 Andreas Lutterotti
Andreas Lutterotti Helen Hayward-Koennecke
Helen Hayward-Koennecke Mireia Sospedra
Mireia Sospedra Roland Martin
Roland Martin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 March 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.640935
This article is part of the Research Topic Emerging Therapeutics for Immune Tolerance View all 24 articles
Antigen-specific tolerance induction aims at treating multiple sclerosis (MS) at the root of its pathogenesis and has the prospect of personalization. Several promising tolerization approaches using different technologies and modes of action have already advanced to clinical testing. The prerequisites for successful tolerance induction include the knowledge of target antigens, core pathomechanisms, and how to pursue a clinical development path that is distinct from conventional drug development. Key aspects including patient selection, outcome measures, demonstrating the mechanisms of action as well as the positioning in the rapidly growing spectrum of MS treatments have to be considered to bring this therapy to patients.
Therapeutic interventions in medicine should provide the highest possible specificity and well-known mechanisms when targeting the pathogenic processes underlying a specific disease. In the evolving era of precision medicine, this aspect has become the most important goal of treatment development and is driven by advances in the understanding of disease etiology and relevant pathomechanisms. Immune-mediated diseases, including autoimmune disease (AIDs) and allergies, which together affect up to 20% of the population in industrialized countries, are important examples, in which the field aims to move from broadly immunomodulatory to highly specific treatments.
Organ-specific AIDs are characterized by acute or chronic inflammation driven by an autoreactive immune response against self-antigens. Although the relative contribution of different cellular and humoral immune effector mechanisms differ between diseases and even individual patients, the selective abrogation of the autoreactive immune response offers the opportunity to specifically treat and potentially cure an AID (1). The concept of reverting autoimmunity by induction of antigen-specific immune tolerance stands in contrast to currently available therapies, which target the inflammatory immune response broadly, often compromise protection against infections and may even lead to secondary autoimmunity. This is particularly relevant in chronic diseases affecting young patients with consequential need for long-term immunosuppression. One such example is multiple sclerosis (MS).
MS is considered a prototypic, organ-specific AID characterized by chronic inflammation of the brain and spinal cord leading to variable neurologic signs and symptoms and often persisting disability (2). Although the development of effective therapies has been very successful over more than two decades, it has come at the cost of sometimes severe safety concerns related to the lack of specificity and global immunosuppression.
Approaching therapeutic immune tolerance in MS requires a sound understanding of its autoimmune pathogenesis including the main target antigens as well as the mechanisms of immune tolerance and suitable methods to assess the effects of a tolerization regimen. The clinical development of tolerization poses several challenges, which are related to the disease itself, the mechanisms of the tolerizing approach and clinical trial design, which all need to be mastered for successfully advancing tolerization to the clinic.
Here, we will provide an overview about the current state of knowledge of target antigens and immune tolerance mechanisms in MS, discuss lessons learned from previous attempts towards tolerization and what we consider the main hurdles during clinical development of antigen-specific therapies (ASTs). While several innovative tolerization approaches are currently in pre-clinical development [reviewed in (3)], we will focus only on those that have already entered clinical phases.
One core prerequisite for developing antigen-specific tolerization is the firm knowledge of the relevant target autoantigens. Although this aspect has received a lot of attention in the past, the antigen specificity of autoreactive T cells and also of autoantibodies in MS has been examined only by a few groups during recent years comprehensive reviews in (1, 4–8). Since demyelination is one hallmark of MS lesions, the search for targets focused initially on myelin proteins such as myelin basic protein (MBP), proteolipid protein (PLP) and myelin oligodendrocyte glycoprotein (MOG), which had been shown to be encephalitogenic in the animal model experimental autoimmune encephalomyelitis (EAE) (7, 8). Several of the encephalitogenic peptides of MBP, PLP and MOG are also immunodominant in MS patients (4, 7, 9), and peptides of other myelin- (2’-3’ cyclic nucleotide phosphodiesterase (CNPase), myelin oligodendrocyte basic protein (MOBP), oligodendrocyte-specific protein (OSP), myelin-associated glycoprotein (MAG)) and non-myelin proteins (alpha-B crystallin, transaldolase H, S100 beta, contactin 2/TAG-1, RAS guanyl-releasing protein 2, RASGRP2, GDP L-fucose synthase, TSTA3, KIR4.1, anoctamin 2) have been described (5, 6, 10–12), but not yet studied to the same extent.
To assess the biological relevance of putative target autoantigens, the criteria outlined in Box 1 can be used, which we have weighed based on current knowledge (see also Table 1). The selection of target antigens is based on the consideration that autoreactive and proinflammatory CD4+ T cells that are restricted by MS-associated HLA-DR molecules are the drivers of the disease, and hence that antigen-specific tolerization should silence/eliminate these cells (4, 7). Table 1 summarizes the antigens that have been identified and which properties support their importance. We consider the following autoantigens most important in the moment due to the fact that: (i) they are targets of high avidity autoreactive T cells in MS [MBP13-32; MBP111-129, MBP146-170; PLP139-154, MOG1-20 and MOG35-55 (14)], (ii) their encephalitogenicity has been shown with humanized mice expressing T cell receptors (TCRs) of MS patient-derived T cell clones (MBP 83-99), or (iii) because they have been shown to be targeted by brain- and CSF-infiltrating T cells of MS patients (GDP L-fucose synthase, TSTA3, and RASGRP2) (11, 12) (for details see Box 1 and Table 1). Reactivity against the above mix of high avidity myelin peptide targets and MBP 83-99 has been examined in MS patients in North America (14), Germany (15), Spain (16) and Switzerland (own unpublished data), and between 74% and 100% of patients have shown reactivity. This cocktail is a good start since the majority of patients reacts to one or more peptides, and it has been used by us (15) and also adopted by other groups (17) for tolerization trials. It is not clear in the moment, how many peptides are ideal, but we assume that including as many relevant target antigens as possible will increase the likelihood of successful tolerization, particularly if the disease is already ongoing for longer time, and hence it is likely that the autoimmune response is directed against multiple peptides of one (intramolecular) or several proteins (intermolecular), known as epitope spreading (18–20). In this context, the ability of the tolerization approach to prevent epitope spreading is crucial. The use of myelin peptide-coupled splenocytes has been very effective in that respect (21), but to our knowledge prevention of epitope spreading are less well or not examined and shown for other tolerization modalities (22).
Box 1. Criteria to judge the relevance of target autoantigens (key criteria in bold).
* Refers to the special case of increased disease activity following vaccination with an altered peptide ligand of MBP 83-99 (13).
