 Isabelle Serr
Isabelle Serr Maria Kral
Maria Kral Martin G. Scherm
Martin G. Scherm Carolin Daniel
Carolin Daniel
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 18 February 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.643544
This article is part of the Research TopicAdvances in Human Immune System (HIS) Mouse Models for Studying Human Hematopoiesis and Cancer ImmunotherapyView all 19 articles
Immunodeficient mice engrafted with a functional human immune system [Human immune system (HIS) mice] have paved the way to major advances for personalized medicine and translation of immune-based therapies. One prerequisite for advancing personalized medicine is modeling the immune system of individuals or disease groups in a preclinical setting. HIS mice engrafted with peripheral blood mononuclear cells have provided fundamental insights in underlying mechanisms guiding immune activation vs. regulation in several diseases including cancer. However, the development of Graft-vs.-host disease restrains relevant long-term studies in HIS mice. Alternatively, engraftment with hematopoietic stem cells (HSCs) enables mimicking different disease stages, however, low frequencies of HSCs in peripheral blood of adults impede engraftment efficacy. One possibility to overcome those limitations is the use of patient-derived induced pluripotent stem cells (iPSCs) reprogrammed into HSCs, a challenging process which has recently seen major advances. Personalized HIS mice bridge research in mice and human diseases thereby facilitating the translation of immunomodulatory therapies. Regulatory T cells (Tregs) are important mediators of immune suppression and thereby contribute to tumor immune evasion, which has made them a central target for cancer immunotherapies. Importantly, studying Tregs in the human immune system in vivo in HIS mice will help to determine requirements for efficient Treg-targeting. In this review article, we discuss advances on personalized HIS models using reprogrammed iPSCs and review the use of HIS mice to study requirements for efficient targeting of human Tregs for personalized cancer immunotherapies.
With the discovery of checkpoint inhibitors and more recently, the use of chimeric antigen-receptor T cells, immunotherapies have taken center stage in oncology. The common goal for cancer immunotherapies is to boost the immune system to attack and destroy cancerous cells. The gold-standard for pre-clinical testing of cancer therapies has long been the use of xenograft models, where human cancer cell lines are engrafted into immunodeficient hosts. However, both development and evaluation of immunotherapies requires the presence of a functional immune system closely mirroring the immune reaction in the human disease. Immunodeficient mice engrafted with a functional human immune system [human immune system (HIS) mice] have opened up new ways of evaluating immunotherapies directly in vivo in a pre-clinical setting. One main obstacle remaining in cancer therapy is intra-tumor heterogeneity. The use of personalized medicine offers a promising approach to tackle this major hurdle and HIS mice engrafted with patient-derived hematopoietic cells are an important tool for the development of personalized medicine.
Since the discovery that severe combined immunodeficiency (Prkcdscid or SCID) mice, engrafted with human hematopoietic stem cells (HSCs) or human peripheral blood mononuclear cells (PBMCs), can develop a human immune system (1), researchers are constantly working on improving xenoengraftment in such HIS mice to study human diseases. Advancements have been made especially regarding background strain, immunodeficiency mutation as well as engraftment method and material, thereby considerably improving engraftment efficacy (2). For example, mice on the NOD background present with the highest engraftment efficacy due to a polymorphism in the gene Sirpa, encoding the signal regulatory protein alpha (Sirpa) that shows enhanced interaction with human CD47 on hematopoietic cells, thereby preventing their phagocytosis by macrophages (3).
Moreover, to overcome human T cell xeno-reactivity directed against murine MHC molecules, strains of immunodeficient mice lacking murine MHC class I and class II were developed (4, 5). We have used murine MHC class II deficient NOD-scid IL2Rgammanull (NSG)-HLA-DQ8 transgenic mice engrafted with fresh human cord blood (CB) HSCs and demonstrated a high engraftment efficacy in peripheral blood of these animals (6). Importantly, we identified autoreactive disease-relevant insulin-specific CD4+T cells, indicating positive selection on HLA-DQ8 molecules in the thymus of humanized mice (6). The development of T cells with a TCR repertoire recapitulating that of the human donor is essential to analyze T cell responses, also in the setting of cancer immunotherapy. In this regard, in a mouse model for prostate adenocarcinoma, tumor-infiltrating regulatory T cells (Tregs) showed a significant enrichment for distinct TCR specificities targeting an antigen present in healthy pancreatic tissue and not specific to the tumor (7). Therefore, the use of HIS mice with transgenic expression of human HLA molecules will be important to permit for accurate representation of TCR repertoires to study T cell responses to the tumor.
In order to study tumor related immune responses in the human immune system in vivo, HIS mice engrafted with cell line-derived (CDX), or patient-derived xenografted tumors (PDXs) have been established (8, 9). Importantly, MART1-TCR-transgenic T cells from humanized mice reconstituted with fetal thymic tissue and human HSCs were able to mount efficient HLA/antigen-dependent anti-melanoma immune responses also after transfer into tumor-bearing recipients (10). Among others, especially the potent checkpoint inhibition with anti-PD1 humanized antibodies has been tested in HIS mice bearing various types of tumors (8, 9, 11), highlighting the value of PDX engrafted HIS models for cancer immunotherapies.
