 Marilia Cascalho
Marilia Cascalho Jeffrey L. Platt
Jeffrey L. Platt
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol. , 17 February 2021
Sec. B Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.634544
This article is part of the Research Topic The B-Side of B Cells View all 10 articles
B cell differentiation and memory are controlled by the transmembrane activator and CAML interactor (TACI), a receptor encoded by TNFRSF13B. TNFRSF13B mutations are frequently found in common variable immunodeficiency (CVID) and in IgA -deficiency; yet, ~98% of those with mutant TNFRSF13B are healthy. Indeed, TNFRSF13B is among the 5% most polymorphic genes in man. Other mammals evidence polymorphism at comparable loci. We hypothesize that TNFRSF13B diversity might promote rather than detract from well-being by controlling key elements of innate immunity. We shall discuss how extraordinary diversity of TNFRSF13B could have evolved and persisted across diverse species of mammals by controlling innate and adaptive B cell responses in apparently paradoxical ways.
B cell responses are often characterized as T cell-independent or T cell-dependent that differ on how T cells are engaged. In “T-independent B cell responses” antigens with repetitive epitopes, such as polysaccharides, engage B cell antigen receptors and/or toll-like receptors and in doing so induce proliferation and plasma cell differentiation. The antibodies produced in T cell-independent responses may appear relatively soon after introduction of antigen, but not immediately as in recall responses. The Ig variable region genes encoding these antibodies typically lack extensive somatic mutation (1). Repeated exposure to antigen that had generated a T cell-independent response does not hasten and amplify the response [i.e., B cell memory is not manifest; (1)]. In contrast, the T-dependent pathway requires T cell help and is associated with responses to protein antigens. Proteins are processed and peptides presented on major histocompatibility class II molecules expressed by antigen presenting cells that activate cognate CD4-positive T cells. These T cells in turn, engage cognate B cells activated by the same antigen by binding peptide MHC class-II complexes and co-receptors such as CD40. T cell-dependent responses induce long-lived memory responses and are associated with high affinity binding antibodies produced by plasma cells descending from the germinal centers in the secondary lymphoid organs (1). Naturally occurring polymorphisms in the TNFRSF13B gene differentially control T-dependent and T-independent pathways of antibody production.
TNFRSF13B encodes the tumor necrosis factor superfamily member 13B, a transmembrane receptor of lymphocytes that recognizes a proliferation induced ligand (APRIL) and B cell activation factor (BAFF), members of the tumor necrosis ligand family (2). TNFRSF13B also binds heparan sulfate chains associated with syndecan-2 and−4 cores (3). The signaling events initiated by TNFRSF13B are complex and intersect with signaling by Toll-Like receptors (TLRs) and will be only briefly summarized here. Binding of BAFF and APRIL to the cysteine rich domain of the receptor closest to the cell membrane (CRD2) engages TNFR—associated factors (TRAF 2, 5, and 6) and activates NF-kB, c-Jun NH2-terminal kinase (4) and activator protein 1 (AP-1) (5). TNFRSF13B interacts with calcium modulator and cyclophilin ligand (CAML), which in turn activates calcineurin and nuclear factor of activated T cells (NFAT) (6). TNFRSF13B is sometimes called “transmembrane activator and CAML interactor” or TACI (6) reflecting this series of interactions. TNFRSF13B potentiates signaling by Toll-like family receptors in B cells (7) and in macrophages (8). Accordingly, TNFRSF13B interacts with MyD88, recruits mechanistic target of rapamycin (mTOR), activates mTORC1 and NF-kB (9–11). TNFRSF13B signaling in B cells generates expression of BLIMP-1, a transcription factor that drives differentiation of B cells into long-lived plasma cells (12).
The importance of TNFRSF13B and BLIMP-1 for development of plasma cells and production of much of the Ig in blood was suggested by investigation of genetic basis of hypogammaglobulinemia, i.e., IgG-deficiency, IgM-deficiency, and IgA-deficiency observed in common variable immunodeficiency (CVID) and in selective IgA deficiency (13, 14). Consistent with this phenotype, TNFRSF13B-deficient mice have few plasma cells in secondary lymphoid organs and in the bone marrow and low concentrations of IgM, IgA, and IgG in serum (12). However, TNFRSF13B governs more than the machinery for long term-Ig production. Human subjects with CVID have an increased risk of lymphoma and gastro-intestinal cancer (15) and a propensity for development of autoimmunity (16). Mice with deficient tnfrsf13b exhibit pronounced expansion of follicular and germinal center B cells, despite hypogammaglobulinemia, suggesting tnfrsf13B may govern B cell differentiation and T and B cell interactions (12, 17, 18).
Although some functions of TNFRSF13B, such as control of plasma cell differentiation are understood, some puzzling contradictions remain. One contradiction concerns the impact of TNFRSF13B on the B cell response to antigen. TACI appears more or less essential for natural immunity because humans and mice lacking TACI (targeted deletion in mouse; expression of dominant-negative variants in humans) have extremely low levels of IgG, IgM, and IgA in blood (19) and produce little antigen specific antibodies after exposure to antigen or foreign organisms (20–22). However, most people with dominant negative TACI variants do not manifest immunodeficiency (23) and TACI knockout mice and mice expressing dominant negative TACI variants corresponding to those in humans mount proficient antibody responses and antibody-mediated defenses against pathogenic bacteria (17). Still more puzzling is the relationship between diversity of TNFRSF13B genotypes and phenotype. We shall describe recent work that may have begun to clarify apparently disparate aspects of the TNFRSF13B phenotype and identify yet unsettled questions we think of importance.
