 Michał Rudnik1
Michał Rudnik1 Amela Hukara1
Amela Hukara1 Ievgeniia Kocherova1Suzana Jordan1
Ievgeniia Kocherova1Suzana Jordan1 Janine Schniering1Vincent Milleret2
Janine Schniering1Vincent Milleret2 Martin Ehrbar2
Martin Ehrbar2 Karin Klingel3
Karin Klingel3 Carol Feghali-Bostwick4
Carol Feghali-Bostwick4 Oliver Distler1
Oliver Distler1 Przemysław Błyszczuk1,5
Przemysław Błyszczuk1,5 Gabriela Kania1*
Gabriela Kania1*- 1Department of Rheumatology, Center of Experimental Rheumatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland
- 2Department of Obstetrics, University Hospital Zurich, Zurich, Switzerland
- 3Department of Molecular Pathology, University Hospital Tuebingen, Tuebingen, Germany
- 4Division of Rheumatology, Medical University of South Carolina, Charleston, SC, United States
- 5Department of Clinical Immunology, Jagiellonian University Medical College, Krakow, Poland
Background: Systemic sclerosis (SSc) is an autoimmune disease characterized by overproduction of extracellular matrix (ECM) and multiorgan fibrosis. Animal studies pointed to bone marrow-derived cells as a potential source of pathological ECM-producing cells in immunofibrotic disorders. So far, involvement of monocytes and macrophages in the fibrogenesis of SSc remains poorly understood.
Methods and Results: Immunohistochemistry analysis showed accumulation of CD14+ monocytes in the collagen-rich areas, as well as increased amount of alpha smooth muscle actin (αSMA)-positive fibroblasts, CD68+ and mannose-R+ macrophages in the heart and lungs of SSc patients. The full genome transcriptomics analyses of CD14+ blood monocytes revealed dysregulation in cytoskeleton rearrangement, ECM remodeling, including elevated FN1 (gene encoding fibronectin) expression and TGF-β signalling pathway in SSc patients. In addition, single cell RNA sequencing analysis of tissue-resident CD14+ pulmonary macrophages demonstrated activated profibrotic signature with the elevated FN1 expression in SSc patients with interstitial lung disease. Peripheral blood CD14+ monocytes obtained from either healthy subjects or SSc patients exposed to profibrotic treatment with profibrotic cytokines TGF-β, IL-4, IL-10, and IL-13 increased production of type I collagen, fibronectin, and αSMA. In addition, CD14+ monocytes co-cultured with dermal fibroblasts obtained from SSc patients or healthy individuals acquired a spindle shape and further enhanced production of profibrotic markers. Pharmacological blockade of the TGF-β signalling pathway with SD208 (TGF-β receptor type I inhibitor), SIS3 (Smad3 inhibitor) or (5Z)-7-oxozeaenol (TGF-β-activated kinase 1 inhibitor) ameliorated fibronectin levels and type I collagen secretion.
Conclusions: Our findings identified activated profibrotic signature with elevated production of profibrotic fibronectin in CD14+ monocytes and CD14+ pulmonary macrophages in SSc and highlighted the capability of CD14+ monocytes to acquire a profibrotic phenotype. Taking together, tissue-infiltrating CD14+ monocytes/macrophages can be considered as ECM producers in SSc pathogenesis.
Introduction
Systemic sclerosis (SSc) is an autoimmune disease, characterized by high morbidity and mortality and a significant reduction in quality of life. Microvascular damages, dysregulation of innate and adaptive immunity and multiorgan fibrosis are implicated in the pathophysiology of SSc (1). SSc is characterized by a patient-to-patient variability in clinical manifestations, autoantibody profiles, extent of disease, treatment response and reduced survival rate (2). Changes in internal organ architecture, leading to pulmonary and cardiac complications and dysfunction remains the major causes of deaths among SSc patients (3).
Fibrogenesis is a multistage process, considered as the result of impaired tissue repair responses, in which abnormal production of cytokines, growth factors and angiogenic factors turn tissue homeostasis towards the excessive accumulation of extracellular matrix (ECM) (4). Fibrosis is usually an outcome of prolonged and exaggerated activation of fibroblasts, which differentiate into myofibroblasts (5). In contrast to physiological wound healing, in which myofibroblast are present only transiently, in fibrosis myofibroblasts become a permanent cellular component of the tissue (6).
Myofibroblasts are characterized by expression of alpha smooth muscle actin (αSMA) forming stress fibers and exhibit an increased capacity to synthesize collagens, fibronectin, and other ECM components. Fibronectin is a high-molecular weight glycoprotein of the extracellular matrix, which binds to integrins and other extracellular matrix proteins such as collagen, fibrin, and heparan sulfate proteoglycans (7). Increased deposition of fibronectin, paralleled to accumulated collagen, has been reported in the SSc skin (8, 9).
The cellular source of myofibroblasts in wound healing and fibrotic lesions is still debatable. Although most evidence point to resident fibroblasts, other tissue-resident cell types or cells recruited from circulation have been shown to acquire a myofibroblast-like phenotype (10). In experimental models of lung and kidney fibrosis, bone marrow-derived fibrocytes have been shown to be recruited into injured or fibrotic tissues in response to chemokine signals, where they differentiate into fibroblasts or myofibroblasts (11). Lineage tracing experiments have provided convincing evidence that pericytes are an important source of myofibroblasts during renal fibrogenesis in animal models, and these cells might also be a source of myofibroblasts in SSc (12, 13). During fibrogenesis, epithelial cells have been hypothesized to undergo epithelial-to-mesenchymal transition and transdifferentiate into myofibroblasts in response to TGFβ and other profibrotic cytokines. Additionally, SSc tissues are chronically hypoxic, which can further promote the formation of myofibroblast-like cells (14). In addition to epithelial cells, endothelial cells have also been hypothesized to transdifferentiate into myofibroblasts through endothelial-mesenchymal transition (15).
The primary mechanism underlying excessive fibrosis in SSc remains unknown; however, TGFβ is one of the best-studied mediators regulating fibrotic processes, including fibroblast differentiation, ECM deposition and tissue contraction (16). Elevated levels of TGFβ-regulated genes (cartilage oligomeric matrix protein, thrombospondin-1) were reported in the skin lesions from SSc patients (17). TGFβ signalling regulates the expression of profibrotic genes mostly via the SMAD-dependent canonical pathway (18). However, the involvement of several non-canonical pathways in fibrotic processes was acknowledged. For example, activation of the ERK-MAPK signalling pathway leads to upregulation of type I collagen in SSc fibroblasts (19). Ultimately, inhibition of TGFβ signalling has been addressed as a potential treatment strategy; however, conflicting results were reported following TGFβ blockade (20).
Myeloid cells, including monocytes, have been shown to be essential regulators of fibrosis as producers of chemokines, inflammatory cytokines and growth factors (21, 22). Our group has previously reported that the microenvironment of inflamed lung induces myeloid progenitors to acquire a myofibroblast phenotype (23). Similarly, we showed that heart-infiltrating myeloid cells served as myofibroblast progenitors in the model of post-inflammatory cardiac fibrosis (24). In the present study, we evaluated the differentiation potential of circulating CD14+ monocytes from healthy controls and SSc patients into a myofibroblast-like phenotype.
Material and Methods
SSc Patients and Healthy Controls
Human blood samples and skin biopsies collection were approved by the local ethics committee of the Canton Zurich (KEK-ZH 515, PB-2016-02014, KEK-Nr 2018-01873). All study subjects provided written informed consent. SSc patients and healthy controls were recruited at the Department of Rheumatology of University Hospital Zurich. All the patients fulfilled the ACR/EULAR 2013 classification criteria for SSc.
The patients diagnosed with SSc by experts with Raynaud syndrome and a least one other SSc characteristic such as: SSc specific antibodies, SSc characteristic capillaroscopy changes and/or puffy fingers. The patients not fulfilling the ACR/EULAR 2013 classification criteria for SSc were grouped as early-SSc. The patients fulfilling the criteria were further divided into lcSSc and dcSSc subgroups, according to Le Roy et al. (25). Detailed demographics and clinical characteristics of SSc patients are included in Table 1. Healthy control group was age- and gender matched (age mean=44.4 ± 10.8, female 17/20 (85%).
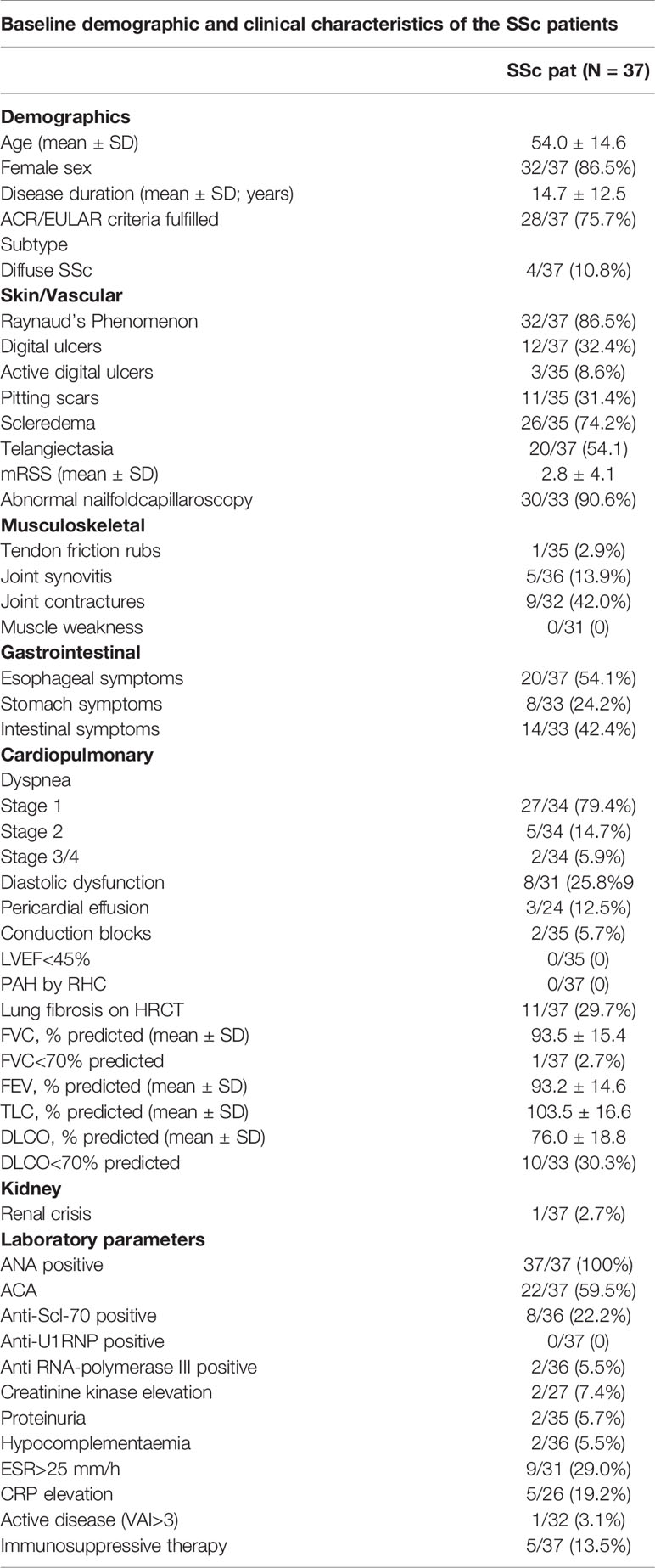
Table 1 Definitions of items and organ manifestation are according to EUSTAR.
