 Lee K. Page
Lee K. Page Karl J. Staples
Karl J. Staples C. Mirella Spalluto1,2
C. Mirella Spalluto1,2 Alastair Watson
Alastair Watson Tom M. A. Wilkinson
Tom M. A. Wilkinson- 1Clinical and Experimental Sciences, University of Southampton Faculty of Medicine, Southampton, United Kingdom
- 2NIHR Southampton Biomedical Research Centre, Southampton Centre for Biomedical Research, Southampton General Hospital, Southampton, United Kingdom
- 3Birmingham Medical School, University of Birmingham, Birmingham, United Kingdom
Under normal physiological conditions, the lung remains an oxygen rich environment. However, prominent regions of hypoxia are a common feature of infected and inflamed tissues and many chronic inflammatory respiratory diseases are associated with mucosal and systemic hypoxia. The airway epithelium represents a key interface with the external environment and is the first line of defense against potentially harmful agents including respiratory pathogens. The protective arsenal of the airway epithelium is provided in the form of physical barriers, and the production of an array of antimicrobial host defense molecules, proinflammatory cytokines and chemokines, in response to activation by receptors. Dysregulation of the airway epithelial innate immune response is associated with a compromised immunity and chronic inflammation of the lung. An increasing body of evidence indicates a distinct role for hypoxia in the dysfunction of the airway epithelium and in the responses of both innate immunity and of respiratory pathogens. Here we review the current evidence around the role of tissue hypoxia in modulating the host-pathogen interaction at the airway epithelium. Furthermore, we highlight the work needed to delineate the role of tissue hypoxia in the pathophysiology of chronic inflammatory lung diseases such as asthma, cystic fibrosis, and chronic obstructive pulmonary disease in addition to novel respiratory diseases such as COVID-19. Elucidating the molecular mechanisms underlying the epithelial-pathogen interactions in the setting of hypoxia will enable better understanding of persistent infections and complex disease processes in chronic inflammatory lung diseases and may aid the identification of novel therapeutic targets and strategies.
Introduction
The airway epithelium is located at the interface between the internal and external environment and is strategically positioned to interact with the environment in a dynamic fashion. The pseudostratified mucosal barrier consisting of multiple cell types, constitutes the lung epithelium (1). In a healthy state the airway epithelium plays an integral role in host defense through a physical and mechanical barrier, innate immune mediator production, and chemokine and cytokine production to recruit inflammatory cells for both propagation and resolution of the immune response (2–4). The importance of the lung epithelium is exemplified in chronic inflammatory lung diseases, where epithelial cell dysfunction is associated with compromised immunity and chronic inflammation in the lung (5–9). Structural and functional abnormalities in both the airway and alveolar epithelium have a significant impact on host defenses, immune/inflammatory response, and the repair process leading to progressive lung damage and impaired lung function.
Although an oxygen rich environment under normal physiological conditions, the lung mucosal surface is susceptible to conditions of oxygen deficiency or tissue hypoxia (10) during infection and inflammation, which occurs when cellular demand exceeds supply. Direct in vivo evidence has demonstrated that pulmonary infection is associated with profound local hypoxia (11–14). The occurrence of hypoxia during infection and associated inflammation is multifaceted and involves increased oxygen demand in order to satisfy the requirements of inflamed resident cells, and in some instances, multiplying pathogens (15–17). Furthermore, infiltrating inflammatory cells such as neutrophils are thought to influence the tissue environment due to their metabolic cost. For example, it has been demonstrated that migration of neutrophils across the epithelium increases the transcriptional activity of hypoxia-inducible genes in epithelial cells, due to localized oxygen depletion, resulting in microenvironmental hypoxia which in turn, influences the resolution of inflammation (18). Furthermore, beyond acute infection, chronic inflammatory respiratory diseases are also commonly associated with mucosal hypoxia. The airways of respiratory disease patients are characterized by chronic inflammation, structural changes and fibrosis, and airways obstruction through excessive mucus accumulation (19–22), which can lead to regions of local tissue hypoxia. Cystic fibrosis (CF) is an autosomal recessive disorder caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. CF is characterized by airway mucus plugging, reduced mucus clearance due CFTR defects which renders the CF airways vulnerable to chronic infection and inflammation. The mucus filled CF airway, infected with P. aeruginosa is extremely hypoxic (23). It is thought that thick stagnant mucus infiltrated with immune cells and multiplying pathogens creates a steep oxygen gradient within the mucus, exposing the underlying epithelial cells to marked hypoxia. Furthermore, the airway epithelium of mouse models of CF stained strongly with the specific hypoxia probe, pimonidazole hydrochloride (Hypoxyprobe, which binds at a threshold of ≤ 10 mmHg O2) (24), confirming that tissue hypoxia is present in this inflamed airway epithelium. Chronic obstructive pulmonary disease (COPD) is characterized by chronic airway inflammation and functional and structural alterations in the lung, primarily caused by long-term inhalation of harmful particles such as cigarette smoke (20, 25, 26). Remodeling in the large airway in COPD, is accompanied by thickening and fibrosis of the subepithelial microvasculature and perivascular fibrosis (27), which may significantly reduce oxygenation of the airway epithelium. Increased expression of hypoxia-inducible factor (HIF)-1α is detected in the bronchial epithelium in COPD in areas of airway remodeling and goblet cell hyperplasia (28–30). Asthma, is another obstructive airway disease that involves chronic airway inflammation of the respiratory tract and excessive mucus production which is triggered by a variety of airborne insults including allergens, dust, smoking and respiratory pathogens. The increased expression of HIF-1α in lung mucosal biopsy specimens from asthmatic patients (31), may also indicate the presence of a tissue hypoxia in the asthmatic airway.
Pulmonary diseases associated with infection, excessive airway inflammation, airway obstruction, airway remodeling and emphysema can lead to decreased blood and also tissue oxygenation and consequently a fall in the partial pressure of oxygen in the arterial blood (10, 32, 33). This is particularly evident in COVID-19 where hypoxia is a major risk factor for pneumonia and respiratory distress following severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (34, 35). Furthermore, in COPD, the progression of the disease increases the risk of alveolar hypoxia and consequent hypoxemia (36). Ventilation/perfusion (V/Q) mismatch resulting from progressive airflow limitation and emphysematous destruction of the pulmonary capillary bed is the main factor contributing to hypoxemia in COPD patients (36). Hypoxemia associated with COPD contributes to reduced quality of life, diminished exercise tolerance, reduced skeletal muscle function, and ultimately increased risk of death (37). Moreover, exacerbations of COPD, which are associated with disease morbidity and mortality (38–41), are also frequently associated with deterioration in gas exchange and associated hypoxemia, due to increased tissue oxygen consumption and V/Q mismatch (42).
Whilst local tissue hypoxia and systemic hypoxia in the lung play a prominent role during infection and is present in chronic inflammatory respiratory diseases, the role of hypoxia at the level of the tissue in shaping the host-pathogen interactions in respiratory diseases is not fully understood. Here we review the current evidence around the role of tissue hypoxia in modulating the host-pathogen interaction at the lung epithelium. Furthermore, we highlight the essential work now needed to outline the role of tissue hypoxia in the pathophysiology of inflammatory lung diseases and emerging lung diseases such as COVID-19 and post-COVID-19 chronic lung disease.
Transcriptional Responses to Hypoxia
For the host to be able respond to pathogens effectively and efficiently in hypoxic conditions, certain hypoxic regulatory mechanisms are essential. Thus, it is important to fully appreciate the significant effect that hypoxia has on downstream signaling pathways in tissue resident and infiltrating immune cells. The immune response under hypoxic conditions is governed by several pathways and metabolic activity, of which hypoxia-inducible factor (HIF), is the best characterized and termed the master regulator of the host response to hypoxia (Figure 1). HIF is a DNA-binding transcription factor that associates with specific nuclear cofactors during hypoxia. HIF regulates hundreds of downstream genes which are involved in diverse biological pathways (43). The regulatory complex is comprised of HIF-1β, a constitutive subunit, and one of the HIF-α isoforms: HIF-1α or HIF-2α (44). In the presence of oxygen, HIF-α subunits undergo hydroxylation by prolyl hydroxylase domain (PHD) proteins and an asparagine hydroxylase known as the factor inhibiting HIF (FIH), in an oxygen and iron-dependent manner (45). Hydroxylated HIF-α is then targeted by the von Hippel-Lindau (VHL) protein, a substrate-recognition subunit of an ubiquitin-protein ligase which subsequently interacts with HIF-α to undergo proteasomal degradation (46). During hypoxia, the activity of hydroxylase is reduced, leading to HIF stabilization, translocating to the nucleus from the cytoplasm, ultimately dimerizing with the HIF-1β subunit, to form the active HIF complex (47). Finally, the active forms of HIF recruit coactivator proteins to the hypoxia-response element, activating the transcription of target genes required for undergoing adaptations to hypoxia. HIF-1α is widely expressed in many innate immune cells including macrophages, neutrophils, dendritic cells and epithelial cells. Integral innate immune functions are preserved in hypoxic conditions through HIF. For example, the survival of neutrophils is extended in hypoxic conditions through HIF-1α and nuclear factor kappa-light- chain-enhancer of activated B cells (NF-κB) activation (48). Furthermore, it has been demonstrated that hypoxia-mediated HIF-1 activation of NF-κB in airway epithelial cells releases greater amounts of proinflammatory cytokines (49). However, hypoxia may also suppress the epithelial inflammatory response (50, 51). Exposure of murine airway epithelial cells to hypoxia has been shown to reduce the expression of key innate immune molecules through reduced NF-κB signaling (51). Moreover, the macrophage response to bacterial lipopolysaccharide (LPS) is enhanced under hypoxia, through HIF-1-mediated, increased toll-like receptor (TLR) gene expression, resulting in increased expression of cyclooxygenase-2, interleukin-6 and Regulated on Activation, Normal T Cell Expressed and Secreted (RANTES) (52, 53), suggesting that hypoxic conditions may contribute to the aggravated inflammatory responses during infection. Hypoxia also regulates other key transcription factors which contribute to the overall cellular response, including the major regulator of immunity NF-κB (54). Hypoxia-mediated activation of NF-κB is through decreased PHD-dependent hydroxylation of inhibitor of nuclear factor kappa B kinase subunit beta (IKKβ), resulting in the phosphorylation-dependent degradation of IκBα and liberation of NF-κB. Therefore, HIF-dependent and HIF-independent regulatory pathways in host cells that are regulated during tissue hypoxia may impact on the progression of chronic inflammatory respiratory diseases. Although hypoxia is essential in driving immunological processes and resolving respiratory infections, the relationship between immunity and hypoxia is delicately balanced. Resultant sustained aberrant inflammation and activity of immune cells leads to tissue damage and is a pathological hallmark of many chronic inflammatory respiratory diseases.