Recent investigations in mice and human subjects have clarified discrepancies concerning the impact of TNFRSF13B on B cell responses to antigen. It is now apparent that stimulation of TNFRSF13B is essential for T-independent- but not for T-cell-dependent B cell responses. The requirement for TNFRSF13B (TACI) function for mounting T-independent antibody responses was first shown by von Bulow et al. (24), who found that TACI-KO mice produce less antibodies in response to immunization with pneumococcus. Failure of T cell-independent responses in tnfrsf13B-mutant mice was confirmed by Wolf et al. (22) and Mantchev et al. (25), who showed the defective T-independent responses were due to a block in plasma cell differentiation. Tsuji et al. (12) found that defective plasma cell differentiation was due to defective Blimp-1 synthesis. Similarly, Grasset et al. (18) reported that production of gut IgA by T-independent response depends on tnfrsf13B. In contrast, human subjects with TNFRSF13B mutations, including those with dominant negative phenotype and mice with tnfrsf13B deficiency or dominant negative variants respond to antigen when T cell help is provided. Tsuji et al. (12, 17) reported that tnfrsf13B-deficient mice are quite proficient in responses to polypeptide antigens associated with enteric organisms (or purified therefrom), generating high affinity antibodies and conferring protection against reinfection. Likewise, Grasset et al. (18) found tnfrsf13B deficient mice quite effectively mount gut IgA responses if T cell help is present.
The differential impact of TNFRSF13B on T cell-independent and T-cell-dependent responses may reflect differences in how Blimp-1 is induced. In T cell-independent responses, induction of Blimp-1 depends absolutely on stimulation of TNFRSF13B by BAFF or APRIL (12, 17). In T cell-dependent responses, double strand DNA breaks generated by class-switch recombination (17) and/or engagement of CD40 and IL21/STAT3 (26) signaling can induce Blimp-1 independently of TNFRSF13B signaling. Although TNFRSF13B may not directly impact on Ig isotype class switching, changes of B cell development post-activation by TNFRSF13B mutation, or deletion, that interfere with signaling change the distribution of Ig isotypes in response to stimulation and in the steady state (27).
How TNFRSF13B signaling contributes to the generation and activation of memory B cells remains incompletely understood. The increase in TNFRSF13B expression by memory B cells suggests that TNFRSF13B signaling is important for memory B cell differentiation, survival or function (28). Indeed memory B cells from CVID subjects with monoallelic C104R or A181E mutations fail to activate in response to BCR, TLR7, or TLR9 stimuli (28). Memory B cells of subjects with TNFRSF13B hemizygosity (one null allele and one WT allele) mount a partial response to activation to BCR, TLR7, or TLR9 stimuli suggesting that signaling intensity is important in determining magnitude of response (28). Whether or not the phenotypic manifestations of monoallelic dominant-negative mutations or haplo-deficiency impact memory B cell responses to T-dependent stimuli and in subjects who are healthy is not known.
The diversity of TNFRSF13B, by some measures, equals or exceeds that of genes encoding the major histocompatibility complex (MHC) but the mechanisms contributing to TNFRSF13B diversity are less apparent. Diversity of MHC is generally ascribed to the function of housing a vast diversity of microbial peptides for presentation to T cells (29) and while some diversification likely reflects specificity for peptides, some may also reflect non-effector functions such as recognition leading to immune regulation for MHC-class II or controlling NK cells for MHC class I (29).
Yet, TNFRSF13B encodes a polypeptide the sole function of which may be recognition of several relatively non-polymorphic agonists, BAFF and APRIL (30). Why then do humans exhibit extreme genetic polymorphism of TNFRSF13B? There are 951 TNFRSF13B missense and only 383 synonymous mutations reported in compilations of more than 100.000 human genomes (https://useast.ensembl.org/index.html). Only 4 of the TNFRSF13B missense alleles are common (freq >0.05). Unlike TNFRSF13B variants, nearly 20% of HLA-A variants are found in 5% or more of the population (Genome Aggregation Database, v2.1.1; http://exac.broadinstitute.org). TNFRSF13B has a greater number of missense variants than it would be predicted by a sequence-context-based mutational model as reflected by a z score of −1.2, and TNFRSF13B has more observed than expected loss of function variants (LoF) (stop gain and splice site variants), which indicate a high tolerance to these types of mutations (pLI = 0.00, where pLI of 1 is the most intolerant) [Genome Aggregation Database, v2.1.1; http://exac.broadinstitute.org; (31)]. Most common variants are phenotypically dominant, either as dominant negatives or causing haplo-insufficiency (32–34) but how exactly expression of mutant alleles results in changes in TNFRSF13B signaling and function is incompletely known (10). In contrast, the number of missense mutations in the HLA-A gene is as expected by a sequence-context-based mutational model as (z score of −0.1 according to the Genome Aggregation Database, v2.1.1; http://exac.broadinstitute.org) and the HLA-A gene is less tolerant than TNFRSF13B to loss of function mutations. Thus, TNFRSF13B missense alleles appear to have been selectively retained and recent analysis by the McDonald-Kreitman neutrality index suggests the locus is under strong positive selection (35) in contrast with prior analysis of smaller cohorts (36). This is in contrast to genes encoding HLA which are under moderate purifying pressure (35). Some TNFRSF13B polymorphisms are conserved across mammalian species. As an example, mice have 17 missense alleles, 2 non-sense alleles, and 2 splice variants (37).
