
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 16 April 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.630938
This article is part of the Research Topic Multiple Implications of the Kynurenine Pathway in Inflammatory Diseases: Diagnostic and Therapeutic Applications View all 14 articles
 Eliseu F. de Araújo
Eliseu F. de Araújo Flávio V. Loures†
Flávio V. Loures† Nycolas W. Preite†
Nycolas W. Preite† Cláudia Feriotti
Cláudia Feriotti Nayane AL Galdino†
Nayane AL Galdino† Tânia A. Costa
Tânia A. Costa Vera L. G. Calich*
Vera L. G. Calich*In agreement with other fungal infections, immunoprotection in pulmonary paracoccidioidomycosis (PCM) is mediated by Th1/Th17 cells whereas disease progression by prevalent Th2/Th9 immunity. Treg cells play a dual role, suppressing immunity but also controlling excessive tissue inflammation. Our recent studies have demonstrated that the enzyme indoleamine 2,3 dioxygenase (IDO) and the transcription factor aryl hydrocarbon receptor (AhR) play an important role in the immunoregulation of PCM. To further evaluate the immunomodulatory activity of AhR in this fungal infection, Paracoccidioides brasiliensis infected mice were treated with two different AhR agonists, L-Kynurenin (L-Kyn) or 6-formylindole [3,2-b] carbazole (FICZ), and one AhR specific antagonist (CH223191). The disease severity and immune response of treated and untreated mice were assessed 96 hours and 2 weeks after infection. Some similar effects on host response were shared by FICZ and L-Kyn, such as the reduced fungal loads, decreased numbers of CD11c+ lung myeloid cells expressing activation markers (IA, CD40, CD80, CD86), and early increased expression of IDO and AhR. In contrast, the AhR antagonist CH223191 induced increased fungal loads, increased number of pulmonary CD11c+ leukocytes expressing activation markers, and a reduction in AhR and IDO production. While FICZ treatment promoted large increases in ILC3, L-Kyn and CH223191 significantly reduced this cell population. Each of these AhR ligands induced a characteristic adaptive immunity. The large expansion of FICZ-induced myeloid, lymphoid, and plasmacytoid dendritic cells (DCs) led to the increased expansion of all CD4+ T cell subpopulations (Th1, Th2, Th17, Th22, and Treg), but with a clear predominance of Th17 and Th22 subsets. On the other hand, L-Kyn, that preferentially activated plasmacytoid DCs, reduced Th1/Th22 development but caused a robust expansion of Treg cells. The AhR antagonist CH223191 induced a preferential expansion of myeloid DCs, reduced the number of Th1, Th22, and Treg cells, but increased Th17 differentiation. In conclusion, the present study showed that the pathogen loads and the immune response in pulmonary PCM can be modulated by AhR ligands. However, further studies are needed to define the possible use of these compounds as adjuvant therapy for this fungal infection.
The aryl hydrocarbon receptor (Ahr), a ligand-dependent transcription factor that resides in the cytoplasm of many cell types, was first described due to its involvement in the metabolism of xenobiotic compounds such as dioxin (1). Currently, it is well known that AhR is activated by a diverse set of endogenous and exogenous ligands (2). At a steady-state, AhR remains in the cytoplasm (3) but translocates to the nucleus after ligand binding. In the nucleus, AhR heterodimerizes with AhR Nuclear Translocator (ARNT) and then interacts with its genomic binding motifs inducing the transcription of its target genes, including detoxifying enzymes of the cytochrome P450 family (4). AhR also interacts with other transcription factors that regulate AhR signaling (3). It was also reported that the ligand structure and affinity control AhR activity (5). Several AhR ligands were described: L-Kynurenines (L-Kyn), products of tryptophan degradation by the enzymatic action of indoleamine 2,3 dioxygenase (IDO), 6-formylindole [3,2-b] carbazole (FICZ), a tryptophan photoproduct, and several microbial and dietary products (5–7). AhR is expressed by innate and adaptive immune cells and influences the development and activation of the immune system. This transcription factor plays an important role in the control of cell differentiation, proliferation, and cytokines production (7–11). Indeed, AhR was shown to exert an important activity on T helper 17 (Th17) and regulatory T cells (Treg) differentiation, influencing the severity of several experimental pathologies (9–11). Innate lymphoid cells (ILCs), a family of immune cells that do not express antigen receptors but exhibit phenotypes that reflect Th cell subpopulations, were also reported to be regulated by the AhR expression. The differentiation of ILC3 and lymphoid tissue inducing lymphocyte (LTi), that secrete IL-17, IL-22, and lymphotoxin is dependent on the transcription factors RORγτ and AhR (12).
The regulatory activity of AhR has been demonstrated in several infectious pathologies (13). However, the effects of AhR activation were not homogeneous due to the various AhR ligand used, type of pathology studied, and treatment protocols employed (3, 13–15).
P. brasiliensis, a fungal pathogen endemic to Latin America, is sensed by a variety of pattern recognition receptors that stimulate the differentiation of a wide range of T cell subpopulations involved in host immunity (16–23). In humans and experimental models of PCM (PCM), Th1/Th17 promote immunoprotection: Th1 by controlling fungal loads via IFN-γ activated macrophages and Th17 by promoting neutrophil recruitment and activation. Th2 and Th9 cells are associated with increased fungal growth, inefficient inflammatory reactions, and disease severity (18, 19, 24, 25). In the human disease, Treg cells are associated with the progressive and severe forms of the disease (25–28), but experimental models of pulmonary PCM clearly showed the dual role of this T cell subset: it is deleterious due to its suppressive effect on protective immunity but it has also a beneficial effect mediated by the inhibition of excessive inflammatory reactions (24, 28, 29).
