 Hanwen Zhang
Hanwen Zhang Zhuonan Li2
Zhuonan Li2 Wei Li
Wei Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 17 March 2021
Sec. Alloimmunity and Transplantation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.648539
This article is part of the Research TopicNew Insights into and Interventions of the Persistent Immune Responses in Chronic Graft RejectionView all 18 articles
Allograft functional failure due to acute or chronic rejection has long been a major concern in the area of solid organ transplantation for decades. As critical component of innate immune system, the macrophages are unlikely to be exclusive for driving acute or chronic sterile inflammation against allografts. Traditionally, macrophages are classified into two types, M1 and M2 like macrophages, based on their functions. M1 macrophages are involved in acute rejection for triggering sterile inflammation thus lead to tissue damage and poor allograft survival, while M2 macrophages represent contradictory features, playing pivotal roles in both anti-inflammation and development of graft fibrosis and resulting in chronic rejection. Macrophages also contribute to allograft vasculopathy, but the phenotypes remain to be identified. Moreover, increasing evidences are challenging traditional identification and classification of macrophage in various diseases. Better understanding the role of macrophage in chronic rejection is fundamental to developing innovative strategies for preventing late graft loss. In this review, we will update the recent progress in our understanding of diversity of macrophage-dominated innate immune response, and reveal the roles of M2 macrophages in chronic allograft rejection as well.
Organ transplantation appears to be the best treatment method for patients who suffered from the end-stage diseases. However, chronic allograft rejection following transplantation remains to be a major challenge to long term allograft survival and functions. Organ allografts are susceptible to ischemia reperfusion injury (IRI) and recipient alloimmunity, suffering from acute and chronic rejection inevitably (1). Much attention has once focused on acute rejection, but nowaday it is no longer the common cause of late graft loss due to the application of systemic immunosuppressants (2). Increasing evidences are focusing on the profound meaning of innate immune cells, such as NK cells, macrophages, and neutrophils, in the late phase change of allografts.
Diversity of innate immune cells is drawing much attention. Neutrophils have been characterized as a major component in driving inflammation for a long time. They are known to be recruited into stressed solid organ and contribute to tissue injury, while recent data have demonstrated their contradictory function in tissue repair (3, 4). Identically, distinctive functional features between two different populations of macrophages are considered as a hot spot to study in varies diseases models. Despite the fact that macrophages are recognized as a key participant in triggering both graft acute inflammatory damage and inflammation resolution in organ transplantation (5). The role of macrophage-mediated late phase graft rejection and corresponding therapies to mitigate allograft dysfunction or loss still remain largely unknown.
Monocyte-derived macrophages infiltrate into allografts after transplantation and then mainly differentiate into two phenotypes: pro-inflammatory M1-like macrophages and anti-inflammatory M2-like type (6), thus represent opposite functions during acute phase in organ transplant. However, M2-like macrophages also show potentials for enabling fibrosis, resulting in poor long-term graft survival. Moreover, macrophages are reported to be related with graft vasculopathy (7), but the data on their heterogeneity are limited. Tissue-resident macrophages are displaying differential features compared with their infiltrating counterpart. Although it has been proved that the replenishment of local macrophage niche in organ under unsteady state, increasing data have shown the differential characteristics and features between embryonic and monocyte-derived populations (8). Diverse macrophages dynamically represent various functions in local immune environment in response to stimuli, stress and tissue injury, while a great deal of work is yet to be done in the area of chronic allograft rejection. An improved understanding of macrophage related cellular immune events which trigger the late phase rejection and ultimately responsible for allograft dysfunction and loss, is fundamental for the development of innovative strategies for the treatment of organ allograft recipients. In this review, we will update the recent progresse of diversity of macrophage dominated innate immune responses and reveal the roles of M2 macrophages in chronic allograft rejection as well.
As we all know so far, both innate and adaptive immune responses are involved in chronic allograft rejection. The key effective innate immune cells, macrophages, play important roles in variety of physiologic and pathologic processes, for instance, host defense, acute and chronic inflammation and tissue repair (9). Macrophages are derived from monocytes in the circulation and then represent resident features in lymphoid and non-lymphoid tissues, including solid organs, involved in tissue homeostasis (10). They were characterized by plasticity and diverse functions. The activated macrophages polarize and exhibit classical M1-like or alternative M2-like phenotypes. M1 macrophages tend to produce reactive oxygen species (ROS), nitric oxide (NO) and pro-inflammatory cytokines such as IL-6, IL-12, and TNF-α that play crucial roles in defense against dead cells, microbial infection and cancer. On the contrary, M2 macrophages tend to express scavenging receptors and produce various anti-inflammatory mediators including IL-10 and TGF-β to promote the inflammation resolution, tissue repair, wound healing and fibrosis (11).