Adding to TNFRSF13B diversity the receptor is expressed as two isoforms that differ by the presence (long, L) or absence (short, S) of exon 2 that is alternatively spliced following B cell activation (38). The short version of the receptor lacks the cysteine rich domain 1 thought to mediate ligand binding (38). However, absence of the CDR1 domain in the short form does not appear to preclude assembly of the receptor trimer or signaling. Garcia-Carmona et al. (10) showed that TNFRSF13B-S and TNFRSF13B-L assembled receptor complexes composed of one single S or L isoform, or of mixed complexes composed of both S and L isoforms. TNFRSF13B-S requires a lower ligand concentration to signal than TNFRSF13B-L, in part owing to increased ligand binding affinity (10). In contrast, certain mutated isoforms, C104R, A181E, and S194X, produced receptors that had impaired or no signaling (10).
TNFRSF13B polymorphisms may be maintained by balancing selection. Balancing selection is thought to occur when multiple alleles (variants) are maintained in the population in an equilibrium, at frequencies more evenly distributed than expected under models of neutral evolution, because selection favors the heterozygote. To measure the likelihood of balancing selection at the TNFRSF13B locus we used a statistic, the β index, which detects clusters in close proximity to a site targeted by balancing selection [https://academic.oup.com/mbe/article/34/11/2996/3988103; (39)]. TNFRSF13B manifests balancing selection in four regions (two of them between exons 2 and 3, and the other two between exons 4 and 5) (39). Research by Jagoda et al. (40) suggests that certain haplotypes of TNFRSF13B may be of Neanderthal in origin and kept in European/Eurasian populations as a result from adaptive introgression. Thus, TNFRSF13B variants may have originated by positive selection of archaic ancestry variants, maintained thereafter by balancing selection.
What the selection pressures are that maintain TNFRSF13B diversity is not known but our research and the research of others connects these properties to the control of T-independent natural IgA antibody production.
TNFRSF13B promotes IgA synthesis. In support, TNFRSF13B-deficient animals and animals with dominant-negative TNFRSF13B alleles are IgA deficient (12, 17, 20) and; selective IgA deficiency in humans is often associated with mutant TNFRSF13B alleles (41). We hypothesize that TNFRSF13B polymorphisms may in part be driven by the receptor impact on secretory IgA (sIgA).
First discovered in 1953, IgA is by some measures the most abundant Ig in the body (42, 43). IgA exists as a monomer in circulation and as a dimer in lumina of the respiratory, intestinal, and genito-urinary system. Secretory IgA makes 2/3 of all IgA produced in the body (44). An IgA monomer is, like other Ig isotypes, a tetramer composed of two identical heavy chains and two identical light chains united by covalent and non-covalent bonds. Each monomer contains two Fab domains and one Fc region that includes C alpha 2 and C alpha 3 exons (44). Humans, express IgA1 and IgA2, the later encoded by several distinct alleles. IgA1 and IgA2 differ on the hinge region that links constant domain alpha 1 to the constant domain alpha 2 of the heavy chain, longer in IgA1 and shorter on IgA2. The significance of the longer hinge region for the function of IgA1 is not completely known but modeling suggests that the longer hinge region may afford greater flexibility to the variable region relative to the Fc region at the cost of increased sensitivity to proteolysis (44).
The secreted form of IgA is a complex comprised of an IgA dimer linked by the joining (J) chain and a secretory component, a fragment of the polymeric Ig receptor (PIgR). Secretion and dimerization of IgA is made possible by two important adaptations. In one, IgA has an 18 aa tail piece at the C-terminus, highly homologous to another found at the C-terminus of IgM. The tail piece allows IgA (and IgM) polymerization because it binds to the J chain through 2 cysteine disulfide bonds (45, 46). The J chain is a 137 amino-acid polypeptide and it is bound to IgA before secretion. The second adaptation is the development of a highly specific IgA transport system across the epithelium and into secretions. The transport of IgA depends on the polymeric Ig receptor (pIgR) that binds only polymeric Ig (IgA or IgM) and is expressed on the basolateral surface of epithelial cells that line mucosal surfaces of the gut, lung biliary tract, and lacrimal glands. The pIgR has a 620 aa extra-cellular domain, a 23 aa transmembrane domain and a 103 aa cytoplasmic domain (47). Transcytosis of IgA from the submucosal region to the lumen requires binding of dimeric IgA C alpha 3 to the pIgR at the basolateral side of epithelial cells followed by internalization and transport to the apical surface of the cell. IgA is released from its attachment by cleavage of the pIgR ectodomain (secretory component) at the apical surface. IgA, covalently bound to the secretory component is released on to the mucosa, forming sIgA. Bacterial and viral products induce heightened pIgR synthesis by epithelial cells and hence increase transcytosis of IgA (47). The secretory component confers resistance to proteolysis and anti-microbial functions. In addition to binding to IgA, the secretory component exists as a free protein and as such it binds directly to a variety of microbes inhibiting adhesion to epithelial cells, binds to mucus mediating immune exclusion of antigens and pathogens, retains IL-8 and in this way, inhibits neutrophil chemotaxis (48).