● Recognized by CSF- and/or brain-infiltrating T cells; recognized by autoproliferating T cell fraction
● Use of the respective peptide or protein or derivative thereof has shown tolerizing activity in tolerance trial in MS
● Immunodominant for (proinflammatory) CD4+ T cells in MS patients in the context of one or several MS-associated HLA-DR molecules
● Recognized with high(er) avidity by T cells of MS patients
● Encephalitogenic in EAE models
● MS patient-derived TCR with specificity for the antigen is encephalitogenic in humanized mouse models; or encephalitogenic in HLA-DR humanized tg mice
● “Encephalitogenic” in MS patients*
● T cell cross-reactivity between autoantigen and MS-associated pathogen/s, e.g. EBV, Akkermansia
● Autoantibody cross-reactivity between autoantigen and MS-associated pathogen/s, e.g. EBV, Akkermansia
● Target of autoantibodies in MS and pathogenicity shown in EAE:
● Exclusive expression in the brain (relative)
● Generation of peptide is independent of antigen processing and mimics naturally occurring sequences
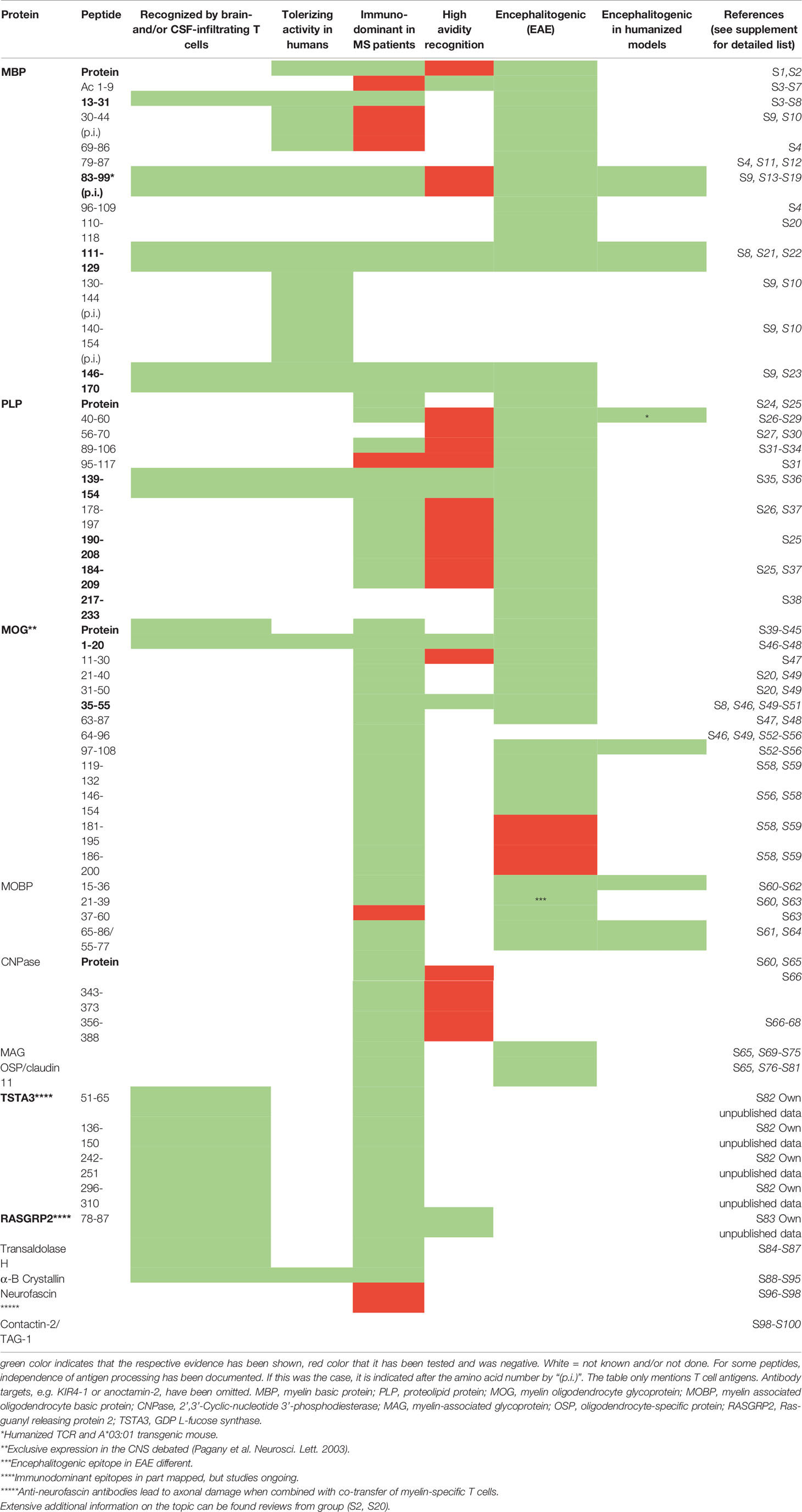
Table 1 Autoreactive T cell targets in multiple sclerosis: evidence for relevance.
An additional criterion to select peptides for tolerization is their independence of antigen processing. During antigen processing, proteins are digested by specific proteases, and it has been demonstrated that peptides that are generated by the naturally occurring processing mechanisms are protected from degradation (23, 24) and that this aspect is relevant for tolerance induction. Peptides MBP30-44, MBP83-99, MBP131-145 and MBP140-154 fulfill these criteria and have been tested clinically (see below).
Regarding targets of autoantibodies identified in MS such as KIR4.1 (25) and anoctamin 2 (26), it will be important to examine whether these autoantigens are also recognized by autoreactive CD4+ T cells and if further evidence supports their pathogenetic relevance before including them in tolerization trials.
In summary, careful examination of disease-relevant target antigens, which shall be used for tolerance induction, is warranted. Based on the criteria outlined in Box 1 and Table 1, we will soon add immunodominant peptides derived from TSTA3 (12) and RASGRP2 (11) to the tolerizing cocktail of high-avidity myelin peptides that we currently use for tolerization.
Unresponsiveness of the adaptive immune system against self-antigens is generated by so-called central tolerance mechanisms in the thymus for T cells and in the bone marrow for B cells. Central tolerance assures that T cells that recognize self-antigens with high avidity are eliminated by apoptosis, a process called negative selection, while T cells that respond with low avidity (that is only at high antigen concentration) are positively selected into the peripheral immune system to protect the host from pathogens. This mechanism destroys potentially dangerous T cells with specificity for most self-antigens, however, it also implies that all peripheral blood T cells are able to recognize autoantigens and are to some extent autoreactive. Peripheral tolerance mechanisms including anergy, a state of functional silence when T cells are stimulated in the absence of costimulatory molecules, non-responsiveness at low antigen concentrations and the deletion of autoreactive T cells by activation-induced cell death (AICD) assure under physiological conditions that pathologic autoreactivity is avoided [reviewed in (27)]. The latter mechanisms are antigen-specific, but not expected to mediate long-lasting non-responsiveness. Further control mechanisms include several types of T regulatory cells (Tregs), most notably natural, thymus-derived Tregs (nTregs), which are characterized among other markers by expression of the transcription factor FoxP3 (28), and so-called induced, IL-10-secreting Tregs (iTregs or Tr1 cells) (29). In the context of therapeutic tolerance induction, the activation and expansion of Tregs is critical for actively controlling autoreactive T cells against multiple antigens. A phase Ib/IIa using autologous Tregs in MS patients has been reported recently with good safety results (30). Different from the above elimination of autoreactive T cells by apoptosis or silencing by anergy, Treg-mediated tolerance is expected to last long(er) and be able to control a broader range of autoreactive T cells. An important aspect that has not been addressed well in humans/MS is, to what extent Tregs need to be antigen-specific. Finally, there are various other immunoregulatory cell populations including B regulatory cells (31), regulatory plasma cells (32), CD56bright natural killer cells (33) and others, which will not be addressed here.
Many modalities to induce immune tolerance have been tested with varying success in animal models (3). These include different routes of administration (RoA) of autoantigens, for instance oral, nasal (generally mucosal), transdermal or intravenous application, coupling of autoantigens (usually peptides) to cells (white blood cells, red blood cells) or other carriers like nanoparticles, but also the intramuscular injection of a plasmid encoding MBP for ectopic expression in muscle. The experimental data, mechanism/s of action and potential caveats have been reviewed (3). Furthermore, not only the RoA, but also the site of degradation of tolerizing peptides and the context of their presentation to the immune system, that is tolerogenic or immunogenic/inflammatory, are critical. The generation and maintenance of peripheral tolerance against proteins that enter the body via the gut or the natural degradation of dying cells in the body occur preferentially in the liver and spleen, while antigen processing and presentation in lymph nodes or in an inflammatory context induce proinflammatory immune responses instead.
For certain tolerization approaches, for instance peptide-coupled fixed white or red blood cells and antigen-coated nanoparticles, the mechanistic data are robust and both prophylactic and long-lasting therapeutic effects have been shown (21, 22). Fixed, peptide-coupled cells induce tolerance by several mechanisms including anergy and the expansion of Tregs, and further they block epitope spreading (34).
The translation of such a therapy to patients poses multiple challenges, particularly to demonstrate that autoreactive T cells are silenced and/or deleted and that the approach is indeed antigen-specific. Different from anergy induction, which will require repeated administration of autoantigen over long/er periods of time, it is desirable and needs to be shown that active peripheral tolerance mechanisms, particularly the induction/expansion of Tregs can be achieved. Ideally, the mechanistic studies that accompany the clinical trials should be able to demonstrate that the putative mechanism(s) of action of the respective approach indeed operate in patients.
The main difficulties are outlined in the following. As described above, autoreactive T cells express low avidity TCRs and are also present in healthy donors (14, 35, 36). Distinguishing pathogenic autoreactive T cells from the “physiological” level of autoreactivity is therefore very difficult. The functional phenotype of CD4+ T cells, which in the case of MS are mainly Th1 and Th1* cells based on certain chemokine receptor profiles, expression of signature transcription factors or cytokines like IFN-γ and IL-17, can in principle be used, but also are not easy to quantitate reliably. Further, pathogenic autoreactive T cells are rare. Depending on the assay that is used, frequencies range between a few percent to 1 in 107 (37, 38). It is therefore difficult to reliably enumerate autoreactive CD4+ T cells with a certain specificity before therapy, but even more to show their reduction or change of phenotype after tolerization. Testing sufficient numbers of cells in vitro and to use a sensitive assay are both important. We have recently employed a protocol modified from Geiger et al. (39), primary proliferation with peripheral blood T cells without pre-selection (15), and a Fluorospot assay with bead-coupled whole myelin proteins (40) to successfully quantify these cells (41). Equally demanding and currently not solved are methods to reliably identify and enumerate the different Treg populations, most importantly nTregs and Tr1 cells. Again, their low frequency is one problem. Further, surface markers of nTregs, CD25 and CD39, are not specific for these cells (42). Intracellular detection of FoxP3 is more demanding and, in order to demonstrate functionally stable Treg differentiation, the methylation status of the FoxP3 locus is better, but not established for easy detection of nTreg numbers. Accurate enumeration of Tr1 cells (CD3+, CD4+, CD45RA-, CD49b+, LAG3+) by flow cytometry (43), is difficult, again due to their low numbers. IL-10, their signature cytokine, may be used as a surrogate for Tr1 function, however, IL-10 is not exclusively produced by Tr1 cells, and serum levels are at the limit for detection. Finally, biomarkers that are related to damage of the target tissue (for example neurofilament light chain) may be used as an indirect measure for a tolerizing effect if their levels drop after treatment (44).