Traditionally, the polarization of macrophage may be defined by M1/M2 markers including Nitric Oxide Synthases (NOSs), arginase family (Arg1/2), CD206 and CD163. In spite of that, macrophages are likely to represent distinctive polarization and differentiation under various circumstances in complex organism. Understanding dynamic reprogramming landscapes of macrophage is important for exploiting the mechanisms of transplant immune responses (Figure 1). Of note, a unique macrophage subset driven by IL-23 has been recently discussed. IL-23, a heterodimeric cytokine essential for expression of IL-17, shares p40 subunit with IL-12 that induces the production of IFN-γ (12). It was reported that IL-23 involved in IL-17 and IFN-γ related tissue inflammatory response by innate lymphoid cells, including macrophages (13, 14). Treated with recombinant IL-23, unpolarized mouse peritoneal macrophages represent significantly increased production of IL-17A and IFN-γ via STAT3-RORγ T pathways, but not M1/M2 related cytokines, and neither polarized M1 nor M2-like macrophages can convert to such an IL-17/IFN-γ high-producing subset: M(IL-23) (15). Thus, M(IL-23) is likely to be defined as a new macrophage subpopulation and is worth discussing since alloreactive production of IL-17/IFN-γ is closely related to allograft rejection (16).
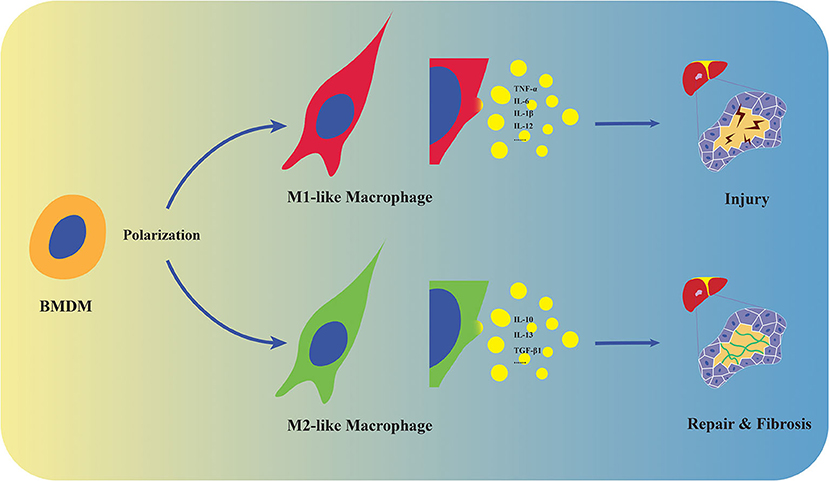
Figure 1. Polarization and distinct functions of macrophages. Macrophages derive from monocytes in the circulation and polarize into M1/M2-like phenotypes. M1-like macrophages promote tissue injury by producing pro-inflammatory cytokines including TNF-α, IL-6, IL-1β, and IL-12, while M2 represent tissue repair and fibrosis by expressing anti-inflammatory and fibrogenic mediators such as IL-10, IL-13, and TGF-β1 in stressed liver.
Tissue resident macrophages are crucial participants in organ homeostasis and are likely to exhibit a M2-like phenotype (17). However, M2-like macrophages could also display pro-fibrogenic feature. In other word, tissue-resident macrophages are double-edged swords that have potential to either alleviate IRI and inflammation, or result in fibrotic rejection. Recently, many studies are focusing on postnatal development of tissue resident macrophages and their functions in metabolism, immune activation and regulation. Unlike primitive tissue-resident macrophages such as Kupffer cells (KCs) in the liver, microglia in brain and renal resident macrophages that arise from yolk sac (18–20), macrophages originate from bone marrow-derived monocytes and then reside in solid organs, serving as executors of innate immunity. Identical to liver parenchyma cells, non-parenchyma cells (NPCs) in the liver suffer from hypoxia, leading to ischemia-induced necrotic activation (21). Although it has been widely acknowledged that immigrant macrophages and tissue-resident macrophages display distinct features and properties in some aspects, niche-specific reprogramming of recruited macrophages with the help of “KC-enhancers” expressed by liver sinusoidal endothelial cells and hepatocytes have been recently discovered on the basis of “resident like” marker V-set immunoglobulin-domain-containing 4 (VSIG4) (22, 23). Tissue resident macrophages are susceptible to endo-/exogenous stimulation and contribute to necrotic or apoptotic depletion. Tremendous findings have demonstrated that the potentials on the transformation of the immigrant populations into the resident are not only compensating impaired resident macrophage pool, but also representing resident-like features and functions that “newcome” macrophages lack in response to stress including surgery-induced IRI, infection and chronic inflammation (17, 22, 24). Despite the fact that limited works have focused on macrophage reprogramming in organ transplantation, progress pertaining to the aforementioned factors suggest the innovative possibility in alleviating macrophage-mediated chronic allograft rejection.
IRI which triggered by activation of innate immune receptors, not only play a key role in graft failure at the early stage, but also result in chronic rejection at the late stage of transplantation (25). This inevitable complication initiates tissue damage by promoting release of danger-associated molecular pattern (DAMP) such as high mobility group box-1 (HMGB1) which derived from necrotic or stressed cells to activate sentinel pattern recognition receptor Toll-like receptor 4 (TLR4), thus mediating sterile inflammation (26, 27). As the major innate immune cells responding to DAMPs, monocyte-derived macrophage are activated and infiltrate into IR-stressed organs, which is closely associated with inflammation and tissue injury. However, the functions of macrophage are different in their population and may represent distinct roles in response to IRI stress. It has been revealed based on the biological phenotype of macrophage that M1-like macrophages are responsible for inducing neutrophil-mediated tissue injury, while M2-like phenotype monocyte-derived macrophages and kupffer cells are likely to represent anti-inflammatory and wound-healing features during liver IRI (28, 29).
