 Xiang-Zhi Fang1,2
Xiang-Zhi Fang1,2 You Shang
You Shang- 1Department of Critical Care Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Institute of Anesthesiology and Critical Care Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
COVID-19 is an acute, complex disorder that was caused by a new β-coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Based on current reports, it was surprising that the characteristics of many patients with COVID-19, who fulfil the Berlin criteria for acute respiratory distress syndrome (ARDS), are not always like those of patients with typical ARDS and can change over time. While the mechanisms of COVID-19–related respiratory dysfunction in COVID-19 have not yet been fully elucidated, pulmonary microvascular thrombosis is speculated to be involved. Considering that thrombosis is highly related to other inflammatory lung diseases, immunothrombosis, a two-way process that links coagulation and inflammation, seems to be involved in the pathophysiology of COVID-19, including respiratory dysfunction. Thus, the current manuscript will describe the proinflammatory milieu in COVID-19, summarize current evidence of thrombosis in COVID-19, and discuss possible interactions between these two.
Introduction
The novel coronavirus disease 2019 (COVID-19) pandemic, which first broke out in Wuhan, China, has now grown into a global pandemic. COVID-19 is an acute, complex disorder that is caused by a new β-coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (1). Similar to other human coronaviruses, SARS-CoV-2 mainly affects the respiratory system. While most people with COVID-19 present with only mild illness (2), approximately 5% to 16% require intensive care (3–5), among whom 50% to 70% rapidly progress to severe fatal respiratory dysfunction requiring mechanical ventilation especially acute respiratory distress syndrome (ARDS) (5, 6). Elderly individuals and persons with comorbidities have higher mortality rates (7). Surprisingly, the characteristics of many patients with COVID-19, who fulfil the Berlin criteria for ARDS, are not always like those of patients with typical ARDS and can change over time (8). Of note, COVID-19 patients develop profound hypoxemia early in their disease course, characterized by better compliance of the respiratory system, a low ventilation-to-perfusion (VA/Q) ratio and low lung recruitability, which is termed L-type ARDS (9). The initial acute respiratory dysfunction of COVID-19 dominantly results from pulmonary microvascular thrombosis, which is supported by pathological reports (10).
Studies have revealed that COVID-19 often causes episodes of thrombosis, and the incidence is higher in severe cases (11, 12). Recently published studies have considered thrombosis an important factor contributing to COVID-19 pathogenesis, especially respiratory dysfunction (11, 13). Additionally, thrombosis appears to be associated with other inflammatory lung diseases, such as acute respiratory distress syndrome and influenza-associated pneumonia (14, 15). Interestingly, inflammation predisposes patients to thrombosis, and conversely, thrombosis is associated with inflammation, a process sometimes known as immunothrombosis (16).
In this review, we attempt to describe the proinflammatory milieu in COVID-19, summarize current evidence of thrombosis in COVID-19, and identify possible interactions between inflammation and thrombosis. Given the relative lack of information in relation to COVID-19 thrombosis, we also summarize information from other similar RNA viral infections and inflammatory disorders.
The Proinflammatory Milieu In Covid-19
The pathogenesis of COVID-19 is associated with a hyperinflammatory response (17, 18). The first step in the pathogenesis of COVID-19 is SARS-CoV-2 entry into target cells through its S proteins outside the viral lipid layer (19, 20). After entering host cells, mainly respiratory epithelial cells, SARS-CoV-2–expressing pathogen-associated molecular pattern molecules (PAMPs) activate a large number of innate and adaptive immune cells (21, 22), leading to the production of inflammatory cytokines and the type I interferons (IFN) IFN-α and IFN-β, which establish a proinflammatory milieu.
The Cytokine Storm in COVID-19
In 1993, Ferrara (23) first proposed the concept of a cytokine storm in acute graft-versus-host disease. Since then, this concept has been further extended to other diseases, such as rheumatological disease and sepsis. Macrophage activation syndrome (MAS) refers to the cytokine storm induced by autoimmune disorders (24). Additionally, the cytokine storm that occurs after chimeric antigen receptor (CAR) T cell therapy is called cytokine release syndrome (CRS) (25). Elevated serum levels of interleukin (IL)-6 IL-7, IL-2, granulocyte colony-stimulating factor (G-CSF), macrophage inflammatory protein 1α (MIP1α), and tumor necrosis factor-α (TNF-α) have been reported in COVID-19 patients (2, 26, 27), which was widely recognized as cytokine storm.
CD8+ and CD4+ T cells are the most dominant cells that participate in the immune responses to SARS-CoV-2 infection (28). Recently, a clinical experiment suggested that SARS-CoV-2–specific CD8+ and CD4+ T cells were present in ∼70% and 100% of COVID-19 convalescent patients, respectively (29). Other immunological cells involved in SARS-CoV-2 infection include B cells, macrophages, T helper (Th) cells, neutrophils, and natural killer/cytotoxic T lymphocytes (CTLs) (21, 30–32). Excessively secreted cytokines attract neutrophils, monocytes, and macrophages to the site of the insult, where they not only clear viral particles but also may cause organ failure (33). Importantly, these cytokines also activate immune cells, further increasing the production of cytokines.
While the specific details and mechanisms of the cytokine storm in COVID-19 remain unclear, researchers suppose that this cytokine storm is connected with a dysfunctional immune response to remove the virus (34, 35). The immune response to SARS-CoV-2 includes two different phases. The first phase is the incubation phase, in which recruited cells and released cytokines fight SARS-CoV-2 infection. In most individuals, the immune response clears SARS-CoV-2, the immune response recedes, and patients recover. However, when there is a failure to fight SARS-CoV-2, the immune response enters the second phase. During this phase, an overactive immune response occurs, compensating for the target clearance failure, whereby clinical manifestations of a cytokine storm are present. Additionally, it is noteworthy that a recent study showed that SARS-CoV-2 coding protein open reading frame 8 (ORF8) activates the IL-17 signaling pathway and promotes the cytokine storm during COVID-19 (36).
Neutrophil Extracellular Traps in COVID-19
Neutrophils are the most abundant white blood cell type circulating in the human bloodstream (37). When a pathogen enters the body, neutrophils, as the key components of the innate immune cell population, are recruited to infection or inflammation sites, where they activate other types of immune cells and eliminate pathogens (38–40). In 2004, Brinkmann et al. (41) first observed that, in response to bacterial endotoxins and inflammatory cytokines or drugs, circulating neutrophils form web-like structures that are commonly referred to as neutrophil extracellular traps (NETs). These NETs are composed of intracellular components released by activated neutrophils, including neutrophil elastase (NE), myeloperoxidase (MPO), histones, defensins, calprotectin matrix metalloproteinase-9, and cathepsin G (42). As part of the innate immune system, the main function of NETs is to trap and possibly even kill microorganisms. However, a large number of studies have confirmed that the activities of NETs is a double-edged sword, because, in addition to their microbicidal activity, NETs have been implicated in various tissue damage and involved in the development of sepsis and influenza pneumonia (43, 44).
An autopsy specimen from a patient who has succumbed to COVID-19 showed neutrophil infiltration in lung tissues (45). Elevated number of peripheral blood neutrophils is considered an early indicator of SARS-CoV-2 infection, associating with severe respiratory dysfunction and worse clinical outcomes (46, 47). Recent study has demonstrated that NET markers including cell-free DNA, myeloperoxidase (MPO)-DNA, and citrullinated histone H3 (Cit-H3) were significantly increased in serum samples from severe COVID-19 patients (48). Other studies found that increased plasma NETs are positively correlated with COVID-19 severity (49, 50). Importantly, SARS-CoV-2 can directly lead to the formation of NETs in healthy neutrophils through ACE2–serine protease axis and PAD-4 signaling (51).