In experimental candidiasis, the enzyme indoleamine 2,3 dioxygenase (IDO), which regulates tryptophan (Trp) degradation, was shown to reduce fungal loads but also to control immunity by reducing Th17 expansion via increased Treg cell proliferation mediated by L-Kyn-activated AhR (30–32). Moreover, AhR was also involved in the protection of Candida albicans infected mucosae (32) due to its regulatory activity on IL-22 production (9, 33). In pulmonary PCM, our recent studies have shown that P. brasiliensis infection induces a vigorous IDO expression that mediates Trp catabolism, resulting in increased L-Kyn production and AhR activation. P. brasiliensis uses two distinct mechanisms to trigger IDO expression. In susceptible (B10.A) mice, IDO is induced by IFN-γ and exhibits a prevalent enzymatic activity whereas in resistant (A/J) mice IDO is TGF-β induced and behaves as a signaling molecule (34–36). Our studies have also demonstrated that IDO and AhR are mutually regulated and control the number of ILCs and the Th17/Treg balance (34–37). Altogether, our findings defined the important regulatory role of the IDO/AhR axis in the immunity and severity of pulmonary PCM leading us to better evaluate the role of AhR in pulmonary PCM. To this aim, P. brasiliensis infected mice were treated with three different AhR ligands, two agonists (L-Kyn and FICZ) and an antagonist (CH223191), and disease severity and immune response assessed 96 hours and 2 weeks after infection. We verified that AhR ligands control fungal burdens, cytokines production, and activation of pulmonary myeloid cells. Importantly, FICZ showed a prevalent effect on the differentiation of Th17 and Th22 cells, L-Kyn on Tregs, and CH223191 on Th17 cells. Altogether, our findings demonstrate that pulmonary PCM can be modulated by AhR ligands that could be used to regulate the differentiation of pro- or anti-inflammatory T cell subsets.
The experiments were performed in strict accordance with the Brazilian Federal Law 11,794 establishing procedures for the scientific use of animals, and the State Law establishing the Animal Protection Code of the State of São Paulo. All efforts were made to minimize animal suffering. The procedures were approved by the Ethics Committee on Animal Experiments of the Institute of Biomedical Sciences of the University of São Paulo (Proc.180/11/CEEA).
C57B/6 SPF male mice, bred at the Isogenic Breeding Unit of the Department of Immunology, Institute of Biomedical Sciences, were used at the age of 6-8 weeks.
The virulent Pb18 isolate from P. brasiliensis was maintained in the yeast form by weekly cultivation in Fava Netto’s semi-solid medium at 36°C and used on days 6–8 of culture. The fungus was collected and washed with phosphate-buffered saline (PBS, pH 7.2). The fungal viability was determined by the Janus Green B vital dye. All experimental procedures were carried out with fungal suspensions presenting viability between 90 and 95%. For i.t. infection, mice were anesthetized with ketamine and xylazine and submitted to i.t. infection with 1x106 yeast cells, contained in 50 µL of PBS as previously described (35).
C57BL/6 mice were infected as described above and treated with two different AhR agonists, 6-formylindol [3,2-b] carbazole (FICZ, Enzo Labs) or L-Kynurenine (L-Kyn, Sigma Aldrich). The drug 2-methyl-2H-pyrazole-3-carboxylic acid-amide (CH223191-Signa Aldrich) was employed as an AhR antagonist. Stock solutions of L-Kyn (20 mg/ml, 96 mM), FICZ (2 mg/ml, 7 mM) and CH223191 (30 mg/ml, 90 mM) were prepared in DMSO. These drugs were properly diluted in phosphate buffered solution (PBS) just before use. After i.t. infection, mice were inoculated intraperitoneally on alternate days with 200 μg of FICZ, or 400 μg of CH223191or 800 μg of L-Kyn per animal, contained in 500 μl of diluent solution. PBS was used in control infected mice. These protocols were adapted from those previously described (38–41).
The disease severity of control and ligand-treated infected mice was assessed after 96 hours and 2 weeks of infection. The analysis was carried out by recovering viable fungal cells from lungs, liver, and spleen, using a BHI medium supplemented with horse serum and a culture filtrate obtained from P. brasiliensis (isolate 192).
Lung cell suspensions were prepared as previously described (34). The lungs were removed and digested for 30 min. in digestion buffer containing collagenase (Sigma). The organs were then macerated in a homogenizer with RPMI 1640 culture medium. The erythrocytes were lysed with lysis buffer, the cells counted, and their viability assessed by Trypan blue dye.
Lung cell suspensions were adjusted to 1x106 cells and suspended in PBS-azide (0.1%) containing fetal bovine serum (SFB, 5%). Fc receptors were blocked with anti-CD16/32 monoclonal antibody and then labeled with fluorophore-conjugated antibodies as previously described (37) Labeled antibodies (BD Biosciences) were used in the appropriate combination for the cell population to be analyzed. For lymphocytes, the following antibodies were used: anti-CD3, CD4, CD25, and Foxp3; for myeloid cells: anti-CD45, CD11b, CD11c, CD40, CD80, CD86, MHC-II, and F4/80. For ILCs characterization, lung leukocytes were first treated with an anti-mouse lineage cocktail (Biolegend) containing antibodies to CD3, Ly6G/Ly6C, CD11b, CD45R/B220, TER 119/erythroid cells, that react with T cells, B cells, monocytes, macrophages, NK cells, and erythrocytes. Intracellular staining was conducted using the eBioscience Transcription Factor staining kit and specific antibodies for IL-17, IL-4, IFN-γ, IL-22, IL-1β, IL-12, TNF-α, IL-6, TGF-β, IL-10, FoxP3, IDO-1, and AhR. Supplementary Table 1 lists the monoclonal antibodies used in flow cytometry assays. Cells were run on FACSCantoII (BD Biosciences) and a minimum of 50,000 events was acquired using FACSDiva software (BD Biosciences). Cells were analyzed using FlowJo software (Tree Star).
The presence and concentration of cytokines (IL-12, TNF-α, IFN-γ, IL-1β, IL-4, IL-10, TGF-β, IL-35, IL-6, IL-23, IL-17, and IL-22 were determined in lung homogenates obtained 96 h and 2 weeks after infection of AhR ligands treated and untreated mice. The methodology used was that recommended by the supplier (EBioscience).