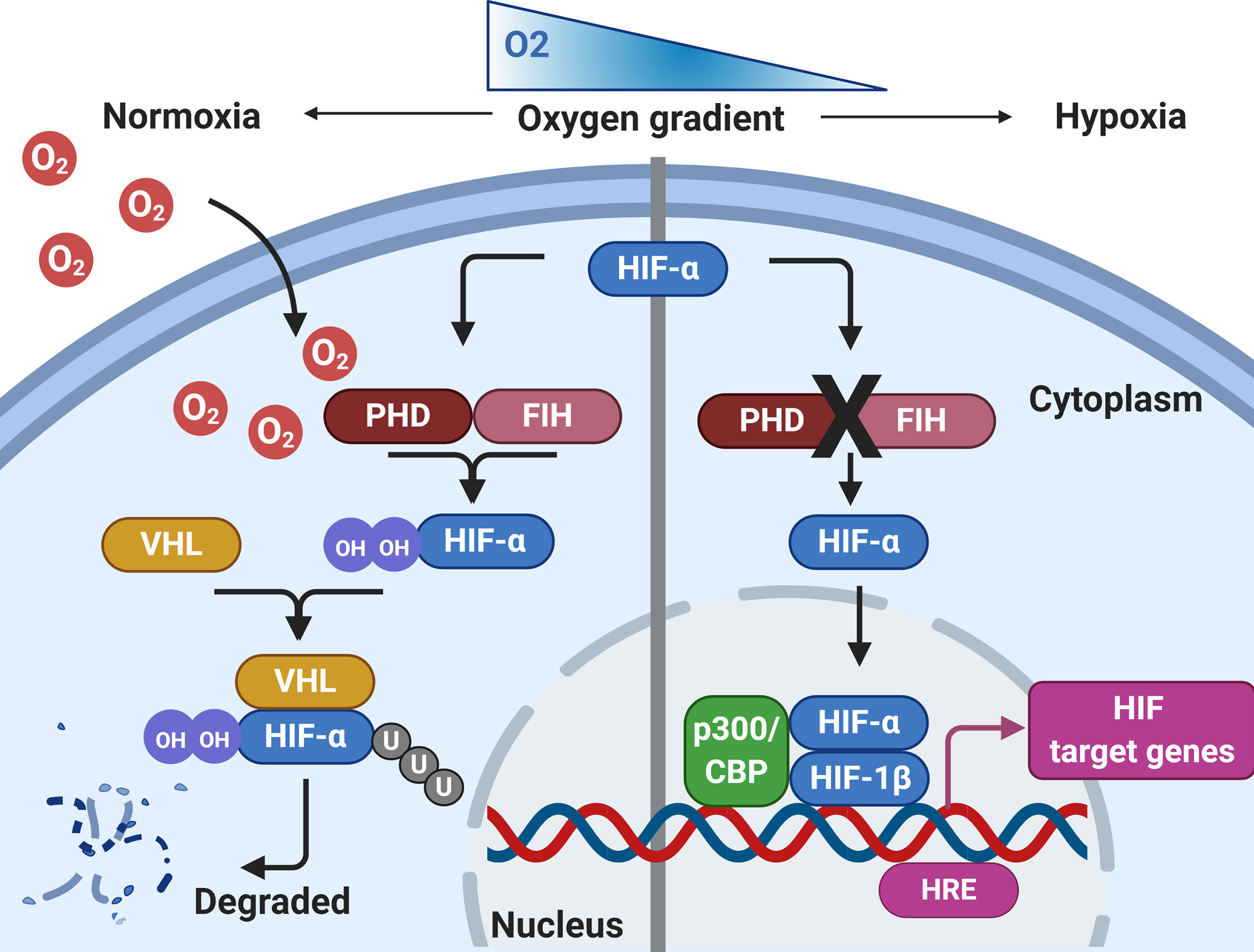
Figure 1 Regulation of HIF by hypoxia. During normoxia, HIF-α is hydroxylated by prolyl hydroxylase domain proteins (PHD) and factor inhibiting HIF (FIH), using molecular oxygen. This leads to HIF-α interacting with Von Hipple-Lindau (VHL), before being targeted for proteasomal degradation. During conditions of hypoxia, HIF-α hydroxylation by PHD/FIH is inhibited and HIF-α is not targeted for proteasomal degradation. HIF-α can translocate to the nucleus, where it binds with HIF-1β and recruits co-activators at the hypoxia response element (HRE) to initiate gene transcription. Created with BioRender.com.
Hypoxia-Epithelial Interactions: Consequences for the Host During Respiratory Infection
The airway epithelium forms the interface between the external environment and the internal milieu, making it a prime target for inhaled pathogens. However, the epithelium is not just a bystander, it forms an integral part of the innate immune system through being a physical barrier as well as releasing effectors to initiate and orchestrate immune and inflammatory responses (1). Furthermore, dysfunction of the airway epithelium is associated with inflammatory lung diseases and increases the susceptibility to infection (55). Here we will discuss how hypoxia influences the epithelial-pathogen interactions in the lung, with important implications for respiratory disease (Figure 2).
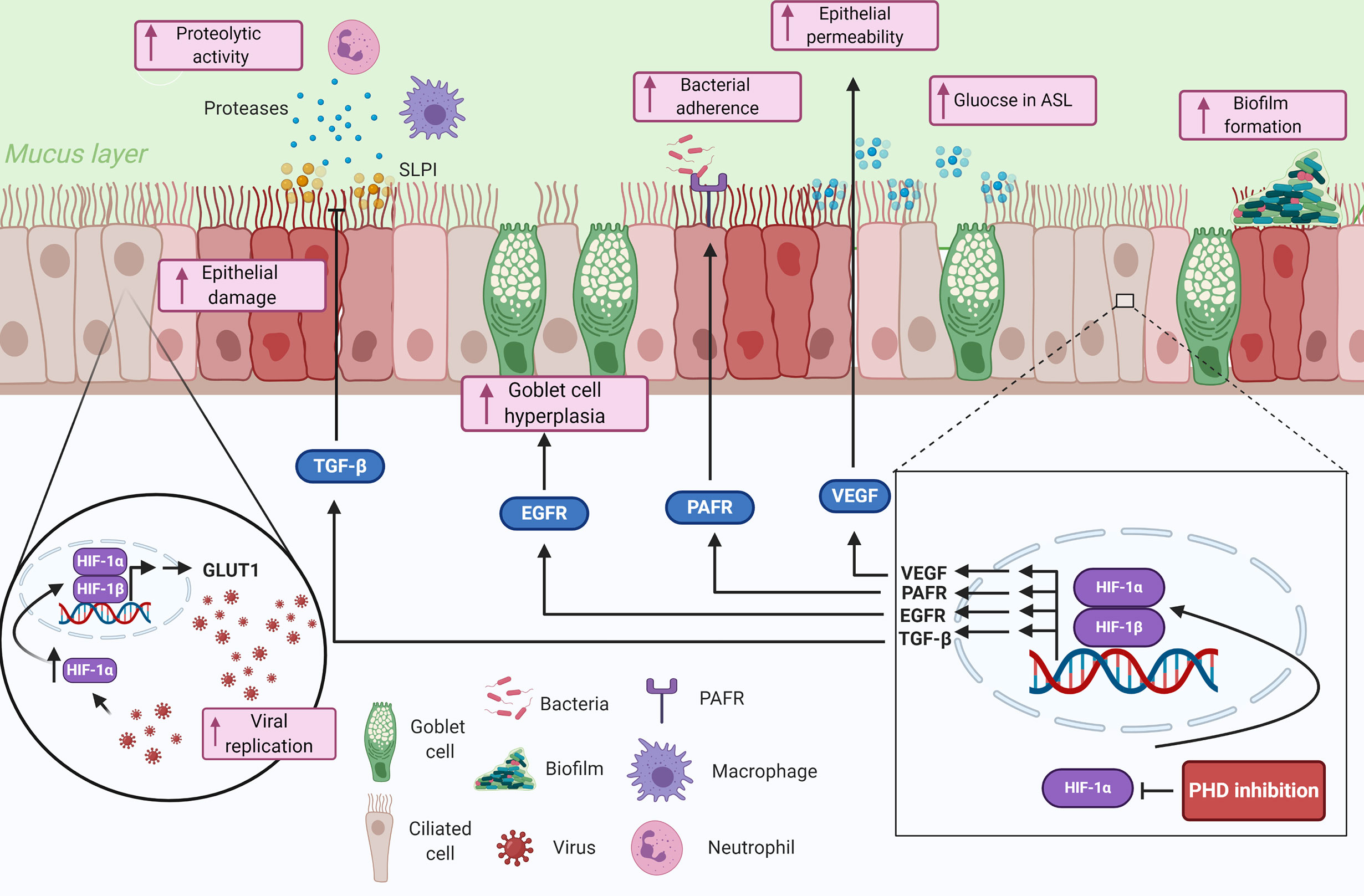
Figure 2 Summary of the potential contributions of tissue hypoxia to the epithelial-pathogen interactions in respiratory diseases. Hypoxia is sensed by airway epithelial cells, that proceed to upregulate genes involved in the response to hypoxia through HIF-1α. The upregulated genes and proteins in response to HIF-1 transcription modulate several immune responses including impairing epithelial barrier function, reducing mucociliary clearance, modulating nutrient availability, and reducing anti-proteolytic enzymes. Hypoxia could also play important roles in pathogen colonization of the lung epithelium by mediating bacterial adherence through HIF-1-dependent mechanisms and enhance biofilm formation through bacterial-adaptive mechanisms. Finally, viral infected epithelial cells can be manipulated to upregulate glycolytic pathways through a HIF-1α-dependent mechanism, which consequently increases viral replication in the cells. Created with BioRender.com.
Mucus Hypersecretion and Reduced Mucociliary Clearance
A fundamental lung mucosal defense mechanism is the secretion of mucus into the bronchial airway lumen to capture and trap invading pathogens, before being mechanically removed via mucociliary clearance (56, 57). The mucosal layer in the conducting airways exhibits a continuous layer of secreted proteins called mucins. Mucins are large glycoproteins formed of O-linked polysaccharides, which form a fluid-like barrier tethered to epithelial cells (57). The mucus-producing goblet cells function as the primary secretory cell, with the more abundant ciliated cells functioning as transporters to move the mucus up the airways for removal (58, 59). The ratio of secretory to ciliated cells is regulated both during normal physiological processes and also during infection to maintain optimal mucociliary function (60). However, chronic respiratory diseases are associated with pathological changes with the development of goblet cell hyperplasia, reduced expression of ciliated cells, mucus hypersecretion and mucus plugging (61–65). Sustained activation of disease associated signals such as from cigarette smoke, allergens and pathogens, promote the excessive differentiation towards goblet cell hyperplasia and mucus hypersecretion (66). Consequently, increased mucus production, dehydration of the airway surface liquid and mucus plugging prevents the mucociliary functions of the innate immune system to adequately clear the airways of mucus and pathogens (67, 68).
The development of mucus plugs has also been associated with cellular hypoxia of epithelial cells lining the airways, creating hypoxic niches within the adherent mucus and subjacent epithelial cells (24). This is exemplified by mucus obstructed airway epithelial samples from COPD patients which are known to be hypoxic, exhibiting an increased expression of HIF-1α in areas of goblet cell hyperplasia, and the hypoxic mucus filled CF airways (23, 69). Furthermore, analysis of CF and COPD lung tissue samples found that hypoxic airway epithelial necrosis in the mucus obstructed airways was a key trigger for neutrophilic inflammation, which may further exacerbate epithelial hypoxia (70). Interestingly, HIF-1α has been shown to induce goblet cell hyperplasia and increase the expression of the mucin MUC5AC, via the epidermal growth factor receptor (EGFR) (71–73). The EGFR is upregulated in the diseased airway epithelium and aberrant EGFR signaling has been implicated in the pathogenesis of asthma, COPD and CF (74, 75). EGFR is an important protein involved in the epithelial repair process via the induction of epithelial migration, proliferation, differentiation, and extracellular matrix synthesis. However, the activation of EGFR, also leads to goblet cell hyperplasia, mucus hypersecretion, and overproduction of MUC5AC, which may exacerbate the pathological changes in respiratory diseases (76–79). Overproduction of the MUC5AC gene is a characteristic of many respiratory diseases including CF, asthma, and COPD (7, 80, 81), creating hyper-concentrated mucus, which is thicker, more adherent and prone to mucus plugging (82). Consequently, thicker and more adherent mucus may create further hypoxic niches within it. This has major implications not only for the progression of diseases but also for the host-pathogen interactions within the airways. Several bacterial pathogens, including non-typeable Haemophilus influenzae (NTHi), P. aeruginosa and Staphylococcus aureus, are able to bind to and utilize mucins as a mechanism for colonization of the lung (83–85). Additionally, the hypoxic niches created in the mucus plugged airway may favour infectious organisms that gain energy efficiently under anaerobic conditions (86, 87). The increased presence of anaerobic bacteria in the airway at more advanced stages of respiratory diseases have been demonstrated in lung microbiome studies (88–90). The changes in microbiota in the diseased airway may negatively impact the progression of the disease (91–93). For example, P. aeruginosa which is found in sputum more frequently in advanced stages of COPD is associated with exacerbations and increased risk of mortality (94, 95). Furthermore the presence of obligate anaerobes have been linked to disease severity and inflammation in CF (96, 97). Although still unknown, it may be postulated that mucolytic therapies which clear the airways of sputum, reduce mucus production and plugging and prevent airway obstruction could also play a role in reducing mucosal hypoxia. Alternatively, interventions which correct local hypoxia e.g. long-term oxygen therapy could hypothetically improve mucosal regulation and mucus hypersecretion and warrants investigation.