Thehemostatic Abnormalities And Pulmonary Microvascular Thrombosis In Covid-19
Ebolavirus, dengue fever, and Lassa virus, similar to SARS-CoV-2 virus, are enveloped, single-stranded RNA viruses that are thought to promote thrombosis (52, 53). To date, the human coronavirus most closely related to SARS-CoV-2 is SARS-CoV-1 (54). SARS-CoV-1 infection has been associated with hematological abnormalities, including thrombocytosis (49%), elevated D-dimer (45.0%), thrombocytopenia (55%), and prolonged activated partial thromboplastin time (aPTT) (63%) (55, 56). Chong et al. also reported that 20.5% of patients infected with SARS-CoV-1 had deep vein thrombosis, and 11.4% of these patients showed clinical evidence of pulmonary embolism (57). Furthermore, in a SARS-CoV-1–infected patient, edema and fibrin thrombi were identified in the pulmonary vasculature (58). Additionally, postmortem examinations from patients infected with SARS-CoV-1 revealed that thrombi were present in pulmonary, bronchial, and small lung veins, which implied a prothrombotic effect in the pulmonary vasculature of SARS-CoV-1–infected patients (59–61).
In addition to systemic hyperinflammation, SARS-CoV-2 infection is associated with coagulation abnormalities, which cause hypoxemic respiratory failure. COVID-19 patients appear to be more susceptible to thrombotic complications. This blood coagulation seems to be not only faster, but also more severe than that observed in life-threatening influenza or sepsis. Purple rashes, swollen legs, and clogged catheters are common clinical manifestations of COVID-19 (62, 63). A study from Wuhan China of critical COVID-19 pneumonia documented acro-ischemia including dry gangrene finger/toe cyanosis, and skin bulla, which accounted for 21% of critically ill patients hospitalized at the same time (62, 64). Recently, Helms and colleagues reported that more pulmonary embolisms were diagnosed in COVID-19 ARDS patients than in patients with non-COVID-19 ARDS (65). Despite widespread use of thromboprophylaxis, incidences of pulmonary embolism have still been reported to be as high as 21% in COVID-19 (66), which was two-fold higher than that in critically ill influenza patients. Importantly, pathological examinations revealed marked lung microvascular congestion in patients who died in early stages of COVID-19 (67).
According to different reports (12, 68, 69), the most common hematological disorder in COVID-19 patients is consistently represented by elevations in D-dimer and prothrombin time, and a relatively modest decrease in platelet count. Of particular note, increased D-dimer concentration was associated with a higher COVID-19 mortality rate. Additionally, the laboratory parameters in COVID-19 patients are distinct from those in patients with sepsis-induced coagulopathy and disseminated intravascular coagulation (DIC), who present with more serious thrombocytopenia and slight D-dimer abnormalities than COVID-19 patients (70). Likewise, COVID-19 patients are predominated by hypercoagulable state, with only 2% to 3% of patients presenting with serious bleeding (65). Collectively, this available evidence implies that hemostatic abnormalities and pulmonary intravascular coagulopathy occur in patients with COVID-19, even in the early course of the disease.
Links Between Inflammation And Thrombosis In Covid-19
It has been shown that the immune response actively participates in the formation of thrombi within blood vessels, particularly in microvessels (71–73). The process, defined as immunothrombosis, accurately describes the intricate network between the coagulation system and the innate immune system (74). Immunothrombosis can locally confine an infection by facilitating the recognition, containment, and destruction of pathogens. Considering that the similar mechanisms exist in COVID-19 and bacterial sepsis, immunothrombosis has been reconsidered in SARS-CoV-2 infection. There are complex interactions between inflammation and thrombosis, involving endothelial cells (ECs), coagulation (activated TF, platelets, and neutrophils), anticoagulation (impaired AT, APC and TFPI systems), and decreased fibrinolysis (Figure 1).
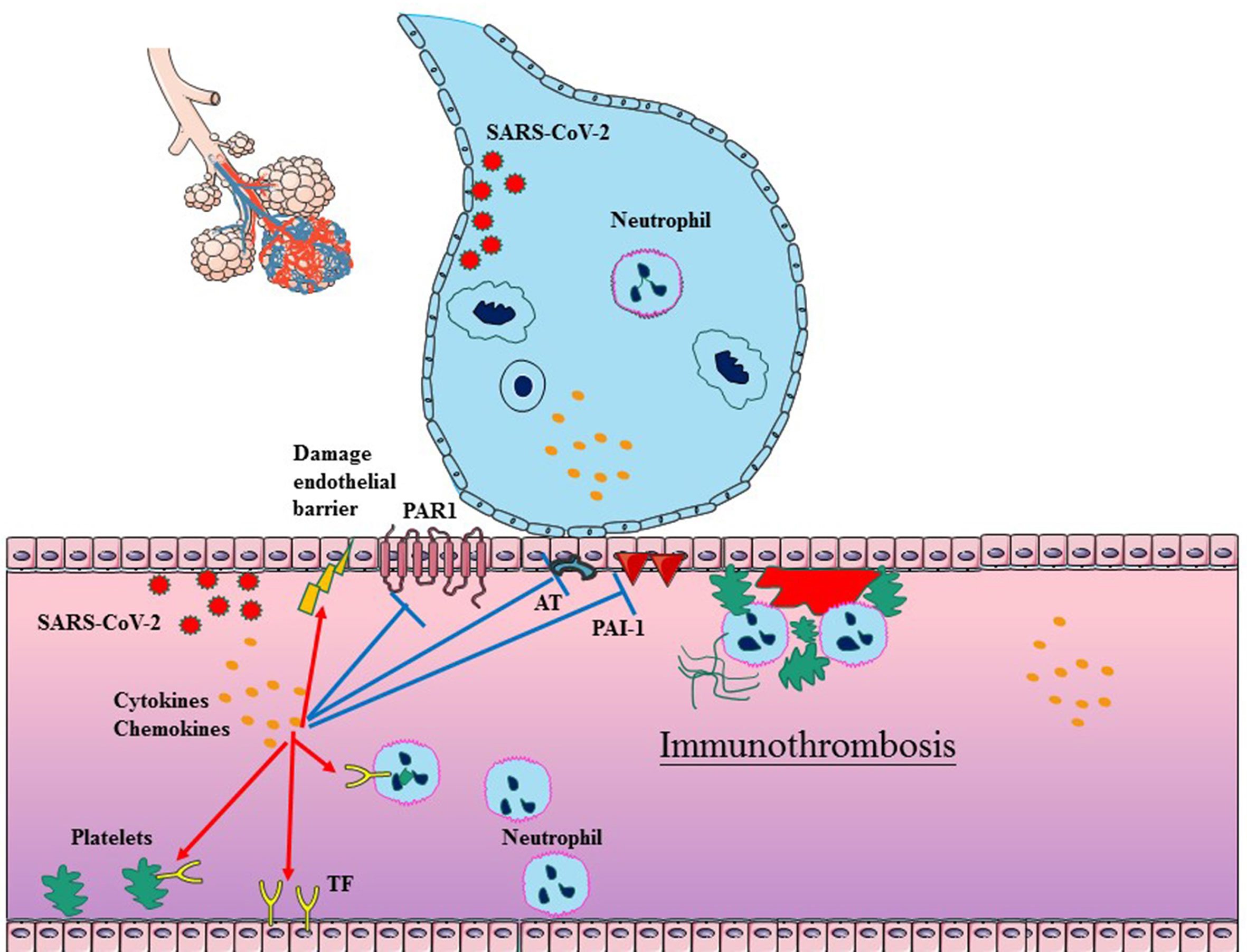
Figure 1 Schematic diagram of immunothrombosis in COVID-19. SARS-CoV-2 infection typically begins in epithelial cells of the respiratory tract, which can further promote the cytokine storm and infiltration of neutrophil. The cytokine storm can result in endothelial dysfunction, tissue factor (TF) overexpression, and the platelets activation, which initiates the TF-dependent coagulation process. The release of NETs promotes platelet–neutrophil aggregation and neutrophil activation in the bloodstream. Furthermore, the cytokine storm also suppresses antithrombin, APC and PAI-1, which impair anticoagulation and decrease fibrinolysis. In addition, SARS-CoV-2 can also directly impair ECs. The above factors collectively facilitate the generation of thrombus in COVID-19.
