 Julianna Blagih
Julianna Blagih Marc Hennequart
Marc Hennequart Fabio Zani
Fabio Zani- The Francis Crick Institute, London, United Kingdom
Regulatory T cells (Tregs) are essential for mitigating inflammation. Tregs are found in nearly every tissue and play either beneficial or harmful roles in the host. The availability of various nutrients can either enhance or impair Treg function. Mitochondrial oxidative metabolism plays a major role in supporting Treg differentiation and fitness. While Tregs rely heavily on oxidation of fatty acids to support mitochondrial activity, they have found ways to adapt to different tissue types, such as tumors, to survive in competitive environments. In addition, metabolic by-products from commensal organisms in the gut also have a profound impact on Treg differentiation. In this review, we will focus on the core metabolic pathways engaged in Tregs, especially in the context of tissue nutrient environments, and how they can affect Treg function, stability and differentiation.
Introduction
The immune system is in a dynamic balance between inflammation and suppression. One critical population in helping maintain this balance are regulatory T cells (Treg), a population first identified in the mid-nineties (1). Humans bearing mutations in the forkhead box P3 (FOXP3) gene, which leads to lethal autoimmunity, helped identify FOXP3 as the signature transcription factor for Treg lineages (2). Tregs are derived from two different origins. Thymic Tregs (tTregs), also known as natural Tregs (nTregs), are selected and educated during thymic development and egress to secondary lymphoid organs (SLO). To further secure against uncontrolled inflammation, Tregs arising from naïve CD4 T cells in the SLO, termed inducible Tregs (iTregs), require cues from IL2 and TGFβ for differentiation (3).
The overarching function of Tregs is to maintain peripheral tolerance, such as in the colon which commensal bacteria live in harmony with the host. Other functions include preventing autoimmunity, limiting inflammation once the infection has been cleared and tissue repair, which all benefit the host (4). However, there are situations where Tregs become harmful, such as in tumors where they tip the balance towards tumor growth (5). There are several methods of suppression that modulate immune responses: cytokine mediated inhibition (i.e. IL10 and TGFβ), IL-2 deprivation, prevention of dendritic cell (DC) maturation, and cytolysis (6–8).
Treg development and suppressive programs depend on signalling downstream of the T cell receptor (TCR), CD28 and the IL2 receptor (CD25). These different inputs work either in tandem or in parallel to regulate FoxP3 expression. TCR engagement induces the activation of Class IA inositide lipid kinases, PI3Ks, to recruit PDK1 and Akt to the plasma membrane. Phosphorylation of Akt by PDK1 maintains suppressive function and response to TCR signal strength in Tregs (9). This signalling axis shapes Treg fate by regulating the FOXO family of transcription factors and the activation of the mammalian target of rapamycin complex I (mTORC1) (10, 11). STAT5, a transcription factor downstream of IL2 signalling, also determines Treg polarization, proliferation, stability and competitive fitness (12–15).
Many of these transduction pathways converge on cellular metabolism. Naïve T cells are in a state of quiescence and their metabolic demand is therefore low; however, once activated T cells upregulate anabolic and catabolic metabolism including glycolysis and amino acid synthesis (16–18). A large number of these anabolic pathways are reliant on adenosine triphosphate (ATP) which is mainly produced from glycolysis and mitochondrial oxidative phosphorylation (OXPHOS). Besides proliferation, defined metabolic changes are required to support specific T cell fates. For example, CD8+ effector T cells transitioning into central memory cells rewire their metabolism from glycolysis towards fatty acid oxidation (FAO) (19). Complex metabolic alterations also support the differentiation of CD4+ T cells towards different Thelper subsets. This topic has been recently reviewed by Shyer et al. and Geltink et al. and will not be the focus of this review (20, 21).
Treg metabolism is a growing area of research in the field of immunometabolism. In this review, we will focus on the metabolic features, metabolites, and metabolic modulators that influence Treg function, homeostasis, and plasticity. While lymphoid organs may perhaps be metabolically rich environments, Tregs extravasating to tissue sites face notably different nutrient availability depending on their location. How different metabolic environments affect Treg cell fate and biology will be further explored in this review.
Fatty Acids and Mitochondrial Metabolism in Tregs
One key organelle that shapes Treg cell fate and function is the mitochondrion. The metabolic pathways that support mitochondrial activity and cell energy production are the tricarboxylic acid cycle (TCA), amino acid and lipid metabolism.
A major source of ATP is derived from mitochondrial oxidative phosphorylation (OXPHOS) which is driven by the electron transport chain (ETC). The ETC and TCA cycle are intrinsically coupled processes. For example, NADH/NAD+ recycling in the mitochondria relies on the TCA cycle regenerating NADH, an electron donor, for the ETC (22). The ETC comprises four subunits (Complex I-IV) and an ATP synthase (Figure 1). Treg-specific deletion of ETC components remarkably impair Treg suppressive function, underpinning a fundamental function for mitochondrial OXPHOS in Tregs (23–25). The TCA cycle is driven by the oxidation of Acetyl CoA, which could be generated from glucose-derived pyruvate through the pyruvate dehydrogenase (PDH) complex (Figure 1). Deletion of the negative regulator of PDH (PDH kinase) increases PDH activity thus favoring oxidative cellular metabolism for iTreg differentiation (26).
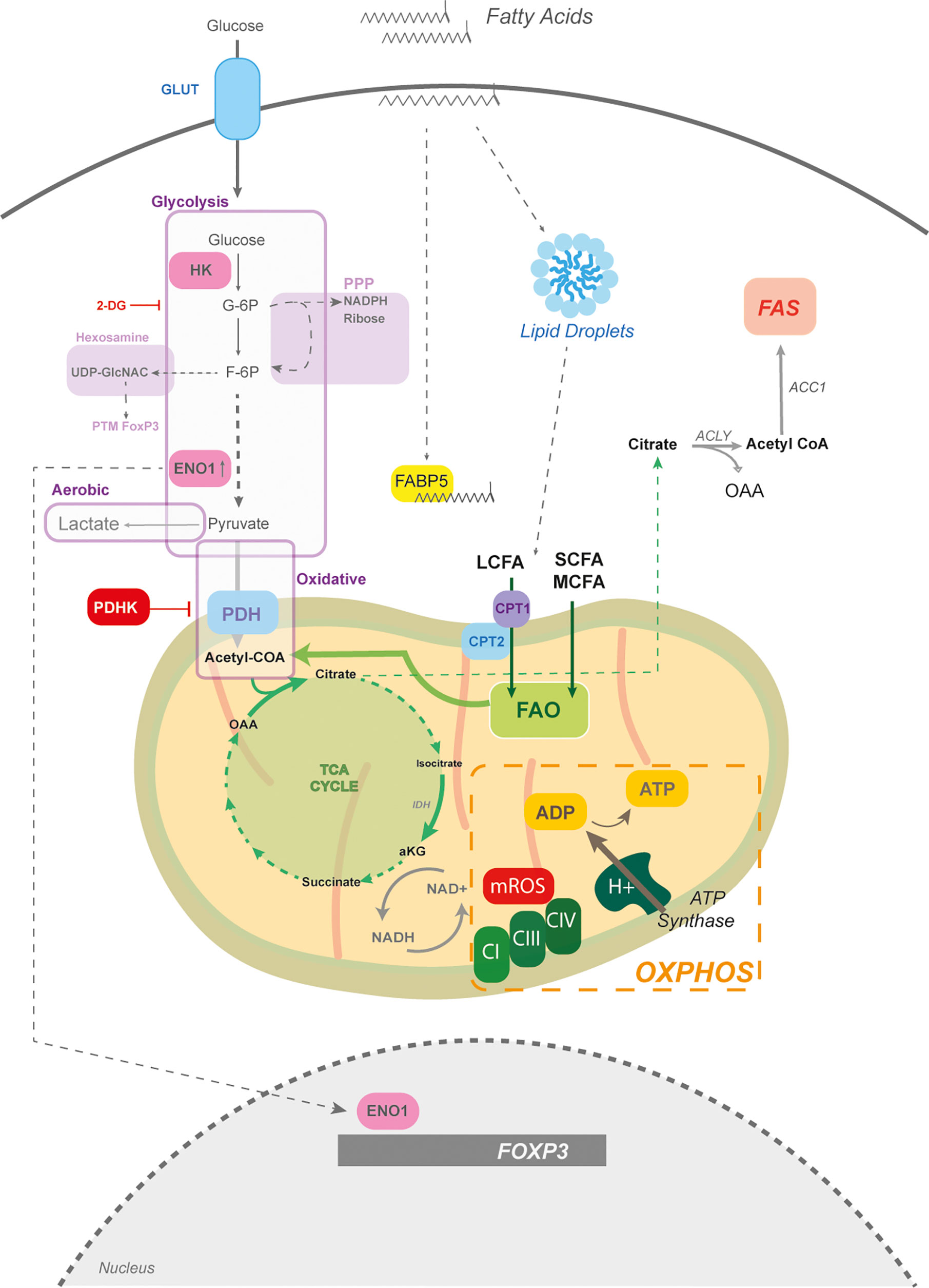
Figure 1 The mitochondria and fatty acid metabolism in Tregs. The core of Treg metabolism centres around mitochondrial activity, which is partially supported by glycolysis. Glycolysis is the breakdown of glucose into pyruvate; however, glycolytic intermediates can feed into other pathways, such as the pentose phosphate pathway (PPP) for NADPH production and the hexosamine pathway for amine sugar production (i.e. UDP-GlcNac) involved in post-translational modifications (PTM). If the end product of glycolysis is pyruvate, which can enter the mitochondria, it is termed oxidative glycolysis. However, if glucose-derived pyruvate is shunted toward lactate production it is termed aerobic glycolysis. Pyruvate enters mitochondria through the pyruvate dehydrogenase (PDH) and generates acetyl CoA to drive the tricarboxylic acid (TCA) cycle. PHD is negatively regulated by PDH kinase. While the TCA cycle can generate amino acids, its function is to regenerate NADH which is critical for electron transport chain (ETC) function. The ETC, composed of complexes I-IV, builds a proton motif force to drive the generation of ATP by ATP synthase. Mitochondrial reactive oxygen species (mROS) are produced at complex I and III. Fatty acids through fatty acid oxidation (FAO) can also provide acetyl CoA units for the TCA cycle. Tregs store fatty acids in the form of lipid droplets, which are partially taken up from the environment. The fatty acid binding protein (FABP5) is a chaperone for long chain fatty acids (LCFAs) and plays a key role in mitochondrial structure. LCFAs require assisted entry into the mitochondria, which is mediated by CPT1 and CPT2. Other sources of fatty acids are the short chain (SCFAs) and medium chain fatty acids (MCFA), that pass freely into the mitochondria. Tregs also perform fatty acid synthesis (FAS) which takes mitochondrial citrate and exports it into the cytoplasm to regenerate acetyl CoA by ATP citrate lyase (ACLY). Acetyl CoA is then committed to fatty acid synthesis by the acetyl CoA carboxylase (ACC). The glycolytic enzyme, enolase (ENO1) can also moonlight as a transcription factor and bind to the FOXP3 promoter to mediate the generation of different splice forms.
