 Sean Robinson
Sean Robinson Ranjeny Thomas
Ranjeny Thomas
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 16 July 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.654701
This article is part of the Research TopicGenerating and Sustaining Stable Autoantigen-specific CD4 and CD8 Regulatory T Cells in LupusView all 11 articles
Systemic lupus erythematosus (SLE) is a chronic complex systemic autoimmune disease characterized by multiple autoantibodies and clinical manifestations, with the potential to affect nearly every organ. SLE treatments, including corticosteroids and immunosuppressive drugs, have greatly increased survival rates, but there is no curative therapy and SLE management is limited by drug complications and toxicities. There is an obvious clinical need for safe, effective SLE treatments. A promising treatment avenue is to restore immunological tolerance to reduce inflammatory clinical manifestations of SLE. Indeed, recent clinical trials of low-dose IL-2 supplementation in SLE patients showed that in vivo expansion of regulatory T cells (Treg cells) is associated with dramatic but transient improvement in SLE disease markers and clinical manifestations. However, the Treg cells that expanded were short-lived and unstable. Alternatively, antigen-specific tolerance (ASIT) approaches that establish long-lived immunological tolerance could be deployed in the context of SLE. In this review, we discuss the potential benefits and challenges of nanoparticle ASIT approaches to induce prolonged immunological tolerance in SLE.
Systemic Lupus Erythematosus (SLE) is a chronic autoimmune inflammatory disease that affects multiple organ systems. Clinical symptoms are heterogenous and range from mild to life threatening. SLE has a significant disease burden worldwide. Mortality in SLE has decreased significantly in the past 50 years (1), attributed to the use of immunosuppressive drugs, better supportive treatments and earlier diagnosis. Acute SLE-related mortality is usually due to uncontrolled inflammation and acute renal failure, while late mortality is linked to cardiovascular complications (2). Since the 1990’s late-phase clinical trials from more than 40 agents have failed in SLE. However, improvement in outcome measures, the efficacy of B cell activating factor (BAFF) and type 1 interferon (IFN) receptor 1 inhibition, and the promise of tolerance restoration, through drugs such as low-dose (LD) IL-2, underpin new optimism for future drug development (3–5). Tolerizing immunotherapies have the potential to revolutionize the treatment of autoimmune diseases by directly impacting adaptive immunity and restricting autoinflammatory responses by inducing peripheral immunological tolerance, either by expanding pre‐existing regulatory T cells (Treg) or by reprogramming autoreactive CD4+ T cells into Treg. While not extensively trialed in SLE yet, promising data in other autoimmune diseases provide learnings that may be applicable in SLE and patients at high-risk. In this review we examine the potential for antigen-specific immunotherapy to restore tolerance in lupus autoimmunity and discuss the advantages and challenges of immunotherapies and tolerizing approaches in SLE.
SLE is 43.9% heritable, and the relative risk for siblings is 23.7. Shared environmental factors - such as infections - account for 25.8% of risk: the relative risk for spouses is 4.4 (6). Although the pathogenesis of SLE is not fully understood, the key elements are: dysregulated immune tolerance towards autologous nucleic acids with concurrent production of autoantibodies and autoreactive T-cells, disrupted clearance of apoptotic debris with increased self-antigen load and presentation to T cells, and interferon-driven inflammatory responses (7). Tissue damage – to skin, respiratory, renal, cardiovascular, central nervous and musculoskeletal systems – results from pathogenic autoantibodies, immune complex deposition and inflammation. SLE-associated environmental stressors, including UV light and infections may increase apoptotic load. With inadequate clearance, Toll Like Receptors (TLRs) recognize cellular debris (through damage associated molecular patterns, DAMPs) and initiate the inflammatory cascade, with pro inflammatory cytokine and type 1 interferon (IFN) production (8, 9). Presentation of nuclear self-antigens, such as dsDNA, chromatin, and RNA-containing antigens, to T and B cells induces the production of nuclear antigen-specific autoantibodies and autoreactive T-cells. There are multiple autoantibodies in SLE, including those directed towards nuclear antigen (ANA), double-stranded DNA (dsDNA), Smith (Sm), Ro, La, antiphospholipid (APL), and ribonucleoproteins (RNP) (10). Multiple lines of enquiry demonstrate loss of T and B cell tolerance in lupus. For example, the study of rare genetic variants associated with familial aggregation of lupus with other rheumatic autoimmune diseases identified regulation of T cell activation and T cell receptor (TCR) signaling as key underlying pathways (11). Furthermore, single cell transcriptomic analysis of peripheral blood (PB) identified antigen presenting cell, B cell and T cell dysregulation (12).
SLE is classified (EULAR/ACR 2019 criteria) by the presence of ANA >1:80 and weighted scores for clinical and serological parameters (13). Some ANA+ individuals with very early disease or disease in evolution may fall below classification threshold. They may progress, to be re-classified as SLE, or may follow a milder and more stable clinical course. By the time of diagnosis, the majority of patients that meet SLE criteria will have some type of irreversible organ damage with clinical complications. The lupus disease course is characterized by flares and ongoing organ damage (14). Therapeutic intervention to a target of low disease activity (LLDAS) or clinical and serological remission reduces lupus-associated flares and organ damage, even when only achieved transiently (15). Typically, phase 3 trials of novel agents in SLE have struggled with small effect sizes due to disease heterogeneity, trial design issues, use of concomitant immunosuppression and endpoint validation (15). Instigation of trials in early disease and high-risk subjects not yet classified as SLE may improve the capacity to discriminate responses in patients with minimal organ damage. In a landmark phase 2 trial, a short course of T cell tolerizing immunotherapy Teplizumab, halved the progression of high-risk individuals to type 1 diabetes (16), while it had failed to meet its primary end-point in a phase 3 trial in recent-onset diabetes (17). Thus, T cell immunotherapy in people at risk (18) may be more effective before substantial organ damage.
SLE is associated with more autoantibodies than any other autoimmune disorder (19). Even before the development of disease pathology and symptoms, the pre-clinical phase is characterized by increased levels of autoantibodies, followed by a shift to multiple pathogenic autoantibodies associated with kidney, joint, heart, brain, skin and hematopoietic damage, including ANA, anti-dsDNA, anti-Sm, anti RNP, anti-APL, anti-Ro and anti-La (10). In general, anti-Ro, anti-La, and APL appear several years before the diagnosis of SLE, even in otherwise healthy individuals (20). In contrast, anti-dsDNA, anti-Sm, and anti–nuclear RNP antibodies usually appear only months before the clinical manifestations of SLE and are rarely present in healthy individuals (21, 22). In a retrospective study of 130 military personnel, use of hydroxychloroquine prior to SLE diagnosis delayed the onset of classified SLE and reduced the number of autoantibody specificities at and after diagnosis (23). At least 80% of individuals in this group met at least one SLE criterion prior to diagnosis. These results support a case for earlier therapeutic intervention with treatments of low toxicity before SLE classification and stratification of patients based on likelihood to respond. For example, current smoking was associated with elevated BAFF and reduced IL-10, particularly in ANA+ women (24). T cell expansion and type 1 IFN signatures were associated with a diagnosis of SLE in ANA+ individuals (25). Longitudinal cohort studies mapping the progression of SLE in auto-antibody positive healthy at-risk subjects will help identify early biomarkers of progression from autoantibodies to SLE, such as markers of functional loss of immune tolerance (26). Furthermore certain immune phenotypes might also be useful response biomarkers in mechanistic trials of immune tolerizing immunotherapies in individuals at high risk or with early disease.
Alternatively tolerizing approaches could be used to increase the likelihood that immunosuppressive drugs can be safely withdrawn without flare. In the BOLD clinical trial, standard immunosuppressive drugs were withdrawn and steroids substituted until flare, followed by reinstatement of standard therapy. During each phase cytokines and gene expression were analyzed to assess drug mechanism of action relative to baseline type 1 IFN transcriptomic signature. The authors identified that IL-17, IL-23 and BLyS pathways were changing with disease state and that IFN signature influenced the response of these pathways to individual drugs (27). This study provides an interesting proof-of-concept for a mechanistic trial of agents, such as tolerizing therapies, that could be introduced to reduce flare upon drug withdrawal. Although no cellular markers were included in this study, it demonstrates the utility of baseline IFN signature to stratify immune biomarker response outcomes. Future trials might also include Treg or T cell proliferation biomarkers. In this regard, a PB single cell transcriptomic resource shows co-clustering of a Treg T cell signature with dendritic cells (DC) lacking IFN-stimulated genes in lupus patients and healthy donors (12).
Immunological tolerance is a vital aspect of a healthy immune system as it allows for appropriate immune responses to infectious and tumor antigens while containing potentially damaging immune responses to self-antigen and healthy tissue. Reviews of B and T cell antigen recognition and maturation can be found here (28, 29). During development, highly self-reactive T cells in the thymus are controlled by deletion (negative selection) of T cells with the highest affinity TCR for self-peptides, and by differentiation into CD4+ CD25+ FOXP3+ Treg cells (for non-deleted autoreactive CD4+ T cells), known as central tolerance. As negative selection depends on a TCR affinity threshold, weakly autoreactive T cells circulate in the periphery (30). Peripheral T cell tolerance mechanisms control autoreactive T cells through anergy (chronic antigen exposure deactivating T cell function), deletion, and regulation by Treg (derived from thymus or generated in the periphery). Antigen-specific Treg cells can suppress activation, proliferation and cytokine production of CD4+ T cells and CD8+ T cells through interaction with APCs, including B cells and dendritic cells (DCs), presenting cognate antigen. Functional antigen-specific peripheral Treg are key to restoration of immunological tolerance with immunotherapy as they can be induced from diverse T cell precursors, and their autoantigen specificity avoids generalized immune suppression (31).
Peripherally derived Treg cells, including IL-10+ type 1 regulatory T (Tr1) cells, are promising targets for immunotherapy to counteract established autoimmune diseases. Tr1 cells are induced in the periphery, predominantly from memory CD4 T cells, and are thus an important potential target for antigen-specific tolerance approaches (32–34). They are characterized by expression of IL-10, IFN-γ and TGF-β, lack of FOXP3 expression, expression of surface markers LAG3 and CD49b, and transcription factors EOMES and Tbet (35–37). With ongoing signaling by tolerogenic APCs presenting cognate peptide. Tr1 cells are long-lived, and associated with prolonged tolerance in multiple human autoimmune conditions (38–41).
DCs comprise a heterogeneous group of phagocytic APCs that sample soluble or apoptotic antigen at skin and mucosal surfaces, and process and present antigenic peptides to T cells in draining lymph nodes in context of MHC molecules. During an inflammatory episode, e.g. driven by infection, adjuvants, or damage, pathogen- or damage-associated molecular patterns (PAMPs, DAMPs) trigger the activation of the NF-κB pathway in DCs, enhancing their capacity to stimulate naïve T cells (42). DCs presenting antigens in the presence of regulatory signals that inhibit NF-κB, such as TGF-β or immunomodulatory drugs, skew antigen-specific T-cells towards regulation (43).
