 Yunxuan Li
Yunxuan Li Yixi Zhang
Yixi Zhang Xun Zeng*
Xun Zeng*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 March 2021
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.656097
This article is part of the Research Topic Friend and Foe: Innate Neuro-Immune Interactions and Inflammation in Development, Homeostasis and Injury in the Central Nervous System View all 9 articles
γδ T cells are distributed in various lymphoid and nonlymphoid tissues, and act as early responders in many conditions. Previous studies have proven their significant roles in infection, cancer, autoimmune diseases and tissue maintenance. Recently, accumulating researches have highlighted the crosstalk between γδ T cells and nervous systems. In these reports, γδ T cells maintain some physiological functions of central nervous system by secreting interleukin (IL) 17, and neurons like nociceptors can in turn regulate the activity of γδ T cells. Moreover, γδ T cells are involved in neuroinflammation such as stroke and multiple sclerosis. This review illustrates the relationship between γδ T cells and nervous systems in physiological and pathological conditions.
γδ T cells are T lymphocytes that express T-cell receptor gamma chain and delta chain to constitute γδ T-cell receptors (TCRs). Like conventional αβ T cells and B cells, γδ T cells also utilize somatic V, D, J gene rearrangement to express various TCRs for antigen recognition. Different from major histocompatibility complex (MHC) restricted manner, γδ TCRs follow antibody-like recognition manner to bind diverse ligands such as small and large, peptidic and non-peptidic, and foreign and self-molecules (1–3). Though γδ T cells contribute a minor population in the blood and lymphoid tissue, they are abundant in barrier tissue and their frequency in the blood can expand dramatically during infection (2). Using different V regions of γδ TCR chains, different subsets of γδ T cells reside in meninges, skins, lungs, livers, peritoneal cavity, adipose tissue, uterus, tongue, gut, blood and secondary lymphoid organs, depending on the different waves of γδ T cell development before and after the birth (4–9). Depending on TCR signaling strength during development, γδ T cells differentiate into two main effector subsets based on the types of cytokines produced: interferon-gamma (IFN-γ) and interleukin17 (IL-17) (γδ T17 cells) (5, 10). γδ T cells can participate in an immediate immune response as their direct antigen recognition, wide distribution, diverse ligands of γδ TCRs and expression of innate receptors. Indeed, tons of evidences have indicated that γδ T cells play pivotal roles in infection, tumor, autoimmunity and immune surveillance (7, 11–13). During the immune responses, γδ T cells can be activated by TCRs and/or by innate signals [e.g. cytokines and natural-killer group 2, member D (NKG2D)] (14). The clear roles of TCR signals and innate signals in regulating the functions of γδ T cells are still under debate. Regardless, the activated γδ T cells can secrete IL-17 to recruit neutrophils to amplify the inflammatory signals, promote the maturation of dendritic cells to prime αβ T cells, eliminate infected cells by directly releasing IFN-γ, perforin, granzyme B, and granulysin after sensing antigen and antibody-dependent cell-mediated cytotoxicity, and present antigen to αβ T cells (11). In addition, γδ T cells can repair damaged tissues by producing cytokines and chemokines, which is crucial for the homeostatic maintenance in the epidermis, intestinal epithelium, and adipose tissues (7). These tissue-resident γδ T cells show more functions than any other immune cells, as they can secrete insulin-like growth factor 1 (IGF-1) to improve epithelial cell survival and produce IL-17F to promote lipolysis and thermogenesis in adipose tissue (15, 16).
γδ T cells are also important for the inflammatory responses in neurological diseases. More importantly, new evidences have indicated that meningeal γδ T cells play important roles in maintaining the homeostasis of nervous system. These findings have suggested the complex role of γδ T cells in neuron-immune interactions. To better understand the functions of γδ T cells in nervous system, it is necessary to summarize the latest progress in the interaction between γδ T cells and nervous system. In this review, we will discuss how γδ T cells interact with nervous system in physiological and pathological conditions.
For past decades, it has been conventionally believed that the central nervous system (CNS) has immune-privileged properties as it is shielded by the blood-brain barrier (BBB) that features with low expression of leukocyte adhesion molecules and tight junctions between brain capillary endothelial cells (17, 18). However, accumulating evidences have suggested that CNS and immune system can directly crosstalk with each other (19). Some immune molecules, such as cytokines, play a role in learning, memory and social behavior (20, 21). Moreover, a triple-layered membrane surrounding brain parenchyma called meninges is confirmed to bypass the BBB, which may be a place for immune surveillance and maintaining homeostasis of CNS (19). Along this line, diverse meningeal immune cells have been described in many articles. Among these immune cells, meningeal T cells were identified to secrete interleukin 4 (IL-4), interleukin 13 (IL-13), and IFN-γ, which correlate with learning, long-term memory and social behavior (20–22). IL-17, a key cytokine for inflammation, has also been discovered to administrate the fetal brain development and behavioral abnormalities (23, 24). Since meningeal γδ T cells have been identified as well, it is interesting to know the role of γδ T cells in regulating brain functions. Recently, two elegant works by Ribot’s group and Kipnis’ group found that meningeal γδ T cells could secrete IL-17 to regulate short-term memory and anxiety-like behavior, which partially addressed this issue (8, 25) (Figure 1).
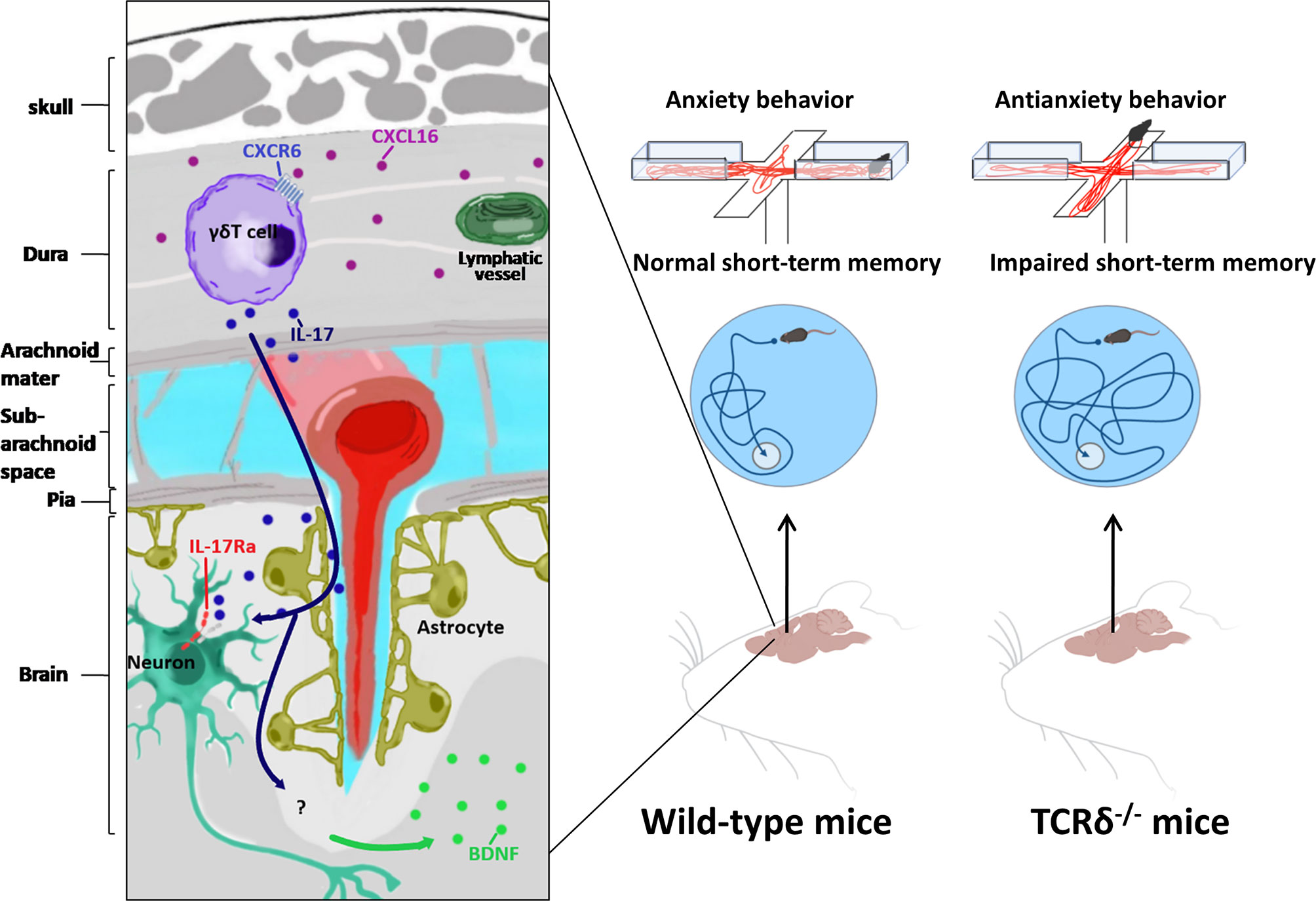
Figure 1 Meningeal γδ T cells involved in behavior regulation. Relying on CXCR6-CXCL16 axis, meningeal γδ T cells migrate to dura mater shortly after birth. Meningeal γδ T cells influence brain by constant secretion of IL-17. Neurons in medial prefrontal cortex(mPFC) and somatosensory cortex (S1DZ) express IL-17Ra in response to IL-17 to regulate anxiety-like behavior of mice. IL-17 induced glial brain-derived neurotrophic factor (BDNF) modulates hippocampal neuronal plasticity to maintain short-term memory.