IgA complexed with J chain and the secretory component is heavily glycosylated and glycosylation is necessary for many of non-specific, variable region-independent IgA functions (49). IgA complexes are modified by N-linked glycosylation at Asn (N) residues and by O-linked sugars. Glycosylation of the secretory component protects IgA from degradation (50) and mediates some IgA immune functions. Glycans enable secretory component binding to adhesins and lectins, to a wide range of bacteria and to toxins [(48), and references there-in]. Glycosylation of the secretory component determines the distribution of IgA to specific areas of the epithelium (51) and the secretory component is needed with IgA to neutralize rotavirus (52). Furthermore, secretory component free or complexed to IgA, binds to the lectin binding domain of Mac-1 (CR3, CD11b/CD18) enhancing IgA receptor Fc alpha signaling and augmenting phagocytosis and inflammation [reviewed in (53)].
J chain glycosylation is necessary for dimerization of IgA (54) and for maintaining the correct conformation to facilitate interaction with pIgR (55, 56). Serum IgA and secretory IgA are differentially glycosylated presumably reflecting differences in the glycosylation machinery in plasma cells of the spleen and bone marrow vis a vis plasma cells in the mucosae (48).
The heavy-chain N-linked glycans help maintain the correct IgA conformation and assist with dimer formation and secretion (57, 58). Although the glycan composition of IgA HCs activate the lectin complement activation pathway by binding mannose binding lectin (MBL), the glycans are shielded by the secretory component at physiologic pH and only become accessible for binding at low pH and perhaps upon engagement with bacterial adhesins which pull the secretory component away from the IgA (48). Once uncovered, IgA HC glycans may also directly bind mannose receptors on phagocytic cells promoting internalization and antigen presentation (59). Secretory IgA HCs are also extensively modified by O-linked glycans which form highly complex structures that in addition to stabilizing the hinge region also interact with bacterial adhesins (60). Glycosylation-dependent IgA functions are likely due to low affinity interactions and therefore critically dependent on IgA concentration. Although there is no evidence in support of TNFRSF13B direct influence on IgA glycosylation, by controlling IgA secretion TNFRSF13B is likely to impact more on the non-specific IgA functions than on the functions of IgA that depend on high affinity interactions between mutated V regions and their targets.
One might expect that a cytokine receptor such TNFRSF13B would exert straight forward functions that promote host defense and/or immune regulation and accordingly polymorphism should be rare and harmful. Yet, TNFRSF13B is as diverse as MHC. Why?
Although the story is yet incomplete, the complexity of TNFRSF13B gene and protein functions has been coming more fully into focus. Although TNFRSF13B does not bind antigen it controls response of B cells to T-independent and to T-dependent antigens differently. TNFRSF13B promotes T-independent antibody production in part by facilitating differentiation of plasma cells. In contrast, TNFRSF13B-deficiency does not inhibit and in some cases enhances IgG or IgA responses to T-dependent antigens.
Most sIgA results from T-independent B cell responses. IgA exerts protective functions by helping to eliminate pathogens directly and/or through maintaining microbial homeostasis and these functions are in a great extent independent of IgA specificity (61–63). The equilibrium established between IgA, microbes and pathogens results from anti-microbial functions of IgA and microbial adaptations to IgA (49, 64). In one example of a remarkable microbial adaptation, Nakajima et al. (49) showed that highly glycosylated IgA of an irrelevant specificity (specific to ovalbumin) bound to a human symbiont Bacteroides thetaiotaomicron (B. theta) changing gene expression. IgA induced transcription of B. theta polysaccharide utilization loci (65) changing polysaccharide utilization and in this way conferring competitive advantage (49). As another example, Bacteroides fragilis capsule induces polysaccharide specific IgA which in turn, increases its adherence to intestinal epithelial cells by binding to mucus as well as to the capsule polysaccharides (62). In these examples, commensal bacteria co-opt IgA to colonize the gut.
We hypothesize TNFRSF13B polymorphisms are maintained at least in part as the result of microbial adaptation to IgA (Figure 1). We offer the following reasoning in support of the hypothesis.
