
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 15 March 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.656039
This article is part of the Research Topic Immune Dysfunction: An Update of New Immune Cell Subsets and Cytokines in Sepsis View all 15 articles
 Jingjing Ji1,2
Jingjing Ji1,2 Jie Fan1,3,4*
Jie Fan1,3,4*Sepsis is life-threatening organ dysfunction caused by a dysregulated host response to infection. During the development and progression of sepsis, polymorphonuclear neutrophils (PMNs) are the most abundantly recruited innate immune cells at sites of infection, playing critical roles in the elimination of local infection and healing of the injury. PMN reverse migration (rM) describes the phenomenon in which PMNs migrate away from the inflammatory site back into the vasculature following the initial PMN infiltration. The functional role of PMN rM within inflammatory scenarios requires further exploration. Current evidence suggests that depending on the context, PMN rM can be both a protective response, by facilitating an efficient resolution to innate immune reaction, and also a tissue-damaging event. In this review, we provide an overview of current advancements in understanding the mechanism and roles of PMN rM in inflammation and sepsis. A comprehensive understanding of PMN rM may allow for the development of novel prophylactic and therapeutic strategies for sepsis.
Polymorphonuclear neutrophils (PMNs) are the first responders in the circulation, playing an important role in defending against invading pathogens (1). Being attracted by a chemokine concentration gradient, PMNs migrate from the bloodstream into the inflamed extravascular tissues (2, 3). In the inflammatory site, PMNs eliminate pathogen through degranulation, phagocytosis, the formation of neutrophil extracellular traps (NETs), and releasing cytokines (3, 4). After PMNs execute their antimicrobial agenda, timely clearance of PMNs is crucial to maintain homeostasis (2, 5, 6). Traditionally, apoptosis or necrosis and subsequent phagocytosis by macrophages were considered as the main ways for PMN clearance (7, 8). However, with the development of imaging technology, it has been found that the recruited PMNs could migrate back to the circulation, which serves as a new way of PMN clearance in inflammatory or injury site (8, 9). In 1997, Hughes et al. using a rat glomerular capillary injury model and radiolabeling of PMN found that over 70% of PMNs that entered inflamed glomerular capillaries were able to return to the main circulation without undergoing apoptosis in the inflammatory site (8, 10). The process of PMN migrating back to circulation has been referred to as “PMN reverse migration (rM)” to describe the general phenomenon of PMN moving in the opposite direction to that expected (11). The in vivo visualization of PMN rM was first carried out on zebrafish larvae in 2006 (12). Mathias et al. found that not all recruited PMNs died at the site of injury, and most PMNs left the site and back to circulation (13). In the same year, Buckley et al. observed in vitro that human PMNs could reverse transmigrate through the tumor necrosis factor-α (TNF-α)-activated endothelial monolayer (14). Further studies showed that reverse migrated PMNs are characterized by high expression of intercellular adhesion molecule-1 (ICAM-1 high) and low expression of C-X-C motif chemokine receptor 1 (CXCR1 low), which are different from the PMN in circulation with ICAM-1 low/CXCR1 high or those resident in tissue with ICAM-1 high/CXCR1 high (15, 16). The markers of ICAM-1 high/CXCR1 low are then found also valid for the reverse migrated PMNs in the peripheral blood of patients with systemic inflammation (14). In 2017, in vivo PMN rM in mice was visualized by Wang et al. in a sterile thermal hepatic injury model, in which reverse migrated PMNs from the inflammatory site were imaged in the lungs and bone marrow (17). The published data support the note that PMN rM is an important biological conservative phenomenon existing in from zebrafish to humans (11, 14, 17–21).
The infiltration of PMNs from circulation to an inflammatory site is a regulated multi-stage process, as summarized in several review articles (2, 6, 22). PMNs first recognize inflammatory signs by sensing chemokines, e.g., macrophage-inflammatory protein-2 (MIP-2) and keratinocyte-derived chemokine (KC) (23, 24), followed by several processes, including capture, rolling, firm adhesion, and transendothelial migration, to reach the site of inflammation (25, 26). Corresponding to these steps, PMN rM has been considered as a continuous multi-stage process as well. Sussan et al. proposed terminologies to describe cell reverse migration, including reverse abluminal crawling (rAC), reverse interstitial migration (rIM), reverse luminal crawling (rLC), and reverse transendothelial cell migration (rTEM) (11). These terminologies imply the mechanisms by which PMN rM occurs.
