pascual DW. Active immunization using a single dose immunotherapeutic abates established EAE via IL-10 and regulatory T cells. Eur J Immunol (2011) 41(2):313–23. doi: 10.1002/eji.201041104
131. Pishesha N, Bilate AM, Wibowo MC, Huang N-J, Li Z, Deshycka R, et al. Engineered erythrocytes covalently linked to antigenic peptides can protect against autoimmune disease. Proc Natl Acad Sci USA (2017) 114(12):3157–62. doi: 10.1073/pnas.1701746114
132. Tan LJ, Kennedy MK, Dal Canto MC, Miller SD. Successful treatment of paralytic relapses in adoptive experimental autoimmune encephalomyelitis via neuroantigen-specific tolerance. J Immunol (1991) 147(6):1797–802.
133. Smith CE, Miller SD. Multi-peptide coupled-cell tolerance ameliorates ongoing relapsing EAE associated with multiple pathogenic autoreactivities. J Autoimmun (2006) 27(4):218–31. doi: 10.1016/j.jaut.2006.12.002
134. Tan LJ, Kennedy MK, Miller SD. Regulation of the effector stages of experimental autoimmune encephalomyelitis via neuroantigen-specific tolerance induction. II. Fine specificity of effector T cell inhibition. J Immunol (1992) 148(9):2748–55.
135. Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, et al. Tolerance Induced by Apoptotic Antigen-Coupled Leukocytes Is Induced by PD-L1+ and IL-10–Producing Splenic Macrophages and Maintained by T Regulatory Cells. J Immunol (2011) 187(5):2405–17. doi: 10.4049/jimmunol.1004175
136. Kennedy KJ, Smith WS, Miller SD, Karpus WJ. Induction of antigen-specific tolerance for the treatment of ongoing, relapsing autoimmune encephalomyelitis: a comparison between oral and peripheral tolerance. J Immunol (1997) 159(2):1036–44.
137. Vandenbark AA, Celnik B, Vainiene M, Miller SD, Offner H. Myelin antigen-coupled splenocytes suppress experimental autoimmune encephalomyelitis in Lewis rats through a partially reversible anergy mechanism. J Immunol (1995) 155(12):5861–7.
138. Spiering R, Margry B, Keijzer C, Petzold C, Hoek A, Wagenaar-Hilbers J, et al. DEC205+ Dendritic Cell-Targeted Tolerogenic Vaccination Promotes Immune Tolerance in Experimental Autoimmune Arthritis. J Immunol (2015) 194(10):4804–13. doi: 10.4049/jimmunol.1400986
139. Bruder D, Westendorf AM, Hansen W, Prettin S, Gruber AD, Qian Y, et al. On the edge of autoimmunity: T-cell stimulation by steady-state dendritic cells prevents autoimmune diabetes. Diabetes (2005) 54(12):3395–401. doi: 10.2337/diabetes.54.12.3395
140. Petzold C, Riewaldt J, Koenig T, Schallenberg S, Kretschmer K. Dendritic cell-targeted pancreatic beta-cell antigen leads to conversion of self-reactive CD4(+) T cells into regulatory T cells and promotes immunotolerance in NOD mice. Rev Diabetic Stud RDS (2010) 7(1):47–61. doi: 10.1900/RDS.2010.7.47
141. Mahnke K, Qian Y, Knop J, Enk AH. Induction of CD4+/CD25+ regulatory T cells by targeting of antigens to immature dendritic cells. Blood (2003) 101(12):4862–9. doi: 10.1182/blood-2002-10-3229
142. Mukhopadhaya A, Hanafusa T, Jarchum I, Chen YG, Iwai Y, Serreze DV, et al. Selective delivery of beta cell antigen to dendritic cells in vivo leads to deletion and tolerance of autoreactive CD8+ T cells in NOD mice. Proc Natl Acad Sci USA (2008) 105(17):6374–9. doi: 10.1073/pnas.0802644105
143. Moorman CD, Curtis AD,2, Bastian AG, Elliott SE, Mannie MD. A GMCSF-Neuroantigen Tolerogenic Vaccine Elicits Systemic Lymphocytosis of CD4(+) CD25(high) FOXP3(+) Regulatory T Cells in Myelin-Specific TCR Transgenic Mice Contingent Upon Low-Efficiency T Cell Antigen Receptor Recognition. Front Immunol (2018) 9:3119. doi: 10.3389/fimmu.2018.03119
144. Islam SMT, Curtis AD,2, Taslim N, Wilkinson DS, Mannie MD. GM-CSF-neuroantigen fusion proteins reverse experimental autoimmune encephalomyelitis and mediate tolerogenic activity in adjuvant-primed environments: association with inflammation-dependent, inhibitory antigen presentation. J Immunol (2014) 193(5):2317–29. doi: 10.4049/jimmunol.1303223
145. Abbott DJ, Blanchfield JL, Martinson DA, Russell SC, Taslim N, Curtis AD, et al. Neuroantigen-specific, tolerogenic vaccines: GM-CSF is a fusion partner that facilitates tolerance rather than immunity to dominant self-epitopes of myelin in murine models of experimental autoimmune encephalomyelitis (EAE). BMC Immunol (2011) 12:72–. doi: 10.1186/1471-2172-12-72
146. Satyaraj E, Rath S, Bal V. Induction of tolerance in freshly isolated alloreactive CD4+ T cells by activated T cell stimulators. Eur J Immunol (1994) 24(10):2457–61. doi: 10.1002/eji.1830241030
147. Bettens F, Frei E, Frutig K, Mauri D, Pichler WJ, Wyss-Coray T. Noncytotoxic Human CD4+ T-Cell Clones Presenting and Simultaneously Responding to an Antigen Die of Apoptosis. Cell Immunol (1995) 161(1):72–8. doi: 10.1006/cimm.1995.1010