DC subsets developing from hematopoietic progenitors in bone marrow include plasmacytoid DC (pDCs), myeloid/conventional DC1 (cDC1) and myeloid/conventional DC2 (cDC2), based on surface markers and immune functions (44, 45). cDC1 and moDCs can cross-present antigens derived from tissues – including viral, tumor and self-antigens – to CD8 and CD4 T cells in context of MHC I and II (46, 47). cDC2 are potent activators of naïve T cells and induce CD4+ Th1, Th2, and Th17 responses (48, 49). pDCs produce high levels of type 1 IFN in response to nucleic acids via TLR7 and TLR9 signaling (50, 51). In SLE, pDCs produce high levels of type 1 IFN in response to nucleic acid and nuclear antigen (52). DCs are potential targets for immunotherapies to restore the dysregulated SLE immune system. For example, crosstalk between Treg cells and DCs through cell and cytokine signaling, controls DC activation and effector T cell activation. The signaling pathways for DC development and activation are crucial when considering drug cargo in the development of novel therapies in lupus.
A basic understanding of the pathogenesis of SLE underpins a discussion on the development and effectiveness of novel immunotherapeutic agents. Here we highlight a few important factors that point to the underlying causes of SLE and that could be targeted in a therapeutic approach. For further reading please refer to the following reviews (53, 54).
SLE has been extensively studied using mouse models, which has helped illuminate pathogenesis. Some mouse models are genetically predisposed to the development of a lupus-like disease. Alternatively, a lupus-like disease may be induced in previously healthy mice. However, although spontaneous SLE models have been used to test potential therapeutics, successes in mouse models have not translated well in human trials. The NZB/NZWF1 (BW) mice and related strains develop spontaneous immune complex-mediated glomerulonephritis and mild vasculitis, with autoantibodies (anti-nuclear antibodies (ANA) and anti-dsDNA predominantly) (55). The MRL/lpr mouse is a unique spontaneous lupus mouse model that produces a variety of autoantibodies (ANA, anti-dsDNA, anti-Sm, anti-Ro and anti-La) and develops arthritis, cerebritis, dermatitis, vasculitis, and glomerulonephritis (56, 57). In induced mouse models of SLE, exogenous irritants or antigens are administered to replicate an environmental trigger (58, 59). Knock-out and knock-in mice backcrossed to lupus-susceptible backgrounds has expanded understanding of signaling cascades crucial for the development of SLE (60). Few antigens have been described in lupus mouse models, hampering the testing of antigen-specific approaches for lupus in general. However, antigen non-specific tolerizing approaches have been demonstrated to improve SLE disease manifestations in mouse models (61).
Hormones, smoking, ultraviolet light, and viral/bacterial infections are classic examples of exposures triggering SLE (62, 63). Estrogen and prolactin have been shown to drive immune responses underpin in part preponderance of women with SLE (64). Viral infection such as Epstein-Barr virus and cytomegalovirus have been suspected to play a triggering role in SLE pathogenesis whereas some pathogens have been linked to a protective role in SLE (65–67). Circulating levels of lipopolysaccharides have been shown to be elevated in SLE patients and to be correlated with disease severity, presumably through cytokine production (68, 69). Recently, alterations in gut microbiome have been linked to SLE disease status (70–72). This review will not thoroughly cover these environmental factors of but we note that they are important considerations when developing therapeutic trials for potential interventions.
Abnormal apoptotic clearance can trigger TLRs and nucleic acid sensors on immune and non-immune cells and produce an immune response with cytokine production (7). Rare hereditary genetic mutations e.g. in DNASE1L3 and PRKCD that lead to abnormal apoptotic pathways provide crucial insight into the role of apoptotic breakdown and debris clearance in SLE (73, 74). DNase I activity degrades chromatin in the apoptotic process and mice with a mutation in this enzyme had increased levels of anti-DNA antibody production (75). Smoking induces cellular damage and promotes cytokine production, and UV light enhances apoptotic turnover, and thus may increase self-antigen burden in susceptible individuals (76, 77)
Nucleic acid sensors are important surveyors of the environment and are specifically able to recognize viral infections and induce type I IFN production. Toll-Like receptors 3, 7, 8, and 9 shape the immune response by sensing cellular debris (78). In a pristane-induced lupus mouse model, TLR7, which senses single stranded RNA, was required for RNA-reactive autoantibodies (8). TLR9 senses unmethylated CpG sequence motifs. SLE patients with active disease have higher level of TLR9+ B cells and monocytes than healthy controls, and TLR9 levels correlated with antibodies to dsDNA (79, 80).
Type I and Type II IFN contribute a large role to the pathogenesis of SLE and become elevated prior to development of autoantibodies (81). Rare single gene disorders, grouped together as Aicardi-Goutiere’s syndrome, display gene defects that cause an overproduction of type I IFN (82). These patients display similar phenotypes to classic SLE, including autoantibodies.
There is a marked imbalance of T cell cytokines in SLE, with low levels of IL-2 accompanied by elevated IL-17 and IL-6 (83). IL-2 is a key cytokine in Treg development, survival and maintenance. It restricts Th17 cell development (84, 85). Elevated levels of IL-17 are thought to induce tissue inflammation and recruitment of immune cells. B cell activating factor (BAFF or BLyS), expressed by stromal and immune cells, promotes B cell activation in SLE and its levels positively correlate with antibody levels (86, 87).
The process of autoimmune disease development can be roughly categorized into three stages: 1) a priming phase that includes an inciting event or accumulation of events in individuals at genetic and environmental risk; 2) the onset of clinical symptoms marked by organ-specific inflammation; and 3) a chronic inflammatory tissue-destructive phase (88). During the transition to clinically significant symptoms, regulatory processes, including Treg cells, fail to control pathological autoreactive B and T cells. This imbalance perpetuates the processes of bystander activation, epitope spreading and uncontrolled cytokine and antibody production. Epitope spreading involves the diversification of epitope specificity from the initial dominant epitope-specific immune response (89). The specificity of the autoimmune response spreads to include additional self-epitopes besides the initiating self-antigens. Chronic inflammation promotes tissue damage and cascading self-antigen presentation, expanding autoreactive T-cell specificities, including cryptic or sequestered epitopes (90). For example, late-stage SLE is characterized by an explosion of autoantibodies, apparently the result of chronic inflammation and epitope spreading (19). Bystander activation occurs with stress, infection or trauma-induced activation of tissue APCs, activating T cells of additional specificities, which further promote inflammation and tissue damage. Bystander T cells can provide help to B cells for autoantibody production, or to cross-presenting DCs presenting tissue-derived self-antigen (91). Treg cells may control bystander T cells and epitope spreading through interaction with cross-presenting DCs. In a rheumatoid arthritis mouse model Treg cell depletion promoted the expansion of pathogenic autoreactive T cells, an increase in inflammatory cytokines, and B‐cell epitope spreading (92).
SLE is marked by abnormal B and T cell interactions and spontaneous germinal centers in secondary lymphoid organs (93–95). In SLE there is loss of functional Treg and induction of effector T cells that produce proinflammatory cytokines and BAFF, which is not normally observed in healthy people (96, 97) (98). Multiple lines of evidence demonstrate the importance of Treg in lupus pre-clinical models. In the NZB/NZWF1 spontaneous model, Treg cell adoptive transfer delayed SLE progression, reduced renal pathology, and improved survival (61), while Treg depletion accelerated disease development (99). In human SLE, most but not all studies demonstrate a reduced frequency of Treg cells (100, 101). Targeted depletion of pDCs decreased SLE-associated glomerulonephritis in mice (102, 103). In human SLE, while pDC are decreased in the blood, they are increased in lupus-affected organs, suggesting their chemo-attraction and possible expansion at these sites (104–106).
There are multiple potential targeted immunotherapies undergoing research and development and early phase clinical trials for SLE (107, 108). Most techniques exploit antigen‐presentation pathways of APCs or attempt to deliver antigenic cargos to locations thought to be involved in regulatory T‐cell formation (109). Other strategies target antigen‐specific T‐cells to re‐program pathogenic autoreactivity into disease‐suppressing autoregulation (110, 111). Table 1 outlines some promising therapeutic directions aiming to enhance immune tolerance by targeting DCs and Treg cells.

Table 1 Treg and DC based Therapies without Autoantigen.
Several groups have developed methods to expand Treg cells ex vivo for reintroduction as an autologous cell therapy product. Treg cells can be isolated from peripheral blood or umbilical cord blood, but must be expanded due to their low frequency. In vitro expansion strategies include anti-CD3/CD28-coated beads, with addition of IL-2 and/or TGF-β and rapamycin (121). Proof of concept experiments in lupus-prone mice showed that ex vivo-expanded Treg cells suppressed glomerulonephritis and prolonged survival (61, 122). Ex vivo-expansion of Treg cells in the presence of immunosuppressive drugs or Treg transfer into patients on immunosuppressants can be challenging, as the drugs may hinder expansion or change function (112). Furthermore, the process requires a good manufacturing practice (GMP) environment, which is challenging and expensive. A clinical trial using ex vivo-expanded autologous polyclonal Treg cells in patients with autoimmune disease was terminated in November 2019 due to screen failures and low enrolment. In a case report, the treatment was shown to be safe and clinical disease activity to be stable in a single SLE patient. Infused labeled Treg cells were transiently observed in PB then in diseased SLE skin, accompanied by skewing from Th1 to Th17 immunity locally (123). Treg are highly plastic and may differentiate to Th17 in inflammatory settings and where IL-2 is limiting (124). Larger studies are needed to understand the impact of Treg therapy on disease severity.
Hematopoietic and/or mesenchymal stem cell transfer (HSCT and MSCT, respectively) have been trialed in patients with severe autoimmune diseases, including SLE, who have failed standard therapy. In SLE patients, HSCT has successfully induced long-term remission (125). In 15 patients with severe SLE evaluated up to 8 years after HSCT, CD4+CD25highFoxp3+ Treg and LAPhighTGF-β+CD8+Foxp3+ cells were restored to levels and function similar to healthy subjects (117). These promising results suggest that HSCT may reestablish immune tolerance by replenishing multiple types of Treg cells. However, as HSCT is associated with significant risks, treatment complications and cost, it is currently reserved for treatment-refractory patients. A 4-year follow-up of an open-label trial of MSCT in 87 treatment-refractory SLE patients found a 28% remission rate post-infusion (118). While double-blind placebo-controlled trials are needed to understand the true benefits of MSCT, these trials provide evidence that tolerance may be successfully re-established in SLE.
IL-2 levels and CD25 expression by Treg are reduced in SLE patients and murine lupus models (126–128). IL-2 plays a pleiomorphic role in the immune system. One of its functions is to expand and promote survival of Treg cells (129, 130). Reduced IL-2 favors the differentiation of IFN-γ-producing Th1 and IL-17 producing Th17 cells and their accumulation in skin and kidneys (131, 132), and is associated with inflammation. In lupus-prone mice, IL-2 treatment increased levels of Treg cells in lymphoid and peripheral organs and protected them from SLE-related organ damage (99, 133). There have been several trials in lupus showing safety and Treg expansion (128, 134). In a recent double-blind placebo-controlled clinical trial in patients with suboptimally controlled SLE, LD IL-2 for 12 weeks (s.c. alternate days for three 2-week cycles), the SLE Responder Index (SRI)-4 response rates at week 12 were 55.17% and 30.00% in LD IL-2 and placebo groups respectively (p=0.052). Although the primary end point was not met, the significantly greater lupus nephritis complete remission rate in the LD IL-2 arm was notable. Immunologically, IL-2 supplementation significantly increased Tregs and NK cells but did not change total CD4+ or CD8+ T cells and there was no increase in viral load of pre-existing viruses (3). While promising, LD IL-2 dosing may be complicated by concomitant expansion of regulatory and cytotoxic cells. Furthermore, development of neutralizing autoantibodies with continued treatment is a potential risk (135). Targeted IL-2 therapies may allow more precise manipulation of the immune response and longer duration of action. For example, anti-CD4 and anti-CD2-coated poly(lactic-co-glycolic) acid (PLGA) nanoparticles loaded with IL-2 and TGFβ expanded Treg cells in vitro and in vivo in the BDF1 lupus pre-clinical model (136). In a recent phase 1b clinical trial of a polyethylene glycol (PEG) conjugate of IL-2 (NKTR-358) in patients with mild to moderate SLE, dose-dependent increases in Tregs (up to 11 fold) were observed, which returned to baseline 20-30 days post-dose (137). Anti-IL-2 antibodies were not reported.
