 Julie Giraud1
Julie Giraud1 Maya Saleh
Maya Saleh- 1University of Bordeaux, CNRS, ImmunoConcEpT, UMR 5164, Bordeaux, France
- 2University of Bordeaux, INSERM UMR 1053, Bordeaux, France
- 3Department of Oncology, CHU Bordeaux, Haut Leveque Hospital, Pessac, France
- 4Department of Medicine, McGill University, Montreal, QC, Canada
Hepatocellular carcinoma (HCC) is the most common liver tumor and among the deadliest cancers worldwide. Advanced HCC overall survival is meager and has not improved over the last decade despite approval of several tyrosine kinase inhibitors (TKi) for first and second-line treatments. The recent approval of immune checkpoint inhibitors (ICI) has revolutionized HCC palliative care. Unfortunately, the majority of HCC patients fail to respond to these therapies. Here, we elaborate on the immune landscapes of the normal and cirrhotic livers and of the unique HCC tumor microenvironment. We describe the molecular and immunological classifications of HCC, discuss the role of specific immune cell subsets in this cancer, with a focus on myeloid cells and pathways in anti-tumor immunity, tumor promotion and immune evasion. We also describe the challenges and opportunities of immunotherapies in HCC and discuss new avenues based on harnessing the anti-tumor activity of myeloid, NK and γδ T cells, vaccines, chimeric antigen receptors (CAR)-T or -NK cells, oncolytic viruses, and combination therapies.
Preface
The liver is a critical hub of metabolism, glucose storage, lipid and cholesterol homeostasis, detoxification and processing of xenobiotics, endocrine regulation of growth signaling, blood volume regulation, and immune surveillance. These essential functions are coordinated by multiple cell types: the hepatocytes, which make up 80% of the liver volume; the cholangiocytes, which line the biliary ducts and are the second most abundant parenchymal cells of the liver; the liver sinusoidal endothelial cells (LSECs), which line the hepatic sinusoidal walls and display specialized functions in scavenging, antigen presentation and leukocyte recruitment [reviewed in (1)]; the hepatic stellate cells (HSCs), the body's largest storage site of vitamin A at quiescent state; and the liver-resident immune cells, which are particularly enriched in this important immune organ. The liver is continuously challenged with microbial- and danger-associated molecular patterns (MAMPs and DAMPs) and non-self-peptides derived from dietary and gut-derived microbial antigens. Its capacity to deal with these insults is reflected by its particular immune environment. Indeed, the liver hosts the largest population of tissue-resident macrophages, known as Kupffer cells (KCs). It also exhibits a high frequency of tissue-resident lymphocytes, namely natural killer (NK) cells, NKT cells, conventional αβ T cells, unconventional γδ T cells and B cells. The liver's diverse immunotolerance mechanisms limit the development of chronic liver diseases, including cirrhosis and liver cancers.
Hepatocellular carcinoma (HCC), which accounts for approximately 90% of the incidence of all primary liver cancers, is the 5th most prevalent cancer worldwide and the 4th leading cause of death globally (2). Both environmental and genetic risk factors contribute to the etiology of HCC. The most notable environmental and potentially preventable risk factors include oncogenic virus infection with hepatitis B virus (HBV) or hepatitis C virus (HCV), alcohol abuse, and the metabolic syndrome related to obesity and diabetes mellitus [reviewed in (3)]. In addition, some rare monogenic diseases and several single nucleotide polymorphisms (SNPs) predispose individuals to HCC [reviewed in (4)] (Figure 1A). HCC incidence has doubled in the last three decades in the US, presumably due to high prevalence of HCV infection in the mid 1900's and increasing obesity-related non-alcoholic fatty liver disease (NAFLD) progressing to non-alcoholic steatohepatitis (NASH). Accordingly, suppression of HBV/HCV infections may improve HCC clinical outcomes, but few patients with HCC are cured of their hepatic infections due to treatment cost, compliance and toxicity issues, and NAFLD is expected to become the major risk factor for developing HCC in developed countries in the near future (5). In very early or early-stage HCC (stage 0/A, according to the Barcelona Clinic Liver Cancer [BCLC] staging system), the most effective therapeutic option remains surgical resection, liver transplantation or percutaneous local ablation. In this early stage, the median overall survival (mOS) is >60 months with a 5-year survival of 60–80%, but the 5-year recurrence rate is up to 70% [reviewed in (6)]. However, the large majority of HCCs are diagnosed at an intermediate (stage B) or an advanced stage (stage C), when the mOS is ~11–20 months with a 5-year survival of 16%.
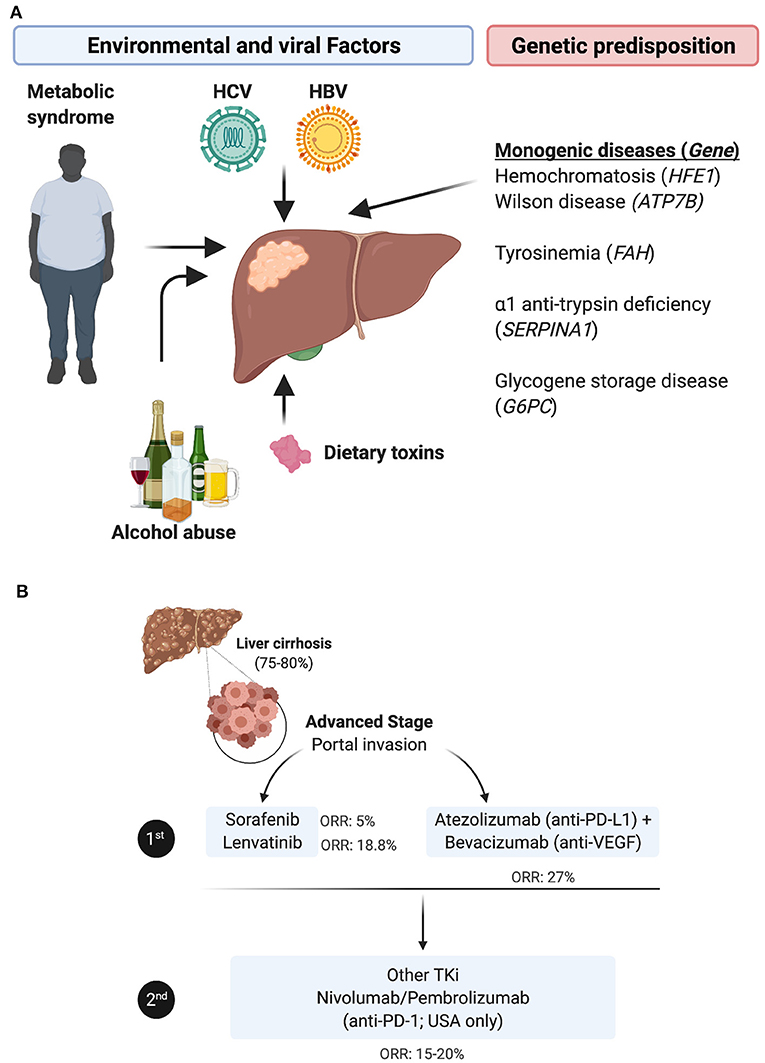
Figure 1. HCC etiologies, genetic predisposition and current standard of care for the advanced stage. (A) HCC etiologies include chronic infection with HBV or HCV, alcohol abuse, dietary toxins and/or the metabolic syndrome linked to obesity and type 2 diabetes. In rare cases, HCC stems from a monogenic disease e.g., hemochromatosis, caused by mutations in the homeostatic iron regulator gene HFE1; Wilson disease involving mutations in the ATPase copper transporting beta gene ATP7B; tyrosinemia, resulting from mutations in the gene encoding fumarylacetoacetate hydrolase FAH, α1-trypsin deficiency caused by mutations in serpin family A member 1 SERPINA1; or glycogen storage disease, in which the glucose-6-phosphatase gene is mutated. (B) The standard of care for treating patients with advanced HCC has been revised with the approval of immune checkpoint inhibitors. In first line, patients are administered TKi, mainly sorafenib or lenvatinib, or given the newly approved combination of bevacizumab (anti-VEGF) + atezolizumab (anti-PD-L1). In second line, patients refractory to TKi are treated with other TKIs, whereas anti-PD-1 ICI, nivolumab or pembrolizumab, have only been approved in the USA as an option for second line (despite the lack of superior efficacity in phase III trials compared to TKi).
The therapeutic options for these stages are limited to locoregional treatments, including transarterial chemoembolization (TACE) or radioembolization with yttrium 90 (90Y)-microspheres, and systemic treatment with multi tyrosine kinase inhibitors (TKi), such as Sorafenib (7) or Lenvatinib (8), according to international guidelines (9). While approved as a first-line therapy, these TKi improve mOS by 3 months (7, 8, 10) and are associated with significant side effects (11). In patients that progress following first line TKi treatment, the second-line options have been, until recently, alternative TKi, primarily regorafenib (12) and cabozantinib (13), or the fully human monoclonal antibody targeting vascular-endothelial growth factor (VEGF) receptor type 2 (VEGF-R2) ramucirumab (14). More recently, immune checkpoint inhibitors (ICI) have emerged as an alternative therapy in HCC and two anti-PD-1 drugs, nivolumab and pembrolizumab, have been approved in the USA based on two trials (15, 16) as a second line treatment for patients with advanced HCC refractory to sorafenib. The overall response rate (ORR) of nivolumab was reported to be 23% in sorafenib-naïve patients and 16-19% in sorafenib-experienced patients, with a mOS of 15 months. However, this did not reproduce in the phase III trial checkmate 459, in which the ORR to nivolumab in sorafenib-naïve patients was 15%, with a mOS of 16 months, i.e., not different from that with sorafenib. Further, in a recent trial, pembrolizumab monotherapy did not statistically impact HCC patients mOS and progression-free survival (PFS), as a second-line treatment (17). The combination of Regorafenib (angiogenesis inhibitor) and nivolumab has next been proposed as a second line treatment in sorafenib non-responders. This year, the combination of atezolizumab (anti-PD-L1) and bevacizumab (anti-VEGF) has obtained approval as a new first line therapy, as it improved mOS > 17 months (18) (Figure 1B). However, despite this therapeutic advance, ~75% of HCC patients do not respond to these immunotherapies for unclear reasons. While there is evidence that boosting the activity of tumor-specific T cells might benefit patients with HCC, the underlying chronic inflammation renders this cancer's tumor microenvironment (TME) somewhat unique, and highlights the urgent need to further explore this organ-specific immunity, identify biomarkers to select patients who are likely to respond to such treatments, and develop new immunotherapies combinations.
