 David E. Ochayon
David E. Ochayon Stephen N. Waggoner
Stephen N. Waggoner- 1Center for Autoimmune Genomics and Etiology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
- 2Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, United States
Cytokines are soluble and membrane-bound factors that dictate immune responses. Dogmatically, cytokines are divided into families that promote type 1 cell-mediated immune responses (e.g., IL-12) or type 2 humoral responses (e.g., IL-4), each capable of antagonizing the opposing family of cytokines. The discovery of additional families of cytokines (e.g., IL-17) has added complexity to this model, but it was the realization that immune responses frequently comprise mixtures of different types of cytokines that dismantled this black-and-white paradigm. In some cases, one type of response may dominate these mixed milieus in disease pathogenesis and thereby present a clear therapeutic target. Alternatively, synergistic or blended cytokine responses may obfuscate the origins of disease and perplex clinical decision making. Most immune cells express receptors for many types of cytokines and can mediate a myriad of functions important for tolerance, immunity, tissue damage, and repair. In this review, we will describe the unconventional effects of a variety of cytokines on the activity of a prototypical type 1 effector, the natural killer (NK) cell, and discuss how this may impact the contributions of these cells to health and disease.
Introduction
Cytokines serve as messengers of the immune system, dictating the functional differentiation of immune cells to suit the nature of threats to health (1). The dichotomy between cell-mediated responses against intracellular viruses or bacteria and the need for humoral responses against parasitic infection is dictated by the cytokine milieu (2). Within these dual arms of the immune response, cytokines fine tune the immune response, for example, by determining isotype class switching (3). The capacity of innate immune cells to recruit effector cells, process and present antigens, trigger tolerogenic responses, and promote tissue repair are all determined by and executed via release of cytokines (4, 5).
The orthodox dogma of cytokine responses suggests a dichotomy between type 1 and type 2 cytokines, meaning that type 1 responses will counteract type 2 responses and vice versa (6, 7). While the prototypical type 1 cytokine IL-12 promotes IFN-γ expression, the type 2 cytokine IL-4 can suppress this response (7). Nevertheless, IL-4 can paradoxically promote IFN-γ expression and memory responses in CD8 cytotoxic T and NK cells (8, 9). In this review, we aim to elucidate some of the complexity of cytokine signaling and its function during viral infection.
Natural killer (NK) cells are part of the innate immune compartment with a fundamental role in combating viral infections and eliminating tumor cells (10). NK cells kill targets rapidly via release of perforin- and granzyme-containing cytolytic granules, or more slowly via death receptor (e.g. FasL) interactions with target cells. At various points during the immune response, NK cells are vital sources of cytokines (11). These include the hallmark cytokine interferon-gamma (IFN-γ) as well as pro-inflammatory mediators like tumor necrosis factor alpha (TNF-α) and granulocyte-macrophage colony-stimulating factor (GM-CSF) (12, 13). There is also evidence that NK cells can produce type 2 (e.g. IL-5, IL-13) and immunoregulatory cytokines (e.g. IL-10, TGF-β). This variety of functional contributions of NK cells is likely dictated by the cytokine milieu to which they are exposed during an immune response. How cells translate signals from this complex cytokine milieu into concerted functional activity remains incompletely defined. The following review aims to shed light on NK-cell responses to unconventional cytokines.
The IL-12 Family of Cytokines
NK cells are highly responsive to IL-12 which triggers IFN-γ production via STAT4 phosphorylation and Tbet transcriptional activity (14, 15). Bioactive IL-12 is heterodimer of the IL-12p40 and IL-12p35 subunits. The role of IL-12 in NK-cell biology has been extensively reviewed elsewhere (11), and will not be discussed in detail in this review.
IL-23 is a closely related IL-12 family member; it is a heterodimer of IL-12p40 and IL-12p19 subunits. IL-23 is released from activated myeloid cells, including dendritic cells and macrophages, which are distributed in peripheral tissues such as skin, lung and intestine (16). IL-23 shares a resemblance with IL-12 in its ability to induce the release of IFN-γ from NK cells. In addition to phosphorylation of STAT4, IL-23 also triggers phosphorylation of STAT3 (16). However, in mucosal tissues IL-23 mediates a wide variety of immune responses (17). In T cells, it supports the proliferation and activation of Th17 cells, which express IL-17 and IL-22 (18). IL-23 plays a similar role in triggering production of these cytokines by group 3 innate lymphoid cells (ILC3) (19).
IL-17 and IL-22 play key roles in mucosal immunology (16).
In humans, CD56bright NK cells exhibit increased expression of IL-23R in comparison to CD56dim NK cells. Consistent with this expression, CD56bright compared to CD56dim NK cells showed superior ability to express IFN-γ in response to IL-23 stimulation (20). In addition, IL-23 is critical during T. gondii infection for promoting NK cell activation and expression of IFN-γ (Figure 1A). With the growing appreciation of IL-23 role in mucosal and autoimmune pathologies (21, 22), additional studies are required to fully understand the role of IL-23 in NK-cell responses.
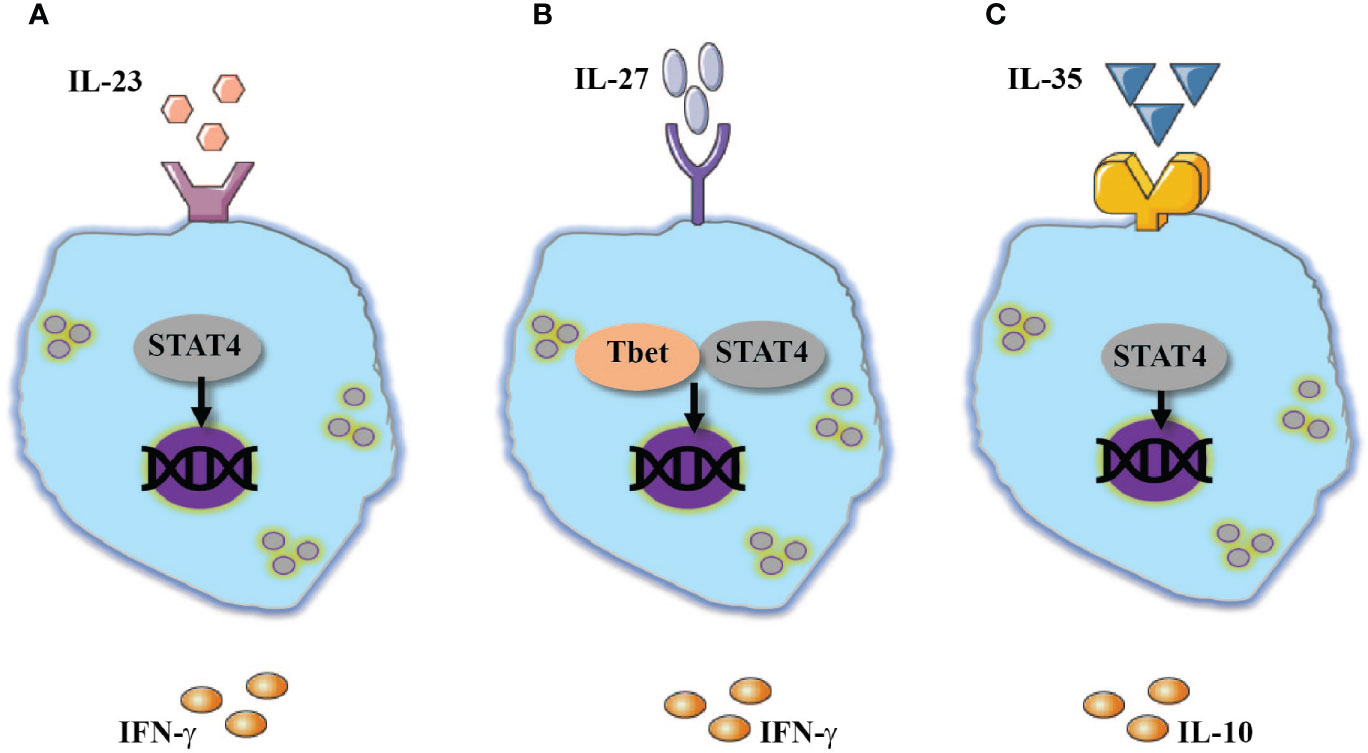
Figure 1 Additional members of IL-12 family cytokines modulate NK-cell responses. Activation of NK cells are mediated by: (A) IL-23 or (B) IL-27 through activation of STAT4 and Tbet which resulted in IFN-γ expression. (C) An opposite effect to the above-mentioned cytokines was documented by IL-35 which also utilize STA4 and promote the expression of IL-10.
Interleukin-27 (IL-27) is a heterodimeric cytokine in the IL-12 family comprised of Epstein-Barr virus–induced gene 3 (EBI3) and IL-27p28. The IL-27 signal is transduced by its receptor which contains gp130 and WSX1 subunits (23). Macrophages, microglia, dendritic cells, and inflammatory monocytes are the main sources of IL-27. Endothelial and epithelial cells also express IL-27 (23). The role of IL-27 in T cell biology is diverse; initial studies showed that IL-27 mediates the expression of Tbet, STAT4 and IFN-γ in naïve T cells (23, 24). However, recent studies have demonstrated that IL-27 can also suppress Th1- inflammatory responses by promoting the survival and proliferation of T regulatory cells and enhancing their ability to express IL-10 (25, 26). In a similar fashion to its effect on Th1 cells, IL-27 is suggested to enhance the inflammatory responses of NK cells. Indeed, human NK cells stimulated with IL-27 showed diverse cytokine release which included increased release of IL-10 and IFN-γ (27). Moreover, IL-27 stimulated NK cells were shown to have increased cytolytic function associated with elevated expression of NKp46 and Tbet (28). Combined Stimulation of NK cells with IL-27 in combination with IL-18 or IL-15 enhanced the effects of IL-27 on NK cell activity (28, 29). In vivo, IL-27 mediates early NK cells responses during viral infection (30, 31). In an experimental model of influenza, deletion of IL-27R or EBI3 resulted in reduced expression of IFN-γ and repressed degranulation responses by NK cells (31). The reduced response of NK cells was linked to increased susceptibility to viral infection. Interestingly and different from human NK cells, in vitro stimulation of mouse NK cells with added IL-27 did not enhance IFN-γ expression in IL-12 and IL-18 stimulated NK cells (31). Nevertheless, IL-27 increased the expression of IFN-γ when murine NK cells were stimulated via NKG2D (31).
In addition to these reported stimulatory effects, IL-27 can also suppress NK-cell inflammatory responses via an indirect mechanism. The interaction of human NK cells with IL-27-stimulated monocytes resulted in reduced expression of IFN-γ but did not alter NK-cell cytotoxic responses (32). The inhibition of IFN-γ release was attributed to increased expression of human leukocyte antigen class I histocompatibility antigen, alpha chain E (HLA-E), which was demonstrated to directly interact with NK cells (33).
The above-mentioned studies demonstrate that on one hand, IL-27 possesses a similar function as IL-12 and can promote NK cells activation and assist in anti-microbial responses (Figure 1B). On the other hand, indirectly, IL-27 can downregulate NK-cell responses. Thus, additional studies should clarify under which circumstances does IL-27 activate or suppress NK-cell responses.
IL-35 is another member of the IL-12 family, with structural similarity to IL-27 and IL-23. IL-35 is a heterodimeric cytokine comprised of the IL-12p35 and EBI3 subunits (34). Several T-cell subsets, but most prominently regulatory T cells, express IL-35 (35), which is suggested to suppress inflammatory responses in a variety of cells (34).