The mechanisms that mediate PMN rM from inflammatory sites remain largely unclear. Many factors, which involve in PMN forward migration, such as chemoattractant and chemotactic repellents, chemokine receptors, the interaction between PMN and endothelia, and alteration of PMN behavior, are also considered as main factors regulating PMN rM (Figure 1).
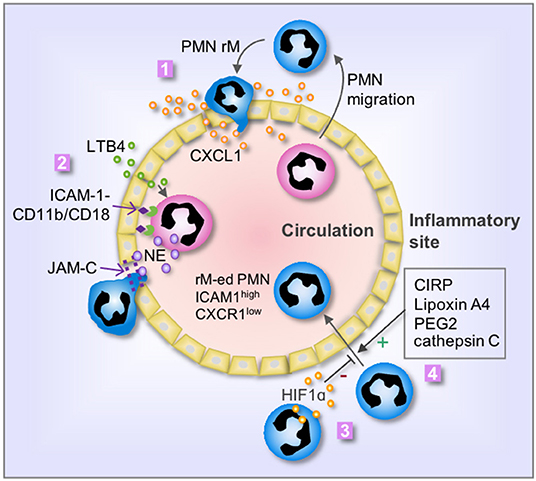
Figure 1. Mechanisms of PMN rM. 1. Breach of endothelium results in leakage of chemokine, i.e., CXCL1, from the inflammatory site into the vasculature, therefore driving PMN to reenter the circulation; 2. LTB4 induces PMN release of neutrophil elastase (NE), which in turn cleaves endothelial JAM-C and subsequent damage of endothelial junction and promotes PMN rM. The binding between PMN CD11b/CD18 and endothelial ICAM-1 retains the PMN on the surface of endothelial cells and secures the cleavage of JAM-C by NE; 3. Activation of HIF1α suppresses PMN rM; and 4. Many factors, including cold-inducible RNA-binding protein (CIRP), Lipoxin A4, PEG2, and cathepsin C, can promote PMN rM. The phenotype of reverse migrated PMN (rM-ed PMN) is ICAM1high CXCR1low.
During the PMN forward migration, PMNs are chemoattracted by chemokines, and the migration direction is controlled by the concentration gradient of the chemoattractant. Endothelium breach is an important event in the PMN migration from circulation to the inflammatory area (27, 28). Similarly, the alteration of endothelial junction and permeability also play an important role in the mechanism of PMN rM. Owen-Woods et al. revealed the importance of endothelial permeability in regulating PMN trafficking (29). Inflammation usually damages the endothelial junctions and increases endothelial permeability, thereby, resulting in leakage of chemokines from the inflammatory site to circulation. This leakage of chemokines may reverse the directional cues of PMN migration across venular walls (30). For example, the leakage of chemokine and PMN chemoattractant, CXCL1, impairs the chemoattractant gradient and confuses PMNs and droves PMNs reenter the systemic circulation (29, 31). Regarding the mechanism underlying endothelial junction damage, Colom et al. reported that neutrophil elastase (NE) cleaves endothelial junctional adhesion molecule C (JAM-C), therefore, promoting PMN rM (32). This process is mediated by leukotriene B4 (LTB4). The study showed that LTB4 exhibits high efficacy in promoting rapid NE release and/or cell-surface expression, which then mediates the cleavage of endothelial JAM-C (32). Decreasing LTB4 concentration or blocking LTB4 receptor could decrease PMN rM in a mouse model of acute pancreatitis (33). Hirano et al. found that inhibition of JAM-C degradation reduces PMN rM in septic mice (34). The study further showed that the soluble JAM-C concentration in circulation is positively correlated with the PMN rM, and thus, increased plasma soluble JAM-C level may serve as an indicator for PMN rM occurring (32).
PMN transepithelial migration is an important event during PMN migration. However, the study on the mechanism of reverse PMN transepithelial migration is lacking in the literature. Future studies are needed to explore the mechanism since reverse PMN transepithelial migration is an indispensable piece of the puzzle for a comprehensive understanding of PMN rM.