148. Sidhu S, Deacock S, Bal V, Batchelor JR, Lombardi G, Lechler RI. Human T cells cannot act as autonomous antigen-presenting cells, but induce tolerance in antigen-specific and alloreactive responder cells. J Exp Med (1992) 176(3):875–80. doi: 10.1084/jem.176.3.875
149. Kasper LH, Reder AT. Immunomodulatory activity of interferon-beta. Ann Clin Transl Neurol (2014) 1(8):622–31. doi: 10.1002/acn3.84
150. Mannie MD, Abbott DJ, Blanchfield JL. Experimental autoimmune encephalomyelitis in Lewis rats: IFN-beta acts as a tolerogenic adjuvant for induction of neuroantigen-dependent tolerance. J Immunol (2009) 182(9):5331–41. doi: 10.4049/jimmunol.0803756
151. Kontos S, Kourtis IC, Dane KY, Hubbell JA. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc Natl Acad Sci USA (2013) 110(1):E60–E8. doi: 10.1073/pnas.1216353110
152. Karpus WJ, Peterson JD, Miller SD. Anergy in vivo: down-regulation of antigen-specific CD4+ Th1 but not Th2 cytokine responses. Int Immunol (1994) 6(5):721–30. doi: 10.1093/intimm/6.5.721
153. Miller SD, Wetzig RP, Claman HN. The induction of cell-mediated immunity and tolerance with protein antigens coupled to syngeneic lymphoid cells. J Exp Med (1979) 149(3):758–73. doi: 10.1084/jem.149.3.758
154. Kennedy MK, Tan LJ, Dal Canto MC, Miller SD. Regulation of the effector stages of experimental autoimmune encephalomyelitis via neuroantigen-specific tolerance induction. J Immunol (1990) 145(1):117–26.
155. Kennedy MK, Tan LJ, Dal Canto MC, Tuohy VK, Lu ZJ, Trotter JL, et al. Inhibition of murine relapsing experimental autoimmune encephalomyelitis by immune tolerance to proteolipid protein and its encephalitogenic peptides. J Immunol (1990) 144(3):909–15.
156. Xiao-min S, Sriram S. Treatment of chronic relapsing experimental allergic encephalomyelitis with the intravenous administration of splenocytes coupled to encephalitogenic peptide 91–103 of myelin basic protein. J Neuroimmunol (1991) 34(2):181–90. doi: 10.1016/0165-5728(91)90128-T
157. Smarr CB, Hsu C-L, Byrne AJ, Miller SD, Bryce PJ. Antigen-Fixed Leukocytes Tolerize Th2 Responses in Mouse Models of Allergy. J Immunol (2011) 187(10):5090–8. doi: 10.4049/jimmunol.1100608
158. Braley-Mullen H, Tompson JG, Sharp GC, Kyriakos M. Suppression of experimental autoimmune thyroiditis in guinea pigs by pretreatment with thyroglobulin-coupled spleen cells. Cell Immunol (1980) 51(2):408–13. doi: 10.1016/0008-8749(80)90272-5
159. Dua HS, Gregerson DS, Donoso LA. Inhibition of experimental autoimmune uveitis by retinal photoreceptor antigens coupled to spleen cells. Cell Immunol (1992) 139(2):292–305. doi: 10.1016/0008-8749(92)90072-W
160. Gregorian SK, Clark L, Heber-Katz E, Amento EP, Rostami A. Induction of Peripheral Tolerance with Peptide-Specific Anergy in Experimental Autoimmune Neuritis. Cell Immunol (1993) 150(2):298–310. doi: 10.1006/cimm.1993.1198
161. Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, et al. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1–PD-L1 pathway. J Exp Med (2006) 203(12):2737–47. doi: 10.1084/jem.20061577
162. Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol (2007) 7(9):665–77. doi: 10.1038/nri2153
163. Kishimoto TK, Maldonado RA. Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Front Immunol (2018) 9:230–. doi: 10.3389/fimmu.2018.00230
164. Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol (2012) 30(12):1217–24. doi: 10.1038/nbt.2434
165. Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci USA (2015) 112(2):E156–65. doi: 10.1073/pnas.1408686111
166. LaMothe RA, Kolte PN, Vo T, Ferrari JD, Gelsinger TC, Wong J, et al. Tolerogenic Nanoparticles Induce Antigen-Specific Regulatory T Cells and Provide Therapeutic Efficacy and Transferrable Tolerance against Experimental Autoimmune Encephalomyelitis. Front Immunol (2018) 9:281. doi: 10.3389/fimmu.2018.00281
167. Casey LM, Pearson RM, Hughes KR, Liu JMH, Rose JA, North MG, et al. Conjugation of Transforming Growth Factor Beta to Antigen-Loaded Poly(lactide- co-glycolide) Nanoparticles Enhances Efficiency of Antigen-Specific Tolerance. Bioconjug Chem (2018) 29(3):813–23. doi: 10.1021/acs.bioconjchem.7b00624
168. Tostanoski Lisa H, Chiu Y-C, Gammon Joshua M, Simon T, Andorko James I, Bromberg Jonathan S, et al. Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific. Cell Rep (2016) 16(11):2940–52. doi: 10.1016/j.celrep.2016.08.033
169. Hess KL, Oh E, Tostanoski LH, Andorko JI, Susumu K, Deschamps JR, et al. Engineering Immunological Tolerance Using Quantum Dots to Tune the Density of Self-Antigen Display. Adv Funct Mater (2017) 27(22):1700290. doi: 10.1002/adfm.201700290
170. Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H, et al. Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J Hepatol (2015) 62(6):1349–56. doi: 10.1016/j.jhep.2015.01.006
171. Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA (2012) 109(28):11270–5. doi: 10.1073/pnas.1120611109
172. Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, et al. Tolerogenic nanoparticles inhibit T cell–mediated autoimmunity through SOCS2. Sci Signal (2016) 9(433):ra61–ra. doi: 10.1126/scisignal.aad0612
173. Umeshappa CS, Mbongue J, Singha S, Mohapatra S, Yamanouchi J, Lee JA, et al. Ubiquitous antigen-specific T regulatory type 1 cells variably suppress hepatic and extrahepatic autoimmunity. J Clin Investig (2020) 130(4):1823–9. doi: 10.1172/JCI130670
174. Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature (2016) 530(7591):434–40. doi: 10.1038/nature16962
175. Pei W, Wan X, Shahzad KA, Zhang L, Song S, Jin X, et al. Direct modulation of myelin-autoreactive CD4(+) and CD8(+) T cells in EAE mice by a tolerogenic nanoparticle co-carrying myelin peptide-loaded major histocompatibility complexes, CD47 and multiple regulatory molecules. Int J Nanomed (2018) 13:3731–50. doi: 10.2147/IJN.S164500
176. Belogurov AA, Jr., Stepanov AV, Smirnov IV, Melamed D, Bacon A, Mamedov AE, et al. Liposome-encapsulated peptides protect against experimental allergic encephalitis. FASEB J Off Publ Fed Am Soc Exp Biol (2013) 27(1):222–31. doi: 10.1096/fj.12-213975
177. Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Shea LD, et al. Tolerogenic Ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells. J Autoimmun (2018) 89:112–24. doi: 10.1016/j.jaut.2017.12.010
178. Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, et al. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano (2014) 8(3):2148–60. doi: 10.1021/nn405033r
179. Smarr CB, Yap WT, Neef TP, Pearson RM, Hunter ZN, Ifergan I, et al. Biodegradable antigen-associated PLG nanoparticles tolerize Th2-mediated allergic airway inflammation pre- and postsensitization. Proc Natl Acad Sci USA (2016) 113(18):5059–64. doi: 10.1073/pnas.1505782113
180. Freitag TL, Podojil JR, Pearson RM, Fokta FJ, Sahl C, Messing M, et al. Gliadin Nanoparticles Induce Immune Tolerance to Gliadin in Mouse Models of Celiac Disease. Gastroenterology (2020) 158(6):1667–81.e12. doi: 10.1053/j.gastro.2020.01.045
181. Howard GP, Verma G, Ke X, Thayer WM, Hamerly T, Baxter VK, et al. Critical Size Limit of Biodegradable Nanoparticles for Enhanced Lymph Node Trafficking and Paracortex Penetration. Nano Res (2019) 12(4):837–44. doi: 10.1007/s12274-019-2301-3
182. Kenison JE, Jhaveri A, Li Z, Khadse N, Tjon E, Tezza S, et al. Tolerogenic nanoparticles suppress central nervous system inflammation. Proc Natl Acad Sci USA (2020) 117(50):32017–28. doi: 10.1073/pnas.2016451117
183. Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, et al. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity (2010) 32(4):568–80. doi: 10.1016/j.immuni.2010.03.015
184. Umeshappa CS, Singha S, Blanco J, Shao K, Nanjundappa RH, Yamanouchi J, et al. Suppression of a broad spectrum of liver autoimmune pathologies by single peptide-MHC-based nanomedicines. Nat Commun (2019) 10(1):2150. doi: 10.1038/s41467-019-09893-5
185. Fissolo N, Montalban X, Comabella M. DNA-based vaccines for multiple sclerosis: Current status and future directions. Clin Immunol (2012) 142(1):76–83. doi: 10.1016/j.clim.2010.11.011
186. Ferrera F, Cava AL, Rizzi M, Hahn BH, Indiveri F, Filaci G. Gene Vaccination for the Induction of Immune Tolerance. Ann New York Acad Sci (2007) 1110(1):99–111. doi: 10.1196/annals.1423.012
187. Tsunoda I, Kuang L-Q, Tolley ND, Whitton JL, Fujinami RS. Enhancement of Experimental Allergic Encephalomyelitis (EAE) by DNA Immunization with Myelin Proteolipid Protein (PLP) Plasmid DNA. J Neuropathol Exp Neurol (1998) 57(8):758–67. doi: 10.1097/00005072-199808000-00005
188. Bourquin C, Iglesias A, Berger T, Wekerle H, Linington C. Myelin oligodendrocyte glycoprotein-DNA vaccination induces antibody-mediated autoaggression in experimental autoimmune encephalomyelitis. Eur J Immunol (2000) 30(12):3663–71. doi: 10.1002/1521-4141(200012)30:12<3663::AID-IMMU3663>3.0.CO;2-7
189. Selmaj K, Kowal C, Walczak A, Nowicka J, Raine CS. Naked DNA vaccination differentially modulates autoimmune responses in experimental autoimmune encephalomyelitis. J Neuroimmunol (2000) 111(1):34–44. doi: 10.1016/S0165-5728(00)00329-5
190. Iruretagoyena MI, Sepúlveda SE, Lezana JP, Hermoso M, Bronfman M, Gutiérrez MA, et al. Inhibition of Nuclear Factor-κB Enhances the Capacity of Immature Dendritic Cells to Induce Antigen-Specific Tolerance in Experimental Autoimmune Encephalomyelitis. J Pharmacol Exp Ther (2006) 318(1):59–67. doi: 10.1124/jpet.106.103259
