
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 22 February 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.643852
This article is part of the Research TopicAdvances in Human Immune System (HIS) Mouse Models for Studying Human Hematopoiesis and Cancer ImmunotherapyView all 19 articles
 Tijana Martinov1†
Tijana Martinov1† Kelly M. McKenna1,2,3†
Kelly M. McKenna1,2,3† Wei Hong Tan1†
Wei Hong Tan1† Emily J. Collins1
Emily J. Collins1 Allie R. Kehret1
Allie R. Kehret1 Jonathan D. Linton1
Jonathan D. Linton1 Tayla M. Olsen1
Tayla M. Olsen1 Nour Shobaki1
Nour Shobaki1 Anthony Rongvaux1,4*
Anthony Rongvaux1,4*Since the late 1980s, mice have been repopulated with human hematopoietic cells to study the fundamental biology of human hematopoiesis and immunity, as well as a broad range of human diseases in vivo. Multiple mouse recipient strains have been developed and protocols optimized to efficiently generate these “humanized” mice. Here, we review three guiding principles that have been applied to the development of the currently available models: (1) establishing tolerance of the mouse host for the human graft; (2) opening hematopoietic niches so that they can be occupied by human cells; and (3) providing necessary support for human hematopoiesis. We then discuss four remaining challenges: (1) human hematopoietic lineages that poorly develop in mice; (2) limited antigen-specific adaptive immunity; (3) absent tolerance of the human immune system for its mouse host; and (4) sub-functional interactions between human immune effectors and target mouse tissues. While major advances are still needed, the current models can already be used to answer specific, clinically-relevant questions and hopefully inform the development of new, life-saving therapies.
Biomedical research aims to provide a platform for the development and testing of new therapies that can reduce human suffering and deaths. In vitro studies using human cells or organoids are useful, but animal models can better elucidate the fundamental principles of complex biological processes in mammals. In particular, laboratory mice are often the model organism of choice, as their small size and short generation time enable extensive genetic engineering and invasive experimentation. Many fundamental characteristics of hematopoietic and immune systems are shared across mice and humans. However, with 91 million years of divergent evolution, differences exist and results from murine studies cannot always be directly translated into clinical applications, driving the development of experimental murine platforms that faithfully model human physiology and diseases. Specifically, mice can be transplanted with a human hemato-lymphoid system (1). Such “humanized mice” (Box 1) have been increasingly used since the late 1980s, and have contributed to major breakthroughs in several research fields, including human hematopoiesis (2), hematologic malignancies (3), and immunity to human-tropic pathogens (4, 5). Successful generation of humanized mice requires: (i) a source of human donor hematopoietic cells, (ii) an effective transplantation protocol, and (iii) an appropriate recipient mouse strain.
Box 1. What is a humanized mouse?
The Wikipedia definition of a humanized mouse is “a mouse carrying functioning human genes, cells, tissues, and/or organs.” This review focuses on mice transplanted with human hematopoietic cells, colloquially referred to as “humanized mice” (6, 7). When used to study immune responses, such mice are better designated as “human(ized) immune system” (HIS) mice (5, 8, 9). However, not all hematopoietic cells contribute to the immune response, and “human(ized) hemato-lymphoid system” (HHLS) mice is a more encompassing term, particularly when human CD34+ HSPCs are transplanted (1, 10, 11).
Many models combine multiple characteristics listed in the definition of humanized mice. In addition to human hematopoietic cell transplantation, diverse human tissues or tumors can be co-transplanted and the genome of the recipient mouse can be engineered to contain human genes.
Human peripheral blood mononuclear cells (PBMCs) can be used as a source of hematopoietic cells for transplantation, but their engraftment favors T cell maintenance, resulting in an incomplete human immune system (7, 12). In contrast, hematopoietic stem and progenitor cells (HSPCs), enriched in the CD34+ cell fraction, give rise to all human hematopoietic lineages upon transplantation in mice (7, 13). Human CD34+ HSPCs can be obtained from different sources. Fetal CD34+ cells, abundant in the liver, very efficiently engraft and undergo multilineage differentiation upon transplantation in mice, but ethical concerns limit access to fetal tissues (14). Other sources of CD34+ cells, such as cord blood, bone marrow (BM) or peripheral blood following granulocyte colony-stimulating factor (G-CSF) mobilization, are more accessible with fewer ethical concerns. However, their stemness declines with the age of the donor (15, 16), resulting in lower engraftment potential (17, 18). Of note, donor cells can be obtained from patients affected by diverse hematopoietic diseases, thereby providing small animal models of human diseases including genetic immune disorders or hematological malignancies (3, 19).
Human hematopoietic cells can be transplanted by systemic intravenous delivery or by orthotopic injection into a site of primary hematopoiesis. In adult mice, hematopoietic cells can be implanted in the BM niche by intrafemoral injection (20). Intrahepatic injection has also become a common route of CD34+ cell implantation in newborn mice; the liver is a site of primary hematopoiesis during embryonic life and continues to be for several days after birth, until the hematopoietic niche is established in the BM and the mouse host naturally supports the expansion and multilineage differentiation of the hematopoietic system (21) (Figure 1).
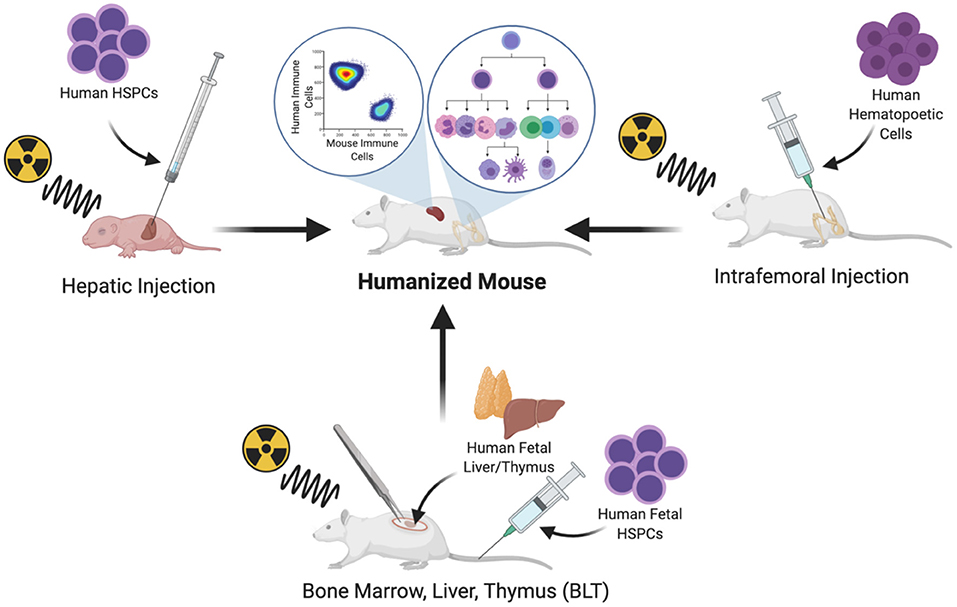
Figure 1. Protocols commonly used for the generation of humanized mice.
The array of recipient mice for hematopoietic humanization has expanded over the past few decades, with advances in mouse genome engineering. This review focuses on these developments, highlighting the genetic engineering of the host, as well as the co-transplantation of human tissues to support human hematopoiesis. We first discuss three guiding principles that have been employed for the development of the currently available recipient mice (Figure 2): (i) preventing rejection of the human graft by the mouse immune system, (ii) opening the niche to make it accessible to human hematopoietic cells, and (iii) supporting human hematopoiesis in the mouse. We then consider how these principles are being applied to the development of newer mouse strains, aiming to resolve four remaining major challenges: (i) the development and function of missing human hematopoietic lineages, (ii) efficient and durable antigen-specific adaptive immunity, (iii) tolerance of the engrafted human immune system for the mouse host and (iv) functional cross-reactivity between the human graft and target tissues.
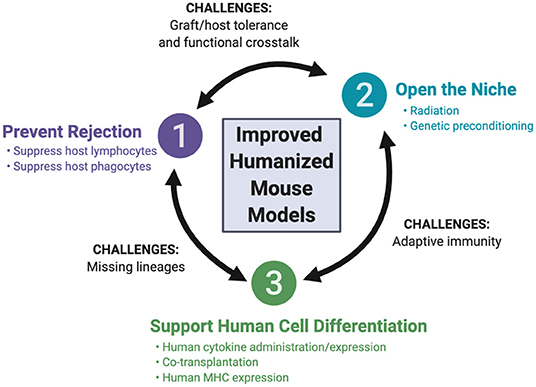
Figure 2. Fundamental principles of mouse humanization and remaining challenges.
The field of humanized mice was launched in the late 1980s, a few years after the discovery of mice with severe combined immunodeficiency (SCID). Prkdcscid (protein kinase, DNA activated, catalytic polypeptide; severe combined immunodeficiency) is a spontaneous mutation identified in a colony of C.B-17 mice (22). The functional inactivation of the PRKDC enzyme in SCID mice leads to defective DNA repair and repair-dependent somatic V(D)J recombination of B and T cell receptor-encoding genes (23). As a result, lymphocyte development is arrested at an early stage and mature B and T lymphocytes are absent in SCID mice. Taking advantage of the severe immunodeficiency of these animals, several groups successfully transplanted human PBMCs (12), human BM cells (24), human fetal tissues (25) or human HSPCs (26) in SCID (12, 25, 26) or equivalent recipient mice (24). Coinciding with the early years of the HIV-1/AIDS epidemic, these pioneering models provided a much-needed tool for in vivo studies of HIV-1 infection (27–29). Recipient mouse strains have since undergone numerous iterative improvements, and this historical perspective has been comprehensively reviewed previously [e.g., (6)]. The most notable modifications include further preventing rejection of the human graft, through the elimination of endogenous natural killer (NK) cells and the induction of phagocytic tolerance, as discussed below.
NK cells are lymphoid cells that eliminate cells lacking major histocompatibility complex class I (MHC-I) molecules (30). Engrafted human cells express human MHC-I molecules that are not recognized by mouse NK cells. Therefore, depletion of mouse NK cells is essential, to prevent them from recognizing and eliminating the graft as “missing self.” The interleukin-2 receptor γ chain (IL-2Rγ, encoded by Il2rg) is shared by multiple cytokines of the IL-2 family, including IL-15 that is essential for NK cell development (31). Consequently, Il2rg deficiency eliminates host NK cells and improves human hematopoietic cell engraftment in immunodeficient recipient mice (21, 32–35).
Transplanted human cells are also rejected through phagocytosis by mouse cells, such as monocytes and macrophages. Because phagocytes are essential for normal development and physiology, they cannot be easily depleted genetically without affecting mouse health and survival (36, 37). An alternative strategy is to alter their functional properties, by inducing phagocytic tolerance through the signal regulatory protein alpha (SIRPα) and CD47 axis (38). The polymorphic Sirpa gene in the non-obese diabetic (NOD) mouse strain encodes a variant of the SIRPα receptor that cross-reacts with the human CD47 ligand. As a result, human cells transplanted in NOD mice can engage the CD47/SIRPα “don't eat me” signal and are protected from phagocytosis by mouse macrophages (39). Consequently, backcrossing the scid mutation, and later the Il2rg deficiency, onto the NOD background significantly increased the efficiency of human cell transplantation (33–35, 40, 41). The resulting strains, NOD SCID Il2rg−/− (NOG and NSG), became very popular as they combine T, B and NK cell deficiencies with SIRPα-mediated phagocytic tolerance (33–35), but multiple other strains are functionally equivalent. These different strains abrogate V(D)J recombination [Prkdcscid mutation (12, 25), or deletion of recombination activating gene (RAG)-1 or RAG-2 (21, 32, 42)] and IL-2Rγ [Il2rg gene deletion or truncation (21, 32–35)]. SIRPα/CD47-dependent cross-species tolerance can be achieved by expressing the mouse SirpaNOD variant (43, 44) or human SIRPA (45–47), or by employing a Cd47 deficiency that produces tolerance by an unknown mechanism (48, 49). These strains are on diverse genetic backgrounds (NOD, BALB/c, C57BL/6), and are known by distinct acronyms, listed in Table 1. Upon transplantation of human CD34+ HSPCs, all of these strains support the differentiation of high levels of human CD45+ cells, reaching about 80% engraftment in the BM and 50% in the periphery. However, immune cell differentiation is disproportionately skewed toward the B and T lymphoid lineages (33, 35, 45). Human myelo-monocytic and NK cells are present only at low frequencies (50, 51), and human circulating red blood cells and platelets are barely detectable (52, 53). Investigators should carefully select the background strain they use, based on their research question, as the specific strain can impact the outcome of experiments. For example, SCID mice are highly susceptible to DNA damage; therefore RAG-deficient mice are the preferred recipient strain when testing chemo- and radiotherapies (42, 54). For studies involving complement-dependent cytotoxicity, the C57BL/6 or BALB/c backgrounds should be preferred, since NOD mice lack hemolytic complement C5 (41, 44).
