 Atefeh Ghorbani
Atefeh Ghorbani Emma M. Quinlan
Emma M. Quinlan Mani Larijani
Mani Larijani
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 May 2021
Sec. Comparative Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.642343
This article is part of the Research Topic Evolution and Comparative Immunology of Immune Systems in Marine Organisms View all 14 articles
The immune system is unique among all biological sub-systems in its usage of DNA-editing enzymes to introduce targeted gene mutations and double-strand DNA breaks to diversify antigen receptor genes and combat viral infections. These processes, initiated by specific DNA-editing enzymes, often result in mistargeted induction of genome lesions that initiate and drive cancers. Like other molecules involved in human health and disease, the DNA-editing enzymes of the immune system have been intensively studied in humans and mice, with little attention paid (< 1% of published studies) to the same enzymes in evolutionarily distant species. Here, we present a systematic review of the literature on the characterization of one such DNA-editing enzyme, activation-induced cytidine deaminase (AID), from an evolutionary comparative perspective. The central thesis of this review is that although the evolutionary comparative approach represents a minuscule fraction of published works on this and other DNA-editing enzymes, this approach has made significant impacts across the fields of structural biology, immunology, and cancer research. Using AID as an example, we highlight the value of the evolutionary comparative approach in discoveries already made, and in the context of emerging directions in immunology and protein engineering. We introduce the concept of 5-dimensional (5D) description of protein structures, a more nuanced view of a structure that is made possible by evolutionary comparative studies. In this higher dimensional view of a protein’s structure, the classical 3-dimensional (3D) structure is integrated in the context of real-time conformations and evolutionary time shifts (4th dimension) and the relevance of these dynamics to its biological function (5th dimension).
The adaptive immune system in its classical mammalian form first appeared in the common ancestor of all jawed vertebrates (gnathostomes), with the cartilaginous fish being the first extant animals to evolve somatically diversified lymphocyte (B and T cell) receptors (BCR or antibodies, and TCR, respectively) (1). However, further study of the earlier-evolved jawless vertebrates revealed that these animals too were capable of adaptive immunity. Instead of B and T cell lymphocytes, their respective humoral and cellular adaptive immune responses are mediated by lymphocyte-like cells with Variable Lymphocyte Receptors (VLRs). Interestingly, these VLRs also appeared to be somatically diversified, highlighting the importance of lymphocyte receptor diversification in the adaptive immune response (2).
Lymphocyte receptors are diversified via purposeful induction of DNA damage in the form of recombination and gene mutation (3). Unlike other genes, in jawed vertebrates, the genes encoding the adaptive immune antigen receptors are segmented. To encode a functional receptor, the variable (V), diversity (D; only in the case of the heavy chain), and joining (J) fragments are assembled by V(D)J recombination, a site-specific recombination process that is lymphocyte-specific and mediated by the recombination-activating gene products 1 and 2 (RAG1/2) co-enzyme complex (4–7). Following binding to recombination signal sequences (RSS) at the ends of V, D, or J gene segments, the RAG1/2 complex introduces double strand breaks (DSBs) at the RSS-coding juncture. Non-homologous end joining (NHEJ) is initiated to repair the DSBs, resulting in ligation and forming the V(D)J-encoding gene.
This primary diversification process that occurs during B and T cell development in the bone marrow and thymus respectively, gives rise to the initial antibody (BCR) or TCR repertoire in B and T lymphocytes. In the case of B lymphocytes, further secondary diversification rounds of the BCR are initiated when a mature peripheral B lymphocytes bind its cognate antigen (8). As a result of secondary diversification, activated B cells, expressing low affinity IgM, give rise to B cells secreting high affinity antibodies of switched isotopes including IgA, IgG and IgE. Secondary antibody diversification in jawed vertebrates includes two processes: affinity maturation (AM) and isotype switching (IS), driven by somatic hypermutation (SHM) and Class Switch Recombination (CSR), respectively. SHM in the antibody V region genes, followed by cellular selection leads to antibodies of higher affinity to the cognate antigen. CSR changes the class of antibody from IgM into other isotypes (i.e., IgA, IgG, or IgE). CSR is mediated by DSBs in the switch (S) regions flanking the heavy chain constant genes (CH) which initiate a NHEJ event resulting in the replacement of CHμ with other CH isotypes, changing the antibody’s effector function (9–11). The outcome of secondary antibody diversification is the production of more effective isotypes of antibodies which also have as much as 1000-fold higher affinity for the antigen. The mutations and DSBs that underlie SHM and CSR are both caused by the enzyme activation-induced cytidine deaminase (AID) (12, 13). AID is a member of the AID/APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) family of cytidine deaminase enzymes that carry out cytidine (dC) to uridine (dU) conversion in single stranded DNA or cytidine (C) to uridine (U) conversion in RNA (14, 15).
The AID/APOBEC family includes 11 family members in humans: AID, APOBEC1, APOBEC2, APOBEC3 (A-H, excluding E), and APOBEC4. APOBEC4 and related enzymes have been found as early as Cnidarian invertebrates but are frequently absent in actinopterygians and present again in all mammals (14, 16) (Figure 1A). The APOBEC3 sub-branch emerged in mammals followed by rapid expansion and diversification in primates (16, 64) (Figure 1A). APOBEC3s function in immune response by acting as restriction factors against viruses. They do so through mediating mutagenesis of viral genomes, or interference with the reverse transcription and integration of the viral DNA (65–68). In addition the adenosine deaminases acting on double-stranded RNA (ADARs) are enzymes that mediate cellular mRNA processing through Adenosine (A) to Inosine (I) conversion; however, they have also been demonstrated to mutate viral RNA and carry out a range of cytoplasmic innate anti-viral functions (69–73).
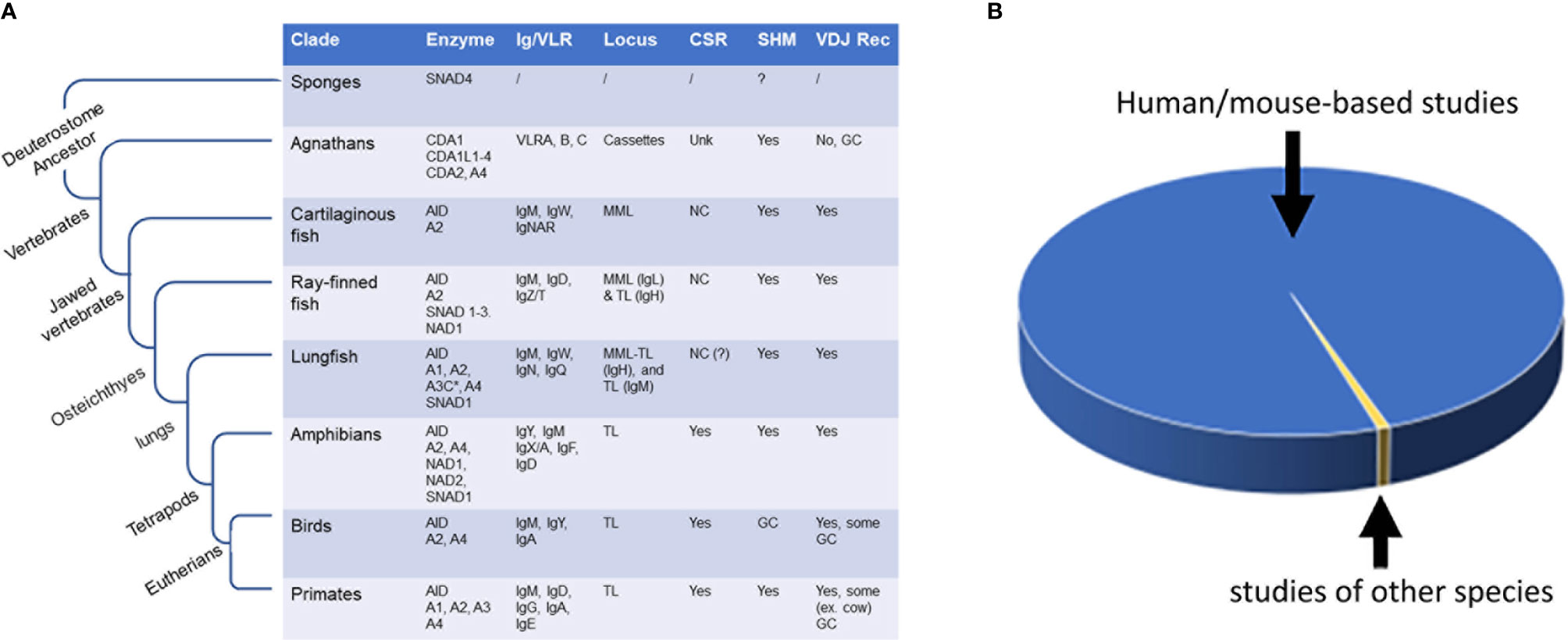
Figure 1 Evolutionary and evolutionary comparative studies of AID/APOBEC and AID/APOBEC-like enzymes. (A) The emergence of AID/APOBEC and AID/APOBEC-like enzymes during evolution and the evolution of antibody genes (Ig/VLR), and occurrence of secondary antibody diversification (i.e., antibody maturation; SHM and CSR) and primary antibody diversification (i.e., V[D]J recombination) within and outside of vertebrate class. (B) Comparison of number of reports examining AID in human and mouse (blue; > 99%) and studies done on other species (yellow; < 0.7%). The total number of published peer-reviewed studies on AID as measured by AID being a keyword in title/abstract (pubmed/scopus search) is 2368. Of these, 49 discussed the topic of AID in species other than human or mice (15–63) and 17 (0.7%) presented experimental data examining evolutionary divergent AID orthologs (17, 20, 21 26, 30–33, 39, 42, 45, 47, 52, 56, 61–63). NC, noncanonical; MML, multiple mini loci; TL, translocon; GC, Gene conversion (or Gene conversion-like); unk, unknown.
In contrast to jawed vertebrates, the jawless vertebrate lamprey lacks a classical antibody and TCR, and their antibody structure is grossly different, both at the genetic and protein levels. Rather than the classical V(D)J recombination-based Ig system of jawed vertebrates, lampreys employ a presumed gene conversion-like process to assemble 8-10 variable leucine rich repeat motifs in between conserved genes that encode N- and C- terminal ends of their antibody protein. Although the jawless vertebrates lack the classical RAG and AID/APOBEC enzymes, the proposed lymphocytes antigen receptor diversification process is thought to be mediated by AID/APOBEC-like cytidine deaminase enzymes denoted CDA (cytosine deaminase) of which there are two sub-types, CDA1 and CDA2, the former group appearing to have multiple enzyme members (2, 17, 74–78).
Though essential for immunity, the DNA-editing enzymes used to diversify antigen receptors also mediate significant off-target genome damage. There are several mechanisms in place to ensure targeting of RAG1/2 to the Ig and T cell receptor (TCR) genes. These mechanisms include precursor lymphocyte-restricted RAG expression, CTCF-binding elements flanking paired RSS sequences, active chromatin markers, active transcription, and stalled RNA polymerase II (79–82). Despite these regulatory mechanisms, RAG is known to cause chromosomal translocation, deletion, and insertions leading to different types of T cell and B cell lymphoid malignancies, and many of these off-target RAG cleavage events are believed to occur through recognition of RSS-like sequences at non-Ig loci, termed cryptic RSSs (83, 84). It has also been shown that the excised signal circle can play the role of RSS and cause RAG-mediated DSBs at a cryptic RSSs in a process termed “cut-and-run” (85). RAG-mediated chromosomal translocations, presumably as a form of mis-targeting of V(D)J recombination are implicated in the etiology of chronic myeloid leukemia (CML), leukemias and lymphomas (79, 86, 87).
Mis-targeted activity of AID also causes genome instability and mutations in B cells (88). For example, in patients with chronic myeloid leukemia (CML), AID-mediated hypermutation of tumor repressor and DNA repair genes have been associated with progression into B lymphoid blast crisis and Imatinib-resistance phenotype (89). In diffuse large B cell lymphomas (DLBCL), somatic hypermutation off-targeting has been reported in proto-oncogenes (90). The IGH/MYC translocation that is signature of Burkitt lymphoma (BL) has a frequency that is correlated with AID activity level (91). AID-induced hypermutations have also been observed in chronic lymphoid leukemia (CLL) (92). There has also been evidence of AID-mediated carcinogenesis in germinal center (GC) B cells as the result of Epstein-Barr virus (EBV)-induced AID expression (93). Interestingly, under strong inflammatory stimuli, the premature expression of AID during B cell development creates an opportunity for cooperation between RAG and AID to drive the clonal evolution of childhood B cell acute lymphoblastic leukemia (B-ALL) (94). The role of AID in tumorigenesis has been conclusively established in several mouse models. In mouse models of IgH/MYC translocation-driven BL, AID has been shown to be directly responsible for this tumor-driving chromosomal translocation (95), and AID transgenic mice are also prone to AID-driven tumorigenesis (96).
In addition to AID, its APOBEC relatives, the APOBEC3 sub-branch of enzymes (A3A, A3B, A3H), which have antiviral properties, are also a significant source of genome damage and mutations implicated in many types of cancers, such as breast, ovarian, and lung cancers, as the driving mutation and cancer progression associated signatures (68, 97–108). Their mutagenic activity in tumors is often the most prevalent mutational signature, and overall, only second to aging-related mutations signatures. In addition to AID/APOBEC cytidine deaminases, recent evidence also implicates ADARs as sources of mRNA mutations in cancer (109–112). Like AID, the role of APOBEC enzymes in tumor initiation has also recently been established in APOBEC-transgenic mouse models (113).
The diversification of the adaptive immune antigen receptors is the only vertebrate example of controlled self-DNA editing and damage in the form of purposeful mutation and rearrangement. The RAG, AID/APOBEC, and ADAR DNA-editing enzymes play important roles in adaptive and innate immunity through the mutagenesis and recombination of the endogenous Ig genes, and the response to viral infection. The importance of these enzymes is underscored by the immunodeficiency disorders caused by their deficiency: severe combined immunodeficiency (SCID) and Hyper IgM syndrome in the case of dysfunctional RAG and AID, respectively (114–121). On the other hand, these enzymes also mediate considerable disease-driving collateral damage to the genome. Given their importance to immunity, infection, and cancer, it is not surprising that the DNA-editing enzymes of the immune system have been the topic of intense study in various fields including immunology, virology, cancer, DNA damage/repair and structural biology. In the next section, we provide an overview of the methodological and model organism landscape of this research area. The central thesis of this review is that the evolutionary aspect of these enzymes, despite being an understudied area, has provided key insights from the basic biological and applied biomedical perspectives.
A survey of published literature on PubMed/Scopus reveals ~5000 articles focusing on the DNA-editing and DNA-damaging enzymes of the immune system (RAGs: 729, AID: 2368, APOBECs: 2628, wherein these enzymes are in the title/abstract), published over the last 3 decades of work on RAGs and 1-2 decades of work on AID/APOBEC/ADARs. In the remainder of this article, because our work has mostly focused on AID, we will use this enzyme as a representative example of a genome-editing enzyme that has been extensively studied for 20 years [since its discovery in 1999 – (12, 13)] in the fields of immunity, cancer, DNA damage/repair, and epigenetics. In the following paragraphs, we examine the themes, experimental approaches and model systems used to study AID. The principles discussed and the conclusions reached at the end of this review in the context of AID apply equally and in the same manner to other DNA/RNA-editing enzymes involved in immunity (APOBECs, RAGs, and others, discussed below), and, for that matter, to the study of all other molecules that play roles in human health and disease.
First, in terms of study themes, topics of investigation include: understanding (1) functions, including “normal” immune functions (antibody diversification), non-immune biological functions (epigenetic regulation of the genome), and deleterious functions as a result of mis-expression or mis-targeted activity (initiation and progression of cancers), (2) regulation, including regulation of expression, interacting partners (protein, DNA or RNA), and regulation of the targeting of these enzymes to specific genes or genomic loci, (3) networks of cellular processes including for instance the DNA repair and damage response pathways activated downstream of these enzymes’ mutational activities, (4) molecular mechanisms, including biochemical analyses, and (5) 3D structure determination.