RNA isolation from lung macerates of AhR ligand-treated and untreated mice was performed as previously described (37). A NanoDrop ND-1000 spectrophotometer was used to determine RNA purity and concentration. The cDNA was synthesized using 1 µg of RNA and the high-capacity RNA-to-cDNA kit (Applied Biosystems) according to the manufacturer’s instructions. The cDNA was amplified using TaqMan Universal PCR Master Mix (Applied Biosystems) and pre-developed TaqMan assay primers and probes (Ifng, Mm001168134_m1, Tnf, Mm99999068_m1, Il6, Mm00446190_m1, Il10, Mm00439614_m1, Tgfb1, Mm00117882_m1, Il17, Mm00439618_m1, Il22, Mm01226722_m1, Tbet, Mm00450960_m1; Gata3, Mm00484683_m1; Rorc, Mm01261022_m1; Foxp3, Mm00475162_m1; Gapdh, Mm99999915_g1a, all from Applied Biosystems.). PCR assays were performed on an MxP3000P QPCR System and data were developed using the MxPro qPCR software (Stratagene). The average threshold cycle (CT) values of samples were normalized to the CT value of the Gapdh gene. The relative expression was determined by the 2-ΔΔCT method.
Data were analyzed as previously described (42) and expressed as the M ± SD. Differences between groups were tested using a one-way analysis of variance (ANOVA) followed by the Dunnet’s post hoc test to compare every mean with a control mean. Data were analyzed using GraphPad Prism 7.03 software (GraphPad Prism Software, Inc.). A P value ≤ 0.05 was considered significant.
C57BL/6 male mice were infected with 1 x 106 P. brasiliensis yeasts and groups of 5 animals were treated with the AhR agonists L-Kyn (800 μg i.p./mice) or FICZ (200 μg i.p./mice) every other day starting at day-1 of fungal infection. Another group was treated with the AhR antagonist CH223191 (400 μg i.p./mice) on alternate days after infection. Control mice were infected and treated with the drug vehicle following the same protocol above described. The AhR ligands treated and untreated infected mice were sacrificed 96 hours and 2 weeks after infection, their lungs and liver macerated, and the presence of viable fungi evaluated by the colony-forming units (CFU) method. Figure 1 shows that there was a significant reduction in pulmonary and hepatic fungal loads in FICZ and L-Kyn treated mice at both assayed periods; in contrast, treatment with CH223191 increased the fungal load of the lungs, but not that of the liver.
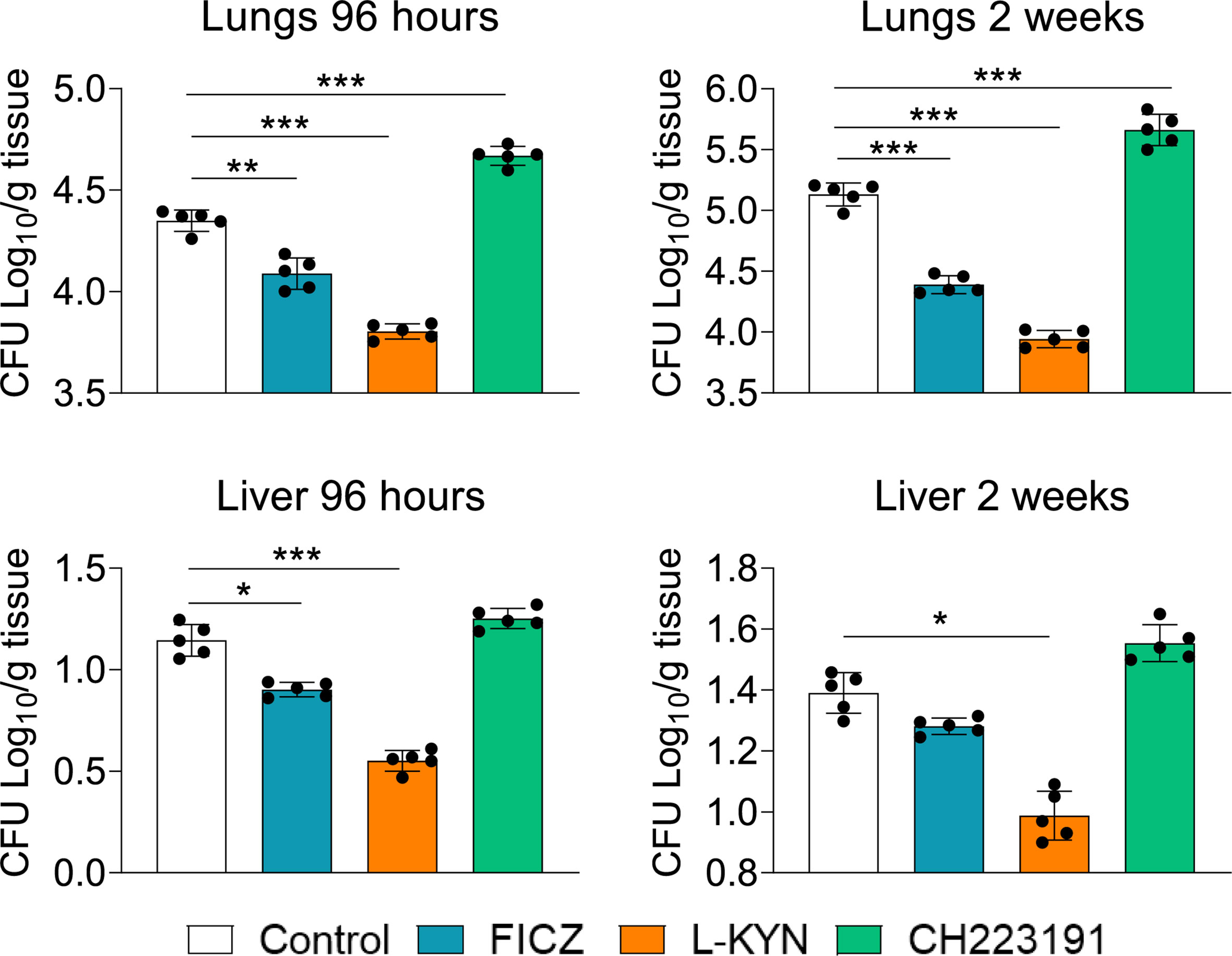
Figure 1 Treatment with AhR agonists (FICZ and L-Kyn) reduces while the antagonist (CH223191) increases the fungal load in mice infected with P. brasiliensis. C57BL/6 mice (n = 5) were infected with 1x106 yeasts of P. brasiliensis and treated by route i,p. on alternate days with FICZ, L-Kyn, or CH22391 at doses of 200 μg or 800 μg or 400 μg/animal, respectively. Control mice were treated with the PBS. The animals were sacrificed 96 hours and 2 weeks after infection, their lungs and liver removed, macerated, and evaluated for the fungal load. The experiment was repeated twice, and data are expressed as M ± SD. P values < 0.05 were considered significant (*p < 0.05; **p < 0.005 and ***p < 0.001).
