 Hazel Morrison
Hazel Morrison Helen McShane
Helen McShane
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 05 March 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.640916
This article is part of the Research Topic Immunological Biomarkers for Tuberculosis View all 30 articles
Regardless of the eventual site of disease, the point of entry for Mycobacterium tuberculosis (M.tb) is via the respiratory tract and tuberculosis (TB) remains primarily a disease of the lungs. Immunological biomarkers detected from the respiratory compartment may be of particular interest in understanding the complex immune response to M.tb infection and may more accurately reflect disease activity than those seen in peripheral samples. Studies in humans and a variety of animal models have shown that biomarkers detected in response to mycobacterial challenge are highly localized, with signals seen in respiratory samples that are absent from the peripheral blood. Increased understanding of the role of pulmonary specific biomarkers may prove particularly valuable in the field of TB vaccines. Here, development of vaccine candidates is hampered by the lack of defined correlates of protection (COPs). Assessing vaccine immunogenicity in humans has primarily focussed on detecting these potential markers of protection in peripheral blood. However, further understanding of the importance of local pulmonary immune responses suggests alternative approaches may be necessary. For example, non-circulating tissue resident memory T cells (TRM) play a key role in host mycobacterial defenses and detecting their associated biomarkers can only be achieved by interrogating respiratory samples such as bronchoalveolar lavage fluid or tissue biopsies. Here, we review what is known about pulmonary specific immunological biomarkers and discuss potential applications and further research needs.
Tuberculosis (TB) remains one of the top ten causes of death worldwide. Around a quarter of the world's population are estimated to be infected with Mycobacterium tuberculosis (M.tb) (1). The World Health Organization's (WHO) End TB Strategy has set the goals of reducing TB incidence by 90% and TB deaths by 95% globally by 2035. If there is any chance of meeting these ambitious targets, new tools to combat this devastating disease will be needed. These include an urgent need for improved diagnostic tests, shorter treatment regimens and more effective vaccines (2).
The range of clinical phenotypes following M.tb exposure spans complete elimination of the pathogen through immunologically contained latent infection to active TB disease (3). This spectrum is governed by complex and incompletely understood interactions between the pathogen and host innate and adaptive immune responses. Innate immune mechanisms within the lung mucosa may be responsible for early clearance of M.tb bacilli prior to T-cell sensitization in exposed individuals who appear to be resistant to M.tb infection (4). Of those who do have presumed latent M.tb infection (LTBI), 5–10% of immunocompetent individuals go on to develop TB disease in their lifetime (5), with the remaining majority achieving immunological equipoise.
Incomplete knowledge of the desired immune responses needed to prevent either active disease or initial infection is one of the key barriers to effective vaccine development (6). It is well-characterized that a T-helper 1 (Th1) cell-mediated adaptive immune response is required, but insufficient, for protection (7, 8). Likewise, whilst antigen-specific interferon gamma (IFN-γ) plays a key role, the level of vaccine-induced IFN-γ in the blood does not correlate with protection (9, 10). Understanding the host immune responses that are needed to confer adequate protection against M.tb would dramatically help in the development and prioritization of vaccines that induce these putative responses.
An immunological biomarker is a measurable characteristic of the immune system that can be assessed as an indicator of normal immune function, disease process, or response to a therapeutic intervention (11). Biomarkers of disease can be used in diagnosis and disease monitoring. Vaccines aim to induce an immunological response to prevent infection or reduce disease severity, termed protection. Biomarkers that are believed to correspond with this effect are termed immune correlates of protection (COP) (12) and form the main focus of biomarkers discussed in this review.
The majority of TB studies looking at biomarkers of protection, both from disease and from infection, have focussed on the peripheral blood compartment in humans and blood and lymphoid organs in animal models. Regardless of the site of active disease, the predominant route via which M.tb bacilli enter the body is via aerosol droplets that are deposited onto alveolar surfaces of the lungs (4). Systemic immunity does not necessarily reflect pulmonary immune responses in the bronchoalveolar spaces at this site of entry for M.tb in humans. Cells of both the innate (such as alveolar macrophages) and adaptive (such as tissue resident memory cells) components of the pulmonary immune system play an increasing recognized role that may be interrogated in the search for markers of protection.
Trained immunity refers to immunological memory within the innate immune system, leading to an augmented response to subsequent, often heterologous insults (13). Innate immune memory is induced in animals after vaccination with BCG (14, 15) although the precise mechanisms via which this occurs are still being studied. Studies of TB contacts show that despite high levels of exposure, up to 30–50% of individuals do not become infected with M.tb, as evidenced by non-reactive tuberculin skin tests and negative IFN-γ release assay (IGRA) testing (16). BCG vaccination correlates with this state of immune protection, suggesting that BCG-potentiated innate immunity may contribute to early M.tb clearance (17).
Given this, it is unclear why, in mice, BCG does not protect against M.tb in the first 14 days post-challenge (18). The kinetics and role of the innate immune response need further study. Controlled human infection models with serial mucosal and systemic sampling allow us to define the kinetics of innate and adaptive immunity and may help us understand this further.
In CD4/CD8 T-cell knock out mice, subcutaneous BCG vaccination induces lasting protective immunity within 7 days, prior to any adaptive immune mechanisms (18). Cells from the lungs of vaccinated mice show a higher proportion of tissue resident macrophages (CD11b+F4/80+) compared to circulating monocytes. Following an infection, or in this case immunization, monocytes may differentiate into interstitial lung macrophages, which then self-perpetuate within the pulmonary compartment. This may represent a mechanism via which BCG induces innate immune memory within the lung (18).
Respiratory viral infection has been found to induce immune memory in lung resident mouse alveolar macrophages (AMs), which go on to produce accelerated levels of detectable neutrophil chemokines, such as CXCL1 and CXCL2 upon restimulation. These trained AMs protect against secondary bacterial infection, with a memory response that is not reliant on circulating monocytes (19).