In response to various stimuli, mouse NK cells were demonstrated to express EBI3 and IL-35, but not p28, an IL-27 subunit (36). A similar phenomenon was observed in the experimental model of murine cytomegalovirus (MCMV). Post infection NK cells were shown to express EBI3 and subunit p35 but not p28 which is associated with IL-27. Moreover, NK cells derived from EBI3-deficient mice, expressed a lesser amount of IL-10 and showed reduced viral latency (36). Although IL-35 appears to be an immunosuppressive agent, reduced activation of cytotoxic cells such as NK cells and CD8 T cells might be beneficial to the host by limiting tissue injury (37, 38) (Figure 1C).
IL-1 Family of Cytokines
IL-18 is a member of the type 1 cytokine family, formerly known as interferon-γ-inducing factor. Indeed, the combination of IL-18 with IL-12 provokes strong IFN-γ expression in both human and murine NK cells (13). IL-18 activates the NF-κB and p38 MAPK pathways, and the latter enhances the stability of IFN-γ transcripts (39, 40). The combined stimulation of IL-18 and IL-12 also mediates release of additional cytokines such as GM-CSF and TNF-α (13). The role of IL-18 in anti-viral responses in vivo includes support for NK cell expansion and activation (41–44).
IL-33 is one of the most recently discovered members of the IL-1 family (45, 46). Previous studies showed that IL-33 has dual functionality. Intracellularly, IL-33 was demonstrated to interact with chromatin with unknown consequences for cellular biology (47). Extracellularly, IL-33 promotes inflammatory responses as a damage associated molecule, released passively from cells after injury (45). Early studies showed the ability of IL-33 to mediate type 2 responses in basophils, T cells, mast cells and ILC2 cells (45). However, IL-33 also mediated type 1 responses in pre-stimulated macrophages, which resulted in increased expression of TNF-α and IL-1β (48–50). During viral infection, IL-33 is vital for Th1 responses and infection resolution (51). In mouse NK cells, IL-33 enhanced the release of IFN-γ in IL-12 stimulated NK cells (52). In addition, in an experimental model which mimics the injurious effect of smoking damage, IL-33 is released from lung tissue and couples with inflammatory signals caused by influenza virus infection to provoke greater IFN-γ expression than seen in the absence of smoke (53). In addition to its direct effects on NK cells, IL-33 can also enhance the expression of IL-12p40 and thereby indirectly enhance IFN-γ production by NK cells (53). Recent reports showed that IFN-γover-production mediates airway hypersensitivity (AHR) and inflammation which is not affiliated with classic AHR inducers such as neutrophils (54). Smokers and patients with chronic obstructive pulmonary disorder (COPD) or severe asthma exhibit increased susceptibility to respiratory viral infections such as influenza virus, respiratory syncytial virus, and rhinovirus (55–60).
Human NK cells showed a similar response to IL-33 (Figure 2A). IL-33 did mediate the release of IFN-γ, however, when combined with IL-12 or IL-23 it mediated significant increase of IFN-γ expression (61). In a similar fashion to IL-18, IL-33 combined with IL-12 mediated the release of inflammatory cytokines such as TNF-α and GM-CSF (62). Mechanistically, IL-33 enhanced the expression of NK-cell inflammatory cytokines via p38 MAPK signaling pathway and stabilization of IFNG transcripts (52, 62). IL-33 also induced activation of a disintegrin and metalloproteinase (ADAM)-17, which enhances the release of TNF-α from NK cells (62).
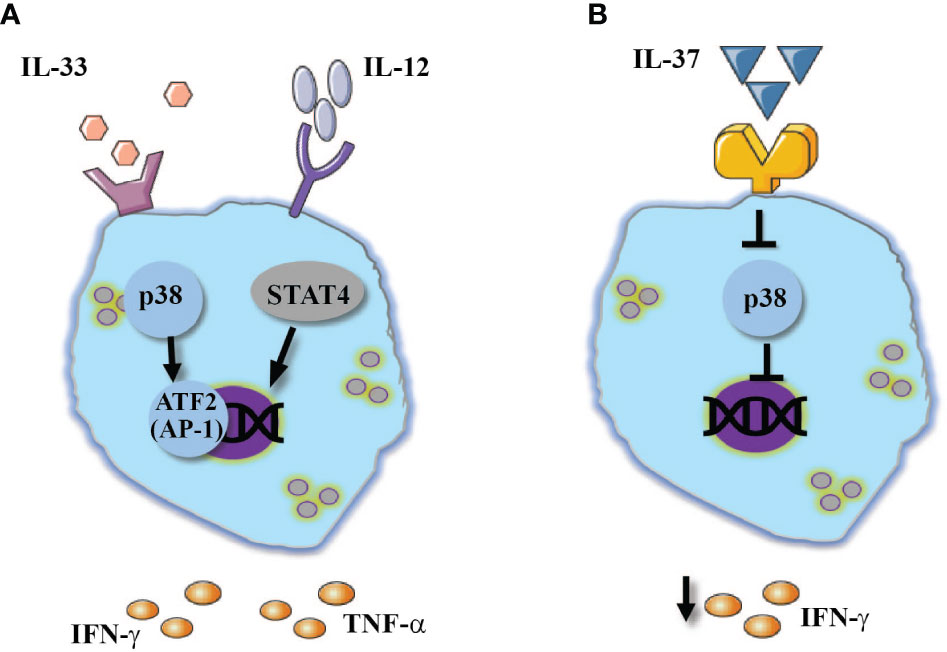
Figure 2 IL-1 family cytokines regulate NK-cell responses. The combined stimulation of IL-12 stimulated NK cell with: (A) IL-33 mediates the activation of p38 pathway which resulted in enhanced IFN-γ and TNF-α expression. (B) In contrast, IL-37 was shown to inhibit p38 MAPK signaling pathway which resulted in reduced IFN-γ expression.
Of note, IL-33 was suggested to have a similar role to IL-18 in the context of modulating NK-cell memory responses. In an experimental model of MCMV infection, an increase in the expression of IL-33 was detected three days after infection (63). Furthermore, the IL-33 receptor ST2 (Il1rl1) was essential for proliferation of Ly49H+ memory NK cells and their ability to eliminate virus-infected cells (63).
In a similar fashion to IL-18 and IL-33, IL-1 also enhances IFN-γ expression in IL-12 stimulated NK cells (64). In humans, CD56bright NK cells exhibit greater sensitivity to IL-1β than CD56dim NK cells (65). Earlier studies showed that IL-1β can stimulate NK cell cytotoxicity (66).
In contrast to the rest of the IL-1 family, the newly discovered IL-37 predominately suppresses innate inflammatory responses (67–69). In hepatocellular carcinoma patients, increased expression of IL-37 within the tumor environment was resulted in increased infiltration of CD57+ NK cells into the tumor as well as a better prognosis (70). A possible explanation of improved NK cell function in the presence of IL-37 could be drawn from Qi et al. (2018). In an experimental model of influenza virus infection, anti-viral treatment combined with IL-37 increased the expression of IL-18 receptor (71), where IL-18 can promote NK-cell cytotoxic responses which in turn contribute to viral clearance (12). Moreover, IL-37 suppresses p38 MAPK signaling pathway (71), which was demonstrated to activate ADAM17 (72). In human NK cells, ADAM17 was demonstrated to shed CD16 from cell membrane (73), thus we can speculate that IL-37 diversely downregulates ADAM17 activation and TNF-α release from NK cells, while assisting in the preservation of CD16 expression, a molecule that plays a key role in ADCC. Thus, it appears that although IL-37 is characterized as an anti-inflammatory cytokine, it promotes NK cells functionality in various pathologies (Figure 2B).
Interferons and IL-15
IL-15 is a common-γ chain cytokine which, unlike other cytokines, can activate NK cells as a trans- or cis-presented membrane bound cytokine (74). IL-15 plays a pivotal role in NK-cell biology (1). The interaction of IL-15 with its receptor via STAT5 phosphorylation mediates the proliferation, differentiation, survival, and activation of NK cells (75). Although IL-15 can activate NK cells on its own, the combination of IL-15 with cytokines such as IL-12, IL-18 or their combination was shown to increase the repertoire of released cytokines and was also demonstrated to mediate cytokine induced memory NK-cell responses (76).
In the context of viral infection, IL-15 is a key mediator in NK cell control of viral replication via cytotoxicity (77). Interestingly, IL-15 serum levels were shown to be associated with pediatric viral bronchiolitis severity, which might indicate that NK cells activated by IL-15 could play a harmful role in the course of viral infection (77).
In a similar fashion to IL-15, type I interferons IFN-α and IFN-β also play key roles in NK cell anti-viral responses. In the absence of type I interferons receptor or its key signaling molecule STAT1, NK cells showed impairment in survival, proliferation and reduced capacity to clear virally infected cells (78). Nonetheless, continuous stimulation with IFN-α or with IL-15 were shown to promote NK-cell exhaustion which resulted in reduced IFN-γ expression and cytotoxicity (78–80).
The IFN family also includes IL-28a/IL-28a and IL-29. The immune responses of these type III interferons are similar to those induced by type I interferons (81). These cytokines signal via IL-28R and IL-10R2 (81). In regard to NK cell responses, type III interferons were shown to activate NK-cell responses in the course of influenza infection and bacterial infection model (82). In the absence of IL-28R, a reduced response of NK cells was detected (83).
Type 2 Cytokines
IL-4 is a common-γ chain cytokine, and it serves as a central cytokine in shaping type 2 responses (84–86). In T cells, it was shown to mediate the differentiation of Naïve T cells to Th2 via signaling activation signal (STAT) 6 and GATA3 (87, 88). In response to IL-4, Th2 cells release additional type 2 cytokines such as IL-13, IL-5 and IL-4 (89, 90). Similarly, IL-4 was shown to attract and activate ILC 2 which resulted in additional release and expression of type 2 cytokines (91). Thus, from the orthodox perspective, IL-4 appears to strictly activate Th2 related responses important in humoral immunity. Yet, IL-4 is also vital for the development of memory CD8 T cells (92, 93) and for enhanced expression of IFN-γ (94).
In vivo, mouse NK cells respond to IL-4 administration with an increased expression of IFN-γ several hours after IL-4 administration. The enhanced expression of IFN-γ levels was related to the expression of STAT6 but not STAT4 (9). In a different study, overexpression of IL-4 by hydrodynamic injection primed splenic and liver NK cells to respond to subsequent stimulation with IL-12 and IL-21, resulting in increased expression of IFN-γ, IL-10 and GMCSF. In addition, NK cells derived from mice that overexpressed IL-4 showed increased levels of granzyme B and elevated cytotoxicity towards YAC-1 cells. Interestingly, overexpression of IL-13, which share similar responses with IL-4, in a similar experimental setting did not show similar results that were induced by IL-4 (95).
In a similar manner to type 1 cytokines, IL-4 was shown to promote increased expression of IFN-γ when it was combined with type 1 cytokines such as IL-12 and IL-2 (96). In this context, the IFN-γ stimulating capacity of IL-4 was independent of STAT6. The ability of IL-4 to support the activation of STAT5 phosphorylation likely mediates IFN-γ expression (97).
In contrast to the aforementioned effects of IL-4 as a NK-cell stimulant, pretreatment of human NK cells with IL-4 prior to stimulation via NKp46 did not enhance cytokine expression (98). Yet, IL-4 stimulation did dampen cytolytic responses of NK cells against targets lacking MHC expression. Lastly, pretreatment of human NK cells with IL-4 prevented their ability to interact with vascular endothelium; this phenomenon is suggested to limit the ability to of NK cells to recruit immune cells upon interaction with endothelial cells (99). The effects of IL-4 on NK cell responses are summarized in Figures 3A, B.
