 Nicholas Stoy
Nicholas Stoy- Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, United Kingdom
Interleukin-1 receptor-associated kinase 4 (IRAK4) and interferon regulatory factor 5 (IRF5) lie sequentially on a signaling pathway activated by ligands of the IL-1 receptor and/or multiple TLRs located either on plasma or endosomal membranes. Activated IRF5, in conjunction with other synergistic transcription factors, notably NF-κB, is crucially required for the production of proinflammatory cytokines in the innate immune response to microbial infection. The IRAK4-IRF5 axis could therefore have a major role in the induction of the signature cytokines and chemokines of the hyperinflammatory state associated with severe morbidity and mortality in COVID-19. Here a case is made for considering IRAK4 or IRF5 inhibitors as potential therapies for the “cytokine storm” of COVID-19.
Introduction
Effective treatments are required for COVID-19 hyperinflammatory syndrome, occurring characteristically 7–14 days after first symptoms (1) and variously described as “macrophage activation syndrome” (2), “cytokine storm” (3) or “acute respiratory distress syndrome” (4). Its immunological hallmarks are excessive elevation of predominantly proinflammatory cytokines, chemokines (5), and other bioactive molecules, such as HMGB1 (6) and reactive oxygen species (7). Upregulated cytokines include IL-6, TNF-α, IFN-γ, IL-1β, IL-15, IL-23, and IL-10, and chemokines, CXCL8(IL-8), CXCL9(MIG), CXCL10(IP10), CCL2(MCP-1), CCL3(MIP-1α), CCL5(RANTES), CCL7(MCP-3), CCL8(MCP-2), CCL11(eotaxin-1), and CCL20(MIP-3α) (1, 2, 8–11). This review examines the role of IRAK4 and IRF5 in the evolution and modulation of the immune response to SARS-CoV-2 and whether IRAK4 or IRF5 inhibitors could have a role in treating the hyperinflammatory phase (12–14).
Overview of IRAK4 and IRF5 Signaling
IRAK4, recruited with other binding partners to MYD88 (Figure 1) forms the myddosome (14–16), which is activated by ligands of the IL-1 receptor or TLRs that bind MYD88 (13, 17, 18). IRAK4 is recruited to the complex with IRAK1 and TRAF6 (19). On receptor activation IRAK4 homodimerizes, autophosphorylates and subsequently phosphorylates IRAK1 (20–22). These kinases are ultimately responsible for activation of IRF5, requiring phosphorylation of critical C-teminal serines (23, 24). Another component of IRF5 activation is its K63polyubiquitination by TRAF6 (25, 26). The inducible (and IRAK1-phosphorylated) ubiquitin ligase Pellino-1, with E2-conjugating enzymes (27), reciprocally K63polyubiquinates both IRF5 and IRAK1/4 but, conversely, kinase-active IRAK1/4 mediates degradative polyubiquitination of Pellino-1 (27–29).
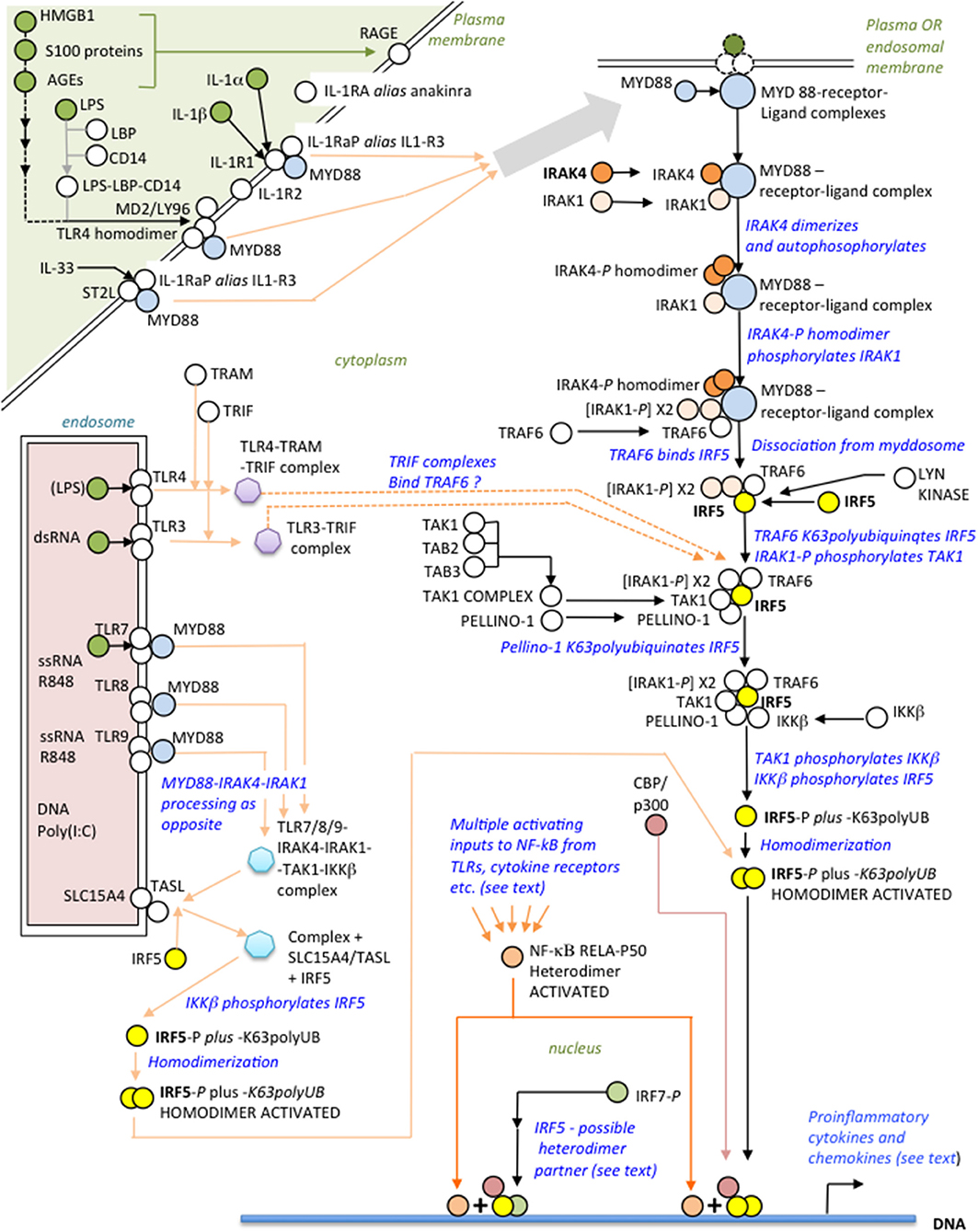
Figure 1. Overview of the IRAK4-IRF5 signaling axis and some ligands of possible relevance to SARS-CoV-2 immunopathogenesis.
Distal to the myddosome the signaling pathway bifurcates into IRF5- and NF-κB-activating branches (30). At commencement of the IRF5 branch, activated IRAK4/IRAK1 phosphorylates the kinase TAK1 (31), which in turn phosphorylates IKKβ (24, 32); finally, IKKβ phosphorylates IRF5 (33, 34) facilitating its dimerization and translocation to the nucleus (35). Whereas, IKKβ is the archetypal kinase activator of NF-κB by phosphorylating IκBα, it is important to appreciate that kinase activity of IRAK4 is not essential for NF-κB activation by the myddosome route (35); however, this does not preclude an IRAK4 scaffolding function (20). Crucially, therefore, blocking IRAK4 with a specific kinase inhibitor abolishes IRF5 activation but still permits NF-κB activation by other means, either by IKKβ itself via this or other signaling pathways (31), or using other kinases such as a MEKK3-dependent pathway (17, 30, 35, 36). Speculatively, endosomal TLR3, responsive to dsRNA, may signal independently of MYD88 to IRF5 through TRAF6 using the adaptor TRIF (as well as to IRF3/7 via TRAF3) and may synergise with other TLRs (37, 38, 183); TLR4, translocated to endosomes, may also signal using TRAM-TRIF instead of MYD88 (19, 39–41, 183). IRF5 homodimers complex with CBP/p300 to initiate the IRF5 transcriptome synergistically with NF-κB (42, 43).
Activation of IRF5 is tightly controlled. Inducible IRAK-M inhibits assembly of the IRAK1-IRAK4-TRAF6 complex both directly, and indirectly by induced negative feedback (44–46); Lyn kinase, in dendritic cells (DCs), binds IRF5, inhibiting its K63polyubiquitination and phosphorylation, but not affecting the NF-κB branch (47); IRF8 competes with IRF5 at promoters, blocking its action (48, 49); and KAP1/TRIM28 is an IRF5 transcriptional co-repressor (50). TLRS 7-9 require at least two adapter proteins, TASL and SLC15A4, at the endosomal membrane, to engage the IRAK4-IRF5 pathway (51).
In responding to viruses, activated IRF5 homodimers bind with low affinity to “viral response elements” inducing primarily IFNA type I interferons (52). However, IRF5 binds strongly to the regulatory loci of other IRF5-targetted genes, such as IFNB, CXCL10, IL-10 (52), IL-12, and IL-23 (53), although in the case of anti-inflammatory IL-10, IRF5 is not directly responsible for its elevation in “cytokine storm,” being inhibitory at the IL-10 promoter (53, 54). Mechanistically, a challenging complexity of variables influence IRAK4-IRF5 pathway activation outcomes (55): these include different IRF5 dimerization partners—including homodimerization and IRF7 (56); functionally different IRF5 isoforms, as investigated in plasmacytoid DCs (pDCs) (57); IRF5 interacting with different transcription factors (17), most critically the NF-κB subunits, p50 (48, 58), and/or p65(RELA) (41, 59, 60); different cellular localizations, notably monocytes, macrophages, pDCs, and B cells (55, 58, 61); different triggers of pathway activation, for e.g., viral infection or autoimmunity; inhibition of the IRF5-mediated activation of IFN-β by the IKKα pathway (62); and differences between murine and human cells (63)—all beyond the scope of this review. Nevertheless, despite these many complicating factors, the IRAK4-IRF5 axis consistently polarizes monocytes/macrophages toward the proinflammatory M1 (49, 53, 64) phenotype, displaying a similar innate cytokine/chemokine profile as in “cyokine storm” and indicating a potential therapeutic role for IRAK4 or IRF5 inhibition.