The Landscape of Parenchymal, Stromal and Immune Cells in the Healthy vs. Cirrhotic Liver
Prior to delving into the immune landscape and immunosuppressive mechanisms of HCC, we briefly overview the architecture of the liver and its immune system under physiological conditions, and highlight specific changes occurring in cirrhosis. Anatomically, the human liver is composed of eight functional segments organized into hepatic lobules containing their portal vein, hepatic artery and bile duct triads (Figure 2A). Around 80% of the blood supply is delivered from the gut via the portal vein, while the remaining 20% flows through the hepatic artery. Upon mixing, the blood equilibrates and drains across the lobule through the hepatic sinusoids into the central veins, while the bile flows in the opposite direction via bile canaliculi. Such an organization creates oxygen and metabolic gradients, referred to as liver zonation, controlled in part by WNT/β-catenin signaling (Figure 2B). Liver sinusoids are lined by a fenestrated monolayer of LSECs that lack a basement membrane, allowing the blood to directly reach the underlying hepatocytes, organized in two-layered plates. The luminal side of LSECs interacts with liver resident immune cells, such as KCs, whereas their basal side, facing the space of Disse, interacts with hepatocytes and HSCs (Figure 2C). The liver has long been considered as a site of immune tolerance. This was based on early findings that transplanted allogeneic liver was significantly better tolerated than other organs, and patients required low levels of immunosuppression [reviewed in (19)]. Liver immune tolerance stems from complex interactions among liver-resident cells and peripheral leukocytes, and involves poor or incomplete activation of CD4+ and CD8+ T cells, elevated expression of immune checkpoints and an immunosuppressive environment mediated by IL-10 and TGFβ [reviewed in (20)]. KCs that function to preserve tissue homeostasis through their phagocytic and antigen presentation activity are important players in maintaining immune tolerance. Interestingly, a recent paper from the Germain group unraveled that microbiota sensing by LSECs imposes a chemokine gradient around the portal triads resulting in discriminate abundance of KCs and other immune cells (e.g., NKT cells) in periportal regions. Functionally, such an “immune zonation” is critical in limiting local infection and associated inflammatory tissue damage and in preventing the systemic spread of bacteria (21). Besides KCs, hepatic NK cells are capable of directly killing stressed cells, and mediate antibody-dependent cell cytotoxicity (ADCC) upon engagement of CD16 (FcγRIIIA). Their activity is regulated by a dynamic equilibrium between activating [NKG2D, NKp46 (NCR1), NKp44 (NCR2), and NKp30 (NCR3)] and inhibitory (KIR and NKG2A) receptors. In addition to producing various cytokines, chemokines, and growth factors, they maintain immune tolerance through expression of immune checkpoints. Liver-resident NK cells (LrNK) differ from conventional NK (cNK) cells with respect to their origin, phenotypes and functions. Notably, LrNK cells share functional properties with innate lymphoid cells (ILCs) commonly found in mucosal tissues. NKT cells, which also express the NK cell marker CD56, actively patrol the liver and contribute to the clearance of pre-malignant senescent hepatocytes (22). They are recruited via the chemokine receptor CXCR6 interacting with CXCL16, secreted by LSECs and KCs, and are activated upon engagement of the glycolipid receptor CD1d. Last, CD19+ B cells exert their functions through antibody production, antigen presentation and immune cell regulation.
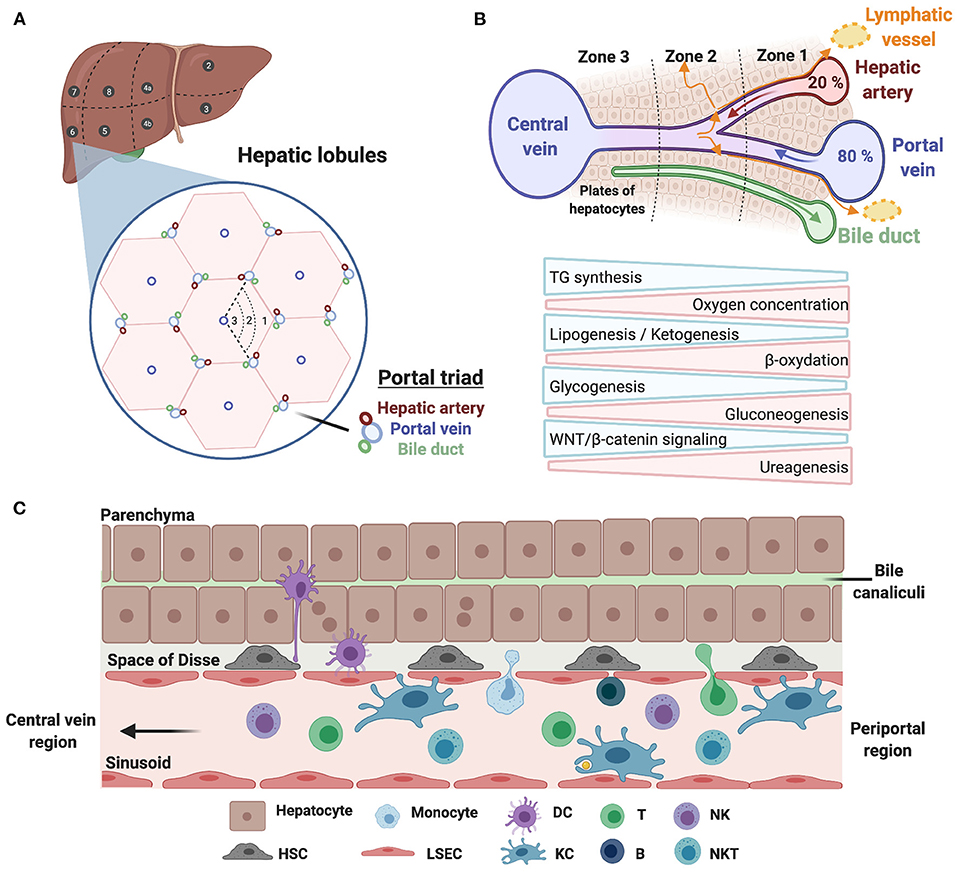
Figure 2. Architecture of the human liver and its immune system. (A) Schematic illustration of the human liver anatomy namely its 8 segments, hepatic lobules, and triads of portal vein/hepatic artery/bile duct. (B) The liver zonation. Oxygen and metabolic gradients define three liver zones with specialized hepatocytes functions. (C) A zoom on hepatic cellular interactions across the sinusoids, the space of Disse and the hepatocyte plates. Liver sinusoidal endothelial cells (LSECs) line the liver sinusoid by forming a fenestrated monolayer. Their basal side interacts with hepatocytes and hepatic stellate cells (HSCs) in the space of Disse, whereas their luminal side interacts with liver-resident leukocytes, including Kupffer cells (KCs).
Liver injury, caused by viral infection or chronic steatohepatitis related to alcohol or metabolic disorders, triggers an inflammatory cell death, leading to DAMP release and the influx of immune cells. Chronic inflammation activates HSCs, the main actors in liver fibrosis that produce extracellular matrix (ECM) components, forming the so-called “scar tissue.” Liver cirrhosis, which affects 1% of the world population, represents the soil where most HCC cases develop. Indeed, continuous cellular stress, repetitive cycles of necrosis and compensatory regeneration of parenchymal cells and chronic inflammation elicit cellular senescence and mutagenesis leading eventually to HCC development. Furthermore, a reduction of sinusoid porosity (defenestration), associated with collagenization of the space of Disse, was shown to impede immunosurveillance [reviewed in (23)].
The recent use of high-dimensional single cell approaches (e.g., mass cytometry and single cell RNA sequencing [scRNAseq]) in humans has unraveled the cellular landscape of the healthy (24, 25) and cirrhotic (26) livers and uncovered subtype heterogeneity for all major liver populations. According to two reports by Aizarani et al. (24) and MacParland et al. (25), in which parenchymal and non-parenchymal cells from dissociated human normal liver tissue were analyzed, the healthy liver is predominantly populated by leukocytes, which make up 45% of all liver cells, out-numbering hepatocytes (ALBhigh) that account for ~35% of the cells in this organ. This is followed by endothelial cells, including LSECs (CD34− CLEC4G+ CLEC4M+) and macrovascular endothelial cells (CD34+ PECAMhigh) that account for ~7.5% and ~2.5% of hepatic cells, respectively. HSCs (RGS5+ ACTA2+) are found at <1% of the cells in this organ whereas cholangiocytes (EPCAM+ KRT19high CTFRhigh ALBlow) occupy ~9% of the liver cellular landscape (24, 25) (Figure 3). Interestingly, among the EpCAM+ cholangiocytes, a putative bipotent liver progenitor population was identified by Aizarani et al. (24) based on the expression of intermediate levels of the intracellular calcium signal transducer TACTSD2/TROP2 (TROP2int). This population was shown to give rise to ASGR1+ hepatocyte-biased cells (TROP2low) or KRT19high CFTRhigh ALBlow cholangiocytes (TROP2hi) (24). Furthermore, to model liver zonation, Aizarani et al. (24) applied diffusion pseudotime analysis and showed that hepatocytes and LSECs gene expression is highly zonated. LSECs in the periportal zone expressed genes involved in hormone signaling and metabolism, whereas pericentral and mid zone LSECs and hepatocytes were enriched in gene expression related to platelet activation, immune regulation and scavenging.
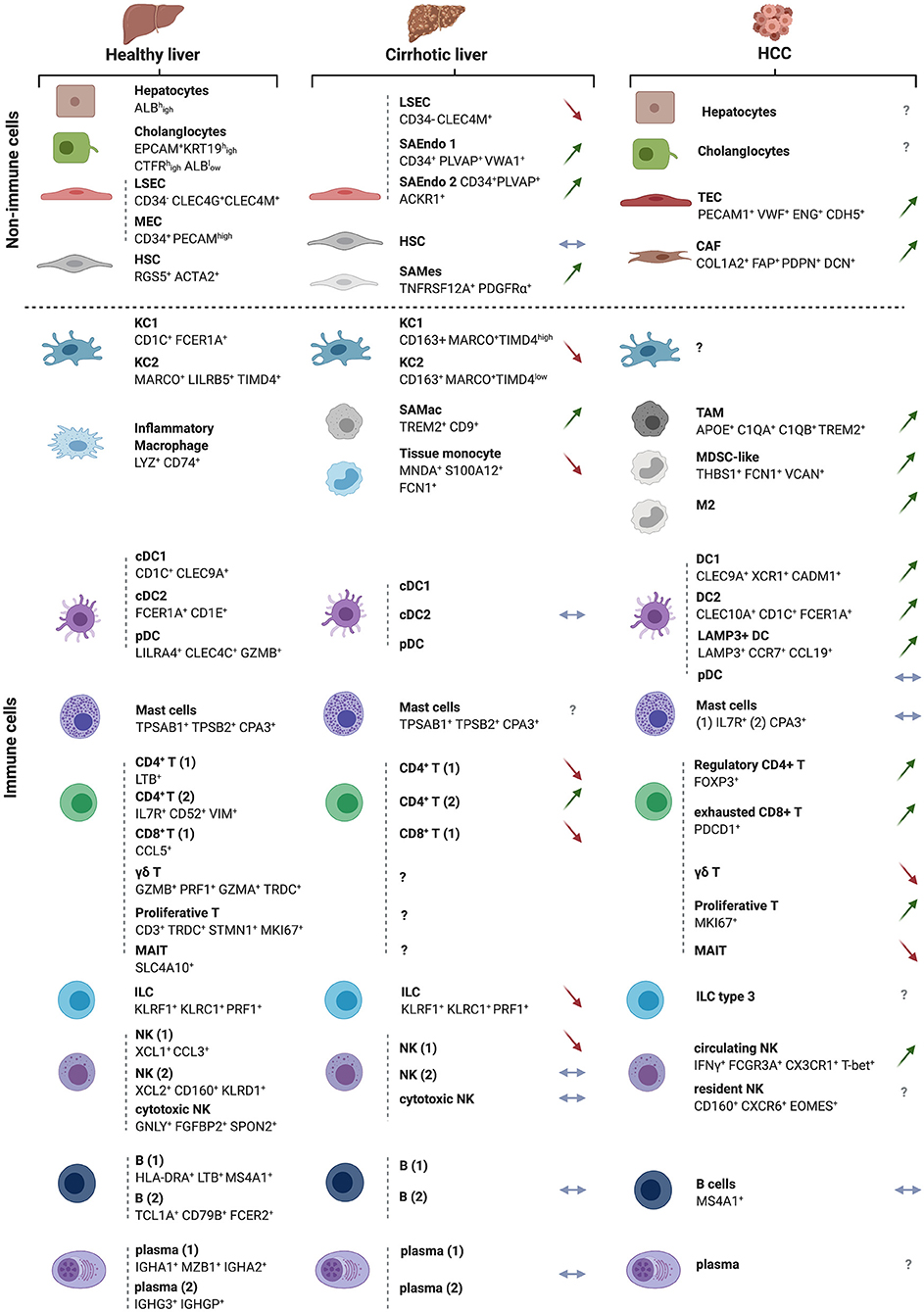
Figure 3. The landscapes of the normal, cirrhotic and HCC-bearing livers. All immune and non-immune cell types identified by high-resolution single cell analyses of the human healthy and cirrhotic livers and of HCC are illustrated along with their discriminatory markers. Arrows depict direction of change in cirrhosis or HCC vs. the normal liver, with green arrows indicating an expansion, red arrows a depletion and blue horizontal arrow no change in the examined cell subset. The cellular landscapes of the healthy and cirrhotic livers were from (26). The information on the HCC landscape was from (27), but with complementary information from the following studies: γδ T cells and M2 macrophages (28), cancer-associated fibroblasts (CAFs) and tumor-endothelial cells (TECs) (29). New subsets of cells arising in the cirrhotic condition are also depicted and labeled as ‘scar-associated’ cells: SAEndo: scar-associated endothelial cells; SAMes: scar-associated mesenchymal cells; SAMac: scar-associated macrophages, as in (26). The HCC analyses were on sorted CD45+ immune cells. Symbols for genes and associated proteins are defined in Supplementary Table 1.