Second, in terms of methodological approaches, studies fall into several categories: (1) whole animal in vivo, (2) mechanistic experiments using primary cells or model cell line ex vivo, (3) genomics or bioinformatics studies examining genome-altering signatures of these enzymes, and association with immunity or cancer, (4) structure determination by crystallography or nucleic magnetic resonance (NMR) or emerging computational methods, (5) “simple” cellular experimental systems such as bacteria or yeast in which the enzyme is exogenously expressed followed by reporter assays, (6) biochemical reductionist cell-free studies of the enzymes as purified molecules, in vitro.
Third, in terms of model organisms, which will be the focus of this review, for DNA/RNA-editing enzymes involved in immunity and cancer, and indeed for most molecules that play roles in human health and disease, the vast majority of research has been focused on human and, to a lesser extent, mice. For the past several decades, cellular and molecular biology approaches for studying molecules involved in human health focused almost entirely on a handful of well-characterized model species, including the fruit fly D. Melanogaster, the worm C. elegans, and rodents, most notably lab mice. There are several reasons for this: first, many disease-related molecules function in similar pathways in humans and these model organisms and their dysfunction in the model species closely mirrors the resulting human condition; second, many of these disease-causing molecular pathways are well understood within the model organisms due to decades of research; and third, the model organisms are easy to grow, observe and manipulate at the cellular and genetic levels. Therefore, the concept of studying a handful of model organisms to glean mechanisms of human disease is logical. Indeed, studying molecular mechanisms of human health/disease-related processes in great depth but in a limited number of model organisms is what has led to an unprecedented pace of generating insights into the molecular basis of human diseases.
The total number of studies with AID as the main, or one of the main topics of study, as of the time of preparing this article, is 2368, of which 49 have discussed the topic of AID in species other than human or mice (15–63). Of these, 14 are literature reviews, and of the remaining 35, only 17 studies have presented primary experimental data wherein activities or functional properties of evolutionary divergent AID orthologs were compared (17, 20, 21, 26, 30–33, 39, 42, 45, 47, 52, 56, 61–63). And, among these 17, only less than a handful of studies had an evolutionary comparison as a main conceptual thrust. Therefore, in terms of effort, this area makes up a minuscule (0.7%) subset of the research devoted to the AID enzyme, with > 99% of studies being restricted to human or mouse AID (Figure 1B).
The goal of this review is two-fold: Our first aim is to make the case that despite this underrepresentation of effort, several important discoveries have been contributed by working on evolutionary distant AID orthologs, with implications across the fields of cancer, immunity, and genetics. Using the example of AID, we aim to highlight the concept that despite being a road infrequently taken, the evolutionary comparative approach to molecules involved in human health and disease provides immense value for fundamental biological discovery, with emerging practical applications in therapeutics and biotechnology. Our second aim is to suggest that considering the scale of the evolutionary diversity of species, there is an immense knowledge gap in our understanding of DNA/RNA-editing enzymes from species other than human and mouse. In the sections below, we first review the evolution of AID and related enzymes, followed by a review of the contributions made by examining AID through a species-comparative and evolutionary lens, and the future potential of such avenues of inquiry.
The AID/APOBEC family is thought to have originated from tRNA adenosine deaminase (Tad)/adenosine deaminase acting on tRNA (ADAT2), the latter of which forms a heterodimer with ADAT3 to deaminate adenosine (A) to inosine (I) in 34 tRNA. These mutated tRNAs can recognize multiple mRNA codons, as I pairs with U, C, or A in the wobble (3rd) position (15, 16). Interestingly, ADAT2 may be able to deaminate cytidine in DNA as well (122) indicating the substrate promiscuity of the AID/APOBEC family may have evolved before the APOBEC family divided into the multiple family members. Other enzymes related to Tad/ADAT2, but not to the AID/APOBEC family, include Tad1p/ADAT1, which deaminates tRNA, adenosine deaminases acting on pre-mRNA (ADARs 1, 2, and 3), which is involved in post-translational modifications of RNA (123–125); and cytosine deaminase, cytidine deaminase, and deoxycytidine monophosphate deaminase (dCMP), members of the pyrimidine salvage pathway which recycles nucleotides (126). These enzymes are found throughout the metazoa phylum (16).
Members of the classical AID/APOBEC family (APOBECs 1, 2, 3, and 4) and their newly discovered sister clades and members are discussed below, in the order in which they likely evolved. It is suggested that the AID/APOBEC family has evolved from the tRNA adenosine deaminases containing the consensus motif (C/H)xExnPCxxC (x is any given amino acid) as their catalytic domain (14, 127). The shift in substrate specificity from adenine to cytidine during the divergence of the AID/APOBEC family from Tad2/TadA deaminases has been attributed to the expansion of the α4-β4 loop (i.e., Loop8) and a conserved tyrosine in this loop. The larger L8 decreases the size of the substrate-binding pocket, and the conserved tyrosine could participate in base-stacking interactions (128). Moreover, the HxExnPCxxC motif is the conserved catalytic domain shared by the AID/APOBEC family in which the glutamate (E) acts as a proton donor and the histidine (H) with two cysteines (C) coordinate a Zn2+ ion with the help of a water molecule (39, 52, 129, 130).
The secreted novel AID/APOBEC-like (SNAD) enzymes belong to a sister clade to the classical AID/APOBEC family, evolving in the first animals to diverge from fungi (sponges, SNAD4) and appearing throughout the vertebrate phylum (SNAD1). SNAD2 and 3 found only in the ray-finned fishes are likely the result of whole genome duplication event and/or subsequent expansion of this class. SNAD enzymes are the only AID-like enzymes in multicellular eukaryotes with a characteristic predicted secretion sequence and have therefore been proposed to be secreted potentially for delivery to virus-infected cells or extracellular parasites; however, their catalytic activity and other biochemical characteristics remain unknown. They may have originated from bacterial toxin proteins (16).
APOBEC4 (A4), a member of the classical AID/APOBEC family, was likely next to evolve, first appearing in the cnidarians (corals), which diverged after sponges (16). The lack of introns in the A4 gene indicates it may be the result of early retrotranspositional events. A4 is present in the first vertebrates, the jawless fish (agnathans), the lobe-finned fish (sarcopterygians), and tetrapods, but is lost in sharks and often lost in ray-finned fishes (actinopterygians). It is expressed in human testes, but its biological role and catalytic activity are unknown. Unlike the other members of the AID/APOBEC family which are known to deaminate polynucleotides, critical amino acids required for polynucleotide deamination (SWS and F in the middle of the deaminase motif HXE….PCXXC) are missing from A4, indicating it may act on other substrates (15, 16, 131).
The next-evolved branch of AID-like enzymes include cytidine deaminase-like 1 (CDA1), CDA1L1, 2, 3, and 4, and CDA2 found in the jawless vertebrates (agnathans). Lampreys lack many canonical “pillars” of the adaptive immune system, such as RAGs and MHC; however, they do have antibody-like proteins (VLRs) that are diversified somatically, which led to the discovery of CDA1-like, and CDA1 and 2 in the freshwater and sea lampreys, respectively. These enzymes will be discussed in detail in a following section (16, 17, 20).
This was followed by the emergence of AID and APOBEC2 (A2) in the common ancestor(s) of jawed vertebrate classes of cartilaginous and bony fish. Hence, A2 and AID are considered the ancestral family members of the classical APOBEC family due to being present in most jawed vertebrates tested to date. They appeared at the same evolutionary juncture where the classical V(D)J-based Ig recombination and canonical heavy/light-chain based antibody structures emerged (16, 57). Interestingly, the involvement of CDA1 in diversifying the lamprey’s immune receptors and the continuing of a similar role for AID in the jawed vertebrate may be an example of convergent evolution in that the acquisition of the lymphocyte receptor diversification role by the AID-like branch had already occurred before the further divergence of this branch within vertebrates. A2 may be the result of early retrotranspositional events, which used AID as a scaffold. Like A4, human A2 does not appear to edit RNA, DNA, or free cytidine in vitro. Its ortholog in zebrafish, which has been implicated in retina and muscle generation, also lacks deaminase activity (132–134). Additionally, A2 seems to inhibit transforming growth factor (TGF)-β in Xenopus (frog, amphibian) and zebrafish (135).
The so-called novel AID/APOBEC-like Deaminases 1 and 2 (NAD1/2), while not being original members of the classical AID/APOBEC family, are closer in sequence to A1, A2, A3 than A4. NAD1 is found in ray-finned fishes, the coelacanth (sarcopterygian), amphibians, lizards, and marsupials; NAD2 is found only in amphibians. Neither NAD has been characterized and their biological relevance remains unknown (16).
APOBEC1 (A1) is the founding member of the AID/APOBEC family (136–138). It was originally thought to be first evolved in mammals due to duplication of AID; however, this duplication likely occurred in or before the lungfish. A1 deaminates the cytosine at position 6666 of Apolipoprotein B mRNA, creating a premature stop codon at this position, altering ApoB100 to ApoB48, which is essential for secretion of triglyceride-rich chylomicrons (139). It was later discovered that like AID and A3s (below), A1 is also quite promiscuous, acting on retroviral substrates and ssDNA (140, 141). As A1 is among the later AID/APOBEC family members to evolve, the RNA-editing capabilities seen in other members of this family may be a late-evolved characteristic. On the other hand, due to the progenitors of the AID/APOBEC family acting on RNA and, in some cases, both RNA and DNA (142), substrate promiscuity may be an original characteristic of the many family members, whose activity has just not yet been fully elucidated. In support of this, changes in substrate binding surface regions of the AID and APOBEC-related deaminases appear to be the most rapidly evolving structural feature of these enzymes, and AID certainly appears to recognize RNA and DNA/RNA hybrids with very high affinity though its catalytic activity is restricted to the ssDNA strand (143).
APOBEC3 (A3) is the last group of AID/APOBEC enzymes to have emerged, likely the result of AID’s gene duplication events. A pronounced expansion has occurred most recently in primates leading to 7 unique primate-specific A3 genes (A3A, A3B, A3C, A3DE, A3F, A3G, and A3H) (64, 144). The expansion of these enzymes has been proposed to be due to an arms race between mammals and the targets of A3, retroviruses. The origin of A3 is not fully clear: the initiating duplication event was thought to take place in the first placental mammal where no A3 ortholog were found in animals that diverged before placental mammals. It is thought that in rodents, pigs, and cattle, two AID-like genes fused to form a single gene; in horses, bats, and felines, one of the two genes repeatedly duplicated leading to an expansion of A3 genes. However, the sequenced lungfish genome appeared to contain a putative A3C gene (145). It is possible that the A3C found in the lungfish was a novel APOBEC-like gene representative of convergent evolution.
Pre-vertebrates (protochordates) lack AID but have AID-like enzymes such as the aforementioned SNADs. While also lacking B cell receptors, these animals have immune receptors belonging to the immunoglobulin superfamily (146–149). It is believed that a type of proto-AID (or AID ortholog) was present in the first vertebrate ancestor, which then diverged to CDA in the lamprey and to AID in the early jawed vertebrates, the shark (17, 18). Similarly, it is hypothesized that the targets of this proto-AID (somatically diversified lymphocyte receptors) diverged into three unique receptors with three different lymphocyte cell lineages: a secreted form (VLRB in the lamprey and BCR in jawed vertebrates in B cell-like cells) and two membrane-bound receptors (VLRA/C in the lamprey and TCR αβ/γδ in jawed vertebrates in T cell-like cells) (18, 150). Due to CDA1/1L genes lacking introns, it has been posited that CDA2 was the original enzyme in all three lamprey lineages, with the ability to somatically diversify all three VLRs, and that CDA1/1L genes were the result of retrotransposon events after which CDA2 was subsequentially silenced in CDA1/1L+ cell lineages. This idea is supported by the fact that in the first-diverged subsequent jawed vertebrate, the shark, AID appears to initiate somatic hypermutation of both B and T cell receptors (19, 35, 151, 152). This suggests that perhaps this broader dual role of AID was lost in subsequent vertebrate lineages and the role became focused on antibody diversification in B cells only but the dual role appears again in limited later-diverged species, such as the Ballan Rasse (ray-finned fish) and in camels (36, 153). Lamprey CDAs have been relatively understudied after their discovery, with their VLR antibodies garnering the most attention as novel non-classical antibody structures that may hold biotechnological and therapeutic potential (154–156).
The first immunoglobulin loci to evolve were those in the elasmobranchs (sharks and skates) that are organized quite differently from the most-studied mammal Ig loci. Shark Ig loci are organized into multiple mini loci (MML) (149, 157), with a mini locus or “cluster” equating to one V region placed next to one or more D regions, followed by one J segment and a single constant region (V-DDD-J-C)n (158). Some MML are rearranged in the germline, while most are rearranged by the RAG recombinase. Shark Ig undergo SHM, with long, tandem substitutions unique to these species and presumed to be due to AID-initiated mutations (57, 158–162). It was initially believed that shark Ig did not undergo CSR; however, though shark Ig sequences lack the conventional switch regions which first appeared in amphibians, recombined VDJ of one cluster can be “switched” with that of another, leading to a different constant region attached to the recombined VDJ region, possibly initiated by AID acting on recombination hotspots in a process that is concomitant with SHM rather than separated as in after the appearance of distinct switch regions (24, 163, 164). The studied Sharks have three types of Ig: IgM, present in almost all vertebrates, IgW (may be a counterpart to IgD), and IgNAR, which is unique to sharks, being made up of only heavy chains (165).
Outside of humans and mice, SHM and CSR have been studied most in ray-finned fish. Poikilotherms such as ray-finned fish have modest changes in antibody affinity, which has been reported in several species to be initiated by SHM (40, 41, 43, 49, 57, 166–168). This is likely due to inefficiencies caused by a lack of organized GCs; instead, ray-finned fish appear to have GC-like clusters of melanomacrophages with AID-producing cells in the centre (41, 169). Teleost fish (ray- and lobe-finned fish) appear to have Ig loci made up of both MML and translocon-type organizations, the latter of which is how most tetrapod Ig loci are arranged. In ray-finned fish, the V, D, and J segments are arranged as in mammalian Ig loci, with the IgM and IgD constant regions at the 3’ end, one after the other. However, the teleost-unique IgZ/T constant region is located further upstream, separated from the IgM and IgD constant regions by D segments (Vn-Dn-Jn-CZ-Dn-CµCδ) (57, 157, 170–172). Lungfish also have IgW and the lungfish-specific IgN and IgQ (173). Though bony fish Ig loci do not undergo CSR which appears first in amphibians, the IgM and IgD isotopes are “switched” via alternative mRNA splicing, while IgZ/T can be expressed after alternative V(D)J rearrangement.
The most emphasis outside human and mouse AID has been on fish, because of the expected level of divergence in the primary sequence, and unique features found in fish AID’s primary structure compared to the very well conserved mammalian counterparts. Due to evidence of SHM in the early-diverging vertebrate fish lineages as discussed in the above section, it was hypothesized that an AID ortholog could be found in bony fish, and it was indeed found in channel catfish (Ip-AID) (40). This was the first non-human/mouse AID ortholog to be identified followed by detailed work on tissue expression patterns and possible roles in SHM. Shortly thereafter, it was determined that zebrafish also has a bona fide AID gene (Dr-AID) and noted that it, along with the predicted AID genes from other ray-finned fish, encodes an additional 9 amino acids (aa) in the cytidine deaminase motif, along with a different N terminal motif compared to tetrapod AIDs (44). In 2004-2006, a series of early studies looked at the functionality of a small number of fish AID alongside Xenopus AID using exogenous expression in bacteria or yeast and measuring mutagenic activity in colony formation reversion assays, or expression in murine or human AID-deficient B cells followed by assaying for CSR (31–33, 49). Even though canonical CSR only occurs in tetrapods (37), multiple fish AID orthologs were able to initiate both mutations in E. coli, S. cerevisiae, and murine cells and CSR when exogenously expressed in AID-deficient B cells, albeit less effectively than mammalian AID (31–33, 49). This suggested that CSR as it occurs in mammals evolved due to the emergence of switch regions within immunoglobulin loci, and not due to adaptations of the different AID orthologs, and that the poikilotherm AID itself is fully capable of mediating CSR. In depth analyses of the regions of human AID required for CSR pointed to the C-terminus raising the possibility that this region of AID may be important in other biological roles prior to the evolutionary emergence of Ig CSR (174). Importantly, these studies also provide strong opposition to the view that CSR mediation by AID requires a specific set of protein co-factors, because early fish AID are presumably not co-evolved with such presumed co-factors required to chaperone AID to switch regions of the Ig genes in mouse cells. These findings are in line with later findings that the role of AID in mediating CSR is simple dC mutation and DSB generation, and that likely AID is targeted to these regions through the abundance of ssDNA structures such as R-loops and DNA/RNA hybrids that are inherently favored by AID (143, 175, 176).