DCs play a critical role in maintaining self-tolerance. Indeed, targeting steady-state skin migratory DC with antigen coupled to DC-selective antibodies induced antigen-specific tolerance (138). Tolerogenic DCs can also be generated in vitro from monocytes or murine bone marrow precursors in the presence of NF-κB inhibitors 1,25 (OH)2 vitamin D3 (calcitriol), rapamycin or glucocorticoids. After proof-of-concept studies in experimental animal models (139, 140), several groups translated antigen-specific immunotherapy using modified or tolerogenic autologous DCs and autoantigenic peptides to clinical trials for MS (113) and RA (114, 115). These trials demonstrate the feasibility and safety of this approach, with preliminary evidence of an immunomodulatory effect in RA. In two pre-clinical lupus models, histone antigen-loaded tolerogenic DCs improved clinical scores, increased Treg in affected skin and reduced anti-histone autoantibodies (141). Tolerogenic DCs exposed to apoptotic cells were generated from PB monocytes derived from lupus patients (142). Other approaches have been developed to target DCs directly in situ, including a PLGA nanogel to deliver the immunomodulator mycophenolic acid (MPA) to DCs (119, 120). DCs took up the PLGA-lipid-MPA nanogel more efficiently and with better DC suppression than a PLGA nanogel. In a murine lupus model, PLGA-MPA nanogel increased median survival by 3 months when given prophylactically and by 2 months when given to mice with advanced renal damage. Consistent with the local effects of MPA on DCs, treated mice had a substantial reduction in DC-derived inflammatory cytokines such as IFN-γ and IL-12. Although not strictly immune tolerance, this approach achieves sustained delivery of MPA to induce a prolonged anti-inflammatory effect.
Lupuzor (rigerimod or IPP-201101) is a 21aa peptide representing residues 131–151 of the 70K spliceosomal protein within the U1 small nuclear RNP, phosphorylated at Ser140. This promiscuous peptide sequence was identified using ex vivo peptide screening techniques (143). This epitope is recognized by IgG antibodies and CD4+ T cells from H‐2k MRL/lpr and H‐2d/z (NZB × NZW)F1 lupus‐prone mice (143, 144). With i.v. delivery, the peptide inhibits chaperone-mediated autophagy and reduces B cell MHC class II expression (145). Two trials of IPP-201101 immunotherapy in SLE demonstrated safety and potential efficacy (146, 147). However, IPP-201101 failed to meet its primary end point of superiority over standard care in phase III clinical trials (148). The peptide seemed to have non antigen-specific immunomodulatory properties, rather than inducing antigen-specific regulation, and this may be why it was not superior to standard care. Standard of care high dose glucocorticoids and immunosuppressive drugs are likely more bioavailable than an immunosuppressive peptide.
These treatment strategies are antigen non-specific and use nanoparticles (NP) to deliver biologics or immunosuppressive drugs. In the following sections we consider antigen-specific tolerizing approaches using NP in SLE.
Antigen-specific therapies for autoimmune diseases involve the delivery of autoantigen in a regulatory context, with or without a delivery vehicle that reprograms APCs by modulating NF-κB, or by antigen delivery to a naturally tolerogenic site e.g. by targeting steady-state skin-draining APCs or the liver tolerogenic environment. Some approaches may directly differentiate Tr1 cells from memory T cells.
Peptide alone, delivered s.c., can be tolerogenic. For example, an islet proinsulin epitope returned promising results in phase 1 trials in T1D (149). Peptides that associate with MHC class II molecules expressed by APCs, without the need for antigen processing, can directly target steady-state DC in vivo. Such antigen processing independent epitopes (“apitopes”) selectively bind steady-state DCs in vivo because steady-state DCs bear peptide receptive/empty MHC II at the cell surface, which is lost upon DC activation (150, 151). Apitopes induce tolerance through induction of anergy and generation of Tr1 cells (152). Tr1 cells selectively express a tolerance-associated set of genes (153, 154). Phase 1 and 2 clinical trials of multiple low dose apitope delivery have been undertaken in Graves’ disease and MS respectively. While low-dose soluble antigen administered s.c. is non-immunogenic, high dose peptide, aggregates or protein complexes can induce an immune response through immune complex formation, macrophage or DC activation and development of autoantibodies.
Liposome formulations loaded with peptide or protein antigens and various NF-κB inhibitors, including curcumin, quecertin and BAY11-7082 induced antigen-specific tolerance in mice with antigen-induced arthritis (155). We also developed and undertook pre-clinical studies of liposomes co-encapsulating calcitriol and peptide. Calcitriol/peptide liposomes promoted the differentiation of antigen-specific Foxp3+ Treg, anergy of Tmem, and IL-10 production upon restimulation with antigen ex vivo (156). Notably, liposomes were preferentially taken up by activated PD-L1+ migratory DCs, and regulation was PD-L1-dependent. We translated this to a phase 1b clinical trial in RA. Other groups have co-encapsulated antigens in NPs with either rapamycin (157) or aryl hydrocarbon receptor (AhR) ligands (158) for in vivo uptake by DC. With substitution of suitable lupus antigenic peptides, these liposome or NP approaches could be adapted to lupus patients.
Other research groups have developed NPs that resemble apoptotic bodies, to promote a tolerogenic response to encapsulated antigen. Specifically, i.v. administration of 500nm PLGA particles encapsulating antigen induced antigen specific tolerance (159, 160). These relatively large, negatively-charged particles are preferentially taken up by DCs and macrophages expressing MARCO, and induce antigen-specific suppression in the absence of an immunomodulatory drug (161). Another strategy to mimic signals from apoptotic bodies uses phosphatidylserine (PS) liposomes. During apoptosis, the PS phospholipid translocates from the inner leaflet to the outer leaflet of the lipid bilayer of the dying cell. PS liposomes suppressed pre-clinical models of T1D and acute EAE in a non-antigen-specific manner (162, 163). It is unclear whether this technique would succeed in SLE, which is characterized by impaired clearance of apoptotic cells.
The TCR may also be directly targeted with NPs coated with peptide loaded onto MHC class I or II, without co-stimulation. After i.v. delivery of iron oxide nanoparticles coated with peptide-MHC class I complexes (pMHC-I) they suppressed autoreactive CD8+ memory T cells and converted them to a regulatory, anergic phenotype (110). Nanoparticles coated with pMHC-II differentiated cognate autoreactive CD4 memory T cells into Tr1 cells producing IL-10 (111, 164). Nanoparticles coated with pMHC-II suppressed autoimmune symptoms in several pre-clinical models in an antigen-specific manner, without compromising systemic immunity (111). To date, this approach has not been translated to clinical trials.
Thus, a wide array of nanoparticle technologies has been developed. Figure 1 describes some of the technologies incorporating autoantigens, immunomodulatory drugs, or targeting strategies, or a combination of strategies. In summary, approaches that promote the expansion of antigen-specific Treg cells, particularly Tr1 cells derived from autoreactive memory T cells, will be required to control bystander cytokine production and epitope spreading in multi-system autoimmune diseases, such as SLE.
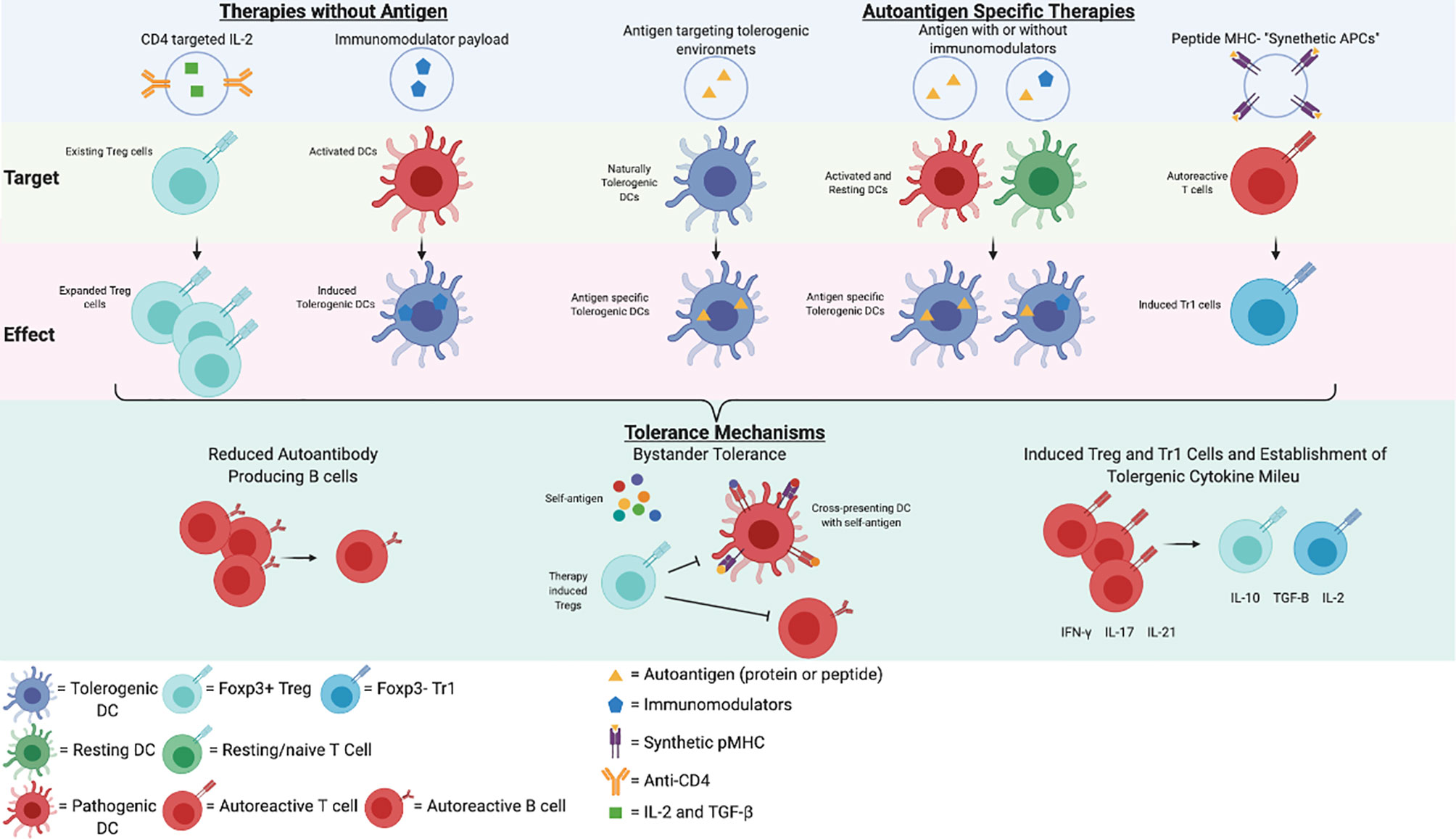
Figure 1 Potential Nanoparticle Therapies in SLE. A representation of potential immunotherapies for SLE and a simplified schematic of their mechanisms of action. This figure was created in BioRender.com.
