
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 05 March 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.639226
 Faris Ghalib Bakri1,2
Faris Ghalib Bakri1,2 Michelle Mollin3Sylvain Beaumel3Bénédicte Vigne3
Michelle Mollin3Sylvain Beaumel3Bénédicte Vigne3 Nathalie Roux-Buisson4,5Adel Mohammed Al-Wahadneh6
Nathalie Roux-Buisson4,5Adel Mohammed Al-Wahadneh6 Raed Mohammed Alzyoud6Wail Ahmad Hayajneh7Ammar Khaled Daoud8
Raed Mohammed Alzyoud6Wail Ahmad Hayajneh7Ammar Khaled Daoud8 Mohammed Elian Abu Shukair9Mansour Fuad Karadshe10Mahmoud Mohammad Sarhan11Jamal Ahmad Wadi Al-Ramahi12Julien Fauré4,5
Mohammed Elian Abu Shukair9Mansour Fuad Karadshe10Mahmoud Mohammad Sarhan11Jamal Ahmad Wadi Al-Ramahi12Julien Fauré4,5 John Rendu4,5
John Rendu4,5 Marie Jose Stasia3,13*
Marie Jose Stasia3,13*Chronic granulomatous Disease (CGD) is a rare innate immunodeficiency disorder caused by mutations in one of the six genes (CYBA, CYBB, NCF1, NCF2, NCF4, and CYBC1/EROS) encoding the superoxide-producing nicotinamide adenine dinucleotide phosphate (NADPH)—oxidase complex in phagocytes. In the Western population, the most prevalent form of CGD (about two-thirds of all cases) is the X-linked form (X-CGD) caused by mutations in CYBB. The autosomal recessive forms (AR-CGD), due to mutations in the other genes, collectively account for the remaining one-third of CGD cases. We investigated the clinical and molecular features of 22 Jordanian, 7 Libyan, and 2 Iraqi CGD patients from 21 different families. In addition, 11 sibling patients from these families were suspected to have been died from CGD as suggested by their familial and clinical history. All patients except 9 were children of consanguineous parents. Most of the patients suffered from AR-CGD, with mutations in CYBA, NCF1, and NCF2, encoding p22phox, p47phox, and p67phox proteins, respectively. AR-CGD was the most frequent form, in Jordan probably because consanguineous marriages are common in this country. Only one patient from non-consanguineous parents suffered from an X910 CGD subtype (0 indicates no protein expression). AR670 CGD and AR220 CGD appeared to be the most frequently found sub-types but also the most severe clinical forms compared to AR470 CGD. As a geographical clustering of 11 patients from eight Jordanian families exhibited the c.1171_1175delAAGCT mutation in NCF2, segregation analysis with nine polymorphic markers overlapping NCF2 indicates that a common ancestor has arisen ~1,075 years ago.
Chronic granulomatous disease (CGD) is a rare congenital immunodeficiency syndrome with an incidence of 1 in ~250,000 individuals. CGD is a genetically heterogeneous disease with all ethnic groups equally affected. The disease results from defects in one of the five components of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex in phagocytes, i.e., the membrane proteins gp91phox (or NOX2) and p22phox (together forming cytochrome b558) and the cytosolic components p47phox, p67phox, and p40phox (1). These proteins are present in phagocytic leukocytes (neutrophils, eosinophils, monocytes, and macrophages). In addition, it was recently demonstrated that mutations in EROS/CYBC1 are responsible for a decrease in NADPH oxidase activity of phagocytes and could lead to the development of CGD (2).
In resting phagocytes, the NADPH oxidase enzyme is dissociated and becomes complexed upon stimulation by specific interactions of opsonized microorganisms with membrane receptors. When the enzyme is assembled, the gp91phox flavin- and heme-containing oxidase element becomes capable of transferring electrons from NADPH in the cytosol to molecular oxygen in the extracellular or intra-phagosomal compartment, while the p22phox subunit stabilizes the expression of gp91phox in phagocytic cells (3). The NADPH oxidase complex catalyzes the conversion of molecular oxygen O2 to superoxide anion . Thus, this enzyme is the key of the generation of toxic reactive oxygen species responsible for the intracellular killing of microorganisms. As a result, CGD patients, with a defect in this system, suffer from recurrent and often life-threatening bacterial and fungal infections (4, 5). In addition, inflammatory manifestations are common in CGD and are often associated with neutrophil and/or granulomatous inflammation. These inflammatory diseases affect the gastrointestinal tract, lung, skin, and genitourinary tract and can cause overt autoimmune disease. Gastrointestinal manifestations or lung granuloma of CGD can precede the onset of infectious symptoms and can mimic symptoms of Crohn's disease or sarcoidosis, respectively (6).
The most frequently found form of CGD is the X-linked form, with mutations in the CYBB gene encoding gp91phox subunit or NOX2 (~70% of CGD cases). The AR-CGD form most frequently encountered is due to mutations in NCF1 encoding p47phox (accounting for ~25% of CGD cases). The often-reported recurrent mutation ΔGT at the beginning of exon 2 is caused by recombination events between NCF1 and two pseudogenes surrounding NCF1 (7). Rare AR subgroups (<5% of CGD cases) are caused by mutations in CYBA, NCF2 or NCF4 genes encoding p22phox, p67phox, or p40phox subunits, respectively (8–10). In very rare cases, the small G protein, Rac2, involved in regulating NADPH oxidase activity could also be mutated leading to a specific innate immunodeficiency characterized by an unresponsiveness to the chemotactic peptide FormylMetLeuPhe (for NADPH oxidase activity and chemotaxis) (11–15).
Based on several cohort studies, it appears that the X-CGD form affecting the membrane redox element of the NADPH oxidase complex is related to the most severe clinical features (4, 5, 16, 17). However, it is important to note that mutations in NADPH oxidase genes leading to residual activity, such as certain X−-CGDvariants or some NCF1 mutations appear to be associated with milder clinical forms (16, 18–20). One way to obtain information on the severity of rare cases of AR670 and AR220CGD is to investigate patients in countries in which these forms predominate. We have previously reported for the first time the clinical and genetic investigations of 15 Jordanian patients from nine different families (21, 22). Our report described the presence of rare mutations and the predominance of AR types. Of note, this predominance of the AR types is in accordance with other Middle Eastern countries (16, 19, 21, 23, 24) in which consanguineous marriages prevail and is in contrast to other countries and regions, such as the United States of America, South America, Europe, China, and Japan, where the X-linked disease is exceedingly more prevalent than the AR types (4, 5, 17, 25–27).
Here, we present the second report of clinical and genetic characteristics of 31 CGD patients suffering mainly from AR-CGD. Only one case of X-CGD was reported. Twenty-two Jordanian, seven Lybian, and two Iraqi CGD patients from 21 different families were investigated. Most of the marriages were consanguineous except five. Most of the patients underwent extensive clinical and laboratory investigations to clarify the relationship between the identification of the mutated protein and the clinical severity in very rare AR-CGD forms affecting p22phox and p67phox mainly and p47phox too, and in one case of X-CGD affecting NOX2. Eleven patients from the same families, who did not have laboratory but only clinical investigations, were suspected to have been died from CGD too. Because of high frequency of the c.1171_ 1175delAAGCT mutation found in NCF2, we report the first founder effect of this mutation in eight unrelated Jordan families by performing segregation analysis with nine polymorphic markers overlapping the NCF2 gene.
Blood samples were collected from healthy volunteers, their patients and relatives after obtaining signed informed consent. Written consents for DNA analysis of samples from patients, their parents and relatives were also obtained. CGD patients were identified from personal communications with relevant specialist physicians during the study period (Jan 2006–Sep 2019). The patients with confirmed diagnosis (or suspected diagnosis) of CGD were referred to the main study center (Division of Infectious Diseases, Jordan University Hospital, Amman, Jordan) for further clinical investigations. Twenty-two Jordanian, seven Libyan, and two Iraqi CGD patients (17 females, 14 males) from 21 different families were investigated. All patients underwent extensive clinical (Jordan University Hospital in Amman, Jordan) and laboratory investigations (CGD Diagnosis and Research Centre, Grenoble, France). All patients underwent extensive clinical and laboratory investigations. Clinical history was taken from medical records and interviewing the families. In addition, we reviewed the clinical characteristics of 15 patients of the same set of families who did not have genetic testing but had either an abnormal Nitroblue Tetrazolium (NBT) test (for NADPH oxidase activity) or clinical suspicion of CGD. Below is a brief description of patients with emphasis on important complications. Clinical data and pedigrees are shown in Table 1 and Figure 1, respectively. All families were Jordanian unless otherwise specified. Follow-up was continued until November 2019 unless mentioned otherwise.
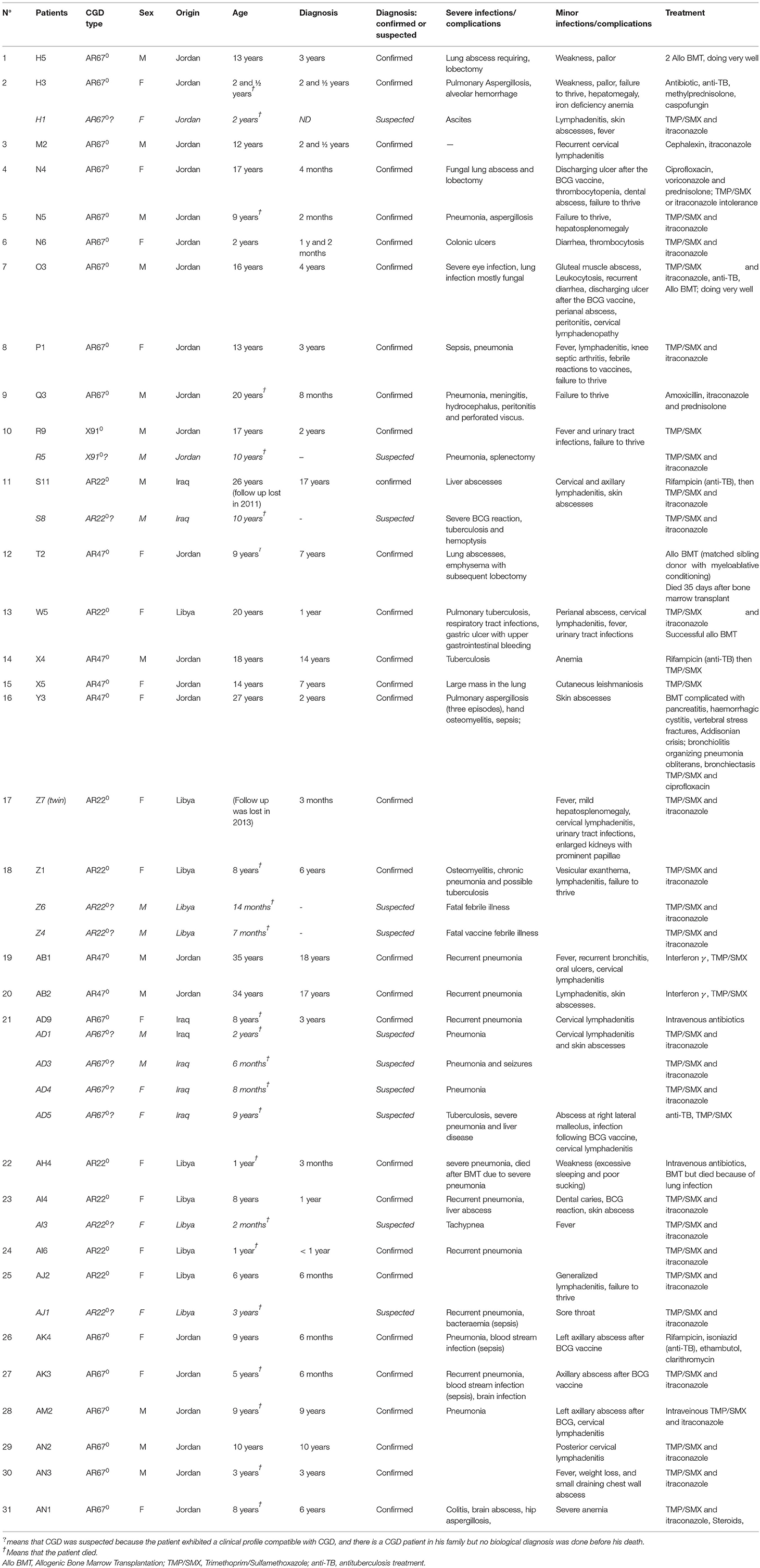
Table 1. Summary of clinical histories of CGD patients.