Studies have shown that some chemokines and chemokine receptors are critically involved in PMN rM. CXCR1 and CXCR2 are the main receptors on PMNs to sense chemokines during cell migration. The study showed that reverse migrated PMNs exhibit decreased CXCR1 (14). Therefore, it was speculated that those PMNs may lose sensitivity to chemokine cues and move in a reverse direction. However, the direct evidence is still missing. Powell et al. reported that CXCL8a and CXCR2 are required for PMN rM in zebrafish, evidenced by that CXCR2 knockout in zebrafish decreased PMN rM (35). Wang et al. reported that the PMNs that reentered into circulation entailed a sojourn in the lungs, where they up-regulated CXCR4 expression followed by homing back to the bone marrow. The PMNs trafficking back to bone marrow exhibit increased Annexin V in the membrane, suggesting that those PMNs may undergo apoptosis in bone marrow (17).
PMN forward migration to the inflammatory site requires an important step of adhesion to endothelium, which is mediated through the binding between PMN Cd18/Cd11b and endothelial ICAM-1. During PMN rM, Colom et al. found that the binding between Cd18/Cd11b and ICAM-1 secures JAM-C cleavage by neutrophil elastase, which subsequently promotes PMN rM (32).
Some studies revealed negative regulation of PMN rM. For example, the role for hypoxia-inducible factor 1 subunit alpha (HIF1α) in suppressing PMN rM in zebrafish has been reported (36). Activation of HIF1α reduced PMN rM and delayed inflammation resolution in zebrafish; whereas, administration of HIF1α inhibitor enhanced PMN rM (36). Besides, Wang et al. found that reducing serine protease activity by using cathepsin C deficient mice led to a profound reduction in PMN rM (17). Considering that PMNs reenter into circulation followed by migration into the lungs and sequentially homing back to bone marrow, PMN rM is more likely a programmed process, rather than a random event.
Chemotactic repellent in the inflammatory site is another mechanistic hypothesis for PMN rM (8). CXCL8 is an effective chemokine in human. It has been found in vitro that CXCL8 functions as a chemoattractant in lower concentrations (at the nM level), whereas, in higher concentrations (at the μM level), CXCL8 plays a role as chemorepellent to promote PMN rM (37). Loynes et al. reported that eicosanoid prostaglandin E2 (PGE2) can also act as a chemorepellent that drives PMN rM in zebrafish (38). In the late stage of inflammation, pathways of production of pro-inflammatory mediators are usually shifted to the production of pro-resolution mediators, i.e. lipoxin A4 (LXA4) (39, 40). It has been found in a microfluidic device that LXA4 enhances human PMN rM, suggesting that PMN rM may serve as a partial mechanism of inflammation resolution (41). Macrophages also play a role in regulating PMN rM (42). It has been observed in zebrafish that macrophage depletion decreased PMN rM and resulted in continuous neutrophilic inflammation (38). Redox-Src family kinase (SFK) signaling in macrophages seems important in mediating macrophage-induced PMN rM, although redox-SFK also involves in regulating PMN forward migration (43).
Collectively, published studies underscore the complexity and diversity of mechanisms driving PMN rM, highlighting the need to better understand PMN rM characteristics, prevalence, and downstream implications.
Both PMN infiltration in tissue and timely clearance are important for maintaining homeostasis, as concluded previously (9, 44). Persistent and excessive PMN infiltration is responsible for many chronic diseases, such as pulmonary fibrosis and rheumatoid arthritis, etc. (45, 46). The functional role of PMN rM in inflammatory scenarios is controversial and requires further exploration. Current evidence suggests that depending on the context, PMN rM can be both a protective response, by facilitating an efficient resolution to innate immune reaction, and also a tissue-damaging event (8, 13, 17, 22, 32, 47–49). Removal of activated PMNs from the inflammatory sites through PMN rM may alleviate the local inflammatory response. On the other aspect, however, activated PMNs that migrate back to circulation may result in dissemination of inflammation (32, 47, 50). The different outcomes may be related to the different disease models, severity, and timing.