Deficiency or Inhibition of IRAK4 or IRF5 and Viral Infections
A proinflammatory response is characteristic of the innate immune system's reaction to microbial infection. Endotoxin tolerance in monocytes blunts this response by interfering with recruitment and activation of IRAK4 at the MYD88 receptor complex, inhibiting K63polyubiquitination of IRAK1 and TRAF6, and compromising IRAK1-TRAF6 function and TAK1 activation (65). Mice lacking IRAK4 exhibit deficient IL-1 and TLR signaling, are resistant to LPS and cannot induce TNF-α or IL-6 (17, 66, 67). The IRAK4 inhibitor, chlorogenic acid, extracted from lonicerae flos, protects mice from endotoxic shock: chlorogenic acid inhibits autophosphorylation of IRAK4 in peritoneal macrophages subjected to various activating stimuli, including ssRNA, IL-1α, or HMGB1 (6, 68, 69). Inhibition of IRAK4 or IRF5 downregulates the proinflammatory IRF5 transcriptome independently of NF-κB activation (35). In the same way that endotoxic shock is abrogated by inhibiting IRF5, “cytokine storm” in viral infection can also be suppressed by IRF5 inhibition, as shown for influenza A (26, 70). Thus, IRF5 inhibition protects from hyperinflammation whether induced by viral or bacterial infection, the latter a common complication of acute respiratory distress syndrome, although its incidence in COVID-19 is only just being investigated (71–73, 184).
The Immunopathogenesis and Clinical Correlates of SARS-CoV-2 Infection
Figure 2 summarizes how SARS-CoV-2 innate immune activation is linked to specific T cell (cytotoxic and memory) and B cell (antibody) adaptive immune responses, comprising the substantive immunological reaction necessary for viral elimination (53, 54). COVID-19 immunopathogenesis divides conveniently into three overlapping interactive phases with sequential involvement of epithelial cells, innate immune cells and adaptive immune cells. Nasal and alveolar type II epithelial cells express high levels of ACE2, the SARS-CoV-2 entry receptor, and respond first. Epithelial immune activation is mediated by IRF3 phospho-dimerisation, with a lesser contribution from IRF5 phospho-dimerization, and NF-κB p65 as coactivator (59, 60, 75). Critically, the type I interferon component of the epithelial cell proinflammatory response is selectively suppressed by proliferating SARS-CoV-2 (74), so disrupting secondary expression of interferon-stimulated genes (ISGs) including the potent IRF3 dimerisation partner IRF7 (75). Epithelial cells favor IFN-λ expression but this is a less effective inducer of ISGs than type I interferons (76). The viral MDA5 RNA-sensor requires kinases TBK1 or IKKε to activate IRF3 (77, 78); the same kinases can activate IRF5. By contrast, IKKβ, a strong activator of IRF5, fails to activate IRF3 (61) In phase two, epithelial chemokines attract a large influx of innate immune cells comprising DCs, natural killer (NK) cells and neutrophils (11, 79–81); IRF5 is considered the main orchestrator of this innate response (61). DCs are pivotal in communicating with the adaptive immune system to initiate phase three: programming of adaptive immunity. Phase three culminates either in viral clearance and COVID-19 resolution or complications such as “cytokine storm,” clotting disorders, cardiovascular complications or multi-organ failure. Involvement of adaptive immunity adds further cytokines to the mix. IL-17, the product of Th-17 cells, is triggered by DCs expressing IL-23 (increased with age). Induction of this and other DC cytokines IL-1β, IL-6, IL-12, and TNF-α is again dependent on IRF5, usually with NF-κB coactivators (53, 58, 59, 61, 82–84). Indeed, recent evidence suggests IRF5 may even vie with IRF7 for the title ‘master regulator’, if not of type I interferons, at least of most other DC cytokines (42, 52, 75, 85–88); furthermore, there is mutual inhibition between these two IRFs (56). Th1 cells are activated by the innate cytokine IL-12 from DCs and secrete IFN-γ, as do NK cells. However, in COVID-19 multifunctional activated T cells secreting two of the three cytokines IFN-γ, IL-2, and TNF-α were reduced whilst T cells producing all three were non-functional (89).
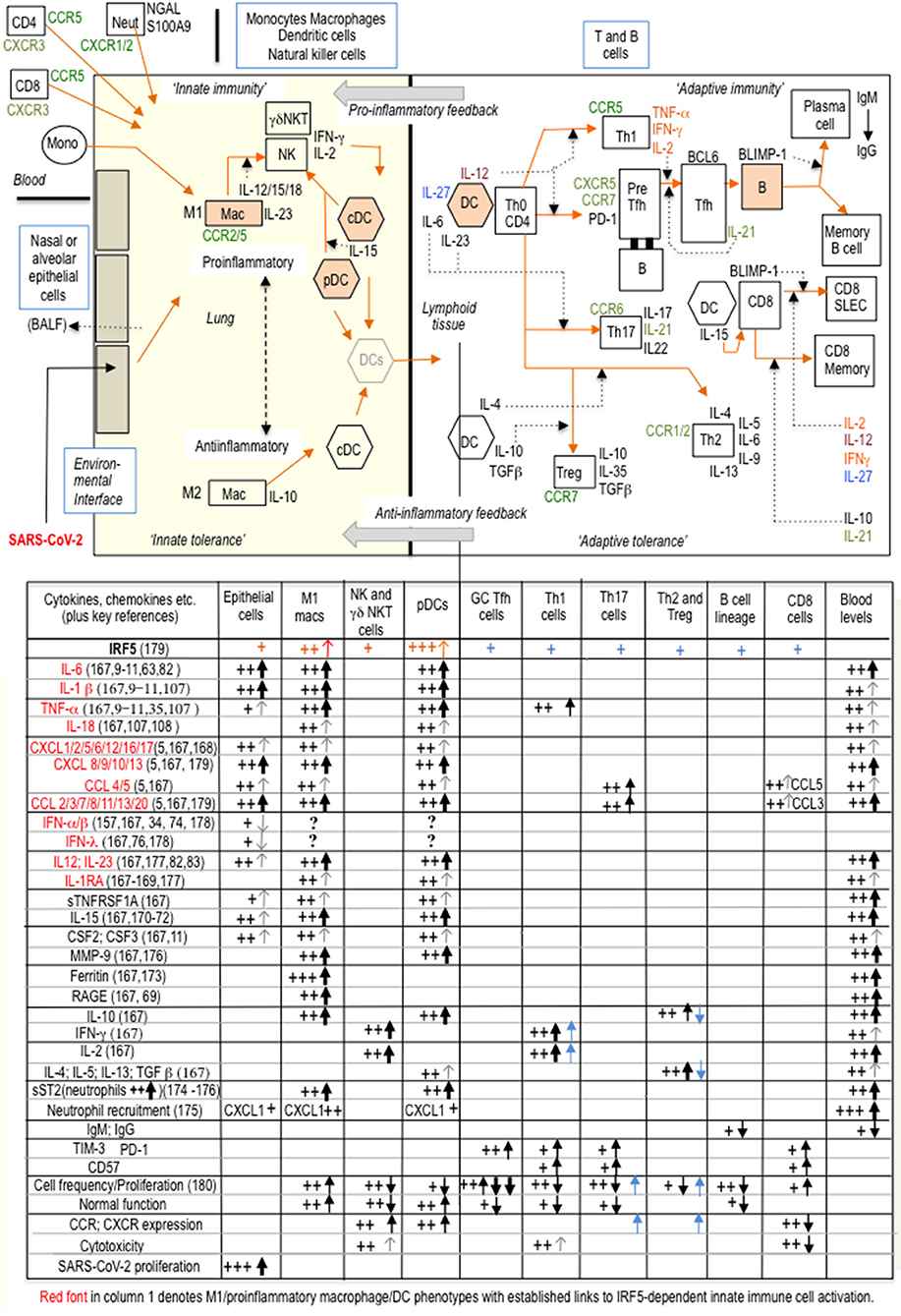
Figure 2. With burgeoning knowledge of immune cell phenotypes in COVID-19, particularly from single-cell transcriptomics, and despite much heterogeneity amongst T-cell clusters, it is now possible to attempt a broad generalization of (at least some) changes in innate and adaptive immunity during COVID-19 progression. In this schematic representation, a suppressed type I interferon response in epithelial cells, IRF5-dependent proinflammatory macrophage and DC polarization, and an inadequate adaptive response are three sequential major drivers of COVID-19 immunopathogenesis. The accompanying table is a tentative interpretation of some of the (sometimes conflicting) features of the immune cell phenotypic landscape of “cytokine storm” (1, 2, 9–11, 157–163). SARS-CoV-2 can be taken up by macrophages and DCs but does not proliferate, whilst the highly variable type I IFN response from each individual cell likely depends on temporal sequencing and integration of inputs both from viral components and from other non-viral inputs (TLR and/or cytokine), either synergistic or separate (179–183). Key: +signs indicate changes in immune cell parameters associated with “cytokine storm” and black vertical arrows indicate changes from the expected normal immune response. IRF5 appears to be widely expressed in most immune cells (164–166) and recently CD4+ and CD8+ intrinsic IRF5 activity has been demonstrated to be responsible for increased secretion of Th1 and Th17 cytokines and for reduced Th2 and T reg cytokines, on T cell activation, as indicated by blue arrows and blue +signs in the table: IRF5 upregulates chemokine receptors CXCR4/5 and CCR6/7/9, on stimulated CD4+ T cells (165, 166); the relevance of these observations to COVID-19 pathogenesis is as yet unknown. SLEC: short-lived effector cells; BALF: bronchoalveolar lavage fluid; GC, germinal center.