Disruption of the Epithelial Barrier
An important function of epithelial cells is to act as a physical barrier towards the outside environment. Tight junctions (TJ), adherens junctions (AJ) and desmosomes form the transcellular junctions (98). These junctions are formed through intercellular junctional proteins including claudins, connexins, paranexins, cadherins, adhesions, and zonula occludins (ZO), which link to the actin cytoskeleton (99, 100). This tightly regulated physical barrier not only controls paracellular ionic movements and non-permeability of the epithelium (101, 102), but also prevents microbial compounds and airborne substances access to the body interior. The epithelial barrier is critical in the innate host defense as a loss of barrier function increases the susceptibility of the host to infection and injury by pathogens and proteases (103). In addition, a pathological hallmark of several chronic inflammatory respiratory diseases, including asthma and COPD, is impaired epithelial barrier function and increased epithelial permeability, which may permit access for pathogens to the underlying submucosa (5, 7, 104–107). A number of factors have been implicated in epithelial barrier dysfunction including infection (108–110) and inhalation of noxious particles such as cigarette smoke (111). Several studies have also demonstrated that exposing airway epithelial cells to hypoxia decreased the expression of apical cytoskeleton proteins actin and α-spectrin (112), and also the expression of the TJ proteins ZO-1, claudin-4, occludin, and E-Cadherin (49, 112, 113), which consequently reduced epithelial barrier function. One possible mechanism is through the HIF-1α-mediated induction of vascular endothelial growth factor (VEGF), during hypoxia, which has been shown to increase epithelial permeability (114). VEGF is a pleiotropic protein that regulates vascular angiogenesis and endothelial permeability and is an important adaptive mechanism to hypoxia, enhancing local vascularization and oxygen transport (115), in addition to being an important mediator of inflammation (116). The expression of VEGF is upregulated in the diseased airway epithelium, and has been implicated in airway remodeling processes (28, 117–119). Additionally, VEGF expression itself in the epithelium is a good indicator of local tissue hypoxia. Several studies have demonstrated that exposing airway epithelial cells to hypoxia, increased the expression of VEGF via HIF-1 (114, 120–122). Though the induction of VEGF may be an adaptive response of angiogenesis when oxygen availability is reduced; paradoxically this may have pathological consequences in terms of epithelial barrier disruption. Thus, the hypoxia-HIF-1α-VEGF axis may be an important mechanism driving epithelial barrier disruption and increased permeability of the epithelium. Regarding the lung mucosal defense mechanisms, vulnerability to adherence and invasion of pathogens is increased by the leaky epithelial barrier, resulting in airway infection and subsequent inflammation. Therefore, hypoxia-mediated epithelial permeability may ultimately facilitate pathogen invasion of the epithelium and persistence in the airway epithelium.
Increased Bacterial Adherence to the Epithelium
A crucial step for effective bacterial colonization and invasion involves adherence to host components in the airways (123, 124). Platelet-activating factor receptor (PAFR) is one of the main epithelial cell-derived adhesion molecules used by both Gram-positive and Gram-negative bacteria (125–129). PAFR is a G-protein coupled epithelial cell membrane receptor that naturally binds the phosphorylcholine (ChoP) ligand on the eukaryotic proinflammatory chemokine PAF (130). Several species of airway bacteria display a host-derived zwitterionic/bipolar molecule ChoP on their bacterial walls, mimicking PAF to facilitate adherence to epithelial cells (131, 132). PAFR is upregulated in the lungs of asthmatic and COPD patients (133–135). The expression of PAFR is upregulated by noxious environmental particles such as cigarette smoke, electronic cigarettes, and biomass smoke exposure and exposure to pathogens (126, 128, 134, 136–139). Additionally, hypoxia prominently induces epithelial PAFR through HIF-dependent mechanisms (140). The upregulation of PAFR in the lungs has been associated with increased adherence to the mucosal surfaces by respiratory pathogens, airway inflammation, speed of lung function decline, and development of pneumonia (126, 136, 138, 141). Ultimately, microbial manipulation of PAFR, may be an important strategy for successful colonization and infection. The importance of ChoP-PAFR mediated bacterial adherence to epithelial cells has been confirmed by the use of PAFR antagonists, which have been shown to reduce bacterial adherence and invasion (127–129, 137). Thus, the dynamic and integral role of HIF-1α in key immune functions opens complex questions regarding HIF-1α. However, there is still much needed research regarding the role of HIF-1α and PAFR.
Enhanced Biofilm Formation
The formation of multicellular microbial communities called biofilms is a critical step for pathogens during colonization of the lung, enabling survival and persistence in the challenging environment by attaching to a living surface (142, 143). Many different microbes reside in biofilms and the majority of persistent infections involve biofilms (144). Notably, biofilm communities enhance antimicrobial resistance to exogenous and endogenous effector molecules (145). There are clear associations between biofilm formation, inflammation, and respiratory disease and biofilms have been implicated in the pathogenesis of COPD and CF (143, 144, 146, 147) and developing bronchopneumonia (148, 149). Hypoxic conditions in the diseased lung may provide prime conditions for biofilm formation by P. aeruginosa which produce greater amounts of alginate under anaerobic conditions, a component involved in biofilm formation and protection against host immune responses (23, 150). Additionally, P. aeruginosa grown under anaerobic or hypoxic conditions yields greater antibiotic resistance and biofilm formation suggesting that hypoxia may be a crucial component in bacterial persistence (151, 152). The increase in antibiotic resistance appears to be due to hypoxia altering the stoichiometry of multidrug efflux pumps (153). An important mechanism of antibiotic resistance is the expulsion of antibiotics through multidrug resistance efflux pump systems belonging to the resistance-nodulation-division family (154). Therefore, these may be important mechanisms facilitating bacterial adaptive responses to hypoxia, increasing virulence and persistence in the diseased airways. There are also suggestions that local tissue hypoxia in the diseased lung is advantageous to anaerobic pathogens such as P. aeruginosa over other pathogens (155). For example, biofilm formation itself contributes to local hypoxia of the diseased CF lung, which correlates with increased dependency on systems that mediate the uptake of reduced ferrous iron (Fe2+) by P. aeruginosa (156). Future studies investigating the role of hypoxia in biofilm formation with the use of ex vivo models would be valuable to understand the mechanisms of biofilm formation in the hypoxic airways (157). Such models could also be used to investigate potential future biofilm-targeting therapeutics.
Dysregulated Proteolytic Activity
During inflammatory processes, a plethora of toxic inflammatory by-products are released by immune cells. For example, neutrophils infiltrating the site of infection have been implicated in causing excessive tissue damage through release of proteases and reactive oxygen species (ROS) (158). Furthermore, macrophages produce matrix metalloproteinases (MMPs) during infectious processes, which can lead to excessive tissue damage (159). Therefore, an adequate protease-antiprotease balance is required to prevent excessive tissue damage and inflammation. Secretory leukocyte protease inhibitor (SLPI) is an important antiprotease, which prevents excessive tissue damage and inflammation (160). This protein also possesses key antimicrobial functions against Gram-negative and Gram-positive bacteria (161, 162). In vivo experiments have demonstrated that SLPI has the ability to dampen the macrophage associated inflammatory burden induced by LPS (163). Furthermore, the pathogenesis of chronic inflammatory respiratory diseases are thought to involve a protease-antiprotease imbalance (164, 165). Sputum samples from COPD patients display lower levels of SLPI during infectious exacerbations (166, 167), and lower levels of SLPI are associated with pronounced airway inflammation, susceptibility to infection, and disease severity (168, 169). It has been demonstrated that hypoxia downregulates the expression of SLPI in airway epithelial cells via the upregulation of transforming growth factor (TGF)-β (170). The expression and function of TGF-β is mediated by HIF-1α during hypoxia to promote cell growth and proliferation (171, 172). Furthermore, TGF-β has been implicated in the vascular remodeling of hypoxia-induced pulmonary hypertension and is also upregulated in the lungs of CF and COPD patients (173–176). Inhibition of SLPI by TGF-β via the SMAD signaling pathway has been demonstrated at both the RNA and protein level (177, 178). This modulation of complementary mechanisms by tissue hypoxia through alterations in TGF-β and thus SLPI expression could accentuate the protease-antiprotease imbalance and exacerbate inflammatory responses leading to increased tissue damage and progression of inflammatory lung diseases.
Disrupted Airway Glucose Homeostasis
In the airways, the composition of the airway surface liquid (ASL) plays a critical role in the first line of defense against infection. In health, glucose concentrations in the fluid lining the ASL are maintained at 0.4 mM, about 12 times lower than the concentration of glucose in the bloodstream (179). This is an important airway defense mechanism against infection, limiting bacterial growth by restricting nutrient availability (180). However, in chronic inflammatory respiratory diseases including CF and COPD, the concentration of glucose in the ASL is increased (179, 181, 182). Disruption of airway glucose homeostasis increases the availability of glucose as a nutrient source in the ASL for bacterial pathogens. Consequently, this has the potential to support proliferation of bacteria able to utilize glucose as a carbon source, increasing bacterial loads and altering bacterial communities. Evidence from in vitro (181, 183–185), animal (186), and human studies (187), indicates that elevated ASL glucose stimulates the proliferation of P. aeruginosa, S. aureus, and other Gram-negative bacteria, which promote bacterial lung infections (188–190). Additionally, elevated levels of glucose in the ASL is associated with exacerbation, inflammatory markers and bacterial load in COPD (181). The principle mechanism thought to be limiting ASL glucose concentration are the epithelial TJs, which restrict paracellular glucose movement (191). It has previously been shown that airway epithelial cell cultures infected with P. aeruginosa, resulted in TJ protein disruption, which was associated with increases in paracellular glucose flux, indicating the importance of epithelial barrier integrity in glucose airway homeostasis (192). It is not yet known if hypoxia impacts ASL glucose concentrations but the presence of hypoxia in chronic inflammatory respiratory diseases and the profound impact of hypoxia on the epithelial barrier and TJ expression may indicate a role for tissue hypoxia and HIF-1. Increased glucose concentrations in the ASL may consequently facilitate nutrient availability for invading pathogens and increase their persistence in the airways. Thus, future experiments assessing the role of hypoxia and glucose airway homeostasis could be explored further in human ex vivo cell cultures.