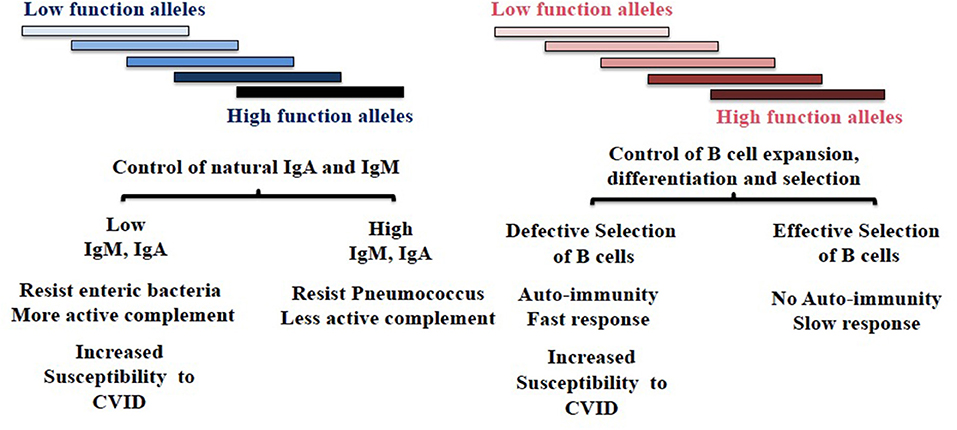
Figure 1. Diversity of TNFRSF13B in host response to environmental challenges. TNFRSF13B encodes a receptor (TACI) through to support the maturation of B cells, yet the gene is among the most polymorphic in the genome and polymorphism is maintained across mammalian species. The authors speculate that polymorphism of TNFRSF13B reflects balancing selection—balancing benefits and disadvantages of signals the receptor delivers but also manifold intermediate functions the protein and its neighbors in cell membrane confer. The figure depicts two hypothetically distinct regions of the protein that confer phenotypically distinct properties. The balancing of complex sets of phenotypic properties that presumably impact all mammals could underlie extraordinary diversity of the gene across species. CVID, Common Variable Immunodeficiency.
In a recent report Grasset et al. (18) showed that TNFRSF13B is necessary for T-independent IgA responses to commensal bacteria. These responses include secretion of polyreactive IgA and represent the majority of secreted IgA. In contrast, T cell-dependent IgA responses occur independently of TNFRSF13B. Thus, varying TNFRSF13B function changes the ratio between highly-specific and polyreactive or non-specific IgA responses which, in turn, may counteract pathogen adaptation to IgA and facilitate specific elimination. In support, Grasset et al. (18) showed that tnfrsf13b-KO mice generated mutated IgA that specifically targeted a restricted subset of microbes. In an earlier publication, Tsuji et al. (17) showed that tnfrsf13b-KO produced highly mutated antibodies that rapidly cleared C. rodentium. Whether changes of the properties and amount of sIgA and/or varying the proportion of mixed polyreactive and specific mono-reactive Ig in individuals or mice expressing TNFRSF13B polymorphisms is protective awaits investigation.
For all the benefits of IgA, selective IgA deficiency is often asymptomatic (41). TNFRSF13B polymorphisms are associated with IgA deficiency (41), the most common immune-deficiency (with frequencies varying between 1:143 in the Arabian peninsula to 1 in 500 Caucasian individuals) (66, 67). The limited morbidity of IgA deficiency, suggests as one possibility that benefits conferred by TNFRSF13B variants may outweigh the detrimental impact of decreased IgA in the gut. Because tnfrsf13b mutants maintain the ability to make “adaptive” IgA it is possible that protective functions of IgA are maintained or even enhanced. In accord, IgA-deficient patients were found to have enhanced adaptive antibody responses to pneumococcal vaccination (68). Perhaps it is this type of response that explains the mild phenotype of many individuals with IgA deficiency and the maintenance of the extreme polymorphism at the TNFRSF13B locus.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Animal studies and human subject research that inspired some of the hypotheses in the manuscript were reviewed and approved by the Institutional Animal Care & Use Committee (IACUC) or by the Institutional Review Board (IRB) at the University of Michigan.
MC and JP wrote the manuscript. MC analyzed data. All authors contributed to the article and approved the submitted version.
This research was supported by NIH grants R01 AI51588 and AI122369 (MC and JP), a grant from the State of Michigan, CASE-283529 UM-MTRAC for Life Sciences (MC and JP), NIH grant R21GM140359 (MC), and UM Michigan Institute for Clinical & Health Research Accelerating Synergy Award.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge the members of their laboratories for valuable discussions and insights, and Drs. Siewert and Voigt at the Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, who helped us interpret their statistics data on the TNFRSF13B locus.
1. Cyster JG, Allen CDC. B cell responses: cell interaction dynamics and decisions. Cell. (2019) 177:524–40. doi: 10.1016/j.cell.2019.03.016
2. Elgueta R, de Vries VC, Noelle RJ. The immortality of humoral immunity. Immunol Rev. (2010) 236:139–50. doi: 10.1111/j.1600-065X.2010.00924.x
3. Bischof D, Elsawa SF, Mantchev G, Yoon J, Michels GE, Nilson A, et al. Selective activation of TACI by syndecan-2. Blood. (2006) 107:3235–42. doi: 10.1182/blood-2005-01-0256
4. Xia XZ, Treanor J, Senaldi G, Khare SD, Boone T, Kelley M, et al. TACI is a TRAF-interacting receptor for TALL-1, a tumor necrosis factor family member involved in B cell regulation. J Exp Med. (2000) 192:137–43. doi: 10.1084/jem.192.1.137
5. Bonilla FA, Geha RS. Common variable immunodeficiency. Pediatr Res. (2009) 65(5 Pt 2):13R−9. doi: 10.1203/PDR.0b013e31819dbf88
6. von Bulow GU, Bram RJ. NF-AT activation induced by a CAML-interacting member of the tumor necrosis factor receptor superfamily. Science. (1997) 278:138–41.