Innate immunity is relatively preserved during aging and constitutively upregulated in many comorbid conditions exacerbating COVID-19, albeit stimulating a defective adaptive response. In aging, pDCs retain most of the proinflammatory phenotype, but type I and type III interferons are impaired (90), as are interactions with T and B cells for antigen presentation, primarily due to T and B cell dysfunction, exacerbated by SARS-CoV-2 (91). IRF5 is constitutively expressed by pDCs, especially in females who produce more IFN-α on TLR stimulation than males, making dysregulation of immune responses in COVID-19 in females less likely (92, 93). IRF5-dependent IFN-β expression in DCs is demonstrated in IRF5-knock-out mice, which exhibit poor interferon responses to TLR stimulation or microbial infection (49, 94). Overall, DCs adopt a proinflammatory phenotype on contact with SARS-CoV-2, a tendency exacerbated by increasing age (91, 95). Cellular correlates of poor outcome in COVID-19 are neutrophilia (3), low CD4+ and CD8+ T cells and general lymphopenia (96, 97), combined with increased markers of T cell exhaustion (PD-1 and TIM-3) and senescence (CD57) and a specific cytokine signature (10, 73, 98–100).
B cells depend on IRF5-induced Blimp-1 for differentiation into plasma cells, responsible for long-lasting antibody immunity (101). In SARS-CoV-2 “cytokine storm,” B cell function is compromised by reduced total circulating B cells, reduced class switching from IgM to IgG and increased plasmablasts and transitional cells, suggestive of rapid B cell proliferation and exhaustion, probably related to excessive IL-6 and TNF-α (102).
Platelets are integral components of the immune system. Viruses can enter platelets, activate endosomal TLRs (TLR7/TLR9) and downstream MYD88-IRAK4-IRAK1-IKKβ (and presumably IRF5), possibly contributing to COVID-19 thrombocytopenia and clotting irregularities (103, 104).
The Metabolic Dimension and COVID-19 Comorbidities
Maintaining the M1 phenotype is energy-consuming and achieved by a “metabolic switch” from oxidative to glycolytic metabolism during M2-to-M1 polarization (41). Viral infections increase glucose metabolism in macrophages, involving activation of the hexosamine biosynthesis pathway and associated enzyme O-GlcNAc transferase, as already proposed for SARS-CoV-2 (105). Thus, increased activation of IRF5 by K63polyubiquitination may turn out to provide an important link between so-called “metabolic inflammation” and increased severity of the cytokine response in COVID-19 (105, 106). Infection with influenza virus markedly increases GlcNAcylation of IRF5 at serine 430 in human macrophages, which is essential for K63polyubiquitination of the same residue that activates IRF5, thus promoting proinflammatory cytokine expression and possibly increased viral replication (26). Inflammation is a well-recognized driver of the metabolic syndrome, manifest clinically in obesity, type II diabetes and other conditions in which insulin resistance occurs. Blood sugar instability associates with IRF5 upregulation and M1 phenotype in adipose tissue macrophages, including elevation of the cardiovascular risk factor, matrix metalloproteinase-9. TLR4 is also upregulated and it has even been hypothesized that increased proinflammatory cytokines could be triggered by endogenous TLR4 ligands, presumably through IRAK4-IRF5 signaling (41). IRF5 knock-out mice exhibit improved glucose tolerance and reduced excess body fat. The M1 macrophage cytokine and chemokine profile of adipose tissue in obesity and diabetes, signifying chronic inflammation, is in many respects similar to a muted version of cytokine storm. Thus, adipose IRF5 transcripts in obesity correlate positively with TNF-α, IL-1β, IL-6, CXCL8/IL-8, CXCL9/MIG, CXCL10/IP10, CCL2/MCP1, CCL5, and CCL7/MCP3, all of which can be elevated in COVID-19 “cytokine storm” (107, 108); positive correlations with IL-2 and IL-12 have been reported by the same group. TLR4, TLR7, and TLR8 are increased in obesity and correlate with IRF5 expression, but whether this occurs in SARS-CoV-2 infection is unknown (109, 110).
There is increased Pellino-1 expression in adipose tissue macrophages in obesity. Pellino-1 exacerbates glucose intolerance in obese mice through K63polyubiquitination of IRF5, promoting M1 macrophage polarization (106); the adverse proinflammatory skewing of innate immunity is further compounded by Pellino-1 inhibition of tolerogenic M2 macrophages by K63polyubiquitination of IRAK1 (111). Correspondingly, in acute viral respiratory infections, there is an association between elevated Pellino-1 and proinflammatory cytokines (112).
Chronic innate proinflammatory drive to the adaptive immune system in metabolic inflammation leads eventual to T cell exhaustion. Changes in T and B cell function in metabolic inflammation, as well as in the elderly and, more acutely, in COVID-19 are all broadly similar, in that innate function is relatively preserved, but T cell and B cell compartments exhibit features of “exhaustion” or “senescence” (73, 113, 114). Therefore, pre-existing metabolic inflammation across a variety of chronic conditions presages an unfavorable course and outcome of COVID-19 (115, 116).
Discussion
The well-established paradigm that innate immunity programs adaptive immunity applies not only in microbial infection (117) but also autoimmunity (118, 119) and cancer, being generally tolerogenic in the latter. “Cytokine storm” of COVID-19 illustrates the dangers of a fundamental mismatch between increased proinflammatory innate signaling and a defective adaptive response, insufficient to kill the virus or prevent spread (105), thus failing to abort innate immune activation. This review has presented evidence that IRF5 is a key “hub” molecule determining the normal balance between innate and adaptive immunity. In what clinical situations, therefore, could IRAK4/IRF5 inhibitors have therapeutic benefit?
Many clinical consequences arise from immune system dysregulation in COVID-19. Importantly, for proposed treatment of “cytokine storm” with an IRAK4 inhibitor, timing, and dose titration are critical—too early, too protracted or too high a dose and the natural host immune response is further blunted. It follows that reliable real–time (blood) biomarkers of IRF5-driven immune activation would be essential to determine the threshold for both commencement and termination of treatment (120, 121). To avoid overdosage, the ideal IRAK4 inhibitor would be quick-acting with short half-life (122, 123). Amongst candidate COVID-19 inflammation biomarkers is IRF5 itself, raising the possibility of studying this key molecule across the whole range of SARS-CoV-2 infection and associated comorbidities (124, 125); already IRF5 is suggested as a novel adipose marker in chronic metabolic inflammation (108) and inflammatory bowel disease (125).
Another practical issue is management of patients on long-term immuno-suppressants: these drugs could be viewed simplistically as raising the threshold for effective adaptive immune activation, particularly with drugs inhibiting T or B cell function. Nevertheless, it is surmised that many patients could reset their immune response appropriately and experience symptomless or mild COVID-19, but in others, nearer the tipping-point, the chances of hyperinflammatory syndome may be significantly increased. As there is no a priori reason in these patients to suppose increased viral uptake at the onset, the advised management of COVID-19 has continued to be on accepted lines and routine immuno-suppressants continued unless “cytokine storm” becomes imminent (126, 127).
Similar reasoning may be applied to initial high viral load or prolonged exposure, which could overwhelm adaptive immunity and push the balance toward increased, but less effective, innate immune activation and “cytokine storm.” A related unresolved difficulty is management of chronic COVID-19 symptoms, especially if associated with identifiable chronic inflammation, including neurological sequelae (115). Indeed, the predisposing conditions for hyperinflammatory COVID-19 are likely to overlap with at least some of those responsible for post-infection sequelae. Whether viral persistence occurs is uncertain, but post-infection inflammatory markers suggest ongoing low-grade innate immune activation linked to adaptive immune dysregulation and/or exhaustion (128). Chronic infection promotes the death of protective CD4+ cells through TLR7 and IRF5 (129). Thus, in so-called “long COVID” the perceived imbalance of innate and adaptive immunity may be finely poised and potentially amenable to favorable manipulation, conceivably using IRAK4 or IRF5 inhibition.
Dexamethasone, anakinra and tocilizumab are amongst anti-inflammatory drugs already repurposed for treatment of “cytokine storm.” Although the extent of dexamethasone interaction with the IRAK4-IRF5 axis is not established, IRAK4/IRF5 inhibitors are still likely to provide a more focused approach than the generalized actions of steroids (130). On the other hand, IRAK4/IRF5 inhibitors would have a wider spectrum of action than the IL-1 receptor antagonist, anakinra (131, 132) or IL-6 receptor blocker, tocilizumab. Predictably, there is concern that overuse/prolonged use of steroids as immuno-suppressants could suppress viral clearance (133): by contrast, IRAK4 inhibition is potentially steroid-sparing (134). Latest data indicates significant benefit in severe COVID-19 from tocilizumab, either alone or with dexamethasone (135–137). CXCL8/IL-8 inhibitors are being trialed to reduce neutrophil recruitment (138, 139). However, as proposed here, a better option might be concurrent suppression by just one drug of multiple innate cytokines and chemokines, including IL-1, IL-6 and neutrophil-attractant chemokines (CXCL8 and CXCL5), as would be achievable by an IRAK4 or IRF5 inhibitor. Indeed, in co-cultured RNA-stimulated pDCs and NK cells, IRAK4 inhibition reduced IL-6, CXCL8, CCL3, CCL4, TNF-α, and IFN-γ (140), whereas, raised expression of IRF5 (but not IRF3 or IRF7) in kupffer cells and neutrophils in experimental cholestatic jaundice correlated with increased IL-6, TLR4, TLR7, TLR9, HMGB1, CXCL8, and CCL2, with some evidence of steroid reversibility (141).