In summary, the mechanistic testing should demonstrate immunosafety, that is the absence of a vaccination response that induces rather than abrogates autoimmune inflammation as most important acute safety concern. With respect to proving the mechanism(s) of action (MoA), the accompanying in vitro testing should query putative peripheral tolerance effects including the reduction or elimination of pathogenic, autoantigen-specific T cells, the induction of regulatory T cells and their cytokines as well as markers that indicate the reduction of inflammation and damage in the target organ (see Box 2). Testing of CSF parameters is highly desirable since they likely better reflect pathogenic immune mechanisms in the target organ, but not possible in larger clinical trials. Besides establishing the MoA of the tolerizing regime and providing indications for its immunological efficacy, these studies are important for finding the best dose and dosing interval. Successful development of tolerization therapies will depend on whether the above described challenges of mechanistic studies can be overcome or not.
Box 2. Goals and assays for testing the mechanism/s of action of tolerance induction.
To assess immunosafety and exclude that the respective approach induces disease activity, loss or increase of immune cells
-Standard hematology and flow cytometry testing (or mass cytometry) for the major populations (CD4+ and CD8+ T cells, B cells, monocytes, dendritic cells, NK cells, NK-T cells)
Assess the loss/decrease of antigen-specific autoreactive T cells and change of phenotype
-Various types of proliferation assays using sufficient numbers of input cells
-Intracellular cytokine staining, chemokine receptor profiles by surface staining
-Combination of the above can be achieved by Fluorospot testing assessing numbers of antigen-specific T cells and their functional phenotype
-Antigen/HLA-DR tetramers for direct enumeration of autoreactive cells
Induction of T regulatory cells
-Flow cytometry testing for nTregs and Tr1 cells
-Support nTreg induction by demonstrating demethylation status of Fox-P3 (quantitative PCR)
-Support Tr1 cell increase by intracellular cytokine staining and/or increase of serum IL-10
-Functional assays
Biomarkers indicating reduced target organ damage and/or reduction of inflammation
-Markers for neuronal/axonal damage or brain inflammation, for example neurofilament light chain (NFL)
The appeal of selectively silencing the autoimmune response without impairing protective immunity has prompted numerous efforts to translate promising results of ASTs from animal models to the clinic. ASTs employed different approaches ranging from the use of whole proteins, peptides in various routes of administration, tolerogenic dendritic cells, DNA, T cell or TCR vaccinations, all operating via different mechanisms and most of them targeting the trimolecular complex between HLA-class II molecule, antigenic peptide and a CD4+ T cell’s TCR (Figure 1 and Table 2). While most of the early tolerization trials failed to reach their clinical endpoints despite promising mechanistic results, some of the recent studies have been encouraging in affecting imaging-based outcomes in early phase clinical trials. Below, we will summarize the most important observations and lessons from tolerance trials in MS. A detailed list of all trials and their main characteristics and findings is given in Table 2.
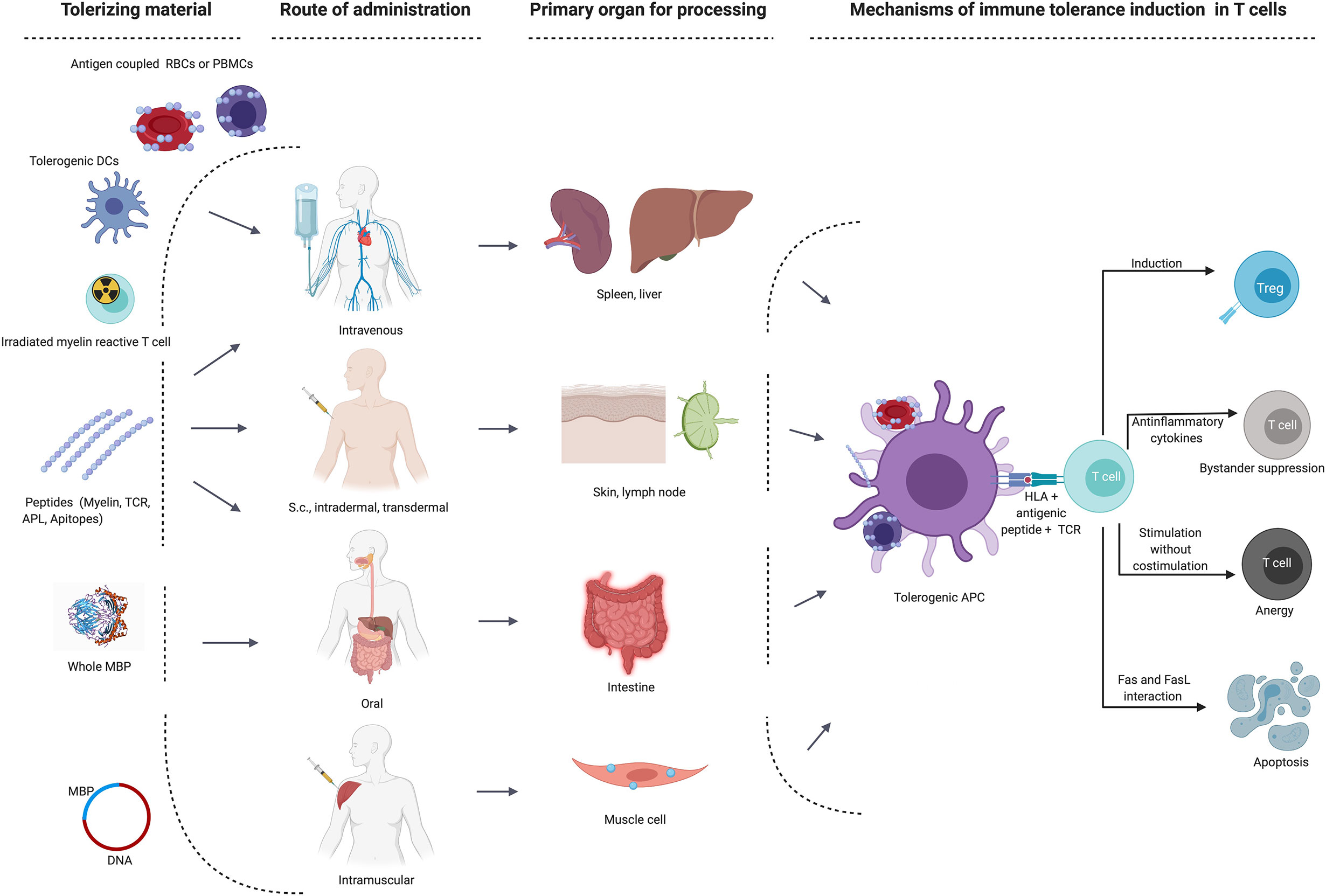
Figure 1 Main target organs and mechanisms of action depending on route of administration in different immune tolerance strategies. APC, antigen-presenting cells; DC, dendritic cell; MBP, myelin basic protein; RBCs, red blood cells; s.c., subcutaneous; TCR, T cell receptor; Treg, regulatory T cell.

Table 2 Antigen-specific tolerization approaches in the clinic.
A pioneering approach aimed at tolerization through oral administration of whole bovine myelin. A randomized, placebo-controlled clinical trial in early relapsing-remitting MS (RRMS) patients failed the primary endpoint of reducing clinical disease activity, including the number of relapses and disability progression, despite promising data on antigen-specific T cells (47). Both gender and HLA haplotypes were unequally distributed between the treatment groups, limiting the interpretation of results and already pointing at the importance of patient stratification in tolerization trials (68). A double-blind, phase 3 clinical trial of a single dose of bovine myelin in 515 MS patients failed to show a reduction in relapses, however, an extraordinarily strong placebo effect was observed, which might have influenced the result (69). The formulation and the dose might have been additional factors leading to the failure of the approach, since the human equivalent dose was lower than the effective dose in mice.
