 Andrea Mancini
Andrea Mancini Veronica Ghiglieri
Veronica Ghiglieri Lucilla Parnetti
Lucilla Parnetti Paolo Calabresi
Paolo Calabresi Massimiliano Di Filippo
Massimiliano Di Filippo- 1Section of Neurology, Department of Medicine and Surgery, Università degli Studi di Perugia, Perugia, Italy
- 2Università Telematica San Raffaele, Rome, Italy
- 3Section of Neurology, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy
- 4Department of Neuroscience, Università Cattolica del Sacro Cuore, Rome, Italy
The basal ganglia network is represented by an interconnected group of subcortical nuclei traditionally thought to play a crucial role in motor learning and movement execution. During the last decades, knowledge about basal ganglia physiology significantly evolved and this network is now considered as a key regulator of important cognitive and emotional processes. Accordingly, the disruption of basal ganglia network dynamics represents a crucial pathogenic factor in many neurological and psychiatric disorders. The striatum is the input station of the circuit. Thanks to the synaptic properties of striatal medium spiny neurons (MSNs) and their ability to express synaptic plasticity, the striatum exerts a fundamental integrative and filtering role in the basal ganglia network, influencing the functional output of the whole circuit. Although it is currently established that the immune system is able to regulate neuronal transmission and plasticity in specific cortical areas, the role played by immune molecules and immune/glial cells in the modulation of intra-striatal connections and basal ganglia activity still needs to be clarified. In this manuscript, we review the available evidence of immune-based regulation of synaptic activity in the striatum, also discussing how an abnormal immune activation in this region could be involved in the pathogenesis of inflammatory and degenerative central nervous system (CNS) diseases.
Highlights
● The basal ganglia network operates for appropriate context-dependent cognitive, behavioral and emotional responses.
● Bidirectional plastic changes of striatal synapses allow input integration and input-output associations in the basal ganglia network.
● Astrocytes gate striatal excitatory synaptic transmission and orchestrate striatal pathways and subnetworks activation.
● Soluble immune molecules may influence striatal glutamatergic transmission acting on both pre- and post-synaptic sites.
● Pathological activation of striatal astrocytes and microglia could influence the synaptic bases of basal ganglia network functioning, leading to cognitive and behavioral abnormalities during neurological disorders.
Introduction
The extensive research performed during the last years has made it clear the crucial role of the immune system in the field of cognitive and behavioral sciences. Human behavioral, cognitive and social traits could be deeply influenced by the activation of immune cells in both physiological and pathological conditions. The classical concept of the central nervous system (CNS) as an immune-privileged site has significantly evolved during the last years, acknowledging the presence of functional meningeal lymphatic vessels and a complex neuro-immune cross-talk involving innate and adaptive immunity, as well as resident immune cells within the CNS (1, 2). Indeed, the release of soluble immune mediators is thought to physiologically tune the activity of neural networks, influencing learning and memory processes through the regulation of synaptic transmission and long-term plasticity (3–5).
A prototypical example of the neuromodulatory role of the immune system is represented by the shift in an individual’s behavior and perceptions frequently accompanying infectious diseases. Indeed, a reduced interest in social interactions and unnecessary physical activity could represent a protective evolutionistic response aimed at limiting pathogen spreading in a social community. These complex cognitive and behavioral responses are thought to be caused by pro-inflammatory mediators released by immune cells counteracting the infection (6). If the immune activation is inappropriate or unabated, this para-physiological process may become pathological. Indeed, many disabling cognitive and behavioral features occurring during neuroinflammatory and neurodegenerative disorders are thought to rely on the detrimental neuronal and synaptic effects triggered by an uncontrolled cerebral inflammatory microenvironment (4, 7). The influence exerted by the immune system on neuronal and synaptic activity has been mainly investigated in cortical areas, such as in the hippocampus (8, 9), while less is known about the neuro-immune cross-talk occurring in subcortical structures such as the basal ganglia, that together with cortical structures mediate cognitive and behavioral functions (10, 11).
The cortico-striato-thalamo-cortical network was originally described as an essential circuit for locomotor activity and movement execution (12). Still, the identification of extensive functional connections between the striatum and non-motor cortical areas (13, 14) raised the hypothesis of an involvement of the basal ganglia also in associative, cognitive and emotional processes. Indeed, thanks to the closed-loop architecture of the basal ganglia network, the striatum can filter and integrate different cortical inputs during goal-directed behavior, decision making and response selection under competition (11).
The complex microstructural organization of the striatum, characterized by multiple inhibitory and excitatory synaptic connections among various neuronal subtypes, highlights the activity of this structure as an input integrator. Functional or structural alterations of such synaptic connections can powerfully influence the final output and tuning of the whole basal ganglia network (15). Despite the essential functions of the basal ganglia, the potential neuro-immune interactions occurring at this level have been less investigated and should be better understood. In this review, we summarize the available evidence suggesting an immune-based regulation of synaptic activity in the striatum during physiological conditions and pathological inflammatory and degenerative processes of the CNS.
The Basal Ganglia Network: From Movements to Emotions
The functional anatomy of the basal ganglia can be described as a closed-loop network with two different pathways canonically considered as parallel and opposed, one favoring (direct) and another inhibiting (indirect) the activation of cortical brain areas. The main input of the basal ganglia network is represented by glutamatergic excitatory projections from cortical and thalamic areas, making synaptic contact with striatal medium spiny neurons (MSNs) and aspiny interneurons (16, 17). Such corticostriatal connections are influenced by dopaminergic projections arising from the substantia nigra pars compacta (SNc) and converging into the dendritic tree of MSNs, which can be distinguished by their dopamine (DA) receptor expression patterns (18, 19). In addition, striatal GABAergic or cholinergic interneurons can act as additional elements for the integration of cortical, thalamic, and dopaminergic afferents and the modulation of neighboring MSNs activation (20).
The extensive net of intra-striatal inter-neuronal connections, integrating various cortical and sub-cortical inputs, makes the striatum a crucial station from which the information is filtered and channeled through the direct and indirect pathways (Figure 1).
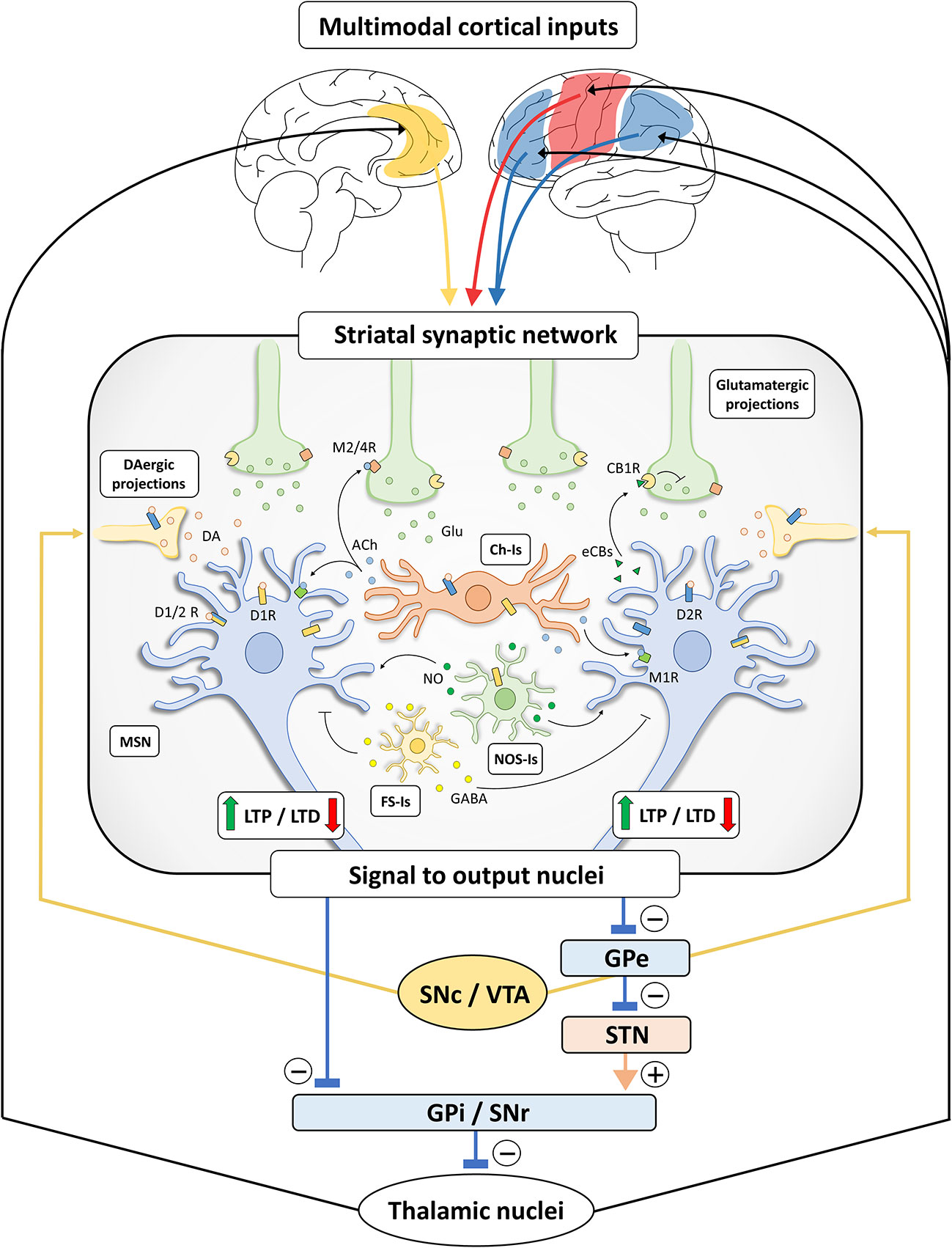
Figure 1 Schematic representation of basal ganglia and striatal synaptic networks. Multimodal inputs are constantly conveyed toward the striatum, including projections arising from sensori-motor cortices (red), limbic structures (yellow) and associative areas (blue) (10, 11, 21). The striatal synaptic network acts as a processing unit through differential signal amplification, output selection and context-dependent input integration. The induction of bidirectional synaptic plastic changes (long-term potentiation, LTP, and long-term depression, LTD) at corticostriatal connections is deeply influenced by DA released by dopaminergic (DAergic) terminals, originating from substantia nigra pars compacta/ventral tegmental area (SNc/VTA). Specifically, LTP of corticostriatal projections is dependent on the activation of D1-like receptors (D1Rs) (22, 23) and under the negative control of D2-like receptors (D2Rs) (24), while the induction of LTD requires the presence of functionally active D1Rs and D2Rs (25–29). These observations are not in line with the classical view of a complete D1- and D2-like receptor functional segregation (30, 31) and may rely on the presence of MSNs expressing both receptor subtypes (32) or membrane heteromeric D1/D2 receptors (D1/2Rs) (33–35). In addition, DA may indirectly act on MSNs through different populations of striatal interneurons (25, 36–38). Striatal cholinergic (Ch-Is), NOS-positive (NOS-Is) and fast-spiking (FS-Is) interneurons exert a feedforward and parallel control of striatal circuit (15). Acetylcholine (Ach) released by Ch-Is can act on M2/4 muscarinic receptors expressed by pre-synaptic glutamatergic terminals and on M1 muscarinic receptors expressed by MSNs. The DA-dependent modulation of Ach release by Ch-Is (expressing both D1Rs and D2Rs) can influence the induction of synaptic LTD in MSNs (39). Nitric oxide (NO) is released by NOS-Is under the control of D1Rs and could act on MSNs facilitating LTD at the post-synaptic level (39). FS-Is releasing GABA represent a parallel inhibitory system. Of note, dopaminergic regulation of LTD induction also relies on the release of retrograde neurotransmitters under the control of different cell-type specific thresholds in D1R- and D2R-expressing MSNs (40). Indeed, the D2R-dependent release of endocannabinoids (eCBs) by MSNs modulates LTD induction through the activation of CB1 cannabinoid receptors (CB1Rs) located on glutamatergic terminals, inhibiting glutamate (Glu) release. Striatal processing of cortical multimodal inputs generated an integrated signal to output nuclei which, in turn, project to thalamic nuclei sending efferents that complete the cortico-basal ganglia-thalamo-cortical loop. Specifically, striatal inhibitory outputs directed toward the GABAergic neurons of substantia nigra pars reticulata (SNr) and globus pallidus pars interna (GPi), which make direct inhibitory synaptic connections with the thalamus, ultimately results in a disinhibition of the thalamic glutamatergic cortical projections (direct pathway). Conversely, the activation of striatal MSNs connected to the globus pallidus pars externa (GPe) results in a disinhibition of the glutamatergic neurons of subthalamic nucleus (STN), leading to a GPi/SNr-dependent inhibition of thalamo-cortical projections (indirect pathway). The presence of bridging collaterals in striatofugal projections ensures signal coordination and mutual inhibition for each pathway and each subnetwork (41, 42). Please, note that the schematic representation of the striatal network does not reflect the effective relative size of the neuronal cells.
The classical “push–pull”, direct–indirect dichotomous view of striatal output pathways has been challenged by the evidence that both pathways are activated during the initiation/stopping of actions or behavioral sequences (11, 43–45) and by the identification of multiple functional and structural connections between the two pathways orchestrating the activity of the whole network (15). The architecture of the basal ganglia network might allow to obtain a simple binary output (go/no-go response) from various cortical and subcortical inputs, perfectly fulfilling their acknowledged role in solving a “selection” problem (46). Accordingly, basal ganglia are considered a phylogenetically conserved network underlying action selection in vertebrates initially devoted to the execution of the previously learned motor plan (47) and subsequently co-opted for other key mammalian superior cortical functions through a process of exaptation, following the evolution of cortical networks (48). Indeed, the striatum receives massive projections from almost all regions of the cortex, acquiring sensorimotor inputs, emotional/motivational information from limbic areas, and multimodal processed data from associative areas (10, 11, 49). This various set of basal ganglia inputs could be considered as a “generator of diversity” (21), from which the striatal filter selects a proper output response which is conveyed through the thalamic nuclei to functionally distinct cortical areas (13, 50). The initially proposed presence of parallel and segregated basal ganglia sub-networks (13), each one processing a different type of input, has been challenged by the description of functional overlap (51–54), allowing the integration of multimodal information in line with the known influence of emotional and motivational state on an individual’s behavior.
Overall, basal ganglia seem to be involved in a wide range of behavioral, cognitive and affective functions, leading to the execution of a specific response out of the different choices continuously arising during daily living. Basal ganglia activity could be involved both in conscious goal-directed behavior and in habitual unconscious actions, representing two possible decision-making performances (21, 55). Ventral and dorsolateral striatal networks seem to be deeply involved in both situations, characterized by the selection of an appropriate action through the evaluation of context-dependent information (21, 55, 56). Extensive afferents from the ventral tegmental area (VTA), the ventral hippocampal subiculum, the prefrontal cortex and the basolateral amygdala converge into the ventral striatum, specifically in the nucleus accumbens (NAc), allowing the integration of contextual/spatial information with affective inputs to select a proper reward-based adaptive action (57–59). In this context, proactive or reactive inhibition of habitual actions is involved not only in motor activity but also in cognitive functions, gating the access to working memory (60, 61), or avoiding the recall of irrelevant information (11) and emotional reactions (like inhibiting context-inappropriate anxious or fear reactions).