191. Zhou Y, Leng X, Luo S, Su Z, Luo X, Guo H, et al. Tolerogenic Dendritic Cells Generated with Tofacitinib Ameliorate Experimental Autoimmune Encephalomyelitis through Modulation of Th17/Treg Balance. J Immunol Res (2016) 2016:5021537. doi: 10.1155/2016/5021537
192. Zhou Y, Leng X, Li H, Yang S, Yang T, Li L, et al. Tolerogenic dendritic cells induced by BD750 ameliorate proinflammatory T cell responses and experimental autoimmune encephalitis in mice. Mol Med (2017) 23:204–14. doi: 10.2119/molmed.2016.00110
193. Lee J-H, Park C-S, Jang S, Kim J-W, Kim S-H, Song S, et al. Tolerogenic dendritic cells are efficiently generated using minocycline and dexamethasone. Sci Rep (2017) 7(1):15087. doi: 10.1038/s41598-017-15569-1
194. Xie Z, Chen J, Zheng C, Wu J, Cheng Y, Zhu S, et al. 1,25-dihydroxyvitamin D3 -induced dendritic cells suppress experimental autoimmune encephalomyelitis by increasing proportions of the regulatory lymphocytes and reducing T helper type 1 and type 17 cells. Immunology (2017) 152(3):414–24. doi: 10.1111/imm.12776
195. Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol (2013) 14(10):1054–63. doi: 10.1038/ni.2695
196. Perona-Wright G, Anderton SM, Howie SEM, Gray D. IL-10 permits transient activation of dendritic cells to tolerize T cells and protect from central nervous system autoimmune disease. Int Immunol (2007) 19(9):1123–34. doi: 10.1093/intimm/dxm084
197. Fissolo N, Costa C, Nurtdinov RN, Bustamante MF, Llombart V, Mansilla MJ, et al. Treatment with MOG-DNA vaccines induces CD4+CD25+FoxP3+ regulatory T cells and up-regulates genes with neuroprotective functions in experimental autoimmune encephalomyelitis. J Neuroinflamm (2012) 9:139. doi: 10.1186/1742-2094-9-139
198. Castor T, Yogev N, Blank T, Barwig C, Prinz M, Waisman A, et al. Inhibition of experimental autoimmune encephalomyelitis by tolerance-promoting DNA vaccination focused to dendritic cells. PloS One (2018) 13(2):e0191927–e. doi: 10.1371/journal.pone.0191927
199. Keeler GD, Kumar S, Palaschak B, Silverberg EL, Markusic DM, Jones NT, et al. Gene Therapy-Induced Antigen-Specific Tregs Inhibit Neuro-inflammation and Reverse Disease in a Mouse Model of Multiple Sclerosis. Mol Ther (2018) 26(1):173–83. doi: 10.1016/j.ymthe.2017.09.001
200. Kang Y, Zhao J, Liu Y, Chen A, Zheng G, Yu Y, et al. FK506 as an adjuvant of tolerogenic DNA vaccination for the prevention of experimental autoimmune encephalomyelitis. J Gene Med (2009) 11(11):1064–70. doi: 10.1002/jgm.1387
201. Kang Y, Sun Y, Zhang J, Gao W, Kang J, Wang Y, et al. Treg cell resistance to apoptosis in DNA vaccination for experimental autoimmune encephalomyelitis treatment. PloS One (2012) 7(11):e49994. doi: 10.1371/journal.pone.0049994
202. Schif-Zuck S, Wildbaum G, Karin N. Coadministration of Plasmid DNA Constructs Encoding an Encephalitogenic Determinant and IL-10 Elicits Regulatory T Cell-Mediated Protective Immunity in the Central Nervous System. J Immunol (2006) 177(11):8241–7. doi: 10.4049/jimmunol.177.11.8241
203. Garren H, Ruiz PJ, Watkins TA, Fontoura P, Nguyen L-VT, Estline ER, et al. Combination of Gene Delivery and DNA Vaccination to Protect from and Reverse Th1 Autoimmune Disease via Deviation to the Th2 Pathway. Immunity (2001) 15(1):15–22. doi: 10.1016/S1074-7613(01)00171-6
204. Krienke C, Kolb L, Diken E, Streuber M, Kirchhoff S, Bukur T, et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science (2021) 371(6525):145–53. doi: 10.1126/science.aay3638
205. Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity (2005) 23(2):165–75. doi: 10.1016/j.immuni.2005.06.008
206. Karikó K, Muramatsu H, Ludwig J, Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res (2011) 39(21):e142–e. doi: 10.1093/nar/gkr695
207. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature (2016) 534(7607):396–401. doi: 10.1038/nature18300
208. van Bekkum DW. Stem Cell Transplantation in Experimental Models of Autoimmune Disease. J Clin Immunol (2000) 20(1):10–6. doi: 10.1023/a:1006682225181
209. Duffy SS, Keating BA, Moalem-Taylor G. Adoptive Transfer of Regulatory T Cells as a Promising Immunotherapy for the Treatment of Multiple Sclerosis. Front Neurosci (2019) 13:1107. doi: 10.3389/fnins.2019.01107
210. Yang M, Rui K, Wang S, Lu L. Regulatory B cells in autoimmune diseases. Cell Mol Immunol (2013) 10(2):122–32. doi: 10.1038/cmi.2012.60
211. Zhang X-M, Lund H, Mia S, Parsa R, Harris RA. Adoptive transfer of cytokine-induced immunomodulatory adult microglia attenuates experimental autoimmune encephalomyelitis in DBA/1 mice. Glia (2014) 62(5):804–17. doi: 10.1002/glia.22643