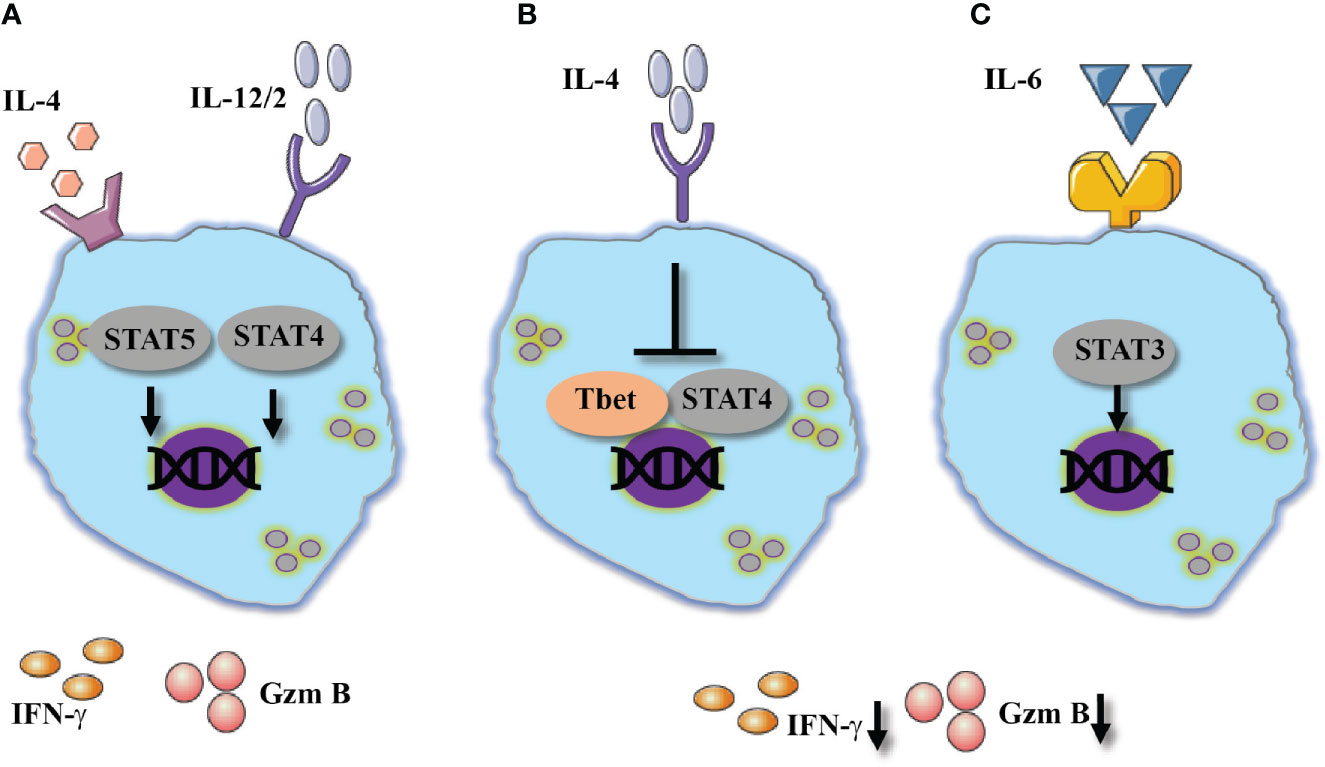
Figure 3 Type 2 cytokines show diverse effect of NK cells activation. Two outcomes in response to IL-4, (A) Alone or combines with 1 cytokines IL-4 mediates NK cells activation which resulted in increased expression of IFN-γ, potentially via STAT5 and STAT4. (B) Suppressive effect of IL-4 resulted in reduced IFN-γ and granzyme B expression suggested by the inhibition of Tbet and STAT4 signaling pathway. (C) IL-6 shows similar suppressive effect via STAT3 signaling pathway.
Of note, a study compared the effect of IL-13 versus IL-4 stimulation on human NK cells. Cells that were stimulated in the presence of IL-13 in comparison to IL-4, showed increased released of IFN-γ (100). No similar effect was detected in model animal, however, the above-mentioned study raised the question whether IL-4 and IL-13 activate similar signaling pathways, since both of these cytokines show a redundant role in pathologies such as asthma and atopic dermatitis.
IL-6 is a pluripotent cytokine that contributes to transcriptional programs for differentiation of regulatory, follicular and IL-17-producing subsets of CD4 T cells (101). Moreover, IL-6 activates a variety of inflammatory responses in hematopoietic cells (102). IL-6 was shown to downregulate IFN-γ expression and cytotoxic responses in CD8 T and NK cells (103, 104). Specifically, human NK cells stimulated in the presence of IL-6 were shown to have reduced expression of perforin and granzyme B, which resulted in reduced cytotoxicity (104). Validation to these findings was observed during the recent COVID-19 pandemic, in which NK cells derived from severe COVID-19 patients, compared to healthy patients showed reduced expression of granzyme A that was associated with IL-6 serum levels, in addition to reduced perforin expression (105).
Mechanistically, IL-6 mediates the phosphorylation of JAK3/STAT3 and the upregulation of suppressor of cytokine signaling 3 (SOCS3) (103). In CD8 T cells, SOCS3 activation was shown to downregulate STAT4 phosphorylation which mediates effector functions (94). Moreover, IL-6 presence was shown to mediate programmed death-ligand (PD-L)1 expression on NK cells, a molecule which is associated with reduced cytotoxicity of NK cells (106). Inhibition of JAK3 or IL-6 blockade (tocilizumab) revoked the suppressive effect of IL-6 and improved NK-cell cytotoxicity. Understanding the kinetics of IL-6 and its full effect of NK cells will provide additional lines of treatment in acute life-threatening viral infections (Figure 3C).
Immunoregulatory Cytokines
IL-10 and TGF-β
Transforming growth factor beta (TGF-β) is a central cytokine in T cell biology. In combination with IL-6, TGF-β activates the transcriptional program for T regulatory and Th17 cells (107). Studies aimed to understand the role of TGF-β in ILC biology showed that TGF-β is also essential for intestinal ILC3 fate and function (108). The interaction of TGF-β with its receptor mediates its signal to mothers against decapentaplegic homolog (SMAD)2 and SMAD3 which in turn promote SMAD4 translocation into the nucleus (109). In the context of cancer immunology, TGF-β was shown to downregulate NK-cell cytotoxic responses in addition to its ability to mediate the release of tumor supporting cytokines such as vascular endothelial growth factor (VEGF)-A (110, 111). Similar results were depicted in human NK cells study, in which the addition of TGF-β to IL-2/15 stimulated NK cells resulted in reduced IFN-γ, granzyme B and CD107a expression, all of which were attributed to the metabolic changes that TGF-β initiates in NK cells. All those effects were reversed when TGF-β receptor was inhibited or knocked down (111, 112).
NK cells are also known to be activated by antibody-dependent cell-mediated cytotoxicity (ADCC) via the interaction of NK cells with antibody-coated target via CD16 (113). In a study performed by Trotta et al., TGF-β was shown to downregulate NK cell activation mediated by CD16 ligation (114). The authors suggested that the inhibitory effect of NK-cell ADCC responses is mediated by SMAD3 which is downstream to TGF-β interaction with its receptor (114).
Thus, it appears that TGF-β possesses the potential of downregulating NK-cell responses. Indeed, high levels of TGF-β were shown in chronic Hepatitis B (HBV) patients. Sun et al. showed NK cells dysfunction and reduced activation markers such as NKG2D and 2B4 were negatively associated with TGF-β levels in HBV patients. The blockade of TGF-β restored the inhibitory effect of TGF-β on NK-cell activation markers and responses (115). TGF-β was shown to protect the host survival in the presence of lymphocytic choriomeningitis virus (LCMV) infection by modulating CD8 T cell responses (116). In the specific case of HBV, although NK cells were shown to assist in viral clearance, they are also suggested to mediate liver injury (117). In this regard, high expression level of TGF-β was reported in COVID-19 patients, and thus it is enticing to examine whether those high levels of TGF-β are harmful or protective. Additional studies will be needed to determine the full spectrum of TGF-β in the context of viral responses.
Interestingly, an unorthodox role of SMAD4 was shown in activating the NK-cell transcriptional program. Specific deletion of SMAD4 (Smad4 f/fNcr1iCre) resulted in increased frequency of ILC1 at the expense of NK cells. Moreover, Smad4 f/fNcr1iCre compared to Smad4 f/f mice showed reduced ability to clear tumor cells and clear virally infected cells (114). The authors associated the important role of SMAD4 in NK cells to TGF-β signaling which was not dependent on TGF-β receptor (114, 118).
A study led by Wang et al. showed that SMAD4 is critical for NK cells homeostasis and maturation, however, the crucial effect of SMAD4 was independent to the role of TGF-β receptor 2 (119). The authors suggested that SMAD4 is mediating its non-canonical functions in cooperation with JunB, as both SMAD4 and JunB were shown to be in interaction with the promoter region of granzyme B (GZMB), which mediates NK cells cytolytic responses (119).
Another cytokine with inhibitory effects is the pleiotropic cytokine IL-10. Numerous studies documented the suppressive effect of IL-10 on inflammatory responses (120). In animal models, IL-10 was shown to modulate T cell responses and its deficiency was resulted in dysregulated T cell responses which led to autoimmunity and intestinal inflammation, demonstrating its critical role in T cell biology (121, 122). On the other hand, IL-10 was shown to moderate harmful inflammatory responses (123). In LCMV infected mice, IL-10 was demonstrated to downregulate the excessive innate and adaptive proinflammatory responses, and thus provides tissue protection at a cost of viral persistence (124).
The specific effect of IL-10 on NK-cell responses shows contradicting evidence. In a similar fashion to the role of IL-10 in T cells, several studies showed that IL-10 suppresses the inflammatory and cytotoxic responses in NK cells. The suppressive role of IL-10 on NK cells effector functions was demonstrated both directly and indirectly. Directly, T regulatory cells were shown to release IL-10 and as a result reduced NK cell cytotoxicity and IFN-γ expression (125). Later it has been demonstrated that IL-10 can downregulate NKG2D expression which mediates NK-cell effector functions (126). Indirectly, viral infections such as MCVM or LCMV were demonstrated to elevate IL-10 expression in myeloid cells like dendritic cells and monocytes (124, 127). As an example, in the experimental model of MCMV, IL-10 prevented NK-cell mediated licensing of dendritic cells, which resulted in reduced CD4 T cell activation and reduced viral clearance (127). Interestingly, NK cells exposure to IL-10 in combination with IL-15, mediated IL-10 release and as a result mediated infection persistence (128).
Surprisingly, IL-10 was also shown to assist in NK-cell functionality and survival during acute MCMV infection. In IL-10Rb deficient mice, reduced frequency of NK cells in the lungs and spleen was observed, which was attributed to elevated apoptosis. Here, IL-10 was shown to prevent NK cell apoptosis which is mediated by acute MCMV infection (129). Moreover, during human HCMV infection, a release of cmvIL‐10, a virokine homologous to human IL-10, was detected. NK cells stimulated with cmvIL‐10 were shown to have induced NK-cell cytotoxic responses and activation receptors expression. cmvIL‐10 was shown to mediate its effect with IL-10a receptor (130).
It appears that both TGF-β and IL-10 play a canonical role in suppressing NK-cell effector responses in order to protect the host from damaging inflammation. However, each of those cytokines was shown to have a non-canonical pathway in which they promote the survival and activation of NK cells. Further studies will clarify the intriguing role of both TGF-β and IL-10 regarding NK cells immune responses (Figures 4 and 5).