Many autoantigens potentially contribute to the development of SLE and it is unclear which antigen(s) should be targeted in antigen-specific immunotherapy. Several promiscuous epitopes have been described across mice and humans. Choosing an antigen is challenging because there are many different pre-clinical lupus models, the disease is highly heterogeneous in humans, and translation of antigen discovery from mouse to human is difficult. However, assays of T cell responses in organ-dominant lupus “endotypes” may offer opportunities to identify relevant skin, joint, renal, neurological and hematologic antigenic epitopes that are suitable for clinical trials with focused outcomes.
Despite these hurdles, antigen-specific immunotherapies with a single strong autoantigen that also promote bystander tolerance could leverage the expansion of antigen-specific Treg cells and the suppression of cross-presenting DCs carrying relevant epitopes from diseased tissue to draining lymph nodes. Bystander tolerance has been demonstrated for several immunotherapies in pre-clinical models, including apitopes, peptide/calcitriol liposomes, and pMHC-NP, associated with the modulation of immune responses other than the epitope included in the immunotherapy (111, 165). For example, in a type 1 diabetes mouse model, calcitriol liposomes encapsulating a single islet CD4 epitope suppressed the disease and bystander islet-reactive CD8+ cytotoxic T cells (166). The advantages of harnessing bystander tolerance mediated by Treg cells, compared to generalized immunosuppression, is that bystander suppression is tissue-restricted, and Treg develop from autoreactive memory T cells. However, suitable antigenic epitopes must be identified.
Haplotypes containing DR2/DQ6, and DR3/DQ2 alleles are associated with SLE (167). DR2/DR3 heterozygosity is associated with anti-Ro, anti-La, anti-Sm, anti-ribosomal-P or anti-ribonuclear protein antibodies, while HLA-DR homozyosity is associated with anti-Sm and anti-dsDNA (167). HLA-restriction poses a potential hurdle for the applicability of peptide-specific immunotherapies, as peptides need to be identified and matched to patient MHC class II. Long antigenic sequences or mixtures of epitopes that cover a large percentage of the diseased population provide potential solutions. HLA-restricted soluble or NP-associated peptide immunotherapy may be a good way to achieve some early positive immune outcomes of antigen-specific immunotherapies, including bystander tolerance in proof-of-concept clinical trials. Subsequently, tolerizing immunotherapies with multiple autoantigens or proteins could be further tested.
Strategies to identify potential self-peptides include: screening autoreactive T cell proliferation or cytokine production ex vivo, peptide elution from MHC II molecules, and autoantibody binding epitopes. Immunization studies in DR3 transgenic mice have been used to map DR3-restricted SmD T cell epitopes (168). Studies investigating apoptotic cell-derived self-epitopes recognized by pathogenic T cells in human and lupus-prone mouse models identified potential histone epitopes, including histone H1’22-42, H416–39, H471–94 and H382–105 (169, 170). These extended epitopes bind multiple HLA-DR allomorphs. Most also bound anti-histone autoantibodies (171, 172). In human PB cultures, these peptides promoted TGF-β secretion and expanded Foxp3+Treg cells in the presence of IL-2 in vitro (170). In SVF1 lupus-prone mice, s.c. administration of H471–94 every 2 weeks induced TGF-β-producing pDCs and Treg cells and protected mice from renal disease (173, 174). A 70K-U1RNP131-151 T helper epitope was identified in NZBxNZW F1 and MRL/Fas(lpr) mice, which led to further identification of SmD1 and hnRNP A2/B1 epitopes in each strain. Of interest the SmD95-119 epitope recognized by anti-Sm antibodies is homologous to an Epstein-Barr virus EBNA I peptide, suggesting a mechanism for epitope spreading through bystander T helper cells (144, 175). Certain nuclear antigens tend to induce epitope spreading to related other nuclear antigens in mouse models (Table 2). If administered as antigen-specific tolerizing immunotherapy, one would therefore predict induction of bystander tolerance (183).
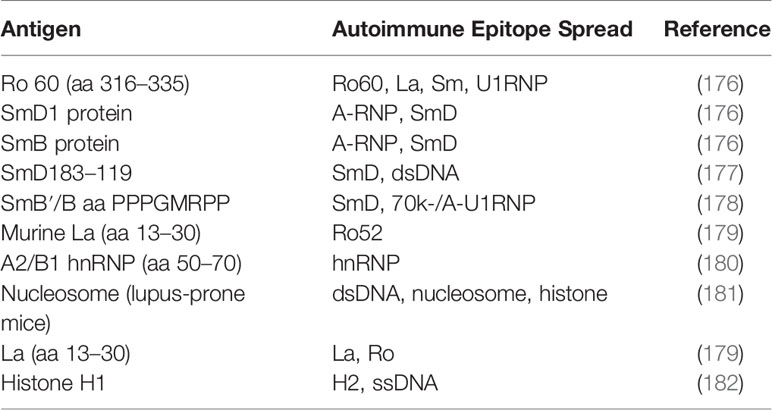
Table 2 Epitope spreading in mouse models after autoantigen immunization.
Further research into SLE immunotherapy would benefit greatly from a humanized model that could better represent the human immune system (184, 185).
SLE is a devastating autoimmune disease with a large unmet need for better therapies. Promising work has identified some immunological markers of immune tolerance in individuals at risk who have not progressed to a diagnosis of SLE, and some nuclear-derived antigenic epitopes that may be presented by multiple MHC II molecules. More work is needed to carefully map the autoantigen specificity and HLA restriction of expanded T cells in patients with recent-onset SLE. The pre-clinical phase and milder organ-specific endotypes of SLE provide potential opportunities to intervene in individuals with a less aggressive or more focused disease processes, associated with lower levels of organ damage. Technological platforms showing promise in early-phase clinical trials or preclinical models in other autoimmune diseases could be adapted for trials in SLE. Given the clinical complexity, sensible beginning strategies would comprise small mechanistic studies with immune biomarker and safety outcomes in well-defined limited disease settings.
SR drafted and edited manuscript, and compiled figure. RT edited manuscript, figure and tables. All authors contributed to the article and approved the submitted version.
This manuscript is part of a project that has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 777357. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA. RT was supported by NHMRC grant 1079238 and by Arthritis Queensland.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bernatsky S, Boivin JF, Joseph L, Manzi S, Ginzler E, Gladman DD, et al. Mortality in Systemic Lupus Erythematosus. Arthritis Rheumatism (2006) 54:2550–7. doi: 10.1002/art.21955
2. Borchers AT, Keen CL, Shoenfeld Y, Gershwin ME. Surviving the Butterfly and the Wolf: Mortality Trends in Systemic Lupus Erythematosus. Autoimmun Rev (2004) 3:423–53. doi: 10.1016/j.autrev.2004.04.002
3. He J, Zhang R, Shao M, Zhao X, Miao M, Chen J, et al. Efficacy and Safety of Low-Dose IL-2 in the Treatment of Systemic Lupus Erythematosus: A Randomised, Double-Blind, Placebo-Controlled Trial. Ann Rheumatic Dis (2020) 79:141–9. doi: 10.1136/annrheumdis-2019-215396
4. Urowitz MB, Ohsfeldt RL, Wielage RC, Dever JJ, Zakerifar M, Asukai Y, et al. Comparative Analysis of Long-Term Organ Damage in Patients With Systemic Lupus Erythematosus Using Belimumab Versus Standard Therapy: A Post Hoc Longitudinal Study. Lupus Sci Med (2020) 7. doi: 10.1136/lupus-2020-000412
5. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med (2020) 382:211–21. doi: 10.1056/NEJMoa1912196
6. Kuo CF, Grainge MJ, Valdes AM, See LC, Luo SF, Yu KH, et al. Familial Aggregation of Systemic Lupus Erythematosus and Coaggregation of Autoimmune Diseases in Affected Families. JAMA Intern Med (2015) 175:1518–26. doi: 10.1001/jamainternmed.2015.3528
7. Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens Targeted in Systemic Lupus Erythematosus Are Clustered in Two Populations of Surface Structures on Apoptotic Keratinocytes. J Exp Med (1994) 179:1317–30. doi: 10.1084/jem.179.4.1317
8. Savarese E, Steinberg C, Pawar RD, Reindl W, Akira S, Anders HJ, et al. Requirement of Toll-Like Receptor 7 for Pristane-Induced Production of Autoantibodies and Development of Murine Lupus Nephritis. Arthritis Rheum (2008) 58:1107–15. doi: 10.1002/art.23407
9. Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, et al. Spatiotemporal Regulation of MyD88–IRF-7 Signalling for Robust Type-I Interferon Induction. Nature (2005) 434:1035–40. doi: 10.1038/nature03547
10. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of Autoantibodies Before the Clinical Onset of Systemic Lupus Erythematosus. New Engl J Med (2003) 349:1526–33. doi: 10.1056/NEJMoa021933
11. Wang CL, Kuo CF, Yeh YH, Hsieh MY, Kuo CT, Chang SH. Familial Aggregation of Myocardial Infarction and Coaggregation of Myocardial Infarction and Autoimmune Disease: A Nationwide Population-Based Cross-Sectional Study in Taiwan. BMJ Open (2019) 9:e023614. doi: 10.1136/bmjopen-2018-023614
12. Nehar-Belaid D, Hong S, Marches R, Chen G, Bolisetty M, Baisch J, et al. Mapping Systemic Lupus Erythematosus Heterogeneity at the Single-Cell Level. Nat Immunol (2020) 21:1094–106. doi: 10.1038/s41590-020-0743-0
13. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Ann Rheum Dis (2019) 78:1151–9. doi: 10.1136/annrheumdis-2019-216700
14. Tselios K, Gladman DD, Touma Z, Su J, Anderson N, Urowitz MB. Disease Course Patterns in Systemic Lupus Erythematosus. Lupus (2019) 28:114–22. doi: 10.1177/0961203318817132
15. Golder V, Tsang A.S.M.W.P.. Treatment Targets in SLE: Remission and Low Disease Activity State. Rheumatol (Oxf) (2020) 59:v19–28. doi: 10.1093/rheumatology/keaa420
16. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med (2019) 381:603–13. doi: 10.1056/NEJMoa1902226
17. Vudattu NK, Herold KC. Treatment of New Onset Type 1 Diabetes With Teplizumab: Successes and Pitfalls in Development. Expert Opin Biol Ther (2014) 14:377–85. doi: 10.1517/14712598.2014.881797
18. Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, et al. Staging Presymptomatic Type 1 Diabetes: A Scientific Statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care (2015) 38:1964–74. doi: 10.2337/dc15-1419
19. Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y. Autoantibody Explosion in Systemic Lupus Erythematosus: More Than 100 Different Antibodies Found in SLE Patients. Semin Arthritis Rheum (2004) 34:501–37. doi: 10.1016/j.semarthrit.2004.07.002
20. Spronk PE, Limburg PC, Kallenberg CGM. Review : Serological Markers of Disease Activity in Systemic Lupus Erythematosus. Lupus (1995) 4:86–94. doi: 10.1177/096120339500400202
21. Gaither KK, Fox OF, Yamagata H, Mamula MJ, Reichlin M, Harley JB. Implications of Anti-Ro/Sjögren's Syndrome A Antigen Autoantibody in Normal Sera for Autoimmunity. J Clin Invest (1987) 79:841–6. doi: 10.1172/JCI112892
22. Ma Z, Rp M, Js M. Anti-dsDNA Antibodies in Laboratory Workers Handling Blood From Patients With Systemic Lupus Erythematosus. J Rheumatol (1992) 19:1380–4.