212. Casacuberta-Serra S, Costa C, Eixarch H, Mansilla MJ, López-Estévez S, Martorell L, et al. Myeloid-derived suppressor cells expressing a self-antigen ameliorate experimental autoimmune encephalomyelitis. Exp Neurol (2016) 286:50–60. doi: 10.1016/j.expneurol.2016.09.012
213. Flórez-Grau G, Zubizarreta I, Cabezón R, Villoslada P, Benitez-Ribas D. Tolerogenic Dendritic Cells as a Promising Antigen-Specific Therapy in the Treatment of Multiple Sclerosis and Neuromyelitis Optica From Preclinical to Clinical Trials. Front Immunol (2018) 9:1169. doi: 10.3389/fimmu.2018.01169
214. Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, et al. In Vitro Generation of Interleukin 10–producing Regulatory CD4+ T Cells Is Induced by Immunosuppressive Drugs and Inhibited by T Helper Type 1 (Th1)– and Th2-inducing Cytokines. J Exp Med (2002) 195(5):603–16. doi: 10.1084/jem.20011629
215. Mansilla MJ, Selles-Moreno C, Fabregas-Puig S, Amoedo J, Navarro-Barriuso J, Teniente-Serra A, et al. Beneficial effect of tolerogenic dendritic cells pulsed with MOG autoantigen in experimental autoimmune encephalomyelitis. CNS Neurosci Ther (2015) 21(3):222–30. doi: 10.1111/cns.12342
216. Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol (2002) 169(9):4712–6. doi: 10.4049/jimmunol.169.9.4712
217. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front Immunol (2019) 10:43. doi: 10.3389/fimmu.2019.00043
218. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol (2014) 153(1):23–30. doi: 10.1016/j.clim.2014.03.016
219. Safinia N, Grageda N, Scottà C, Thirkell S, Fry LJ, Vaikunthanathan T, et al. Cell Therapy in Organ Transplantation: Our Experience on the Clinical Translation of Regulatory T Cells. Front Immunol (2018) 9:354. doi: 10.3389/fimmu.2018.00354
220. Tarbell KV, Petit L, Zuo X, Toy P, Luo X, Mqadmi A, et al. Dendritic cell-expanded, islet-specific CD4+ CD25+ CD62L+ regulatory T cells restore normoglycemia in diabetic NOD mice. J Exp Med (2007) 204(1):191–201. doi: 10.1084/jem.20061631
221. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199(11):1455–65. doi: 10.1084/jem.20040139
222. Stephens LA, Malpass KH, Anderton SM. Curing CNS autoimmune disease with myelin-reactive Foxp3+ Treg. Eur J Immunol (2009) 39(4):1108–17. doi: 10.1002/eji.200839073
223. Yu P, Gregg RK, Bell JJ, Ellis JS, Divekar R, Lee HH, et al. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J Immunol (2005) 174(11):6772–80. doi: 10.4049/jimmunol.174.11.6772
224. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Investig (2016) 126(4):1413–24. doi: 10.1172/JCI82771
225. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MAA, Hannen RF, Cooper D, et al. Expression of a Chimeric Antigen Receptor Specific for Donor HLA Class I Enhances the Potency of Human Regulatory T Cells in Preventing Human Skin Transplant Rejection. Am J Transplant (2017) 17(4):931–43. doi: 10.1111/ajt.14185
226. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of Allograft Rejection by Use of Regulatory T Cells With an MHC-Specific Chimeric Antigen Receptor. Am J Transplant (2017) 17(4):917–30. doi: 10.1111/ajt.14175
227. De Paula Pohl A, Schmidt A, Zhang A-H, Maldonado T, Königs C, Scott DW. Engineered regulatory T cells expressing myelin-specific chimeric antigen receptors suppress EAE progression. Cell Immunol (2020) 358:104222. doi: 10.1016/j.cellimm.2020.104222
228. Elinav E, Adam N, Waks T, Eshhar Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology (2009) 136(5):1721–31. doi: 10.1053/j.gastro.2009.01.049
229. Yoon J, Schmidt A, Zhang A-H, Königs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood (2017) 129(2):238–45. doi: 10.1182/blood-2016-07-727834
230. Fu RY, Chen AC, Lyle MJ, Chen C-Y, Liu CL, Miao CH. CD4+ T cells engineered with FVIII-CAR and murine Foxp3 suppress anti-factor VIII immune responses in hemophilia a mice. Cell Immunol (2020) 358:104216. doi: 10.1016/j.cellimm.2020.104216
231. Skuljec J, Chmielewski M, Happle C, Habener A, Busse M, Abken H, et al. Chimeric Antigen Receptor-Redirected Regulatory T Cells Suppress Experimental Allergic Airway Inflammation, a Model of Asthma. Front Immunol (2017) 8:1125. doi: 10.3389/fimmu.2017.01125
232. Koristka S, Kegler A, Bergmann R, Arndt C, Feldmann A, Albert S, et al. Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J Autoimmun (2018) 90:116–31. doi: 10.1016/j.jaut.2018.02.006
233. Pierini A, Iliopoulou BP, Peiris H, Pérez-Cruz M, Baker J, Hsu K, et al. T cells expressing chimeric antigen receptor promote immune tolerance. JCI Insight (2017) 2(20):e92865. doi: 10.1172/jci.insight.92865
234. Bézie S, Charreau B, Vimond N, Lasselin J, Gérard N, Nerrière-Daguin V, et al. Human CD8+ Tregs expressing a MHC-specific CAR display enhanced suppression of human skin rejection and GVHD in NSG mice. Blood Adv (2019) 3(22):3522–38. doi: 10.1182/bloodadvances.2019000411