In experiments wherein fish AIDs were exogenously expressed in murine AID-deficient B cells, zebrafish AID and mouse AID could mediate equally efficient CSR, with fugu AID and catfish AID being respectively 4- and 7-fold less efficient than these. Nuclear cytoplasmic shuttling of AID has been shown to be a key regulator of its activity and catfish and fugu AIDs appear to have nuclear export and localization domains conserved with other non-mammalian vertebrate domains with expectant results upon their mutation and it was shown that removal of this domain results in accumulation of AID in the nucleus, confirming its functionality. However, generation of hybrid AID with interchanged NES domains demonstrated that the aforementioned difference in their ability to mediate CSR was not due to different NES sequences, suggesting that fish AIDs may have different inherent catalytic robustness (31, 32, 49, 177). In the same set of experiments, the functionality of Xenopus AID was also confirmed for the first time.
Another property of fish AID that was examined in these early studies was temperature sensitivity. It was found that incubating the cells in which the fish AID are being expressed at lower temperatures than the typical 37°C (18°C for bacteria, 30°C for yeast, and 26°C for mammalian cell lines), yielded generally more AID activity in the bacterial colony count, yeast-null mutation, and GFP reversion based assays employed in bacteria, yeast, and cell lines, respectively (15, 31, 32, 49). The lamprey CDA1-class deaminases were also shown in bacterial and yeast-based expression assays to be active cytidine deaminases. Another example of a structure:function insight was the example of using zebrafish AID to propose a role for S38 phosphorylation-dependent interaction of AID with replication protein A (RPA) and its role in mediating CSR. Since zebrafish AID lacks this residue but contains D44 which can act as a phosphomimetic residue, it was proposed that S38 phosphorylation dependent Replication protein A (PRA) interaction is essential for CSR, though another study using a zebrafish AID with a D44 mutation found that this residue is not critical for CSR (30, 42, 62, 178); therefore the importance of this axis of S38 phosphorylation-AID-RPA remains uncertain, as the early view that specific cofactors chaperone AID to the Ig locus ought to be considered in balance with the various explanations that it may be the process of transcription and its unique features at the Ig loci including robust and bidirectional transcription, and unique DNA or RNA secondary structures (e.g. G quadruplexes) are the determinants that recruit AID to the Ig loci to carry out SHM and CSR (179–182).
As the first tetrapods, amphibian (Xenopus) antibodies undergo SHM and CSR; however, the switch regions in Xenopus are AT-rich compared to GC-rich, which may affect switching efficiency (183, 184). Xenopus AID has been shown to demonstrate CSR activity, and is expressed in hematopoietic tissues, hinting at a role in ontogeny (31, 51). Neither Xenopus nor other amphibian AIDs have been biochemically characterized. Avian Ig loci, at least the ones sequenced (duck, chicken, and ostrich) are unique among the higher vertebrates in that there is a single functional germline locus (V-Dn-J or V-J) that is recombined via V(D)J recombination; further diversification occurs via AID-mediated gene recombination (similar to how VLRs are recombined), initiated by avian AID (185–188). Aside from experiments demonstrating that bovine AID can demethylate DNA via deaminase activity (61), no other non-human, non-mouse AID has been characterized in the higher vertebrates, and its targets (Ig) and activity (SHM and CSR) in many non-human animals remain unstudied.
Over the last decade, we have pursued a comparative enzymology approach to study the biochemical properties and structure:function aspects of purified AID from divergent orthologs. The initial goal of this effort was to gain insights into the 3D structure of human AID. Given AID/APOBECs’ involvement in immunity and cancer, intense research has been dedicated to solving their 3D structures. Unfortunately, AID/APOBECs proved to be problematic subjects for X-ray or NMR because they are difficult to make in large quantities due to host cell toxicity, and they form extensive non-specific interactions with other molecules making them hard to purify and insoluble. Hence, > 90% of the 40 reported AID/APOBEC structures are of partial or significantly altered versions, quite a few with < 50% identity to the native protein (PDB databank: https://www.rcsb.org/) (53). These alterations were necessary to enable crystal formation for X-ray crystallography or enhance solubility for NMR. AID is a small (only 198 aa) protein but it has by far the most positively charged surface amongst the AID/APOBEC family, which underlies its exceptionally high binding affinity (~nM-range) for its negatively charged ss-DNA substrate (189). Partially because of this, it has not been possible to obtain a native AID crystal or NMR structure despite intense attempts for 20 years since its discovery in 1999.
Based on the initial insights from the cell-based assays that revealed differences in functional efficiencies of orthologous AIDs and the relatively high divergence among mammalian and fish AID, we posited that AID from more distantly evolved species, might have distinct properties and that discovering the basis of their differences would shed light on AID’s inner workings. We began studying AID from key evolutionary points. Fish were of great interest because they are the most evolutionarily divergent species known to have AID, and their AID sequences exhibit the highest degree of primary sequence divergence. Parallel to the evolutionary approach, several partial X-ray or NMR structures of APOBECs were utilized in computational modelling to generate thousands of predicted AID 3D structures. Through this computational modelling and evolutionary approach, hereafter referred to as the “computational-biochemical-evolutionary” method, parts of AID were predicted to have a specific function. A library of different AID versions (mutants, chimeras with exchanged domains, fish orthologs) was generated, purified, and subjected to functional biochemical enzyme assays (e.g., enzyme kinetics, substrate binding, and optimal temperature determination) to verify whether a motif predicted by the modelling indeed mediated the supposed function. The experimental results were cross-referenced with the evolutionary/computational predictions, in order to refine a functional map of AID’s structure, first published in 2015 through this approach (52). This functional map of AID was later confirmed independently by an X-ray crystal of a near-native AID in 2017 containing 20 aa truncations and a handful of residues mutations which altered the surface charge of AID from ~+10 to +3 (52, 129). In the following paragraphs, we review the insights gained through the computational-biochemical-evolutionary method.
In 2012, by comparing the enzymatic activity and predicted structure of Hs-AID with bony fish AIDs (i.e., zebrafish [Dr-AID] and catfish AID [Ip-AID]), we demonstrated that different AID orthologs present diverse biochemical properties, such as catalytic rate and optimal temperature, which are governed by a single amino acid in their C terminus (26). The difference in the optimal temperature mirrored the ambient temperature of each organism. We observed that Dr-AID was several fold more active than Hs-AID while Ip-AID was significantly less active than Hs-AID, in line with the previous observations of its lesser ability to mediate CSR when deployed in an AID-deficient B cell (26). The different catalytic rates amongst AID orthologs may reflect the different evolutionary paths taken by each species’ immune system. The computational modelling of the surface charge and topology, and functional ssDNA binding assays of bony fish and human AIDs, also led to an early picture of AID’s ssDNA binding grooves. The width of this groove is ~ 10 Å. Given the width of ds-DNA helix (~ 20 Å), the identified DNA binding groove on AID explained its substrate specificity for acting only on ssDNA and not dsDNA (190–193). The presence of this DNA binding groove has been confirmed upon crystallization of Hs-AID with ssDNA (129, 194).
In 2013, we demonstrated that zebrafish AID, unlike its human counterpart and several other bony fish AIDs had the unique enzymatic ability to mutate 5-methyl dC (5mC) in addition to regular dC (39). Soon after its discovery, a possible role of AID in genome methylation and epigenetic reprogramming was suggested where AID demethylation activity in the CpG motifs would convert 5mC to deoxythymidine (dT) (195). Supporting evidence came from the fact that the AICDA gene is located in a cluster with other pluripotency genes and is expressed in oocytes and primordial germ cells (196). Soon after this initial report, AID-mediated deamination of 5-mC was reported in induced pluripotent stem (iPS) cells, primordial germ cells, B cells, cancerous cell lines, and bovine and zebrafish embryo (60, 61, 197–201). Regarding the enzymatic activity of AID on 5-mC, initially, it was claimed that Hs-AID has comparable activity on 5-mC as well (196). However, soon after, several reports showed that although Hs-AID can indeed deaminate 5-mC, its activity on this substrate and on other cytidine derivatives with bulky adducts is many folds less than on dC (39, 202–204). This is a key aspect of AID activity since AID-mediated CpG demethylation through a C to T mutation could be a mutagenic process. Given the importance of CpG motifs in gene expression and epigenetics, one would expect to avoid efficient activity of AID on 5-mC. In fact, methylation has been proposed as a protective mechanism against undesirable AID activity (202). We then used our comparative computational approach and reported that unlike Hs-AID, Ip-AID, medaka AID (Ol-AID), and tetraodon AID (Tn-AID), the zebrafish AID exhibits more efficient activity on 5m-C, deaminating it more efficiently than many other orthologs deaminate regular dC and significantly more efficient as normalized to its own activity on dC (39). From a biological standpoint, these results explained why in zebrafish, AID was uniquely involved in embryonic development and its knockdown resulted in genomes with hypermethylated CpG motifs. From an AID structure:function standpoint, modeling predictions of human and zebrafish AID catalytic pockets docked with dC showed that both AIDs are predicted to form catalytic pockets with the classical triad of Zn-coordinating residues (C87, C92, and H56 in human AID) and catalytic glutamic acid (E58 in human AID) that can accommodate a dC residue in orientations that support the 4-stage deamination chemistry common to cytidine and cytosine deaminase. Importantly, the catalytic pocket of zebrafish AID was predicted to have one of its composing loops extended and more flexible as compared to that of human AID, thus providing more space for a 5mC substrate that is bulkier than a dC (204). In this manner, the computational-biochemical-evolutionary method not only solved a biological puzzle about the role of AID in zebrafish, but it also made a key structural biology contribution by providing the first detailed maps of AID’s catalytic pocket through predictive modelling corroborated with functional enzymology.
In 2015, using our computational-biochemical-evolutionary method, we mapped a network of primary and secondary catalytic residues that either contact and/or stabilize the dC in a catalytic pocket (52). This network of amino acids consists of G23, R24, R25, E26, T27, L29, N51, K52, N53, G54, C55, V57, T82, W84, S85, P86, D89, Y114, F115, C116, and E122 in human AID (52). These residues form the “walls” and “floors” of the catalytic pocket and interact with substrate dC in several predicted protein conformations through hydrogen bonding, electrostatic interactions, and aromatic base stacking. The importance of direct interactions between some of the secondary catalytic residues and substrate DNA was validated when the crystal structure of a partially truncated and mutated but relatively near native AID was published (129). Given the importance of proper positioning of dC inside the active site for efficient deamination activity, defining the secondary catalytic pocket residues was a step forward in solving the functional structure of AID. In the same work, we also described a novel structural regulatory mechanism of AID/APOBEC activity in that the majority of Hs-AID conformations at any given time contain catalytic pockets that are closed and inaccessible for accommodating a dC for deamination (194). Furthermore, we observed that the majority of ssDNA:AID binding events result in ssDNA bound non-productively on the surface in conformations that do not pass over the catalytic pocket, presumably due to the highly positively charged surface of AID (+11, the highest surface positive charge amongst AID/APOBECs) (52, 194). Taken together, the frequent catalytic closure and sporadic ssDNA binding are significant bottlenecks for AID activity such that < 1% of all ssDNA:AID binding events translate into a cytidine deamination event. We then proposed that due to the potential danger of AID/APOBEC activity for genomic DNA, this inherent structural regulatory mechanism is in place as a safe-guard mechanism in AID and in the tumorigenic A3 family members; the main pillar of this hypothesis was that the open:closed dynamic ratio in AID, A3A and A3B correlated with their catalytic rates and with their relative responsibility for mediating tumorigenic mutations in cancers. We termed this novel mechanism Schrödinger’s CATalytic Pocket (53). Here again, the computational-biochemical-evolutionary method was key in providing the functional proof for the existence and regulatory role of Schrödinger’s CATalytic Pocket. A panel of chimeric AID enzymes, including bony fish-human chimeras, was generated since certain fish AID (e.g., the aforementioned zebrafish AID) have catalytic pockets are composed of loops of different lengths and hence different breathing dynamics compared to human AID. The demonstration that the AID chimeras (e.g., human/zebrafish catalytic pocket chimera AID, or AID/A3 chimera) predicted to spend more time in the open conformation also have higher catalytic rates, provided functional proof of the concept for Schrödinger’s CATalytic Pocket. First revealed by the computational-biochemical-evolutionary method, the pocket dynamic has since been independently confirmed by structural analyses of A3s.
In a study in 2017, to examine whether Hs-AID’s unique biochemical properties (i.e., low catalytic rate and high affinity for its substrate) were conserved across vertebrates, we compared the enzymatic activity of Hs-AID to that of sea lamprey, nurse shark, and coelacanth. These species were chosen to represent key points of evolution, lamprey being a jawless vertebrate, shark being the first jawed vertebrate with the classical Ig system, and coelacanth being the “fossil fish” lobe fined fish thought to be the closest fish ancestor of tetrapods (21). We found that despite the biochemical variability amongst these enzymes in substrate sequence preference (WRC vs. non-WRC motifs) and optimal temperatures, the key defining enzymatic characteristics of AID (lethargic catalytic rate and high nM range affinity for ssDNA binding) were maintained (205). This finding suggests that these unique biochemical regulatory features of low catalytic rate and high ssDNA binding affinity in AID are evolutionary conserved and thus important for its function, for instance the balance between making SHM and CSR mutations while protecting the genome from rampant promiscuous mutagenesis. Furthermore, using computational modelling, we showed that all of the above-mentioned AIDs are predicted to exhibit the Schrödinger’s CATalytic Pocket phenomenon, revealing the importance of this intrinsic structural regulatory mechanism for AID activity throughout the vertebrate class (205). Importantly, this was also the first study to show that two species, key in the evolution of adaptive immunity in its classical mammalian form, the shark and the coelacanth, do indeed have a functional AID enzyme.
In a more recent study, colleagues and we turned our focus to the extant agnathan the sea lamprey, in which thus far two AID-like cytidine deaminases (CDA1 and CDA2) have been found (20). Genetic analyses revealed that CDA1 and CDA2 were found in both the sea and freshwater lampreys, along with, unexpectedly, multiple CDA1-like genes that could be divided into two distinct groups (CDA1L1_1, _2, _3, _4 and CDA1L2_1, _2). Genomic DNA from other individuals were searched for homologs of these new CDA1-like genes, which were found, along with splice variants of CDA1L1_1 and CDA1L1_3. When their amino acid sequences were compared with those of other AID orthologs, these novel CDA-like proteins were found to contain the conserved deaminase core catalytic motif (HxExnPCxxC), suggesting they could be active cytidine deaminase. In silico modeling of each CDA ortholog also demonstrated the high likelihood of catalytic cytidine deaminase activity, as each protein formed a putative cytidine deaminase catalytic site, and catalytic activity was demonstrated by expressing these enzymes in 293T cells and assaying the extracts for cytidine deaminase activity. The enzymes exhibited cold adaptation, with optimal temperatures being between 14-22°C, and most had an acid pH-adapted activity profile, reminiscent of the human A3 branch enzymes (A3A, A3B, A3G, A3F) rather than human AID, and commensurate with structural modeling showing that these proteins have a lower surface charge than human AID. These results showed for the first time that lamprey has more than just one version of a CDA1 enzyme, and remarkably, that these are variably expressed in individuals of the same species, a novel biological phenomenon the mechanism and importance of which is yet to be discovered.
In the above sections, we reviewed the insights relevant to structural biology, immunology, and cancer research that have been brought forth by comparative studies of AID from non-human/mouse species. This section highlights the future potential of comparative evolutionary studies for impacting emerging approaches in structural biology, base-editing, and protein engineering. The concept of how evolutionary studies illuminate each of these three arenas is illustrated in Figure 2.