Compared with monocyte-derived macrophages originated from bone-marrow progenitor cells and take time to be recruited from circulation, tissue-resident macrophages represent rapid response. They sense neighboring cells death and react to DAMPs as soon as they are released by dead organ parenchymal cells, directly function on cellular debris degradation and cytokines secretion against injury (25). More notably, they are stimulated and activated during ischemic stage before the recruitment of monocyte-derived macrophages into stressed organ. Thus, diverse macrophage subsets and complex innate immune activation demonstrate a complicated scenario, converting immunologically quiescent milieu into an inflammatory activation vs. resolution condition and resulting in tissue damage, injury repair and fibrosis.
M1-like macrophage-dominated innate immune response is one of the major sources of acute neutrophil-mediated inflammation in transplant recipients. Indeed, study using transgenic cre-inducible diphtheria toxin receptor (DTR) system that specifically targets CD11b+ (a surface marker expressed on macrophages) cells in vivo to deplete infiltrating macrophages demonstrated the alleviative effect on liver IRI in mice (30, 31). At early stage of post-transplant, overwhelming innate immune response dominates acute rejection, while T-cell mediated adaptive allograft rejection has not yet been established. M1-like macrophages are inclined to produce pro-inflammatory cytokines TNF-α, IL-6, and IL-1β, mediating injury. As pro-/anti- inflammatory activation and tissue repair co-exist, implants undergo dynamic change. Failing to resolve acute inflammation may result in chronic inflammation, attributing to establishment of allograft inflammatory fibrosis and gradual deterioration in functions (32).
Upon injuries or diseases, there is an impressive repertoire of machinery in place for self-preservation and healing. One of the most important protective mechanisms for wound healing and tissue regeneration is the formation of extracellular matrix (ECM). As a highly dynamic construction, ECM undergoes an independent remodeling process, contributes to restructuring of tissue architecture in regarding to maintenance of stable organ structure and function (33, 34). ECM unbalance caused by endo-/exogenous risk factors is likely to result in disorganization or dysfunction. Fibroblasts, one of the most abundant cell types are widely distributed in the connective tissue throughout the body. They are acknowledged to compose the basic framework for tissues and organs by producing, maintaining and reabsorption of ECM, but act as circumstantial effectors that unfrequently function expression and remodeling of ECM (35). Under homeostasis, fibroblasts remain relatively quiescent, while in response to stress and stimuli, they adapt to microenvironment and are activated in coordination with inflammation related cytokines, especially TGF-β1, by differentiating into their apex stage, myofibroblasts (36). In response to tissue injury or chronic inflammation, myofibroblasts with a terminally differentiated phenotype not only extensively produce ECM, but also inherit a contractile apparatus in order to manipulate ECM fibers physically that fill the wounds and attribute to acute or chronic inflammatory damage (37).
Fibrosis is a shared and central part of numerous pathologies including autoimmunity, metabolic disorders and graft rejection (38–40). Tissue fibrosis is a dangerous condition and pathological manifestations characterized by an excessive accumulation of ECM in response to acute/chronic injuries. In addition, interstitial fibrosis and tubular atrophy (IF/TA) of kidney cortex is best known as its paramount importance in development of poorer renal function and outcome in chronic kidney disease (CKD) and kidney transplantation (41, 42). In spite of the fact that emphasizing the predominated effects of adaptive antibody-mediated rejection (AMR) in loss of implants, innate-immune-dominated fibrotic rejection triggered by macrophages should not be neglected (43). Diverse populations of macrophage represent functionally distinction during early and late phases post-transplantation. When it comes to fibrosis, they controversially drive spectrum of phenotypes that display both pro- and anti-fibrotic functions.
In the process of chronic rejection, imbalance between pro- and anti-fibrosis play an important role. Wound healing M2-like macrophages not only play critical roles in anti-inflammation in the very early stage of IRI, but also regulate tissue inflammation resolution and promote fibrotic processes via producing TGF-β1 and VEGF (44). Infiltrating macrophages differentiate into M2-like feature is likely to produce IL-10, which is crucial for suppressing pro-inflammatory gene programs. A clinical study obtained renal allograft biopsies from 12 month post-transplant, demonstrating the strong relationship between area of fibrosis and numbers of CD206+ M2-like macrophages (45). Smad-3 is an intracellular molecule facilitates transmit chemical signals from plasma membrane to the nucleus, operating downstream of growth/differentiation factors including TGF-β, activin, and myostatin (46). In the development of chronic kidney allograft rejection, Smad-3-dependent macrophage-myofibroblast transition (MMT) process, which occurs predominantly within M2-like macrophages, resulting in renal interstitial fibrosis (47, 48). Moreover, a novel adenosine triphosphate (ATP)-gated ion channel protein P2X7, initially reported to function in fast synaptic transmission and lysis of antigen-presenting cells (APCs), is recently found to play important roles in neutrophil adhesion, contributes to acute/chronic inflammation and M2-like macrophages-mediated chronic heart fibrotic rejection (33, 49–51). During the progression of fibrotic rejection, macrophages derived TGF-β undoubtedly play a pivotal role in differentiation of fibroblasts into myofibroblasts, but whether and how adaptive immune system affect this process remain worth studying. T helper type 1 cells (Th1 cells), defined by their secretion of IFN-γ, have potential to disrupt TGF-β/Smad3 signaling pathway-mediated fibrosis (52, 53). On the contrary, T helper type 2 cells (Th2 cells) is likely to promote the TGF-βmediated fibrosis via the production of IL-4 and IL-13 (54). Thus, further investigating the interaction between innate and adaptive immune systems in fibrotic rejection might be of great benefits (Figure 2).