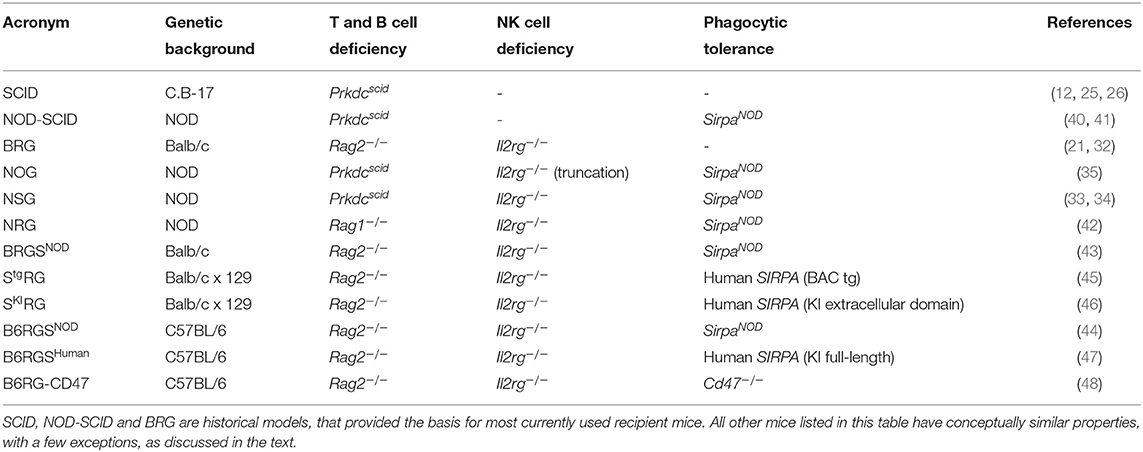
Table 1. List of immunodeficient mice used as recipients for transplantation of human hemato-lymphoid system.
Hematopoiesis is a complex and tightly regulated process during which hematopoietic progenitors undergo expansion and multilineage differentiation (2, 13). This process occurs primarily in the BM that uniquely provides supporting factors, such as cytokines at local physiological concentrations, and also provides a distinct microenvironment for developing cells (55). Accessibility of the transplanted human CD34+ HSPCs to this niche is required for efficient engraftment in the mouse host. Reducing cellularity in the mouse BM creates the needed physical space, described as “opening the niche.” Traditional protocols rely on irradiation as a preconditioning regimen (56–60), typically achieved using sub-lethal X-ray or 137Cs irradiation to kill most hematopoietic cells while limiting toxicity. When no irradiator is available, alternative pre-conditioning protocols can be used, such as the myeloablative drug busulfan (61–63).
Recently, the requirement for radiation preconditioning has been alleviated with recipient mice engineered to have a less populated BM niche, thereby achieving a form of “genetic preconditioning.” An example is provided by the KitW41 mutation, in an NSG-derived mouse strain known as NSGW41 (64) (Table 2). The receptor tyrosine kinase Kit (also known as c-Kit or CD117, encoded by the Kit gene) is the receptor for the cytokine stem cell factor (or SCF, also known as steel factor or Kit ligand). SCF increases HSPC retention in the BM niche by increasing their adhesion to neighboring stromal cells and proteins in the extracellular matrix (75). The KitW41 allelic variant encodes a protein with partially impaired kinase activity (76–78), producing functionally defective hematopoietic stem cells in KitW41/W41 mutant mice. In addition, because SCF is conserved between species [over 82% amino acid identity between mouse and human (1)], mouse SCF cross-reacts on the corresponding human Kit receptor. Consequently, when transplanted into NSGW41 mice, human HSPCs find a partially open BM niche and effectively compete for SCF against the mouse HSPCs that express the impaired Kit receptor. The impact of the KitW41/W41 mutation on mouse humanization is threefold: human CD34+ HSPCs efficiently engraft without radiation preconditioning; even without radiation, engraftment levels in NSGW41 are higher than in irradiated NSG recipients; and better maintenance of functional human HSPCs favors their multilineage differentiation, including into the erythro-megakaryocytic lineage (64, 79).
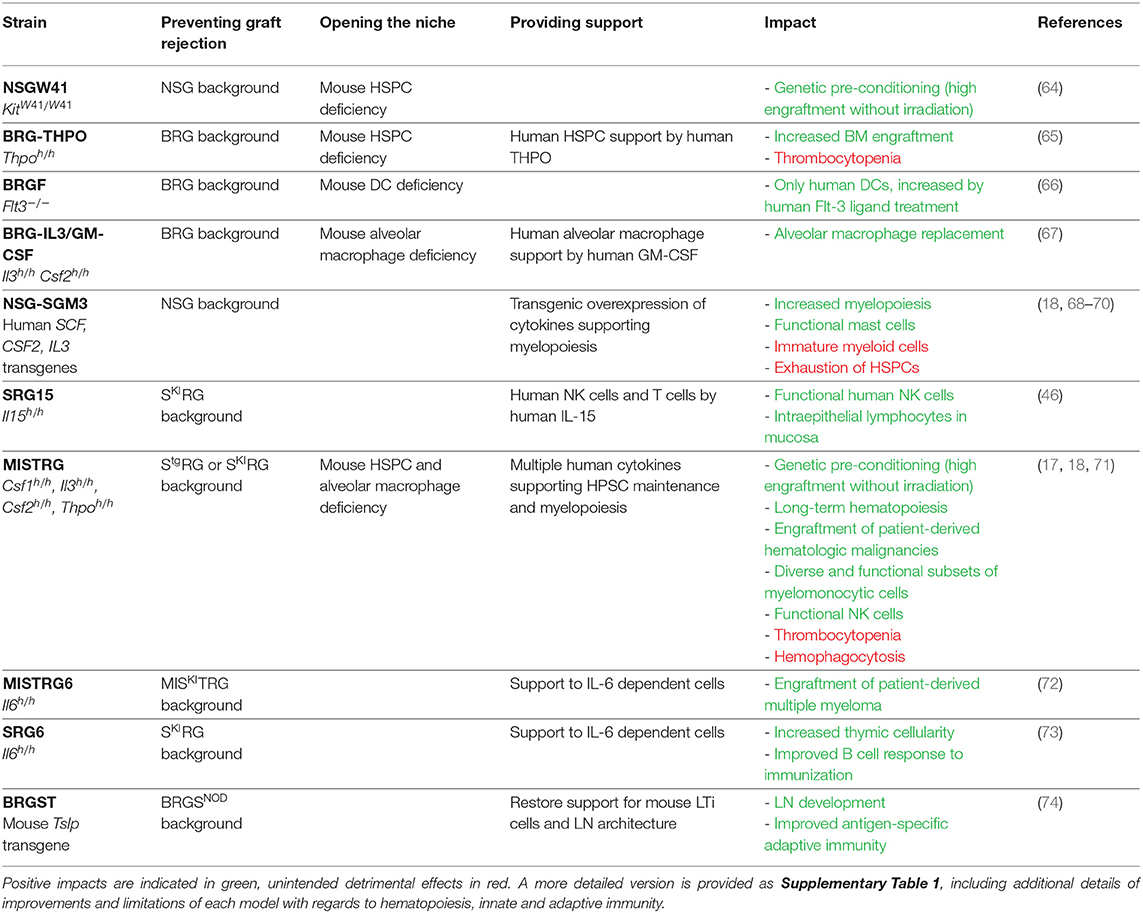
Table 2. List of genetically engineered mice, in the order discussed in this review.
Other genes can be inactivated to open the BM niche. Thpo encodes the cytokine, thrombopoietin, which is essential for the maintenance of quiescent and self-renewing HSPCs (80–83). Mouse Thpo gene inactivation reduces frequencies of mouse HSPCs, thereby opening the niche for transplanted human HSPCs (65). The concept of genetic preconditioning also applies to non-HSPC cell types, and to niches other than the BM. Fms-like tyrosine kinase 3 (Flt-3) ligand is essential to the differentiation of dendritic cells (DCs) (84–87), while granulocyte-macrophage colony stimulating factor (GM-CSF) is required for the maturation of lung alveolar macrophages (AM) (88–90). Genetic inactivation of Flt3 (encoding the receptor for Flt-3 ligand) or of Csf2 (encoding GM-CSF) eliminates mouse DCs or AMs, respectively, thereby opening the niche for the development of the corresponding human cell lineages (66, 67). In these three cases (Thpo, Ftl3, or Csf2 gene deficiencies), the elimination of mouse cell populations was supplemented with provision of the corresponding human cytokines (65–67), as discussed next.
The spatial microenvironment of the BM niche includes diverse cell types, including endothelial cells and mesenchymal stromal/stem cells (MSCs) that are known to release cytokines, signaling mediators and growth factors, such as SCF, IL-3, IL-6, THPO, and GM-CSF (91–93). These molecules are important for the maintenance of human HSPCs and their differentiation into all hematopoietic and immune cell lineages (1, 2). Some of the cytokines supporting hematopoiesis are poorly conserved (e.g., 29% amino acid identity between human and mouse IL-3) and do not cross-react from mouse to human, while others are highly conserved and largely cross-reactive (e.g., SCF) (1). However, even when amino acid identity is high, cross-reactivity of cytokines is not always complete in local microenvironments at physiological concentrations. To account for this incomplete cross-reactivity, various protocols were developed to promote the differentiation of a more complete human immune system upon human CD34+ cell transplantation in mice.
The simplest method of supplying graft-supporting factors is the repeated injection of recombinant human cytokines. This approach was used in an early-generation SCID model and the injection of cytokines, including human SCF, IL-3, and GM-CSF, supported enhanced engraftment levels of human BM cells as well as their myeloid differentiation (26). Because human NK cell differentiation is limited in mice, humanized BRG mice were treated with recombinant human IL-15 coupled to IL-15Rα, which is an essential growth factor for NK cell development and homeostasis. This treatment resulted in a significant increase in the differentiation and homeostasis of human NK cells in mice transplanted with human CD34+ cells (94).
A second cytokine delivery approach relies on hydrodynamic injection of a human cytokine-encoding DNA plasmid, resulting in the in vivo “transfection” of hepatocytes. In turn, hepatocytes produce high levels of the encoded cytokine and release it into the circulation for up to 5 days (95, 96). This method was used to demonstrate that human cytokines, such as GM-CSF and M-CSF, support myeloid differentiation of human CD34+ cells in NSG mice, while IL-15 and Flt-3 ligand supported NK cell differentiation (96, 97).
The administration of human cytokines can be combined with genetic opening of the niche. In BRGF mice, which lack the Flt3 gene, injection of human recombinant Flt-3 ligand boosts the development of human dendritic cells, in the absence of mouse dendritic cells (66).
Recombinant cytokine injection and hydrodynamic plasmid delivery are easy to implement, irrespective of the recipient mouse and protocol of human cell transplantation used. However, they both result in transient, systemic and generally supra-physiological cytokine expression.
To circumvent the requirement for repeated cytokine administrations, the genome of the recipient mouse can be engineered to express human cytokines (Table 2). The initial method relied on transgenic overexpression of a human cytokine-encoding cDNA, under the control of a strong promoter. Such transgenic mice are still in use, based on NSG or similar genetic backgrounds. However, results obtained with these mice need to be interpreted cautiously as the systemic overexpression of human cytokines frequently results in non-physiological hematopoiesis. For example, the transgenic pCMV promoter-driven overexpression of human SCF, GM-CSF, and IL-3 results in the mobilization of CD34+ HSPCs in the NSG-SGM3 mouse and the loss of their functional properties (68). As a consequence, although high-level human hematopoietic engraftment is achieved in NSG-SGM3 mice, hematopoietic progenitors lose their stemness and long-term hematopoiesis is deficient, as demonstrated by their inability to serially engraft (18, 69). Nevertheless, transgenic overexpression of cytokines can support the development of specific cell lineages and provide useful models to study those cells. In NSG-SGM3, the overexpression of human SCF results in the development of abundant and functional human mast cells, which are effective in models of passive cutaneous and systemic anaphylaxis (98). Similarly, human IL-15 overexpression in NOG mice supports the maintenance and function of human NK cells isolated from human peripheral blood (99, 100). Such models can provide useful experimental systems to evaluate the effect of candidate drugs on specific human immune cell populations.