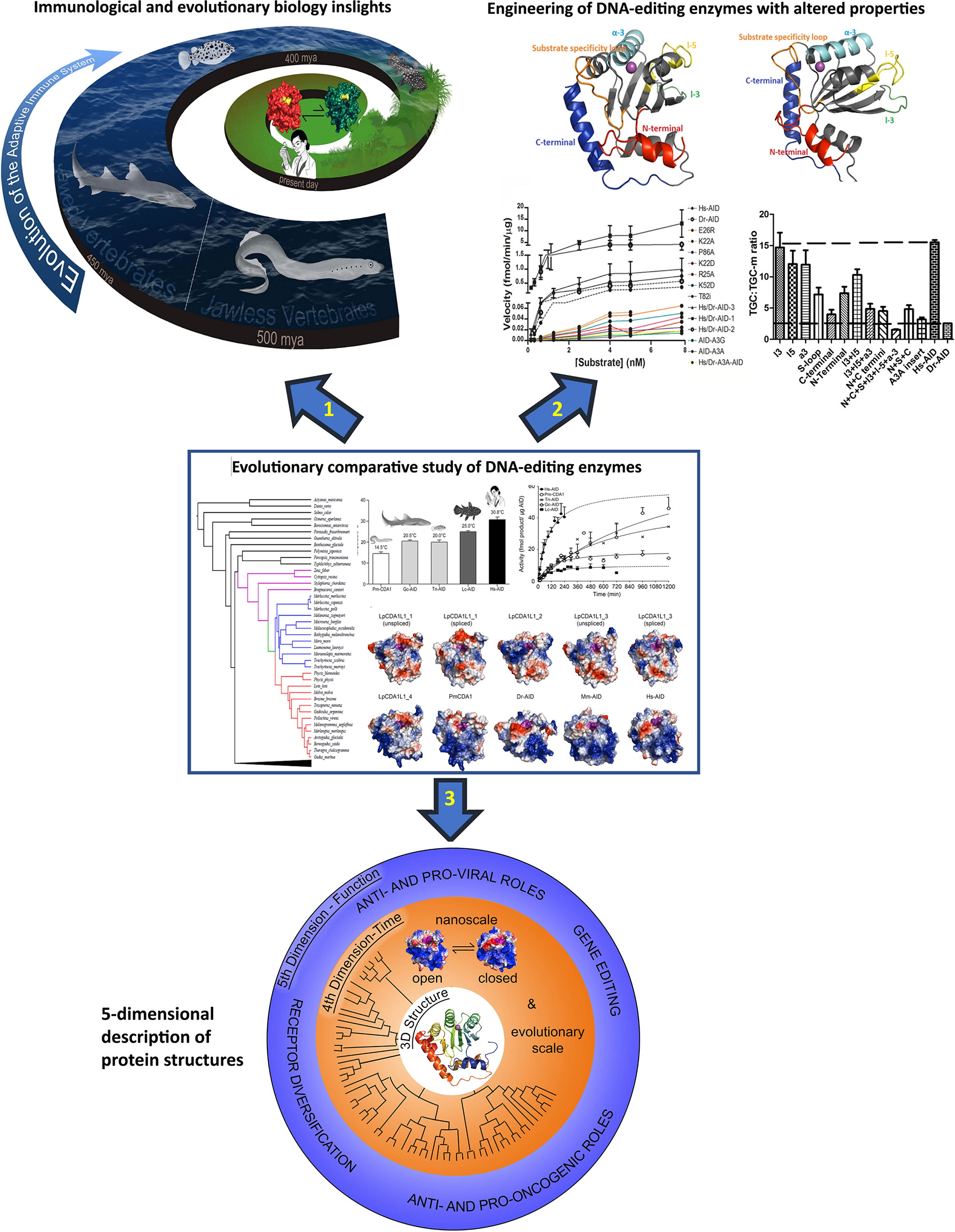
Figure 2 The concept and three main applications of the evolutionary-biochemical-computational approach to studying DNA-editing enzymes. The evolutionary comparative approach is shown in the middle with 3 arrows each pointing to an area wherein this approach can make significant impact. The evolutionary comparative approach shown in the middle panel consists of comparing biochemical properties (Michaelis-Menten kinetics, substrate binding kinetics, optimal temperature, optimal pH, substrate sequence or shape specificity, etc.) of the enzymes using enzyme assays and considering insights in the context of their 3D solved structures or computational predicted models as shown in this figure. Due to vast biochemical diversity observed amongst various AID orthologs, examining the biochemical properties of divergent AID orthologs has shed light on many structure:function aspects of AID/APOBEC enzymes. Arrow 1: the evolutionary comparative study of DNA-editing enzymes can provide insights into the evolution of the immune system, for instance on whether the immune systems use active deaminases and how/if they have gene sequences or other immune genes that have co-evolved with their deaminases. Arrow 2: using different orthologs allows for generation of libraries of mutants and chimeric enzymes which can have diverse biochemical properties such as DNA/RNA-targeting profiles and sequence specificities, and these can be used for applications such as base editing. Arrow 3: the most important highlight of the evolutionary-biochemical-computational approach is the birth of the concept of 5-dimentional (5D) structural description, proposed in this article. The 5D description integrates the classical 3D structure of a protein with dynamic changes in time (4th dimension) and the relevance of these to function (5th dimension). The middle panel contains reproduced figures from previous publications. The thermosensitivity and enzyme velocity plots are from our previous work Quinlan EM et al. (21). Biochemical regulatory features of activation-induced cytidine deaminase remain conserved from lampreys to humans. Mol Cell Biol 37:e00077-17. https://doi.org/10.1128/MCB.00077-17. Copyright © 2017 American Society for Microbiology. The computational models are adapted from our previous work Holland et al. (20). Expansions, diversification, and interindividual copy number variations of AID/APOBEC family cytidine deaminase genes in lampreys. 2018 Apr 3;115(14):E3211-E3220. doi: 10.1073/pnas.1720871115 Copyright (2018) National Academy of Sciences.
First, with respect to structural biology (Figure 2 arrow 3, bottom panel, and Figure 3), the significance of this computational-biochemical-evolutionary approach for AID is evident by its track record of providing the first 3D map of AID structure and revealing the concept of Schrödinger’s CATalytic Pocket in the AID/APOBEC family, both of which have subsequently been confirmed by independent studies employing the traditional structure solution methods of crystallography and NMR. Thus, in the case of AID, not only did the evolutionary-biochemical-computational approach for solving its structure prove to be quicker, it was also the only approach able to deal with AID in an unaltered native state, as the only way to crystalize AID has been to alter it, with the most near-native crystal structure still containing 20 aa truncation and multiple surface mutations that change the charge of native AID drastically (from +11 to +3) (52, 129). Furthermore, the evolutionary-biochemical-computational method also revealed additional time/space dimensions of the structure that are not normally probed through the traditional methodologies (53). For this reason, we termed this type of computational-biochemical-evolutionary structure a five-dimensional (5D) description of a 3D structure. In the 5D structure of a protein, as opposed to the classical protein structure which has always been viewed as a 3D shape, the structure’s dynamics are further explored through time (4th dimension) dimensions of ‘tempus’ and ‘aevum’. The ‘tempus’ analysis is the studying of a protein structure in a real time manner where one can examine/predict the ‘protein breathing’ on the time scale of fractions of a second, while the ‘aevum’ sub-dimension is one wherein dynamic change is compared throughout ortholog evolution from both closely- and remotely-related species, on the time scale of hundreds of millions of years. The 5th dimension, which is “function”, then explores the understanding of how these dynamic 3D and 4D structures relate to the biological function of the protein, including functions in human health/disease. Figure 3 illustrates the concept of how a 5D structure description contains orders of magnitude more information than the conventional 3D picture.
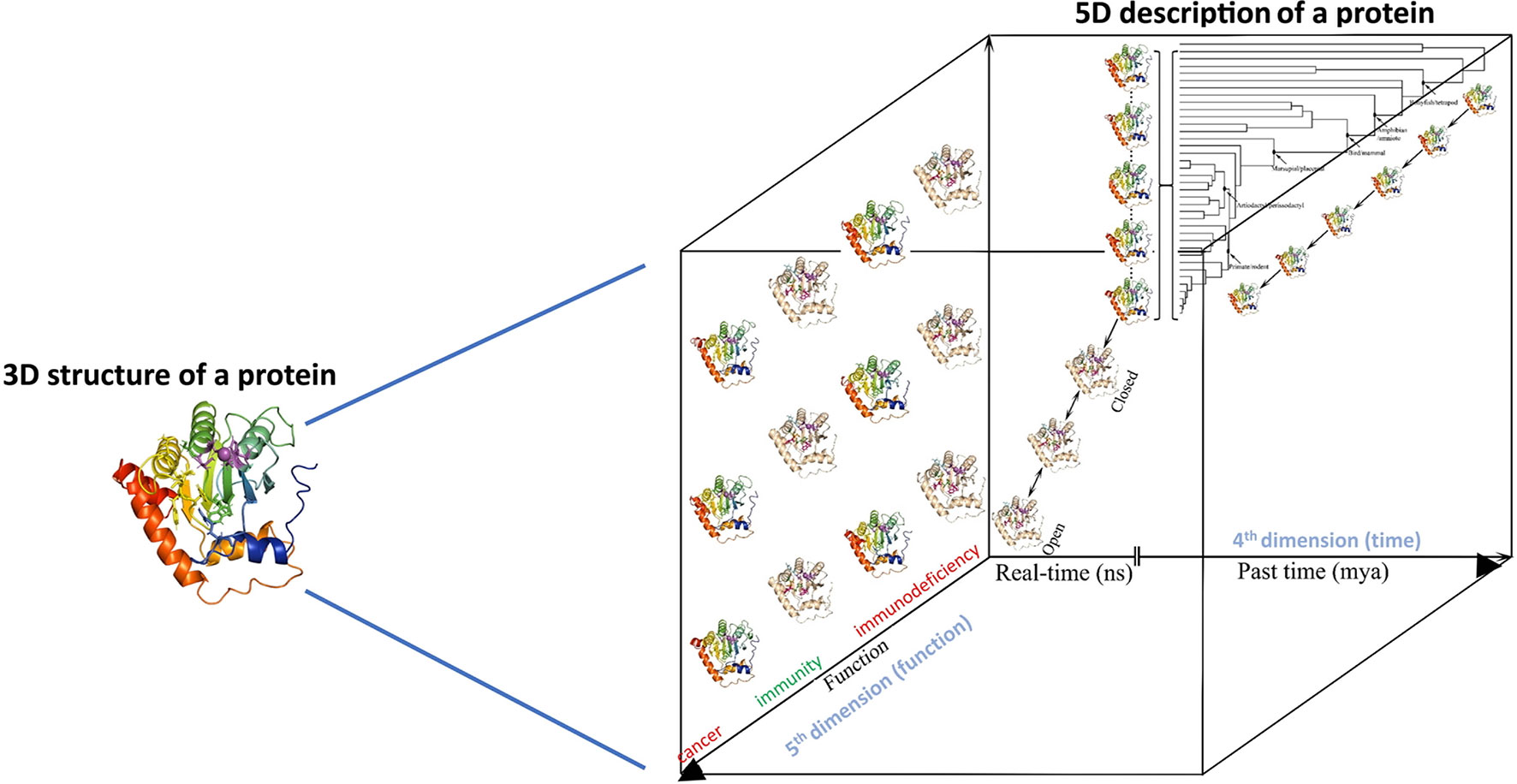
Figure 3 5-dimensional description of biological molecules. In the 5D structure description proposed here, the information from the traditional 3D structure is combined with the structure dynamics in time (4th dimension = time, in real time measured in fractions of a second, and evolutionary time measured in millions of years) and integrated with how these real-time and evolutionary dynamic structural changes impact the biological function of the protein (5th dimension = biological function as dependent on 3D and 4D descriptions of a protein’s structure).
Others and we have shown that AID orthologs exhibit a vast diversity in many of their biochemical properties such as catalytic rate, optimal temperature, optimal pH, and substrate sequence specificity. Indeed, the catalytic rate varies over 3 orders of magnitude, temperature optima vary from very cold to human body temperature, and pH optima vary over a range of nearly 2 units. Firstly, this is indeed a remarkable range of variation for evolutionary closely related versions of the same enzyme, given that a large portion of the enzyme’s primary sequence and its overall 3D structural architecture are conserved. Secondly, each of these biochemical characteristics is an indicator of a specific structural aspect of a protein. For instance, variations in catalytic rate can be due to differences in substrate binding or differential dynamics of the catalytic pocket as dictated by breathing loops that compose the catalytic pocket. Variations in optimal pH are largely owing to the surface charge of the protein, which in AID can vary from only slight positive in some bony fish (e.g., +3 in Salmo Salar) to extremely positive (e.g., +11 in human and mouse). Substrate specificity differences are mediated by a well-defined substrate specificity loop which is one of the more variable structural regions among the AID/APOBEC family members, causing different surface binding pockets next to the catalytic pocket that underlie differential preference for the -2 and -1 base positions next to the target dC that is positioned in the catalytic pocket. For temperature sensitivity, proteins may increase their thermoresistance using several strategies. In the first mechanism, the enthalpy change (ΔHs) measured at the temperature of maximum stability (Ts) becomes more negative, causing ΔG for all temperatures to decrease. This strategy can be seen as a stability curve to be shifted downward. The second strategy is to increase (less negative) the change in the heat capacity upon folding (ΔCp) which causes Tm to increase. In this case, the stability curve would broaden. The last approach is to increase Ts which shifts the curve to the right. Proteins may apply one, two, or all of these strategies to improve their thermal resistance, and this is dictated by differences in the secondary structures employed in various parts of the protein and/or overall flexibility of the structure. Thus, not only is each of an enzyme’s biochemical properties reflective of a structural trait in terms of the 3D folding of the structure, but the relationship between properties (e.g., between catalytic rate and optimal temperature) itself can also provide finer level information into the subtle differences of 3D folding of the enzyme’s protein structure.
Modelling of proteins with no known related structure is a long-standing challenge in the field of structural biology where the recent breakthrough of AlpahFold has gained considerable attention (206). The AlphaFold algorithm, a learned-based method in contrast to knowledge- and physics-based ones, uses co-evolution methods and deep convolutional neural networks. Remarkably, combining the deep-learning methods such as AlphaFold with molecular dynamics stimulations has improved the accuracy of protein structural prediction even further (207). However, to achieve an accurate result using learned-based methods, access to a large dataset (e.g., multiple sequence alignment [MSA] of 105 to 106 sequences) of evolutionarily diverse sequences is necessary (208). Co-evolution-derived contact methods are based on the idea that the residues in close contact (< 8 Å considering the Cα) in the 3D structure, which define the local secondary structural features, co-evolve while the residues with medium- and long-range contact specify the overall 3D structure of a protein. In fact, the evolutionarily conserved dynamical/functional domains (termed evolutionary domains [ED]) have been predicted by coevolutionary coupling analysis of co-evolving residues (209). The contact map of the protein can be retrieved either through the evolutionary coupling analysis (ECA) or supervised machine learning (SML). ECA relies on a high-quality large MSA (with at least 64 times the square root of the length of the target protein) while SML methods are capable of retrieving the contact map even in the case of smaller MSA by combining the sequence-dependent and independent information (210). Therefore, the approach of studying a family of proteins from many orthologs that cover a large range of biochemical properties, coupled with artificial intelligence (AI) learning, will pave the road for even more refinement of such AI-based computational approaches to protein folding, and especially so in the field of enzymology. Given that this methodology may be nearing the accuracy of experimental structure determination, as announced recently, and the applicability of enzyme (e.g., virus polymerases) structure prediction and engineering for treatment of emerging pathogens, the evolutionary comparative study of enzymes can make a critical contribution in this domain.