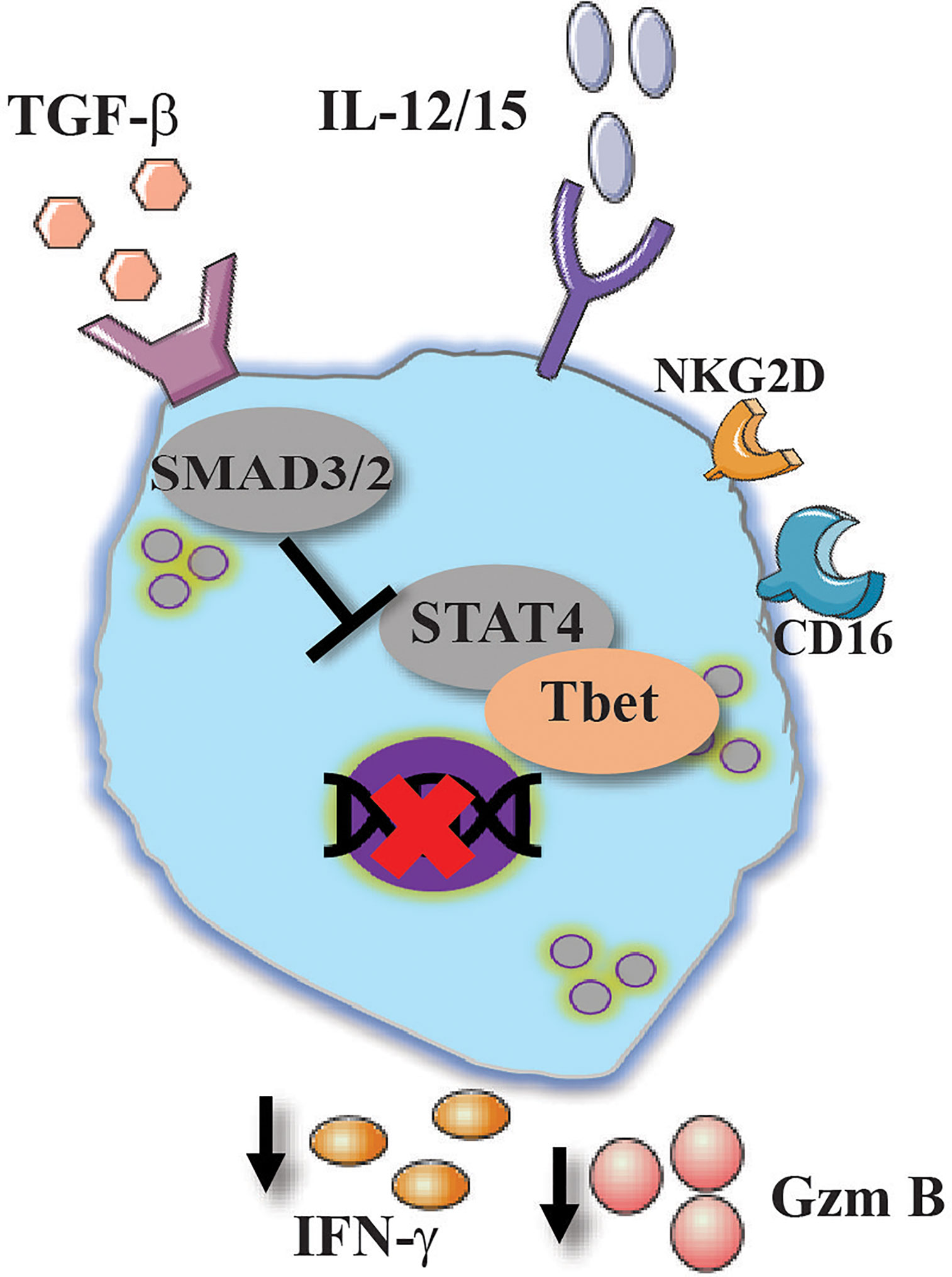
Figure 4 Conventional and unconventional effect of TGF-β on NK cells activation. TGF-β suppress NK cells activation by reducing the expression of CD16, NKG2D, IFN-γ and granzyme B via the canonical pathway by SMAD2/3.
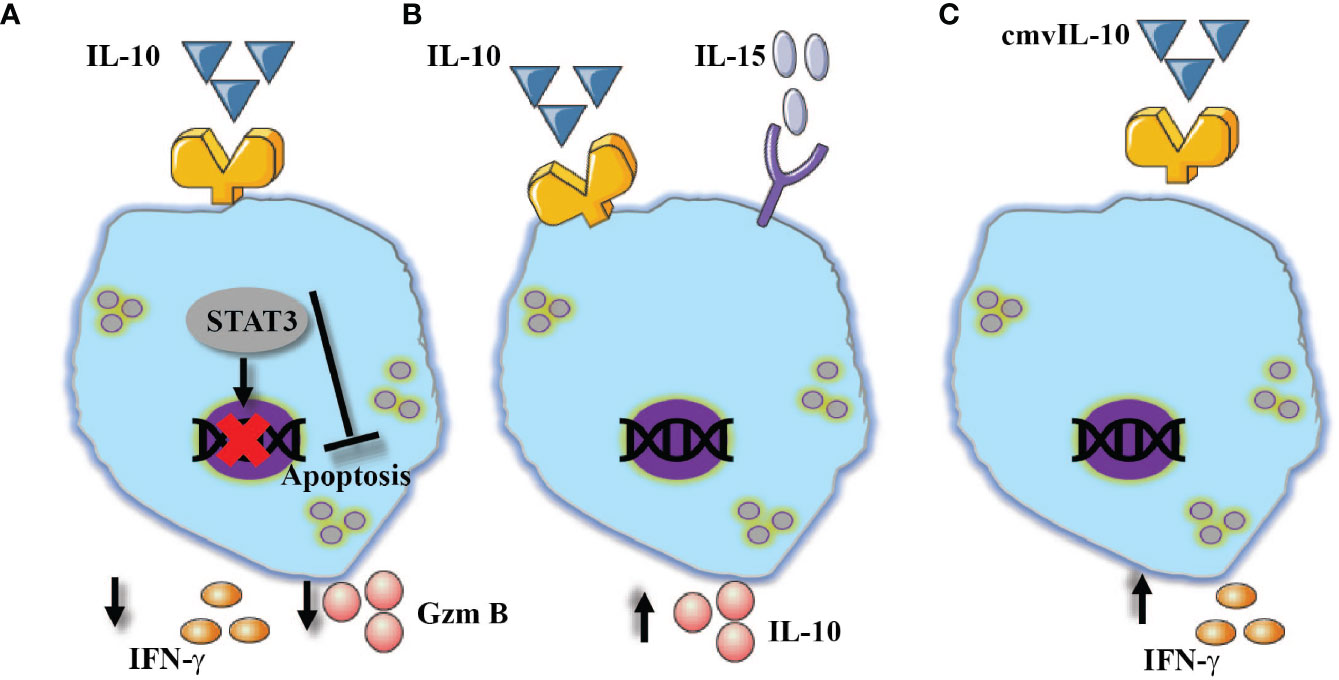
Figure 5 NK cells show diversified responses in the presence of IL-10. (A) Alone, reduced expression of IFN-γ and granzyme B, in addition to its ability to prevent apoptosis via STAT3 signaling pathway. (B) In combination with IL-15, increased expression of IL-10. (C) The virokine cmvIL-10 mediates NK cells activation which resulted in enhanced IFN-γ expression.
Mixed Cytokine Milieus
The majority of pathologies are categorized as type 1, 2 or 3 diseases and patients are being treated according to the dominant phenotype of the disease (131–133). It appears that numerous pathologies which progress from an acute form, transition into a chronic stage and have the potential to aggravate and develop into subsequent pathologies, are characterized by a complex inflammatory milieu. For example, in COPD caused by patient’s smoking and additional environmental factors, the lung tissue is going through injury and remodeling, processes which are characterized by continuous release of IL-33 and IL-4 and TGF-β respectively (134–136). COPD patients suffer from malfunctioning epithelial tissue and thus, are more susceptible to microbial infections (137, 138). COPD exacerbation is mediated by respiratory microbial insults which modulate the release of cytokines such as IL-1β, IL-12 (139). COPD provides an example for a pathology which contains a mixed inflammatory environment.
Another example is atopic dermatitis (AD), which presents a diversity of phenotypes (140, 141). In atopic patients, the acute phase is mediated by type 2 cytokines such as IL-4 and IL-13 and leads to skin barrier impairment (141, 142). The modulation of the skin tissue by the acute phase of AD promotes tissue injury and mediates the release of IL-33 and microbes’ infiltration (e.g., staphylococcus aureus) resulting in release of type 1 cytokines such as IL-12 (143). Thus, the immune system of AD patients is exposed to a complex inflammatory milieu which dictates dysfunctional immune responses, suggested to lead to food allergy, asthma or chronic rhinitis (144). Indeed, in a subsequent disease to AD, asthma (145), which is characterized by mixed inflammatory milieu, NK cells demonstrated a dysregulated phenotype (146). NK cells derived from asthmatic compared to normal patients demonstrated reduced cytotoxicity while showing an increased expression of inflammatory cytokines. dysregulated phenotype, on one hand reduced cytotoxicity while expressing high levels of inflammatory cytokine, a phenomenon that might provide an explanation to the increased susceptibility of asthma patients to viral infection (147), and thus pose NK cells as having a pivotal role in mixed cytokines pathologies. The next section will bring several examples of how NK cells function in a complex inflammatory milieu (Figure 6).
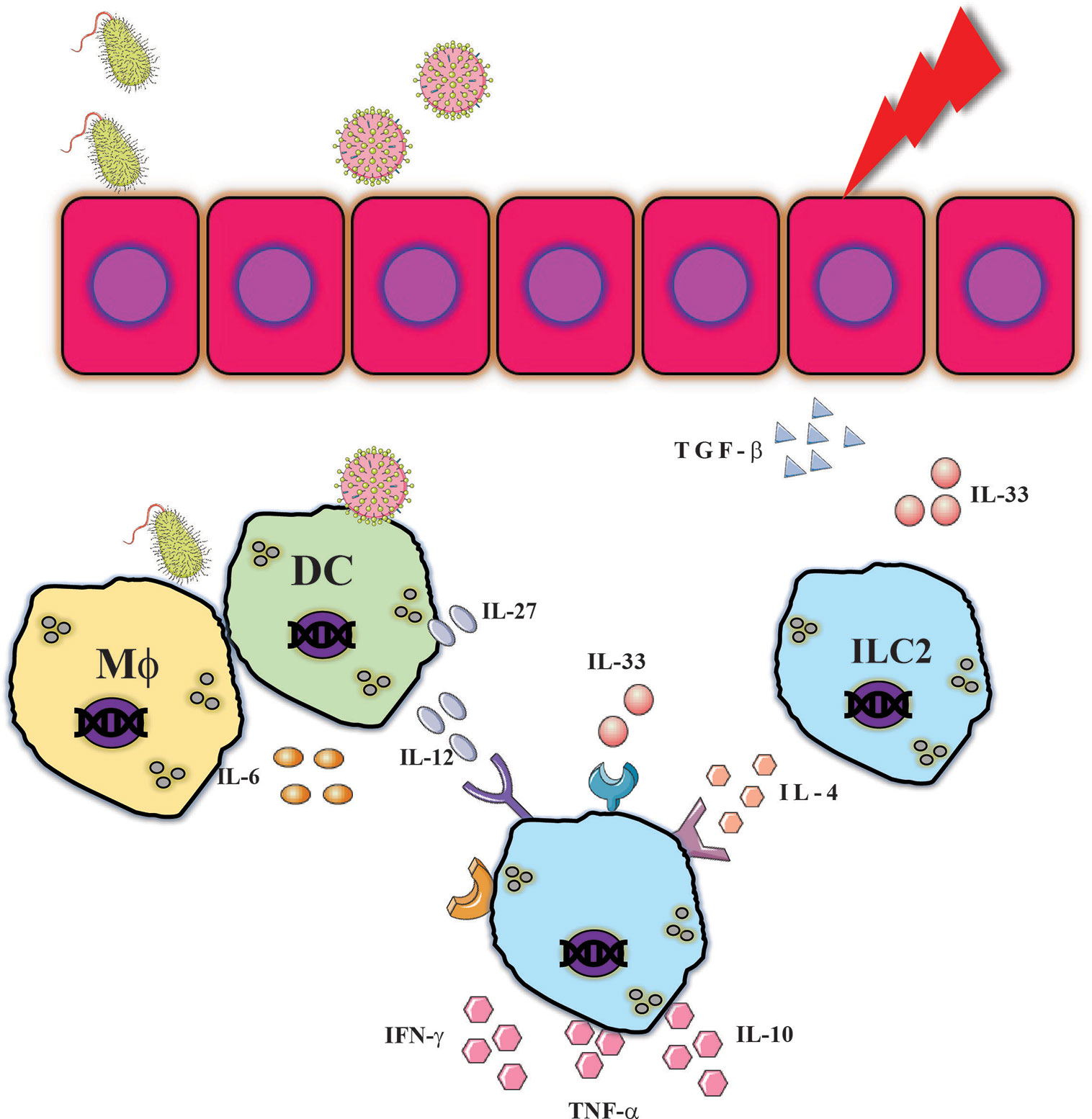
Figure 6 NK cells are exposed to complex inflammatory milieu. Various insults can provide innate immune responses which resulted in release of type 1 and type 2 cytokines which resulted in cytokines such as. IFN-γ, TNF-α or IL-10 which could assist in combating pathogens but can also mediate tissue injury.