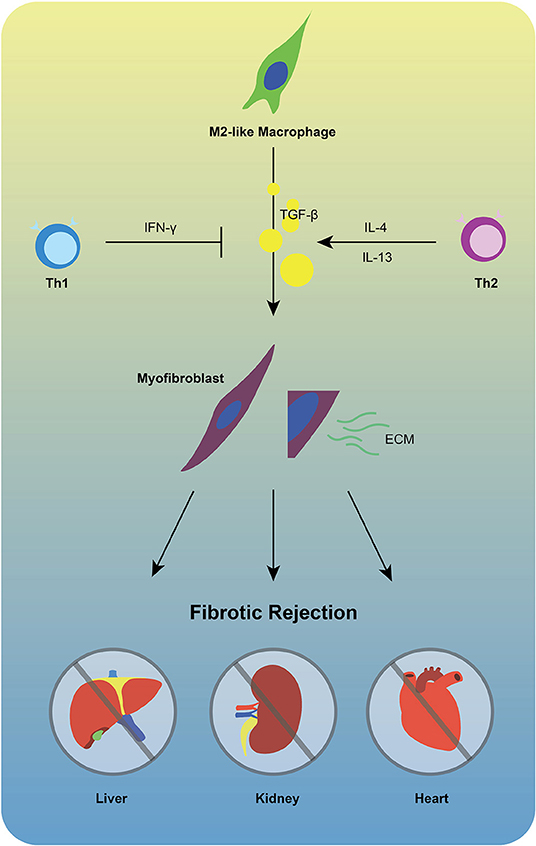
Figure 2. M2-like macrophages are unlikely to be exclusive for fibrotic rejection. TGF-β1 produced by M2-like macrophages facilitates differentiation of fibroblasts into myofibroblasts that extensively produce ECM, promoting chronic fibrotic allograft rejection. During this process, Th1 cells may inhibit the activation of TGF-β by producing IFN-γ, while Th2 cells represent opposite feature via secretion of IL-4/IL-13.
In spite of progresses in immunosuppressive therapy, long-term success of allogeneic organ transplantation is limited by the establishment of allograft vasculopathy. Vasculopathy is another dangerous risk factor and hallmark of chronic allograft rejection triggered by multifactorial immunologic events, characterized by endothelial injury and dysfunction, myointimal hyperplasia and extracellular matrix synthesis (55, 56). Transplant vasculopathy inevitably occurs in ~90% of allografts, developing severe intimal hyperplastic lesions that eventually result in luminal flow restriction and graft failure during long-term follow-up (57). In line with this scenario, cellular and antibody-mediated rejection processes and anti-HLA antibodies against implant play crucial roles in mediating inflammation that drive the development of vasculopathy-induced chronic allograft failure (58). Transplant vasculopathy has long been considered to consequent to chronic allograft rejection, especially kidney and heart (59, 60). Nearly one-third of patients developed cardiac allograft vasculopathy (CAV) by 5 years post-transplant, leading to 12.5% CAV-induced death rate (61). Moreover, it is demonstrated that the renal vasculopathy characterized by perivascular leukocyte infiltration and neointimal hyperplasia is unlikely to be exclusive for affecting the intrarenal blood vessels (62).
It has been reported by Croker et al. (63) that infiltrating macrophage-based post-transplant inflammation played a crucial role in chronic allograft nephropathy and dysfunction. Transplant vasculopathy is referred to as an accelerated pathologically fibroproliferative process characterized by smooth muscle proliferation and lipid deposition-induced circumferential intimal thickening (61, 64). Studies define macrophage as predominant cell type in the intimal of renal allograft arteries and demonstrate their unfavorably effects on graft survival in both acute and chronic rejection (65, 66). Depleting or inhibiting infiltrating macrophages by using λ-carrageenan type IV or a small molecule β2 integrin agonist leukadherin-1 (LA1) significantly suppressed cardiac and renal vasculopathy, alleviated chronic tissue injury and dysfunction (67, 68). Although increasing evidence suggest that macrophage-mediated vasculopathy is an integral part of the chronic allograft rejection process, the underlying mechanisms have not been well-defined. Distinct macrophage populations and functions in the process of allograft vasculopathy still remain to be further studied.