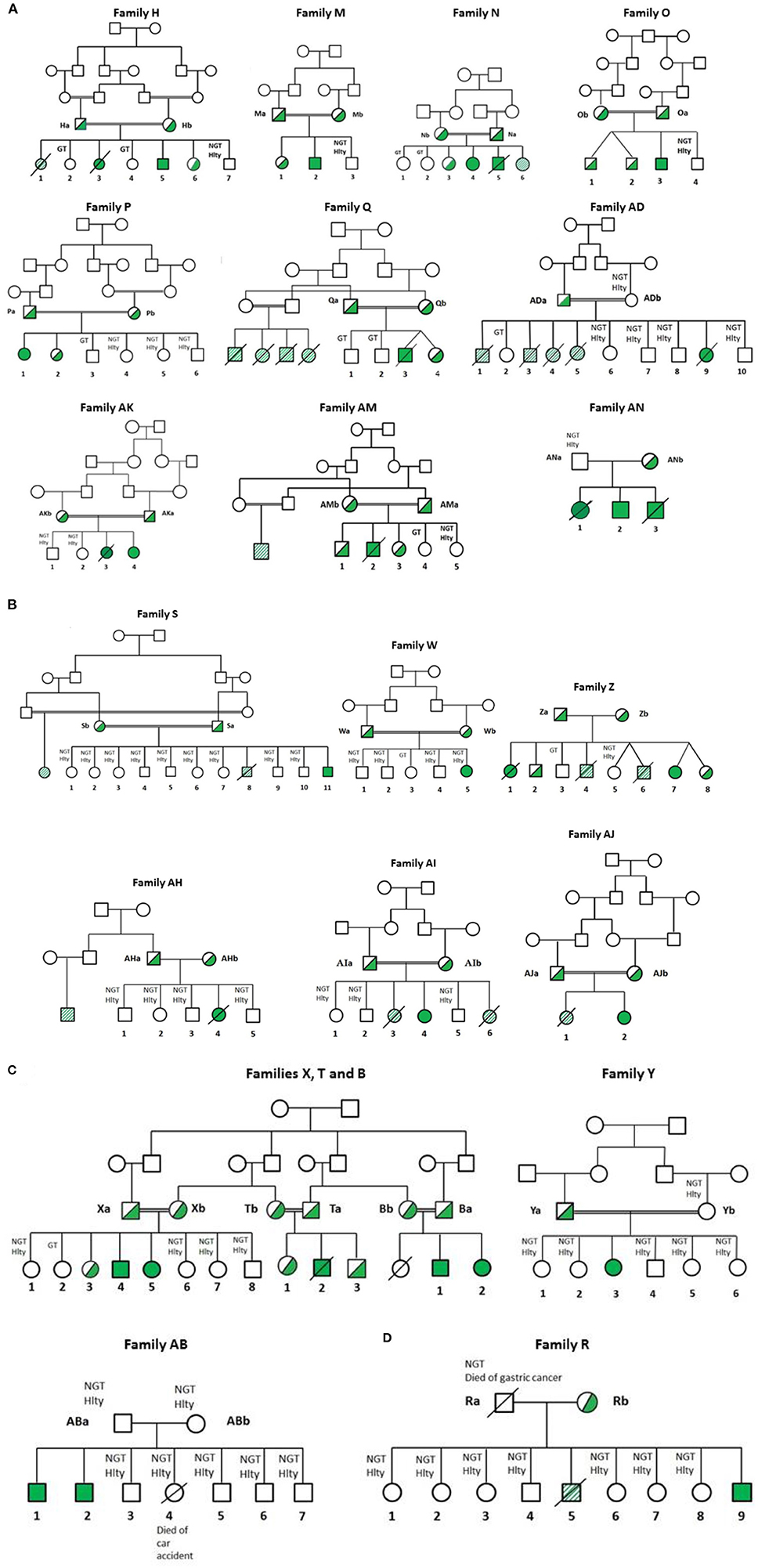
Figure 1. Pedigree of Jordanian, Lybian, and Iraqi families. CGD was diagnosed by measurement of NADPH oxidase activity in neutrophils as described in Patients and Methods. All families contained consanguineous marriages, except families R, Z, AB, AH, and AN. Diagonal shaded box: highly suspicious but functionally and genetically not tested (H1, R5, S8, Z6, Z4, AD1, AD3, AD4 and AD5, AI3 and AJ1). Hlty, healthy; NGT, not genetically tested; GT, genetically tested; Open symbols: no mutation in CGD genes among the tested members; filled symbols: homozygous for the CGD mutations; half-filled symbols: heterozygous for the mutation; cross-out symbols: deceased patients. (A) CGD patients from families H, M, N, O, P, Q, AK, AM, AN, and AD suffering from AR670 CGD. (B) CGD patients from families S, W, Z, AH, AI, and AJ suffering from AR220 CGD. (C) CGD patients from families T, Y, X, and AB suffering from AR470 CGD. T and X are related families. These families were also related to family B (21). (D) CGD patients from family R suffering from X0 CGD.
Blood samples were obtained from the CGD patients with appropriate institutional consent. Neutrophils were purified by Ficoll isolation and isotonic lysis of red blood cells (28). Epstein Barr virus-immortalized B-lymphocytes were obtained as described previously (29).
For the CGD diagnosis, NADPH oxidase activity was measured in purified neutrophils using the classical test of reduction of NBT by superoxide () release from neutrophils during phagocytosis of opsonized latex particules (30). Superoxide was sometimes measured in an SOD-sensitive cytochrome c reduction test (31). Reactive oxygen species produced by neutrophils after phorbol-myristate acetate (PMA) stimulation, were also measured by flow cytometry in the presence of dihydrorhodamine-1,2,3 (DHR) (20).
Expression of gp91phox (NOX2), p22phox, p47phox, p67phox, and p40phox in a 1% Triton X100 soluble extract prepared from human neutrophils was examined by Western blot analysis (50 μg of protein in each lane) (32) using monoclonal antibodies anti-NOX2 48 and anti-p22phox 449 (33, 34) and polyclonal antibodies anti-p67phox (C19, SC7662, Santa Cruz), anti-p47phox and anti-p40phox (35, 36). Polyclonal anti-IgG-HRP was used as a second antibody and the immune complexes were detected by chemiluminescence in an ECL kit Femtomax (Rockland Immunochemicals, Limerick, PA, USA). Protein concentration was determined by Pierce BCA Protein AssayTM Kit (ThermoFisher, Illkirsh, France) (37).
Monoclonal antibody (mab) 7D5 directed against external epitopes of gp91phox (NOX2) (D162-3, Clinisciences, Nanterre, France), monoclonal anti-p22phox (SC-130550, Santa Cruz Biotechnologies Inc, Heidelberg, Germany), monoclonal anti-p67phox (AB109523/EPR5065, Abcam, Paris, France) and monoclonal anti-p47phox (AB179457/EPR 13134, Abcam, Paris, France) with secondary antibody conjugated with Alexa-Fluor 488 (A11070, Invitrogen Life technologies, Villebon sur Yvette, France) or PE (A10543, Invitrogen Life Technologies, Villebon sur Yvette, France), were used for analysis of NADPH oxidase subunit expression in phagocytic cells (2 × 105 cells). In case of p22phox, p47 phox, or p67 phox expression analysis, neutrophils were previously fixed in PFA 2% (v/v) and then incubated with specific antibodies directed against p22phox, p47phox, or p67phox diluted in PBS, BSA 0.2% (w/v), saponin 0.01% (v/v) (38). Control staining with appropriate isotype-matched control antibodies was included to establish thresholds for positive staining. Cell fluorescence was quantified with a FACS Canto II (BD Biosciences). Data were collected and analyzed with the FACS DIVA software and FlowJo software-Tree Star (BD Biosciences, Pont de Claix, France).
Total RNA was isolated from either mononuclear leukocytes or EBV-transformed B-lymphocytes of CGD patients and healthy individuals, using a modified single-step method (39). Genomic DNA was purified with a purification kit (ref 51206 Flexigene DNA kit, Qiagen, Hilden, Germany).
When possible, first-strand cDNA was synthesized from total RNA by reverse transcriptase reaction according to the manufacturer's instructions (ref 11EMAMV203, MP Biomedicals Santa Ana, CA, USA). Total cDNA was immediately amplified by PCR with appropriate primers and separated by gel electrophoresis. All PCR products were sequenced with an ABI 3730 XL 96 capillary sequencer (Perkin Elmer, Foster City, CA, USA). More than 500 samples of control cDNA of gp91phox (NOX2), p22phox, p67phox, and p47phox were analyzed to rule out the possibility of polymorphisms. In all cases, location of mutations found in cDNA was verified in genomic DNA after PCR amplification of each exon and flanking intron regions with appropriate forward and backward primers, followed by Sanger sequencing (21). Aliquots of all PCR products in bromophenol blue solution were run together with a DNA ladder (ref R0211, ThermoFisher Scientific, Illkirch, France) on 1.5% (wt/vol) agarose containing Gel RedTM nucleic acid stain (ref 41003, Biotium, Inc, Fremont, CA, USA) in parallel with a negative control (PCR products amplified without DNA) and a positive control (PCR amplification of control DNA) to analyze size and purity. In some cases, PCR products were purified from agarose gel according to manufacturer instruction (QIAquick Gel Extraction kit, ref 28704, Qiagen, Courtaboeuf, France).