From a basic evolutionary immunology perspective (Figure 2, arrow 1, top left), the comparative enzymology approach also has brought forth meaningful insights and points for further research. For instance, the discovery that AID’s catalytic pocket has evolved in one fish species to be significant more active, or capable of carrying out genome demethylation, speaks to issues of DNA-repair and genome demethylation that provide hints that in some instances in evolution, AID may indeed have had a significantly higher weight of non-immune based physiological functions as compared to the case in mammals where it plays a strictly immune role. Though other roles such as epigenetic remodelling have been proposed for human and mouse AID, the fact that AID-deficient mice appear only to suffer from Hyper IgM and no other perturbations suggest that any non-immune functions of AID in mammals are either marginal or highly redundant. This in turn suggests that perhaps AID initially emerged for other functions in the fish and was later co-opted by the immune system, a familiar pattern, that has already been shown for other DNA-damaging enzymes used by the immune system, namely the RAG recombinases. Demonstrating that AID is an active deaminase in species like sharks and coelacanth, which are key fishes in the evolution of vertebrates, also shed light on AID’s role in earlier-evolved immune systems. Lastly, the unexpected and novel expansion and inter-individual copy number variation of the AID-like CDA1 enzymes in the lamprey speaks to the intriguing possibility that somehow the enzymes themselves may be the subject of an as-yet-undiscovered type of genetic diversification or environmental response.
From the perspective of protein biotechnological advancements in protein engineering (Figure 2, arrow 2, top right), the comparative evolutionary enzymology method is also of value for emerging biotechnological applications, such as in the emerging field of base-editing. DNA base editing is a new genome editing tool, introduced in 2016, based on the clustered regularly interspaced short palindromic repeats (CRISPR) associated (Cas) system of bacterial adaptive immunity, where a point mutation is precisely introduced into the genomic DNA (211–214). This tool is comprised of a guide RNA, a catalytically impaired Cas nuclease coupled to a ssDNA mutating enzyme. There are two different classes of ssDNA base editors, the cytidine base editors (CBEs) and adenine base editors (ABEs), where different deaminases are used as the ssDNA mutating enzyme (215). CBEs accomplish the conversion of C:G to T:A using cytidine deaminases (i.e., AID/APOBEC family members) while ABEs perform the reverse mutation using adenine deaminases (e.g., TadA). The specificity of the CBE complexes is defined by the protospacer adjacent motif (PAM) which is recognized by the Cas enzyme, the activity-window which is defined by the target sequence incorporated into the single-guide RNA (sg-RNA), and the substrate specificity of the ssDNA mutating enzyme. Since the sequence content of the target dC is defined by the target genomic regions, diversifying the substrate specificity of the ssDNA editing enzymes are of a great interest. To accomplish this goal, different members of the AID/APOBEC family, such as AID, APOBEC1, A3A, 3B, 3C, 3D, 3F, 3G, 3H, and CDA1 from human, rat, and sea lamprey, and their variants have been tethered to Cas. Deamination of methylated dC was also accomplished by using A3A variants as the ssDNA editing enzyme (216). Given the observed diversity in the biochemical properties of AID orthologs, using AID from different species, especially bony fish, would assist in expanding the specificity of the CBEs arsenal. A recent study acts as evidence for this; the study did a screen of 153 in vitro-evolved cytidine deaminases (APOBECs, AIDs, CDAs, etc.), led to ones that exhibited the lowest unguided off-target DNA and cellular RNA deamination events along with the highest on-target deamination events. Using this screening approach to choosing a ssDNA editing enzyme, it became possible to reduce the unguided off-target DNA deamination events by 45-fold and transcriptome-wide deamination events by 12- to 69-fold, all while maintaining a similar DNA on-target editing frequency (212). Others and we who have been studying cytidine deaminase structure:function and evolution have also generated libraries of chimeric and mutant enzymes, bearing different motifs exchanged between orthologs in order to pinpoint enzyme functionality to structural parts (Figure 2, arrow 2, top right). In so doing, these libraries often contain engineered enzymes with variable targeting and substrate specificity profiles that could also prove as useful tools in the field of base-editing.
In conclusion, molecules involved in human health and disease are typically studied in only a handful of well-characterized model species. Here, using the example AID, a DNA-editing enzyme involved in immunity and cancer, we have reviewed how the few studies that have examined this molecule in evolutionarily distant species have brought forth important and unexpected insights in structural biology, immunology, and cancer research. For other DNA-damaging enzymes involved in immunity and cancer, such as RAGs, the case is parallel, with less than a handful of hundreds of studies probing non-mouse/human species; however, the studies that have ventured into the evolutionary past have brought forth intriguing ideas that have changed our understanding of RAG function and evolution (217–221). This, taken together with the fact that by far the greatest window of evolutionary diversity in these DNA-editing proteins, and indeed in all proteins, lies in earlier-evolved species that have remained unstudied, would make it reasonable to conclude that much fundamental and applicable biological insights can be uncovered by large-scale evolutionary studies. The case of understudied orthologs discussed here (Figure 1B) is made even more glaring considering that unlike the field of evolutionary immunology which is a recognized subfield of immunology with its own research groups, journals and scientific meetings, other disciplines such as DNA repair, cancer research, neurodegenerative diseases, and many others do not have a formal evolutionary sub-discipline. We have also discussed how, in addition to generating novel fundamental knowledge on biology, the evolutionary comparative approach for studying protein structure:function is a valuable tool to complement the emerging AI-guided protein folding methodologies as well as protein engineering in the field of base-editing and beyond.
All authors contributed to the preparation of the text and figures. All authors contributed to the article and approved the submitted version.
Natural Sciences and Engineering Research Council of Canada (NSERC), Grant/Award Number: 2015-047960.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Flajnik MF, Kasahara M. Origin and Evolution of the Adaptive Immune System: Genetic Events and Selective Pressures. Nat Rev Genet (2010) 11(1):47–59. doi: 10.1038/nrg2703
2. Pancer Z, Amemiya CT, Ehrhardt GR, Ceitlin J, Gartland GL, Cooper MD, et al. Somatic Diversification of Variable Lymphocyte Receptors in the Agnathan Sea Lamprey. Nature (2004) 430(6996):174–80. doi: 10.1038/nature02740
3. Lewis SM, Wu GE. The Origins of V(D)J Recombination. Cell (1997) 88(2):159–62. doi: 10.1016/S0092-8674(00)81833-4
4. Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, Adjacent Genes That Synergistically Activate V(D)J Recombination. Science (1990) 248(4962):1517–23. doi: 10.1126/science.2360047
5. Schatz DG, Oettinger MA, Baltimore D. The V(D)J Recombination Activating Gene, RAG-1. Cell (1989) 59(6):1035–48. doi: 10.1016/0092-8674(89)90760-5
6. Gellert M. V(D)J Recombination: RAG Proteins, Repair Factors, and Regulation. Annu Rev Biochem (2002) 71:101–32. doi: 10.1146/annurev.biochem.71.090501.150203
7. Schatz DG, Swanson PC. V(D)J Recombination: Mechanisms of Initiation. Annu Rev Genet (2011) 45:167–202. doi: 10.1146/annurev-genet-110410-132552
8. Honjo T, Kinoshita K, Muramatsu M. Molecular Mechanisms of Class Switch Recombination: Linkage With Somatic Hypermutation. Annu Rev Immunol (2002) 20:165–96. doi: 10.1146/annurev.immunol.20.090501.112049
9. Methot SP, Di Noia JM. Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. Adv Immunol (2017) 133:37–87. doi: 10.1016/bs.ai.2016.11.002
10. Min IM, Selsing E. Antibody Class Switch Recombination: Roles for Switch Sequences and Mismatch Repair Proteins. Adv Immunol (2005) 87:297–328. doi: 10.1016/S0065-2776(05)87008-7
11. Stavnezer J, Guikema JE, Schrader CE. Mechanism and Regulation of Class Switch Recombination. Annu Rev Immunol (2008) 26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248
12. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T, et al. Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell (2000) 102(5):553–63. doi: 10.1016/S0092-8674(00)00078-7
13. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-Induced Cytidine Deaminase (AID) Deficiency Causes the Autosomal Recessive Form of the Hyper-IgM Syndrome (HIGM2). Cell (2000) 102(5):565–75. doi: 10.1016/S0092-8674(00)00079-9
14. Conticello SG. The AID/APOBEC Family of Nucleic Acid Mutators. Genome Biol (2008) 9(6):229. doi: 10.1186/gb-2008-9-6-229
15. Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS. Evolution of the AID/APOBEC Family of Polynucleotide (Deoxy)Cytidine Deaminases. Mol Biol Evol (2005) 22(2):367–77. doi: 10.1093/molbev/msi026
16. Krishnan A, Iyer A, Holland SJ, Boehm T, Aravind L. Diversification of AID/APOBEC-like Deaminases in Metazoa: Multiplicity of Clades and Widespread Roles in Immunity. Proc Natl Acad Sci USA (2018) 115(14):E3201–10. doi: 10.1073/pnas.1720897115
17. Rogozin IB, Iyer LM, Liang L, Glazko GV, Liston VG, Pavlov YI, et al. Evolution and Diversification of Lamprey Antigen Receptors: Evidence for Involvement of an AID-APOBEC Family Cytosine Deaminase. Nat Immunol (2007) 8(6):647–56. doi: 10.1038/ni1463
18. Trancoso I, Morimoto R, Boehm T. Co-Evolution of Mutagenic Genome Editors and Vertebrate Adaptive Immunity. Curr Opin Immunol (2020) 65:32–41. doi: 10.1016/j.coi.2020.03.001
19. Ott JA, Castro CD, Deiss TC, Ohta Y, Flajnik MF, Criscitiello MF. Somatic Hypermutation of T Cell Receptor Alpha Chain Contributes to Selection in Nurse Shark Thymus. Elife (2018) 7. doi: 10.7554/eLife.28477
20. Holland SJ, Berghuis LM, King JJ, Iyer LM, Sikora K, Fifield H, et al. Expansions, Diversification, and Interindividual Copy Number Variations of AID/APOBEC Family Cytidine Deaminase Genes in Lampreys. Proc Natl Acad Sci USA (2018) 115(14):E3211–20. doi: 10.1073/pnas.1720871115
21. Quinlan EM, King JJ, Amemiya CT, Hsu E, Larijan M. Biochemical Regulatory Features of Activation-Induced Cytidine Deaminase Remain Conserved From Lampreys to Humans. Mol Cell Biol (2017) 37(20):00077–17. doi: 10.1128/MCB.00077-17
22. Zhang ZZ, Pannunzio NR, Lu Z, Hsu E, Yu K, Lieber MR. The Repetitive Portion of the Xenopus Igh Mu Switch Region Mediates Orientation-Dependent Class Switch Recombination. Mol Immunol (2015) 67(2 Pt B):524–31. doi: 10.1016/j.molimm.2015.07.039
23. Hirano M, Das S, Guo P, Cooper MD. The Evolution of Adaptive Immunity in Vertebrates. Adv Immunol (2015) 109:125–57. doi: 10.1016/B978-0-12-387664-5.00004-2
24. Magor BG. Antibody Affinity Maturation in Fishes-Our Current Understanding. Biol (Basel) (2015) 4(3):512–24. doi: 10.3390/biology4030512
25. Kato L, Stanlie A, Begum NA, Kobayashi M, Aida M, Honjo T, et al. An Evolutionary View of the Mechanism for Immune and Genome Diversity. J Immunol (2012) 188(8):3559–66. doi: 10.4049/jimmunol.1102397
26. Dancyger AM, King JJ, Quinlan MJ, Fifield H, Tucker S, Sauders HL. Differences in the Enzymatic Efficiency of Human and Bony Fish AID are Mediated by a Single Residue in the C Terminus Modulating Single-Stranded DNA Binding. FASEB J (2012) 26(4):1517–25. doi: 10.1096/fj.11-198135
27. Zhu C, Hsu E. Error-Prone DNA Repair Activity During Somatic Hypermutation in Shark B Lymphocytes. J Immunol (2010) 185(9):5336–47. doi: 10.4049/jimmunol.1000779
28. Basu U, Chaudhuri J, Phan RT, Datta A. Regulation of Activation Induced Deaminase Via Phosphorylation. Adv Exp Med Biol (2007) 596:129–37. doi: 10.1007/0-387-46530-8_11
29. Arakawa H, Buerstedde JM. Activation-Induced Cytidine Deaminase-Mediated Hypermutation in the DT40 Cell Line. Philos Trans R Soc Lond B Biol Sci (2009) 364(1517):639–44. doi: 10.1098/rstb.2008.0202
30. Basu U, Wang Y, Alt FW. Evolution of Phosphorylation-Dependent Regulation of Activation-Induced Cytidine Deaminase. Mol Cell (2008) 32(2):285–91. doi: 10.1016/j.molcel.2008.08.019
31. Ichikawa HT, Sowden MP, Torelli AT, Bachl J, Huang P, Dance GS, et al. Structural Phylogenetic Analysis of Activation-Induced Deaminase Function. J Immunol (2006) 177(1):355–61. doi: 10.4049/jimmunol.177.1.355
32. Wakae K, Magor BG, Saunders H, Nagaoka H, Kawamura A, Kinoshita K, et al. Evolution of Class Switch Recombination Function in Fish Activation-Induced Cytidine Deaminase, AID. Int Immunol (2006) 18(1):41–7. doi: 10.1093/intimm/dxh347
33. Barreto VM, Pan-Hammarstrom Q, Zhao Y, Hammarstrom L, Misulovin Z, Nussenzweig MC. AID From Bony Fish Catalyzes Class Switch Recombination. J Exp Med (2005) 202(6):733–8. doi: 10.1084/jem.20051378
34. Stavnezer J, Amemiya CT. Evolution of Isotype Switching. Semin Immunol (2004) 16(4):257–75. doi: 10.1016/j.smim.2004.08.005
35. Ott JA, Harrison J, Flajnik MF, Criscitiello MF. Nurse Shark T-cell Receptors Employ Somatic Hypermutation Preferentially to Alter Alpha/Delta Variable Segments Associated With Alpha Constant Region. Eur J Immunol (2020) 50(9):1307–20. doi: 10.1002/eji.201948495
36. Bilal S, Lie KK, Saele O, Hord I. T Cell Receptor Alpha Chain Genes in the Teleost Ballan Wrasse (Labrus Bergylta) Are Subjected to Somatic Hypermutation. Front Immunol (2018) 9:1101. doi: 10.3389/fimmu.2018.01101
37. Zhu C, Lee V, Finn A, Senger KT, Zarrin AA, Du Pasquier L, et al. Origin of Immunoglobulin Isotype Switching. Curr Biol (2012) 22(10):872–80. doi: 10.1016/j.cub.2012.03.060