235. Mekala DJ, Alli RS, Geiger TL. IL-10-dependent infectious tolerance after the treatment of experimental allergic encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Proc Natl Acad Sci USA (2005) 102(33):11817–22. doi: 10.1073/pnas.0505445102
236. Abdeladhim M, Zhang AH, Kropp LE, Lindrose AR, Venkatesha SH, Mitre E, et al. Engineered ovalbumin-expressing regulatory T cells protect against anaphylaxis in ovalbumin-sensitized mice. Clin Immunol (2019) 207:49–54. doi: 10.1016/j.clim.2019.07.009
237. Zhang AH, Yoon J, Kim YC, Scott DW. Targeting Antigen-Specific B Cells Using Antigen-Expressing Transduced Regulatory T Cells. J Immunol (2018) 201(5):1434–41. doi: 10.4049/jimmunol.1701800
238. Wright GP, Notley CA, Xue S-A, Bendle GM, Holler A, Schumacher TN, et al. Adoptive therapy with redirected primary regulatory T cells results in antigen-specific suppression of arthritis. Proc Natl Acad Sci USA (2009) 106(45):19078–83. doi: 10.1073/pnas.0907396106
239. Kim YC, Zhang AH, Yoon J, Culp WE, Lees JR, Wucherpfennig KW, et al. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J Autoimmun (2018) 92:77–86. doi: 10.1016/j.jaut.2018.05.003
240. Kim YC, Zhang A-H, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood (2015) 125(7):1107–15. doi: 10.1182/blood-2014-04-566786
241. Kansal R, Richardson N, Neeli I, Khawaja S, Chamberlain D, Ghani M, et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci Transl Med (2019) 11(482):eaav1648. doi: 10.1126/scitranslmed.aav1648
242. Ward DE, Fay BL, Adejuwon A, Han H, Ma Z. Chimeric Antigen Receptors Based on Low Affinity Mutants of FcϵRI Re-direct T Cell Specificity to Cells Expressing Membrane IgE. Front Immunol (2018) 9(2231). doi: 10.3389/fimmu.2018.02231
243. Zhang L, Sosinowski T, Cox AR, Cepeda JR, Sekhar NS, Hartig SM, et al. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J Autoimmun (2019) 96:50–8. doi: 10.1016/j.jaut.2018.08.004
244. Fishman S, Lewis MD, Siew LK, De Leenheer E, Kakabadse D, Davies J, et al. Adoptive Transfer of mRNA-Transfected T Cells Redirected against Diabetogenic CD8 T Cells Can Prevent Diabetes. Mol Ther (2017) 25(2):456–64. doi: 10.1016/j.ymthe.2016.12.007
245. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science (2016) 353(6295):179–84. doi: 10.1126/science.aaf6756
246. Oh S, O’Connor K, Payne A. MuSK Chimeric Autoantibody Receptor (CAAR) T Cells for Antigen-specific Cellular Immunotherapy of Myasthenia Gravis (2769). Neurology (2020) 94(15 Supplement):2769.
247. Dawson NAJ, Rosado-Sánchez I, Novakovsky GE, Fung VCW, Huang Q, McIver E, et al. Functional effects of chimeric antigen receptor co-receptor signaling domains in human regulatory T cells. Sci Transl Med (2020) 12(557):eaaz3866. doi: 10.1126/scitranslmed.aaz3866
248. Lacroix-Desmazes S, Voorberg J, Lillicrap D, Scott DW, Pratt KP. Tolerating Factor VIII: Recent Progress. Front Immunol (2020) 10:2991. doi: 10.3389/fimmu.2019.02991
249. Yu Y, Ma X, Gong R, Zhu J, Wei L, Yao J. Recent advances in CD8(+) regulatory T cell research. Oncol Lett (2018) 15(6):8187–94. doi: 10.3892/ol.2018.8378
250. Flippe L, Bézie S, Anegon I, Guillonneau C. Future prospects for CD8(+) regulatory T cells in immune tolerance. Immunol Rev (2019) 292(1):209–24. doi: 10.1111/imr.12812
251. Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, et al. Generation of human islet-specific regulatory T cells by TCR gene transfer. J Autoimmun (2017) 79:63–73. doi: 10.1016/j.jaut.2017.01.001
252. Lee DSW, Rojas OL, Gommerman JL. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discovery (2020) 20:179–99. doi: 10.1038/s41573-020-00092-2
253. Gallo P, Centonze D, Marrosu MG. Alemtuzumab for multiple sclerosis: the new concept of immunomodulation. Mult Scler Demyelinating Disord (2017) 2(1):7. doi: 10.1186/s40893-017-0024-4
254. Zhang L, Crawford F, Yu L, Michels A, Nakayama M, Davidson HW, et al. Monoclonal antibody blocking the recognition of an insulin peptide–MHC complex modulates type 1 diabetes. Proc Natl Acad Sci USA (2014) 111(7):2656–61. doi: 10.1073/pnas.1323436111
255. Chen Y, Sun J, Liu H, Yin G, Xie Q. Immunotherapy Deriving from CAR-T Cell Treatment in Autoimmune Diseases. J Immunol Res (2019) 2019:5727516. doi: 10.1155/2019/5727516
256. Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity (2002) 17(2):167–78. doi: 10.1016/S1074-7613(02)00367-9
257. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol (2005) 6(11):1142–51. doi: 10.1038/ni1263
258. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol (2016) 17(11):1322–33. doi: 10.1038/ni.3540