According to the critical roles played by the basal ganglia circuit in brain physiology, its dysfunctional activity could lead to a wide range of behavioral/cognitive/emotional consequences. The selection process mediated by the circuit could become altered, since basal ganglia malfunction could be followed by an excessive impulsivity in high-conflict decisions (62), lowering the information threshold required for a selection (63) and influencing the balance between speed and accuracy of performance (64). In line with this view, some clinical characteristics of different neuropsychiatric disorders are thought to rely on an alteration of basal ganglia activity. These include, but are not limited to, bradykinesia, apathy and abulia in Parkinson’s disease (PD); motor or verbal urges in Tourette’s Syndrome (TS); impulsivity and lack of attention in attention-deficit hyperactivity disorder; intrusive thoughts and compulsive behaviors in obsessive-compulsive disorder (OCD); hyperactivity in Huntington’s disease (HD) (11, 65, 66). The pathophysiology of these conditions could rely on the alteration of cellular and synaptic mechanisms underlying the context-dependent selection operated by the basal ganglia network.
Striatal Synaptic Plasticity and Information Processing in the Basal Ganglia
Since their discovery, long-lasting and activity-dependent plastic changes of synaptic transmission have been considered a plausible biological process underlying brain ability to translate experiences into memories (67–70). Synaptic long-term potentiation (LTP) might enhance the synaptic weight of specific neuronal connections, increasing input specificity of neural network and lasting sufficiently long to induce the formation of stable memories (69, 70). On the other side, long-term depression (LTD) of synaptic connections may enhance input divergences, inhibiting competitive connections or reversing a previous synaptic potentiation due to bidirectional synaptic changes (39, 69, 70).
In this scenario, the synaptic plastic changes described at excitatory corticostriatal connections (15, 39, 71–73) are deeply influenced by the activation of both D1- and D2-like DA receptors (39) and by an extensive net of parallel connections, involving interneurons, such as fast-spiking GABA-releasing cells, large cholinergic neurons, and NO synthase (NOS)-positive interneurons (20, 36, 39, 74, 75) (Figure 1).
A fine coordination of striatal direct/indirect MSNs synaptic activity is thought to be crucial for the execution of a specific task (76–80), especially considering that the in vivo activation of the two striatal pathways was found to be concurrent (43) and complementary (81, 82) during the execution of motor and behavioral sequences. In addition, learning and refinement of actions seem to require parallel but dissociable input processing within associative and sensorimotor striatal subnetworks, implying a learning-related in vivo modulation of corticostriatal synaptic transmission (83) and a dynamic filtering of cortical inputs (84–86).
Behaviorally relevant reinforcement signals might influence striatal synaptic plasticity through short-latency and phasic release of DA from the ascending midbrain projections (87–89). These dopaminergic inputs are thought to play a key role in prediction/learning of reward-related processes by reinforcing causal relationships and input-outcome association during the execution of novel actions (90). This hypothesis is supported by the evidence that appropriately timed dopaminergic reinforcement signals are required to induce corticostriatal bidirectional plasticity, with divergent outcomes depending on the intensity and timing of MSNs activation by cortical/thalamic projections (39, 91). Specifically, it has been shown in intact animals that behaviorally relevant reinforcement signals, inducing a phasic release of DA in the striatum, are required for corticostriatal potentiation, and this occurs only if the electrical stimulation of the motor cortex precedes the depolarization of striatal MSNs (positive paring). Conversely, the same dopaminergic reinforcement is able to induce corticostriatal depression when cortical activation occurs after MSNs membrane depolarization (negative pairing) (91). This form of bidirectional synaptic plasticity, named spike-timing-dependent plasticity (STDP), is considered as a synaptic Hebbian learning paradigm (92, 93) and is deeply influenced by striatal eCBs release, serotonergic transmission, and stimulation of dopaminergic receptors (94–97).
In this scenario, it has been proposed that the input component from the cortex represents an ongoing behavior/action (98), and a positive pairing would arise when a specific cortical projection has directly contributed to MSNs depolarization (91). This striatal synaptic pairing could enhance input specificity and input divergence for a proper behavior/action selection allowing action-outcome association and context-dependent positive selection of satisfactory actions. In parallel, the divergent depressive changes of negatively-paired connections can refine striatal habit formation, lowering the strength of corticostriatal connections not contributing to action yielding reward and increasing signal-to-noise ratio (91).
Of note, the detection of the temporal contingency between two consecutive stimuli requires a balanced removal/reuptake of neurotransmitters previously released in the synaptic cleft. Astroglial cells could be deeply involved in these processes, and an alteration of their homeostatic functions can disrupt the induction of Hebbian synaptic plastic changes, leading to aberrant non-timing-dependent plasticity for uncorrelated events or precluding STDP expression (99).
Overall, the emerging picture of the basal ganglia network organization is more dynamic and fluid than that previously established. Cortical and thalamic inputs can be filtered and integrated in the striatum by the intrinsic membrane plastic properties of MSNs, fluctuating between an “up” or “down” state depending on the firing frequency of cortical inputs (100–102). Input signal specificity and input divergence may be guaranteed and enhanced by bidirectional plastic changes of corticostriatal synapses, under the control of parallel intra-striatal connections among MSNs and interneuronal cells and vertical dopaminergic projections arising from the midbrain, influencing motivational behavior and reward-related learning.
Such a functional view gets away from the simplistic dichotomous model of direct/indirect pathways and focuses on the plastic properties of corticostriatal connections as the core processing units for basal ganglia activity. In this scenario, growing evidence suggests the involvement of glial cells in synaptic transmission, synaptic plasticity, and synaptic remodeling, both in the post-natal and adult brain (103–106). Glial cells, including astrocytes, microglia, oligodendrocytes, and other specialized cells, appear as a highly represented cellular population throughout the CNS (107–109). The functional architecture of the neuronal-glial network has been deeply investigated during the last years in different brain structures, including basal ganglia (110, 111). Glia/neuron ratio was found to vary in the human brain in relation to neuronal density and the numerical relationship between these cellular elements was found to be remarkably conserved among different species, as if a proper balance is essential for the physiological brain activity (107, 112). It has been shown that the overall ratio between non-neuronal/neuronal cells in the whole human brain is close to 1, varying from a value of 1.48 in the gray matter of the cerebral cortex to 11.35 in basal ganglia/diencephalon/mesencephalon/pons (113). Accordingly, an updated view of the striatal network function should necessarily take into account the contribution of glial cells during both physiological and pathological conditions.
Neuron-Astrocyte Interactions in the Striatum
Astrocytes are widely represented in the brain, counting approximately 19-40% of total brain cells (112) and exerting multiple homeostatic functions through thousands of fine processes, creating “bushy” territories around neuronal somata, dendrites, and blood vessels (110). The functional view of these cells has significantly changed after the discovery that astrocytes can display a form of cellular excitability based on variations of intracellular calcium ion (Ca2+) concentration (114, 115), occurring spontaneously and in response to neurotransmitter release by neighboring synaptic connections (116, 117). The identification of a bidirectional neuron-astrocyte communication led to identify synaptic connections as “tripartite” elements, where astrocytes represent cellular processors of information with selective responses to specific synaptic inputs and integrative abilities due to cell-intrinsic properties and nonlinear input-output relationships (104). A recent study has identified, through cortical live-cell 3D-STED microscopy in mice, astrocytic Ca2+ signals at the level of bulbous enlargements localized along the thin astrocytic processes (118). Such “nodes” have been found to be in tight contact with dendritic spines, suggesting the presence of specific signaling domains tailored for neuron-astrocyte communication (118). The astrocytic processes can contact neighboring synapses and create an “astroglial cradle” essential for synaptic maturation and isolation (119), influencing neuron transmission through different mechanisms, including vesicular gliotransmission, release of neuroactive substances, potassium buffering, and neurotransmitter recycling (103, 120–122).
It has been shown that astrocytes may influence synapse structure and function through several contact-mediated and soluble synaptogenic cues (123). Specifically, astrocytes may regulate cortical synaptogenesis through the secretion of thrombospondins (TSP1 and 2) (124) or through cell adhesion proteins like gamma protocadherins (125). Other astrocyte-derived soluble mediators may modulate the expression of neurotransmitter receptors at synaptic sites since heparan sulfate proteoglycans glypican 4 and 6 (Gpc4 and 6) were linked to an increased expression of GluA1 AMPA receptor (AMPAR) subunit at the post-synaptic level (126) and tumor necrosis factor α (TNF-α) was associated with enhanced surface expression of AMPARs in hippocampal neurons (127).
Interestingly, CNS astrocytes are not a homogeneous cell population, displaying different region-specific functions to optimize local neural network activity (110, 128, 129). Transcriptomic and proteomic analysis revealed significant differences in gene expression patterns between striatal and hippocampal astrocytes (111). Moreover, from a morphological point of view, it has been shown in murine tissues that striatal and hippocampal astrocytes are characterized by equivalent somatic volumes, number of primary branches, and cell volumes, but striatal astrocytes displayed larger territory volumes impinging upon greater numbers of neurons (129). In line with these findings, other authors have recently found, through a genetically targeted neuron-astrocyte proximity assay (NAPA), that murine striatal astrocytes tightly interact with cortical, thalamic, and nigral projections (130). Interestingly, no substantial anatomical difference was found in astrocyte-synapse proximity for D1- and D2-like receptor-expressing MSNs (130). Overall, it has been estimated that each striatal astrocyte could make contact with an average number of ~11 MSNs, sampling D1- and D2-like receptor-expressing MSNs in an almost equivalent way (130), and could interact with approximately 50,700 excitatory synapses (129).
This extensive net of neuron-astrocyte interactions could exert a key role in the regulation of striatal network function. Indeed, it has been hypothesized that homogeneously distributed striatal astrocytes could display different patterns of activation in order to sustain and modulate the coordinated activity of direct and indirect striatal pathways (131). It has been shown that homotypic (D1-D1 or D2-D2) but not heterotypic MSNs stimulation is characterized by an endocannabinoid-dependent activation of astrocytic CB1 receptors (CB1Rs), leading to glutamate release upon elevating their Ca2+ levels (131). Of note, glutamate released by corticostriatal projections and by astrocytes could directly stimulate glutamatergic NMDA receptors (NMDARs) expressed at the synaptic cleft by MSNs (131), but it could also act on metabotropic receptor subtype 5 (mGluR5) expressed by astrocytes (132). The activation of these astrocytic receptors could lead to an additional Ca2+-dependent release of glutamate, triggering a stimulation of MSNs through GluN2B containing NMDARs which could last for minutes beyond the initial stimulus (133). In this scenario, the selective reinforcement of homotypic synapses supports the presence of specific astrocyte subpopulations enhancing striatal pathways divergence and coordination during behavior/action execution (131).
Astrocytes can also influence the induction of corticostriatal synaptic plastic changes, such as LTD (134) or STDP (99), through neurotransmitter release or regulation of glutamate reuptake. Astrocytes ensure a proper striatal signal-to-noise ratio, regulating glutamate concentration in the synaptic cleft (135), and this activity could act as a gatekeeper for the induction of corticostriatal synaptic plasticity. It has been hypothesized that the proper activation of astrocytic excitatory amino-acid transporter type-2/glutamate transporter 1 (EAAT2/GLT-1) could represent a key element for ensuring the detection of the temporal contingency required for Hebbian synaptic plastic changes like STDP, since the blockade or the overexpression of this astrocytic protein could lead to aberrant synaptic plastic changes (99).
Moreover, other authors proposed a model in which up-state MSNs could stimulate neighboring astrocytes through dendritic GABA release, leading to a GABAB receptor- and Gi-dependent release of Ca2+ from intracellular stores (136). The activation of this transduction pathway is supposed to upregulate the synaptogenic cue thrombospondin-1 (TSP1) in astrocytes, boosting excitatory synapse formation, fast excitatory synaptic transmission and MSNs firing frequency in the striatum (136). The abnormal activation of this Gi-dependent astrocytic pathway could pathologically enhance corticostriatal transmission leading to behavioral hyperactivity and impaired attention in mice (136). In line with these observations, an alteration of astrocytic Ca2+ dynamics has been linked with abnormal MSNs activity and excessive self-grooming behavior, as assessed with in vivo electrophysiological recordings in mice (137).
Overall, the astrocytic modulation of STDP through reinforcement signals and the maintenance of a proper signal-to-noise ratio, allowing the detection of the temporal relationship between two paired stimuli (negative or positive pairing), could deeply influence the basal ganglia action-outcome synaptic associations. Accordingly, altered astrocytic activity has been linked to enhanced reward-seeking behavior and to the pathological intake of drugs of abuse (138). In rodent models, methamphetamine and cocaine assumption has been associated with a reduction in the contacts between astrocytes and synapses in the NAc (139, 140), while cocaine and heroin seem to be linked to reduced expression of the glutamate transporter EAAT2/GLT-1, mainly located in astrocytes (141, 142). A recent study showed that DA in the NAc could directly stimulate astrocytes through D1-like receptors (D1Rs), with a subsequent astrocytic release of ATP/adenosine and inhibition of excitatory transmission through the stimulation of pre-synaptic A1 receptors expressed by glutamatergic projections (143). This evidence suggests new possible astrocyte- and dopamine-dependent pathways regulating reward-related behaviors.
Lastly, the neuron-astrocyte net could represent an anatomical “track” facilitating microglial cells movements and synaptic surveillance (130). Indeed, astrocytes may participate in network modeling and synapse elimination via direct synaptic phagocytosis and in cooperation with microglial cells, tagging synapses for elimination through the secretion of transforming growth factor-β (TGF-β) (144, 145).
Microglial Cells as Modulators of Striatal Synaptic Function
Microglia can account for 5%–12% of total cellular elements in the CNS, representing the main element of the resident CNS immune system with critical roles in organizing rapid responses against different kinds of tissue injury (146–148). Activated microglial cells can produce soluble chemotactic and pro-inflammatory molecules orchestrating inflammatory responses within the CNS, and these cells can assume a phagocytic profile aimed at clearing cellular debris (146–148). During the last decades, it has been clearly established that microglial cells are physiologically involved in synaptic transmission, plasticity and structural remodeling during CNS development and adult life, dynamically interacting with synapses as “synaptic sensors” (105, 149, 150).
Several high-resolution imaging studies have shown that immature, redundant, or dysfunctional axonal terminals and dendritic spines can be engulfed by microglial cells as a mechanism to refine brain networks (149, 151). Such a dynamic process of synaptic pruning was found to be dependent on several potential ‘find-me’ and ‘eat-me’ neuro-immune pathways (152). The chemokine CX3CL1, which could be secreted or expressed as a membrane-tethered protein, is thought to represent a synaptic tagging mechanism through which neuronal cells may attract resident CX3CR1-expressing microglia (153, 154). Accordingly, the genetic ablation of CX3CL1 was associated with an increased density of immature synapses in cortical areas (151, 155) and impaired cortical synaptic remodeling (154). Another potential ‘eat-me’ signal may be represented by the classical complement proteins C1q and C3, which can be expressed in an activity-dependent manner by neuronal cells in less active or immature synapses, flagging them for removal by microglia (149, 152, 156). Of note, the expression of complement proteins at synaptic sites is influenced by astrocyte-derived TGF-β (144), suggesting a cooperation between these glial elements in the process of synaptic pruning. Such active neural network refinement is thought to be involved in learning and memory processes, mediating the removal of specific synaptic connections as a way of active forgetting (157–159). The microglia- and complement-dependent process of synaptic shaping could be crucial in maintaining a physiological balance between retaining relevant memory engrams and removing irrelevant ones (160), and it could be pathologically enhanced in various CNS diseases characterized by learning/memory deficits, including Alzheimer’s disease (158, 159).