NK cells were shown to have a crucial role in the clearance of RSV infection (148). A recent study highlighted the importance of NK cells in disease resolution in the presence of neutralizing antibodies and by performing ADCC (149). However, in the absence of neutralizing antibodies, RSV triggers NK cells inflammatory responses via IL-12, which in turn mediate the release of IFN-γ (150). Excessive expression of IFN-γ was shown to mediate lung injury and inhibit efficient clearance of RSV (151, 152). RSV was demonstrated to trigger the expression of both type 1 (e.g. IL-12) and type 2 (e.g. IL-4) cytokines (153). Enhanced expression of IL-4 in experimental RSV infection enhanced disease phenotype with increased IFN-γ expression but with reduced cytotoxicity (154).
The harmful effect of RSV is not limited to neonates, which lack the humoral response towards RSV. Studies have shown that RSV infection promotes the exacerbation of COPD and asthma (155, 156), which suggests that NK cells fail to perform efficient cytotoxic responses while maintaining high expression of IFN-γ and additional inflammatory cytokines, and thus contribute to tissue injury. However, the specific molecular mechanism which mediates those responses is in need to be clarified.
In contrast to RSV infection, Influenza infection was shown to be controlled on several levels; the activation receptors NKp46 and NKp44 were demonstrated to recognize viral hemagglutinin (HA) which results in viral neutralization (157). In addition, NK cells were shown to clear virally infected cells by cytotoxic responses. As previously mentioned, and as demonstrated by other studies, IL-27 meditates NK-cell viral clearance of influenza (158). In contrast, influenza infection in mice exposed to cigarette smoke, led to increased expression of IL-33 and hyper-activated the NK cells which contributed to excessive tissue damage (53). In addition, IL-4 was shown to reduce the cellular cytotoxicity in response to influenza infection (159), however additional studies are needed to elucidate the role of IL-4 on NK cell responses in the course of influenza infection.
Although the role of TGF-β during respiratory viral infection is suggested to be inhibitory by promoting viral evasion on one hand (160), while on the other hand impairing NK cells responses, an interesting beneficiary effect for the host was observed (161). In an experimental model of asthma, TGF-β was shown to downregulate NK-cell responses during influenza infection which resulted in reduced tissue injury (161). Additional studies should be conducted to examine the beneficial and harmful effects of TGF-β in the presence of respiratory viral infections, and at which time points post infection it can play each of the above-mentioned roles.
In summary, it appears that respiratory viral infections might take advantage of a situation of a mixed inflammatory milieu. In this type of environment, NK cells were shown to have reduced cytotoxic responses, however, when examined in the presence of non-type 1 cytokines, NK cells have the potential to increase the release of IFN-γ as well as additional cytokines, which in turn might enhance tissue damage or recovery from the disease.
Concluding Remarks
Conventional rationale will suggest that type 2 cytokines (e.g., IL-33, IL-4) should counteract type 1 (e.g. IL-12) responses, and thus, NK cells should not be activated in the presence of type 2 cytokines. However, in the present review there are evidences which show unorthodox responses of NK cells to non-type 1 cytokines. In fact, in the majority of the discussed cases, NK cells were shown to express the NK-cell hallmark cytokine IFN-γ, in response to those cytokines alone, or in combination with type 1 cytokines such as IL-12 (11–13).
The common approach to research pathologies is to focus on one molecule, receptor, signaling pathway in one or several disease models. However, pathologies such as asthma, COPD, atopic dermatitis and even the 2020 pandemic COVID-19 are characterized by a heterogenous inflammatory environment which contains diversity of cytokines (143, 162–165), and dictates NK-cell immune responses. For example, according to the conservative cytokine response, NK-cell responses should be suppressed in conditions such as asthma or smoking, however, surprisingly it appears that in the presence of a mixed inflammatory environment NK cells fail to perform cytotoxic cell death, but remain fantastic cytokine producers, which results in exacerbated conditions (146).
A mixed inflammatory milieu is not limited to pathological conditions; at homeostasis, NK cells which are distributed in the blood as well as in tissues, are exposed to tissue remodeling, turnover and injury, all are mediated by the release of heterogenous cytokines which should be assumed to modulate NK cell responses. However, how NK cells function, express cytokines and which cellular or molecular mechanisms govern those responses, needs additional clarification.
Enhanced understanding of how complex inflammatory environments affect NK cells will assist us in developing and utilizing therapeutic approaches, which will modulate NK cells responses in a variety of pathologies in which NK cells play significant roles.
Author Contributions
DO: Conceptualization, investigation, and writing—original draft. SNW: conceptualization, writing—original draft, and study supervision. All authors contributed to the article and approved the submitted version.
Funding
This project was supported in part by Arnold W. Strauss Fellow Award (DO) and other support from the Cincinnati Children’s Research Foundation, a postdoctoral fellowship form American Heart Association (DO), and NIH awards DA038017, AI148080, AR073228, and AI145304.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ADAM, A disintegrin and metalloprotease; COPD, Chronic obstructive pulmonary disorder; GM-CSF, Granulocyte macrophage colony stimulating factor; IFN, Interferon; IL, Interleukin; ILC, innate lymphoid cells; MAPK, Mitogen-activated protein kinase; NK cells, Natural killer cells; STAT, Signal transducer and activator of transcription; RSV, Respiratory Syncytial Virus; TGF, Tumor transforming growth factor; TNF, Tumor Necrosis Factor; VEGF, Vascular endothelial growth factor.
References
1. Wu Y, Tian Z, Wei H. Developmental and Functional Control of Natural Killer Cells by Cytokines. Front Immunol (2017) 8:930:930. doi: 10.3389/fimmu.2017.00930
2. Anthony RM, Rutitzky LI, Urban JF Jr., Stadecker MJ, Gause WC. Protective immune mechanisms in helminth infection. Nat Rev Immunol (2007) 7(12):975–87. doi: 10.1038/nri2199
3. Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol (2008) 26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248
4. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol (2016) 17(7):758–64. doi: 10.1038/ni.3482
5. Gasteiger G, D’Osualdo A, Schubert DA, Weber A, Bruscia EM, Hartl D. Cellular Innate Immunity: An Old Game with New Players. J Innate Immun (2017) 9(2):111–25. doi: 10.1159/000453397
6. Gor DO, Rose NR, Greenspan NS. TH1-TH2: a procrustean paradigm. Nat Immunol (2003) 4(6):503–5. doi: 10.1038/ni0603-503
7. O’Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity (1998) 8(3):275–83. doi: 10.1016/s1074-7613(00)80533-6
8. Oliver JA, Stolberg VR, Chensue SW, King PD. IL-4 acts as a potent stimulator of IFN-gamma expression in CD8+ T cells through STAT6-dependent and independent induction of Eomesodermin and T-bet. Cytokine (2012) 57(1):191–9. doi: 10.1016/j.cyto.2011.10.006
9. Morris SC, Orekhova T, Meadows MJ, Heidorn SM, Yang J, Finkelman FD. IL-4 induces in vivo production of IFN-gamma by NK and NKT cells. J Immunol (2006) 176(9):5299–305. doi: 10.4049/jimmunol.176.9.5299
10. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol (2016) 17(9):1025–36. doi: 10.1038/ni.3518
11. Fehniger TA, Cooper MA. Harnessing NK Cell Memory for Cancer Immunotherapy. Trends Immunol (2016) 37(12):877–88. doi: 10.1016/j.it.2016.09.005
12. Lauwerys BR, Garot N, Renauld JC, Houssiau FA. Cytokine production and killer activity of NK/T-NK cells derived with IL-2, IL-15, or the combination of IL-12 and IL-18. J Immunol (2000) 165(4):1847–53. doi: 10.4049/jimmunol.165.4.1847
13. Peritt D, Robertson S, Gri G, Showe L, Aste-Amezaga M, Trinchieri G. Differentiation of human NK cells into NK1 and NK2 subsets. J Immunol (1998) 161(11):5821–4.