Endotheliitis and Endothelial Dysfunction
Endothelial cells (ECs) maintain the balance between the coagulation and anticoagulation systems of blood by expressing several mediators that prevent platelet activation and suppress coagulation and thrombus formation (75). Under normal conditions, ECs provide an environment with a slight tendency to promote anticoagulation in blood vessels (76). Therefore, dysfunctional ECs may contribute to the pathogenesis of thrombosis by altering the expression of pro- and antithrombotic factors.
Both autopsy findings and clinical observations have described the coexistence of vascular damage and thrombotic complications in a wide range of organs (77, 78), which provides indirect evidence to support that endotheliitis has an important role in immunothrombosis processes. In addition, COVID-19 patients have a higher circulating endothelial cell (CEC) count, suggesting pronounced endothelial injury in COVID-19 (79). A single-center study found that endotheliopathy might be an important factor in the pathophysiology of COVID-19–associated coagulopathy (80).
Transmission electron microscopy of the endothelium in COVID-19 showed ultrastructural damage, as well as the presence of intracellular and extracellular SARS-CoV-2 (81, 82). Perivascular inflammatory cell infiltration was observed in COVID-19 patients (82, 83). Thus, endotheliitis occurs in several organs, especially in the lung, as a consequence of direct SARS-CoV-2 infection and an overactive host immune response. Among the various inflammatory cytokines, IL-6 is one of the most major cytokines involved in cytokine storms in COVID-19 patients. L-6 has been confirmed to activate ECs, thus resulting in endothelial dysfunction, further activation of the coagulation cascades (84). Further larger studies are required to provide more direct evidence for the link between cytokine storm and thrombus formation in COVID-19.
Tissue Factor
As the key element in the initiation of the extrinsic pathway of the blood coagulation cascade, tissue factor (TF) is constitutively expressed in perivascular cells, including adventitial fibroblasts, pericytes, and epithelial cells of the lung, brain, skin and kidney (85–87). Interestingly, vascular cells in contact with the blood, such as smooth muscle cells (SMCs), endothelial cells (ECs), alveolar epithelial cells, macrophages and neutrophils, do not express considerable amounts of TF in a quiescent state but can be transiently induced to do so under inflammatory conditions (88, 89). TF is encrypted under normal physiological conditions (90). In response to injury and inflammation, TF in vascular endothelial cells is released into the bloodstream and becomes exposed to circulating FVII/FVIIa. After the binding and activation of FVII, the TF-activated VII (FVIIa) complex consequently activates FX to FXa, which then interacts with its cofactor, factor FVa, phospholipids, and calcium to constitute a prothrombinase complex (91, 92). The prothrombinase complex subsequently converts prothrombin into proteolytically active thrombin, leading to fibrin deposition and microthrombi formation. Although microthrombi act as a barrier against the invasion of pathogens in the initial defense procedure, they recruit and activate innate immune cells that generate the inflammatory response and further enhance TF expression (93). Xue et al. (94) reported that the increased plasma levels of TF are associated with the outcome in sepsis-induced ARDS. Additionally, blocking TF activity dramatically reduces physiological and histological lung injury and improves survival (95).
Three recent reviews speculate that TF may be involved in COVID-19–related thrombosis (96–98). Furthermore, Hotz and colleagues found increased expression of TF in monocytes in critically ill COVID-19 patients (99). Higher levels of TF+ platelets, TF+ granulocytes, and TF+ procoagulant microvesicles (MVs) are reported in COVID-19 patients (100). The active form of TF can disseminate around the body through the extracellular vesicles (EVs), which participate in sepsis-induced coagulopathy and thrombus formation (101, 102). Recent study has shown that EV-TF activity increased in COVID-19 patients, associating with elevated thrombotic risk (103). However, another study found that TF levels were not increased in BALF from COVID-19 patients (104). This contradictory result may be due to differences in sample and detection methods. These studies indicate that TF appears to be a critical determinant of the inflammatory thrombosis process in COVID-19. However, direct evidence that the proinflammatory milieu of COVID-19 induces TF overexpression is still lacking.
Neutrophils and NETs
Neutrophils contribute to immunothrombosis depending on the formation of NETs. NETs were originally observed in patients’ thrombi (105). Researchers have gradually realized that NETs provide a scaffold to recruit red blood cells, platelets, and white blood cells and to bind plasma proteins (106). Ischemic stroke thrombi were found to be positive for DNA, MPO, and citrullinated histone H4 upon triple containment, and NETs were abundant in different types of stroke thrombi (107). Perhaps NETs facilitate the development of venous thrombosis because NETs can directly result in fiber remodeling. Moreover, intravascular thrombosis induced by NETs led to extensive microvascular obliteration and multiple organ failure in sepsis (108).
NETs have been shown to colocalize with thrombi in COVID-19 (49). In addition, a recent study demonstrated a strong correlation between markedly elevated NETs and D-dimer levels, a degradation product of fibrin (109). Interestingly, serum samples from patients with progressive/severe COVID-19 cause abundant formation of NETs in healthy donors, which suggests that patients with COVID-19 have circulating factors that induce NETs (110). Dismantling or inhibiting NETs in COVID-19 could ameliorate thrombotic tissue damage associated with ARDS and higher mortality rates (45). These data suggest a clinical link between NET formation and immunothrombosis, which may thereby contribute to the COVID-19 prothrombotic state.
Platelets
The adhesion and activation of platelets are essential events of pathological thrombosis and inflammation during ARDS pathogenesis, as they participate in not only hemostasis but also the infectious response. During endothelial damage, platelets adhere and aggregate to the site of vascular injury via subendothelial collagen and von Willebrand factor (VWF) bound to glycoprotein receptors (GPs), such as GPVI, GPIa/IIa, and GPIb/IX/V, on the platelet surface (111). This initial platelet adhesion to the subendothelial matrix at the site of vascular injury promotes a series of downstream signaling responses, which switch platelets from an inactivated to an activated state. The change in platelet shape and platelet dense granule secretion is a characteristic feature of the activated state of platelets (112, 113). Equally notable is that dense granules are composed of multiple cytokines and other bioactive molecules, which are critical mediators in the complex microenvironment of blood coagulation but also take part in the inflammatory reaction process (114). In addition, platelets and their released products facilitate neutrophil aggregation and activation in damaged endothelial cells (115–117)
In patients with severe COVID-19, the activation of platelets during SARS-CoV-2 infection has been observed (99, 100, 118). A temporal trend of dropping platelet counts in COVID-19 patients could suggest a worsening thrombotic state, while an increase in platelet count was associated with improved survival and reduced thrombotic risk in COVID-19 (119). Furthermore, plasma VWF protein levels are consistently elevated in patients with severe COVID-19 and correlate with adverse outcomes, suggesting enhanced adhesive interactions between circulating platelets and the damaged vessel wall (120). Interestingly, platelets incubated in plasma from COVID-19 induced platelet activation. These data indicate that the inflammatory milieu is at least in part accountable for increased platelet activation in severe COVID-19.
Antithrombin
Antithrombin (AT) is a plasma glycoprotein produced mainly by the liver, and AT has anticoagulant characteristics. The anticoagulant effect of AT is achieved through the inhibition of thrombin, plasmin, FIXa, Xa, XIa, and XII (121). Free plasma AT neutralizes coagulation enzymes in a slow, progressive manner since binding is very slow. When AT bind to heparin sulfate molecules on the vascular endothelial surface, a conformational change occurs to results in a ≥1,000-fold enhancement of AT activity. This suggests that heparin may be ineffective in patients with low AT levels. It has already been shown that AT induces endothelial cell release of prostacyclin, a molecule that prohibits platelet aggregation and activation and neutrephil infiltration (122). Furthermore, AT can interact directly with leukocytes and lymphocytes, inhibiting their interaction with endothelial cells and alleviating the severity of capillary leakage and subsequent organ damage (123).