Fragments of genomic DNA from AR470 CGD patients and their relatives were amplified with primers that anneal with regions in NCF1 as well as in ψNCF1 regions around the GTGT sequence at the start of exon 2, with PCR conditions according to Dekker et al. (40). The mixture of NCF1 and ψNCF1 products was analyzed in a sequencer ABI 3730 XL 96 capillary sequencer (Perkin Elmer, Foster City, CA, USA) to determine the ratio between the number of NCF1 and ψNCF1 genes present (40, 41).
Homozygosity mapping was performed with genomic DNA from eight patients of eight different Jordan families (H, N, O, P, Q, AK, AM, and AN). We used a set of nine microsatellite markers (D1S212, D1S2751, D1S2640, D1S2619, D1S2623, D1S2701, D1S2711, D1S202, and D1S238) previously described (42). Amplification of the microsatellite markers were performed by multiplex PCR. Sizes were determined with Genemapper® Software. The estimation of the most recent ancestor (MRCA) was realized with the algorithm developed by Gandolfo et al. (43), which is based on the shared haplotypes length between patients with the same mutation.
Clinical and genetic characteristics of CGD were analyzed in 22 Jordanian, seven Libyan and, two Iraqi patients from 21 families (Tables 1, 2); 14 families were from Jordan (H, M, N, O, P, Q, R, T, X, Y, AB, AK, AM, and AN), five from Libya (W, Z, AH, AI, and AJ), and two from Iraq (S and AD). All families were consanguineous except 5 (R, Z, AB, AH, and AN). Genetic results are presented from a total of 27 patients; 26 were tested in this study (H5, H3, M2, N4, N5, O3, P1, Q3, R9, S11, T2, W5, X4, X5, Y3, Z7, AB1, AB2, AD9, AH4, AI4, AJ2, AK4, AM2, AN2, and AN3) and one patient had abnormal NBT test and genetic testing in Germany before she died (Z1). NBT testing alone was performed in a total of 4 patients (N6, AI6, AK3, and AN1). Clinical suspicion for CGD was present in 11 patients (H1, R5, S8, Z4, Z6, AD1, AD3, AD4, AD5, AI3, and AJ1). The overall mortality among the tested families was 3/4 (75%) among those who had abnormal NBT but no genetic testing. The overall mortality was 11/11 (100%) among those who were only clinically suspected. Furthermore, there were seven affected relatives; two relatives with abnormal NBT test (a cousin in the AH family and a cousin in the AM family), five with clinical suspicion (four cousins in the Q family and a cousin in the S family). All patients suffered from AR-CGD except one patient in the R family who suffered from X-CGD. A relationship was established between families T and X. These families were also related to family B described in our previous article (21). The diagnosis of CGD was suspected on clinical grounds and confirmed by functional and genetic studies of the patients.
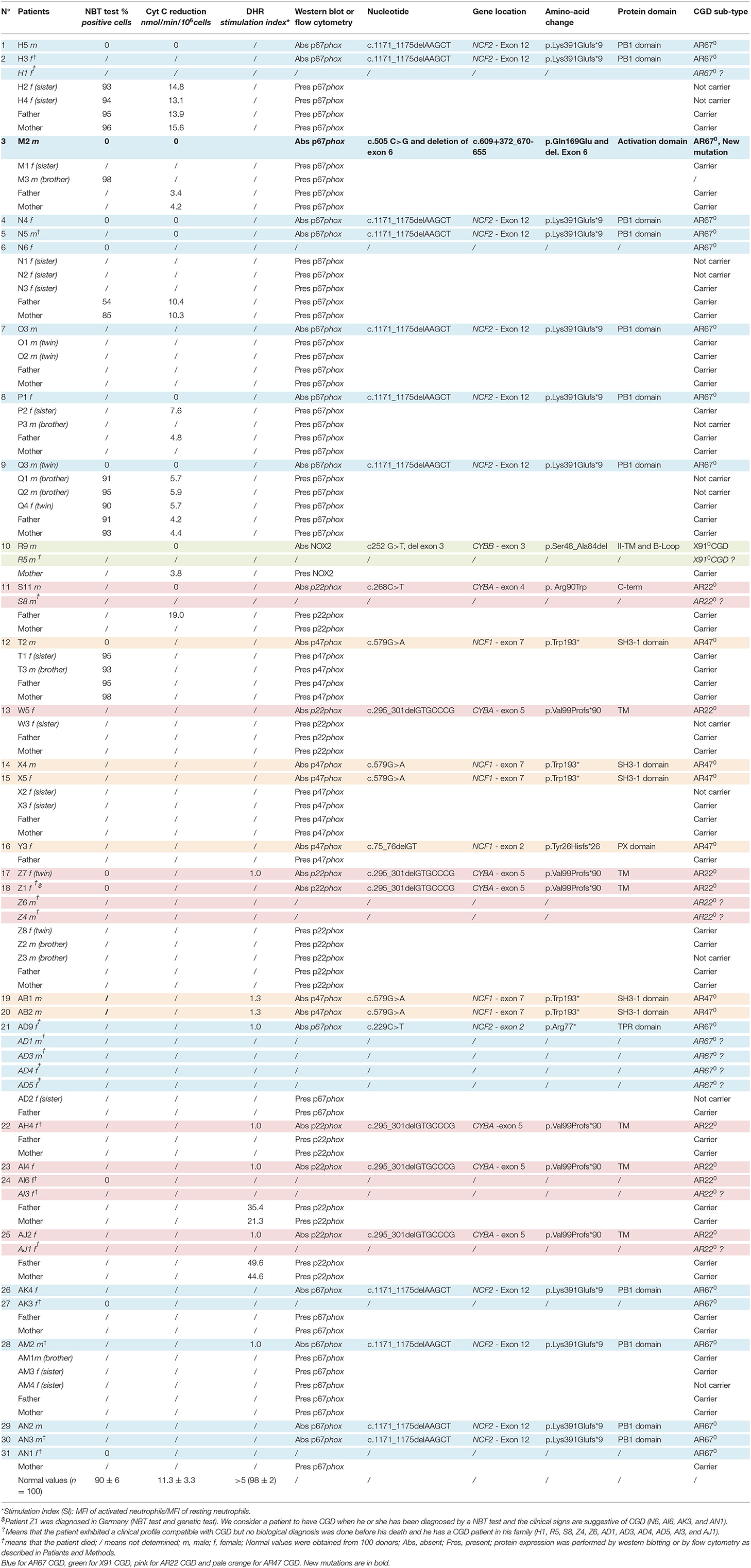
Table 2. Phenotype and genotype of CGD patients and their relatives.
Our analysis will focus mainly on comparing the different forms of AR-CGD because there is only one diagnosed case of X910 CGD. One criterial aspect to judge the severity of this disease is to focus on the age of diagnosis (Figure 2). As can be seen AR670 CGD and AR220 CGD were diagnosed at the earliest time. Patients S11 and Z1 clinically suffered from AR220 CGD at an early stage, although the diagnostic NBT test was done later. This is the same for patients AM2, AN1, and AN2 with AR670 CGD (see Patients and Methods). On the other hand, the late age of diagnosis (18, 17, and 14 years) for patients with p47phox deficiency (AB1, AB2, and X4) confirmed that this type of CGD is less severe than the other subtypes. Patient R9 who suffered from X910 CGD was diagnosed early (at the age of 2, Table 1).
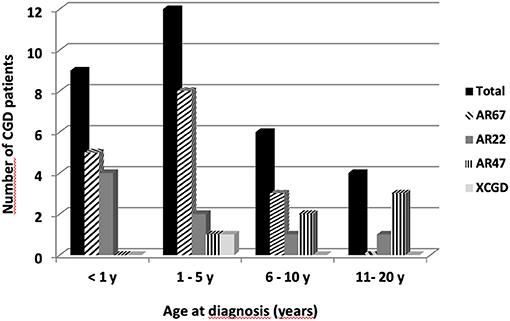
Figure 2. Age at diagnosis of the 31 CGD patients from Jordan, Iraq, and Lybian according to the CGD types. Twenty-two Jordanian, seven Lybian, and two Iraqi CGD patients from 21 different families were investigated. Sixteen patients suffered from AR670 CGD, eight patients from AR220 CGD, six from AR470CGD, and only one patient had X-CGD.
To analyze the relative severity of these different CGD forms, the diverse infections and complications of each patient were classified into severe or minor infections and complications (Table 3). The severe infections and complications are mainly located in the lungs, with pneumonia and lung abscesses by far the most common severe complications, regardless of the type of CGD (69 and 63% in AR670 CGD and AR220 CGD patients, respectively, and 100% in AR470 CGD patients). The X910 CGD patient R9 did not present any severe infections or complications although he was diagnosed at the age of 2 years. Until then, he had only growth retardation, anemia, and urinary tract infection, despite a total lack of NADPH oxidase activity (Table 2). However, his brother R5 had a splenectomy and died from pneumonia at the age of 10. Gastrointestinal complications occurred in approximately 13% of all AR-CGD patients, and sepsis was found also in the same proportion, except for AR220 CGD patients who did not have sepsis (Table 3). Brain infections were found in 3 AR670 CGD patients (Q3, AK3, and AN1), liver abscesses in 2 AR220 CGD patients (S11 and AI4), osteomyelitis in one AR220 CGD patient (Z1), and in one AR470 CGD (Y3). Regarding the minor infections and complications, lymphadenitis and skin infections were by far the most common severe complications regardless of the type of AR-CGD (Table 3). Failure to thrive was also found relatively frequent in AR-CGD patients (38 and 25% in AR670 CGD and AR220 CGD patients, respectively). BCG vaccine reaction was also frequently observed in AR-CGD patients (31 and 13% in AR670 CGD and in AR220 CGD patients, respectively). Hepatomegaly was observed only in 2 AR670 CGD patients (H3 and N5) and in 1 AR220 CGD patient (Z7). Finally, urinary tract infections were rare and only observed in 2 AR220 CGD patients (W5 and Z7). Other minor infections and complications were observed in only one or two patients (Table 3).