Data are presented as number (n)/total valid cases (N) (%). Disease duration was calculated as the difference between the date of the baseline visit and the date of the first non-Raynaud’s symptom of the disease as reported by the patient. Pulmonary hypertension was judged on RHC. Active disease was defined as a score >3 by calculating European Scleroderma Study Group disease activity indices for systemic sclerosis proposed by Valentini. Immunosuppressive therapy was defined as treatment with corticosteroids (prednisone dose ≥10 mg/day or other dosage forms in equal dose) or any immunosuppressant.
ACA, anti-centromere antibody; ANA, antinuclear antibody; Anti-Scl-70, anti-topoisomerase antibody; CRP, C reactive protein; HRCT, computed tomography; DLCO, diffusing capacity for carbon monoxide; ESR, erythrocyte sedimentation rate; FEV1, forced expiratory volume in 1 sec; FVC, forced vital capacity; LVEF, left ventricular ejection fraction; mRSS, modified Rodnan skin score; NYHA, New York Heart Association; TLC, total lung capacity; VAI, Valentini activity index.
Human endomyocardial biopsies were provided by the University Hospital Tubingen, Germany. Samples were obtained from SSc patients with cardiac involvement (inflammatory dilated cardiomyopathy) and controls (patients with healed myocarditis). Lung biopsies were provided by the Division of Rheumatology, Medical University of South Carolina, Charleston, USA. The samples were obtained from SSc-related interstitial lung disease (SSc-ILD). Sample tissue from downsized lung transplants from healthy individuals served as controls. The experiments with re-use of human material were approved by Swissethics (KEK-Nr 2019-00058, KEK-Nr 2018-01873) and were performed in conformity with the principles outlined in the Declaration of Helsinki. Transcriptomic analysis of an already published dataset of human lung tissues has been included. For this, the University of Pittsburgh Institutional Review Board approved ethics of use the human lung samples as previously described (26).
CD14+ Monocytes Isolation and Differentiation
Blood samples were collected in EDTA tubes (BD Vacutainer) and processed within 24h. Peripheral blood mononuclear cells (PBMCs) were isolated by gradient centrifugation on cell separation medium (Lympholyte, Cedarlane), followed by magnetic-activated cell sorting for CD14 using human microbeads and AutoMACS Pro device (Miltenyi Biotec). Cells were cultured in DMEM low glucose medium (Sigma) with the addition of 10% Foetal Bovine Serum (FBS) and 1% penicillin/streptomycin (both Gibco). To differentiate into myofibroblast-like cells, CD14+ monocytes were stimulated with 10 ng/ml TGFβ (PeproTech), 10 ng/ml IL-4, 10 ng/ml IL-10 and 10 ng/ml IL-13 (Immunotools) for 7 days.
RNA Extraction
Total RNA was isolated using the Quick-RNA Microprep isolation kit (Zymo Research). Directly after monocyte treatment, cells were washed with PBS and lysed in RNA lysis buffer (Zymo Research). An equal volume of absolute ethanol (Millipore) was added and mixed. Lysates were further processed on the columns. Genomic DNA was removed by DNase I treatment. RNA was washed twice and eluted in 10-15 μl of nuclease-free water (Promega). RNA concentration and purity were assessed on NanoDrop 1000 (Thermo Fisher Scientific).
Bulk and Single Cell (sc) RNA Sequencing and Data Analysis
For RNA sequencing, RNA was isolated from SSc patients and healthy controls [as described previously in Supplementary Table 1 in (27)] as described above, and RNA Integrity Number (RIN) was assessed by Tape Station (Agilent). Samples with RIN≥8 were further processed. RNA sequencing was performed by the Functional Genomics Centre Zurich. From 100 ng of total RNA, polyA libraries were prepared using the Illumina TruSeq RNA Stranded mRNA library Kit. Sequencing was performed on the Illumina HiSeq 4000 platform. Quality of the sequencing was controlled by FastQC package. Reads were aligned to the genome using the STAR algorithm. Gene expression profiles were next calculated by the FeatureCount algorithm. For differentially expressed genes, the DeSEQ2 algorithm was used with a threshold of minimum 10 reads for a transcript to be considered as present. Pathway enrichment analysis of differentially expressed genes (p ≤ 0.01, |log2ratio| ≥0.5) was performed by the Metacore software, as described previously (27).
Human explanted lung tissues were digested and scRNA-sequencing was performed as previously described (26).
ScRNAseq Analysis of Each Sample
Raw count matrices produced from CellRanger were loaded from the dataset GSE128169. Empty droplets were distinguished from droplet-containing cells by using the emptyDrops function from the R package DropletUtils (28, 29). Only droplets that obtained an FDR <= 0.001 were called as cells. Doublets were also discarded from further analysis using the R package scDblFinder. Low quality cells were identified based on the number of reads, genes and mitochondrial content using the R package scuttle. We excluded cells that were outliers according to at least one of the following thresholds: number of reads < 3 MADs, number of gene < 3 MADs, mitochondrial content > 3 MADs. Additionally, we discarded genes with < 1 UMI count in < 0.01 of the remaining cells. We used the SCTransform (30) method from the R package Seurat (31) to normalize and scale the data.
Integration of Multiple Samples
Multiple samples were combined using the Seurat integration workflow (32). After integration we reduced the dimensionality of the data using Principal Component Analysis (PCA). The main 30 principal components were used to perfor unsupervised clustering with a resolution value of 0.8. After clustering and visualization with Uniform Manifold Approximation and Projection (UMAP), cell populations were identified through examination of gene markers in the associated transcriptome.
Cells that had > 0 counts for the CD14 gene were called as CD14+ cells. All plots and downstream analysis were performed using Seurat.
RT-qPCR
For reverse transcription, 200-300ng of total RNA was used. The reaction was performed using MultiScribe reverse transcriptase (Thermo Fisher Scientific), random hexamers and RNAase inhibitor (both Roche). Subsequently, the qPCR reaction was performed using the SYBR green GoTaq qPCR master mix (Promega) on Agilent Stratagene Mx3005P qPCR instrument. Sequences of primers are listed in Supplementary Table 1. Relative gene expression was calculated using the 2-ΔΔCt method. GAPDH was used as the reference gene.
Protein Extraction and Immunoblotting
After the stimulation with cytokines, cells were washed once with ice-cold PBS, collected, centrifuged and lysed for 30 minutes on ice in RIPA buffer (Sigma) supplemented with proteases and phosphatases inhibitors (Roche). An equal amount of the protein was loaded and separated by SDS-PAGE electrophoresis, followed by wet transfer on the nitrocellulose membrane (GE Healthcare). The membrane was further incubated for 1 hour in blocking buffer (5% BSA in TBS-T). Further, membranes were probed overnight with primary antibodies, listed in Supplementary Table 2, in blocking buffer at 4°C. Further, membranes were incubated for 1 hour at room temperature with secondary HRP-conjugated antibodies. Signal was developed with ECL substrate (SuperSignal West Pico PLUS, Thermo Scientific) and acquired on the Fusion fx (Vilber) device.
ELISA
For the detection of human pro-collagen 1α1 DuoSet ELISA (RnD Systems) was used according to the manufacturer’s protocol. Briefly, 96-well plates were coated with capture antibodies overnight in room temperature and further blocked with 2% BSA in phosphate-buffered saline (PBS) supplemented with 0.05%Tween 20. Between each step, plates were washed three times with PBS. The protein standards and samples were applied and incubated for 2 hours. Next, plates were incubated with biotin-conjugated detection antibodies for 2 hours and streptavidin-HRP for 30 minutes in room temperature. Signal was developed with TMB substrate (Thermo Scientific), and 450 nm absorbance was measured on a BioTEK HT plate reader (Tecan). Concentrations were calculated according to the respective standard curves.
Treatment With Pharmacological Inhibitors and Cytotoxicity Assessment
SD208 [1 μM], SIS3 [2 μM], (5Z)-7-Oxozeaenol (OXO) [1 μM] pharmacological inhibitors used in the project were purchased from Tocris Biosciences. To determine optimal non-toxic concentrations, we performed toxicity tests. CD14+ monocytes were incubated with 2-fold dilutions of inhibitors starting from 5 or 10 µM. Cells were incubated for 24h, stained with propidium iodide (Biolegend), and cytotoxicity was evaluated by flow cytometry. The highest non-toxic concentration was used in further experiments.
2D and 3D Co-Culture With Dermal Fibroblasts
To distinguish cells in both systems, monocytes were stained with Cell Trace Violet (Thermo Scientific), and fibroblasts were stained with CFSE (Biolegend) according to the manufacturer’s protocol. For 2D co-cultures, fibroblasts were plated 24h prior to the addition of monocytes. Cells were cultured for 7 days and sorted using FACSAria III cell sorter.
For the 3D co-culture model, 3DProSeed® hydrogel microtiter plate (Ectica Technologies) were used. Firstly, labelled fibroblasts were plated and allowed to penetrate hydrogels for 24h. Next, monocytes were added. Plates were incubated for 7 days, and anti-αSMA/phalloidin (both Sigma) staining was performed. Co-cultures were visualized using Leica SP8 confocal microscope.
Immunohistochemistry, Imaging and Quantification
Collected tissue samples were washed in PBS and fixed for 16 hours in 4% paraformaldehyde in PBS. Next, tissues were rinsed in distilled water and transferred to 50% ethanol. Biopsies were then dehydrated (three incubations in 80% ethanol for 1 hour, three incubations in 96% ethanol for 1 hour, two incubations in 100% ethanol for 1 hour). Tissues were cleared twice in xylene for 1 hour and subsequently incubated twice in a 56°C paraffin bath for 3 hours. After paraffin embedding, 4 μm thick sections were placed on Superfrost Plus slides (Thermo Scientific) and dried overnight. Lung sections were cut at a thickness of 4 μm and stained with hematoxylin and eosin (HE) for analysis of the lung architecture and the presence of cellular infiltrates, and with Picrosirius Red to detect collagen deposition using standard protocols (33).
For immunohistochemistry sections were deparaffinized in xylene for 10 minutes (3 times) and rehydrated by sequential incubations in ethanol solutions (100%, 100%, 96% and 80%) for 3 minutes each. Sections were eventually washed for 5 minutes in distilled water. Antigen retrieval was performed in citrate buffer (10 mM citrate, 0.05% Tween, pH=6), and incubated at 95°C for 15 minutes. Endogenous peroxidases were blocked by 3% H2O2 solution for 15 minutes. Unspecific antibody binding was blocked with 10% goat serum in Background Reducing Antibody Diluent (Dako). Endogenous biotin was blocked by Avidin-Biotin Block kit (Vector Laboratories). Sections were incubated with primary antibodies, listed in the Supplementary Table 2, in 4°C overnight. The appropriate biotinylated secondary antibodies (Vector Laboratories) were incubated for 30 minutes at room temperature, followed by 30 minutes incubation with VECTASTAIN Elite ABC kit (Vector Laboratories). Staining was developed using Vector DAB or Vector Red (Vector Laboratories) followed by a counterstaining of nuclei for 1 minute in Mayer’s hematoxylin solution (J.T Baker). All sections were mounted using Pertex mounting medium (Dako). CD14-collagen-1 was performed by Sophistolab AG.