Hyperinflammation can markedly decrease AT levels and glycosaminoglycan synthesis, which is associated with coagulation (124). Additionally, several recent studies have shown that plasma antithrombin values are significantly decreased in COVID-19, which is strongly associated with mortality in COVID-19 (125–127). At supplementation, using fresh frozen plasma (FFP) in COVID-19 patients may improve thrombosis prophylaxis and thus have an impact on their survival (126). Together, the results of these studies imply that AT may be the link between inflammation and thrombosis in COVID-19. However, further studies are needed to clarify the anti-inflammatory properties of AT in COVID-19.
Activated Protein C
Protein C is a plasma serine protease zymogen with 419 amino acids. Upon thrombin is bound to thrombomodulin on the vascular endothelial surface, protein C is converted to activated protein C (APC), which exerts potent anticoagulant activity through its irreversible proteolytic inactivation of activated FV and neutralizes PAI-1 (128). The cleavage of activated FVIII is also strengthened by APC (129). Changes in levels of APC or protein C (PC) are associated with modifications in the risk of thrombosis. In addition to its anticoagulant activity, APC reduces the inflammatory response, inhibits apoptosis and protects the endothelial cell barrier. APC not only suppresses proinflammatory and proapoptotic signals but also enhances anti-inflammatory and antiapoptotic pathways (130, 131).
Since APC plays an essential role in coagulation and the immune response, it is possible that APC is involved in immunothrombosis in COVID-19 (132, 133). This concept is further supported by a trial study to derive and validate a predictive score for disease worsening in patients with COVID-19 (134). COVID-19 patients admitted to the intensive care unit (ICU) had lower levels of antithrombin activity and protein C activity as well as higher D-dimer and fibrinogen levels than COVID-19 patients admitted to a conventional ward. It is noteworthy that multivariate analysis identified decreased activity of protein C as significant predictors of worsening disease.
Decreased Fibrinolysis
The fibrinolytic system, controlled by coagulation itself, removes fibrin from the vascular system, preventing increased amounts of clots in the microcirculation. Plasminogen is converted to plasmin by the action of urokinase (u-PA) and tissue plasminogen activator (t-PA), which is the central link in the fibrinolytic system. Therefore, PAI-1, a rapidly acting inhibitor of t-PA and u-PA is the main inhibitor of fibrinolysis (135).
When the level of PAI-1 in the circulation is elevated, fibrinolysis is impeded by inhibitory function of t-PA and u-PA, leading to failed removal of thrombi from the vascular wall. Binding PAI-1 to t-PA or u-PA forms an inactive complex, thus negatively mediating fibrinolysis in the vascular wall. It has been shown that fibrinolysis is impaired in sepsis, primarily due to an exaggerated release of PAI-1 as a result of endothelial dysfunction, given the coexistence of an inflammatory response and endothelial dysfunction (136). In addition, increased activated platelets may also release large amounts of PAI-1, as platelets are the major circulating pool of PAI-1 that can contribute to a high local concentration of PAI-1 at the site of a growing fibrin clot (137).
Impaired fibrinolysis has been suggested in COVID-19 patients, which could further heighten their thrombotic risk. This has been evidenced by markedly reduced clot lysis at 30 min via thromboelastography (TEG) in COVID-19 patients (138). Elevated levels of t-PA and PAI-1 were observed in patients during COVID-19, further suggesting impaired fibrinolytic ability (139, 140). IL-6 is the most clinically suitable biomarker for COVID-19. The latest research (141) found that the blockade of IL-6 signaling using tocilizumab treatment significantly decreases serum PAI-1 levels in patients with severe COVID-19.
Summary And Conclusions
Emerging evidence has suggested that COVID-19 patients suffer from pulmonary microvascular thrombosis, which may explain the rapid deterioration and pulmonary collapse that is observed in patients who suddenly progress to ARDS. Immunothrombosis may be a key link between COVID-19 and thrombosis. The process involves a highly coordinated and mutual regulating process of joint participation of multiple factors, such as inflammatory cells, TF, endothelial dysfunction, and platelets. However, the pathophysiology of COVID-19 and thrombotic complications is complex, and the roles of many of the important factors, such as tissue factor pathway inhibitor and protease-activated receptors, are not fully understood. Accordingly, much attention should be directed to a deeper understanding of the pathogenesis of COVID-19.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
X-ZF and YS conceptualized and wrote the original draft of the manuscript. J-QX and Y-XW drew diagrams. Z-KP and Y-JH reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kannan S, Shaik Syed Ali P, Sheeza A, Hemalatha K. Covid-19 (Novel Coronavirus 2019) - Recent Trends. Eur Rev Med Pharmacol Sci (2020) 24:2006–11. doi: 10.26355/eurrev_202002_20378
2. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
3. Grasselli G, Pesenti A, Cecconi M. Critical Care Utilization for the COVID-19 Outbreak in Lombardy, Italy: Early Experience and Forecast During an Emergency Response. JAMA (2020) 323:1545–6. doi: 10.1001/jama.2020.4031
4. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med (2020) 383:1708–20. doi: 10.1056/NEJMoa2002032
5. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical Course and Outcomes of Critically Ill Patients With SARS-CoV-2 Pneumonia in Wuhan, China: A Single-Centered, Retrospective, Observational Study. Lancet Respir Med (2020) 8:475–81. doi: 10.1016/S2213-2600(20)30079-5
6. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
7. Burki TK. Coronavirus in China. Lancet Respir Med (2020) 8(3):238. doi: 10.1016/S2213-2600(20)30056-4
8. Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, Chiumello D. Covid-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med (2020) 201:1299–300. doi: 10.1164/rccm202003-0817LE
9. Mauri T, Spinelli E, Scotti E, Colussi G, Basile MC, Crotti S, et al. Potential for Lung Recruitment and Ventilation-Perfusion Mismatch in Patients With the Acute Respiratory Distress Syndrome From Coronavirus Disease 2019. Crit Care Med (2020) 48:1129–34. doi: 10.1097/CCM.0000000000004386
10. Lax SF, Skok K, Zechner P. Pulmonary Arterial Thrombosis in COVID-19 With Fatal Outcome: Results From a Prospective, Single-Center, Clinicopathologic Case Series. Ann Intern Med (2020) 173:350–61. doi: 10.7326/M20-2566
11. Ciceri F, Beretta L, Scandroglio AM, Colombo S, Landoni G, Ruggeri A, et al. Microvascular COVID-19 Lung Vessels Obstructive Thromboinflammatory Syndrome (MicroCLOTS): An Atypical Acute Respiratory Distres. Crit Care Resusc (2020) 22:95–7. doi: 10.3316/informit.196503333515429
12. Tang N, Li D, Wang X, Sun Z. Abnormal Coagulation Parameters Are Associated With Poor Prognosis in Patients With Novel Coronavirus Pneumonia. J Thromb Haemost (2020) 18(4):844–7. doi: 10.1111/jth.14768
13. Parker S, Mahomed O. Hypoxia and Thrombosis in COVID-19: New Considerations for Air Passengers. J Travel Med (2020) 27:taaa122. doi: 10.1093/jtm/taaa122
14. Frantzeskaki F, Armaganidis A, Orfanos SE. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks Between Inflammation and Coagulation. Respiration (2017) 93:212–25. doi: 10.1159/000453002
15. Ishiguro T, Matsuo K, Fujii S, Takayanagi N. Acute Thrombotic Vascular Events Complicating Influenza-Associated Pneumonia. Respir Med Case Rep (2019) 28:100884. doi: 10.1016/j.rmcr.2019.100884
16. Grover SP, Mackman N. Neutrophils, NETs, and Immunothrombosis. Blood (2018) 132:1360–1. doi: 10.1182/blood-2018-08-868067
17. Payen D, Cravat M, Maadadi H, Didelot C, Prosic L, Dupuis C, et al. A Longitudinal Study of Immune Cells in Severe Covid-19 Patients. Front Immunol (2020) 11:580250. doi: 10.3389/fimmu.2020.580250
18. Petrey AC, Qeadan F, Middleton EA, Pinchuk IV, Campbell RA, Beswick EJ. Cytokine Release Syndrome in COVID-19: Innate Immune, Vascular, and Platelet Pathogenic Factors Differ in Severity of Disease and Sex. J Leukoc Biol (2021) 109:55–66. doi: 10.1002/JLB.3COVA0820-410RRR
19. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A Pneumonia Outbreak Associated With a New Coronavirus of Probable Bat Origin. Nature (2020) 579(7798):270–3. doi: 10.1038/s41586-020-2012-7
20. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-Cov-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
21. Carsetti R, Zaffina S, Piano Mortari E, Terreri S, Corrente F, Capponi C, et al. Different Innate and Adaptive Immune Responses to SARS-CoV-2 Infection of Asymptomatic, Mild, and Severe Cases. Front Immunol (2020) 11:610300. doi: 10.3389/fimmu.2020.610300.