In addition, beyond synaptic removal, microglial cells may participate in the functional modulation of synaptic transmission and plasticity by producing soluble immune mediators, including the pro-inflammatory cytokine interleukin-1β (IL-1β) (161) or neurotrophic factors like brain-derived neurotrophic factor (BDNF) (162). Moreover, inflammatory processes of the CNS may be accompanied by a microglia-dependent disruption of neuronal plastic properties, relying on the microglial over-expression of reactive oxygen species (ROS)-producing enzymes such as NADPH oxidase (163). In this scenario, the molecular mechanisms underlying microglia-centered neuro-immune pathways have been mostly investigated in cortical and hippocampal areas, with few reports regarding its involvement in the striatal network (152).
Murine microglial cells display a spectrum of distinct anatomical features, lysosome content and membrane properties across basal ganglia nuclei, suggesting that region-specific local cues could shape the functional state of these cells (164). A recent study described a role for microglia and complement in sex-specific synaptic shaping in the NAc during rat adolescence, with potential key consequences on social behavior (165). Specifically, in adolescent male rats, D1Rs in the NAc were found to be downregulated and degraded through a microglial and C3R-dependent engulfment of C3-tagged D1Rs. Interestingly, the reduced expression of this receptor at synaptic sites in the NAc was correlated with social play behavior in male rats, and the pharmacological interference of C3–C3R interactions was able to increase social play in a D1R-dependent manner (165). This evidence raises the hypothesis that an immune-mediated shaping of striatal synapses may regulate behavioral responses in an age-related way (165).
Moreover, microglial cells could modulate goal-directed and drug-seeking behavior through a molecular pathway, yet to be fully characterized, involving a Toll-like receptor 4 (TLR4)-induced modulation of NMDAR-dependent synaptic plasticity in MSNs of NAc (166), or through the secretion of soluble inflammatory mediators exerting neuromodulatory effects on excitatory striatal transmission, such as TNF-α (167). Interestingly, it has been shown that microglial cells express dopaminergic receptors and DA can modulate the activation of these cells (168–170). Indeed, the release of TNF-α can be induced by the activation of microglial DA D2 receptors (D2Rs) (167). A proper investigation of the molecular pathways linking microglia and striatal synaptic transmission will help understand the puzzling neuro-glial interactions within the basal ganglia network.
Soluble Immune Molecules as Striatal Neuromodulators
Increasing evidence suggests that soluble products of inflammation can influence learning/memory processes and human behavior through the modulation of synaptic transmission and plasticity in different neural networks (2, 3, 5, 6, 171, 172). In this context, the effects exerted by pro-inflammatory cytokines have been mainly investigated in hippocampal and cortical areas, suggesting that some of these molecules could play a key physiological role in memory formation, storage and retrieval (3, 172). The connection between IL-1β production and hippocampal synaptic plasticity induction can be considered as paradigmatic in the field. Synaptic LTP in hippocampal areas was found to be followed by IL-1β gene expression and the genetic ablation or the pharmacological blockade of the IL-1β axis were found to alter the induction and maintenance of hippocampal synaptic long-term changes, together with the execution of hippocampal-dependent memory tasks (173–176). Interestingly, IL-1β-dependent modulation of synaptic plasticity seems to rely on several mechanisms, including the modulation of NMDARs and AMPARs phosphorylation, synaptic localization, and calcium conductance (177–180). Similarly, other cytokines like interleukin-6 (IL-6) (181–184), interleukin-18 (IL-18) (185–187), interferon-γ (IFN-γ) (188, 189), TNF-α and -β (127, 190–196) and, more recently, interleukin-17 (IL-17) (197–199) have been described as key cortical neuromodulators influencing synaptic transmission and plasticity during physiological and pathological conditions, with relevant behavioral and cognitive implications.
On the other side, less is known about the potential effects of such molecules on subcortical network activity. It has been suggested that IL-1β could be involved in the modulation of striatal neurotransmission during both physiological and pathological conditions. Indeed, the exposure of murine corticostriatal slices to IL-1β is followed by an enhanced frequency of spontaneous excitatory transmission, an effect potentially caused by the activation of transient receptor potential vanilloid 1 (TRPV1) channels located in striatal pre-synaptic terminals (200) together with a reduced sensitivity of CB1Rs controlling glutamate release (201). Interestingly, exposure to IL-1β could induce an hyperactivation of MSNs not only through the enhancement of glutamatergic neurotransmission but also by lowering GABAergic inhibition of MSNs (202). In this case too, the modulation of pre-synaptic TRPV1 channels and CB1Rs seems to mediate the IL-1β-dependent modulation of inhibitory terminals (203, 204). The disruption of striatal glutamatergic/GABAergic balance through the IL1β-CB1R axis could play a key role in triggering altered motivated behavior during pathological neuroinflammation, with a possible additional effect linked to an IL-1β-dependent alteration of dopaminergic transmission or DA-induced release of neurotrophic factors (205, 206). Of note, nigral dopaminergic neurons were found to express IL-1 receptors (207), thus suggesting a potential direct modulation of striatal dopaminergic projections.
Another pro-inflammatory cytokine, TNF-α, has been hypothesized to modulate glutamatergic neurotransmission in the striatum. Specifically, exposure of murine brain slices to TNF-α was able to induce an alteration of spontaneous excitatory synaptic currents in MSNs, with an increased duration and decay time that could be reversed by the application of anti-TNF receptor (TNFR)–antibodies (208). Interestingly, the same study has shown that pathological neuroinflammation in mice is characterized by similar striatal synaptic changes in association with an abnormal microglial release of this cytokine and an increased expression and phosphorylation of AMPARs in MSNs (208). These results suggest a potential TNF-α-dependent modulation of glutamatergic neurotransmission, in line with what has been described in other brain areas (209–211). In support of this hypothesis, intracerebroventricular (icv) injections of TNF-α were able to enhance striatal glutamatergic transmission, mimicking the synaptic alterations observed during pathological neuroinflammation (212). Of note, such modifications in striatal excitatory neurotransmission were paralleled by behavioral abnormalities that could be reversed by the icv administration of anti-TNF-α drugs (212).
On the other hand, it has been proposed that TNF-α could play a role in the homeostatic maintenance of excitatory synaptic weights around a firing set point, exerting a physiological and adaptive role aimed at limiting the corticostriatal drive during pathological conditions (213). Specifically, Lewitus and coworkers have shown that TNF-α is upregulated after the prolonged disinhibition of MSNs through the blockade of D2Rs, and can drive AMPARs internalization, DARPP-32 and GluA1 dephosphorylation in these cells (213) collectively reducing corticostriatal synaptic input as an adaptive homeostatic response. Taking into account that this cytokine was found to induce an opposite and rapid exocytosis of AMPARs in hippocampal, motor and visual cortex neurons (214–217), it could be hypothesized that TNF-α could exert region- and neuronal type-specific modulations of activity-dependent synaptic upscaling or downscaling (218). Moreover, considering that the stimulation of microglial D2Rs was found to induce the production of TNF-α by these cells (167), this TNF-α-centered neuro-immune interaction could be involved in the well-known D2R-dependent inhibition of corticostriatal transmission. An abnormal striatal release of this cytokine during neurodegenerative and neuroinflammatory disorders could impair the physiological tuning of corticostriatal inputs, altering basal ganglia activity with potential cognitive and behavioral abnormalities.
Lastly, interferons (IFNs) represent a family of soluble immune mediators exerting pleiotropic immune-modulating effects, with particular regard to immune-surveillance processes against viral infections (219, 220). Interestingly, IFNs can be produced by different cellular subtypes in the CNS and can directly modulate neuronal function and synaptic transmission leading to cognitive and psychiatric disturbances during infectious or inflammatory disorders of the CNS (189, 221–223). Several drugs have been designed to mimic the immunomodulating effects of IFNs, in order to treat human disorders characterized by a pathological immune system activation (219). In this scenario, it has been shown that IFN-β1a is able to reduce the amplitude of excitatory synaptic currents in MSNs, suggesting an inhibitory effect on glutamatergic transmission in the striatum (224). In particular, this cytokine was found to specifically influence NMDAR-mediated synaptic currents in MSNs, interacting with the GluN2A subunit of this receptor, with no effect on the AMPAR-dependent component of striatal excitatory transmission (224). Interestingly, it has been hypothesized that the effect of IFN-β1a on synaptic transmission relies on the activation of post-synaptic Ca2+/Calmodulin(CaM)-dependent protein kinase II (CaMKII) (224), known to strictly interact with NMDAR and GluN2A subunit (225, 226). Of note, the exposure of murine brain slices to IFN-β1a was found to reduce the detrimental consequences induced by mitochondrial complex I inhibition in the striatum, through the modulation of the IFN-activated intracellular JAK-STAT1 pathway (227).
A Synaptocentric Perspective: Striatal Neuroinflammation and Neurological Disorders
The last decades have been characterized by an intense investigation of the crucial role played by neuroinflammation in the pathogenesis of several neurodegenerative disorders (228, 229), including the prototypical disorder of the basal ganglia network: Parkinson’s disease (PD) (230). Mounting evidence suggests that an aberrant immune system activation and a chronic inflammatory process within the CNS may contribute to the progressive loss of midbrain dopaminergic neurons characterizing PD (231–234). Pathological studies showed that midbrain infiltrating T cells (235) and increased basal ganglia levels of proinflammatory cytokines, such as TNF-α, IL-1β and IL-6 (236, 237), can be found in post-mortem brains of PD patients. Moreover, several studies have reported increased levels of pro-inflammatory cytokines in serum and cerebrospinal fluid (CSF) of PD patients (238–242). In this scenario, brain infiltrating immune cells could represent autoreactive T lymphocytes targeting alpha-synuclein (α-syn) aggregates, as suggested by a recent study (243), or different and still unknown neuronal antigens, orchestrating a pathological inflammatory reaction through the production of chemo-active and pro-inflammatory molecules. Several studies performed in experimental PD models have led to hypothesize that such abnormal immune activation in the basal ganglia and the midbrain could act as a co-factor in PD-associated neurodegeneration, by triggering cell-to-cell death signals or because of the toxic damage induced by soluble pro-inflammatory cytokines (235, 244–250).
Resident immune cells are thought to be involved in these pathological processes. Early reports showed high levels of activated microglia in the midbrain and in the striatum of PD brains (251, 252), and the temporal relationship between the presence of inflammation with activated microglia and the emergence of α-syn pathology has been recently investigated in dopaminergic neuronal grafts implanted in the striatum of PD patients (253). Interestingly, the authors have found evidence of inflammation long before the accumulation of α-syn, supporting the concept that microglia plays an integral role in the propagation and spread of α-syn pathology (253). Studies performed in experimental models supported the hypothesis of a microglial-driven degeneration of dopaminergic neurons during the disease, probably due to an increased production of reactive oxygen species and soluble mediators like TNF-α and IL-1β (254–260). Similar neurotoxic effects have been described for chemokines and cytokines released by the reactive and dysfunctional astrocytes that have been identified in human and experimental PD (261, 262) since the early phases of the disease (263). However, most of the studies have focused on the pathological neuro-immune interactions potentially triggering progressive dopaminergic neuronal death during PD, overlooking the possible detrimental influence exerted by activated immune cells on synaptic connections within the basal ganglia network, which could also anticipate irreversible cell loss.
As introduced above, the abnormal release of pro-inflammatory soluble mediators within the striatum, together with the loss of the physiological supporting functions exerted by glial cells, could alter the function of the striatal synaptic network during PD development, long before the occurrence of neurodegenerative features (Figure 2). Accordingly, it has been hypothesized that a striatal immune-mediated synaptopathy could account for disabling cognitive, motor and behavioral abnormalities in PD patients (264), which can be highlighted in disease stages characterized by a still partial dopaminergic cell loss (265). In this scenario, several studies performed in experimental models of PD have highlighted that synaptic dysfunction can be considered as an early event in the pathogenesis of the disease, altering the ability of corticostriatal connections to express short- and long-term plastic changes (266–268). The disruption of the physiological filtering activity of the basal ganglia network could induce an extensive reorganization of the overall architecture of brain node connectivity since early disease stages, as shown by functional imaging studies (269, 270).
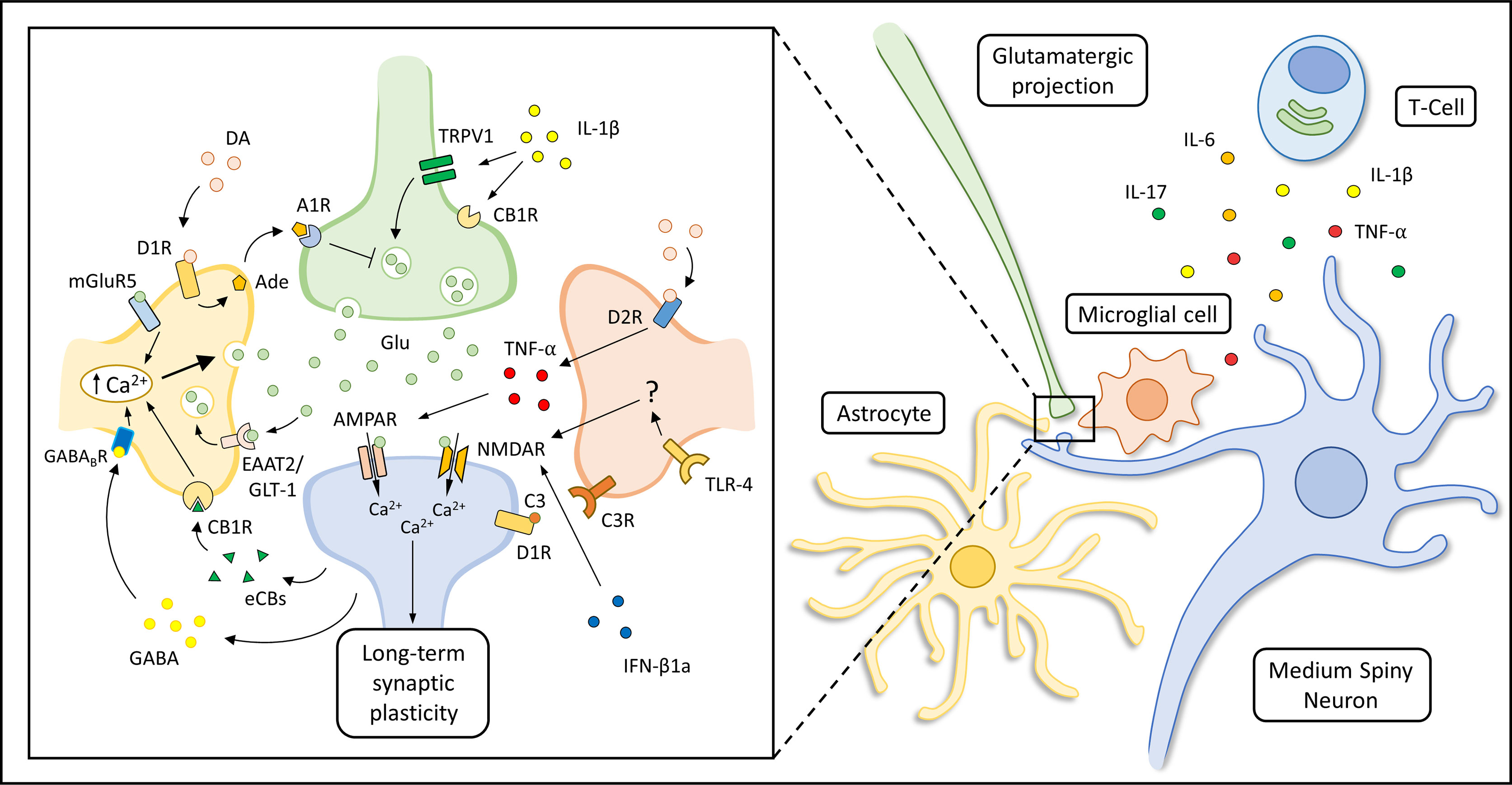
Figure 2 Immune modulation of striatal synaptic transmission. Suggested mechanisms underlying astrocytic, microglial and immune modulation of corticostriatal synaptic transmission are represented in the box on the left. The production of soluble immune mediators (like IL-1β, TNF-α, IL-6 and IL-17) by activated T-cells, astrocytes and microglial cells can influence striatal transmission during the course of neuro-psychiatric disorders. Specifically, IL-1β can enhance striatal excitatory transmission activating transient receptor potential vanilloid 1 (TRPV1) channels (200) and reducing CB1 receptors (CB1Rs) activation (201) at pre-synaptic glutamatergic terminals. In addition, it has been shown that TNF-α can increase the decay time and duration of spontaneous striatal excitatory transmission during pathological neuroinflammation (208) or induce AMPAR internalization as an adaptive response to prolonged MSNs disinhibition (167, 213). Microglial cells can also regulate dopamine D1R expression through a complement (C3-C3R) dependent internalization and degradation of this receptor (165). Other authors have shown that IFN-β1a can inhibit NMDAR-mediated glutamatergic transmission interacting with NMDAR subunit and CaMKII (224). Glutamate (Glu) released in the synaptic cleft could activate AMPARs and NMDARs of MSNs, but could also act on metabotropic receptor subtype 5 (mGluR5) expressed by astrocytes triggering a Ca2+-dependent release of Glu, sustaining MSNs activation for minutes after the initial stimulus (133). Depolarization of MSNs is associated with endocannabinoids (eCBS) release which can activate astrocytic CB1Rs leading to an increase of intracellular Ca2+ levels and glutamate (Glu) release (131). Moreover, up-state MSNs could lead to Gi-coupled GABAB receptor activation in neighboring astrocytes through dendritic GABA release (Nagai et al., 2019). The activation of this astrocytic receptor is thought to induce astrocytic Ca2+ release from cellular stores and influence striatal excitatory transmission through the production of the synaptogenic cue TSP1 (not shown in the figure). The astrocytic expression of EAAT2/GLT-1 is thought to be required for the maintenance of a proper Glu concentration in the synaptic cleft. Astrocytic Glu reuptake allows the detection of the temporal contingency of synaptic stimuli, modulating the induction of corticostriatal synaptic plasticity (99). In addition, dopamine (DA) could trigger the release of ATP/adenosine (Ade) by astrocytes through D1Rs activation, leading to A1 receptor (A1R)-dependent inhibition of striatal excitatory transmission (143). Microglial cells can modulate NMDAR-dependent synaptic plasticity in MSNs through a still not fully characterized Toll-like receptor 4 (TLR4)-dependent mechanism (166), or influence glutamatergic transmission through the secretion of tumor necrosis factor α (TNF-α), which could be also induced by the activation of microglial D2Rs (167).