Genetic perturbations in regulators involved in maintaining mitochondrial oxidative activity, such as the mitochondrial transcription factor TFAM, reduce Treg differentiation, function and result in lethal systemic autoimmunity (25, 27–29). Other sources of acetyl CoA that can enter the TCA cycle come from fatty acid oxidation (FAO), which catabolizes fatty acids within the mitochondria into acetyl CoA units. Tregs take up fatty acids and store them in the form of lipid droplets (30, 31) (Figure 1). Surprisingly, in the context of cancer, human and mouse Tregs engage in fatty acid synthesis (FAS) to support their functional maturation (32, 33) (Figure 1).
FAS is an ATP-consuming process and relies on the rate limiting enzyme acetyl CoA carboxylase (ACC) (Figure 1). Inhibition of this rate-limiting step increases FAO and preferentially skews naïve CD4+ T cells towards Tregs, delaying disease onset in experimental models of autoimmunity (34). Directing cells to engage in FAO, by replacing glucose with galactose, enhances expression of FoxP3 in Tregs (30). Mitochondrial activity and oxidation of fats are supported in Tregs by fatty acid uptake (35). Fatty acids are characterized by their carbon length – less than 4C are short chain FA (SCFA), medium chain (MCFA) and greater than 12C are long chain FA (LCFA). LCFAs require carnitine palmitoyl transferase 1 (CPT1) to ease mitochondrial transportation by attaching carnitine (e.g. palmitoyl-carnitine), while SCFAs and MCFAs move freely across the membrane (Figure 1).
CPT1a loss, the predominant isoform in lymphocytes, inhibits oxidation of LCFA in Tregs. However, this does not affect the development of tissue resident Tregs, de novo polarization, or suppression, suggesting that Tregs most likely depend on SCFA and MCFA for FAO (23, 36). Nevertheless, this does not mean that LCFAs do not modulate Treg metabolism. The fatty acid binding protein 5 (FABP5), the dominant isoform in Tregs, binds to LCFAs and negatively regulates Treg function, suggesting an unexpected role for LCFAs in Tregs (37) (Figure 1).
Tregs more often rewire their metabolism towards mitochondrial oxidative metabolism and rely less on glycolysis, which is in part mediated through the transcriptional activity of FoxP3 (32, 38, 39). It is important to note that glucose is absolutely required for Treg generation and that Tregs transcriptionally express the glucose transporters GLUT1,3,6, and 8. While GLUT1 is expressed by Tregs, it is dispensable for differentiation, suggesting a role for other isoforms (Figure 1) (25, 40). In addition, iTregs increase their glycolytic rate, albeit at a lower extent than other CD4 lineages. Tregs generally prefer oxidative glycolysis, which is characterized by glucose being broken down into pyruvate, rather than anaerobic glycolysis where pyruvate is shunted towards lactate production (38, 41) (Figure 1).
A common pharmacological approach for disrupting glycolysis is with 2-deoxyglucose (2-DG) which inhibits both phosphoglucose isomerase (PGI) and hexokinases (HK) (42, 43). Tregs express HKI and HKII and 2-DG exposure promotes their differentiation and function by limiting glycolysis (44–46). Inhibition of PGI by 2-DG leads to diversion of glucose 6-phosphate into the pentose phosphate pathway (PPP). Products of the PPP are NADPH, ribose for ribonucleotides and glycolytic intermediates (Figure 1) (47, 48). Although the PPP has yet to be investigated in Tregs, 2-DG data suggests that it may play an important role.
Another branching pathway from glycolysis is the hexosamine pathway where fructose-6-phosphate produces glucosamines, primarily UDP-GlcNAC (Figure 1). These sugar amines play a critical role in maintaining FoxP3 stabilization and suppressive function through post-translational modifications (49–51).
It is worthy to note that Tregs are not restricted in their ability to engage in aerobic glycolysis. In fact, proliferating and expanding Tregs in response to multiple receptor stimuli, such as Toll Like Receptors (TLR), TCR and CD28, metabolically switch towards aerobic glycolysis (Figure 1). In many cases, increasing aerobic glycolysis does not weaken their suppressive activity; on the contrary, glycolysis reinforces suppressive function in human and mouse Tregs (30, 35, 45, 52–55). Interestingly, the glycolytic enzyme, Enolase 1 (ENO1) has a secondary function in human Tregs. ENO1 binds to the FOXP3 gene as means to regulate FOXP3 splice forms (De Rosa et al., 2015) (Figure 1). Overall, the relationship with glycolysis in Tregs is a complex one. On the one hand its restricted function allows for increased differentiation and on the other hand glycolysis is integral to Treg expansion and suppressive activity.
Mitochondrial ROS
The function of mitochondria expands beyond that of mere ATP generation. Mitochondria are powerful signalling organelles, including mitochondrial production of reactive oxygen species (mROS) and programmed cell death (56). For example, TCR-activated Tregs couple downstream signalling – namely the T-cell specific tyrosine kinase Lck – with mitochondrial function by mobilizing Lck to the mitochondria (57). Tregs show a clear dependence on mitochondrial activity and as a consequence of increased ETC function, Tregs display enhanced mROS compared to activated effector CD4+ T cells (25) (Figure 1). Impeding mROS production at complex III (CIII) by genetic deletion of the CIII ROS-producing subunit in Tregs severely reduces suppressive capacity and drives lethal multiorgan inflammation (24). However, mROS can also arise from inhibition of complex I (CI). Inhibition or mutation in CI reduces Treg differentiation and suppressive function in vivo (25, 58). Levels of mROS require fine tuning since too much of it becomes damaging to the cell (59). In cases of chronic inflammation, such as autoimmunity, circulating Treg numbers decline and bear hallmarks of damaging mROS leading to cell death (60). Collectively, Tregs are highly dependent on mitochondrial metabolism for their survival, function, and differentiation. Any mitochondrial dysregulation leads to deleterious effects in Tregs.
Lipids in Transcriptional Regulation
Peroxisome Proliferator Activated Receptors - PPARs
Fatty acids and lipid-derivatives can also act as signalling molecules which converge on transcriptional reprograming. The family of peroxisome proliferator activated receptors (PPARs) are part of a super family of nuclear hormone receptors that bind to natural and synthetic lipophilic acids. There are four types of PPARs: PPAR α, β, γ, and δ. Acting as lipid sensors, PPARs heterodimerize with the retinoid receptor RXR within the nucleus and bind to DNA at PPAR response elements (PPRE) (Figure 2). For example, endogenous ligands for PPARs are polyunsaturated fatty acids (PUFAs) and eicosanoids that drive transcriptional regulation of glycolytic, lipid and mitochondrial genes (61). The visceral adipose tissue (VAT), an abundant site of lipid metabolism, harbors a distinctive set of Tregs called VAT Tregs that express high levels of PPARγ (Figure 2). Indeed, Treg-specific ablation of PPARγ selectively reduces homeostatic levels of VAT Tregs (62). Agonist activation of PPARα and γ with natural and synthetic lipophilic compounds act through transcriptional activity not only to increase iTreg polarization, but also to boost their function in vitro and in T-cell mediated colitis (63–65) (Figure 2).
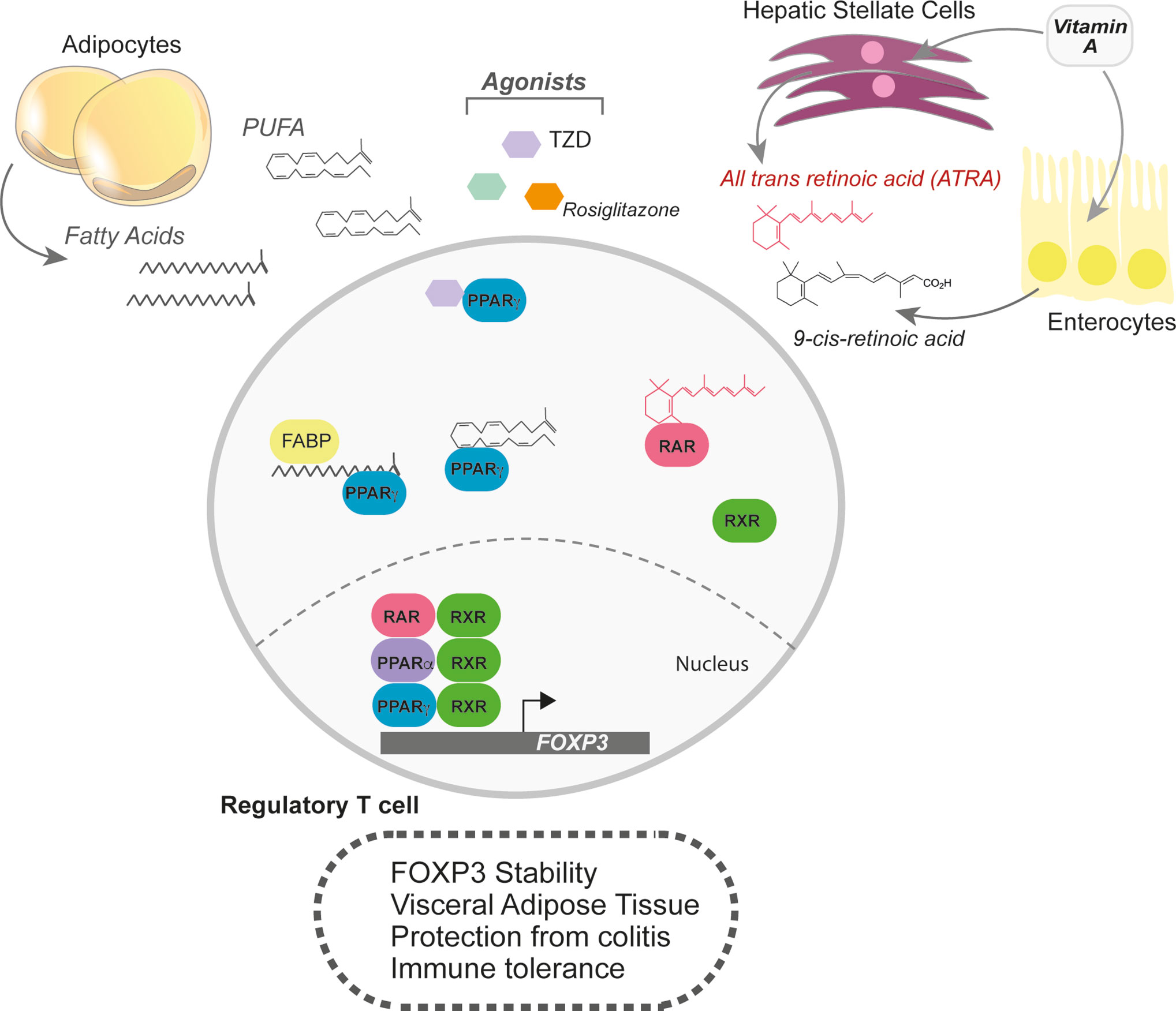
Figure 2 Lipophilic sensors in shaping the transcriptional landscape of Tregs. Fatty acids are also powerful signalling molecules that can change the transcriptional profile of cells. In Tregs, the lipophilic sensors, PPARα, PPARγ, and the retinoic acid receptor (RAR) support Treg differentiation and functional programs. The PPAR family of transcription factors respond to broad range of fatty acids, but quite potently to polyunsaturated fatty acids (PUFAs). Agonists for the PPARs, such as rosiglitazone and thiazolidinedione (TZD) help boost Treg differentiation, FoxP3 stability and function. Visceral Adipose tissue (VAT) Tregs are in constant supply of lipid ligands and upregulate PPARγ. RARα also responds to lipophilic ligands, but only those derived from retinol or retinoic acid. Two key sites of retinoic acid storage and production of all-trans retinoic acid (ATRA) are the liver and enterocytes. In addition, hepatic stellate cells can store retinoids and, when activated, produce ATRA.