In a recent model using T-cell depleted mice, mucosal, but not intramuscular, vaccination with an adenoviral-vectored vaccine expressing the M.tb antigen 85A resulted in upregulation of activation markers, such as MHC II, on alveolar and pulmonary interstitial macrophages. This corresponded with reduced M.tb burden after challenge and suggests that activated airway macrophages may play an important role in early M.tb control (20). Debate is ongoing about the precise role AM play in M.tb control. AMs do not readily express pro-inflammatory genes until 10 days after host M.tb infection, which may allow early mycobacterial replication (21). AM-depleted mice show defective granuloma formation, but increased recruitment of other phagocytic and cytotoxic cells to the lungs, with corresponding improved M.tb clearance (22).
AM have been shown to leave the alveolar space and transport M.tb to the lung interstitium in an IL-1 dependent manner, proliferating within the lung to form aggregates (23). Whether this represents the initiation of effective immunological control or the first step in M.tb dissemination is not clear. Systemic BCG immunization in mice has been shown to hasten this egress of M.tb-infected AM from the alveoli into the lung interstititum, increase attraction of monocyte-derived macrophages to the site of infection and promote the early transfer of M.tb from AM to other phagocytic cells (24). In humans, infant AM are less able to control M.tb replication in vitro than adult AM, which may partly explain their susceptibility to more severe, disseminated forms of TB disease. Infant AM were found to express lower levels of chemotactic cytokines including chemokine (C-X-C motif) ligand 9 (CXCL9), suggesting that failure of AM to recruit additional mononuclear cells to the site of infection may result in failure of initial M.tb control (25).
Innate lymphoid cells (ILCS) mediate protective immunity in a variety of tissues, including the lungs. Activated ILCs proliferate in the lungs of mice following mucosal BCG vaccination and lead to increased levels of IFN-γ production (26).
Group 3 ILCs (ILC3s) have similar functionality to Th-17 cells, including production of Il-17 and Il-22. In a human lung tissue explant model, ILC3s upregulate IL-22 and GM-CSF following ex vivo M.tb infection (27) and IL-22 producing ILCs have been shown to enhance phagolysosomal fusion leading to mycobacterial growth inhibition (28). Inhibition of phagolysosomal fusion is one of the key immune mechanisms whereby M.tb evades host immunity.
ILC3s proliferate in the lungs of M.tb infected mice, leading to early alveolar macrophage accumulation. ILC knockout mice showed loss of early AM-mediated M.tb control, which could be rescued by adoptive cell transfer (ACT) of lung ILCs from M.tb-infected control mice (29). ACT of ILC3s also prolonged the survival of diabetic M.tb-infected mice, with increased IL-22 production resulting in reduced lung epithelial damage (30). Loss of ILCs, in particular ILC3, leads to a decrease in AM recruitment within the lung and subsequent higher mycobacterial burden during M.tb infection (29).
Distinct populations of CD103-expressing ILC2 and ILC3s and CXCR5-expressing ILC3s have been identified in human M.tb-infected lung tissue (29). CXCR5 signaling is essential in the formation of inducible bronchus associated lymphoid tissue (iBALT). iBALT is seen surrounding granuloma formation in non-human primate (NHP) and humans with LTBI, but not TB disease (31). iBALT proliferation in the lungs of mice lacking lymph nodes and spleen may be sufficient to control M.tb infection (32).
These studies suggest that ILC3s in particular may have a protective role in early M.tb control, via CXCR5-dependant iBALT formation and the production of IL-22 and IL-17. Mouse models of intranasal BCG vaccination have shown a correlation between protection and levels of IL-17 producing cells within the lungs following M.tb challenge (33).
Mucosal-associated invariant T (MAIT) cells preferentially reside in mucosal tissues, including the pulmonary mucosa. They express pattern recognition receptors, conferring innate immune function, and secrete IFN-γ following stimulation. In humans and NHPs, MAIT cells are enriched in the lungs and BAL fluid following M.tb infection and NHP MAITs express activation markers such as CD69 following both M.tb challenge and intradermal (ID) BCG vaccination (34, 35). In rhesus macaques, intravenous (IV) BCG vaccination induces pulmonary MAIT expansion, which corresponds with subsequent protection against M.tb challenge (36). Following M.bovis infection, MAIT cell deficient mice show higher bacterial colony forming units (CFUs) at early time points compared to wild-type mice (37), highlighting a potential role for MAITs in early mycobacterial clearance.
γδ T-cells are defined by heterodimeric T-cell receptors (TCRs) composed of γ and δ chains and are enriched in epithelial and mucosal tissues, including lung alveoli. The majority are activated in an MHC-independent manner and produce cytotoxic granules and canonical pro-inflammatory cytokines, including IFN-γ, TNF-α, and IL-17. Their activation results in killing of M.tb infected macrophages (38). Following bacterial infection, lung γδ T-cells in mice exhibit increased expression of activation markers such as CD69 and CD25, and proliferate by local expansion rather than recruitment from the periphery (39). In NHPs, expansion of lung γδ T-cells by selective vaccination reduces disease pathology and dissemination following M.tb challenge (40).
Tissue resident memory cells (TRM) represent a distinct subset of lymphocytes. They share functional similarities with central and effector memory T-cells, but remain situated within localized tissue compartments and do not recirculate into the blood stream. They have been demonstrated at sites including the skin, intestines, urogenital tract, and lung mucosa (41–44). This positioning at key anatomical barrier sites means that TRM can respond rapidly to potential infective stimuli and lung TRM may signify the first line of adaptive cellular defense against specific respiratory pathogens, including M.tb.