Nonetheless, clearance of infiltrated PMNs from the injury or infection sites is essential for inflammation resolution. Most studies in zebrafish found that PMN rM plays a protective role in promoting inflammation resolution. Several interventions aiming to inhibit PMN rM, such as macrophage depletion and activation of HIF1α, aggravated the damage of the wound due to the persistent neutrophilic infiltration (36, 38). Similar results have also been observed in a mouse model of sterile thermal hepatic injury. Using photoactivatable-GFP transgenic mice, Wang et al. visualized the migration of PMNs back into the vasculature in a sterile thermal hepatic injury. They found that reducing serine protease activity by using cathepsin C–deficient mice (Ctsc−/−) led to a profound reduction in PMNs reentering healthy patent vessels, and further resulted in delayed revascularization. Though the mechanism was unclear, Ctsc deficiency only decreased PMN rM, but did not affect PMN infiltration, suggesting that Ctsc involved in inflammation resolution (17).
On the other aspect, data from mammal experiments showed that activated PMNs migrating back to circulation contributes to inflammation dissemination and distant organ dysfunction. In a murine model of cremaster muscle ischemia-reperfusion, inhibition of PMN rM by knockout NE or JAM-C resulted in the alleviation of the remote lung, heart, and liver injury, suggesting that those reverse migrated PMNs may contribute to the distant organ injury (32, 47). The mechanism of how the reverse migrated PMNs lead to distant organ damage remains unclear. This may relate to the interaction between reverse migrated PMNs and circulating cells. Studies have shown that the PMN subsets, which present similar phenotype with reverse migrated PMNs, modulate T cell functions, as summarized by Hirano et al. (51). PMNs with increased cell surface expression of CD11b, CD11c, CD16, and CD54 demonstrate the ability to suppress T cell proliferation in a Cd18/Cd11b dependent manner (51).
It is noticeable that reverse migrated PMNs have an altered phenotype consisting of increased expression of ICAM-1 and effector functions, e.g., increased ROS generation (14, 47). Expression of ICAM-1 on the surface of inactivated PMNs is usually low. PMN expression of ICAM-1 can be induced by chemokines, and thus, infiltrated PMNs in the site of injury or infection present upregulated expression of ICAM-1 (49, 50). Whether the high expressed ICAM-1 involves in the mechanism of PMN rM is under investigation in the authors' laboratory.
Sepsis is life-threatening organ dysfunction caused by a dysregulated host response to infection (52). In the progression of sepsis, PMNs are the most abundantly recruited immune cells at sites of infection, playing critical roles in the elimination of local infection and the healing of injury (53–55). Although the mechanisms underlying sepsis development and progression remain to be fully addressed, it is well-accepted that immune imbalance serves as a critical mechanism of the development and progression of sepsis, in which PMNs play important roles (56, 57). PMNs execute their functions to eliminate the pathogen mainly through degranulation, phagocytosis, the formation of neutrophil extracellular traps (NETs), and release of cytokines. These mechanisms, however, may induce tissue damage as well (54, 55).
Theoretically, timely removal of PMNs in a resolution phase from the inflamed site should benefit local inflammation. However, active PMNs that migrate back to systemic circulation may disseminate inflammatory responses to remote organs and tissue. In the development of sepsis, pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) are the early initiators; and PMNs play an important role in further amplifying the inflammation during the progression of sepsis (8, 58). Inflammatory mediators released by PMNs can exaggerate other innate immune cell activation, including PMN itself (8, 59). Reverse migrated PMNs present a pro-inflammatory phenotype, including increased production of superoxide and high membrane expression of ICAM-1 (47, 60). Ode et al found that ICAM-1 positive PMNs express higher levels of inducible nitric oxide synthase (iNOS) and NETs, suggesting that reverse migrated PMNs are still highly active (60, 61). In addition, reverse migrated PMNs show prolonged lifespan and delayed apoptosis (62, 63), which might contribute to persistent and amplified inflammation (64). Studies using the mouse abdominal infection model revealed that the ratio of reverse migrated PMNs in circulation shows a positive correlation with acute lung injury (33, 50, 60). A study using the cecal ligation and puncture (CLP) mouse model showed that cold-inducible RNA-binding protein (CIRP) knockout mice exhibited decreased PMN rM and lung injury. After administration of recombinant CIRP, the reverse migrated PMNs significantly increased in the blood in the time- and dose-dependent manner, and NE expression was upregulated, while JAM-C expression was downregulated in the lungs. These results suggest that CIRP promotes PMN rM and subsequent acute lung injury by increasing NE and decreasing JAM-C (50). Interestingly, the CIRP-induced PMN rM occurred in septic mice can be suppressed by neutralizing antibody against TLR4 and inhibitor for NF-κB. In CLP mice, CIRP-TLR4 interaction in PMNs leads to increased PMN rM through the NF-κB pathway. The reverse migrated PMNs produce excessive iNOS and NETs and promote tissue inflammation and injury (60). Li et al. using another sepsis model, the acute pancreatitis model, revealed that LTB4 production promoted PMN rM, and this effect was mediated by substance P. Substance P treatment increase phosphorylation of protein kinase C (PKC) α and mitogen- activated protein kinases (MAPKs), which further promoted LTB4 production. Blocking the leukotriene B4 receptor 1(LTB4R1) resulted in the decreased PMN rM into the circulation and alleviated the severity of acute lung injury (33).