Another RoA was explored by intravenous administration of a soluble MBP82-98 peptide, which was well tolerated and showed a beneficial effect on disease progression in HLA DR2+ secondary-progressive MS (SPMS) patients (56, 70). An increase of regulatory T cells up until 6 months post treatment was shown, and interestingly a reversal of the T cell anergic state was seen in the high dose group (71). However, a phase 3 trial failed with no significant benefit over placebo with respect to reducing disease progression (57). The results of the trial emphasize the importance of choosing the optimal disease stage for tolerization approaches. At later stages like SPMS, it is probably not only challenging to curb a long-lasting autoimmune response, but also much more difficult to prove an effect in clinical trials.
An important lesson came from a clinical trial with an altered peptide ligand (APL) of the immunodominant MBP83-99 peptide, which led to induction of new disease activity in RRMS patients. APLs are generated through modification of amino acids in TCR contact positions, which can block or alter T cell responses through serving as partial agonist and antagonist. Despite compelling evidence from pre-clinical studies (72) a phase 2a trial using MBP83-99-derived APL was halted due to induction of MS disease activity through stimulation of encephalitogenic MBP83-99 reactive T cells (13). Thus far, this is the only evidence in humans that a MBP-specific immune response can trigger inflammatory lesions and relapses in MS patients. Further, the study demonstrated the importance of thorough safety monitoring by clinical and imaging measures in early phase trials. Whether the unusually low number of DR15+ MS patients contributed to the outcome is currently not clear, but possible. Hypersensitivity reactions led to discontinuation of a second and larger phase 2 trial with the same APL given at three doses, although there were signs of a beneficial effect on the number of contrast-enhancing lesions (54, 73).
A mix of four processing-independent MBP peptides (ATX-MS-1467) for subcutaneous or intradermal application, was safe and well tolerated in a phase 1 and successful in a phase 2 trial in RRMS patients showing a significant reduction of new and total contrast-enhancing lesions (24, 59). However, the trial also demonstrated the limitations of an antigen-specific therapy in patients with very high disease activity. Further trials are warranted to confirm the beneficial effect of the approach and to assess whether it might lead to a long-lasting therapeutic effect, that is persisting immune tolerance, or may need continuous administration of the AST.
Different from the above modalities, BHT-3009 builds on ectopic expression of a myelin protein via intramuscular injection of a plasmid encoding full length MBP, which leads to muscle cells expressing sustained low levels of MBP. BHT-3009 demonstrated promising effects on radiological disease activity in active MS patients in a phase 1/2 study, a reduction of MBP-specific CD4+ T cells with a Th1 phenotype in peripheral blood and a decrease of myelin-specific auto-antibody titers in CSF (45). A subsequent phase 2 trial did not meet the primary endpoint in reducing the number of new contrast-enhancing lesions (46). Overall, the approach remains promising and is currently followed in type 1 diabetes and neuromyelitis optica (NMO) (https://tolerion.bio/pipeline/).
Inducing immune tolerance to peptides of different myelin proteins simultaneously, including MBP, MOG and PLP promises to increase the efficacy of the treatment. Transdermal administration of three myelin peptides (MBP85-99, MOG35-55 and PLP139-155) via skin patches was one of the first ASTs in MS to demonstrate efficacy in reducing clinical- and MRI disease activity in RRMS patients (58). Peptide application led to local activation of Langerhans cells, reduced myelin peptide-specific T cell responses and increases of IL-10-secreting T cells (74).
Our group employed chemical coupling of seven myelin peptides from MOG, MBP and PLP (MOG1-20, MOG35-55, MBP13-32, MBP83-99, MBP111-129, MBP146-170, PLP139-154), (see above) to autologous peripheral blood mononuclear cells (PBMC) (14, 15). The approach targets the highest number of antigens based on the above considerations (see Table 1, Box 1) and was safe and well tolerated in a phase 1b study in RRMS and SPMS patients with T cell reactivity against at least one of the myelin peptides (15). Mechanistic studies including immunophenotyping of immune cell populations, cytokine responses and both anti-myelin and -non-myelin autoantibodies did not show any signs of induction of autoreactivity (15, 75). In patients receiving high doses a reduction of myelin peptide-specific T cell responses was observed after treatment. To improve the tolerization regimen by targeting both liver and spleen as important tolerogenic organs, we switched to autologous red blood cells (RBC) as carrier cells and assessed the safety and feasibility of autologous myelin peptide-coupled RBCs in a phase 1b trial (41). Following these promising results, a phase 2 study is currently in preparation.
Besides peptide-coupled fixed carrier cells, several groups explore antigen-loaded dendritic cells (DCs) with a good safety and tolerability profile in a phase 1b study in MS (seven myelin peptides) and NMO (aquaporin-4 peptides) patients (17). Increased production of IL-10 and of Tr1 cell numbers were observed. Currently, three different open label phase 1 studies are conducted to evaluate myelin peptide-pulsed DCs in different immature/tolerogenic states and given by different routes (intravenous, intradermally and intranodally; NCT02618902, NCT02903537, NCT02283671).
An alternative AST strategy aims to induce an immune response against important effector mechanisms of the autoreactive immune system, for example the autoreactive T cell clone or its TCRs. The appeal is that induction versus abrogation of immune responses might be easier to achieve. There have been various promising studies with T cell- or T cell receptor (TCR) vaccination, which were well tolerated and led to a reduction of myelin-reactive and IL-2- or IFN-γ secreting T cells (62–67, 76–79). Renewed disease progression 12 months after the last vaccination indicated that refresher injections are needed (65), as did reappearing MBP-specific TCC, which could be eliminated by additional vaccination (78).
MBP-specific T cells from MS patients frequently express specific TCR variable chains Vβ5.2 and Vβ6.1 (80, 81). Several trials in MS patients using intradermal or intramuscular injections of synthetic TCR Vβ5.2 and/or and Vβ6.1 peptides reported clinical improvements and reduced frequency of MBP-specific T cells and the induction of TCR peptide-specific T cells (48–50). The administration of a trivalent TCR vaccine induced TCR-peptide specific T cells secreting IL-10 and increased the expression of FoxP3 in Tregs, which was paralleled by a reduction in MOG145-160 specific T cells, suggesting the induction of a regulatory network by the vaccine (51, 52) (NCT02057159).
Several tolerization strategies in MS were safe and feasible in phase 1 studies but the consecutive phase 2 and -3 trials remained unsatisfying. It has become clear that the clinical development of antigen-specific therapies poses distinct challenges that need to be tackled along the clinical trial program. Several aspects of the disease like inflammatory activity, progression or disease stage, patient selection, characteristics of the tolerizing product such as RoA, dose and interval of administration, concomitant therapies need to be considered, and each step is difficult (Figure 2). Developing an optimal trial design therefore remains very demanding.