23. Dema B, Charles N. Autoantibodies in SLE: Specificities, Isotypes and Receptors. Antibodies (Basel) (2016) 5:2. doi: 10.3390/antib5010002
24. Hahn J, Leatherwood C, Malspeis S, Liu X, Lu B, Roberts AL, et al. Associations Between Smoking and Systemic Lupus Erythematosus (SLE)-Related Cytokines and Chemokines Among US Female Nurses. Arthritis Care Res (Hoboken) (2020). doi: 10.1002/acr.24370
25. Slight-Webb S, Smith M, Bylinska A, Macwana S, Guthridge C, Lu R, et al. Autoantibody-Positive Healthy Individuals With Lower Lupus Risk Display a Unique Immune Endotype. J Allergy Clin Immunol (2020) 146:1419–33. doi: 10.1016/j.jaci.2020.04.047
26. Li Q-Z, Karp DR, Quan J, Branch VK, Zhou J, Lian Y, et al. Risk Factors for ANA Positivity in Healthy Persons. Arthritis Res Ther (2011) 13:R38–8. doi: 10.1186/ar3271
27. Merrill JT, Immermann F, Whitley M, Zhou T, Hill A, O’Toole M, et al. The Biomarkers of Lupus Disease Study: A Bold Approach May Mitigate Interference of Background Immunosuppressants in Clinical Trials. Arthritis Rheumatol (2017) 69:1257–66. doi: 10.1002/art.40086
28. Aster JC. Normal B and T Lymphocyte Development. In: Rosmarin AG, editor. (2021). UpToDate, UpToDate, UpToDate.
29. Monroe JG, Rothenberg E, eds. Molecular Biology of B-Cell and T-Cell Development. Springer (1998).
30. Anderton SM, Wraith DC. Selection and Fine-Tuning of the Autoimmune T-Cell Repertoire. Nat Rev Immunol (2002) 2:487–98. doi: 10.1038/nri842
31. Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, et al. A Requisite Role for Induced Regulatory T Cells in Tolerance Based on Expanding Antigen Receptor Diversity. Immunity (2011) 35:109–22. doi: 10.1016/j.immuni.2011.03.029
32. Meiron M, Zohar Y, Anunu R, Wildbaum G, Karin N. CXCL12 (SDF-1α) Suppresses Ongoing Experimental Autoimmune Encephalomyelitis by Selecting Antigen-Specific Regulatory T Cells. J Exp Med (2008) 205:2643–55. doi: 10.1084/jem.20080730
33. Bollyky PL, Wu RP, Falk BA, Lord JD, Long SA, Preisinger A, et al. ECM Components Guide IL-10 Producing Regulatory T-Cell (TR1) Induction From Effector Memory T-Cell Precursors. PNAS (2011) 108:7938–43. doi: 10.1073/pnas.1017360108
34. Yao Y, Vent-Schmidt J, McGeough MD, Wong M, Hoffman HM, Steiner TS, et al. Tr1 Cells, But Not Foxp3+ Regulatory T Cells, Suppress NLRP3 Inflammasome Activation via an IL-10–Dependent Mechanism. J Immunol (2015) 195:488–97. doi: 10.4049/jimmunol.1403225
35. Bluestone JA, Abbas AK. Natural Versus Adaptive Regulatory T Cells. Nat Rev Immunol (2003) 3:253–7. doi: 10.1038/nri1032
36. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M, Gagliani N. The Biology of T Regulatory Type 1 Cells and Their Therapeutic Application in Immune-Mediated Diseases. Immunity (2018) 49:1004–19. doi: 10.1016/j.immuni.2018.12.001
37. Zhang P, Lee JS, Gartlan KH, Schuster IS, Comerford I, Varelias A, et al. Eomesodermin Promotes the Development of Type 1 Regulatory T (TR1) Cells. Sci Immunol (2017) 2. doi: 10.1126/sciimmunol.aah7152
38. Bacchetta R, Bigler M, Touraine JL, Parkman R, Tovo PA, Abrams J, et al. High Levels of Interleukin 10 Production In Vivo Are Associated With Tolerance in SCID Patients Transplanted With HLA Mismatched Hematopoietic Stem Cells. J Exp Med (1994) 179:493–502. doi: 10.1084/jem.179.2.493
39. Gianfrani C, Levings MK, Sartirana C, Mazzarella G, Barba G, Zanzi D, et al. Gliadin-Specific Type 1 Regulatory T Cells From the Intestinal Mucosa of Treated Celiac Patients Inhibit Pathogenic T Cells. J Immunol (2006) 177:4178–86. doi: 10.4049/jimmunol.177.6.4178
40. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 Cells and the Counter-Regulation of Immunity: Natural Mechanisms and Therapeutic Applications. In: Fillatreau S, O'Garra A, editors. Interleukin-10 in Health and Disease. Berlin, Heidelberg: Springer (2014). p. 39–68.
41. Głobińska A, Boonpiyathad T, Satitsuksanoa P, Kleuskens M, van de Veen W, Sokolowska M, et al. Mechanisms of Allergen-Specific Immunotherapy: Diverse Mechanisms of Immune Tolerance to Allergens. Ann Allergy Asthma Immunol (2018) 121:306–12. doi: 10.1016/j.anai.2018.06.026
42. Mellman I. Dendritic Cells: Master Regulators of the Immune Response. Cancer Immunol Res (2013) 1:145–9. doi: 10.1158/2326-6066.CIR-13-0102
43. Martin E, O'Sullivan B, Low P, Thomas R. Antigen-Specific Suppression of a Primed Immune Response by Dendritic Cells Mediated by Regulatory T Cells Secreting Interleukin-10. Immunity (2003) 18:155–67. doi: 10.1016/S1074-7613(02)00503-4
44. Granot T, Senda T, Carpenter DJ, Matsuoka N, Weiner J, Gordon CL, et al. Dendritic Cells Display Subset and Tissue-Specific Maturation Dynamics Over Human Life. Immunity (2017) 46:504–15. doi: 10.1016/j.immuni.2017.02.019
45. Guilliams M, Dutertre C-A, Scott CL, McGovern N, Sichien D, Chakarov S, et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells Across Tissues and Species. Immunity (2016) 45:669–84. doi: 10.1016/j.immuni.2016.08.015
46. Australian Government Department of Health. The Pharmaceutical Benefits Scheme: Expenditure and Prescriptions Twelve Months to 30 June 2018 (2018). Available at: https://www.pbs.gov.au/info/statistics/expenditure-prescriptions/expenditure-prescriptions-twelve-months-to-30-june-2018.
47. Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, et al. Human Tissues Contain CD141hi Cross-Presenting Dendritic Cells With Functional Homology to Mouse CD103+ Nonlymphoid Dendritic Cells. Immunity (2012) 37:60–73. doi: 10.1016/j.immuni.2012.04.012
48. Villani A-C, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science (2017) 356:eaah4573. doi: 10.1126/science.aah4573
49. Di Blasio S, Wortel IMN, van Bladel DAG, de Vries LE, Duiveman-de Boer T, Worah K, et al. Human CD1c+ DCs Are Critical Cellular Mediators of Immune Responses Induced by Immunogenic Cell Death. OncoImmunology (2016) 5:e1192739. doi: 10.1080/2162402X.2016.1192739
50. Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood. Science (1999) 284:1835. doi: 10.1126/science.284.5421.1835
51. Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-Dependent Viral Recognition by Plasmacytoid Dendritic Cells. Science (2007) 315:1398. doi: 10.1126/science.1136880
52. Liu Y-J. IPC: Professional Type 1 Interferon-Producing Cells and Plasmacytoid Dendritic Cell Precursors. Annu Rev Immunol (2004) 23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633
53. Tsokos GC, Lo MS, Reis PC, Sullivan KE. New Insights Into the Immunopathogenesis of Systemic Lupus Erythematosus. Nat Rev Rheumatol (2016) 12:716–30. doi: 10.1038/nrrheum.2016.186
54. Mok CC, Lau CS. Pathogenesis of Systemic Lupus Erythematosus. J Clin Pathol (2003) 56:481–90. doi: 10.1136/jcp.56.7.481
55. Dixon FJ, Andrews BS, Eisenberg RA, McConahey PJ, Theofilopoulos AN, Wilson CB. Etiology and Pathogenesis of a Spontaneous Lupus-Like Syndrome in Mice. Arthritis Rheum (1978) 21:S64–7. doi: 10.1002/art.1780210909
56. Rauch J, Murphy E, Roths JB, Stollar BD, Schwartz RS. A High Frequency Idiotypic Marker of Anti-DNA Autoantibodies in MRL-Ipr/Ipr Mice. J Immunol (1982) 129:236–41.