259. Toomer KH, Lui JB, Altman NH, Ban Y, Chen X, Malek TR. Essential and non-overlapping IL-2Ralpha-dependent processes for thymic development and peripheral homeostasis of regulatory T cells. Nat Commun (2019) 10(1):1037. doi: 10.1038/s41467-019-08960-1
260. Sharfe N, Dadi HK, Shahar M, Roifman CM. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc Natl Acad Sci USA (1997) 94(7):3168–71. doi: 10.1073/pnas.94.7.3168
261. Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, et al. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol (2006) 177(5):2770–4. doi: 10.4049/jimmunol.177.5.2770
262. Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol (2007) 119(2):482–7. doi: 10.1016/j.jaci.2006.10.007
263. Humrich JY, Morbach H, Undeutsch R, Enghard P, Rosenberger S, Weigert O, et al. Homeostatic imbalance of regulatory and effector T cells due to IL-2 deprivation amplifies murine lupus. Proc Natl Acad Sci USA (2010) 107(1):204–9. doi: 10.1073/pnas.0903158107
264. Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Ciullini Mannurita S, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol (2013) 146(3):248–61. doi: 10.1016/j.clim.2013.01.004
265. Long SA, Buckner JH, Greenbaum CJ. IL-2 therapy in type 1 diabetes: “Trials” and tribulations. Clin Immunol (2013) 149(3):324–31. doi: 10.1016/j.clim.2013.02.005
266. Hull CM, Peakman M, Tree TIM. Regulatory T cell dysfunction in type 1 diabetes: what’s broken and how can we fix it? Diabetologia (2017) 60(10):1839–50. doi: 10.1007/s00125-017-4377-1
267. Malek TR, Yu A, Zhu L, Matsutani T, Adeegbe D, Bayer AL. IL-2 family of cytokines in T regulatory cell development and homeostasis. J Clin Immunol (2008) 28(6):635–9. doi: 10.1007/s10875-008-9235-y
268. Abbas AK, Trotta E, RS D, Marson A, Bluestone JA. Revisiting IL-2: Biology and therapeutic prospects. Sci Immunol (2018) 3(25):eaat1482. doi: 10.1126/sciimmunol.aat1482
269. Burchill MA, Yang J, Vang KB, Farrar MA. Interleukin-2 receptor signaling in regulatory T cell development and homeostasis. Immunol Lett (2007) 114(1):1–8. doi: 10.1016/j.imlet.2007.08.005
270. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol (2014) 192(12):5451–8. doi: 10.4049/jimmunol.1490019
271. Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity (2009) 30(2):204–17. doi: 10.1016/j.immuni.2008.11.014
272. Yu A, Snowhite I, Vendrame F, Rosenzwajg M, Klatzmann D, Pugliese A, et al. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes (2015) 64(6):2172–83. doi: 10.2337/db14-1322
273. Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol (2004) 22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122
274. McNally A, Hill GR, Sparwasser T, Thomas R, Steptoe RJ. CD4+CD25+ regulatory T cells control CD8+ T-cell effector differentiation by modulating IL-2 homeostasis. Proc Natl Acad Sci USA (2011) 108(18):7529–34. doi: 10.1073/pnas.1103782108
275. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med (2013) 5(179):179ra43. doi: 10.1126/scitranslmed.3005265
276. Koreth J, Kim HT, Jones KT, Lange PB, Reynolds CG, Chammas MJ, et al. Efficacy, durability, and response predictors of low-dose interleukin-2 therapy for chronic graft-versus-host disease. Blood (2016) 128(1):130–7. doi: 10.1182/blood-2016-02-702852
277. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med (2011) 365(22):2067–77. doi: 10.1056/NEJMoa1105143
278. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med (2016) 22(9):991–3. doi: 10.1038/nm.4148
279. von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis (2016) 75(7):1407–15. doi: 10.1136/annrheumdis-2015-207776
280. Humrich JY, Riemekasten G. Low-dose interleukin-2 therapy for the treatment of systemic lupus erythematosus. Curr Opin Rheumatol (2019) 31(2):208–12. doi: 10.1097/BOR.0000000000000575
281. Koreth J, Matsuoka K-I, Kim HT, McDonough SM, Bindra B, Alyea EP, et al. Interleukin-2 and Regulatory T Cells in Graft-versus-Host Disease. N Engl J Med (2011) 365(22):2055–66. doi: 10.1056/NEJMoa1108188
282. Mitra S, Ring AM, Amarnath S, Spangler JB, Li P, Ju W, et al. Interleukin-2 activity can be fine tuned with engineered receptor signaling clamps. Immunity (2015) 42(5):826–38. doi: 10.1016/j.immuni.2015.04.018
283. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science (2006) 311(5769):1924–7. doi: 10.1126/science.1122927
284. Trotta E, Bessette PH, Silveria SL, Ely LK, Jude KM, Le DT, et al. A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat Med (2018) 24(7):1005–14. doi: 10.1038/s41591-018-0070-2
285. Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to Interleukin-2 Elicit Selective T Cell Subset Potentiation through Distinct Conformational Mechanisms. Immunity (2015) 42(5):815–25. doi: 10.1016/j.immuni.2015.04.015