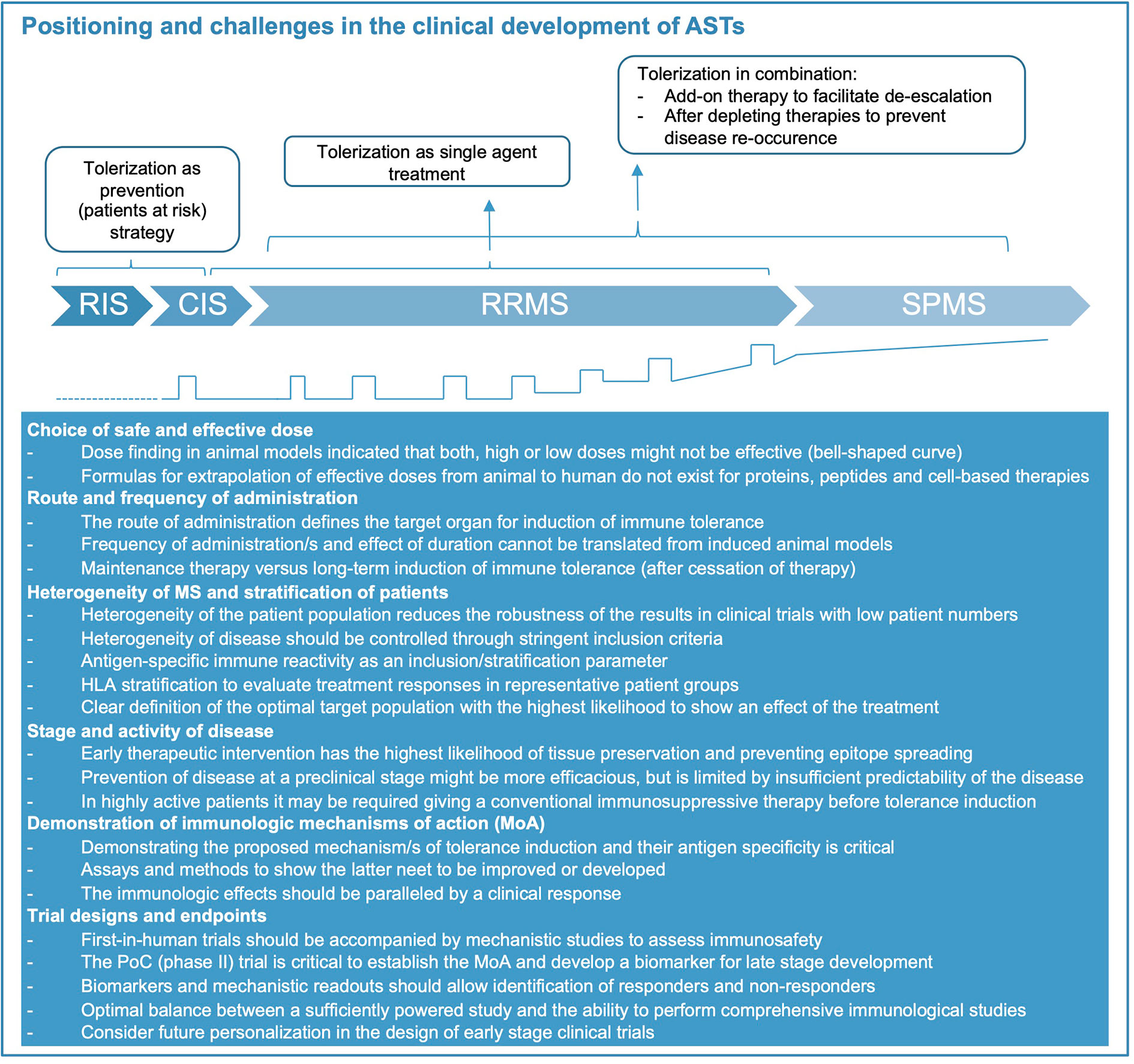
Figure 2 Positioning of immune tolerance in disease stages of MS and key challenges for treatment development. Considerations how tolerization appears most meaningful during the different disease stages of MS (represented also graphically at bottom of figure, relapses indicated by open squares). Abbreviations: CIS, clinically isolated syndrome; RIS, radiologically isolated syndrome; RRMS, relapsing-remitting MS; SPMS, secondary-progressive MS. During RIS and CIS, as well as following highly active immunomodulatory therapy tolerization aims at preventing further evolution or re-occurrence of disease. As single agent treatment during RRMS, tolerization aims at replacing currently approved therapies. Its role in SPMS and primary progressive (PP) MS (not shown) is speculative at present.
Certainly, an important challenge in the early proof-of-concept studies is the heterogeneity of the disease with regard to genetic background, main pathomechanisms, and clinical aspects including disease form, course and response to treatments. The HLA DR-15 haplotype is by far the most important susceptibility gene and key in shaping antigen-specific immune responses. Consequently, the individual HLA background might contribute to heterogeneity of antigen-specific immune responses and influence the efficacy of ASTs. The individual’s HLA type therefore needs to be considered as already demonstrated in the early oral tolerance trial (47), and particularly during early clinical development, one should assure that a representative population of patients is included.
Ideally, the tolerization approach should be administered early in the disease and block epitope spreading (Figure 2). Although several studies have reported promising immunological effects of tolerization in SPMS patients, they failed to show efficacy on clinical and imaging measures of inflammatory disease activity and disability progression. Thus, both disease duration and stage should be considered, and ideally biomarkers including antigen-specific immune responses should be used for stratification of patients. The intensity and extent of inflammatory disease activity, which is often higher during the early stage of the disease is another important factor. Since it may take some time until tolerization becomes effective, it may be necessary to start the tolerizing therapy in combination with an effective anti-inflammatory treatment to decrease disease activity and provide the optimal environment for induction of tolerance. Future trials should consider this aspect particularly in highly active patients and explore the optimal duration for a combination therapy before switching to monotherapy with the tolerizing agent (conceptual considerations shown in Figure 2). For such sequential application or combined use of tolerizing therapies the specific immunologic effects of the conventional immune therapy, its durability and how this might interfere with the main mechanisms of immune tolerance induced by the AST are important aspects to keep in mind. Immune therapies leading to a broad reduction or even long-term depletion of several or single lymphocyte subsets might also impede the generation of regulatory cell populations and thus dampen the tolerizing effects. More specific inhibition of immune cell activation/proliferation or trafficking of autoreactive lymphocytes to the CNS is less likely to interfere with the induction of immune tolerance and in case of the latter might even act synergistically by providing better exposure of autoreactive T and B cells to the tolerizing agent (11). The choice of a combination therapy will also depend on the timing of the treatment, i.e. whether the AST is applied early in the disease (for example as first-line therapy) or as part of a de-escalation strategy where the conventional immune therapy is already established.
Establishing the dose and frequency of administration of an AST are important aspects during clinical development. Prior studies have shown that the antigen dose might influence the mechanism of immune tolerance induced by the treatment (82). Extrapolation of an effective dose from rodents to humans is difficult, since, different from small molecules, accepted formulas do not exist for cell-based therapies or other novel strategies (e.g. nanoparticle-based approaches, DNA vaccination, and others). In addition, almost all animal models are induced and have a monophasic disease course, or the AST shows long-lasting, sometimes life-long, efficacy even after a single treatment (21), thus providing little guidance with respect to frequency of administration in patients. Consequently, there is a strong need for biomarkers that can be employed for dose finding and assessing the duration of the tolerization effect in AST trials.
As mentioned above the RoA of the target antigen is critical. It influences cell type and organ that take up the tolerizing peptide, nanoparticle or cell product and affects the type of immune response that the respective tolerizing approach produces. Oral application has long been considered ideal for the induction of tolerance due to the important role of the mucosal immune system, which assures tolerance to food antigens. However, clinical studies of oral tolerance did not yet show efficacy in MS, which may be related to the formulation, dose and choice of antigen. Subcutaneous (s.c.) administration is usually associated with immune activation (APL trial) as opposed to intravenous (i.v.) application (iv MBP 83-99), intradermal (i.d.) and transdermal (t.d.) application of peptides (59). Hypersensitivity reactions were an important issue in a phase 2 trial of subcutaneous administration of an APL (13, 54). The spleen is important for degradation of aged cells, and particularly the liver plays a role in immune tolerance to blood borne antigens (83). Therefore, i.v. application of antigens, peptide-coupled cells or nanoparticles have been considered most effective in targeting these organs and to be safe although hypersensitivity reactions have been observed in animal models (84).
Phase 1 testing of ASTs need to establish safety and tolerability and exclude proinflammatory activation of immune responses to the target antigens. Finding the right starting dose is critical for gene and cell-based approaches, but generally for ASTs (see above). The efficacy outcomes for early phase 2 clinical trials in MS are well established and mainly use MRI as a surrogate for inflammatory disease activity (85). Documenting efficacy on the surrogate outcome (MRI) should be accompanied by mechanistic studies, which support the putative MoA of the AST, and may identify subgroups of patients with strong or poor responsiveness and the optimal dose range (Figure 2). As outlined above, there is a need for a consensus on suitable MoA-oriented outcome parameters for tolerizing therapies. Approaching this goal, requires coordination and collaboration between research groups, which could build on ongoing initiatives like the Immune Tolerance Network in the US and dedicated scientific networks in Europe (86, 87), ideally with further involvement of competent authorities. Current highly active therapies reduce inflammatory MRI activity by 90% or more, which is unlikely to be improved by ASTs. However, since tolerance induction would be a completely new treatment modality and is expected to be superior with respect to safety and tolerability over short and particularly longer treatment courses, these aspects should be built into the clinical development strategy. Experiences in the past have shown that following the path of clinical testing that is well established for small molecules and biologics may not be ideal for ASTs. The current treatment landscape of MS offers several approved therapies for patients with high disease activity, but there is a lack of therapies that are safe, do not pose problems for women, who wish to get pregnant, and do not increase the risk for infections or damage organs. These considerations are particularly relevant for the increasing number of patients with low disease activity and those who are in the very early stages of the disease. Induction of immune tolerance potentially fills an unmet medical need for therapies that provide an ideal balance between efficacy and safety (Figure 2). Furthermore, a group of patients, which is currently not treated, are patients with radiological evidence for MS prior to any clinical symptom, i.e. radiologically isolated syndrome (RIS). RIS patients would greatly benefit from a therapy that does not lead to unspecific immunosuppression. Thus, tolerance-inducing therapies may fit best for patients at early or preclinical stages of the disease or as a sequential therapy after induction with highly effective immunomodulatory treatments in patients with high disease activity (Figure 2). Depending on how the identification of genetic risk profiles and biomarkers evolves, it can even be envisioned that that tolerance induction may be used prophylactically to prevent the onset of MS in the future.