It has been widely recognized that adaptive immune response mediated by T and B lymphocytes, and donor-specific antibodies (DSA) are all involved in culminating chronic allograft rejection (69). In consideration of important roles of macrophages in the development of transplant rejection, questions concerning the link between macrophages and adaptive immune system need to be addressed. Despite the potential effects of CD4+ T cells in macrophage-mediated fibrotic rejection, which we have mentioned above, interestingly, recent arguments are challenging the traditional perspective on macrophage as innate immune cells. Chu et al. have advocated the concept that macrophage is potential to represent adaptive immune feature in allograft rejection, stating that CD4+T cells facilitate the acquisition of long-term specific memory of macrophages against skin allograft (70, 71). Moreover, another study demonstrates that macrophages can produce B-cell activating factor (BAFF), resulting in graft damage during antibody mediated rejection (72). Thus, in spite of the innate immune features of macrophages, mechanistic appreciation of their roles in connecting with adaptive immune system will be of interest in the future.
Lots of efforts have been made to dissect the mechanisms of both innate and adaptive immune response so as to prevent chronic rejection and improve allograft survival (73). At present, a remarkable progress has been made in the study of macrophage-related immunoregulation and immunosuppression for the treatment of organ transplant rejection. Inhibiting infiltration or activation of monocytes/macrophages by mycophenolate mofetil (MMF) seems to be a promising therapeutic strategy (74). Indeed, depletion of monocytes/macrophages attenuate neutrophils-related tissue damage at the early stage of transplant (75–77), while it may also prevent tissue repair and induction of tolerance at late stage due to loss of reprogrammed macrophages that participate in reconstitution of homeostasis (17, 78). Moreover, chronic rejection mediated by M2-like macrophages is also an ineligible issue. Manipulating macrophage activities and polarization will be of significant challenge in against chronic allograft rejection.
HZ initially collected literatures and primarily drafted the article. ZL performed an intensive revision of the manuscript. WL contributed to the conceptual design and carried out critical revision and finalization of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledged the Nature Science Foundation of the Science and Technology Bureau of Jilin Province (Li 20190201227JC), the innovation capacity building fund of The Development and Reform Commission of Jilin Province (Li 2019C015), and NSFC regional innovation and Development Fund (U20A20360).
1. Hirao H, Dery KJ, Kageyama S, Nakamura K, Kupiec-Weglinski JW. Heme oxygenase-1 in liver transplant ischemia-reperfusion injury: from bench-to-bedside. Free Radic Biol Med. (2020) 157:75–82. doi: 10.1016/j.freeradbiomed.2020.02.012
2. Loupy A, Lefaucheur C. Antibody-mediated rejection of solid-organ allografts. N Engl J Med. (2018) 379:1150–60. doi: 10.1056/NEJMra1802677
3. Schofield ZV, Woodruff TM, Halai R, Wu MC, Cooper MA. Neutrophils–a key component of ischemia-reperfusion injury. Shock. (2013) 40:463–70. doi: 10.1097/SHK.0000000000000044
4. Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat Commun. (2019) 10:1076. doi: 10.1038/s41467-019-09046-8
5. Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant. (2011) 11:1563–9. doi: 10.1111/j.1600-6143.2011.03579.x
6. Yue S, Rao J, Zhu J, Busuttil RW, Kupiec-Weglinski JW, Lu L, et al. Myeloid PTEN deficiency protects livers from ischemia reperfusion injury by facilitating M2 macrophage differentiation. J Immunol. (2014) 192:5343–53. doi: 10.4049/jimmunol.1400280
7. Khan SQ, Guo L, Cimbaluk DJ, Elshabrawy H, Faridi MH, Jolly M, et al. A small molecule β2 integrin agonist improves chronic kidney allograft survival by reducing leukocyte recruitment and accompanying vasculopathy. Front Med. (2014) 1:45. doi: 10.3389/fmed.2014.00045
8. Blériot C, Chakarov S, Ginhoux F. Determinants of resident tissue macrophage identity and function. Immunity. (2020) 52:957–70. doi: 10.1016/j.immuni.2020.05.014
9. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. (2008) 13:453–61. doi: 10.2741/2692
10. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. (2010) 327:656–61. doi: 10.1126/science.1178331
11. Luo Y, Shao L, Chang J, Feng W, Liu YL, Cottler-Fox MH, et al. M1 and M2 macrophages differentially regulate hematopoietic stem cell self-renewal and ex vivo expansion. Blood Adv. (2018) 2:859–70. doi: 10.1182/bloodadvances.2018015685
12. Husted TL, Blanchard J, Schuster R, Shen H, Lentsch AB. Potential role for IL-23 in hepatic ischemia/reperfusion injury. Inflamm Res. (2006) 177-178. doi: 10.1007/s00011-006-0073-1
13. Li L, Huang L, Vergis AL, Ye H, Bajwa A, Narayan V, et al. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest. (2010) 120:331–42. doi: 10.1172/JCI38702