Due to the highly vascular nature of the lungs, distinguishing genuine TRM, truly resident in the lung mucosa, from blood lymphocytes that egress from the vasculature following a stimulus such as infection, is difficult. Mouse models, using techniques such as parabiosis and in vivo intravascular staining, have confirmed that true lung TRM cells are identifiable and do not re-enter the peripheral circulation, in comparison to lymphoid memory T cells.
Many of the techniques employed in animal models to delineate TRM from pulmonary vascular lymphocytes are not feasible in humans but have been crucial in confirming that biomarkers seen in humans correspond to TRM specific markers identified in animals. Upregulation of CD69 is a key marker of TRM activation at a variety of sites including the lung and results in inhibition of sphingosine 1-phosphate-meditated lymphocyte migration (45). Additionally, CD8+ TRM cells express the αEβ7 integrin heterodimer, identified by CD103 marker staining (46). Other significant markers of lung TRM in both human and animal models include PD-1, CD44, CXCR3, and integrins including CD49a, CD11a, and VLA-4 (45), with KLRG-1 and CD62L downregulated (47). CD4+ TRM form a heterogeneous group, with some displaying an effector profile (T-bet+) and others appearing more regulatory (Foxp3hi IL-10hi]. In contrast, pulmonary CD8+ TRM cells appear more homogenous, expressing predominantly Th1 cytokines (48).
The key importance of these cells in animal models of respiratory infection has been shown in several studies. In murine adoptive transfer studies, CXCR3hiCD4+ T-cells preferentially localize to the lung parenchyma and are better at controlling M.tb infection than their CX3CR1hiKLRG1hi equivalents which remain within the vasculature (47). Intranasal immunization of mice with a recombinant influenza A vaccine expressing the PR8.p25 Ag85B epitope led to the development CD4+ TRM throughout the lung parenchyma. Persistence of these cells following FTY720-induced intravascular lymphopaenia indicates true tissue-resident memory status, without reliance on circulating cells, and was sufficient for protection against subsequent M.tb challenge (49).
Route of vaccination may alter the magnitude and character of the adaptive pulmonary immune response, but it is unclear if this will necessarily lead to improved overall protective efficacy. For example, airway mucosal boosting following parental priming with the subunit vaccine candidate H56:CAF01 results in a significant increase in pulmonary TRM and early local T-cell responses, without conferring any additional protection against M.tb challenge (50). Intramuscular vaccination of mice with the adjuvanted subunit TB vaccine candidate ID-93 results in a systemic, TH1-dominated immune response. In contrast, following ID-93 intranasal immunization, a predominantly IL-17A-producing, TH-17 response is seen; with an increase in antigen specific CD4+ TRM in the lung and BAL fluid. Despite these differences, the level of protection conferred was equal across the different delivery methods (51). In a recent study, protection conferred by intra-tracheal administration of the fusion protein TB vaccine candidate, CysVac2, was associated with the induction of higher levels of antigen-specific CD4+ lung TRM, expressing IL-17, and RORγT (52).
While intradermal BCG vaccination is able to generate antigen-specific pulmonary TRM in mice, mucosal BCG vaccination produces increased numbers of both CD4+ and CD8+ TRM and this corresponds with subsequent enhanced protection against M.tb challenge (48, 53). Mucosal transfer of sorted airway resident T-cells, in particular CD8+ TRM, from mucosally BCG-vaccinated mice provided increased protection against M.tb challenge in recipient mice (48). Non-human primates immunized with intravenous BCG were found to have significantly higher levels of CD69+ (with a subset of CD103+) lung parenchymal CD4+ T-cells than intradermal or aerosol immunized animals and this was associated with sterilizing immunity against M.tb challenge (36).
These findings suggest that vaccination routes and strategies which induce pulmonary CD4+ and CD8+ TRM may result in superior levels of protection. This may be one reason why levels of peripheral circulating antigen-specific T-cells do not adequately correlate with protection. Biomarkers of TRM may be useful as correlates of vaccine induced protection, but would require a significant change in sampling methods to assess vaccine efficacy.
The role of the humoral immune system in TB control is uncertain. In humans, M.tb infection induces M.tb-specific IgA, as well as IgG, antibodies in BAL fluid, but their precise role and level of interaction with M.tb at the mucosal level remains unknown (54, 55).
Secretory Immunoglobulin A (sIgA) is the predominant isotype in mucosal secretions and may contribute to protection. Intranasal administration of purified human sIgA to mice leads to increased M.tb clearance and improved disease control (56). Knockout mice lacking the polymeric IgR receptor necessary for IgA transport to the respiratory mucosa are more susceptible to M.tb infection than wild-type mice (57). In a BCG challenge model, IgA deficient mice are more susceptible to infection than wild-type (58).
A functional mycobacterial growth inhibition assay (MGIA), which measures the sum of the parts of the innate and adaptive immune response, may be a useful tool to facilitate vaccine development. Such a tool could also allow the interrogation of potential COP by depletion studies using serum, peripheral blood mononuclear cells (PBMCs) or other specific cell types. To date such an assay has been optimized for use in whole blood and PBMC (59, 60). Using mucosal samples in such an assay may further identify lung specific protective mechanisms in future.
Animal and human studies that focus on sampling the lung mucosal compartment will improve our understanding of lung mucosal immunity to M.tb. Parallel animal and human studies would allow more detailed interrogation of these processes. Delivery of vaccine candidates via aerosol routes has been shown to induce specific mucosal immune components that can be compared across species, with BAL samples from macaques and humans following aerosol MVA85A showing increased levels of antigen-specific cellular immune responses compared to peripheral blood (61–63). Further detailed mechanistic interrogation of lung-specific immunity is possible in the more tractable murine model (64, 65).
The study of human lung immunity gives the opportunity to interrogate the interaction between the host and M.tb bacilli at the site of natural infection. However, significant barriers exist. Obtaining high quality respiratory samples for immunological analysis generally requires invasive sampling (see Figure 1). The scarcity of these resources in areas with the highest burdens of TB disease, coupled with the costs and ethical considerations of invasive sampling, may explain the relative lack of immunological studies focussing on the human pulmonary microenvironment (4).