Although developed recently, IRAK4 inhibitors are under assessment in psoriasis, whilst in rheumatoid arthritis a completed phase II clinical trial has demonstrated clinical improvement (142). Interestingly, dimethyl fumarate, already of proven clinical efficacy in treating both multiple sclerosis and psoriasis, is not only a direct inhibitor of IRAK4 but also suppresses innate proinflammatory cytokines in pDCs, providing a strong mechanistic rationale for its recently proposed repurposing for COVID-19 “cytokine storm” (143, 144). Low-grade inflammation is common in autoimmunity (145), with an inflammatory signature similar to COVID-19 (146). The therapeutic usefulness of IRF5 inhibitors is yet to be determined (13, 123, 145–148, 175).
Finally, in SARS-CoV-2 vaccine development, an adjuvant stimulating the evolutionary-conserved, IRAK4-IRF5 pathway should be an ideal partner for a SARS-CoV-2 vaccine. IRAK4-IRF5 pathway activators could be included in multi-epitope vaccines (149). Such formulations should promote optimum immune responses and immunological memory (150). Suitable targets would be TLR3, TL7, TLR8, or TLR9 (151–153). Paradoxically, even with highly potent vaccines, the adaptive immune system in vulnerable groups may still fail to respond appropriately because risk factors predicting a poor adaptive immune response to vaccination could be the same as those predisposing to COVID-19 “cytokine storm,” although it is yet to be determined whether this will account for a significant fraction of vaccine failures.
In conclusion a caveat: given that IRF5 is essential for normal immunity and that “cytokine storm” in SARS-CoV-2 infection indicates a failure of adaptive immunity to respond appropriately to enhanced (IRF5-mediated) innate signals, it follows that attempts to stop “cytokine storm” by damping down innate immunity should be combined with, or ideally replaced by, effective SARS-CoV-2 virucidal drugs, another high priority in COVID-19 research (154–156).
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I should like to thank Andrezj Kierzek, Graham Stewart, and David Lewis for their encouragement when I was a Research Fellow at University of Surrey, 2012 to 2019, supported by a grant from BioVacSafe (project grant code RR0D29A); and to Denis Noble who recently accepted me as Visitor at the Department of Physiology, Anatomy and Genetics, University of Oxford.
References
1. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
2. Booz GW, Altara R, Eid AH, Wehbe Z, Fares S, Zaraket H, et al. Macrophage responses associated with COVID-19: a pharmacological perspective. Eur J Pharmacol. (2020) 887:173547. doi: 10.1016/j.ejphar.2020.173547
3. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol. (2020) 108:17–41. doi: 10.1002/JLB.3COVR0520-272R
4. Navas-Blanco JR, Dudaryk R. Management of respiratory distress syndrome due to COVID-19 infection. BMC Anesthesiol. (2020) 20:177. doi: 10.1186/s12871-020-01095-7
5. Khalil BA, Elemam NM, Maghazachi AA. Chemokines and chemokine receptors during COVID-19 infection. Comput Struct Biotechnol J. (2021) 19:976–88. doi: 10.1016/j.csbj.2021.01.034
6. Andersson U, Ottestad W, Tracey KJ. Extracellular HMGB1: a therapeutic target in severe pulmonary inflammation including COVID-19? Mol Med. (2020) 26:42. doi: 10.1186/s10020-020-00172-4
7. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 Infection and cytokine shock syndromes. Cell. (2021) 184:149–168.e17. doi: 10.1016/j.cell.2020.11.025
8. Najm A, Alunno A, Mariette X, Terrier B, De Marco G, Emmel J, et al. Pathophysiology of acute respiratory syndrome coronavirus 2 infection: a systematic literature review to inform EULAR points to consider. RMD Open. (2021) 7:e001549. doi: 10.1136/rmdopen-2020-001549
9. Chi Y, Ge Y, Wu B, Zhang W, Wu T, Wen T, et al. Serum cytokine and chemokine profile in relation to the severity of coronavirus disease 2019 in China. J Infect Dis. (2020) 222:746–54. doi: 10.1093/infdis/jiaa363
10. Akbari H, Tabrizi R, Lankarani KB, Aria H, Vakili S, Asadian F, et al. The role of cytokine profile and lymphocyte subsets in the severity of coronavirus disease 2019 (COVID-19): a systematic review and meta-analysis. Life Sci. (2020) 258:118167. doi: 10.1016/j.lfs.2020.118167
11. Allegra A, Di Gioacchino M, Tonacci A, Musolino C, Gangemi S. Immunopathology of SARS-CoV-2 infection: immune cells and mediators, prognostic factors, and immune-therapeutic implications. Int J Mol Sci. (2020) 21:4782. doi: 10.3390/ijms21134782
12. Bahia MS, Kaur M, Silakari P, Silakari O. Interleukin-1 receptor associated kinase inhibitors: potential therapeutic agents for inflammatory- and immune-related disorders. Cell Signal. (2015) 27:1039–55. doi: 10.1016/j.cellsig.2015.02.025
13. Thompson CD, Matta B, Barnes BJ. Therapeutic targeting of IRFs: pathway-dependence or structure-based? Front Immunol. (2018) 9:2622. doi: 10.3389/fimmu.2018.02622
14. Almuttaqi H, Udalova IA. Advances and challenges in targeting IRF5, a key regulator of inflammation. FEBS J. (2019) 286:1624–37. doi: 10.1111/febs.14654
15. Zarrin AA, Bao K, Lupardus P, Vucic D. Kinase inhibition in autoimmunity and inflammation. Nat Rev Drug Discov. (2020) 20:39–63. doi: 10.1038/s41573-020-0082-8
16. Ryzhakov G, Eames HL, Udalova IA. Activation and function of interferon regulatory factor 5. J Interferon Cytokine Res. (2015) 35:71–8. doi: 10.1089/jir.2014.0023
17. Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. (2005) 434:243–9. doi: 10.1038/nature03308
18. Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem Pharmacol. (2010) 80:1981–91. doi: 10.1016/j.bcp.2010.06.020
19. Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal. (2013) 8:7. doi: 10.1186/1750-2187-8-7
20. Cushing L, Stochaj W, Siegel M, Czerwinski R, Dower K, Wright Q, et al. Interleukin 1/Toll-like receptor-induced autophosphorylation activates interleukin 1 receptor-associated kinase 4 and controls cytokine induction in a cell type-specific manner. J Biol Chem. (2014) 289:10865–75. doi: 10.1074/jbc.M113.544809
21. Gottipati S, Rao NL, Fung-Leung WP. IRAK1: a critical signaling mediator of innate immunity. Cell Signal. (2008) 20:269–76. doi: 10.1016/j.cellsig.2007.08.009
22. Wang L, Qiao Q, Ferrao R, Shen C, Hatcher JM, Buhrlage, et al. Crystal structure of human IRAK1. Proc Natl Acad Sci USA. (2017) 114:13507–12. doi: 10.1073/pnas.1714386114
23. Chang Foreman HC, Van Scoy S, Cheng TF, Reich NC. Activation of interferon regulatory factor 5 by site specific phosphorylation. PLoS ONE. (2012) 7:e33098. doi: 10.1371/journal.pone.0033098
24. Ren J, Chen X, Chen ZJ. IKKbeta is an IRF5 kinase that instigates inflammation. Proc Natl Acad Sci USA. (2014) 111:17438–43. doi: 10.1073/pnas.1418516111
25. Balkhi MY, Fitzgerald KA, Pitha PM. Functional regulation of MyD88-activated interferon regulatory factor 5 by K63-linked polyubiquitination. Mol Cell Biol. (2008) 28:7296–308. doi: 10.1128/MCB.00662-08
26. Wang Q, Fang P, He R, Li M, Yu H, Zhou L, et al. O-GlcNAc transferase promotes influenza A virus-induced cytokine storm by targeting interferon regulatory factor-5. Sci Adv. (2020) 6:eaaz7086. doi: 10.1126/sciadv.aaz7086
27. Smith H, Peggie M, Campbell DG, Vandermoere F, Carrick E, Cohen P. Identification of the phosphorylation sites on the E3 ubiquitin ligase pellino that are critical for activation by IRAK1 and IRAK4. Proc Natl Acad Sci USA. (2009) 106:4584–90. doi: 10.1073/pnas.0900774106
28. Butler MP, Hanly JA, Moynagh PN. Kinase-active interleukin-1 receptor-associated kinases promote polyubiquitination and degradation of the pellino family: direct evidence for PELLINO proteins being ubiquitin-protein isopeptide ligases. J Biol Chem. (2007) 282:29729–37. doi: 10.1074/jbc.M704558200
29. Cohen P, Strickson S. The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death Differ. (2017) 24:1153–59. doi: 10.1038/cdd.2017.17
30. Yao J, Kim TW, Qin J, Jiang Z, Qian Y, Xiao H, et al. Interleukin-1 (IL-1)-induced TAK1-dependent Versus MEKK3-dependent NF-κB activation pathways bifurcate at IL-1 receptor-associated kinase modification. J Biol Chem. (2007) 282:6075–89. doi: 10.1074/jbc.M609039200
31. Scarneo SA, Hughes PF, Yang KW, Carlson DA, Gurbani D, Westover KD, et al. A highly selective inhibitor of interleukin-1 receptor-associated kinases 1/4 (IRAK-1/4) delineates the distinct signaling roles of IRAK-1/4 and the TAK1 kinase. J Biol Chem. (2020) 295:1565–74. doi: 10.1074/jbc.RA119.011857