Treated and untreated infected mice were sacrificed 96 hours and 2 weeks after infection, their lungs removed, macerated and CD11c+ lung myeloid cells analyzed by flow cytometry for the expression of activation markers (IAb, CD40, CD80, and CD86). As can be seen in Figure 2, the number of activated CD11c+ myeloid cells was present in reduced numbers in the lungs of mice treated with AhR agonists. In contrast, treatment with the CH223191 antagonist increased the number of activated CD11c+ cells in the lungs of infected mice.
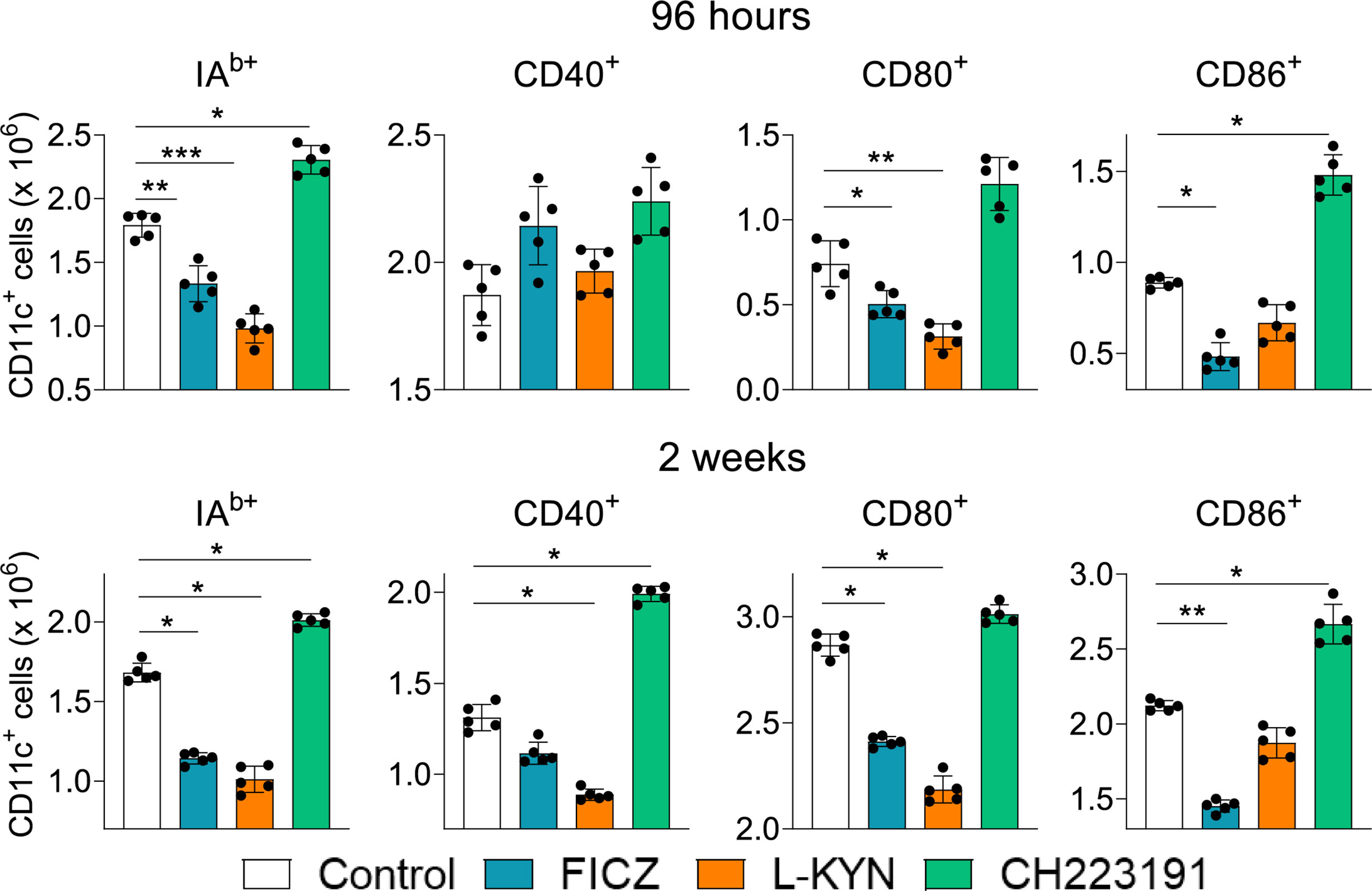
Figure 2 Treatment with AhR agonists (FICZ and L-Kyn) reduces, while the antagonist (CH223191) increases the number of activated myeloid CD11c+ cells in the lungs of P. brasiliensis infected mice. C57BL/6 mice (n = 5) were infected with 1 x 106 P. brasiliensis yeasts and treated by route i,p. on alternate days with FICZ, L-Kyn, or CH22 according to the protocol previously described. Control mice were treated with PBS. The animals were sacrificed 96 hours and 2 weeks after infection, their lungs removed, macerated, and CD11c+ leukocytes analyzed by flow cytometry for the expression of activation markers (IAb, CD40, CD80, and CD86). The experiment was repeated twice, and data are expressed as M ± SD. P values < 0.05 were considered significant (*p < 0.05; **p < 0.005 and ***p < 0.001).