To date, PD-related synaptopathy has been mainly linked to the synaptic and molecular effects of pathological α-syn aggregates (271–278). However, an involvement of the immune system in the synaptic dysfunction triggered by α-syn accumulation cannot be ruled out since α-syn can activate different subsets of T-cells (243) and oligomeric or fibrillary α-syn can induce a pro-inflammatory activation of microglia through the interaction with toll-like receptor 2 (TLR2) (279) or the activation of NF-κB pathway (262). Moreover, it has been suggested that astrocytes can uptake and accumulate the pathological α-syn released by neighboring neuronal cells (280), triggering a pro-inflammatory astrocytic reaction with the production of soluble molecules such as IL-1β, IL-6 and TNF-α (281). Collectively, accumulating evidence suggests that striatal neurons and synapses could be submerged in an inflammatory micro-environment linked to α-syn aggregation (282), with the exposure to soluble immune molecules with demonstrated modulatory effects on corticostriatal terminals. Such abnormal immune influence on striatal transmission could also account for late disease complications, like L-DOPA-induced dyskinesia (283), which pathogenesis is thought to rely on an abnormal corticostriatal synaptic plasticity (284–286).
The potential relevance of pathological neuro-immune interactions during PD is supported by a recent study showing that corticostriatal synaptic plasticity can be rescued through the modulation of astrocytic and microglial activation by transcranial magnetic stimulation (TMS) (287). Specifically, it has been shown that the loss of LTD and LTP of corticostriatal projections accompanying striatal dopaminergic denervation can be restored by TMS treatment in an experimental model of PD (287). The beneficial effects of TMS on synaptic function were paralleled by an increase in striatal DA levels and an amelioration of PD-related deficits in motor behavior. Interestingly, such a therapeutic protocol was also associated with a significant reduction of astrocytic and microglial pro-inflammatory responses in the striatum (287). This result is of particular relevance since glial cells have been proposed as key targets and effectors of TMS protocols, potentially mediating widespread effects in neural networks through extensive connections and cell type-specific modulation of neuronal firing (288). The TMS-dependent reduction of pathological glial activation in the striatum could lower the production of soluble pro-inflammatory mediators and lead to the recovery of glial supporting functions relevant for neurons, like the modulation of DA and glutamate metabolism, reuptake, or release (138, 289). In line with these findings, another research group has shown that the modulation of astrocytic glutamate content and reuptake in the globus pallidus pars externa (GPe) is able to restore the proper pre-synaptic tuning of striato-pallidal input in an experimental model of PD (290). This result is interesting since the hyperactivity of striato-pallidal pathway is thought to underlie hypokinetic features of PD patients (291, 292) and the astrocytic gating of these synapses, which is disrupted during PD, can represent an alternative therapeutic strategy (290).
Further investigations are required to clearly decipher the potential dysfunction of striatal astrocytes during the course of PD, but findings obtained in research studies on other basal ganglia disorders like HD seem to suggest that the loss of physiological astrocytic properties could be associated with altered action selection, habit formation, impulse inhibition and motor behavior (111, 293). HD is a genetic disorder primarily affecting cortico-basal ganglia-thalamo-cortical network, linked to a pathological expansion of a polyglutamine-encoding CAG repeat in the huntingtin gene (HTT) and characterized by progressive motor hyperactivity with psychiatric and cognitive disturbances (294, 295). Accumulating evidence, both clinical and pre-clinical, suggests that an alteration in astrocytic activity could be deeply involved in the pathogenesis of HD (261, 293). In post-mortem tissues obtained from HD patients, the striatum was characterized by a significant astrocytic reaction (296) with an altered expression of the transporter EAAT2/GLT-1, potentially triggered by mutant HTT (mHTT) (297–299). Interestingly, the delivery of mHTT-expressing human glial cells to the striatum was able to cause an HD-like phenotype in mice, with the evidence of MSNs hyperexcitability and striatum-dependent motor impairment (300). Conversely, the selective deletion of mHTT in astrocytes was found to be protective in an experimental model of HD, with beneficial effects on motor and psychiatric-like disturbances (301). Overall, it has been hypothesized that an altered expression of astrocytic proteins, such as Kir4.1 and EAAT2/GLT-1, together with an impairment in astrocytic Ca2+ signaling, could alter MSNs membrane excitability and disrupt cortico-striatal glutamatergic transmission (111, 293). Such disease-related synaptic and neuronal abnormalities could influence the physiological mechanisms underlying basal ganglia’s ability to inhibit context-inappropriate actions, leading to the typical excessive and uncontrolled motor behavior of HD patients. In line with this hypothesis, the pathological activation of striatal glial cells has been proposed as a key pathogenic factor in neuropsychiatric disorders characterized by repetitive and impulsive behaviors like OCD, TS and “pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections” (PANDAS) (302, 303), potentially inducing a disruption of synaptic tuning in the striatal network. In this scenario, the neuronal and synaptic consequences triggered by the acquisition of a pro-inflammatory phenotype by striatal astrocytes and the role of microglial cells during HD are still under investigation (261, 304). Overall, interventions aimed at lowering the immune system activation and restoring the physiological glial functions could counteract the synaptic imbalance characterizing basal ganglia disorders, limiting early clinical features, late disease complications and, potentially, disease progression in several neurological and psychiatric diseases.
Concluding Remarks
The functional view of the basal ganglia network slowly moved from the brain motor control station to the decision-maker of appropriate context-dependent cognitive, behavioral and emotional responses. In parallel, the physiological processes underlying the integrative and filtering activity of the basal ganglia started to be deciphered during the last decades. Plastic properties of striatal synaptic connections have been demonstrated as crucial for the integration of multimodal cortical inputs and for conveying a proper basal ganglia output driving an individual’s action selection/inhibition and habit formation. In this scenario, the current evidence on the neuro-modulatory role played by immuno-glial cells in cortical areas suggests that corticostriatal projections and subcortical networks can be influenced by the immune system as well.
The characterization of the neuro-immune interactions taking place in the striatum, both in its dorsal and ventral areas, could help to decipher the molecular mechanisms underlying the previously underscored effects of the immune system on motivated and context-dependent human behavior. The identification of cells and soluble immune mediators involved in the striatal neuro-immune cross-talk could lead to a new approach to basal ganglia disorders, disclosing a novel pathophysiological view for motor, behavioral, cognitive and emotional abnormalities accompanying neurological and psychiatric disorders.
Author Contributions
MDF conceived the review. AM performed the literature review, wrote the manuscript draft and prepared the figures. MDF, VG, LP, and PC reviewed and integrated the manuscript draft and the figures. All authors contributed to the article and approved the submitted version.
Funding
This work has been supported by the Marlene and Paolo Fresco Institute for Parkinson’s and Movement Disorder, Fresco Parkinson Institute, New York University School of Medicine.
Conflict of Interest
AM received travel grants from Biogen, Novartis, Merck, Teva, Almirall and Sanofi to attend national and international conferences. MDF participated on advisory boards for and received speaker or writing honoraria and funding for traveling from Bayer, Biogen Idec, Sanofi, Merck, Mylan, Novartis, Roche and Teva. PC participated on advisory boards for and received funding for traveling, speaker honoraria and research support from AbbVie, Biogen Idec, Merck, Genzyme, Novartis, Prexton, Teva, UCB and Zambon.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Louveau A, Harris TH, Kipnis J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol (2015) 36:569–77. doi: 10.1016/j.it.2015.08.006
2. Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Sci (80 ) (2016) 353:766–71. doi: 10.1126/science.aag2638
3. Di Filippo M, Sarchielli P, Picconi B, Calabresi P. Neuroinflammation and synaptic plasticity: theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends Pharmacol Sci (2008) 29:402–12. doi: 10.1016/j.tips.2008.06.005
4. Di Filippo M, Portaccio E, Mancini A, Calabresi P. Multiple sclerosis and cognition: synaptic failure and network dysfunction. Nat Rev Neurosci (2018) 19:599–609. doi: 10.1038/s41583-018-0053-9
5. Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun (2011) 25:181–213. doi: 10.1016/j.bbi.2010.10.015
6. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci (2008) 9:46–56. doi: 10.1038/nrn2297
7. Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci (2017) 20:136–44. doi: 10.1038/nn.4475
8. Di Filippo M, de Iure A, Durante V, Gaetani L, Mancini A, Sarchielli P, et al. Synaptic plasticity and experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Brain Res (2015) 1621:205–13. doi: 10.1016/j.brainres.2014.12.004
9. Mancini A, Gaetani L, Di Gregorio M, Tozzi A, Ghiglieri V, Calabresi P, et al. Hippocampal neuroplasticity and inflammation: relevance for multiple sclerosis. Mult Scler Demyelinating Disord (2017) 2:2. doi: 10.1186/s40893-017-0019-1
10. Bostan AC, Strick PL. The basal ganglia and the cerebellum: nodes in an integrated network. Nat Rev Neurosci (2018) 19:338–50. doi: 10.1038/s41583-018-0002-7
11. Obeso I, Wilkinson L, Casabona E, Bringas ML, Álvarez M, Álvarez L, et al. Deficits in inhibitory control and conflict resolution on cognitive and motor tasks in Parkinson’s disease. Exp Brain Res (2011) 212:371–84. doi: 10.1007/s00221-011-2736-6
12. DeLong MR, Alexander GE, Georgopoulos AP, Crutcher MD, Mitchell SJ, Richardson RT. Role of basal ganglia in limb movements. Hum Neurobiol (1984) 2:235–44.
13. Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci (1986) 9:357–81. doi: 10.1146/annurev.ne.09.030186.002041
14. Middleton FA. Basal-ganglia “Projections” to the Prefrontal Cortex of the Primate. Cereb Cortex (2002) 12:926–35. doi: 10.1093/cercor/12.9.926
15. Calabresi P, Picconi B, Tozzi A, Ghiglieri V, Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat Neurosci (2014) 17:1022–30. doi: 10.1038/nn.3743
16. Dubé L, Smith AD, Bolam JP. Identification of synaptic terminals of thalamic or cortical origin in contact with distinct medium-size spiny neurons in the rat neostriatum. J Comp Neurol (1988) 267:455–71. doi: 10.1002/cne.902670402
17. Lapper SR, Bolam JP. Input from the frontal cortex and the parafascicular nucleus to cholinergic interneurons in the dorsal striatum of the rat. Neuroscience (1992) 51:533–45. doi: 10.1016/0306-4522(92)90293-B
18. Pickel VM, Chan J, Sesack SR. Cellular basis for interactions between catecholaminergic afferents and neurons containing leu-enkephalin-like immunoreactivity in rat caudate-putamen nuclei. J Neurosci Res (1992) 31:212–30. doi: 10.1002/jnr.490310203
19. Bouyer JJ, Park DH, Joh TH, Pickel VM. Chemical and structural analysis of the relation between cortical inputs and tyrosine hydroxylase-containing terminals in rat neostriatum. Brain Res (1984) 302:267–75. doi: 10.1016/0006-8993(84)90239-7
20. Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci (1995) 18:527–35. doi: 10.1016/0166-2236(95)98374-8
21. Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci (2006) 7:464–76. doi: 10.1038/nrn1919
22. Calabresi P, Gubellini P, Centonze D, Picconi B, Bernardi G, Chergui K, et al. Dopamine and cAMP-Regulated Phosphoprotein 32 kDa Controls Both Striatal Long-Term Depression and Long-Term Potentiation, Opposing Forms of Synaptic Plasticity. J Neurosci (2000) 20:8443–51. doi: 10.1523/JNEUROSCI.20-22-08443.2000
23. Kerr JND, Wickens JR. Dopamine D-1/D-5 Receptor Activation Is Required for Long-Term Potentiation in the Rat Neostriatum In Vitro. J Neurophysiol (2001) 85:117–24. doi: 10.1152/jn.2001.85.1.117
24. Calabresi P, Saiardi A, Pisani A, Baik J-H, Centonze D, Mercuri NB, et al. Abnormal Synaptic Plasticity in the Striatum of Mice Lacking Dopamine D2 Receptors. J Neurosci (1997) 17:4536–44. doi: 10.1523/JNEUROSCI.17-12-04536.1997
25. Tozzi A, de Iure A, Di Filippo M, Tantucci M, Costa C, Borsini F, et al. The Distinct Role of Medium Spiny Neurons and Cholinergic Interneurons in the D2/A2A Receptor Interaction in the Striatum: Implications for Parkinson’s Disease. J Neurosci (2011) 31:1850–62. doi: 10.1523/JNEUROSCI.4082-10.2011
26. Calabresi P, Maj R, Pisani A, Mercuri N, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci (1992) 12:4224–33. doi: 10.1523/JNEUROSCI.12-11-04224.1992
27. Lovinger DM, Tyler EC, Merritt A. Short- and long-term synaptic depression in rat neostriatum. J Neurophysiol (1993) 70:1937–49. doi: 10.1152/jn.1993.70.5.1937
28. Walsh JP. Depression of excitatory synaptic input in rat striatal neurons. Brain Res (1993) 608:123–8. doi: 10.1016/0006-8993(93)90782-I
29. Bagetta V, Picconi B, Marinucci S, Sgobio C, Pendolino V, Ghiglieri V, et al. Dopamine-dependent long-term depression is expressed in striatal spiny neurons of both direct and indirect pathways: implications for Parkinson’s disease. J Neurosci (2011) 31:12513–22. doi: 10.1523/JNEUROSCI.2236-11.2011
30. Gerfen C, Engber T, Mahan L, Susel Z, Chase T, Monsma F, et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Sci (80 ) (1990) 250:1429–32. doi: 10.1126/science.2147780
31. Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci (1992) 15(4):133–9. doi: doi: 10.1016/0166-2236(92)90355-c
32. Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci (1996) 16:6579–91. doi: 10.1523/jneurosci.16-20-06579.1996
33. Rico AJ, Dopeso-Reyes IG, Martínez-Pinilla E, Sucunza D, Pignataro D, Roda E, et al. Neurochemical evidence supporting dopamine D1–D2 receptor heteromers in the striatum of the long-tailed macaque: changes following dopaminergic manipulation. Brain Struct Funct (2017) 222:1767–84. doi: 10.1007/s00429-016-1306-x
34. Hasbi A, Fan T, Alijaniaram M, Nguyen T, Perreault ML, O’Dowd BF, et al. Calcium signaling cascade links dopamine D1–D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc Natl Acad Sci (2009) 106:21377–82. doi: 10.1073/pnas.0903676106
35. Perreault ML, Hasbi A, O’Dowd BF, George SR. Heteromeric Dopamine Receptor Signaling Complexes: Emerging Neurobiology and Disease Relevance. Neuropsychopharmacology (2014) 39:156–68. doi: 10.1038/npp.2013.148
36. Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, et al. Dopaminergic Control of Corticostriatal Long-Term Synaptic Depression in Medium Spiny Neurons Is Mediated by Cholinergic Interneurons. Neuron (2006) 50:443–52. doi: 10.1016/j.neuron.2006.04.010
37. Augustin SM, Chancey JH, Lovinger DM. Dual Dopaminergic Regulation of Corticostriatal Plasticity by Cholinergic Interneurons and Indirect Pathway Medium Spiny Neurons. Cell Rep (2018) 24:2883–93. doi: 10.1016/j.celrep.2018.08.042
38. Centonze D, Grande C, Usiello A, Gubellini P, Erbs E, Martín AB, et al. Receptor Subtypes Involved in the Presynaptic and Postsynaptic Actions of Dopamine on Striatal Interneurons. J Neurosci (2003) 23:6245–54. doi: 10.1523/JNEUROSCI.23-15-06245.2003
39. Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci (2007) 30:211–9. doi: 10.1016/j.tins.2007.03.001
40. Trusel M, Cavaccini A, Gritti M, Greco B, Saintot P-P, Nazzaro C, et al. Coordinated Regulation of Synaptic Plasticity at Striatopallidal and Striatonigral Neurons Orchestrates Motor Control. Cell Rep (2015) 13:1353–65. doi: 10.1016/j.celrep.2015.10.009
41. Cazorla M, de Carvalho FD, Chohan MO, Shegda M, Chuhma N, Rayport S, et al. Dopamine D2 Receptors Regulate the Anatomical and Functional Balance of Basal Ganglia Circuitry. Neuron (2014) 81:153–64. doi: 10.1016/j.neuron.2013.10.041
42. Nadjar A. Phenotype of Striatofugal Medium Spiny Neurons in Parkinsonian and Dyskinetic Nonhuman Primates: A Call for a Reappraisal of the Functional Organization of the Basal Ganglia. J Neurosci (2006) 26:8653–61. doi: 10.1523/JNEUROSCI.2582-06.2006
43. Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, et al. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature (2013) 494:238–42. doi: 10.1038/nature11846
44. Jin X, Tecuapetla F, Costa RM. Basal ganglia subcircuits distinctively encode the parsing and concatenation of action sequences. Nat Neurosci (2014) 17:423–30. doi: 10.1038/nn.3632
45. Schmidt R, Leventhal DK, Mallet N, Chen F, Berke JD. Canceling actions involves a race between basal ganglia pathways. Nat Neurosci (2013) 16:1118–24. doi: 10.1038/nn.3456
46. Redgrave P, Prescott TJ, Gurney K. The basal ganglia: a vertebrate solution to the selection problem? Neuroscience (1999) 89:1009–23. doi: 10.1016/s0306-4522(98)00319-4
47. Marsden CD. The mysterious motor function of the basal ganglia: the Robert Wartenberg Lecture. Neurology (1982) 32:514–39. doi: 10.1212/wnl.32.5.514
48. Stephenson-Jones M, Samuelsson E, Ericsson J, Robertson B, Grillner S. Evolutionary Conservation of the Basal Ganglia as a Common Vertebrate Mechanism for Action Selection. Curr Biol (2011) 21:1081–91. doi: 10.1016/j.cub.2011.05.001
49. Kemp JM, Powell TP. The connexions of the striatum and globus pallidus: synthesis and speculation. Philos Trans R Soc London B Biol Sci (1971) 262:441–57. doi: 10.1098/rstb.1971.0106
50. Bostan AC, Dum RP, Strick PL. Functional Anatomy of Basal Ganglia Circuits with the Cerebral Cortex and the Cerebellum. Prog Neurol Surg (2018) 33:50–61. doi: 10.1159/000480748
51. Joel D, Weiner I. The organization of the basal ganglia-thalamocortical circuits: Open interconnected rather than closed segregated. Neuroscience (1994) 63:363–79. doi: 10.1016/0306-4522(94)90536-3
52. Haynes WIA, Haber SN. The Organization of Prefrontal-Subthalamic Inputs in Primates Provides an Anatomical Substrate for Both Functional Specificity and Integration: Implications for Basal Ganglia Models and Deep Brain Stimulation. J Neurosci (2013) 33:4804–14. doi: 10.1523/JNEUROSCI.4674-12.2013
53. Hunnicutt BJ, Jongbloets BC, Birdsong WT, Gertz KJ, Zhong H, Mao T. A comprehensive excitatory input map of the striatum reveals novel functional organization. Elife (2016) 5:e19103. doi: 10.7554/eLife.19103
54. Hintiryan H, Foster NN, Bowman I, Bay M, Song MY, Gou L, et al. The mouse cortico-striatal projectome. Nat Neurosci (2016) 19:1100–14. doi: 10.1038/nn.4332
55. Balleine BW, O’Doherty JP. Human and rodent homologies in action control: corticostriatal determinants of goal-directed and habitual action. Neuropsychopharmacology (2010) 35:48–69. doi: 10.1038/npp.2009.131
56. Graybiel AM. Habits, Rituals, and the Evaluative Brain. Annu Rev Neurosci (2008) 31:359–87. doi: 10.1146/annurev.neuro.29.051605.112851
57. Yu J, Ishikawa M, Wang J, Schlüter OM, Sesack SR, Dong Y. Ventral Tegmental Area Projection Regulates Glutamatergic Transmission in Nucleus Accumbens. Sci Rep (2019) 9:18451. doi: 10.1038/s41598-019-55007-y
58. Schultz W. Dopamine reward prediction-error signalling: a two-component response. Nat Rev Neurosci (2016) 17:183–95. doi: 10.1038/nrn.2015.26
59. Sesack SR, Grace AA. Cortico-Basal Ganglia Reward Network: Microcircuitry. Neuropsychopharmacology (2010) 35:27–47. doi: 10.1038/npp.2009.93
60. McNab F, Klingberg T. Prefrontal cortex and basal ganglia control access to working memory. Nat Neurosci (2008) 11:103–7. doi: 10.1038/nn2024
61. O’Reilly RC, Frank MJ. Making Working Memory Work: A Computational Model of Learning in the Prefrontal Cortex and Basal Ganglia. Neural Comput (2006) 18:283–328. doi: 10.1162/089976606775093909
62. Frank MJ, Samanta J, Moustafa AA, Sherman SJ. Hold Your Horses: Impulsivity, Deep Brain Stimulation, and Medication in Parkinsonism. Sci (80 ) (2007) 318:1309–12. doi: 10.1126/science.1146157
63. Cavanagh JF, Wiecki TV, Cohen MX, Figueroa CM, Samanta J, Sherman SJ, et al. Subthalamic nucleus stimulation reverses mediofrontal influence over decision threshold. Nat Neurosci (2011) 14:1462–7. doi: 10.1038/nn.2925
64. Obeso I, Wilkinson L, Casabona E, Speekenbrink M, Luisa Bringas M, Álvarez M, et al. The subthalamic nucleus and inhibitory control: impact of subthalamotomy in Parkinson’s disease. Brain (2014) 137:1470–80. doi: 10.1093/brain/awu058
65. Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Prim (2015) 1:15005. doi: 10.1038/nrdp.2015.5
66. Burguière E, Monteiro P, Mallet L, Feng G, Graybiel AM. Striatal circuits, habits, and implications for obsessive-compulsive disorder. Curr Opin Neurobiol (2015) 30:59–65. doi: 10.1016/j.conb.2014.08.008
67. Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (1973) 232:331–56. doi: 10.1113/jphysiol.1973.sp010273
68. Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature (1993) 361:31–9. doi: 10.1038/361031a0
69. Titley HK, Brunel N, Hansel C. Toward a Neurocentric View of Learning. Neuron (2017) 95:19–32. doi: 10.1016/j.neuron.2017.05.021
70. Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron (2004) 44:5–21. doi: 10.1016/j.neuron.2004.09.012
71. Mahon S, Deniau J-M, Charpier S. Corticostriatal plasticity: life after the depression. Trends Neurosci (2004) 27:460–7. doi: 10.1016/j.tins.2004.06.010
72. Calabresi P, Pisani A, Mercuri NB, Bernardi G. The corticostriatal projection: from synaptic plasticity to dysfunctions of the basal ganglia. Trends Neurosci (1996) 19:19–24. doi: 10.1016/0166-2236(96)81862-5
73. Di Filippo M, Picconi B, Tantucci M, Ghiglieri V, Bagetta V, Sgobio C, et al. Short-term and long-term plasticity at corticostriatal synapses: implications for learning and memory. Behav Brain Res (2009) 199:108–18. doi: 10.1016/j.bbr.2008.09.025
74. Deffains M, Bergman H. Striatal cholinergic interneurons and cortico-striatal synaptic plasticity in health and disease. Mov Disord (2015) 30:1014–25. doi: 10.1002/mds.26300
75. Tepper JM, Bolam JP. Functional diversity and specificity of neostriatal interneurons. Curr Opin Neurobiol (2004) 14:685–92. doi: 10.1016/j.conb.2004.10.003
76. Shipp S. The functional logic of corticostriatal connections. Brain Struct Funct (2017) 222:669–706. doi: 10.1007/s00429-016-1250-9
77. Sippy T, Lapray D, Crochet S, Petersen CCH. Cell-Type-Specific Sensorimotor Processing in Striatal Projection Neurons during Goal-Directed Behavior. Neuron (2015) 88:298–305. doi: 10.1016/j.neuron.2015.08.039
78. O’Hare JK, Ade KK, Sukharnikova T, Van Hooser SD, Palmeri ML, Yin HH, et al. Pathway-Specific Striatal Substrates for Habitual Behavior. Neuron (2016) 89:472–9. doi: 10.1016/j.neuron.2015.12.032
79. Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci (2012) 15:816–8. doi: 10.1038/nn.3100
80. Tecuapetla F, Matias S, Dugue GP, Mainen ZF, Costa RM. Balanced activity in basal ganglia projection pathways is critical for contraversive movements. Nat Commun (2014) 5:4315. doi: 10.1038/ncomms5315
81. Klaus A, Martins GJ, Paixao VB, Zhou P, Paninski L, Costa RM. The Spatiotemporal Organization of the Striatum Encodes Action Space. Neuron (2017) 95:1171–80.e7. doi: 10.1016/j.neuron.2017.08.015
82. Markowitz JE, Gillis WF, Beron CC, Neufeld SQ, Robertson K, Bhagat ND, et al. The Striatum Organizes 3D Behavior via Moment-to-Moment Action Selection. Cell (2018) 174:44–58.e17. doi: 10.1016/j.cell.2018.04.019
83. Kupferschmidt DA, Juczewski K, Cui G, Johnson KA, Lovinger DM. Parallel, but Dissociable, Processing in Discrete Corticostriatal Inputs Encodes Skill Learning. Neuron (2017) 96:476–89.e5. doi: 10.1016/j.neuron.2017.09.040
84. Yin HH, Mulcare SP, Hilário MRF, Clouse E, Holloway T, Davis MI, et al. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat Neurosci (2009) 12:333–41. doi: 10.1038/nn.2261
85. Thorn CA, Graybiel AM. Differential Entrainment and Learning-Related Dynamics of Spike and Local Field Potential Activity in the Sensorimotor and Associative Striatum. J Neurosci (2014) 34:2845–59. doi: 10.1523/JNEUROSCI.1782-13.2014
86. Thorn CA, Atallah H, Howe M, Graybiel AM. Differential Dynamics of Activity Changes in Dorsolateral and Dorsomedial Striatal Loops during Learning. Neuron (2010) 66:781–95. doi: 10.1016/j.neuron.2010.04.036
87. Yagishita S, Hayashi-Takagi A, Ellis-Davies GCR, Urakubo H, Ishii S, Kasai H. A critical time window for dopamine actions on the structural plasticity of dendritic spines. Sci (80 ) (2014) 345:1616–20. doi: 10.1126/science.1255514
88. Garris PA, Christensen JRC, Rebec GV, Wightman RM. Real-Time Measurement of Electrically Evoked Extracellular Dopamine in the Striatum of Freely Moving Rats. J Neurochem (2002) 68:152–61. doi: 10.1046/j.1471-4159.1997.68010152.x
89. Reynolds JNJ, Hyland BI, Wickens JR. A cellular mechanism of reward-related learning. Nature (2001) 413:67–70. doi: 10.1038/35092560
90. Redgrave P, Vautrelle N, Reynolds JNJ. Functional properties of the basal ganglia’s re-entrant loop architecture: selection and reinforcement. Neuroscience (2011) 198:138–51. doi: 10.1016/j.neuroscience.2011.07.060
91. Fisher SD, Robertson PB, Black MJ, Redgrave P, Sagar MA, Abraham WC, et al. Reinforcement determines the timing dependence of corticostriatal synaptic plasticity in vivo. Nat Commun (2017) 8:334. doi: 10.1038/s41467-017-00394-x
92. Feldman DE. The Spike-Timing Dependence of Plasticity. Neuron (2012) 75:556–71. doi: 10.1016/j.neuron.2012.08.001
93. Perrin E, Venance L. Bridging the gap between striatal plasticity and learning. Curr Opin Neurobiol (2019) 54:104–12. doi: 10.1016/j.conb.2018.09.007
94. Xu H, Perez S, Cornil A, Detraux B, Prokin I, Cui Y, et al. Dopamine–endocannabinoid interactions mediate spike-timing-dependent potentiation in the striatum. Nat Commun (2018) 9:4118. doi: 10.1038/s41467-018-06409-5
95. Cui Y, Paillé V, Xu H, Genet S, Delord B, Fino E, et al. Endocannabinoids mediate bidirectional striatal spike-timing-dependent plasticity. J Physiol (2015) 593:2833–49. doi: 10.1113/JP270324
96. Cui Y, Prokin I, Xu H, Delord B, Genet S, Venance L, et al. Endocannabinoid dynamics gate spike-timing dependent depression and potentiation. Elife (2016) 5:e13185. doi: 10.7554/eLife.13185
97. Cavaccini A, Gritti M, Giorgi A, Locarno A, Heck N, Migliarini S, et al. Serotonergic Signaling Controls Input-Specific Synaptic Plasticity at Striatal Circuits. Neuron (2018) 98:801–16.e7. doi: 10.1016/j.neuron.2018.04.008
98. Reiner A, Jiao Y, Del Mar N, Laverghetta AV, Lei WL. Differential morphology of pyramidal tract-type and intratelencephalically projecting-type corticostriatal neurons and their intrastriatal terminals in rats. J Comp Neurol (2003) 457:420–40. doi: 10.1002/cne.10541