32. Hayden MS, Ghosh S. Innate sense of purpose for IKKβ. Proc Natl Acad Sci USA. (2014) 111:17348–9. doi: 10.1073/pnas.1419689111
33. Lopez-Pelaez M, Lamont DJ, Peggie M, Shpiro N, Gray NS, Cohen P. Protein kinase IKKβ-catalyzed phosphorylation of IRF5 at Ser462 induces its dimerization and nuclear translocation in myeloid cells. Proc Natl Acad Sci USA. (2014) 111:17432–7. doi: 10.1073/pnas.1418399111
34. Bergstrøm B, Aune MH, Awuh JA, Kojen JF, Blix KJ, Ryan L, et al. TLR8 senses Staphylococcus aureus RNA in human primary monocytes and macrophages and induces IFN-β Production via a TAK1-IKKβ-IRF5 signaling pathway. J Immunol. (2015) 195:1100–11. doi: 10.4049/jimmunol.1403176
35. Cushing L, Winkler A, Jelinsky SA, Lee K, Korver W, Hawtin R, et al. IRAK4 kinase activity controls toll-like receptor-induced inflammation through the transcription factor IRF5 in primary human monocytes. J Biol Chem. (2017) 292:18689–98. doi: 10.1074/jbc.M117.796912
36. Yamazaki K, Gohda J, Kanayama A, Miyamoto Y, Sakurai H, Yamamoto M, et al. Two mechanistically and temporally distinct NF-κB activation pathways in IL-1 signaling. Sci Signal. (2009) 2:ra66. doi: 10.1126/scisignal.2000387
37. Kim S, Jin Y, Choi Y, Park T. Resveratrol exerts anti-obesity effects via mechanisms involving down-regulation of adipogenic and inflammatory processes in mice. Biochem Pharmacol. (2011) 81:1343–51. doi: 10.1016/j.bcp.2011.03.012
38. Ouyang X, Negishi H, Takeda R, Fujita Y, Taniguchi T, Honda K. Cooperation between MyD88 and TRIF pathways in TLR synergy via IRF5activation. Biochem Biophys Res Commun. (2007) 354:1045–51. doi: 10.1016/j.bbrc.2007.01.090
39. Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF-κB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-β. Proc Natl Acad Sci USA. (2004) 101:3533–8. doi: 10.1073/pnas.0308496101
40. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. (2003) 301:640–3. doi: 10.1126/science.1087262
41. Al-Rashed F, Sindhu S, Arefanian H, Al Madhoun A, Kochumon S, Thomas R, et al. Repetitive intermittent hyperglycemia drives the M1 Polarization and inflammatory responses in THP-1 macrophages through the mechanism involving the TLR4-IRF5 Pathway. Cells. (2020) 9:1892. doi: 10.3390/cells9081892
42. Csumita M, Csermely A, Horvath A, Nagy G, Monori F, Göczi L, et al. Specific enhancer selection by IRF3, IRF5 and IRF9 is determined by ISRE half-sites, 5' and 3' flanking bases, collaborating transcription factors and the chromatin environment in a combinatorial fashion. Nucleic Acids Res. (2020) 48:589–604. doi: 10.1093/nar/gkz1112
43. Chen W, Royer WE Jr. Structural insights into interferon regulatory factor activation. Cell Signal. (2010) 22:883–7. doi: 10.1016/j.cellsig.2009.12.005
44. Hubbard LL, Moore BB. IRAK-M regulation and function in host defense and immune homeostasis. Infect Dis Rep. (2010) 2:e9. doi: 10.4081/idr.2010.e9
45. Kobayashi K, Hernandez LD, Galán JE, Janeway CA Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of toll-like receptor signalling. Cell. (2002) 110:191–202. doi: 10.1016/S0092-8674(02)00827-9
46. Zhou H, Yu M, Fukuda K, Im J, Yao P, Cui W, et al. IRAK-M mediates toll-like receptor/IL-1R-induced NF-κB activation and cytokine production. EMBO J. (2013) 32:583–96. doi: 10.1038/emboj.2013.2
47. Ban T, Sato GR, Nishiyama A, Akiyama A, Takasuna M, Umehara M, et al. Lyn kinase suppresses the transcriptional activity of IRF5 in the TLR-MyD88 pathway to restrain the development of autoimmunity. Immunity. (2016) 45:319–32. doi: 10.1016/j.immuni.2016.07.015
48. Steinhagen F, Rodriguez LG, Tross D, Tewary P, Bode C, Klinman DM. IRF5 and IRF8 modulate the CAL-1 human plasmacytoid dendritic cell line response following TLR9 ligation. Eur J Immunol. (2016) 46:647–55. doi: 10.1002/eji.201545911
49. Kaur A, Lee LH, Chow SC, Fang CM. IRF5-mediated immune responses and its implications in immunological disorders. Int Rev Immunol. (2018) 37:229–48. doi: 10.1080/08830185.2018.1469629
50. Eames HL, Saliba DG, Krausgruber T, Lanfrancotti A, Ryzhakov G, Udalova IA. KAP1/TRIM28: an inhibitor of IRF5 function in inflammatory macrophages. Immunobiology. (2012) 217:1315–24. doi: 10.1016/j.imbio.2012.07.026
51. Heinz LX, Lee J, Kapoor U, Kartnig F, Sedlyarov V, Papakostas K, et al. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7-9. Nature. (2020) 581:316–22. doi: 10.1038/s41586-020-2282-0
52. Andrilenas KK, Ramlall V, Kurland J, Leung B, Harbaugh AG, Siggers T. DNA-binding landscape of IRF3, IRF5 and IRF7 dimers: implications for dimer-specific gene regulation. Nucleic Acids Res. (2018) 46:2509–20. doi: 10.1093/nar/gky002
53. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. (2011) 12:231–8. doi: 10.1038/ni.1990
54. Inoue M, Arikawa T, Chen YH, Moriwaki Y, Price M, Brown M, et al. T cells down-regulate macrophage TNF production by IRAK1-mediated IL-10 expression and control innate hyperinflammation. Proc Natl Acad Sci USA. (2014) 111:5295–300. doi: 10.1073/pnas.1321427111
55. Matta B, Barnes BJ. Coordination between innate immune cells, type I IFNs and IRF5 drives SLE pathogenesis. Cytokine. (2020) 132:154731. doi: 10.1016/j.cyto.2019.05.018
56. Barnes BJ, Field AE, Pitha-Rowe PM. Virus-induced heterodimer formation between IRF-5 and IRF-7 modulates assembly of the IFNA enhanceosome in vivo and transcriptional activity of IFNA genes. J Biol Chem. (2003) 278:16630–41. doi: 10.1074/jbc.M212609200
57. Mancl ME, Hu G, Sangster-Guity N, Olshalsky SL, Hoops K, Fitzgerald-Bocarsly P, et al. Two discrete promoters regulate the alternatively spliced human interferon regulatory factor-5 isoforms. Multiple isoforms with distinct cell type-specific expression, localization, regulation, and function. J Biol Chem. (2005) 280:21078–90. doi: 10.1074/jbc.M500543200
58. Steinhagen F, McFarland AP, Rodriguez LG, Tewary P, Jarret A, Savan R, et al. IRF-5 and NF-κB p50 co-regulate IFN-β and IL-6 expression in TLR9-stimulated human plasmacytoid dendritic cells. Eur J Immunol. (2013) 43:1896–906. doi: 10.1002/eji.201242792
59. Stein T, Wollschlegel A, Te H, Weiss J, Joshi K, Kinzel B, et al. Interferon regulatory factor 5 and nuclear factor kappa-B exhibit cooperating but also divergent roles in the regulation of pro-inflammatory cytokines important for the development of TH1 and TH17 responses. FEBS J. (2018) 285:3097–113. doi: 10.1111/febs.14600
60. Saliba DG, Heger A, Eames HL, Oikonomopoulos S, Teixeira A, Blazek K, et al. IRF5:RelA interaction targets inflammatory genes in macrophages. Cell Rep. (2014) 8:1308–17. doi: 10.1016/j.celrep.2014.07.034
61. Chow KT, Wilkins C, Narita M, Green R, Knoll M, Loo YM, et al. Differential and overlapping immune programs regulated by IRF3 and IRF5 in plasmacytoid dendritic cells. J Immunol. (2018) 201:3036–50. doi: 10.4049/jimmunol.1800221
62. Balkhi MY, Fitzgerald KA, Pitha PM. IKKα negatively regulates IRF-5 function in a MyD88-TRAF6 pathway. Cell Signal. (2010) 22:117–27. doi: 10.1016/j.cellsig.2009.09.021
63. Pelka K, Latz E. IRF5, IRF8, and IRF7 in human pDCs - the good, the bad, and the insignificant? Eur J Immunol. (2013) 43:1693–7. doi: 10.1002/eji.201343739
64. Schneider A, Weier M, Herderschee J, Perreau M, Calandra T, Roger T, et al. IRF5 is a key regulator of macrophage response to lipopolysaccharide in newborns. Front Immunol. (2018) 9:1597. doi: 10.3389/fimmu.2018.01597
65. Xiong Y, Qiu F, Piao W, Song C, Wahl LM, Medvedev AE. Endotoxin tolerance impairs IL-1 receptor-associated kinase (IRAK) 4 and TGF-beta-activated kinase 1 activation, K63-linked polyubiquitination and assembly of IRAK1, TNF receptor-associated factor 6, and IκB kinase gamma and increases A20 expression. J Biol Chem. (2011) 286:7905–16. doi: 10.1074/jbc.M110.182873
66. Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, et al. Severe impairment of interleukin-1 and toll-like receptor signalling in mice lacking IRAK-4. Nature. (2002) 416:750–6. doi: 10.1038/nature736
67. Suzuki N, Suzuki S, Eriksson U, Hara H, Mirtosis C, Chen NJ, et al. IL-1R-associated kinase 4 is required for lipopolysaccharide-induced activation of APC. J Immunol. (2003) 171:6065–71. doi: 10.4049/jimmunol.171.11.6065
68. Park SH, Baek SI, Yun J, Lee S, Yoon DY, Jung JK, et al. IRAK4 as a molecular target in the amelioration of innate immunity-related endotoxic shock and acute liver injury by chlorogenic acid. J Immunol. (2015) 194:1122–30. doi: 10.4049/jimmunol.1402101