Meningeal γδ T cells are tissue-resident cells expressing C-X-C Motif Chemokine Receptor 6 (CXCR6) and are attracted by chemokine C-X-C Motif Chemokine Ligand 16 (CXCL16), which is highly expressed in dura-resident myeloid cells (25). They migrate to meninges shortly after birth and are prevalent in dura mater. More importantly, they are the major source of IL-17 (8, 25). The majority of these meningeal γδ T17 cells are fetal derived Vγ6Vδ1 (the V region of TCR γ chain uses TRGV6 gene and the δ chain uses TRDV1 gene) T cells with canonical identical Vγ6-Jγ1 and Vδ1-Dδ2-Jδ2 chains that can be found in various non-lymphoid tissues (8, 9, 26, 27). Mice deficient γδ T cells and IL-17 showed impairments in short-term memory in tests of Y-maze and the Morris water maze. γδ T cell derived IL-17 can modulate the expression of neurotrophic factor (BDNF) in the hippocampus, which is able to regulate synaptic plasticity of neurons required for short-term memory (8). On the other hand, compared to WT mice, TCRδ-/- mice and WT mice with the presence of anti-TCRδ antibodies in the Cerebrospinal fluid (CSF) showed severe anxiety-like behavior in the elevated plus maze and the open field (25). Collectively, these data suggested that meningeal γδ T17 cells played a key role in short-term memory and anxiety-like behavior. In this scenario, it is important to figure out the target of IL-17. Since IL-17 receptor A (IL-17RA) is expressed not only on astrocytes and microglial cells, but also on neurons throughout all cortical layers of the medial prefrontal cortex (mPFC) and somatosensory cortex (S1DZ), the observation that conditional knockout of IL-17RA on astrocytes and microglial cells did not disturb short-term memory suggested that IL-17 could directly affect neurons (8, 25, 28). Indeed, IL-17 signaling affected mPFC neurons by down-regulating the activity of gamma-aminobutyric acid (GABA)-benzodiazepine, the prototypical pathway for anxiolytic drugs (25). The further detailed molecular mechanism of how IL-17 signaling regulates short-term memory and anxiety-like behavior requires further study. Furthermore, unlike lungs, skins and guts, meninges do not have pathogenic or inflammatory stimuli in steady state (18, 29). How could meningeal γδ T cells continuously produce IL-17? It is found that the IL-17 production of meningeal γδ T cells is irrelative with pro-inflammatory cytokines interleukin 1 (IL-1) and interleukin 23 (IL-23), and pathogen-associated molecular pattern signals (8). In addition, it is still inconclusive whether components of commensal microbiota regulate IL-17 production of meningeal γδ T cells (8, 25). Therefore, the detailed mechanisms of continuous production of IL-17 by meningeal γδ T cells are still unclear.
As one of the most essential protective mechanisms of human body, nociceptive pain responds to chemical, mechanical, and thermal stimuli and can be detected by nociceptors around the body in peripheral nervous system (PNS) (30). As a particular subset of primary sensory neurons, nociceptors can respond to pain stimuli and subsequently convert the stimuli into nerve impulses to inform brain to produce the sensation of pain (31). Once receiving the stimuli, nociceptors can regulate the immune cell response activity at the tissue by releasing neuropeptides which were stored at the dense-core vesicles both in nociceptors’ synaptic terminals at the CNS and in the nerve endings within the peripheral tissues (32). The transducers of noxious stimuli are voltage-gated and ligand-gated ion channels expressed on the nociceptor nerve terminals, such as transient receptor potential vanilloid subfamily member 1 (TRPV1), transient receptor potential ankyrin 1 (TRPA1), Nav (Voltage-gated sodium channels)1.7, Nav1.8, and Nav1.9 (33).
Among various immune cells, γδ T cells are also regulated b nociceptors. It has been reported that TRPV1+ and Nav1.8+ nociceptors were necessary factors to drive imiquimod (IMQ) induced psoriasis-like inflammation in skin by promoting dermal dendritic cells (dDCs) to produce Interleukin 23 (IL-23). IL-23-producing dDCs could activate IL-23 receptor positive (IL-23R+) dermal γδ T cells to secrete IL-17A, IL-17F and IL-22, which resulted in the recruitment of neutrophils to skin and hyperproliferation of keratinocytes (34, 35). Extended studies provided more details of how TRPV1+ nociceptors, dDCs and dermal γδ T cells interacted with each other in a fungus infection mouse model. TRPV1+ neurons were activated through Dectin-1 by sensing the β-glucan of Candida albicans, a kind of sugars on cell walls of fungus. Activated TRPV1+ neurons released neuropeptide calcitonin gene–related peptide (CGRP) to drive dDCs to produce IL-23, which could promote dermal γδ T cells to produce IL-17 and subsequently active downstream pathways to inhibit C. albicans infection (36–38). Ablating TRPV1+ nociceptors by resiniferatoxin (RTX) could reduce the numbers of IL-17 producing γδ T cells and the efficiency of C. albicans elimination (36). This finding, together with other findings that α-hemolysin of Staphylococcus aureus (S. aureus) and streptolysin S of Streptococcus pyogenes (S. pyogenes) can activate TRPV1+ nociceptors to secrete CGRP, indicated that some pathogen related molecules were sufficient for TRPV1+nociceptors activation and CGRP secretion as a consequence (39, 40). Therefore, Kaplan and colleagues tried to figure out whether the activation of TRPV1+ neurons alone could trigger γδ T17 response in a pathogenic molecule-free condition. By using optogenetic mouse model, they found that activation of TRPV1+ neurons alone sufficiently induced IL-17 production by γδ T cells in skin via releasing CGRP. More importantly, activated TRPV1+ neurons provided signals through nerve reflex arc that could induce γδ T17 response at adjacent, unstimulated skin (37). Therefore, neurons secreted molecules are perfectly capable of activating skin innate immune response, not only at the stimulated skin, but also at adjacent unstimulated skin, which may be benefit for limiting the infection. In addition, γδ T cells can cooperate with TRPA1+ nociceptors to promote systemic skin regeneration. Leung’s group found that in IMQ induced inflammation mouse model, TRPA1+ neurons, but not NLR family pyrin domain containing 3 (NLRP3), Toll like receptor 7 (TLR7) and TRPV1+ neurons, stimulated local IL-23 production by dDCs, thereby activating γδ T17 for tissue regeneration. Although the details of how TRPA1+ neurons promote dDCs to secrete IL-23 still need to be clarified, the results in mice with genetically defective of TRPA1+ neurons, γδ T cells and chemical removing dDCs suggested that none of these cells were redundant in skin wound healing (41). However, since both TRPA+ and TRPV+ neurons can activate γδ T17 cells via IL-23 secreted by dDCs, the reason why only TRPA+ neurons can promote γδ T17 cells for wound healing needs to be clarified. Other potential mechanisms may exist in wound healing by TRPA+ neurons regulated γδ T17 cells and are required for further investigation. Taken together, the axis of nociceptors-dDCs-γδ T17 cells has been discovered and plays a key role in defense against pathogen invasion and skin wound healing (Figure 2).
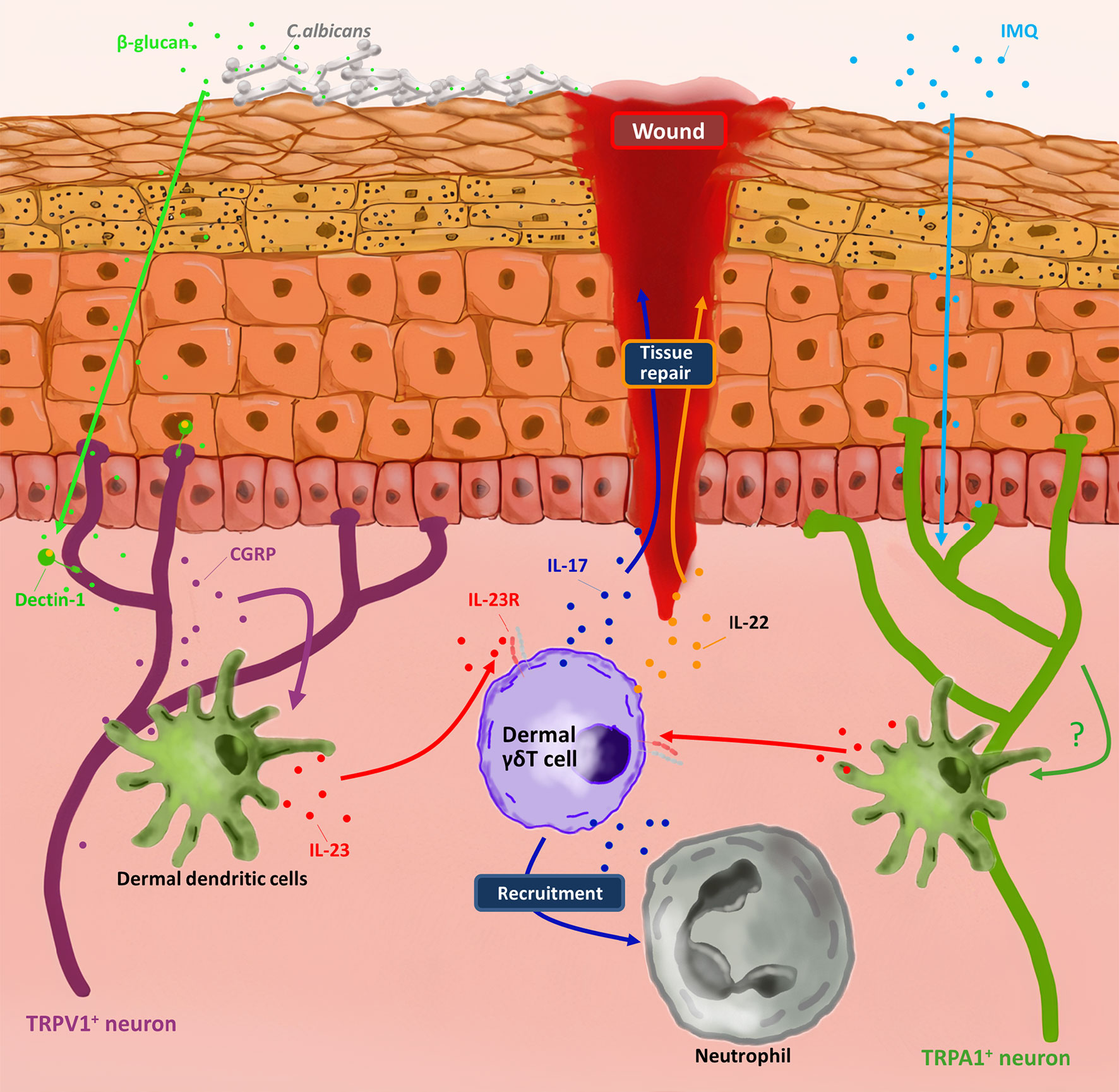
Figure 2 Dermal γδ T cells involved in nociceptors-induced skin protection. By sensing molecules such as C. albicans-derived soluble β-glucan and imiquimod (IMQ) respectively, TRPV1+ neuron and TRPA1+ neuron active dermal dendritic cells produce IL-23. In response to IL-23, dermal γδ T cells produce IL-17 and IL-22 to repair the wound and recruit neutrophils for pathogen clearance.