Natural PPAR agonists, such as omega-3 fatty acids (Ω-3-FA), which are a class of PUFAs, display immunomodulatory effects (Figure 2). Elevating dietary Ω-3-FA or genetically manipulating their levels reduces severity in experimental models of autoimmunity, such as T-cell induced colitis (66–68). In dietary induced obesity, Ω-3-FA supplementation dampens adipose tissue inflammation, in part by promoting de novo Treg differentiation (68). In cases of cardiac allografts and milk protein allergies, Ω-3-FA increase peripheral Tregs to achieve immune tolerance (69, 70).
Retinoic Acid Receptors - RARs
Other nuclear hormone receptors responding to lipophilic metabolites also play key roles in regulating Treg biology. While PPARγ heterodimerizes with RXR, RXR also heterodimerizes with the retinoic acid receptor alpha (RARα). The RAR and RXR family of transcription factors bind to metabolites of retinol (lipid soluble vitamin A), such as all-trans retinoic acid (ATRA) or 9-cis-RA (71). Vitamin A is absorbed in the small intestine and is either catabolized in enterocytes or in the liver (Figure 2). ATRA exposure during de novo Treg differentiation promotes the expression of gut-homing receptors and RA supplementation increases Tregs at the mucosal interface in experimental models of colitis (72, 73). Pro-inflammatory cytokines weaken Treg stability; however, RA can protect Tregs by preferentially enhancing FoxP3 expression and stabilization (74–79) (Figure 2).
Another site of Vitamin A metabolism is the liver. Naïve hepatic stellate cells (HSCs) store retinoids, which can account for nearly 50% of all RA in the body, and produce ATRA upon activation in the local environment (80). In addition to ATRA, activated HSCs can produce TGFβ, a potent combination for Treg differentiation. Co-culture of HSCs with DCs and naïve CD4+ T cells skewed differentiation towards Tregs in RARα-dependent manner (81) (Figure 2). In conclusion, retinoic acid metabolism, either at mucosal sites or in the liver, can promote an immunosuppressive environment mediated through FoxP3 induction and stabilization.
Of note, other essential vitamins can influence Treg biology. It has been shown that folic acid (Vitamin B9) is necessary for maintenance of Tregs in the colon; mice fed a diet depleted of folic acid were more susceptible to gut inflammation (82). Both vitamin C and D have also been shown to enhance de novo Treg induction; however, only vitamin D has demonstrated reduced severity in experimental models of autoimmunity (83–87).
Oxygen Poor Environments and Treg Adaptation
Tumors have been thought of as “wounds that do not heal”, with many similarities in stromal reorganization reminiscent of wound healing (88). One of the hallmarks of cancer is tissue reorganization which leads to changes in local supply of nutrients and oxygen surrounding the cells (89, 90). One prominent cell type found in cancer are Tregs, which adapt to these metabolically competitive environments.
Changes in oxygen levels within tissues are sensed intracellularly by prolyl hydroxylases (PHD). When oxygen levels are within the normoxic range (20% in tissue culture and around 5% in tissues), PHDs hydroxylate the hypoxia inducible factor 1 α (HIF1α) and target it for proteasomal degradation (91, 92). Under low oxygen levels, HIF1α is released from PHD-dependent degradation and heterodimerizes with HIF1β to translocate to the nucleus. HIF1α upregulates genes involved in aerobic glycolysis, angiogenesis, erythropoiesis, and cell survival as a mechanism for cellular adaptation under hypoxia (93).
Treg migration and survival in inflamed peripheral tissues and tumors depend on metabolic adaptation to support these processes. One major metabolic change in migrating Tregs is their switch towards aerobic glycolysis, which is mediated by glucokinase (GCK) and HIF1α (53, 94). Once within the tissue, Tregs are exposed and adapt to different O2 environments. Localized tissue hypoxia can induce the differentiation of Tregs through the direct upregulation of FoxP3 by HIF1α (95). Hypoxia-induced Tregs may help limit tissue damage under states of hypoxic inflammation. The gut has been documented to function under a physiologically hypoxic environment in comparison to other tissues (96). In fact, HIF1α-expressing Tregs repress T-cell induced colitis, suggesting a key role for hypoxia and aerobic glycolysis in mitigating inflammation in oxygen-poor tissues (95). However, under conditions of normoxia, HIF1α negatively regulates Treg differentiation, suggesting that oxygen availability shapes Treg fate (44).
Tumor oxygenation is generally much lower than that of normal tissues, yet Tregs readily populate tumors to provide additional immune tolerance to support tumor growth (97, 98). For instance, the brain is highly sensitive to oxygen variability and functions around 4.4%pO2, yet Tregs manage to adapt in order to survive under oxygen competitive environments (99, 100). Tregs in glioma use HIF1α to support this metabolic adaptation by increasing aerobic glycolysis for migration and depend mainly on fatty acid uptake to support OXPHOS-mediated immune suppression (94, 101). However, other tissues have different levels of O2 supply and degrees of O2 sensitivity. Oxygen ranges from 5.6-14.5% pO2 in the lung (100) suggest that Tregs may adapt accordingly. In the case of lung metastasis, PHD, the negative regulator of HIF1α is activated in Tregs and drives their accumulation in the lung to create an immune-tolerant metastatic niche (102).
In response to low O2 levels, the acidity of the local milieu within tumors increases compared to normal tissue (103). This increase in acidity imposes harsh conditions for surrounding T cells. One main metabolite that could regulate local acidity is lactate; the major transporters for lactate in T cells are SLC5a12 and MCT1 (41, 104–106) (Figure 3). Tregs within the tumor interstitial space are able to outcompete other T cell subsets by surviving under low glucose and high lactate conditions. Intratumoral Tregs upregulate the SLC5a12 and MCT1 allowing lactate to fuel the TCA-cycle for OXPHOS-mediated immune suppression and to support gluconeogenic pathways for survival (41, 106) (Figure 3). Limiting tumor-derived lactate in the context of anti-CTLA4 blockade increases glucose consumption in intratumoral Tregs and skews the balance towards IFN-γ producing CD4+ T cells (107).
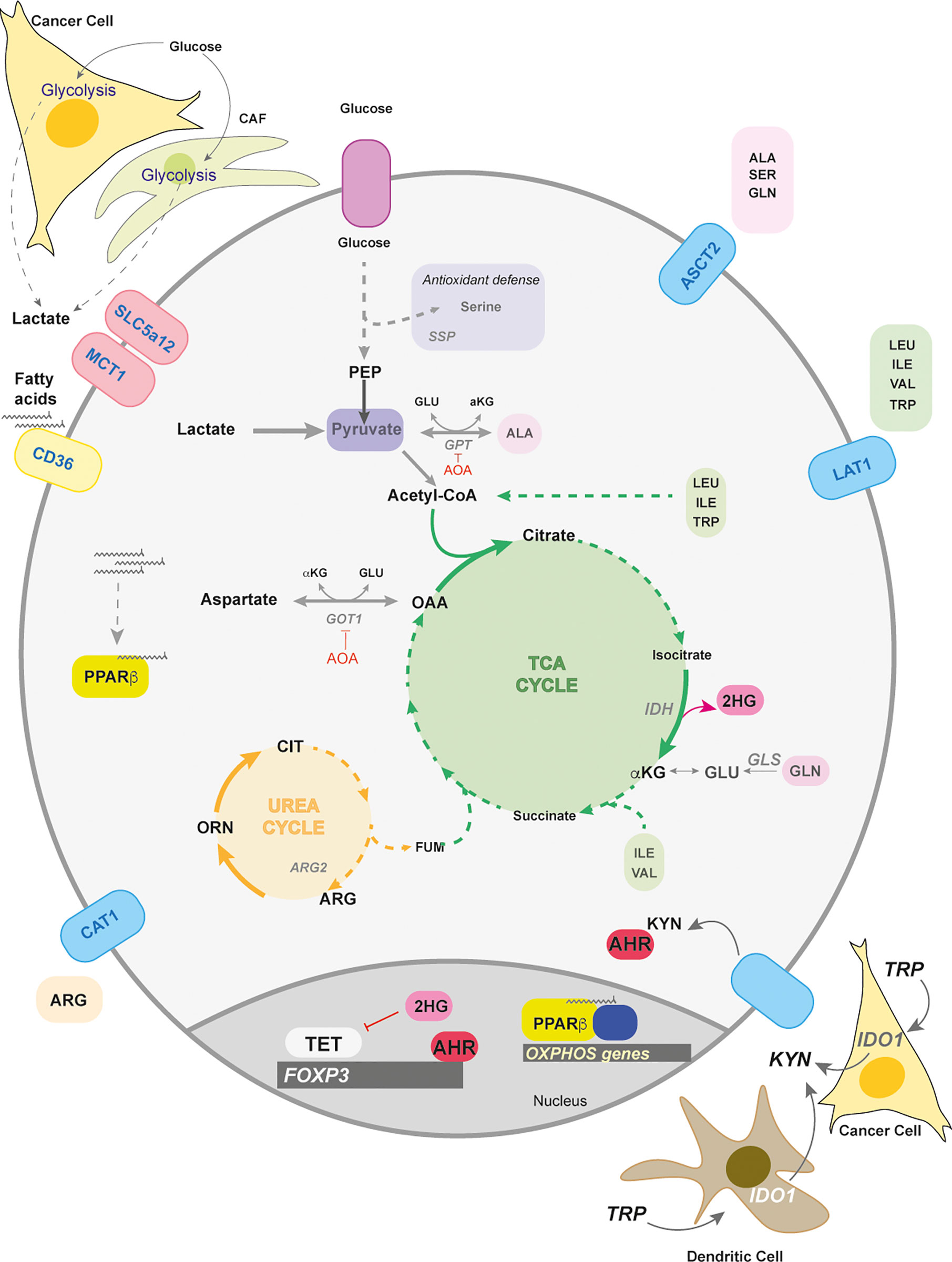
Figure 3 Lactate and amino acid metabolism in Tregs. Lactate and amino acid metabolism in Tregs play important roles in their differentiation and function. Lactate can arise in the tumors and from cancer associated fibroblasts (CAFs) as a consequence of their highly glycolytic nature. The transporters for lactate are SLC5a12 and MCT1. Tregs metabolically adapt to a low glucose/high lactate environment by using lactate to fuel the TCA cycle that supports oxidative phosphorylation in the mitochondria. Intra-tumoral Tregs increase the fatty acid receptor CD36 when extracellular lactate concentrations increase. The import of fatty acids activates the transcription factor PPARβ to maintain mitochondrial fitness. Other transported nutrients are amino acids. ASCT2, a neutral amino acid transporter, is responsible for the intake of amino acids such as alanine, serine, glutamine. Serine uptake and biosynthesis (serine synthesis pathway – SSP) are involved in antioxidant defence in Tregs. The amino acids, alanine and aspartate, can enter central carbon metabolism through transamination by glutamate pyruvate transaminase (GPT) and glutamate oxaloacetate transaminase (GOT), both of which are negative regulators of FoxP3 induction. Both GOT and GPT are inhibited by the transaminase inhibitor, aminooxyacetate (AOA). Another negative regulator is alpha-ketoglutarate (αKG) which can be converted by the error-prone isocitrate dehydrogenase (IDH) to 2-hydroxyglutarate (2-HG). 2-HG inhibits the family of DNA demethylases – ten eleven translocase (TET) 1-3. Another amino acid that can enter the TCA cycle is arginine which is transported by the cationic transporter (CAT1). Arginine helps fuel the urea cycle, where arginase (ARG2) converts arginine to ornithine and later citrulline. The transport of branched chain amino acids (BCAA), and aromatic amino acids, such as isoleucine and tryptophan, use the L-type amino acid transporter (LAT1). BCAA can help fuel the TCA either through acetyl CoA or succinate generation. Tryptophan (TRP) can be converted into kynurenine by the Indoleamine 2,3-dioxygenase (IDO1) in dendritic or tumor cells, which activates the ligand receptor, Aryl hydrocarbon Receptor (AhR). Ligand-bound AhR translocates to the nucleus and upregulates Foxp3 expression.