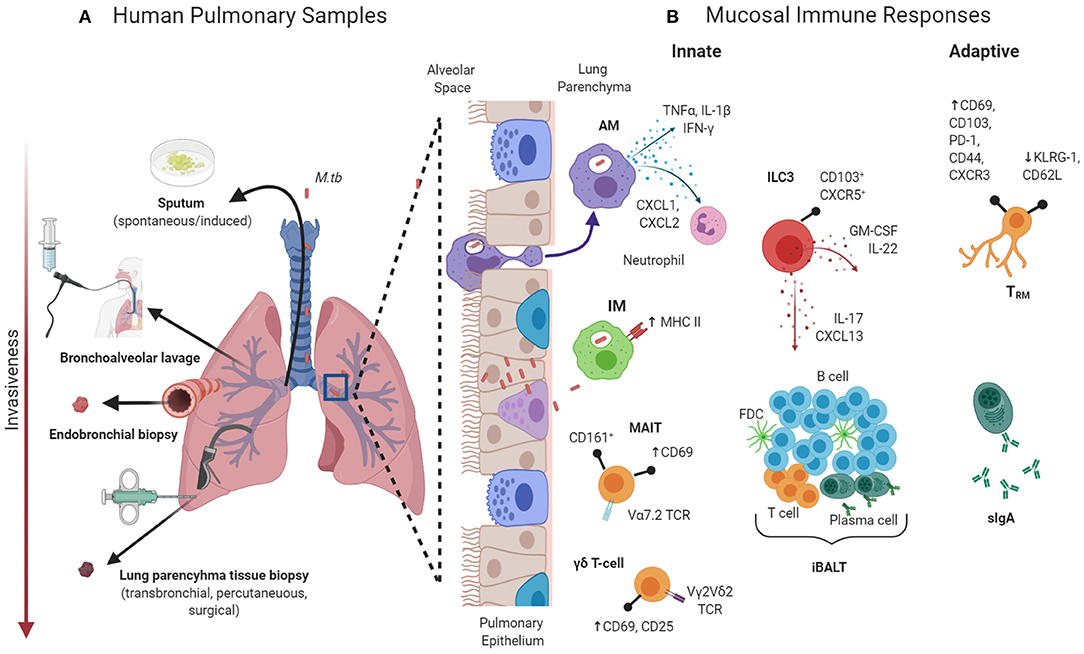
Figure 1. (A) Samples that can be obtained from the human pulmonary compartment from immunological interrogation. (B) Selected components of the pulmonary mucosal immune system that may be involved in protection against Mycobacterium tuberculosis (M.tb). AM, alveolar macrophages; CD, cluster of differentiation; CXCR, C-X-C chemokine receptor; FBC, follicular dendritic cell; GM-CSF, granulocyte-macrophage colony-stimulating factor; iBALT, inducible bronchus-associated lymphoid tissue; IFN, interferon; ILC, innate lymphoid cells; IM, interstitial macrophages; IL, interleukin; KLRG, killer-cell lectin like receptor G; MAIT, mucosal-associated invariant T-cells; PD, programmed cell death protein; sIgA, secretory immunoglobulin A; TCR, T-cell receptor; TNF, tumor necrosis factor; TRM, tissue-resident memory T-cell. Created with BioRender.com.
How best to sample the human pulmonary compartment remains the subject of debate. Sputum (induced or spontaneous) is often too contaminated, for example with upper airway epithelial cells and microbes, to provide detailed immunological analysis of the lower respiratory tract. Bronchoalveolar lavage (BAL) can be used to obtain bronchoalveolar cells (66). Studies comparing BAL cells to lung tissue biopsies in healthy controls and TB patients suggest that BAL cells are a reasonable representation of lung cellular composition (67). However, this assumption may not hold true for HIV infected individuals, where significant depletion of lung interstial CD4+ T cells may occur despite relatively normal CD4+ T cells levels in bronchoalveolar cells (68).
Where comparative data does exist, it suggest there is a significant difference in immunological activity and therefore possible biomarkers of disease and protection in the lungs compared to the peripheral blood. Schwander et al. found compartmentalized markers of active TB disease, with significantly increased levels of activated T lymphocytes (CD69+ HLA DR +) seen in bronchoalveolar cells of patients with active TB compared to healthy controls, whereas in PBMCs there was no difference across groups (69). PBMC in TB patients are hyporesponsive, with respect to both frequency of IFN-γ producing cells and DNA synthesis, to both mycobacterial and non-mycobacterial antigens compared to healthy subjects. Conversely, bronchoalveolar cells from affected lung segments in TB patients show increased responses to mycobacterial antigens, suggesting significant localization of antigen–specific cells within the affected lungs during active pulmonary TB (70).
Pulmonary TB disease is characterized by an enhancement of local Th1-mediated immunity, with increased IL-12 and IFN-γ production within affected lung segments (70). Despite this apparently functional local Th1-mediated immune response, there is clearly failure to control M.tb in those with active disease. Suppressive cytokines, including IL-4, TGF-β and IL-10, are increased in bronchoalveolar cell samples of active TB compared with healthy controls (71) and may represent distinct local immunosuppressive mechanisms that interfere with Th1-mediated effectors in the bronchoalveolar environment. One difficulty in studying the respiratory mucosal immune response to M.tb infection in humans is the inability to define precisely the time of infection. Due to the varying clinical course and potential for latency, active disease may only be diagnosed months or years after the point of infection. In other diseases, such as influenza, malaria and typhoid, controlled human infection models (CHIMs) have been used to interrogate the immune response and can also be used to evaluate vaccine efficacy. Treatment for active TB disease requires a lengthy combination of potentially toxic medications, and proof of definitive cure may not be possible. For these reasons, a CHIM with M.tb would not be ethical. However, use of alternative mycobacterial models to mimic M.tb infection are being explored. For example, BCG may be used as a surrogate, as it does not cause active disease in immunocompetent humans but is a live replicating mycobacteria that stimulates an immune response. Interrogation of the pulmonary mucosal immune response following a defined time point infection with BCG may lead to greater understanding of key immunological mechanisms, in particular in the early stages of infection.