The crosstalk of nociceptors and γδ T cells is not always to protect the host against infection. In lethal S. aureus pneumonia mouse model, Chiu’s group found that TRPV1+neurons downregulated lung γδ T cells, resulting in a decrease in the recruitment of neutrophils that are essential for bacterial clearance. Mice ablating TRPV1+ nociceptors by RTX showed better survival rate and could increase lung γδ T cell number, in which the major increased γδ T cell subsets were Vγ1+ and Vγ1-Vγ2- subtypes (42). Although the blockade of CGRP antagonist and the ablation of TPRV1+nociceptors have the similar phenotypes in the regulation of S. aureus pneumonia, the direct evidence of how CGRP regulates γδ T cells is still missing. Moreover, the specific role of IL-17 in this model has not been well described, even though the dynamic changes of neutrophils in TPRV1+nociceptors ablated mice have indicated that IL-17 played a pivotal role during the infection. In addition, Ghasemlou’s group tried to decipher the function of γδ T cells in different pain mouse models. Compared with WT mice, TCRδ-/- mice had no differences in baseline sensitivity and mechanical or thermal hypersensitivity after injury, but with higher numbers of myeloid cells and monocytes. This finding suggested that γδ T cells did not contribute to the sensitization of inflammatory pain, but were involved in regulating the recruitment of myeloid cells and monocytes (43). Regardless, these data suggested that the mechanism of nociceptors regulating immune cells may be very complicated in different organs. A comprehensive study of how nociceptors regulate the whole immune system is required.
Neuroinflammation happens in the nervous system especially in the CNS, and is associated with most neurodegenerative disease (such as Parkinson’s disease (PD), multiple sclerosis (MS) and amyotrophic lateral sclerosis) (44–46). Several factors, such as autoimmunity, infection, injury and aging, may induce the incidence of neuroinflammation (47–49). At the beginning of neuroinflammation in the damaged tissue, recruited immune cells help to reconstruct tissue and repair neurons (50, 51). The persistent neuroinflammation results in chronic inflammation and neuronal death (52). As kick-starters of inflammation, γδ T cells participate in many neuroinflammation related diseases (53) (Table 1).
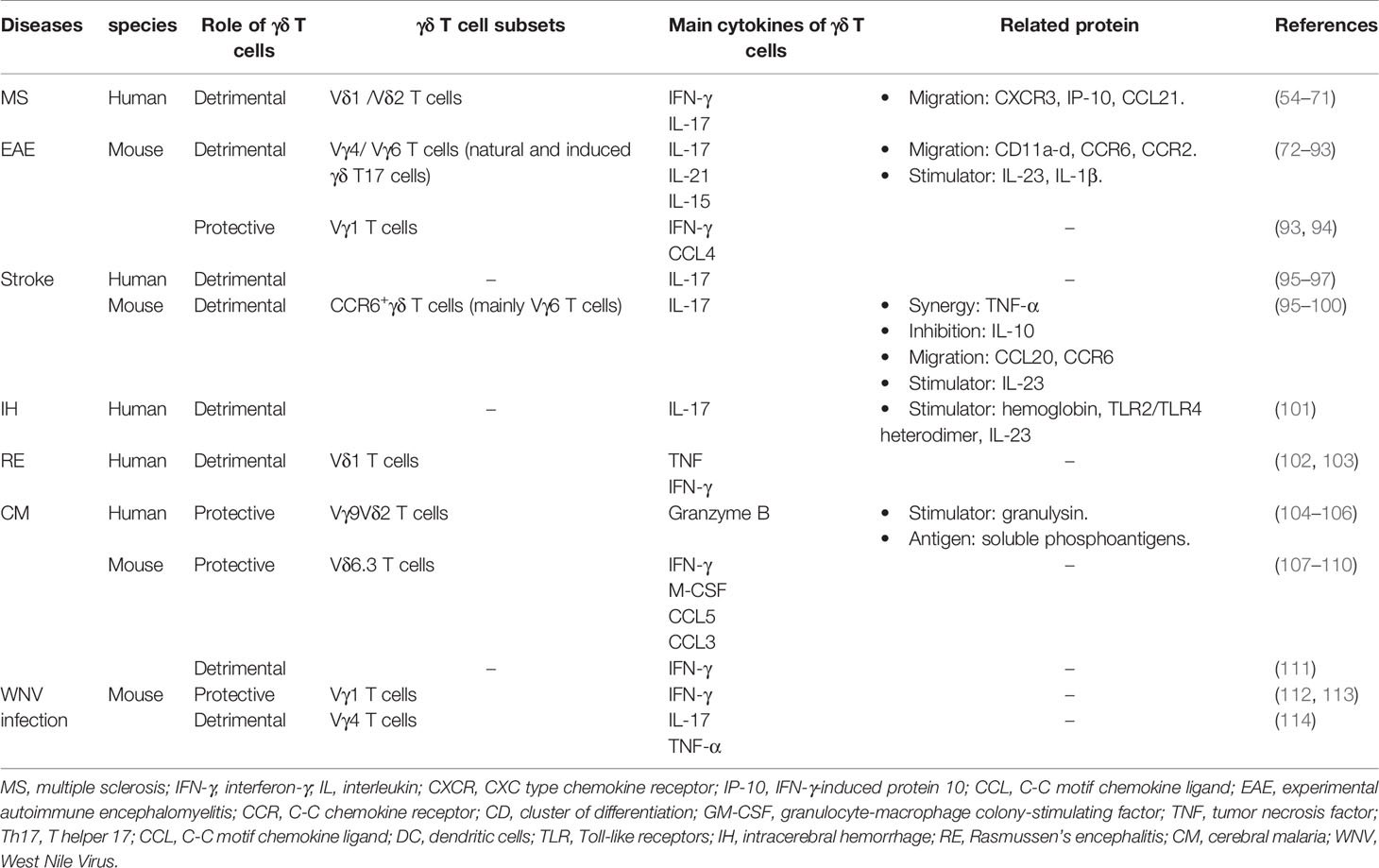
Table 1 The role of γδ T cells in neurological diseases.
As a potentially disabling disease of the CNS, multiple sclerosis (MS) is caused by the immune system attacking on protective myelin sheaths that cover neurons, eventually disabling the communication between brain and the rest of the body (45). Though many comparisons among patients with primary progressive MS, patients with relapsing-remitting MS (RRMS), healthy controls and patients with other neurological diseases have been done and the differences of the frequency of γδ T cells in the peripheral blood in patients with MS remains contradictory (54–58).While the percentage of Vδ2 T cells decreased and the percentage of Vδ1 T cells increased in peripheral blood of MS patients (58, 59), γδ T cells in MS patients expressed higher level of C-X-C Motif Chemokine Receptor 3 (CXCR3) that was related to the migration of T cells to MS plaques (60). As one of the two ligands of CXCR3, IFN-γ-induced protein 10 (IP-10) was elevated in both primary progressive MS and RRMS (61). Another ligand Chemokine (C-C motif) ligand 21 (CCL21) was remarkably reduced in the CSF during remission (62). For a better understanding of infiltrated γδ T cells in CNS, TCR repertoire analyzes were performed and the data revealed an oligo clonal expansion of γδ T cells in CNS of MS patients, suggesting these γδ T cells responded to common antigens (63–65). Along this line, the non-classical major histocompatibility complex (MHC) molecule CD1d, which could express lipid antigens to T cells, was found to be able to present myelin-derived glycosphingolipid antigen sulfatide in MS and recognized by γδ TCRs in sulfatide-specific manner (66). CD1d immunoreactivity was increased in MS, which suggested that one of early events in active phases of demyelination might be the lipid antigen presentation to γδ T cells (67). Furthermore, another potential γδ TCRs ligand, heat-shock protein 65 (HSP65), could induce γδ T cells expansion (68). Vδ1T cells could co-localize with HSP65+ oligodendrocytes within the sites of remyelination in MS lesions (69, 70). Studies further revealed that Vδ1 T cells expressed high level of IFN-γ that correlated with inflammation and nerve damage in newly diagnosed MS, whereas CD161high CCR6+ γδ T cells (a γδ T cell subset expressing IL-17 in human) were enriched and produced IL-17 in the CSF of patients during relapse (58, 71). The component and function of γδ T cells have been extensively studied in EAE mouse model.