14. Koch A, Raidl M, Lux M, Muller K, Buning H, Humme S, et al. IL-12-induced T-bet expression and IFNgamma release in lymphocytes from asthmatics–role of MAPkinases ERK-1/-2, p38(MAPK) and effect of dexamethasone. Respir Med (2007) 101(6):1321–30. doi: 10.1016/j.rmed.2006.10.010
15. Morinobu A, Gadina M, Strober W, Visconti R, Fornace A, Montagna C, et al. STAT4 serine phosphorylation is critical for IL-12-induced IFN-gamma production but not for cell proliferation. Proc Natl Acad Sci U S A (2002) 99(19):12281–6. doi: 10.1073/pnas.182618999
16. Tang C, Chen S, Qian H, Huang W. Interleukin-23: as a drug target for autoimmune inflammatory diseases. Immunology (2012) 135(2):112–24. doi: 10.1111/j.1365-2567.2011.03522.x
17. Croxford AL, Mair F, Becher B. IL-23: one cytokine in control of autoimmunity. Eur J Immunol (2012) 42(9):2263–73. doi: 10.1002/eji.201242598
18. Puig L. The role of IL 23 in the treatment of psoriasis. Expert Rev Clin Immunol (2017) 13(6):525–34. doi: 10.1080/1744666X.2017.1292137
19. Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med (2011) 208(6):1127–33. doi: 10.1084/jem.20101712
20. Ziblat A, Nunez SY, Raffo Iraolagoitia XL, Spallanzani RG, Torres NI, Sierra JM, et al. Interleukin (IL)-23 Stimulates IFN-gamma Secretion by CD56(bright) Natural Killer Cells and Enhances IL-18-Driven Dendritic Cells Activation. Front Immunol (2017) 8:1959. doi: 10.3389/fimmu.2017.01959
21. Sands BE, Sandborn WJ, Panaccione R, O’Brien CD, Zhang H, Johanns J, et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med (2019) 381(13):1201–14. doi: 10.1056/NEJMoa1900750
22. Gelfand JM, Shin DB, Alavi A, Torigian DA, Werner T, Papadopoulos M, et al. Randomized, Double-Blind, Placebo-Controlled Crossover Study of the Effects of Ustekinumab on Vascular Inflammation in Psoriasis (the VIP-U Trial). J Invest Dermatol (2020) 140(1):85–93 e2. doi: 10.1016/j.jid.2019.07.679
23. Yoshida H, Hunter CA. The immunobiology of interleukin-27. Annu Rev Immunol (2015) 33:417–43. doi: 10.1146/annurev-immunol-032414-112134
24. Iwasaki Y, Fujio K, Okamura T, Yamamoto K. Interleukin-27 in T cell immunity. Int J Mol Sci (2015) 16(2):2851–63. doi: 10.3390/ijms16022851
25. Moon SJ, Park JS, Heo YJ, Kang CM, Kim EK, Lim MA, et al. In vivo action of IL-27: reciprocal regulation of Th17 and Treg cells in collagen-induced arthritis. Exp Mol Med (2013) 45:e46. doi: 10.1038/emm.2013.89
26. Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, et al. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity (2012) 37(3):511–23. doi: 10.1016/j.immuni.2012.06.014
27. Laroni A, Gandhi R, Beynon V, Weiner HL. IL-27 imparts immunoregulatory function to human NK cell subsets. PloS One (2011) 6(10):e26173. doi: 10.1371/journal.pone.0026173
28. Choi YH, Lim EJ, Kim SW, Moon YW, Park KS, An HJ. IL-27 enhances IL-15/IL-18-mediated activation of human natural killer cells. J Immunother Cancer (2019) 7(1):168. doi: 10.1186/s40425-019-0652-7
29. Ziblat A, Domaica CI, Spallanzani RG, Iraolagoitia XL, Rossi LE, Avila DE, et al. IL-27 stimulates human NK-cell effector functions and primes NK cells for IL-18 responsiveness. Eur J Immunol (2015) 45(1):192–202. doi: 10.1002/eji.201444699
30. Harker JA, Wong KA, Dallari S, Bao P, Dolgoter A, Jo Y, et al. Interleukin-27R Signaling Mediates Early Viral Containment and Impacts Innate and Adaptive Immunity after Chronic Lymphocytic Choriomeningitis Virus Infection. J Virol (2018) 92(12):1–18. doi: 10.1128/JVI.02196-17
31. Kumar P, Rajasekaran K, Nanbakhsh A, Gorski J, Thakar MS, Malarkannan S. IL-27 promotes NK cell effector functions via Maf-Nrf2 pathway during influenza infection. Sci Rep (2019) 9(1):4984. doi: 10.1038/s41598-019-41478-6
32. Morandi F, Airoldi I, Pistoia V. IL-27 driven upregulation of surface HLA-E expression on monocytes inhibits IFN-gamma release by autologous NK cells. J Immunol Res (2014) 2014:938561. doi: 10.1155/2014/938561
33. Braud VM, Allan DS, O’Callaghan CA, Soderstrom K, D’Andrea A, Ogg GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature (1998) 391(6669):795–9. doi: 10.1038/35869
34. Sawant DV, Hamilton K, Vignali DA. Interleukin-35: Expanding Its Job Profile. J Interferon Cytokine Res (2015) 35(7):499–512. doi: 10.1089/jir.2015.0015
35. Kourko O, Seaver K, Odoardi N, Basta S, Gee K. IL-27, IL-30, and IL-35: A Cytokine Triumvirate in Cancer. Front Oncol (2019) 9:969. doi: 10.3389/fonc.2019.00969
36. Jensen H, Chen SY, Folkersen L, Nolan GP, Lanier LL. EBI3 regulates the NK cell response to mouse cytomegalovirus infection. Proc Natl Acad Sci U.S.A. (2017) 114(7):1625–30. doi: 10.1073/pnas.1700231114
37. Shao X, Ma J, Jia S, Yang L, Wang W, Jin Z. Interleukin-35 Suppresses Antiviral Immune Response in Chronic Hepatitis B Virus Infection. Front Cell Infect Microbiol (2017) 7:472:472. doi: 10.3389/fcimb.2017.00472
38. Liu S, Zhang Q, Shao X, Wang W, Zhang C, Jin Z. An immunosuppressive function of interleukin-35 in chronic hepatitis C virus infection. Int Immunopharmacol (2017) 50:87–94. doi: 10.1016/j.intimp.2017.06.015
39. Mavropoulos A, Sully G, Cope AP, Clark AR. Stabilization of IFN-gamma mRNA by MAPK p38 in IL-12- and IL-18-stimulated human NK cells. Blood (2005) 105(1):282–8. doi: 10.1182/blood-2004-07-2782
40. Yasuda K, Nakanishi K, Tsutsui H. Interleukin-18 in Health and Disease. Int J Mol Sci (2019) 20(3):6–54. doi: 10.3390/ijms20030649
41. Madera S, Sun JC. Cutting edge: stage-specific requirement of IL-18 for antiviral NK cell expansion. J Immunol (2015) 194(4):1408–12. doi: 10.4049/jimmunol.1402001
42. Hammer Q, Ruckert T, Dunst J, Romagnani C. Adaptive Natural Killer Cells Integrate Interleukin-18 during Target-Cell Encounter. Front Immunol (2017) 8:1976:1976. doi: 10.3389/fimmu.2017.01976
43. Lee AJ, Chen B, Chew MV, Barra NG, Shenouda MM, Nham T, et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J Exp Med (2017) 214(4):1153–67. doi: 10.1084/jem.20160880
44. Carson WE, Dierksheide JE, Jabbour S, Anghelina M, Bouchard P, Ku G, et al. Coadministration of interleukin-18 and interleukin-12 induces a fatal inflammatory response in mice: critical role of natural killer cell interferon-gamma production and STAT-mediated signal transduction. Blood (2000) 96(4):1465–73. doi: 10.1182/blood.V96.4.1465.h8001465_1465_1473
45. Cayrol C, Girard JP. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol Rev (2018) 281(1):154–68. doi: 10.1111/imr.12619
46. Girard JP, Moussion C, Forster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol (2012) 12(11):762–73. doi: 10.1038/nri3298
47. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol (2016) 16(11):676–89. doi: 10.1038/nri.2016.95
48. Gopfert C, Andreas N, Weber F, Hafner N, Yakovleva T, Gaestel M, et al. The p38-MK2/3 Module Is Critical for IL-33-Induced Signaling and Cytokine Production in Dendritic Cells. J Immunol (2018) 200(3):1198–206. doi: 10.4049/jimmunol.1700727
49. Ohno T, Oboki K, Morita H, Kajiwara N, Arae K, Tanaka S, et al. Paracrine IL-33 stimulation enhances lipopolysaccharide-mediated macrophage activation. PloS One (2011) 6(4):e18404. doi: 10.1371/journal.pone.0018404
50. Espinassous Q, Garcia-de-Paco E, Garcia-Verdugo I, Synguelakis M, von Aulock S, Sallenave JM, et al. IL-33 enhances lipopolysaccharide-induced inflammatory cytokine production from mouse macrophages by regulating lipopolysaccharide receptor complex. J Immunol (2009) 183(2):1446–55. doi: 10.4049/jimmunol.0803067
51. Baumann C, Bonilla WV, Frohlich A, Helmstetter C, Peine M, Hegazy AN, et al. T-bet- and STAT4-dependent IL-33 receptor expression directly promotes antiviral Th1 cell responses. Proc Natl Acad Sci U S A (2015) 112(13):4056–61. doi: 10.1073/pnas.1418549112
52. Bourgeois E, Van LP, Samson M, Diem S, Barra A, Roga S, et al. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur J Immunol (2009) 39(4):1046–55. doi: 10.1002/eji.200838575
53. Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N, et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity (2015) 42(3):566–79. doi: 10.1016/j.immuni.2015.02.011
54. Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, et al. High IFN-gamma and low SLPI mark severe asthma in mice and humans. J Clin Invest (2015) 125(8):3037–50. doi: 10.1172/JCI80911
55. Sajjan US. Susceptibility to viral infections in chronic obstructive pulmonary disease: role of epithelial cells. Curr Opin Pulm Med (2013) 19(2):125–32. doi: 10.1097/MCP.0b013e32835cef10
56. Yamaya M. Virus infection-induced bronchial asthma exacerbation. Pulm Med (2012) 2012:834826. doi: 10.1155/2012/834826
57. Busse WW, Lemanske RF Jr, Gern JE. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet (2010) 376(9743):826–34. doi: 10.1016/S0140-6736(10)61380-3
58. Motz GT, Eppert BL, Wortham BW, Amos-Kroohs RM, Flury JL, Wesselkamper SC, et al. Chronic cigarette smoke exposure primes NK cell activation in a mouse model of chronic obstructive pulmonary disease. J Immunol (2010) 184(8):4460–9. doi: 10.4049/jimmunol.0903654
59. Wedzicha JA. Role of viruses in exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc (2004) 1(2):115–20. doi: 10.1513/pats.2306030
60. Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med (2004) 164(20):2206–16. doi: 10.1001/archinte.164.20.2206
61. Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol (2008) 20(8):1019–30. doi: 10.1093/intimm/dxn060
62. Ochayon DE, Ali A, Alarcon PC, Krishnamurthy D, Kottyan LC, Borchers MT, et al. IL-33 promotes type 1 cytokine expression via p38 MAPK in human NK cells. J Leukoc Biol (2020) 107(4):663–71. doi: 10.1002/JLB.3A0120-379RR
63. Nabekura T, Girard JP, Lanier LL. IL-33 receptor ST2 amplifies the expansion of NK cells and enhances host defense during mouse cytomegalovirus infection. J Immunol (2015) 194(12):5948–52. doi: 10.4049/jimmunol.1500424
64. Hunter CA, Timans J, Pisacane P, Menon S, Cai G, Walker W, et al. Comparison of the effects of interleukin-1 alpha, interleukin-1 beta and interferon-gamma-inducing factor on the production of interferon-gamma by natural killer. Eur J Immunol (1997) 27(11):2787–92. doi: 10.1002/eji.1830271107
65. Cooper MA, Fehniger TA, Ponnappan A, Mehta V, Wewers MD, Caligiuri MA. Interleukin-1beta costimulates interferon-gamma production by human natural killer cells. Eur J Immunol (2001) 31(3):792–801. doi: 10.1002/1521-4141(200103)31:3<792::aid-immu792>3.0.co;2-u
66. Dinarello CA, Conti P, Mier JW. Effects of human interleukin-1 on natural killer cell activity: is fever a host defense mechanism for tumor killing? Yale J Biol Med (1986) 59(2):97–106.