Among the leukocytes, the ratio of lymphocytes to mononuclear phagocytes (MNPs) is 3:1, with the former occupying ~35% and the latter 10% of total liver cells. The lymphocytic compartment includes ~11% αβ T cells, ~6.7% γδ T cells, ~12.3% NK + NKT cells, and ~ 5% B cells (25). Among the innate immune cells, NK cells cluster in three groups, NK1 (XCL1+ CCL3+), NK2 (XCL2+ CD160+ KLRD1+) and cytotoxic NKs (GNLY+ FGFBP2+ SPON2+), whereas MNPs consist of three subsets, including two CD68+ KC clusters, KC1 (CD1C+ FCER1A+) and KC2 (MARCO+ LILRB5+TIMD4+) and a liver resident inflammatory macrophage subset (LYZ+ CD74+). The 3 classical dendritic cell (DC) subsets were also identified, namely conventional DCs, cDC1 (CD1C+ CLEC9A+) and cDC2 (FCER1A+ CD1E+), and plasmacytoid DCs (LILRA4+ CLEC4C+ GZMB+) (Figure 3).
In the cirrhotic liver, scRNAseq uncovered all major immune cell populations and revealed a decrease in CD8+ T cells, associated with an increase in CD4+ T cells, as compared to the healthy liver. Re-clustering of MNPs identified four subgroups, annotated as KC1 (CD163+ MARCO+ TIMD4hi), KC2 (CD163+ MARCO+ TIMD4low), scar-associated macrophages (TREM2+ CD9+), tissue monocytes (MNDA+ S100A12+ FCN1+) (Figure 3). The MARCO+ population decreases in cirrhosis compared to the healthy liver while TREM2+ CD9+ scar-associated macrophages, derived from circulating monocytes, expand early in the course of the disease. This latter population of cells is conserved in humans and mice and displays pro-fibrogenic properties (26). Deep clustering of mesenchymal cell populations uncovered a cluster of PDGFα+ cells that also expand in cirrhosis, expressing high levels of fibrillar collagens and pro-fibrogenic genes. RNA velocity experiments indicated a trajectory from human HSCs to these scar-associated mesenchymal cells, and ligand/cognate receptors analysis combined with functional studies, pointed to TNFRSF12A, PDGFRA, and Notch signaling as important regulators of mesenchymal cell function in the human liver fibrotic niche (26).
Collectively, these single cell analyses revealed context-dependent cellular phenotypic diversity, opening the field to exploring potential mechanisms involved in HCC progression from cirrhosis. For instance, the fibrotic context is associated with the emergence of scar-associated mesenchymal cells and scar-associated macrophages with pro-fibrogenic properties. Future functional studies are needed to determine the value of targeting these cell subsets or specific molecular effectors therein as therapeutic strategies in HCC.
HCC Subtypes According to Molecular and Immune Classifications
Molecular Classification of HCC
Progression from cirrhosis to HCC is mediated by a step-wise accumulation of somatic mutations and copy number variations in driver genes (30). The most frequent alteration is the reactivation of the telomerase reverse transcriptase (TERT), a key event observed in 20% of high-grade dysplastic lesions and up to 60% of early HCC (31). Besides TERT promoter mutations that impact telomere maintenance, 10 pathways were found to be recurrently altered in HCC, including pathways involved in cell cycle control (TP53, CDKNA2, CCND1), oxidative stress (NFE2L2, KEAP1), and chromatin modification (ARID1A, ARID2), but also the Wnt/β-catenin pathway (CTNNB1, AXIN1) and the RTK/RAS/PI3K pathway (RPS6KA3, PIK3CA, KRAS, NRAS, FGF19, VEGFA) (32) (Figure 4). The TGFβ pathway is additionally involved in HCC progression, with some tumors presenting aberrant activation of this pathway, whereas others harboring inactivating mutations in genes required for TGFβ signal transduction e.g., the SPTBN1 gene (33). Last, ~20% of HCC express markers of progenitor cells, e.g., epithelial cell adhesion molecule (EpCAM) and cytokeratin 19 (CK19) and arise from either progenitors or dedifferentiated hepatocytes (12).
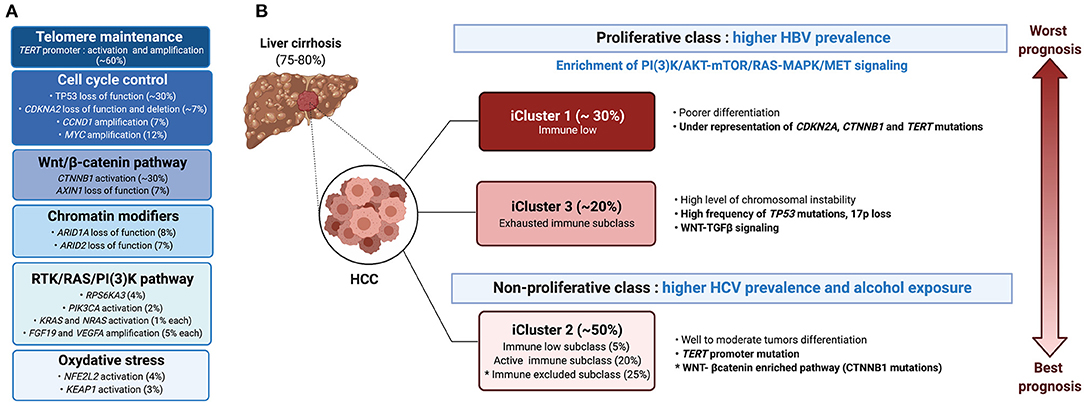
Figure 4. Molecular and immunological classifications of HCC. (A) The main oncogenic events associated with HCC are presented in descending order of incidence, the most common being defects in telomere maintenance, followed by alterations in cell cycle control, and activation of the WNT/β-catenin pathway. (B) Based on molecular features, HCC patients can be grouped into either the proliferative or non-proliferative class, the first with higher prevalence of HBV infection and a bad prognosis, whereas the second including cases with HCV infection or alcohol abuse and having a better prognosis. Integration of genomic, expression and epigenetic data by the TCGA research network (32) identified a different classification, namely iClusters 1-3. iCluster 2 which represents 50% of the patients and is related to the non-proliferative class, includes three sub-classes: an “active immune subclass,” an “immune excluded” subclass and an “immune low” subclass. An “immune exhausted” subclass is found within iCluster 3 of the proliferative class, whereas iCluster 1 is characterized by an “immune low” signature.
Earlier studies classified HCC into two main transcriptome-based classes, based on genetic, epigenetic and phenotypic features: HCC of the proliferative class, which displayed a poor clinical outcome, and HCC of the non-proliferative class, with a better outcome. The proliferative class was associated with the HBV etiology, and characterized by the activation of PI3K–AKT–mTOR, RAS–MAPK, and MET signaling along with chromosomal instability (34). The non-proliferative class, which is more prevalent in alcohol- or HCV-related HCC, regrouped heterogeneous tumors, including a subclass characterized by mutations in CTNNB1, the gene encoding β-catenin. More recent classification by Schulze et al. (35) and the Cancer Genome Atlas (TCGA) network (32) revised the molecular landscape of HCC (Figure 4). Three integrative clusters were identified: whereas, iCluster 1 and iCluster 3 distinguished two subclasses of the proliferative class, iCluster 2 overlapped with the non-proliferative class. iCluster 1 is associated with poorer differentiation, higher tumor grade, the presence of macrovascular invasion and overexpression of proliferation (PLK1, MKI67) and progenitor cells (EPCAM and AFP) gene markers, while iCluster 3 is characterized by high frequency of TP53 mutation, 17p loss and activation of WNT-TGFβ signaling. On the other end, iCluster 2 regroups heterogenous moderately differentiated tumors characterized by TERT promotor mutations. Identification of the mutational landscape of HCC unveiled several druggable targets in >25% of the cases (35). However, a potential limitation of tumor cell-based therapy is a notable inter- and intra-tumoral heterogeneity, mediated in part by non-neutral selection of mutations conferring a selective advantage (30) and subclone evolution (36). Using scRNAseq of liver cancers, Ma et al. (29) identified links between intra-tumoral heterogeneity (ITH), tumor micro-environment (TME) and survival outcome. They discriminated ITH according to the average expression of 10 cancer stemness genes, namely EPCAM, CD24, CD44, CD47, KRT19, PROM1, ALDH1A1, ANPEP, ICAM1 and SOX9. This allowed them to derive diversity scores based on transcriptomic profiles, grouping the tumors into Div-high and Div-low groups. The Div-high group displayed poorer mOS and PFS, expressed higher levels of hypoxia-inducible factor 1-alpha (HIF1α)-dependent VEGFA and displayed a marked TME reprogramming. Concordantly, NOTCH and VEGF signaling, together with fetal-associated endothelial cells (PLVAP+ VEGFR2+) found in tumors, have been demonstrated to reprogram the CD14+ monocytes into fetal-like immunosuppressive tumor-associated macrophages (TAMs (FOLR2high CD163high) (37).