Experimental autoimmune encephalomyelitis (EAE) is one of widely used MS animal models that shares the same pathological feature including inflammation, demyelination, axonal loss and gliosis (115). Several immune response–modifying therapies have been successfully translated from EAE studies to clinical practice for MS treatment (116). IL-17 is indicated as a key pro-inflammatory cytokine in EAE, which is secreted by T helper 17 (Th17) cells and γδ T17 cells (72–74).The two cell types cannot be replaced by one another, as a reduced EAE severity was observed in either Th17 depletion or γδ T cell deficient mice (74, 75). In the process of EAE, γδ T cells expressed CD11a-d that might be essential for γδ T cell trafficking to the CNS, as indicated by the fact that deletion three out of four CD11 molecules dramatically reduced the severity of EAE (76–78). Moreover, γδ T17 cells with a downregulation CCR6 and an upregulation C-C Motif Chemokine Receptor 2 (CCR2) promoted the migration of γδ T cells to CNS in EAE (79). Therefore, these molecules promoted the rapid infiltration of γδ T17 cells into CNS and enabled them to be involved in early inflammation in EAE. Notably, a dynamic γδ TCR repertoire analysis indicated that most of infiltrated γδ T cells at the early phase of EAE were Vγ4Vδ6 and Vγ6Vδ1 with a highly focused γδ TCR repertoire, which has been reported as natural γδ T17 cells (nγδ T17) (74, 80–82). This data was consistent with early studies that the majority of infiltrated γδ T cells in the brain and spinal cord expressed Vγ1, Vγ4 and Vγ6 at the onset of EAE, while the majority of Vγ transcripts could be detected at the later phase, suggesting that different γδ T subsets participate the process of EAE (80). In addition to thymic-derived nγδ T17, peripheral γδ T cells, especiallyVγ4+T cells, can be induced to differentiate and produce IL-17 upon IL-23 stimulation in EAE (83). Both Vγ4 and Vγ6 T cells could produce high expression levels of IL-1 receptor (IL-1R) and IL-23R to bind activated monocytes and dendritic cells secreted IL-1β and IL-23 to release IL-17 and interleukin 21 (IL-21), which could facilitate Th17 cells to produce IL-17, IL-22 and granulocyte-macrophage colony stimulating factor (GM-CSF) to exacerbate neuroinflammation (74, 83–85). IL-17 could also stimulate BBB endothelial cells, microglia and astrocytes to release multiple cytokines and chemokines to recruit neutrophils to breakdown BBB, and finally, to attract various leukocytes into the CNS (86–89). IL-23-activated γδ T cells could not only promote Th17 cells function, but also restrained the conversion of naïve T cells to Tregs and suppressed the Treg responses to enhance inflammation (74, 90). In addition, a subset of interleukin 15 (IL-15)-secreting γδ T cells was found to induce CD44high memory T cells by releasing IL-15 and help to switch memory T cells to Th17 cells to induce EAE (91). However, not all γδ T cells were inflammatory signals promoter. IFN-γ producing γδ T cells, majority of which were Vγ1 T cells, induced IFN-γ expression by encephalitogenic T cells, suppressed the activity of Th17 and released Chemokine (C-C motif) ligand 4 (CCL4) to recruit C-C Motif Chemokine Receptor 5 (CCR5) + Tregs to reduce the inflammatory signals (92, 93). Additionally, γδ T cells regulated inflammation through Fas/Fas ligand, which could induce encephalitogenic T cells apoptosis and facilitate the recovery from EAE (94). Regardless, given that the infiltrated γδ T cells highly expressed IL-17 in the CNS and amplified Th17 responses, it was recognized that γδ T cells were more pathogenic than protective, especially in the early stage of the diseases (74). Therefore, Therapies targeting IL-17, IL-17 receptor (IL-17R) or upstream cytokines IL-1β or IL-23 would not only suppress Th17 and γδ T17 cells function, but also blocked the positive feedback loop between Th17 and γδ T17 cells (117–119). Indeed, several clinical trials targeting IL-17 have already shown encouraging results in relapsing remitting MS patients (117, 120, 121). As a potential therapeutic target, γδ T cells are required for more detailed investigation.
Stroke ranks second as the leading cause of death and third as the cause of disability all over the world. As a main kind of stroke, Ischemic stroke results from the middle cerebral artery occlusion, followed by brain tissue damage in the affected territory, which is caused by inflammatory response (122). Pathogenic mechanisms of γδ T cells in stroke are mainly due to the production of IL-17 (123). In human brain tissues, immunohistochemistry staining for γδ T cells and IL-17 showed the presence of γδ T cells and the production of IL-17 shortly after stroke (95). In addition, compared to healthy control, patients with stroke have increased level of IL-17 in peripheral blood (96). In rodent models, γδ T cells, rather than Th17 cells, was found as the major IL-17 producers in ischemia-reperfusion (I/R) injury (123). In this scenario, IL-23 is found to be a key cytokine to induce IL-17 production by γδ T cells during the delayed phase of ischemia. The mice with the deficiency of IL-23 or IL-17 had significantly reduced infarct size, whereas mice treated with IL-17 neutralizing antibodies within 3 hours of stroke had a better prognosis (95, 123). Further studies illustrated that interferon regulatory factor 4 (IRF4) +/CD172a+ conventional type 2 DCs infiltrate into the ischemic brain rapidly and became the major source of IL-23 within 24 hours to stimulate CCR6+ γδ T cells (mainly Vγ6 T cells) to express IL-17 (97, 98). The absence of CD11c+ cells or the impaired IL-23 signaling could abrogate the production of IL-17 by γδ T cells (97). Genetic deficiency in Ccr6 significantly diminished the infiltration of γδ T cells, highlighting the important role that chemokine (C-C motif) ligand 20 (CCL20)/CCR6 axis plays for γδ T cell migration in stroke (98). γδ T17 cells are not the only source of IL-17, astrocyte-derived IL-17 A facilitates survival and neuronal differentiation of neural precursor cells in the recovery phase of stroke (99). After synergistic stimulation of IL-17 produced by γδ T cells and TNF-α produced by macrophages, astrocytes secrete chemokines, such as CXCL-1, to facilitate the infiltration of neutrophils, thereby inducing matrix metalloproteinase 3 (MMP3) and MMP9, which were involved in the destruction of the BBB (95, 98). Blocking the signal of IL-17 or CXCL-1/CXCR2-axis could inhibit the invasion of neutrophils and improve neurological prognosis (95). It is worth noting that intestinal γδ T17 cells could migrate to the meninges to induce ischemic neuroinflammation by producing IL-17 after stroke. Intestinal dysbiosis affected stroke through γδ T cells by inhibiting intestinal γδ T17 cells trafficking from gut to meninges (100). After the treatment of antibiotics, the altered intestinal commensal bacteria activated CD103+ DCs in mesenteric lymph node, thereby inducing Tregs expansion and secreting the anti-inflammatory cytokine interleukin 10 (IL-10), which could suppress the differentiation of γδ T17 cells in lamina propria of the small intestine (100). Interestingly, as aforementioned, most of meningeal γδ T17 cells were Vγ6Vδ1T cells and the secretion of IL-17 contributed to the physiological functions of the brain (8). Moreover, commensal microbiota might conduce to IL-17 production of meningeal γδ T cells (25). Therefore, it is interesting to know whether meningeal γδ T17 cells are the main source of infiltrated γδ T cells into the ischemic brain and how commensal microbiota affect IL-17 production of meningeal γδ T cells directly or indirectly. Moreover, since IL-17 plays a key role in the progression of stroke, it can be a therapeutic target to reduce the severity of stroke (99). Controlling commensal microbiota may also benefit for the prognosis of stroke.
The mechanical injury induced neuroinflammation in CNS is normally the outcome of BBB breakdown and inflammatory immune cells infiltration. For example, during intracerebral hemorrhage, hemoglobin from the hematoma can activate macrophages via Toll like receptor 2 (TLR2)/Toll like receptor 4(TLR4) heterodimer, which can secrete IL-23 to induce γδ T cells to produce IL-17 to aggravate secondary damage (124). Brain damage of Periventricular leukomalacia is also partially attributed to γδ T cells through the IL-17/IL-22 unrelated signaling pathways (101). While in Spinal cord injury, γδ T cells are sources of producing IFN-γ to aggravate lesions in the early phase (125). Moreover, traumatic brain injury has been linked with γδ T cells in the gut, for their increasing frequency after fluid percussion injury (126). Notably, most of mechanical injury induced neuroinflammation is pathogen free in CNS, suggesting that pattern recognition receptors (PRRs) expressed on γδ T cells are important signals for their activation. Therefore, further investigation is required to reveal how the respective and/or integrated TCRs and PRRs signals regulate γδ T cells function in CNS.
PD is a chronic neurodegenerative disease that leads to a detrimental result of the CNS, especially the motor nervous system. The most important pathological features of PD are the degeneration of dopaminergic neurons in the substantia nigra and the accumulation of unique cytoplasmic inclusions (Lewis bodies) containing α-synuclein (44). A few preliminary correlations between PD and γδ T cells have been documented clinically. Compared to some other neurological diseases and tension headache, a higher proportion of γδ T cells was observed in the CSF in patients with PD (127). The frequencies and total numbers of γδ T cells were significantly decreased in the blood of PD group than that in healthy control group (128). In addition, γδ T cells partially expressed CD25 in the CSF of PD patients whereas they hardly expressed CD25 in blood, indicating a preferential activation of γδ T cells in the CSF (127). The relation between γδ T cells and PD might rely on microglia, which serve as tissue-resident macrophages within the brain. Stimulated through TLR2, TLR4, TLR7 or TLR9, microglia can release IL-1β and IL-23 to active γδ T cells to produce IL-17 in vitro (129). And neuron-released α-synuclein could directly bind TLR2 and trigger inflammatory responses in the microglia (130). TLR2 was additionally expressed on γδ T cells and exhibited co-stimulatory effects for activated γδ T cells (131). α-synuclein may be important for γδ T cells to participate in the PD. In addition, as another main neurodegenerative disease, Alzheimer’s disease (AD) is also connected with γδT cells. Clonotypes of TCR γ chain are more specific in patients with AD and in the brain compared with that in peripheral blood (132).
Rasmussen’s encephalitis (RE), especially occurring in children under the age of 10, is a rare chronic inflammatory neurological disease without the involvement of pathogenic microorganisms that feature with progress local atrophy of the cerebral cortex on unilateral cerebral hemisphere, refractory epilepsy and cognitive impairment (133). The majority of infiltrated T cells are cytotoxic CD8+ T cells and CD4+ T cells. γδ T cells can also be found in brain and they contribute to the secretion of TNF and IFN-γ. The ratio of γδ T cells to αβ T cells is obviously higher in brain-infiltrating lymphocytes than that in peripheral blood (134). The same TCRδ1 chain with the identical third complementarity determining region (CDR3) sequences was found in the brains of RE patients, suggesting that γδ T cells might respond to the same antigen(s) and be clonally expanded. What’s more, the same γδ TCR clones were found in focal cortical dysplasia (FCD), a disease with congenital abnormality of brain development, implying that the ligands recognized by γδ TCRs were more likely to come from self-antigens rather than foreign antigens (102). Identifying the potential γδ TCR ligands may be benefit for investigating the function of γδ T cells in RE or related diseases in CNS.