69. Liu T, Xiang A, Peng T, Doran AC, Tracey KJ, Barnes BJ, et al. HMGB1-C1q complexes regulate macrophage function by switching between leukotriene and specialized proresolving mediator biosynthesis. Proc Natl Acad Sci USA. (2019) 116:23254–263. doi: 10.1073/pnas.1907490116
70. Chen X, Zhou L, Peng N, Yu H, Li M, Cao Z, et al. MicroRNA-302a suppresses influenza A virus-stimulated interferon regulatory factor-5 expression and cytokine storm induction. J Biol Chem. (2017) 292:21291–303. doi: 10.1074/jbc.M117.805937
71. Hedl M, Yan J, Witt H, Abraham C. IRF5 is required for bacterial clearance in human M1-polarized macrophages, and IRF5 immune-mediated disease risk variants modulate this outcome. J Immunol. (2019) 202:920–30. doi: 10.4049/jimmunol.1800226
72. Wang X, Guo J, Wang Y, Xiao Y, Wang L, Hua S. Expression levels of interferon regulatory factor 5 (IRF5) and related inflammatory cytokines associated with severity, prognosis, and causative pathogen in patients with community-acquired pneumonia. Med Sci Monit. (2018) 24:3620–30. doi: 10.12659/MSM.910756
73. De Biasi S, Meschiari M, Gibellini L, Bellinazzi C, Borella R, Fidanza L, et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat Commun. (2020) 11:3434. doi: 10.1038/s41467-020-17292-4
74. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. (2020) 181:1036–45.e9. doi: 10.1016/j.cell.2020.04.026
75. Honda K, Taniguchi T. IRFs: master regulators of signalling by toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. (2006) 6:644–58. doi: 10.1038/nri1900
76. Andreakos E, Tsiodras S. COVID-19: lambda interferon against viral load and hyperinflammation. EMBO Mol Med. (2020) 12:e12465. doi: 10.15252/emmm.202012465
77. Yin X, Riva L, Pu Y, Martin-Sancho L, Kanamune J, Yamamoto Y, et al. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep. (2021) 34:108628. doi: 10.1016/j.celrep.2020.108628
78. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. (2003) 4:491–6. doi: 10.1038/ni921
79. Chen N, Xia P, Li S, Zhang T, Wang TT, Zhu J. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life. (2017) 69:297–304. doi: 10.1002/iub.1625
80. Diaz-Salazar C, Sun JC. Natural killer cell responses to emerging viruses of zoonotic origin. Curr Opin Virol. (2020) 44:97–111. doi: 10.1016/j.coviro.2020.07.003
81. Jewett A. The potential effect of novel coronavirus SARS-CoV-2 on NK Cells; a perspective on potential therapeutic interventions. Front Immunol. (2020) 11:1692. doi: 10.3389/fimmu.2020.01692
82. Yan J, Hedl M, Abraham C. Myeloid cell-Intrinsic IRF5 promotes T cell responses through multiple distinct checkpoints in vivo, and IRF5 immune-mediated disease risk variants modulate these myeloid cell functions. J Immunol. (2020) 205:1024–38. doi: 10.4049/jimmunol.1900743
83. El Mezayen R, El Gazzar M, Myer R, High KP. Aging-dependent upregulation of IL-23p19 gene expression in dendritic cells is associated with differential transcription factor binding and histone modifications. Aging Cell. (2009) 8:553–65. doi: 10.1111/j.1474-9726.2009.00502.x
84. Oriss TB, Raundhal M, Morse C, Huff RE, Das S, Hannum R, et al. IRF5 distinguishes severe asthma in humans and drives Th1 phenotype and airway hyperreactivity in mice. JCI Insight. (2017) 2:e91019. doi: 10.1172/jci.insight.91019
85. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. (2005) 434:772–7. doi: 10.1038/nature03464
86. Yasuda K, Richez C, Maciaszek JW, Agrawal N, Akira S, Marshak-Rothstein A, et al. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. J Immunol. (2007) 178:6876–85. doi: 10.4049/jimmunol.178.11.6876
87. Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. (2013) 9:e1003118. doi: 10.1371/journal.ppat.1003118
88. Barnes BJ, Moore PA, Pitha PM. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon α genes. J Biol Chem. (2001) 276:23382–90. doi: 10.1074/jbc.M101216200
89. Zheng HY, Zhang M, Yang CX, Zhang N, Wang XC, Yang XP, et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell Mol Immunol. (2020) 17:541–3. doi: 10.1038/s41423-020-0401-3
90. Prakash S, Agrawal S, Cao JN, Gupta S, Agrawal A. Impaired secretion of interferons by dendritic cells from aged subjects to influenza: role of histone modifications. Age. (2013) 35:1785–97. doi: 10.1007/s11357-012-9477-8
91. Borges RC, Hohmann MS, Borghi SM. Dendritic cells in COVID-19 immunopathogenesis: insights for a possible role in determining disease outcome. Int Rev Immunol. (2020) 1–18. doi: 10.1080/08830185.2020.1844195
92. Griesbeck M, Ziegler S, Laffont S, Smith N, Chauveau L, Tomezsko P, et al. Sex differences in plasmacytoid dendritic cell levels of IRF5 drive higher IFN-alpha production in women. J Immunol. (2015) 195:5327–36. doi: 10.4049/jimmunol.1501684
93. Klein SL, Dhakal S, Ursin RL, Deshpande S, Sandberg K, Mauvais-Jarvis F. Biological sex impacts COVID-19 outcomes. PLoS Pathog. (2020) 16:e1008570. doi: 10.1371/journal.ppat.1008570
94. Gratz N, Hartweger H, Matt U, Kratochvill F, Janos M, Sigel S, et al. Type I interferon production induced by Streptococcus pyogenes-derived nucleic acids is required for host protection. PLoS Pathog. (2011) 7:e1001345. doi: 10.1371/journal.ppat.1001345
95. Agrawal A, Sridharan A, Prakash S, Agrawal H. Dendritic cells and aging: consequences for autoimmunity. Expert Rev Clin Immunol. (2012) 8:73–80. doi: 10.1586/eci.11.77
96. Zhao Q, Meng M, Kumar R, Wu Y, Huang J, Deng Y, et al. Lymphopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a systemic review and meta-analysis. Int J Infect Dis. (2020) 96:131–5. doi: 10.1016/j.ijid.2020.04.086
97. Omarjee L, Janin A, Perrot F, Laviolle B, Meilhac O, Mahe G. Targeting T-cell senescence and cytokine storm with rapamycin to prevent severe progression in COVID-19. Clin Immunol. (2020) 216:108464. doi: 10.1016/j.clim.2020.108464
98. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. (2020) 11:827. doi: 10.3389/fimmu.2020.00827
99. Bouadma L, Wiedemann A, Patrier J, Surénaud M, Wicky PH, Foucat E, et al. Immune alterations in a patient with SARS-CoV-2-related acute respiratory distress syndrome. J Clin Immunol. (2020) 40:1082–92. doi: 10.1007/s10875-020-00839-x
100. Angioni R, Sánchez-Rodríguez R, Munari F, Bertoldi N, Arcidiacono D, Cavinato S, et al. Age-severity matched cytokine profiling reveals specific signatures in Covid-19 patients. Cell Death Dis. (2020) 11:957. doi: 10.1038/s41419-020-03151-z
101. Lien C, Fang CM, Huso D, Livak F, Lu R, Pitha PM. Critical role of IRF-5 in regulation of B-cell differentiation. Proc Natl Acad Sci USA. (2010) 107:4664–8. doi: 10.1073/pnas.0911193107
102. De Biasi S, Lo Tartaro D, Meschiari M, Gibellini L, Bellinazzi C, Borella R, et al. Expansion of plasmablasts and loss of memory B cells in peripheral blood from COVID-19 patients with pneumonia. Eur J Immunol. (2020) 50:1283–94. doi: 10.1002/eji.202048838
103. Banerjee M, Huang Y, Joshi S, Popa GJ, Mendenhall MD, Wang QJ, et al. Platelets endocytose viral particles and are activated via TLR (toll-like receptor) signaling. Arterioscler Thromb Vasc Biol. (2020) 40:1635–50. doi: 10.1161/ATVBAHA.120.314180
104. Liu Y, Gao W, Guo W, Guo Y, Shi M, Dong G, et al. Prominent coagulation disorder is closely related to inflammatory response and could be as a prognostic indicator for ICU patients with COVID-19. J Thromb Thrombolysis. (2020) 50:825–32. doi: 10.1007/s11239-020-02174-9
105. Laviada-Molina HA, Leal-Berumen I, Rodriguez-Ayala E, Bastarrachea RA. Working hypothesis for glucose metabolism and SARS-CoV-2 replication: interplay between the hexosamine pathway and interferon RF5 triggering hyperinflammation. Role of BCG vaccine? Front Endocrinol. (2020) 11:514. doi: 10.3389/fendo.2020.00514
106. Kim D, Lee H, Koh J, Ko JS, Yoon BR, Jeon YK, et al. Cytosolic Pellino-1-mediated K63-linked ubiquitination of IRF5 in M1 macrophages regulates glucose intolerance in obesity. Cell Rep. (2017) 20:832–45. doi: 10.1016/j.celrep.2017.06.088
107. Sindhu S, Thomas R, Kochumon S, Wilson A, Abu-Farha M, Bennakhi A, et al. Increased adipose tissue expression of interferon regulatory factor (IRF)-5 in obesity: association with metabolic inflammation. Cells. (2019) 8:1418. doi: 10.3390/cells8111418
108. Sindhu S, Kochumon S, Thomas R, Bennakhi A, Al-Mulla F, Ahmad R. Enhanced adipose expression of interferon regulatory factor (IRF)-5 associates with the signatures of metabolic inflammation in diabetic obese patients. Cells. (2020) 9:730. doi: 10.3390/cells9030730