Despite the therapeutic advances indicated above, some hurdles remain which hinder efficient and safe implementation of cancer immunotherapies to the patient. During disease progression, genomic instability leads to genetic variations within tumor cells resulting in various distinct populations of cancer cells (12). This intra-tumor heterogeneity is a major obstacle for the success of efficient immunotherapies since it fosters resistance to therapy and leads to highly variable treatment outcomes thereby calling for personalized strategies.
The potential of personalized HIS mouse models for the analysis of patient specific immune responses was demonstrated in a PDX-bearing HIS mouse model based on NSG mice engrafted with autologous tumor-infiltrating T cells and tumor cells from the same patient. Importantly, the autologous T cell transfer was successful in eradicating tumors only in mice generated with material from patients that show a positive autologous cell transfer response in the clinic (13).
To investigate complex immune responses more closely in a preclinical personalized setting, the engraftment of HIS mice with autologous hematopoietic cells and tumor cells from the same patient is required. This can be achieved by engraftment with patient-derived PBMCs. However, such mice are not useful for long-term studies as they can develop severe Graft-vs.-Host-Disease (GvHD) within few weeks upon engraftment. Alternatives for long-term studies are the engraftment with patient-derived HSCs or HSCs originating from fetal liver or CB. Here, engraftment with patient-derived HSCs allows for positive selection of T and B cells based on murine antigens in the thymus and bone marrow, therefore limiting GvHD reactions and further enabling MHC compatibility with the autologous tumor tissue. One drawback of HSC engraftment, however, are low frequencies of HSCs in the peripheral blood of adults without mobilization, which impede an effective reconstitution of the human immune system in HIS mice. Of note, in 2006, a group led by Shinya Yamanaka generated pluripotent stem cells from adult cells. The discovery of these induced pluripotent stem cells (iPSCs) has thenceforward substantially revolutionized the field of translational research and personalized medicine (14) and enabled an unlimited source for the derivation of HSCs (15, 16).
The discovery that iPSCs are able to self-renew and differentiate into any cell type by introducing the reprogramming factors Oct4, Sox2, cMyc, and Klf4 (14, 17) has enabled the development of novel therapeutic strategies in personalized and regenerative medicine. Noteworthy, iPSCs can be derived from different sources of somatic cells, thereby evading the limitations of using primary, patient-derived disease affected cells for e.g., disease modeling (18) (Figure 1).
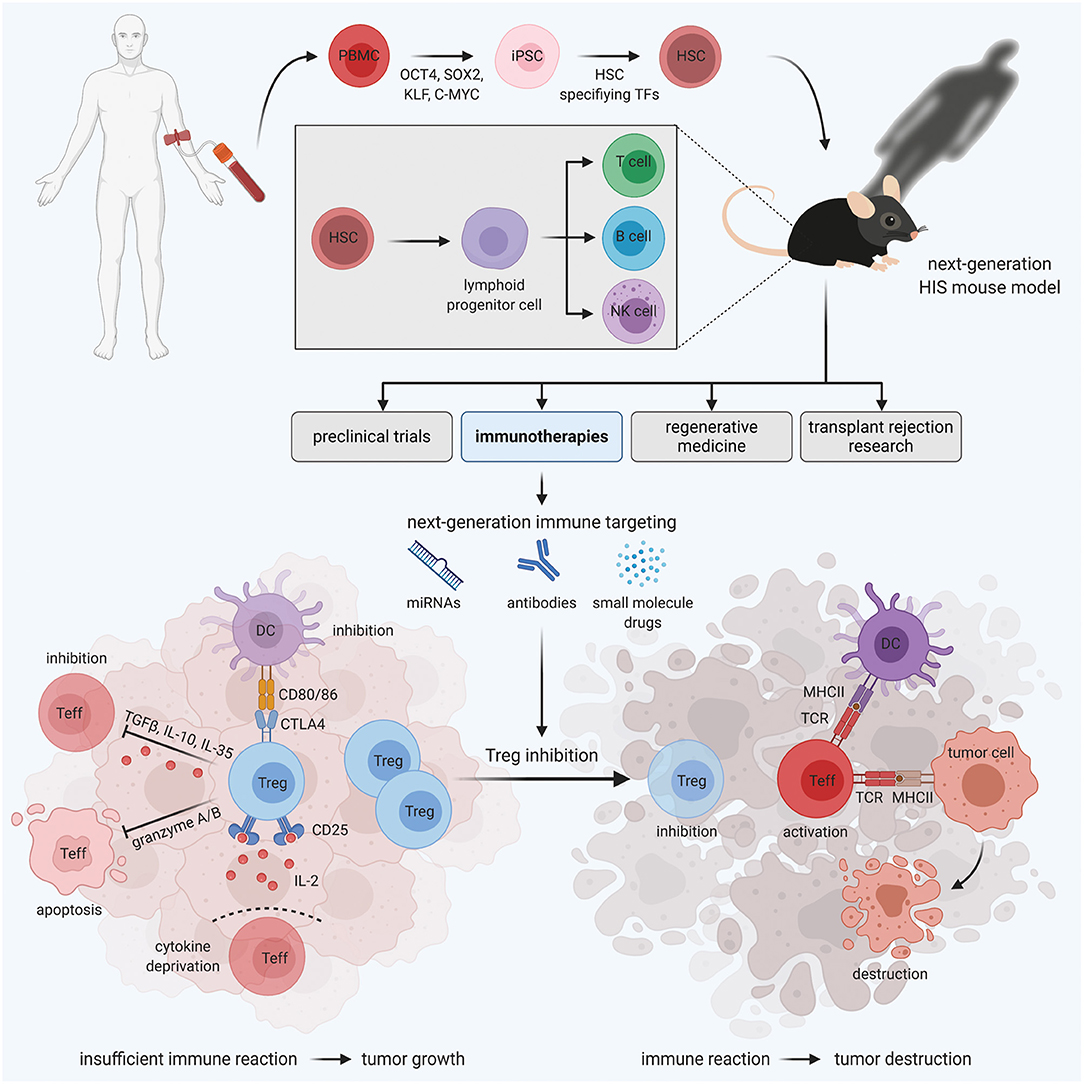
Figure 1. HIS mouse models for personalized Treg-based cancer immunotherapies. Immunodeficient mice engrafted with human PBMC-derived HSCs develop a functional human immune system. These HIS mice are valuable tools for various areas of medical research, including preclinical trials, regenerative medicine, transplant rejection research, and immunotherapies. In the field of immunotherapies, next-generation HIS mouse models enable the development and testing of novel immune targeting strategies, aiming at Treg inhibition to boost the immune reaction against the tumor, in a human immune system in vivo.