More physiological expression of human cytokines can be achieved by bacterial artificial chromosome (referred to as “BAC”) transgenesis, inserting an entire human gene, including its regulatory elements, into the mouse genome. This method has been used to express human IL-6 and human IL-7 in NSG mice (101, 102). In another strategy, the mouse gene can be eliminated and corresponding human gene inserted in its place, including introns and exons from the start to the stop codon (Figure 3A) (103). This knock-in strategy has been used for a number of cytokine-encoding genes, including Csf1, Csf2, Il3, Il6, Il15, and Thpo (46, 65, 67, 72, 104). A slightly different approach was used for replacement of the Il7, Il15, and Tnfsf13b genes, through insertion of a human cDNA in frame with the start codon of the corresponding mouse gene, followed by a poly-adenylation signal (Figure 3B) (101, 105).
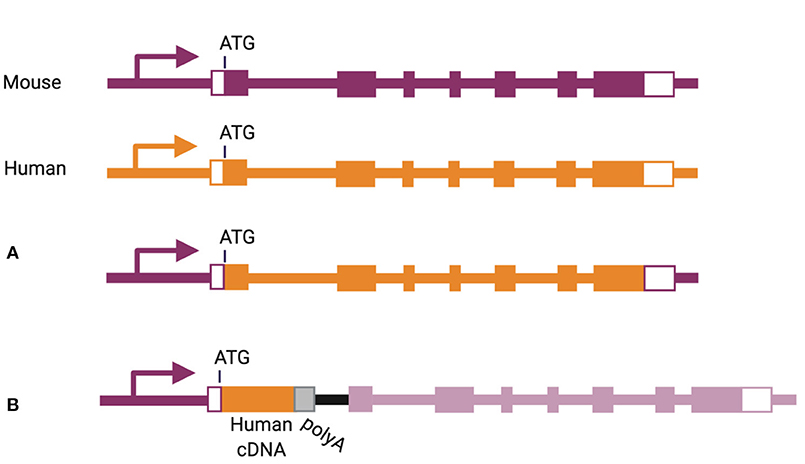
Figure 3. Methods of knock-in gene humanization by (A) gene replacement or (B) cDNA insertion.
Because of its role in supporting NK cell development, human IL-15 is highly featured in the humanized mouse literature, and it has been delivered to recipient mice by each of the methods described above (46, 94, 96, 99, 101). Since these approaches have been reported by different groups, their direct comparison and the evaluation of respective merits and limitations are difficult. However, the SRG-15 recipient mouse stands out as a definitive solution to the limited development of human NK cells in humanized mice, owing to its simplicity of use (no cytokine administration is needed), the physiological expression of the cytokine by knock-in gene replacement, the comprehensive phenotypic comparison to human peripheral blood cells and rigorous functional in vivo characterization (46).
In addition to expressing a human cytokine, knock-in gene replacement abrogates expression of the corresponding mouse cytokine (Figure 3). If the human cytokine is not fully cross-reactive, this can result in the absence of the cytokine-dependent mouse cell population and niche opening (103). This is best illustrated in the case of GM-CSF (encoded by Csf2/CSF2), where only 56% of amino acids are shared between human and mouse (1, 67). In mice with homozygous humanization of the Csf2 locus, the absence of mouse GM-CSF induces alveolar macrophage deficiency in the lung and a resulting pathology described as pulmonary alveolar proteinosis (67), recapitulating the phenotype of Csf2 knockout mice (88–90). Upon human CD34+ cell transplantation, human GM-CSF supports the development of human alveolar macrophages and partial rescue of the proteinosis pathology (67).
Importantly, because hematopoiesis is a stepwise process in which stem cells gradually differentiate toward more committed progenitors and multiple lineages of mature cells (1, 2), humanization of several cytokines is required to support the maintenance and multilineage differentiation of human HSPCs. This is exemplified in the MISTRG mouse in which M-CSF, IL-3, GM-CSF and THPO are humanized by knock-in replacement, on the SRG genetic background (17, 71). Niche opening and four human cytokines synergize to support the engraftment of human CD34+ cells without radiation preconditioning (17, 18). The long-term maintenance of functional HSPCs is demonstrated by their serial transplantation for up to four generations of MISTRG recipients (18). These mice also support multilineage differentiation of human B, T and dendritic cells (similarly to NSG mice) as well as different subsets of functional myelo-monocytic cells in lymphoid and non-lymphoid tissues (17, 18). The myelo-monocytic cells themselves express human IL-15/IL-15Rα, which in turn supports the development and function of human NK cells (17). Overall, MISTRG mice emphasize the benefits of combining multiple strategies for the provision of human cytokines in the development of improved recipient mice by: niche opening by elimination of the corresponding mouse cytokines; physiological expression of each cytokine; synergy between multiple cytokines; and, indirectly, cross-support between different human immune cell lineages.
Although providing specific cytokines can be effective, it would be a long and arduous journey to humanize the entire spectrum of hematopoiesis-supporting cytokines (and possibly other required factors). Therefore, co-transplantation of supporting human cells or tissues, naturally found in the BM niche or in other primary lymphoid organs, can be used to provide all required factors and more fully recapitulate human hematopoietic development and homeostasis.
A well-established protocol of such co-transplantation is the BM, liver, thymus (BLT) humanized mouse model (106, 107). BLT mice are generated by co-transplanting human fetal liver and thymus under the murine renal capsule in preconditioned adult immunodeficient mice, before injecting syngeneic CD34+ HSPCs intravenously. The liver and thymus implants form a liver-thymus organoid that contains stromal microenvironments and provides cytokines at local physiological concentrations, requisite for the differentiation of functional B, T, and NK cells, dendritic cells and monocytes/macrophages, as well as long-term maintenance of human hematopoiesis and lymphopoiesis (106–108). Because human T cells develop in the context of thymic epithelial cells, the human thymus organoid contains significantly higher absolute numbers of thymocytes, compared to mouse thymus (109) and a repertoire of immunocompetent human leukocyte antigen (HLA) class I- and class II-restricted T lymphocytes is selected (106, 107, 110). The immune cell distribution in BLT humanized mice is well-described for both primary and secondary lymphoid organs, including BM, human and mouse thymus, intestines (lamina propria), mesenteric lymph nodes, vaginal tissues, liver and lungs (106, 107, 109, 111–113). Although the BLT protocol was first described using the NOD-SCID background (106, 107), this technique can be applied to any recipient mouse, and has been used successfully in NSG (109, 113), NSG-SGM3 (98), and B6RG-CD47 (48) mice. Thus, investigators can use the BLT protocol to enhance human T cell development in a mouse strain with immune characteristics that are appropriate for their study.
Intravenous or intrafemoral co-transplantation of human BM-derived MSCs along with CD34+ cord blood cells is another strategy to improve engraftment of CD34+ and CD45+ cells in immunodeficient mice (114–117). Overall, co-transplantation of the BM-derived MSCs into humanized mice can improve human HSPC maintenance and expansion, and improve reconstitution of the human hemato-lymphoid system in humanized mice. Furthermore, recent studies have used genetically engineered MSCs to deliver additional human factors to support human hematopoiesis, or to facilitate the expansion and maintenance of HSPCs to allow serial transplantation and generation of larger quantities of humanized mice (118, 119).
Recent studies have used human bone organoid (ossicles) as a method to add human BM microenvironment for engrafted HSPCs to occupy (120, 121). Ossicles are often generated by seeding 3D polymer scaffolds with human BM-derived MSCs. These are then implanted subcutaneously into NSG mice and become colonized with subsequently injected human HSPCs, enabling their expansion and differentiation (122–128). Humanized ossicles contain increased human immature and mature hematopoietic cells as compared to the bones of host mice implanted with only human CD34+ cells, indicating homing of HSPCs (122, 128). Additionally, self-renewing HSPCs from humanized ossicles can reconstitute hematopoiesis in secondary recipient mice, demonstrating maintenance of their functional properties (129). Most importantly, high engraftment levels of hCD45+ cells were measured in the blood, spleen and mouse bone (130, 131). High numbers of human erythroid lineage cells and robust differentiation of mature myeloid cells were also detected (132).
With the development of MISTRG and MISTRG6 mouse strains that express essential human cytokines, and protocols for co-transplantation of human fetal bone chips or ossicles, major progress has been made in transplanting patient-derived hematologic malignancies into humanized BM niches. Samples from patients with myelodysplastic syndromes, myeloproliferative neoplasms, low risk acute myeloid leukemia, diffuse large B cell lymphoma or multiple myeloma have been successfully engrafted, using diverse recipient mice and protocols (72, 123, 125, 133–136).
Provision of specific human cytokines significantly improved the development, homeostasis and function of human NK cells and myelo-monocytic cells in humanized mice. But several lineages remain defective. For example, human neutrophils are generally present in the BM of humanized mice, but their frequency is negligible in the periphery (50). Cytokine overexpression in NSG-SGM3 mice increases the frequency of granulocytic CD33+ cells in the BM and the periphery (18, 69, 70), but these cells display the morphology and cell surface phenotype of immature cells (18). Human cytokine knock-in MISTRG mice also do not have improved mature neutrophil numbers in the periphery (17, 18). Therefore, the differentiation, egress, maturation and/or survival of human neutrophils likely requires additional factors.
Human red blood cells (RBCs) and platelets are probably the most challenging hematopoietic cells to develop in mice. They illustrate that additional strategies, beyond the provision of human cytokines, will likely be needed to support the complete spectrum of human hematopoietic lineages in mice. In the BM of humanized NSG mice, human erythroid (CD235a+) and megakaryocytic (CD41+CD61+) progenitors are extremely rare. Their frequency is increased by at least an order of magnitude in NSGW41 and MISTRG recipient mice but surprisingly, overexpression of human erythropoietin (EPO) did not further improve human erythropoiesis (79, 133). The increase in genetically preconditioned mice is likely due to the better competition of human progenitors against mouse progenitors in the open hematopoietic niche, and/or support from knocked-in human factors. However, erythropoiesis is arrested at an immature (CD71+ CD235a+) stage. As a result, few mature CD71−CD235a+ human reticulocytes are detectable in the BM and human RBCs rarely exceed 1% in peripheral blood (79).
Several lines of evidence demonstrate that this deficiency is due to a developmental defect as well as impaired survival. Indeed, human RBCs are highly susceptible to destruction in mice. Expression of human SIRPα in BRG mice, or depletion of macrophages by clodronate treatment in NOD SCID mice, extends the lifespan of adoptively transferred human RBCs (45). However, in both cases, the half-life of human RBCs does not exceed ~16 h, which is much shorter than the 10–20-days half-life of mouse RBCs in similar transfer experiments (137). Accordingly, clodronate treatment results in significant but transient and incomplete increase in human RBCs and platelets in mice humanized by CD34+ cell transplantation (52, 53, 79). In those conditions, injection of the human cytokines, IL-3 and EPO, promotes an increase in peripheral RBC counts (52). But, because clodronate targets both mouse and human cells, the treatment results in a humanized mouse entirely lacking human phagocytic cells, which limits the applicability of the model for studies of human immunity.
Therefore, the entire panel of mechanisms limiting the half-life of human RBCs and platelets in mice will need to be identified and resolved. In addition, the adequate combination of human cytokines will have to be provided, to enable the mice to live with primarily human platelets and RBCs. Such a model would be highly useful for studying diseases caused by pathogens with exclusive tropism for human RBCs [e.g., malaria caused by Plasmodium falciparum (138)] or diseases in which platelets contribute to pathogenesis [e.g., dengue fever, autoimmune thrombocytopenia (139, 140)]. In the meantime, current models (such as MISTRG) have already demonstrated their utility for modeling the early BM stages of human erythropoiesis and thrombopoiesis, as well as for studying drug responses in pathologies, including myelodysplastic syndromes and myeloproliferative neoplasms (133, 134).
As a result of advances discussed above, we now have humanized mice in which both cellular and humoral adaptive immune responses can be elicited. However, these responses are largely modest in magnitude, quality and duration, as outlined below.
Humanized mice generated by transplantation of CD34+ cells, or following the BLT protocol, can mount antigen-specific adaptive immune responses (21, 107, 141, 142). Evidence of protective immunity, capable of controlling pathogen replication and resulting disease, was provided in the context of Epstein-Barr virus (EBV), dengue virus (DENV), and human immunodeficiency virus-1 (HIV-1) infection. Indeed, in CD34+-humanized NSG mice, antibody-mediated depletion of T cells led to the development of EBV-associated tumors, suggesting that T cell-mediated immunity can control EBV infection in this setting (143). Robust CD4+ and CD8+ T cell responses were also detected after DENV and HIV-1 infection. Notably, in HIV-1-infected BLT mice, CD8+ T cell responses were HLA-restricted and directed against epitopes previously described as immunogenic in humans. Furthermore, CD8+ T cell-mediated viral recognition led to viral epitope evolution, closely resembling clinical observations (142).