Bronchoscopic instillation of BCG into lung segments of healthy, HIV-negative participants in South Africa with a range of TB phenotypes was shown to be safe and resulted in changes to differentially expressed genes and proteomics in the BAL fluid which were not detectable in the blood, suggesting a highly localized response (72). Studies in our group are ongoing to define the human innate and adaptive immune response to a defined time point challenge with aerosol BCG, and specifically comparing the peripheral and pulmonary compartment (Clinical trials.gov/NCT03912207). Parallel ongoing studies in non-human primates will add value to this work.
Growing evidence shows that immunological responses are compartmentalized and biomarkers present in the peripheral blood may be poorly representative of important, local effects within the lungs. Innate, trained and adaptive components of the pulmonary immune system are likely to play an interconnected role in protection, with distinct features of lung mucosal immunity such as alveolar macrophages, BALT and TRM all warranting further investigation. The characterization of these immunological responses at the natural site of M.tb infection is of paramount importance, both in to increase our understanding of pathogenesis and more specifically to aid rational vaccine development.
Pre-clinical animal models play a key role in defining the pulmonary immune response to both M.tb and systemic and mucosally-delivered TB vaccines. Carefully designed small studies in humans can complement and add to these pre-clinical studies. Interrogating the initial stages of M.tb immunity in human lungs, for example in healthy household contacts, would have the potential to distinguish biomarkers of protective immunity (COP) at the site of initial host contact with M.tb. Logistical and ethical difficulties in obtaining invasive human pulmonary sample in these circumstances mean that more novel investigative strategies may be needed.
Vaccine development in TB faces a paradox—a vaccine-induced COP can only be validated in large field trials of an effective vaccine. However, selection of which candidate vaccines to take forward for such costly trials requires some level of discrimination. As evidenced by the compartmentalized nature of immunological biomarkers in both human and animal models, peripheral blood biomarkers, whilst easier to obtain, may not be the best choice of read-out for rational vaccine selection.
CHIMs in healthy volunteers, either with BCG or potentially in future with rationally attenuated M.tb strains, may prove an alternative strategy to delineate human immunological response to a defined time point infection (73). Davids et al. human lung challenge model showed that responses to in-vitro and in-vivo PPD and BCG stimulation were significantly different, raising the prospect that the study of vaccine-induced immune biomarkers of protection may need to focus more on lung mycobacterial challenges and sampling, rather than peripheral blood (72). Given the considerable technical obstacles this approach faces, it could prove more likely that if pulmonary correlates of protection (or disease) are identified, systemic surrogate markers may be identifiable that can then be more easily appraised in future studies.
HMo wrote the first draft of the manuscript. HMo and HMc contributed to manuscript revision and read and approved the submitted version.
HMc is a Wellcome Trust Investigator (WT 206331/Z/17/Z). The views expressed are those of the author(s) and not necessarily those of the Wellcome Trust.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Houben RM, Dodd PJ. The global burden of latent tuberculosis infection: a re-estimation using mathematical modelling. PLoS Med. (2016) 13:e1002152. doi: 10.1371/journal.pmed.1002152
3. Basaraba RJ, Hunter RL. Pathology of tuberculosis: how the pathology of human tuberculosis informs and directs animal models. Microbiol Spectr. (2017) 5. doi: 10.1128/microbiolspec.TBTB2-0029-2016
4. Schwander S, Dheda K. Human lung immunity against Mycobacterium tuberculosis: insights into pathogenesis and protection. Am J Respir Crit Care Med. (2011) 183:696–707. doi: 10.1164/rccm.201006-0963PP
5. Comstock GW, Livesay VT, Woolpert SF. The prognosis of a positive tuberculin reaction in childhood and adolescence. Am J Epidemiol. (1974) 99:131–8. doi: 10.1093/oxfordjournals.aje.a121593
6. Wilkie MEM, McShane H. TB vaccine development: where are we and why is it so difficult? Thorax. (2015) 70:299–301. doi: 10.1136/thoraxjnl-2014-205202
7. Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J Exp Med. (2001) 193:271–80. doi: 10.1084/jem.193.3.271
8. Lyadova IV, Panteleev AV. Th1 and Th17 cells in tuberculosis: protection, pathology, and biomarkers. Mediator Inflammat. (2015) 2015:854507. doi: 10.1155/2015/854507
9. Leal IS, Smedegård B, Andersen P, Appelberg R. Failure to induce enhanced protection against tuberculosis by increasing T-cell-dependent interferon-gamma generation. Immunology. (2001) 104:157–61. doi: 10.1046/j.1365-2567.2001.01305.x