Immunological Classification of HCC
Immunological classification of HCC has been proposed by different groups using gene expression profiling (38) and protein level approaches based on multiplex immunohistochemistry (IHC) analysis (38) and mass cytometry (CyTOF) (28). Using deconvolution of 8 datasets, Llovet and colleagues analyzed a total of 956 HCC samples and reported that ~25% of HCC cases expressed an immune gene signature (39). Such an “immune class” was found to be associated with better mOS, and expressed PD-1 and PD-L1, tertiary lymphoid structures (TLS) markers and determinants of cytolytic T cells activity e.g., an IFNγ signature. Further stratification identified two TME-based sub-classes within the immune class, dubbed the “active immune” and the “exhausted immune” subclasses. The “active immune” sub-class was enriched in T cell response effectors (IFNγ and granzyme B signatures), whereas the “exhausted immune” sub-class included signatures of T cell exhaustion, immunosuppressive macrophages and TGFβ signaling. A third immunological class, referred to as “immune excluded” was distinguished in ~25% of HCC patients, based on the expression of immune genes, particularly an immunosuppressive signature, in the tissue surrounding the tumor, but with little immune gene expression in the tumor core. Such an “immune excluded” class was associated with a bad prognosis and overlapped with a subset of tumors in TCGA iCluster 2 with an activated WNT-β-catenin pathway (39) (Figure 4B). The immunological environment of HCC and its association with the molecular classification was further analyzed by Kurebayashi et al. (38) using multiplex immunohistochemistry. The authors classified HCC into three immune-subtypes based on the numbers of infiltrating immune cells: “Immune-high,” “immune-mid” and “immune-low.” Consistent with Sia et al. (39), the “immune-high” subtype, which was enriched in T cells and B-/plasma cells, was associated with a good prognosis (38). Zhang et al. (28) expanded this analysis and defined three HCC groups, namely the “immunocompetent,” “immunosuppressive,” and “immunodeficient” subtypes. The immunocompetent subtype, characterized as CD45high FOXP3low, had normal T cell infiltration including high infiltration of γδ T cells. On the contrary, the immunosuppressive subtype, marked by a CD45high FOXP3high staining, exhibited high frequencies of immunosuppressive cells (regulatory T and B cells and immunosuppressive macrophages) and molecules (PD-1, PD-L1, TIM-3, CTLA-4, VEGF, TGFβ, and IL-10). Finally, the CD45low immunodeficient subtype showed a reduced infiltration of lymphocytes (28). While these studies demonstrated marked heterogeneity in HCC tumors and their associated TME with broad classification of patients, in depth characterization of the immune landscape of HCC at high resolution is expected to refine patients stratification and identify putative immune-therapeutic targets.
The Immune Landscape of HCC
The immune landscape of HCC has been more recently explored using single cell approaches. In general, a progressive depletion of intrahepatic LrNK cells, cytolytic T cells and γδ T cells and an enrichment of regulatory T cells (Treg) and macrophages occur in HCC (28, 28, 32, 40–42) (Figures 3, 5). While tumor-infiltrating CD8+ T cells are significantly correlated with better prognosis (38, 43), Treg are associated with a poorer mOS (44). RNA velocity analysis indicated a directional flow from proliferative to exhausted CD8+ T cells in HCC (27). Exhaustion is characterized by the expression of a range of inhibitory receptors, including PD-1, TIM-3, LAG3, TIGIT, and LAYN [reviewed in (45)], and with reduced effector functions via TOX-mediated epigenetic and transcriptional alterations (46–48). However, not all exhausted CD8+ T cells are the same, as two subsets can be discriminated: PD-1+ TCF1+ “precursors” that self-renew and give rise to PD-1+ TCF1− “terminally differentiated” exhausted T cells (49–51). Notably, the presence of the precursors, but not the terminally differentiated exhausted T cells, is associated with a better response to anti-PD-1. Similarly, NK cells display an exhausted phenotype, expressing high levels of immune checkpoints such as PD-L1, PD-1, LAG3, TIM-3, CD155, and CD96 (52, 53). Further, they produce immunosuppressive cytokines such as TGFβ and IL-10 and less IFN-γ (52–56). The role of B lymphocytes in the development of HCC and their prognostic value is still debated. Their ADCC and antigen-presentation functions are countered by their ability to induce immunosuppression. In surgically resected HCC, CD20+ B cells are associated with a better prognosis (38, 57), especially when they are in close proximity of tumor-infiltrating T cells (58). However, their prognostic value in the context of TLS depends on whether these are found intra-tumorally or in the surrounding tissue [reviewed in (59)]. Notably, TLS presence in the adjacent non-tumoral liver tissue was associated with an increased risk for late recurrence and a poor mOS in 82 patients with surgically resected HCC (60). Mechanistically, such ectopic TLS harbored progenitors/cancer stem cells (expressing CD44v6) and a tumor-promoting environment characterized by a persistent NF-κB activation favoring tumor outgrowth, as demonstrated in a mouse model (60). Concordantly, alymphoid conditions suppressed CD44v6+ HCC-initiating cells and prevented hepatocarcinogenesis (60, 61). Additionally, B cells favored HCC progression by limiting the resolution of senescence-mediated liver fibrosis (62). In contrast, it was recently shown that intra-tumoral TLS were predictive of a lower risk of early relapse after surgical resections, as analyzed in 273 patients (63). In contrast, CD68+ CD163+ TAMs, which accumulate at the tumor margin, and CCR1+ monocytes, are associated with bad mOS (64–66). TAMs are recruited to HCC in response to CCL2. The expression of the CCL2 gene is controlled by diverse mechanisms including through APOBEC3B-mediated de-repression of epigenetic marks in its promoter (67), and through Yes-associated protein (YAP) transcriptional regulation, as shown in tumor-initiating cells (TICs) (68). Together with IL-13, CCL2 drives the metastasis of MYC/Twist1 tumors (69). TAMs contribute to HCC malignant progression and metastasis through diverse mechanisms, e.g., via the production of cytokines such as IL-6 (70) and hepatocyte compensatory proliferation (71), immunosuppression and induction of epithelial-to-mesenchymal transition (EMT) [reviewed in (72)]. The immune checkpoint TIM-3, induced by TGFβ in the tumor microenvironment (TME), is implicated in the pro-tumoral effects of TAMs in HCC (70). Similarly, the receptors Triggering Receptor Expressed on Myeloid cells (TREM)1 (73) and TREM2 (74) have been demonstrated to promote the dysfunction and apoptosis of cytotoxic CD8+ T cells in HCC, while enhancing the recruitment of CCR6+ Foxp3+ Tregs. Platelets, key effectors of immune-mediated tissue damage, have also been implicated in HCC. Using different mouse models of dietary-inducing NASH and data from human patients, Malehmir et al. (75) demonstrated enhanced platelets influx, aggregation and activation in liver sinusoids in NASH. This was mediated by their interaction with KCs, involving hyaluronic acid/CD44 binding and the platelet receptor glycoprotein 1b alpha (GPIbα). Anti-platelet treatments, including aspirin, or specific blockade of GPIbα blunted the development of NASH, through limiting CD8 T lymphocytes, NKT cells and KC recruitment. In the HBsAg transgenic mouse model of HCC, platelets were similarly shown to promote the recruitment of HBsAg-specific CD8 T cells that elicit cycles of hepatocyte killing and inflammation leading to fibrosis. Inhibition of platelet activation potently reduced the development of HCC in this model (76).
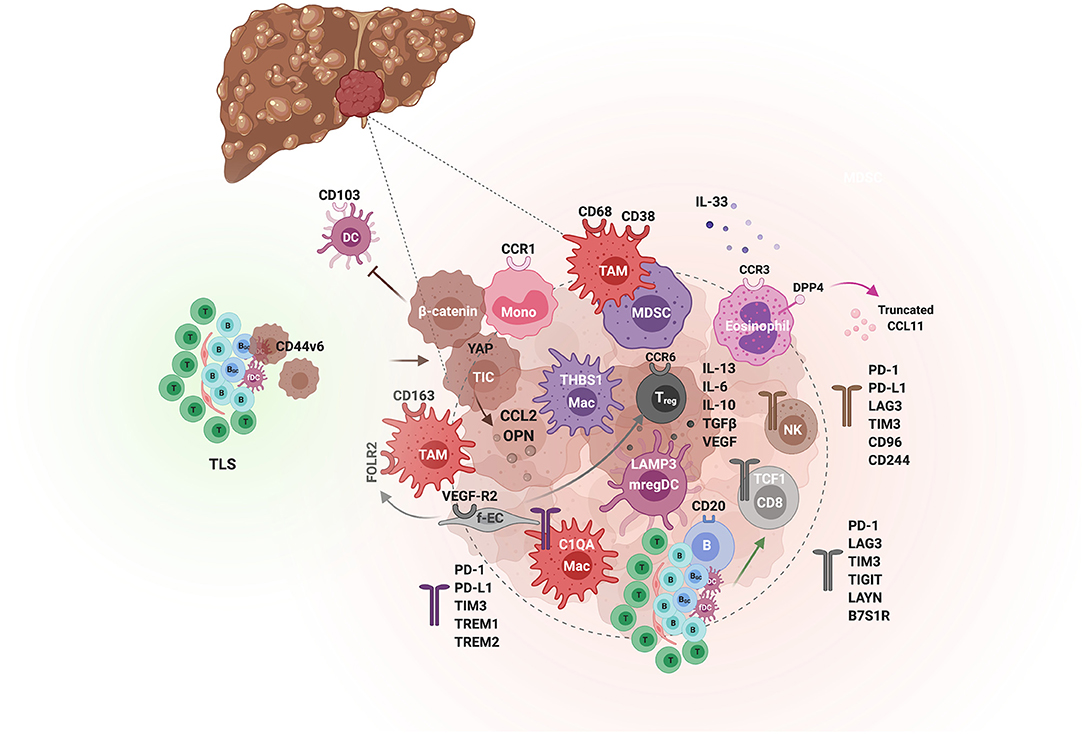
Figure 5. The tumor immunological microenvironment of HCC. Schematic illustration of the different actors demonstrated to contribute to immunosuppression or immune activity in the TME of HCC. Tumor-initiating cells (TIC) and tumor cells (hues of brown) orchestrate the immunological environment by secreting inflammatory cytokines e.g., osteopontin (OPN) and chemokines e.g., the monocyte chemoattractant CCL2, which promote tumorigenesis through the recruitment of monocytes and their differentiation to tumor-associated macrophages (TAMs). In addition, β-catenin-activated tumor cells can inhibit anti-tumor immunity by blocking CD103+ DCs tumor infiltration. Spatially, TAMs are enriched in the peri-tumoral area, express CD68, CD163, CD38, and the folate receptor (FOLR2), induced by fetal-associated endothelial cells (fEC). Two subsets of tumor-enriched macrophages can be distinguished by single cell analyses, THBS1+ macrophages and C1QA+ macrophages, with an MDSC and TAM signature, respectively. The latter is associated with a poor prognosis in HCC. The immunosuppressive activity of TAMs is mediated by various immune checkpoints, including PD-1, PD-L1, TIM-3, TREM1 and TREM2. Besides TAMs, the TME is enriched in Treg but depleted of other T cells and NK cells, which when present exhibit an exhausted phenotype. Among the CD8 T cells, The PD-1+ TCF1+ “precursors” are associated with a good response to anti-PD-1. TLS found in the adjacent non-tumoral liver tissue are associated with a poor prognosis as they can harbor progenitor/cancer stem cells (expressing CD44v6) and promote tumorigenesis by impairing the clearance of senescent hepatocytes. In contrast, intratumoral TLS and CD20+ B cells are predictive of a lower risk of relapse and a better CD8 T cell anti-tumor activity. Eosinophils also confer a tumorilytic activity, and enhancing their recruitment with inhibitors of the CCL11 peptidase DPP4 improved the efficacy of anti-PD-1+anti-CTLA4 immunotherapy.
scRNAseq of sorted CD45+ cells from the tumor, adjacent liver, hepatic lymph nodes, blood, and ascites of 16 treatment-naive HCC patients recovered all of the major cell populations such as T, B, NK and myeloid cells, but also few minor cell populations including mast cells and ILCs (27) (Figure 3). All types of T cells (including Treg, exhausted T cells [Tex] and proliferative T cells) were enriched in the tumors, as previously reported (77). Four clusters of NK cells, enriched in the tumor, were identified, including two circulating NK clusters (IFNγ+ FCGR3A+ CX3CR1+ T-bet+) and two LrNK clusters (CD160+ CXCR6+ EOMES+). However, their respective roles in tumorigenesis and patients prognosis have not been addressed. A diverse repertoire of functionally distinct myeloid cells were identified, particularly, two subsets of macrophages within the tumors: THBS1+ macrophages enriched in myeloid-derived suppressor cell (MDSC) genes (S100A genes, FCN1 and VCAN) and C1QA+ macrophages, enriched in tumor associated macrophage (TAM) genes APOE, C1QB and TREM2 (Figures 3, 5). Only the latter was associated with a poor prognosis in the TCGA liver hepatocellular carcinoma (LIHC) cohort. In parallel, 3 intra-tumoral clusters of DCs were distinguished, namely cDC2 (highly expressing CD1C, FCER1A, and CLEC10A), cDC1 (highly expressing CLEC9A, XCR1 and CADM1) and a non-classical LAMP3+ DCs (highly expressing CCR7, LAMP3, CD80 and CCL19) with migration capacity toward the lymph nodes. Interestingly, ligand-receptor pairs analysis indicated that the LAMP3+ DCs are the subset that would interact with Tex cells and Tregs. This LAMP3 population seems to correspond to mregDCs, a DC cluster annotated in human lung cancer as a population involved in tumor antigen uptake and expressing immunoregulatory molecules (78, 79).