67. Li S, Amo-Aparicio J, Neff CP, Tengesdal IW, Azam T, Palmer BE, et al. Role for nuclear interleukin-37 in the suppression of innate immunity. Proc Natl Acad Sci U.S.A. (2019) 116(10):4456–61. doi: 10.1073/pnas.1821111116
68. Cavalli G, Dinarello CA. Suppression of inflammation and acquired immunity by IL-37. Immunol Rev (2018) 281(1):179–90. doi: 10.1111/imr.12605
69. Boraschi D, Lucchesi D, Hainzl S, Leitner M, Maier E, Mangelberger D, et al. IL-37: a new anti-inflammatory cytokine of the IL-1 family. Eur Cytokine Netw (2011) 22(3):127–47. doi: 10.1684/ecn.2011.0288
70. Zhao JJ, Pan QZ, Pan K, Weng DS, Wang QJ, Li JJ, et al. Interleukin-37 mediates the antitumor activity in hepatocellular carcinoma: role for CD57+ NK cells. Sci Rep (2014) 4:5177. doi: 10.1038/srep05177
71. Qi F, Liu M, Li F, Lv Q, Wang G, Gong S, et al. Interleukin-37 Ameliorates Influenza Pneumonia by Attenuating Macrophage Cytokine Production in a MAPK-Dependent Manner. Front Microbiol (2019) 10:2482. doi: 10.3389/fmicb.2019.02482
72. Xu P, Derynck R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol Cell (2010) 37(4):551–66. doi: 10.1016/j.molcel.2010.01.034
73. Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood (2013) 121(18):3599–608. doi: 10.1182/blood-2012-04-425397
74. Becknell B, Caligiuri MA. Interleukin-2, interleukin-15, and their roles in human natural killer cells. Adv Immunol (2005) 86:209–39. doi: 10.1016/S0065-2776(04)86006-1
75. Marcais A, Viel S, Grau M, Henry T, Marvel J, Walzer T. Regulation of mouse NK cell development and function by cytokines. Front Immunol (2013) 4:450. doi: 10.3389/fimmu.2013.00450
76. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med (2016) 8(357):357ra123. doi: 10.1126/scitranslmed.aaf2341
77. Perera PY, Lichy JH, Waldmann TA, Perera LP. The role of interleukin-15 in inflammation and immune responses to infection: implications for its therapeutic use. Microbes Infect (2012) 14(3):247–61. doi: 10.1016/j.micinf.2011.10.006
78. Stackaruk ML, Lee AJ, Ashkar AA. Type I interferon regulation of natural killer cell function in primary and secondary infections. Expert Rev Vaccines (2013) 12(8):875–84. doi: 10.1586/14760584.2013.814871
79. Frutoso M, Mortier E. NK Cell Hyporesponsiveness: More Is Not Always Better. Int J Mol Sci (2019) 20(18):2–20. doi: 10.3390/ijms20184514
80. Felices M, Lenvik AJ, McElmurry R, Chu S, Hinderlie P, Bendzick L, et al. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight (2018) 3(3):1–14. doi: 10.1172/jci.insight.96219
81. Zhou JH, Wang YN, Chang QY, Ma P, Hu Y, Cao X. Type III Interferons in Viral Infection and Antiviral Immunity. Cell Physiol Biochem (2018) 51(1):173–85. doi: 10.1159/000495172
82. Souza-Fonseca-Guimaraes F, Young A, Mittal D, Martinet L, Bruedigam C, Takeda K, et al. NK cells require IL-28R for optimal in vivo activity. Proc Natl Acad Sci U S A (2015) 112(18):E2376–84. doi: 10.1073/pnas.1424241112
83. Wang Y, Li T, Chen Y, Wei H, Sun R, Tian Z. Involvement of NK Cells in IL-28B-Mediated Immunity against Influenza Virus Infection. J Immunol (2017) 199(3):1012–20. doi: 10.4049/jimmunol.1601430
84. Pelly VS, Kannan Y, Coomes SM, Entwistle LJ, Ruckerl D, Seddon B, et al. IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal Immunol (2016) 9(6):1407–17. doi: 10.1038/mi.2016.4
85. Chen L, Grabowski KA, Xin JP, Coleman J, Huang Z, Espiritu B, et al. IL-4 induces differentiation and expansion of Th2 cytokine-producing eosinophils. J Immunol (2004) 172(4):2059–66. doi: 10.4049/jimmunol.172.4.2059
86. Seder RA, Paul WE, Davis MM, Fazekas de St Groth B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J Exp Med (1992) 176(4):1091–8. doi: 10.1084/jem.176.4.1091
87. Maier E, Duschl A, Horejs-Hoeck J. STAT6-dependent and -independent mechanisms in Th2 polarization. Eur J Immunol (2012) 42(11):2827–33. doi: 10.1002/eji.201242433
88. Zhu J, Yamane H, Cote-Sierra J, Guo L, Paul WE. GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res (2006) 16(1):3–10. doi: 10.1038/sj.cr.7310002
89. Gieseck RL,3, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol (2018) 18(1):62–76. doi: 10.1038/nri.2017.90
90. Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol (2015) 15(5):271–82. doi: 10.1038/nri3831
91. Symowski C, Voehringer D. Th2 cell-derived IL-4/IL-13 promote ILC2 accumulation in the lung by ILC2-intrinsic STAT6 signaling in mice. Eur J Immunol (2019) 49(9):1421–32. doi: 10.1002/eji.201948161
92. Rolot M, Dougall AM, Chetty A, Javaux J, Chen T, Xiao X, et al. Helminth-induced IL-4 expands bystander memory CD8(+) T cells for early control of viral infection. Nat Commun (2018) 9(1):4516. doi: 10.1038/s41467-018-06978-5
93. Renkema KR, Lee JY, Lee YJ, Hamilton SE, Hogquist KA, Jameson SC. IL-4 sensitivity shapes the peripheral CD8+ T cell pool and response to infection. J Exp Med (2016) 213(7):1319–29. doi: 10.1084/jem.20151359
94. Ventre E, Brinza L, Schicklin S, Mafille J, Coupet CA, Marcais A, et al. Negative regulation of NKG2D expression by IL-4 in memory CD8 T cells. J Immunol (2012) 189(7):3480–9. doi: 10.4049/jimmunol.1102954
95. Kiniwa T, Enomoto Y, Terazawa N, Omi A, Miyata N, Ishiwata K, et al. NK cells activated by Interleukin-4 in cooperation with Interleukin-15 exhibit distinctive characteristics. Proc Natl Acad Sci U.S.A. (2016) 113(36):10139–44. doi: 10.1073/pnas.1600112113
96. Bream JH, Curiel RE, Yu CR, Egwuagu CE, Grusby MJ, Aune TM, et al. IL-4 synergistically enhances both IL-2- and IL-12-induced IFN-gamma expression in murine NK cells. Blood (2003) 102(1):207–14. doi: 10.1182/blood-2002-08-2602
97. Herr F, Lemoine R, Gouilleux F, Meley D, Kazma I, Heraud A, et al. IL-2 phosphorylates STAT5 to drive IFN-gamma production and activation of human dendritic cells. J Immunol (2014) 192(12):5660–70. doi: 10.4049/jimmunol.1300422
98. Marcenaro E, Della Chiesa M, Bellora F, Parolini S, Millo R, Moretta L, et al. IL-12 or IL-4 prime human NK cells to mediate functionally divergent interactions with dendritic cells or tumors. J Immunol (2005) 174(7):3992–8. doi: 10.4049/jimmunol.174.7.3992
99. Paganin C, Matteucci C, Cenzuales S, Mantovani A, Allavena P. IL-4 inhibits binding and cytotoxicity of NK cells to vascular endothelium. Cytokine (1994) 6(2):135–40. doi: 10.1016/1043-4666(94)90034-5
100. Yu CR, Kirken RA, Malabarba MG, Young HA, Ortaldo JR. Differential regulation of the Janus kinase-STAT pathway and biologic function of IL-13 in primary human NK and T cells: a comparative study with IL-4. J Immunol (1998) 161(1):218–27.
101. Dienz O, Rincon M. The effects of IL-6 on CD4 T cell responses. Clin Immunol (2009) 130(1):27–33. doi: 10.1016/j.clim.2008.08.018
102. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol (2014) 6(10):a016295. doi: 10.1101/cshperspect.a016295
103. Wu W, Dietze KK, Gibbert K, Lang KS, Trilling M, Yan H, et al. TLR ligand induced IL-6 counter-regulates the anti-viral CD8(+) T cell response during an acute retrovirus infection. Sci Rep (2015) 5:10501. doi: 10.1038/srep10501
104. Cifaldi L, Prencipe G, Caiello I, Bracaglia C, Locatelli F, De Benedetti F, et al. Inhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol (2015) 67(11):3037–46. doi: 10.1002/art.39295
105. Mazzoni A, Salvati L, Maggi L, Capone M, Vanni A, Spinicci M, et al. Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J Clin Invest (2020) 130(9):4694–703. doi: 10.1172/JCI138554
106. Xu L, Chen X, Shen M, Yang DR, Fang L, Weng G, et al. Inhibition of IL-6-JAK/Stat3 signaling in castration-resistant prostate cancer cells enhances the NK cell-mediated cytotoxicity via alteration of PD-L1/NKG2D ligand levels. Mol Oncol (2018) 12(3):269–86. doi: 10.1002/1878-0261.12135
107. Wan YY, Flavell RA. ‘Yin-Yang’ functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol Rev (2007) 220:199–213. doi: 10.1111/j.1600-065X.2007.00565.x
108. Viant C, Rankin LC, Girard-Madoux MJ, Seillet C, Shi W, Smyth MJ, et al. Transforming growth factor-beta and Notch ligands act as opposing environmental cues in regulating the plasticity of type 3 innate lymphoid cells. Sci Signal (2016) 9(426):ra46. doi: 10.1126/scisignal.aaf2176
109. Batlle E, Massague J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity (2019) 50(4):924–40. doi: 10.1016/j.immuni.2019.03.024
110. Hawke LG, Whitford MKM, Ormiston ML. The Production of Pro-angiogenic VEGF-A Isoforms by Hypoxic Human NK Cells Is Independent of Their TGF-beta-Mediated Conversion to an ILC1-Like Phenotype. Front Immunol (2020) 11:1903. doi: 10.3389/fimmu.2020.01903
111. Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal (2016) 9(415):ra19. doi: 10.1126/scisignal.aad1884
112. Zaiatz-Bittencourt V, Finlay DK, Gardiner CM. Canonical TGF-beta Signaling Pathway Represses Human NK Cell Metabolism. J Immunol (2018) 200(12):3934–41. doi: 10.4049/jimmunol.1701461
113. Lucar O, Reeves RK, Jost S. A Natural Impact: NK Cells at the Intersection of Cancer and HIV Disease. Front Immunol (2019) 10:1850. doi: 10.3389/fimmu.2019.01850
114. Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol (2008) 181(6):3784–92. doi: 10.4049/jimmunol.181.6.3784
115. Sun C, Fu B, Gao Y, Liao X, Sun R, Tian Z, et al. TGF-beta1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PloS Pathog (2012) 8(3):e1002594. doi: 10.1371/journal.ppat.1002594
116. Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity (2009) 31(1):145–57. doi: 10.1016/j.immuni.2009.06.015
117. Zheng Q, Zhu YY, Chen J, Ye YB, Li JY, Liu YR, et al. Activated natural killer cells accelerate liver damage in patients with chronic hepatitis B virus infection. Clin Exp Immunol (2015) 180(3):499–508. doi: 10.1111/cei.12597
118. Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, et al. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nat Immunol (2017) 18(9):995–1003. doi: 10.1038/ni.3809
119. Wang Y, Chu J, Yi P, Dong W, Saultz J, Wang Y, et al. SMAD4 promotes TGF-beta-independent NK cell homeostasis and maturation and antitumor immunity. J Clin Invest (2018) 128(11):5123–36. doi: 10.1172/JCI121227
120. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210-101312
121. Leon LR, Kozak W, Kluger MJ. Role of IL-10 in inflammation. Studies using cytokine knockout mice. Ann N Y Acad Sci (1998) 856:69–75. doi: 10.1111/j.1749-6632.1998.tb08314.x
122. Beebe AM, Cua DJ, de Waal Malefyt R. The role of interleukin-10 in autoimmune disease: systemic lupus erythematosus (SLE) and multiple sclerosis (MS). Cytokine Growth Factor Rev (2002) 13(4-5):403–12. doi: 10.1016/s1359-6101(02)00025-4
123. Sun K, Torres L, Metzger DW. A detrimental effect of interleukin-10 on protective pulmonary humoral immunity during primary influenza A virus infection. J Virol (2010) 84(10):5007–14. doi: 10.1128/JVI.02408-09
124. Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med (2006) 12(11):1301–9. doi: 10.1038/nm1492
125. Littwitz-Salomon E, Malyshkina A, Schimmer S, Dittmer U. The Cytotoxic Activity of Natural Killer Cells Is Suppressed by IL-10(+) Regulatory T Cells During Acute Retroviral Infection. Front Immunol (2018) 9:1947:1947. doi: 10.3389/fimmu.2018.01947
126. Geng X, Li M, Cui B, Lu C, Liu X, Zhang P, et al. CD4+CD25+Foxp3+ regulatory T cells suppress NKG2D-mediated NK cell cytotoxicity in peripheral blood. Med (Baltimore) (2019) 98(22):e15722. doi: 10.1097/MD.0000000000015722
127. Mandaric S, Walton SM, Rulicke T, Richter K, Girard-Madoux MJ, Clausen BE, et al. IL-10 suppression of NK/DC crosstalk leads to poor priming of MCMV-specific CD4 T cells and prolonged MCMV persistence. PloS Pathog (2012) 8(8):e1002846. doi: 10.1371/journal.ppat.1002846
128. Clark SE, Burrack KS, Jameson SC, Hamilton SE, Lenz LL. NK Cell IL-10 Production Requires IL-15 and IL-10 Driven STAT3 Activation. Front Immunol (2019) 10:2087:2087. doi: 10.3389/fimmu.2019.02087
129. Stacey MA, Marsden M, Wang EC, Wilkinson GW, Humphreys IR. IL-10 restricts activation-induced death of NK cells during acute murine cytomegalovirus infection. J Immunol (2011) 187(6):2944–52. doi: 10.4049/jimmunol.1101021
130. Holder KA, Grant MD. Human cytomegalovirus IL-10 augments NK cell cytotoxicity. J Leukoc Biol (2019) 106(2):447–54. doi: 10.1002/JLB.2AB0418-158RR
131. Fargnoli MC. Secukinumab: The Anti-IL-17A Biologic for the Treatment of Psoriasis. Case Rep Dermatol (2019) 11(Suppl 1):1–3. doi: 10.1159/000501991. PubMed PMID: 31662731; PubMed Central PMCID: PMCPMC6816127 far as she knows, the studies were conducted in accordance with the World Medical Association Declaration of Helsinki and that the patients have given their written informed consent to publish their case, including images.