As a lethal neurological complication of Plasmodium infection, cerebral malaria (CM) is responsible for the majority of child mortality (103). γδT cells can protect against Plasmodium infection by killing extracellular merozoites and intracellular late-stage parasites and regulating other lymphocytes such as αβ T cells and dendritic cells in both human and Plasmodium infection mouse model (104–109, 135). However, IFN‐γ producing γδ T cells in the liver stage of infection are responsible for experimental cerebral malaria (ECM). This proportion of liver γδ T cells promote a proinflammatory microenvironment to activate CD4+ and CD8+ T cells (110). These functional CD4+ and CD8+ T cells subsequently migrate to the brain and cause neuroinflammation resulting in ECM (110, 111). Besides, the parasites become more virulent in the presence of liver IFN‐γ producing γδ T cells to induce more pathogenic inflammation causing ECM development (110). In TCRδ-/- mice or mice injected antibody to deplete γδ T cells, the CM development can be partially inhibited (110, 136).
West Nile Virus (WNV) infection is lethal for the induction of encephalitis (137). In WNV infected mice, γδ T cells play a dual role (138). On the one hand, γδ T cells can eliminate infected cells and contribute to the maturation of DCs to prime αβ T cells (112, 114). In this case, IFN-γ producing Vγ1+ T cells are able to limit the dissemination of WNV and prevent mortal WNV encephalitis (114, 138). On the other hand, γδ T17 cells (mainly Vγ4+T cells) can suppress the proliferation of Vγ1+ T cells, produce IL-17 and TNF-α to enhance BBB permeability and finally induce encephalitis (138). Similarly, γδ T17 cells have the same detrimental effects in the infection induced neuroinflammation in the mouse model of Angiostrongylus cantonensis infection. Among them, γδ T17 cells contributed to demyelination of the brain (113).
Perinatal infection can cause cerebral white matter injury in infants. In LPS-induced sepsis of postnatal days’ mice, it was γδ T cells, rather than αβ T cells, that contributed to white matter injury and subsequent abnormal motor function (139). Taken together, as one of the earliest immune responders, γδ T cells secret variety of cytokines to defend or exacerbate the infection in CNS.
Most studies are focused on the detrimental or protective effects of γδ T cells in the diseases of nervous system. Here, we have also reviewed that the meningeal γδ T17 cells can support the short-term memory and anxiety-like behavior of mice, and the nociceptors induced activating or suppressing reactivity on dermal or lung γδ T cells. However, it is just a beginning. The reactions between γδ T cells and nervous system are far more than what these studies have reported. For example, meningeal γδ T cells will increase the expression of IL-17A to modulate anxiety-like behavior after the injection of LPS, indicating the possible link between meningeal γδ T cells and microbiota (25). It is interesting to know the role of meningeal γδ T cells play in gut-brain axis crosstalk. In addition, nociceptors regulate immune system (including γδ T cells) to respond infection (32). In this condition, the details of how neurons interact with immune cells are still missing. Therefore, further investigations about γδ T cells and behaviors, neuron-immune interactions in various disease models are required. More systematic researches need to be performed to reveal their relationships.
YL and XZ drafted the main body of this manuscript. YZ modified the manuscript. XZ takes primary responsibility for this paper as the corresponding author. All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China General Program (31870899).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
TCR, T-cell receptors; MHC, major histocompatibility complex; IFN, interferon; IL, interleukin; IGF, insulin-like growth factor; CXCR, CXC type chemokine receptor; CXCL, C-X-C Motif Chemokine Ligand; CCL, C-C motif chemokine ligand; CCR, C-C chemokine receptor; IP-10, IFN-γ-induced protein 10; CD, cluster of differentiation; GM-CSF, granulocyte-macrophage colony-stimulating factor; TNF, tumor necrosis factor; BBB, blood-brain barrier; BDNF, neurotrophic factor; WT, wild type; mPFC, medial prefrontal cortex; S1DZ, somatosensory cortex; GABA, gamma-aminobutyric acid; PNS, peripheral nervous system; TRPV1, transient receptor potential vanilloid subfamily member 1; TRPA1, transient receptor potential ankyrin 1; NLRP3, NLR family pyrin domain containing 3; IRF, interferon regulatory factor; IMQ, imiquimod; DC, dendritic cells; RTX, resiniferatoxin; CGRP, calcitonin gene–related peptide; PD, Parkinson’s disease; MS, multiple sclerosis; RRMS, relapsing-remitting MS; EAE, experimental autoimmune encephalomyelitis; Th17, T helper 17; CSF, Cerebrospinal fluid; HSP, heat-shock protein; CNS, central nervous system; FCD, focal cortical dysplasia; CDR3, complementarity determining region; IH, intracerebral hemorrhage; CM, cerebral malaria; ECM, experimental cerebral malaria; RE, Rasmussen’s encephalitis; TLR, Toll-like receptors; PPR, pattern recognition receptor; MMP, matrix metalloproteinase; WNV, West Nile Virus; LPS, lipopolysaccharide.
1. Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature (1988) 334(6181):395–402. doi: 10.1038/334395a0
2. Chien YH, Meyer C, Bonneville M. gammadelta T cells: first line of defense and beyond. Annu Rev Immunol (2014) 32:121–55. doi: 10.1146/annurev-immunol-032713-120216
3. Born WK, Kemal Aydintug M, O’Brien RL. Diversity of gammadelta T-cell antigens. Cell Mol Immunol (2013) 10(1):13–20. doi: 10.1038/cmi.2012.45
4. Borst J, Vroom TM, Bos JD, Van Dongen JJM. Tissue Distribution and Repertoire Selection of Human γδT Cells: Comparison With the Murine System. In: Pfeffer K, Heeg K, Wagner H, Riethmüller G, editors. Function and Specificity of γ/δ T Cells: International Workshop, Schloß Elmau, Bavaria, FRG October 14–16, 1990. Berlin, Heidelberg: Springer Berlin Heidelberg (1991). p. 41–6.
5. Munoz-Ruiz M, Sumaria N, Pennington DJ, Silva-Santos B. Thymic Determinants of gammadelta T Cell Differentiation. Trends Immunol (2017) 38(5):336–44. doi: 10.1016/j.it.2017.01.007
6. Ohga S, Yoshikai Y, Takeda Y, Hiromatsu K, Nomoto K. Sequential appearance of gamma/delta- and alpha/beta-bearing T cells in the peritoneal cavity during an i.p. infection with Listeria monocytogenes. Eur J Immunol (1990) 20(3):533–8. doi: 10.1002/eji.1830200311
7. Johnson MD, Witherden DA, Havran WL. The Role of Tissue-resident T Cells in Stress Surveillance and Tissue Maintenance. Cells (2020) 9(3):686. doi: 10.3390/cells9030686
8. Ribeiro M, Brigas HC, Temido-Ferreira M, Pousinha PA, Regen T, Santa C, et al. Meningeal γδ T cell-derived IL-17 controls synaptic plasticity and short-term memory. Sci Immunol (2019) 4(40):eaay5199. doi: 10.1126/sciimmunol.aay5199
9. Carding SR, Egan PJ. Gammadelta T cells: functional plasticity and heterogeneity. Nat Rev Immunol (2002) 2(5):336–45. doi: 10.1038/nri797
10. Muro R, Takayanagi H, Nitta T. T cell receptor signaling for γδT cell development. Inflamm Regen (2019) 39:6. doi: 10.1186/s41232-019-0095-z
11. Zhao Y, Lin L, Xiao Z, Li M, Wu X, Li W, et al. Protective Role of gammadelta T Cells in Different Pathogen Infections and Its Potential Clinical Application. J Immunol Res (2018) 2018:5081634. doi: 10.1155/2018/5081634
12. Sebestyen Z, Prinz I, Dechanet-Merville J, Silva-Santos B, Kuball J. Translating gammadelta (gammadelta) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov (2020) 19(3):169–84. doi: 10.1038/s41573-019-0038-z
13. Shiromizu CM, Jancic CC. gammadelta T Lymphocytes: An Effector Cell in Autoimmunity and Infection. Front Immunol (2018) 9:2389. doi: 10.3389/fimmu.2018.02389
14. Hayday AC. gammadelta T Cell Update: Adaptate Orchestrators of Immune Surveillance. J Immunol (2019) 203(2):311–20. doi: 10.4049/jimmunol.1800934
15. Sharp LL, Jameson JM, Cauvi G, Havran WL. Dendritic epidermal T cells regulate skin homeostasis through local production of insulin-like growth factor 1. Nat Immunol (2005) 6(1):73–9. doi: 10.1038/ni1152
16. Hu B, Jin C, Zeng X, Resch JM, Jedrychowski MP, Yang Z, et al. γδ T cells and adipocyte IL-17RC control fat innervation and thermogenesis. Nature (2020) 578(7796):610–4. doi: 10.1038/s41586-020-2028-z
17. Rössler K, Neuchrist C, Kitz K, Scheiner O, Kraft D, Lassmann H. Expression of leucocyte adhesion molecules at the human blood-brain barrier (BBB). J Neurosci Res (1992) 31(2):365–74. doi: 10.1002/jnr.490310219
18. Bauer H-C, Krizbai IA, Bauer H, Traweger A. “You Shall Not Pass”-tight junctions of the blood brain barrier. Front Neurosci (2014) 8:392. doi: 10.3389/fnins.2014.00392
19. Rustenhoven J, Kipnis J. Bypassing the blood-brain barrier. Science (American Association for the Advancement of Science) (2019) 366: (6472):1448–9. doi: 10.1126/science.aay0479
20. Brombacher TM, Nono JK, De Gouveia KS, Makena N, Darby M, Womersley J, et al. IL-13-Mediated Regulation of Learning and Memory. J Immunol (2017) 198(7):2681–8. doi: 10.4049/jimmunol.1601546
21. Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, et al. Unexpected role of interferon-gamma in regulating neuronal connectivity and social behaviour. Nature (2016) 535(7612):425–9. doi: 10.1038/nature18626
22. Maher FO, Nolan Y, Lynch MA. Downregulation of IL-4-induced signalling in hippocampus contributes to deficits in LTP in the aged rat. Neurobiol Aging (2005) 26(5):717–28. doi: 10.1016/j.neurobiolaging.2004.07.002