Enhanced Viral Replication in Epithelial Cells
Switching to glycolytic pathways may also be an important mechanism for viral replication in the lungs. During hypoxic stress, the rate of oxidative phosphorylation is reduced, switching cellular metabolism to the use of anaerobic glycolytic pathways via HIF-1α transcriptional activity (193). Airway epithelial cells respond in a similar fashion, displaying both aerobic and anaerobic glycolytic capacities (194). Ouiddir et al., demonstrated that airway epithelial cells exposed to hypoxia induced a three-fold increase in the expression of the glucose transporter GLUT1, at both the mRNA and protein level (195). The authors concluded that the epithelial cells ability to sustain ATP production during hypoxia was due to an increase in anaerobic glycolysis and increased glucose transport at the membrane level (195). Additionally, hypoxia has been shown to induce the expression and activation of key glycolytic enzymes important for the breakdown of glucose, including pyruvate kinase, lactate dehydrogenase (194), and glyceraldehyde phosphate dehydrogenase (GAPDH) (196). More recently it was discovered that airway epithelial cells exposed to hypoxia resulted in an increase in HIF-1α (197). In these hypoxic cells mitochondrial respiration was subsequently reduced as well as the rate of protein synthesis and demands for ATP, and the activity of GAPDH was increased (197). The influenza H1N1 virus has been found to exploit this mechanism, mimicking the hypoxic response to stabilize HIF-1α during infection of airway epithelial cells (198). This in turn, upregulates the expression of GLUT1, which may reprogram the host cellular glucose metabolism towards enhanced glycolysis to support nucleotide biosynthesis and lipogenesis for efficient viral replication. Rhinovirus infection has also been shown to induce metabolic alterations in host cells by increasing GLUT1 expression with increased glucose uptake and enhanced viral replication (199). More recently, it was demonstrated that monocytes infected with SARS-CoV-2 resulted in mitochondrial ROS-mediated stabilization of HIF-1α and increased glycolysis (200). The increase in glycolysis consequently promoted SARS-CoV-2 replication and cytokine expression. It is not yet known if similar processes take place in airway epithelial cells. However, it can be postulated that hypoxia in the airway during SARS-CoV-2 infection may increase the use of glycolytic enzymes through HIF-1 pathways which may support SARS-CoV-2 replication. In summary, tissue hypoxia and HIF-1 may play an important role in the pathogenesis of viral infections through the switching of glycolytic pathways which can support viral replication. This could be of particular importance in COVID-19 and respiratory diseases such as asthma and COPD where viral infections play a key role in exacerbations and disease progression (201, 202).
Conclusions and Future Directions
Our understanding of the role of tissue hypoxia in mediating the epithelial-pathogen interactions in respiratory diseases is increasing. Hypoxia modulates several innate immune responses including impairing epithelial barrier function, reducing mucociliary clearance, modulating nutrient availability, and reducing protease inhibitors. Hypoxia could also play an important role in pathogen colonization of the lung epithelium through mediating bacterial adherence, internalization, biofilm formation, and viral replication. This review has highlighted potential future experimental work that may aid identification of new drug targets and the development of novel therapeutics. Developing a deeper understanding of the oxygen microenvironment within the lung is now key to create appropriate models and more accurately delineate mucosal host-pathogen interactions. Furthermore, understanding the impact of hypoxia-mediated modulation of transcription factors and the potential numerous implications for respiratory disease is essential. A greater understanding of the balance between beneficial and detrimental HIF-1 activation is also needed. Indeed, HIF-1 activity is critical for host response to pathogens and helps shape the innate and adaptive response. Thus, not all HIF-1 activation can be considered harmful and the therapeutic inhibition of this pathway must be balanced against its beneficial contribution. Nonetheless, targeting the HIF signaling pathway in chronic respiratory disease may still hold promise in effectively managing or delaying the progression of disease (203). Novel therapeutics could be developed which specifically interfere with mRNA expression, protein synthesis, protein degradation, protein dimerization, DNA binding or transcriptional activity of HIF-1. Several in vitro studies have used various methods to inhibit or silence HIF-1 and have ameliorated its deleterious effects in disease models (73, 114, 140, 204). Furthermore, the use of HIF-1α inhibitors for the treatment of patients infected with COVID-19 has recently been highlighted (205). These potential strategies provide interesting opportunities to potentially modulate tissue hypoxia and lung mucosal host-pathogen interactions.
Author Contributions
LP: conceptualization, investigation, literature searching, analysis, project administration, writing original draft, reviewing, and editing. KS: supervision, conceptualization, reviewing, and editing. MS: supervision, conceptualization, reviewing, and editing. AW: supervision, reviewing, and editing. TW: supervision, conceptualization, reviewing, and editing. All authors contributed to the article and approved the submitted version.
Funding
LP is funded by a BBSRC-GSK iCASE studentship.
Conflict of Interest
TW reports grants and personal fees from AstraZeneca, personal fees and other from MMH, grants and personal fees from GSK, personal fees from BI and grants and personal fees from Synairgen, outside the submitted work. KS reports grants from AstraZeneca outside the submitted work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol (2011) 45(2):189–201. doi: 10.1165/rcmb.2011-0011RT
2. Weitnauer M, Mijošek V, Dalpke AH. Control of local immunity by airway epithelial cells. Mucosal Immunol (2016) 9(2):287–98. doi: 10.1038/mi.2015.126
3. Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol (2015) 16(1):27–35. doi: 10.1038/ni.3045
4. Watson A, Spalluto CM, McCrae C, Cellura D, Burke H, Cunoosamy D, et al. Dynamics of IFN-β Responses During Respiratory Viral Infection: Insights for Therapeutic Strategies. Am J Respir Crit Care Med (2019) 201:83–94. doi: 10.1164/rccm.201901-0214OC
5. Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol (2011) 128(3):549–56. doi: 10.1016/j.jaci.2011.05.038
6. Hochberg CH, Sidhaye VK. The Respiratory Epithelium in COPD. In: Lung Epithelial Biology in the Pathogenesis of Pulmonary Disease. Academic Press (2017) 165–84. doi: 10.1016/B978-0-12-803809-3.00009-9
7. De Rose V, Molloy K, Gohy S, Pilette C, Greene CM. Airway Epithelium Dysfunction in Cystic Fibrosis and COPD. Mediators Inflamm (2018) 2018:1–20. doi: 10.1155/2018/1309746
8. Hurst JR, Perera WR, Wilkinson TMA, Donaldson GC, Wedzicha JA. Systemic and upper and lower airway inflammation at exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2006) 173(1):71–8. doi: 10.1164/rccm.200505-704OC
9. Staples KJ, Taylor S, Thomas S, Leung S, Cox K, Pascal TG, et al. Relationships between mucosal antibodies, non-typeable Haemophilus influenzae (NTHi) infection and airway inflammation in COPD. PloS One (2016) 11(11):1–17. doi: 10.1371/journal.pone.0167250
10. Chen T, Yang C, Li M, Tan X. Alveolar Hypoxia-Induced Pulmonary Inflammation: From Local Initiation to Secondary Promotion by Activated Systemic Inflammation. J Vasc Res (2017) 53(5–6):317–29. doi: 10.1159/000452800
11. Harper J, Skerry C, Davis SL, Tasneen R, Weir M, Kramnik I, et al. Mouse model of necrotic tuberculosis granulomas develops hypoxic lesions. J Infect Dis (2012) 205(4):595–602. doi: 10.1093/infdis/jir786
12. Grahl N, Puttikamonkul S, Macdonald JM, Gamcsik MP, Ngo LY, Hohl TM, et al. In vivo hypoxia and a fungal alcohol dehydrogenase influence the pathogenesis of invasive pulmonary aspergillosis. PloS Pathog (2011) 7(7):e1002145. doi: 10.1371/journal.ppat.1002145
13. Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Seok YE, et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun (2008) 76(6):2333–40. doi: 10.1128/IAI.01515-07
14. Belton M, Brilha S, Manavaki R, Mauri F, Nijran K, Hong YT, et al. Hypoxia and tissue destruction in pulmonary TB. Thorax (2016) 71(12):1145–53. doi: 10.1136/thoraxjnl-2015-207402
15. Schaffer K, Taylor CT. The impact of hypoxia on bacterial infection. FEBS J (2015) 282(12):2260–6. doi: 10.1111/febs.13270
16. Schaible B, McClean S, Selfridge A, Broquet A, Asehnoune K, Taylor CT, et al. Hypoxia Modulates Infection of Epithelial Cells by Pseudomonas aeruginosa. PloS One (2013) 8(2):1–11. doi: 10.1371/journal.pone.0056491
17. Devraj G, Beerlage C, Brüne B, Kempf VAJ. Hypoxia and HIF-1 activation in bacterial infections. Microbes Infect (2017) 19(3):144–56. doi: 10.1016/j.micinf.2016.11.003
18. Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity (2014) 40(1):66–77. doi: 10.1016/j.immuni.2013.11.020
19. Ostridge K, Williams N, Kim V, Harden S, Bourne S, Coombs NA, et al. Distinct emphysema subtypes defined by quantitative CT analysis are associated with specific pulmonary matrix metalloproteinases. Respir Res (2016) 17(1):1–8. doi: 10.1186/s12931-016-0402-z
20. Donaldson GC, Seemungal TAR, Patel IS, Bhowmik A, Wilkinson TMA, Hurst JR, et al. Airway and systemic inflammation and decline in lung function in patients with COPD. 2005. Chest (2009) 136(5 Suppl):1995–2004. doi: 10.1378/chest.128.4.1995
21. Williams NP, Ostridge K, Devaster JM, Kim V, Coombs NA, Bourne S, et al. Impact of radiologically stratified exacerbations: Insights into pneumonia aetiology in COPD. Respir Res (2018) 19(1):1–12. doi: 10.1186/s12931-018-0842-8
22. Ostridge K, Williams N, Kim V, Bennett M, Harden S, Welch L, et al. Relationship between pulmonary matrix metalloproteinases and quantitative CT markers of small airways disease and emphysema in COPD. Thorax (2016) 71(2):126–32. doi: 10.1136/thoraxjnl-2015-207428
23. Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest (2002) 109(3):317–25. doi: 10.1172/JCI0213870
24. Mall MA, Harkema JR, Trojanek JB, Treis D, Livraghi A, Schubert S, et al. Development of chronic bronchitis and emphysema in β-epithelial Na+ channel-overexpressing mice. Am J Respir Crit Care Med (2008) 177(7):730–42. doi: 10.1164/rccm.200708-1233OC
25. Kim VL, Coombs NA, Staples KJ, Ostridge KK, Williams NP, Wootton SA, et al. Impact and associations of eosinophilic inflammation in COPD: analysis of the AERIS cohort. Eur Respir J (2017) 50(4):1700853. doi: 10.1183/13993003.00853-2017
26. Day K, Ostridge K, Conway J, Cellura D, Watson A, Spalluto CM, et al. Interrelationships Among Small Airways Dysfunction, Neutrophilic Inflammation, and Exacerbation Frequency in COPD. Chest (2020) S0012-3692(20)35299-5. doi: 10.1016/j.chest.2020.11.018