7. Ozcan E, Rauter I, Garibyan L, Dillon SR, Geha RS. Toll-like receptor 9, transmembrane activator and calcium-modulating cyclophilin ligand interactor, and CD40 synergize in causing B-cell activation. J Allergy Clin Immunol. (2011) 128:601–9.e1-4. doi: 10.1016/j.jaci.2011.04.052
8. Allman WR, Dey R, Liu L, Siddiqui S, Coleman AS, Bhattacharya P, et al. TACI deficiency leads to alternatively activated macrophage phenotype and susceptibility to Leishmania infection. Proc Natl Acad Sci USA. (2015) 112:E4094–103. doi: 10.1073/pnas.1421580112
9. He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol. (2010) 11:836–45. doi: 10.1038/ni.1914
10. Garcia-Carmona Y, Ting AT, Radigan L, Athuluri Divakar SK, Chavez J, Meffre E, et al. TACI isoforms regulate ligand binding and receptor function. Front Immunol. (2018) 9:2125. doi: 10.3389/fimmu.2018.02125
11. Sintes J, Gentile M, Zhang S, Garcia-Carmona Y, Magri G, Cassis L, et al. mTOR intersects antibody-inducing signals from TACI in marginal zone B cells. Nat Commun. (2017) 8:1462. doi: 10.1038/s41467-017-01602-4
12. Tsuji S, Cortesao C, Bram RJ, Platt JL, Cascalho M. TACI deficiency impairs sustained Blimp-1 expression in B cells decreasing long-lived plasma cells in the bone marrow. Blood. (2011) 118:5832–9. doi: 10.1182/blood-2011-05-353961
13. Salzer U, Chapel HM, Webster AD, Pan-Hammarstrom Q, Schmitt-Graeff A, Schlesier M, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. (2005) 37:820–8. doi: 10.1038/ng1600
14. Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. (2005) 37:829–34. doi: 10.1038/ng1601
15. Kralickova P, Milota T, Litzman J, Malkusova I, Jilek D, Petanova J, et al. CVID-associated tumors: czech nationwide study focused on epidemiology, immunology, and genetic background in a cohort of patients with CVID. Front Immunol. (2018) 9:3135. doi: 10.3389/fimmu.2018.03135
16. Romberg N, Chamberlain N, Saadoun D, Gentile M, Kinnunen T, Ng YS, et al. CVID-associated TACI mutations affect autoreactive B cell selection and activation. J Clin Invest. (2013) 123:4283–93. doi: 10.1172/JCI69854
17. Tsuji S, Stein L, Kamada N, Nunez G, Bram R, Vallance BA, et al. TACI deficiency enhances antibody avidity and clearance of an intestinal pathogen. J Clin Invest. (2014) 124:4857–66. doi: 10.1172/JCI74428
18. Grasset EK, Chorny A, Casas-Recasens S, Gutzeit C, Bongers G, Thomsen I, et al. Gut T cell-independent IgA responses to commensal bacteria require engagement of the TACI receptor on B cells. Sci Immunol. (2020) 5:eaat7117. doi: 10.1126/sciimmunol.aat7117
19. Kanswal S, Katsenelson N, Selvapandiyan A, Bram RJ, Akkoyunlu M. Deficient TACI expression on B lymphocytes of newborn mice leads to defective Ig secretion in response to BAFF or APRIL. J Immunol. (2008) 181:976–90. doi: 10.4049/jimmunol.181.2.976
20. Jabara HH, Lee JJ, Janssen E, Ullas S, Liadaki K, Garibyan L, et al. Heterozygosity for transmembrane activator and calcium modulator ligand interactor A144E causes haploinsufficiency and pneumococcal susceptibility in mice. J Allergy Clin Immunol. (2017) 139:1293–301.e4. doi: 10.1016/j.jaci.2016.07.028
21. Roque R, Ponce R, Burleson F, Cabrit M, Broly H, Rogge M. Influenza virus host response of C57Bl/6 mice treated with TACI-Ig. Immunopharmacol Immunotoxicol. (2006) 28:13–32. doi: 10.1080/08923970600623541
22. Wolf AI, Mozdzanowska K, Quinn WJ, 3rd, Metzgar M, Williams KL, Caton AJ, et al. Protective antiviral antibody responses in a mouse model of influenza virus infection require TACI. J Clin Invest. (2011) 121:3954–64. doi: 10.1172/JCI57362DS1
23. Salzer U, Bacchelli C, Buckridge S, Pan-Hammarstrom Q, Jennings S, Lougaris V, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. (2009) 113:1967–76. doi: 10.1182/blood-2008-02-141937
24. von Bulow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity. (2001) 14:573–82. doi: 10.1016/S1074-7613(01)00130-3
25. Mantchev GT, Cortesao C, Rebrovich M, Cascalho M, Bram RJ. TACI is required for efficient plasma cell differentiation in response to T-independent type 2 antigens. J Immunol. (2007) 179:2282–8. doi: 10.4049/jimmunol.179.4.2282
26. Ding BB, Bi E, Chen H, Yu JJ, Ye BH. IL-21 and CD40L synergistically promote plasma cell differentiation through upregulation of Blimp-1 in human B cells. J Immunol. (2013) 190:1827–36. doi: 10.4049/jimmunol.1201678
27. Sakurai D, Kanno Y, Hase H, Kojima H, Okumura K, Kobata T. TACI attenuates antibody production costimulated by BAFF-R and CD40. Eur J Immunol. (2007) 37:110–8. doi: 10.1002/eji.200636623