Despite having detectable anti-viral T cells, CD34+-humanized and BLT mice generally exhibit weak humoral responses after viral infection (144, 145). While neutralizing antibodies were detected in a subset of DENV-infected mice, in both CD34+-only and BLT humanized mice, the anti-EBV or anti-HIV-1 antibody responses were either weak, equivocal or delayed (111, 141). This was also the case in humanized MISTRG mice, where enhanced innate immune cell engraftment led to improved T cell function after Listeria monocytogenes infection, but humoral immunity remained weak (17). Across these models, immunoglobulin class-switching and somatic hypermutation are rarely achieved, likely because most B cells fail to reach a fully mature phenotype in the periphery, T cell repertoire is selected on mouse and human MHC and is suboptimal, and recipient mice have disorganized lymph nodes and little capacity for germinal center formation (146).
While human B cells are detectable in high frequencies in humanized mice, most exhibit an immature phenotype, and remain blocked at the transitional stage of B cell development (1, 146). Since immature B cells have a reduced capacity to respond to antigen (147), several groups sought to improve B cell maturation and by extension, their function by knocking-in human cytokines that are known to support B cell development. Surprisingly, human B cell activation factor (BAFF, encoded by TNFSF13B) knock-in resulted in reduced numbers of mature naïve B cells, and reduced antibody production after immunization (105). Human IL-7 knock-in had no effect on B cell numbers and any effect on B cell function remains to be determined (101). In contrast, human IL-6 positively impacted B cell differentiation into plasmablasts and memory cells after immunization with model antigen, and promoted somatic hypermutation and class switching, albeit to a lower level than that observed in humans (73). IL-6 knock-in mice also had improved T cell development; therefore, it is possible that enhanced B cell responses were in part due to increased T cell help.
Thymic lymphopoiesis is suboptimal in mice humanized by transplantation of CD34+ HSPCs, in part due to a lack of thymopoiesis-supporting human cytokines. As a result, thymic cellularity is extremely low (only a few million cells) and CD4+ CD8+ double positive cells, which represent the vast majority of thymocytes in a healthy thymus, are frequently underrepresented. Human IL-6 knock-in produces increased thymic cellularity (73), but additional cytokines are likely required to restore the normal size of a mouse thymus, populated by human thymocytes. While a human IL-7 knock-in recipient mouse has been developed, a thorough characterization of thymic cellularity upon HSPC engraftment has not been reported to date (101).
Human T cell repertoire selection in the mouse thymus involves interactions with both mouse MHC and human HLA-expressing cells-expressing cells (1, 10, 21). Specifically, developing human T cells are positively selected by mouse epithelial cells and negatively selected by both mouse epithelial and mouse and human hematopoietic cells (148). The resulting T cell receptor (TCR) repertoire is weakly reactive to autologous human leukocyte antigen (HLA) class I and II, and tolerant to mouse MHC. Indeed, in in vitro re-stimulation assays, T cells from humanized mice were shown to proliferate better in response to allogeneic human DCs compared to autologous human DCs or mouse DCs (21). To improve the repertoire and function of human T cells, several recipient strains have been engineered to express HLA class I and class II (143, 144, 149–154). HLA-restricted T cell responses have been reported in these mice (143, 149). Additional characterization and comparison of these mice are required to rigorously evaluate how transgenic HLA expression in recipient HSPC-humanized mice impacts human T cell selection and function.
In BLT mice, T cells develop in the human thymus organoid and are positively and negatively selected on human autologous HLA molecules (106, 107, 155). Consequently, BLT mice are generally accepted as having a more diverse T cell repertoire, capable of mounting more robust adaptive immune responses, as discussed above. However, because the induction of tolerance for mouse MHC and mouse tissue-restricted antigens is incomplete, BLT mice are prone to the development of xenogeneic graft-vs.-host disease (xGvHD) (156, 157), as discussed below.
Few direct comparisons between BLT and HSPC-engrafted mice have been reported to date (144, 145). In both models, it is apparent that thymic lymphopoiesis and the selection of a diverse and tolerant T cell repertoire is a complex issue, and additional work is needed.
Secondary lymphoid organs including lymph nodes (LNs), spleen, Peyer's patches, and mucosa-associated lymphoid tissues normally provide niche microenvironments where T and B cells interact with hematopoietic antigen-presenting cells and stromal follicular dendritic cells (FDCs) to initiate the adaptive immune response (158, 159). LN formation requires IL-7- and IL-2Rγ-dependent lymphoid tissue inducer (LTi) cells (160). However, because most immunodeficient recipient mice lack Il2rg to promote tolerance to the human graft through NK cell depletion, they also lack LTi cells and defined LN structures. This represents a major obstacle in recapitulating the human adaptive immune response in mice. While engraftment with human CD34+ cells can partially rescue the LN anlagen, T and B cell zones remain poorly organized compared to those in normal human or normal mouse LNs. Furthermore, the germinal center formation is impaired, in part because it involves human B cells interacting with stromal FDCs of mouse origin (1, 159).
Given that the germinal center is the primary site of B cell-T cell collaboration, antibody affinity maturation and class switching, multiple groups sought to improve LN formation and germinal center development (158). Adoptive transfer of autologous human DCs lentivirally transduced to express GM-CSF, IFNα, and human cytomegalovirus (HCMV) viral antigen enhances LN development in humanized NRG mice, and by extension promotes T cell-B cell collaboration and the production of neutralizing anti-HCMV antibodies (161, 162). Human fetal organ co-transplantation also partially rescues secondary lymphoid organ development. BLT mice have improved lymphoid structure development in the spleen and lymph nodes compared to CD34+-only engrafted mice, and as a result, they also have more robust antigen-specific adaptive immunity (106, 107). However, a comparison of IL-2Rγ-sufficient NOD-SCID BLT humanized mice and IL-2Rγ-deficient NSG BLT humanized mice showed that only NOD-SCID BLT mice contain mucosal tissue-associated T cells (109). This result emphasizes the critical nature of IL-2Rγ in the development of LN and other secondary lymphoid organs that are essential for adaptive immune responses and mucosal immunity. The importance of structured secondary lymphoid organs for adaptive immunity is further demonstrated by the addition of fetal spleen to BLT mice (163). Human fetal spleen implants grow into spleen organoids with prominent follicular lymphoid structure; implanted mice have improved B cell and T cell engraftment, compared to control mice without human spleen implants, and can mount antigen-specific responses to immunization (163).
Another strategy to overcome the near-absence of LNs in Il2rg−/− humanized mice involves transgenic overexpression of murine thymic stromal lymphopoietin (TSLP). TSLP is another cytokine of the IL-2 family, its receptor is independent of the IL-2Rγ chain and, when overexpressed under a keratin 14 promoter (specific for epithelial/mesenchymal cells), it rescues LN development in Il7 deficient mice (164, 165). Crossing the transgene to BRGS resulted in the BRGST model, in which mouse LTi cells and LN structures are restored (74). Upon transplantation of CD34+ HSPCs, BRGST mice support the development of a human immune system, including sizeable LNs with compartmentalized human T and B cell zones. BRGST mice also have more mature B cells and IL-21 producing follicular helper T cells, essential to promoting adaptive immunity. Consequently, BRGST mice mounted enhanced antigen-specific humoral immune responses upon immunization with an experimental antigen (74). Overall, this novel model successfully addresses a major limitation that had hampered immune function in most humanized mouse models to date.
The transplantation of human cells into recipient mice is feasible because the recipient mice used are immunologically tolerant to the graft. As these mice support an increasingly functional human immune system, immunologic tolerance of the graft for its host can become an issue.
Adoptive transfer of human T cells from donor PBMCs into immunodeficient recipient mice results in xGvHD, thereby limiting the potential duration of experiments with these mice. To prevent xGvHD, two strains of immunodeficient mice lacking mouse MHC-I and MHC-II have been developed, which prevented the onset of xGvHD, while retaining the functional properties of human T cells (166, 167). But, as discussed above, PBMC transfer results in an incomplete human hematopoietic system in the mice.
When mice are humanized by transplantation of CD34+ HSPCs, developing human T cells undergo positive and negative selection in the mouse thymus. Consequently, they are tolerized for mouse MHC-I and MHC-II, and xGvHD is not a limitation in this model (1, 10, 21).
Finally, in BLT mice, T cells are educated in the human thymus organoid and once they reach the periphery, they are allogeneic to the mouse MHC molecules and can induce xGvHD (156, 157). Different levels of xGvHD are reported by different groups, suggesting that the disease could be affected by subtle differences in protocols, the microbiota of the mice, or the recipient strain used (48). The development of xGvHD in the BLT model is attributed to residual mature T cells present in the fetal human thymus grafts, and these passenger T cells can be removed by treating the thymic implants with 2′-deoxyguanosine, or by treating the mice with anti-human CD2 antibody post-surgery (168).
The efficient development of human phagocytic cells of the myelo-monocytic lineage in MISTRG creates a new challenge; i.e., the absence of phagocytic tolerance from the human graft toward the mouse host. Mouse red blood cells are particularly susceptible to destruction by human phagocytic cells, and highly engrafted MISTRG mice develop lethal anemia (17). Consequently, the transplantation protocol in MISTRG needs to be optimized so that engraftment allows mouse erythropoiesis in the BM (or extramedullary erythropoiesis) to sufficiently compensate for the loss of mouse red blood cells by phagocytosis, as long as a specific experiment requires (17, 18, 58, 133). Long-term solutions will need to be implemented, either to establish human-to-mouse phagocytic tolerance or to enable human erythropoiesis to reach healthy human RBC counts and maintain mouse homeostasis, as discussed above.
Xenogeneic GvHD and hemophagocytosis remind us that activation and tolerance are two equally important features of the immune system and are inseparable. As human immune function improves in mouse recipients, it is likely that parallel strategies will need to be developed to maintain tolerance of the human immune graft for the mouse host.
Immune responses require functional interactions between immune cells and their target, either through direct cell-cell contact or via soluble factors. In humanized mice generated by transplantation of human HSPCs, this requirement is fulfilled when the target of the immune response is also a human hematopoietic cell. Accordingly, T and B cell-mediated immunity have been demonstrated in the context of infection by pathogens with tropism for human hematopoietic cells, such as EBV, HIV-1, or dengue virus (141, 143, 169). But, for pathogens that infect non-hematopoietic tissues, or for inflammatory mediators that induce systemic responses, the cross-reactivity between human immune effector mechanisms and mouse target tissues may be incomplete. Humanizing cytokine receptors or other factors, such as adhesion molecules, could improve the responsiveness of mouse tissues to human immune cells and soluble mediators.
Co-transplantation of the human target tissue along with the human hematopoietic system has been performed in the context of cancer and infectious diseases. Implantation of human tumors, either from an established cell line or from a “patient-derived xenograft” (PDX), in mice already repopulated with a human immune system, provides useful models for immuno-oncology and immunotherapy studies (170–173). However, developing such “immuno-PDX” models can be challenging as each patient-derived tumor or cell line has different growth characteristics in mice, and matching patient's HLA to the HLA of the HSPC donor is not always feasible. Additionally, many components of the tumor microenvironment (e.g., vasculature) remain of mouse origin. Consequently, antitumoral immunity, or the response to immunotherapies, can be highly variable from experiment to experiment (171). In the case of hematologic malignancies, new strains of recipient mice (i.e., MISTRG and MISTRG6) extend the range of transplantable diseases (72, 133–136) and provide new opportunities to evaluate candidate immunotherapies, such as adoptive T cell therapies (174, 175).
Transplantation of human liver tissue enables infection of the mouse host by human hepatotropic viruses. Because implantation of a liver fragment at an ectopic site does not fully recapitulate the architecture and function of the liver, protocols of orthotopic implantation have been developed. These methods follow the same principles as for the transplantation of human hematopoiesis (176): an immunodeficient strain to prevent immune rejection of the graft; engineering of the mouse to induce mouse liver injury and open the niche for human hepatocytes; and, in some instances, support from human growth factors (177). Several methods have been developed to eliminate mouse hepatocytes, relying on the expression of cytolytic proteins [e.g., overexpression of urokinase plasminogen activator under an albumin promoter in uPA mice (178, 179)] and/or inactivation of metabolic enzymes essential for hepatocyte homeostasis [e.g., inactivation of the fumarylacetoacetate hydrolase in Fah−/− mice (180, 181)]. Upon injection of human hepatocytes, these mice support high levels of liver chimerism and are permissive for infection by the human hepatotropic viruses, HBV and HCV. To study human immune responses to these pathogens, mice can be dually transplanted with human hepatocytes and CD34+ HSPCs; human immune cells are recruited to the human liver in these chimeric mice and immune responses are triggered upon viral infections (177, 182, 183).