Mice were infected and treated as above described. The animals were sacrificed 96 hours and 2 weeks after infection, their lungs removed, macerated and the leukocytes analyzed by flow cytometry for the intracellular expression of IDO, AhR, and cytokines (IL-12, TNF-α, IL-1β, IL-6, TGB-β, and IL-10) by CD11c+ myeloid cells. Figure 3A shows the gating strategy used to characterize these cells. As can be seen in Figure 3B, at 96 hours and 2 weeks after infection both agonists increased while the antagonist reduced the number of CD11c+ cells expressing intracellular IDO and AhR. As for cytokine expression, a general view suggests that FICZ led to increased, while CH223191 to a reduced number of CD11c+ cells expressing intracellular cytokines. Interestingly, all treatments at both periods assayed caused a robust reduction in IL-1β+ CD11c+ cells. At both post-infection periods, FICZ increased the numbers of CD11c+ cells expressing IL-12, IL-6, TGF-β and IL-10. L-Kyn reduced the number of TNF-α+ and IL-1β+ CD11c+ cells at both infection periods but increased the number of IL-12 and TGF-β expressing CD11c+ cells by 96 hours of infection. On the other hand, treatment with CH223191increased the number of CD11c+ myeloid cells expressing TNF-α but reduced those producing IL-12, IL-1β, and IL-6 at both time points assayed.

Figure 3 Treatment with AhR ligands (FICZ, L-Kyn, and CH2231991) alters the intracellular expression of IDO, AhR, and cytokines by pulmonary myeloid CD11c+ cells from P. brasiliensis infected mice. C57BL/6 mice (n = 5) were infected with 1 x 106 P. brasiliensis yeasts and treated by route i,p. on alternate days with FICZ, L-Kyn, or CH223191 at doses of 200, 800, or 400 μg/animal, respectively. Control mice were treated with PBS. The animals were sacrificed 96 hours and 2 weeks after infection, their lungs removed, macerated and CD11c+ leukocytes analyzed by flow cytometry for the intracellular expression of the enzyme IDO, the AhR transcription factor, and cytokines (IL-12, TNF-α, IL -1β, IL-6, TGB-β and IL-10). (A) Gate strategy to define CD11c+ expressing IDO, AhR, and cytokines. (B) Number of pulmonary CD11c+ cells expressing AhR, IDO and cytokines detected at 96 hours and 2 weeks after infection. The experiment was repeated twice, and data are expressed as M ± SD. P values < 0.05 were considered significant (* p < 0.05; **p < 0.005 and ***p < 0.001).
Mice were treated as previously described and analyzed two weeks post-infection by flow cytometry regarding the presence of myeloid (CD11c+CD11b+), lymphoid (CD11c+CD8+), and plasmacytoid (CD11c+mPDCA+) DCs in the lungs of infected mice. Figure 4A shows the gating strategy used to define DCs subpopulations. The number of CD11c+ cells increased in the lungs of mice receiving all three treatments and at both time points assayed (Figure 4B). All DC subpopulations were found in higher numbers in FICZ treated mice at both post-infection periods. L-Kyn also increased all DC subsets by 96 hours after infection but only plasmacytoid DCs appeared in higher number at week 2. The AhR antagonist CH223191 preferentially augmented the migration of myeloid DCs to the lungs of infected mice.
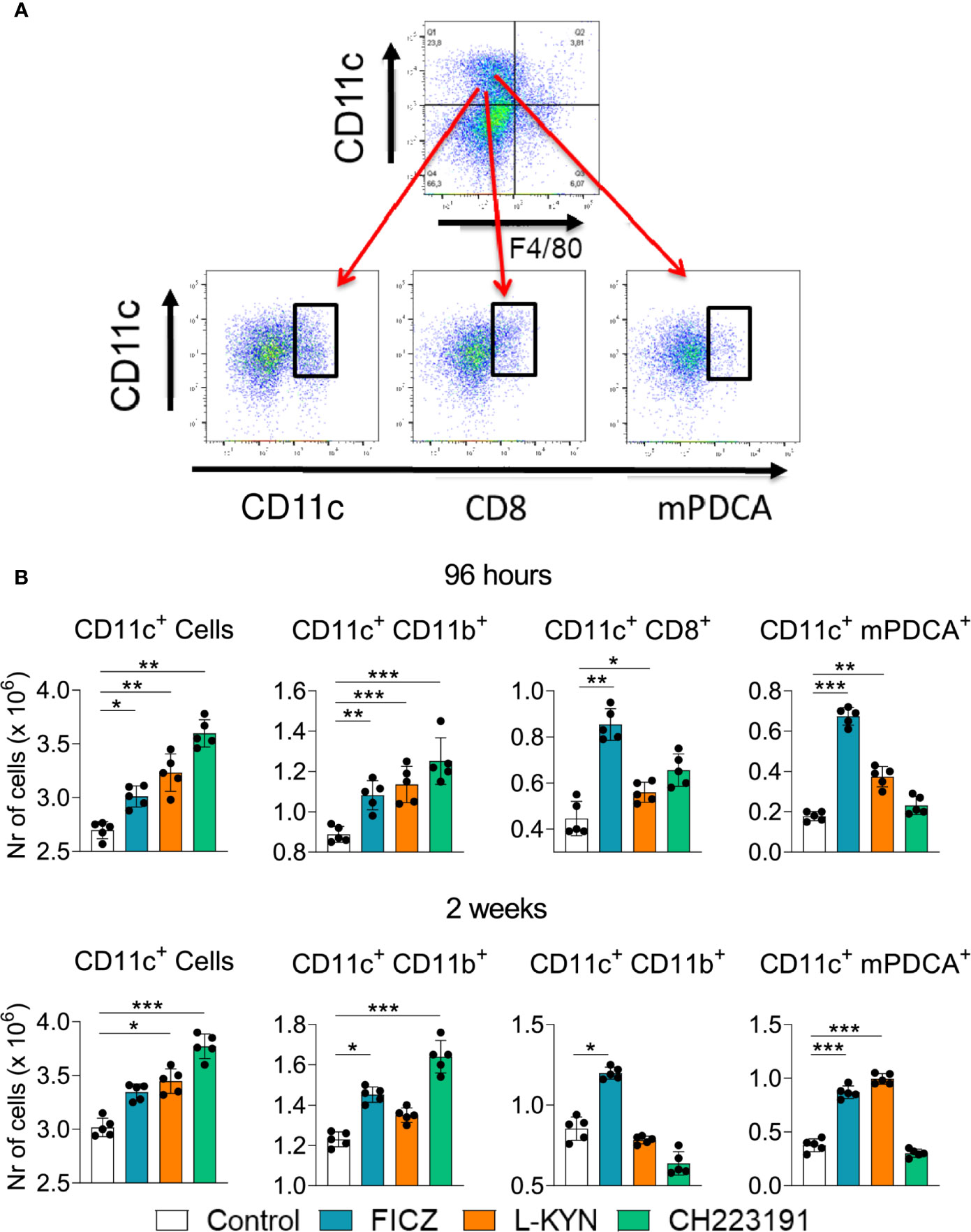
Figure 4 Treatment with AhR agonists (FICZ and L-Kyn) and antagonist (CH223191) increases the migration of dendritic cells (DCs) to the lungs of P. brasiliensis infected mice. C57BL/6 mice (n = 5) were infected with 1 x 106 P. brasiliensis yeasts and treated by route i,p. on alternate days with FICZ, L-Kyn, or CH223191 as previously described. Control mice were treated with PBS. The animals were sacrificed 2 weeks after infection, their lungs removed, macerated and the number total CD11c+, myeloid (CD11c+CD11b +), lymphoid (CD11c+CD8+), and plasmacytoid (CD11c+mPDCA+) dendritic cells analyzed by flow cytometry. (A) Gate strategy to define DCs subsets. (B) Number of total pulmonary CD11c+ cells and DCs subsets observed 2 weeks after infection. The experiment was repeated twice, and data are expressed as M ± SD. P values < 0.05 were considered significant (* p < 0.05; **p < 0.005 and ***p < 0.001).