23. Reed MD, Yim YS, Wimmer RD, Kim H, Ryu C, Welch GM, et al. IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature (2020) 577(7789):249–53. doi: 10.1038/s41586-019-1843-6
24. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science (2016) 351(6276):933–9. doi: 10.1126/science.aad0314
25. Alves de Lima K, Rustenhoven J, Da Mesquita S, Wall M, Salvador AF, Smirnov I, et al. Meningeal gammadelta T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat Immunol (2020) 21(11):1421–9. doi: 10.1038/s41590-020-0776-4
26. Haas JD, Ravens S, Duber S, Sandrock I, Oberdorfer L, Kashani E, et al. Development of interleukin-17-producing gammadelta T cells is restricted to a functional embryonic wave. Immunity (2012) 37(1):48–59. doi: 10.1016/j.immuni.2012.06.003
27. Hatano S, Tun X, Noguchi N, Yue D, Yamada H, Sun X, et al. Development of a new monoclonal antibody specific to mouse Vgamma6 chain. Life Sci Alliance (2019) 2(3):e201900363. doi: 10.26508/lsa.201900363
28. Sarma JD, Ciric B, Marek R, Sadhukhan S, Caruso ML, Shafagh J, et al. Functional interleukin-17 receptor A is expressed in central nervous system glia and upregulated in experimental autoimmune encephalomyelitis. J Neuroinflamm (2009) 6(1):14. doi: 10.1186/1742-2094-6-14
29. Kemter AM, Nagler CR. Influences on allergic mechanisms through gut, lung, and skin microbiome exposures. J Clin Invest (2019) 129(4):1483–92. doi: 10.1172/JCI124610
30. Ellison DL. Physiology of Pain. Crit Care Nurs Clin North Am (2017) 29(4):397–406. doi: 10.1016/j.cnc.2017.08.001
31. Dubin AE, Patapoutian A. Nociceptors: the sensors of the pain pathway. J Clin Invest (2010) 120(11):3760–72. doi: 10.1172/JCI42843
32. Chu C, Artis D, Chiu IM. Neuro-immune Interactions in the Tissues. Immunity (2020) 52(3):464–74. doi: 10.1016/j.immuni.2020.02.017
33. Pinho-Ribeiro FA, Verri WA, Jr, Chiu IM. Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol (2017) 38(1):5–19. doi: 10.1016/j.it.2016.10.001
34. Lowes MA, Russell CB, Martin DA, Towne JE, Krueger JG. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol (2013) 34(4):174–81. doi: 10.1016/j.it.2012.11.005
35. Riol-Blanco L, Ordovas-Montanes J, Perro M, Naval E, Thiriot A, Alvarez D, et al. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature (2014) 510(7503):157–61. doi: 10.1038/nature13199
36. Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L, Kaplan DH. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity (2015) 43(3):515–26. doi: 10.1016/j.immuni.2015.08.016
37. Cohen JA, Edwards TN, Liu AW, Hirai T, Jones MR, Wu J, et al. Cutaneous TRPV1(+) Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell (2019) 178(4):919–32.e14. doi: 10.1016/j.cell.2019.06.022
38. Maruyama K, Takayama Y, Kondo T, Ishibashi KI, Sahoo BR, Kanemaru H, et al. Nociceptors Boost the Resolution of Fungal Osteoinflammation via the TRP Channel-CGRP-Jdp2 Axis. Cell Rep (2017) 19(13):2730–42. doi: 10.1016/j.celrep.2017.06.002
39. Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature (2013) 501(7465):52–7. doi: 10.1038/nature12479
40. Pinho-Ribeiro FA, Baddal B, Haarsma R, O’Seaghdha M, Yang NJ, Blake KJ, et al. Blocking Neuronal Signaling to Immune Cells Treats Streptococcal Invasive Infection. Cell (2018) 173(5):1083–97.e22. doi: 10.1016/j.cell.2018.04.006
41. Wei JJ, Kim HS, Spencer CA, Brennan-Crispi D, Zheng Y, Johnson NM, et al. Activation of TRPA1 nociceptor promotes systemic adult mammalian skin regeneration. Sci Immunol (2020) 5: (50):eaba5683. doi: 10.1126/sciimmunol.aba5683
42. Baral P, Umans BD, Li L, Wallrapp A, Bist M, Kirschbaum T, et al. Nociceptor sensory neurons suppress neutrophil and gammadelta T cell responses in bacterial lung infections and lethal pneumonia. Nat Med (2018) 24(4):417–26. doi: 10.1038/nm.4501
43. Petrovic J, Silva JR, Bannerman CA, Segal JP, Marshall AS, Haird CM, et al. gammadelta T Cells Modulate Myeloid Cell Recruitment but Not Pain During Peripheral Inflammation. Front Immunol (2019) 10:473. doi: 10.3389/fimmu.2019.00473
44. De Virgilio A, Greco A, Fabbrini G, Inghilleri M, Rizzo MI, Gallo A, et al. Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun Rev (2016) 15(10):1005–11. doi: 10.1016/j.autrev.2016.07.022
45. Bjelobaba I, Savic D, Lavrnja I. Multiple Sclerosis and Neuroinflammation: The Overview of Current and Prospective Therapies. Curr Pharm Des (2017) 23(5):693–730. doi: 10.2174/1381612822666161214153108
46. Liu J, Wang F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front Immunol (2017) 8:1005. doi: 10.3389/fimmu.2017.01005
47. Gendelman HE. Neural immunity: Friend or foe? J NeuroVirol (2002) 8(6):474–9. doi: 10.1080/13550280290168631
48. Licastro F, Candore G, Lio D, Porcellini E, Colonna-Romano G, Franceschi C, et al. Innate immunity and inflammation in ageing: a key for understanding age-related diseases. Immun Ageing (2005) 2(1):8. doi: 10.1186/1742-4933-2-8
49. Tohidpour A, Morgun AV, Boitsova EB, Malinovskaya NA, Martynova GP, Khilazheva ED, et al. Neuroinflammation and Infection: Molecular Mechanisms Associated with Dysfunction of Neurovascular Unit. Front Cell Infect Microbiol (2017) 7:276. doi: 10.3389/fcimb.2017.00276
50. Shaked I, Porat Z, Gersner R, Kipnis J, Schwartz M. Early activation of microglia as antigen-presenting cells correlates with T cell-mediated protection and repair of the injured central nervous system. J Neuroimmunol (2004) 146(1):84–93. doi: 10.1016/j.jneuroim.2003.10.049
51. Subhramanyam CS, Wang C, Hu Q, Dheen ST. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol (2019) 94:112–20. doi: 10.1016/j.semcdb.2019.05.004
52. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms Underlying Inflammation in Neurodegeneration. Cell (2010) 140(6):918–34. doi: 10.1016/j.cell.2010.02.016
53. Papotto PH, Ribot JC, Silva-Santos B. IL-17+ γδ T cells as kick-starters of inflammation. Nat Immunol (2017) 18(6):604–11. doi: 10.1038/ni.3726
54. Stinissen P, Vandevyver C, Medaer R, Vandegaer L, Nies J, Tuyls L, et al. Increased frequency of gamma delta T cells in cerebrospinal fluid and peripheral blood of patients with multiple sclerosis. Reactivity, cytotoxicity, and T cell receptor V gene rearrangements. J Immunol (1995) 154(9):4883–94.
55. Nick S, Pileri P, Tongiani S, Uematsu Y, Kappos L, De Libero G. T cell receptor gamma delta repertoire is skewed in cerebrospinal fluid of multiple sclerosis patients: molecular and functional analyses of antigen-reactive gamma delta clones. Eur J Immunol (1995) 25(2):355–63. doi: 10.1002/eji.1830250208
56. Paź A, Fiszer U, Zaborski J, Korlak J, Członkowski A, Członkowska A. Phenotyping analysis of peripheral blood leukocytes in patients with multiple sclerosis. Eur J Neurol (1999) 6(3):347–52. doi: 10.1046/j.1468-1331.1999.630347.x
57. Ramos S, Brenu E, Broadley S, Kwiatek R, Ng J, Nguyen T, et al. Regulatory T, natural killer T and γδ T cells in multiple sclerosis and chronic fatigue syndrome/myalgic encephalomyelitis: a comparison. Asian Pac J Allergy Immunol (2016) 34(4):300–5. doi: 10.12932/AP0733
58. Singh AK, Novakova L, Axelsson M, Malmeström C, Zetterberg H, Lycke J, et al. High Interferon-γ Uniquely in Vδ1 T Cells Correlates with Markers of Inflammation and Axonal Damage in Early Multiple Sclerosis. Front Immunol (2017) 8:260. doi: 10.3389/fimmu.2017.00260
59. Maimaitijiang G, Shinoda K, Nakamura Y, Masaki K, Matsushita T, Isobe N, et al. Association of Decreased Percentage of Vδ2(+)Vγ9(+) γδ T Cells With Disease Severity in Multiple Sclerosis. Front Immunol (2018) 9:748. doi: 10.3389/fimmu.2018.00748
60. Murzenok PP, Matusevicius D, Freedman MS. gamma/delta T cells in multiple sclerosis: chemokine and chemokine receptor expression. Clin Immunol (Orlando Fla) (2002) 103(3 Pt 1):309–16. doi: 10.1006/clim.2001.5213
61. Scarpini E, Galimberti D, Baron P, Clerici R, Ronzoni M, Conti G, et al. IP-10 and MCP-1 levels in CSF and serum from multiple sclerosis patients with different clinical subtypes of the disease. J Neurol Sci (2002) 195(1):41–6. doi: 10.1016/S0022-510X(01)00680-3
62. Villoslada P, Edwards KR, Goyal J, Plavina T, Czerkowicz J, Goelz S, et al. Feasibility of the Use of Combinatorial Chemokine Arrays to Study Blood and CSF in Multiple Sclerosis. PLoS One (2013) 8(11):e81007. doi: 10.1371/journal.pone.0081007
63. Bieganowski P, Bieganowska K, Zaborski J, Członkowska A. Oligoclonal expansion of gamma delta T cells in cerebrospinal fluid of multiple sclerosis patients. Mult Scler (Houndmills Basingstoke England) (1996) 2(2):78–82. doi: 10.1177/135245859600200203
64. Shimonkevitz R, Colburn C, Burnham JA, Murray RS, Kotzin BL. Clonal expansions of activated gamma/delta T cells in recent-onset multiple sclerosis. Proc Natl Acad Sci USA (1993) 90(3):923–7. doi: 10.1073/pnas.90.3.923
65. Liedtke W, Meyer G, Faustmann PM, Warnatz H, Raine CS. Clonal expansion and decreased occurrence of peripheral blood gamma delta T cells of the V delta 2J delta 3 lineage in multiple sclerosis patients. Int Immunol (1997) 9(7):1031–41. doi: 10.1093/intimm/9.7.1031
66. Bai L, Picard D, Anderson B, Chaudhary V, Luoma A, Jabri B, et al. The majority of CD1d-sulfatide-specific T cells in human blood use a semiinvariant Vδ1 TCR. Eur J Immunol (2012) 42(9):2505–10. doi: 10.1002/eji.201242531
67. Muir FGW, Samadi-Bahrami Z, Moore GRW, Quandt JA. Expression of CD1d by astrocytes corresponds with relative activity in multiple sclerosis lesions. Brain Pathol (2020) 30(1):26–35. doi: 10.1111/bpa.12733
68. Hisaeda H, Nagasawa H, Maeda K, Maekawa Y, Ishikawa H, Ito Y, et al. Gamma delta T cells play an important role in hsp65 expression and in acquiring protective immune responses against infection with Toxoplasma gondii. J Immunol (1995) 155(1):244–51.