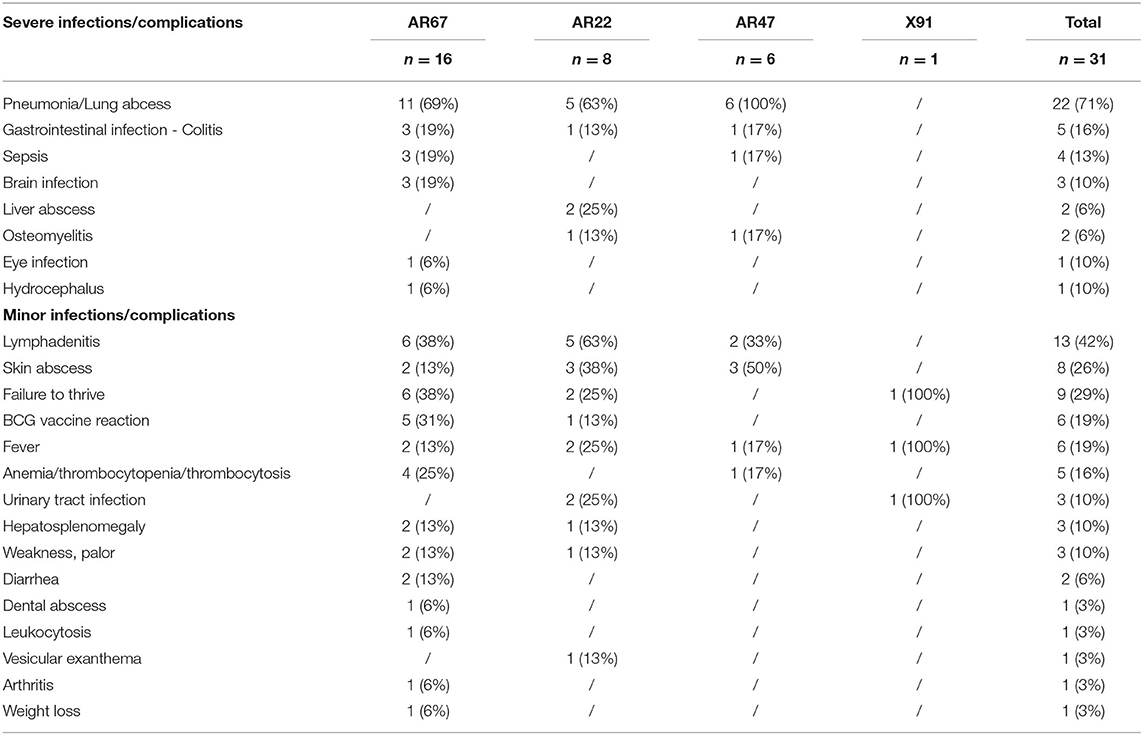
Table 3. Summary of the most common infections and complications in the CGD patients.
Looking at the major severe complications suffered by the 11 potentially CGD patients who died before their laboratory diagnosis has been made, it is obvious that pneumonia was the most frequently occurring clinical event in AR-CGD patients (R5, AD1, AD3, AD4, AD5, AJ1). Sepsis (patients Z4, Z6, and AJ1), lymphadenitis (patients H1, AD1, and AD5), skin abscesses (patients H1 and AD1), and BCG vaccine reaction (patients AD5 and S8) were also frequently observed (Table 1).
Treatment of the CGD patients is outlined in Table 1. All patients had a prophylactic life-long treatment (most of the time TMP/SMX in combination with itraconazole). Patients H3, O3, S11, X4 AD5, and AK4 received in addition to TMP/SMX an anti-TB treatment to treat a suspected TB infection. When severe inflammatory syndrome was associated with infections, steroid therapy was given to patients H3, N4, Q3, and AN1. IFNγ was given to two patients for prophylaxis (AB1 and AB2). IFNγ was given to these two patients for only 1 year when it was available at that time. Six patients received allogeneic bone marrow transplantation (allo BMT) (H5, O3, W5, T2, Y3, and AH4). Three patients have complete chimerism and are doing well (H5, O3, W5). Patient T2 died 35 days after transplantation because of complications. Patient AH4 died after allo-BMT because of severe lung infection. Patient Y3 had severe complications such as pancreatitis, hemorrhagic cystitis, vertebral stress fractures and Addisonian crisis, but she is now maintained on TMP/SMX and ciprofloxacin.
To conclude from the AR-CGD patients' cohort studied, AR670 CGD and AR220 CGD appear to be equally severe clinical forms of the disease, whereas AR470 CGD appears to have a milder clinical form. One exception is patient M2, who suffered from AR670 CGD with mild clinical signs. Finally, our unique X910 CGD patient suffered from a mild clinical form too, but this form cannot be considered as mild regarding the death of his brother from pneumonia at the age of 10.
X-CGD with defects in the CYBB gene was detected only in patient R9. Although its clinical form was not the most aggressive, we found no NADPH oxidase activity in his neutrophils. In his mother's neutrophils an intermediate value was measured (Figure 3A). No expression of gp91phox and p22phox in R9's neutrophils was detected (Figure 3B). This is understandable, since these two proteins need association for stable expression. His mother's neutrophils exhibited a diminished expression of both proteins. We amplified cDNA from gp91phox mRNA in R9's leukocytes and found absence of exon 3 (confirmed by sequencing) in patient R9 (Figure 3C). Then exon 3 from the CYBB gene was sequenced and we found a silent hemizygous missense mutation c252 G>A at the end of exon 3, explaining skipping of exon 3 in the mRNA. The same mutation in one allele of CYBB was present in his mother, confirming her carrier status (Figure 3D). The mild clinical form of R9 cannot be explained by residual NADPH oxidase activity measured by cytochrome c reduction assay. In addition, his brother R5, suspected to have had CGD, died at the age of 10 years from pneumonia (Table 1).
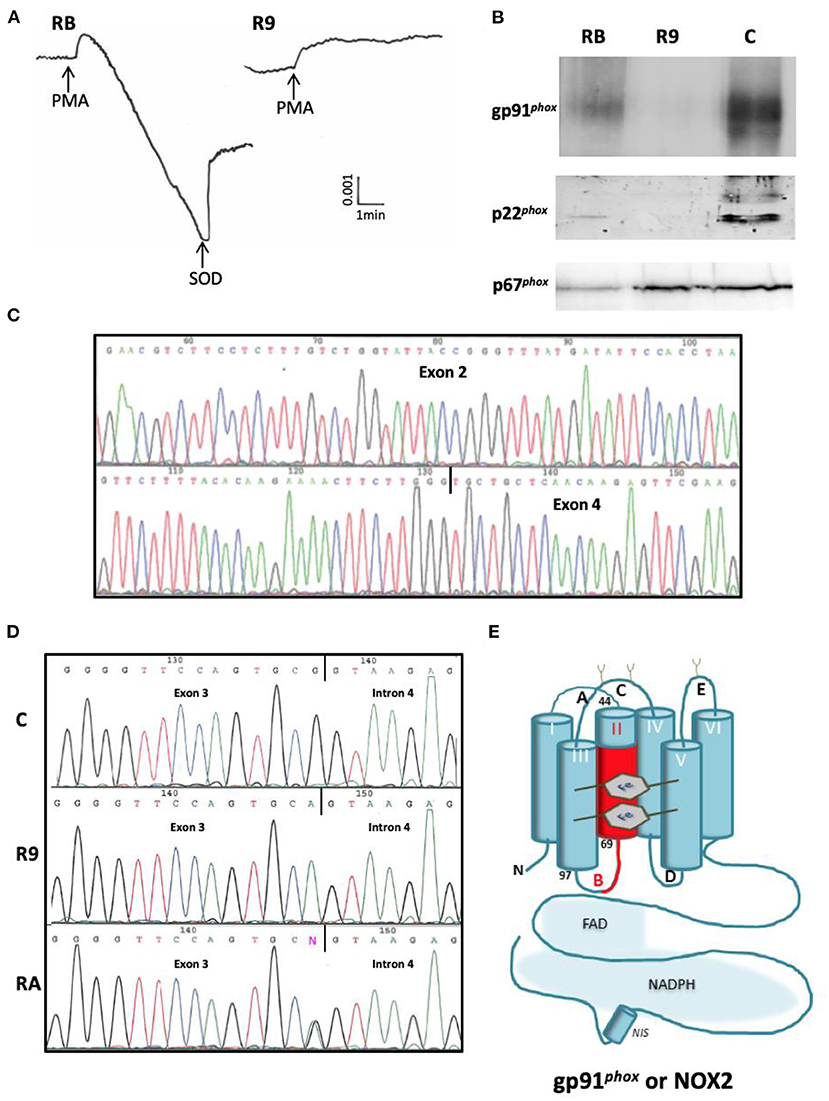
Figure 3. Phenotypic and genotypic characterization of the missense mutation c.252 G>T at the 3' end of exon 3 of CYBB in the X910 CGD patient from family R. (A) NADPH oxidase activity measured by SOD sensitive-cytochrome c reduction with 5 × 105 purified neutrophils stimulated by PMA (80 ng/mL). RB = mother, R9 = CGD patient. (B) Western-blot of p22phox and gp91phox analysis of soluble extracts (50 μg of protein in each lane) from purified neutrophils of Family R's members (RB mother, R9 CGD patient). The p67phox expression was used to control the protein load. (C) Analysis of mutated p67phox mRNA. (D) Analysis of mutated NCF2 gene. (E) Consequence on the sequence of gp91phox protein. The absence of exon 3 corresponds to the amino acid deletion p.Ser48_Ala84del in the second transmembrane domain of gp91phox with a probable structural disorganization of the protein.
AR-CGD with defects in the NCF1 gene were detected in 4 families (19%) T, X, Y, and AB and in six patients (19%) (T2, X4, X5, Y3, AB1, and AB2) (Table 2). Families T, X (related families), and AB exhibited the same mutation. Affected patients (T2, X4, X5, AB1, and AB2) were homozygous for the G579A mutation in codon Trp193 in exon 7 of NCF1. The parents of T2, his sister T1, and his brother T3 were all heterozygous for this mutation. The parents of X4 and X5, and their sister X3 were also heterozygous for this mutation. However, their sister X2 was unaffected. For all these patients CGD was discovered as a result of pneumonia (Tables 1, 3). The residual NADPH oxidase activity measured by flow cytometry (DHR probe) for patients AB1 and AB2 can explain the partial protection of these patients and the delay of the CGD diagnosis (Table 2). In addition, patients AB1 and AB2 are the oldest CGD patients of the cohort (34 and 35 years old, respectively) (Table 1). Unfortunately, the NADPH oxidase activity for patients T2, X4, and X5 was either not measured or measured by the NBT test, which is not sensitive enough to evaluate a residual NADPH oxidase activity, in contrast to flow cytometry (DHR). Patient Y3 exhibited the classical GT deletion (c.75_76delGT) in a GTGT repeat sequence in exon 2 of NCF1, which predicts a frameshift and a premature stop codon at residue 51. Her father was heterozygous for this mutation. We were not able to evaluate her mother. This mutation was also described in our previous report, in which all AR470 CGD patients exhibited this mutation (21). The manifestation of the disease of patient Y3 was the most severe of all AR470 CGD cases in this study, as she suffered from three episodes of lung infections, a sepsis and a hand osteomyelitis. In addition, she had severe complications after bone marrow transplant (BMT), but now she is maintained on TMP/SMX and ciprofloxacin. She's now 28 years old.