Full slide images were acquired using a Zeiss Axio Scan Z1 slide scanner. ImageJ software was used for relative quantification of the signal in the sections. For CD45, mannose-R, CD86, iNOS and arginase-1 staining analysis, the “HDAB” plug-in was applied, while for Picrosirius Red and α-SMA “Fast Red, Fast Blue” plug-in was used. Deconvoluted images were used to calculate the area of the nuclear staining and the area of the specific signal. The value for each section was calculated as the ratio of the specific signal to nuclei signal.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 8 software. Data distribution was calculated using the Shapiro-Wilk test. Normally distributed data were presented as mean ± standard deviation and analyzed by unpaired two-tailed parametric t-test. For non-normally distributed data, unpaired non-parametric Mann-Whitney U test was used, and medians were presented. For comparisons of more than two groups, two-way ANOVA test with multiple comparisons (normally distributed data) and Kruskal-Wallis test with multiple comparisons (non-normally distributed data) were used. Differences were considered statistically significant for p<0.05. n refers to the number of biological replicates.
Additional methods are described in the Supplementary Material.
Results
Infiltration of CD14+ Monocytes Into the Organs Co-Localizes With Fibrosis in SSc
In line with data published previously (34), we observed increased infiltration of CD14+ cells into the heart and lungs of SSc patients compared to control tissues (Figures 1A, B). Moreover, CD14 signal co-localized with collagen-rich lesions in different SSc tissues (Figure 1C). This data suggests that monocytes may participate in fibrotic processes in many organs during pathological tissue remodeling in SSc. Furthermore, heart and lungs of SSc patients showed the increased levels of αSMA+ (myo)fibroblasts, CD68+ pro-inflammatory macrophages and mannose-R+ (CD206) alternatively activated macrophages (Figures 2A–C) that reflects inflammatory and fibrotic microenvironment in different SSc tissues.
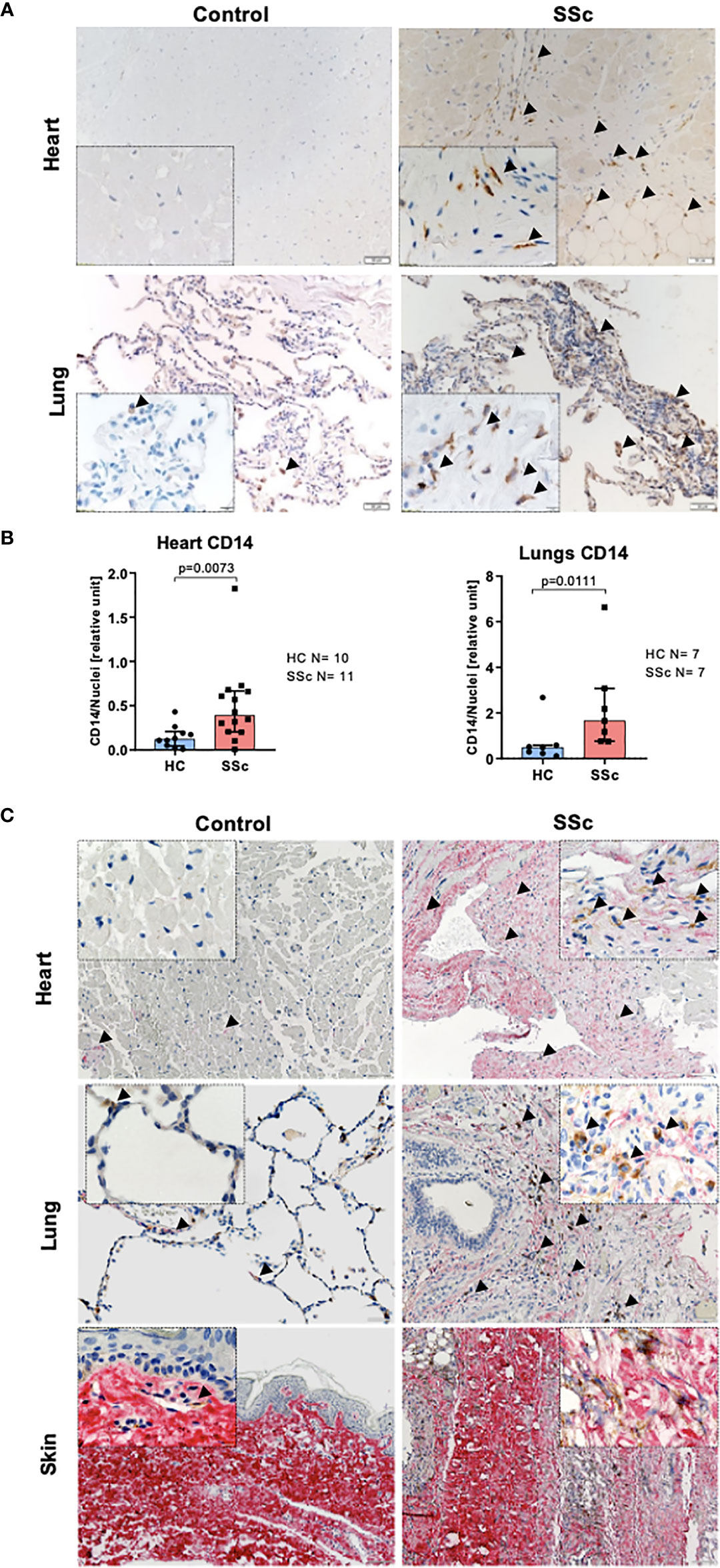
Figure 1 Monocyte infiltration into the fibrotic lesion in SSc tissues. (A) Representative images of paraffin-embedded sections of the heart and lungs, and (B) corresponding quantification of IHC staining for CD14 (lungs HC N=7, SSc N=7, heart HC N=10, SSc N=11, unpaired t-test). (C) Representative images of paraffin-embedded sections of the heart, lungs and skin co-stained for pro-collagen I (red) and CD14 (brown). (A, C) Scale bar 100μm, scale bar in insert 20μm.
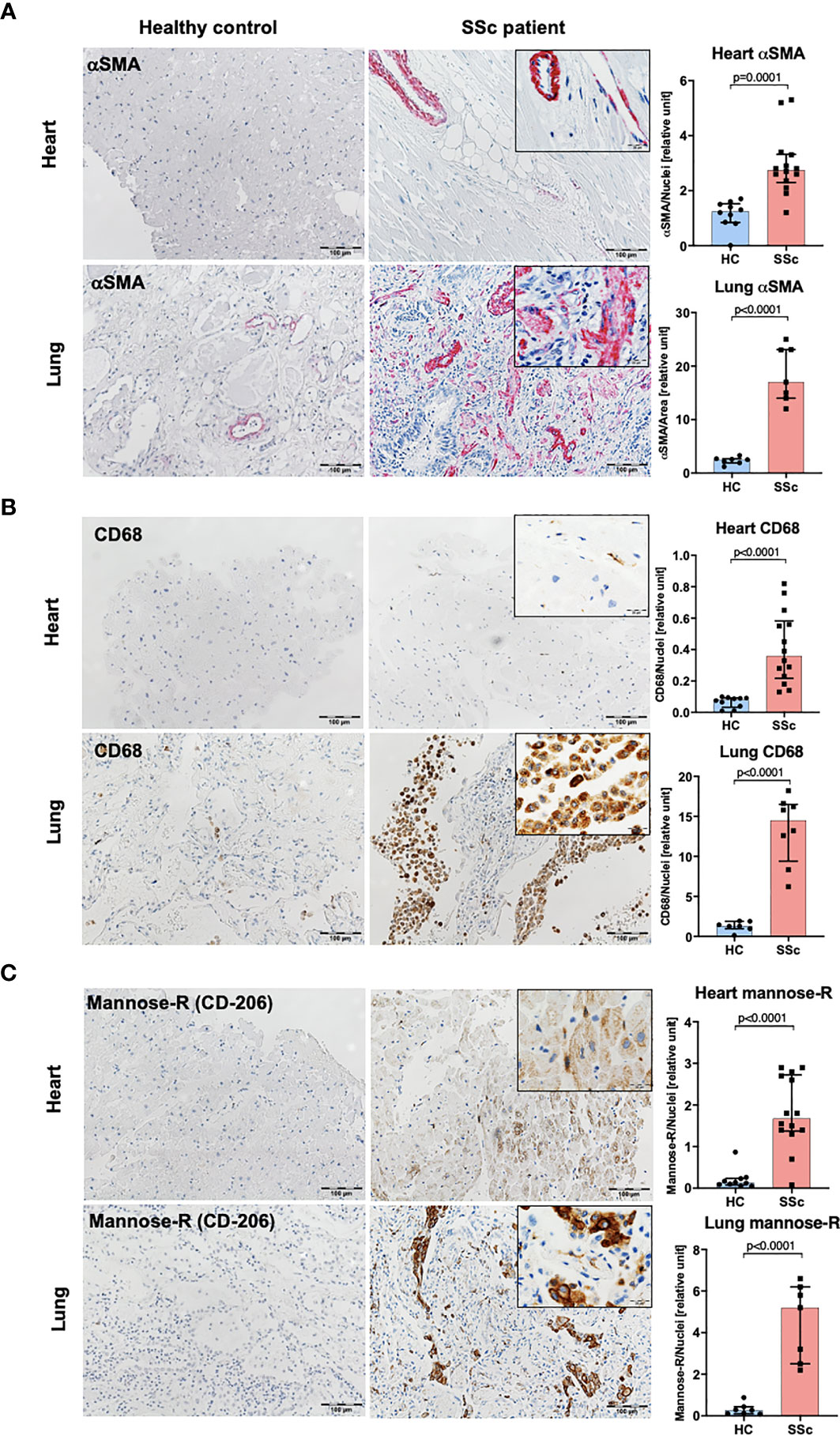
Figure 2 Characteristic of human SSc lung and heart tissues. Representative images of paraffin-embedded sections of the heart and lungs and corresponding quantification of IHC staining for αSMA (A), CD68 (B) and mannose-R (CD206) (C) (lung HC N=7, lung SSc N=7, heart HC N=10, heart SSc N=11, unpaired t-test; scale bar 100μm, scale bar in insert 20μm).
Activation of Profibrotic Pathways in CD14+ Blood Monocytes in SSc
We compared the transcriptome profiles of the total pool of SSc CD14+ monocytes in relation to CD14+ monocytes obtained from healthy controls (HC). Pathway analysis revealed enrichment of SSc and fibrotic-associated processes, such as “TGF-beta signalling pathway”, “Toll-like Receptor Signalling Pathway”, “Angiogenesis” and “VEGF signalling pathway” in SSc CD14+ monocytes (Figure 3A). At the transcription level, we observed dysregulated expression of essential profibrotic genes: TGFBR2, MMP9, JUN, COL9A3, COL18A1, FN1 and WNT5B (Figures 3B, C). Further, we demonstrated that SSc CD14++CD16- monocyte population showed significantly higher FN1 expression than SSc CD14+CD16+ and CD14lowCD16+ monocyte populations (Figure 3D). Of note, we did not notice any difference in FN1 expression between these three monocyte populations in HC monocytes (Figure 3D). Importantly, SSc CD14++CD16- and CD14+CD16+ monocyte populations revealed profibrotic features by expressing upregulated FN1 levels compared to HC monocytes (Figure 3D).