HCC Patients Response to ICIs
PD-1 is primarily expressed on the surface of activated T cells, but also on NK/NKT cells (54), B cells (80) and myeloid cells including monocytes, DCs, MDSCs and TAMs (81). Its induction in response to cytokine signaling is tightly regulated at the epigenetic and post-transcriptional levels (48, 81, 82). Recently, two studies used multiparametric flow cytometry and multiplex IHC to show that higher intratumoral frequency of PD-1high CD8+ T cells (83) and CD38+ CD68+ macrophages (84) was strongly associated with improved response to ICI in patients with advanced HCC. PD-L1 is expressed by DCs, monocytes, macrophages, B cells, NK cells, LSECs, and tumor cells. Its expression is induced by hypoxia (73), among other mechanisms. PD-L1 expression on tumor-infiltrating immune cells is associated with a better prognosis while the prognostic value of its expression on neoplastic cells is controversial (39, 43). Further, the response of patients with HCC to Nivolumab (anti-PD1) was not found to be associated with PD-L1 expression on tumor cells, implicating other PD-L1 expressing cells in this response (15). In murine models with transplantable HCC, PD-L1 expression on myeloid cells mediated the anti-PD-L1 response (85).
Previous studies in melanoma and non-small cell lung cancer (NSCLC) have attributed the response to ICIs to tumor mutational burden (86, 87), levels of neo-antigens (88) or tumor-specific antigens (89, 90), the presence of TLS [reviewed in (59)] or specific oncogenic pathways (91, 92). Nonetheless, the mechanisms involved in patients response to ICIs, particularly in HCC, remain for the most part unclear. For example, the mutational burden did not correlate with ICI response in HCC (93), and neither the mutational load nor the presence of neoantigens was associated with the immune class, which predicted a favorable response to ICI therapy (39). Instead, the activation of β-catenin was associated with resistance to ICI, as demonstrated in a mouse model (94) (Figure 4). Using a MYC;p53−/− HCC mouse model, Ruiz de Galarreta et al. (94) demonstrated that β-catenin promoted immune escape by preventing the recruitment of CD103+ DCs, impairing antigen-specific T cells-mediated anti-tumor immunity. Accordingly, activating mutations in CTNNB1 correlate with resistance to ICI monotherapy with either anti-PD-1 or anti-PD-L1, as shown in a prospective sequencing analysis of 27 evaluable advanced HCC patients, in which none of the 10 patients with WNT pathway alterations achieved clinical benefit, whereas around half of the non-WNT pathway–altered patients showed durable stable disease (93). Nevertheless, these results also show that 50% of ICI non-responders harbor mechanisms unrelated to β-catenin activation. Treating fibrosis using a TGFβ neutralizing antibody in the STAM™ mouse model fibrosis-associated HCC, triggered a redistribution of CD8+ lymphocytes into the tumors, which re-invigorated anti-tumor response (95). These results are consistent with those of Mariathasan et al. (96), who reported that TGFβ attenuated the response to PD-L1 blockade by restricting intra-tumoral T-cell infiltration. Since TGFβ alterations are found in a subset of HCC patients (33), agents that block this pathway should be tested in this group, highlighting the need for personalized medicine. Similarly, the immunosuppressive molecule VEGF was found to be enriched in a subset of HCC patients, particularly those with Div-high tumors, supporting the use of the anti-PD-L1 (Atezolizumab) + anti-VEGF (Bevacizumab) combination. However, the cellular subsets, putative signaling pathways, and associated biomarkers required for an effective patient's response to this new combination therapy require further exploration. The etiology of HCC might contribute to the heterogeneity in patients' response to immunotherapy. Indeed, Lim et al. (44) reported that the TME of HBV-related HCC is more immunosuppressive than that of non-viral HCC. Particularly, PD-1high Tregs and PD-1+ CD8+ resident memory T cells were more prominent in HBV-related HCC, suggesting that PD-1 blockade might be a suited strategy for this etiology. In contrast, immunotherapies that target CD244+ NK cells and Tim-3+ CD8+ T cells, enriched in non-viral HCC, may be more effective in those patients (44).
ICI Combination Therapies
Several pre-clinical studies and ongoing clinical trials (Table 1) are exploring the potential of combining different ICIs. For e.g., a phase III trial is currently testing the combination of durvalumab (anti-PD-L1) and tremelimumab (anti-CTLA-4) as a first line therapy (NCT03298451). The combinations of ICIs together with ablation (97), chemo-radioembolization or targeted therapies (TKi or anti-VEGF) in the adjuvant or neoadjuvant setting are also being explored (Table 1). For e.g., two trials are testing the combination of pembrolizumab + Lenvatinib (NCT 03006926) or of pembrolizumab + regorafenib (NCT03347292) for first line therapies. Radioembolization was reported to elicit an immune response, both locally and systemically, leading to enhanced infiltration of TIM-3+ tumor-infiltrating lymphocytes (TILs), NK, and NKT cells (98). It is thus plausible that an ICI targeting TIM-3 might enhance the clinical response of radioembolization or other interventions in HCC patients. A phase II trial is currently testing cobolimab, a TIM-3 binding antibody, in combination with anti-PD-1 on the response of patients with locally advanced or metastatic liver cancer (NCT03680508). Similarly, combining multiple strategies targeting inhibitory receptors (PD-1, TIM-3, LAG3, CTLA4, TREM1, TREM2) and/or their ligands (PD-L1, B7 superfamily member1 [B7S1]) have shown synergistic effects in restoring TILs anti-tumoral immune responses in pre-clinical studies (73, 74, 99–102) and enhancing NK cell infiltration and activity (52–56, 103). Additional strategies include the inhibition of TAM recruitment, their polarization to an immunosuppressive phenotype or their function in hampering anti-tumor immunity or promoting tumorigenesis. The pro-inflammatory protein osteopontin (OPN) produced by cancer cells has been implicated in cancer promotion and metastasis, through the stimulation of CSF1 signaling in TAMs. Blockade of the CSF1/CSF1R pathway enhanced the efficacy of anti PD-L1 in OPN-overexpressing HCC, by reducing macrophage recruitment (102). Blockade of the CCL2/CCR2 axis was also shown to inhibit the recruitment of TAMs leading to enhanced infiltration of CD8+ T cells and improved anti-tumor immunity (104). However, this approach should be considered with caution as some macrophages exert anti-tumoral activity. Indeed, Eggert et al. (105) reported that the CCL2-CCR2 axis promotes the clearance of senescent hepatocytes preventing HCC outgrowth in mice. Among the TAM targets that recently surfaced as critical inhibitors of anti-tumor immunity are the receptors TREM1 and TREM2. Blockade of TREM1 (73) or TREM2 (74) attenuated immunosuppression and CD8+ T cell dysfunction boosting the efficacy of anti-PD-1/PD-L1 immunotherapy. An alternative approach to skew TAM functions is through vaccination. Using a mouse model, a recent study demonstrated that a Listeria-based HCC vaccine enhanced the efficacy of PD-1 blockade by skewing the TAMs to an anti-tumoral phenotype (106). Consistent with the improved patients response to the anti-PD-L1 + anti-VEGF combination therapy, it has been recently demonstrated using murine models of HCC that this approach fortified hepatic vessels and overcame resistance to either monotherapy (107). Last, a less studied immune cell population in the context of anti-tumor immunity are the eosinophils, which were recently shown in a murine model of HCC to promote tumor-cell killing through degranulation and contribute to the efficacy of the anti-PD1 + anti-CTLA4 combination immunotherapy. Their recruitment in response to the cancer-cell secreted alarmin IL-33, is mediated by the chemokine CCL11, and enhanced with the administration of sitagliptin, an inhibitor of dipeptidyl peptidase DPP4 (CD26) that cleaves CCL11. These results suggest that combined modulation of both type 1 and 2 immune responses may improve therapeutic management of HCC (108).
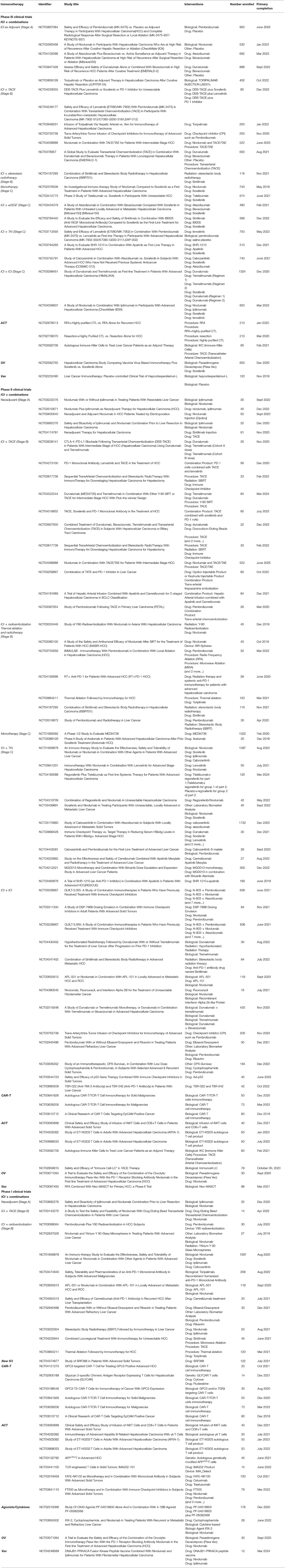
Table 1. Clinical trials of immunotherapies for HCC.