69. Selmaj K, Brosnan CF, Raine CS. Colocalization of lymphocytes bearing gamma delta T-cell receptor and heat shock protein hsp65+ oligodendrocytes in multiple sclerosis. Proceedings of the National Academy of Sciences - PNAS (1991) 88: (15):6452–6. doi: 10.1073/pnas.88.15.6452
70. Selmaj K, Brosnan CF, Raine CS. Expression of heat shock protein-65 by oligodendrocytes in vivo and in vitro: implications for multiple sclerosis. Neurology (1992) 42(4):795–800. doi: 10.1212/WNL.42.4.795
71. Schirmer L, Rothhammer V, Hemmer B, Korn T. Enriched CD161high CCR6+ γδ T cells in the cerebrospinal fluid of patients with multiple sclerosis. JAMA Neurol (2013) 70(3):345–51. doi: 10.1001/2013.jamaneurol.409
72. Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol (2006) 177(1):566–73. doi: 10.4049/jimmunol.177.1.566
73. Yan Y, Ding X, Li K, Ciric B, Wu S, Xu H, et al. CNS-specific therapy for ongoing EAE by silencing IL-17 pathway in astrocytes. Mol Ther J Am Soc Gene Ther (2012) 20(7):1338–48. doi: 10.1038/mt.2012.12
74. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 Induce Innate IL-17 Production from γδ T Cells, Amplifying Th17 Responses and Autoimmunity. Immunity (2009) 31(2):331–41. doi: 10.1016/j.immuni.2009.08.001
75. Liu X, Lee YS, Yu CR, Egwuagu CE. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol (2008) 180(9):6070–6. doi: 10.4049/jimmunol.180.9.6070
76. Smith SS, Barnum SR. Differential expression of beta 2-integrins and cytokine production between gammadelta and alphabeta T cells in experimental autoimmune encephalomyelitis. J Leuk Biol (2008) 83(1):71–9. doi: 10.1189/jlb.0407263
77. Bullard DC, Hu X, Adams JE, Schoeb TR, Barnum SR. p150/95 (CD11c/CD18) Expression Is Required for the Development of Experimental Autoimmune Encephalomyelitis. Am J Pathol (2007) 170(6):2001–8. doi: 10.2353/ajpath.2007.061016
78. Gordon EJ, Myers KJ, Dougherty JP, Rosen H, Ron Y. Both anti-CD11a (LFA-1) and anti-CD11b (MAC-1) therapy delay the onset and diminish the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol (1995) 62(2):153–60. doi: 10.1016/0165-5728(95)00120-2
79. McKenzie DR, Kara EE, Bastow CR, Tyllis TS, Fenix KA, Gregor CE, et al. IL-17-producing γδ T cells switch migratory patterns between resting and activated states. Nat Commun (2017) 8(1):15632. doi: 10.1038/ncomms15632
80. Olive C. γδ T cell receptor variable region usage during the development of experimental allergic encephalomyelitis. J Neuroimmunol (1995) 62(1):1–8. doi: 10.1016/0165-5728(95)00081-C
81. Saligrama N, Zhao F, Sikora MJ, Serratelli WS, Fernandes RA, Louis DM, et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature (2019) 572(7770):481–7. doi: 10.1038/s41586-019-1467-x
82. Wei Y-L, Han A, Glanville J, Fang F, Zuniga LA, Lee JS, et al. A Highly Focused Antigen Receptor Repertoire Characterizes γδ T Cells That are Poised to Make IL-17 Rapidly in Naive Animals. Front Immunol (2015) 6:118. doi: 10.3389/fimmu.2015.00118
83. Papotto PH, Gonçalves-Sousa N, Schmolka N, Iseppon A, Mensurado S, Stockinger B, et al. IL-23 drives differentiation of peripheral γδ17 T cells from adult bone marrow-derived precursors. EMBO Rep (2017) 18(11):1957–67. doi: 10.15252/embr.201744200
84. Akitsu A, Ishigame H, Kakuta S, Chung S-H, Ikeda S, Shimizu K, et al. IL-1 receptor antagonist-deficient mice develop autoimmune arthritis due to intrinsic activation of IL-17-producing CCR2(+)Vγ6(+)γδ T cells. Nat Commun (2015) 6:7464. doi: 10.1038/ncomms8464
85. Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti T-D. Inflammasome-Derived IL-1β Regulates the Production of GM-CSF by CD4+ T Cells and γδ T Cells. J Immunol(1950) (2012) 188: (7):3107–15. doi: 10.4049/jimmunol.1103308
86. Setiadi AF, Abbas AR, Jeet S, Wong K, Bischof A, Peng I, et al. IL-17A is associated with the breakdown of the blood-brain barrier in relapsing-remitting multiple sclerosis. J Neuroimmunol (2019) 332:147–54. doi: 10.1016/j.jneuroim.2019.04.011
87. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J (2010) 24(4):1023–34. doi: 10.1096/fj.09-141978
88. Zimmermann J, Krauthausen M, Hofer MJ, Heneka MT, Campbell IL, Müller M. CNS-targeted production of IL-17A induces glial activation, microvascular pathology and enhances the neuroinflammatory response to systemic endotoxemia. PLoS One (2013) 8(2):e57307–e. doi: 10.1371/journal.pone.0057307
89. Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med (2008) 205(7):1535–41. doi: 10.1084/jem.20080159
90. Petermann F, Rothhammer V, Claussen MC, Haas JD, Blanco LR, Heink S, et al. γδ T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity (2010) 33(3):351–63. doi: 10.1016/j.immuni.2010.08.013
91. Wang X, Wei Y, Liu X, Xing C, Han G, Chen G, et al. IL-15-secreting γδT cells induce memory T cells in experimental allergic encephalomyelitis (EAE) mice. Mol Immunol (2015) 66(2):402–8. doi: 10.1016/j.molimm.2015.04.021
92. Blink SE, Caldis MW, Goings GE, Harp CT, Malissen B, Prinz I, et al. γδ T cell subsets play opposing roles in regulating experimental autoimmune encephalomyelitis. Cell Immunol (2014) 290(1):39–51. doi: 10.1016/j.cellimm.2014.04.013
93. Ponomarev ED, Novikova M, Yassai M, Szczepanik M, Gorski J, Dittel BN. γδ T Cell Regulation of IFN-γ Production by Central Nervous System-Infiltrating Encephalitogenic T Cells: Correlation with Recovery from Experimental Autoimmune Encephalomyelitis. J Immunol (1950) (2004) 173: (3):1587–95. doi: 10.4049/jimmunol.173.3.1587
94. Ponomarev ED, Dittel BN. Gamma delta T cells regulate the extent and duration of inflammation in the central nervous system by a Fas ligand-dependent mechanism. J Immunol (2005) 174(8):4678–87. doi: 10.4049/jimmunol.174.8.4678
95. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood (2012) 120(18):3793–802. doi: 10.1182/blood-2012-02-412726
96. Hu Y, Zheng Y, Wu Y, Ni B, Shi S. Imbalance between IL-17A-producing cells and regulatory T cells during ischemic stroke. Mediators Inflamm (2014) 2014:813045. doi: 10.1155/2014/813045
97. Gelderblom M, Gallizioli M, Ludewig P, Thom V, Arunachalam P, Rissiek B, et al. IL-23 (Interleukin-23)-Producing Conventional Dendritic Cells Control the Detrimental IL-17 (Interleukin-17) Response in Stroke. Stroke (2018) 49(1):155–64. doi: 10.1161/STROKEAHA.117.019101
98. Arunachalam P, Ludewig P, Melich P, Arumugam TV, Gerloff C, Prinz I, et al. CCR6 (CC Chemokine Receptor 6) Is Essential for the Migration of Detrimental Natural Interleukin-17-Producing gammadelta T Cells in Stroke. Stroke (2017) 48(7):1957–65. doi: 10.1161/STROKEAHA.117.016753
99. Lin Y, Zhang JC, Yao CY, Wu Y, Abdelgawad AF, Yao SL, et al. Critical role of astrocytic interleukin-17 A in post-stroke survival and neuronal differentiation of neural precursor cells in adult mice. Cell Death Dis (2016) 7(6):e2273–e. doi: 10.1038/cddis.2015.284
100. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med (2016) 22(5):516–23. doi: 10.1038/nm.4068
101. Albertsson AM, Zhang X, Vontell R, Bi D, Bronson RT, Supramaniam V, et al. gammadelta T Cells Contribute to Injury in the Developing Brain. Am J Pathol (2018) 188(3):757–67. doi: 10.1016/j.ajpath.2017.11.012
102. Owens GC, Erickson KL, Malone CC, Pan C, Huynh MN, Chang JW, et al. Evidence for the involvement of gamma delta T cells in the immune response in Rasmussen encephalitis. J Neuroinflamm (2015) 12:134. doi: 10.1186/s12974-015-0352-2
103. Luzolo AL, Ngoyi DM. Cerebral malaria. Brain Res Bull (2019) 145:53–8. doi: 10.1016/j.brainresbull.2019.01.010
104. Hernández-Castañeda MA, Happ K, Cattalani F, Wallimann A, Blanchard M, Fellay I, et al. γδ T Cells Kill Plasmodium falciparum in a Granzyme- and Granulysin-Dependent Mechanism during the Late Blood Stage. J Immunol (2020) 204(7):1798–809. doi: 10.4049/jimmunol.1900725
105. Howard J, Loizon S, Tyler CJ, Duluc D, Moser B, Mechain M, et al. The Antigen-Presenting Potential of Vγ9Vδ2 T Cells During Plasmodium falciparum Blood-Stage Infection. J Infect Dis (2017) 215(10):1569–79. doi: 10.1093/infdis/jix149
106. Mamedov MR, Scholzen A, Nair RV, Cumnock K, Kenkel JA, Oliveira JHM, et al. A Macrophage Colony-Stimulating-Factor-Producing γδ T Cell Subset Prevents Malarial Parasitemic Recurrence. Immunity (2018) 48(2):350–63.e7. doi: 10.1016/j.immuni.2018.01.009
107. Kopacz J, Kumar N. gamma delta T-cells may interfere with a productive immune response in Plasmodium yoelii infections. Int J Parasitol (1999) 29(5):737–42. doi: 10.1016/S0020-7519(99)00026-0
108. Seixas E, Fonseca L, Langhorne J. The influence of gammadelta T cells on the CD4+ T cell and antibody response during a primary Plasmodium chabaudi chabaudi infection in mice. Parasit Immunol (2002) 24(3):131–40. doi: 10.1046/j.1365-3024.2002.00446.x
109. Inoue S, Niikura M, Takeo S, Mineo S, Kawakami Y, Uchida A, et al. (2012). Enhancement of dendritic cell activation via CD40 ligand-expressing γδ T cells is responsible for protective immunity to Plasmodium parasites, in: Proceedings of the National Academy of Sciences of the United States of America, , Vol. 109. pp. 12129–34.