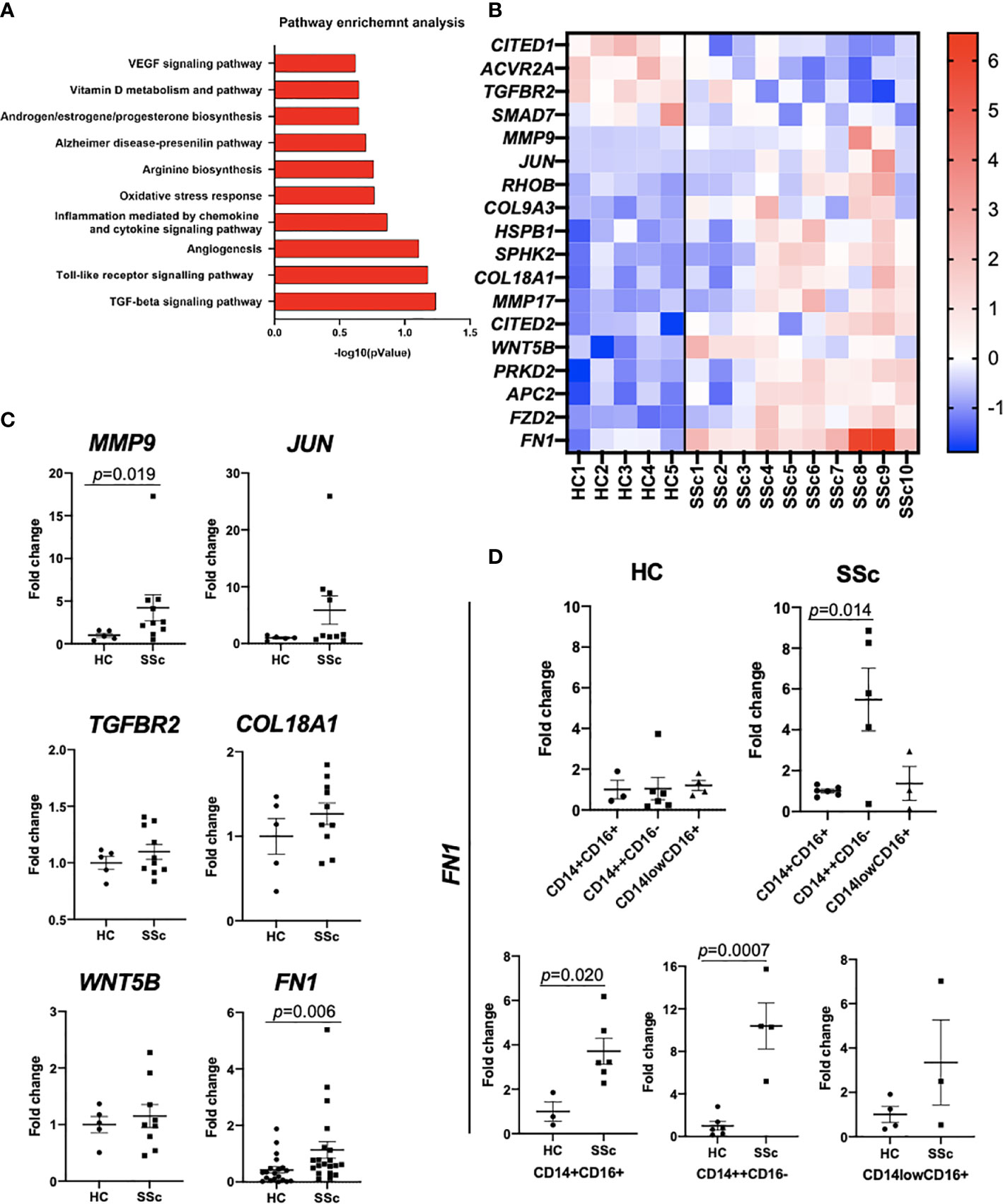
Figure 3 Transcriptomic analysis of CD14+ monocytes from SSc patients and HC. (A) Pathway enrichment analysis of SSc-related biological processes calculated based on differentially expressed gene sets (Metacore software). (B) Differentially expressed genes involved in fibrosis from RNAseq data. (C) qPCR confirmatory analyses of selected genes from RNAseq data (N=5-20, Mann Whitney-U-test). (D) qPCR analysis of FN1 mRNA levels in sorted CD14++CD16-, CD14lowCD16+ and CD14+CD16+ monocyte from SSc patients and HC (n=3-6, Mann Whitney-U-test).
This result indicated an activated, profibrotic phenotype of SSc CD14+ monocytes in the circulation, which might predispose them to acquire pathogenic phenotype in the fibrotic tissue.
Activation of Profibrotic Pathways in CD14+ Pulmonary Macrophages in SSc
Transcriptomic analysis of an already published dataset of lung tissue from 4 HC and 4 SSc-ILD patients has been performed (26). CD14+ cells were visualized in all macrophage clusters (FCN1hi, SPP1hi, FABP4hi and proliferating macrophages) (Figure 4A), determined as previously shown (26). Pathway analysis revealed enrichment of fibrosis-associated processes, such as “Extracellular matrix disassembly”, “Extracellular matrix organization”, “Cellular response to cytokine stimulus” and “Cytokine-mediated signalling pathway” in SSc CD14+ cells (Figure 4B). At the transcription level, we noticed upregulation expression of several profibrotic genes, including TGFβ1, TIPM1, TIPM2, FN1 and ADAM10 (Figure 4C). Importantly, in all macrophage clusters in SSc lungs, we observed upregulated expression of FN1 in CD14+ cells (Figures 4D, E).
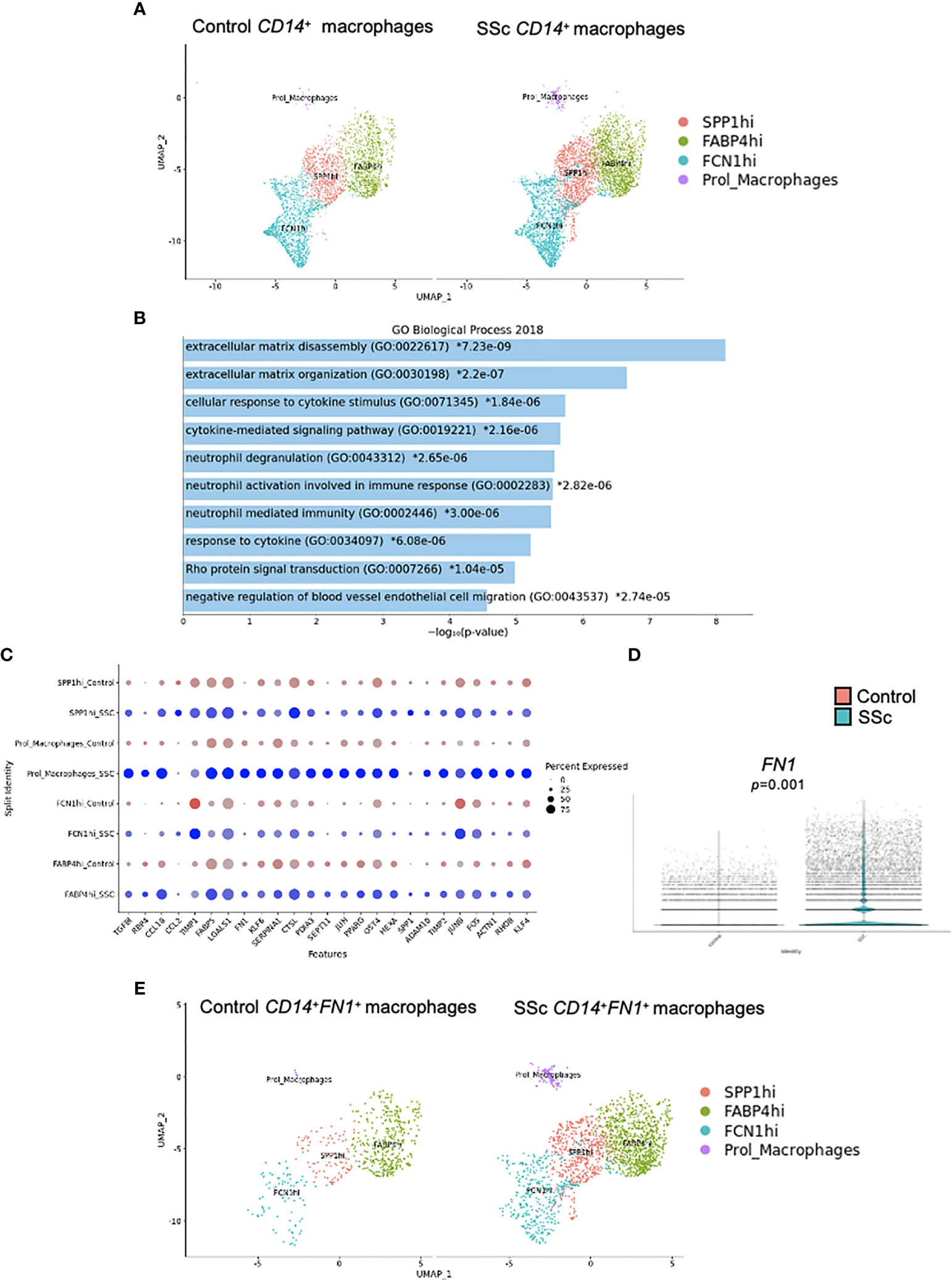
Figure 4 Characteristic of CD14+ macrophages in SSc-ILD lung tissues. Four human healthy control (HC) and 4 SSc-ILD lung tissue samples were used for a single-cell (sc) RNA-sequencing analysis according to Valenzi et al. (26). (A) UMAP plot visualization of CD14+ cells in macrophage clusters in HC and SSc-ILD samples. (B) Pathway expression in the specific GO biological process (Enrichr 2018) and the expression of selected fibrotic-related genes (C) upregulated in SSc-ILD CD14+fibronectin (FN1)+ cells compared to HC CD14+ cells in lungs. (D) FN1 expression in SSc-ILD CD14+ cells compared to HC CD14+ cells in lungs (N=4, Mann Whitney-U-test). (E) UMAP plot visualization of CD14+ cells in macrophage clusters in HC and SSc-ILD samples. UMAP, uniform manifold approximation and projection; SSc-ILD, systemic sclerosis-associated interstitial lung disease.
Profibrotic Cytokine Stimulation Induces Differentiation of Monocytes Into Myofibroblast-Like Cells
Bone marrow-derived cells have been proposed as one of the sources of myofibroblasts. Therefore, next, we evaluated the potential of CD14+ monocytes to differentiate into myofibroblast-like cells in response to profibrotic stimulation with TGFβ, IL-4, IL-10 and IL-13 cytokines. We observed the induction of profibrotic gene expression including ACTA2, COL1A1 and FN1 (Figure 5A). Additionally, stimulated monocyte-derived cells secreted ECM components: type I collagen and fibronectin (Figures 5B, C). Time-dependent profibrotic stimulation with TGFβ, IL-4, IL-10 and IL-13 cytokines revealed that CD14+ monocytes upregulated profibrotic gene expression already after 24h and sustain these levels up to 7 days (Figure 5D). Of note, we did not observe any difference in the differentiation potential between CD14+ monocytes obtained from SSc patients and healthy controls.