Finally, subcutaneous implantation of a fragment of human fetal lung contains all cell types naturally present in this tissue, and extends the permissiveness of the host mouse to respiratory pathogens with human tropism, including respiratory syncytial virus, Middle East respiratory syndrome coronavirus, and HCMV. This was recently accomplished by surgical implantation of a fragment of human lung tissue in BLT mice, resulting in the “BLT-L” model (184). Upon infection with HCMV, by direct injection in the lung implant, BLT-L mounted an antigen specific adaptive immune response, capable of controlling virus replication (184).
Thus, co-transplantation models greatly broaden the spectrum of human immune responses that can be studied in humanized mice. However, current approaches rely on highly specialized protocols and recipient mice, and should still be considered as prototypes under development.
Tremendous progress has been accomplished since pioneering humanized mouse models were developed in the late 1980s. In the past decade, a flurry of new opportunities have been enabled by the optimization of recipient mouse strains and humanization protocols. The most advanced models support long-term multilineage human hematopoiesis, and recapitulate essential aspects of innate immunity and antigen-specific adaptive immunity. Furthermore, numerous hematologic diseases can now be modeled by xenotransplantation of primary patient-derived samples.
Despite this progress, limitations remain. Rigorous evaluation and comparison of the new models is needed, while supporting additional innovations that might drive transformative advances. Until one or more humanized mouse model fully recapitulates human immunity, we can enthusiastically but critically use the currently available strains to answer specific, clinically-relevant questions and hopefully inform the development of new, life-saving therapies (Box 2).
Box 2. Which humanized mouse model is best for your studies?
There is no one-size-fits-all model. Here are a few practical considerations:
(1) Which mice and human cells/tissue do you have access to?
(2) What question are you trying to answer?
(3) Which cell types are important to answer your question?
In addition to considering these practical questions, it is important to know what has been done before you. Take time to read the literature and critically analyze the experiments. If you are able to talk to the people who developed the model or to someone who has recently published on the model of interest, you may receive invaluable unpublished data and advice.
Finally, applying these criteria, we provide our own selection, subjective and probably slightly biased, of the models that we consider most suitable for specific applications.
Hematopoiesis
• HSPC biology: NSGW41, MISTRG, MISTRG6
• Erythropoiesis and thrombopoiesis: NSGW41, MISTRG, MISTRG6 (BM only, limited)
• Myelopoiesis: MISTRG, MISTRG6
• Hematologic malignancies: MISTRG, MISTRG6
Innate immunity
• Monocytes/macrophages: MISTRG, MISTRG6
• DCs: NSG (or any other strain, depending on additional requirements)
• Mast cells: NSG-SGM3
• Neutrophils: no suitable model available
• NK cells: SRG15, MISTRG
• Innate lymphoid cells: NSG (or any other strain, depending on additional requirements)
Adaptive immunity
BLT mice are generally considered as supporting more robust adaptive immunity than mice transplanted with HSPCs only. However, few direct comparisons have been reported and antigen-specific immune responses remain relatively weak or delayed, even in BLT mice. Importantly, the BLT protocol can be applied with any recipient mice.
Expression of HLA molecules by the mouse host qualitatively favors HLA-restricted immune responses. But the impact on the amplitude of responses remains to be rigorously quantified.
• B cells: BLT, SRG6, MISTRG6
• T cells: BLT, BRGST.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Research in our lab was supported by the NIH/NCI CA234720, NIH/NIAID AI138011, The Hartwell Foundation, The Emerson Collective, the Seattle Translational Tumor Research, the University of Washington Center for AIDS Research, and the Immunotherapy Integrated Research Center at Fred Hutch. TM was supported by a Parker Institute for Cancer Immunotherapy Scholar Award. EC was supported by a postdoctoral fellowship from Fred Hutch's Immunotherapy Integrated Research Center.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Deborah Banker for manuscript editing, and the Guest Editors for inviting us to submit this manuscript, which we wrote as an educational work from home activity. Figures created with BioRender.com.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.643852/full#supplementary-material
AM, Alveolar macrophages; BLT, bone marrow, liver, thymus; BM, Bone marrow; BRG, BALB/c Rag2−/− Il2g−/−; BRGF, BALB/c Rag2−/− Il2g−/− Flt3−/−; DCs, Dendritic cells; EBV, Epstein–Barr virus; EPO, Erythropoietin; G-CSF, Granulocyte colony-stimulating factor; GM-CSF, Granulocyte-macrophage colony-stimulating factor; GvHD, Graft-vs.-host disease; HCMV, Human Cytomegalovirus; HIV-1, human immunodeficiency virus 1; HLA, Human leukocyte antigen; HSPCs, Hematopoietic stem and progenitor cells; IL, Interleukin; MSCs, Mesenchymal stromal cells; MHC-I, Major histocompatibility complex class I; MHC-II, Major histocompatibility complex class II; MISTRG, M-CSFh/h IL-3/GM-CSFh/h SIRPαh/m THPOh/h RAG2−/− IL2Rγ−/−; MISTRG6, M-CSFh/h IL-3/GM-CSFh/h SIRPαh/m THPOh/h RAG2−/− IL2Rγ−/− IL6h/h; NK, Natural killer; NOD, Non-obese diabetic; NSG, NOD/SCID/Il2rg−/−; NSGW41, NOD/SCID/Il2rg−/− KitW41/W41; PBMCs, peripheral blood mononuclear cells; PDX, Patient derived xenograft; RBCs, Red blood cells; SCF, Stem cell factor; SCID, severe-combined immunodeficient; TCR, T cell receptor; xGvHD, xenogeneic graft-vs.-host disease.
1. Rongvaux A, Takizawa H, Strowig T, Willinger T, Eynon EE, Flavell RA, et al. Human hemato-lymphoid system mice: current use and future potential for medicine. Annu Rev Immunol. (2013) 31:635–74. doi: 10.1146/annurev-immunol-032712-095921
2. Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. (2012) 10:120–36. doi: 10.1016/j.stem.2012.01.006
3. Gbyli R, Song Y, Halene S. Humanized mice as preclinical models for myeloid malignancies. Biochem Pharmacol. (2020) 174:113794. doi: 10.1016/j.bcp.2020.113794
4. Douam F., Ploss A, The use of humanized mice for studies of viral pathogenesis and immunity. Curr Opin Virol. (2018) 29:62–71. doi: 10.1016/j.coviro.2018.03.003
5. Li Y, Di Santo JP. Modeling infectious diseases in mice with a “humanized” immune system. Microbiol Spectr. (2019) 7:BAI-0019-2019. doi: 10.1128/microbiolspec.BAI-0019-2019
6. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. (2007) 7:118–30. doi: 10.1038/nri2017
7. Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. (2012) 12:786–98. doi: 10.1038/nri3311
8. Huntington ND, Di Santo JP. Humanized immune system (HIS) mice as a tool to study human NK cell development. Curr Top Microbiol Immunol. (2008) 324:109–24. doi: 10.1007/978-3-540-75647-7_7
9. Li Y, Di Santo JP. Probing human NK cell biology using human immune system (HIS) Mice. Curr Top Microbiol Immunol. (2016) 395:191–208. doi: 10.1007/82_2015_488
10. Manz MG. Human-hemato-lymphoid-system mice: opportunities and challenges. Immunity. (2007). 26:537-41. doi: 10.1016/j.immuni.2007.05.001
11. Theocharides AP, Rongvaux A, Fritsch K, Flavell RA, Manz MG. Humanized hemato-lymphoid system mice. Haematologica. (2016) 101:5–19. doi: 10.3324/haematol.2014.115212
12. Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature. (1988) 335:256–9. doi: 10.1038/335256a0
13. Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, et al. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. (2003) 21:759–806. doi: 10.1146/annurev.immunol.21.120601.141007
14. Allen TM, Brehm MA, Bridges S, Ferguson S, Kumar P, Mirochnitchenko O, et al. Humanized immune system mouse models: progress, challenges and opportunities. Nat Immunol. (2019) 20:770–4. doi: 10.1038/s41590-019-0416-z
15. Holyoake TL, Nicolini FE, Eaves CJ. Functional differences between transplantable human hematopoietic stem cells from fetal liver, cord blood, adult marrow. Exp Hematol. (1999) 27:1418–27. doi: 10.1016/S0301-472X(99)00078-8
16. Nicolini FE, Holyoake TL, Cashman JD, Chu PP, Lambie K, Eaves CJ. Unique differentiation programs of human fetal liver stem cells shown both in vitro and in vivo in NOD/SCID mice. Blood. (1999) 94:2686–95. doi: 10.1182/blood.V94.8.2686.420k15_2686_2695
17. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. (2014). 32:364–72. doi: 10.1038/nbt.2858
18. Sippel TR, Radtke S, Olsen TM, Kiem HP, Rongvaux A. Human hematopoietic stem cell maintenance and myeloid cell development in next-generation humanized mouse models. Blood Adv. (2019) 3:268–274. doi: 10.1182/bloodadvances.2018023887
19. Tyagi RK, Li J, Jacobse J, Snapper SB, Shouval DS, Goettel JA. Humanized mouse models of genetic immune disorders and hematological malignancies. Biochem Pharmacol. (2020) 174:113671. doi: 10.1016/j.bcp.2019.113671
20. Mazurier F, Doedens M, Gan OI, Dick JE. Rapid myeloerythroid repopulation after intrafemoral transplantation of NOD-SCID mice reveals a new class of human stem cells. Nat Med. (2003) 9:959–63. doi: 10.1038/nm886
21. Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. (2004) 304:104–7. doi: 10.1126/science.1093933
22. Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. (1983) 301:527–30. doi: 10.1038/301527a0
23. Blunt T, Finnie NJ, Taccioli GE, Smith GC, Demengeot J, Gottlieb TM, et al. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell. (1995) 80:813–23. doi: 10.1016/0092-8674(95)90360-7
24. Kamel-Reid S, Dick JE. Engraftment of immune-deficient mice with human hematopoietic stem cells. Science. (1988) 242:1706–9. doi: 10.1126/science.2904703
25. McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science. (1988) 241:1632–9. doi: 10.1126/science.2971269
26. Lapidot T, Pflumio F, Doedens M, Murdoch B, Williams DE, Dick JE. Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID mice. Science. (1992) 255:1137–41. doi: 10.1126/science.1372131
27. Namikawa R, Kaneshima H, Lieberman M, Weissman IL, McCune JM. Infection of the SCID-hu mouse by HIV-1. Science. (1988) 242:1684–6. doi: 10.1126/science.3201256
28. Mosier DE, Gulizia RJ, Baird SM, Wilson DB, Spector DH, Spector SA. Human immunodeficiency virus infection of human-PBL-SCID mice. Science. (1991) 251:791–4. doi: 10.1126/science.1990441
29. McCune J, Kaneshima H, Krowka J, Namikawa R, Outzen H, Peault B, et al. The SCID-hu mouse: a small animal model for HIV infection and pathogenesis. Annu Rev Immunol. (1991) 9:399–429. doi: 10.1146/annurev.iy.09.040191.002151
30. Miller JS, Lanier LL. Natural killer cells in cancer immunotherapy. Annu Rev Cancer Biol. (2019) 3:77–103. doi: 10.1146/annurev-cancerbio-030518-055653
31. Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol. (2006) 24:657–79. doi: 10.1146/annurev.immunol.24.021605.090727
32. Mazurier F, Fontanellas A, Salesse S, Taine L, Landriau S, Moreau-Gaudry F, et al. de Verneuil, A novel immunodeficient mouse model–RAG2 x common cytokine receptor gamma chain double mutants–requiring exogenous cytokine administration for human hematopoietic stem cell engraftment. J Interferon Cytokine Res. (1999) 19:533–41. doi: 10.1089/107999099313983
33. Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood. (2005) 106:1565–73. doi: 10.1182/blood-2005-02-0516
34. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. (2005) 174:6477–89. doi: 10.4049/jimmunol.174.10.6477
35. Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. (2002) 100:3175–82. doi: 10.1182/blood-2001-12-0207
36. Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. (1990) 345:442–4. doi: 10.1038/345442a0
37. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, reproductive defects. Blood. (2002) 99:111–20. doi: 10.1182/blood.V99.1.111
38. Barclay A.N., Brown MH, The SIRP family of receptors and immune regulation. Nat Rev Immunol. (2006) 6:457–64. doi: 10.1038/nri1859
39. Takenaka K, Prasolava TK, Wang JC, Mortin-Toth SM, Khalouei S, Gan OI, et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol. (2007) 8:1313–23. doi: 10.1038/ni1527
40. Hesselton RM, Greiner DL, Mordes JP, Rajan TV, Sullivan JL, Shultz LD. High levels of human peripheral blood mononuclear cell engraftment and enhanced susceptibility to human immunodeficiency virus type 1 infection in NOD/LtSz-scid/scid mice. J Infect Dis. (1995) 172:974–82. doi: 10.1093/infdis/172.4.974
41. Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. (1995) 154:180–91.