Although the data on the role of PMN rM in sepsis are limited and it is too early to conclude, the published studies tend to suggest a detrimental effect of PMN rM on the development of remote organ injury in sepsis.
The mechanism of sepsis remains to be fully elucidated, which results in poor therapeutic outcomes for septic patients. PMNs have become an important target for preventive and therapeutic interventions. Despite advances in understanding PMN biology over the last several decades, there remains a significant gap in our knowledge regarding various PMN functions and behavior in sepsis. PMN rM describes the phenomenon in which PMNs migrate away from the inflammatory site back into the vasculature following the initial PMN infiltration. The functional role of PMN rM within inflammatory scenarios, particularly in sepsis, requires further exploration. Current evidence suggests that depending on the context, PMN rM can be both a protective response, by facilitating an efficient resolution to innate immune reaction, and also a tissue-damaging event through the dissemination of inflammation. The investigation on the mechanism of PMN rM is still in a premature stage. Numerous questions are still open in the research of PMN rM. For example, what are the mediators in the circulation that chemoattract the PMNs leaving from the inflammatory sites? Whether the reverse migrated PMNs are a particular subset of PMNs? What are the different functions between the infiltrating PMNs and reverse migrated PMNs? Where is the final destination and fate of the reverse migrated PMNs? Nonetheless, the potential physiological and pathological roles of PMN rM emphasize the importance of gaining more in-depth insight into these phenomena, since these may serve as novel means of modulating inflammation and treatment of sepsis.
JJ collected the data and drafted the manuscript. JJ and JF conceived and designed the study. JF reviewed and finalized the manuscript. Both authors read and approved the final manuscript.
This work was supported by the USA National Institutes of Health Grant R01-HL-079669 (JF), USA National Institutes of Health Grant R01HL076179 (JF), USA National Institutes of Health Grant R01-HL-139547 (JF), VA Merit Award 1I01BX002729 (JF), and VA BLR&D Award 1IK6BX004211 (JF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
CIRP, Cold-inducible RNA-binding protein; CLP, Cecal ligation and puncture; Ctsc, Cathepsin C; CXCL1, C-X-C motif chemokine ligand 1; CXCL8a, Chemokine (C-X-C motif) ligand 8a; CXCR1, C-X-C motif chemokine receptor 1; CXCR2, C-X-C motif chemokine receptor 2; CXCR4, C-X-C motif chemokine receptor 4; DAMPs, Damage associated molecular patterns; HIF1α, Hypoxia inducible factor 1 subunit alpha; ICAM1, Intercellular adhesion molecule-1; iNOS, inducible nitric oxide synthase; JAM-C, Junctional adhesion molecule C; KC, Keratinocyte-derived chemokine; LTB4, leukotriene B4; LTB4R1, leukotriene B4 receptor 1; LXA4, lipoxin A4; MAPKs, Mitogen activated protein kinases; MIP-2, Macrophage-inflammatory protein-2; NE, Neutrophil elastase; NETs, Neutrophil Extracellular Traps; NF-κB, Nuclear factor kappa B subunit 1; PAMPs, Pathogen-associated molecular patterns; PGE2, Prostaglandin E2; PKC, Phosphorylation of protein kinase C; PMN, Polymorphonuclear neutrophils; rAC, reverse abluminal crawling; rIM, reverse interstitial migration; rLC, reverse luminal crawling; rM, reverse migration; rTEM, reverse transendothelial-cell migration; SFK, Src family kinase; TLR4, Toll-like receptor 4; TNF-α, Tumor necrosis factor-α.
1. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
2. Langereis JD. Neutrophil integrin affinity regulation in adhesion, migration, and bacterial clearance. Cell Adh Migr. (2013) 7:476–81. doi: 10.4161/cam.27293
3. Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol. (2011) 32:452–60. doi: 10.1016/j.it.2011.06.008
4. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
5. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. (2010) 31:318–24. doi: 10.1016/j.it.2010.05.006
6. Injarabian L, Devin A, Ransac S, Marteyn BS. Neutrophil metabolic shift during their lifecycle: impact on their survival and activation. Int J Mol Sci. (2019) 21:287. doi: 10.3390/ijms21010287
7. Pittman K, Kubes P. Damage-associated molecular patterns control neutrophil recruitment. J Innate Immun. (2013) 5:315–23. doi: 10.1159/000347132
8. de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. (2016) 16:378–91. doi: 10.1038/nri.2016.49
9. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
10. Hughes J, Johnson RJ, Mooney A, Hugo C, Gordon K, Savill J. Neutrophil fate in experimental glomerular capillary injury in the rat. Emigration exceeds in situ clearance by apoptosis. Am J Pathol. (1997) 150:223–34.
11. Nourshargh S, Renshaw SA, Imhof BA. Reverse migration of neutrophils: where, when, how, and why? Trends Immunol. (2016) 37:273–86. doi: 10.1016/j.it.2016.03.006
12. Starnes TW, Huttenlocher A. Neutrophil reverse migration becomes transparent with zebrafish. Adv Hematol. (2012) 2012:398640. doi: 10.1155/2012/398640
13. Mathias JR, Perrin BJ, Liu T-X, Kanki J, Look AT, Huttenlocher A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol. (2006) 80:1281–8. doi: 10.1189/jlb.0506346
14. Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol. (2006) 79:303–11. doi: 10.1189/jlb.0905496
15. Silvestre-Roig C, Hidalgo A, Soehnlein O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood. (2016) 127:2173–81. doi: 10.1182/blood-2016-01-688887
16. Powell DR, Huttenlocher A. Neutrophils in the tumor microenvironment. Trends Immunol. (2016) 37:41–52. doi: 10.1016/j.it.2015.11.008
17. Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. (2017) 358:111–6. doi: 10.1126/science.aam9690
18. Yoo SK, Huttenlocher A. Spatiotemporal photolabeling of neutrophil trafficking during inflammation in live zebrafish. J Leukoc Biol. (2011) 89:661–7. doi: 10.1189/jlb.1010567
19. Hall C, Flores MV, Chien A, Davidson A, Crosier K, Crosier P. Transgenic zebrafish reporter lines reveal conserved Toll-like receptor signaling potential in embryonic myeloid leukocytes and adult immune cell lineages. J Leukoc Biol. (2009) 85:751–65. doi: 10.1189/jlb.0708405
20. Burn T, Alvarez JI. Reverse transendothelial cell migration in inflammation: to help or to hinder? Cell Mol Life Sci. (2017) 74:1871–81. doi: 10.1007/s00018-016-2444-2
21. Wu D, Zeng Y, Fan Y, Wu J, Mulatibieke T, Ni J, et al. Reverse-migrated neutrophils regulated by JAM-C are involved in acute pancreatitis-associated lung injury. Sci Rep. (2016) 6:20545. doi: 10.1038/srep20545
22. Hind LE, Huttenlocher A. Neutrophil reverse migration and a chemokinetic resolution. Dev Cell. (2018) 47:404–5. doi: 10.1016/j.devcel.2018.11.004
23. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
24. Li L, Chen K, Xiang Y, Yoshimura T, Su S, Zhu J, et al. New development in studies of formyl-peptide receptors: critical roles in host defense. J Leukoc Biol. (2016) 99:425–35. doi: 10.1189/jlb.2RI0815-354RR