132. Kau AL, Korenblat PE. Anti-interleukin 4 and 13 for asthma treatment in the era of endotypes. Curr Opin Allergy Clin Immunol (2014) 14(6):570–5. doi: 10.1097/ACI.0000000000000108
133. Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discovery (2012) 11(8):633–52. doi: 10.1038/nrd3800
134. Caramori G, Adcock IM, Di Stefano A, Chung KF. Cytokine inhibition in the treatment of COPD. Int J Chron Obstruct Pulmon Dis (2014) 9:397–412. doi: 10.2147/COPD.S42544
135. Verhamme FM, Bracke KR, Joos GF, Brusselle GG. Transforming growth factor-beta superfamily in obstructive lung diseases. more suspects than TGF-beta alone. Am J Respir Cell Mol Biol (2015) 52(6):653–62. doi: 10.1165/rcmb.2014-0282RT
136. Gabryelska A, Kuna P, Antczak A, Bialasiewicz P, Panek M. IL-33 Mediated Inflammation in Chronic Respiratory Diseases-Understanding the Role of the Member of IL-1 Superfamily. Front Immunol (2019) 10:692:692. doi: 10.3389/fimmu.2019.00692
137. Wang H, Anthony D, Selemidis S, Vlahos R, Bozinovski S. Resolving Viral-Induced Secondary Bacterial Infection in COPD: A Concise Review. Front Immunol (2018) 9:2345:2345. doi: 10.3389/fimmu.2018.02345
138. Beasley V, Joshi PV, Singanayagam A, Molyneaux PL, Johnston SL, Mallia P. Lung microbiology and exacerbations in COPD. Int J Chron Obstruct Pulmon Dis (2012) 7:555–69. doi: 10.2147/COPD.S28286
139. Barnes PJ. The cytokine network in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol (2009) 41(6):631–8. doi: 10.1165/rcmb.2009-0220TR
140. Bieber T, D’Erme AM, Akdis CA, Traidl-Hoffmann C, Lauener R, Schappi G, et al. Clinical phenotypes and endophenotypes of atopic dermatitis: Where are we, and where should we go? J Allergy Clin Immunol (2017) 139(4S):S58–64. doi: 10.1016/j.jaci.2017.01.008
141. Mack MR, Brestoff JR, Berrien-Elliott MM, Trier AM, Yang TB, McCullen M, et al. Blood natural killer cell deficiency reveals an immunotherapy strategy for atopic dermatitis. Sci Transl Med (2020) 12(532):1–14. doi: 10.1126/scitranslmed.aay1005
142. Sehra S, Yao Y, Howell MD, Nguyen ET, Kansas GS, Leung DY, et al. IL-4 regulates skin homeostasis and the predisposition toward allergic skin inflammation. J Immunol (2010) 184(6):3186–90. doi: 10.4049/jimmunol.0901860
143. Klonowska J, Glen J, Nowicki RJ, Trzeciak M. New Cytokines in the Pathogenesis of Atopic Dermatitis-New Therapeutic Targets. Int J Mol Sci (2018) 19(10):1–12. doi: 10.3390/ijms19103086
144. Leung DY, Boguniewicz M, Howell MD, Nomura I, Hamid QA. New insights into atopic dermatitis. J Clin Invest (2004) 113(5):651–7. doi: 10.1172/JCI21060
145. Spergel JM. From atopic dermatitis to asthma: the atopic march. Ann Allergy Asthma Immunol (2010) 105(2):99–106; quiz 7-9, 17. doi: 10.1016/j.anai.2009.10.002
146. Duvall MG, Barnig C, Cernadas M, Ricklefs I, Krishnamoorthy N, Grossman NL, et al. Natural killer cell-mediated inflammation resolution is disabled in severe asthma. Sci Immunol (2017) 2(9):1–23. doi: 10.1126/sciimmunol.aam5446
147. Wark PAB, Ramsahai JM, Pathinayake P, Malik B, Bartlett NW. Respiratory Viruses and Asthma. Semin Respir Crit Care Med (2018) 39(1):45–55. doi: 10.1055/s-0037-1617412
148. Bhat R, Farrag MA, Almajhdi FN. Double-edged role of natural killer cells during RSV infection. Int Rev Immunol (2020) 39(5):233–44. doi: 10.1080/08830185.2020.1770748
149. van Erp EA, Lakerveld AJ, de Graaf E, Larsen MD, Schepp RM, Hipgrave Ederveen AL, et al. Natural killer cell activation by respiratory syncytial virus-specific antibodies is decreased in infants with severe respiratory infections and correlates with Fc-glycosylation. Clin Transl Immunol (2020) 9(2):e1112. doi: 10.1002/cti2.1112
150. van Erp EA, Feyaerts D, Duijst M, Mulder HL, Wicht O, Luytjes W, et al. Respiratory Syncytial Virus Infects Primary Neonatal and Adult Natural Killer Cells and Affects Their Antiviral Effector Function. J Infect Dis (2019) 219(5):723–33. doi: 10.1093/infdis/jiy566
151. Long X, Xie J, Zhao K, Li W, Tang W, Chen S, et al. NK cells contribute to persistent airway inflammation and AHR during the later stage of RSV infection in mice. Med Microbiol Immunol (2016) 205(5):459–70. doi: 10.1007/s00430-016-0459-9
152. Tregoning JS, Wang BL, McDonald JU, Yamaguchi Y, Harker JA, Goritzka M, et al. Neonatal antibody responses are attenuated by interferon-gamma produced by NK and T cells during RSV infection. Proc Natl Acad Sci U S A (2013) 110(14):5576–81. doi: 10.1073/pnas.1214247110
153. Tang YW, Graham BS. Anti-IL-4 treatment at immunization modulates cytokine expression, reduces illness, and increases cytotoxic T lymphocyte activity in mice challenged with respiratory syncytial virus. J Clin Invest (1994) 94(5):1953–8. doi: 10.1172/JCI117546
154. Bukreyev A, Belyakov IM, Prince GA, Yim KC, Harris KK, Berzofsky JA, et al. Expression of interleukin-4 by recombinant respiratory syncytial virus is associated with accelerated inflammation and a nonfunctional cytotoxic T-lymphocyte response following primary infection but not following challenge with wild-type virus. J Virol (2005) 79(15):9515–26. doi: 10.1128/JVI.79.15.9515-9526.2005
155. Mehta J, Walsh EE, Mahadevia PJ, Falsey AR. Risk factors for respiratory syncytial virus illness among patients with chronic obstructive pulmonary disease. COPD (2013) 10(3):293–9. doi: 10.3109/15412555.2012.744741
156. Westerly BD, Peebles RS Jr. Respiratory syncytial virus infections in the adult asthmatic–mechanisms of host susceptibility and viral subversion. Immunol Allergy Clin North Am (2010) 30(4):523–39, vi-vii. doi: 10.1016/j.iac.2010.08.006
157. Arnon TI, Lev M, Katz G, Chernobrov Y, Porgador A, Mandelboim O. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur J Immunol (2001) 31(9):2680–9. doi: 10.1002/1521-4141(200109)31:9<2680::aid-immu2680>3.0.co;2-a
158. Liu FD, Kenngott EE, Schroter MF, Kuhl A, Jennrich S, Watzlawick R, et al. Timed action of IL-27 protects from immunopathology while preserving defense in influenza. PloS Pathog (2014) 10(5):e1004110. doi: 10.1371/journal.ppat.1004110
159. Moran TM, Isobe H, Fernandez-Sesma A, Schulman JL. Interleukin-4 causes delayed virus clearance in influenza virus-infected mice. J Virol (1996) 70(8):5230–5. doi: 10.1128/JVI.70.8.5230-5235.1996
160. Denney L, Branchett W, Gregory LG, Oliver RA, Lloyd CM. Epithelial-derived TGF-beta1 acts as a pro-viral factor in the lung during influenza A infection. Mucosal Immunol (2018) 11(2):523–35. doi: 10.1038/mi.2017.77
161. Furuya Y, Furuya AK, Roberts S, Sanfilippo AM, Salmon SL, Metzger DW. Prevention of Influenza Virus-Induced Immunopathology by TGF-beta Produced during Allergic Asthma. PloS Pathog (2015) 11(9):e1005180. doi: 10.1371/journal.ppat.1005180
162. Costela-Ruiz VJ, Illescas-Montes R, Puerta-Puerta JM, Ruiz C, Melguizo-Rodriguez L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev (2020) 54:62–75. doi: 10.1016/j.cytogfr.2020.06.001
163. Fahy JV. Type 2 inflammation in asthma–present in most, absent in many. Nat Rev Immunol (2015) 15(1):57–65. doi: 10.1038/nri3786
164. Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest (2008) 118(11):3546–56. doi: 10.1172/JCI36130
Keywords: innate lymphoid cell, NK cell, interleukin, transforming growth factor, interferon gamma
Citation: Ochayon DE and Waggoner SN (2021) The Effect of Unconventional Cytokine Combinations on NK-Cell Responses to Viral Infection. Front. Immunol. 12:645850. doi: 10.3389/fimmu.2021.645850
Received: 24 December 2020; Accepted: 01 March 2021;
Published: 19 March 2021.
Edited by:
Kristina De Paris (Abel), University of North Carolina at Chapel Hill, United StatesReviewed by:
Zvi Gershon Grossman, National Institute of Allergy and Infectious Diseases (NIH), United StatesMarina Cella, Washington University School of Medicine in St. Louis, United States
Copyright © 2021 Ochayon and Waggoner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David E. Ochayon, ZGF2aWRvY0Bwb3N0LmJndS5hYy5pbA==