The Future of Immunotherapies in HCC Beyond ICI
Besides ICI, several immunotherapeutic strategies for HCC patients are emerging, such as targeted therapies promoting ADCC, adoptive cell therapy (ACT), including the transfer of autologous CD8 T cells, iNKT cells, γδ T cells, cytokine-induced immune killer cells (IKC), chimeric antigen receptor (CAR)-T cells, oncolytic viruses and vaccines (Figure 6). For a number of these strategies, tumor-specific antigens (TSA) or tumor-associated antigens (TAA) are targeted. In HCC, these include α-fetoprotein (109–111), hTERT (112), glypican-3 (GPC3) (113–115), p53 (116), melanoma antigen gene A (MAGE-A) (117), squamous cell carcinoma antigen recognized by T cells (SART) (118), and NY-ESO-1 (119). More recently, the oncogenic phosphatase PRL3 was confirmed as a TAA, as it was shown to be expressed in tumors, but not in patient-matched normal tissue, across 11 cancers. A humanized antibody targeting this TAA, PRL3-zumab, was shown to enhance the intra-tumoral recruitment of B cells, NK cells and macrophages, suggesting that this antibody might promote tumor killing by ADCC (120). Similarly, the elevated expression of GPC3 in >70% of HCC, and its association with poor prognosis (121), has led to the development of several immunotherapeutic strategies, including the humanized monoclonal antibody codrituzumab (122), bi-specific antibodies (123), CAR-T cells (124), antibody-drug conjugates (125), and vaccines (126). GPC3-CAR-T cells have been shown to be polyfunctional and capable of eliminating HCC in a transplantable orthotopic mouse model (127), and there are currently at least 5 phase I clinical trials recruiting patients with HCC to test GPC3-CAR-T cells (Table 1; ClinicalTrials.gov, December 2020). Another CAR-T cell tested in multiple solid tumors including HCC is the EpCAM-CAR-T, as registered in a phase I/II trial (NCT03013712). In a similar approach, a phase I trial is testing the transfer of autologous genetically modified AFPc332T cells, T cells expressing an enhanced TCR Specific for α-fetoprotein in HLA-A2 positive patients with advanced HCC (NCT03132792). In the oncolytic viruses sphere, a phase III trial (NCT02562755) is testing a vaccinia virus-based immunotherapy, Pexastimogene Devacirepvec (Pexa-Vec), in patients with advanced HCC, based on promising results from the phase IIb trial TRAVERSE (128). Pexa-Vec is also being tested in combination with nivolumab in the first-line treatment of advanced HCC in a phase II trial (NCT03071094). As for vaccines, a phase III trial (NCT02232490) is evaluating the benefit of hepcortespenlisimut-L (Hepko-V5), an oral allogeneic vaccine derived from patients' blood, given in an experimental arm vs. placebo, to patients with advanced HCC. Similarly, a phase II trial (NCT03067493) is testing the Neoantigen Multiple Target Antigen Stimulating Cell Therapy (Neo-MASCT) vaccine, which consists of 18 cycles, each including one DC subcutaneous injection and one CTL infusion.
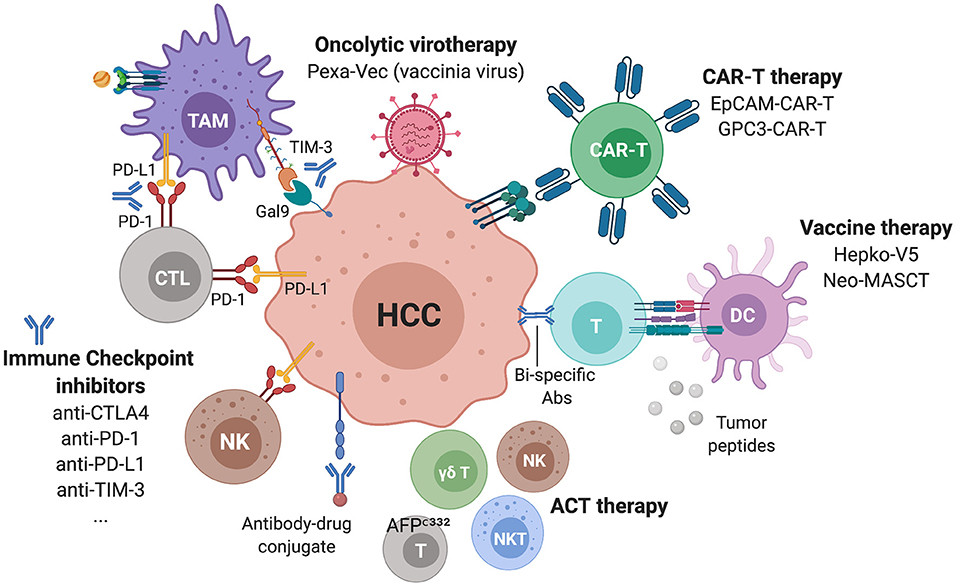
Figure 6. Immunotherapies in ongoing clinical trials for advanced HCC. Several ICIs targeting checkpoints on lymphocytes but also NK and myeloid cells are currently being assessed as monotherapies or in combinations. Additional strategies include CAR-T cells, oncolytic viruses, vaccines, antibody-drug conjugates and bi-specific antibodies.
Conclusions
HCC comprises a heterogeneous set of cancers with different etiologies, mutations and immune microenvironments, as demonstrated by broad molecular and immunological classifications. The advent of recent technologies including single cell approaches is now allowing high resolution characterization of the immune landscapes of HCC and is expected to uncover novel immunotherapeutic targets and approaches tailored to patients. ICI combination therapies are expected to dramatically improve the systemic therapy of advanced HCC. However, the prioritization of different combinations requires additional understanding of liver-specific immunity and the validation of therapeutic targets in suitable pre-clinical models of HCC taking into consideration the genetic heterogeneity of tumor cells and the cirrhotic or NASH environments. Further, an in-depth characterization of the biomarkers leading to improved patients' response to the various such combinations will contribute to better selection of patients and ameliorate the outcome. Last, a critical issue not discussed here is the management of immune-related adverse events (irAEs) often elicited by immunotherapies and that should be considered in designing and implementing immunotherapies. It is hoped that with the rapidly evolving field of oncoimmunology and trials in different cancer types, we will learn valuable lessons for future drug discovery in HCC.
Author Contributions
JG and MS conceived the structure of this review. DC contributed to the description of the immune landscape of the liver and of HCC and prepared Figure 3. J-FB contributed to the review of clinical activity in HCC. All authors revised the manuscript and approved the final version.
Funding
MS is funded by grants from the ARC foundation, IDEX Bordeaux, SIRIC BRIO and The New Aquitaine Region.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.655697/full#supplementary-material
Abbreviations
ACT, adoptive cell therapy; ADCC, antibody-dependent cell cytotoxicity; BCLC, Barcelona Clinic Liver Cancer; CAR, chimeric antigen receptors; DAMPs, danger-associated molecular patterns; EC, endothelial cells; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; fDC, follicular dendritic cell; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HSC, hepatic stellate cells; ICI, immune checkpoint inhibitor; IFNg, interferon gamma; IHC, immunohistochemistry; ILCs, innate lymphoid cells; IKC, immune killer cells; irAEs, immune-related adverse events; KC, Kupffer cells; LIHC, liver hepatocellular carcinoma; LSEC, liver sinusoidal endothelial cells; MAGE-A, melanoma antigen gene A; MAMPs, microbial-associated molecular patterns; MAPK, mitogen-activated protein kinase; MDSCs, myeloid-derived suppressor cells; MNPs, mononuclear phagocytes; mOS, median overall survival; mregDCs, mature DCs enriched in immunoregulatory molecules; mTOR, mammalian target of rapamycin; NK cells, natural killer cells; cNK cells, conventional NK cells; LrNK cells, liver-resident NK cells; NKT cells, natural killer T cells; NSCLC, non-small cell lung cancer; ORR, overall response rate; pDC, plasmacytoïd dentritic cells; PFS, progression-free survival; PI(3)K, Phosphoinositide 3-kinase; RTK, receptor tyrosine kinase; SART, squamous cell carcinoma antigen recognized by T cells; scRNAseq, single cell RNA sequencing; SNPs, single nucleotide polymorphisms; TAA, tumor-associated antigens; TACE, transarterial chemo-embolization; TAMs, tumor-associated macrophages; TCR, T-cell receptor; Tex, exhausted T cells; TIC, tumor-initiating cells; TILs, tumor-infiltrating lymphocytes; TKi, tyrosine kinase inhibitors; TLS, tertiary lymphoid structures; TME, tumor microenvironment; Treg, regulatory T cells; TSA, tumor-specific antigens.
References
1. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol. (2018) 15:555–67. doi: 10.1038/s41575-018-0020-y
2. Global Burden of Disease Cancer C. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-Adjusted life-Years for 29 cancer groups:1990 to 2017: a Systematic analysis for the global burden of disease study. JAMA Oncol. (2019) 5:1749–68. doi: 10.1001/jamaoncol.2019.2996
3. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. (2019) 16:589–604. doi: 10.1038/s41575-019-0186-y
4. Muller M, Bird TG, Nault JC. The landscape of gene mutations in cirrhosis and hepatocellular carcinoma. J Hepatol. (2020) 72:990–1002. doi: 10.1016/j.jhep.2020.01.019
5. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From nASH to hCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. (2019) 16:411–28. doi: 10.1038/s41575-019-0145-7
6. Bruix J, Reig M, Sherman M. Evidence-Based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology. (2016) 150:835–53. doi: 10.1053/j.gastro.2015.12.041
7. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. (2008) 359:378–90. doi: 10.1056/NEJMoa0708857
8. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. (2018) 391:1163–73. doi: 10.1016/S0140-6736(18)30207-1
9. Vogel A, Cervantes A, Chau I, Daniele B, Llovet JM, Meyer T, et al. Hepatocellular carcinoma: eSMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2018) 29:iv238–55. doi: 10.1093/annonc/mdy308
10. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. (2009) 10:25–34. doi: 10.1016/S1470-2045(08)70285-7
11. Reig M, Torres F, Rodriguez-Lope C, Forner A, N LL, Rimola J, et al. Early dermatologic adverse events predict better outcome in hCC patients treated with sorafenib. J Hepatol. (2014) 61:318–24. doi: 10.1016/j.jhep.2014.03.030
12. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2017) 389:56–66. doi: 10.1016/S0140-6736(16)32453-9
13. Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med. (2018) 379:54–63. doi: 10.1056/NEJMoa1717002
14. Spratlin JL, Cohen RB, Eadens M, Gore L, Camidge DR, Diab S, et al. Phase I pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J Clin Oncol. (2010) 28:780–7. doi: 10.1200/JCO.2009.23.7537
15. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. (2017) 389:2492–502. doi: 10.1016/S0140-6736(17)31046-2
16. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. (2018) 19:940–952. doi: 10.1016/S1470-2045(18)30351-6
17. Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab as second-Line therapy in patients with advanced hepatocellular carcinoma in kEYNOTE-240: a Randomized, double-Blind, phase III trial. J Clin Oncol. (2020) 38:193–202. doi: 10.1200/JCO.19.01307
18. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. (2020) 382:1894–905. doi: 10.1056/NEJMoa1915745
19. Crispe IN. Immune tolerance in liver disease. Hepatology. (2014) 60:2109–17. doi: 10.1002/hep.27254
20. Robinson MW, Harmon C, O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. (2016) 13:267–76. doi: 10.1038/cmi.2016.3
21. Gola A, Dorrington MG, Speranza E, Sala C, Shih RM, Radtke AJ, et al. Commensal-driven immune zonation of the liver promotes host defence. Nature. (2020) doi: 10.1038/s41586-020-2977-2
22. Mossanen JC, Kohlhepp M, Wehr A, Krenkel O, Liepelt A, Roeth AA, et al. CXCR6 inhibits hepatocarcinogenesis by promoting natural killer t- and cD4(+) t-Cell-Dependent control of senescence. Gastroenterology. (2019) 156:1877–89.e1874. doi: 10.1053/j.gastro.2019.01.247
23. Benechet AP, Iannacone M. Determinants of hepatic effector cD8(+) t cell dynamics. J Hepatol. (2017) 66:228–33. doi: 10.1016/j.jhep.2016.07.011
24. Aizarani N, Saviano A, Sagar Mailly L, Durand S, Herman JS, Pessaux P, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. (2019) 572:199–204. doi: 10.1038/s41586-019-1373-2
25. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell rNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. (2018) 9:4383. doi: 10.1038/s41467-018-06318-7
26. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. (2019) 575:512–518. doi: 10.1038/s41586-019-1631-3
27. Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. (2019) 179:829–45.e820. doi: 10.1016/j.cell.2019.10.003
28. Zhang Q, Lou Y, Yang J, Wang J, Feng J, Zhao Y, et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut. (2019) 68:2019–31. doi: 10.1136/gutjnl-2019-318912
29. Ma L, Hernandez MO, Zhao Y, Mehta M, Tran B, Kelly M, et al. Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell. (2019) 36:418–30.e416. doi: 10.1016/j.ccell.2019.08.007
30. Brunner SF, Roberts ND, Wylie LA, Moore L, Aitken SJ, Davies SE, et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature. (2019) 574:538–42. doi: 10.1038/s41586-019-1670-9
31. Nault JC, Calderaro J, Di Tommaso L, Balabaud C, Zafrani ES, Bioulac-Sage P, et al. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology. (2014) 60:1983–92. doi: 10.1002/hep.27372
32. Cancer Genome Atlas Research Network. Electronic address WBE, Cancer Genome Atlas Research N. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. (2017) 169:1327–41.e1323. doi: 10.1016/j.cell.2017.05.046
33. Chen J, Zaidi S, Rao S, Chen JS, Phan L, Farci P, et al. Analysis of genomes and transcriptomes of hepatocellular carcinomas identifies mutations and gene expression changes in the transforming growth factor-beta pathway. Gastroenterology. (2018) 154:195–210. doi: 10.1053/j.gastro.2017.09.007
34. Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of vEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. (2008) 68:6779–88. doi: 10.1158/0008-5472.CAN-08-0742
35. Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nature genetics. (2015) 47:505–11. doi: 10.1038/ng.3252
36. Zhu M, Lu T, Jia Y, Luo X, Gopal P, Li L, et al. Somatic mutations increase hepatic clonal fitness and regeneration in chronic liver disease. Cell. (2019) 177:608–21.e612. doi: 10.1016/j.cell.2019.03.026
37. Sharma A, Seow JJW, Dutertre CA, Pai R, Bleriot C, Mishra A, et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. (2020) 183:377–94.e321. doi: 10.1016/j.cell.2020.08.040
38. Sia D, Jiao Y, Martinez-Quetglas I, Kuchuk O, Villacorta-Martin C, Castro de Moura M, et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology. (2017) 153:812–26. doi: 10.1053/j.gastro.2017.06.007
39. Kurebayashi Y, Ojima H, Tsujikawa H, Kubota N, Maehara J, Abe Y, et al. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology. (2018) 68:1025–41. doi: 10.1002/hep.29904
40. Rohr-Udilova N, Klinglmuller F, Schulte-Hermann R, Stift J, Herac M, Salzmann M, et al. Deviations of the immune cell landscape between healthy liver and hepatocellular carcinoma. Sci Rep. (2018) 8:6220. doi: 10.1038/s41598-018-24437-5
41. Foerster F, Hess M, Gerhold-Ay A, Marquardt JU, Becker D, Galle PR, et al. The immune contexture of hepatocellular carcinoma predicts clinical outcome. Sci Rep. (2018) 8:5351. doi: 10.1038/s41598-018-21937-2
42. Chew V, Lai L, Pan L, Lim CJ, Li J, Ong R, et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc Natl Acad Sci USA. (2017) 114:E5900–909. doi: 10.1073/pnas.1706559114
43. Itoh S, Yoshizumi T, Yugawa K, Imai D, Yoshiya S, Takeishi K, et al. Impact of immune response on outcomes in hepatocellular carcinoma: association with vascular formation. Hepatology. (2020) doi: 10.1002/hep.31206
44. Lim CJ, Lee YH, Pan L, Lai L, Chua C, Wasser M, et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis b virus-related hepatocellular carcinoma. Gut. (2019) 68:916–27. doi: 10.1136/gutjnl-2018-316510
45. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining 'T cell exhaustion'. Nature reviews. (2019) 19:665–74. doi: 10.1038/s41577-019-0221-9
46. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. TOX transcriptionally and epigenetically programs cD8(+) t cell exhaustion. Nature. (2019) 571:211–8. doi: 10.1038/s41586-019-1325-x
47. Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX is a critical regulator of tumour-specific t cell differentiation. Nature. (2019) 571:270–4. doi: 10.1038/s41586-019-1324-y
48. Wang X, He Q, Shen H, Xia A, Tian W, Yu W, et al. TOX promotes the exhaustion of antitumor cD8(+) t cells by preventing pD1 degradation in hepatocellular carcinoma. J Hepatol. (2019) 71:731–41. doi: 10.1016/j.jhep.2019.05.015
49. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted cD8(+) t cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
50. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Author correction: subsets of exhausted cD8(+) t cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. (2019) 20:1556. doi: 10.1038/s41590-019-0528-5
51. Siddiqui I, Schaeuble K, Chennupati V, Marraco Fuertes SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral tcf1(+)PD-1(+)CD8(+) T Cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. (2019) 50:195–211.e110. doi: 10.1016/j.immuni.2018.12.021
52. Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human cD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. (2019) 70:168–83. doi: 10.1002/hep.30347
53. Zhou J, Peng H, Li K, Qu K, Wang B, Wu Y, et al. Liver-Resident nK cells control antiviral activity of hepatic T Cells via the pD-1-PD-L1 axis. Immunity. (2019) 50:403–17.e404. doi: 10.1016/j.immuni.2018.12.024
54. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased expression of programmed cell death protein 1 on nK cells inhibits nK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene. (2017) 36:6143–53. doi: 10.1038/onc.2017.209
55. Sun H, Xu J, Huang Q, Huang M, Li K, Qu K, et al. Reduced cD160 expression contributes to impaired nK-cell function and poor clinical outcomes in patients with hCC. Cancer Res. (2018) 78:6581–93. doi: 10.1158/0008-5472.CAN-18-1049
56. Sun H, Xu J, Huang Q, Huang M, Li K, Qu K, et al. Correction: reduced cD160 expression contributes to impaired nK-cell function and poor clinical outcomes in patients with hCC. Cancer Res. (2019) 79:1714. doi: 10.1158/0008-5472.CAN-19-0630
57. Zhang Z, Ma L, Goswami S, Ma J, Zheng B, Duan M, et al. Landscape of infiltrating b cells and their clinical significance in human hepatocellular carcinoma. Oncoimmunology. (2019) 8:e1571388. doi: 10.1080/2162402X.2019.1571388
58. Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, et al. Interaction between tumour-infiltrating b cells and t cells controls the progression of hepatocellular carcinoma. Gut. (2017) 66:342–51. doi: 10.1136/gutjnl-2015-310814
59. Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. (2019) 19:307–25. doi: 10.1038/s41568-019-0144-6
60. Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol. (2015) 16:1235–44. doi: 10.1038/ni.3290
61. Endig J, Buitrago-Molina LE, Marhenke S, Reisinger F, Saborowski A, Schutt J, et al. Dual role of the adaptive immune system in liver injury and hepatocellular carcinoma development. Cancer Cell. (2016) 30:308–23. doi: 10.1016/j.ccell.2016.06.009
62. Faggioli F, Palagano E, Di Tommaso L, Donadon M, Marrella V, Recordati C, et al. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology. (2018) 67:1970–85. doi: 10.1002/hep.29636
63. Calderaro J, Petitprez F, Becht E, Laurent A, Hirsch TZ, Rousseau B, et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J Hepatol. (2019) 70:58–65. doi: 10.1016/j.jhep.2018.09.003
64. Yeung OW, Lo CM, Ling CC, Qi X, Geng W, Li CX, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol. (2015) 62:607–16. doi: 10.1016/j.jhep.2014.10.029
65. Yeung OW, Lo CM, Ling CC, Qi X, Geng W, Li CX, et al. Corrigendum to “Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma” [J hepatol 2015;62:607-616]. J Hepatol. (2016) 64:1461. doi: 10.1016/j.jhep.2016.02.038
66. Chen DP, Ning WR, Jiang ZZ, Peng ZP, Zhu LY, Zhuang SM, et al. Glycolytic activation of peritumoral monocytes fosters immune privilege via the pFKFB3-PD-L1 axis in human hepatocellular carcinoma. J Hepatol. (2019) 71:333–43. doi: 10.1016/j.jhep.2019.04.007
67. Wang D, Li X, Li J, Lu Y, Zhao S, Tang X, et al. APOBEC3B interaction with pRC2 modulates microenvironment to promote hCC progression. Gut. (2019) 68:1846–57. doi: 10.1136/gutjnl-2018-317601
68. Guo X, Zhao Y, Yan H, Yang Y, Shen S, Dai X, et al. Single tumor-initiating cells evade immune clearance by recruiting type iI macrophages. Genes Dev. (2017) 31:247–59. doi: 10.1101/gad.294348.116
69. Dhanasekaran R, Baylot V, Kim M, Kuruvilla S, Bellovin DI, Adeniji N, et al. MYC and twist1 cooperate to drive metastasis by eliciting crosstalk between cancer and innate immunity. Elife. (2020) 9:e50731. doi: 10.7554/eLife.50731
70. Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, et al. Tim-3 fosters hCC development by enhancing tGF-beta-mediated alternative activation of macrophages. Gut. (2015) 64:1593–604. doi: 10.1136/gutjnl-2014-307671
71. Lanaya H, Natarajan A, Komposch K, Li L, Amberg N, Chen L, et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat Cell Biol. (2014) 16:972–7. doi: 10.1038/ncb3031
72. Wan S, Kuo N, Kryczek I, Zou W, Welling TH. Myeloid cells in hepatocellular carcinoma. Hepatology. (2015) 62:1304–12. doi: 10.1002/hep.27867
73. Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, et al. Blocking triggering receptor expressed on myeloid cells-1-Positive tumor-Associated macrophages induced by hypoxia reverses immunosuppression and anti-Programmed cell death ligand 1 resistance in liver cancer. Hepatology. (2019) 70:198–214. doi: 10.1002/hep.30593
74. Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell. (2020) 182:886–900.e817. doi: 10.1016/j.cell.2020.07.013
75. Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, et al. Platelet gPIbalpha is a mediator and potential interventional target for nASH and subsequent liver cancer. Nat Med. (2019) 25:641–55. doi: 10.1055/s-0038-1677172
76. Sitia G, Aiolfi R, Di Lucia P, Mainetti M, Fiocchi A, Mingozzi F, et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc Natl Acad Sci USA. (2012) 109:E2165–72. doi: 10.1073/pnas.1209182109
77. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T Cells in liver cancer revealed by single-Cell sequencing. Cell. (2017) 169:1342–56.e1316. doi: 10.1016/j.cell.2017.05.035
78. Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, et al. Author correction: a conserved dendritic-cell regulatory program limits antitumour immunity. Nature. (2020) 582:E17. doi: 10.1038/s41586-020-2326-5
79. Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature. (2020) 580:257–62. doi: 10.1038/s41586-020-2134-y
80. Xiao X, Lao XM, Chen MM, Liu RX, Wei Y, Ouyang FZ, et al. PD-1hi identifies a novel regulatory b-cell population in human hepatoma that promotes disease progression. Cancer Discov. (2016) 6:546–59. doi: 10.1158/2159-8290.CD-15-1408
81. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. (2017) 545:495–9. doi: 10.1038/nature22396