22. Bolouri H, Speake C, Skibinski D, Long SA, Hocking AM, Campbell DJ, et al. The COVID-19 Immune Landscape Is Dynamically and Reversibly Correlated With Disease Severity. J Clin Invest (2021) 131(3):e143648. doi: 10.1172/JCI143648
23. Ferrara JL. Cytokine Dysregulation as a Mechanism of Graft Versus Host Disease. Curr Opin Immunol (1993) 5(5):794–9. doi: 10.1016/0952-7915(93)90139-j
24. Carter SJ, Tattersall RS, Ramanan AV. Macrophage Activation Syndrome in Adults: Recent Advances in Pathophysiology, Diagnosis and Treatment. Rheumatol (Oxford) (2019) 58(1):5–17. doi: 10.1093/rheumatology/key006
25. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. N Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
26. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical Predictors of Mortality Due to COVID-19 Based on an Analysis of Data of 150 Patients From Wuhan, China. Intensive Care Med (2020) 46:846–8. doi: 10.1007/s00134-020-05991-x
27. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. Covid-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet (2020) 395(10229):1033–4. doi: 10.1016/S0140-6736(20)30628-0
28. Sekine T, Perez-Potti A, Rivera-Ballesteros O, Strålin K, Gorin JB, Olsson A, et al. Robust T Cell Immunity in Convalescent Individuals With Asymptomatic or Mild Covid-19. Cell (2020) 183(1):158–68.e14. doi: 10.1016/j.cell.2020.08.017
29. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans With COVID-19 Disease and Unexposed Individuals. Cell (2020) 181(7):1489–501.e15. doi: 10.1016/j.cell.2020.05.015.
30. Sosa-Hernández VA, Torres-Ruíz J, Cervantes-Díaz R, Romero-Ramírez S, Páez-Franco JC, Meza-Sánchez DE, et al. B Cell Subsets as Severity-Associated Signatures in COVID-19 Patients. Front Immunol (2020) 11:611004. doi: 10.3389/fimmu.2020.611004
31. Ueland T, Heggelund L, Lind A, Holten AR, Tonby K, Michelsen AE, et al. Elevated Plasma sTIM-3 Levels in Patients With Severe COVID-19. J Allergy Clin Immunol (2021) 147(1):92–8. doi: 10.1016/j.jaci.2020.09.007
32. Ronit A, Berg RMG, Bay JT, Haugaard AK, Ahlström MG, Burgdorf KS, et al. Compartmental Immunophenotyping in COVID-19 Ards: A Case Series. J Allergy Clin Immunol (2021) 147(1):81–91. doi: 10.1016/j.jaci.2020.09.009
33. Peddapalli A, Gehani M, Kalle AM, Peddapalli SR, Peter AE, Sharad S. Demystifying Excess Immune Response in COVID-19 to Reposition an Orphan Drug for Down-Regulation of NF-κb: A Systematic Review. Viruses (2021) 13(3):378. doi: 10.3390/v13030378
34. McGonagle D, Sharif K, O’Regan A, Bridgewood C. The Role of Cytokines Including Interleukin-6 in COVID-19 Induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun Rev (2020) 19(6):102537. doi: 10.1016/j.autrev.2020.102537.
35. Ye Q, Wang B, Mao J. The Pathogenesis and Treatment of the `Cytokine Storm’ in COVID-19. J infect J Infect (2020) 80(6):607–13. doi: 10.1016/j.jinf.2020.03.037.
36. Lin X, Fu B, Yin S, Li Z, Liu H, Zhang H, et al. ORF8 Contributes to Cytokine Storm During SARS-CoV-2 Infection by Activating IL-17 Pathway. iScience (2021) 24(4):102293. doi: 10.1016/j.isci.2021.102293
37. Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JA, et al. In Vivo Labeling With 2H2O Reveals a Human Neutrophil Lifespan of 5.4 Days. Blood (2010) 116(4):625–7. doi: 10.1182/blood-2010-01-259028.
38. Ingelsson B, Söderberg D, Strid T, Söderberg A, Bergh AC, Loitto V, et al. Lymphocytes Eject Interferogenic Mitochondrial DNA Webs in Response to CpG and Non-CpG Oligodeoxynucleotides of Class C. Proc Natl Acad Sci USA (2018) 115(3):E478–87. doi: 10.1073/pnas.1711950115.
39. Nakazawa D, Shida H, Kusunoki Y, Miyoshi A, Nishio S, Tomaru U, et al. The Responses of Macrophages in Interaction With Neutrophils That Undergo Netosis. J Autoimmun (2016) 67:19–28. doi: 10.1016/j.jaut.2015.08.018
40. Fang C, Wei X, Wei Y. Mitochondrial DNA in the Regulation of Innate Immune Responses. Rotein Cell (2016) 7(1):11–6. doi: 10.1007/s13238-015-0222-9
41. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
42. Jeong JH, Choi SJ, Ahn SM, Oh JS, Kim YG, Lee CK, et al. Neutrophil Extracellular Trap Clearance by Synovial Macrophages in Gout. Arthritis Res Ther (2021) 23(1):88. doi: 10.1186/s13075-021-02472-4
43. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular Neutrophil Extracellular Traps Capture Bacteria From the Bloodstream During Sepsis. Cell Host Microbe (2012) 12(3):324–33. doi: 10.1016/j.chom.2012.06.011
44. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Am J Pathol (2011) 179(1):199–210. doi: 10.1016/j.ajpath.2011.03.013
45. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J Exp Med (2020) 217(6):e20200652. doi: 10.1084/jem.20200652
46. Zhang B, Zhou X, Zhu C, Song Y, Feng F, Qiu Y, et al. Immune Phenotyping Based on Neutrophil-to-Lymphocyte Ratio and IgG Predicts Disease Severity and Outcome for Patients With COVID-19. Front Mol Biosci (2020) 7:157. doi: 10.3389/fmolb.2020.00157
47. Song CY, Xu J, He JQ, Lu YQ. Covid-19 Early Warning Score: A Multi-Parameter Screening Tool to Identify Highly Suspected Patients. medRxiv (2020). doi: 10.1101/2020.03.05.20031906
48. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight (2020) 5(11):e138999. doi: 10.1172/jci.insight.138999
49. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood (2020) 136(10):1169–79. doi: 10.1182/blood.2020007008
50. Godement M, Zhu J, Cerf C, Vieillard-Baron A, Maillon A, Zuber B, et al. Neutrophil Extracellular Traps in SARS-CoV2 Related Pneumonia in ICU Patients: The Netcov2 Study. Front Med (Lausanne) (2021) 8:615984. doi: 10.3389/fmed.2021.615984.
51. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. Sars-CoV-2–Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J Exp Med (2020) 217(12):e20201129. doi: 10.1084/jem.20201129
52. Paessler S, Walker DH. Pathogenesis of the Viral Hemorrhagic Fevers. Annu Rev Pathol (2013) 8:411–40. doi: 10.1146/annurev-pathol-020712-164041
53. Falasca L, Agrati C, Petrosillo N, Di Caro A, Capobianchi MR, Ippolito G, et al. Molecular Mechanisms of Ebola Virus Pathogenesis: Focus on Cell Death. Cell Death Differ (2015) 22(8):1250–9. doi: 10.1038/cdd.2015.67
54. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-nCoV and Naming it SARS-Cov-2. Nat Microbiol (2020) 5(4):536–44. doi: 10.1038/s41564-020-0695-z
55. Wong RS, Wu A, To KF, Lee N, Lam CW, Wong CK, et al. Haematological Manifestations in Patients With Severe Acute Respiratory Syndrome: Retrospective Analysis. BMJ (2003) 326:1358–62. doi: 10.1136/bmj.326.7403.1358
56. Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, et al. A Major Outbreak of Severe Acute Respiratory Syndrome in Hong Kong. N Engl J Med (2003) 348:1986–9. doi: 10.1056/NEJMoa030685
57. Chong PY, Chui P, Ling AE, Franks TJ, Tai DY, Leo YS, et al. Analysis of Deaths During the Severe Acute Respiratory Syndrome (SARS) Epidemic in Singapore: Challenges in Determining a SARS Diagnosis. Arch Pathol Lab Med (2004) 128:195–204. doi: 10.1043/1543-2165(2004)128<195:AODDTS>2.0.CO;2
58. Nicholls JM, Poon LL, Lee KC, Ng WF, Lai ST, Leung CY, et al. Lung Pathology of Fatal Severe Acute Respiratory Syndrome. Lancet (2003) 361:1773–8. doi: 10.1016/s0140-6736(03)13413-7
59. Ng KHL, Wu AKL, Cheng VCC, Chan Y, Yuan CY, Luk SH, et al. Pulmonary Artery Thrombosis in a Patient With Severe Acute Respiratory Syndrome. Postgrad Med J (2005) 81:e3. doi: 10.1136/pgmj.2004.030049
60. Lang Z, Zhang L, Zhang S, Meng X, Li J, Song C, et al. Pathological Study on Severe Acute Respiratory Syndrome. Chin Med J (Engl) (2003) 116:976–80.
61. Ding Y, Wang H, Shen H, Li Z, Geng J, Han H, et al. The Clinical Pathology of Severe Acute Respiratory Syndrome (SARS): A Report From China. J Pathol (2003) 200:282–9. doi: 10.1002/path.1440
62. Nirenberg MS, Herrera MaríaDMR. Foot Manifestations in a COVID-19 Positive Patient A Case Study. J Am Podiatr Med Assoc (2020). doi: 10.7547/20-088
63. Recalcati S. Cutaneous Manifestations in COVID-19: A First Perspective. J Eur Acad Dermatol Venereol (2020) 34(5):e212–3. doi: 10.1111/jdv.16387
64. Zhang Y, Cao W, Xiao M, Li YJ, Yang Y, Zhao J, et al. Clinical and Coagulation Characteristics of 7 Patients With Critical COVID-2019 Pneumonia and Acro-Ischemia. Zhonghua Xue Ye Xue Za Zhi (2020) 28:41: E006. doi: 10.3760/cma.j.issn.0253-2727.2020.0006
65. Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. High Risk of Thrombosis in Patients With Severe Sars-CoV-2 Infection: A Multicenter Prospective Cohort Study. Intensive Care Med (2020) 4:1–10. doi: 10.1007/s00134-020-06062-x
66. Poissy J, Goutay J, Caplan M, Parmentier E, Duburcq T, Lassalle F, et al. Pulmonary Embolism in COVID-19 Patients: Awareness of an Increased Prevalence. Circulation (2020) 142(2):184–6. doi: 10.1161/CIRCULATIONAHA.120.047430
67. Tian S, Hu W, Niu L, Liu H, Xu H, Xiao S. Pulmonary Pathology of Early-Phase 2019 Novel Coronavirus (Covid-19) Pneumonia in Two Patients With Lung Cancer. J Thorac Oncol (2020) 15(5):700–4. doi: 10.1016/j.jtho.2020.02.010
68. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet (2020) 395:507–13. doi: 10.1016/S0140-6736(20)30211-7
69. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical Characteristics of 138hospitalized Patients With 2019 Novel Coronavirus−Infected Pneumonia in Wuhan, China. JAMA (2020) 323(11):1061–9. doi: 10.1001/jama.2020.1585
70. Levi M, Thachil J, Iba T, Levy JH. Coagulation Abnormalities and Thrombosis in Patients With Covid-19. Lancet Haematol (2020) 7(6):e438–40. doi: 10.1016/S2352-3026(20)30145-9
71. Yi X, Zhou H, Chao Y, Xiong S, Zhong J, Chai Z, et al. Bacteria-Triggered Tumor-Specific Thrombosis to Enable Potent Photothermal Immunotherapy of Cancer. Sci Adv (2020) 6(33):eaba3546. doi: 10.1126/sciadv.aba3546
72. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome Activation Triggers Blood Clotting and Host Death Through Pyroptosis. Immunity (2019) 50(6):1401–11.e4. doi: 10.1016/j.immuni.2019.04.003
73. Skjeflo EW, Christiansen D, Fure H, Ludviksen JK, Woodruff TM, Espevik T, et al. Staphylococcus Aureus-Induced Complement Activation Promotes Tissue Factor-Mediated Coagulation. J Thromb Haemost (2018) 16(5):905–18. doi: 10.1111/jth.13979
74. Engelmann B, Massberg S. Thrombosis as an Intravascular Effector of Innate Immunity. Nat Rev Immunol (2013) 13(1):34–45. doi: 10.1038/nri3345
75. Bochenek ML, Schäfer K. Role of Endothelial Cells in Acute and Chronic Thrombosis. Hamostaseologie (2019) 39(2):128–39. doi: 10.1055/s-0038-1675614
76. Machado F, Silva E. Coagulation and Sepsis. Endocr Metab Immune Disord Drug Targets (2006) 6(2):175–82. doi: 10.2174/187153006777442369
77. Price LC, McCabe C, Garfield B, Wort SJ. Thrombosis and COVID-19 Pneumonia: The Clot Thickens. Eur Respir J (2020) 56(1):2001608. doi: 10.1183/13993003.01608-2020.
78. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological Findings of COVID-19 Associated With Acute Respiratory Distress Syndrome. Lancet Respir Med (2020) 8(4):420–2. doi: 10.1016/S2213-2600(20)30076-X
79. Guervilly C, Burtey S, Sabatier F, Cauchois R, Lano G, Abdili E, et al. Circulating Endothelial Cells as a Marker of Endothelial Injury in Severe Covid-19. J Infect Dis (2020) 222(11):1789–93. doi: 10.1093/infdis/jiaa528
80. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19 Associated Coagulopathy: Evidence From a Single-Centre, Cross-Sectional Study. Lancet Haematol (2020) 7(8):e575–82. doi: 10.1016/S2352-3026(20)30216-7
81. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med (2020) 383(2):120–8. doi: 10.1056/NEJMoa2015432
82. Colmenero I, Santonja C, Alonso-Riaño M, Noguera-Morel L, Hernández-Martín A, Andina D, et al. Sars-CoV-2 Endothelial Infection Causes COVID-19 Chilblains: Histopathological, Immunohistochemical and Ultrastructural Study of Seven Paediatric Cases. Br J Dermatol (2020) 183(4):729–37. doi: 10.1111/bjd.19327.
83. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet (2020) 395(10234):1417–8. doi: 10.1016/S0140-6736(20)30937-5.
84. Desai TR, Leeper NJ, Hynes KL, Gewertz BL. Interleukin-6 Causes Endothelial Barrier Dysfunction Via the Protein Kinase C Pathway. J Surg Res (2002) 104:118–23. doi: 10.1006/jsre.2002.6415
85. Fleck RA, Rao LV, Rapaport SI, Varki N. Localization of Human Tissuefactor Antigen by Immunostaining With Monospecific, Polyclonal Anti-Human Tissue Factor Antibody. Thromb Res (1990) 59:421–37. doi: 10.1016/0049-3848(90)90148-6
86. Drake TA, Morrissey JH, Edgington TS. Selective Cellular Expressionof Tissue Factor in Human Tissues. Implications for Disorders of Hemostasis and Thrombosis. Am J Pathol (1989) 134(05):1087–97.