27. Polosukhin VV, Lawson WE, Milstone AP, Egunova SM, Kulipanov AG, Tchuvakin SG, et al. Association of progressive structural changes in the bronchial epithelium with subepithelial fibrous remodeling: A potential role for hypoxia. Virchows Arch (2007) 451(4):793–803. doi: 10.1007/s00428-007-0469-5
28. Lee SH, Lee SH, Kim CH, Yang KS, Lee EJ, Min KH, et al. Increased expression of vascular endothelial growth factor and hypoxia inducible factor-1α in lung tissue of patients with chronic bronchitis. Clin Biochem (2014) 47(7–8):552–9. doi: 10.1016/j.clinbiochem.2014.01.012
29. Fu X, Zhang F. Role of the hif-1 signaling pathway in chronic obstructive pulmonary disease. Exp Ther Med (2018) 16(6):4553–61. doi: 10.3892/etm.2018.6785
30. Tzouvelekis A, Harokopos V, Paparountas T, Oikonomou N, Chatziioannou A, Vilaras G, et al. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1α in disease pathogenesis. Am J Respir Crit Care Med (2007) 176(11):1108–19. doi: 10.1164/rccm.200705-683OC
31. Huerta-Yepez S, Baay-Guzman GJ, Bebenek IG, Hernandez-Pando R, Vega MI, Chi L, et al. Hypoxia Inducible Factor promotes murine allergic airway inflammation and is increased in asthma and rhinitis. Allergy Eur J Allergy Clin Immunol (2011) 66(7):909–18. doi: 10.1111/j.1398-9995.2011.02594.x
32. Kim V, Benditt JO, Wise RA, Sharafkhaneh A. Oxygen therapy in chronic obstructive pulmonary disease. Proc Am Thorac Soc (2008) 5(4):513–8. doi: 10.1513/pats.200708-124ET
33. Xie J, Covassin N, Fan Z, Singh P, Gao W, Li G, et al. Association Between Hypoxemia and Mortality in Patients With COVID-19.. Mayo Clin Proc (2020) 95: (6):1138–47. doi: 10.1016/j.mayocp.2020.04.006
34. Kashani KB. Hypoxia in COVID-19: Sign of Severity or Cause for Poor Outcomes. Mayo Clin Proc (2020) 95(6):1094–6. doi: 10.1016/j.mayocp.2020.04.021
35. Sivaloganathan AA, Nasim-Mohi M, Brown MM, Abdul N, Jackson A, Fletcher SV, et al. Noninvasive ventilation for COVID-19-associated acute hypoxaemic respiratory failure: experience from a single centre. Br J Anaesth (2020) 125(4):e368–71. doi: 10.1016/j.bja.2020.07.008
36. Kent BD, Mitchell PD, Mcnicholas WT. Hypoxemia in patients with COPD: Cause, effects, and disease progression. Int J COPD (2011) 6(1):199–208. doi: 10.2147/COPD.S10611
37. Martinez FJ, Foster G, Curtis JL, Criner G, Weinmann G, Fishman A, et al. Predictors of mortality in patients with emphysema and severe airflow obstruction. Am J Respir Crit Care Med (2006) 173(12):1326–34. doi: 10.1164/rccm.200510-1677OC
38. Wilkinson TMA, Donaldson GC, Hurst JR, Seemungal TAR, Wedzicha JA. Early therapy improves outcomes of exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2004) 169(12):1298–303. doi: 10.1164/rccm.200310-1443OC
39. Wedzicha JA, Wilkinson T. Impact of chronic obstructive pulmonary disease exacerbations on patients and payers. Proc Am Thorac Soc (2006) 3(3):218–21. doi: 10.1513/pats.200510-114SF
40. Donaldson GC, Seemungal TAR, Patel IS, Lloyd-Owen SJ, Wilkinson TMA, Wedzicha JA. Longitudinal changes in the nature, severity and frequency of COPD exacerbations. Eur Respir J (2003) 22(6):931–6. doi: 10.1183/09031936.03.00038303
41. Hurst JR, Perera WR, Wilkinson TMA, Donaldson GC, Wedzicha JA. Exacerbation of chronic obstructive pulmonary disease: Pan-airway and systemic inflammatory indices. Proc Am Thorac Soc (2006) 3(6):481–2. doi: 10.1513/pats.200603-059MS
42. Barberà JA, Roca J, Ferrer A, Félez MA, Díaz O, Roger N, et al. Mechanisms of worsening gas exchange during acute exacerbations of chronic obstructive pulmonary disease. Eur Respir J (1997) 10(6):1285–91. doi: 10.1183/09031936.97.10061285
43. Colgan SP, Furuta GT, Taylor CT. Hypoxia and Innate Immunity: Keeping Up with the HIFsters. Annu Rev Immunol (2020) 38(1):341–63. doi: 10.1146/annurev-immunol-100819-121537
44. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA (1995) 92(12):5510–4. doi: 10.1073/pnas.92.12.5510
45. Peyssonnaux C, Nizet V, Johnson RS. Role of the hypoxia inducible factors in iron metabolism. Cell Cycle (2008) 7(1):28–32. doi: 10.4161/cc.7.1.5145
46. Semenza GL. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology (2004) 19(4):176–82. doi: 10.1152/physiol.00001.2004
47. Semenza GL. Regulation of Oxygen Homeostasis by Hypoxia-Inducible Factor 1. Physiology (2009) Apr24(2):97–106. doi: 10.1152/physiol.00045.2008
48. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-κB activity. J Exp Med (2005) 201(1):105–15. doi: 10.1084/jem.20040624
49. Titto M, Ankit T, Saumya B, Gausal A, Sarada S. Curcumin prophylaxis refurbishes alveolar epithelial barrier integrity and alveolar fluid clearance under hypoxia. Respir Physiol Neurobiol (2020) 274:103336. doi: 10.1016/j.resp.2019.103336
50. Polke M, Seiler F, Lepper PM, Kamyschnikow A, Langer F, Monz D, et al. Hypoxia and the hypoxia-regulated transcription factor HIF-1α suppress the host defence of airway epithelial cells. Innate Immun (2017) 23(4):373–80. doi: 10.1177/1753425917698032
51. Sturrock A, Woller D, Freeman A, Sanders K, Paine R. Consequences of Hypoxia for the Pulmonary Alveolar Epithelial Cell Innate Immune Response. J Immunol (2018) 201(11):3411–20. doi: 10.4049/jimmunol.1701387
52. Kim SY, Choi YJ, Joung SM, Lee BH, Jung YS, Lee JY. Hypoxic stress up-regulates the expression of Toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology (2010) 129(4):516–24. doi: 10.1111/j.1365-2567.2009.03203.x
53. Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-κB. Biochem J (2006) 396(3):517–27. doi: 10.1042/BJ20051839
54. Bartels K, Grenz A, Eltzschig HK. Hypoxia and inflammation are two sides of the same coin. Proc Natl Acad Sci USA (2013) 110(46):18351–2. doi: 10.1073/pnas.1318345110
55. Steelant B. Epithelial dysfunction in chronic respiratory diseases, a shared endotype? Curr Opin Pulm Med (2020) 26(1):20–6. doi: 10.1097/MCP.0000000000000638
56. Pavia D. Lung mucociliary clearance. In: Clarke SW, Pavia D, ed. Aerosols and the Lung: clinical and experimental aspects. Boston: Butterworths (1984). p. 127–55.
57. Bustamante-Marin XM, Ostrowski LE. Cilia and mucociliary clearance. Cold Spring Harb Perspect Biol (2017) 9(4):1–18. doi: 10.1101/cshperspect.a028241
58. Ma J, Rubin BK, Voynow JA. Mucins, Mucus, and Goblet Cells. Chest [Internet] (2018) 154(1):169–76. doi: 10.1016/j.chest.2017.11.008
59. Satir P, Christensen ST. Overview of Structure and Function of Mammalian Cilia. Annu Rev Physiol (2007) 69(1):377–400. doi: 10.1146/annurev.physiol.69.040705.141236
60. Houtmeyers E, Gosselink R, Decramer M. Regulation of mucociliary clearance in health and disease. Eur Respir J (1999) 13(5):1177–88. doi: 10.1034/j.1399-3003.1999.13e39.x
61. Prescott E, Lange P, Vestbo J. Chronic mucus hypersecretion in COPD and death from pulmonary infection. Eur Respir J (1995) 8(8):1333–8. doi: 10.1183/09031936.95.08081333
62. Kim V, Han MLK, Vance GB, Make BJ, Newell JD, Hokanson JE, et al. The chronic bronchitic phenotype of COPD: An analysis of the COPDGene study. Chest (2011) 140(3):626–33. doi: 10.1378/chest.10-2948
63. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The Nature of Small-Airway Obstruction in Chronic Obstructive Pulmonary Disease. N Engl J Med (2004) 350(26):2645–53. doi: 10.1056/NEJMoa032158
64. Puchelle E, Zahm JM, Tournier JM, Coraux C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc Am Thorac Soc (2006) 3(8):726–33. doi: 10.1513/pats.200605-126SF
65. Mall MA. Unplugging mucus in cystic fibrosis and chronic obstructive pulmonary disease. Ann Am Thorac Soc (2016) 13:S177–85. doi: 10.1513/AnnalsATS.201509-641KV
66. Shaykhiev R. Emerging biology of persistent mucous cell hyperplasia in COPD. Thorax (2019) 74(1):4–6. doi: 10.1136/thoraxjnl-2018-212271
67. Zalacain R, Sobradillo V, Amilibia J, Barrón J, Achótegui V, Pijoan JI, et al. Predisposing factors to bacterial colonization in chronic obstructive pulmonary disease. Eur Respir J (1999) 13(2):343–8. doi: 10.1034/j.1399-3003.1999.13b21.x
68. Munkholm M, Mortensen J. Mucociliary clearance: Pathophysiological aspects. Clin Physiol Funct Imaging (2014) 34(3):171–7. doi: 10.1111/cpf.12085
69. Polosukhin VV, Cates JM, Lawson WE, Milstone AP, Matafonov AG, Massion PP, et al. Hypoxia-inducible factor-1 signalling promotes goblet cell hyperplasia in airway epithelium. J Pathol (2011) 224(2):203–11. doi: 10.1002/path.2863
70. Fritzsching B, Zhou-Suckow Z, Trojanek JB, Schubert SC, Schatterny J, Hirtz S, et al. Hypoxic epithelial necrosis triggers neutrophilic inflammation via IL-1 receptor signaling in cystic fibrosis lung disease. Am J Respir Crit Care Med (2015) 191(8):902–13. doi: 10.1164/rccm.201409-1610OC
71. Yu H, Li Q, Kolosov VP, Perelman JM, Zhou X. Regulation of cigarette smoke-mediated mucin expression by hypoxia-inducible factor-1α via epidermal growth factor receptor-mediated signaling pathways. J Appl Toxicol (2012) 32(4):282–92. doi: 10.1002/jat.1679
72. Zhou X, Tu J, Li Q, Kolosov VP, Perelman JM. Hypoxia induces mucin expression and secretion in human bronchial epithelial cells. Transl Res (2012) 160(6):419–27. doi: 10.1016/j.trsl.2012.08.001
73. Wu S, Li H, Yu L, Wang N, Li X, Chen W. IL-1β upregulates Muc5ac expression via NF-κB-induced HIF-1α in asthma. Immunol Lett (2017) 192:20–6. doi: 10.1016/j.imlet.2017.10.006
74. De Boer WI, Hau CM, Van Schadewijk A, Stolk J, Van Krieken JHJM, Hiemstra PS. Expression of epidermal growth factors and their receptors in the bronchial epithelium off subjects with chronic obstructive pulmonary disease. Am J Clin Pathol (2006) 125(2):184–92. doi: 10.1309/W1AXKGT7UA37X257
75. Vallath S, Hynds RE, Succony L, Janes SM, Giangreco A. Targeting EGFR signalling in chronic lung disease: Therapeutic challenges and opportunities. Eur Respir J (2014) 44(2):513–22. doi: 10.1183/09031936.00146413
76. Takeyama K, Dabbagh K, Lee HM, Agustí C, Lausier JA, Ueki IF, et al. Epidermal growth factor system regulates mucin production in airways. Proc Natl Acad Sci USA (1999) 96(6):3081–6. doi: 10.1073/pnas.96.6.3081
77. Takeyama K, Dabbagh K, Jeong Shim J, Dao-Pick T, Ueki IF, Nadel JA. Oxidative Stress Causes Mucin Synthesis Via Transactivation of Epidermal Growth Factor Receptor: Role of Neutrophils. J Immunol (2000) 164(3):1546–52. doi: 10.4049/jimmunol.164.3.1546
78. Aarbiou J, Verhoosel RM, Van Wetering S, De Boer WI, Van Krieken JH, Litvinov SV, et al. Neutrophil Defensins Enhance Lung Epithelial Wound Closure and Mucin Gene Expression in Vitro. Am J Respir Cell Mol Biol (2004) 30(2):193–201. doi: 10.1165/rcmb.2002-0267OC
79. Kato K, Chang EH, Chen Y, Lu W, Kim MM, Niihori M, et al. MUC1 contributes to goblet cell metaplasia and MUC5AC expression in response to cigarette smoke in vivo. Am J Physiol Cell Mol Physiol (2020) 0903702(2):ajplung.00049.2019. doi: 10.1152/ajplung.00049.2019
80. Lo Bello F, Ieni A, Hansbro PM, Ruggeri P, Di Stefano A, Nucera F, et al. Role of the mucins in pathogenesis of COPD: implications for therapy. Expert Rev Respir Med (2020) 14(5):465–83. doi: 10.1016/j.jallcom.2011.09.092