We have also characterized the influence of the AhR ligands on the differentiation of pulmonary ILCs. These cells represent a new family of lymphocytes that do not express receptors for antigens, produce significant amounts of cytokines, and can be cytotoxic when activated. In ILCs, T and B cell receptors are absent, and their development is independent of RAG genes. The different subpopulations of ILCs exhibit transcription factors and cytokines that are prototypical of CD4 + T cell subsets. These characteristics include the shared expression of Tbet and IFN-γ by ILC1 and Th1, GATA-3, IL-5 and IL-13 by Th2 and ILC2; RORC, IL-17, and IL-22 by ILC3 and Th17/Th22 cells, as well as Eomes, IFN-γ and cytotoxic molecules by CD8+ T cells and conventional NK cells (43). Figure 5A depicts the gating strategy used to define ILCs subsets. We could demonstrate (Figure 5B) that FICZ treatment induced a great expansion of ILC3 but reduced the number of NK1.1 and ILC1 cells. L-Kyn induced the expansion of ILC1 but reduced ILC3. On the other hand, the AhR antagonist CH223191 caused only a profound reduction in the presence of ILC3 lymphocytes in the lungs of P. brasiliensis infected mice.
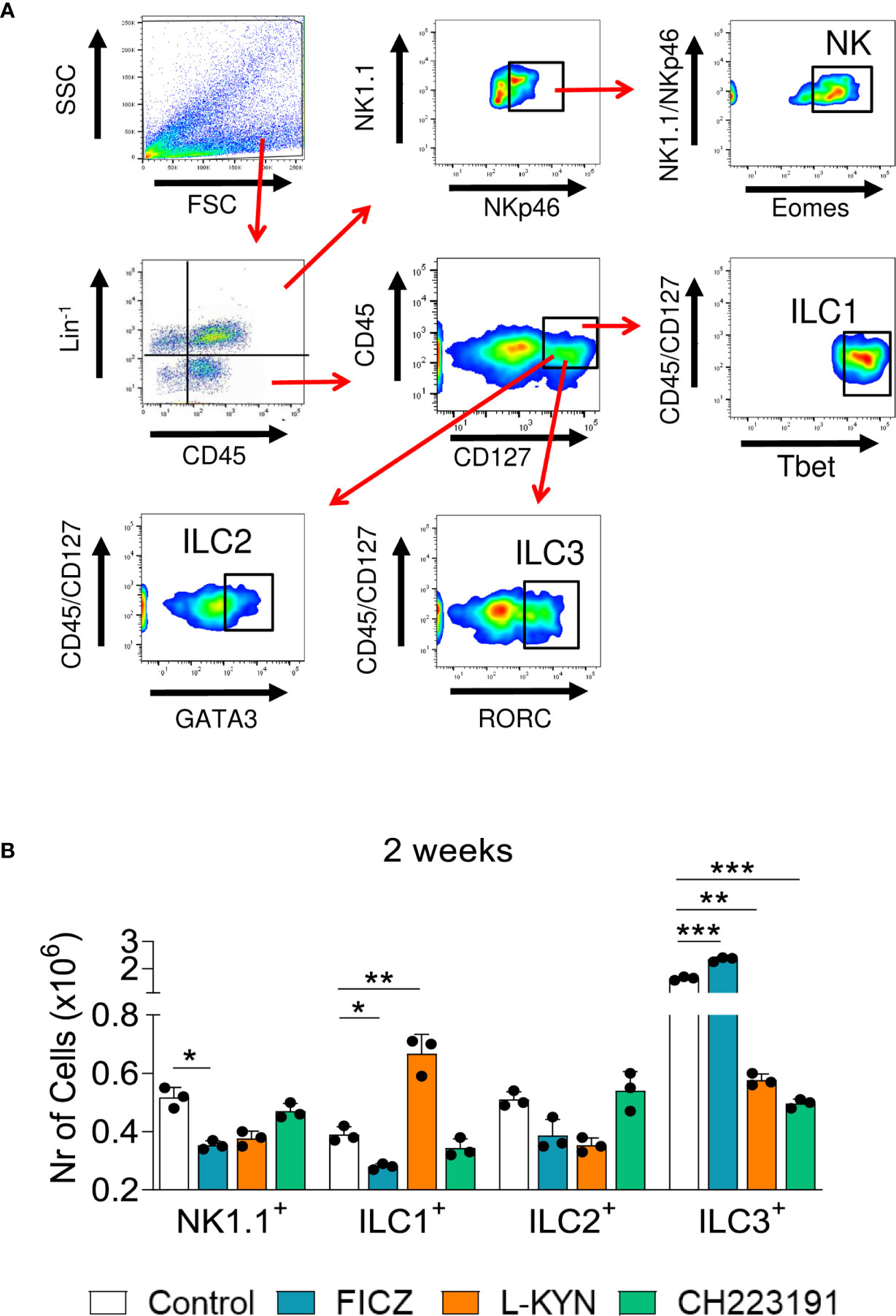
Figure 5 Treatment with AhR ligands alters the expansion and presence of Innate Lymphoid Cells (ILCs) in the lungs of P. brasiliensis infected mice. C57BL/6 mice were infected with 1 x 106 P. brasiliensis yeasts and treated with FICZ, L-Kyn, CH223191 as previously described. Control mice were treated with PBS. The animals were sacrificed 96 hours and 2 weeks after infection, their lungs removed, macerated, the leukocytes obtained, and analyzed by flow cytometry for ILCs phenotypes (NK 1.1, ILC1, ILC2, and ILC3). (A) Gate strategy to define ILCs subsets. Lung leukocytes were first treated with an anti-mouse lineage cocktail (Biolegend) containing antibodies to CD3, Ly6G/Ly6C, CD11b, CD45R/B220, TER 119/erythroid cells, that react with T cells, B cells, monocytes, macrophages, NK cells, and erythrocytes. NK cells were then classified as Lin+CD45+NK1.1+ NKp46+Eomes+, ILC1 as CD45+Lin-CD127+Tbet+, ILC2 as CD45+Lin-CD127+Gata3+, and ILC3 as CD45+Lin-CD127+RORC+. The cell surface and intracellular markers were measured by flow cytometry and 50.000 cells were counted. (B) Number of NK1.1, ILC1, ILC2, and ILC3 positive cells detected in the lungs of mice at 96 hours and 2 weeks after infection. The experiment was repeated twice and data are expressed as M ± SD. P values < 0.05 were considered significant (*p < 0.05; **p < 0.005 and ***p < 0.001).