Yet, de novo generation of functional HSCs from iPSCs for efficient in vivo engraftment and multi-lineage potential remains challenging due to the complex nature of hematopoietic ontogeny (19). Recently, methods for the generation of self-renewing hematopoietic stem and progenitor cells with multi-lineage potential after engraftment into immunodeficient hosts have been developed. Although analysis was largely limited to the bone marrow, T and B cells, as well as myeloid cells were also detected in spleen, thymus and peripheral blood, showing a diverse TCR repertoire and functional antibody responses (16, 20). These initial studies highlight the potential use for the generation of HIS mice with iPSC-derived HSCs (20). Furthermore, hPSCs generated from iPSCs from patients with Diamond-Blackfan anemia (DBA) were used to enable a new therapeutic pathway for the treatment of DBA (21) and immunodeficient mice reconstituted with patient-derived iPSCs were used to develop a drug-testing system for chronic myelomonocytic leukemia (22). Even though still low in number, such studies highlight the vast opportunities and potential therapeutic applications that are offered by iPSC technology, which have to be further explored and improved for future investigations.
Personalized HIS mice reconstituted with tumor cells and autologous patient-derived hematopoietic cells from the same donor are especially important, since they will help to mirror the immunological tumor microenvironment. Importantly, the tumor microenvironment contains immunosuppressive immune subsets [reviewed in (23)], including Tregs that promote tumor immune evasion and hence impede effective antitumor immune responses [reviewed in (24)].
Tregs are a specialized suppressive subtype of CD4+T cells that controls the immune response in the periphery and prevents adverse immune reactions and autoimmunity (25). They are characterized by the high expression of the high-affinity interleukin-2 receptor alpha-chain (CD25) and the expression of the transcription factor Foxp3, which is the master regulator of their phenotype and function (26–28). Mutations in the Foxp3 gene lead to defects in Treg development and function as illustrated by severe multiorgan autoimmunity in patients with the Immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome (29) and mice with Scurfy mutations (30). While defects in Treg function and reduced frequencies are associated with autoimmune disorders, increased Treg infiltration into solid tumors and the resulting suppression of effector immune cells are associated with poor prognosis in several types of cancer (31–35). Accordingly, the manipulation of Tregs has gained interest in the field of cancer immunotherapy. However, many aspects of Treg-mediated immune suppression in tumors remain to be determined. Importantly, HIS mice reconstituted with CB HSCs or PBMCs display comparable frequencies of Tregs in various tissues as non-humanized mice (6, 11, 36–40), making them a suitable tool to study human Tregs in vivo in a preclinical model (Figure 1). Notably, the development of immune cells in NSG mice is skewed to the lymphoid lineage, with cells of the myeloid lineage being under represented, due to limited cross-reactivity of murine cytokines with human hematopoietic cells [reviewed in (41)]. Therefore, novel humanized mouse strains such as NSG-SGM3 mice (42) and others (43) that transgenically express human cytokines and thereby support myeloid cell development have been developed. Importantly, it was demonstrated that NSG-SGM3 mice further support the development of Foxp3+Tregs (44), thereby highlighting the importance of considering different HIS mouse models as basis for pre-clinical studies.