38. Zhu C, Feng W, Weedon J, Hua P, Stefanov D, Ohta Y, et al. The Multiple Shark Ig H Chain Genes Rearrange and Hypermutate Autonomously. J Immunol (2011) 187(5):2492–501. doi: 10.4049/jimmunol.1101671
39. Abdouni H, King JJ, Suliman M, Quinlan M, Fifield H, Larijani M. Zebrafish AID is Capable of Deaminating Methylated Deoxycytidines. Nucleic Acids Res (2013) 41(10):5457–68. doi: 10.1093/nar/gkt212
40. Saunders HL, Magor BG. Cloning and Expression of the AID Gene in the Channel Catfish. Dev Comp Immunol (2004) 28(7–8):657–63. doi: 10.1016/j.dci.2004.01.002
41. Saunders HL, Oko AL, Scott AN, Fan CW, Magor BG. The Cellular Context of AID Expressing Cells in Fish Lymphoid Tissues. Dev Comp Immunol (2010) 34(6):669–76. doi: 10.1016/j.dci.2010.01.013
42. Chatterji M, Unniraman S, McBride KM, Schatz DG. Role of Activation-Induced Deaminase Protein Kinase A Phosphorylation Sites in Ig Gene Conversion and Somatic Hypermutation. J Immunol (2007) 179(8):5274–80. doi: 10.4049/jimmunol.179.8.5274
43. Yang F, Waldbieser GC, Lobb CJ. The Nucleotide Targets of Somatic Mutation and the Role of Selection in Immunoglobulin Heavy Chains of a Teleost Fish. J Immunol (2006) 176(3):1655–67. doi: 10.4049/jimmunol.176.3.1655
44. Zhao Y, Pan-Hammarstr√∂m Q, Zhao Z, Hammarstr√∂m L. Identification of the Activation-Induced Cytidine Deaminase Gene From Zebrafish: An Evolutionary Analysis. Dev Comp Immunol (2005) 29(1):61–71. doi: 10.1016/j.dci.2004.05.005
45. Lada AG, Dhar A, Boissy RJ, Hirano M, Rubel AA, Rogozin IB, et al. AID/APOBEC Cytosine Deaminase Induces Genome-Wide Kataegis. Biol Direct (2012) 7:47. doi: 10.1186/1745-6150-7-47
46. Kasahara M. Variable Lymphocyte Receptors: A Current Overview. Results Probl Cell Differ (2015) 57:175–92. doi: 10.1007/978-3-319-20819-0_8
47. Lada AG, Stepchenkova EI, Waisertreiger IS, Noskov VN, Dhar A, Eudy JD, et al. Genome-Wide Mutation Avalanches Induced in Diploid Yeast Cells by a Base Analog or an APOBEC Deaminase. PloS Genet (2013) 9(9):e1003736. doi: 10.1371/journal.pgen.1003736
48. Bajoghli B, et al. A Thymus Candidate in Lampreys. Nature (2011) 470(7332):90–4. doi: 10.1038/nature09655
49. Barreto VM, Magor BG. Activation-Induced Cytidine Deaminase Structure and Functions: A Species Comparative View. Dev Comp Immunol (2011) 35(9):991–1007. doi: 10.1016/j.dci.2011.02.005
50. Bascove M, Frippiat JP. Molecular Characterization of Pleurodeles Waltl Activation-Induced Cytidine Deaminase. Mol Immunol (2010) 47(7-8):1640–9. doi: 10.1016/j.molimm.2010.01.005
51. Marr S, Morales H, Bottaro A, Cooper M, Flajnik M, Robert J, et al. Localization and Differential Expression of Activation-Induced Cytidine Deaminase in the Amphibian Xenopus Upon Antigen Stimulation and During Early Development. J Immunol (2007) 179(10):6783–9. doi: 10.4049/jimmunol.179.10.6783
52. King JJ, Manuel CA, Barrett CV, Raber S, Lucas H, Sutter P, et al. Catalytic Pocket Inaccessibility of Activation-Induced Cytidine Deaminase is a Safeguard Against Excessive Mutagenic Activity. Structure (2015) 23(4):615–27. doi: 10.1016/j.str.2015.01.016
53. King JJ LM. A Novel Intrinsic Regulator of AID/APOBECs: Schrödinger’s CATalytic Pocket. Front Immunol (2017) 8:351. doi: 10.3389/fimmu.2017.00351
54. Morimoto R, O'Meara CP, Holland SJ, Trancoso I, Souissi A, Schorpp M, et al. Cytidine Deaminase 2 is Required for VLRB Antibody Gene Assembly in Lampreys. Sci Immunol (2020) 5(45). doi: 10.1126/sciimmunol.aba0925
55. Costello R, Cantillo JF, Kenter AL. Chicken MBD4 Regulates Immunoglobulin Diversification by Somatic Hypermutation. Front Immunol (2019) 10:2540. doi: 10.3389/fimmu.2019.02540
56. Liu MC, Liao WY, Buckley KM, Yang SY, Rast JP, Fugmann SD. AID/APOBEC-Like Cytidine Deaminases are Ancient Innate Immune Mediators in Invertebrates. Nat Commun (2018) 9(1):1948. doi: 10.1038/s41467-018-04273-x
57. Patel B, Banerjee R, Samanta M, Das S. Diversity of Immunoglobulin (Ig) Isotypes and the Role of Activation-Induced Cytidine Deaminase (AID) in Fish. Mol Biotechnol (2018) 60(6):435–53. doi: 10.1007/s12033-018-0081-8
58. Villota-Herdoiza D, Pila EA, Quiniou S, Waldbieser GC, Magor BG. Transcriptional Regulation of Teleost Aicda Genes. Part 1 - Suppressors of Promiscuous Promoters. Fish Shellfish Immunol (2013) 35(6):1981–7. doi: 10.1016/j.fsi.2013.09.035
59. Verma S, Goldammer T, Aitken R. Cloning and Expression of Activation Induced Cytidine Deaminase From Bos Taurus. Vet Immunol Immunopathol 134(3-4):151–9. doi: 10.1016/j.vetimm.2009.08.016
60. Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR, et al. DNA Demethylation in Zebrafish Involves the Coupling of a Deaminase, a Glycosylase, and Gadd45. Cell (2008) 135(7):1201–12. doi: 10.1016/j.cell.2008.11.042
61. Moon SY, Eun HJ, Baek SK, Jin SJ, Kim TS, Kim SW, et al. Activation-Induced Cytidine Deaminase Induces Dna Demethylation of Pluripotency Genes in Bovine Differentiated Cells. Cell Reprogram (2016) 18(5):298–308. doi: 10.1089/cell.2015.0076
62. Basu U, Franklin A, Schwer B, Cheng HL, Chaudhuri J, Alt FW. Regulation of Activation-Induced Cytidine Deaminase DNA Deamination Activity in B-cells by Ser38 Phosphorylation. Biochem Soc Trans (2009) 37(Pt 3):561–8. doi: 10.1042/BST0370561
63. Methot SP, et al. Consecutive Interactions With HSP90 and eEF1A Underlie a Functional Maturation and Storage Pathway of AID in the Cytoplasm. J Exp Med (2015) 212(4):581–96. doi: 10.1084/jem.20141157
64. Zhang J, Webb DM. Rapid Evolution of Primate Antiviral Enzyme APOBEC3G. Hum Mol Genet (2004) 13(16):1785–91. doi: 10.1093/hmg/ddh183
65. Harris RS, Liddament MT. Retroviral Restriction by APOBEC Proteins. Nat Rev Immunol (2004) 4(11):868–77. doi: 10.1038/nri1489
66. Smith HC. APOBEC3G: A Double Agent in Defense. Trends Biochem Sci (2011) 36(5):239–44. doi: 10.1016/j.tibs.2010.12.003
67. Monajemi M, Woodworth CF, Benkaroun J, Grant M, Larijani M. Emerging Complexities of APOBEC3G Action on Immunity and Viral Fitness During HIV Infection and Treatment. Retrovirology (2012) 9:35. doi: 10.1186/1742-4690-9-35
68. Borzooee F, Asgharpour M, Quinlan E, Grant MD, Larijani M. Viral Subversion of APOBEC3s: Lessons for Anti-Tumor Immunity and Tumor Immunotherapy. Int Rev Immunol (2018) 37(3):151–64. doi: 10.1080/08830185.2017.1403596
69. Pujantell M, Riveira-Munoz E, Badia R, Castellvi M, Garcia-Vidal E, Sirera G, et al. RNA Editing by ADAR1 Regulates Innate and Antiviral Immune Functions in Primary Macrophages. Sci Rep (2017) 7(1):13339. doi: 10.1038/s41598-017-13580-0
70. Lamers MM, van den Hoogen BG, Haagmans BL. Adar1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front Immunol (2019) 10:1763. doi: 10.3389/fimmu.2019.01763
71. Sabag O, Zamir A, Keshet I, Hecht M, Ludwig G, Tabib A, et al. Establishment of Methylation Patterns in ES Cells. Nat Struct Mol Biol (2014) 21(1):110–2. doi: 10.1038/nsmb.2734
72. Samuel CE. Adenosine Deaminases Acting on RNA (Adars) are Both Antiviral and Proviral. Virology (2011) 411(2):180–93. doi: 10.1016/j.virol.2010.12.004
73. Samuel CE. Adars: Viruses and Innate Immunity. Curr Top Microbiol Immunol (2012) 353:163–95. doi: 10.1007/82_2011_148
74. Alder MN, et al. Antibody Responses of Variable Lymphocyte Receptors in the Lamprey. Nat Immunol (2008) 9(3):319–27. doi: 10.1038/ni1562
75. Das S, Sutoh Y, Hirano M, Han Q, Li J, Cooper MD, et al. Characterization of Lamprey BAFF-Like Gene: Evolutionary Implications. J Immunol (2016) 197(7):2695–703. doi: 10.4049/jimmunol.1600799
76. Holland SJ, Gao M, Hirano M, Iyer LM, Luo M, Schorpp M, et al. Selection of the Lamprey VLRC Antigen Receptor Repertoire. Proc Natl Acad Sci USA (2014) 111(41):14834–9. doi: 10.1073/pnas.1415655111
77. Pancer Z, Amemiya CT, Ehrhardt GRA, Ceitlin J, Gartland GL, Cooper MD. Pillars Article: Somatic Diversification of Variable Lymphocyte Receptors in the Agnathan Sea Lamprey. Nature. 2004. 430: 174-180. J Immunol (2018) 201(5):1336–42. doi: 10.1038/nature02740
78. Pancer Z, Mayer WE, Klein J, Cooper MD. Prototypic T Cell Receptor and CD4-like Coreceptor are Expressed by Lymphocytes in the Agnathan Sea Lamprey. Proc Natl Acad Sci USA (2004) 101(36):13273–8. doi: 10.1073/pnas.0405529101
79. Thomson DW, Shahrin NH, Wang PPS, Wadham C, Shanmuganathan N, Scott HS, et al. Aberrant RAG-mediated Recombination Contributes to Multiple Structural Rearrangements in Lymphoid Blast Crisis of Chronic Myeloid Leukemia. Leukemia (2020). doi: 10.1038/s41375-020-0751-y
80. Hu J, Zhang Y, Zhao L, Frock RL, Du Z, Meyers RM, et al. Chromosomal Loop Domains Direct the Recombination of Antigen Receptor Genes. Cell (2015) 163(4):947–59. doi: 10.1016/j.cell.2015.10.016
81. Heinaniemi M, Vuorenmaa T, Teppo S, Kaikkonen MU, Bouvy-Liivrand M, Mehtonen J, et al. Transcription-Coupled Genetic Instability Marks Acute Lymphoblastic Leukemia Structural Variation Hotspots. Elife (2016) 5. doi: 10.7554/eLife.13087
82. Shimazaki N, Tsai AG, Lieber MR. H3k4me3 Stimulates the V(D)J RAG Complex for Both Nicking and Hairpinning in Trans in Addition to Tethering in Cis: Implications for Translocations. Mol Cell (2009) 34(5):535–44. doi: 10.1016/j.molcel.2009.05.011
83. Mijuskovic M, Chou YF, Gigi V, Lindsay CR, Shestova O, Lewis SM, et al. Off-Target V(D)J Recombination Drives Lymphomagenesis and Is Escalated by Loss of the Rag2 C Terminus. Cell Rep (2015) 12(11):1842–52. doi: 10.1016/j.celrep.2015.08.034
84. Schlissel MS, Kaffer CR, Curry JD. Leukemia and Lymphoma: A Cost of Doing Business for Adaptive Immunity. Genes Dev (2006) 20(12):1539–44. doi: 10.1101/gad.1446506
85. Kirkham CM, Scott JNF, Wang X, Smith AL, Kupinski AP, Ford AM, et al. Cut-and-Run: A Distinct Mechanism by Which V(D)J Recombination Causes Genome Instability. Mol Cell (2019) 74(3):584–97.e9. doi: 10.1016/j.molcel.2019.02.025
86. Walker BA, Wardell CP, Johnson DC, Kaiser MF, Begum DB, Dahir NB, et al. Characterization of IGH Locus Breakpoints in Multiple Myeloma Indicates a Subset of Translocations Appear to Occur in Pregerminal Center B Cells. Blood (2013) 121(17):3413–9. doi: 10.1182/blood-2012-12-471888
87. Vaandrager JW, Schuuring E, Philippo K, Kluin PM. V(D)J Recombinase-Mediated Transposition of the BCL2 Gene to the IGH Locus in Follicular Lymphoma. Blood (2000) 96(5):1947–52. doi: 10.1182/blood.V96.5.1947
88. Choudhary M, Tamrakar A, Singh AK, Jain M, Jaiswal A, Kodgire P. Aid Biology: A Pathological and Clinical Perspective. Int Rev Immunol (2018) 37(1):37–56. doi: 10.1080/08830185.2017.1369980
89. Klemm L, Duy C, Iacobucci I, Kuchen S, von Levetzow G, Feldhahn N, et al. The B Cell Mutator AID Promotes B Lymphoid Blast Crisis and Drug Resistance in Chronic Myeloid Leukemia. Cancer Cell (2009) 16(3):232–45. doi: 10.1016/j.ccr.2009.07.030
90. Seifert M, Scholtysik R, Kuppers R. Origin and Pathogenesis of B Cell Lymphomas. Methods Mol Biol (2019) 1956:1–33. doi: 10.1007/978-1-4939-9151-8_1
91. Takizawa M, Tolarova H, Li Z, Dubois W, Lim S, Callen E, et al. AID Expression Levels Determine the Extent of cMyc Oncogenic Translocations and the Incidence of B Cell Tumor Development. J Exp Med (2008) 205(9):1949–57. doi: 10.1084/jem.20081007
92. Burns A, Alsolami R, Becq J, Timbs A, Bruce D, Robbe P, et al. Whole-Genome Sequencing of Chronic Lymphocytic Leukaemia Reveals Distinct Differences in the Mutational Landscape Between IgHVmut and IgHVunmut Subgroups. Leukemia (2017). doi: 10.1038/leu.2017.311
93. Mohri T, Nagata K, Kuwamoto S, Matsushita M, Sugihara H, Kato M, et al. Aberrant Expression of AID and AID Activators of NF-kappaB and PAX5 is Irrelevant to EBV-associated Gastric Cancers, But is Associated With Carcinogenesis in Certain EBV-non-associated Gastric Cancers. Oncol Lett (2017) 13(6):4133–40. doi: 10.3892/ol.2017.5978
94. Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon SM, et al. Mechanisms of Clonal Evolution in Childhood Acute Lymphoblastic Leukemia. Nat Immunol (2015) 16(7):766–74. doi: 10.1038/ni.3160
95. Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, et al. AID is Required for Germinal Center-Derived Lymphomagenesis. Nat Genet (2008) 40(1):108–12. doi: 10.1038/ng.2007.35
96. Kotani A, Kakazu N, Tsuruyama T, Okazaki IM, Muramatsu M, Kinoshita K, et al. Activation-Induced Cytidine Deaminase (AID) Promotes B Cell Lymphomagenesis in Emu-cmyc Transgenic Mice. Proc Natl Acad Sci USA (2007) 104(5):1616–20. doi: 10.1073/pnas.0610732104
97. Lindley RA, Humbert P, Larner C, Akmeemana EH, Pendlebury CR. Association Between Targeted Somatic Mutation (TSM) Signatures and HGS-OvCa Progression. Cancer Med (2016) 5(9):2629–40. doi: 10.1002/cam4.825
98. Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, et al. APOBEC3B Upregulation and Genomic Mutation Patterns in Serous Ovarian Carcinoma. Cancer Res (2013) 73(24):7222–31. doi: 10.1158/0008-5472.CAN-13-1753
99. Ruder U, Denkert C, Kunze CA, Jank P, Lindner J, Johrens K, et al. APOBEC3B Protein Expression and mRNA Analyses in Patients With High-Grade Serous Ovarian Carcinoma. Histol Histopathol (2019) 34(4):405–17.
100. Zou J, Wang C, Ma X, Wang E, Peng G. APOBEC3B, a Molecular Driver of Mutagenesis in Human Cancers. Cell Biosci (2017) 7:29. doi: 10.1186/s13578-017-0156-4
101. Sasaki H, Suzuki A, Tatematsu T, Shitara M, Hikosaka Y, Okud K, et al. APOBEC3B Gene Overexpression in non-Small-Cell Lung Cancer. BioMed Rep (2014) 2(3):392–5. doi: 10.3892/br.2014.256
102. Swanton C, McGranahan N, Starrett GJ, Harris RS. Apobec Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discovery (2015) 5(7):704–12. doi: 10.1158/2159-8290.CD-15-0344
103. Siriwardena SU, Chen K, Bhagwat AS. Functions and Malfunctions of Mammalian Dna-Cytosine Deaminases. Chem Rev (2016) 116(20):12688–710. doi: 10.1021/acs.chemrev.6b00296
104. Burns MB, et al. APOBEC3B is an Enzymatic Source of Mutation in Breast Cancer. Nature (2013) 494(7437):366–70. doi: 10.1038/nature11881
105. Roberts SA, Gordenin DA. Hypermutation in Human Cancer Genomes: Footprints and Mechanisms. Nat Rev Cancer (2014) 14(12):786–800. doi: 10.1038/nrc3816
106. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of Mutational Processes in Human Cancer. Nature (2013) 500(7463):415–21.