Infected control and AhR ligands treated mice were sacrificed, their lungs removed, macerated, and the relative expression of mRNA for cytokines analyzed by RT-PCR. The results obtained at the two infection periods studied were similar (Figure 6). FICZ enhanced the expression of il-17, il-22, il-10, and tgf-b mRNAs at both periods but reduced il-6 levels at 96 hours of infection. L-Kyn increased the expression of ifn-γ and tgf-β but reduced the levels of il-6, il-17, and il-22 mRNAs. CH223191, on the other hand, increased the synthesis of il-6 but reduced il-17 and il-22 mRNA at both post-infection periods, but tgf-β only at 96 hours after infection.
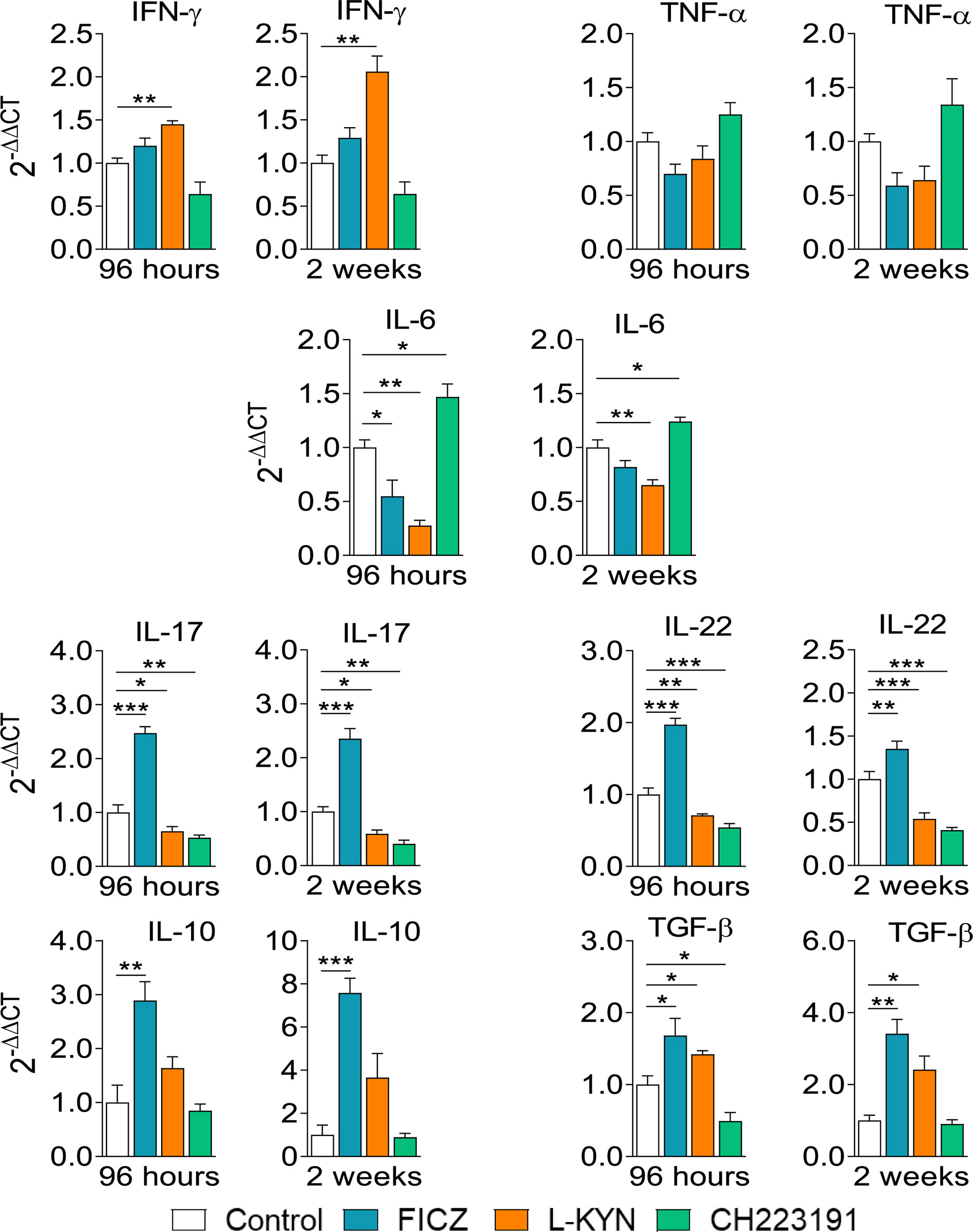
Figure 6 Treatment with AhR ligands alters mRNA expression for pro- and anti-inflammatory cytokines in the lungs of P. brasiliensis infected mice. C57BL/6 mice (n = 5) were infected with 1 x 106 P. brasiliensis yeasts and treated with FICZ, L-Kyn, CH223191 as previously described. Control mice were treated with PBS. The animals were sacrificed 96 hours and 2 weeks after infection, their lungs removed, macerated, and the RNA obtained analyzed by RT-PCR as described in M&M. The experiment was repeated twice, and data are expressed as M ± SD. P values < 0.05 were considered significant (*p < 0.05; **p < 0.005 and ***p < 0.001).