81. Kim SH, Pei QM, Jiang P, Liu J, Sun RF, Qian XJ, et al. Upregulation of MUC5AC by VEGF in human primary bronchial epithelial cells: implications for asthma. Respir Res (2019) 20(1):282. doi: 10.1186/s12931-019-1245-1
82. Lin VY, Kaza N, Birket SE, Kim H, Edwards LJ, LaFontaine J, et al. Excess mucus viscosity and airway dehydration impact COPD airway clearance. Eur Respir J (2020) 55(1):1900419. doi: 10.1183/13993003.00419-2019
83. Shuter J, Hatcher VB, Lowy FD. Staphylococcus aureus binding to human nasal mucin. Infect Immun (1996) 64(1):310–8. doi: 10.1128/IAI.64.1.310-318.1996
84. Moghaddam SJ, Clement CG, De La Garza MM, Zou X, Travis EL, Young HWJ, et al. Haemophilus influenzae lysate induces aspects of the chronic obstructive pulmonary disease phenotype. Am J Respir Cell Mol Biol (2008) 38(6):629–38. doi: 10.1165/rcmb.2007-0366OC
85. Hoffman CL, Lalsiamthara J, Aballay A. Host Mucin Is Exploited by Pseudomonas aeruginosa To Provide Monosaccharides Required for a Successful Infection. MBio (2020) 11(2):1–15. doi: 10.1128/mBio.00060-20
86. Cowley ES, Kopf SH, Lariviere A, Ziebis W, Newman DK. Pediatric cystic fibrosis sputum can be chemically dynamic, anoxic, and extremely reduced due to hydrogen sulfide formation. MBio (2015) 6(4):1–15. doi: 10.1128/mBio.00767-15
87. Depas WH, Starwalt-lee R, Sambeek LV, Kumar R, Gradinaru V, Newman K. Exposing the Three-Dimensional Biogeography and Metabolic States. mBio (2016) 7:(5):1–11. doi: 10.1128/mBio.00796-16
88. Millares L, Ferrari R, Gallego M, Garcia-Nuñez M, Pérez-Brocal V, Espasa M, et al. Bronchial microbiome of severe COPD patients colonised by Pseudomonas aeruginosa. Eur J Clin Microbiol Infect Dis (2014) 33(7):1101–11. doi: 10.1007/s10096-013-2044-0
89. Madan JC, Koestle DC, Stanton BA, Davidson L, Moulton LA, Housman ML, et al. Serial analysis of the gut and respiratory microbiome in cystic fibrosis in infancy: Interaction between intestinal and respiratory tracts and impact of nutritional exposures. MBio (2012) 3(4):1–10. doi: 10.1128/mBio.00251-12
90. Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. The Lung Microbiome in Moderate and Severe Chronic Obstructive Pulmonary Disease. PloS One (2012) 7(10):e47305. doi: 10.1371/journal.pone.0047305
91. Hurst JR, Wilkinson TMA, Perera WR, Donaldson GC, Wedzicha JA. Relationships among bacteria, upper airway, lower airway, and systemic inflammation in COPD. Chest (2005) 127(4):1219–26. doi: 10.1016/S0012-3692(15)34470-6
92. Wilkinson TMA, Schembri S, Brightling C, Bakerly ND, Lewis K, MacNee W, et al. Non-typeable Haemophilus influenzae protein vaccine in adults with COPD: A phase 2 clinical trial. Vaccine (2019) 37(41):6102–11. doi: 10.1016/j.vaccine.2019.07.100
93. Wilkinson TMA, Patel IS, Wilks M, Donaldson GC, Wedzicha JA. Airway bacterial load and FEV1 decline in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2003) 167(8):1090–5. doi: 10.1164/rccm.200210-1179OC
94. Almagro P, Salvad M, Garcia-Vidal C, Rodrguez-Carballeira M, Cuchi E, Torres J, et al. Pseudomonas aeruginosa and mortality after hospital admission for chronic obstructive pulmonary disease. Respiration (2012) 84(1):36–43. doi: 10.1159/000331224
95. Murphy TF. Pseudomonas aeruginosa in adults with chronic obstructive pulmonary disease. Curr Opin Pulm Med (2009) 15(2):138–42. doi: 10.1097/MCP.0b013e328321861a
96. Harris JK, De Groote MA, Sagel SD, Zemanick ET, Kapsner R, Penvari C, et al. Molecular identification of bacteria in bronchoalveolar lavage fluid from children with cystic fibrosis. Proc Natl Acad Sci USA (2007) 104(51):20529–33. doi: 10.1073/pnas.0709804104
97. Sherrard LJ, Bell SC, Tunney MM. The role of anaerobic bacteria in the cystic fibrosis airway. Curr Opin Pulm Med (2016) 22(6):637–43. doi: 10.1097/MCP.0000000000000299
98. Koval M. Junctional Interplay in Lung Epithelial Barrier Function. In: Lung Epithelial Biology in the Pathogenesis of Pulmonary Disease. Elsevier Inc: Academic Press (2017). p. 1–20. doi: 10.1016/B978-0-12-803809-3.00001-4
99. Karhausen J, Haase VH, Colgan SP. Inflammatory hypoxia: Role of hypoxia-inducible factor. Cell Cycle (2005) 4(2):256–8. doi: 10.4161/cc.4.2.1407
100. Tam A, Wadsworth S, Dorscheid D, Man S f P, Sin DD. The airway epithelium: More than just a structural barrier. Ther Adv Respir Dis (2011) 5(4):255–73. doi: 10.1177/1753465810396539
101. Schneeberger EE, Lynch RD. The tight junction: A multifunctional complex. Am J Physiol - Cell Physiol (2004) 286(6 55-6):C1213–28. doi: 10.1152/ajpcell.00558.2003
102. Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, Ishitani R, et al. Crystal structure of a claudin provides insight into the architecture of tight junctions. Sci (80- ) (2014) 344(6181):304–7. doi: 10.1126/science.1248571
103. LaFemina MJ, Sutherland KM, Bentley T, Gonzales LW, Allen L, Chapin CJ, et al. Claudin-18 deficiency results in alveolar barrier dysfunction and impaired alveologenesis in mice. Am J Respir Cell Mol Biol (2014) 51(4):550–8. doi: 10.1165/rcmb.2013-0456OC
104. Heijink IH, Noordhoek JA, Timens W, van Oosterhout AJM, Postma DS. Abnormalities in Airway Epithelial Junction Formation in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2014) 189(11):1439–42. doi: 10.1164/rccm.201311-1982LE
105. Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: Role of cigarette smoke exposure. Am J Respir Cell Mol Biol (2018) 58(2):157–69. doi: 10.1165/rcmb.2017-0200TR
106. Gindele JA, Kiechle T, Benediktus K, Birk G, Brendel M, Heinemann F, et al. Intermittent exposure to whole cigarette smoke alters the differentiation of primary small airway epithelial cells in the air-liquid interface culture. Sci Rep (2020) 10(1):6257. doi: 10.1038/s41598-020-63345-5
107. Schamberger AC, Staab-Weijnitz CA, Mise-Racek N, Eickelberg O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci Rep (2015) 5:1–9. doi: 10.1038/srep08163
108. Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod Pathol (2007) 20(1):108–19. doi: 10.1038/modpathol.3800725
109. Bagga B, Cehelsky JE, Vaishnaw A, Tomwilkinson T, Meyers R, Harrison LM, et al. Effect of Preexisting Serum and Mucosal Antibody on Experimental Respiratory Syncytial Virus (RSV) Challenge and Infection of Adults. J Infect Dis (2015) 212(10):1719–25. doi: 10.1093/infdis/jiv281
110. Kaufhold I, Osbahr S, Shima K, Marwitz S, Rohmann K, Drömann D, et al. Nontypeable Haemophilus influenzae (NTHi) directly interfere with the regulation of E-cadherin in lung epithelial cells. Microbes Infect (2017) 19(11):560–6. doi: 10.1016/j.micinf.2017.07.002
111. Guan R, Wang J, Li D, Li Z, Liu H, Ding M, et al. Hydrogen sulfide inhibits cigarette smoke-induced inflammation and injury in alveolar epithelial cells by suppressing PHD2/HIF-1α/MAPK signaling pathway. Int Immunopharmacol (2019) 81:105979. doi: 10.1016/j.intimp.2019.105979
112. Bouvry D, Planès C, Malbert-Colas L, Escabasse V, Clerici C. Hypoxia-induced cytoskeleton disruption in alveolar epithelial cells. Am J Respir Cell Mol Biol (2006) 35(5):519–27. doi: 10.1165/rcmb.2005-0478OC
113. Min HJ, Kim TH, Yoon JH, Kim CH. Hypoxia increases epithelial permeability in human nasal epithelia. Yonsei Med J (2015) 56(3):825–31. doi: 10.3349/ymj.2015.56.3.825
114. Song HA, Kim YS, Cho HJ, Kim SI, Kang MJ, Kim JH, et al. Hypoxia modulates epithelial permeability via regulation of vascular endothelial growth factor in airway epithelia. Am J Respir Cell Mol Biol (2017) 57(5):527–35. doi: 10.1165/rcmb.2016-0080OC
115. Zarember KA, Malech HL, Zarember KA, Malech HL. HIF-1 a: a master regulator of innate host defenses? J Clin Invest (2005) 115(7):1702–4. doi: 10.1172/JCI25740
116. Lee CG, Link H, Baluk P, Homer RJ, Chapoval S, Bhandari V, et al. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat Med (2004) 10(10):1095–103. doi: 10.1038/nm1105
117. Kähler CM, Prior C, Gunsilius E, McColley SA, Crawford SE. Serum vascular endothelial growth factor is elevated in cystic fibrosis and decreases with treatment of acute pulmonary exacerbation [2]. Am J Respir Crit Care Med (2001) 163(4):1030–1. doi: 10.1164/ajrccm.163.4.correspondence_b
118. Lee SY, Kwon S, Kim KH, Moon HS, Song JS, Park SH, et al. Expression of vascular endothelial growth factor and hypoxia-inducible factor in the airway of asthmatic patients. Ann Allergy Asthma Immunol (2006) 97(6):794–9. doi: 10.1016/S1081-1206(10)60971-4
119. Kranenburg AR, De Boer WI, Alagappan VKT, Sterk PJ, Sharma HS. Enhanced bronchial expression of vascular endothelial growth factor and receptors (Flk-1 and Flt-1) in patients with chronic obstructive pulmonary disease. Thorax (2005) 60(2):106–13. doi: 10.1136/thx.2004.023986
120. Pham I, Uchida T, Planes C, Ware LB, Kaner R, Matthay MA, et al. Hypoxia upregulates VEGF expression in alveolar epithelial cells in vitro and in vivo. Am J Physiol - Lung Cell Mol Physiol (2002) 283(5 27-5):1133–42. doi: 10.1152/ajplung.00464.2001
121. Tuder RM, Flook BE, Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia: Modulation of gene expression by nitric oxide. J Clin Invest (1995) 95(4):1798–807. doi: 10.1172/JCI117858
122. Boussat S, Eddahibi S, Coste A, Fataccioli V, Gouge M, Housset B, et al. Expression and regulation of vascular endothelial growth factor in human pulmonary epithelial cells. Am J Physiol - Lung Cell Mol Physiol (2000) 279(2 23-2):1–4. doi: 10.1152/ajplung.2000.279.2.L371
123. Duell BL, Su YC, Riesbeck K. Host–pathogen interactions of nontypeable Haemophilus influenzae: from commensal to pathogen. FEBS Lett (2016) 590(21):3840–53. doi: 10.1002/1873-3468.12351
124. Heath CJ, Cendra MDM, Watson A, Auger JP, Pandey A, Tighe P, et al. Co-transcriptomes of initial interactions in vitro between streptococcus pneumoniae and human pleural mesothelial cells. PloS One (2015) 10(11):1–26. doi: 10.1371/journal.pone.0142773
125. Shukla SD, Sohal SS, O’Toole RF, Eapen MS, Walters EH. Platelet activating factor receptor: Gateway for bacterial chronic airway infection in chronic obstructive pulmonary disease and potential therapeutic target. Expert Rev Respir Med (2015) 9(4):473–85. doi: 10.1586/17476348.2015.1070673
126. Miyashita L, Suri R, Dearing E, Mudway I, Dove RE, Neill DR, et al. E-cigarette vapour enhances pneumococcal adherence to airway epithelial cells. Eur Respir J (2018) 51(2):1701592. doi: 10.1183/13993003.01592-2017
127. Shukla SD, Fairbairn RL, Gell DA, Latham RD, Sohal SS, Walters EH, et al. An antagonist of the platelet-activating factor receptor inhibits adherence of both nontypeable Haemophilus influenzae and Streptococcus pneumoniae to cultured human bronchial epithelial cells exposed to cigarette smoke. Int J COPD (2016) 11(1):1647–55. doi: 10.2147/COPD.S108698
128. Barbier M, Oliver A, Rao J, Hanna SL, Goldberg JB, Albertí S. Novel Phosphorylcholine-Containing Protein of Pseudomonas aeruginosa Chronic Infection Isolates Interacts with Airway Epithelial Cells. J Infect Dis (2008) 197(3):465–73. doi: 10.1086/525048
129. Liu J, Chen X, Zhou J, Ye L, Yang D. Song Y. Particulate matter exposure promotes Pseudomonas aeruginosa invasion into airway epithelia by upregulating PAFR via the ROS − mediated PI3K pathway. Hum Cell (2020) 33(4):963–73. doi: 10.1007/s13577-020-00378-y