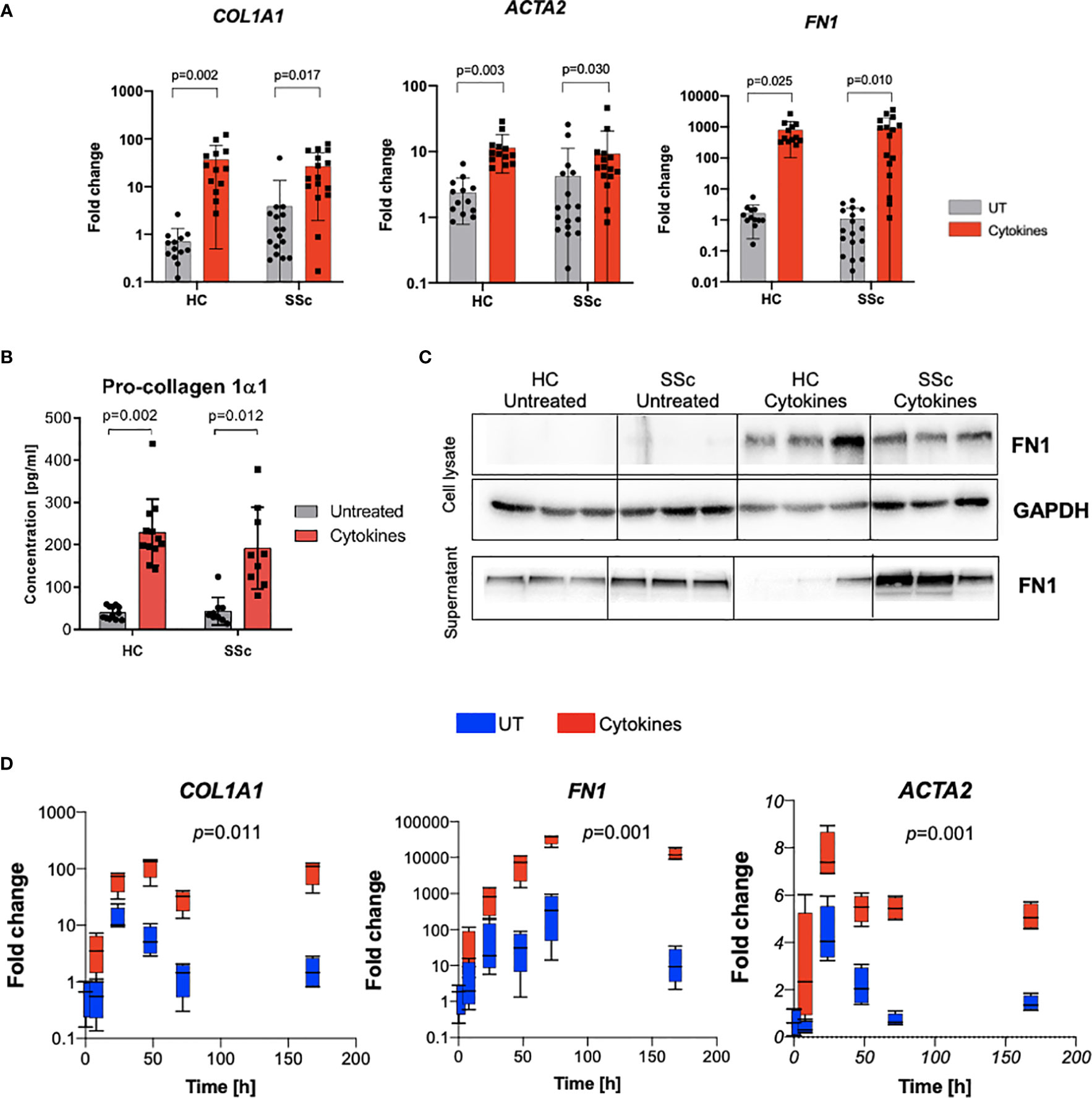
Figure 5 Profibrotic stimulation induces differentiation of monocytes into fibroblast-like cells. (A) mRNA expression of COL1A1, ACTA2 and FN1 after profibrotic cytokines stimulation (TGFβ, IL-4, IL-10, IL-13 [10 ng/ml each]) of HC and SSc monocytes (N=13, two-way ANOVA with Benjamini, Krieger and Yekutieli post-hoc test). (B) ELISA measurement of Pro-collagen 1α1 concentration in supernatants from CD14+ monocytes after profibrotic cytokines stimulation (TGFβ, IL-4, IL-10, IL-13, [10 ng/ml each]) (N=13, two-way ANOVA with Benjamini, Krieger and Yekutieli post-hoc test). (C) Western blot assessment of fibronectin level in cell lysates and supernatants from CD14+ monocytes stimulated with profibrotic cytokines (TGFβ, IL-4, IL-10, IL-13, [10 ng/ml each]) (N=3). (D) Time-dependent mRNA expression of COL1A1, ACTA2 and FN1 after profibrotic cytokines stimulation (TGFβ, IL-4, IL-10, IL-13 [10 ng/ml each]) of HC monocytes at following time points: 0, 8, 24, 47, 72 and 168h (N=4, two-way ANOVA with Tukey’s multiple comparison test, p values for row factor).
Co-Culture With Dermal Fibroblasts Enhances Profibrotic Phenotype of CD14+ Monocytes
We hypothesized that the microenvironment might play a determinant role in the profibrotic response of CD14+ monocytes. To test this hypothesis, we co-cultured CD14+ monocytes with side-matched dermal fibroblast from SSc patients and HCs for 7 days. To analyze gene expression, cell types were separated using FACS sorting. We observed that co-culture with dermal fibroblasts induced expression of profibrotic genes ACTA2, COL1A1 and FN1 in both HC and SSc monocytes. Importantly, we noticed a significant induction of gene expression by SSc fibroblasts (Figure 6A).
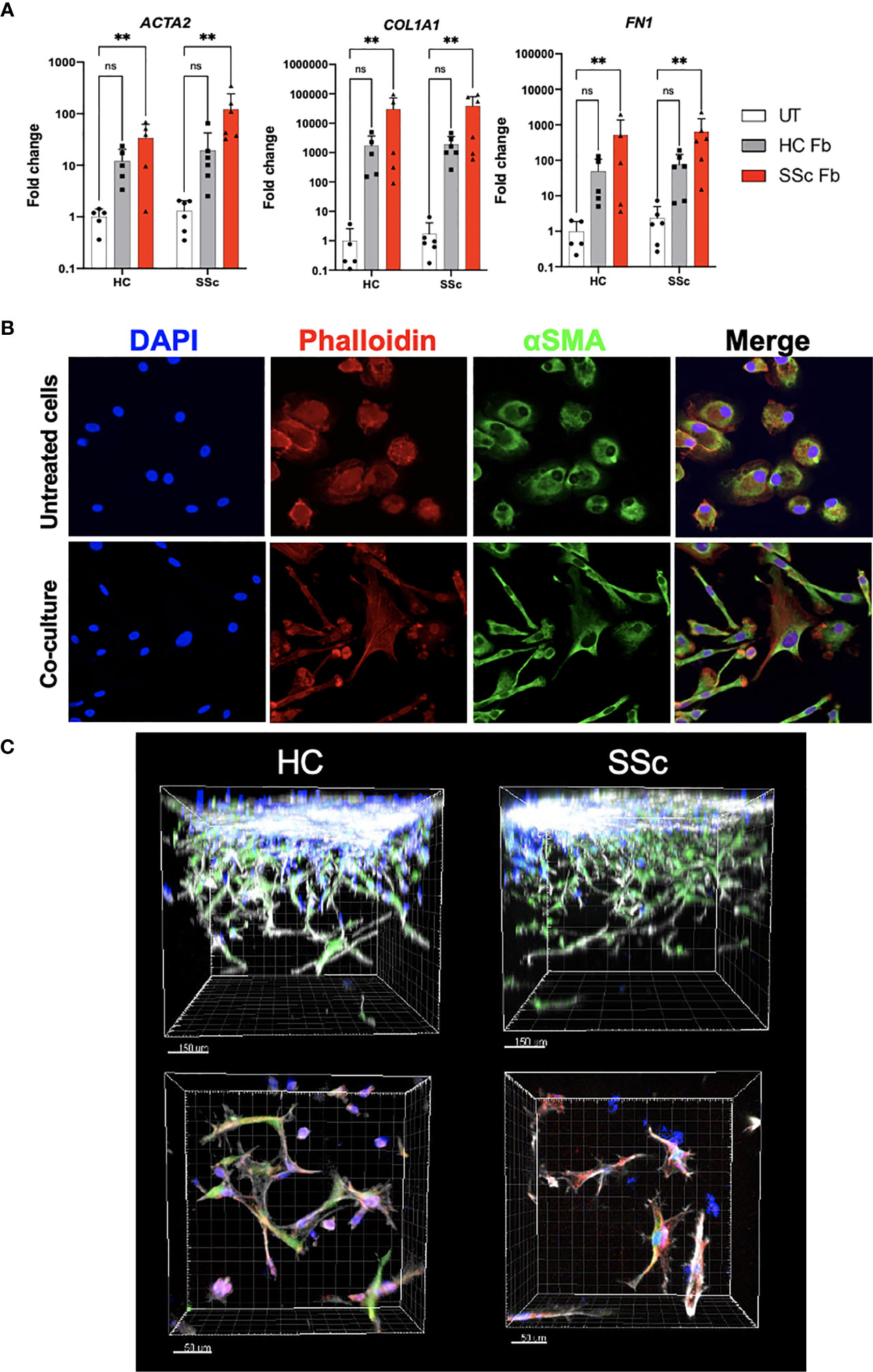
Figure 6 Profibrotic microenvironment induces differentiation of monocytes into fibroblast-like cells. (A) mRNA expression of ACTA2, COL1A1 and FN1 in CD14+ monocytes from HC and SSc patients after co-culture with HC and SSc skin fibroblasts (HC N=5, SSc N=6, two-way ANOVA with Uncorrected Fischer’s LSD post-hoc test, **p < 0.005). (B) Representative images of immunofluorescence staining of CD14+ monocytes with and without co-culture with fibroblasts (blue– DAPI nuclear staining, red– Phalloidin staining, green- α-SMA staining). (C) Representative images of immunofluorescence staining of HC and SSc CD14+ monocytes co-cultured with fibroblasts in 3D hydrogel model (blue– monocytes stained with Cell Trace Violet, green– fibroblasts stained with CFSE, red– α-SMA staining, grey- Phalloidin staining). ns, not significant.
Further, we aimed to observe phenotypic changes of CD14+ monocytes after co-cultures in 2D and 3D models. Monocytes, which were sorted out after the 2D co-culture and re-plated in a chamber slide, were stained with phalloidin and anti-αSMA antibody. Compared to monocytes cultured alone, cells after the co-culture acquired spindle shape, however, they failed to form stress fibers (Figure 6B). Similar characteristics were observed for monocytes in 3D co-culture (Figure 6C). Taken together, the fibroblast microenvironment induced a change in monocyte gene expression and morphology towards myofibroblast-like cells.
TGFβ Is a Key Regulator of Profibrotic Gene Expression in Monocyte-Derived Myofibroblast-Like Cells
Fibrotic processes might be driven by various stimuli; however, TGFβ is regarded as a pivotal mediator during SSc. To analyze the role of TGFβ signalling in monocyte-to-myofibroblast-like cell differentiation, we inhibited selected TGFβ downstream pathways in CD14+ monocytes treated with the profibrotic cytokine cocktail (TGFβ, IL-4, IL-10 and IL-13). Treatment with the TGFBR1 kinase inhibitor SD-208 completely abolished expression of the ECM components collagen 1 and fibronectin on both mRNA and protein levels (Figure 7). Interestingly, TGFβ-induced expression of COL1A1 was suppressed by the inhibitor of canonical SMAD-dependent pathway SIS3, but not by inhibition of TAK1-dependent non-canonical pathway with 5z-7-oxozeaenol (OXO). On the other hand, blocking of canonical and non-canonical TGFβ downstream pathways was sufficient to decrease expression and secretion of fibronectin (Figure 7B).