10. Mittrücker HW, Steinhoff U, Köhler A, Krause M, Lazar D, Mex P, et al. Poor correlation between BCG vaccination-induced T cell responses and protection against tuberculosis. Proc Natl Acad Sci USA. (2007) 104:12434–9. doi: 10.1073/pnas.0703510104
11. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. (2001) 69:89–95. doi: 10.1067/mcp.2001.113989
12. Plotkin SA. Correlates of protection induced by vaccination. Clin Vaccine Immunol. (2010) 17:1055–65. doi: 10.1128/CVI.00131-10
13. Netea MG, van der Meer JW. Trained Immunity: an Ancient Way of Remembering. Cell Host Microbe. (2017) 21:297–300. doi: 10.1016/j.chom.2017.02.003
14. Sakuma T, Suenaga T, Yoshida I, Azuma M. Mechanisms of enhanced resistance of Mycobacterium bovis BCG-treated mice to ectromelia virus infection. Infect Immun. (1983) 42:567–73. doi: 10.1128/IAI.42.2.567-573.1983
15. van 't Wout JW, Poell R, van Furth R. The role of BCG/PPD-activated macrophages in resistance against systemic candidiasis in mice. Scand J Immunol. (1992) 36:713–9. doi: 10.1111/j.1365-3083.1992.tb03132.x
16. Morrison J, Pai M, Hopewell PC. Tuberculosis and latent tuberculosis infection in close contacts of people with pulmonary tuberculosis in low-income and middle-income countries: a systematic review and meta-analysis. Lancet Infect Dis. (2008) 8:359–68. doi: 10.1016/S1473-3099(08)70071-9
17. Verrall AJ, Alisjahbana B, Apriani L, Novianty N, Nurani AC, van Laarhoven A, et al. Early clearance of Mycobacterium tuberculosis: the INFECT case contact cohort study in Indonesia. J Infect Dis. (2020) 221:1351–60. doi: 10.1093/infdis/jiz168
18. Bickett TE, McLean J, Creissen E, Izzo L, Hagan C, Izzo AJ, et al. Characterizing the BCG induced macrophage and neutrophil mechanisms for defense against Mycobacterium tuberculosis. Front Immunol. (2020) 11:1202. doi: 10.3389/fimmu.2020.01202
19. Yao Y, Jeyanathan M, Haddadi S, Barra NG, Vaseghi-Shanjani M, Damjanovic D, et al. Induction of autonomous memory alveolar macrophages requires T cell help and is critical to trained immunity. Cell. (2018) 175:1634–50.e17. doi: 10.1016/j.cell.2018.09.042
20. D'Agostino MR, Lai R, Afkhami S, Khera A, Yao Y, Vaseghi-Shanjani M, et al. Airway macrophages mediate mucosal vaccine–induced trained innate immunity against Mycobacterium tuberculosis in early stages of infection. J Immunol. (2020) 205:2750–62. doi: 10.4049/jimmunol.2000532
21. Rothchild AC, Olson GS, Nemeth J, Amon LM, Mai D, Gold ES, et al. Alveolar macrophages generate a noncanonical NRF2-driven transcriptional response to Mycobacterium tuberculosis in vivo. Sci Immunol. (2019) 4:eaaw6693. doi: 10.1126/sciimmunol.aaw6693
22. Leemans JC, Juffermans NP, Florquin S, van Rooijen N, Vervoordeldonk MJ, Verbon A, et al. Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J Immunol. (2001) 166:4604. doi: 10.4049/jimmunol.166.7.4604
23. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar macrophages provide an early Mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. (2018) 24:439–46.e4. doi: 10.1016/j.chom.2018.08.001
24. Delahaye JL, Gern BH, Cohen SB, Plumlee CR, Shafiani S, Gerner MY, et al. Cutting edge: bacillus calmette–guérin–induced T cells shape Mycobacterium tuberculosis infection before reducing the bacterial burden. J Immunol. (2019) 203:807. doi: 10.4049/jimmunol.1900108
25. Goenka A, Prise IE, Connolly E, Fernandez-Soto P, Morgan D, Cavet JS, et al. Infant alveolar macrophages are unable to effectively contain Mycobacterium tuberculosis. Front Immunol. (2020) 11:486. doi: 10.3389/fimmu.2020.00486
26. Steigler P, Daniels NJ, McCulloch TR, Ryder BM, Sandford SK, Kirman JR. BCG vaccination drives accumulation and effector function of innate lymphoid cells in murine lungs. Immunol Cell Biol. (2018) 96:379–89. doi: 10.1111/imcb.12007
27. Maertzdorf J, Tönnies M, Lozza L, Schommer-Leitner S, Mollenkopf H, Bauer TT, et al. Mycobacterium tuberculosis Invasion of the human lung: first contact. Front Immunol. (2018) 9:1346. doi: 10.3389/fimmu.2018.01346
28. Dhiman R, Indramohan M, Barnes PF, Nayak RC, Paidipally P, Rao LV, et al. IL-22 produced by human NK cells inhibits growth of Mycobacterium tuberculosis by enhancing phagolysosomal fusion. J Immunol. (2009) 183:6639–45. doi: 10.4049/jimmunol.0902587
29. Ardain A, Domingo-Gonzalez R, Das S, Kazer SW, Howard NC, Singh A, et al. Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature. (2019) 570:528–32. doi: 10.1038/s41586-019-1276-2
30. Tripathi D, Radhakrishnan RK, Sivangala Thandi R, Paidipally P, Devalraju KP, Neela VSK, et al. IL-22 produced by type 3 innate lymphoid cells (ILC3s) reduces the mortality of type 2 diabetes mellitus (T2DM) mice infected with Mycobacterium tuberculosis. PLoS Pathog. (2019) 15:e1008140. doi: 10.1371/journal.ppat.1008140
31. Stylianou E, Paul MJ, Reljic R, McShane H. Mucosal delivery of tuberculosis vaccines: a review of current approaches and challenges. Exp Rev Vaccines. (2019) 18:1271–84. doi: 10.1080/14760584.2019.1692657