107. Starrett GJ, Luengas EM, McCann JL, Ebrahimi D, Temiz NA, Love RP, et al. The DNA Cytosine Deaminase APOBEC3H Haplotype I Likely Contributes to Breast and Lung Cancer Mutagenesis. Nat Commun (2016) 7:12918. doi: 10.1038/ncomms12918
108. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences. Nature (2016) 534(7605):47–54.
109. Chan TH, Qamra A, Tan KT, Guo J, Yang H, Qi L, et al. Adar-Mediated RNA Editing Predicts Progression and Prognosis of Gastric Cancer. Gastroenterology (2016) 151(4):637–50.e10. doi: 10.1053/j.gastro.2016.06.043
110. Wang C, Zou J, Ma X, Wang E, Peng G. Mechanisms and Implications of ADAR-mediated RNA Editing in Cancer. Cancer Lett (2017) 411:27–34. doi: 10.1016/j.canlet.2017.09.036
111. Roberts JT, Patterson DG, King VM, Amin SV, Polska CJ, Houserova D, et al. Adar Mediated Rna Editing Modulates MicroRNA Targeting in Human Breast Cancer. Processes (Basel) (2018) 6(5). doi: 10.3390/pr6050042
112. Jiang Q, Isquith J, Ladel L, Mark A, Holm F, Mason C, et al. Inflammation-Driven Deaminase Deregulation Fuels Human Pre-Leukemia Stem Cell Evolution. Cell Rep (2021) 34(4):108670. doi: 10.1016/j.celrep.2020.108670
113. Law EK, Levin-Klein R, Jarvis MC, Kim H, Argyris PP, Carpenter MA, et al. APOBEC3A Catalyzes Mutation and Drives Carcinogenesis in Vivo. J Exp Med (2020) 217(12). doi: 10.1084/jem.20200261
114. Durandy A. Mini-Review Activation-induced Cytidine Deaminase: A Dual Role in Class-Switch Recombination and Somatic Hypermutation. Eur J Immunol (2003) 33(8):2069–73. doi: 10.1002/eji.200324133
115. Durandy A. Activation-Induced Cytidine Deaminase: A Dual Role in Class-Switch Recombination and Somatic Hypermutation. Eur J Immunol (2003) 33(8):2069–73. doi: 10.1002/eji.200324133
116. Durandy A, Peron S, Fischer A. Hyper-IgM Syndromes. Curr Opin Rheumatol (2006) 18(4):369–76. doi: 10.1097/01.bor.0000231905.12172.b5
117. Gennery A. Recent Advances in Understanding RAG Deficiencies. F1000Res (2019) 8. doi: 10.12688/f1000research.17056.1
118. Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG Mutations in Human B Cell-Negative SCID. Science (1996) 274(5284):97–9. doi: 10.1126/science.274.5284.97
119. Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, et al. Rag-2-deficient Mice Lack Mature Lymphocytes Owing to Inability to Initiate V(D)J Rearrangement. Cell (1992) 68(5):855–67. doi: 10.1016/0092-8674(92)90029-C
120. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE, et al. Rag-1-deficient Mice Have No Mature B and T Lymphocytes. Cell (1992) 68(5):869–77. doi: 10.1016/0092-8674(92)90030-G
121. Lada AG, Iyer LM, Rogozin IB, Aravind L, Pavlov Iu I. Vertebrate Immunity: Mutator Proteins and Their Evolution. Genetika (2007) 43(10):1311–27.
122. Rubio MA, Pastar I, Gaston KW, Ragone FL, Janzen CJ, Cross GA, et al. An Adenosine-to-Inosine tRNA-editing Enzyme That can Perform C-to-U Deamination of DNA. Proc Natl Acad Sci USA (2007) 104(19):7821–6. doi: 10.1073/pnas.0702394104
123. Gerber A, Grosjean A, Melcher T, Keller W. Tad1p, a Yeast tRNA-specific Adenosine Deaminase, is Related to the Mammalian pre-mRNA Editing Enzymes ADAR1 and ADAR2. EMBO J (1998) 17(16):4780–9. doi: 10.1093/emboj/17.16.4780
124. Gerber AP, Keller W. An Adenosine Deaminase That Generates Inosine At the Wobble Position of Trnas. Science (1999) 286(5442):1146–9. doi: 10.1126/science.286.5442.1146
125. Zhou W, Karcher D, Bock R. Importance of Adenosine-to-Inosine Editing Adjacent to the Anticodon in an Arabidopsis Alanine tRNA Under Environmental Stress. Nucleic Acids Res (2013) 41(5):3362–72. doi: 10.1093/nar/gkt013
126. Conticello SG, Langlois MA, Yang Z, Neuberger MS. DNA Deamination in Immunity: AID in the Context of its APOBEC Relatives. Adv Immunol (2007) 94:37–73. doi: 10.1016/S0065-2776(06)94002-4
127. Torres AG, Pineyro D, Filonava L, Stracker TH, Batlle E, Ribas de Pouplana L. A-to-I Editing on tRNAs: Biochemical, Biological and Evolutionary Implications. FEBS Lett (2014) 588(23):4279–86. doi: 10.1016/j.febslet.2014.09.025
128. Iyer LM, Zhang D, Rogozin IB, Aravind L. Evolution of the Deaminase Fold and Multiple Origins of Eukaryotic Editing and Mutagenic Nucleic Acid Deaminases From Bacterial Toxin Systems. Nucleic Acids Res (2011) 39(22):9473–97. doi: 10.1093/nar/gkr691
129. Qiao Q, Wang L, Meng FL, Hwang JK, Alt FW, Wu H. Aid Recognizes Structured DNA for Class Switch Recombination. Mol Cell (2017) 67(3):361–73.e4. doi: 10.1016/j.molcel.2017.06.034
130. Silvas TV, Schiffer CA. Apobec3s: DNA-editing Human Cytidine Deaminases. Protein Sci (2019) 28(9):1552–66. doi: 10.1002/pro.3670
131. Rogozin IB, Basu MK, Jordan IK, Pavlov YI, Koonin EV. APOBEC4, a New Member of the AID/APOBEC Family of Polynucleotide (Deoxy)Cytidine Deaminases Predicted by Computational Analysis. Cell Cycle (2005) 4(9):1281–5. doi: 10.4161/cc.4.9.1994
132. Liao W, Hong SH, Chan BH, Rudolph FB, Clark SC, Chan L. Apobec-2, a Cardiac- and Skeletal Muscle-Specific Member of the Cytidine Deaminase Supergene Family. Biochem Biophys Res Commun (1999) 260(2):398–404. doi: 10.1006/bbrc.1999.0925
133. Mikl MC, Watt IN, Lu M, Reik W, Davies SL, Neuberger MS, et al. Mice Deficient in APOBEC2 and APOBEC3. Mol Cell Biol (2005) 25(16):7270–7. doi: 10.1128/MCB.25.16.7270-7277.2005
134. Sato Y, Probst HC, Tatsumi R, Ikeuchi Y, Neuberger MS, Rad C. Deficiency in APOBEC2 Leads to a Shift in Muscle Fiber Type, Diminished Body Mass, and Myopathy. J Biol Chem (2010) 285(10):7111–8. doi: 10.1074/jbc.M109.052977
135. Vonica A, Rosa A, Arduini BL, Brivanlou AH. APOBEC2, a Selective Inhibitor of TGFbeta Signaling, Regulates Left-Right Axis Specification During Early Embryogenesis. Dev Biol (2011) 350(1):13–23. doi: 10.1016/j.ydbio.2010.09.016
136. Fujino T, Navaratnam N, Scott J. Human Apolipoprotein B RNA Editing Deaminase Gene (APOBEC1). Genomics (1998) 47(2):266–75. doi: 10.1006/geno.1997.5110
137. Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA Editing Enzyme APOBEC1 and Some of its Homologs can Act as DNA Mutators. Mol Cell (2002) 10(5):1247–53. doi: 10.1016/S1097-2765(02)00742-6
138. Nakamuta M, Oka K, Krushkal J, Kobayashi K, Yamamoto M, Li WH, et al. Alternative mRNA Splicing and Differential Promoter Utilization Determine Tissue-Specific Expression of the Apolipoprotein B mRNA-editing Protein (Apobec1) Gene in Mice. Structure and Evolution of Apobec1 and Related Nucleoside/Nucleotide Deaminases. J Biol Chem (1995) 270(22):13042–56. doi: 10.1074/jbc.270.22.13042
139. Nakamuta M, Chang BH, Zsigmond E, Kobayashi K, Lei H, Ishida BY, et al. Complete Phenotypic Characterization of Apobec-1 Knockout Mice With a Wild-Type Genetic Background and a Human Apolipoprotein B Transgenic Background, and Restoration of Apolipoprotein B mRNA Editing by Somatic Gene Transfer of Apobec-1. J Biol Chem (1996) 271(42):25981–8. doi: 10.1074/jbc.271.42.25981
140. Petersen-Mahrt SK, Neuberger MS. In Vitro Deamination of Cytosine to Uracil in Single-Stranded DNA by Apolipoprotein B Editing Complex Catalytic Subunit 1 (APOBEC1). J Biol Chem (2003) 278(22):19583–6. doi: 10.1074/jbc.C300114200
141. Petit V, Guetard D, Renard M, Keriel A, Sitbon M, Wain-Hobson S, et al. Murine APOBEC1 is a Powerful Mutator of Retroviral and Cellular RNA In Vitro and In Vivo. J Mol Biol (2009) 385(1):65–78. doi: 10.1016/j.jmb.2008.10.043
142. Sharma S, Patnaik SK, Taggart RT, Kannisto ED, Enriquez SM, Gollnick P, et al. APOBEC3A Cytidine Deaminase Induces RNA Editing in Monocytes and Macrophages. Nat Commun (2015) 6:6881. doi: 10.1038/ncomms7881
143. Abdouni HS, King JJ, Ghorbani A, Fifield H, Berghuis L, Larijani M. DNA/RNA Hybrid Substrates Modulate the Catalytic Activity of Purified AID. Mol Immunol (2018) 93:94–106. doi: 10.1016/j.molimm.2017.11.012
144. Sawyer SL, Emerman M, Malik HS. Ancient Adaptive Evolution of the Primate Antiviral DNA-editing Enzyme APOBEC3G. PloS Biol (2004) 2(9):E275. doi: 10.1371/journal.pbio.0020275
145. Tacchi L, Larragoite ET, Munoz P, Amemiya CT, Salinas I. African Lungfish Reveal the Evolutionary Origins of Organized Mucosal Lymphoid Tissue in Vertebrates. Curr Biol (2015) 25(18):2417–24. doi: 10.1016/j.cub.2015.07.066
146. Flajnik MF. Primitive Vertebrate Immunity: What is the Evolutionary Derivative of Molecules That Define the Adaptive Immune System? Ciba Found Symp (1994) 186:224–32; discussion 233-6. doi: 10.1002/9780470514658.ch13
147. Flajnik MF. The Immune System of Ectothermic Vertebrates. Vet Immunol Immunopathol (1996) 54(1-4):145–50. doi: 10.1016/S0165-2427(96)05685-1
148. Du Pasquier L. The Phylogenetic Origin of Antigen-Specific Receptors. Curr Top Microbiol Immunol (2000) 248:160–85. doi: 10.1007/978-3-642-59674-2_8
149. Hsu E, Pulham N, Rumfelt LL, Flajnik MF. The Plasticity of Immunoglobulin Gene Systems in Evolution. Immunol Rev (2006) 210:8–26. doi: 10.1111/j.0105-2896.2006.00366.x
150. Hirano M, Guo P, McCurley N, Schorpp M, Das S, Boehm T, et al. Evolutionary Implications of a Third Lymphocyte Lineage in Lampreys. Nature (2013) 501(7467):435–8. doi: 10.1038/nature12467
151. Chen H, Kshirsagar S, Jensen I, Lau K, Covarrubias R, Schluter SF, et al. Characterization of Arrangement and Expression of the T Cell Receptor Gamma Locus in the Sandbar Shark. Proc Natl Acad Sci USA (2009) 106(21):8591–6. doi: 10.1073/pnas.0811283106
152. Chen H, Kshirsagar S, Jensen I, Lau K, Simonson C, Schluter SF, et al. Characterization of Arrangement and Expression of the Beta-2 Microglobulin Locus in the Sandbar and Nurse Shark. Dev Comp Immunol (2010) 34(2):189–95. doi: 10.1016/j.dci.2009.09.008
153. Ciccarese S, Vaccarelli G, Lefranc MP, Tasco G, Consiglio A, Casadio R, et al. Characteristics of the Somatic Hypermutation in the Camelus Dromedarius T Cell Receptor Gamma (TRG) and Delta (TRD) Variable Domains. Dev Comp Immunol (2014) 46(2):300–13. doi: 10.1016/j.dci.2014.05.001
154. Tasumi S, Velikovsky CA, Xu G, Gai SA, Wittrup KD, Flajnik MF, et al. High-Affinity Lamprey VLRA and VLRB Monoclonal Antibodies. Proc Natl Acad Sci USA (2009) 106(31):12891–6. doi: 10.1073/pnas.0904443106
155. Waters EA, Shusta EV. The Variable Lymphocyte Receptor as an Antibody Alternative. Curr Opin Biotechnol (2018) 52:74–9. doi: 10.1016/j.copbio.2018.02.016
156. Moot R, Moot SS, Moot L, Moot M, Moot DE, Moot H, et al. Genetic Engineering of Chimeric Antigen Receptors Using Lamprey Derived Variable Lymphocyte Receptors. Mol Ther Oncolytics (2016) 3:16026. doi: 10.1038/mto.2016.26
157. Ghaffari SH, Lobb CJ. Structure and Genomic Organization of a Second Cluster of Immunoglobulin Heavy Chain Gene Segments in the Channel Catfish. J Immunol (1999) 162(3):1519–29.