Other stromal components, such as cancer associated fibroblasts (CAF), contribute to lactate production that preferentially selects for intratumoral CD4+ Foxp3+ populations through a NF-kB-FoxP3 axis (108) (Figure 3). Another method of survival under lactic acid rich environments is achieved by upregulating the fatty acid receptor CD36. Intratumoral Tregs particularly express CD36 in order to enhance PPARβ expression as a means to support mitochondrial metabolism in Tregs and their survival (109) (Figure 3). High levels of environing lactate due to the highly glycolytic nature of cancer cells and CAFs could be part of a strategy employed by tumors to promote immune suppression.
Tissues and Amino Acid Metabolism in Tregs
Other immunomodulatory nutrients found in inflammatory sites and tumor lesions are amino acids and their catabolic by-products (110, 111). General amino acid uptake and breakdown is imperative for T cell proliferation and CD4+ Thelper differentiation. Known amino acid (AA) transporters expressed on the surface of T cells are the SLC7A5/SLC3A2 (LAT1), and SLC1A5 (ACST2) (112, 113).
Non-Essential Amino Acids
Glutamine
Typical amino acids transported by ASCT2 are alanine, serine, cysteine, methionine and glutamine (114). Interestingly, ASCT2 is dispensable for thymic development of nTregs and de novo polarization (112). However, glutamine deprivation increases FoxP3 expression upon TCR engagement and also increases in a TGFβ-dependent manner (115, 116). Glutamine restriction has also been shown to enhance suppressive function in a T-cell mediated colitis model (115), which could also be a method for tumors to promote immune suppression (Figure 3).
How glutamine negatively regulates Treg induction and activity is still poorly understood. Interestingly, deletion of glutaminase (GLS), an amidohydrolase enzyme that converts glutamine to glutamate, does not affect homeostatic Treg frequencies nor their differentiation in vitro (117) (Figure 3). This would suggest that Tregs do not depend on glutamine to fuel the TCA cycle and further supports the notion that Tregs utilize other carbon sources, such as fatty acids (Figure 3).
Unexpectedly, increased transamination - the transfer of amine groups either from glutamine or glutamate that generates glutamate or αKG respectively – negatively impacts Treg biology. Aminooxyacetate (AOA), a global transaminase inhibitor, increases FoxP3 expression both in Treg and Th17 polarization conditions (118) (Figure 3). Deletion of GOT1, which transfers amino groups between aspartate and glutamate interchangeably to produce OAA and αKG, rewires Th17 cells towards Tregs in vitro and in experimental autoimmune encephalomyelitis (EAE). This effect was partially due to the promiscuous nature of dehydrogenases (e.g. isocitrate dehydrogenase – IDH) leading to conversion of αKG into 2-hydroxyglutarate (2-HG). Elevated levels of 2-HG inhibit the DNA demethylating enzymes TET1-3 and this inhibition keeps the Foxp3 promoter silenced (118) (Figure 3).
Arginine
While the majority of amino acids are neutral, basic amino acids, such as arginine and to a lesser extent histidine, require cationic amino acid transporters (CAT) (119). CAT1 is highly expressed on proliferating T cells and transports arginine downstream of TCR engagement (120) (Figure 3). In cases of muscle wound healing, a specific Treg population infiltrates the site for damage resolution as well as causes a local increase in arginine metabolism (110, 121). Intracellular arginine can be broken down in the urea cycle by arginase (ARG1 and ARG2) into ornithine and citrulline (122). Activated Tregs found in the skin enhance ARG2 expression to outcompete and deprive effector T cells of environmental arginine (123) (Figure 3). Tregs can also utilize citrulline, a component of the urea cycle, for enhancing FoxP3 induction, thus potentially still surviving in arginine competitive environments, such as in arginine-addicted tumors (124, 125) (Figure 3).
Serine
Serine is a non-essential amino acid implicated in numerous processes such as glycine synthesis, purine synthesis, anti-oxidant defence and the methylation cycle whose overall function is critical for T effector cell function (126). Anti-oxidant programs in T cells include the de novo generation of glutathione from glutamate, cysteine and glycine by glutamate cysteine ligase (Gclc) (127). Gclc-loss in Tregs drives ROS-dependent serine uptake and de novo serine synthesis leading to multiorgan autoimmunity (128). Impeding serine metabolism in Gclc-null Tregs rescues Treg suppressive activity, suggesting that reduced glutathione is crucial for Treg function by restraining serine metabolism (Figure 3).
Essential Amino Acids
Essential amino acids also require transporters for uptake; for example, LAT1 imports branched chain amino acids (BCAA) and aromatic amino acids (e.g. Phenylalanine, histidine, leucine and tryptophan) at the expense of exporting glutamine (129). Interestingly, BCAA bypass first-pass hepatic metabolism, thus allowing for the muscle (primarily) and other organs to immediately acquire BCAA (130). In addition to being essential to protein synthesis, BCAA help replenish the TCA cycle and provide carbon units for fatty acids synthesis (131). Disrupting BCAA metabolism by limiting dietary isoleucine diminishes homeostatic numbers of Tregs and weakens their suppressive function. Disrupting part of the BCAA transporter, SLC3A2, similarly reduces Treg homeostatic survival and their suppressive activity in experimental colitis models (132). In some cases of cancer, such as pancreatic cancer, there is an early rise of serum BCAA (133), which may perhaps support Treg function in limiting anti-tumor immunity, but this is yet to be explored.
Tryptophan and Kynurenine
One well described mechanism of tumor evasion is the expression by cancer cells or tolerogenic DCs of tryptophan-degrading enzymes such as indoleamine-pyrrole 2,3-dioxygenase 1 (IDO1) (Figure 3). This enzyme catalyzes the conversion of tryptophan into kynurenine in a pathway that leads to the production of kynurenic acid, quinolinic acid and NAD. Both the reduced presence of tryptophan and the accumulation of kynurenine have been shown to potentiate an immunosuppressive cancer microenvironment (134–136).
One IDO-dependent mechanism of promoting immune suppression is the generation of Tregs. It has been shown that under low-tryptophan/high kynurenine or IDO1-expressing DC’s, naïve CD4+ T cells enhance FoxP3 expression (137, 138) (Figure 3). In a mouse model of B16-OVA tumor cells implanted in FoxP3-GFP reporter mice, IDO1 inhibition led to increased IL-17 expression in Tregs cells suggesting that tryptophan and kynurenine metabolism play an important role in Th17/Treg plasticity in vivo (139).
Intracellular Sensing of Amino Acids by mTOR
Amino acids play a critical role in modulating Treg homeostasis, polarization, and function; however, T cells require intracellular nitrogen sensors to couple availability with function. One major nitrogen sensor is the mammalian target of rapamycin (mTOR) Ser/Thr kinase. mTOR assembles into two distinct complexes termed complex 1 (mTORC1) and complex 2 (mTORC2), with mTOR harboring the catalytic function for both complexes. Amino acids, growth factors, and glucose can stimulate mTORC1 activation for global protein synthesis (140). Translation consumes around 25% of cellular ATP in lymphocytes and requires charged amino acids as building blocks for protein synthesis (141), thus providing mTORC1 with the responsibility of sensing amino acid availability to keep translation in check.
Amino acids promote the Rag GTPases to recruit mTORC1 to the lysosomal membrane and bring mTORC1 in close proximity to the small GTPase Rheb (142). GTP-bound Rheb then activates mTORC1 (143). The many functions of mTORC1 include downstream targets involved in protein synthesis (e.g. S6 ribosomal protein) and lipid synthesis (144). Loss of the mTOR catalytic subunit in T cells and Treg-specific loss increases FoxP3 induction, in part mediated by the mTORC2 complex. However, mTOR catalytic activity and mTORC1 are indispensable for suppression by regulating transcripts involved in mitochondrial oxidative and lipid metabolism (28, 145–148). Foxp3-specific ablation of upstream mTORC1 activators, RagA/B and Rheb, severely impair Treg suppressive function leading not only to uncontrolled systemic inflammation, but also enhanced anti-tumor immunity (147–149). Interestingly, activation of human Tregs has identified an oscillatory role for mTORC1 activity in controlling metabolic plasticity (150).
Amino acids are essential for T cell proliferation and Tregs are no exception. While glutamine and serine metabolic pathways are critical to T effector cells, they seem to reduce Treg differentiation and activity. Tregs rely on BCAA and arginine metabolism potentially as means to support the TCA cycle for OXPHOS. The activation of the amino acid sensor, mTORC1, is essential for the suppressive fitness of Tregs. These data collectively suggest that amino acid metabolism and sensing are key regulators of Treg homeostasis, lineage polarization, and function.