It is now established, that Tregs take residence in various tissues, where they express tissue specific signature genes and exert key non-canonical functions (45–49). Notably, a tissue specific signature has been likewise identified for tumor-infiltrating Tregs. Importantly, the investigation of gene expression of Tregs isolated from breast cancer, colorectal cancer, non-small cell lung cancer and hepatocellular carcinoma in comparison to Tregs from adjacent healthy tissue or peripheral blood identified a tumor-specific Treg gene expression signature (50–52). Some of these signature genes (e.g., CTLA4, GITR, CCR4) are likewise present on peripheral Tregs, whereas their expression is increased in tumor-infiltrating Tregs. Other tumor-specific Treg signature genes (e.g., CCR8, FCRL3, IL1R2) are exclusively expressed on tumor-infiltrating Tregs and absent in their peripheral counterparts. However, the functional consequence of this gene signature in tumor-infiltrating Tregs remains to be determined.
Checkpoint inhibitors have taken center stage in cancer immunotherapy research, since they block signaling through immune inhibitory molecules, especially programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA4) (53, 54). Many tumors promote immune evasion by expression of PD-L1, a ligand for PD-1, which is in turn expressed on activated T cells. The engagement of PD-1 by PD-L1 is highly immunosuppressive and leads to T cell anergy and apoptosis. Accordingly, blocking PD-1/PD-L1 signaling has shown great efficacy in various cancers, however in a large proportion of patients PD-1/PD-L1 blockade shows limited success, highlighting the importance of understanding underlying mechanisms of checkpoint inhibitor blockade as well as defining biomarkers to predict treatment success (39). In addition to effector T cells, PD-1 is likewise expressed on Tregs and multiple studies indicate, that PD-1 blockade can increase Treg proliferation and suppression, which could account for the differences in treatment outcome (53). In this regard, the ratio of PD-1+CD8+T cells/PD-1+Tregs in the tumor microenvironment can predict the efficacy of PD-1 blocking therapy (39). While PD-1 blockade has been studied extensively in HIS mice, only few studies investigated the specific effect on Tregs. In line with highly variable treatment outcomes in patients, two studies did not observe changes in tumor-infiltrating Treg frequencies (11, 38), while one study using a combination of anti-PD-1 antibody and CD137 antibody therapies reported an increase in CD8/Treg ratio in tumors of HIS mice xenotransplanted with gastric carcinoma and engrafted with autologous PBMCs from the same donor (55). These studies highlight the strength of personalized HIS mice using xenotransplanted tumors and autologous immune cells for the assessment of treatment outcome.
The identification of tumor-related Treg signature genes is an important step toward more specific targeting of tumor Tregs without affecting the peripheral Treg pool which could potentially lead to autoimmune reactions. CTLA4 mediated tolerization of antigen-presenting cells is an important mechanism of suppression by Tregs. Importantly, CTLA4 blockade has been studied extensively as checkpoint inhibitor in cancer immunotherapy, since CTLA4 is often upregulated by tumor cells for immune evasion (54). However, the anti-tumor effects of anti-CTLA4 treatment largely rely on the antibody-dependent cellular cytotoxicity (ADCC) mediated depletion of tumor-infiltrating Tregs (56, 57). Here, HIS mice offer an important advantage over classical non-humanized mouse models in the pre-clinical assessment of depleting antibodies. ADCC relies on the interaction of the human IgG Fc region on depleting antibodies with human immune cells and can therefore be studied accurately only in the presence of these cells (58, 59). Anti-CTLA4 antibodies were used to successfully deplete Tregs in fetal liver engrafted HIS mice treated with low-dose IL2 to demonstrate that critical side effects and toxicity of high-dose IL2 treatment are largely dependent on the depletion of Tregs (40). This study highlights the usefulness of HIS mice to study not only treatment efficacy, but also critical side effects of cancer immunotherapy.
Like CTLA4 the chemokine receptor type 4 (CCR4), albeit being expressed on peripheral Tregs, is further upregulated on tumor-infiltrating Tregs, while additionally being expressed on different types of cancer cells. To test anti-CCR4 treatment in a pre-clinical setting, a humanized mouse model for lymphoma based on CCR4+ lymphoma bearing NOG mice reconstituted with human PBMCs was used (60). The treatment with an anti-CCR4 antibody induced robust ADCC leading to reduced tumor mass, accompanied by decreased Treg frequencies in the tumor (60). The latter finding suggests that anti-CCR4 treatment might be more generally applicable also to CCR4-negative tumors, by targeting tumor-infiltrating Tregs. Accordingly, a Phase Ia clinical trial demonstrated efficient Treg depletion by an anti-CCR4 antibody in patients with CCR4-negative solid tumors resulting in stable disease in half of the patients 12 weeks after treatment start (61). However, CCR4 is likewise expressed on peripheral Tregs, albeit at lower levels, leading to the simultaneous depletion of peripheral Tregs by anti-CCR4 treatment. Tumor cells and cells in the tumor microenvironment secrete high amounts of CCL22, a ligand for CCR4, in various cancers (32, 62, 63). Therefore, small molecule CCR4 antagonists might be a safer alternative to block Treg migration to the tumor microenvironment without Treg depletion.