158. Dooley H, Flajnik MF. Antibody Repertoire Development in Cartilaginous Fish. Dev Comp Immunol (2006) 30(1-2):43–56. doi: 10.1016/j.dci.2005.06.022
159. Hinds-Frey KR, Nishikata H, Litman RT, Litman GW, et al. Somatic Variation Precedes Extensive Diversification of Germline Sequences and Combinatorial Joining in the Evolution of Immunoglobulin Heavy Chain Diversity. J Exp Med (1993) 178(3):815–24. doi: 10.1084/jem.178.3.815
160. Malecek K, Brandman J, Brodsky JE, Ohta Y, Flajnik MF, Hsu E, et al. Somatic Hypermutation and Junctional Diversification At Ig Heavy Chain Loci in the Nurse Shark. J Immunol (2005) 175(12):8105–15. doi: 10.4049/jimmunol.175.12.8105
161. Dooley H, Flajnik MF. Shark Immunity Bites Back: Affinity Maturation and Memory Response in the Nurse Shark, Ginglymostoma Cirratum. Eur J Immunol (2005) 35(3):936–45. doi: 10.1002/eji.200425760
162. Diaz M, Greenberg AS, Flajnik MF. Somatic Hypermutation of the New Antigen Receptor Gene (NAR) in the Nurse Shark Does Not Generate the Repertoire: Possible Role in Antigen-Driven Reactions in the Absence of Germinal Centers. Proc Natl Acad Sci USA (1998) 95(24):14343–8. doi: 10.1073/pnas.95.24.14343
163. Eason DD, Cannon JP, Haire RN, Rast JP, Ostrov DA, Litman GW, et al. Mechanisms of Antigen Receptor Evolution. Semin Immunol (2004) 16(4):215–26. doi: 10.1016/j.smim.2004.08.001
164. Eason DD, Litman RT, Luer CA, Kerr W, Litman GW, et al. Expression of Individual Immunoglobulin Genes Occurs in an Unusual System Consisting of Multiple Independent Loci. Eur J Immunol (2004) 34(9):2551–8. doi: 10.1002/eji.200425224
165. Greenberg AS, Avila D, Hughes M, Hughes A, McKinney EC, Flajnik MF, et al. A New Antigen Receptor Gene Family That Undergoes Rearrangement and Extensive Somatic Diversification in Sharks. Nature (1995) 374(6518):168–73. doi: 10.1038/374168a0
166. Kaattari SL, Zhang HL, Khor IW, Kaattari IM, Shapiro DA, et al. Affinity Maturation in Trout: Clonal Dominance of High Affinity Antibodies Late in the Immune Response. Dev Comp Immunol (2002) 26(2):191–200. doi: 10.1016/S0145-305X(01)00064-7
167. Cain KD, Jones DR, Raison RL. Antibody-Antigen Kinetics Following Immunization of Rainbow Trout (Oncorhynchus Mykiss) With a T-cell Dependent Antigen. Dev Comp Immunol (2002) 26(2):181–90. doi: 10.1016/S0145-305X(01)00063-5
168. Marianes AE, Zimmerman AM. Targets of Somatic Hypermutation Within Immunoglobulin Light Chain Genes in Zebrafish. Immunology (2011) 132(2):240–55. doi: 10.1111/j.1365-2567.2010.03358.x
169. Neely HR, Flajnik MF. Emergence and Evolution of Secondary Lymphoid Organs. Annu Rev Cell Dev Biol (2016) 32:693–711. doi: 10.1146/annurev-cellbio-111315-125306
170. Bengten E, Clem LW, Miller NW, Warr GW, Wilson M, et al. Channel Catfish Immunoglobulins: Repertoire and Expression. Dev Comp Immunol (2006) 30(1-2):77–92. doi: 10.1016/j.dci.2005.06.016
171. Danilova N, Bussmann J, Jekosch K, Steiner LA, et al. The Immunoglobulin Heavy-Chain Locus in Zebrafish: Identification and Expression of a Previously Unknown Isotype, Immunoglobulin Z. Nat Immunol (2005) 6(3):295–302. doi: 10.1038/ni1166
172. Hansen JD, Landis ED, Phillips RB. Discovery of a Unique Ig Heavy-Chain Isotype (IgT) in Rainbow Trout: Implications for a Distinctive B Cell Developmental Pathway in Teleost Fish. Proc Natl Acad Sci USA (2005) 102(19):6919–24. doi: 10.1073/pnas.0500027102
173. Zhang T, Tacchi L, Wei Z, Zhao Y, Salinas I, et al. Intraclass Diversification of Immunoglobulin Heavy Chain Genes in the African Lungfish. Immunogenetics (2014) 66(5):335–51. doi: 10.1007/s00251-014-0769-2
174. Zahn A, Eranki AK, Patenaude AM, Methot SP, Fifield H, Cortizas EM, et al. Activation Induced Deaminase C-terminal Domain Links DNA Breaks to End Protection and Repair During Class Switch Recombination. Proc Natl Acad Sci USA (2014) 111(11):E988–97. doi: 10.1073/pnas.1320486111
175. Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR, et al. R-Loops At Immunoglobulin Class Switch Regions in the Chromosomes of Stimulated B Cells. Nat Immunol (2003) 4(5):442–51. doi: 10.1038/ni919
176. Zarrin AA, Del Vecchio C, Tseng E, Gleason M, Zarin P, Tian M, et al. Antibody Class Switching Mediated by Yeast Endonuclease-Generated DNA Breaks. Science (2007) 315(5810):377–81. doi: 10.1126/science.1136386
177. Patenaude AM, Orthwein A, Hu Y, Campo VA, Kavli B, Buschiazzo A, et al. Active Nuclear Import and Cytoplasmic Retention of Activation-Induced Deaminase. Nat Struct Mol Biol (2009) 16(5):517–27. doi: 10.1038/nsmb.1598
178. Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath G, et al. The AID Antibody Diversification Enzyme is Regulated by Protein Kinase A Phosphorylation. Nature (2005) 438(7067):508–11. doi: 10.1038/nature04255
179. Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, et al. B Cell Super-Enhancers and Regulatory Clusters Recruit AID Tumorigenic Activity. Cell (2014) 159(7):1524–37. doi: 10.1016/j.cell.2014.11.013
180. Meng FL, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon KR, et al. Convergent Transcription At Intragenic Super-Enhancers Targets AID-initiated Genomic Instability. Cell (2014) 159(7):1538–48.
181. Branton SA, Ghorbani A, Bolt BN, Fifield H, Berghuis LM, Larijani M, et al. Activation-Induced Cytidine Deaminase can Target Multiple Topologies of Double-Stranded DNA in a Transcription-Independent Manner. FASEB J (2020) 34(7):9245–68. doi: 10.1096/fj.201903036RR
182. Zheng S, Vuong BQ, Vaidyanathan B, Lin JY, Huang FT, Chaudhuri J, et al. Non-Coding RNA Generated Following Lariat Debranching Mediates Targeting of AID to DNA. Cell (2015) 161(4):762–73. doi: 10.1016/j.cell.2015.03.020
183. Zarrin AA, Alt FW, Chaudhuri J, Stokes N, Kaushal D, Du Pasquier L, et al. An Evolutionarily Conserved Target Motif for Immunoglobulin Class-Switch Recombination. Nat Immunol (2004) 5(12):1275–81. doi: 10.1038/ni1137
184. Mussmann R, Courtet M, Schwager J, Du Pasquier L, et al. Microsites for Immunoglobulin Switch Recombination Breakpoints From Xenopus to Mammals. Eur J Immunol (1997) 27(10):2610–9. doi: 10.1002/eji.1830271021
185. Lundqvist ML, Middleton DL, Radford C, Warr GW, Magor KE, et al. Immunoglobulins of the non-Galliform Birds: Antibody Expression and Repertoire in the Duck. Dev Comp Immunol (2006) 30(1-2):93–100. doi: 10.1016/j.dci.2005.06.019
186. Huang T, Zhang M, Wei Z, Wang P, Sun Y, Hu X, et al. Analysis of Immunoglobulin Transcripts in the Ostrich Struthio Camelus, a Primitive Avian Species. PloS One (2012) 7(3):e34346. doi: 10.1371/journal.pone.0034346
187. Reynaud CA, Anquez V, Dahan A, Weill JC, et al. A Single Rearrangement Event Generates Most of the Chicken Immunoglobulin Light Chain Diversity. Cell (1985) 40(2):283–91. doi: 10.1016/0092-8674(85)90142-4
188. Arakawa H, Hauschild J, Buerstedde JM. Requirement of the Activation-Induced Deaminase (Aid) Gene for Immunoglobulin Gene Conversion. Science (2002) 295(5558):1301–6. doi: 10.1126/science.1067308
189. Larijani M, Petrov AP, Kolenchenko O, Berru M, Krylov SN, Martin A, et al. AID Associates With Single-Stranded DNA With High Affinity and a Long Complex Half-Life in a Sequence-Independent Manner. Mol Cell Biol (2007) 27(1):20–30. doi: 10.1128/MCB.00824-06
190. Bransteitter R, Pham P, Scharff MD, Goodman MF, et al. Activation-Induced Cytidine Deaminase Deaminates Deoxycytidine on Single-Stranded DNA But Requires the Action of Rnase. Proc Natl Acad Sci USA (2003) 100(7):4102–7. doi: 10.1073/pnas.0730835100
191. Dickerson SK, Market E, Besmer E, Papavasiliou FN, et al. Aid Mediates Hypermutation by Deaminating Single Stranded Dna. J Exp Med (2003) 197(10):1291–6. doi: 10.1084/jem.20030481
192. Sohail A, Klapacz J, Samaranayake M, Ullah A, Bhagwat AS, et al. Human Activation-Induced Cytidine Deaminase Causes Transcription-Dependent, Strand-Biased C to U Deaminations. Nucleic Acids Res (2003) 31(12):2990–4. doi: 10.1093/nar/gkg464
193. Larijani M, Martin A. Single-Stranded DNA Structure and Positional Context of the Target Cytidine Determine the Enzymatic Efficiency of AID. Mol Cell Biol (2007) 27(23):8038–48. doi: 10.1128/MCB.01046-07
194. King JJ, Larijani M. Structural Plasticity of Substrate Selection by Activation-Induced Cytidine Deaminase as a Regulator of its Genome-Wide Mutagenic Activity. FEBS Lett (2020) 595(1):3–13. doi: 10.1002/1873-3468.13962
195. Ramiro AR, Barreto VM. Activation-Induced Cytidine Deaminase and Active Cytidine Demethylation. Trends Biochem Sci (2015) 40(3):172–81. doi: 10.1016/j.tibs.2015.01.006
196. Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-Induced Cytidine Deaminase Deaminates 5-Methylcytosine in DNA and is Expressed in Pluripotent Tissues: Implications for Epigenetic Reprogramming. J Biol Chem (2004) 279(50):52353–60. doi: 10.1074/jbc.M407695200
197. Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM, et al. Reprogramming Towards Pluripotency Requires AID-dependent DNA Demethylation. Nature (2010) 463(7284):1042–7. doi: 10.1038/nature08752
198. Dominguez PM, Teater M, Chambwe N, Kormaksson M, Redmond D, Ishii J, et al. Dna Methylation Dynamics of Germinal Center B Cells Are Mediated by AID. Cell Rep (2015) 12(12):2086–98. doi: 10.1016/j.celrep.2015.08.036
199. Munoz DP, Lee EL, Takayama S, Coppe JP, Heo SJ, Boffelli D, et al. Activation-Induced Cytidine Deaminase (AID) is Necessary for the Epithelial-Mesenchymal Transition in Mammary Epithelial Cells. Proc Natl Acad Sci USA (2013) 110(32):E2977–86. doi: 10.1073/pnas.1301021110
200. Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, et al. Genome-Wide Erasure of DNA Methylation in Mouse Primordial Germ Cells is Affected by AID Deficiency. Nature (2010) 463(7284):1101–5. doi: 10.1038/nature08829
201. Kumar R, DiMenna L, Schrode N, Liu TC, Franck P, Munoz-Descalzo S, et al. AID Stabilizes Stem-Cell Phenotype by Removing Epigenetic Memory of Pluripotency Genes. Nature (2013) 500(7460):89–92. doi: 10.1038/nature12299
202. Larijani M, Frieder D, Sonbuchner TM, Bransteitter R, Goodman MF, Bouhassira EE, et al. Methylation Protects Cytidines From AID-mediated Deamination. Mol Immunol (2005) 42(5):599–604. doi: 10.1016/j.molimm.2004.09.007
203. Wijesinghe P, Bhagwat AS. Efficient Deamination of 5-Methylcytosines in DNA by Human APOBEC3A, But Not by AID or APOBEC3G. Nucleic Acids Res (2012) 40(18):9206–17. doi: 10.1093/nar/gks685
204. Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, et al. AID/APOBEC Deaminases Disfavor Modified Cytosines Implicated in DNA Demethylation. Nat Chem Biol (2012) 8(9):751–8. doi: 10.1038/nchembio.1042
205. Quinlan EM, King JJ, Amemiya CT, Hsu E, Larijani M. Biochemical Regulatory Features of AID Remain Conserved From Lamprey to Humans. Mol Cell Biol (2017) 37(15). doi: 10.1128/MCB.00077-17
206. Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, et al. Improved Protein Structure Prediction Using Potentials From Deep Learning. Nature (2020) 577(7792):706–10. doi: 10.1038/s41586-019-1923-7
207. Heo L, Feig M. High-Accuracy Protein Structures by Combining Machine-Learning With Physics-Based Refinement. Proteins (2020) 88(5):637–42. doi: 10.1002/prot.25847
208. AlQuraishi M. AlphaFold At CASP13. Bioinformatics (2019) 35(22):4862–5. doi: 10.1093/bioinformatics/btz422
209. Granata D, Ponzoni L, Micheletti C, Carnevale V, et al. Patterns of Coevolving Amino Acids Unveil Structural and Dynamical Domains. Proc Natl Acad Sci USA (2017) 114(50):E10612–21. doi: 10.1073/pnas.1712021114
210. Feng J, Shukla D. Fingerprintcontacts: Predicting Alternative Conformations of Proteins From Coevolution. J Phys Chem B (2020) 124(18):3605–15. doi: 10.1021/acs.jpcb.9b11869
211. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR, et al. Programmable Editing of a Target Base in Genomic DNA Without Double-Stranded DNA Cleavage. Nature (2016) 533(7603):420–4. doi: 10.1038/nature17946
212. Yu Y, Leete TC, Born DA, Young L, Barrera LA, Lee SJ, et al. Cytosine Base Editors With Minimized Unguided DNA and RNA Off-Target Events and High on-Target Activity. Nat Commun (2020) 11(1):2052. doi: 10.1038/s41467-020-15887-5
213. Shimatani Z, Kashojiya S, Takayama M, Terada R, Arazoe T, Ishii H, et al. Targeted Base Editing in Rice and Tomato Using a CRISPR-Cas9 Cytidine Deaminase Fusion. Nat Biotechnol (2017) 35(5):441–3. doi: 10.1038/nbt.3833
214. Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, et al. Targeted Nucleotide Editing Using Hybrid Prokaryotic and Vertebrate Adaptive Immune Systems. Science (2016) 353(6305). doi: 10.1126/science.aaf8729
215. Rees HA, Liu DR. Base Editing: Precision Chemistry on the Genome and Transcriptome of Living Cells. Nat Rev Genet (2018) 19(12):770–88. doi: 10.1038/s41576-018-0059-1
216. Wang X, Li J, Wang Y, Yang B, Wei J, Wu J, et al. Efficient Base Editing in Methylated Regions With a Human APOBEC3A-Cas9 Fusion. Nat Biotechnol (2018) 36(10):946–9. doi: 10.1038/nbt.4198
217. Fugmann SD, Messier C, Novack LA, Cameron RA, Rast JP, et al. An Ancient Evolutionary Origin of the Rag1/2 Gene Locus. Proc Natl Acad Sci USA (2006) 103(10):3728–33. doi: 10.1073/pnas.0509720103
218. Martin EC, Vicari C, Tsakou-Ngouafo L, Pontarotti P, Petrescu AJ, Schatz DG, et al. Identification of RAG-like Transposons in Protostomes Suggests Their Ancient Bilaterian Origin. Mob DNA (2020) 11:17. doi: 10.1186/s13100-020-00214-y
219. Teng G, Schatz DG. Regulation and Evolution of the RAG Recombinase. Adv Immunol (2015) 128:1–39. doi: 10.1016/bs.ai.2015.07.002
220. Zhang Y, Cheng TC, Huang G, Lu Q, Surleac MD, Mandell JD, et al. Transposon Molecular Domestication and the Evolution of the RAG Recombinase. Nature (2019) 569(7754):79–84. doi: 10.1038/s41586-019-1093-7
Keywords: DNA-editing enzyme, immune response, cancer, gene mutations, cytidine deaminase, AID/APOBEC and ADAR deaminases, protein structure/folding, evolutionary immunology
Citation: Ghorbani A, Quinlan EM and Larijani M (2021) Evolutionary Comparative Analyses of DNA-Editing Enzymes of the Immune System: From 5-Dimensional Description of Protein Structures to Immunological Insights and Applications to Protein Engineering. Front. Immunol. 12:642343. doi: 10.3389/fimmu.2021.642343
Received: 15 December 2020; Accepted: 06 April 2021;
Published: 31 May 2021.
Edited by:
Gyri T. Haugland, University of Bergen, NorwayReviewed by:
Hao-Ching Wang, Taipei Medical University, TaiwanCopyright © 2021 Ghorbani, Quinlan and Larijani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atefeh Ghorbani, QXRlZmVoX0dob3JiYW5pQHNmdS5jYQ==; Emma M. Quinlan, bXE2MzBAbW9uLmNh; Mani Larijani, bWFuaV9sYXJpamFuaUBzZnUuY2E=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.