99. Valtcheva S, Venance L. Astrocytes gate Hebbian synaptic plasticity in the striatum. Nat Commun (2016) 7:13845. doi: 10.1038/ncomms13845
100. Wilson C, Kawaguchi Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci (1996) 16:2397–410. doi: 10.1523/JNEUROSCI.16-07-02397.1996
101. Stern EA, Kincaid AE, Wilson CJ. Spontaneous Subthreshold Membrane Potential Fluctuations and Action Potential Variability of Rat Corticostriatal and Striatal Neurons In Vivo. J Neurophysiol (1997) 77:1697–715. doi: 10.1152/jn.1997.77.4.1697
102. Stern EA, Jaeger D, Wilson CJ. Membrane potential synchrony of simultaneously recorded striatal spiny neurons in vivo. Nature (1998) 394:475–8. doi: 10.1038/28848
103. Araque A, Carmignoto G, Haydon PG, Oliet SHR, Robitaille R, Volterra A. Gliotransmitters travel in time and space. Neuron (2014) 81:728–39. doi: 10.1016/j.neuron.2014.02.007
104. Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci (2009) 32:421–31. doi: 10.1016/j.tins.2009.05.001
105. Schafer DP, Lehrman EK, Stevens B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia (2013) 61:24–36. doi: 10.1002/glia.22389
106. Mederos S, González-Arias C, Perea G. Astrocyte–Neuron Networks: A Multilane Highway of Signaling for Homeostatic Brain Function. Front Synaptic Neurosci (2018) 10:45. doi: 10.3389/fnsyn.2018.00045
107. Herculano-Houzel S. The glia/neuron ratio: How it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia (2014) 62:1377–91. doi: 10.1002/glia.22683
108. Allen NJ, Lyons DA. Glia as architects of central nervous system formation and function. Science (2018) 362:181–5. doi: 10.1126/science.aat0473
109. Allen NJ, Barres BA. Neuroscience: Glia - more than just brain glue. Nature (2009) 457:675–7. doi: 10.1038/457675a
110. Khakh BS, Sofroniew MV. Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci (2015) 18:942–52. doi: 10.1038/nn.4043
111. Khakh BS. Astrocyte–Neuron Interactions in the Striatum: Insights on Identity, Form, and Function. Trends Neurosci (2019) 42:617–30. doi: 10.1016/j.tins.2019.06.003
112. von Bartheld CS, Bahney J, Herculano-Houzel S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J Comp Neurol (2016) 524:3865–95. doi: 10.1002/cne.24040
113. Azevedo FAC, Carvalho LRB, Grinberg LT, Farfel JM, Ferretti REL, Leite REP, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol (2009) 513:532–41. doi: 10.1002/cne.21974
114. Charles AC, Merrill JE, Dirksen ER, Sandersont MJ. Intercellular signaling in glial cells: Calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron (1991) 6:983–92. doi: 10.1016/0896-6273(91)90238-U
115. Cornell-Bell A, Finkbeiner S, Cooper M, Smith S. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Sci (80 ) (1990) 247:470–3. doi: 10.1126/science.1967852
116. Verkhratsky A, Kettenmann H. Calcium signalling in glial cells. Trends Neurosci (1996) 19:346–52. doi: 10.1016/0166-2236(96)10048-5
117. Perea G, Araque A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci (2005) 25:2192–203. doi: 10.1523/JNEUROSCI.3965-04.2005
118. Arizono M, Inavalli VVGK, Panatier A, Pfeiffer T, Angibaud J, Levet F, et al. Structural basis of astrocytic Ca2+ signals at tripartite synapses. Nat Commun (2020) 11:1906. doi: 10.1038/s41467-020-15648-4
119. Verkhratsky A, Nedergaard M. Astroglial cradle in the life of the synapse. Philos Trans R Soc Lond B Biol Sci (2014) 369:20130595. doi: 10.1098/rstb.2013.0595
120. Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron (2008) 60:430–40. doi: 10.1016/j.neuron.2008.10.013
121. Oliveira JF, Sardinha VM, Guerra-Gomes S, Araque A, Sousa N. Do stars govern our actions? Astrocyte involvement in rodent behavior. Trends Neurosci (2015) 38:535–49. doi: 10.1016/j.tins.2015.07.006
122. Nedergaard M, Verkhratsky A. Artifact versus reality–how astrocytes contribute to synaptic events. Glia (2012) 60:1013–23. doi: 10.1002/glia.22288
123. Allen NJ, Eroglu C. Cell Biology of Astrocyte-Synapse Interactions. Neuron (2017) 96:697–708. doi: 10.1016/j.neuron.2017.09.056
124. Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell (2005) 120:421–33. doi: 10.1016/j.cell.2004.12.020
125. Garrett AM, Weiner JA. Control of CNS synapse development by {gamma}-protocadherin-mediated astrocyte-neuron contact. J Neurosci (2009) 29:11723–31. doi: 10.1523/JNEUROSCI.2818-09.2009
126. Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature (2012) 486:410–4. doi: 10.1038/nature11059
127. Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFalpha. Science (2002) 295:2282–5. doi: 10.1126/science.1067859
128. Ben HL, Rowitch DH. Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci (2017) 18:31–41. doi: 10.1038/nrn.2016.159
129. Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, et al. Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron (2017) 95:531–49.e9. doi: 10.1016/j.neuron.2017.06.029
130. Octeau JC, Chai H, Jiang R, Bonanno SL, Martin KC, Khakh BS. An Optical Neuron-Astrocyte Proximity Assay at Synaptic Distance Scales. Neuron (2018) 98:49–66.e9. doi: 10.1016/j.neuron.2018.03.003
131. Martin R, Bajo-Graneras R, Moratalla R, Perea G, Araque A. Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Sci (80 ) (2015) 349:730–4. doi: 10.1126/science.aaa7945
132. Panatier A, Robitaille R. Astrocytic mGluR5 and the tripartite synapse. Neuroscience (2016) 323:29–34. doi: 10.1016/j.neuroscience.2015.03.063
133. D’Ascenzo M, Fellin T, Terunuma M, Revilla-Sanchez R, Meaney DF, Auberson YP, et al. mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci (2007) 104:1995–2000. doi: 10.1073/pnas.0609408104
134. Cavaccini A, Durkee C, Kofuji P, Tonini R, Araque A. Astrocyte Signaling Gates Long-Term Depression at Corticostriatal Synapses of the Direct Pathway. J Neurosci (2020) 40:5757–68. doi: 10.1523/JNEUROSCI.2369-19.2020
135. Goubard V, Fino E, Venance L. Contribution of astrocytic glutamate and GABA uptake to corticostriatal information processing. J Physiol (2011) 589:2301–19. doi: 10.1113/jphysiol.2010.203125
136. Nagai J, Rajbhandari AK, Gangwani MR, Hachisuka A, Coppola G, Masmanidis SC, et al. Hyperactivity with Disrupted Attention by Activation of an Astrocyte Synaptogenic Cue. Cell (2019) 177:1280–92.e20. doi: 10.1016/j.cell.2019.03.019
137. Yu X, Taylor AMW, Nagai J, Golshani P, Evans CJ, Coppola G, et al. Reducing Astrocyte Calcium Signaling In Vivo Alters Striatal Microcircuits and Causes Repetitive Behavior. Neuron (2018) 99:1170–87.e9. doi: 10.1016/j.neuron.2018.08.015
138. Scofield MD. Exploring the Role of Astroglial Glutamate Release and Association With Synapses in Neuronal Function and Behavior. Biol Psychiatry (2018) 84:778–86. doi: 10.1016/j.biopsych.2017.10.029
139. Siemsen BM, Reichel CM, Leong KC, Garcia-Keller C, Gipson CD, Spencer S, et al. Effects of Methamphetamine Self-Administration and Extinction on Astrocyte Structure and Function in the Nucleus Accumbens Core. Neuroscience (2019) 406:528–41. doi: 10.1016/j.neuroscience.2019.03.040
140. Scofield MD, Li H, Siemsen BM, Healey KL, Tran PK, Woronoff N, et al. Cocaine Self-Administration and Extinction Leads to Reduced Glial Fibrillary Acidic Protein Expression and Morphometric Features of Astrocytes in the Nucleus Accumbens Core. Biol Psychiatry (2016) 80:207–15. doi: 10.1016/j.biopsych.2015.12.022
141. Knackstedt LA, Melendez RI, Kalivas PW. Ceftriaxone Restores Glutamate Homeostasis and Prevents Relapse to Cocaine Seeking. Biol Psychiatry (2010) 67:81–4. doi: 10.1016/j.biopsych.2009.07.018
142. Shen H-W, Scofield MD, Boger H, Hensley M, Kalivas PW. Synaptic Glutamate Spillover Due to Impaired Glutamate Uptake Mediates Heroin Relapse. J Neurosci (2014) 34:5649–57. doi: 10.1523/JNEUROSCI.4564-13.2014
143. Corkrum M, Covelo A, Lines J, Bellocchio L, Pisansky M, Loke K, et al. Dopamine-Evoked Synaptic Regulation in the Nucleus Accumbens Requires Astrocyte Activity. Neuron (2020) 105:1036–47.e5. doi: 10.1016/j.neuron.2019.12.026
144. Bialas AR, Stevens B. TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci (2013) 16:1773–82. doi: 10.1038/nn.3560
145. Chung W-S, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature (2013) 504:394–400. doi: 10.1038/nature12776
146. Gomez-Nicola D, Perry VH. Microglial Dynamics and Role in the Healthy and Diseased Brain. Neurosci (2015) 21:169–84. doi: 10.1177/1073858414530512
147. Hanisch U-K, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci (2007) 10:1387–94. doi: 10.1038/nn1997
148. Ransohoff RM, Perry VH. Microglial Physiology: Unique Stimuli, Specialized Responses. Annu Rev Immunol (2009) 27:119–45. doi: 10.1146/annurev.immunol.021908.132528
149. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
150. Werneburg S, Feinberg PA, Johnson KM, Schafer DP. A microglia-cytokine axis to modulate synaptic connectivity and function. Curr Opin Neurobiol (2017) 47:138–45. doi: 10.1016/j.conb.2017.10.002
151. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science (2011) 333:1456–8. doi: 10.1126/science.1202529
152. Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol (2015) 36:605–13. doi: 10.1016/j.it.2015.08.008
153. Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci USA (1998) 95:10896–901. doi: 10.1073/pnas.95.18.10896
154. Gunner G, Cheadle L, Johnson KM, Ayata P, Badimon A, Mondo E, et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat Neurosci (2019) 22:1075–88. doi: 10.1038/s41593-019-0419-y
155. Hoshiko M, Arnoux I, Avignone E, Yamamoto N, Audinat E. Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J Neurosci (2012) 32:15106–11. doi: 10.1523/JNEUROSCI.1167-12.2012
156. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
157. Wang C, Yue H, Hu Z, Shen Y, Ma J, Li J, et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Sci (80 ) (2020) 367:688–94. doi: 10.1126/science.aaz2288
158. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Sci (80 ) (2016) 352:712–6. doi: 10.1126/science.aad8373
159. Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature (2016) 534:538–43. doi: 10.1038/nature18283
160. Klein RS. On Complement, Memory, and Microglia. N Engl J Med (2020) 382:2056–8. doi: 10.1056/NEJMcibr2002480
161. Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci (2011) 31:16241–50. doi: 10.1523/JNEUROSCI.3667-11.2011
162. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell (2013) 155:1596–609. doi: 10.1016/j.cell.2013.11.030
163. Di Filippo M, de Iure A, Giampà C, Chiasserini D, Tozzi A, Orvietani PL, et al. Erratum: Persistent activation of microglia and NADPH oxidase drive hippocampal dysfunction in experimental multiple sclerosis. Sci Rep (2016) 6:23855. doi: 10.1038/srep23855
164. De Biase LM, Schuebel KE, Fusfeld ZH, Jair K, Hawes IA, Cimbro R, et al. Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron (2017) 95:341–56.e6. doi: 10.1016/j.neuron.2017.06.020
165. Kopec AM, Smith CJ, Ayre NR, Sweat SC, Bilbo SD. Microglial dopamine receptor elimination defines sex-specific nucleus accumbens development and social behavior in adolescent rats. Nat Commun (2018) 9:3769. doi: 10.1038/s41467-018-06118-z
166. Kashima DT, Grueter BA. Toll-like receptor 4 deficiency alters nucleus accumbens synaptic physiology and drug reward behavior. Proc Natl Acad Sci (2017) 114:8865–70. doi: 10.1073/pnas.1705974114
167. Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, Stellwagen D. Microglial TNF-α Suppresses Cocaine-Induced Plasticity and Behavioral Sensitization. Neuron (2016) 90:483–91. doi: 10.1016/j.neuron.2016.03.030
168. Kettenmann H, Hanisch U-K, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev (2011) 91:461–553. doi: 10.1152/physrev.00011.2010
169. Färber K, Pannasch U, Kettenmann H. Dopamine and noradrenaline control distinct functions in rodent microglial cells. Mol Cell Neurosci (2005) 29:128–38. doi: 10.1016/j.mcn.2005.01.003
170. Huck JHJ, Freyer D, Böttcher C, Mladinov M, Muselmann-Genschow C, Thielke M, et al. De Novo Expression of Dopamine D2 Receptors on Microglia after Stroke. J Cereb Blood Flow Metab (2015) 35:1804–11. doi: 10.1038/jcbfm.2015.128
171. Haroon E, Raison CL, Miller AH. Psychoneuroimmunology Meets Neuropsychopharmacology: Translational Implications of the Impact of Inflammation on Behavior. Neuropsychopharmacology (2012) 37:137–62. doi: 10.1038/npp.2011.205
172. Donzis EJ, Tronson NC. Modulation of learning and memory by cytokines: Signaling mechanisms and long term consequences. Neurobiol Learn Mem (2014) 115:68–77. doi: 10.1016/j.nlm.2014.08.008
173. Schneider H, Pitossi F, Balschun D, Wagner A, del Rey A, Besedovsky HO. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci USA (1998) 95:7778–83. doi: 10.1073/pnas.95.13.7778
174. Avital A, Goshen I, Kamsler A, Segal M, Iverfeldt K, Richter-Levin G, et al. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus (2003) 13:826–34. doi: 10.1002/hipo.10135
175. Coogan AN, O’Neill LAJ, O’Connor JJ. The p38 mitogen-activated protein kinase inhibitor SB203580 antagonizes the inhibitory effects of interleukin-1β on long-term potentiation in the rat dentate gyrus in vitro. Neuroscience (1999) 93:57–69. doi: 10.1016/S0306-4522(99)00100-1
176. Schmid AW, Lynch MA, Herron CE. The effects of IL-1 receptor antagonist on beta amyloid mediated depression of LTP in the rat CA1 in vivo. Hippocampus (2009) 19:670–6. doi: 10.1002/hipo.20542
177. Lai AY, Swayze RD, El-Husseini A, Song C. Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J Neuroimmunol (2006) 175:97–106. doi: 10.1016/j.jneuroim.2006.03.001
178. Viviani B, Gardoni F, Bartesaghi S, Corsini E, Facchi A, Galli CL, et al. Interleukin-1β Released by gp120 Drives Neural Death through Tyrosine Phosphorylation and Trafficking of NMDA Receptors. J Biol Chem (2006) 281:30212–22. doi: 10.1074/jbc.M602156200
179. Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1β Enhances NMDA Receptor-Mediated Intracellular Calcium Increase through Activation of the Src Family of Kinases. J Neurosci (2003) 23:8692–700. doi: 10.1523/JNEUROSCI.23-25-08692.2003
180. Yang S, Liu Z-W, Wen L, Qiao H-F, Zhou W-X, Zhang Y-X. Interleukin-1β enhances NMDA receptor-mediated current but inhibits excitatory synaptic transmission. Brain Res (2005) 1034:172–9. doi: 10.1016/j.brainres.2004.11.018
181. Braida D, Sacerdote P, Panerai AE, Bianchi M, Aloisi AM, Iosuè S, et al. Cognitive function in young and adult IL (interleukin)-6 deficient mice. Behav Brain Res (2004) 153:423–9. doi: 10.1016/j.bbr.2003.12.018
182. Wei H, Chadman KK, McCloskey DP, Sheikh AM, Malik M, Brown WT, et al. Brain IL-6 elevation causes neuronal circuitry imbalances and mediates autism-like behaviors. Biochim Biophys Acta Mol Basis Dis (2012) 1822:831–42. doi: 10.1016/j.bbadis.2012.01.011
183. Tancredi V, D’Antuono M, Cafè C, Giovedì S, Buè MC, D’Arcangelo G, et al. The Inhibitory Effects of Interleukin-6 on Synaptic Plasticity in the Rat Hippocampus Are Associated with an Inhibition of Mitogen-Activated Protein Kinase ERK. J Neurochem (2002) 75:634–43. doi: 10.1046/j.1471-4159.2000.0750634.x
184. Balschun D, Wetzel W, Del Rey A, Pitossi F, Schneider H, Zuschratter W, et al. Interleukin-6: a cytokine to forget. FASEB J (2004) 18:1788–90. doi: 10.1096/fj.04-1625fje
185. Curran B, O’Connor J. The pro-inflammatory cytokine interleukin-18 impairs long-term potentiation and NMDA receptor-mediated transmission in the rat hippocampus in vitro. Neuroscience (2001) 108:83–90. doi: 10.1016/S0306-4522(01)00405-5
186. Cumiskey D, Pickering M, O’Connor JJ. Interleukin-18 mediated inhibition of LTP in the rat dentate gyrus is attenuated in the presence of mGluR antagonists. Neurosci Lett (2007) 412:206–10. doi: 10.1016/j.neulet.2006.11.007
187. Cumiskey D, Curran BP, Herron CE, O’Connor JJ. A role for inflammatory mediators in the IL-18 mediated attenuation of LTP in the rat dentate gyrus. Neuropharmacology (2007) 52:1616–23. doi: 10.1016/j.neuropharm.2007.03.006
188. Monteiro S, Ferreira FM, Pinto V, Roque S, Morais M, de Sá-Calçada D, et al. Absence of IFNγ promotes hippocampal plasticity and enhances cognitive performance. Transl Psychiatry (2016) 6:e707. doi: 10.1038/tp.2015.194
189. Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature (2016) 535:425–9. doi: 10.1038/nature18626
190. Belarbi K, Jopson T, Tweedie D, Arellano C, Luo W, Greig NH, et al. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J Neuroinflamm (2012) 9:23. doi: 10.1186/1742-2094-9-23
191. Butler MP, O’Connor JJ, Moynagh PN. Dissection of tumor-necrosis factor-alpha inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early-but not late-phase LTP. Neuroscience (2004) 124:319–26. doi: 10.1016/j.neuroscience.2003.11.040
192. Paredes D, Acosta S, Gemma C, Bickford PC. Role of TNFα Induced Inflammation in Delay Eyeblink Conditioning in Young and Aged Rats. Aging Dis (2010) 1:191–8.