32. Kashino SS, Vallerskog T, Martens G, Troudt J, Keyser A, Taylor J, et al. Initiation of acquired immunity in the lungs of mice lacking lymph nodes after infection with aerosolized Mycobacterium tuberculosis. Am J Pathol. (2010) 176:198–204. doi: 10.2353/ajpath.2010.090446
33. Uranga S, Marinova D, Martin C, Aguilo N. Protective efficacy and pulmonary immune response following subcutaneous and intranasal BCG administration in mice. J Vis Exp. (2016) 19115. doi: 10.3791/54440
34. Gold MC, Napier RJ, Lewinsohn DM. MR1-restricted mucosal associated invariant T (MAIT) cells in the immune response to Mycobacterium tuberculosis. Immunol Rev. (2015) 264:154–66. doi: 10.1111/imr.12271
35. Greene JM, Dash P, Roy S, McMurtrey C, Awad W, Reed JS, et al. MR1-restricted mucosal-associated invariant T (MAIT) cells respond to mycobacterial vaccination and infection in nonhuman primates. Mucosal Immunol. (2017) 10:802–13. doi: 10.1038/mi.2016.91
36. Darrah PA, Zeppa JJ, Maiello P, Hackney JA, Wadsworth MH, 2nd, Hughes TK, et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature. (2020) 577:95–102. doi: 10.1038/s41586-019-1817-8
37. Chua WJ, Truscott SM, Eickhoff CS, Blazevic A, Hoft DF, Hansen TH. Polyclonal mucosa-associated invariant T cells have unique innate functions in bacterial infection. Infect Immun. (2012) 80:3256–67. doi: 10.1128/IAI.00279-12
38. Meraviglia S, El Daker S, Dieli F, Martini F, Martino A. γδ T cells cross-link innate and adaptive immunity in Mycobacterium tuberculosis infection. Clin Dev Immunol. (2011) 2011:587315. doi: 10.1155/2011/587315
39. Kirby AC, Newton DJ, Carding SR, Kaye PM. Evidence for the involvement of lung-specific gammadelta T cell subsets in local responses to Streptococcus pneumoniae infection. Eur J Immunol. (2007) 37:3404–13. doi: 10.1002/eji.200737216
40. Shen L, Frencher J, Huang D, Wang W, Yang E, Chen CY, et al. Immunization of Vγ2Vδ2 T cells programs sustained effector memory responses that control tuberculosis in nonhuman primates. Proc Natl Acad Sci USA. (2019) 116:6371–8. doi: 10.1073/pnas.1811380116
41. Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. (2010) 207:553–64. doi: 10.1084/jem.20090858
42. Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrançois L, Farber DL. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. (2011) 187:5510–4. doi: 10.4049/jimmunol.1102243
43. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. (2013) 14:1294–301. doi: 10.1038/ni.2744
44. Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci USA. (2012) 109:7037–42. doi: 10.1073/pnas.1202288109
45. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
46. Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol. (2014) 95:215–24. doi: 10.1189/jlb.0313180
47. Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer-Barber KD, Masopust D, et al. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol. (2014) 192:2965–9. doi: 10.4049/jimmunol.1400019
48. Perdomo C, Zedler U, Kühl AA, Lozza L, Saikali P, Sander LE, et al. Mucosal BCG vaccination induces protective lung-resident memory T cell populations against tuberculosis. mBio. (2016) 7:e01686–16. doi: 10.1128/mBio.01686-16
49. Flórido M, Muflihah H, Lin LCW, Xia Y, Sierro F, Palendira M, et al. Pulmonary immunization with a recombinant influenza A virus vaccine induces lung-resident CD4(+) memory T cells that are associated with protection against tuberculosis. Mucosal Immunol. (2018) 11:1743–52. doi: 10.1038/s41385-018-0065-9
50. Woodworth JS, Christensen D, Cassidy JP, Agger EM, Mortensen R, Andersen P. Mucosal boosting of H56:CAF01 immunization promotes lung-localized T cells and an accelerated pulmonary response to Mycobacterium tuberculosis infection without enhancing vaccine protection. Mucosal Immunol. (2019) 12:816–26. doi: 10.1038/s41385-019-0145-5
51. Orr MT, Beebe EA, Hudson TE, Argilla D, Huang PW, Reese VA, et al. Mucosal delivery switches the response to an adjuvanted tuberculosis vaccine from systemic TH1 to tissue-resident TH17 responses without impacting the protective efficacy. Vaccine. (2015) 33:6570–8. doi: 10.1016/j.vaccine.2015.10.115
52. Counoupas C, Ferrell KC, Ashhurst A, Bhattacharyya ND, Nagalingam G, Stewart EL, et al. Mucosal delivery of a multistage subunit vaccine promotes development of lung-resident memory T cells and affords interleukin-17-dependent protection against pulmonary tuberculosis. NPJ Vaccines. (2020) 5:105. doi: 10.1038/s41541-020-00255-7
53. Bull NC, Stylianou E, Kaveh DA, Pinpathomrat N, Pasricha J, Harrington-Kandt R, et al. Enhanced protection conferred by mucosal BCG vaccination associates with presence of antigen-specific lung tissue-resident PD-1(+) KLRG1(-) CD4(+) T cells. Mucosal Immunol. (2019) 12:555–64. doi: 10.1038/s41385-018-0109-1
54. Demkow U, Białas-Chromiec B, Filewska M, Sobiecka M, Kuś J, Szturmowicz M, et al. Humoral immune response against mycobacterial antigens in bronchoalveolar fluid from tuberculosis patients. J Physiol Pharmacol. (2005) 56(Suppl 4):79–84.