Induction of antigen-specific immune tolerance is an attractive treatment goal for MS and other autoimmune diseases, and different strategies are in pre-clinical and clinical stages. We outline key points that should be considered to improve the development of tolerizing therapies and to find the best way how to fit them into our current treatment algorithms. These include validation of biomarkers to measure induction of immune tolerance, definition of relevant MoA of each strategy and documentation of long-term reduction in antigen-specific immune responses parallel to effects on clinical outcome parameters. Clinical trial designs need to be improved and tailored to the specific challenges that AST pose. Consensus criteria for AST trials should be developed. If the above challenges can be mastered, the successful application of AST in any autoimmune disease would represent a major breakthrough in medicine and enter a new treatment era that aims at treating autoimmunity with high specificity and minimal side effects or even preventing its development.
All authors declare that they have substantially participated in the preparation and writing of the manuscript and have taken due care regarding their contribution to ensure the integrity of the work. All authors contributed to the article and approved the submitted version.
HH-K, AL, RM, and MS are supported by the Clinical Research Priority Program of the University of Zurich (PrecisionMS) and the Wyss Zurich. Swiss National Fund Project#310030_197652 (A Lutterotti).
AL received financial compensation and/or travel support for lectures and advice from Biogen, Merck, Novartis, Teva, Genzyme, Bayer, Celgene and he is a co-founder and co-owner of Cellerys, a company which pursues antigen-specific tolerization. He is co-inventor on a patent held by the University of Zurich on the use of peptide-coupled cells for treatment of MS. HH-K received compensation for advice, lecturing or travel support from Biogen, Genzyme, Merck, Novartis, and Teva. MS is a co-founder and co-owner of Cellerys, which pursues antigen-specific tolerization. Together with RM, she is an inventor on a Univ. Zurich-held patent of target antigens GDP L-fucose synthase and RAS guanyl-releasing protein 2 for multiple sclerosis. RM received unrestricted grant support from Biogen and Novartis, and compensation for advice or lecturing by Biogen, Novartis, Sanofi Genzyme, Merck, Hoffmann La Roche, Teva, Neuway, CellProtect, and Third Rock Ventures. He is a co-founder and co-owner of Cellerys, which pursues antigen-specific tolerization. Together with MS, he is an inventor on a Univ. Zurich-held patent of target antigens GDP L-fucose synthase and RAS guanyl-releasing protein 2 for multiple sclerosis.
Figure 1 was created with BioRender.com.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.640935/full#supplementary-material
1. Steinman L, Ho PP, Robinson WH, Utz PJ, Villoslada P. Antigen-specific tolerance to self-antigens in protein replacement therapy, gene therapy and autoimmunity. Curr Opin Immunol (2019) 61:46–53. doi: 10.1016/j.coi.2019.07.011
2. Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. New Engl J Med (2018) 378:169–80. doi: 10.1056/NEJMra1401483
3. Serra P, Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nat Biotechnol (2019) 37:238–51. doi: 10.1038/s41587-019-0015-4
4. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol (2015) 15:545–58. doi: 10.1038/nri3871
5. Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 1: autoreactive CD4+ T lymphocytes as pathogenic effectors and therapeutic targets. Lancet Neurol (2016) 15:198–209. doi: 10.1016/S1474-4422(15)00334-8
6. Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 2: CD8+ T cells, B cells, and antibodies in the focus of reverse-translational research. Lancet Neurol (2016) 15:317–31. doi: 10.1016/S1474-4422(15)00313-0
7. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol (2005) 23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707
8. Sospedra M, Martin R. Immunology of Multiple Sclerosis. Semin Neurol (2016) 36:115–27. doi: 10.1055/s-0036-1579739
9. Baxter AG. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol (2007) 7:904–12. doi: 10.1038/nri2190
10. Ayoglu B, Mitsios N, Kockum I, Khademi M, Zandian A, Sjoberg R, et al. Anoctamin 2 identified as an autoimmune target in multiple sclerosis . P Natl Acad Sci USA (2016) 113:2188–93. doi: 10.1073/pnas.1518553113
11. Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory B Cells Activate Brain-Homing, Autoreactive CD4(+) T Cells in Multiple Sclerosis. Cell (2018) 175:85–100 e23. doi: 10.1016/j.cell.2018.08.011
12. Planas R, Santos R, Tomas-Ojer P, Cruciani C, Lutterotti A, Faigle W, et al. GDP-L-fucose synthase is a CD4(+) T cell-specific autoantigen in DRB3*02:02 patients with multiple sclerosis. Sci Transl Med (2018) 10(462):eaat4301. doi: 10.1126/scitranslmed.aat4301
13. Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand (vol 6, pg 1167, 2000). Nat Med (2000) 6:1412–2. doi: 10.1038/80516
14. Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol (2004) 172:3893–904. doi: 10.4049/jimmunol.172.6.3893
15. Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, et al. Antigen-Specific Tolerance by Autologous Myelin Peptide-Coupled Cells: A Phase 1 Trial in Multiple Sclerosis. Sci Transl Med (2013) 5(188):188ra75. doi: 10.1126/scitranslmed.3006168
16. Grau-Lopez L, Raich D, Ramo-Tello C, Naranjo-Gomez M, Davalos A, Pujol-Borrell R, et al. Specific T-cell proliferation to myelin peptides in relapsing-remitting multiple sclerosis. Eur J Neurol (2011) 18:1101–4. doi: 10.1111/j.1468-1331.2010.03307.x
17. Zubizarreta I, Florez-Grau G, Vila G, Cabezon R, Espana C, Andorra M, et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. P Natl Acad Sci USA (2019) 116:8463–70. doi: 10.1073/pnas.1820039116
18. Tuohy VK, Yu M, Yin L, Kawczak JA, Kinkel RP. Regression and spreading of self-recognition during the development of autoimmune demyelinating disease. J Autoimmun (1999) 13:11–20. doi: 10.1006/jaut.1999.0293
19. Tuohy VK, Yu M, Weinstock-Guttman B, Kinkel RP. Diversity and plasticity of self recognition during the development of multiple sclerosis. J Clin Invest (1997) 99:1682–90. doi: 10.1172/JCI119331
20. Goebels N, Hofstetter H, Schmidt S, Brunner C, Wekerle H, Hohlfeld R. Repertoire dynamics of autoreactive T cells in multiple sclerosis patients and healthy subjects: epitope spreading versus clonal persistence. Brain (2000) 123 Pt 3:508–18. doi: 10.1093/brain/123.3.508
21. Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol (2007) 7:665–77. doi: 10.1038/nri2153
22. Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, et al. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano (2014) 8:2148–60. doi: 10.1021/nn405033r
23. Manoury B, Mazzeo D, Fugger L, Viner N, Ponsford M, Streeter H, et al. Destructive processing by asparagine endopeptidase limits presentation of a dominant T cell epitope in MBP. Nat Immunol (2002) 3:169–74. doi: 10.1038/ni754
24. Streeter HB, Rigden R, Martin KF, Scolding NJ, Wraith DC. Preclinical development and first-in-human study of ATX-MS-1467 for immunotherapy of MS. Neurol-Neuroimmunol (2015) 2(3):e93. doi: 10.1212/NXI.0000000000000093
25. Srivastava R, Aslam M, Kalluri SR, Schirmer L, Buck D, Tackenberg B, et al. Potassium Channel KIR4.1 as an Immune Target in Multiple Sclerosis. New Engl J Med (2012) 367:115–23. doi: 10.1056/NEJMoa1110740
26. Ayoglu B, Kockum I, Olsson T, Nilsson P. Anoctamin 2 identified as an autoimmune target in multiple sclerosis. Mult Scler J (2016) 22:10–0. doi: 10.1073/pnas.1518553113
27. Walker LSK, Abbas AK. The enemy within: Keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol (2002) 2:11–9. doi: 10.1038/nri701
28. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
29. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 Cells and the Counter-Regulation of Immunity: Natural Mechanisms and Therapeutic Applications. Curr Top Microbiol (2014) 380:39–68. doi: 10.1007/978-3-662-43492-5_3
30. Chwojnicki K, Iwaszkiewicz-Grzes D, Jankowska A, Zielinski M, Lowiec P, Gliwinski M, et al. Administration of CD4(+)CD25(high)CD127(-)FoxP3(+) Regulatory T Cells for Relapsing-Remitting Multiple Sclerosis: A Phase 1 Study. BioDrugs (2021) 35:47–60. doi: 10.1007/s40259-020-00462-7
31. Dang VD, Hilgenberg E, Ries S, Shen P, Fillatreau S. From the regulatory functions of B cells to the identification of cytokine-producing plasma cell subsets. Curr Opin Immunol (2014) 28:77–83. doi: 10.1016/j.coi.2014.02.009
32. Fillatreau S. Regulatory plasma cells. Curr Opin Pharmacol (2015) 23:1–5. doi: 10.1016/j.coph.2015.04.006
33. Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci USA (2006) 103:5941–6. doi: 10.1073/pnas.0601335103
34. Turley DM, Miller SD. Peripheral tolerance induction using ethylenecarbodiimide-fixed APCs uses both direct and indirect mechanisms of antigen presentation for prevention of experimental autoimmune encephalomyelitis. J Immunol (2007) 178:2212–20. doi: 10.4049/jimmunol.178.4.2212
35. Pette M, Fujita K, Kitze B, Whitaker JN, Albert E, Kappos L, et al. Myelin basic protein-specific T lymphocyte lines from MS patients and healthy individuals. Neurology (1990) 40:1770–6. doi: 10.1212/WNL.40.11.1770
36. Martin R, Jaraquemada D, Flerlage M, Richert J, Whitaker J, Long EO, et al. Fine specificity and HLA restriction of myelin basic protein-specific cytotoxic T cell lines from multiple sclerosis patients and healthy individuals. J Immunol (1990) 145:540–8.