28. Romberg N, Virdee M, Chamberlain N, Oe T, Schickel JN, Perkins T, et al. TNF receptor superfamily member 13b (TNFRSF13B) hemizygosity reveals transmembrane activator and CAML interactor haploinsufficiency at later stages of B-cell development. J Allergy Clin Immunol. (2015) 136:1315–25. doi: 10.1016/j.jaci.2015.05.012
29. Radwan J, Babik W, Kaufman J, Lenz TL, Winternitz J. Advances in the evolutionary understanding of MHC polymorphism. Trends Genet. (2020) 36:298–311. doi: 10.1016/j.tig.2020.01.008
30. Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, et al. TACI and BAFF-R mediate isotype switching in B cells. J Exp Med. (2005) 201:35–9. doi: 10.1084/jem.20032000
31. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536:285–91. doi: 10.1038/nature19057
32. Garibyan L, Lobito AA, Siegel RM, Call ME, Wucherpfennig KW, Geha RS. Dominant-negative effect of the heterozygous C104R TACI mutation in common variable immunodeficiency (CVID). J Clin Invest. (2007) 117:1550–7. doi: 10.1172/JCI31023
33. Lee JJ, Jabara HH, Garibyan L, Rauter I, Sannikova T, Dillon SR, et al. The C104R mutant impairs the function of transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) through haploinsufficiency. J Allergy Clin Immunol. (2010) 126:1234–41.e2. doi: 10.1016/j.jaci.2010.08.017
34. Lee JJ, Rauter I, Garibyan L, Ozcan E, Sannikova T, Dillon SR, et al. The murine equivalent of the A181E TACI mutation associated with common variable immunodeficiency severely impairs B-cell function. Blood. (2009) 114:2254–62. doi: 10.1182/blood-2008-11-189720
35. Quintana-Murci L. Human immunology through the lens of evolutionary genetics. Cell. (2019) 177:184–99. doi: 10.1016/j.cell.2019.02.033
36. Sazzini M, Zuntini R, Farjadian S, Quinti I, Ricci G, Romeo G, et al. An evolutionary approach to the medical implications of the tumor necrosis factor receptor superfamily member 13B (TNFRSF13B) gene. Genes Immunity. (2009) 10:566–78. doi: 10.1038/gene.2009.43
37. Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl (2018). Nucleic Acids Res. (2018) 46:D754–61. doi: 10.1093/nar/gkx1098
38. Garcia-Carmona Y, Cols M, Ting AT, Radigan L, Yuk FJ, Zhang L, et al. Differential induction of plasma cells by isoforms of human TACI. Blood. (2015) 125:1749–58. doi: 10.1182/blood-2014-05-575845
39. Siewert KM, Voight BF. Detecting long-term balancing selection using allele frequency correlation. Mol Biol Evol. (2017) 34:2996–3005. doi: 10.1093/molbev/msx209
40. Jagoda E, Lawson DJ, Wall JD, Lambert D, Muller C, Westaway M, et al. Disentangling Immediate adaptive introgression from selection on standing introgressed variation in humans. Mol Biol Evol. (2018) 35:623–30. doi: 10.1093/molbev/msx314
41. Vo Ngoc DT, Krist L, van Overveld FJ, Rijkers GT. The long and winding road to IgA deficiency: causes and consequences. Expert Rev Clin Immunol. (2017) 13:371–82. doi: 10.1080/1744666X.2017.1248410
42. Cunningham-Rundles C. Physiology of IgA and IgA deficiency. J Clin Immunol. (2001) 21:303–9. doi: 10.1023/A:1012241117984
43. Grabar P, Williams CA. [Method permitting the combined study of the electrophoretic and the immunochemical properties of protein mixtures; application to blood serum]. Biochim Biophys Acta. (1953) 10:193–4. doi: 10.1016/0006-3002(53)90233-9
44. de Sousa-Pereira P, Woof JM. IgA: structure, function, and developability. Antibodies. (2019) 8:57. doi: 10.3390/antib8040057
45. Frutiger S, Hughes GJ, Paquet N, Luthy R, Jaton JC. Disulfide bond assignment in human J chain and its covalent pairing with immunoglobulin M. Biochemistry. (1992) 31:12643–7. doi: 10.1021/bi00165a014
46. Hamburger AE, Bjorkman PJ, Herr AB. Structural insights into antibody-mediated mucosal immunity. Curr Top Microbiol Immunol. (2006) 308:173–204. doi: 10.1007/3-540-30657-9_8
47. Kaetzel CS. The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol Rev. (2005) 206:83–99. doi: 10.1111/j.0105-2896.2005.00278.x
48. Royle L, Roos A, Harvey DJ, Wormald MR, van Gijlswijk-Janssen D, Redwanel RM, et al. Secretory IgA N- and O-glycans provide a link between the innate and adaptive immune systems. J Biol Chem. (2003) 278:20140–53. doi: 10.1074/jbc.M301436200
49. Nakajima A, Vogelzang A, Maruya M, Miyajima M, Murata M, Son A, et al. IgA regulates the composition and metabolic function of gut microbiota by promoting symbiosis between bacteria. J Exp Med. (2018) 215:2019–34. doi: 10.1084/jem.20180427
50. Crottet P, Corthesy B. Secretory component delays the conversion of secretory IgA into antigen-binding competent F(ab')2: a possible implication for mucosal defense. J Immunol. (1998) 161:5445–53.