AR-CGD with defects in the CYBA gene were detected in six families (19%) S, W, Z, AH, AI, and AJ and in eight patients (26%) (S11, W5, Z1, Z7, AH4, AI4, AI6, and AJ2) (Table 2). Patients W5, Z7, AH4, AI4, and AJ2 were all from Libya and showed a seven-basepair deletion c.295_301delGTGCCG in exon 5 of CYBA. This mutation was previously found in our first report (21). Patient AH4 died because of lung infection after BMT. Among these families, only the parents in family Z were not consanguineous. Patient Z1, and patients Z4, and Z6 from this family—not genetically tested—died, probably because they all suffered from AR220 CGD. In addition, patients AI3, AI6 (NBT tested), and AJ1 (all not genetically tested) passed away too, confirming the severity of this AR220 CGD type.
Patient S11 presented with a missense mutation c.268C>T in exon 4 of CYBA, changing Arg90 to Trp and leading to the absence of p22phox expression. This mutation is rare and was reported only once, but was not functionally characterized (44). His brother S8 (not genetically tested) died at the age of 10 after a severe BCG reaction and hemoptysis. The NADPH oxidase activity, p22phox and gp91phox (NOX2) expression were abolished in the neutrophils of all AR220 CGD patients (Table 2). This probably explains the severity of the clinical symptoms developed by these patients.
AR-CGD with defects in the NCF2 gene were detected in 10 families (48%) H, M, N, O, P, Q, AD, AK, AM, and AN corresponding to 16 patients (52%) (H3, H5, M2, N4, N5, N6, O3, P1, Q3, AD9, AK3, AK4, AM2, AN1, AN2, and AN3) including 8 who died (H3, N5, Q3, AD9, AK3, AM2, AN1, and AN3) (Tables 1, 2). In addition, 7 patients (H1, AD1, AD3, AD4, AD5, AK3, and AN1) died probably because they suffered from AR670 CGD but they were not genetically tested (N6, AK3, and AN1 diagnosed with an NBT test). These numerous deaths confirm that the AR670 CGD is one of the most severe forms of the disease. Whenever possible, the NADPH oxidase activity of patients' neutrophils was measured and was shown to be totally abolished, as well as the expression of p67phox (Table 2). An exception for disease severity may be made for patient M2 exhibiting a new mutation in NCF2 (Figure 4). The NADPH oxidase measured in his parents' and sister's neutrophils was normal but null in the patient's neutrophils (cytochrome c reduction and NBT test) (Figure 4A, Table 2). Unfortunately, the NADPH oxidase activity could not be measured by the DHR oxidation in flow cytometry to detect a possible residual activity that could have explained the mild clinical form of this AR670 CGD (Table 1). We found a very faint band of p67phox by Western blotting in the M2 neutrophils (Figure 4B), and by amplifying the cDNA from the p67phox mRNA, we found absence of exon 6 (confirmed by sequencing) (Figure 4C). We could not amplify exon 6 in the NCF2 gene. After several attempts of amplification and sequencing we found a large deletion from part of the intron 5 to part of intron 6 (2,974 pb) (Figure 4D). His parents and his sister M1 were carriers of the mutation (Table 2). His brother M3 is in good health with a normal NBT test but his carrier status has not been determined.
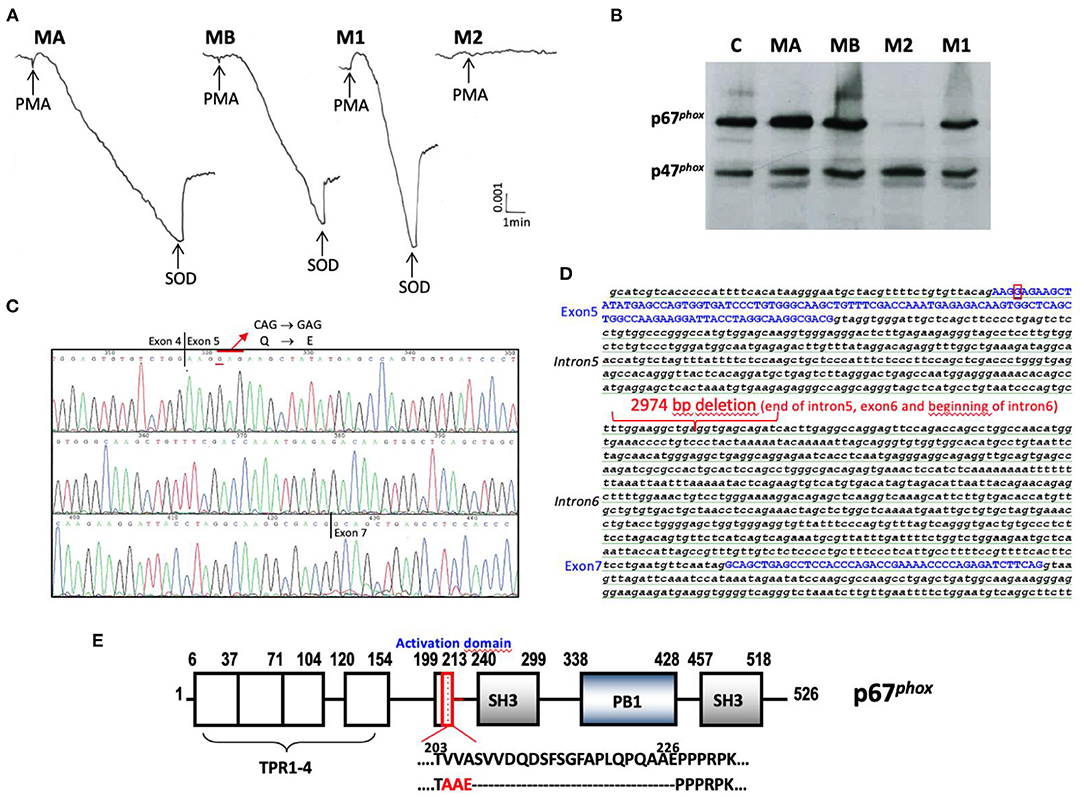
Figure 4. Phenotypic and genotypic characterization of the new mutation of the AR670CGD patient from family M. (A) NADPH oxidase activity measured by SOD sensitive-cytochrome c reduction with 5 × 105 purified neutrophils stimulated by PMA (80 ng/mL). (B) Western blot of cytosolic factors p47phox and p67phox analysis of soluble extracts (50 μg of protein in each lane) from purified neutrophils of family M (MA father, MB mother, M2 CGD patient, M1 sister). The p47phox expression was used to control the protein load. (C) Analysis of mutated p67phox mRNA. (D) Analysis of mutated NCF2 gene. (E) Consequence on the sequence of p67phox protein. The molecular consequence is an amino acid change p.ValValAla204-206AlaAlaGlu and p.Ser207-Glu226del in the activation domain of p67phox protein that could be a potential interacting domain with NOX2.
Patient AD9 had a non-sense mutation in exon 2 of NCF2 c.229C>T, changing the CGA codon for Arg77 into the TGA stop codon (p.Arg77*). This mutation was previously reported but is quite rare (45). The functional consequence of this mutation seems to be severe, because all patients (patients AD1, 3, 4, 5, and 9) from this family died, although AD1, 3, 4, and 5 were not genetically tested (Table 1). In addition, the NADPH oxidase activity measured in the neutrophils from patient AD9 by DHR oxidation in flow cytometry was null (Table 2). The father of these patients was carrier of the mutation whereas one of his daughters AD2 was unaffected. Unfortunately, the mother could not be tested.
All families (H, N, O, P, Q, AK, AM, and AN), except families M and AD, suffered from the same mutation, viz. a deletion of 5-bp AAGCT at position 1171 in exon 12 of the p67phox cDNA (c.1171_1175delAAGCT). The molecular consequence is a frameshift starting at Lys391 and the introduction of a stop codon at Ser399 in the PB1 domain of p67phox. This mutation was reported previously (21, 46, 47). Like patient AD, all patients from these different families suffered from a severe clinical form of CGD (Table 1) with absence of NADPH oxidase activity and p67phox expression (Table 2). Seven among 15 patients from these families died (Table 1). Because of the high frequency of this mutation in families H, N, O, P, Q, AK, AM, and AN, we decided to determine the founder effect of the c.1171_1175delAAGCT mutation using microsatellites markers to estimate the time of living of the most recent ancestor. Family AM was excluded from analysis since AM2 did not share any consecutive marker from the haplotype. The shared haplotype was estimated to 2.3 mega base between family H, N, O, P, Q, AK, and AN (Table 4). Assuming an independent genealogy the most recent common ancestor (MRCA) of the eight families was estimated to be 42.9 generations in the past [95% confidence intervals (CI): 21.9–63.5]. Assuming an independent genealogy, the MRCA of the eight families was estimated to have lived 37.2 generations ago (95% CI: 21.9– 63.5). Considering a generation time of 25 years, the common ancestor would have appeared 1,075 years ago (CI 550–1,600 years). A geographical clustering of the Jordanian AR670 CGD families sharing the c.1171_1175delAAGCT mutation in NCF2 can be seen in Figure 5.

Table 4. Haplotype study.
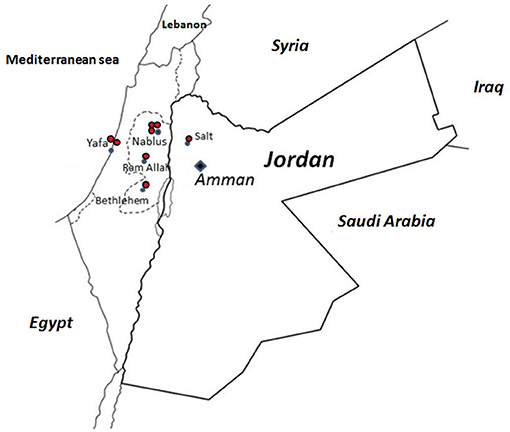
Figure 5. Geographical clustering of the Jordanian AR67 families sharing the c.1171_1175delAAGCT mutation in NCF2. The ancestor origin of each family is marked on the Jordan and Palestine map by a red dot.
The genetic analysis in the Arab cohort allows us to conclude that the major inheritance was AR with a predominance of AR670 CGD (16 patients) over AR220CGD (eight patients) and AR470CGD (six patients). Overall, in the 31 patients, eight different mutations were found; three in NCF2 including a new one, two in CYBA, two in NCF1, and 1 in CYBB. All these mutations were homozygous, indicating that both parents contributed an identical, mutated allele that caused the disease. Contrary to what is found in the Western population, only one male patient (R9) suffered from X-CGD, in a non-consanguineous family. This report confirms our previous finding that the most common form of CGD in Jordan is the AR type (21). The high frequency of AR-CGD appears also in Tunisia, Egypt, Turkey, Iran, Oman, Saudi Arabia, and Israel because of the high rate of consanguineous marriage in these countries (16, 19, 23, 24, 48–51) and it is in contrast to the published literature from USA, Europe, South America, China, and Japan, in which X-CGD is the main genetic form (4, 5, 17, 25–27, 52–54). The population in Jordan amounted from 5.3 million in 2004 to 10.2 million in 2020, and is composed of a variety of ethnic groups, the majority being Arabs. Indeed, many Arab countries display a long tradition of consanguinity due to socio-cultural factors. In Jordan 20–30% of all marriages occur between first cousins, and are strongly associated with the appearance of AR diseases (55, 56). This is also true for other Arab countries such as Iraq and Libya that are close to Jordan (57). In our study, only four families out of 14 Jordanian families (R, AB, AH, and AN) and one out of five Libyan families (Z) did not result from consanguineous marriages. The two Iraqi families (S and AD) were also consanguineous. No publication related to CGD in Iraq was found except in the Israeli cohort of 84 CGD patients comprising two Iraqi Jewish patients suffering from X-CGD (16).