37. Raddassi K, Kent SC, Yang JB, Bourcier K, Bradshaw EM, Seyfert-Margolis V, et al. Increased Frequencies of Myelin Oligodendrocyte Glycoprotein/MHC Class II-Binding CD4 Cells in Patients with Multiple Sclerosis. J Immunol (2011) 187:1039–46. doi: 10.4049/jimmunol.1001543
38. Martin R, Voskuhl R, Flerlage M, Mcfarlin DE, Mcfarland HF. Myelin Basic Protein-Specific T-Cell Responses in Identical-Twins Discordant or Concordant for Multiple-Sclerosis. Ann Neurol (1993) 34:524–35. doi: 10.1002/ana.410340405
39. Geiger R, Duhen T, Lanzavecchia A, Sallusto F. Human naive and memory CD4(+) T cell repertoires specific for naturally processed antigens analyzed using libraries of amplified T cells. J Exp Med (2009) 206:1525–34. doi: 10.1084/jem.20090504
40. Bronge M, Kaiser A, Carvalho-Queiroz C, Nilsson OB, Ruhrmann S, Holmgren E, et al. Sensitive detection of antigen-specific T-cells using bead-bound antigen for in vitro re-stimulation. Methodsx (2019) 6:1635–41. doi: 10.1016/j.mex.2019.07.004
41. Lutterotti A, Ludersdorfer TH, Docampo MJ, Hohmann M, Sellés-Moreno C, Hayward-Koennecke H, et al. Establish Tolerance in MS with myelin-peptide coupled red blood cells – the Phase Ib ETIMSred trial. Mult Scler J (2019) 25:890–938. doi: 10.1177/1352458519869496
42. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3(+) Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110:1225–32. doi: 10.1182/blood-2006-12-064527
43. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M, Gagliani N. The Biology of T Regulatory Type 1 Cells and Their Therapeutic Application in Immune-Mediated Diseases. Immunity (2018) 49:1004–19. doi: 10.1016/j.immuni.2018.12.001
44. Canto E, Barro C, Zhao C, Caillier SJ, Michalak Z, Bove R, et al. Association Between Serum Neurofilament Light Chain Levels and Long-term Disease Course Among Patients With Multiple Sclerosis Followed up for 12 Years. JAMA Neurol (2019) 76(11):1359–66. doi: 10.1001/jamaneurol.2019.2137
45. Bar-Or A, Vollmer T, Antel J, Arnold DL, Bodner CA, Campagnolo D, et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch Neurol-Chicago (2007) 64:1407–15. doi: 10.1001/archneur.64.10.nct70002
46. Garren H, Robinson WH, Krasulova E, Havrdova E, Nadj C, Selmaj K, et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann Neurol (2008) 63:611–201. doi: 10.1002/ana.21370
47. Weiner HL, Mackin GA, Matsui M, Orav EJ, Khoury SJ, Dawson DM, et al. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science (1993) 259:1321–4. doi: 10.1126/science.7680493
48. Bourdette DN, Whitham RH, Chou YK, Morrison WJ, Atherton J, Kenny C, et al. Immunity to TCR peptides in multiple sclerosis. I. Successful immunization of patients with synthetic V beta 5.2 and V beta 6.1 CDR2 peptides. J Immunol (1994) 152:2510–9.
49. Vandenbark AA, Chou YK, Whitham R, Mass M, Buenafe A, Liefeld D, et al. Treatment of multiple sclerosis with T-cell receptor peptides: Results of a double-blind pilot trial. Nat Med (1996) 2:1109–15. doi: 10.1038/nm1096-1109
50. Gold DP, Smith RA, Golding AB, Morgan EE, Dafashy T, Nelson J, et al. Results of a phase I clinical trial of a T-cell receptor vaccine in patients with multiple sclerosis. II. Comparative analysis of TCR utilization in CSF T-cell populations before and after vaccination with a TCRV beta 6 CDR2 peptide. J Neuroimmunol (1997) 76:29–38. doi: 10.1016/S0165-5728(97)00029-5
51. Bourdette DN, Edmonds E, Smith C, Bowen JD, Guttmann CR, Nagy ZP, et al. A highly immunogenic trivalent T cell receptor peptide vaccine for multiple sclerosis. Mult Scler (2005) 11:552–61. doi: 10.1191/1352458505ms1225oa
52. Vandenbark AA, Culbertson NE, Bartholomew RM, Huan J, Agotsch M, LaTocha D, et al. Therapeutic vaccination with a trivalent T-cell receptor (TCR) peptide vaccine restores deficient FoxP3 expression and TCR recognition in subjects with multiple sclerosis. Immunology (2008) 123:66–78. doi: 10.1111/j.1365-2567.2007.02703.x
53. Yadav V, Bourdette D, Bowen JD, Lynch SG, Mattson D, Preiningerova J, et al. Recombinant T Cell Receptor Ligand (RTL) for the Treatment of Multiple Sclerosis: Report of a Phase I Clinical Trial. Neurology (2010) 74:A293–4. doi: 10.1155/2012/954739
54. Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. Altered Pept Ligand Relapsing MS Study Group Nat Med (2000) 6:1176–82. doi: 10.1038/80525
55. Goodkin DE, Shulman M, Winkelhake MJ, Waubant E, Andersson PB, Stewart T, et al. A phase I trial of solubilized DR2 : MBp(84-102) (AG284) in multiple sclerosis. Neurology (2000) 54:1414–20. doi: 10.1212/WNL.54.7.1414
56. Warren KG, Catz I, Wucherpfennig KW. Tolerance induction to myelin basic protein by intravenous synthetic peptides containing epitope P85 VVHFFKNIVTP96 in chronic progressive multiple sclerosis. J Neurol Sci (1997) 152:31–8. doi: 10.1016/S0022-510X(97)00130-5
57. Freedman MS, Bar-Or A, Oger J, Traboulsee A, Patry D, Young C, et al. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology (2011) 77:1551–60. doi: 10.1212/WNL.0b013e318233b240
58. Walczak A, Siger M, Ciach A, Szczepanik M, Selmaj K. Transdermal Application of Myelin Peptides in Multiple Sclerosis Treatment. JAMA Neurol (2013) 70:1105–9. doi: 10.1001/jamaneurol.2013.3022
59. Chataway J, Martin K, Barrell K, Sharrack B, Stolt P, Wraith DC, et al. Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology (2018) 90:E955–504. doi: 10.1212/WNL.0000000000005118
60. Belogurov A Jr., Zakharov K, Lomakin Y, Surkov K, Avtushenko S, Kruglyakov P, et al. CD206-Targeted Liposomal Myelin Basic Protein Peptides in Patients with Multiple Sclerosis Resistant to First-Line Disease-Modifying Therapies: A First-in-Human, Proof-of-Concept Dose-Escalation Study. Neurotherapeutics (2016) 13:895–904. doi: 10.1007/s13311-016-0448-0
61. Lomakin Y, Belogurov A Jr., Glagoleva I, Stepanov A, Zakharov K, Okunola J, et al. Administration of Myelin Basic Protein Peptides Encapsulated in Mannosylated Liposomes Normalizes Level of Serum TNF-alpha and IL-2 and Chemoattractants CCL2 and CCL4 in Multiple Sclerosis Patients. Mediators Inflammation (2016) 2016:2847232. doi: 10.1155/2016/2847232
62. Medaer R, Stinissen P, Truyen L, Raus J, Zhang J. Depletion of myelin-basic-protein autoreactive T cells by T-cell vaccination: pilot trial in multiple sclerosis. Lancet (1995) 346:807–8. doi: 10.1016/S0140-6736(95)91622-9