42. Pearson T, Shultz LD, Miller D, King M, Laning J, Fodor W, et al. Non-obese diabetic-recombination activating gene-1 (NOD-Rag1 null) interleukin (IL)-2 receptor common gamma chain (IL2r gamma null) null mice: a radioresistant model for human lymphohaematopoietic engraftment. Clin Exp Immunol. (2008) 154:270–84. doi: 10.1111/j.1365-2249.2008.03753.x
43. Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, Strick-Marchand H, et al. Functional CD47/signal regulatory protein alpha (SIRPα) interaction is required for optimal human T- and natural killer- (NK) cell homeostasis in vivo. Proc Natl Acad Sci USA. (2011) 108:13224–9. doi: 10.1073/pnas.1101398108
44. Yamauchi T, Takenaka K, Urata S, Shima T, Kikushige Y, Tokuyama T, et al. Polymorphic Sirpa is the genetic determinant for NOD-based mouse lines to achieve efficient human cell engraftment. Blood. (2013) 121:1316–25. doi: 10.1182/blood-2012-06-440354
45. Strowig T, Rongvaux A, Rathinam C, Takizawa H, Borsotti C, Philbrick W, et al. Transgenic expression of human signal regulatory protein alpha in Rag2-/-gamma(c)-/- mice improves engraftment of human hematopoietic cells in humanized mice. Proc Natl Acad Sci USA. (2011) 108:13218–23. doi: 10.1073/pnas.1109769108
46. Herndler-Brandstetter D, Shan L, Yao Y, Stecher C, Plajer V, Lietzenmayer M, et al. Humanized mouse model supports development, function, and tissue residency of human natural killer cells. Proc Natl Acad Sci USA. (2017) 114:E9626–34. doi: 10.1073/pnas.1705301114
47. Jinnouchi F, Yamauchi T, Yurino A, Nunomura T, Nakano M, Iwamoto C, et al. A human SIRPA knock-in xenograft mouse model to study human hematopoietic and cancer stem cells. Blood. (2020) 135:1661–72. doi: 10.1182/blood.2019002194
48. Lavender KJ, Pang WW, Messer RJ, Duley AK, Race B, Phillips K, et al. BLT-humanized C57BL/6 Rag2-/-γc-/-CD47-/- mice are resistant to GVHD and develop B- and T-cell immunity to HIV infection. Blood. (2013) 122:4013–20. doi: 10.1182/blood-2013-06-506949
49. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. (2000) 288:2051–4. doi: 10.1126/science.288.5473.2051
50. Tanaka S, Saito Y, Kunisawa J, Kurashima Y, Wake T, Suzuki N, et al. Development of mature and functional human myeloid subsets in hematopoietic stem cell-engrafted NOD/SCID/IL2rgammaKO mice. J Immunol. (2012) 188:6145–55. doi: 10.4049/jimmunol.1103660
51. Strowig T, Chijioke O, Carrega P, Arrey F, Meixlsperger S, Ramer PC, et al. Human NK cells of mice with reconstituted human immune system components require preactivation to acquire functional competence. Blood. (2010) 116:4158–67. doi: 10.1182/blood-2010-02-270678
52. Hu Z, Van Rooijen N, Yang YG. Macrophages prevent human red blood cell reconstitution in immunodeficient mice. Blood. (2011) 118:5938–46. doi: 10.1182/blood-2010-11-321414
53. Hu Z, Yang YG. Full reconstitution of human platelets in humanized mice after macrophage depletion. Blood. (2012) 120:1713–6. doi: 10.1182/blood-2012-01-407890
54. Wunderlich M, Manning N, Sexton C, Sabulski A, Byerly L, O'Brien E, et al. Improved chemotherapy modeling with RAG-based immune deficient mice. PLoS ONE. (2019) 14:e0225532. doi: 10.1371/journal.pone.0225532
55. Crane GM, Jeffery E, Morrison SJ. Adult haematopoietic stem cell niches. Nat Rev Immunol. (2017) 17:573–590. doi: 10.1038/nri.2017.53
56. Saito Y, Ellegast JM, Manz MG. Generation of humanized mice for analysis of human dendritic cells. Methods Mol Biol. (2016) 1423:309–20. doi: 10.1007/978-1-4939-3606-9_22
57. Legrand N, Weijer K, Spits H. Experimental model for the study of the human immune system: production and monitoring of “human immune system” Rag2-/-gamma c-/- mice. Methods Mol Biol. (2008) 415:65–82. doi: 10.1007/978-1-59745-570-1_4
58. Song Y, Gbyli R, Fu X, Halene S. Functional analysis of human hematopoietic stem cells in vivo in humanized mice. Methods Mol Biol. (2020) 2097:273–89. doi: 10.1007/978-1-0716-0203-4_18
59. Hasgur S, Aryee KE, Shultz LD, Greiner DL, Brehm MA. Generation of immunodeficient mice bearing human immune systems by the engraftment of hematopoietic stem cells. Methods Mol Biol. (2016) 1438:67–78. doi: 10.1007/978-1-4939-3661-8_4
60. Vatakis DN, Bristol GC, Kim SG, Levin B, Liu W, Radu CG, et al. Using the BLT humanized mouse as a stem cell based gene therapy tumor model. J Vis Exp. (2012) 2012:e4181. doi: 10.3791/4181
61. Choi B, Chun E, Kim M, Kim ST, Yoon K, Lee KY, et al. Human B cell development and antibody production in humanized NOD/SCID/IL-2Rγ(null) (NSG) mice conditioned by busulfan. J Clin Immunol. (2011) 31:253–64. doi: 10.1007/s10875-010-9478-2
62. Hayakawa J, Hsieh MM, Uchida N, Phang O, Tisdale JF. Busulfan produces efficient human cell engraftment in NOD/LtSz-Scid IL2Rgamma(null) mice. Stem Cells. (2009) 27:175–82. doi: 10.1634/stemcells.2008-0583
63. Singh M, Singh P, Gaudray G, Musumeci L, Thielen C, Vaira D, et al. An improved protocol for efficient engraftment in NOD/LTSZ-SCIDIL-2Rγnull mice allows HIV replication and development of anti-HIV immune responses. PLoS ONE. (2012) 7:e38491. doi: 10.1371/journal.pone.0038491
64. Cosgun KN, Rahmig S, Mende N, Reinke S, Hauber I, Schafer C, et al. Kit regulates HSC engraftment across the human-mouse species barrier. Cell Stem Cell. (2014) 15:227–38. doi: 10.1016/j.stem.2014.06.001
65. Rongvaux A, Willinger T, Takizawa H, Rathinam C, Auerbach W, Murphy AJ, et al. Human thrombopoietin knockin mice efficiently support human hematopoiesis in vivo. Proc Natl Acad Sci USA. (2011) 108:2378–83. doi: 10.1073/pnas.1019524108
66. Li Y, Mention JJ, Court N, Masse-Ranson G, Toubert A, Spits H, et al. A novel Flt3-deficient HIS mouse model with selective enhancement of human DC development. Eur J Immunol. (2016) 46:1291–9. doi: 10.1002/eji.201546132
67. Willinger T, Rongvaux A, Takizawa H, Yancopoulos GD, Valenzuela DM, Murphy AJ, et al. Human IL-3/GM-CSF knock-in mice support human alveolar macrophage development and human immune responses in the lung. Proc Natl Acad Sci USA. (2011) 108:2390–5. doi: 10.1073/pnas.1019682108
68. Nicolini FE, Cashman JD, Hogge DE, Humphries RK, Eaves CJ. NOD/SCID mice engineered to express human IL-3, GM-CSF and Steel factor constitutively mobilize engrafted human progenitors and compromise human stem cell regeneration. Leukemia. (2004) 18:341–7. doi: 10.1038/sj.leu.2403222
69. Wunderlich M, Chou FS, Sexton C, Presicce P, Chougnet CA, Aliberti J, et al. Improved multilineage human hematopoietic reconstitution and function in NSGS mice. PLoS ONE. (2018) 13:e0209034. doi: 10.1371/journal.pone.0209034
70. Billerbeck E, Barry WT, Mu K, Dorner M, Rice CM, Ploss A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rgamma(null) humanized mice. Blood. (2011) 117:3076–86. doi: 10.1182/blood-2010-08-301507
71. Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. (2015) 517:381–5. doi: 10.1038/nature14053
72. Das R, Strowig T, Verma R, Koduru S, Hafemann A, Hopf S, et al. Microenvironment-dependent growth of preneoplastic and malignant plasma cells in humanized mice. Nat Med. (2016) 22:1351–7. doi: 10.1038/nm.4202
73. Yu H, Borsotti C, Schickel JN, Zhu S, Strowig T, Eynon EE, et al. A novel humanized mouse model with significant improvement of class-switched, antigen-specific antibody production. Blood. (2017) 129:959–69. doi: 10.1182/blood-2016-04-709584
74. Li Y, Masse-Ranson G, Garcia Z, Bruel T, Kök A, Strick-Marchand H, et al. A human immune system mouse model with robust lymph node development. Nat Methods. (2018) 15:623–30. doi: 10.1038/s41592-018-0071-6
75. Broudy VC. Stem cell factor and hematopoiesis. Blood. (1997). 90:1345–64. doi: 10.1182/blood.V90.4.1345
76. Geissler EN, McFarland EC, Russell ES. Analysis of pleiotropism at the dominant white-spotting (W) locus of the house mouse: a description of ten new W alleles. Genetics. (1981) 97:337–61.