An alternative to Treg depletion is the manipulation of their functional properties. In this regard, glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR) has been studied extensively as a target for cancer immunotherapies. GITR is a coreceptor, which is expressed at low levels on naïve CD4 and CD8 T cells and is upregulated upon activation with Foxp3+Tregs harboring the highest levels of GITR (64, 65). Importantly, targeting GITR for cancer immunotherapy became of interest with the discovery that an agonist anti-GITR antibody could break self-tolerance in mice by making effector T cells resistant to Treg suppression (66). Accordingly, anti-GITR antibody treatment could induce strong anti-tumor immunity in mice (67–69). While these studies demonstrated that the anti-GITR antibody acts mainly by enhancing effector T cell frequencies through avoidance of Treg mediated suppression, the exact mechanism remains unclear. In addition to rendering effector T cells resistant to Treg mediated suppression, agonist anti-GITR treatment was suggested to likewise enhance the effector T cell: Treg ratio in the tumor (67, 70). This change was attributed to the agonist anti-GITR antibody rendering Tregs unstable, causing them to lose Foxp3 expression (67), or to Treg depletion (70) in different studies. In NSG mice reconstituted with human hCD34+HSCs and grafted with melanoma tumors the treatment increased the effector T cell: Treg ratio in the spleen and the tumor, while remaining tumor-infiltrating Tregs showed reduced expression of activation markers such as ICOS (70). Overall, although the treatment was not able to clear the tumor completely, treated mice exhibited significantly reduced tumor growth (70). The exact mechanisms leading to the enhanced effector T cell: Treg ratio in this particular study remain to be defined. The loss of activation markers such as ICOS observed in the anti-GITR study in HIS mice could indicate that, in addition to Treg depletion, remaining Tregs are rendered phenotypically unstable by the treatment.
Destabilization of Tregs is a promising approach for Treg-based immunotherapy and mechanisms of Treg destabilization have been studied in the setting of autoimmune diseases. Treg stability is mainly mediated by epigenetic mechanisms, most importantly the demethylation of the conserved non-coding sequence 2 (CNS2) in the Foxp3 locus (71). We were able to show that microRNA (miRNA) 142-3p contributes to Treg instability during progression of Type 1 Diabetes (T1D) by targeting TET2, a molecule that can actively demethylate DNA (37, 72, 73). MiRNAs are small non-coding RNAs that can sequence-specifically inhibit their target mRNAs, thereby regulating complex cellular states, such as T cell activation, which makes them important targets for immunotherapy (74). Importantly, miRNA modulation has been studied extensively in the setting of autoimmunity and infection. Accordingly, a miR122 inhibitor is currently being tested in clinical trials for hepatitis C virus (HCV) infections, highlighting the feasibility of targeting miRNAs for immune modulation in human diseases (75). Regarding miRNA modulation for Treg targeting, we used NSG mice reconstituted with PBMCs and were able to demonstrate that the blockade of miRNAs that impact Treg induction or stability in vivo enhances human Treg frequencies both in the periphery and in the pancreas (36, 37, 76). Lessons learned from the autoimmune setting could be used for cancer immunotherapy. Enhancing the function of these miRNAs using miRNA mimics could induce immune responses against cancer cells by reducing Treg-mediated suppression mechanisms. Importantly, miRNAs that induce autoimmunity when overexpressed in lymphocytes, can still have tumor-promoting properties because of their expression in other cell types (e.g., miR17~92 cluster) (77, 78). Therefore, the implementation of miRNA modulation for immune targeting in cancer therapy relies on the development of targeted approaches that allow the modulation of these miRNAs specifically in immune cells or even specific subsets. Importantly, miRNA delivery systems are currently intensively studied [reviewed in (79)], as miRNA instability and degradation still hinder the development of a successful system. To improve the delivery of miRNAs in a preclinical setting that also enables the testing of drug safety and efficiency, HIS mouse models offer a suitable platform prior the translation to humans.
The perpetual improvements of HIS mouse models have enabled the design of personalized immunotherapies for various diseases including cancer and the evaluation of their efficacy prior to the translation to the human setting. Using patient-specific HIS mice will enable us to better understand the heterogeneity of immune responses and advance the development of novel personalized immunotherapies for the treatment of cancer. Even though still in its infancy, the use of iPSC-derived HSCs for engraftment could be an important step toward personalized HIS models.
Additionally, the immunosuppressive tumor microenvironment influences antitumor responses and the efficiency of cancer immunotherapies. Targeting Tregs has therefore become an important tool in cancer immunotherapy and has been studied extensively in murine models. In order to target Tregs most efficiently in human diseases the development of novel models depicting the immune reaction of the human disease is pivotal. HIS mouse models offer the opportunity to study the interaction of human immune cells, including Tregs, with human tumors directly in vivo.
Overall, current advances in establishing a “truly” humanized mouse model for the development of immunotherapies and drug delivery systems will guide personalized medicine approaches further to the translation to the clinic.
IS and MK reviewed the literature and wrote the manuscript. MGS reviewed the manuscript and prepared the figure using BioRender. CD reviewed the literature and contributed to the conceptualization of the manuscript. All authors contributed to the article and approved the submitted version.
CD holds a professorship grant from the Excellence Program for Outstanding Female Scientists from the Helmholtz Association, was supported by a Research Group at Helmholtz Zentrum München, the German Center for Diabetes Research (DZD), through a membership in the CRC1054 of the Deutsche Forschungsgemeinschaft (B11), and is funded through an EFSD award supported by EFSD/JDRF/Lilly European Programme in Type 1 Diabetes Research. IS was supported by a Research Grant of the Deutsche Forschungsgemeinschaft (DFG, SE 3036/2-1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank all former and current members of our group for valuable input and helpful discussion.
1. Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature. (1988) 335:256–9. doi: 10.1038/335256a0
2. Shultz LD, Keck J, Burzenski L, Jangalwe S, Vaidya S, Greiner DL, et al. Humanized mouse models of immunological diseases and precision medicine. Mamm Genome. (2019) 30:123–42. doi: 10.1007/s00335-019-09796-2
3. Kwong LS, Brown MH, Barclay AN, Hatherley D. Signal-regulatory protein alpha from the NOD mouse binds human CD47 with an exceptionally high affinity–implications for engraftment of human cells. Immunology. (2014) 143:61–7. doi: 10.1111/imm.12290
4. Brehm MA, Kenney LL, Wiles MV, Low BE, Tisch RM, Burzenski L, et al. Lack of acute xenogeneic graft- versus-host disease, but retention of T-cell function following engraftment of human peripheral blood mononuclear cells in NSG mice deficient in MHC class I and II expression. FASEB J. (2019) 33:3137–51. doi: 10.1096/fj.201800636R
5. Yaguchi T, Kobayashi A, Inozume T, Morii K, Nagumo H, Nishio H, et al. Human PBMC-transferred murine MHC class I/II-deficient NOG mice enable long-term evaluation of human immune responses. Cell Mol Immunol. (2018) 15:953–62. doi: 10.1038/cmi.2017.106
6. Serr I, Furst RW, Achenbach P, Scherm MG, Gokmen F, Haupt F, et al. Type 1 diabetes vaccine candidates promote human Foxp3(+)Treg induction in humanized mice. Nat Commun. (2016) 7:10991. doi: 10.1038/ncomms10991
7. Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, et al. Aire-dependent thymic development of tumor-associated regulatory T cells. Science. (2013) 339:1219–24. doi: 10.1126/science.1233913
8. Choi B, Lee JS, Kim SJ, Hong D, Park JB, Lee KY. Anti-tumor effects of anti-PD-1 antibody, pembrolizumab, in humanized NSG PDX mice xenografted with dedifferentiated liposarcoma. Cancer Lett. (2020) 478:56–69. doi: 10.1016/j.canlet.2020.02.042
9. Wang M, Yao LC, Cheng M, Cai D, Martinek J, Pan CX, et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. (2018) 32:1537–49. doi: 10.1096/fj.201700740R
10. Hu Z, Xia J, Fan W, Wargo J, Yang YG. Human melanoma immunotherapy using tumor antigen-specific T cells generated in humanized mice. Oncotarget. (2016) 7:6448–59. doi: 10.18632/oncotarget.7044
11. Capasso A, Lang J, Pitts TM, Jordan KR, Lieu CH, Davis SL, et al. Characterization of immune responses to anti-PD-1 mono and combination immunotherapy in hematopoietic humanized mice implanted with tumor xenografts. J Immunother Cancer. (2019) 7:37. doi: 10.1186/s40425-019-0518-z
12. Dagogo-Jack I, Shaw AT, Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. (2018) 15:81–94. doi: 10.1038/nrclinonc.2017.166
13. Jespersen H, Lindberg MF, Donia M, Soderberg EMV, Andersen R, Keller U, et al. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat Commun. (2017) 8:707. doi: 10.1038/s41467-017-00786-z
14. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
15. Ferreira AF, Calin GA, Picanco-Castro V, Kashima S, Covas DT, de Castro FA. Hematopoietic stem cells from induced pluripotent stem cells-considering the role of microRNA as a cell differentiation regulator. J Cell Sci. (2018) 131:jcs203018. doi: 10.1242/jcs.203018
16. Tan YT, Ye L, Xie F, Beyer AI, Muench MO, Wang J, et al. Respecifying human iPSC-derived blood cells into highly engraftable hematopoietic stem and progenitor cells with a single factor. Proc Natl Acad Sci USA. (2018) 115:2180–5. doi: 10.1073/pnas.1718446115
17. Takahashi K, Okita K, Nakagawa M, Yamanaka S. Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc. (2007) 2:3081–9. doi: 10.1038/nprot.2007.418
18. Elitt MS, Barbar L, Tesar PJ. Drug screening for human genetic diseases using iPSC models. Hum Mol Genet. (2018) 27:R89–98. doi: 10.1093/hmg/ddy186
19. Vo LT, Daley GQ. De novo generation of HSCs from somatic and pluripotent stem cell sources. Blood. (2015) 125:2641–8. doi: 10.1182/blood-2014-10-570234
20. Sugimura R, Jha DK, Han A, Soria-Valles C, da Rocha EL, Lu YF, et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature. (2017) 545:432–8. doi: 10.1038/nature22370
21. Doulatov S, Vo LT, Chou SS, Kim PG, Arora N, Li H, et al. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell. (2013) 13:459–70. doi: 10.1016/j.stem.2013.09.002
22. Taoka K, Arai S, Kataoka K, Hosoi M, Miyauchi M, Yamazaki S, et al. Using patient-derived iPSCs to develop humanized mouse models for chronic myelomonocytic leukemia and therapeutic drug identification, including liposomal clodronate. Sci Rep. (2018) 8:15855. doi: 10.1038/s41598-018-34193-1
23. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. (2008) 27:5904–12. doi: 10.1038/onc.2008.271
24. Tormoen GW, Crittenden MR, Gough MJ. Role of the immunosuppressive microenvironment in immunotherapy. Adv Radiat Oncol. (2018) 3:520–6. doi: 10.1016/j.adro.2018.08.018
25. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. (1995) 155:1151–64.
26. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
27. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
28. Roncador G, Brown PJ, Maestre L, Hue S, Martinez-Torrecuadrada JL, Ling KL, et al. Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single-cell level. Eur J Immunol. (2005) 35:1681–91. doi: 10.1002/eji.200526189
29. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27:20–21. doi: 10.1038/83713
30. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko S-A, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. (2001) 27:68–73. doi: 10.1038/83784
31. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol. (2006) 24:5373–80. doi: 10.1200/JCO.2006.05.9584
32. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. (2004) 10:942–9. doi: 10.1038/nm1093
33. Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. (2007) 132:2328–39. doi: 10.1053/j.gastro.2007.03.102
34. Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. (2009) 69:2000–9. doi: 10.1158/0008-5472.CAN-08-2360
35. Shou J, Zhang Z, Lai Y, Chen Z, Huang J, Worse outcome in breast cancer with higher tumor-infiltrating FOXP3+ Tregs: a systematic review and meta-analysis. BMC Cancer. (2016) 16:687. doi: 10.1186/s12885-016-2732-0
36. Serr I, Scherm MG, Zahm AM, Schug J, Flynn VK, Hippich M, et al. A miRNA181a/NFAT5 axis links impaired T cell tolerance induction with autoimmune type 1 diabetes. Sci Transl Med. (2018) 10:eaag1782. doi: 10.1126/scitranslmed.aag1782
37. Scherm MG, Serr I, Zahm AM, Schug J, Bellusci S, Manfredini R, et al. miRNA142-3p targets Tet2 and impairs Treg differentiation and stability in models of type 1 diabetes. Nat Commun. (2019) 10:5697. doi: 10.1038/s41467-019-13587-3
38. Ma SD, Xu X, Jones R, Delecluse HJ, Zumwalde NA, Sharma A, et al. PD-1/CTLA-4 blockade inhibits epstein-barr virus-induced lymphoma growth in a cord blood humanized-mouse model. PLoS Pathog. (2016) 12:e1005642. doi: 10.1371/journal.ppat.1005642
39. Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. (2020) 21:1346–58. doi: 10.1038/s41590-020-0769-3
40. Li Y, Strick-Marchand H, Lim AI, Ren J, Masse-Ranson G, Dan L, et al. Regulatory T cells control toxicity in a humanized model of IL-2 therapy. Nat Commun. (2017) 8:1762. doi: 10.1038/s41467-017-01570-9
41. Theocharides AP, Rongvaux A, Fritsch K, Flavell RA, Manz MG. Humanized hemato-lymphoid system mice. Haematologica. (2016) 101:5–19. doi: 10.3324/haematol.2014.115212
42. Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF, and IL-3. Leukemia. (2010) 24:1785–8. doi: 10.1038/leu.2010.158
43. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. (2014) 32:364–72. doi: 10.1038/nbt.2858
44. Billerbeck E, Barry WT, Mu K, Dorner M, Rice CM, Ploss A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rγ(null) humanized mice. Blood. (2011) 117:3076–86. doi: 10.1182/blood-2010-08-301507
45. Becker M, Serr I, Salb VK, Ott VB, Mengel L, Bluher M, et al. Short-term cold exposure supports human Treg induction in vivo. Mol Metab. (2019) 28:73–82. doi: 10.1016/j.molmet.2019.08.002
46. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
47. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. (2012) 486:549–53. doi: 10.1038/nature11132
48. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. (2009) 15:930–9. doi: 10.1038/nm.2002
49. Kalin S, Becker M, Ott VB, Serr I, Hosp F, Mollah MMH, et al. A Stat6/Pten axis links regulatory T cells with adipose tissue function. Cell Metab. (2017) 26:475–92.e7. doi: 10.1016/j.cmet.2017.08.008
50. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. (2016) 45:1122–34. doi: 10.1016/j.immuni.2016.10.032
51. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. (2016) 45:1135–47. doi: 10.1016/j.immuni.2016.10.021
52. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. (2017) 169:1342–56.e16. doi: 10.1016/j.cell.2017.05.035
53. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression—implications for anticancer therapy. Nat Rev Clin Oncol. (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
54. Tanaka A, Sakaguchi S. Targeting Treg cells in cancer immunotherapy. Eur J Immunol. (2019) 49:1140–6. doi: 10.1002/eji.201847659
55. Sanmamed MF, Rodriguez I, Schalper KA, Onate C, Azpilikueta A, Rodriguez-Ruiz ME, et al. Nivolumab and urelumab enhance antitumor activity of human T lymphocytes engrafted in Rag2-/-IL2Rgammanull immunodeficient mice. Cancer Res. (2015) 75:3466–78. doi: 10.1158/0008-5472.CAN-14-3510
56. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. (2013) 1:32–42. doi: 10.1158/2326-6066.CIR-13-0013
57. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. (2018) 33:649–63.e4. doi: 10.1016/j.ccell.2018.02.010
58. Kang TH, Jung ST. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp Mol Med. (2019) 51:1–9. doi: 10.1038/s12276-019-0345-9
59. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. (2000) 6:443–46. doi: 10.1038/74704
60. Ito A, Ishida T, Yano H, Inagaki A, Suzuki S, Sato F, et al. Defucosylated anti-CCR4 monoclonal antibody exercises potent ADCC-mediated antitumor effect in the novel tumor-bearing humanized NOD/Shi-scid, IL-2Rgamma(null) mouse model. Cancer Immunol Immunother. (2009) 58:1195–206. doi: 10.1007/s00262-008-0632-0
61. Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, et al. Phase ia study of FoxP3+ CD4 Treg depletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res, (2015) 21:4327–36. doi: 10.1158/1078-0432.CCR-15-0357
62. Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer. (2008) 122:2286–93. doi: 10.1002/ijc.23392
63. Ménétrier-Caux C, Faget J, Biota C, Gobert M, Blay J-Y, Caux C. Innate immune recognition of breast tumor cells mediates CCL22 secretion favoring Treg recruitment within tumor environment. Oncoimmunology. (2012) 1:759–61. doi: 10.4161/onci.19680
64. Ronchetti S, Nocentini G, Riccardi C, Pandolfi PP. Role of GITR in activation response of T lymphocytes. Blood. (2002) 100:350–2. doi: 10.1182/blood-2001-12-0276
65. Tone M, Tone Y, Adams E, Yates SF, Frewin MR, Cobbold SP, et al. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc Natl Acad Sci USA. (2003) 100:15059–64. doi: 10.1073/pnas.2334901100
66. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. (2002) 3:135–42. doi: 10.1038/ni759
67. Cohen AD, Schaer DA, Liu C, Li Y, Hirschhorn-Cymmerman D, Kim SC, et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS ONE. (2010) 5:e10436. doi: 10.1371/journal.pone.0010436
68. Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med. (2005) 202:885–91. doi: 10.1084/jem.20050940
69. Ramirez-Montagut T, Chow A, Hirschhorn-Cymerman D, Terwey TH, Kochman AA, Lu S, et al. Glucocorticoid-induced TNF receptor family related gene activation overcomes tolerance/ignorance to melanoma differentiation antigens and enhances antitumor immunity. J Immunol. (2006) 176:6434–42. doi: 10.4049/jimmunol.176.11.6434
70. Mahne AE, Mauze S, Joyce-Shaikh B, Xia J, Bowman EP, Beebe AM, et al. Dual roles for regulatory T-cell depletion and costimulatory signaling in agonistic GITR targeting for tumor immunotherapy. Cancer Res. (2017) 77:1108–18. doi: 10.1158/0008-5472.CAN-16-0797
71. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. (2008) 38:1654–63. doi: 10.1002/eji.200838105
72. Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of tet proteins in 5 mC to 5 hmC conversion, ES-cell self-renewal, and inner cell mass specification. Nature. (2010) 466:1129–33. doi: 10.1038/nature09303
73. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. (2009) 324:930–5. doi: 10.1126/science.1170116
74. Cobb BS, Nesterova TB, Thompson E, Hertweck A, O'Connor E, Godwin J, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. (2005) 201:1367–73. doi: 10.1084/jem.20050572
75. van der Ree MH, de Vree JM, Stelma F, Willemse S, van der Valk M, Rietdijk S, et al. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial. Lancet. (2017) 389:709–17. doi: 10.1016/S0140-6736(16)31715-9
76. Serr I, Furst RW, Ott VB, Scherm MG, Nikolaev A, Gokmen F, et al. miRNA92a targets KLF2 and the phosphatase PTEN signaling to promote human T follicular helper precursors in T1D islet autoimmunity. Proc Natl Acad Sci USA. (2016) 113:E6659–68. doi: 10.1073/pnas.1606646113
77. He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature. (2005) 435:828–33. doi: 10.1038/nature03552
78. Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. (2008) 9:405–14. doi: 10.1038/ni1575
Keywords: HIS mice, Treg, cancer immunotherapy, personalized medicine, iPSC-derived HSCs, microRNA
Citation: Serr I, Kral M, Scherm MG and Daniel C (2021) Advances in Human Immune System Mouse Models for Personalized Treg-Based Immunotherapies. Front. Immunol. 12:643544. doi: 10.3389/fimmu.2021.643544
Received: 18 December 2020; Accepted: 26 January 2021;
Published: 18 February 2021.
Edited by:
Tim Willinger, Karolinska Institutet, SwedenReviewed by:
Takeshi Takahashi, Central Institute for Experimental Animals, JapanCopyright © 2021 Serr, Kral, Scherm and Daniel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolin Daniel, Y2Fyb2xpbi5kYW5pZWxAaGVsbWhvbHR6LW11ZW5jaGVuLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.