The Gut and Microbiome Metabolite By-Products
The colon is an extraordinary organ of the mammalian body, as it holds a large mass of bacterial species. It has been documented that 10-20% of faecal weight is contributed by bacteria and >90% are obligate anaerobes (151). Most importantly, these anaerobic bacteria further metabolize unabsorbed peptides, fats and oligosaccharides, reaching a metabolic activity equivalent to that of the liver (152, 153) (Figure 4). The by-products of anaerobic fermentation in the gut are organic amines from peptides, ammonia from peptides and nitrogen-based metabolites, and SCFAs (i.e. acetate, butyrate, and propionate) from dietary fibre (153). SCFAs are water soluble and allow for easy absorption. It has been observed since the 1970’s that germ-free mice suffer from chronic diarrhoea, which could be remedied by providing SCFAs, suggesting that bacterial SCFAs are responsible for maintaining colonic homeostasis (154)
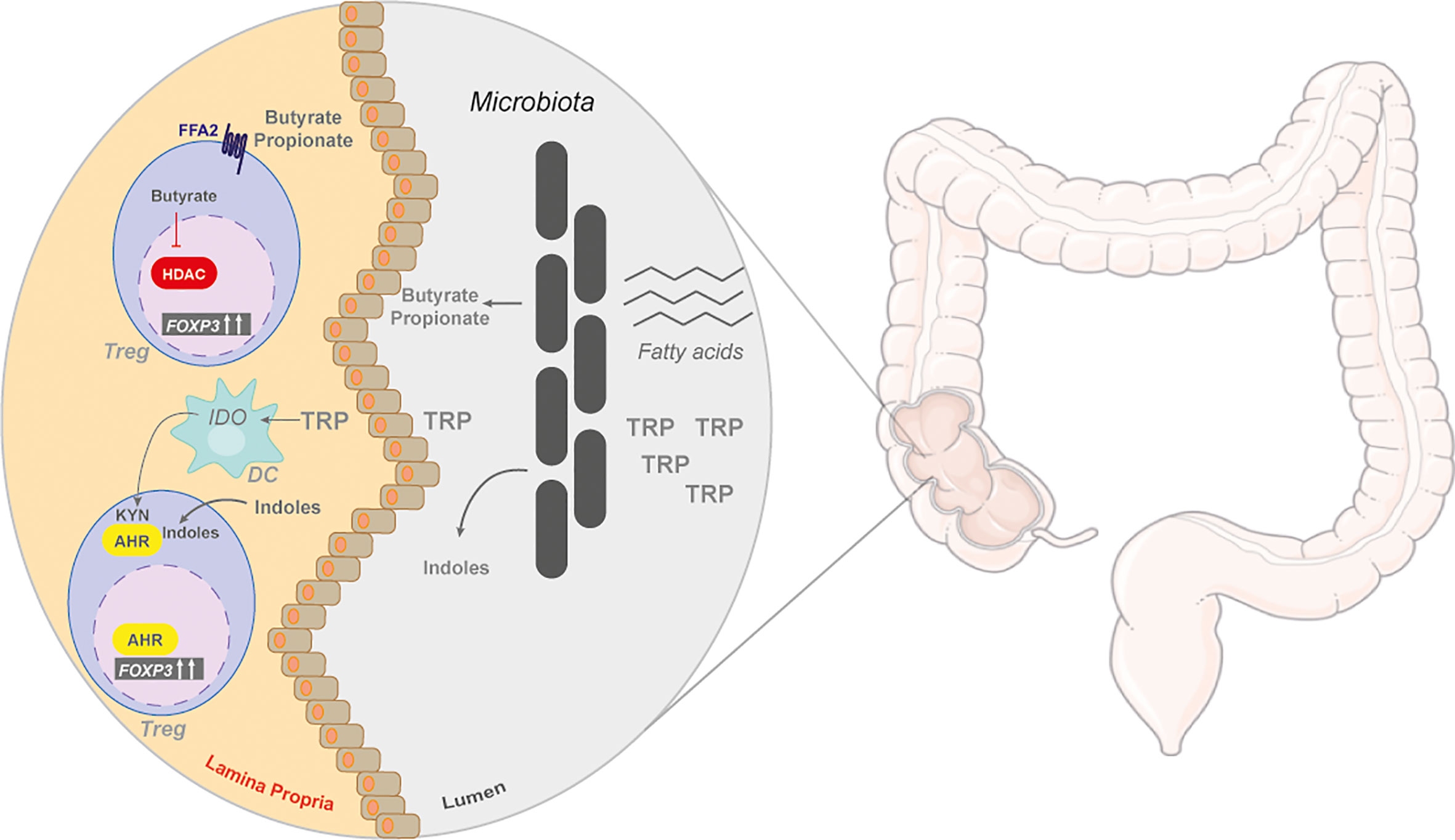
Figure 4 The gut and microbiome by-products in regulating Tregs. The colon is a metabolically active site, with many unabsorbed nutrients, such as amino acids and fats. These nutrients are metabolized by the gut commensal bacteria. Unabsorbed fatty acids can be converted to short chain fatty acids (SCFAs), butyrate and propionate, that are released and enter the lamina propria. In the colonic lamina propria, SCFAs can induce the differentiation of naïve T cells into iTregs through the G protein-coupled receptor (GPCR), free fatty acid receptor 2 (FFA2), or through its inhibitory action on histone deacetylases (HDACs). Tryptophan (TRP) is also catabolized by gut symbionts into indoles or can be catabolized by dendritic cells (DC) into kynurenine (KYN) by indoleamine-pyrrole 2,3-dioxygenase 1 (IDO), both of metabolites activate AhR.
Tregs are a key modulator of colonic homeostasis with luminal concentrations of SCFAs positively correlating with colonic Treg frequencies (155) (Figure 4). Germ-free mice have reduced colonic Tregs that can be rescued by butyrate supplementation, suggesting that bacterial fermentation of dietary fibre into SCFAs helps maintain colonic Treg pools (156). Butyrate, and to a lesser extent propionate, can induce Tregs in a TGFβ-dependent manner in vitro and in models of T-cell-induced colitis (155, 156). Since SCFAs are water soluble, Treg accumulation in other organs can also occur, providing beneficial outcomes in experimental models of type I diabetes and kidney allograft transplantation (157, 158).
SCFAs can act in two ways to promote Treg accumulation: G protein-coupled receptor (GPCR) and histone deacetylases (HDAC) (Figure 4). Loss of the free fatty acid receptor, FFA2, obliterates any Treg-inducing effect of SCFAs in the colon and in allograft transplantation, suggesting that signalling through the GPCR regulates FoxP3 induction (156, 157). Butyrate treatment also increases histone H3 acetylation on the promoter of the FoxP3 locus by inhibiting class I and class II HDACs (155, 159, 160). These two mechanisms employed by SCFAs are perhaps linked, but this has yet to be investigated.
Another abundant dietary nutrient catabolized by the gut and microbiome is tryptophan, an aromatic amino acid with an indole side chain. Tryptophan can be broken down by both the host and commensal bacteria in the gut. Host degradation of tryptophan is used as a precursor to serotonins and niacin (vitamin B3) biosynthesis (161). A few commensal bacterial species in the gut, such as Escherichia coli, metabolize tryptophan and release indoles and indole-derivatives into circulation (162) (Figure 4). Dietary administration of indoles, such as 2-(1H-Indol-3-ylcarbonyl)-4-thiazolecarboxylic acid methyl ester (ITE), protect against experimental models of colitis, EAE, and type I diabetes (159, 163–166). Indoles and their derivatives were found to activate the Aryl hydrocarbon receptor (AhR). AhR is a ligand-activated transcription factor with a promiscuous binding site recognizing synthetic and natural ligands (167, 168). Indole-derived ligands enhance TGFβ-induced Treg differentiation and increase IL10 production in activated T cells in vitro (163, 169) (Figure 4). Dietary indole supplementation promotes Treg frequencies in mice by directly activating AhR or by modifying the microbiota towards butyrate-producing species (170, 171). However, it should be noted that not all protective qualities of indoles act through enhancing Treg activity in models of autoimmunity (165).
The intestinal tissue also contributes to ligand activation of AhR through the degradation of tryptophan to NAD by the kynurenine pathway (167). The by-products of this pathway (e.g. kynurenine) activate AhR (167) (Figure 3). Other sources of kynurenine are gut-specific CD11c+CD103+ DCs expressing IDO (Figure 4). This DC subset enhances Treg differentiation as a means to mediate host-microbe homeostasis in the gut (172). Overall, tryptophan metabolism either through host or microbial breakdown can have a profound impact on Treg homeostasis and differentiation in the host.
Conclusion
Nutrient availability and the production of various intracellular metabolites can shape Treg cell fate and function (Table 1). The key metabolic cues governing Tregs are driven by mitochondrial oxidative metabolism, mainly the TCA cycle and FAO, along with support from glycolytic intermediates. However, in the process of trafficking within the host and reaching new tissue sites, Tregs quickly adapt to new carbon sources and oxygen levels through metabolic rewiring in accordance with the microenvironment. While tissue resident Tregs have been identified in nearly every organ, the type of metabolism that is required for them to survive in these environments is only starting to be understood.

Table 1 Metabolites in shaping Treg biology. Tregs are sensitive to various metabolites. The presence of certain metabolites can increase Treg differentiation and suppressive function. However, in some cases, as with 2-hydroxyglutarate, limit Treg differentiation.
In recent years, the democratisation of single cell RNA sequencing has led to numerous studies aiming to understand the transcriptome of Tregs and their potential metabolic landscape (173, 174). However, cellular metabolism is per definition a dynamic process that cannot be fully encapsulated by transcriptomics. In fact, further proteomic studies of Tregs would greatly benefit the field of Treg immunometabolism (175). While 2-NBDG, a fluorescent analogue of 2-DG, has helped in the field of immunometabolism and tissue Tregs to measure glucose uptake, a recent study has suggested it as an unreliable tool as it retains intracellular transport in the presence of multiple GLUT inhibitors (176). The most promising technology available to help disentangle the complex metabolic interface between Tregs and tissues is mass spectrometry imaging. Techniques such as MALDI or DESI-MSI in combination with classical immunohistochemistry confers a spatial-temporal resolution which would provide a more accurate picture of Treg metabolism within tissues (177).
On a final note, our understanding of Treg metabolism has mainly been a blend of findings from both mice and humans. However, it is important to underscore that human Tregs are distinct from their mouse counterparts both metabolically and phenotypically (178, 179). For example, one metabolic difference is that ex-vivo human Tregs are glycolytic in nature, while ex-vivo mouse Tregs are not (35, 178). A feature that may explain some of the metabolic differences between human and mouse Tregs could lie in the functional heterogeneity of circulating human Tregs – an aspect that is absent in mice (179). While mouse FoxP3+ Tregs show functional homogeneity, human FOXP3+ Tregs are a pool of Tregs with various phenotypes and different levels of suppressive activity (180). These phenotypic differences will inevitably complicate the understanding of the metabolic landscape of human Tregs and underline a limitation of mouse-to-human Treg metabolism translation.
The nutrient environment in each tissue – be it mouse or human- is a complex formula, yet Tregs find ways to metabolically adapt to these new environments. By understanding these nodes of adaptation, new therapies can emerge to either promote their function in autoimmunity or limit their efficacy in cancer.
Author Contributions
JB, MH, and FZ conceptualized the review. JB and MH wrote the review with significant input from FZ. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by Cancer Research UK grant C596/A26855 and supported by the Francis Crick Institute which receives its core funding from Cancer Research UK (FC0010557), the UK Medical Research Council (FC0010557) and the Wellcome Trust (FC0010557).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64.