87. McVey JH. Tissue Factor Pathway. Baillieres Clin Haematol (1994) 7(3):469–84. doi: 10.1016/s0950-3536(05)80094-0
88. Eisenreich A, Rauch U. Regulation and Differential Role of the Tissue Factor Isoforms in Cardiovascular Biology. Trends CardiovascMed (2010) 20(6):199–203. doi: 10.1016/j.tcm.2011.08.001
89. Pawlinski R, Mackman N. Cellular Sources of Tissue Factor in Endotoxemia and Sepsis. Thromb Res (2010) 125(Suppl 1):S70–3. doi: 10.1016/j.thromres.2010.01.042.
90. DelGiudice LA, White GA. The Role of Tissue Factor and Tissue Factor Pathway Inhibitor in Health and Disease States. J Vet Emerg Crit Care (San Antonio) (2009) 19(1):23–9. doi: 10.1111/j.1476-4431.2008.00380.x
91. Rauch U, Nemerson Y. Tissue Factor, the Blood, and the Arterial Wall. Trends Cardiovasc Med (2000) 10(4):139–43. doi: 10.1016/s1050-1738(00)00049-9
92. Sebag SC, Bastarache JA, Ware LB. Therapeutic Modulation of Coagulation and Fibrinolysis in Acute Lung Injury and the Acute Respiratory Distress Syndrome. Curr Pharm Biotechnol (2011) 12(9):1481–96. doi: 10.2174/138920111798281171
93. Kirschenbaum LA, McKevitt D, Rullan M, Reisbeck B, Fujii T, Astiz ME. Importance of Platelets and Fibrinogen in Neutrophil-Endothelial Cell Interactions in Septic Shock. Crit Care Med (2004) 32(9):1904–9. doi: 10.1097/01.ccm.0000139918.80602.57
94. Xue M, Sun Z, Shao M, Yin J, Deng Z, Zhang J, et al. Diagnostic and Prognostic Utility of Tissue Factor for Severe Sepsis and Sepsis-Induced Acute Lung Injury. J Transl Med (2015) 13:172. doi: 10.1186/s12967-015-0518-9
95. Carraway MS, Welty-Wolf KE, Miller DL, Ortel TL, Idell S, Ghio AJ, et al. Blockade of Tissue Factor: Treatment for Organ Injury in Established Sepsis. Am J Respir Crit Care Med (2003) 167:1200–9. doi: 10.1164/rccm.200204-287OC
96. Eslamifar Z, Behzadifard M, Soleimani M, Behzadifard S. Coagulation Abnormalities in SARS-CoV-2 Infection: Overexpression Tissue Factor. Thromb J (2020) 18(1):38. doi: 10.1186/s12959-020-00250-x
97. Bautista-Vargas M, Bonilla-Abadía F, Cañas CA. Potential Role for Tissue Factor in the Pathogenesis of Hypercoagulability Associated With in COVID-19. J Thromb Thrombolysis (2020) 50(3):479–83. doi: 10.1007/s11239-020-02172-x
98. DiNicolantonio JJ, McCarty M. Thrombotic Complications of COVID-19 May Reflect an Upregulation of Endothelial Tissue Factor Expression That Is Contingent on Activation of Endosomal NADPH Oxidase. Open Heart (2020) 7(1):e001337. doi: 10.1136/openhrt-2020-001337
99. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, et al. Platelet Activation and Platelet-Monocyte Aggregate Formation Trigger Tissue Factor Expression in Patients With Severe COVID-19. Blood (2020) 136(11):1330–41. doi: 10.1182/blood.2020007252
100. Canzano P, Brambilla M, Porro B, Cosentino N, Tortorici E, Vicini S, et al. Platelet and Endothelial Activation as Potential Mechanisms Behind the Thrombotic Complications of COVID-19 Patients. JACC Basic Transl Sci (2021) 6(3):202–18. doi: 10.1016/j.jacbts.2020.12.009
101. Iba T, Ogura H. Role of Extracellular Vesicles in the Development of Sepsis-Induced Coagulopathy. J Intensive Care (2018) 6:68. doi: 10.1186/s40560-018-0340-6.
102. Grover SP, Mackman N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler Thromb Vasc Biol (2018) 38(4):709–25. doi: 10.1161/ATVBAHA.117.309846
103. Rosell A, Havervall S, von Meijenfeldt F, Hisada Y, Aguilera K, Grover SP, et al. Dissemination of Extreme Levels of Extracellular Vesicles: Tissue Factor Activity in Patients With Severe COVID-19. Arterioscler Thromb Vasc Biol (2020) 41(2):ATVBAHA120315547. doi: 10.1161/ATVBAHA.120.315547
104. Mast AE, Wolberg AS, Gailani D, Garvin MR, Alvarez C, Miller JI, et al. SARS-Cov-2 Suppresses Anticoagulant and Fibrinolytic Gene Expression in the Lung. Elife (2021) 10:e64330. doi: 10.7554/eLife.64330
105. Chen Z, Zhang H, Qu M, Nan K, Cao H, Cata JP, et al. The Emerging Role of Neutrophil Extracellular Traps in Sepsis and Sepsis-Associated Thrombosis. Front Cell Infect Microbiol (2021) 11:653228. doi: 10.3389/fcimb.2021.653228
106. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA Traps Promote Thrombosis. Proc Natl Acad Sci USA (2010) 107(36):15880–5. doi: 10.1073/pnas.1005743107
107. Ducroux C, Di Meglio L, Loyau S, Delbosc S, Boisseau W, Deschildre C, et al. Thrombus Neutrophil Extracellular Traps Content Impair Tpa-Induced Thrombolysis in Acute Ischemic Stroke. Stroke (2018) 49(3):754–7. doi: 10.1161/STROKEAHA.117.019896
108. Chrysanthopoulou A, Mitroulis I, Apostolidou E, Arelaki S, Mikroulis D, Konstantinidis T, et al. Neutrophil Extracellular Traps Promote Differentiation and Function of Fibroblasts. J Pathol (2014) 233(3):294–307. doi: 10.1002/path.4359.
109. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil Extracellular Traps and Thrombosis in COVID-19. JCI Insight (2020) 5(11):e138999. doi: 10.1172/jci.insight.138999
110. Busch MH, Timmermans SAMEG, Nagy M, Visser M, Huckriede J, Aendekerk JP, et al. Neutrophils and Contact Activation of Coagulation as Potential Drivers of COVID-19. Circulation (2020) 142(18):1787–90. doi: 10.1161/CIRCULATIONAHA.120.050656.
111. Matsuno H, Kozawa O, Nagashima S, Kanamaru M, Uematsu T. Comparative Antiplatelet Effects of Aspirin, Vapiprost and GR144053, a GPIIb/IIIa Antagonist, With a Special Reference to the Role of Platelet Microaggregates. Br J Pharmacol (1999) 127(5):1129–34. doi: 10.1038/sj.bjp.0702651
112. Otterdal K, Pedersen TM, Solum NO. Platelet Shape Change Induced by the Peptide Yfllrnp. Thromb Res (2001) 103(5):411–20. doi: 10.1016/s0049-3848(01)00334-6
113. Chang H, Yanachkov IB, Michelson AD, Li Y, Barnard MR, Wright GE, et al. Agonist and Antagonist Effects of Diadenosine Tetraphosphate, a Platelet Dense Granule Constituent, on Platelet P2Y1, P2Y12 and P2X1 Receptors. Thromb Res (2010) 125(2):159–65. doi: 10.1016/j.thromres.2009.11.006
114. Youssefian T, Massé JM, Rendu F, Guichard J, Cramer EM. Platelet and Megakaryocyte Dense Granules Contain Glycoproteins Ib and Iib-Iiia. Blood (1997) 89(11):4047. doi: 10.1182/blood.V89.11.4047
115. Yadav H, Kor DJ. Platelets in the Pathogenesis of Acute Respiratory Distress Syndrome. Am J Physiol Lung Cell Mol Physiol (2015) 309:L915–23. doi: 10.1152/ajplung.00266.2015
116. Li J, Kim K, Jeong SY, Chiu J, Xiong B, Petukhov PA, et al. Platelet Protein Disulfide Isomerase Promotes Glycoprotein Ibα-Mediated Platelet-Neutrophil Interactions Under Thromboinflammatory Conditions. Circulation (2018) 139(10):1300–19. doi: 10.1161/CIRCULATIONAHA.118.036323
117. Kornerup KN, Salmon GP, Pitchford SC, Liu WL, Page CP. Circulating Platelet-Neutrophil Complexes are Important for Subsequent Neutrophil Activation and Migration. J Appl Physiol (2010) 109(3):758–67. doi: 10.1152/japplphysiol.01086.2009.
118. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet Gene Expression and Function in Patients With COVID-19. Blood (2020) 136(11):1317–29. doi: 10.1182/blood.2020007214
119. Sjöström A, Wersäll J, Warnqvist A, Farm M, Magnusson M, Oldner A, et al. Platelet Count Rose While D-dimer Levels Dropped as Deaths and Thrombosis Declined, an Observational Study on Anticoagulation Shift in COVID-19. Thromb Haemost (2021). doi: 10.1055/a-1477-3829
120. Turecek PL, Peck RC, Rangarajan S, Reilly-Stitt C, Laffan MA, Kazmi R, et al. Recombinant ADAMTS13 Reduces Abnormally Up-Regulated Von Willebr and Factor in Plasma From Patients With Severe COVID-19. Thromb Res (2021) 201:100–12. doi: 10.1016/j.thromres.2021.02.012
121. White B, Perry D. Acquired Antithrombin Deficiency in Sepsis. Br J Haematol (2001) 112(1):26–31. doi: 10.1046/j.1365-2141.2001.02396.x
122. Oelschläger C, Römisch J, Staubitz A, Stauss H, Leithäuser B, Tillmanns H, et al. Antithrombin III Inhibits Nuclear Factor Kappab Activation in Human Monocytes and Vascular Endothelial Cells. Blood (2002) 99:4015–20. doi: 10.1182/blood.v99.11.4015
123. Kaneider NC, Reinisch CM, Dunzendorfer S, Römisch J, Wiedermann CJ. Syndecan-4 Mediates Antithrombin-Induced Chemotaxis of Human Peripheral Blood Lymphocytes and Monocytes. J Cell Sci (2002) 115(Pt 1):227–36. doi: 10.1242/jcs.115.1.227
124. Levy JH, Sniecinski RM, Welsby IJ, Levi M. Antithrombin: Anti-Inflammatory Properties and Clinical Applications. Thromb Haemostasis (2016) 115(4):712–28. doi: 10.1046/j.1365-2141.2001.02396.x
125. Anaklı İ, Ergin Özcan P, Polat Ö, Orhun G, Alay GH, Tuna V, et al. Prognostic Value of Antithrombin Levels in COVID-19 Patients and Impact of Fresh Frozen Plasma Treatment: A Retrospective Study. Turk J Haematol (2021) 38(1):15–21. doi: 10.4274/tjh.galenos.2021.2020.0695
126. Tabatabai A, Rabin J, Menaker J, Madathil R, Galvagno S, Menne A, et al. Factor VIII and Functional Protein C Activity in Critically Ill Patients With Coronavirus Disease 2019: A Case Series. A A Pract (2020) 14(7):e01236. doi: 10.1213/XAA.0000000000001236
127. Lippi G, Henry BM, Sanchis-Gomar F. Plasma Antithrombin Values Are Significantly Decreased in Coronavirus Disease 2019 (Covid-19) Patients With Severe Illness. Semin Thromb Hemost (2020). doi: 10.1055/s-0040-1716873
128. Griffin JH, Fernández JA, Gale AJ, Mosnier LO. Activated Protein C. J Thromb Haemost (2007) 5:73–80. doi: 10.1111/j.1538-7836.2007.02491.x
130. Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene Expression Profile of Antithrombotic Protein Cdefines New Mechanisms Modulating Inflammation and Apoptosis. J Biol Chem (2001) 276:11199–203. doi: 10.1074/jbc.C100017200
131. Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of Endothelial Cell Protease Activated Receptor 1 by the Protein C Pathway. Science (2002) 296(5574):1880–2. doi: 10.1126/science.1071699
132. Pestka SB. Old Drug, New Trick? The Rationale for the Treatment of COVID-19 With Activated Protein C. Med Hypotheses (2021) 149:110537. doi: 10.1016/j.mehy.2021.110537
133. Griffin JH, Lyden P. Covid-19 Hypothesis: Activated Protein C for Therapy of Virus-Induced Pathologic Thromboinflammation. Res Pract Thromb Haemost (2020) 4(4):506–9. doi: 10.1002/rth2.12362
134. Gerotziafas GT, Sergentanis TN, Voiriot G, Lassel L, Papageorgiou C, Elabbadi A, et al. Derivation and Validation of a Predictive Score for Disease Worsening in Patients With COVID-19. Thromb Haemost (2020) 120(12):1680–90. doi: 10.1055/s-0040-1716544
135. Ghosh AK, Vaughan DE. PAI-1 in Tissue Fibrosis. J Cell Physiol (2012) 227(2):493–507. doi: 10.1002/jcp.22783
136. Katayama S, Koyama K, Shima J, Tonai K, Goto Y, Koinuma T, et al. Thrombomodulin, Plasminogen Activator Inhibitor-1 and Protein C Levels, and Organ Dysfunction in Sepsis. Crit Care Explor (2019) 1(5):e0013. doi: 10.1097/CCE.0000000000000013
137. Brogren H, Wallmark K, Deinum J, Karlsson L, Jern S. Platelets Retain High Levels of Active Plasminogen Activator Inhibitor 1. PloS One (2011) 6(11):e26762. doi: 10.1371/journal.pone.0026762
138. Wright FL, Vogler TO, Moore EE, Moore HB, Wohlauer MV, Urban S, et al. Fibrinolysis Shutdown Correlation With Thromboembolic Events in Severe Covid-19 Infection. J Am Coll Surg (2020) 231(2):193–203.e1. doi: 10.1016/j.jamcollsurg.2020.05.007.
139. Zuo Y, Warnock M, Harbaugh A, Yalavarthi S, Gockman K, Zuo M, et al. Plasma Tissue Plasminogen Activator and Plasminogen Activator Inhibitor-1 in Hospitalized COVID-19 Patients. Sci Rep (2021) 11(1):1580. doi: 10.1038/s41598-020-80010-z
140. Nougier C, Benoit R, Simon M, Desmurs-Clavel H, Marcotte G, Argaud L, et al. Hypofibrinolytic State and High Thrombin Generation May Play a Major Rolein SARS-COV2 Associated Thrombosis. J Thromb Haemost (2020) 18(9):2215–9. doi: 10.1111/jth.15016
Keywords: COVID-19, inflammation, thrombosis, respiratory dysfunction, therapeutic
Citation: Fang X-Z, Wang Y-X, Xu J-Q, He Y-J, Peng Z-K and Shang Y (2021) Immunothrombosis in Acute Respiratory Dysfunction of COVID-19. Front. Immunol. 12:651545. doi: 10.3389/fimmu.2021.651545
Received: 10 January 2021; Accepted: 12 May 2021;
Published: 02 June 2021.
Edited by:
Pei-Hui Wang, Shandong University, ChinaReviewed by:
Yun Cheng, The University of Hong Kong, ChinaXin Yin, Chinese Academy of Agricultural Sciences, China
Copyright © 2021 Fang, Wang, Xu, He, Peng and Shang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: You Shang, eW91X3NoYW5naHVzdEAxNjMuY29t