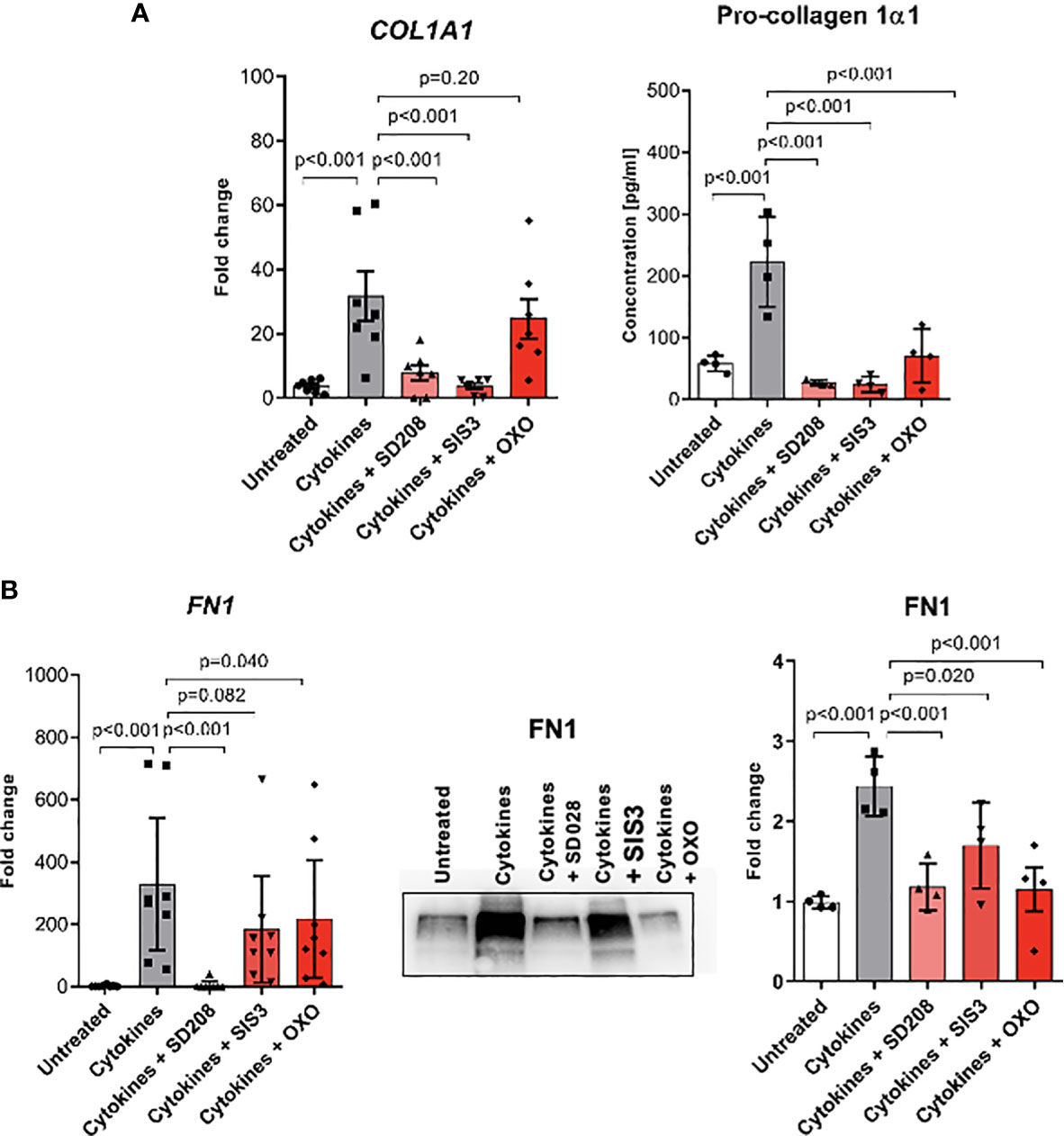
Figure 7 Monocyte-to-fibroblast-like cells differentiation is dependent on TGFβ signalling pathways. (A) COL1A1 mRNA expression level and ELISA measurements of Pro-collagen 1α1 protein level in supernatants after profibrotic cytokine stimulation (TGFβ, IL-4, IL-10, IL-13 [10 ng/ml each]) and treated with TGFβ signalling pathway inhibitors [SD208 and (5Z)-7-Oxozeaenol (OXO) 1 μM, SIS3 2 μM] (N=4-7, one-way ANOVA with Benjamini, Krieger and Yekutieli post-hoc test). (B) FN1 mRNA expression level and Western Blot analysis, and corresponding densitometry of Fibronectin protein level in the supernatants after profibrotic cytokine stimulation (TGFβ, IL-4, IL-10, IL-13 [10 ng/ml each]) and treated with TGFβ signalling pathway inhibitors [SD208 and (5Z)-7-Oxozeaenol (OXO) 1 μM, SIS3 2 μM] (N=4-7, one-way ANOVA with Benjamini, Krieger and Yekutieli post-hoc test).
Discussion
Our data demonstrated that human peripheral blood circulating CD14+ monocytes exhibited profibrotic phenotype in SSc patients. Similarly, several reports indicated fibroblast-like/circulating Collagen I-producing CD14+ monocytes or CD14- fibrocytes as profibrotic cell progeny in SSc (35). Likewise, the alternatively activated macrophages derived from circulating CD14+ monocytes have also been considered as profibrotic cells in SSc (36). SSc patients have higher levels of circulating CD34+CD14+ cells, Collagen-I-producing CD14+ monocytes and CD163+ monocytes (36). Importantly, monocyte count could be incorporated into the clinical assessment of patients with fibro-proliferative disorders (37), including SSc patients with interstitial lung disease (ILD). Accordingly, the elevated numbers of Collagen-I-producing fibrocytes and profibrotic monocytes in SSc patients were associated with ILD progression and implicated in the pathogenesis of SSc-ILD (36). Circulating Collagen-I-producing fibrocytes have also been coupled with a growing repertoire of human diseases including renal fibrosis (38), cirrhosis (39), nephrogenic systemic fibrosis (40), pulmonary fibrosis (41–43), and asthma (44).
It has been reported that the fibronectin splice variant- fibronectin extra domain A (FnEDA) is an endogenous TLR4 ligand. Importantly, elevated levels of FnEDA were demonstrated in the serum and skin lesions from SSc patients and in mouse model of cutaneous fibrosis (45). In chronic inflammatory diseases and during the healing phase of acute inflammatory reactions, the alternatively activated M(IL-4) macrophages showed significantly upregulated levels of fibronectin that suggest an active role of fibronectin+ macrophages in the ECM deposition and tissue remodeling (46). Furthermore, the alveolar macrophages from lungs of SSc-ILD patients displayed elevated levels of fibronectin (47). These reports are in line with our results, which presented the elevated fibronectin levels in circulating CD14+ monocytes and CD14+ pulmonary macrophages in SSc patients and highlighted the capability of CD14+ monocytes to acquire a profibrotic phenotype. Of note, monocyte-to-macrophage differentiation has been linked with the increase production of fibronectin (48). We, therefore, concluded that tissue-infiltrating CD14+ monocytes/macrophages can be considered as ECM producers in the pathogenesis of fibrosis in SSc. All these data point to fibronectin as a potential target in therapeutic strategy in SSc.
Monocyte infiltration into the tissue affects the initiation of fibrotic processes in the skin and internal organs in SSc (49–51). Our data clearly presented infiltrating CD14+ monocytes in the collagen-rich area in SSc-ILD lungs, myocardium of SSc patients with inflammatory dilated cardiomyopathy (iDCM) and in SSc skin (27). Increased numbers of spindle-shaped CD14+CD34+collagen-I+ cells were found in the lungs of SSc-ILD patients (36). The bone marrow origin of fibroblasts or myofibroblasts in different tissues under homeostasis and disease condition has been debatable for many years now. In vivo data, with the use of cutaneous wound mouse model, confirmed that at least a part (15%-20%) of the spindle-shaped dermal fibroblasts were bone marrow origin (52). In contrast, another report suggested that bone marrow-derived progenitors contributed to the inflammatory cell pool infiltrating the wound area; however, they did not differentiate into dermal (myo)fibroblasts at the wound site (53, 54). In the bleomycin-induced skin fibrosis model, a significant number of CD45-positive collagen-producing cells of bone marrow origin contributed to collagen production during dermal fibrogenesis but not under homeostasis, indicating bone marrow originated cells as a cell source for (myo)fibroblasts under disease condition (55). Similarly, in bleomycin-induced lung fibrosis mouse model bone marrow-derived progenitors gave rise to collagen-producing cells but not to αSMA+ myofiboblasts in the inflamed/fibrotic lungs (56, 57). Oppositely, in the renal fibrosis the inflammatory macrophages, characterized by alternatively activated macrophage markers, acquired fibroblast phenotype by collagen I and αSMA expression and actively contributed to the fibrogenesis (58).
Nevertheless, the transition of bone marrow-derived monocytes or macrophages (mainly alternatively activated macrophages) into functional myofibroblasts has been mediated by canonical SMAD2/3-dependent or non-canonical TAK-1-dependent TGFβ signal (24, 59–61). Accordingly, our results confirmed that both canonical and non-canonical TGFβ pathways were important to obtain the fibroblast-like phenotype of circulating CD14+ monocytes, indicating the broad treatment options.
The pathophysiology of SSc is closely related to the activated TGFβ-dependent pathway. The levels of a latent, not-active form of TGFβ is similar in SSc and healthy serum samples. Active TGFβ serum levels were significantly higher in SSc, mainly in dcSSc, patients and correlated with clinical manifestations (digital ulcers, lung fibrosis, positive antitopoisomerase I and higher modified Rodnan score) (62). These results indicate TGFβ as a potential marker of fibrotic and vascular involvement in SSc on one side, and on the other side, it reflects an altered microenvironment, which may predispose circulating monocytes towards an activated and profibrotic phenotype. Accordingly, we used this cytokine, among others, to activate circulating monocytes towards a profibrotic phenotype. As expected, CD14+ monocytes stimulated with TGFβ, IL-4, IL-10 and IL-13 or co-cultured in 2D and 3D model with human dermal fibroblasts were able to adopt a functional myofibroblast-like phenotype that may indicate these cells as one of the cellular sources of profibrotic cells upon their entrance into the tissues. The previous report indeed showed that GM-CSF, IL-4 and endothelin-1 alone or in combination induced myofibroblast-like differentiation of circulating SSc and healthy monocytes (63). Additionally, the profibrotic phenotype of circulating CD14+ monocytes from SSc-ILD patients was confirmed by the expression of CD163 and boosted secretion of CCL18 and IL-10 in response to pro-inflammatory stimuli (36). However, as in our stimulation conditions, monocytes from SSc patients showed no differences in acquiring more pronounced profibrotic phenotype compared to monocytes from healthy donors. Therefore, we assume that under inflammatory conditions (i.e. in SSc) more circulating already activated monocytes are recruited to the injury site, where they play a profibrotic role, either by the transition into fibroblast-like cells or by the production of profibrotic stimuli. In line with this hypothesis, Bhandari et al. demonstrated that soluble factors in the local milieu are crucial for pro-fibrotic activation of SSc macrophages (measured as secretion of CCL2, IL-6, TGFβ), arisen from circulating monocytes either stimulated with SSc sera or conditioned media from indirect co-culture with dermal SSc fibroblasts (64). These results point to monocytes and monocyte-derived macrophages as probable key players in fibrogenesis in SSc, suggesting targeted cell therapeutic options as feasible and favorable in SSc.