110. Ribot JC, Neres R, Zuzarte-Luís V, Gomes AQ, Mancio-Silva L, Mensurado S, et al. (2019). γδ-T cells promote IFN-γ-dependent Plasmodium pathogenesis upon liver-stage infection, in: Proceedings of the National Academy of Sciences of the United States of America, , Vol. 116. pp. 9979–88.
111. Villegas-Mendez A, Greig R, Shaw TN, de Souza JB, Gwyer Findlay E, Stumhofer JS, et al. IFN-γ-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J Immunol (2012) 189(2):968–79. doi: 10.4049/jimmunol.1200688
112. Fang H, Welte T, Zheng X, Chang GJ, Holbrook MR, Soong L, et al. gammadelta T cells promote the maturation of dendritic cells during West Nile virus infection. FEMS Immunol Med Microbiol (2010) 59(1):71–80. doi: 10.1111/j.1574-695X.2010.00663.x
113. Feng Y, Zhou Z, Zheng C, Feng F, Xie F, Wu Z. Interleukin 17-Producing γδ T Cell Induced Demyelination of the Brain in Angiostrongylus Cantonensis Infection. Res Square (2020). doi: 10.21203/rs.3.rs-19711/v1
114. Wang T, Scully E, Yin Z, Kim JH, Wang S, Yan J, et al. IFN-gamma-producing gamma delta T cells help control murine West Nile virus infection. J Immunol (2003) 171(5):2524–31. doi: 10.4049/jimmunol.171.5.2524
115. Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol (2011) 164(4):1079–106. doi: 10.1111/j.1476-5381.2011.01302.x
116. Baker D, Amor S. Mouse models of multiple sclerosis: lost in translation? Curr Pharm Des (2015) 21(18):2440–52. doi: 10.2174/1381612821666150316122706
117. Havrdová E, Belova A, Goloborodko A, Tisserant A, Wright A, Wallstroem E, et al. Activity of secukinumab, an anti-IL-17A antibody, on brain lesions in RRMS: results from a randomized, proof-of-concept study. J Neurol (2016) 263(7):1287–95. doi: 10.1007/s00415-016-8128-x
118. Campa M, Mansouri B, Warren R. Menter A. A Review of Biologic Therapies Targeting IL-23 and IL-17 for Use in Moderate-to-Severe Plaque Psoriasis. Dermatol Ther (2016) 6(1):1–12. doi: 10.1007/s13555-015-0092-3
119. Chevalier X, Eymard F. Anti-IL-1 for the treatment of OA: dead or alive? Nat Rev Rheumatol (2019) 15(4):191–2. doi: 10.1038/s41584-019-0185-y
120. Bittner S, Wiendl H. Neuroimmunotherapies Targeting T Cells: From Pathophysiology to Therapeutic Applications. Neurotherapeutics (2016) 13(1):4–19. doi: 10.1007/s13311-015-0405-3
121. Orthmann-Murphy JL, Calabresi PA. Therapeutic Application of Monoclonal Antibodies in Multiple Sclerosis. Clin Pharmacol Ther (2017) 101(1):52–64. doi: 10.1002/cpt.547
122. Campbell BCV, De Silva DA, Macleod MR, Coutts SB, Schwamm LH, Davis SM, et al. Ischaemic stroke. Nat Rev Dis Primers (2019) 5(1):70. doi: 10.1038/s41572-019-0118-8
123. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med (2009) 15(8):946–50. doi: 10.1038/nm.1999
124. Zhong Q, Zhou K, Liang QL, Lin S, Wang YC, Xiong XY, et al. Interleukin-23 Secreted by Activated Macrophages Drives gammadeltaT Cell Production of Interleukin-17 to Aggravate Secondary Injury After Intracerebral Hemorrhage. J Am Heart Assoc (2016) 5(10):e004340. doi: 10.1161/JAHA.116.004340
125. Sun G, Yang S, Cao G, Wang Q, Hao J, Wen Q, et al. gammadelta T cells provide the early source of IFN-gamma to aggravate lesions in spinal cord injury. J Exp Med (2018) 215(2):521–35. doi: 10.1084/jem.20170686
126. Newell-Rogers MK, Rogers SK, Tobin RP, Mukherjee S, Shapiro LA. Antagonism of Macrophage Migration Inhibitory Factory (MIF) after Traumatic Brain Injury Ameliorates Astrocytosis and Peripheral Lymphocyte Activation and Expansion. Int J Mol Sci (2020) 21(20):7448. doi: 10.3390/ijms21207448
127. Fiszer U, Mix E, Fredrikson S, Kostulas V, Olsson T, Link H. gamma delta+ T cells are increased in patients with Parkinson’s disease. J Neurol Sci (1994) 121(1):39–45. doi: 10.1016/0022-510X(94)90154-6
128. Zhou C, Zhou X, He D, Li Z, Xie X, Ren Y. Reduction of Peripheral Blood iNKT and gammadeltaT Cells in Patients With Parkinson’s Disease: An Observational Study. Front Immunol (2020) 11:1329. doi: 10.3389/fimmu.2020.01329
129. Derkow K, Kruger C, Dembny P, Lehnardt S. Microglia Induce Neurotoxic IL-17+ gammadelta T Cells Dependent on TLR2, TLR4, and TLR9 Activation. PlLoS One (2015) 10(8):e0135898. doi: 10.1371/journal.pone.0135898
130. Kim C, Ho D-H, Suk J-E, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun (2013) 4:1562–. doi: 10.1038/ncomms2534
131. Zhang J, Wang J, Pang L, Xie G, Welte T, Saxena V, et al. The co-stimulatory effects of MyD88-dependent Toll-like receptor signaling on activation of murine γδ T cells. PLoS One (2014) 9(9):e108156. doi: 10.1371/journal.pone.0108156
132. Aliseychik M, Patrikeev A, Gusev F, Grigorenko A, Andreeva T, Biragyn A, et al. Dissection of the Human T-Cell Receptor γ Gene Repertoire in the Brain and Peripheral Blood Identifies Age- and Alzheimer’s Disease-Associated Clonotype Profiles. Front Immunol (2020) 11:12. doi: 10.3389/fimmu.2020.00012
133. Varadkar S, Bien CG, Kruse CA, Jensen FE, Bauer J, Pardo CA, et al. Rasmussen’s encephalitis: clinical features, pathobiology, and treatment advances. Lancet Neurol (2014) 13(2):195–205. doi: 10.1016/S1474-4422(13)70260-6
134. Al Nimer F, Jelcic I, Kempf C, Pieper T, Budka H, Sospedra M, et al. Phenotypic and functional complexity of brain-infiltrating T cells in Rasmussen encephalitis. Neurol Neuroimmunol Neuroinflamm (2018) 5(1):e419. doi: 10.1212/NXI.0000000000000419
135. Costa G, Loizon S, Guenot M, Mocan I, Halary F, de Saint-Basile G, et al. Control of Plasmodium falciparum erythrocytic cycle: γδ T cells target the red blood cell-invasive merozoites. Blood (2011) 118(26):6952–62. doi: 10.1182/blood-2011-08-376111
136. Yañez DM, Batchelder J, van der Heyde HC, Manning DD, Weidanz WP. Gamma delta T-cell function in pathogenesis of cerebral malaria in mice infected with Plasmodium berghei ANKA. Infect Immun (1999) 67(1):446–8. doi: 10.1128/IAI.67.1.446-448.1999
137. Rossi SL, Ross TM, Evans JD. West Nile virus. Clinics Lab Med (2010) 30(1):47–65. doi: 10.1016/j.cll.2009.10.006
138. Welte T, Lamb J, Anderson JF, Born WK, O’Brien RL, Wang T. Role of two distinct γδ T cell subsets during West Nile virus infection. FEMS Immunol Med Microbiol (2008) 53(2):275–83. doi: 10.1111/j.1574-695X.2008.00430.x
Keywords: γδ T cells, neuroinflammation, nociceptors, short-term memory, anxiety, IL-17
Citation: Li Y, Zhang Y and Zeng X (2021) γδ T Cells Participating in Nervous Systems: A Story of Jekyll and Hyde. Front. Immunol. 12:656097. doi: 10.3389/fimmu.2021.656097
Received: 20 January 2021; Accepted: 15 March 2021;
Published: 31 March 2021.
Edited by:
Jennifer K. Dowling, Royal College of Surgeons in Ireland, IrelandReviewed by:
Michael Levy, Massachusetts General Hospital and Harvard Medical School, United StatesCopyright © 2021 Li, Zhang and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xun Zeng, eHVuemVuZ0B6anUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.