14. Jing X, Korchagina AA, Shein SA, Muraoka WT, Koroleva E, Tumanov AV. IL-23 contributes to Campylobacter jejuni-induced intestinal pathology via promoting IL-17 and IFNγ responses by innate lymphoid cells. Front Immunol. (2021) 11:579615. doi: 10.3389/fimmu.2020.579615
15. Hou Y, Zhu L, Tian H, Sun HX, Wang R, Zhang L, et al. IL-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein Cell. (2018) 9:1027–38. doi: 10.1007/s13238-018-0505-z
16. Chen X, Huang Y, Wang D, Dong N, Du X. PJ34, a PARP1 inhibitor, attenuates acute allograft rejection after murine heart transplantation via regulating the CD4+ T lymphocyte response. Transpl Int. (2020) doi: 10.1111/tri.13809
17. Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity. (2015) 42:145–58. doi: 10.1016/j.immuni.2014.12.020
18. Kierdorf K, Prinz M, Geissmann F, Gomez Perdiguero E. Development and function of tissue resident macrophages in mice. Semin Immunol. (2015) 27:369–78. doi: 10.1016/j.smim.2016.03.017
19. Cole AP, Hoffmeyer E, Chetty SL, Cruz-Cruz J, Hamrick F, Youssef O, et al. Microglia in the brain tumor microenvironment. Adv Exp Med Biol. (2020) 1273:197–208. doi: 10.1007/978-3-030-49270-0_11
20. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
21. Yue S, Zhou H, Wang X, Busuttil RW, Kupiec-Weglinski JW, Zhai Y. Prolonged ischemia triggers necrotic depletion of tissue-resident macrophages to facilitate inflammatory immune activation in liver ischemia reperfusion injury. J Immunol. (2017) 198:3588–95. doi: 10.4049/jimmunol.1601428
22. Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain Kupffer cell identity. Immunity. (2019) 51:655–70.e8. doi: 10.1016/j.immuni.2019.09.002
23. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gélineau A, et al. Impaired Kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity. (2020) 53:627–40.e5. doi: 10.1016/j.immuni.2020.06.003
24. Devisscher L, Scott CL, Lefere S, Raevens S, Bogaerts E, Paridaens A, et al. Non-alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell Immunol. (2017) 322:74–83. doi: 10.1016/j.cellimm.2017.10.006
25. Rao J, Yue S, Fu Y, Zhu J, Wang X, Busuttil RW, et al. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am J Transplant. (2014) 14:1552–61. doi: 10.1111/ajt.12711
26. Sosa RA, Terry AQ, Kaldas FM, Jin YP, Rossetti M, Ito T, et al. Disulfide high-mobility group box 1 drives ischemia-reperfusion injury in human liver transplantation. Hepatology. (2020) doi: 10.1002/hep.31324. [Epub ahead of print].
27. Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: toll-like receptors. Free Radic Biol Med. (2010) 48:1121–32. doi: 10.1016/j.freeradbiomed.2010.01.006
28. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91 doi: 10.1016/j.immuni.2012.12.001
29. Jin D, Lu T, Ni M, Wang H, Zhang J, Zhong C, et al. Farnesoid X receptor activation protects liver from ischemia/reperfusion injury by up-regulating small heterodimer partner in Kupffer cells. Hepatol Commun. (2020) 4:540–54. doi: 10.1002/hep4.1478
30. Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, et al. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods. (2005) 2:419–26. doi: 10.1038/nmeth762
31. Devey L, Ferenbach D, Mohr E, Sangster K, Bellamy CO, Hughes J, et al. Tissue-resident macrophages protect the liver from ischemia reperfusion injury via a heme oxygenase-1-dependent mechanism. Mol Ther. (2009) 17:65–72. doi: 10.1038/mt.2008.237
32. Khan F, Sar A, Gonul I, Benediktsson H, Doulla J, Yilmaz S, et al. Graft inflammation and histologic indicators of kidney chronic allograft failure: low-expressing interleukin-10 genotypes cannot be ignored. Transplantation. (2010) 90:630–8. doi: 10.1097/TP.0b013e3181ea391e
33. Nakamura K, Kageyama S, Kupiec-Weglinski JW. The evolving role of neutrophils in liver transplant ischemia-reperfusion injury. Curr Transplant Rep. (2019) 6:78–89. doi: 10.1007/s40472-019-0230-4
34. Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. (2011) 3:a005058. doi: 10.1101/cshperspect.a005058
35. Dick MK, Miao JH, Limaiem F. Histology, Fibroblast. Treasure Island, FL: StatPearls Publishing. (2020). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK541065/
36. Feghali CA, Wright TM. Cytokines in acute and chronic inflammation. Front Biosci. (1997) 2:d12–26. doi: 10.2741/A171
37. Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. (2014) 5:123. doi: 10.3389/fphar.2014.00123
38. Janik MK, Kruk B, Szczepankiewicz B, Kostrzewa K, Raszeja-Wyszomirska J, Górnicka B, et al. Measurement of liver and spleen stiffness as complementary methods for assessment liver fibrosis in autoimmune hepatitis. Liver Int. (2020) 41:348–56. doi: 10.1016/S0168-8278(20)31449-5
39. Iritani S, Akuta N, Kawamura Y, Kajiwara A, Kasuya K, Fujiyama S, et al. Non-invasive predictors of prognosis of Asian patients with histopathologically-confirmed lean nonalcoholic fatty liver disease. BMC Gastroenterol. (2020) 20:368. doi: 10.1186/s12876-020-01509-3
40. Helgeson ES, Mannon R, Grande J, Gaston RS, Cecka MJ, Kasiske BL, et al. i-IFTA and chronic active T cell-mediated rejection: a tale of 2 (DeKAF) cohorts. Am J Transplant. (2020) doi: 10.1111/ajt.16352. [Epub ahead of print].