Besides a known and active role of IL-6 and TGF-β signalling in the pathogenesis of SSc, there is increasing evidence that also Th-2 cytokines: IL-4 and IL-13, are involved in the pathology of SSc (65, 66). Both cytokines relate to profibrotic responses, including connection with increased expression of novel myofibroblast marker: periostin, a matricellular protein important in fibrogenesis (67). Based on our data, the combination of TGFβ, IL-4, IL-10 and IL-13 was sufficient to obtain a fibroblast-like phenotype by circulating CD14+ monocytes, indicating that IL-4/IL-13 targeted therapy might be an attractive option against fibrogenesis in SSc (68). Indeed, a randomized, double-blind, placebo-controlled, 24-week, phase II, proof-of-concept study (trial registration number: NCT02921971) with romilkimab (a bispecific immunoglobulin-G4 antibody that binds and neutralizes IL-4/IL-13; SAR156597) in early dcSSc patients revealed a beneficial effect of this treatment on skin disease changes, i.e. statistically significant decrease in mRSS from baseline to week 24 versus placebo (69).
Given the alerted activation status of circulating CD14+ monocytes and their ability to accumulate in the affected organs in SSc, we believe that further research should focus on finding even more targeted therapies, which may invert the activation status of monocytes in the circulation.
Data Availability Statement
The RNAseq datasets for this study can be found in the Gene Expression Omnibus under the accession numbers: GSE157840, GSE128169.
Ethics Statement
Human blood samples and skin biopsies collection were approved by the local ethics committee of the Canton Zurich (KEK-ZH 515, PB-2016-02014, KEK-Nr 2018-01873). All study subjects provided written informed consent. The experiments with re-use of human material were approved by Swissethics (KEK-Nr 2019-00058, KEK-Nr 2018-01873) and were performed in conformity with the principles outlined in the Declaration of Helsinki. The animal study was reviewed and approved by Commission on Animal Experimentation of the Canton Zurich.
Author Contributions
GK directed the project and obtained funding. MR, AH, IK, SJ, and GK designed, analyzed, and interpreted experiments. MR, AH, IK, JS, and VM performed the experiments. CF-B and KK provided heart and lungs biopsies. MR, GK, and PB wrote the manuscript. OD, CF-B, ME contributed to final corrections of the drafted manuscript. All authors contributed to the article and approved the submitted version.
Funding
GK acknowledges support from the Swiss National Science Foundation (310030_152876/1; 310030_175663), Swiss Life Foundation and Swiss Heart Foundation.
Conflict of Interest
OD had consultancy relationship and/or has received research funding from Actelion, Acceleron Pharma, AnaMar, Bayer, Baecon Discovery, Blade Therapeutics, Boehringer, CSL Behring, ChemomAb, Curzion Pharmaceuticals, Ergonex, Galapagos NV, GSK, Glenmark Pharmaceuticals, Inventiva, Italfarmaco, iQvia, medac, Medscape, Mitsubishi Tanabe Pharma, MSD, Roche, Sanofi, UCBin the area of potential treatments of scleroderma and its complications. In addition, OD has a patent mir-29 for the treatment of systemic sclerosis issued (US8247389, EP2331143).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Maria Comazzi-Fornallaz and Dr. Magdalena Diaz-Ovalle for performing immunohistochemistry, Functional Genomic Center University of Zurich for RNA sequencing and The Center for Microscopy and Image Analysis of University of Zurich for image scanning.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.642891/full#supplementary-material
References
1. Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic Sclerosis. Nat Rev Dis Primers (2015) 1:15002. doi: 10.1038/nrdp.2015.2
2. Mayes MD, Lacey JV Jr, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, Incidence, Survival, and Disease Characteristics of Systemic Sclerosis in a Large US Population. Arthritis Rheum (2003) 48(8):2246–55. doi: 10.1002/art.11073
3. Elhai M, Meune C, Boubaya M, Avouac J, Hachulla E, Balbir-Gurman A, et al. Mapping and Predicting Mortality From Systemic Sclerosis. Ann Rheum Dis (2017) 76(11):1897–905. doi: 10.1136/annrheumdis-2017-211448
4. Bhattacharyya S, Wei J, Varga J. Understanding Fibrosis in Systemic Sclerosis: Shifting Paradigms, Emerging Opportunities. Nat Rev Rheumatol (2011) 8(1):42–54. doi: 10.1038/nrrheum.2011.149
5. Garrett SM, Baker Frost D, Feghali-Bostwick C. The Mighty Fibroblast and Its Utility in Scleroderma Research. J Scleroderma Relat Disord (2017) 2(2):69–134. doi: 10.5301/jsrd.5000240
6. van Caam A, Vonk M, van den Hoogen F, van Lent P, van der Kraan P. Unraveling SSc Pathophysiology; The Myofibroblast. Front Immunol (2018) 9:2452. doi: 10.3389/fimmu.2018.02452
7. Pankov R, Yamada KM. Fibronectin at a Glance. J Cell Sci (2002) 115(Pt 20):3861–3. doi: 10.1242/jcs.00059
8. Xu WD, Leroy EC, Smith EA. Fibronectin Release by Systemic Sclerosis and Normal Dermal Fibroblasts in Response to TGF-Beta. J Rheumatol (1991) 18(2):241–6.
9. Cooper SM, Keyser AJ, Beaulieu AD, Ruoslahti E, Nimni ME, Quismorio FP Jr. Increase in Fibronectin in the Deep Dermis of Involved Skin in Progressive Systemic Sclerosis. Arthritis Rheum (1979) 22(9):983–7. doi: 10.1002/art.1780220906
10. Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ. Diverse Origins of the Myofibroblast-Implications for Kidney Fibrosis. Nat Rev Nephrol (2015) 11(4):233–44. doi: 10.1038/nrneph.2014.246
11. Reilkoff RA, Bucala R, Herzog EL. Fibrocytes: Emerging Effector Cells in Chronic Inflammation. Nat Rev Immunol (2011) 11(6):427–35. doi: 10.1038/nri2990
12. Chang FC, Chou YH, Chen YT, Lin SL. Novel Insights Into Pericyte-Myofibroblast Transition and Therapeutic Targets in Renal Fibrosis. J Formos Med Assoc (2012) 111(11):589–98. doi: 10.1016/j.jfma.2012.09.008
13. Wilson CL, Stephenson SE, Higuero JP, Feghali-Bostwick C, Hung CF, Schnapp LM. Characterization of Human PDGFR-Beta-Positive Pericytes From IPF and Non-IPF Lungs. Am J Physiol Lung Cell Mol Physiol (2018) 315(6):L991–L1002. doi: 10.1152/ajplung.00289.2018
14. Hill C, Jones MG, Davies DE, Wang Y. Epithelial-Mesenchymal Transition Contributes to Pulmonary Fibrosis via Aberrant Epithelial/Fibroblastic Cross-Talk. J Lung Health Dis (2019) 3(2):31–5. doi: 10.29245/2689-999X/2019/2.1149
15. Jimenez SA, Piera-Velazquez S. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Systemic Sclerosis-Associated Pulmonary Fibrosis and Pulmonary Arterial Hypertension. Myth or Reality? Matrix Biol (2016) 51:26–36. doi: 10.1016/j.matbio.2016.01.012
16. Ayers NB, Sun CM, Chen SY. Transforming Growth Factor-Beta Signaling in Systemic Sclerosis. J BioMed Res (2018) 32(1):3–12. doi: 10.7555/JBR.31.20170034
17. Farina G, Lafyatis D, Lemaire R, Lafyatis R. A Four-Gene Biomarker Predicts Skin Disease in Patients With Diffuse Cutaneous Systemic Sclerosis. Arthritis Rheum (2010) 62(2):580–8. doi: 10.1002/art.27220
18. Walton KL, Johnson KE, Harrison CA. Targeting TGF-Beta Mediated SMAD Signaling for the Prevention of Fibrosis. Front Pharmacol (2017) 8:461. doi: 10.3389/fphar.2017.00461
19. Leask A. Scar Wars: Is TGFbeta the Phantom Menace in Scleroderma? Arthritis Res Ther (2006) 8(4):213. doi: 10.1186/ar1976
20. Varga J, Pasche B. Transforming Growth Factor Beta as a Therapeutic Target in Systemic Sclerosis. Nat Rev Rheumatol (2009) 5(4):200–6. doi: 10.1038/nrrheum.2009.26
21. Shi C, Pamer EG. Monocyte Recruitment During Infection and Inflammation. Nat Rev Immunol (2011) 11(11):762–74. doi: 10.1038/nri3070
22. Kania G, Rudnik M, Distler O. Involvement of the Myeloid Cell Compartment in Fibrogenesis and Systemic Sclerosis. Nat Rev Rheumatol (2019) 15(5):288–302. doi: 10.1038/s41584-019-0212-z
23. Blyszczuk P, Germano D, Stein S, Moch H, Matter CM, Beck-Schimmer B, et al. Profibrotic Potential of Prominin-1+ Epithelial Progenitor Cells in Pulmonary Fibrosis. Respir Res (2011) 12:126. doi: 10.1186/1465-9921-12-126
24. Kania G, Blyszczuk P, Stein S, Valaperti A, Germano D, Dirnhofer S, et al. Heart-Infiltrating Prominin-1+/CD133+ Progenitor Cells Represent the Cellular Source of Transforming Growth Factor Beta-Mediated Cardiac Fibrosis in Experimental Autoimmune Myocarditis. Circ Res (2009) 105(5):462–70. doi: 10.1161/CIRCRESAHA.109.196287
25. LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA Jr, et al. Scleroderma (Systemic Sclerosis): Classification, Subsets and Pathogenesis. J Rheumatol (1988) 15(2):202–5.
26. Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Trejo Bittar H, et al. Single-Cell Analysis Reveals Fibroblast Heterogeneity and Myofibroblasts in Systemic Sclerosis-Associated Interstitial Lung Disease. Ann Rheum Dis (2019) 78(10):1379–87. doi: 10.1136/annrheumdis-2018-214865
27. Rudnik M, Rolski F, Jordan S, Mertelj T, Stellato M, Distler O, et al. CD52 Regulates Monocyte Adhesion and Interferon Type I Signalling in Systemic Sclerosis Patients. Arthritis Rheumatol (2021). doi: 10.1002/art.41737
28. Lun ATL, Riesenfeld S, Andrews T, Dao TP, Gomes T, participants in the 1st Human Cell Atlas J, et al. EmptyDrops: Distinguishing Cells From Empty Droplets in Droplet-Based Single-Cell RNA Sequencing Data. Genome Biol (2019) 20(1):63. doi: 10.1186/s13059-019-1662-y
29. Griffiths JA, Richard AC, Bach K, Lun ATL, Marioni JC. Detection and Removal of Barcode Swapping in Single-Cell RNA-Seq Data. Nat Commun (2018) 9(1):2667. doi: 10.1038/s41467-018-05083-x
30. Hafemeister C, Satija R. Normalization and Variance Stabilization of Single-Cell RNA-Seq Data Using Regularized Negative Binomial Regression. Genome Biol (2019) 20(1):296. doi: 10.1186/s13059-019-1874-1
31. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating Single-Cell Transcriptomic Data Across Different Conditions, Technologies, and Species. Nat Biotechnol (2018) 36(5):411–20. doi: 10.1038/nbt.4096
32. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive Integration of Single-Cell Data. Cell (2019) 177(7):1888–902 e21. doi: 10.1016/j.cell.2019.05.031
33. Schniering J, Benesova M, Brunner M, Haller S, Cohrs S, Frauenfelder T, et al. Visualisation of Interstitial Lung Disease by Molecular Imaging of Integrin Alphavbeta3 and Somatostatin Receptor 2. Ann Rheum Dis (2019) 78(2):218–27. doi: 10.1136/annrheumdis-2018-214322
34. Asano Y. The Pathogenesis of Systemic Sclerosis: An Understanding Based on a Common Pathologic Cascade Across Multiple Organs and Additional Organ-Specific Pathologies. J Clin Med (2020) 9(9):2687. doi: 10.3390/jcm9092687
35. Brunasso AM, Massone C. Update on the Pathogenesis of Scleroderma: Focus on Circulating Progenitor Cells. F1000Res (2016) 5:F1000 Faculty Rev-723. doi: 10.12688/f1000research.7986.1
36. Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, et al. Circulating Monocytes From Systemic Sclerosis Patients With Interstitial Lung Disease Show an Enhanced Profibrotic Phenotype. Lab Invest (2010) 90(6):812–23. doi: 10.1038/labinvest.2010.73
37. Scott MKD, Quinn K, Li Q, Carroll R, Warsinske H, Vallania F, et al. Increased Monocyte Count as a Cellular Biomarker for Poor Outcomes in Fibrotic Diseases: A Retrospective, Multicentre Cohort Study. Lancet Respir Med (2019) 7(6):497–508. doi: 10.1016/S2213-2600(18)30508-3
38. Sakai N, Wada T, Yokoyama H, Lipp M, Ueha S, Matsushima K, et al. Secondary Lymphoid Tissue Chemokine (SLC/CCL21)/CCR7 Signaling Regulates Fibrocytes in Renal Fibrosis. Proc Natl Acad Sci USA (2006) 103(38):14098–103. doi: 10.1073/pnas.0511200103
39. Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, et al. Bone Marrow-Derived Fibrocytes Participate in Pathogenesis of Liver Fibrosis. J Hepatol (2006) 45(3):429–38. doi: 10.1016/j.jhep.2006.04.014
40. Bucala R. Circulating Fibrocytes: Cellular Basis for NSF. J Am Coll Radiol (2008) 5(1):36–9. doi: 10.1016/j.jacr.2007.08.016
41. Andersson-Sjoland A, de Alba CG, Nihlberg K, Becerril C, Ramirez R, Pardo A, et al. Fibrocytes Are a Potential Source of Lung Fibroblasts in Idiopathic Pulmonary Fibrosis. Int J Biochem Cell Biol (2008) 40(10):2129–40. doi: 10.1016/j.biocel.2008.02.012
42. Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating Peripheral Blood Fibrocytes in Human Fibrotic Interstitial Lung Disease. Biochem Biophys Res Commun (2007) 353(1):104–8. doi: 10.1016/j.bbrc.2006.11.149
43. Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, et al. Circulating Fibrocytes Are an Indicator of Poor Prognosis in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2009) 179(7):588–94. doi: 10.1164/rccm.200810-1534OC
44. Wang CH, Huang CD, Lin HC, Lee KY, Lin SM, Liu CY, et al. Increased Circulating Fibrocytes in Asthma With Chronic Airflow Obstruction. Am J Respir Crit Care Med (2008) 178(6):583–91. doi: 10.1164/rccm.200710-1557OC
45. Bhattacharyya S, Tamaki Z, Wang W, Hinchcliff M, Hoover P, Getsios S, et al. FibronectinEDA Promotes Chronic Cutaneous Fibrosis Through Toll-Like Receptor Signaling. Sci Transl Med (2014) 6(232):232ra50. doi: 10.1126/scitranslmed.3008264
46. Gratchev A, Guillot P, Hakiy N, Politz O, Orfanos CE, Schledzewski K, et al. Alternatively Activated Macrophages Differentially Express Fibronectin and Its Splice Variants and the Extracellular Matrix Protein betaIG-H3. Scand J Immunol (2001) 53(4):386–92. doi: 10.1046/j.1365-3083.2001.00885.x
47. Kinsella MB, Smith EA, Miller KS, LeRoy EC, Silver RM. Spontaneous Production of Fibronectin by Alveolar Macrophages in Patients With Scleroderma. Arthritis Rheum (1989) 32(5):577–83. doi: 10.1002/anr.1780320511
48. Sudhakaran PR, Radhika A, Jacob SS. Monocyte Macrophage Differentiation In Vitro: Fibronectin-Dependent Upregulation of Certain Macrophage-Specific Activities. Glycoconj J (2007) 24(1):49–55. doi: 10.1007/s10719-006-9011-2
49. Gu YS, Kong J, Cheema GS, Keen CL, Wick G, Gershwin ME. The Immunobiology of Systemic Sclerosis. Semin Arthritis Rheum (2008) 38(2):132–60. doi: 10.1016/j.semarthrit.2007.10.010
50. Bosello S, Angelucci C, Lama G, Alivernini S, Proietti G, Tolusso B, et al. Characterization of Inflammatory Cell Infiltrate of Scleroderma Skin: B Cells and Skin Score Progression. Arthritis Res Ther (2018) 20(1):75. doi: 10.1186/s13075-018-1569-0
51. Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, et al. Systemic and Cell Type-Specific Gene Expression Patterns in Scleroderma Skin. Proc Natl Acad Sci USA (2003) 100(21):12319–24. doi: 10.1073/pnas.1635114100
52. Fathke C, Wilson L, Hutter J, Kapoor V, Smith A, Hocking A, et al. Contribution of Bone Marrow-Derived Cells to Skin: Collagen Deposition and Wound Repair. Stem Cells (2004) 22(5):812–22. doi: 10.1634/stemcells.22-5-812
53. Barisic-Dujmovic T, Boban I, Clark SH. Fibroblasts/myofibroblasts That Participate in Cutaneous Wound Healing Are Not Derived From Circulating Progenitor Cells. J Cell Physiol (2010) 222(3):703–12. doi: 10.1002/jcp.21997
54. Ishii G, Sangai T, Sugiyama K, Ito T, Hasebe T, Endoh Y, et al. In Vivo Characterization of Bone Marrow-Derived Fibroblasts Recruited Into Fibrotic Lesions. Stem Cells (2005) 23(5):699–706. doi: 10.1634/stemcells.2004-0183
55. Higashiyama R, Nakao S, Shibusawa Y, Ishikawa O, Moro T, Mikami K, et al. Differential Contribution of Dermal Resident and Bone Marrow-Derived Cells to Collagen Production During Wound Healing and Fibrogenesis in Mice. J Invest Dermatol (2011) 131(2):529–36. doi: 10.1038/jid.2010.314
56. Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone Marrow-Derived Progenitor Cells in Pulmonary Fibrosis. J Clin Invest (2004) 113(2):243–52. doi: 10.1172/JCI200418847
57. Habiel DM, Hogaboam CM. Heterogeneity of Fibroblasts and Myofibroblasts in Pulmonary Fibrosis. Curr Pathobiol Rep (2017) 5(2):101–10. doi: 10.1007/s40139-017-0134-x
58. Meng XM, Wang S, Huang XR, Yang C, Xiao J, Zhang Y, et al. Inflammatory Macrophages can Transdifferentiate Into Myofibroblasts During Renal Fibrosis. Cell Death Dis (2016) 7(12):e2495. doi: 10.1038/cddis.2016.402
59. Blyszczuk P, Muller-Edenborn B, Valenta T, Osto E, Stellato M, Behnke S, et al. Transforming Growth Factor-Beta-Dependent Wnt Secretion Controls Myofibroblast Formation and Myocardial Fibrosis Progression in Experimental Autoimmune Myocarditis. Eur Heart J (2017) 38(18):1413–25. doi: 10.1093/eurheartj/ehw116
60. Wang S, Meng XM, Ng YY, Ma FY, Zhou S, Zhang Y, et al. TGF-Beta/Smad3 Signalling Regulates the Transition of Bone Marrow-Derived Macrophages Into Myofibroblasts During Tissue Fibrosis. Oncotarget (2016) 7(8):8809–22. doi: 10.18632/oncotarget.6604
61. Lu J, Liu Q, Wang L, Tu W, Chu H, Ding W, et al. Increased Expression of Latent TGF-Beta-Binding Protein 4 Affects the Fibrotic Process in Scleroderma by TGF-Beta/SMAD Signaling. Lab Invest (2017) 97(9):1121. doi: 10.1038/labinvest.2017.43
62. Dziadzio M, Smith RE, Abraham DJ, Black CM, Denton CP. Circulating Levels of Active Transforming Growth Factor Beta1 Are Reduced in Diffuse Cutaneous Systemic Sclerosis and Correlate Inversely With the Modified Rodnan Skin Score. Rheumatol (Oxford) (2005) 44(12):1518–24. doi: 10.1093/rheumatology/kei088
63. Binai N, O’Reilly S, Griffiths B, van Laar JM, Hugle T. Differentiation Potential of CD14+ Monocytes Into Myofibroblasts in Patients With Systemic Sclerosis. PloS One (2012) 7(3):e33508. doi: 10.1371/journal.pone.0033508
64. Bhandari R, Ball MS, Martyanov V, Popovich D, Schaafsma E, Han S, et al. Profibrotic Activation of Human Macrophages in Systemic Sclerosis. Arthritis Rheumatol (2020) 72(7):1160–9. doi: 10.1002/art.41243
65. Jakubzick C, Choi ES, Joshi BH, Keane MP, Kunkel SL, Puri RK, et al. Therapeutic Attenuation of Pulmonary Fibrosis via Targeting of IL-4- and IL-13-Responsive Cells. J Immunol (2003) 171(5):2684–93. doi: 10.4049/jimmunol.171.5.2684
66. Hasegawa M, Fujimoto M, Kikuchi K, Takehara K. Elevated Serum Levels of Interleukin 4 (IL-4), IL-10, and IL-13 in Patients With Systemic Sclerosis. J Rheumatol (1997) 24(2):328–32. doi: 10.1016/0923-1811(96)89424-2
67. Okamoto M, Izuhara K, Ohta S, Ono J, Hoshino T. Ability of Periostin as a New Biomarker of Idiopathic Pulmonary Fibrosis. Adv Exp Med Biol (2019) 1132:79–87. doi: 10.1007/978-981-13-6657-4_9
68. Gasparini G, Cozzani E, Parodi A. Interleukin-4 and Interleukin-13 as Possible Therapeutic Targets in Systemic Sclerosis. Cytokine (2020) 125:154799. doi: 10.1016/j.cyto.2019.154799
69. Allanore Y, Wung P, Soubrane C, Esperet C, Marrache F, Bejuit R, et al. A Randomised, Double-Blind, Placebo-Controlled, 24-Week, Phase II, Proof-Of-Concept Study of Romilkimab (SAR156597) in Early Diffuse Cutaneous Systemic Sclerosis. Ann Rheum Dis (2020) 79(12):1600–7. doi: 10.1136/annrheumdis-2020-218447
Keywords: systemic sclerosis, CD14+ monocytes, CD14+ macrophages, fibrosis, fibronectin, TGF-β
Citation: Rudnik M, Hukara A, Kocherova I, Jordan S, Schniering J, Milleret V, Ehrbar M, Klingel K, Feghali-Bostwick C, Distler O, Błyszczuk P and Kania G (2021) Elevated Fibronectin Levels in Profibrotic CD14+ Monocytes and CD14+ Macrophages in Systemic Sclerosis. Front. Immunol. 12:642891. doi: 10.3389/fimmu.2021.642891
Received: 17 December 2020; Accepted: 21 July 2021;
Published: 24 August 2021.
Edited by:
Carmelo Carmona-Rivera, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), United StatesReviewed by:
José Jiram Torres-Ruiz, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), MexicoEduardo Patino Martinez, National Institutes of Health Clinical Center (NIH), United States
Yudong Liu, Peking University People’s Hospital, China
Copyright © 2021 Rudnik, Hukara, Kocherova, Jordan, Schniering, Milleret, Ehrbar, Klingel, Feghali-Bostwick, Distler, Błyszczuk and Kania. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela Kania, Z2FicmllbGEua2FuaWFAdXpoLmNo