55. Raja A, Baughman RP, Daniel TM. The detection by immunoassay of antibody to mycobacterial antigens and mycobacterial antigens in bronchoalveolar lavage fluid from patients with tuberculosis and control subjects. Chest. (1988) 94:133–7. doi: 10.1378/chest.94.1.133
56. Alvarez N, Otero O, Camacho F, Borrero R, Tirado Y, Puig A, et al. Passive administration of purified secretory IgA from human colostrum induces protection against Mycobacterium tuberculosis in a murine model of progressive pulmonary infection. BMC Immunol. (2013) 14(Suppl 1):S3. doi: 10.1186/1471-2172-14-S1-S3
57. Tjärnlund A, Rodríguez A, Cardona PJ, Guirado E, Ivanyi J, Singh M, et al. Polymeric IgR knockout mice are more susceptible to mycobacterial infections in the respiratory tract than wild-type mice. Int Immunol. (2006) 18:807–16. doi: 10.1093/intimm/dxl017
58. Rodríguez A, Tjärnlund A, Ivanji J, Singh M, García I, Williams A, et al. Role of IgA in the defense against respiratory infections IgA deficient mice exhibited increased susceptibility to intranasal infection with Mycobacterium bovis BCG. Vaccine. (2005) 23:2565–72. doi: 10.1016/j.vaccine.2004.11.032
59. Fletcher HA, Tanner R, Wallis RS, Meyer J, Manjaly ZR, Harris S, et al. Inhibition of mycobacterial growth in vitro following primary but not secondary vaccination with Mycobacterium bovis BCG. Clin Vaccine Immunol. (2013) 20:1683–9. doi: 10.1128/CVI.00427-13
60. Tanner R, O'Shea MK, White AD, Müller J, Harrington-Kandt R, Matsumiya M, et al. The influence of haemoglobin and iron on in vitro mycobacterial growth inhibition assays. Sci Rep. (2017) 7:43478. doi: 10.1038/srep43478
61. Satti I, Meyer J, Harris SA, Manjaly Thomas ZR, Griffiths K, Antrobus RD, et al. Safety and immunogenicity of a candidate tuberculosis vaccine MVA85A delivered by aerosol in BCG-vaccinated healthy adults: a phase 1, double-blind, randomised controlled trial. Lancet Infect Dis. (2014) 14:939–46. doi: 10.1016/S1473-3099(14)70845-X
62. White AD, Sibley L, Dennis MJ, Gooch K, Betts G, Edwards N, et al. Evaluation of the safety and immunogenicity of a candidate tuberculosis vaccine, MVA85A, delivered by aerosol to the lungs of macaques. Clin Vaccine Immunol. (2013) 20:663–72. doi: 10.1128/CVI.00690-12
63. Manjaly Thomas ZR, Satti I, Marshall JL, Harris SA, Lopez Ramon R, Hamidi A, et al. Alternate aerosol and systemic immunisation with a recombinant viral vector for tuberculosis, MVA85A: a phase I randomised controlled trial. PLoS Med. (2019) 16:e1002790. doi: 10.1371/journal.pmed.1002790
64. Stylianou E, Harrington-Kandt R, Beglov J, Bull N, Pinpathomrat N, Swarbrick GM, et al. Identification and evaluation of novel protective antigens for the development of a candidate tuberculosis subunit vaccine. Infect Immun. (2018) 86:1–16. doi: 10.1128/IAI.00014-18
65. Kaveh DA, Garcia-Pelayo MC, Bull NC, Sanchez-Cordon PJ, Spiropoulos J, Hogarth PJ. Airway delivery of both a BCG prime and adenoviral boost drives CD4 and CD8 T cells into the lung tissue parenchyma. Sci Rep. (2020) 10:18703. doi: 10.1038/s41598-020-75734-x
66. Helmers RA, Dayton CS, Floerchinger C, Hunninghake GW. Bronchoalveolar lavage in interstitial lung disease: effect of volume of fluid infused. J Appl Physiol. (1989) 67:1443–6. doi: 10.1152/jappl.1989.67.4.1443
67. Law KF, Jagirdar J, Weiden MD, Bodkin M, Rom WN. Tuberculosis in HIV-positive patients: cellular response and immune activation in the lung. Am J Respir Crit Care Med. (1996) 153(4 Pt 1):1377–84. doi: 10.1164/ajrccm.153.4.8616569
68. Corleis B, Bucsan AN, Deruaz M, Vrbanac VD, Lisanti-Park AC, Gates SJ, et al. HIV-1 and SIV infection are associated with early loss of lung interstitial CD4+ T cells and dissemination of pulmonary tuberculosis. Cell Rep. (2019) 26:1409–18.e5. doi: 10.1016/j.celrep.2019.01.021
69. Schwander SK, Sada E, Torres M, Escobedo D, Sierra JG, Alt S, et al. T Lymphocytic and immature macrophage alveolitis in active pulmonary tuberculosis. J Infect Dis. (1996) 173:1267–72. doi: 10.1093/infdis/173.5.1267
70. Schwander SK, Torres M, Sada E, Carranza C, Ramos E, Tary-Lehmann M, et al. Enhanced responses to Mycobacterium tuberculosis antigens by human alveolar lymphocytes during active pulmonary tuberculosis. J Infect Dis. (1998) 178:1434–45. doi: 10.1086/314454
71. Herrera MT, Torres M, Nevels D, Perez-Redondo CN, Ellner JJ, Sada E, et al. Compartmentalized bronchoalveolar IFN-gamma and IL-12 response in human pulmonary tuberculosis. Tuberculosis. (2009) 89:38–47. doi: 10.1016/j.tube.2008.08.002
72. Davids M, Pooran A, Hermann C, Mottay L, Thompson F, Cardenas J, et al. A Human lung challenge model to evaluate the safety and immunogenicity of PPD and live bacillus Calmette-Guérin. Am J Respir Crit Care Med. (2020) 201:1277–91. doi: 10.1164/rccm.201908-1580OC
Keywords: tuberculosis, biomarkers, pulmonary, mucosal, vaccines
Citation: Morrison H and McShane H (2021) Local Pulmonary Immunological Biomarkers in Tuberculosis. Front. Immunol. 12:640916. doi: 10.3389/fimmu.2021.640916
Received: 12 December 2020; Accepted: 10 February 2021;
Published: 05 March 2021.
Edited by:
Adam Penn-Nicholson, Foundation for Innovative New Diagnostics, SwitzerlandReviewed by:
Warwick Britton, The University of Sydney, AustraliaCopyright © 2021 Morrison and McShane. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hazel Morrison, aGF6ZWwubW9ycmlzb25AbmRtLm94LmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.