130. Ishii S, Nagase T, Shimizu T. Platelet-activating factor receptor. Prostaglandins Other Lipid Mediat (2002) 68–69:599–609. doi: 10.1016/S0090-6980(02)00058-8
131. Swords WE, Ketterer MR, Shao J, Campbell CA, Weiser JN, Apicella MA. Binding of the non-typeable Haemophilus influenzae lipooligosaccharide to the PAF receptor initiates host cell signalling. Cell Microbiol (2001) 3(8):525–36. doi: 10.1046/j.1462-5822.2001.00132.x
132. Hergott CB, Roche AM, Naidu NA, Mesaros C, Blair IA, Weiser JN. Bacterial exploitation of phosphorylcholine mimicry suppresses inflammation to promote airway infection. J Clin Invest (2015) 125(10):3878–90. doi: 10.1172/JCI81888
133. Shukla SD, Sohal SS, Mahmood MQ, Reid D, Muller HK, Walters EH. Airway epithelial platelet-activating factor receptor expression is markedly upregulated in chronic obstructive pulmonary disease. Int J COPD (2014) 9:853–61. doi: 10.2147/COPD.S67044
134. Suri R, Mallia P, Martin JE, Footitt J, Zhu J, Trujillo-Torralbo MB, et al. Bronchial platelet-activating factor receptor in chronic obstructive pulmonary disease. Respir Med (2014) 108(6):898–904. doi: 10.1016/j.rmed.2014.03.003
135. Shirasaki H, Nishikawa M, Adcock IM, Mak JC, Sakamoto T, Shimizu T, et al. Expression of platelet-activating factor receptor mRNA in human and guinea pig lung. Am J Respir Cell Mol Biol (1994) 10(5):533–7. doi: 10.1165/ajrcmb.10.5.8179916
136. Grigg J, Miyashita L, Suri R. Pneumococcal infection of respiratory cells exposed to welding fumes; Role of oxidative stress and HIF-1 alpha. PloS One (2017) 12(3):1–16. doi: 10.1371/journal.pone.0173569
137. Rajendra KC, Hyland IK, Smith JA, Shukla SD, Hansbro PM, Zosky GR, et al. Cow Dung Biomass Smoke Exposure Increases Adherence of Respiratory Pathogen Nontypeable Haemophilus influenzae to Human Bronchial Epithelial Cells. Expo Heal (2020) 12:883–95.. doi: 10.1007/s12403-020-00351-y
138. Gou X, Zhang Q, More S, Bamunuarachchi G, Liang Y, Haider Khan F, et al. Repeated Exposure to Streptococcus pneumoniae Exacerbates Chronic Obstructive Pulmonary Disease. Am J Pathol (2019) 189(9):1711–20. doi: 10.1016/j.ajpath.2019.05.012
139. Greiller CL, Suri R, Jolliffe DA, Kebadze T, Hirsman AG, Griffiths CJ, et al. Vitamin D attenuates rhinovirus-induced expression of intercellular adhesion molecule-1 (ICAM-1) and platelet-activating factor receptor (PAFR) in respiratory epithelial cells. J Steroid Biochem Mol Biol (2019) 187:152–9. doi: 10.1016/j.jsbmb.2018.11.013
140. Keely S, Glover LE, Weissmueller T, MacManus CF, Fillon S, Fennimore B, et al. Hypoxia-inducible Factor-dependent Regulation of Platelet-activating Factor Receptor as a Route for Gram-Positive Bacterial Translocation across Epithelia. Nusrat A, editor. Mol Biol Cell (2010) 21(4):538–46. doi: 10.1091/mbc.e09-07-0573
141. Ishizuka S, Yamaya M, Suzuki T, Takahashi H, Ida S, Sasaki T, et al. Effects of Rhinovirus Infection on the Adherence of Streptococcus pneumoniae to Cultured Human Airway Epithelial Cells. J Infect Dis (2003) 188(12):1928–39. doi: 10.1086/379833
142. Domingue JC, Drewes JL, Merlo CA, Housseau F, Sears CL. Host responses to mucosal biofilms in the lung and gut. Mucosal Immunol (2020) 13(3):413–22. doi: 10.1038/s41385-020-0270-1
143. Swords WE. Nontypeable Haemophilus influenzae biofilms: role in chronic airway infections. Front Cell Infect Microbiol (2012) 2:97. doi: 10.3389/fcimb.2012.00097
144. Jamal M, Ahmad W, Andleeb S, Jalil F, Imran M, Nawaz MA, et al. Bacterial biofilm and associated infections. J Chin Med Assoc (2018) 81(1):7–11. doi: 10.1016/j.jcma.2017.07.012
145. Ahearn CP, Gallo MC, Murphy TF. Insights on persistent airway infection by non-typeable Haemophilus influenzae in chronic obstructive pulmonary disease. Pathog Dis (2017) 75:42. doi: 10.1093/femspd/ftx042
146. Murphy TF, Kirkham C, Sethi S, Lesse AJ. Expression of a peroxiredoxin-glutaredoxin by Haemophilus influenzae in biofilms and during human respiratory tract infection. FEMS Immunol Med Microbiol (2005) 44(1):81–9. doi: 10.1016/j.femsim.2004.12.008
147. Frick AG, Joseph TD, Pang L, Rabe AM, St. Geme JW, Look DC. Haemophilus influenzae Stimulates ICAM-1 Expression on Respiratory Epithelial Cells. J Immunol (2000) 164(8):4185–96. doi: 10.4049/jimmunol.164.8.4185
148. Bjarnsholt T, Jensen PØ, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, et al. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr Pulmonol (2009) 44(6):547–58. doi: 10.1002/ppul.21011
149. Williams NP, Coombs NA, Johnson MJ, Josephs LK, Rigge LA, Staples KJ, et al. Seasonality, risk factors and burden of community-acquired pneumonia in COPD patients: A population database study using linked health care records. Int J COPD (2017) 12:313–22. doi: 10.2147/COPD.S121389
150. Hassett DJ. Anaerobic production of alginate by Pseudomonas aeruginosa: Alginate restricts diffusion of oxygen. J Bacteriol (1996) 178(24):7322–5. doi: 10.1128/JB.178.24.7322-7325.1996
151. Hill D, Rose B, Pajkos A, Robinson M, Bye P, Bell S, et al. Antibiotic susceptibilities of Pseudomonas aeruginosa isolates derived from patients with cystic fibrosis under aerobic, anaerobic, and biofilm conditions. J Clin Microbiol (2005) 43(10):5085–90. doi: 10.1128/JCM.43.10.5085-5090.2005
152. Ghotaslou R, Behnaz S. Effects of oxygen on in-vitro biofilm formation and antimicrobial resistance of Pseudomonas aeruginosae. Biol (Basel) (2013) 19(3):96–9.
153. Schaible B, Taylor CT, Schaffer K. Hypoxia increases antibiotic resistance in Pseudomonas aeruginosa through altering the composition of multidrug efflux pumps. Antimicrob Agents Chemother (2012) 56(4):2114–8. doi: 10.1128/AAC.05574-11
154. Piddock LJV. Multidrug-resistance efflux pumps - Not just for resistance. Nat Rev Microbiol (2006) 4(8):629–36. doi: 10.1038/nrmicro1464
155. Nguyen AT, Oglesby-Sherrouse AG. Interactions between Pseudomonas aeruginosa and Staphylococcus aureus during co-cultivations and polymicrobial infections. Appl Microbiol Biotechnol (2016) 100(14):6141–8. doi: 10.1007/s00253-016-7596-3
156. Hunter RC, Asfour F, Dingemans J, Osuna BL, Samad T, Malfroot A, et al. Ferrous iron is a significant component of bioavailable iron in cystic fibrosis airways. MBio (2013) 4(4):1–8. doi: 10.1128/mBio.00557-13
157. Nicholas B, Staples KJ, Moese S, Meldrum E, Ward J, Dennison P, et al. A Novel Lung Explant Model for the Ex Vivo Study of Efficacy and Mechanisms of Anti-Influenza Drugs. J Immunol (2015) 194(12):6144–54. doi: 10.4049/jimmunol.1402283
158. Worthen GS, Haslett C, Rees AJ, Gumbay RS, Henson JE, Henson PM. Neutrophil-mediated pulmonary vascular injury. Synergistic effect of trace amounts of lipopolysaccharide and neutrophil stimuli on vascular permeability and neutrophil sequestration in the lung. Am Rev Respir Dis (1987) 136(1):19–28. doi: 10.1164/ajrccm/136.1.19
159. Gibbs DF, Shanley TP, Warner RL, Murphy HS, Varani J, Johnson KJ. Role of matrix metalloproteinases in models of macrophage-dependent acute lung injury: Evidence for alveolar macrophage as source of proteinases. Am J Respir Cell Mol Biol (1999) 20(6):1145–54. doi: 10.1165/ajrcmb.20.6.3482
160. Doumas S, Kolokotronis A, Stefanopoulos P. Anti-inflammatory and antimicrobial roles of secretory leukocyte protease inhibitor. Infect Immun (2005) 73(3):1271–4. doi: 10.1128/IAI.73.3.1271-1274.2005
161. Hiemstra PS, Maassen RJ, Stolk J, Heinzel-Wieland R, Steffens GJ, Dijkman JH. Antibacterial activity of antileukoprotease. Infect Immun (1996) 64(11):4520–4. doi: 10.1128/IAI.64.11.4520-4524.1996
162. Wiedow O, Harder J, Bartels J, Streit V, Christophers E. Antileukoprotease in human skin: An antibiotic peptide constitutively produced by keratinocytes. Biochem Biophys Res Commun (1998) 248(3):904–9. doi: 10.1006/bbrc.1998.9069
163. Nakamura A, Mori Y, Hagiwara K, Suzuki T, Sakakibara T, Kikuchi T, et al. Increased susceptibility to LPS-induced endotoxin shock in secretory leukoprotease inhibitor (SLPI)-deficient mice. J Exp Med (2003) 197(5):669–74. doi: 10.1084/jem.20021824
164. Birrer P, Mcelvaney NG, Rüdeberg A, Wirz Sommer C, Liechti-Gallati S, Kraemer R, et al. Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am J Respir Crit Care Med (1994) 150(1):207–13. doi: 10.1164/ajrccm.150.1.7912987
165. Fischer BM, Pavlisko E, Voynow JA. Pathogenic triad in COPD: Oxidative stress, protease-antiprotease imbalance, and inflammation. Int J COPD (2011) 6(1):413–21. doi: 10.2147/COPD.S10770
166. Parameswaran GI, Wrona CT, Murphy TF, Sethi S. Moraxella catarrhalis acquisition, airway inflammation and protease-antiprotease balance in chronic obstructive pulmonary disease. BMC Infect Dis (2009) 9:1–10. doi: 10.1186/1471-2334-9-178
167. Parameswaran GI, Sethi S, Murphy TF. Effects of bacterial infection on airway antimicrobial peptides and proteins in COPD. Chest (2011) 140(3):611–7. doi: 10.1378/chest.10-2760
168. Persson LJP, Aanerud M, Hardie JA, Nilsen RM, Bakke PS, Eagan TM, et al. Antimicrobial peptide levels are linked to airway inflammation, bacterial colonisation and exacerbations in chronic obstructive pulmonary disease. Eur Respir J (2017) 49(3):1601328. doi: 10.1183/13993003.01328-2016
169. Sibila O, Perea L, Cantó E, Shoemark A, Cassidy D, Smith AH, et al. Antimicrobial peptides, disease severity and exacerbations in bronchiectasis. Thorax (2019) 74(9):835–42. doi: 10.1136/thoraxjnl-2018-212895
170. Påhlman LI, Jögi A, Gram M, Mori M, Egesten A. Hypoxia down-regulates expression of secretory leukocyte protease inhibitor in bronchial epithelial cells via TGF-β1. BMC Pulm Med (2015) 15(1):19. doi: 10.1186/s12890-015-0016-0
171. Copple BL. Hypoxia stimulates hepatocyte epithelial to mesenchymal transition by hypoxia-inducible factor and transforming growth factor-β-dependent mechanisms. Liver Int (2010) 30(5):669–82. doi: 10.1111/j.1478-3231.2010.02205.x
172. Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K, Matsui H, Hara K, et al. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am J Physiol - Lung Cell Mol Physiol (2011) 300(5):740–52. doi: 10.1152/ajplung.00146.2010
173. De Boer WI, Van Schadewijk A, Sont JK, Sharma HS, Stolk J, Hiemstra PS, et al. Transforming growth factor β1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. Am J Respir Crit Care Med (1998) 158(6):1951–7. doi: 10.1164/ajrccm.158.6.9803053
174. Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, Satoh M, et al. Increased expression of transforming growth factor-β1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med (2001) 163(6):1476–83. doi: 10.1164/ajrccm.163.6.9908135
175. Harris WT, Kelly DR, Zhou Y, Wang D, Macewen M, Hagood JS, et al. Myofibroblast Differentiation and Enhanced Tgf-B Signaling in Cystic Fibrosis Lung Disease. PloS One (2013) 8(8):2–9. doi: 10.1371/annotation/d0132de3-56ca-4258-9119-bdab0ceb2cff
176. Qian F, He M, Duan W, Mao L, Li Q, Yu Z, et al. Cross regulation between hypoxia-inducible transcription factor-1α (HIF-1α) and transforming growth factor (TGF)-ß1 mediates nickel oxide nanoparticles (NiONPs)-induced pulmonary fibrosis. Am J Transl Res (2015) 7(11):2364–78.