77. Geissler EN, Ryan MA, Housman DE. The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell. (1988) 55:185–92. doi: 10.1016/0092-8674(88)90020-7
78. Nocka K, Tan JC, Chiu E, Chu TY, Ray P, Traktman P, et al. Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41 and W. Embo j. (1990) 9:1805–13. doi: 10.1002/j.1460-2075.1990.tb08305.x
79. Rahmig S, Kronstein-Wiedemann R, Fohgrub J, Kronstein N, Nevmerzhitskaya A, Bornhauser M, et al. Improved Human Erythropoiesis and Platelet Formation in Humanized NSGW41 Mice. Stem Cell Reports. (2016) 7:591–601. doi: 10.1016/j.stemcr.2016.08.005
80. Fox N, Priestley G, Papayannopoulou T, Kaushansky K. Thrombopoietin expands hematopoietic stem cells after transplantation. J Clin Invest. (2002) 110:389–94. doi: 10.1172/JCI0215430
81. Kirito K, Fox N, Kaushansky K. Thrombopoietin stimulates Hoxb4 expression: an explanation for the favorable effects of TPO on hematopoietic stem cells. Blood. (2003) 102:3172–8. doi: 10.1182/blood-2003-03-0944
82. Qian H, Buza-Vidas N, Hyland CD, Jensen CT, Antonchuk J, Mansson R, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell. (2007) 1:671–84. doi: 10.1016/j.stem.2007.10.008
83. Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. (2007) 1:685–97. doi: 10.1016/j.stem.2007.10.020
84. Lyman SD, James L, Vanden Bos T, de Vries P, Brasel K, Gliniak B, et al. Molecular cloning of a ligand for the flt3/flk-2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell. (1993) 75:1157–67. doi: 10.1016/0092-8674(93)90325-K
85. Maraskovsky E, Brasel K, Teepe M, Roux ER, Lyman SD, Shortman K, et al. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J Exp Med. (1996) 184:1953–62. doi: 10.1084/jem.184.5.1953
86. McKenna HJ, Stocking KL, Miller RE, Brasel K, De Smedt T, Maraskovsky E, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. (2000) 95:3489–97. doi: 10.1182/blood.V95.11.3489
87. Waskow C, Liu K, Darrasse-Jèze G, Guermonprez P, Ginhoux F, Merad M, et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol. (2008) 9:676–83. doi: 10.1038/ni.1615
88. Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. (1994) 264:713–6. doi: 10.1126/science.8171324
89. Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA. (1994) 91:5592–6. doi: 10.1073/pnas.91.12.5592
90. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. (2001) 15:557–67. doi: 10.1016/s1074-7613(01)00218-7
91. Ramasamy SK, Kusumbe AP, Itkin T, Gur-Cohen S, Lapidot T, Adams RH. Regulation of hematopoiesis and osteogenesis by blood vessel-derived signals. Annu Rev Cell Dev Biol. (2016) 32:649–75. doi: 10.1146/annurev-cellbio-111315-124936
92. Rafii S, Shapiro F, Pettengell R, Ferris B, Nachman R, Moore M, et al. Human bone marrow microvascular endothelial cells support long-term proliferation and differentiation of myeloid and megakaryocytic progenitors. Blood. (1995) 86:3353–63. doi: 10.1182/blood.V86.9.3353.bloodjournal8693353
93. Majumdar MK, Thiede MA, Haynesworth SE, Bruder SP, Gerson SL. Human marrow-derived mesenchymal stem cells (MSCs) express hematopoietic cytokines and support long-term hematopoiesis when differentiated toward stromal and osteogenic lineages. J Hematother Stem Cell Res. (2000) 9:841–8. doi: 10.1089/152581600750062264
94. Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med. (2009) 206:25–34. doi: 10.1084/jem.20082013
95. Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. (1999) 6:1258–66. doi: 10.1038/sj.gt.3300947
96. Chen Q, Khoury M, Chen J. Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice. Proc Natl Acad Sci USA. (2009) 106:21783–8. doi: 10.1073/pnas.0912274106
97. Li Y, Chen Q, Zheng D, Yin L, Chionh YH, Wong LH, et al. Induction of functional human macrophages from bone marrow promonocytes by M-CSF in humanized mice. J Immunol. (2013) 191:3192–9. doi: 10.4049/jimmunol.1300742
98. Bryce PJ, Falahati R, Kenney LL, Leung J, Bebbington C, Tomasevic N, et al. Humanized mouse model of mast cell-mediated passive cutaneous anaphylaxis and passive systemic anaphylaxis. J Allergy Clin Immunol. (2016) 138:769–79. doi: 10.1016/j.jaci.2016.01.049
99. Katano I, Nishime C, Ito R, Kamisako T, Mizusawa T, Ka Y, et al. Long-term maintenance of peripheral blood derived human NK cells in a novel human IL-15- transgenic NOG mouse. Sci Rep. (2017) 7:17230. doi: 10.1038/s41598-017-17442-7
100. Katano I, Ito R, Kawai K, Takahashi T. Improved detection of in vivo human Nk cell-mediated antibody-dependent cellular cytotoxicity using a novel NOG-FcγR-Deficient Human IL-15 transgenic mouse. Front Immunol. (2020) 11:532684. doi: 10.3389/fimmu.2020.532684
101. Matsuda M, Ono R, Iyoda T, Endo T, Iwasaki M, Tomizawa-Murasawa M, et al. Human NK cell development in hIL-7 and hIL-15 knockin NOD/SCID/IL2rgKO mice. Life Sci Alliance. (2019) 2:e201800195. doi: 10.26508/lsa.201800195
102. Ono R, Watanabe T, Kawakami E, Iwasaki M, Tomizawa-Murasawa M, Matsuda M, et al. Co-activation of macrophages and T cells contribute to chronic GVHD in human IL-6 transgenic humanised mouse model. EBioMedicine. (2019) 41:584–596. doi: 10.1016/j.ebiom.2019.02.001
103. Willinger T, Rongvaux A, Strowig T, Manz MG, Flavell RA. Improving human hemato-lymphoid-system mice by cytokine knock-in gene replacement. Trends Immunol. (2011) 32:321–7. doi: 10.1016/j.it.2011.04.005
104. Rathinam C, Poueymirou WT, Rojas J, Murphy AJ, Valenzuela DM, Yancopoulos GD, et al. Efficient differentiation and function of human macrophages in humanized CSF-1 mice. Blood. (2011) 118:3119–28. doi: 10.1182/blood-2010-12-326926
105. Lang J, Zhang B, Kelly M, Peterson JN, Barbee J, Freed BM, et al. Replacing mouse BAFF with human BAFF does not improve B-cell maturation in hematopoietic humanized mice. Blood Adv. (2017) 1:2729–41. doi: 10.1182/bloodadvances.2017010090
106. Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. (2006) 108:487–92. doi: 10.1182/blood-2005-11-4388
107. Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med. (2006) 12:1316–22. doi: 10.1038/nm1431
108. Smith DJ, Lin LJ, Moon H, Pham AT, Wang X, Liu S, et al. Propagating Humanized BLT Mice for the Study of Human Immunology and Immunotherapy. Stem Cells Dev. (2016) 25:1863–73. doi: 10.1089/scd.2016.0193
109. Denton PW, Nochi T, Lim A, Krisko JF, Martinez-Torres F, Choudhary SK, et al. IL-2 receptor gamma-chain molecule is critical for intestinal T-cell reconstitution in humanized mice. Mucosal Immunol. (2012) 5:555–66. doi: 10.1038/mi.2012.31
110. Rajesh D, Zhou Y, Jankowska-Gan E, Roenneburg DA, Dart ML, Torrealba J, et al. Th1 and Th17 immunocompetence in humanized NOD/SCID/IL2rgammanull mice. Hum Immunol. (2010) 71:551–9. doi: 10.1016/j.humimm.2010.02.019
111. Brainard DM, Seung E, Frahm N, Cariappa A, Bailey CC, Hart WK, et al. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J Virol. (2009) 83:7305–21. doi: 10.1128/JVI.02207-08
112. Denton PW, Estes JD, Sun Z, Othieno FA, Wei BL, Wege AK, et al. Antiretroviral pre-exposure prophylaxis prevents vaginal transmission of HIV-1 in humanized BLT mice. PLoS Med. (2008) 5:e16. doi: 10.1371/journal.pmed.0050016
113. Stoddart CA, Maidji E, Galkina SA, Kosikova G, Rivera JM, Moreno ME, et al. Superior human leukocyte reconstitution and susceptibility to vaginal HIV transmission in humanized NOD-scid IL-2Rgamma(-/-) (NSG) BLT mice. Virology. (2011) 417:154–60. doi: 10.1016/j.virol.2011.05.013
114. Lee ST, Maeng H, Chwae YJ, Oh DJ, Kim YM, Yang WI. Effect of mesenchymal stem cell transplantation on the engraftment of human hematopoietic stem cells and leukemic cells in mice model. Int J Hematol. (2008) 87:327–37. doi: 10.1007/s12185-008-0041-3
115. Fernandez-Garcia M, Yanez RM, Sanchez-Dominguez R, Hernando-Rodriguez M, Peces-Barba M, Herrera G, et al. Mesenchymal stromal cells enhance the engraftment of hematopoietic stem cells in an autologous mouse transplantation model. Stem Cell Res Ther. (2015) 6:165. doi: 10.1186/s13287-015-0155-5
116. Metheny L III, Eid S, Lingas K, Reese J, Meyerson H, Tong A, et al. Intra-osseous Co-transplantation of CD34-selected umbilical cord blood and mesenchymal stromal cells. Hematol Med Oncol. (2016) 1:25–9. doi: 10.15761/HMO.1000105
117. Morton JJ, Keysar SB, Perrenoud L, Chimed TS, Reisinger J, Jackson B, et al. Dual use of hematopoietic and mesenchymal stem cells enhances engraftment and immune cell trafficking in an allogeneic humanized mouse model of head and neck cancer. Mol Carcinog. (2018) 57:1651–63. doi: 10.1002/mc.22887
118. Khoury M, Drake A, Chen Q, Dong D, Leskov I, Fragoso MF, et al. Mesenchymal stem cells secreting angiopoietin-like-5 support efficient expansion of human hematopoietic stem cells without compromising their repopulating potential. Stem Cells Dev. (2011) 20:1371–81. doi: 10.1089/scd.2010.0456
119. Yin X, Hu L, Zhang Y, Zhu C, Cheng H, Xie X, et al. PDGFB-expressing mesenchymal stem cells improve human hematopoietic stem cell engraftment in immunodeficient mice. Bone Marrow Transplant. (2020) 55:1029–1040. doi: 10.1038/s41409-019-0766-z
120. Abarrategi A, Mian SA, Passaro D, Rouault-Pierre K, Grey W, Bonnet D. Modeling the human bone marrow niche in mice: from host bone marrow engraftment to bioengineering approaches. J Exp Med. (2018) 215:729–43. doi: 10.1084/jem.20172139
121. Sommerkamp P, Mercier FE, Wilkinson AC, Bonnet D, Bourgine PE. Engineering human hematopoietic environments through ossicle and bioreactor technologies exploitation. Exper Hematol. (2020) 94:1–68. doi: 10.1016/j.exphem.2020.11.008
122. Antonelli A, Noort WA, Jaques J, de Boer B, de Jong-Korlaar R, Brouwers-Vos AZ, et al. Establishing human leukemia xenograft mouse models by implanting human bone marrow-like scaffold-based niches. Blood. (2016) 128:2949–59. doi: 10.1182/blood-2016-05-719021
123. Reinisch A, Thomas D, Corces MR, Zhang X, Gratzinger D, Hong WJ, et al. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat Med. (2016) 22:812–21. doi: 10.1038/nm.4103
124. Reinisch A, Hernandez DC, Schallmoser K, Majeti R. Generation and use of a humanized bone-marrow-ossicle niche for hematopoietic xenotransplantation into mice. Nat Protoc. (2017). 12:2169–88. doi: 10.1038/nprot.2017.088
125. Abarrategi A, Foster K, Hamilton A, Mian SA, Passaro D, Gribben J, et al. Versatile humanized niche model enables study of normal and malignant human hematopoiesis. J Clin Invest. (2017) 127:543–8. doi: 10.1172/JCI89364
126. Vaiselbuh SR, Edelman M, Lipton JM, Liu JM. Ectopic human mesenchymal stem cell-coated scaffolds in NOD/SCID mice: an in vivo model of the leukemia niche. Tissue Eng Part C Methods. (2010) 16:1523–31. doi: 10.1089/ten.tec.2010.0179
127. Bourgine PE, Fritsch K, Pigeot S, Takizawa H, Kunz L, Kokkaliaris KD, et al. Fate distribution and regulatory role of human mesenchymal stromal cells in engineered hematopoietic bone organs. iScience. (2019) 19:504–513. doi: 10.1016/j.isci.2019.08.006
128. Fritsch K, Pigeot S, Feng X, Bourgine PE, Schroeder T, Martin I, et al. Engineered humanized bone organs maintain human hematopoiesis in vivo. Exp Hematol. (2018) 61:45–51 e5. doi: 10.1016/j.exphem.2018.01.004
129. Scotti C, Piccinini E, Takizawa H, Todorov A, Bourgine P, Papadimitropoulos A, et al. Engineering of a functional bone organ through endochondral ossification. Proc Natl Acad Sci USA. (2013) 110:3997–4002. doi: 10.1073/pnas.1220108110
130. Martine LC, Holzapfel BM, McGovern JA, Wagner F, Quent VM, Hesami P, et al. Engineering a humanized bone organ model in mice to study bone metastases. Nat Protoc. (2017) 12:639–63. doi: 10.1038/nprot.2017.002
131. Lee J, Li M, Milwid J, Dunham J, Vinegoni C, Gorbatov R, et al. Implantable microenvironments to attract hematopoietic stem/cancer cells. Proc Natl Acad Sci USA. (2012) 109:19638–43. doi: 10.1073/pnas.1208384109
132. Reinisch A, Etchart N, Thomas D, Hofmann NA, Fruehwirth M, Sinha S, et al. Epigenetic and in vivo comparison of diverse MSC sources reveals an endochondral signature for human hematopoietic niche formation. Blood. (2015) 125:249–60. doi: 10.1182/blood-2014-04-572255
133. Song Y, Rongvaux A, Taylor A, Jiang T, Tebaldi T, Balasubramanian K, et al. A highly efficient and faithful MDS patient-derived xenotransplantation model for pre-clinical studies. Nat Commun. (2019) 10:366. doi: 10.1038/s41467-018-08166-x
134. Lysenko V, Wildner-Verhey van Wijk N, Zimmermann K, Weller MC, Bühler M, Wildschut MHE, et al. Enhanced engraftment of human myelofibrosis stem and progenitor cells in MISTRG mice. Blood Adv. (2020) 4:2477–88. doi: 10.1182/bloodadvances.2019001364
135. Ellegast JM, Rauch PJ, Kovtonyuk LV, Muller R, Wagner U, Saito Y, et al. Minv(16) G, and NPM1mut AMLs engraft human cytokine knock-in mice. Blood. (2016) 128:2130–4. doi: 10.1182/blood-2015-12-689356
136. Hashwah H, Bertram K, Stirm K, Stelling A, Wu CT, Kasser S, et al. The IL-6 signaling complex is a critical driver, negative prognostic factor, and therapeutic target in diffuse large B-cell lymphoma. EMBO Mol Med. (2019) 11:e10576. doi: 10.15252/emmm.201910576
137. Lindsey ES, Donaldson GW, Woodruff MF. Erythrocyte survival in normal mice and in mice with autoimmune haemolytic anaemia. Clin Exp Immunol. (1966) 1:85–98.