2. d’Hennezel E, Bin Dhuban K, Torgerson T, Piccirillo CA. The immunogenetics of immune dysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet (2012) 49(5):291–302. doi: 10.1136/jmedgenet-2012-100759
3. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
4. Plitas G, Rudensky AY. Regulatory T Cells: Differentiation and Function. Cancer Immunol Res (2016) 4(9):721–5. doi: 10.1158/2326-6066.CIR-16-0193
5. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol (2019) 16(6):356–71. doi: 10.1038/s41571-019-0175-7
6. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T Cells and Human Disease. Annu Rev Immunol (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
7. Campbell DJ. Control of Regulatory T Cell Migration, Function, and Homeostasis. J Immunol (2015) 195(6):2507–13. doi: 10.4049/jimmunol.1500801
8. Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
9. Crellin NK, Garcia RV, Levings MK. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood (2007) 109(5):2014–22. doi: 10.1182/blood-2006-07-035279
10. Harada Y, Harada Y, Elly C, Ying G, Paik JH, DePinho RA, et al. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J Exp Med (2010) 207(7):1381–91. doi: 10.1084/jem.20100004
11. Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature (2012) 491(7425):554–9. doi: 10.1038/nature11581
12. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol (2005) 6(11):1142–51. doi: 10.1038/ni1263
13. Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM. IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. J Immunol (2011) 186(11):6329–37. doi: 10.4049/jimmunol.1100061
14. Passerini L, Allan SE, Battaglia M, Di Nunzio S, Alstad AN, Levings MK, et al. STAT5-signaling cytokines regulate the expression of FOXP3 in CD4+CD25+ regulatory T cells and CD4+CD25- effector T cells. Int Immunol (2008) 20(3):421–31. doi: 10.1093/intimm/dxn002
15. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol (2016) 17(11):1322–33. doi: 10.1038/ni.3540
16. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35(6):871–82. doi: 10.1016/j.immuni.2011.09.021
17. Ma EH, Verway MJ, Johnson RM, Roy DG, Steadman M, Hayes S, et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8(+) T Cells. Immunity (2019) 51(5):856–70 e5. doi: 10.1016/j.immuni.2019.09.003
18. Saravia J, Raynor JL, Chapman NM, Lim SA, Chi H. Signaling networks in immunometabolism. Cell Res (2020) 30(4):328–42. doi: 10.1038/s41422-020-0301-1
19. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi: 10.1038/nature08097
20. Shyer JA, Flavell RA, Bailis W. Metabolic signaling in T cells. Cell Res (2020) 30(8):649–59. doi: 10.1038/s41422-020-0379-5
21. Geltink RIK, Kyle RL, Pearce EL. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol (2018) 36:461–88. doi: 10.1146/annurev-immunol-042617-053019
22. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342(6155):1242454. doi: 10.1126/science.1242454
23. Saravia J, Zeng H, Dhungana Y, Bastardo Blanco D, Nguyen TM, Chapman NM, et al. Homeostasis and transitional activation of regulatory T cells require c-Myc. Sci Adv (2020) 6(1):eaaw6443. doi: 10.1126/sciadv.aaw6443
24. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature (2019) 565(7740):495–9. doi: 10.1038/s41586-018-0846-z
25. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, et al. Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J (2015) 29(6):2315–26. doi: 10.1096/fj.14-268409
26. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest (2015) 125(1):194–207. doi: 10.1172/JCI76012
27. Yang K, Blanco DB, Neale G, Vogel P, Avila J, Clish CB, et al. Homeostatic control of metabolic and functional fitness of Treg cells by LKB1 signalling. Nature (2017) 548(7669):602–6. doi: 10.1038/nature23665
28. Chapman NM, Zeng H, Nguyen TM, Wang Y, Vogel P, Dhungana Y, et al. mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat Commun (2018) 9(1):2095. doi: 10.1038/s41467-018-04392-5
29. Fu Z, Ye J, Dean JW, Bostick JW, Weinberg SE, Xiong L, et al. Requirement of Mitochondrial Transcription Factor A in Tissue-Resident Regulatory T Cell Maintenance and Function. Cell Rep (2019) 28(1):159–71 e4. doi: 10.1016/j.celrep.2019.06.024
30. Cluxton D, Petrasca A, Moran B, Fletcher JM. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front Immunol (2019) 10:115. doi: 10.3389/fimmu.2019.00115
31. Howie D, Ten Bokum A, Cobbold SP, Yu Z, Kessler BM, Waldmann H. A Novel Role for Triglyceride Metabolism in Foxp3 Expression. Front Immunol (2019) 10:1860. doi: 10.3389/fimmu.2019.01860
32. Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci USA (2018) 115(28):E6546–E55. doi: 10.1073/pnas.1720113115
33. Lim SA, Wei J, Nguyen T-LM, Shi H, Su W, Palacios G, et al. Lipid signalling enforces functional specialization of Treg cells in tumours. Nature (2021) 591:306–11. doi: 10.1038/s41586-021-03235-6
34. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20(11):1327–33. doi: 10.1038/nm.3704
35. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity (2016) 44(3):712. doi: 10.1016/j.immuni.2016.02.022
36. Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, et al. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab (2018) 28(3):504–15 e7. doi: 10.1016/j.cmet.2018.06.002
37. Rao E, Singh P, Li Y, Zhang Y, Chi YI, Suttles J, et al. Targeting epidermal fatty acid binding protein for treatment of experimental autoimmune encephalomyelitis. BMC Immunol (2015) 16:28. doi: 10.1186/s12865-015-0091-2
38. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
39. Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight (2017) 2(3):e89160. doi: 10.1172/jci.insight.89160
40. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi: 10.1016/j.cmet.2014.05.004
41. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab (2017) 25(6):1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
42. Chen W, Gueron M. The inhibition of bovine heart hexokinase by 2-deoxy-D-glucose-6-phosphate: characterization by 31P NMR and metabolic implications. Biochimie (1992) 74(9-10):867–73. doi: 10.1016/0300-9084(92)90070-U
43. Wick AN, Drury DR, Morita TN. 2-Deoxyglucose; a metabolic block for glucose. Proc Soc Exp Biol Med (1955) 89(4):579–82. doi: 10.3181/00379727-89-21881
44. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
45. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol (2016) 17(12):1459–66. doi: 10.1038/ni.3577
46. Mehta MM, Weinberg SE, Steinert EM, Chhiba K, Martinez CA, Gao P, et al. Hexokinase 2 is dispensable for T cell-dependent immunity. Cancer Metab (2018) 6:10. doi: 10.1186/s40170-018-0184-5
47. Pusapati RV, Daemen A, Wilson C, Sandoval W, Gao M, Haley B, et al. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell (2016) 29(4):548–62. doi: 10.1016/j.ccell.2016.02.018
48. Ralser M, Wamelink MM, Struys EA, Joppich C, Krobitsch S, Jakobs C, et al. A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth. Proc Natl Acad Sci USA (2008) 105(46):17807–11. doi: 10.1073/pnas.0803090105
49. Liu B, Salgado OC, Singh S, Hippen KL, Maynard JC, Burlingame AL, et al. The lineage stability and suppressive program of regulatory T cells require protein O-GlcNAcylation. Nat Commun (2019) 10(1):354. doi: 10.1038/s41467-019-08300-3
50. Hao X, Li Y, Wang J, Ma J, Zhao S, Ye X, et al. Deficient O-GlcNAc Glycosylation Impairs Regulatory T Cell Differentiation and Notch Signaling in Autoimmune Hepatitis. Front Immunol (2018) 9:2089. doi: 10.3389/fimmu.2018.02089
51. Sasawatari S, Okamoto Y, Kumanogoh A, Toyofuku T. Blockade of N-Glycosylation Promotes Antitumor Immune Response of T Cells. J Immunol (2020) 204(5):1373–85. doi: 10.4049/jimmunol.1900937
52. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol (2015) 16(11):1174–84. doi: 10.1038/ni.3269
53. Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D, et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity (2017) 47(5):875–89.e10. doi: 10.1016/j.immuni.2017.10.017
54. Carbone F, De Rosa V, Carrieri PB, Montella S, Bruzzese D, Porcellini A, et al. Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat Med (2014) 20(1):69–74. doi: 10.1038/nm.3411
55. Atretkhany KN, Mufazalov IA, Dunst J, Kuchmiy A, Gogoleva VS, Andruszewski D, et al. Intrinsic TNFR2 signaling in T regulatory cells provides protection in CNS autoimmunity. Proc Natl Acad Sci USA (2018) 115(51):13051–6. doi: 10.1073/pnas.1807499115
56. Chandel NS. Evolution of Mitochondria as Signaling Organelles. Cell Metab (2015) 22(2):204–6. doi: 10.1016/j.cmet.2015.05.013
57. Wedel J, Stack MP, Seto T, Sheehan MM, Flynn EA, Stillman IE, et al. T Cell-Specific Adaptor Protein Regulates Mitochondrial Function and CD4(+) T Regulatory Cell Activity In Vivo following Transplantation. J Immunol (2019) 203(8):2328–38. doi: 10.4049/jimmunol.1801604
58. Kunisada Y, Eikawa S, Tomonobu N, Domae S, Uehara T, Hori S, et al. Attenuation of CD4(+)CD25(+) Regulatory T Cells in the Tumor Microenvironment by Metformin, a Type 2 Diabetes Drug. EBioMedicine (2017) 25:154–64. doi: 10.1016/j.ebiom.2017.10.009
59. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev (2014) 94(3):909–50. doi: 10.1152/physrev.00026.2013
60. Alissafi T, Kalafati L, Lazari M, Filia A, Kloukina I, Manifava M, et al. Mitochondrial Oxidative Damage Underlies Regulatory T Cell Defects in Autoimmunity. Cell Metab (2020) 32(4):591–604.e7. doi: 10.1016/j.cmet.2020.07.001
61. Dubois V, Eeckhoute J, Lefebvre P, Staels B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J Clin Invest (2017) 127(4):1202–14. doi: 10.1172/JCI88894
62. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature (2012) 486(7404):549–53. doi: 10.1038/nature11132
63. Hontecillas R, Bassaganya-Riera J. Peroxisome proliferator-activated receptor gamma is required for regulatory CD4+ T cell-mediated protection against colitis. J Immunol (2007) 178(5):2940–9. doi: 10.4049/jimmunol.178.5.2940
64. Bassaganya-Riera J, Reynolds K, Martino-Catt S, Cui Y, Hennighausen L, Gonzalez F, et al. Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology (2004) 127(3):777–91. doi: 10.1053/j.gastro.2004.06.049
65. Lei J, Hasegawa H, Matsumoto T, Yasukawa M. Peroxisome proliferator-activated receptor alpha and gamma agonists together with TGF-beta convert human CD4+CD25- T cells into functional Foxp3+ regulatory T cells. J Immunol (2010) 185(12):7186–98. doi: 10.4049/jimmunol.1001437