109. Kochumon S, Madhoun AA, Al-Rashed F, Azim R, Al-Ozairi E, Al-Mulla F, et al. Adipose tissue gene expression of CXCL10 and CXCL11 modulates inflammatory markers in obesity: implications for metabolic inflammation and insulin resistance. Ther Adv Endocrinol Metab. (2020) 11:2042018820930902. doi: 10.1177/2042018820930902
110. Kochumon S, Al Madhoun A, Al-Rashed F, Thomas R, Sindhu S, Al-Ozairi E, et al. Elevated adipose tissue associated IL-2 expression in obesity correlates with metabolic inflammation and insulin resistance. Sci Rep. (2020) 10:16364. doi: 10.1038/s41598-020-73347-y
111. Kim D, Koh J, Ko JS, Kim HY, Lee H, Chung DH. Ubiquitin E3 ligase pellino-1 inhibits IL-10-mediated M2c polarization of macrophages, thereby suppressing tumor growth. Immune Netw. (2019) 19:e32. doi: 10.4110/in.2019.19.e32
112. Marsh EK, Prestwich EC, Williams L, Hart AR, Muir CF, Parker LC, et al. Pellino-1 regulates the responses of the airway to viral infection. Front Cell Infect Microbiol. (2020) 10:456. doi: 10.3389/fcimb.2020.00456
113. Pangrazzi L, Naismith E, Miggitsch C, Carmona Arana JA, Keller M, Grubeck-Loebenstein B, et al. The impact of body mass index on adaptive immune cells in the human bone marrow. Immun Ageing. (2020) 17:15. doi: 10.1186/s12979-020-00186-w
114. Zheng Y, Liu X, Le W, Xie L, Li H, Wen W, et al. A human circulating immune cell landscape in aging and COVID-19. Protein Cell. (2020) 11:740–70. doi: 10.1007/s13238-020-00762-2
115. Bossù P, Toppi E, Sterbini V, Spalletta G. Implication of aging related chronic neuroinflammation on COVID-19 pandemic. J Pers Med. (2020) 10:102. doi: 10.3390/jpm10030102
116. Mauvais-Jarvis F. Aging, male sex, obesity, and metabolic inflammation create the perfect storm for COVID-19. Diabetes. (2020) 69:1857–63. doi: 10.2337/dbi19-0023
117. Chau AS, Weber AG, Maria NI, Narain S, Liu A, Hajizadeh N, et al. The longitudinal immune response to Coronavirus Disease 2019: chasing the cytokine storm. Arthritis Rheumatol. (2020) 73:23-35. doi: 10.1002/art.41526
118. Stoy NS. Innate origins of multiple sclerosis pathogenesis: implications for computer-assisted design of disease-modifying therapy. Drug Dev Res. (2011) 72:674–88. doi: 10.1002/ddr.20477
119. Lamagna C, Hu Y, DeFranco AL, Lowell CA. B cell-specific loss of lyn kinase leads to autoimmunity. J Immunol. (2014) 192:919–28. doi: 10.4049/jimmunol.1301979
120. Blot M, Jacquier M, Aho Glele LS, Beltramo G, Nguyen M, Bonniaud P, et al. CXCL10 could drive longer duration of mechanical ventilation during COVID-19 ARDS. Crit Care. (2020) 24:632. doi: 10.1186/s13054-020-03328-0
121. Chen Y, Wang J, Liu C, Su L, Zhang D, Fan J, et al. IP-10 and MCP-1 as biomarkers associated with disease severity of COVID-19. Mol Med. (2020) 26:97. doi: 10.1186/s10020-020-00230-x
122. Danto SI, Shojaee N, Singh RSP, Li C, Gilbert SA, Manukyan Z, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of PF-06650833, a selective interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitor, in single and multiple ascending dose randomized phase 1 studies in healthy subjects. Arthritis Res Ther. (2019) 21:269. doi: 10.1186/s13075-019-2008-6
123. Banga J, Srinivasan D, Sun CC, Thompson CD, Milletti F, Huang KS, et al. Inhibition of IRF5 cellular activity with cell-penetrating peptides that target homodimerization. Sci Adv. (2020) 6:eaay1057. doi: 10.1126/sciadv.aay1057
124. Weiss M, Blazek K, Byrne AJ, Perocheau DP, Udalova IA. IRF5 is a specific marker of inflammatory macrophages in vivo. Mediators Inflamm. (2013) 2013:245804. doi: 10.1155/2013/245804
125. Yang Y, Zhang C, Jing D, He H, Li X, Wang Y, et al. IRF5 acts as a potential therapeutic marker in inflammatory bowel diseases. Inflamm Bowel Dis. (2021) 27:407–17. doi: 10.1093/ibd/izaa200
126. Schett G, Sticherling M, Neurath MF. COVID-19: risk for cytokine targeting in chronic inflammatory diseases? Nat Rev Immunol. (2020) 20:271–2. doi: 10.1038/s41577-020-0312-7
127. Ward M, Gooderham M. Asymptomatic SARS-CoV2 infection in a patient receiving risankizumab, an inhibitor of IL-23. JAAD Case Rep. (2020) 7:60–1. doi: 10.1016/j.jdcr.2020.10.032
128. Files JK, Boppana S, Perez MD, Sarkar S, Lowman KE, Qin K, et al. Sustained cellular immune dysregulation in individuals recovering from SARS-CoV-2 infection. J Clin Invest. (2020) 29:140491. doi: 10.1101/2020.07.30.20165175
129. Fabié A, Mai LT, Dagenais-Lussier X, Hammami A, van Grevenynghe J, Stäger S. IRF-5 promotes cell death in CD4 T cells during chronic infection. Cell Rep. (2018) 24:1163–75. doi: 10.1016/j.celrep.2018.06.107
130. Kow CS, Hasan SS. Dexamethasone or hydrocortisone in COVID-19? Cleve Clin J Med. (2020) 87:715. doi: 10.3949/ccjm.87c.12005
131. van de Veerdonk FL, Netea MG. Blocking IL-1 to prevent respiratory failure in COVID-19. Crit Care. (2020) 24:445. doi: 10.1186/s13054-020-03166-0
132. Mehta P, Cron RQ, Hartwell J, Manson JJ, Tattersall RS. Silencing the cytokine storm: the use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. (2020) 2:e358–67. doi: 10.1016/S2665-9913(20)30096-5
133. Wang M, Liao Z. SARS-CoV-2 and COVID-19: how much do we know? Acta Virol. (2020) 64:288–96. doi: 10.4149/av_2020_301
134. Dudhgaonkar S, Ranade S, Nagar J, Subramani S, Prasad DS, Karunanithi P, et al. Selective IRAK4 inhibition attenuates disease in murine lupus models and demonstrates steroid sparing activity. J Immunol. (2017) 198:1308–19. doi: 10.4049/jimmunol.1600583
135. Horby PW, Campbell M, Staplin N, Spata E, Emberson JR, Pessoa-Amorim G, et al. Tocilizumab in patients admitted to hospital with covid-19 (Recovery): preliminary results of a randomised, controlled, open-label, platform trial. medRxiv [Preprint]. (2021) 1–30. doi: 10.1101/2021.02.11.21249258
136. Liu B, Li M, Zhou Z, Guan X, Xiang Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun. (2020) 111:102452. doi: 10.1016/j.jaut.2020.102452
137. Chen JJ, Zhang LN, Hou H, Xu L, Ji K. Interleukin-6 signaling blockade treatment for cytokine release syndrome in COVID-19 (review). Exp Ther Med. (2021) 21:24. doi: 10.3892/etm.2020.9456
138. Park JH, Lee HK. Re-analysis of single cell transcriptome reveals that the NR3C1-CXCL8-neutrophil axis determines the severity of COVID-19. Front Immunol. (2020) 11:2145. doi: 10.3389/fimmu.2020.02145
139. Liang Y, Li H, Li J, Yang ZN, Li JL, Zheng HW, et al. Role of neutrophil chemoattractant CXCL5 in SARS-CoV-2 infection-induced lung inflammatory innate immune response in an in vivo hACE2 transfection mouse model. Zool Res. (2020) 41:621–31. doi: 10.24272/j.issn.2095-8137.2020.118
140. Hjorton K, Hagberg N, Israelsson E, Jinton L, Berggren O, Sandling JK, et al. Cytokine production by activated plasmacytoid dendritic cells and natural killer cells is suppressed by an IRAK4 inhibitor. Arthritis Res Ther. (2018) 20:238. doi: 10.1186/s13075-018-1702-0
141. Huang YH, Wang PW, Tiao MM, Chou MH, Du YY, Huang CC, et al. Glucocorticoid modulates high-mobility group box 1 expression and toll-like receptor activation in obstructive jaundice. J Surg Res. (2011) 170:e47–55. doi: 10.1016/j.jss.2011.05.033
142. Wiese MD, Manning-Bennett AT, Abuhelwa AY. Investigational IRAK-4 inhibitors for the treatment of rheumatoid arthritis. Expert Opin Investig Drugs. (2020) 29:475–82. doi: 10.1080/13543784.2020.1752660
143. Zaro BW, Vinogradova EV, Lazar DC, Blewett MM, Suciu RM, Takaya J, et al. Dimethyl fumarate disrupts human innate immune signaling by targeting the IRAK4-MyD88 complex. J Immunol. (2019) 202:2737–46. doi: 10.4049/jimmunol.1801627
144. Timpani CA, Rybalka E. Calming the (cytokine) storm: dimethyl fumarate as a therapeutic candidate for COVID-19. Pharmaceuticals. (2020) 14:15. doi: 10.3390/ph14010015
145. Ban T, Sato GR, Tamura T. Regulation and role of the transcription factor IRF5 in innate immune responses and systemic lupus erythematosus. Int Immunol. (2018) 30:529–36. doi: 10.1093/intimm/dxy032
146. Eames HL, Corbin AL, Udalova IA. Interferon regulatory factor 5 in human autoimmunity and murine models of autoimmune disease. Transl Res. (2016) 167:167–82. doi: 10.1016/j.trsl.2015.06.018
147. Kim B, Yang Q, Chan LW, Bhatia SN, Ruoslahti E, Sailor MJ. Fusogenic porous silicon nanoparticles as a broad-spectrum immunotherapy against bacterial infections. Nanoscale Horiz. (2021). doi: 10.1039/D0NH00624F. [Epub ahead of print].