286. Karakus U, Sahin D, Mittl PRE, Mooij P, Koopman G, Boyman O. Receptor-gated IL-2 delivery by an anti-human IL-2 antibody activates regulatory T cells in three different species. Sci Transl Med (2020) 12(574):eabb9283. doi: 10.1126/scitranslmed.abb9283
287. DeOca KB, Moorman CD, Garcia BL, Mannie MD. Low-Zone IL-2 Signaling: Fusion Proteins Containing Linked CD25 and IL-2 Domains Sustain Tolerogenic Vaccination in vivo and Promote Dominance of FOXP3(+) Tregs in vitro. Front Immunol (2020) 11:541619–. doi: 10.3389/fimmu.2020.541619
288. Ward NC, Yu A, Moro A, Ban Y, Chen X, Hsiung S, et al. IL-2/CD25: A Long-Acting Fusion Protein That Promotes Immune Tolerance by Selectively Targeting the IL-2 Receptor on Regulatory T Cells. J Immunol (2018) 201(9):2579–92. doi: 10.4049/jimmunol.1800907
289. Sabapathy V, Cheru NT, Corey R, Mohammad S, Sharma R. A Novel Hybrid Cytokine IL233 Mediates regeneration following Doxorubicin-Induced Nephrotoxic Injury. Sci Rep (2019) 9(1):3215. doi: 10.1038/s41598-019-39886-9
290. Padutsch T, Sendetski M, Huber C, Peters N, Pfizenmaier K, Bethea JR, et al. Superior Treg-Expanding Properties of a Novel Dual-Acting Cytokine Fusion Protein. Front Pharmacol (2019) 10:1490. doi: 10.3389/fphar.2019.01490
291. Belizaire R, Kim HT, Poryanda SJ, Mirkovic NV, Hipolito E, Savage WJ, et al. Efficacy and immunologic effects of extracorporeal photopheresis plus interleukin-2 in chronic graft-versus-host disease. Blood Adv (2019) 3(7):969–79. doi: 10.1182/bloodadvances.2018029124
292. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum Dis (2019) 78(2):209–17. doi: 10.1136/annrheumdis-2018-214229
293. Peterson LB, Bell CJM, Howlett SK, Pekalski ML, Brady K, Hinton H, et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J Autoimmun (2018) 95:1–14. doi: 10.1016/j.jaut.2018.10.017
294. Varma R, Liu K, Avery K, Rashid R, Schubbert S, Leung I, et al. Regulatory T Cell Selective IL-2-Fc Fusion Proteins for the Treatment of Autoimmune Diseases. Blood (2018) 132(Supplement 1):3709–. doi: 10.1182/blood-2018-99-117622
295. Tchao N, Gorski K, Yuraszeck T, Sohn S, Ishida K, Wong H, et al. PS7:135 Amg 592 is an investigational il-2 mutein that induces highly selective expansion of regulatory t cells. Lupus Sci Med (2018) 5(Suppl 1):A102–A. doi: 10.1136/lupus-2018-abstract.178
296. Ghelani A, Bates D, Conner K, Wu M-Z, Lu J, Hu Y-L, et al. Defining the Threshold IL-2 Signal Required for Induction of Selective Treg Cell Responses Using Engineered IL-2 Muteins. Front Immunol (2020) 11:1106. doi: 10.3389/fimmu.2020.01106
297. Ito S, Bollard CM, Carlsten M, Melenhorst JJ, Biancotto A, Wang E, et al. Ultra-low dose interleukin-2 promotes immune-modulating function of regulatory T cells and natural killer cells in healthy volunteers. Mol Ther (2014) 22(7):1388–95. doi: 10.1038/mt.2014.50
298. Lu DR, Wu H, Driver I, Ingersoll S, Sohn S, Wang S, et al. Dynamic changes in the regulatory T-cell heterogeneity and function by murine IL-2 mutein. Life Sci Alliance (2020) 3(5):e201900520. doi: 10.26508/lsa.201900520
299. Khoryati L, Pham MN, Sherve M, Kumari S, Cook K, Pearson J, et al. Regulatory T cell expansion by a highly CD25-dependent IL-2 mutein arrests ongoing autoimmunity. bioRxiv (2019) 862789. doi: 10.1101/862789
300. Chen AC, Cai X, Li C, Khoryati L, Gavin MA, Miao CH. A Treg-Selective IL-2 Mutein Prevents the Formation of Factor VIII Inhibitors in Hemophilia Mice Treated With Factor VIII Gene Therapy. Front Immunol (2020) 11:638. doi: 10.3389/fimmu.2020.00638
301. Van Gool F, Molofsky AB, Morar MM, Rosenzwajg M, Liang HE, Klatzmann D, et al. Interleukin-5-producing group 2 innate lymphoid cells control eosinophilia induced by interleukin-2 therapy. Blood (2014) 124(24):3572–6. doi: 10.1182/blood-2014-07-587493
302. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med (2012) 209(2):243–50. doi: 10.1084/jem.20111174
Keywords: tolerogenic vaccine, autoimmune disease, CAR-Tregs, IL-2 mutein, low-dose IL-2 therapy, antigen-specific tolerance, immune tolerance
Citation: Moorman CD, Sohn SJ and Phee H (2021) Emerging Therapeutics for Immune Tolerance: Tolerogenic Vaccines, T cell Therapy, and IL-2 Therapy. Front. Immunol. 12:657768. doi: 10.3389/fimmu.2021.657768
Received: 24 January 2021; Accepted: 04 March 2021;
Published: 29 March 2021.
Edited by:
David William Scott, Uniformed Services University of the Health Sciences, United StatesReviewed by:
Alexander Steinkasserer, University Hospital Erlangen, GermanyCopyright © 2021 Moorman, Sohn and Phee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyewon Phee, aHlld29ucEBhbWdlbi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.