57. Eisenberg RA, Tan EM, Dixon FJ. Presence of Anti-Sm Reactivity in Autoimmune Mouse Strains. J Exp Med (1978) 147:582–7. doi: 10.1084/jem.147.2.582
58. Satoh M, Reeves WH. Induction of Lupus-Associated Autoantibodies in BALB/c Mice by Intraperitoneal Injection of Pristane. J Exp Med (1994) 180:2341–6. doi: 10.1084/jem.180.6.2341
59. Yokogawa M, Takaishi M, Nakajima K, Kamijima R, Fujimoto C, Kataoka S, et al. Epicutaneous Application of Toll-Like Receptor 7 Agonists Leads to Systemic Autoimmunity in Wild-Type Mice: A New Model of Systemic Lupus Erythematosus. Arthritis Rheumatol (2014) 66:694–706. doi: 10.1002/art.38298
60. Reilly CM, Gilkeson GS. Use of Genetic Knockouts to Modulate Disease Expression in a Murine Model of Lupus, MRL/lpr Mice. Immunol Res (2002) 25:143–53. doi: 10.1385/IR:25:2:143
61. Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI. Suppression of Disease in New Zealand Black/New Zealand White Lupus-Prone Mice by Adoptive Transfer of Ex Vivo Expanded Regulatory T Cells. J Immunol (2006) 177:1451–9. doi: 10.4049/jimmunol.177.3.1451
62. Grimaldi CM. Sex and Systemic Lupus Erythematosus: The Role of the Sex Hormones Estrogen and Prolactin on the Regulation of Autoreactive B Cells. Curr Opin Rheumatol (2006) 18:456–61. doi: 10.1097/01.bor.0000240354.37927.dd
63. Stannard JN, Kahlenberg JM. Cutaneous Lupus Erythematosus: Updates on Pathogenesis and Associations With Systemic Lupus. Curr Opin Rheumatol (2016) 28:453–9. doi: 10.1097/BOR.0000000000000308
64. Jara LJ, Medina G, Saavedra MA, Vera-Lastra O, Navarro C. Prolactin and Autoimmunity. Clin Rev Allergy Immunol (2011) 40:50–9. doi: 10.1007/s12016-009-8185-3
65. Nelson P, Rylance P, Roden D, Trela M, Tugnet N. Viruses as Potential Pathogenic Agents in Systemic Lupus Erythematosus. Lupus (2014) 23:596–605. doi: 10.1177/0961203314531637
66. Chen M, Aosai F, Norose K, Mun HS, Ishikura H, Hirose S, et al. Toxoplasma Gondii Infection Inhibits the Development of Lupus-Like Syndrome in Autoimmune (New Zealand Black X New Zealand White) F1 Mice. Int Immunol (2004) 16:937–46. doi: 10.1093/intimm/dxh095
67. Ram M, Anaya JM, Barzilai O, Izhaky D, Porat Katz BS, Blank M, et al. The Putative Protective Role of Hepatitis B Virus (HBV) Infection From Autoimmune Disorders. Autoimmun Rev (2008) 7:621–5. doi: 10.1016/j.autrev.2008.06.008
68. Nockher WA, Wigand R, Schoeppe W, Scherberich JE. Elevated Levels of Soluble CD14 in Serum of Patients With Systemic Lupus Erythematosus. Clin Exp Immunol (1994) 96:15–9. doi: 10.1111/j.1365-2249.1994.tb06222.x
69. Shi L, Zhang Z, Yu AM, Wang W, Wei Z, Akhter E, et al. The SLE Transcriptome Exhibits Evidence of Chronic Endotoxin Exposure and Has Widespread Dysregulation of Non-Coding and Coding RNAs. PloS One (2014) 9:e93846. doi: 10.1371/journal.pone.0093846
70. Belkaid Y, Hand TW. Role of the Microbiota in Immunity and Inflammation. Cell (2014) 157:121–41. doi: 10.1016/j.cell.2014.03.011
71. Hevia A, Milani C, López P, Cuervo A, Arboleya S, Duranti S, et al. Intestinal Dysbiosis Associated With Systemic Lupus Erythematosus. mBio (2014) 5:e01548–14. doi: 10.1128/mBio.01548-14
72. Johnson BM, Gaudreau MC, Gudi R, Brown R, Gilkeson G, Vasu C. Gut Microbiota Differently Contributes to Intestinal Immune Phenotype and Systemic Autoimmune Progression in Female and Male Lupus-Prone Mice. J Autoimmun (2020) 108:102420. doi: 10.1016/j.jaut.2020.102420
73. Belot A, Kasher PR, Trotter EW, Foray AP, Debaud AL, Rice GI, et al. Protein Kinase Cδ Deficiency Causes Mendelian Systemic Lupus Erythematosus With B Cell-Defective Apoptosis and Hyperproliferation. Arthritis Rheum (2013) 65:2161–71. doi: 10.1002/art.38008
74. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-Of-Function Variant in DNASE1L3 Causes a Familial Form of Systemic Lupus Erythematosus. Nat Genet (2011) 43:1186–8. doi: 10.1038/ng.975
75. Shi G, Abbott KN, Wu W, Salter RD, Keyel PA. Dnase1L3 Regulates Inflammasome-Dependent Cytokine Secretion. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00522
76. Hardy CJ, Palmer BP, Muir KR, Sutton AJ, Powell RJ. Smoking History, Alcohol Consumption, and Systemic Lupus Erythematosus: A Case-Control Study. Ann Rheumatic Dis (1998) 57:451. doi: 10.1136/ard.57.8.451
77. Casciola-Rosen L, Rosen A. Ultraviolet Light-Induced Keratinocyte Apoptosis: A Potential Mechanism for the Induction of Skin Lesions and Autoantibody Production in LE. Lupus (1997) 6:175–80. doi: 10.1177/096120339700600213
78. Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ. Inhibition of Toll-Like Receptor-7 (TLR-7) or TLR-7 Plus TLR-9 Attenuates Glomerulonephritis and Lung Injury in Experimental Lupus. J Am Soc Nephrol (2007) 18:1721–31. doi: 10.1681/ASN.2006101162
79. Papadimitraki ED, Choulaki C, Koutala E, Bertsias G, Tsatsanis C, Gergianaki I, et al. Expansion of Toll-Like Receptor 9-Expressing B Cells in Active Systemic Lupus Erythematosus: Implications for the Induction and Maintenance of the Autoimmune Process. Arthritis Rheum (2006) 54:3601–11. doi: 10.1002/art.22197
80. Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-Like Receptor 9 Controls Anti-DNA Autoantibody Production in Murine Lupus. J Exp Med (2005) 202:321–31. doi: 10.1084/jem.20050338
81. Munroe ME, Lu R, Zhao YD, Fife DA, Robertson JM, Guthridge JM, et al. Altered Type II Interferon Precedes Autoantibody Accrual and Elevated Type I Interferon Activity Prior to Systemic Lupus Erythematosus Classification. Ann Rheum Dis (2016) 75:2014–21. doi: 10.1136/annrheumdis-2015-208140
82. Crow YJ, Manel N. Aicardi–Goutières Syndrome and the Type I Interferonopathies. Nat Rev Immunol (2015) 15:429–40. doi: 10.1038/nri3850
83. Talaat RM, Mohamed SF, Bassyouni IH, Raouf AA. Th1/Th2/Th17/Treg Cytokine Imbalance in Systemic Lupus Erythematosus (SLE) Patients: Correlation With Disease Activity. Cytokine (2015) 72:146–53. doi: 10.1016/j.cyto.2014.12.027
84. de la Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin-2 Is Essential for CD4+CD25+ Regulatory T Cell Function. Eur J Immunol (2004) 34:2480–8. doi: 10.1002/eji.200425274
85. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 Signaling via STAT5 Constrains T Helper 17 Cell Generation. Immunity (2007) 26:371–81. doi: 10.1016/j.immuni.2007.02.009
86. Duan JH, Jiang Y, Mu H, Tang ZQ. Expression of BAFF and BR3 in Patients With Systemic Lupus Erythematosus. Braz J Med Biol Res (2016) 49:e4853. doi: 10.1590/1414-431X20154853
87. Pers JO, Daridon C, Devauchelle V, Jousse S, Saraux A, Jamin C, et al. BAFF Overexpression Is Associated With Autoantibody Production in Autoimmune Diseases. Ann N Y Acad Sci (2005) 1050:34–9. doi: 10.1196/annals.1313.004
88. Holmdahl R, Malmström V, Burkhardt H. Autoimmune Priming, Tissue Attack and Chronic Inflammation - The Three Stages of Rheumatoid Arthritis. Eur J Immunol (2014) 44:1593–9. doi: 10.1002/eji.201444486
89. Paul V. Lehmann TF, Alexander Miller EES. Spreading of T-Cell Autoimmunity to Cryptic Determinants of an Autoantigen. Nature (1992) 358:155–7. doi: 10.1038/358155a0
90. Vanderlugt CL, Miller SD. Epitope Spreading in Immune-Mediated Diseases: Implications for Immunotherapy. Nat Rev Immunol (2002) 2:85–95. doi: 10.1038/nri724
91. Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular Mimicry, Bystander Activation, or Viral Persistence: Infections and Autoimmune Disease. Clin Microbiol Rev (2006) 19:80–94. doi: 10.1128/CMR.19.1.80-94.2006
92. Yang M, Klocke K, Hernandez CM, Xu B, Gjertsson I, Wing K, et al. Regulatory T Cells Control Epitope Spreading in Autoimmune Arthritis Independent of Cytotoxic T-Lymphocyte Antigen-4. Immunology (2018) 155:446–57. doi: 10.1111/imm.12983
93. Mandik-Nayak L, Seo SJ, Sokol C, Potts KM, Bui A, Erikson J. MRL-Lpr/Lpr Mice Exhibit a Defect in Maintaining Developmental Arrest and Follicular Exclusion of Anti-Double-Stranded DNA B Cells. J Exp Med (1999) 189:1799–814. doi: 10.1084/jem.189.11.1799
94. Sinai P, Dozmorov IM, Song R, Schwartzberg PL, Wakeland EK, Wülfing C. T/B-Cell Interactions Are More Transient in Response to Weak Stimuli in SLE-Prone Mice. Eur J Immunol (2014) 44:3522–31. doi: 10.1002/eji.201444602
95. Woods M, Zou Y-R, Davidson A. Defects in Germinal Center Selection in SLE. Front Immunol (2015) 6:425–5. doi: 10.3389/fimmu.2015.00425
96. Morimoto S, Nakano S, Watanabe T, Tamayama Y, Mitsuo A, Nakiri Y, et al. Expression of B-Cell Activating Factor of the Tumour Necrosis Factor Family (BAFF) in T Cells in Active Systemic Lupus Erythematosus: The Role of BAFF in T Cell-Dependent B Cell Pathogenic Autoantibody Production. Rheumatology (2007) 46:1083–6. doi: 10.1093/rheumatology/kem097
97. Bradley SJ, Suarez-Fueyo A, Moss DR, Kyttaris VC, Tsokos GC. T Cell Transcriptomes Describe Patient Subtypes in Systemic Lupus Erythematosus. PloS One (2015) 10:e0141171–e0141171. doi: 10.1371/journal.pone.0141171
98. Wing K, Sakaguchi S. Regulatory T Cells Exert Checks and Balances on Self Tolerance and Autoimmunity. Nat Immunol (2010) 11:7–13. doi: 10.1038/ni.1818
99. Humrich JY, Morbach H, Undeutsch R, Enghard P, Rosenberger S, Weigert O, et al. Homeostatic Imbalance of Regulatory and Effector T Cells Due to IL-2 Deprivation Amplifies Murine Lupus. PNAS (2010) 107:204–9. doi: 10.1073/pnas.0903158107
100. Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, et al. Global Natural Regulatory T Cell Depletion in Active Systemic Lupus Erythematosus. J Immunol (2005) 175:8392–400. doi: 10.4049/jimmunol.175.12.8392
101. Crispin JC, Martı́nez A, Alcocer-Varela J. Quantification of Regulatory T Cells in Patients With Systemic Lupus Erythematosus. J Autoimmun (2003) 21:273–6. doi: 10.1016/S0896-8411(03)00121-5
102. Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, et al. Early, Transient Depletion of Plasmacytoid Dendritic Cells Ameliorates Autoimmunity in a Lupus Model. J Exp Med (2014) 211:1977–91. doi: 10.1084/jem.20132620
103. Davison LM, Jørgensen TN. Sialic Acid-Binding Immunoglobulin-Type Lectin H-Positive Plasmacytoid Dendritic Cells Drive Spontaneous Lupus-Like Disease Development in B6.Nba2 Mice. Arthritis Rheumatol (2015) 67:1012–22. doi: 10.1002/art.38989
104. Blomberg S, Eloranta ML, Cederblad B, Nordlin K, Alm GV, Rönnblom L. Presence of Cutaneous Interferon-Alpha Producing Cells in Patients With Systemic Lupus Erythematosus. Lupus (2001) 10:484–90. doi: 10.1191/096120301678416042
105. Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid Dendritic Cells (Natural Interferon- Alpha/Beta-Producing Cells) Accumulate in Cutaneous Lupus Erythematosus Lesions. Am J Pathol (2001) 159:237–43. doi: 10.1016/S0002-9440(10)61689-6
106. Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of Dendritic Cell Differentiation by IFN-α in Systemic Lupus Erythematosus. Science (2001) 294:1540. doi: 10.1126/science.1064890
107. Tawbi HA. NCT03816345: Nivolumab in Treating Patients With Autoimmune Disorders or Advanced, Metastatic, or Unresectable Cancer. (2019). ClinicalTrials.gov.
108. Smith G. NCT03866317: A Study to Assess the Safety and Efficacy of Secukinumab in Alleviating Symptoms of Discoid Lupus Erythematosus. (2020), ClinicalTrials.gov.