148. McHugh J. IRF5 inhibitor shows promise in mouse models of SLE. Nat Rev Rheumatol. (2020) 16:667. doi: 10.1038/s41584-020-00525-7
149. Kumar J, Qureshi R, Sagurthi SR, Qureshi IA. Designing of nucleocapsid protein based novel multi-epitope vaccine against SARS-COV-2 using immunoinformatics approach. Int J Pept Res Ther. (2020) 1–16. doi: 10.1007/s10989-020-10140-5. [Epub ahead of print].
150. Reed SG, Tomai M, Gale MJ Jr. New horizons in adjuvants for vaccine development. Curr Opin Immunol. (2020) 65:97–101. doi: 10.1016/j.coi.2020.08.008
151. Liang Z, Zhu H, Wang X, Jing B, Li Z, Xia X, et al. Adjuvants for coronavirus vaccines. Front Immunol. (2020)11:589833. doi: 10.3389/fimmu.2020.589833
152. Kayraklioglu N, Horuluoglu B, Klinman DM. CpG oligonucleotides as vaccine adjuvants. Methods Mol Biol. (2021) 2197:51–85. doi: 10.1007/978-1-0716-0872-2_4
153. Dowling DJ. Recent advances in the discovery and delivery of TLR7/8 agonists as vaccine adjuvants. Immunohorizons. (2018) 2:185–97. doi: 10.4049/immunohorizons.1700063
154. Al-Motawa MS, Abbas H, Wijten P, de la Fuente A, Xue M, Rabbani N, et al. Vulnerabilities of the SARS-CoV-2 virus to proteotoxicity - opportunity for repurposed chemotherapy of COVID-19 infection. Front Pharmacol. (2020) 11:585408. doi: 10.3389/fphar.2020.585408
155. Gao Y, Wang C, Kang K, Peng Y, Luo Y, Liu H, et al. Cytokine storm may not be the chief culprit for the deterioration of COVID-19. Viral Immunol. (2020). doi: 10.1089/vim.2020.0243. [Epub ahead of print].
156. Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El Jammal T, et al. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun Rev. (2020) 19:102567. doi: 10.1016/j.autrev.2020.102567
157. Zhang JY, Wang XM, Xing X, Xu Z, Zhang C, Song JW, et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat Immunol. (2020) 21:1107–18. doi: 10.1038/s41590-020-0762-x
158. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
159. Xu G, Qi F, Li H, Yang Q, Wang H, Wang X, et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov. (2020) 6:73. doi: 10.1038/s41421-020-00225-2
160. Yan L, Cai B, Li Y, Wang MJ, An YF, Deng R, et al. Dynamics of NK, CD8 and Tfh cell mediated the production of cytokines and antiviral antibodies in Chinese patients with moderate COVID-19. J Cell Mol Med. (2020). 24:14270–9. doi: 10.1111/jcmm.16044
161. Meckiff BJ, Ramírez-Suástegui C, Fajardo V, Chee SJ, Kusnadi A, Simon H, et al. Imbalance of regulatory and cytotoxic SARS-CoV-2-reactive CD4(+) T cells in COVID-19. Cell. (2020) 183:1340–53.e16. doi: 10.1016/j.cell.2020.10.001
162. Duan YQ, Xia MH, Ren L, Zhang YF, Ao QL, Xu SP, et al. Deficiency of Tfh cells and germinal center in deceased COVID-19 patients. Curr Med Sci. (2020) 40:618–24. doi: 10.1007/s11596-020-2225-x
163. Kaneko N, Kuo HH, Boucau J, Farmer JR, Allard-Chamard H, Mahajan VS, et al. Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell. (2020) 183:143–57.e13. doi: 10.1016/j.cell.2020.08.025
164. Song S, De S, Nelson V, Chopra S, LaPan M, Kampta K, et al. Inhibition of IRF5 hyperactivation protects from lupus onset and severity. J Clin Invest. (2020) 130:6700–17. doi: 10.1172/JCI120288
165. Yan J, Pandey SP, Barnes BJ, Turner JR, Abraham C. T Cell-intrinsic IRF5 regulates T cell signaling, migration, and differentiation and promotes intestinal inflammation. Cell Rep. (2020) 31:107820. doi: 10.1016/j.celrep.2020.107820
166. Brune Z, Rice MR, Barnes BJ. Potential T cell-Intrinsic regulatory roles for IRF5 via cytokine modulation in T helper subset differentiation and function. Front Immunol. (2020) 11:1143. doi: 10.3389/fimmu.2020.01143
167. Abers MS, Delmonte OM, Ricotta EE, Fintzi J, Fink DL, de Jesus AAA, et al. An immune-based biomarker signature is associated with mortality in COVID-19 patients. JCI Insight. (2021) 6:e144455. doi: 10.1172/jci.insight.144455
168. Balnis J, Adam AP, Chopra A, Chieng HC, Drake LA, Martino N, et al. Unique inflammatory profile is associated with higher SARS-CoV-2 acute respiratory distress syndrome (ARDS) mortality. Am J Physiol Regul Integr Comp Physiol. (2021) 320:R250–7. doi: 10.1101/2020.05.21.20051300
169. Chan JY, Lee K, Maxwell EL, Liang C, Laybutt DR. Macrophage alterations in islets of obese mice linked to beta cell disruption in diabetes. Diabetologia. (2019) 62:993–9. doi: 10.1007/s00125-019-4844-y
170. Azimi N, Shiramizu KM, Tagaya Y, Mariner J, Waldmann TA. Viral activation of interleukin-15 (IL-15): characterization of a virus-inducible element in the IL-15 promoter region. J Virol. (2000) 74:7338–48. doi: 10.1128/JVI.74.16.7338-7348.2000
171. Colpitts SL, Stoklasek TA, Plumlee CR, Obar JJ, Guo C, Lefrançois L. Cutting edge: the role of IFN-alpha receptor and MyD88 signaling in induction of IL-15 expression in vivo. J Immunol. (2012) 188:2483–7. doi: 10.4049/jimmunol.1103609
172. Yan W, Fan W, Chen C, Wu Y, Fan Z, Chen J, et al. IL-15 up-regulates the MMP-9 expression levels and induces inflammatory infiltration of macrophages in polymyositis through regulating the NF-κB pathway. Gene. (2016) 591:137–47. doi: 10.1016/j.gene.2016.06.055
173. Rosário C, Zandman-Goddard G, Meyron-Holtz EG, D'Cruz DP, Shoenfeld Y. The hyperferritinemic syndrome: macrophage activation syndrome, Still'sdisease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. (2013) 11:185. doi: 10.1186/1741-7015-11-185
174. Ramezani F, Babaie F, Aslani S, Hemmatzadeh M, Mohammadi FS, Gowhari-Shabgah A, et al. The role of the IL-33/ST2 immune pathway in autoimmunity: new insights and perspectives. Immunol Invest. (2021) 1–27. doi: 10.1080/08820139.2021.1878212. [Epub ahead of print].
175. Weiss M, Byrne AJ, Blazek K, Saliba DG, Pease JE, Perocheau D, et al. IRF5 controls both acute and chronic inflammation. Proc Natl Acad Sci USA. (2015) 112:11001–6. doi: 10.1073/pnas.1506254112
176. Akiyama M, Zeisbrich M, Ibrahim N, Ohtsuki S, Berry GJ, Hwang PH, et al. Neutrophil extracellular traps induce tissue-invasive monocytes in granulomatosis with polyangiitis. Front Immunol. (2019) 10:2617. doi: 10.3389/fimmu.2019.02617
177. Corbin AL, Gomez-Vazquez M, Berthold DL, Attar M, Arnold IC, Powrie FM, et al. IRF5 guides monocytes toward an inflammatory CD11c(+) macrophage phenotype and promotes intestinal inflammation. Sci Immunol. (2020) 5:eaax6085. doi: 10.1126/sciimmunol.aax6085
178. Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I, et al. Viral infection and toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol. (2004) 34:796–805. doi: 10.1002/eji.200324610
179. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. (2020) 369:718–24. doi: 10.1126/science.abc6027
180. Laing AG, Lorenc A, Del Molino Del Barrio I, Das A, Fish M, Monin L, et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat Med. (2020) 26:1623–35. doi: 10.1038/s41591-020-1038-6
181. Boumaza A, Gay L, Mezouar S, Bestion E, Diallo AB, Michel M, et al. Monocytes and macrophages, targets of SARS-CoV-2: the clue for Covid-19 immunoparalysis. J Infect Dis. (2021) jiab044. doi: 10.1093/infdis/jiab044. [Epub ahead of print].
182. Wang C, Xie J, Zhao L, Fei X, Zhang H, Tan Y, et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine. (2020) 57:102833. doi: 10.1016/j.ebiom.2020.102833
183. Pradhan P, Toy R, Jhita N, Atalis A, Pandey B, Beach A, et al. TRAF6-IRF5 kinetics, TRIF, and biophysical factors drive synergistic innate responses to particle-mediated MPLA-CpG co-presentation. Sci Adv. (2021) 7:eabd4235. doi: 10.1126/sciadv.abd4235
Keywords: COVID-19, IRAK4, IRF5, M1 macrophages, cytokine storm, Pellino-1, innate immunity, adaptive immunity
Citation: Stoy N (2021) Involvement of Interleukin-1 Receptor-Associated Kinase 4 and Interferon Regulatory Factor 5 in the Immunopathogenesis of SARS-CoV-2 Infection: Implications for the Treatment of COVID-19. Front. Immunol. 12:638446. doi: 10.3389/fimmu.2021.638446
Received: 06 December 2020; Accepted: 24 February 2021;
Published: 07 April 2021.
Edited by:
Pei-Hui Wang, Shandong University, ChinaReviewed by:
Yi Zheng, Shandong University, ChinaQi Wang, Harbin Veterinary Research Institute (CAAAS), China
Copyright © 2021 Stoy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas Stoy, bmljaG9sYXMuc3RveUBkcGFnLm94LmFjLnVr