177. Jaumann F, Elssner A, Mazur G, Dobmann S, Vogelmeier C. Transforming growth factor-β1 is a potent inhibitor of secretory leukoprotease inhibitor expression in a brochial epithelial cell line. Eur Respir J (2000) 15(6):1052–7. doi: 10.1034/j.1399-3003.2000.01513.x
178. Luo BL, Niu RC, Feng JT, Hu CP, Xie XY, Ma LJ. Downregulation of Secretory Leukocyte Proteinase Inhibitor in Chronic Obstructive Lung Disease: The Role Of TGF-β/Smads Signaling Pathways. Arch Med Res (2008) 39(4):388–96. doi: 10.1016/j.arcmed.2008.02.002
179. Baker EH, Clark N, Brennan AL, Fisher DA, Gyi KM, Hodson ME, et al. Hyperglycemia and cystic fibrosis alter respiratory fluid glucose concentrations estimated by breath condensate analysis. J Appl Physiol (2007) 102(5):1969–75. doi: 10.1152/japplphysiol.01425.2006
180. Baker EH, Baines DL. Airway Glucose Homeostasis: A New Target in the Prevention and Treatment of Pulmonary Infection. Chest (2018) 153(2):507–14. doi: 10.1016/j.chest.2017.05.031
181. Mallia P, Webber J, Gill SK, Trujillo-Torralbo MB, Calderazzo MA, Finney L, et al. Role of airway glucose in bacterial infections in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol (2018) 142(3):815–23.e6. doi: 10.1016/j.jaci.2017.10.017
182. Qin C, Liu F, Yen TC, Lan X. 18F-FDG PET/CT findings of COVID-19: a series of four highly suspected cases. Eur J Nucl Med Mol Imaging (2020) 47(5):1281–6. doi: 10.1007/s00259-020-04734-w
183. Gill SK, Hui K, Farne H, Garnett JP, Baines DL, Moore LSP, et al. Increased airway glucose increases airway bacterial load in hyperglycaemia. Sci Rep (2016) 6(May):1–10. doi: 10.1038/srep27636
184. Garnett JP, Baker EH, Naik S, Lindsay JA, Knight GM, Gill S, et al. Metformin reduces airway glucose permeability and hyperglycaemia-induced Staphylococcus aureus load independently of effects on blood glucose. Thorax (2013) 68(9):835–45. doi: 10.1136/thoraxjnl-2012-203178
185. Garnett JP, Gray MA, Tarran R, Brodlie M, Ward C, Baker EH, et al. Elevated Paracellular Glucose Flux across Cystic Fibrosis Airway Epithelial Monolayers Is an Important Factor for Pseudomonas aeruginosa Growth. PloS One (2013) 8(10):2036–45. doi: 10.1371/journal.pone.0076283
186. Åstrand A, Wingren C, Benjamin A, Tregoning JS, Garnett JP, Groves H, et al. Dapagliflozin-lowered blood glucose reduces respiratory Pseudomonas aeruginosa infection in diabetic mice. Br J Pharmacol (2017) 174(9):836–47. doi: 10.1111/bph.13741
187. Philips BJ, Redman J, Brennan A, Wood D, Holliman R, Baines D, et al. Glucose in bronchial aspirates increases the risk of respiratory MRSA in intubated patients. Thorax (2005) 60(9):761–4. doi: 10.1136/thx.2004.035766
188. Brennan AL, Gyi KM, Wood DM, Johnson J, Holliman R, Baines DL, et al. Airway glucose concentrations and effect on growth of respiratory pathogens in cystic fibrosis. J Cyst Fibros (2007) 6(2):101–9. doi: 10.1016/j.jcf.2006.03.009
189. Pezzulo AA, Gutiérrez J, Duschner KS, McConnell KS, Taft PJ, Ernst SE, et al. Glucose depletion in the airway surface liquid is essential for sterility of the airways. PloS One (2011) 6(1):1–8. doi: 10.1371/journal.pone.0016166
190. López-López N, Euba B, Hill J, Dhouib R, Caballero L, Leiva J, et al. Haemophilus influenzae Glucose Catabolism Leading to Production of the Immunometabolite Acetate Has a Key Contribution to the Host Airway-Pathogen Interplay. ACS Infect Dis (2020) 6(3):406–21. doi: 10.1021/acsinfecdis.9b00359
191. Garnett JP, Nguyen TT, Moffatt JD, Pelham ER, Kalsi KK, Baker EH, et al. Proinflammatory Mediators Disrupt Glucose Homeostasis in Airway Surface Liquid. J Immunol (2012) 189(1):373–80. doi: 10.4049/jimmunol.1200718
192. Patkee WRA, Carr G, Baker EH, Baines DL, Garnett JP. Metformin prevents the effects of Pseudomonas aeruginosa on airway epithelial tight junctions and restricts hyperglycaemia-induced bacterial growth. J Cell Mol Med (2016) 20(4):758–64. doi: 10.1111/jcmm.12784
193. Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol (2011) 76:347–53. doi: 10.1101/sqb.2011.76.010678
194. Simon LM, Robin ED, Raffin T, Theodore J, Douglas WHJ. Bioenergetic Pattern of Isolated Type II Pneumocytes in Air and during Hypoxia. J Clin Invest (1978) 61(5):1232–9. doi: 10.1172/JCI109039
195. Ouiddir A, Planès C, Fernandes I, Vanhesse A, Clerici C. Hypoxia upregulates activity and expression of the glucose transporter GLUT1 in alveolar epithelial cells. Am J Respir Cell Mol Biol (1999) 21(6):710–8. doi: 10.1165/ajrcmb.21.6.3751
196. Escoubet B, Planès C, Clerici C. Hypoxia increases glyceraldehyde-3-phosphate dehydrogenase transcription in rat alveolar epithelial cells. Biochem Biophys Res Commun (1999) 266(1):156–61. doi: 10.1006/bbrc.1999.1798
197. Heerlein K, Schulze A, Hotz L, Bärtsch P, Mairbäurl H. Hypoxia decreases cellular ATP demand and inhibits mitochondrial respiration of A549 cells. Am J Respir Cell Mol Biol (2005) 32(1):44–51. doi: 10.1165/rcmb.2004-0202OC
198. Ren L, Zhang W, Han P, Zhang J, Zhu Y, Meng X, et al. Influenza A virus (H1N1) triggers a hypoxic response by stabilizing hypoxia-inducible factor-1α via inhibition of proteasome. Virology (2019) 530:51–8. doi: 10.1016/j.virol.2019.02.010
199. Gualdoni GA, Mayer KA, Kapsch AM, Kreuzberg K, Puck A, Kienzl P, et al. Rhinovirus induces an anabolic reprogramming in host cell metabolism essential for viral replication. Proc Natl Acad Sci USA (2018) 115(30):E7158–65. doi: 10.1073/pnas.1800525115
200. Codo AC, Davanzo GG, Monteiro L de B, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab (2020) 32:1–10. doi: 10.2139/ssrn.3606770
201. Wilkinson TMA, Donaldson GC, Johnston SL, Openshaw PJM, Wedzicha JA. Respiratory syncytial virus, airway inflammation, and FEV1 decline in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2006) 173(8):871–6. doi: 10.1164/rccm.200509-1489OC
202. Sanei F, Wilkinson T. Influenza vaccination for patients with chronic obstructive pulmonary disease: Understanding immunogenicity, efficacy and effectiveness. Ther Adv Respir Dis (2016) 10(4):349–67. doi: 10.1177/1753465816646050
203. Hammond FR, Lewis A, Elks PM. If it’s not one thing, HIF’s another: immunoregulation by hypoxia inducible factors in disease. FEBS J (2020) 13(3):febs.15476. doi: 10.1111/febs.15476
204. Eurlings IMJ, Reynaert NL, Van Den Beucken T, Gosker HR, De Theije CC, Verhamme FM, et al. Cigarette smoke extract induces a phenotypic shift in epithelial cells; involvement of HIF1α in mesenchymal transition. PloS One (2014) 9(10):1–11. doi: 10.1371/journal.pone.0107757
Keywords: epithelial cells, hypoxia, hypoxia-inducible factor (HIF)-1, host-pathogen interactions, innate immunity, respiratory disease
Citation: Page LK, Staples KJ, Spalluto CM, Watson A and Wilkinson TMA (2021) Influence of Hypoxia on the Epithelial-Pathogen Interactions in the Lung: Implications for Respiratory Disease. Front. Immunol. 12:653969. doi: 10.3389/fimmu.2021.653969
Received: 15 January 2021; Accepted: 09 March 2021;
Published: 24 March 2021.
Edited by:
Mats Bemark, University of Gothenburg, SwedenReviewed by:
Adam Byrne, Imperial College London, United KingdomChristoph Beisswenger, Saarland University, Germany
Copyright © 2021 Page, Staples, Spalluto, Watson and Wilkinson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lee K. Page, TC5LLlBhZ2VAc290b24uYWMudWs=