109. Serra P, Santamaria P. Nanoparticle-Based Approaches to Immune Tolerance for the Treatment of Autoimmune Diseases. Eur J Immunol (2018) 48:751–6. doi: 10.1002/eji.201747059
110. Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, et al. Reversal of Autoimmunity by Boosting Memory-Like Autoregulatory T Cells. Immunity (2010) 32:568–80. doi: 10.1016/j.immuni.2010.03.015
111. Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding Antigen-Specific Regulatory Networks to Treat Autoimmunity. Nature (2016) 530:434–40. doi: 10.1038/nature16962
112. Atif M, Conti F, Gorochov G, Oo YH, Miyara M. Regulatory T Cells in Solid Organ Transplantation. Clin Transl Immunol (2020) 9. doi: 10.1002/cti2.1099
113. Weigert O, von Spee C, Undeutsch R, Kloke L, Humrich JY, Riemekasten G. CD4+Foxp3+ Regulatory T Cells Prolong Drug-Induced Disease Remission in (NZBxNZW) F1 Lupus Mice. Arthritis Res Ther (2013) 15:R35. doi: 10.1186/ar4188
114. Scottà C, Fanelli G, Hoong SJ, Romano M, Lamperti EN, Sukthankar M, et al. Impact of Immunosuppressive Drugs on the Therapeutic Efficacy of Ex Vivo Expanded Human Regulatory T Cells. 1 (2016) 101:91–100.
115. Dall’Era M, Pauli ML, Remedios K, Taravati K, Sandova PM, Putnam AL, et al. Adoptive Treg Cell Therapy in a Patient With Systemic Lupus Erythematosus. Arthritis Rheumatol (2019) 71:431–40. doi: 10.1002/art.40737
116. Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T Cell Lineage Differentiation. Immunity (2009) 30:646–55. doi: 10.1016/j.immuni.2009.05.001
117. Alexander T, Thiel A, Rosen O, Massenkeil G, Sattler A, Kohler S, et al. Depletion of Autoreactive Immunologic Memory Followed by Autologous Hematopoietic Stem Cell Transplantation in Patients With Refractory SLE Induces Long-Term Remission Through De Novo Generation of a Juvenile and Tolerant Immune System. Blood (2009) 113:214–23. doi: 10.1182/blood-2008-07-168286
118. Zhang L, Bertucci AM, Ramsey-Goldman R, Burt RK, Datta SK. Regulatory T Cell (Treg) Subsets Return in Patients With Refractory Lupus Following Stem Cell Transplantation, and TGF-Beta-Producing CD8+ Treg Cells Are Associated With Immunological Remission of Lupus. J Immunol (Baltimore Md. (2009) 1950) 183:6346–58. doi: 10.4049/jimmunol.0901773
119. Wang D, Zhang H, Liang J, Li X, Feng X, Wang H, et al. Allogeneic Mesenchymal Stem Cell Transplantation in Severe and Refractory Systemic Lupus Erythematosus: 4 Years of Experience. Cell Transplant (2013) 22:2267–77. doi: 10.3727/096368911X582769c
120. Dauphinée MJ, Kipper SB, Wofsy D, Talal N. Interleukin 2 Deficiency is a Common Feature of Autoimmune Mice. J Immunol (1981) 127:2483–7.
121. Linker-Israeli M, Bakke AC, Kitridou RC, Gendler S, Gillis S, Horwitz DA. Defective Production of Interleukin 1 and Interleukin 2 in Patients With Systemic Lupus Erythematosus (SLE). J Immunol (1983) 130:2651–5.
122. Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic Maintenance of Natural Foxp3+ CD25+ CD4+ Regulatory T Cells by Interleukin (IL)-2 and Induction of Autoimmune Disease by IL-2 Neutralization. J Exp Med (2005) 201:723–35. doi: 10.1084/jem.20041982
123. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A Function for Interleukin 2 in Foxp3-Expressing Regulatory T Cells. Nat Immunol (2005) 6:1142–51. doi: 10.1038/ni1263
124. Gómez-Martín D, Díaz-Zamudio M, Crispín JC, Alcocer-Varela J. Interleukin 2 and Systemic Lupus Erythematosus: Beyond the Transcriptional Regulatory Net Abnormalities. Autoimmun Rev (2009) 9:34–9. doi: 10.1016/j.autrev.2009.02.035
125. Crispín JC, Tsokos GC. IL-17 in Systemic Lupus Erythematosus. J Biomed Biotechnol (2010). doi: 10.1155/2010/943254
126. Mizui M, Koga T, Lieberman LA, Beltran J, Yoshida N, Johnson MC, et al. IL-2 Protects Lupus-Prone Mice From Multiple End-Organ Damage by Limiting CD4–CD8– IL-17–Producing T Cells. J Immunol (2014) 193:2168–77. doi: 10.4049/jimmunol.1400977
127. Meropol NJ, Porter M, Blumenson LE, Lindemann MJ, Perez RP, Vaickus L, et al. Daily Subcutaneous Injection of Low-Dose Interleukin 2 Expands Natural Killer Cells In Vivo Without Significant Toxicity. Clin Cancer Res (1996) 2:669–77.
128. von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-Dose Interleukin-2 Selectively Corrects Regulatory T Cell Defects in Patients With Systemic Lupus Erythematosus. Ann Rheum Dis (2016) 75(7):1407–15. doi: 10.1136/annrheumdis-2015-207776
129. McHugh MD, Park J, Uhrich R, Gao W, Horwitz DA, Fahmy TM. Paracrine Co-Delivery of TGF-β and IL-2 Using CD4-Targeted Nanoparticles for Induction and Maintenance of Regulatory T Cells. Biomaterials (2015) 59:172–81. doi: 10.1016/j.biomaterials.2015.04.003
130. Siddhanti S, Fanton C, Dixit N, Lu L, Chindalore V, Levin R, et al. THU0054 NKTR-358, a Novel IL-2 Conjugate, Stimulates High Levels of Regulatory T Cells in Patients With Systemic Lupus Erythematosus. Ann Rheum Dis (2020) 79. doi: 10.1136/annrheumdis-2020-eular.3165
131. Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, et al. Dendritic Cells Induce Peripheral T Cell Unresponsiveness Under Steady State Conditions in Vivo. J Exp Med (2001) 194:769–80. doi: 10.1084/jem.194.6.769
132. Mansilla MJ, Sellès-Moreno C, Fàbregas-Puig S, Amoedo J, Navarro-Barriuso J, Teniente-Serra A, et al. Beneficial Effect of Tolerogenic Dendritic Cells Pulsed With MOG Autoantigen in Experimental Autoimmune Encephalomyelitis. CNS Neurosci Ther (2014) 21:222–30. doi: 10.1111/cns.12342
133. Van Brussel I, Lee WP, Rombouts M, Nuyts AH, Heylen M, De Winter BY, et al. Tolerogenic Dendritic Cell Vaccines to Treat Autoimmune Diseases: Can the Unattainable Dream Turn Into Reality? Autoimmun Rev (2014) 13:138–50. doi: 10.1016/j.autrev.2013.09.008
134. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and Clinical Effects of Low-Dose Interleukin-2 Across 11 Autoimmune Diseases in a Single, Open Clinical Trial. Ann Rheum Dis (2019) 78(2):209–17. doi: 10.1136/annrheumdis-2018-214229
135. Willekens B, Presas-Rodríguez S, Mansilla MJ, Derdelinckx J, Lee W-P, Nijs G, et al. Tolerogenic Dendritic Cell-Based Treatment for Multiple Sclerosis (MS): A Harmonised Study Protocol for Two Phase I Clinical Trials Comparing Intradermal and Intranodal Cell Administration. BMJ Open (2019) 9:e030309. doi: 10.1136/bmjopen-2019-030309
136. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated Peptide Dendritic Cell Immunotherapy in HLA Risk Genotype-Positive Rheumatoid Arthritis Patients. Sci Transl Med (2015) 7:290ra87. doi: 10.1126/scitranslmed.aaa9301
137. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous Tolerogenic Dendritic Cells for Rheumatoid and Inflammatory Arthritis. Ann Rheumatic Dis (2017) 76:227–34. doi: 10.1136/annrheumdis-2015-208456
138. Funes SC, Rios M, Gomez-Santander F, Fernandez-Fierro A, Altamirano-Lagos MJ, Rivera-Perez D, et al. Tolerogenic Dendritic Cell Transfer Ameliorates Systemic Lupus Erythematosus in Mice. Immunology (2019) 158:322–39. doi: 10.1111/imm.13119
139. Obreque J, Vega F, Torres A, Cuitino L, Mackern-Oberti JP, Viviani P, et al. Autologous Tolerogenic Dendritic Cells Derived From Monocytes of Systemic Lupus Erythematosus Patients and Healthy Donors Show a Stable and Immunosuppressive Phenotype. Immunology (2017) 152:648–59. doi: 10.1111/imm.12806
140. Look M, Stern E, Wang QA, DiPlacido LD, Kashgarian M, Craft J, et al. Nanogel-Based Delivery of Mycophenolic Acid Ameliorates Systemic Lupus Erythematosus in Mice. J Clin Invest (2013) 123:1741–9. doi: 10.1172/JCI65907
141. Look M, Saltzman WM, Craft J, Fahmy TM. The Nanomaterial-Dependent Modulation of Dendritic Cells and Its Potential Influence on Therapeutic Immunosuppression in Lupus. Biomaterials (2014) 35:1089–95. doi: 10.1016/j.biomaterials.2013.10.046
142. Monneaux F, Briand J-P, Muller S. B and T Cell Immune Response to Small Nuclear Ribonucleoprotein Particles in Lupus Mice: Autoreactive CD4+ T Cells Recognize a T Cell Epitope Located Within the RNP80 Motif of the 70K Protein. Eur J Immunol (2000) 30:2191–200. doi: 10.1002/1521-4141(2000)30:8<2191::AID-IMMU2191>3.0.CO;2-R
143. Monneaux F, Dumortier H, Steiner G, Briand J-P, Muller S. Murine Models of Systemic Lupus Erythematosus: B and T Cell Responses to Spliceosomal Ribonucleoproteins in MRL/Faslpr and (NZB × NZW)F1 Lupus Mice. Int Immunol (2001) 13:1155–63. doi: 10.1093/intimm/13.9.1155
144. Wang F, Tasset I, Cuervo AM, Muller S. In Vivo Remodeling of Altered Autophagy-Lysosomal Pathway by a Phosphopeptide in Lupus. Cells (2020) 9. doi: 10.3390/cells9102328
145. Muller S, Monneaux F, Schall N, Rashkov RK, Oparanov BA, Wiesel P, et al. Spliceosomal Peptide P140 for Immunotherapy of Systemic Lupus Erythematosus: Results of an Early Phase II Clinical Trial. Arthritis Rheumatism (2008) 58:3873–83. doi: 10.1002/art.24027
146. Zimmer R, Scherbarth HR, Rillo OL, Gomez-Reino JJ, Muller S. Lupuzor/P140 Peptide in Patients With Systemic Lupus Erythematosus: A Randomised, Double-Blind, Placebo-Controlled Phase IIb Clinical Trial. Ann Rheumatic Dis (2013) 72:1830. doi: 10.1136/annrheumdis-2012-202460