193. Lou CM, Corrigan F, Jaehne EJ, Jawahar MC, Anscomb H, Koerner H, et al. TNF-α and its receptors modulate complex behaviours and neurotrophins in transgenic mice. Psychoneuroendocrinology (2013) 38:3102–14. doi: 10.1016/j.psyneuen.2013.09.010
194. Cartford MC, Gemma C, Bickford PC. Eighteen-Month-Old Fischer 344 Rats Fed a Spinach-Enriched Diet Show Improved Delay Classical Eyeblink Conditioning and Reduced Expression of Tumor Necrosis Factor α (TNFα) and TNFβ in the Cerebellum. J Neurosci (2002) 22:5813–6. doi: 10.1523/JNEUROSCI.22-14-05813.2002
195. Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse (2000) 35:151–9. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P
196. Cumiskey D, Butler MP, Moynagh PN, O’Connor JJ. Evidence for a role for the group I metabotropic glutamate receptor in the inhibitory effect of tumor necrosis factor-α on long-term potentiation. Brain Res (2007) 1136:13–9. doi: 10.1016/j.brainres.2006.12.019
197. Chen C, Itakura E, Nelson GM, Sheng M, Laurent P, Fenk LA, et al. IL-17 is a neuromodulator of Caenorhabditis elegans sensory responses. Nature (2017) 542:43–8. doi: 10.1038/nature20818
198. Reed MD, Yim YS, Wimmer RD, Kim H, Ryu C, Welch GM, et al. IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature (2020) 577:249–53. doi: 10.1038/s41586-019-1843-6
199. Ribeiro M, Brigas HC, Temido-Ferreira M, Pousinha PA, Regen T, Santa C, et al. Meningeal γδ T cell-derived IL-17 controls synaptic plasticity and short-term memory. Sci Immunol (2019) 4(40):eaay5199. doi: 10.1126/sciimmunol.aay5199
200. Rossi S, Furlan R, De Chiara V, Motta C, Studer V, Mori F, et al. Interleukin-1β causes synaptic hyperexcitability in multiple sclerosis. Ann Neurol (2012) 71:76–83. doi: 10.1002/ana.22512
201. De Chiara V, Motta C, Rossi S, Studer V, Barbieri F, Lauro D, et al. Interleukin-1β alters the sensitivity of cannabinoid CB1 receptors controlling glutamate transmission in the striatum. Neuroscience (2013) 250:232–9. doi: 10.1016/j.neuroscience.2013.06.069
202. Rossi S, Studer V, Motta C, De Chiara V, Barbieri F, Bernardi G, et al. Inflammation inhibits GABA transmission in multiple sclerosis. Mult Scler J (2012) 18:1633–5. doi: 10.1177/1352458512440207
203. Musumeci G, Grasselli G, Rossi S, De Chiara V, Musella A, Motta C, et al. Transient receptor potential vanilloid 1 channels modulate the synaptic effects of TNF-α and of IL-1β in experimental autoimmune encephalomyelitis. Neurobiol Dis (2011) 43:669–77. doi: 10.1016/j.nbd.2011.05.018
204. Rossi S, Sacchetti L, Napolitano F, De Chiara V, Motta C, Studer V, et al. Interleukin-1β causes anxiety by interacting with the endocannabinoid system. J Neurosci (2012) 32:13896–905. doi: 10.1523/JNEUROSCI.1515-12.2012
205. Gentile A, Fresegna D, Musella A, Sepman H, Bullitta S, De Vito F, et al. Interaction between interleukin-1β and type-1 cannabinoid receptor is involved in anxiety-like behavior in experimental autoimmune encephalomyelitis. J Neuroinflamm (2016) 13:231. doi: 10.1186/s12974-016-0682-8
206. Gentile A, Fresegna D, Federici M, Musella A, Rizzo FR, Sepman H, et al. Dopaminergic dysfunction is associated with IL-1β-dependent mood alterations in experimental autoimmune encephalomyelitis. Neurobiol Dis (2015) 74:347–58. doi: 10.1016/j.nbd.2014.11.022
207. Ho A, Blum M. Induction of Interleukin-1 Associated with Compensatory Dopaminergic Sprouting in the Denervated Striatum of Young Mice: Model of Aging and Neurodegenerative Disease. J Neurosci (1998) 18:5614–29. doi: 10.1523/JNEUROSCI.18-15-05614.1998
208. Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A, et al. Inflammation Triggers Synaptic Alteration and Degeneration in Experimental Autoimmune Encephalomyelitis. J Neurosci (2009) 29:3442–52. doi: 10.1523/JNEUROSCI.5804-08.2009
209. Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci (2005) 25:3219–28. doi: 10.1523/JNEUROSCI.4486-04.2005
210. Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature (2006) 440:1054–9. doi: 10.1038/nature04671
211. Leonoudakis D, Zhao P, Beattie EC. Rapid Tumor Necrosis Factor -Induced Exocytosis of Glutamate Receptor 2-Lacking AMPA Receptors to Extrasynaptic Plasma Membrane Potentiates Excitotoxicity. J Neurosci (2008) 28:2119–30. doi: 10.1523/JNEUROSCI.5159-07.2008
212. Haji N, Mandolesi G, Gentile A, Sacchetti L, Fresegna D, Rossi S, et al. TNF-α-mediated anxiety in a mouse model of multiple sclerosis. Exp Neurol (2012) 237:296–303. doi: 10.1016/j.expneurol.2012.07.010
213. Lewitus GM, Pribiag H, Duseja R, St-Hilaire M, Stellwagen D. An adaptive role of TNFα in the regulation of striatal synapses. J Neurosci (2014) 34:6146–55. doi: 10.1523/JNEUROSCI.3481-13.2014
214. He P, Liu Q, Wu J, Shen Y. Genetic deletion of TNF receptor suppresses excitatory synaptic transmission via reducing AMPA receptor synaptic localization in cortical neurons. FASEB J (2012) 26:334–45. doi: 10.1096/fj.11-192716
215. Ogoshi F, Yin HZ, Kuppumbatti Y, Song B, Amindari S, Weiss JH. Tumor necrosis-factor-alpha (TNF-α) induces rapid insertion of Ca2+-permeable α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp Neurol (2005) 193:384–93. doi: 10.1016/j.expneurol.2004.12.026
216. Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, et al. Cell Death after Spinal Cord Injury Is Exacerbated by Rapid TNF -Induced Trafficking of GluR2-Lacking AMPARs to the Plasma Membrane. J Neurosci (2008) 28:11391–400. doi: 10.1523/JNEUROSCI.3708-08.2008
217. Kaneko M, Stellwagen D, Malenka RC, Stryker MP. Tumor Necrosis Factor-α Mediates One Component of Competitive, Experience-Dependent Plasticity in Developing Visual Cortex. Neuron (2008) 58:673–80. doi: 10.1016/j.neuron.2008.04.023
218. Rizzo FR, Musella A, De Vito F, Fresegna D, Bullitta S, Vanni V, et al. Tumor Necrosis Factor and Interleukin-1 β Modulate Synaptic Plasticity during Neuroinflammation. Neural Plast (2018) 2018:1–12. doi: 10.1155/2018/8430123
219. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5:375–86. doi: 10.1038/nri1604
220. Lee AJ, Ashkar AA. The Dual Nature of Type I and Type II Interferons. Front Immunol (2018) 9:2061. doi: 10.3389/fimmu.2018.02061
221. Reyes-Vázquez C, Prieto-Gómez B, Dafny N. Interferon modulates central nervous system function. Brain Res (2012) 1442:76–89. doi: 10.1016/j.brainres.2011.09.061
222. Owens T, Khorooshi R, Wlodarczyk A, Asgari N. Interferons in the central nervous system: a few instruments play many tunes. Glia (2014) 62:339–55. doi: 10.1002/glia.22608
223. Dafny N, Prieto-Gomez B, Dong WQ, Reyes-Vazquez C. Interferon modulates neuronal activity recorded from the hypothalamus, thalamus, hippocampus, amygdala and the somatosensory cortex. Brain Res (1996) 734:269–74. doi: 10.1016/0006-8993(96)00650-6
224. Di Filippo M, Tozzi A, Arcangeli S, de Iure A, Durante V, Di Gregorio M, et al. Interferon-β1a modulates glutamate neurotransmission in the CNS through CaMKII and GluN2A-containing NMDA receptors. Neuropharmacology (2016) 100:98–105. doi: 10.1016/j.neuropharm.2015.06.009
225. Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci (2012) 13:169–82. doi: 10.1038/nrn3192
226. Gardoni F, Caputi A, Cimino M, Pastorino L, Cattabeni F, Di Luca M. Calcium/calmodulin-dependent protein kinase II is associated with NR2A/B subunits of NMDA receptor in postsynaptic densities. J Neurochem (1998) 71:1733–41. doi: 10.1046/j.1471-4159.1998.71041733.x
227. Di Filippo M, Tozzi A, Tantucci M, Arcangeli S, Chiasserini D, Ghiglieri V, et al. Interferon-β1a protects neurons against mitochondrial toxicity via modulation of STAT1 signaling: Electrophysiological evidence. Neurobiol Dis (2014) 62:387–93. doi: 10.1016/j.nbd.2013.09.022
228. Amor S, Peferoen LAN, Vogel DYS, Breur M, van der Valk P, Baker D, et al. Inflammation in neurodegenerative diseases–an update. Immunology (2014) 142:151–66. doi: 10.1111/imm.12233
229. Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Sci (80 ) (2016) 353:777–83. doi: 10.1126/science.aag2590
230. Tan E-K, Chao Y-X, West A, Chan L-L, Poewe W, Jankovic J. Parkinson disease and the immune system - associations, mechanisms and therapeutics. Nat Rev Neurol (2020) 16:303–18. doi: 10.1038/s41582-020-0344-4
231. Kannarkat GT, Boss JM, Tansey MG. The Role of Innate and Adaptive Immunity in Parkinson’s Disease. J Parkinsons Dis (2013) 3:493–514. doi: 10.3233/JPD-130250
232. Tansey MG, Goldberg MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis (2010) 37:510–8. doi: 10.1016/j.nbd.2009.11.004
233. Gelders G, Baekelandt V, Van der Perren A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J Immunol Res (2018) 2018:1–12. doi: 10.1155/2018/4784268
234. Chen Z, Chen S, Liu J. The role of T cells in the pathogenesis of Parkinson’s disease. Prog Neurobiol (2018) 169:1–23. doi: 10.1016/j.pneurobio.2018.08.002
235. Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest (2009) 119:182–92. doi: 10.1172/JCI36470
236. Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, et al. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci Lett (1994) 180:147–50. doi: 10.1016/0304-3940(94)90508-8
237. Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett (1994) 165:208–10. doi: 10.1016/0304-3940(94)90746-3
238. Blum-Degen D, Müller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett (1995) 202:17–20. doi: 10.1016/0304-3940(95)12192-7
239. Müller T, Blum-Degen D, Przuntek H, Kuhn W. Interleukin-6 levels in cerebrospinal fluid inversely correlate to severity of Parkinson’s disease. Acta Neurol Scand (1998) 98:142–4. doi: 10.1111/j.1600-0404.1998.tb01736.x
240. Brodacki B, Staszewski J, Toczyłowska B, Kozłowska E, Drela N, Chalimoniuk M, et al. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFα, and INFγ concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci Lett (2008) 441:158–62. doi: 10.1016/j.neulet.2008.06.040
241. Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. Interleukin (IL)-1β, IL-2, IL-4, IL-6 and transforming growth factor-α levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci Lett (1996) 211:13–6. doi: 10.1016/0304-3940(96)12706-3
242. Lin C-H, Chen C-C, Chiang H-L, Liou J-M, Chang C-M, Lu T-P, et al. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J Neuroinflamm (2019) 16:129. doi: 10.1186/s12974-019-1528-y
243. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature (2017) 546:656–61. doi: 10.1038/nature22815
244. Liu Z, Huang Y, Cao B-B, Qiu Y-H, Peng Y-P. Th17 Cells Induce Dopaminergic Neuronal Death via LFA-1/ICAM-1 Interaction in a Mouse Model of Parkinson’s Disease. Mol Neurobiol (2017) 54:7762–76. doi: 10.1007/s12035-016-0249-9
245. Cebrián C, Zucca FA, Mauri P, Steinbeck JA, Studer L, Scherzer CR, et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat Commun (2014) 5:3633. doi: 10.1038/ncomms4633
246. Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, et al. MHCII Is Required for -Synuclein-Induced Activation of Microglia, CD4 T Cell Proliferation, and Dopaminergic Neurodegeneration. J Neurosci (2013) 33:9592–600. doi: 10.1523/JNEUROSCI.5610-12.2013
247. Dutta D, Kundu M, Mondal S, Roy A, Ruehl S, Hall DA, et al. RANTES-induced invasion of Th17 cells into substantia nigra potentiates dopaminergic cell loss in MPTP mouse model of Parkinson’s disease. Neurobiol Dis (2019) 132:104575. doi: 10.1016/j.nbd.2019.104575
248. Chandra G, Roy A, Rangasamy SB, Pahan K. Induction of Adaptive Immunity Leads to Nigrostriatal Disease Progression in MPTP Mouse Model of Parkinson’s Disease. J Immunol (2017) 198:4312–26. doi: 10.4049/jimmunol.1700149