The most frequent mutation found in CYBA (c.295_301delGTGCCCG) was found in 5 patients all originating from Libya (W, Z, AH, AI, and AJ families). This mutation was also found in five parents and relatives of these Libyan families who died. Whereas data on CGD from Libya are scarce (58, 59) one case of a child with the c.295_301delGTGCCCG mutation in CYBA has been described (60). Of note, this mutation was previously found in two patients from one Jordanian family (family J) and here in the Libyan patient AH4 (8, 21, 23, 44). In addition, this mutation was reported in one patient from a French cohort (without any nationality identification) (44) and in two patients out of 15 patients of a Tunisian cohort (23). This mutation was also present in 11 patients out of 28 Egyptian patients, highlighting that AR220 CGD is the commonest form among the Egyptian population (49). Tunisia, Libya, and Egypt are three countries with common frontiers and not too far from Jordan. Our findings argue in favor of a possible founder effect since the single c.295_301 delGTGCCCG mutation in CYBA was detected mainly in patients from these countries. The molecular consequence of this mutation is the change of Val99 to Pro and the introduction of a stop codon in a potential transmembrane domain of p22phox, leading to the disorganization of the protein (21). The second mutation found in the Jordanian patient S11 is a missense mutation c.268C>T in exon 4 of CYBA, changing Arg90 to Trp and leading to the absence of p22phox expression. This mutation was previously reported but not fully characterized (44). Arg90 is located in a potential transmembrane domain of p22phox whose function is not very well-characterized but probably involved in the structural stability of the protein (61). AR220 CGD with mutations c.268C>G, c.269G>A, and c.269G>C corresponding to the missense mutations Arg90Gly, Arg90Gln, and Arg90Pro in p22phox, respectively, were also previously reported, indicating the important structural role of this residue (8).
Six patients out of 31 from four Jordan families (T, Y, X, and AB) suffered from an AR470 CGD. In patient Y3, the classical deletion (c.75_76delGT) in the GTGT repeat sequence at the beginning of exon 2 of NCF1 gene leads to a frameshift and the introduction of a stop codon p.Tyr26Hisfs*26 in p47phox protein. This mutation is located in the PX domain of p47phox involved in the binding to phospholipids of the plasma membrane during the assembly of the activated NADPH oxidase complex (62, 63). This is a very common complication in CGD because of the presence of two pseudogenes close to the NCF1 gene and crossing-over events in chromatin (7, 8). The most common AR form of CGD worldwide is caused by this mutation which represents 25% of total CGD cases and about 60% of all AR-CGD cases. In the first report of CGD characterization in Jordanian families, five patients out of 15 presented with this mutation (21). The c.75_76delGT mutation is also the most commonly reported NCF1 mutation in Tunisia (23), Turkey (19), Egypt (49), Saudi Arabia (51), and India (64, 65). Then, the nonsense mutation c.579G>A in exon 7 of NCF1 was found in five Jordan patients (families T, X, and AB). This mutation causes a change of the TGG codon for Trp193 into the TGA stop codon (41, 46, 66). It is situated in the first SH3 (SRC homology 3) domain of p47phox involved in the binding with p22phox during the NADPH oxidase assembly for activation (67). It is a quite worldwide rare mutation except in Israel (16, 46) and it is also the predominant NCF1 mutation found in AR470 CGD patients in Oman (48). In Israel population, the Trp193* mutation is predominantly found in Kavkazi Jewish patients. The c.579G>A mutation in NCF1 was introduced about 1,200–2,300 years ago in the Kavkazi Jewish population but was present in surrounding populations already for more than 5,000 years (68). Indeed, this mutation was found not only in Jordan (this report) but also in Turkish patients (19). In addition, the c.579G>A mutation in Kavkazi CGD patients is associated with a heterogeneous clinical phenotype (69).
The AR670 CGD form was by far the most frequently found form of CGD found in the Jordan population. Mutations in NCF2 were detected in nine out of 22 families, corresponding to 16 patients including eight deceased patients. All of the patients were from Jordan except patient AD9 who was from Iraq. He presented with a non-sense mutation in exon 2 of NCF2, leading to the introduction of a stop codon and predicting p.Arg77*. Two patients in a Turkish cohort of 89 CGD patients (19) and three patients in a Mexican cohort (17) exhibited the same mutation. The missense mutation Arg77Gln was reported too (70). Arg77 is localized in the tetratricopeptide (TPR) domain of p67phox, which is able to bind the G-protein rac2 during the activation process of the NADPH oxidase (45, 71). The functional consequence is a total absence of NADPH oxidase activity, which explains the severe phenotype found in family AD. Patient M2 exhibits a new deletion mutation in one allele of NCF2 (Table 2). The molecular consequence is an amino acid change and a deletion in the activation domain of p67phox protein, a potential interacting domain with gp91phox (NOX2) (Figure 4E) (72). A missense mutation C.505 C>G was also found in the second allele of NCF2. This last mutation predicted the change p.Gln169Glu in p67phox and has been found in both alleles of NCF2. However, the AR67 CGD subtype was not determined (8). He is the unique AR670 CGD Jordanian patient suffering from a mild form of CGD. By Western blotting it seems that a faint band of p67phox was present. At that time, it was not possible to measure the NADPH oxidase activity by flow cytometry (DHR), which is the proper means to detect residual activity that might explain his mild CGD clinical form. Besides these two rare NCF2 mutations, eight Jordanian families (H, N, O, P, Q, AK, AM, and AN) with 11 CGD patients exhibited the same mutation c.1171_1175delAAGCT in exon 12 of NCF2, leading to a frameshift and the introduction of a stop codon p.Lys391Glufs*9 in the p67phox protein. This mutation was the unique one found in all three families with a defect in NCF2 described in our first report (21). It was also found in two patients from Jordan and Palestine (47). Indeed, geographical clustering of the AR670 CGD Jordanian families sharing the c.1171_1175delAAGCT mutation in NCF2 is in a region of Jordan close to Palestine. Assuming independent genealogy from eight Jordan families, we estimate the most recent common ancestor ~1,075 years ago between families sharing the haplotype. However, due to the absence of shared haplotype between the patient AM2 with the others, it is possible that the mutation is older. A founder effect of the c.2547+2T mutation in NCF2 was also studied in 11 patients from six families in the West of Tunisia (42).
Finally, only one case of X-CGD was found here in the group of 31 CGD Jordanian patients. The splice mutation c.252G>T in exon 3 of CYBB as the origin of his disease was previously reported (9, 73). Skipping of exon 3 in gp91phox cDNA correspond to the amino acid deletion p.Ser48_Ala84del in the second transmembrane domain of gp91phox with a probable structural disorganization of the protein (Figure 3E). Kannengiesser et al. found no NADPH oxidase activity measured by the NBT test, chemiluminescence, and DCFH assays but did not analyze the gp91phox (NOX2) expression in neutrophils (44). We also found no NADPH oxidase activity in the neutrophils of patient R9 measured by the SOD-sensitive cytochrome c reduction test. In addition, we did not find any gp91phox (NOX2) and p22phox expression by Western-blot analysis. This phenotype cannot explain the mild CGD clinical form observed in this patient but is in accordance with the severe clinical form of his brother R5 who died of pneumonia at the age of 10. Brunner et al. studied the functional impact of the c.252G>A mutation, which also induces the outsplicing of exon 3 (74). However, these authors showed that this mutation leads to the presence of few percent of normal mRNA next to the misspliced mRNA, explaining the residual NADPH oxidase activity which they measured in neutrophils from the patient by the DHR oxidation assay. Unfortunately, the authors did not measure gp91phox (NOX2) protein expression. However, they explained the mild clinical form of the patient by the residual NADPH oxidase activity in his neutrophils (74).
A striking fact is that the forms most frequently found in Jordan and Libya are the AR670 and AR220 CGD. It is also the case in Egypt and Tunisia (23, 49). However, in Israel, Saudi Arabia, Libya, Oman, Iran, and India, the AR470 CGD form prevails (16, 24, 48, 51, 59, 64, 65). However, in Turkey about 30–40% of the CGD patients have X-CGD, but AR470 CGD represents about 30–50% of AR-CGD too (19, 75, 76). We have no clear explanation yet for this discrepancy apart from the appearance of ancestral mutations such as the c.1171_1175delAAGCT mutation in NCF2 in Jordan and close countries and probably the same is true for the c.295_301 delGTGCCCG mutation in CYBA.
According to several clinical criteria, it appears clear that AR670 CGD and AR220CGD are by far the most severe clinical forms in the Jordanian CGD patients. AR470 CGD appears to manifest much milder, except for the severe clinical form of AR470 CGD in patient Y3 and the mild clinical form of AR670 CGD in patient M2. Mild clinical CGD forms are certainly due to residual NADPH oxidase activity in CGD patients' neutrophils, as can be observed in AR470 CGD (77) or “variants” of X-CGD such as X91− CGD (18). The most sensitive method of measuring this activity is by far flow cytometry with the highly sensitive DHR fluorescent probe. Unfortunately, we were not always able to measure the NADPH oxidase activity by this method, but for sure this residual oxidant production may account for the better prognosis in AR470 CGD, as in patients AB1 and AB2. Indeed, regarding the stimulation index (SI) of DHR oxidation values measured in the neutrophils of 89 CGD Turkish patients, CGD forms caused by mutations in CYBA and NCF2 were as severe as the X0CGD subtype (19). This was previously observed in studies of large CGD patients' cohorts in Europe and USA (4, 5). In the Israeli experience with 84 CGD patients, the SI levels were significantly higher in the AR470 CGD subtype with a better clinical score compared to other AR-CGD patients (16). However, the analysis of AR470 CGD in the Kavkazi population reveals phenotypic heterogeneity in patients with the same NCF1 mutation c.573G>A (69). Thus, we must remain cautious and bear in mind that other criteria influence the expression of the disease, such as early diagnosis, and how patient care and follow-up is provided for in referral centers.