66. Unoda K, Doi Y, Nakajima H, Yamane K, Hosokawa T, Ishida S, et al. Eicosapentaenoic acid (EPA) induces peroxisome proliferator-activated receptors and ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol (2013) 256(1-2):7–12. doi: 10.1016/j.jneuroim.2012.12.003
67. Kim JY, Lim K, Kim KH, Kim JH, Choi JS, Shim SC. N-3 polyunsaturated fatty acids restore Th17 and Treg balance in collagen antibody-induced arthritis. PloS One (2018) 13(3):e0194331. doi: 10.1371/journal.pone.0194331
68. Onodera T, Fukuhara A, Shin J, Hayakawa T, Otsuki M, Shimomura I. Eicosapentaenoic acid and 5-HEPE enhance macrophage-mediated Treg induction in mice. Sci Rep (2017) 7(1):4560. doi: 10.1038/s41598-017-04474-2
69. Ye P, Li J, Wang S, Xie A, Sun W, Xia J. Eicosapentaenoic acid disrupts the balance between Tregs and IL-17+ T cells through PPARgamma nuclear receptor activation and protects cardiac allografts. J Surg Res (2012) 173(1):161–70. doi: 10.1016/j.jss.2010.08.052
70. van den Elsen LW, van Esch BC, Hofman GA, Kant J, van de Heijning BJ, Garssen J, et al. Dietary long chain n-3 polyunsaturated fatty acids prevent allergic sensitization to cow’s milk protein in mice. Clin Exp Allergy (2013) 43(7):798–810. doi: 10.1111/cea.12111
71. Giguere V, Ong ES, Segui P, Evans RM. Identification of a receptor for the morphogen retinoic acid. Nature (1987) 330(6149):624–9. doi: 10.1038/330624a0
72. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (2007) 317(5835):256–60. doi: 10.1126/science.1145697
73. Kang SG, Wang C, Matsumoto S, Kim CH. High and low vitamin A therapies induce distinct FoxP3+ T-cell subsets and effectively control intestinal inflammation. Gastroenterology (2009) 137(4):1391–402 e1-6. doi: 10.1053/j.gastro.2009.06.063
74. Wang J, Huizinga TW, Toes RE. De novo generation and enhanced suppression of human CD4+CD25+ regulatory T cells by retinoic acid. J Immunol (2009) 183(6):4119–26. doi: 10.4049/jimmunol.0901065
75. Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, et al. Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc Natl Acad Sci USA (2014) 111(33):E3432–40. doi: 10.1073/pnas.1408780111
76. Nolting J, Daniel C, Reuter S, Stuelten C, Li P, Sucov H, et al. Retinoic acid can enhance conversion of naive into regulatory T cells independently of secreted cytokines. J Exp Med (2009) 206(10):2131–9. doi: 10.1084/jem.20090639
77. Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol (2008) 181(4):2277–84. doi: 10.4049/jimmunol.181.4.2277
78. Zhou X, Kong N, Wang J, Fan H, Zou H, Horwitz D, et al. Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J Immunol (2010) 185(5):2675–9. doi: 10.4049/jimmunol.1000598
79. Schambach F, Schupp M, Lazar MA, Reiner SL. Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur J Immunol (2007) 37(9):2396–9. doi: 10.1002/eji.200737621
80. Carmona R, Barrena S, Munoz-Chapuli R. Retinoids in Stellate Cells: Development, Repair, and Regeneration. J Dev Biol (2019) 7(2):10. doi: 10.3390/jdb7020010
81. Dunham RM, Thapa M, Velazquez VM, Elrod EJ, Denning TL, Pulendran B, et al. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J Immunol (2013) 190(5):2009–16. doi: 10.4049/jimmunol.1201937
82. Kinoshita M, Kayama H, Kusu T, Yamaguchi T, Kunisawa J, Kiyono H, et al. Dietary folic acid promotes survival of Foxp3+ regulatory T cells in the colon. J Immunol (2012) 189(6):2869–78. doi: 10.4049/jimmunol.1200420
83. Lu D, Lan B, Din Z, Chen H, Chen G. A vitamin D receptor agonist converts CD4+ T cells to Foxp3+ regulatory T cells in patients with ulcerative colitis. Oncotarget (2017) 8(32):53552–62. doi: 10.18632/oncotarget.18614
84. Gregori S, Giarratana N, Smiroldo S, Uskokovic M, Adorini L. A 1alpha,25-dihydroxyvitamin D(3) analog enhances regulatory T-cells and arrests autoimmune diabetes in NOD mice. Diabetes (2002) 51(5):1367–74. doi: 10.2337/diabetes.51.5.1367
85. Oyarce K, Campos-Mora M, Gajardo-Carrasco T, Pino-Lagos K. Vitamin C Fosters the In Vivo Differentiation of Peripheral CD4(+) Foxp3(-) T Cells into CD4(+) Foxp3(+) Regulatory T Cells but Impairs Their Ability to Prolong Skin Allograft Survival. Front Immunol (2018) 9:112. doi: 10.3389/fimmu.2018.00112
86. Nair VS, Song MH, Ko M, Oh KI. DNA Demethylation of the Foxp3 Enhancer Is Maintained through Modulation of Ten-Eleven-Translocation and DNA Methyltransferases. Mol Cells (2016) 39(12):888–97. doi: 10.14348/molcells.2016.0276
87. Terrier B, Derian N, Schoindre Y, Chaara W, Geri G, Zahr N, et al. Restoration of regulatory and effector T cell balance and B cell homeostasis in systemic lupus erythematosus patients through vitamin D supplementation. Arthritis Res Ther (2012) 14(5):R221. doi: 10.1186/ar4060
88. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med (1986) 315(26):1650–9. doi: 10.1056/NEJM198612253152606
89. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
90. Brauker J, Martinson LA, Young SK, Johnson RC. Local inflammatory response around diffusion chambers containing xenografts. Nonspecific destruction of tissues and decreased local vascularization. Transplantation (1996) 61(12):1671–7. doi: 10.1097/00007890-199606270-00002
91. Min JH, Yang H, Ivan M, Gertler F, Kaelin WG Jr., Pavletich NP. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science (2002) 296(5574):1886–9. doi: 10.1126/science.1073440
92. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature (1999) 399(6733):271–5. doi: 10.1038/20459
93. Masoud GN, Li W. HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B (2015) 5(5):378–89. doi: 10.1016/j.apsb.2015.05.007
94. Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, et al. HIF-1alpha Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep (2019) 27(1):226–37 e4. doi: 10.1016/j.celrep.2019.03.029
95. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA (2012) 109(41):E2784–93. doi: 10.1073/pnas.1202366109
96. Shepherd AP. Metabolic control of intestinal oxygenation and blood flow. Fed Proc (1982) 41(6):2084–9.
97. Blagih J, Zani F, Chakravarty P, Hennequart M, Pilley S, Hobor S, et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep (2020) 30(2):481–96.e6. doi: 10.1016/j.celrep.2019.12.028
98. McKeown SR. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br J Radiol (2014) 87(1035):20130676. doi: 10.1259/bjr.20130676
99. Leach RM, Treacher DF. Oxygen transport-2. Tissue hypoxia. BMJ (1998) 317(7169):1370–3. doi: 10.1136/bmj.317.7169.1370
100. Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med (2011) 15(6):1239–53. doi: 10.1111/j.1582-4934.2011.01258.x
101. Muroski ME, Miska J, Chang AL, Zhang P, Rashidi A, Moore H, et al. Fatty Acid Uptake in T Cell Subsets Using a Quantum Dot Fatty Acid Conjugate. Sci Rep (2017) 7(1):5790. doi: 10.1038/s41598-017-05556-x
102. Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, et al. Oxygen Sensing by T Cells Establishes an Immunologically Tolerant Metastatic Niche. Cell (2016) 166(5):1117–31. doi: 10.1016/j.cell.2016.07.032
103. Garcia-Canaveras JC, Chen L, Rabinowitz JD. The Tumor Metabolic Microenvironment: Lessons from Lactate. Cancer Res (2019) 79(13):3155–62. doi: 10.1158/0008-5472.CAN-18-3726
104. Pucino V, Certo M, Bulusu V, Cucchi D, Goldmann K, Pontarini E, et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4(+) T Cell Metabolic Rewiring. Cell Metab (2019) 30(6):1055–74.e8. doi: 10.1016/j.cmet.2019.10.004
105. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D’Acquisto F, et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PloS Biol (2015) 13(7):e1002202. doi: 10.1371/journal.pbio.1002202
106. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature (2021). doi: 10.1038/s41586-020-03045-2
107. Zappasodi R, Serganova I, Cohen IJ, Maeda M, Shindo M, Senbabaoglu Y, et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature (2021). doi: 10.1038/s41586-021-03326-4
108. Comito G, Iscaro A, Bacci M, Morandi A, Ippolito L, Parri M, et al. Lactate modulates CD4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene (2019) 38(19):3681–95. doi: 10.1038/s41388-019-0688-7
109. Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol (2020) 21(3):298–308. doi: 10.1038/s41590-019-0589-5
110. Albina JE, Mills CD, Barbul A, Thirkill CE, Henry WL Jr., Mastrofrancesco B, et al. Arginine metabolism in wounds. Am J Physiol (1988) 254(4 Pt 1):E459–67. doi: 10.1152/ajpendo.1988.254.4.E459
111. Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife (2019) 8:e44235. doi: 10.7554/eLife.44235
112. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40(5):692–705. doi: 10.1016/j.immuni.2014.04.007
113. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14(5):500–8. doi: 10.1038/ni.2556
114. Utsunomiya-Tate N, Endou H, Kanai Y. Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J Biol Chem (1996) 271(25):14883–90. doi: 10.1074/jbc.271.25.14883
115. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal (2015) 8(396):ra97. doi: 10.1126/scisignal.aab2610
116. Metzler B, Gfeller P, Guinet E. Restricting Glutamine or Glutamine-Dependent Purine and Pyrimidine Syntheses Promotes Human T Cells with High FOXP3 Expression and Regulatory Properties. J Immunol (2016) 196(9):3618–30. doi: 10.4049/jimmunol.1501756
117. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell (2018) 175(7):1780–95.e19. doi: 10.1016/j.cell.2018.10.001
118. Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature (2017) 548(7666):228–33. doi: 10.1038/nature23475
119. Closs EI, Simon A, Vekony N, Rotmann A. Plasma membrane transporters for arginine. J Nutr (2004) 134(10 Suppl):2752S–9S; discussion 65S-67S. doi: 10.1093/jn/134.10.2752S
120. Werner A, Amann E, Schnitzius V, Habermeier A, Luckner-Minden C, Leuchtner N, et al. Induced arginine transport via cationic amino acid transporter-1 is necessary for human T-cell proliferation. Eur J Immunol (2016) 46(1):92–103. doi: 10.1002/eji.201546047
121. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell (2013) 155(6):1282–95. doi: 10.1016/j.cell.2013.10.054
122. Albaugh VL, Pinzon-Guzman C, Barbul A. Arginine-Dual roles as an onconutrient and immunonutrient. J Surg Oncol (2017) 115(3):273–80. doi: 10.1002/jso.24490
123. Lowe MM, Boothby I, Clancy S, Ahn RS, Liao W, Nguyen DN, et al. Regulatory T cells use arginase 2 to enhance their metabolic fitness in tissues. JCI Insight (2019) 4(24):e129756. doi: 10.1172/jci.insight.129756