82. Meng X, Liu X, Guo X, Jiang S, Chen T, Hu Z, et al. FBXO38 mediates pD-1 ubiquitination and regulates anti-tumour immunity of t cells. Nature. (2018) 564:130–5. doi: 10.1038/s41586-018-0756-0
83. Macek Jilkova Z, Aspord C, Kurma K, Granon A, Sengel C, Sturm N, et al. Immunologic features of patients with advanced hepatocellular carcinoma before and during sorafenib or anti-programmed death-1/Programmed death-L1 treatment. Clin Transl Gastroenterol. (2019) 10:e00058. doi: 10.14309/ctg.0000000000000058
84. Ng HHM, Lee RY, Goh S, Tay ISY, Lim X, Lee B, et al. Immunohistochemical scoring of CD38 in the tumor microenvironment predicts responsiveness to anti-PD-1/PD-L1 immunotherapy in hepatocellular carcinoma. J Immunother Cancer. (2020) 8:987. doi: 10.1136/jitc-2020-000987
85. Tang H, Liang Y, Anders RA, Taube JM, Qiu X, Mulgaonkar A, et al. PD-L1 on host cells is essential for pD-L1 blockade-mediated tumor regression. J Clin Invest. (2018) 128:580–8. doi: 10.1172/JCI96061
86. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to cTLA-4 blockade in melanoma. N Engl J Med. (2014) 371:2189–99. doi: 10.1056/NEJMoa1406498
87. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
88. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit t cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. (2016) 351:1463–9. doi: 10.1038/s41586-018-0756-0.96
89. DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. (2012) 482:405–9. doi: 10.1038/nature10803
90. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to pD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
91. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of pTEN promotes resistance to T Cells-Mediated immunotherapy. Cancer Discov. (2016) 6:202–16. doi: 10.1158/1538-7445.AM2016-4363
92. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. (2015) 523:231–5. doi: 10.1038/nature14404
93. Harding JJ, Nandakumar S, Armenia J, Khalil DN, Albano M, Ly M, et al. Prospective genotyping of hepatocellular carcinoma: clinical implications of next-Generation sequencing for matching patients to targeted and immune therapies. Clin Cancer Res. (2019) 25:2116–26. doi: 10.1158/1078-0432.CCR-18-2293
94. Ruiz de Galarreta M, Bresnahan E, Molina-Sanchez P, Lindblad KE, Maier B, Sia D, et al. beta-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. (2019) 9:1124–41. doi: 10.1158/2159-8290.CD-19-0074
95. Okrah K, Tarighat S, Liu B, Koeppen H, Wagle MC, Cheng G, et al. Transcriptomic analysis of hepatocellular carcinoma reveals molecular features of disease progression and tumor immune biology. NPJ Precis Oncol. (2018) 2:25. doi: 10.1038/s41698-018-0068-8
96. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to pD-L1 blockade by contributing to exclusion of t cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
97. Duffy AG, Ulahannan SV, Makorova-Rusher O, Rahma O, Wedemeyer H, Pratt D, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol. (2017) 66:545–51. doi: 10.1016/j.jhep.2016.10.029
98. Chew V, Lee YH, Pan L, Nasir NJM, Lim CJ, Chua C, et al. Immune activation underlies a sustained clinical response to Yttrium-90 radioembolisation in hepatocellular carcinoma. Gut. (2019) 68:335–46. doi: 10.1136/gutjnl-2017-315485
99. Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al. Antibodies against immune checkpoint molecules restore functions of tumor-Infiltrating T Cells in hepatocellular carcinomas. Gastroenterology. (2017) 153:1107–19.e1110. doi: 10.1053/j.gastro.2017.06.017
100. Li J, Lee Y, Li Y, Jiang Y, Lu H, Zang W, et al. Co-inhibitory molecule b7 superfamily member 1 expressed by tumor-Infiltrating myeloid cells induces dysfunction of anti-tumor cD8(+)T Cells. Immunity. (2018) 48:773–86.e775. doi: 10.1016/j.immuni.2018.03.018
101. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of lAG-3. Cell. (2019) 176:334–47.e312. doi: 10.1016/j.cell.2018.11.010
102. Zhu Y, Yang J, Xu D, Gao XM, Zhang Z, Hsu JL, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. (2019) 68:1653–66. doi: 10.1136/gutjnl-2019-318419
103. Tan S, Xu Y, Wang Z, Wang T, Du X, Song X, et al. Tim-3 hampers tumor surveillance of liver-Resident and conventional nK cells by disrupting pI3K signaling. Cancer Res. (2020) 80:1130–42. doi: 10.1158/0008-5472.CAN-19-2332
104. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via cCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. (2017) 66:157–67. doi: 10.1136/gutjnl-2015-310514
105. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence-Associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. (2016) 30:533–47. doi: 10.1016/j.ccell.2016.09.003
106. Xu G, Feng D, Yao Y, Li P, Sun H, Yang H, et al. Listeria-based hepatocellular carcinoma vaccine facilitates anti-PD-1 therapy by regulating macrophage polarization. Oncogene. (2020) 39:1429–44. doi: 10.1038/s41388-019-1072-3
107. Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A, et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology. (2020) 71:1247–61. doi: 10.1002/hep.30889
108. Hollande C, Boussier J, Ziai J, Nozawa T, Bondet V, Phung W, et al. Inhibition of the dipeptidyl peptidase dPP4 (CD26) reveals iL-33-dependent eosinophil-mediated control of tumor growth. Nat Immunol. (2019) 20:257–64. doi: 10.1038/s41590-019-0321-5
109. Vollmer CM, Jr Eilber FC, Butterfield LH, Ribas A, Dissette VB, Koh A, et al. Alpha-fetoprotein-specific genetic immunotherapy for hepatocellular carcinoma. Cancer Res. (1999) 59:3064–7.
110. Thimme R, Neagu M, Boettler T, Neumann-Haefelin C, Kersting N, Geissler M, et al. Comprehensive analysis of the alpha-fetoprotein-specific cD8+ t cell responses in patients with hepatocellular carcinoma. Hepatology. (2008) 48:1821–33. doi: 10.1002/hep.22535
111. Zhu W, Peng Y, Wang L, Hong Y, Jiang X, Li Q, et al. Identification of alpha-fetoprotein-specific t-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology. (2018) 68:574–89. doi: 10.1002/hep.29844
112. Mizukoshi E, Nakamoto Y, Marukawa Y, Arai K, Yamashita T, Tsuji H, et al. Cytotoxic t cell responses to human telomerase reverse transcriptase in patients with hepatocellular carcinoma. Hepatology. (2006) 43:1284–94. doi: 10.1002/hep.21203
113. Nakatsura T, Yoshitake Y, Senju S, Monji M, Komori H, Motomura Y, et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem Biophys Res Commun. (2003) 306:16–25. doi: 10.1016/S0006-291X(03)00908-2
114. Capurro M, Wanless IR, Sherman M, Deboer G, Shi W, Miyoshi E, et al. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. (2003) 125:89–97. doi: 10.1016/S0016-5085(03)00689-9
115. Komori H, Nakatsura T, Senju S, Yoshitake Y, Motomura Y, Ikuta Y, et al. Identification of hLA-A2- or hLA-A24-restricted cTL epitopes possibly useful for glypican-3-specific immunotherapy of hepatocellular carcinoma. Clin Cancer Res. (2006) 12:2689–97. doi: 10.1158/1078-0432.CCR-05-2267
116. Cicinnati VR, Zhang X, Yu Z, Ferencik S, Schmitz KJ, Dworacki G, et al. Increased frequencies of cD8+ t lymphocytes recognizing wild-type p53-derived epitopes in peripheral blood correlate with presence of epitope loss tumor variants in patients with hepatocellular carcinoma. Int J Cancer. (2006) 119:2851–60. doi: 10.1002/ijc.22251
117. Zerbini A, Pilli M, Soliani P, Ziegler S, Pelosi G, Orlandini A, et al. Ex vivo characterization of tumor-derived melanoma antigen encoding gene-specific cD8+cells in patients with hepatocellular carcinoma. J Hepatol. (2004) 40:102–9. doi: 10.1016/S0168-8278(03)00484-7
118. Kaji K, Mizukoshi E, Yamashita T, Arai K, Sunagozaka H, Fushimi K, et al. Cellular immune responses for squamous cell carcinoma antigen recognized by T Cells 3 in patients with hepatocellular carcinoma. PLoS ONE. (2017) 12:e0170291. doi: 10.1371/journal.pone.0170291
119. Korangy F, Ormandy LA, Bleck JS, Klempnauer J, Wilkens L, Manns MP, et al. Spontaneous tumor-specific humoral and cellular immune responses to nY-ESO-1 in hepatocellular carcinoma. Clin Cancer Res. (2004) 10:4332–41. doi: 10.1158/1078-0432.CCR-04-0181
120. Thura M, Al-Aidaroos AQ, Gupta A, Chee CE, Lee SC, Hui KM, et al. PRL3-zumab as an immunotherapy to inhibit tumors expressing pRL3 oncoprotein. Nat Commun. (2019) 10:2484. doi: 10.1038/s41467-019-10127-x
121. Shirakawa H, Suzuki H, Shimomura M, Kojima M, Gotohda N, Takahashi S, et al. Glypican-3 expression is correlated with poor prognosis in hepatocellular carcinoma. Cancer Sci. (2009) 100:1403–7. doi: 10.1111/j.1349-7006.2009.01206.x
122. Abou-Alfa GK, Puig O, Daniele B, Kudo M, Merle P, Park JW, et al. Randomized phase II placebo controlled study of codrituzumab in previously treated patients with advanced hepatocellular carcinoma. J Hepatol. (2016) 65:289–95. doi: 10.1016/j.jhep.2016.04.004
123. Hashimoto K. A phase I dose escalation and cohort expansion study of T-cell redirecting bispecific antibody against glypican 3 in patients with advanced solid tumors. J Clin Oncol. (2016) 34:2592. doi: 10.1200/JCO.2016.34.15_suppl.TPS2592
124. Jiang Z, Jiang X, Chen S, Lai Y, Wei X, Li B, et al. Anti-GPC3-CAR T Cells suppress the growth of tumor cells in patient-Derived xenografts of hepatocellular carcinoma. Front Immunol. (2016) 7:690. doi: 10.3389/fimmu.2016.00690
125. Fu Y, Urban DJ, Nani RR, Zhang YF, Li N, Fu H, et al. Glypican-3-Specific antibody drug conjugates targeting hepatocellular carcinoma. Hepatology. (2019) 70:563–76. doi: 10.1002/hep.30326
126. Wu Q, Pi L, Le Trinh T, Zuo C, Xia M, Jiao Y, et al. A novel vaccine targeting glypican-3 as a treatment for hepatocellular carcinoma. Mol Ther. (2017) 25:2299–308. doi: 10.1016/j.ymthe.2017.08.005
127. Li D, Li N, Zhang YF, Fu H, Feng M, Schneider D, et al. Persistent polyfunctional chimeric antigen receptor T Cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology. (2020) 158:2250–65.e2220. doi: 10.1053/j.gastro.2020.02.011
128. Moehler M, Heo J, Lee HC, Tak WY, Chao Y, Paik SW, et al. Vaccinia-based oncolytic immunotherapy pexastimogene devacirepvec in patients with advanced hepatocellular carcinoma after sorafenib failure: a randomized multicenter phase IIb trial (TRAVERSE). Oncoimmunology. (2019) 8:1615817. doi: 10.1080/2162402X.2019.1615817
Keywords: immunotherapy, immune checkpoint inhibitors, tumor microenvironment, tumor-associated macrophages, immunosuppression, inflammation, cirrhosis, NASH
Citation: Giraud J, Chalopin D, Blanc J-F and Saleh M (2021) Hepatocellular Carcinoma Immune Landscape and the Potential of Immunotherapies. Front. Immunol. 12:655697. doi: 10.3389/fimmu.2021.655697
Received: 19 January 2021; Accepted: 22 February 2021;
Published: 18 March 2021.
Edited by:
Frank Tacke, Charité–Universitätsmedizin Berlin, GermanyReviewed by:
Wiebke Werner, Charité–Universitätsmedizin Berlin, GermanySuchira Gallage, German Cancer Research Center (DKFZ), Germany
Copyright © 2021 Giraud, Chalopin, Blanc and Saleh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maya Saleh, bWF5YS5zYWxlaEB1LWJvcmRlYXV4LmZy