148. Alhadj Ali M, Liu Y-F, Arif S, Tatovic D, Shariff H, Gibson VB, et al. Metabolic and Immune Effects of Immunotherapy With Proinsulin Peptide in Human New-Onset Type 1 Diabetes. Sci Transl Med (2017) 9. doi: 10.1126/scitranslmed.aaf7779
149. Santambrogio L, Sato AK, Fischer FR, Dorf ME, Stern LJ. Abundant Empty Class II MHC Molecules on the Surface of Immature Dendritic Cells. Proc Natl Acad Sci USA (1999) 96:15050–5. doi: 10.1073/pnas.96.26.15050
150. Santambrogio L, Strominger JL. The Ins and Outs of MHC Class II Proteins in Dendritic Cells. Immunity (2006) 25:857–9. doi: 10.1016/j.immuni.2006.11.007
151. Burton BR, Britton GJ, Fang H, Verhagen J, Smithers B, Sabatos-Peyton CA, et al. Sequential Transcriptional Changes Dictate Safe and Effective Antigen-Specific Immunotherapy. Nat Commun (2014) 5:4741. doi: 10.1038/ncomms5741
152. Bevington SL, Ng STH, Britton GJ, Keane P, Wraith DC, Cockerill PN. Chromatin Priming Renders T Cell Tolerance-Associated Genes Sensitive to Activation Below the Signaling Threshold for Immune Response Genes. Cell Rep (2020) 31:107748. doi: 10.1016/j.celrep.2020.107748
153. Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and Transcriptional Regulation of the Co-Inhibitory Gene Module in T Cells. Nature (2018) 558:454–9. doi: 10.1038/s41586-018-0206-z
154. Capini C, Jaturanpinyo M, Chang H-I, Mutalik S, McNally A, Street S, et al. Antigen-Specific Suppression of Inflammatory Arthritis Using Liposomes. J Immunol (2009) 182:3556–65. doi: 10.4049/jimmunol.0802972
155. Galea R, Nel HJ, Talekar M, Liu X, Ooi JD, Huynh M, et al. PD-L1– and Calcitriol-Dependent Liposomal Antigen-Specific Regulation of Systemic Inflammatory Autoimmune Disease. JCI Insight (2019) 4:e126025. doi: 10.1172/jci.insight.126025
156. Maldonado RA, LaMothe RA, Ferrari JD, Zhang A-H, Rossi RJ, Kolte PN, et al. Polymeric Synthetic Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. PNAS (2015) 112:E156–65. doi: 10.1073/pnas.1408686111
157. Gutiérrez-Vázquez C, Quintana FJ. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity (2018) 48:19–33. doi: 10.1016/j.immuni.2017.12.012
158. Hlavaty KA, McCarthy DP, Saito E, Yap WT, Miller SD, Shea LD. Tolerance Induction Using Nanoparticles Bearing HY Peptides in Bone Marrow Transplantation. Biomaterials (2016) 76:1–10. doi: 10.1016/j.biomaterials.2015.10.041
159. Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, et al. Microparticles Bearing Encephalitogenic Peptides Induce T-Cell Tolerance and Ameliorate Experimental Autoimmune Encephalomyelitis. Nat Biotechnol (2012) 30:1217–24. doi: 10.1038/nbt.2434
160. Getts DR, Shea LD, Miller SD, King NJC. Harnessing Nanoparticles for Immune Modulation. Trends Immunol (2015) 36:419–27. doi: 10.1016/j.it.2015.05.007
161. Pujol-Autonell I, Serracant-Prat A, Cano-Sarabia M, Ampudia RM, Rodriguez-Fernandez S, Sanchez A, et al. Use of Autoantigen-Loaded Phosphatidylserine-Liposomes to Arrest Autoimmunity in Type 1 Diabetes. PloS One (2015) 10:e0127057. doi: 10.1371/journal.pone.0127057
162. Roberts RA, Eitas TK, Byrne JD, Johnson BM, Short PJ, McKinnon KP, et al. Towards Programming Immune Tolerance Through Geometric Manipulation of Phosphatidylserine. Biomaterials (2015) 72:1–10. doi: 10.1016/j.biomaterials.2015.08.040
163. Singha S, Shao K, Yang Y, Clemente-Casares X, Solé P, Clemente A, et al. Peptide–MHC-Based Nanomedicines for Autoimmunity Function as T-Cell Receptor Microclustering Devices. Nat Nanotechnol (2017) 12:701–10. doi: 10.1038/nnano.2017.56
164. Anderton SM, Wraith DC. Hierarchy in the Ability of T Cell Epitopes to Induce Peripheral Tolerance to Antigens From Myelin. Eur J Immunol (1998) 28:1251–61. doi: 10.1002/(SICI)1521-4141(199804)28:04<1251::AID-IMMU1251>3.0.CO;2-O
165. Bergot A-S, Buckle I, Cikaluru S, Naranjo JL, Wright CM, Zheng G, et al. Regulatory T Cells Induced by Single-Peptide Liposome Immunotherapy Suppress Islet-Specific T Cell Responses to Multiple Antigens and Protect From Autoimmune Diabetes. J Immunol (2020), ji1901128. doi: 10.4049/jimmunol.1901128
166. Graham RR, Ortmann WA, Langefeld CD, Jawaheer D, Selby SA, Rodine PR, et al. Visualizing Human Leukocyte Antigen Class II Risk Haplotypes in Human Systemic Lupus Erythematosus. Am J Hum Genet (2002) 71:543–53. doi: 10.1086/342290
167. Deshmukh US, Sim DL, Dai C, Kannapell CJ, Gaskin F, Rajagopalan G, et al. HLA-DR3 Restricted T Cell Epitope Mimicry in Induction of Autoimmune Response to Lupus-Associated Antigen SmD. J Autoimmun (2011) 37:254–62. doi: 10.1016/j.jaut.2011.07.002
168. Kaliyaperumal A, Mohan C, Wu W, Datta SK. Nucleosomal Peptide Epitopes for Nephritis-Inducing T Helper Cells of Murine Lupus. J Exp Med (1996) 183:2459–69. doi: 10.1084/jem.183.6.2459
169. Zhang L, Bertucci AM, Ramsey-Goldman R, Harsha-Strong ER, Burt RK, Datta SK. Major Pathogenic Steps in Human Lupus can be Effectively Suppressed by Nucleosomal Histone Peptide Epitope-Induced Regulatory Immunity. Clin Immunol (2013) 149:365–78. doi: 10.1016/j.clim.2013.08.008
170. Stemmer C, Briand J-P, Muller S. Mapping of Linear Histone Regions Exposed at the Surface of the Nucleosome in Solution11edited by T. Richmond J Mol Biol (1997) 273:52–60. doi: 10.1006/jmbi.1997.1270
171. Muller S. BD, Thiry M, Van Regenmortel MHV. Reactivity of Autoantibodies in Systemic Lupus Etythematosus With Synthetic Core Histone Peptides. Int Arch Allergy Immunol (1989) 89:288–96. doi: 10.1159/000234962
172. Kang HK, Liu M, Datta SK. Low-Dose Peptide Tolerance Therapy of Lupus Generates Plasmacytoid Dendritic Cells That Cause Expansion of Autoantigen-Specific Regulatory T Cells and Contraction of Inflammatory Th17 Cells. J Immunol (2007) 178:7849–58. doi: 10.4049/jimmunol.178.12.7849
173. Kang HK, Chiang MY, Liu M, Ecklund D, Datta SK. The Histone Peptide H4 71-94 Alone Is More Effective Than a Cocktail of Peptide Epitopes in Controlling Lupus: Immunoregulatory Mechanisms. J Clin Immunol (2011) 31:379–94. doi: 10.1007/s10875-010-9504-4
174. Sabbatini A, Bombardieri S, Migliorini P. Autoantibodies From Patients With Systemic Lupus Erythematosus Bind a Shared Sequence of SmD and Epstein-Barr Virus-Encoded Nuclear Antigen EBNA I. Eur J Immunol (1993) 23:1146–52. doi: 10.1002/eji.1830230525
175. Nel HJ, Malmström V, Wraith DC, Thomas R. Autoantigens in Rheumatoid Arthritis and the Potential for Antigen-Specific Tolerising Immunotherapy. Lancet Rheumatol (2020), e712–23. doi: 10.1016/S2665-9913(20)30344-1
176. Gunawan M, Her Z, Liu M, Tan SY, Chan XY, Tan WWS, et al. A Novel Human Systemic Lupus Erythematosus Model in Humanised Mice. Sci Rep (2017) 7:16642. doi: 10.1038/s41598-017-16999-7
177. Mihaylova N, Chipinski P, Bradyanova S, Velikova T, Ivanova-Todorova E, Chausheva S, et al. Suppression of Autoreactive T and B Lymphocytes by Anti-Annexin A1 Antibody in a Humanized NSG Murine Model of Systemic Lupus Erythematosus. Clin Exp Immunol (2020) 199:278–93. doi: 10.1111/cei.13399
178. Maria Dall'Era AH, Bluestone J, Rosenblum M, Wofsy D. NCT02428309: Autologous Polyclonal Tregs for Lupus. (2015), ClinicalTrials.gov.
179. Deshmukh US, Gaskin F, Lewis JE, Kannapell CC, Fu SM. Mechanisms of Autoantibody Diversification to SLE-Related Autoantigens. Ann NY Acad Sci (2003) 987:91–8. doi: 10.1111/j.1749-6632.2003.tb06036.x
180. Riemekasten G, Langnickel D, Ebling FM, Karpouzas G, Kalsi J, Herberth G, et al. Identification and Characterization of SmD183-119-Reactive T Cells That Provide T Cell Help for Pathogenic Anti-Double-Stranded DNA Antibodies. Arthritis Rheum (2003) 48:475–85. doi: 10.1002/art.10762
181. James JA, Gross T, Scofield RH, Harley JB. Immunoglobulin Epitope Spreading and Autoimmune Disease After Peptide Immunization: Sm B/B'-Derived PPPGMRPP and PPPGIRGP Induce Spliceosome Autoimmunity. J Exp Med (1995) 181:453–61. doi: 10.1084/jem.181.2.453
182. Reynolds P, Gordon TP, Purcell AW, Jackson DC, McCluskey J. Hierarchical Self-Tolerance to T Cell Determinants Within the Ubiquitous Nuclear Self-Antigen La (SS-B) Permits Induction of Systemic Autoimmunity in Normal Mice. J Exp Med (1996) 184:1857–70. doi: 10.1084/jem.184.5.1857
183. Dumortier H, Monneaux F, Jahn-Schmid B, Briand J-P, Skriner K, Cohen PL, et al. B and T Cell Responses to the Spliceosomal Heterogeneous Nuclear Ribonucleoproteins A2 and B1 in Normal and Lupus Mice. J Immunol (2000) 165:2297. doi: 10.4049/jimmunol.165.4.2297
184. Mohan C, Adams S, Stanik V, Datta SK. Nucleosome: A Major Immunogen for Pathogenic Autoantibody-Inducing T Cells of Lupus. J Exp Med (1993) 177:1367–81. doi: 10.1084/jem.177.5.1367
Keywords: systemic lupus erythematosis, tolerance, dendritic cells, antigen (Ag), immunotherapies and vaccines
Citation: Robinson S and Thomas R (2021) Potential for Antigen-Specific Tolerizing Immunotherapy in Systematic Lupus Erythematosus. Front. Immunol. 12:654701. doi: 10.3389/fimmu.2021.654701
Received: 17 January 2021; Accepted: 25 June 2021;
Published: 16 July 2021.
Edited by:
Ciriaco A Piccirillo, McGill University, CanadaReviewed by:
Matthew Cook, Australian National University, AustraliaCopyright © 2021 Robinson and Thomas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ranjeny Thomas, cmFuamVueS50aG9tYXNAdXEuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.