25. Finger EB, Puri KD, Alon R, Lawrence MB, von Andrian UH, Springer TA. Adhesion through L-selectin requires a threshold hydrodynamic shear. Nature. (1996) 379:266–9. doi: 10.1038/379266a0
26. Lawrence MB, Kansas GS, Kunkel EJ, Ley K. Threshold levels of fluid shear promote leukocyte adhesion through selectins (CD62L,P,E). J Cell Biol. (1997) 136:717–27. doi: 10.1083/jcb.136.3.717
27. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. (2015) 15:692–704. doi: 10.1038/nri3908
28. Weninger W, Biro M, Jain R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nat Rev Immunol. (2014) 14:232–46. doi: 10.1038/nri3641
29. Owen-Woods C, Joulia R, Barkaway A, Rolas L, Ma B, Nottebaum AF, et al. Local microvascular leakage promotes trafficking of activated neutrophils to remote organs. J Clin Invest. (2020) 130:2301–18. doi: 10.1172/JCI133661
30. Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J Cell Biol. (1993) 120:1371–80. doi: 10.1083/jcb.120.6.1371
31. Marki A, Ley K. Leaking chemokines confuse neutrophils. J Clin Invest. (2020) 130:2177–9. doi: 10.1172/JCI136259
32. Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, et al. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity. (2015) 42:1075–86. doi: 10.1016/j.immuni.2015.05.010
33. Li B, Han X, Ye X, Ni J, Wu J, Dai J, et al. Substance P-regulated leukotriene B4 production promotes acute pancreatitis-associated lung injury through neutrophil reverse migration. Int Immunopharmacol. (2018) 57:147–56. doi: 10.1016/j.intimp.2018.02.017
34. Hirano Y, Ode Y, Ochani M, Wang P, Aziz M. Targeting junctional adhesion molecule-C ameliorates sepsis-induced acute lung injury by decreasing CXCR4+ aged neutrophils. J Leukoc Biol. (2018) 104:1159–71. doi: 10.1002/JLB.3A0218-050R
35. Powell D, Tauzin S, Hind LE, Deng Q, Beebe DJ, Huttenlocher A. Chemokine signaling and the regulation of bidirectional leukocyte migration in interstitial tissues. Cell Rep. (2017) 19:1572–85. doi: 10.1016/j.celrep.2017.04.078
36. Elks PM, van Eeden FJ, Dixon G, Wang X, Reyes-Aldasoro CC, Ingham PW, et al. Activation of hypoxia-inducible factor-1α (Hif-1α) delays inflammation resolution by reducing neutrophil apoptosis and reverse migration in a zebrafish inflammation model. Blood. (2011) 118:712–22. doi: 10.1182/blood-2010-12-324186
37. Tharp WG, Yadav R, Irimia D, Upadhyaya A, Samadani A, Hurtado O, et al. Neutrophil chemorepulsion in defined interleukin-8 gradients in vitro and in vivo. J Leukoc Biol. (2006) 79:539–54. doi: 10.1189/jlb.0905516
38. Loynes CA, Lee JA, Robertson AL, Steel MJ, Ellett F, Feng Y, et al. PGE2 production at sites of tissue injury promotes an anti-inflammatory neutrophil phenotype and determines the outcome of inflammation resolution in vivo. Sci Adv. (2018) 4:eaar8320. doi: 10.1126/sciadv.aar8320
39. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. (2001) 2:612–9. doi: 10.1038/89759
40. Serhan CN, Chiang N, Dalli J, Levy BD. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol. (2014) 7:a016311. doi: 10.1101/cshperspect.a016311
41. Hamza B, Wong E, Patel S, Cho H, Martel J, Irimia D. Retrotaxis of human neutrophils during mechanical confinement inside microfluidic channels. Integr Biol (Camb). (2014) 6:175–83. doi: 10.1039/C3IB40175H
42. Bystrom J, Evans I, Newson J, Stables M, Toor I, van Rooijen N, et al. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood. (2008) 112:4117–27. doi: 10.1182/blood-2007-12-129767
43. Tauzin S, Starnes TW, Becker FB, Lam P, Huttenlocher A. Redox and Src family kinase signaling control leukocyte wound attraction and neutrophil reverse migration. J Cell Biol. (2014) 207:589–98. doi: 10.1083/jcb.201408090
44. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. (2005) 6:1191–7. doi: 10.1038/ni1276
45. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol. (2014) 10:593–601. doi: 10.1038/nrrheum.2014.80
46. Cantin AM, Hartl D, Konstan MW, Chmiel JF. Inflammation in cystic fibrosis lung disease: pathogenesis and therapy. J Cyst Fibros. (2015) 14:419–30. doi: 10.1016/j.jcf.2015.03.003
47. Woodfin A, Voisin M-B, Beyrau M, Colom B, Caille D, Diapouli F-M, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. (2011) 12:761–9. doi: 10.1038/ni.2062
48. Robertson AL, Holmes GR, Bojarczuk AN, Burgon J, Loynes CA, Chimen M, et al. A zebrafish compound screen reveals modulation of neutrophil reverse migration as an anti-inflammatory mechanism. Sci Transl Med. (2014) 6:225ra29. doi: 10.1126/scitranslmed.3007672
49. Thompson AAR, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood. (2014) 123:366–76. doi: 10.1182/blood-2013-05-500207
50. Jin H, Aziz M, Ode Y, Wang P. CIRP induces neutrophil reverse transendothelial migration in sepsis. Shock. (2019) 51:548–56. doi: 10.1097/SHK.0000000000001257
51. Hirano Y, Aziz M, Wang P. Role of reverse transendothelial migration of neutrophils in inflammation. Biol Chem. (2016) 397:497–506. doi: 10.1515/hsz-2015-0309
52. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
53. Seymour CW, Rosengart MR. Septic shock: advances in diagnosis and treatment. JAMA. (2015) 314:708–17. doi: 10.1001/jama.2015.7885
54. Shen X-F, Cao K, Jiang J-P, Guan W-X, Du J-F. Neutrophil dysregulation during sepsis: an overview and update. J Cell Mol Med. (2017) 21:1687–97. doi: 10.1111/jcmm.13112
55. Zhang F, Liu A-L, Gao S, Ma S, Guo S-B. Neutrophil dysfunction in sepsis. Chin Med J. (2016) 129:2741–4. doi: 10.4103/0366-6999.193447
56. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. (2016) 126:23–31. doi: 10.1172/JCI82224
57. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ. (2016) 353:i1585. doi: 10.1136/bmj.i1585
58. Lämmermann T. In the eye of the neutrophil swarm-navigation signals that bring neutrophils together in inflamed and infected tissues. J Leukoc Biol. (2016) 100:55–63. doi: 10.1189/jlb.1MR0915-403
59. Sadik CD, Luster AD. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukoc Biol. (2012) 91:207–15. doi: 10.1189/jlb.0811402
60. Ode Y, Aziz M, Wang P. CIRP increases ICAM-1+ phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J Leukoc Biol. (2018) 103:693–707. doi: 10.1002/JLB.3A0817-327RR
61. Hesse A-K, Dörger M, Kupatt C, Krombach F. Proinflammatory role of inducible nitric oxide synthase in acute hyperoxic lung injury. Respir Res. (2004) 5:11. doi: 10.1186/1465-9921-5-11
62. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil ageing is regulated by the microbiome. Nature. (2015) 525:528–32. doi: 10.1038/nature15367
63. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. (2019) 19:255–65. doi: 10.1038/s41577-019-0141-8
Keywords: PMN, sepsis, inflammation, rerverse migration, infection
Citation: Ji J and Fan J (2021) Neutrophil in Reverse Migration: Role in Sepsis. Front. Immunol. 12:656039. doi: 10.3389/fimmu.2021.656039
Received: 20 January 2021; Accepted: 17 February 2021;
Published: 15 March 2021.
Edited by:
Yong Ming Yao, First Affiliated Hospital of Chinese PLA General Hospital, ChinaReviewed by:
Jennifer Catherine Brazil, University of Michigan, United StatesCopyright © 2021 Ji and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Fan, amlmN0BwaXR0LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.