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
The studies involving human participants were reviewed and approved by the Faculty of Medicine and deanship of research at The University of Jordan. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
FB did the clinical diagnosis, ensured the care and follow-up of patients and their families, collected the clinical data from all hospitals and medical centers that cared for CGD patients and their families, wrote the clinical description of the patients and their relatives, revised the manuscript, and drew Figures 1, 5 and Table 1. MM, SB, and BV performed the functional and molecular analysis. NR-B, JR, and JF performed the DNA sequencing. JR performed the segregating analysis. AA-W, RA, WH, AD, MEAS, MK, MMS, and JA-R did the clinical diagnosis, ensured the care and follow-up of patients and their families, and collected the clinical data. JF revised the manuscript. MJS carried out the conception, design of the study, acquisition, analysis, interpretation of data, drew Figures 2–4 and Table 2, and the drafting of the article. All authors contributed to the article and approved the submitted version.
MJS is grateful for supports from the Université Grenoble Alpes (UGA) (AGIR program 2014), the Interreg France-Suisse (Programme de Cooperation Territoriale Européenne, Fond Européen de Dévelopement Regional (FEDER), 2017–2020). This work was also supported by the Delegation for Clinical Research and Innovations (DRCI, Rementips project 2014), Centre Hospitalier Universitaire Grenoble Alpes (CHUGA). FB is grateful for supports from the University of Jordan, Amman, Jordan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Prof. Dirk Roos and Dr. Martin de Boer for the gift of control mixture of NCF1 gene and ΦNCF1 genes to standardize the Gene-Scan method in our laboratory. I would also like to thank Prof. Dirk Roos for his careful rereading of the manuscript and for the help and kindness he always gave me. This manuscript is dedicated to the memory of Cécile Martel, an exceptional technician from our laboratory.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.639226/full#supplementary-material
2. Thomas DC, Charbonnier LM, Schejtman A, Aldhekri H, Coomber EL, Dufficy ER, et al. EROS/CYBC1 mutations: decreased NADPH oxidase function and chronic granulomatous disease. J Allergy Clin Immunol. (2019) 143:782–5 e1. doi: 10.1016/j.jaci.2018.09.019
3. Brandes RP, Weissmann N, Schroder K. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med. (2014) 76:208–26. doi: 10.1016/j.freeradbiomed.2014.07.046
4. van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS ONE. (2009) 4:e5234. doi: 10.1371/journal.pone.0005234
5. Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine. (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
6. Dinauer MC. Inflammatory consequences of inherited disorders affecting neutrophil function. Blood. (2019) 133:2130–9. doi: 10.1182/blood-2018-11-844563
7. Roesler J, Curnutte JT, Rae J, Barrett D, Patino P, Chanock SJ, et al. Recombination events between the p47-phox gene and its highly homologous pseudogenes are the main cause of autosomal recessive chronic granulomatous disease. Blood. (2000) 95:2150–6. doi: 10.1182/blood.V95.6.2150
8. Roos D, Kuhns DB, Maddalena A, Bustamante J, Kannengiesser C, de Boer M, et al. Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol Dis. (2010) 44:291–9. doi: 10.1016/j.bcmd.2010.01.009
9. Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis. (2010) 45:246–65. doi: 10.1016/j.bcmd.2010.07.012
10. van de Geer A, Nieto-Patlan A, Kuhns DB, Tool AT, Arias AA, Bouaziz M, et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J Clin Invest. (2018) 128:3957–75. doi: 10.1172/JCI97116
11. Roberts AW, Kim C, Zhen L, Lowe JB, Kapur R, Petryniak B, et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. (1999) 10:183–96. doi: 10.1016/S1074-7613(00)80019-9
12. Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci USA. (2000) 97:4654–9. doi: 10.1073/pnas.080074897
13. Gu Y, Williams DA. RAC2 GTPase deficiency and myeloid cell dysfunction in human and mouse. J Pediatr Hematol Oncol. (2002) 24:791–4. doi: 10.1097/00043426-200212000-00027
14. Sharapova SO Haapaniemi E Sakovich IS Kostyuchenko LV Donko A Dulau-Florea A . Heterozygous activating mutation in RAC2 causes infantile-onset combined immunodeficiency with susceptibility to viral infections. Clin Immunol. (2019) 205:1–5. doi: 10.1016/j.clim.2019.05.003
15. Hsu AP, Donko A, Arrington ME, Swamydas M, Fink D, Das A, et al. Dominant activating RAC2 mutation with lymphopenia, immunodeficiency, and cytoskeletal defects. Blood. (2019) 133:1977–88. doi: 10.1182/blood-2018-11-886028
16. Wolach B, Gavrieli R, de Boer M, van Leeuwen K, Berger-Achituv S, Stauber T, et al. Chronic granulomatous disease: clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients. Am J Hematol. (2017) 92:28–36. doi: 10.1002/ajh.24573
17. Blancas-Galicia L, Santos-Chavez E, Deswarte C, Mignac Q, Medina-Vera I, Leon-Lara X, et al. Genetic, immunological, and clinical features of the first Mexican cohort of patients with chronic granulomatous disease. J Clin Immunol. (2020) 40:475–93. doi: 10.1007/s10875-020-00750-5
18. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. (2010) 363:2600–10. doi: 10.1056/NEJMoa1007097
19. Koker MY, Camcioglu Y, van Leeuwen K, Kilic SS, Barlan I, Yilmaz M, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol. (2013) 132:1156–63 e5. doi: 10.1016/j.jaci.2013.05.039
20. Defendi F, Decleva E, Martel C, Dri P, Stasia MJ. A novel point mutation in the CYBB gene promoter leading to a rare X minus chronic granulomatous disease variant—impact on the microbicidal activity of neutrophils. Biochim Biophys Acta. (2009) 1792:201–10. doi: 10.1016/j.bbadis.2009.01.005
21. Bakri FG, Martel C, Khuri-Bulos N, Mahafzah A, El-Khateeb MS, Al-Wahadneh AM, et al. First report of clinical, functional, and molecular investigation of chronic granulomatous disease in nine Jordanian families. J Clin Immunol. (2009) 29:215–30. doi: 10.1007/s10875-008-9243-y
22. Dar-Odeh NS, Hayajneh WA, Abu-Hammad OA, Hammad HM, Al-Wahadneh AM, Bulos NK, et al. Orofacial findings in chronic granulomatous disease: report of twelve patients and review of the literature. BMC Res Notes. (2010) 3:37. doi: 10.1186/1756-0500-3-37
23. El Kares R, Barbouche MR, Elloumi-Zghal H, Bejaoui M, Chemli J, Mellouli F, et al. Genetic and mutational heterogeneity of autosomal recessive chronic granulomatous disease in Tunisia. J Human Genet. (2006) 51:887–95. doi: 10.1007/s10038-006-0039-8
24. Fattahi F, Badalzadeh M, Sedighipour L, Movahedi M, Fazlollahi MR, Mansouri SD, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol. (2011) 31:792–801. doi: 10.1007/s10875-011-9567-x
25. Ishibashi F, Nunoi H, Endo F, Matsuda I, Kanegasaki S. Statistical and mutational analysis of chronic granulomatous disease in Japan with special reference to gp91-phox and p22-phox deficiency. Hum Genet. (2000) 106:473–81. doi: 10.1007/s004390000288
26. Di Matteo G, Giordani L, Finocchi A, Ventura A, Chiriaco M, Blancato J, et al. Molecular characterization of a large cohort of patients with Chronic Granulomatous Disease and identification of novel CYBB mutations: an Italian multicenter study. Mol Immunol. (2009) 46:1935–41. doi: 10.1016/j.molimm.2009.03.016
27. Wu J, Wang WF, Zhang YD, Chen TX. Clinical features and genetic analysis of 48 patients with chronic granulomatous disease in a single center study from Shanghai, China (2005–2015): new studies and a literature review. J Immunol Res. (2017) 2017:8745254. doi: 10.1155/2017/8745254
28. Boyum A. A one-stage procedure for isolation of granulocytes and lymphocytes from human blood. General sedimentation properties of white blood cells in a 1g gravity field. Scand J Clin Lab Investig. (1968) 97:51–76.