51. Phalipon A, Cardona A, Kraehenbuhl JP, Edelman L, Sansonetti PJ, Corthesy B. Secretory component: a new role in secretory IgA-mediated immune exclusion in vivo. Immunity. (2002) 17:107–15. doi: 10.1016/S1074-7613(02)00341-2
52. Burns JW, Siadat-Pajouh M, Krishnaney AA, Greenberg HB. Protective effect of rotavirus VP6-specific IgA monoclonal antibodies that lack neutralizing activity. Science. (1996) 272:104–7. doi: 10.1126/science.272.5258.104
53. van Egmond M, Damen CA, van Spriel AB, Vidarsson G, van Garderen E, van de Winkel JG. IgA and the IgA Fc receptor. Trends Immunol. (2001) 22:205–11. doi: 10.1016/S1471-4906(01)01873-7
54. Krugmann S, Pleass RJ, Atkin JD, Woof JM. Structural requirements for assembly of dimeric IgA probed by site-directed mutagenesis of J chain and a cysteine residue of the alpha-chain CH2 domain. J Immunol. (1997) 159:244–9.
55. Vaerman JP, Langendries A, Giffroy D, Brandtzaeg P, Kobayashi K. Lack of SC/pIgR-mediated epithelial transport of a human polymeric IgA devoid of J chain: in vitro and in vivo studies. Immunology. (1998) 95:90–6.
56. Vaerman JP, Langendries AE, Giffroy DA, Kaetzel CS, Fiani CM, Moro I, et al. Antibody against the human J chain inhibits polymeric Ig receptor-mediated biliary and epithelial transport of human polymeric IgA. Eur J Immunol. (1998) 28:171–82.
57. Taylor AK, Wall R. Selective removal of alpha heavy-chain glycosylation sites causes immunoglobulin A degradation and reduced secretion. Mol Cell Biol. (1988) 8:4197–203.
58. Atkin JD, Pleass RJ, Owens RJ, Woof JM. Mutagenesis of the human IgA1 heavy chain tailpiece that prevents dimer assembly. J Immunol. (1996) 157:156–9.
59. Heystek HC, Moulon C, Woltman AM, Garonne P, van Kooten C. Human immature dendritic cells efficiently bind and take up secretory IgA without the induction of maturation. J Immunol. (2002) 168:102–7. doi: 10.4049/jimmunol.168.1.102
60. Bos NA, Bun JC, Popma SH, Cebra ER, Deenen GJ, van der Cammen MJ, et al. Monoclonal immunoglobulin A derived from peritoneal B cells is encoded by both germ line and somatically mutated VH genes and is reactive with commensal bacteria. Infect Immun. (1996) 64:616–23. doi: 10.1128/IAI.64.2.616-623.1996
61. Catanzaro JR, Strauss JD, Bielecka A, Porto AF, Lobo FM, Urban A, et al. IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Sci Rep. (2019) 9:13574. doi: 10.1038/s41598-019-49923-2
62. Donaldson GP, Ladinsky MS, Yu KB, Sanders JG, Yoo BB, Chou WC, et al. Gut microbiota utilize immunoglobulin A for mucosal colonization. Science. (2018) 360:795–800. doi: 10.1126/science.aaq0926
63. Macpherson AJ, Yilmaz B, Limenitakis JP, Ganal-Vonarburg SC. IgA function in relation to the intestinal microbiota. Annu Rev Immunol. (2018) 36:359–81. doi: 10.1146/annurev-immunol-042617-053238
64. Mathias A, Corthesy B. Recognition of gram-positive intestinal bacteria by hybridoma- and colostrum-derived secretory immunoglobulin A is mediated by carbohydrates. J Biol Chem. (2011) 286:17239–47. doi: 10.1074/jbc.M110.209015
65. Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. (2008) 4:447–57. doi: 10.1016/j.chom.2008.09.007
67. Wang N, Hammarstrom L. IgA deficiency: what is new? Curr Opin Allergy Clin Immunol. (2012) 12:602–8. doi: 10.1097/ACI.0b013e3283594219
Keywords: B-lymphocyte, TNFRSF13B, antibodies, T cell-dependent antibody response, T cell-independent antibody responses
Citation: Cascalho M and Platt JL (2021) TNFRSF13B Diversification Fueled by B Cell Responses to Environmental Challenges—A Hypothesis. Front. Immunol. 12:634544. doi: 10.3389/fimmu.2021.634544
Received: 28 November 2020; Accepted: 21 January 2021;
Published: 17 February 2021.
Edited by:
Alessandro Camponeschi, University of Gothenburg, SwedenReviewed by:
Paolo Casali, University of Texas Health Science Center at San Antonio, United StatesCopyright © 2021 Cascalho and Platt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marilia Cascalho, bWFyaWxpYUB1bWljaC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.