41. Coelho S, Ortíz F, Gelpi R, Koskinen P, Porta N, Bestard O, et al. Sterile leukocyturia is associated with interstitial fibrosis and tubular atrophy in kidney allograft protocol biopsies. Am J Transplant. (2014) 14:908–15. doi: 10.1111/ajt.12639
42. Denic A, Elsherbiny H, Mullan AF, Leibovich BC, Thompson RH, Ricaurte Archila L, et al. Larger nephron size and nephrosclerosis predict progressive CKD and mortality after radical nephrectomy for tumor and independent of kidney function. J Am Soc Nephrol. (2020) 31:2642–52. doi: 10.1681/ASN.2020040449
43. Clotet-Freixas S, McEvoy CM, Batruch I, Pastrello C, Kotlyar M, Van JAD, et al. Extracellular matrix injury of kidney allografts in antibody-mediated rejection: a proteomics study. J Am Soc Nephrol. (2020) 31:2705–24. doi: 10.1681/ASN.2020030286
44. Kaul AM, Goparaju S, Dvorina N, Iida S, Keslar KS, de la Motte CA, et al. Acute and chronic rejection: compartmentalization and kinetics of counterbalancing signals in cardiac transplants. Am J Transplant. (2015) 15:333–45. doi: 10.1111/ajt.13014
45. Toki D, Zhang W, Hor KL, Liuwantara D, Alexander SI, Yi Z, et al. The role of macrophages in the development of human renal allograft fibrosis in the first year after transplantation. Am J Transplant. (2014) 14:2126–36. doi: 10.1111/ajt.12803
46. Itoh Y, Saitoh M, Miyazawa K. Smad3-STAT3 crosstalk in pathophysiological contexts. Acta Biochim Biophys Sin. (2018) 50:82–90. doi: 10.1093/abbs/gmx118
47. Wang S, Meng XM, Ng YY, Ma FY, Zhou S, Zhang Y, et al. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget. (2016) 7:8809–22. doi: 10.18632/oncotarget.6604
48. Wang YY, Jiang H, Pan J, Huang X-R, Wang Y-C, Huang H-F, et al. Macrophage-to-myofibroblast transition contributes to interstitial fibrosis in chronic renal allograft injury. J Am Soc Nephrol. (2017) 28:2053–67. doi: 10.1681/ASN.2016050573
49. Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science. (1996) 272:735–8. doi: 10.1126/science.272.5262.735
50. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. (2012) 143:1158–72. doi: 10.1053/j.gastro.2012.09.008
51. Wu C, Zhao Y, Xiao X, Fan Y, Kloc M, Liu W, et al. Graft-infiltrating macrophages adopt an M2 phenotype and are inhibited by purinergic receptor P2X7 antagonist in chronic rejection. Am J Transplant. (2016) 16:2563–73. doi: 10.1111/ajt.13808
52. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. (2012) 18:1028–40. doi: 10.1038/nm.2807
53. Ulloa L, Doody J, Massagué J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. (1999) 397:710–3. doi: 10.1038/17826
54. Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. (1999) 104:777–85. doi: 10.1172/JCI7325
55. Rahmani M, Cruz RP, Granville DJ, McManus BM. Allograft vasculopathy versus atherosclerosis. Circ Res. (2006) 99:801–15. doi: 10.1161/01.RES.0000246086.93555.f3
56. Luo Z, Liao T, Zhang Y, Zheng H, Sun Q, Han F, et al. Triptolide attenuates transplant vasculopathy through multiple pathways. Front Immunol. (2020) 11:612. doi: 10.3389/fimmu.2020.00612
57. Mitchell RN, Libby P. Vascular remodeling in transplant vasculopathy. Circ Res. (2007) 100:967–78. doi: 10.1161/01.RES.0000261982.76892.09
58. Shetty M, Chowdhury YS. Heart Transplantation Allograft Vasculopathy. Treasure Island, FL: StatPearls Publishing (2020).
59. Lin FY, Shih CM, Huang CY, Tsai YT, Loh SH, Li CY, et al. Dipeptidyl peptidase-4 inhibitor decreases allograft vasculopathy via regulating the functions of endothelial progenitor cells in normoglycemic rats. Cardiovasc Drugs Ther. (2020) doi: 10.1007/s10557-020-07013-w. [Epub ahead of print].