29. Batot G, Paclet MH, Doussiere J, Vergnaud S, Martel C, Vignais PV, et al. Biochemical and immunochemical properties of B lymphocyte cytochrome b558. Biochim Biophys Acta. (1998) 1406:188–202. doi: 10.1016/S0925-4439(98)00004-0
30. Segal AW. Nitroblue-tetrazolium tests. Lancet. (1974) 2:1248–52. doi: 10.1016/S0140-6736(74)90758-2
31. Martel C, Mollin M, Beaumel S, Brion JP, Coutton C, Satre V, et al. Clinical, functional and genetic analysis of 24 patients with chronic granulomatous disease—identification of eight novel mutations in CYBB and NCF2 genes. J Clin Immunol. (2012) 32:942–58. doi: 10.1007/s10875-012-9713-0
32. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. (1970) 227:680–5. doi: 10.1038/227680a0
33. Verhoeven AJ, Bolscher BG, Meerhof LJ, van Zwieten R, Keijer J, Weening RS, et al. Characterization of two monoclonal antibodies against cytochrome b558 of human neutrophils. Blood. (1989) 73:1686–94. doi: 10.1182/blood.V73.6.1686.1686
34. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. (1979) 76:4350–4. doi: 10.1073/pnas.76.9.4350
35. Vergnaud S, Paclet MH, El Benna J, Pocidalo MA, Morel F. Complementation of NADPH oxidase in p67-phox-deficient CGD patients p67-phox/p40-phox interaction. Eur J Biochem. (2000) 267:1059–67. doi: 10.1046/j.1432-1327.2000.01097.x
36. Stasia MJ, Mollin M, Martel C, Satre V, Coutton C, Amblard F, et al. Functional and genetic characterization of two extremely rare cases of Williams-Beuren syndrome associated with chronic granulomatous disease. Eur J Human Genet. (2013) 21:1079–84. doi: 10.1038/ejhg.2012.310
37. Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. (1985) 150:76–85. doi: 10.1016/0003-2697(85)90442-7
38. Mollin M, Beaumel S, Vigne B, Brault J, Roux-Buisson N, Rendu J, et al. Clinical, functional and genetic characterization of 16 patients suffering from chronic granulomatous disease variants - identification of 11 novel mutations in CYBB. Clin Exp Immunol. (2020) 203:247–66. doi: 10.1111/cei.13520
39. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. (1987) 162:156–9. doi: 10.1016/0003-2697(87)90021-2
40. Dekker J, de Boer M, Roos D. Gene-scan method for the recognition of carriers and patients with p47(phox)-deficient autosomal recessive chronic granulomatous disease. Exp Hematol. (2001) 29:1319–25. doi: 10.1016/S0301-472X(01)00731-7
41. de Boer M, Singh V, Dekker J, Di Rocco M, Goldblatt D, Roos D. Prenatal diagnosis in two families with autosomal, p47(phox)-deficient chronic granulomatous disease due to a novel point mutation in NCF1. Prenatal Diagnosis. (2002) 22:235–40. doi: 10.1002/pd.296
42. Ben-Farhat K, Ben-Mustapha I, Ben-Ali M, Rouault K, Hamami S, Mekki N, et al. A founder effect of c.257 + 2T > C mutation in NCF2 gene underlies severe chronic granulomatous disease in eleven patients. J Clin Immunol. (2016) 36:547–54. doi: 10.1007/s10875-016-0299-9
43. Gandolfo LC, Bahlo M, Speed TP. Dating rare mutations from small samples with dense marker data. Genetics. (2014) 197:1315–27. doi: 10.1534/genetics.114.164616
44. Kannengiesser C, Gerard B, El Benna J, Henri D, Kroviarski Y, Chollet-Martin S, et al. Molecular epidemiology of chronic granulomatous disease in a series of 80 kindreds: identification of 31 novel mutations. Hum Mutat. (2008) 29:E132–49. doi: 10.1002/humu.20820
45. Koker MY, Sanal O, van Leeuwen K, de Boer M, Metin A, Patiroglu T, et al. Four different NCF2 mutations in six families from Turkey and an overview of NCF2 gene mutations. Eur J Clin Investig. (2009) 39:942–51. doi: 10.1111/j.1365-2362.2009.02195.x
46. Wolach B, Gavrieli R, de Boer M, Gottesman G, Ben-Ari J, Rottem M, et al. Chronic granulomatous disease in Israel: clinical, functional and molecular studies of 38 patients. Clin Immunol. (2008) 129:103–14. doi: 10.1016/j.clim.2008.06.012
47. Patino PJ, Rae J, Noack D, Erickson R, Ding J, de Olarte DG, et al. Molecular characterization of autosomal recessive chronic granulomatous disease caused by a defect of the nicotinamide adenine dinucleotide phosphate (reduced form) oxidase component p67-phox. Blood. (1999) 94:2505–14. doi: 10.1182/blood.V94.7.2505.419k10_2505_2514
48. Al-Riyami AZ, Al-Zadjali S, Al-Mamari S, Al-Said B, Al-Qassabi J, Al-Tamemi S. Correlation between flow cytometry and molecular findings in autosomal recessive chronic granulomatous disease: a cohort study from Oman. Int J Lab Hematol. (2018) 40:592–6. doi: 10.1111/ijlh.12873
49. El Hawary R, Meshaal S, Deswarte C, Galal N, Abdelkawy M, Alkady R, et al. Role of flow cytometry in the diagnosis of chronic granulomatous disease: the Egyptian experience. J Clin Immunol. (2016) 36:610–8. doi: 10.1007/s10875-016-0297-y
50. Al-Muhsen S, Al-Hemidan A, Al-Shehri A, Al-Harbi A, Al-Ghonaium A, Al-Saud B, et al. Ocular manifestations in chronic granulomatous disease in Saudi Arabia. J AAPOS. (2009) 13:396–9. doi: 10.1016/j.jaapos.2009.05.011
51. Suliaman ANF, Sheikh S, Almuhsen S, Alsmani O. Epidemiology of CHronic granulomatous disease of childhood in Eastern Province, Saudi Arabia (2009). Pediatric Asthma Allergy Immunol. (2009) 22:21–25. doi: 10.1089/pai.2008.0513
52. Gao LW, Yin QQ, Tong YJ, Gui JG, Liu XY, Feng XL, et al. Clinical and genetic characteristics of Chinese pediatric patients with chronic granulomatous disease. Pediatric Allergy Immunol. (2019) 30:378–86. doi: 10.1111/pai.13033
53. de Oliveira-Junior EB, Zurro NB, Prando C, Cabral-Marques O, Pereira PV, Schimke LF, et al. Clinical and genotypic spectrum of chronic granulomatous disease in 71 Latin American patients: first report from the LASID registry. Pediatric Blood Cancer. (2015) 62:2101–7. doi: 10.1002/pbc.25674
54. Jones LB, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, et al. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol. (2008) 152:211–8. doi: 10.1111/j.1365-2249.2008.03644.x
55. Hamamy HA, Masri AT, Al-Hadidy AM, Ajlouni KM. Consanguinity and genetic disorders. Profile from Jordan. Saudi Med J. (2007) 28:1015–7.
56. Hamamy H, Al-Hait S, Alwan A, Ajlouni K. Jordan: communities and community genetics. Community Genet. (2007) 10:52–60. doi: 10.1159/000096282
57. Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and reproductive health among Arabs. Reproductive Health. (2009) 6:17. doi: 10.1186/1742-4755-6-17
58. Matoug ESI, Bujallawi K, Elter A, Elsalheen H. Granulomatose chronique en Lybie: étude clinique et de laboratoire de 35 patients pédiatriques. La Tunisie Médicale (2015).
59. Elfaituri S, Matoug I. Chronic granulomatous disease in Libya. Int J Med Sci Dent Res. (2020) 3:1–7.
60. Al-Bousafy A, Al-Tubuly A, Dawi E, Zaroog S, Schulze I. Libyan boy with autosomal recessive trait (P22-phox defect) of chronic granulomatous disease. Libyan J Med. (2006) 1:162–71. doi: 10.3402/ljm.v1i2.4675
61. Stasia MJ. CYBA encoding p22(phox), the cytochrome b558 alpha polypeptide: gene structure, expression, role and physiopathology. Gene. (2016) 586:27–35. doi: 10.1016/j.gene.2016.03.050
62. Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, et al. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol. (2001) 3:675–8. doi: 10.1038/35083070
63. Song X, Xu W, Zhang A, Huang G, Liang X, Virbasius JV, et al. Phox homology domains specifically bind phosphatidylinositol phosphates. Biochemistry. (2001) 40:8940–4. doi: 10.1021/bi0155100
64. Rawat A, Singh S, Suri D, Gupta A, Saikia B, Minz RW, et al. Chronic granulomatous disease: two decades of experience from a tertiary care centre in North West India. J Clin Immunol. (2014) 34:58–67. doi: 10.1007/s10875-013-9963-5
65. Kulkarni M, Desai M, Gupta M, Dalvi A, Taur P, Terrance A, et al. Clinical, immunological, and molecular findings of patients with p47(phox) defect chronic granulomatous disease (CGD) in Indian Families. J Clin Immunol. (2016) 36:774–84. doi: 10.1007/s10875-016-0333-y
66. Roos D, de Boer M, Koker MY, Dekker J, Singh-Gupta V, Ahlin A, et al. Chronic granulomatous disease caused by mutations other than the common GT deletion in NCF1, the gene encoding the p47phox component of the phagocyte NADPH oxidase. Hum Mutat. (2006) 27:1218–29. doi: 10.1002/humu.20413
67. Sumimoto H, Hata K, Mizuki K, Ito T, Kage Y, Sakaki Y, et al. Assembly and activation of the phagocyte NADPH oxidase. Specific interaction of the N-terminal Src homology 3 domain of p47phox with p22phox is required for activation of the NADPH oxidase. J Biol Chem. (1996) 271:22152–8. doi: 10.1074/jbc.271.36.22152
68. de Boer M, Tzur S, van Leeuwen K, Dencher PC, Skorecki K, Wolach B, et al. A founder effect for p47(phox)Trp193Ter chronic granulomatous disease in Kavkazi Jews. Blood Cells Mol Dis. (2015) 55:320–7. doi: 10.1016/j.bcmd.2015.07.014
69. Wolach B, Gavrieli R, de Boer M, van Leeuwen K, Wolach O, Grisaru-Soen G, et al. Analysis of chronic granulomatous disease in the Kavkazi population in Israel reveals phenotypic heterogeneity in patients with the same NCF1 mutation (c.579G>A). J Clin Immunol. (2018) 38:193–203. doi: 10.1007/s10875-018-0475-1
70. Noack D, Rae J, Cross AR, Munoz J, Salmen S, Mendoza JA, et al. Autosomal recessive chronic granulomatous disease caused by novel mutations in NCF-2, the gene encoding the p67-phox component of phagocyte NADPH oxidase. Hum Genet. (1999) 105:460–7. doi: 10.1007/s004399900152
71. Koga H, Terasawa H, Nunoi H, Takeshige K, Inagaki F, Sumimoto H. Tetratricopeptide repeat (TPR) motifs of p67(phox) participate in interaction with the small GTPase Rac and activation of the phagocyte NADPH oxidase. J Biol Chem. (1999) 274:25051–60. doi: 10.1074/jbc.274.35.25051
72. Maehara Y, Miyano K, Yuzawa S, Akimoto R, Takeya R, Sumimoto H. A conserved region between the TPR and activation domains of p67phox participates in activation of the phagocyte NADPH oxidase. J Biol Chem. (2010) 285:31435–45. doi: 10.1074/jbc.M110.161166
73. Conti F, Lugo-Reyes SO, Blancas Galicia L, He J, Aksu G, Borges de Oliveira E Jr, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. (2016) 138:241–8 e3. doi: 10.1016/j.jaci.2015.11.041
74. Brunner J, Dockter G, Rosen-Wolff A, Roesler J. X-linked chronic granulomatous disease (CGD) caused by an intra-exonic splice mutation (CYBB exon 3, c.262G->A) is mimicking juvenile sarcoidosis. Clin Exp Rheumatol. (2007) 25:336–8.
75. Kutukculer N, Aykut A, Karaca NE, Durmaz A, Aksu G, Genel F, et al. Chronic granulamatous disease: two decades of experience from a paediatric immunology unit in a country with high rate of consangineous marriages. Scand J Immunol. (2019) 89:e12737. doi: 10.1111/sji.12737
76. Aygun D, Koker MY, Nepesov S, Koker N, van Leeuwen K, de Boer M, et al. Genetic characteristics, infectious, and non-infectious manifestations of 32 patients with chronic granulomatous disease. Int Archiv Allergy Immunol. (2020) 181:540–50. doi: 10.1159/000507366
Keywords: innate immunodeficiency, Jordan, chronic granulomatous disease, autosomal recessive, NADPH oxidase, founder mutation
Citation: Bakri FG, Mollin M, Beaumel S, Vigne B, Roux-Buisson N, Al-Wahadneh AM, Alzyoud RM, Hayajneh WA, Daoud AK, Shukair MEA, Karadshe MF, Sarhan MM, Al-Ramahi JAW, Fauré J, Rendu J and Stasia MJ (2021) Second Report of Chronic Granulomatous Disease in Jordan: Clinical and Genetic Description of 31 Patients From 21 Different Families, Including Families From Lybia and Iraq. Front. Immunol. 12:639226. doi: 10.3389/fimmu.2021.639226
Received: 08 December 2020; Accepted: 08 February 2021;
Published: 05 March 2021.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Amit Rawat, Post Graduate Institute of Medical Education and Research (PGIMER), IndiaCopyright © 2021 Bakri, Mollin, Beaumel, Vigne, Roux-Buisson, Al-Wahadneh, Alzyoud, Hayajneh, Daoud, Shukair, Karadshe, Sarhan, Al-Ramahi, Fauré, Rendu and Stasia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie Jose Stasia, bWpzdGFzaWFAY2h1LWdyZW5vYmxlLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.