138. Phillips MA, Burrows JN, Manyando C, van Huijsduijnen RH, Van Voorhis WC, Wells TNC. Malaria Nat Rev Dis Primers. (2017) 3:17050. doi: 10.1038/nrdp.2017.50
139. Guzman MG, Gubler DJ, Izquierdo A, Martinez E, Halstead SB. Dengue infection. Nat Rev Dis Primers. (2016) 2:16055. doi: 10.1038/nrdp.2016.55
140. Semple JW, Rebetz J, Maouia A, Kapur R. An update on the pathophysiology of immune thrombocytopenia. Curr Opin Hematol. (2020) 27:423–429. doi: 10.1097/MOH.0000000000000612
141. Baenziger S, Tussiwand R, Schlaepfer E, Mazzucchelli L, Heikenwalder M, Kurrer MO, et al. Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2-/-gamma c-/- mice. Proc Natl Acad Sci USA. (2006) 103:15951–6. doi: 10.1073/pnas.0604493103
142. Dudek TE, No DC, Seung E, Vrbanac VD, Fadda L, Bhoumik P, et al. Rapid evolution of HIV-1 to functional CD8+ T cell responses in humanized BLT mice. Sci Transl Med. (2012) 4:143ra98. doi: 10.1126/scitranslmed.3003984
143. Strowig T, Gurer C, Ploss A, Liu YF, Arrey F, Sashihara J, et al. Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J Exp Med. (2009) 206:1423–34. doi: 10.1084/jem.20081720
144. Jaiswal S, Pazoles P, Woda M, Shultz LD, Greiner DL, Brehm MA, et al. Enhanced humoral and HLA-A2-restricted dengue virus-specific T-cell responses in humanized BLT NSG mice. Immunology. (2012) 136:334–43. doi: 10.1111/j.1365-2567.2012.03585.x
145. Seung E, Tager AM, Humoral immunity in humanized mice: a work in progress. J Infect Dis. (2013) 208 (Suppl. 2):S155–9. doi: 10.1093/infdis/jit448
146. Lang J, Kelly M, Freed BM, McCarter MD, Kedl RM, Torres RM, et al. Studies of lymphocyte reconstitution in a humanized mouse model reveal a requirement of T cells for human B cell maturation. J Immunol. (2013) 190:2090–101. doi: 10.4049/jimmunol.1202810
147. Cuss AK, Avery DT, Cannons JL, Yu LJ, Nichols KE, Shaw PJ, et al. Expansion of functionally immature transitional B cells is associated with human-immunodeficient states characterized by impaired humoral immunity. J Immunol. (2006) 176:1506–16. doi: 10.4049/jimmunol.176.3.1506
148. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol. (2014) 14:377–91. doi: 10.1038/nri3667
149. Shultz LD, Saito Y, Najima Y, Tanaka S, Ochi T, Tomizawa M, et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc Natl Acad Sci USA. (2010) 107:13022–7. doi: 10.1073/pnas.1000475107
150. Danner R, Chaudhari SN, Rosenberger J, Surls J, Richie TL, Brumeanu TD, et al. Expression of HLA class II molecules in humanized NOD.Rag1KO.IL2RgcKO mice is critical for development and function of human T and B cells. PLoS ONE. (2011) 6:e19826. doi: 10.1371/journal.pone.0019826
151. Billerbeck E, Horwitz JA, Labitt RN, Donovan BM, Vega K, Budell WC, et al. Characterization of human antiviral adaptive immune responses during hepatotropic virus infection in HLA-transgenic human immune system mice. J Immunol. (2013) 191:1753–64. doi: 10.4049/jimmunol.1201518
152. Najima Y, Tomizawa-Murasawa M, Saito Y, Watanabe T, Ono R, Ochi T, et al. Induction of WT1-specific human CD8+ T cells from human HSCs in HLA class I Tg NOD/SCID/IL2rgKO mice. Blood. (2016) 127:722–34. doi: 10.1182/blood-2014-10-604777
153. Majji S, Wijayalath W, Shashikumar S, Pow-Sang L, Villasante E, Brumeanu TD, et al. Differential effect of HLA class-I versus class-II transgenes on human T and B cell reconstitution and function in NRG mice. Sci Rep. (2016) 6:28093. doi: 10.1038/srep28093
154. Masse-Ranson G, Dusséaux M, Fiquet O, Darche S, Boussand M, Li Y, et al. Accelerated thymopoiesis and improved T-cell responses in HLA-A2/-DR2 transgenic BRGS-based human immune system mice. Eur J Immunol. (2019) 49:954–65. doi: 10.1002/eji.201848001
155. Karpel ME, Boutwell CL, Allen TM. BLT humanized mice as a small animal model of HIV infection. Curr Opin Virol. (2015) 13:75–80. doi: 10.1016/j.coviro.2015.05.002
156. Greenblatt MB, Vrbanac V, Tivey T, Tsang K, Tager AM, Aliprantis AO. Graft versus host disease in the bone marrow, liver and thymus humanized mouse model. PLoS One. (2012) 7:e44664. doi: 10.1371/journal.pone.0044664
157. Covassin L, Jangalwe S, Jouvet N, Laning J, Burzenski L, Shultz LD, et al. Human immune system development and survival of NOD-scid IL2rgamma (NSG) mice engrafted with human thymus and autologous hematopoietic stem cells. Clin Exp Immunol. (2013) 174:372–88. doi: 10.1111/cei.12180
158. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
159. Aguzzi A, Kranich J, Krautler NJ. Follicular dendritic cells: origin, phenotype, and function in health and disease. Trends Immunol. (2014) 35:105–13. doi: 10.1016/j.it.2013.11.001
160. Randall TD, Carragher DM, Rangel-Moreno J. Development of secondary lymphoid organs. Ann Rev Immunol. (2008) 26:627–50. doi: 10.1146/annurev.immunol.26.021607.090257
161. Salguero G, Daenthanasanmak A, Münz C, Raykova A, Guzmán CA, Riese P, et al. Dendritic cell-mediated immune humanization of mice: implications for allogeneic and xenogeneic stem cell transplantation. J Immunol. (2014) 192:4636–47. doi: 10.4049/jimmunol.1302887
162. Theobald SJ, Kreer C, Khailaie S, Bonifacius A, Eiz-Vesper B, Figueiredo C, et al. Repertoire characterization and validation of gB-specific human IgGs directly cloned from humanized mice vaccinated with dendritic cells and protected against HCMV. PLoS Pathog. (2020) 16:e1008560. doi: 10.1371/journal.ppat.1008560
163. Chung YS, Son JK, Choi B, Park JB, Chang J, Kim SJ. Transplantation of human spleen into immunodeficient NOD/SCID IL2Rγ(null) mice generates humanized mice that improve functional B cell development. Clin Immunol. (2015) 161:308–15. doi: 10.1016/j.clim.2015.09.001
164. Chappaz S, Flueck L, Farr AG, Rolink AG, Finke D. Increased TSLP availability restores T- and B-cell compartments in adult IL-7–deficient mice. Blood. (2007) 110:3862–70. doi: 10.1182/blood-2007-02-074245
165. Chappaz S, Finke D, The IL-7 signaling pathway regulates lymph node development independent of peripheral lymphocytes. J Immunolo. (2010) 184:3562–9. doi: 10.4049/jimmunol.0901647
166. Yaguchi T, Kobayashi A, Inozume T, Morii K, Nagumo H, Nishio H, et al. Human PBMC-transferred murine MHC class I/II-deficient NOG mice enable long-term evaluation of human immune responses. Cell Mol Immunol. (2018) 15:953–62. doi: 10.1038/cmi.2017.106
167. Brehm MA, Kenney LL, Wiles MV, Low BE, Tisch RM, Burzenski L, et al. Lack of acute xenogeneic graft- versus-host disease, but retention of T-cell function following engraftment of human peripheral blood mononuclear cells in NSG mice deficient in MHC class I and II expression. Faseb j. (2019) 33:3137–51. doi: 10.1096/fj.201800636R
168. Kalscheuer H, Danzl N, Onoe T, Faust T, Winchester R, Goland R, et al. A model for personalized in vivo analysis of human immune responsiveness. Sci Transl Med. (2012) 4:125ra30. doi: 10.1126/scitranslmed.3003481
169. Kuruvilla JG, Troyer RM, Devi S, Akkina R. Dengue virus infection and immune response in humanized RAG2(-/-)gamma(c)(-/-) (RAG-hu) mice. Virology. (2007) 369:143–52. doi: 10.1016/j.virol.2007.06.005
170. Aspord C, Pedroza-Gonzalez A, Gallegos M, Tindle S, Burton EC, Su D, et al. Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J Exp Med. (2007) 204:1037–47. doi: 10.1084/jem.20061120
171. Wang M, Yao LC, Cheng M, Cai D, Martinek J, Pan CX, et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. Faseb J. (2018) 32:1537–1549. doi: 10.1096/fj.201700740R
172. Nguyen R, Patel AG, Griffiths LM, Dapper J, Stewart EA, Houston J, et al. Next-generation humanized patient-derived xenograft mouse model for pre-clinical antibody studies in neuroblastoma. Cancer Immunol Immunother. (2020) doi: 10.1007/s00262-020-02713-6. [Epub ahead of print].
173. Byrne AT, Alferez DG, Amant F, Annibali D, Arribas J, Biankin AV, et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer. (2017) 17:254–268. doi: 10.1038/nrc.2016.140
174. Myburgh R, Kiefer JD, Russkamp NF, Magnani CF, Nuñez N, Simonis A, et al. Anti-human CD117 CAR T-cells efficiently eliminate healthy and malignant CD117-expressing hematopoietic cells. Leukemia. (2020) 34:2688–703. doi: 10.1038/s41375-020-0818-9
175. Biernacki MA, Foster KA, Woodward KB, Coon ME, Cummings C, Cunningham TM, et al. CBFB-MYH11 fusion neoantigen enables T cell recognition and killing of acute myeloid leukemia. J Clin Invest. (2020) 130:5127–41. doi: 10.1172/JCI137723
176. Grompe M, Strom S. Mice with human livers. Gastroenterology. (2013) 145:1209–14. doi: 10.1053/j.gastro.2013.09.009
177. Billerbeck E, Mommersteeg MC, Shlomai A, Xiao JW, Andrus L, Bhatta A, et al. Humanized mice efficiently engrafted with fetal hepatoblasts and syngeneic immune cells develop human monocytes and NK cells. J Hepatol. (2016) 65:334–43. doi: 10.1016/j.jhep.2016.04.022
178. Dandri M, Burda MR, Török E, Pollok JM, Iwanska A, Sommer G, et al. Repopulation of mouse liver with human hepatocytes and in vivo infection with hepatitis B virus. Hepatology. (2001) 33:981–8. doi: 10.1053/jhep.2001.23314
179. Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A, et al. Hepatitis C virus replication in mice with chimeric human livers. Nat Med. (2001) 7:927–33. doi: 10.1038/90968
180. Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, et al. Robust expansion of human hepatocytes in Fah-/-/Rag2-/-/Il2rg-/- mice. Nat Biotechnol. (2007) 25:903–10. doi: 10.1038/nbt1326
181. Bissig KD, Le TT, Woods NB, Verma IM. Repopulation of adult and neonatal mice with human hepatocytes: a chimeric animal model. Proc Natl Acad Sci USA. (2007) 104:20507–11. doi: 10.1073/pnas.0710528105
182. Bility MT, Cheng L, Zhang Z, Luan Y, Li F, Chi L, et al. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog. (2014) 10:e1004032. doi: 10.1371/journal.ppat.1004032
183. Dusséaux M, Masse-Ranson G, Darche S, Ahodantin J, Li Y, Fiquet O, et al. viral load affects the immune response to HBV in mice with humanized immune system and liver. Gastroenterology. (2017) 153:1647–61.e9. doi: 10.1053/j.gastro.2017.08.034
Keywords: humanized mice, immunity, hematopoiesis, infectious diseases, cancer, immunotherapies
Citation: Martinov T, McKenna KM, Tan WH, Collins EJ, Kehret AR, Linton JD, Olsen TM, Shobaki N and Rongvaux A (2021) Building the Next Generation of Humanized Hemato-Lymphoid System Mice. Front. Immunol. 12:643852. doi: 10.3389/fimmu.2021.643852
Received: 18 December 2020; Accepted: 27 January 2021;
Published: 22 February 2021.
Edited by:
Tim Willinger, Karolinska Institutet, SwedenReviewed by:
Christian Muenz, University of Zurich, SwitzerlandCopyright © 2021 Martinov, McKenna, Tan, Collins, Kehret, Linton, Olsen, Shobaki and Rongvaux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony Rongvaux, cm9uZ3ZhdXhAZnJlZGh1dGNoLm9yZw==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.