63. Correale J, Lund B, McMillan M, Ko DY, McCarthy K, Weiner LP. T cell vaccination in secondary progressive multiple sclerosis. J Neuroimmunol (2000) 107:130–9. doi: 10.1016/S0165-5728(00)00235-6
64. Zhang JZ, Rivera VM, Tejada-Simon MV, Yang D, Hong J, Li S, et al. T cell vaccination in multiple sclerosis: results of a preliminary study. J Neurol (2002) 249:212–8. doi: 10.1007/PL00007867
65. Van der Aa A, Hellings N, Medaer R, Gelin G, Palmers Y, Raus J, et al. T cell vaccination in multiple sclerosis patients with autologous CSF-derived activated T cells: results from a pilot study. Clin Exp Immunol (2003) 131:155–68. doi: 10.1046/j.1365-2249.2003.02019.x
66. Loftus B, Newsom B, Montgomery M, Von Gynz-Rekowski K, Riser M, Inman S, et al. Autologous attenuated T-cell vaccine (Tovaxin) dose escalation in multiple sclerosis relapsing-remitting and secondary progressive patients nonresponsive to approved immunomodulatory therapies. Clin Immunol (2009) 131:202–15. doi: 10.1016/j.clim.2009.01.005
67. Karussis D, Shor H, Yachnin J, Lanxner N, Amiel M, Baruch K, et al. T cell vaccination benefits relapsing progressive multiple sclerosis patients: a randomized, double-blind clinical trial. PloS One (2012) 7:e50478. doi: 10.1371/journal.pone.0050478
68. Hohol MJ, Khoury SJ, Cook SL, Orav EJ, Hafler DA, Weiner HL. Three-year open protocol continuation study of oral tolerization with myelin antigens in multiple sclerosis and design of a phase III pivotal trial. Ann N Y Acad Sci (1996) 778:243–50. doi: 10.1111/j.1749-6632.1996.tb21132.x
69. Faria AM, Weiner HL. Oral tolerance. Immunol Rev (2005) 206:232–59. doi: 10.1111/j.0105-2896.2005.00280.x
70. Warren KG, Catz I, Ferenczi LZ, Krantz MJ. Intravenous synthetic peptide MBP8298 delayed disease progression in an HLA Class II-defined cohort of patients with progressive multiple sclerosis: results of a 24-month double-blind placebo-controlled clinical trial and 5 years of follow-up treatment. Eur J Neurol (2006) 13:887–95. doi: 10.1111/j.1468-1331.2006.01533.x
71. Loo EW, Krantz MJ, Agrawal B. High dose antigen treatment with a peptide epitope of myelin basic protein modulates T cells in multiple sclerosis patients. Cell Immunol (2012) 280:10–5. doi: 10.1016/j.cellimm.2012.11.004
72. Brocke S, Gijbels K, Allegretta M, Ferber I, Piercy C, Blankenstein T, et al. Treatment of experimental encephalomyelitis with a peptide analogue of myelin basic protein. Nature (1996) 379:343–6. doi: 10.1038/379343a0
73. Crowe PD, Qin Y, Conlon PJ, Antel JP. NBI-5788, an altered MBP83-99 peptide, induces a T-helper 2-like immune response in multiple sclerosis patients. Ann Neurol (2000) 48:758–65. doi: 10.1002/1531-8249(200011)48:5<758::AID-ANA9>3.0.CO;2-2
74. Jurynczyk M, Walczak A, Jurewicz A, Jesionek-Kupnicka D, Szczepanik M, Selmaj K. Immune Regulation of Multiple Sclerosis by Transdermally Applied Myelin Peptides. Ann Neurol (2010) 68:593–601. doi: 10.1002/ana.22219
75. Peschl P, Reindl M, Schanda K, Sospedra M, Martin R, Lutterotti A. Antibody responses following induction of antigen-specific tolerance with antigen-coupled cells. Mult Scler J (2015) 21:651–5. doi: 10.1177/1352458514549405
76. Hafler DA, Cohen I, Benjamin DS, Weiner HL. T cell vaccination in multiple sclerosis: a preliminary report. Clin Immunol Immunopathol (1992) 62:307–13. doi: 10.1016/0090-1229(92)90108-Z
77. Zhang J, Medaer R, Stinissen P, Hafler D, Raus J. MHC-restricted depletion of human myelin basic protein-reactive T cells by T cell vaccination. Science (1993) 261:1451–4. doi: 10.1126/science.7690157
78. Hermans G, Medaer R, Raus J, Stinissen P. Myelin reactive T cells after T cell vaccination in multiple sclerosis: cytokine profile and depletion by additional immunizations. J Neuroimmunol (2000) 102:79–84. doi: 10.1016/S0165-5728(99)00157-5
79. Achiron A, Mandel M. T-cell vaccination in multiple sclerosis. Autoimmun Rev (2004) 3:25–32. doi: 10.1016/S1568-9972(03)00060-0
80. Oksenberg JR, Panzara MA, Begovich AB, Mitchell D, Erlich HA, Murray RS, et al. Selection for T-Cell Receptor V-Beta-D-Beta-J-Beta Gene Rearrangements with Specificity for a Myelin Basic-Protein Peptide in Brain-Lesions of Multiple-Sclerosis. Nature (1993) 362:68–70. doi: 10.1038/362068a0
81. Kotzin BL, Karuturi S, Chou YK, Lafferty J, Forrester JM, Better M, et al. Preferential T-Cell Receptor Beta-Chain Variable Gene Use in Myelin Basic Protein-Reactive T-Cell Clones from Patients with Multiple-Sclerosis. P Natl Acad Sci USA (1991) 88:9161–5. doi: 10.1073/pnas.88.20.9161
82. Weiner HL, Friedman A, Miller A, Khoury SJ, al-Sabbagh A, Santos L, et al. Oral tolerance: immunologic mechanisms and treatment of animal and human organ-specific autoimmune diseases by oral administration of autoantigens. Annu Rev Immunol (1994) 12:809–37. doi: 10.1146/annurev.iy.12.040194.004113
83. Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol (2010) 10:753–66. doi: 10.1038/nri2858
84. Smith CE, Eagar TN, Strominger JL, Miller SD. Differential induction of IgE-mediated anaphylaxis after soluble vs. cell-bound tolerogenic peptide therapy of autoimmune encephalomyelitis. Proc Natl Acad Sci USA (2005) 102:9595–600. doi: 10.1073/pnas.0504131102
85. Frank JA, Stone LA, Smith ME, Albert PS, Maloni H, McFarland HF. Serial contrast-enhanced magnetic resonance imaging in patients with early relapsing-remitting multiple sclerosis: implications for treatment trials. Ann Neurol (1994) 36 Suppl:S86–90. doi: 10.1002/ana.410360719
86. Ten Brinke A, Martinez-Llordella M, Cools N, Hilkens CMU, van Ham SM, Sawitzki B, et al. Ways Forward for Tolerance-Inducing Cellular Therapies- an AFACTT Perspective. Front Immunol (2019) 10:181. doi: 10.3389/fimmu.2019.00181
Keywords: multiple sclerosis, immune tolerance, therapy, antigen-specific, target antigens
Citation: Lutterotti A, Hayward-Koennecke H, Sospedra M and Martin R (2021) Antigen-Specific Immune Tolerance in Multiple Sclerosis—Promising Approaches and How to Bring Them to Patients. Front. Immunol. 12:640935. doi: 10.3389/fimmu.2021.640935
Received: 12 December 2020; Accepted: 26 February 2021;
Published: 22 March 2021.
Edited by:
Hyewon Phee, Amgen, United StatesReviewed by:
Hartmut Wekerle, Max-Planck-Gesellschaft (MPG), GermanyCopyright © 2021 Lutterotti, Hayward-Koennecke, Sospedra and Martin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roland Martin, cm9sYW5kLm1hcnRpbkB1c3ouY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.