60. Merola J, Jane-Wit DD, Pober JS. Recent advances in allograft vasculopathy. Curr Opin Organ Transplant. (2017) 22:1–7. doi: 10.1097/MOT.0000000000000370
61. Chih S, Chong AY, Mielniczuk LM, Bhatt DL, Beanlands RS. Allograft vasculopathy: the Achilles' heel of heart transplantation. J Am Coll Cardiol. (2016) 68:80–91. doi: 10.1016/j.jacc.2016.04.033
62. Zarjou A, Guo L, Sanders PW, Mannon RB, Agarwal A, George JF. A reproducible mouse model of chronic allograft nephropathy with vasculopathy. Kidney Int. (2012) 82:1231–5. doi: 10.1038/ki.2012.277
63. Croker BP, Clapp WL, Abu Shamat AR, Kone BC, Peterson JC. Macrophages and chronic renal allograft nephropathy. Kidney Int Suppl. (1996) 57:S42–9.
64. Valantine HA. Cardiac allograft vasculopathy: central role of endothelial injury leading to transplant “atheroma”. Transplantation. (2003) 76:891–9. doi: 10.1097/01.TP.0000080981.90718.EB
65. Sun HJ, Zhou T, Wang Y, Fu YW, Jiang YP, Zhang LH, et al. Macrophages and T lymphocytes are the predominant cells in intimal arteritis of resected renal allografts undergoing acute rejection. Transpl Immunol. (2011) 25:42–8. doi: 10.1016/j.trim.2011.04.002
66. Matheson PJ, Dittmer ID, Beaumont BW, Merrilees MJ, Pilmore HL. The macrophage is the predominant inflammatory cell in renal allograft intimal arteritis. Transplantation. (2005) 79:1658–62. doi: 10.1097/01.TP.0000167099.51275.EC
67. Kozakowski N, Böhmig GA, Exner M, Soleiman A, Huttary N, Nagy-Bojarszky K, et al. Monocytes/macrophages in kidney allograft intimal arteritis: no association with markers of humoral rejection or with inferior outcome. Nephrol Dial Transplant. (2009) 24:1979–86. doi: 10.1093/ndt/gfp045
68. Kitchens WH, Chase CM, Uehara S, Cornell LD, Colvin RB, Russell PS, et al. Macrophage depletion suppresses cardiac allograft vasculopathy in mice. Am J Transplant. (2007) 7:2675–82. doi: 10.1111/j.1600-6143.2007.01997.x
69. Haas M, Loupy A, Lefaucheur C, Mengel M, et al. The Banff 2017 Kidney Meeting Report: revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant. (2018) 18:293–307. doi: 10.1111/ajt.14625
70. Chu Z, Sun C, Sun L, Feng C, Yang F, Xu Y, et al. Primed macrophages directly and specifically reject allografts. Cell Mol Immunol. (2020) 17:237–46. doi: 10.1038/s41423-019-0226-0
71. Chu Z, Feng C, Sun C, Xu Y, Zhao Y. Primed macrophages gain long-term specific memory to reject allogeneic tissues in mice. Cell Mol Immunol. (2020) doi: 10.1038/s41423-020-00521-7. [Epub ahead of print].
72. Peng X, Yong Z, Xiaoyan W, Yuanshan C, Guangzhu W, Xuehuan L. Mechanism of graft damage caused by NTPDase1-activated Macrophages in acute antibody-mediated rejection. Transplant Proc. (2021) 53:436–42. doi: 10.1016/j.transproceed.2020.06.033
73. Moreau A, Varey E, Anegon I, Cuturi MC. Effector mechanisms of rejection. Cold Spring Harb Perspect Med. (2013) 3:a015461. doi: 10.1101/cshperspect.a015461
74. Allison AC, Eugui EM. Mechanisms of action of mycophenolate mofetil in preventing acute and chronic allograft rejection. Transplantation. (2005) 80(Suppl. 2):S181–90. doi: 10.1097/01.tp.0000186390.10150.66
75. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
76. Broichhausen C, Riquelme P, Geissler EK, Hutchinson JA. Regulatory macrophages as therapeutic targets and therapeutic agents in solid organ transplantation. Curr Opin Organ Transplant. (2012) 17:332–42. doi: 10.1097/MOT.0b013e328355a979
77. Jose MD, Ikezumi Y, van Rooijen N, Atkins RC, Chadban SJ. Macrophages act as effectors of tissue damage in acute renal allograft rejection. Transplantation. (2003) 76:1015–22. doi: 10.1097/01.TP.0000083507.67995.13
Keywords: solid organ transplantation, chronic allograft rejection, macrophage, IRI, inflammation, ECM, fibrosis, vasculopathy
Citation: Zhang H, Li Z and Li W (2021) M2 Macrophages Serve as Critical Executor of Innate Immunity in Chronic Allograft Rejection. Front. Immunol. 12:648539. doi: 10.3389/fimmu.2021.648539
Received: 31 December 2020; Accepted: 10 February 2021;
Published: 17 March 2021.
Edited by:
Hao Wang, Tianjin Medical University General Hospital, ChinaReviewed by:
Shintaro Yagi, Kanazawa University, JapanCopyright © 2021 Zhang, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Li, d2VpbGk4ODhAamx1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.