124. Lee YC, Su YT, Liu TY, Tsai CM, Chang CH, Yu HR. L-Arginine and L-Citrulline Supplementation Have Different Programming Effect on Regulatory T-Cells Function of Infantile Rats. Front Immunol (2018) 9:2911. doi: 10.3389/fimmu.2018.02911
125. Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature (2015) 527(7578):379–83. doi: 10.1038/nature15529
126. Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab (2017) 25(2):482. doi: 10.1016/j.cmet.2017.01.014
127. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity (2017) 46(6):1089–90. doi: 10.1016/j.immuni.2017.06.009
128. Kurniawan H, Franchina DG, Guerra L, Bonetti L, Baguet LS, Grusdat M, et al. Glutathione Restricts Serine Metabolism to Preserve Regulatory T Cell Function. Cell Metab (2020) 31(5):920–36.e7. doi: 10.1016/j.cmet.2020.03.004
129. Bhutia YD, Babu E, Ramachandran S, Ganapathy V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res (2015) 75(9):1782–8. doi: 10.1158/0008-5472.CAN-14-3745
130. Stoll B, Henry J, Reeds PJ, Yu H, Jahoor F, Burrin DG. Catabolism dominates the first-pass intestinal metabolism of dietary essential amino acids in milk protein-fed piglets. J Nutr (1998) 128(3):606–14. doi: 10.1093/jn/128.3.606
131. Brosnan JT, Brosnan ME. Branched-chain amino acids: enzyme and substrate regulation. J Nutr (2006) 136(1 Suppl):207S–11S. doi: 10.1093/jn/136.1.207S
132. Ikeda K, Kinoshita M, Kayama H, Nagamori S, Kongpracha P, Umemoto E, et al. Slc3a2 Mediates Branched-Chain Amino-Acid-Dependent Maintenance of Regulatory T Cells. Cell Rep (2017) 21(7):1824–38. doi: 10.1016/j.celrep.2017.10.082
133. Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med (2014) 20(10):1193–8. doi: 10.1038/nm.3686
134. Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med (2002) 196(4):447–57. doi: 10.1084/jem.20020052
135. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9(10):1269–74. doi: 10.1038/nm934
136. Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-gamma-induced immunologic dormancy of tumor-repopulating cells. Nat Commun (2017) 8:15207. doi: 10.1038/ncomms15207
137. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. Tryptophan catabolism generates autoimmune-preventive regulatory T cells. Transpl Immunol (2006) 17(1):58–60. doi: 10.1016/j.trim.2006.09.017
138. Chen W, Liang X, Peterson AJ, Munn DH, Blazar BR. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J Immunol (2008) 181(8):5396–404. doi: 10.4049/jimmunol.181.8.5396
139. Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, et al. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood (2009) 113(24):6102–11. doi: 10.1182/blood-2008-12-195354
140. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev (2004) 18(16):1926–45. doi: 10.1101/gad.1212704
141. Buttgereit F, Brand MD. A hierarchy of ATP-consuming processes in mammalian cells. Biochem J (1995) 312(Pt 1):163–7. doi: 10.1042/bj3120163
142. Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell (2010) 141(2):290–303. doi: 10.1016/j.cell.2010.02.024
143. Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet (2005) 14(Spec No. 2):R251–8. doi: 10.1093/hmg/ddi260
144. Roux PP, Topisirovic I. Regulation of mRNA translation by signaling pathways. Cold Spring Harb Perspect Biol (2012) 4(11):a012252. doi: 10.1101/cshperspect.a012252
145. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30(6):832–44. doi: 10.1016/j.immuni.2009.04.014
146. Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity (2010) 32(6):743–53. doi: 10.1016/j.immuni.2010.06.002
147. Do MH, Wang X, Zhang X, Chou C, Nixon BG, Capistrano KJ, et al. Nutrient mTORC1 signaling underpins regulatory T cell control of immune tolerance. J Exp Med (2020) 217(1):e20190848. doi: 10.1084/jem.20190848
148. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature (2013) 499(7459):485–90. doi: 10.1038/nature12297
149. Shi H, Chapman NM, Wen J, Guy C, Long L, Dhungana Y, et al. Amino Acids License Kinase mTORC1 Activity and Treg Cell Function via Small G Proteins Rag and Rheb. Immunity (2019) 51(6):1012–27 e7. doi: 10.1016/j.immuni.2019.10.001
150. Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity (2010) 33(6):929–41. doi: 10.1016/j.immuni.2010.11.024
151. Moore WE, Cato EP, Holdeman LV. Anaerobic bacteria of the gastrointestinal flora and their occurrence in clinical infections. J Infect Dis (1969) 119(6):641–9. doi: 10.1093/infdis/119.6.641
152. Hill MJ. Diet and the human intestinal bacterial flora. Cancer Res (1981) 41(9 Pt 2):3778–80. doi: 10.1007/BF01295996
153. Roediger WE. The colonic epithelium in ulcerative colitis: an energy-deficiency disease? Lancet (1980) 2(8197):712–5. doi: 10.1016/S0140-6736(80)91934-0
154. Gordon HA, Nakamura S. Elevated levels of colloid osmotic pressure in cecal contents of germfree animals. Proc Soc Exp Biol Med (1975) 149(1):46–9. doi: 10.3181/00379727-149-38740
155. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504(7480):446–50. doi: 10.1038/nature12721
156. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341(6145):569–73. doi: 10.1126/science.1241165
157. Wu H, Singer J, Kwan TK, Loh YW, Wang C, Tan J, et al. Gut Microbial Metabolites Induce Donor-Specific Tolerance of Kidney Allografts through Induction of T Regulatory Cells by Short-Chain Fatty Acids. J Am Soc Nephrol (2020) 31(7):1445–61. doi: 10.1681/ASN.2019080852
158. Marino E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol (2017) 18(5):552–62. doi: 10.1038/ni.3713
159. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504(7480):451–5. doi: 10.1038/nature12726
160. Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr (2003) 133(7 Suppl):2485S–93S. doi: 10.1093/jn/133.7.2485S
161. Davis I, Liu A. What is the tryptophan kynurenine pathway and why is it important to neurotherapeutics? Expert Rev Neurother (2015) 15(7):719–21. doi: 10.1586/14737175.2015.1049999
162. Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun (2018) 9(1):3294. doi: 10.1038/s41467-018-05470-4
163. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453(7191):65–71. doi: 10.1038/nature06880
164. Goettel JA, Gandhi R, Kenison JE, Yeste A, Murugaiyan G, Sambanthamoorthy S, et al. AHR Activation Is Protective against Colitis Driven by T Cells in Humanized Mice. Cell Rep (2016) 17(5):1318–29. doi: 10.1016/j.celrep.2016.09.082
165. Ehrlich AK, Pennington JM, Wang X, Rohlman D, Punj S, Lohr CV, et al. Activation of the Aryl Hydrocarbon Receptor by 10-Cl-BBQ Prevents Insulitis and Effector T Cell Development Independently of Foxp3+ Regulatory T Cells in Nonobese Diabetic Mice. J Immunol (2016) 196(1):264–73. doi: 10.4049/jimmunol.1501789
166. Aoki R, Aoki-Yoshida A, Suzuki C, Takayama Y. Indole-3-Pyruvic Acid, an Aryl Hydrocarbon Receptor Activator, Suppresses Experimental Colitis in Mice. J Immunol (2018) 201(12):3683–93. doi: 10.4049/jimmunol.1701734
167. Noakes R. The aryl hydrocarbon receptor: a review of its role in the physiology and pathology of the integument and its relationship to the tryptophan metabolism. Int J Tryptophan Res (2015) 8:7–18. doi: 10.4137/IJTR.S19985
168. Quintana FJ. The aryl hydrocarbon receptor: a molecular pathway for the environmental control of the immune response. Immunology (2013) 138(3):183–9. doi: 10.1111/imm.12046
169. Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol (2010) 11(9):846–53. doi: 10.1038/ni.1915
170. Busbee PB, Menzel L, Alrafas HR, Dopkins N, Becker W, Miranda K, et al. Indole-3-carbinol prevents colitis and associated microbial dysbiosis in an IL-22-dependent manner. JCI Insight (2020) 5(1):e127551. doi: 10.1172/jci.insight.127551
171. Singh NP, Singh UP, Rouse M, Zhang J, Chatterjee S, Nagarkatti PS, et al. Dietary Indoles Suppress Delayed-Type Hypersensitivity by Inducing a Switch from Proinflammatory Th17 Cells to Anti-Inflammatory Regulatory T Cells through Regulation of MicroRNA. J Immunol (2016) 196(3):1108–22. doi: 10.4049/jimmunol.1501727
172. Matteoli G, Mazzini E, Iliev ID, Mileti E, Fallarino F, Puccetti P, et al. Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut (2010) 59(5):595–604. doi: 10.1136/gut.2009.185108
173. Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, et al. Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity (2019) 50(2):493–504 e7. doi: 10.1016/j.immuni.2019.01.001
174. Xiao Z, Dai Z, Locasale JW. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat Commun (2019) 10(1):3763. doi: 10.1038/s41467-019-11738-0
175. Cuadrado E, van den Biggelaar M, de Kivit S, Chen YY, Slot M, Doubal I, et al. Proteomic Analyses of Human Regulatory T Cells Reveal Adaptations in Signaling Pathways that Protect Cellular Identity. Immunity (2018) 48(5):1046–59 e6. doi: 10.1016/j.immuni.2018.04.008
176. Sinclair LV, Barthelemy C, Cantrell DA. Single Cell Glucose Uptake Assays: A Cautionary Tale. Immunometabolism (2020) 2(4):e200029. doi: 10.20900/immunometab20200029
177. Hulme HE, Meikle LM, Wessel H, Strittmatter N, Swales J, Thomson C, et al. Mass spectrometry imaging identifies palmitoylcarnitine as an immunological mediator during Salmonella Typhimurium infection. Sci Rep (2017) 7(1):2786. doi: 10.1038/s41598-017-03100-5
178. Newton R, Priyadharshini B, Turka LA. Immunometabolism of regulatory T cells. Nat Immunol (2016) 17(6):618–25. doi: 10.1038/ni.3466
179. Wing JB, Tanaka A, Sakaguchi S. Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity (2019) 50(2):302–16. doi: 10.1016/j.immuni.2019.01.020
Keywords: regulatory T cells, metabolism, tissues, metabolic adaptation, nutrients
Citation: Blagih J, Hennequart M and Zani F (2021) Tissue Nutrient Environments and Their Effect on Regulatory T Cell Biology. Front. Immunol. 12:637960. doi: 10.3389/fimmu.2021.637960
Received: 04 December 2020; Accepted: 08 March 2021;
Published: 02 April 2021.
Edited by:
Daniel Puleston, Max Planck Institute for Immunobiology and Epigenetics, GermanyReviewed by:
Hongbo Chi, St. Jude Children’s Research Hospital, United StatesGiuseppe Matarese, University of Naples Federico II, Italy
Copyright © 2021 Blagih, Hennequart and Zani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julianna Blagih, anVsaWFubmEuYmxhZ2loQGNyaWNrLmFjLnVr; Marc Hennequart, bWFyYy5oZW5uZXF1YXJ0QGNyaWNrLmFjLnVr