249. González H, Contreras F, Prado C, Elgueta D, Franz D, Bernales S, et al. Dopamine Receptor D3 Expressed on CD4 + T Cells Favors Neurodegeneration of Dopaminergic Neurons during Parkinson’s Disease. J Immunol (2013) 190:5048–56. doi: 10.4049/jimmunol.1203121
250. Sommer A, Marxreiter F, Krach F, Fadler T, Grosch J, Maroni M, et al. Th17 Lymphocytes Induce Neuronal Cell Death in a Human iPSC-Based Model of Parkinson’s Disease. Cell Stem Cell (2018) 23:123–31.e6. doi: 10.1016/j.stem.2018.06.015
251. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology (1988) 38:1285–5. doi: 10.1212/WNL.38.8.1285
252. Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol (2003) 106:518–26. doi: 10.1007/s00401-003-0766-2
253. Olanow CW, Savolainen M, Chu Y, Halliday GM, Kordower JH. Temporal evolution of microglia and α-synuclein accumulation following foetal grafting in Parkinson’s disease. Brain (2019) 142:1690–700. doi: 10.1093/brain/awz104
254. Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, et al. Blockade of Microglial Activation Is Neuroprotective in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson Disease. J Neurosci (2002) 22:1763–71. doi: 10.1523/JNEUROSCI.22-05-01763.2002
255. Peng Z, Luchtman DW, Wang X, Zhang Y, Song C. Activation of microglia synergistically enhances neurodegeneration caused by MPP+ in human SH-SY5Y cells. Eur J Pharmacol (2019) 850:64–74. doi: 10.1016/j.ejphar.2019.01.024
256. Liu Z, Qiu A-W, Huang Y, Yang Y, Chen J-N, Gu T-T, et al. IL-17A exacerbates neuroinflammation and neurodegeneration by activating microglia in rodent models of Parkinson’s disease. Brain Behav Immun (2019) 81:630–45. doi: 10.1016/j.bbi.2019.07.026
257. Liu Z, Chen H-Q, Huang Y, Qiu Y-H, Peng Y-P. Transforming growth factor-β1 acts via TβR-I on microglia to protect against MPP+-induced dopaminergic neuronal loss. Brain Behav Immun (2016) 51:131–43. doi: 10.1016/j.bbi.2015.08.006
258. Kostuk EW, Cai J, Iacovitti L. Regional microglia are transcriptionally distinct but similarly exacerbate neurodegeneration in a culture model of Parkinson’s disease. J Neuroinflamm (2018) 15:139. doi: 10.1186/s12974-018-1181-x
259. Subbarayan MS, Hudson C, Moss LD, Nash KR, Bickford PC. T cell infiltration and upregulation of MHCII in microglia leads to accelerated neuronal loss in an α-synuclein rat model of Parkinson’s disease. J Neuroinflamm (2020) 17:242. doi: 10.1186/s12974-020-01911-4
260. Duffy MF, Collier TJ, Patterson JR, Kemp CJ, Luk KC, Tansey MG, et al. Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflamm (2018) 15:129. doi: 10.1186/s12974-018-1171-z
261. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
262. Yun SP, Kam T-I, Panicker N, Kim S, Oh Y, Park J-S, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med (2018) 24:931–8. doi: 10.1038/s41591-018-0051-5
263. Booth HDE, Hirst WD, Wade-Martins R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci (2017) 40:358–70. doi: 10.1016/j.tins.2017.04.001
264. Schapira AHV, Chaudhuri KR, Jenner P. Non-motor features of Parkinson disease. Nat Rev Neurosci (2017) 18:435–50. doi: 10.1038/nrn.2017.62
265. Hilker R, Schweitzer K, Coburger S, Ghaemi M, Weisenbach S, Jacobs AH, et al. Nonlinear Progression of Parkinson Disease as Determined by Serial Positron Emission Tomographic Imaging of Striatal Fluorodopa F 18 Activity. Arch Neurol (2005) 62:378. doi: 10.1001/archneur.62.3.378
266. Schirinzi T, Madeo G, Martella G, Maltese M, Picconi B, Calabresi P, et al. Early synaptic dysfunction in Parkinson’s disease: Insights from animal models. Mov Disord (2016) 31:802–13. doi: 10.1002/mds.26620
267. Imbriani P, Schirinzi T, Meringolo M, Mercuri NB, Pisani A. Centrality of Early Synaptopathy in Parkinson’s Disease. Front Neurol (2018) 9:103. doi: 10.3389/fneur.2018.00103
268. Ghiglieri V, Calabrese V, Calabresi P. Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front Neurol (2018) 9:295. doi: 10.3389/fneur.2018.00295
269. Tuovinen N, Seppi K, de Pasquale F, Müller C, Nocker M, Schocke M, et al. The reorganization of functional architecture in the early-stages of Parkinson’s disease. Parkinsonism Relat Disord (2018) 50:61–8. doi: 10.1016/j.parkreldis.2018.02.013
270. Sang L, Zhang J, Wang L, Zhang J, Zhang Y, Li P, et al. Alteration of Brain Functional Networks in Early-Stage Parkinson’s Disease: A Resting-State fMRI Study. PloS One (2015) 10:e0141815. doi: 10.1371/journal.pone.0141815
271. Costa C, Sgobio C, Siliquini S, Tozzi A, Tantucci M, Ghiglieri V, et al. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Brain (2012) 135:1884–99. doi: 10.1093/brain/aws101
272. Tozzi A, de Iure A, Bagetta V, Tantucci M, Durante V, Quiroga-Varela A, et al. Alpha-Synuclein Produces Early Behavioral Alterations via Striatal Cholinergic Synaptic Dysfunction by Interacting With GluN2D N -Methyl-D-Aspartate Receptor Subunit. Biol Psychiatry (2016) 79:402–14. doi: 10.1016/j.biopsych.2015.08.013
273. Durante V, de Iure A, Loffredo V, Vaikath N, De Risi M, Paciotti S, et al. Alpha-synuclein targets GluN2A NMDA receptor subunit causing striatal synaptic dysfunction and visuospatial memory alteration. Brain (2019) 142:1365–85. doi: 10.1093/brain/awz065
274. Giordano N, Iemolo A, Mancini M, Cacace F, De Risi M, Latagliata EC, et al. Motor learning and metaplasticity in striatal neurons: relevance for Parkinson’s disease. Brain (2018) 141:505–20. doi: 10.1093/brain/awx351
275. Tozzi A, Costa C, Siliquini S, Tantucci M, Picconi B, Kurz A, et al. Mechanisms underlying altered striatal synaptic plasticity in old A53T-α synuclein overexpressing mice. Neurobiol Aging (2012) 33:1792–9. doi: 10.1016/j.neurobiolaging.2011.05.002
276. Diogenes MJ, Dias RB, Rombo DM, Vicente Miranda H, Maiolino F, Guerreiro P, et al. Extracellular Alpha-Synuclein Oligomers Modulate Synaptic Transmission and Impair LTP Via NMDA-Receptor Activation. J Neurosci (2012) 32:11750–62. doi: 10.1523/JNEUROSCI.0234-12.2012
277. Chen Y, Yang W, Li X, Li X, Yang H, Xu Z, et al. α-Synuclein-induced internalization of NMDA receptors in hippocampal neurons is associated with reduced inward current and Ca2+ influx upon NMDA stimulation. Neuroscience (2015) 300:297–306. doi: 10.1016/j.neuroscience.2015.05.035
278. Ferreira DG, Temido-Ferreira M, Vicente Miranda H, Batalha VL, Coelho JE, Szegö ÉM, et al. α-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci (2017) 20:1569–79. doi: 10.1038/nn.4648
279. Kim C, Ho D-H, Suk J-E, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun (2013) 4:1562. doi: 10.1038/ncomms2534
280. Lee H-J, Suk J-E, Patrick C, Bae E-J, Cho J-H, Rho S, et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem (2010) 285:9262–72. doi: 10.1074/jbc.M109.081125
281. Lee H-J, Kim C, Lee S-J. Alpha-Synuclein Stimulation of Astrocytes: Potential Role for Neuroinflammation and Neuroprotection. Oxid Med Cell Longev (2010) 3:283–7. doi: 10.4161/oxim.3.4.12809
282. Allen Reish HE, Standaert DG. Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J Parkinsons Dis (2015) 5:1–19. doi: 10.3233/JPD-140491
283. Del-Bel E, Bortolanza M, Dos-Santos-Pereira M, Bariotto K, Raisman-Vozari R. l-DOPA-induced dyskinesia in Parkinson’s disease: Are neuroinflammation and astrocytes key elements? Synapse (2016) 70:479–500. doi: 10.1002/syn.21941
284. Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA–induced dyskinesia. Nat Neurosci (2003) 6:501–6. doi: 10.1038/nn1040
285. Calabresi P, Di Filippo M, Ghiglieri V, Picconi B. Molecular mechanisms underlying levodopa-induced dyskinesia. Mov Disord (2008) 23:S570–9. doi: 10.1002/mds.22019
286. Calabresi P, Pisani A, Rothwell J, Ghiglieri V, Obeso JA, Picconi B. Hyperkinetic disorders and loss of synaptic downscaling. Nat Neurosci (2016) 19:868–75. doi: 10.1038/nn.4306
287. Cacace F, Mineo D, Viscomi MT, Latagliata EC, Mancini M, Sasso V, et al. Intermittent theta-burst stimulation rescues dopamine-dependent corticostriatal synaptic plasticity and motor behavior in experimental parkinsonism: Possible role of glial activity. Mov Disord (2017) 32:1035–46. doi: 10.1002/mds.26982
288. Cullen CL, Young KM. How Does Transcranial Magnetic Stimulation Influence Glial Cells in the Central Nervous System? Front Neural Circuits (2016) 10:26. doi: 10.3389/fncir.2016.00026
289. Oliva I, Fernández M, Martín ED. Dopamine release regulation by astrocytes during cerebral ischemia. Neurobiol Dis (2013) 58:231–41. doi: 10.1016/j.nbd.2013.06.007
290. Cui Q, Pitt JE, Pamukcu A, Poulin J-F, Mabrouk OS, Fiske MP, et al. Blunted mGluR Activation Disinhibits Striatopallidal Transmission in Parkinsonian Mice. Cell Rep (2016) 17:2431–44. doi: 10.1016/j.celrep.2016.10.087
291. Kravitz AV, Freeze BS, Parker PRL, Kay K, Thwin MT, Deisseroth K, et al. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature (2010) 466:622–6. doi: 10.1038/nature09159
292. Lemos JC, Friend DM, Kaplan AR, Shin JH, Rubinstein M, Kravitz AV, et al. Enhanced GABA Transmission Drives Bradykinesia Following Loss of Dopamine D2 Receptor Signaling. Neuron (2016) 90:824–38. doi: 10.1016/j.neuron.2016.04.040
293. Khakh BS, Beaumont V, Cachope R, Munoz-Sanjuan I, Goldman SA, Grantyn R. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci (2017) 40:422–37. doi: 10.1016/j.tins.2017.05.002
295. Eidelberg D, Surmeier DJ. Brain networks in Huntington disease. J Clin Invest (2011) 121:484–92. doi: 10.1172/JCI45646
296. Rüb U, Vonsattel JPV, Heinsen H, Korf H-W. The Neuropathology of Huntington´s disease: classical findings, recent developments and correlation to functional neuroanatomy. Adv Anat Embryol Cell Biol (2015) 217:1–146. doi: 10.1007/978-3-319-19285-7
297. Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum Mol Genet (2010) 19:3053–67. doi: 10.1093/hmg/ddq212
298. Shin J-Y, Fang Z-H, Yu Z-X, Wang C-E, Li S-H, Li X-J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol (2005) 171:1001–12. doi: 10.1083/jcb.200508072
299. Arzberger T, Krampfl K, Leimgruber S, Weindl A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington’s disease–an in situ hybridization study. J Neuropathol Exp Neurol (1997) 56:440–54. doi: 10.1097/00005072-199704000-00013
300. Benraiss A, Wang S, Herrlinger S, Li X, Chandler-Militello D, Mauceri J, et al. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat Commun (2016) 7:11758. doi: 10.1038/ncomms11758
301. Wood TE, Barry J, Yang Z, Cepeda C, Levine MS, Gray M. Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum Mol Genet (2019) 28:487–500. doi: 10.1093/hmg/ddy363
302. Frick L, Pittenger C. Microglial Dysregulation in OCD, Tourette Syndrome, and PANDAS. J Immunol Res (2016) 2016:8606057. doi: 10.1155/2016/8606057
303. Hsu C-J, Wong L-C, Lee W-T. Immunological Dysfunction in Tourette Syndrome and Related Disorders. Int J Mol Sci (2021) 22:853. doi: 10.3390/ijms22020853
Keywords: basal ganglia, nucleus striatum, immune system, synaptic transmission, synaptic plasticity
Citation: Mancini A, Ghiglieri V, Parnetti L, Calabresi P and Di Filippo M (2021) Neuro-Immune Cross-Talk in the Striatum: From Basal Ganglia Physiology to Circuit Dysfunction. Front. Immunol. 12:644294. doi: 10.3389/fimmu.2021.644294
Received: 20 December 2020; Accepted: 16 March 2021;
Published: 19 April 2021.
Edited by:
Antonietta Gentile, San Raffaele Pisana (IRCCS), ItalyReviewed by:
Marc Vincent Fuccillo, University of Pennsylvania, United StatesKenji Tanaka, Keio University, Japan
Copyright © 2021 Mancini, Ghiglieri, Parnetti, Calabresi and Di Filippo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Massimiliano Di Filippo, bWFzc2ltaWxpYW5vLmRpZmlsaXBwb0B1bmlwZy5pdA==
†These authors have contributed equally to this work