Mice were treated as previously described. Supernatants from lung macerates obtained 96 hours and 2 weeks post-infection were analyzed by ELISA for the presence of pro- and anti-inflammatory cytokines. As can be seen in Figure 7, IL-1β was the only cytokine that appeared in increased levels at both periods assayed and treatments used. In contrast, TNF-α and cytokines involved in Th1 (IL-12, IFN-γ) and Th2 (IL-4, IL-10) differentiation or activity appeared in reduced levels in almost all treatments and time points studied. As expected, AhR ligands have also altered the levels of cytokines involved in Th17 and Treg cells differentiation and activity (Figure 8). IL-6, IL-23, and TGF-β and were seen in reduced levels at least in one post-infection period after L-Kyn and CH223191 treatments and this was accompanied by reduced levels of IL-17 (week 2) and IL-22 (both infection periods). On the other hand, FICZ increased the levels of IL-17 (96 hours) and IL-22 (96 hours and 2 weeks), whereas CH223191 caused increased IL-17 production only in the first period assayed. Besides, all employed AhR ligands caused at the late time point assayed a reduction in TGF-β and IL-35, two suppressive cytokines involved in Treg cells activity.
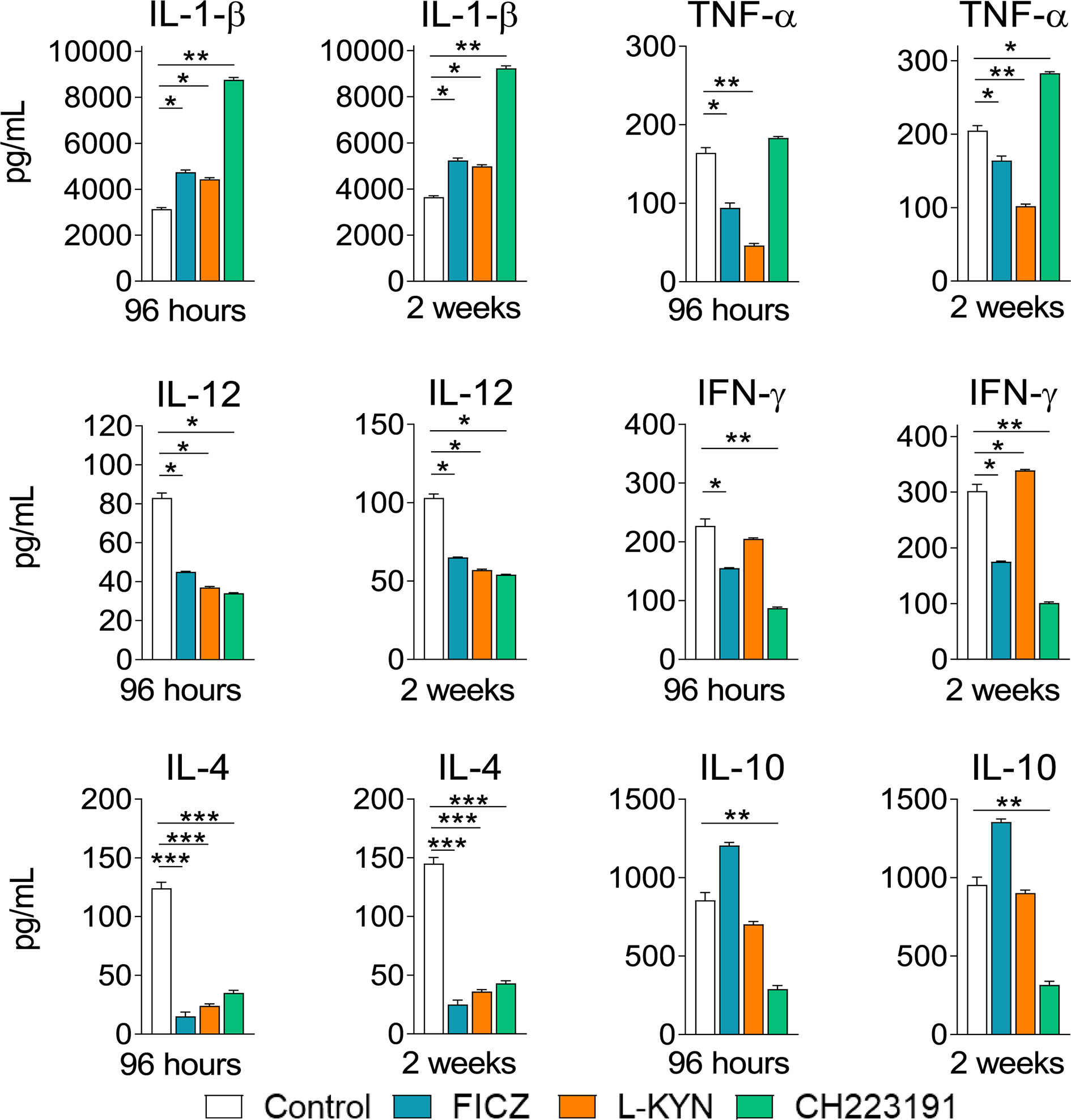
Figure 7 Treatment with AhR ligands increases the levels of pulmonary IL-1β but reduces the levels of cytokines involved in the development or activity of Th1 and Th2 cells. C57BL/6 mice (n = 5) were infected with 1 x 106 P. brasiliensis yeasts and treated with FICZ, L-Kyn, CH223191 as previously described. Control mice were treated with PBS. The animals were sacrificed 96 hours and 2 weeks after infection, their lungs removed, macerated and the supernatants analyzed for the presence of cytokines by ELISA. The experiment was repeated twice, and data are expressed as M ± SD. P values < 0.05 were considered significant (*p < 0.05; **p < 0.005 and ***p < 0.001).
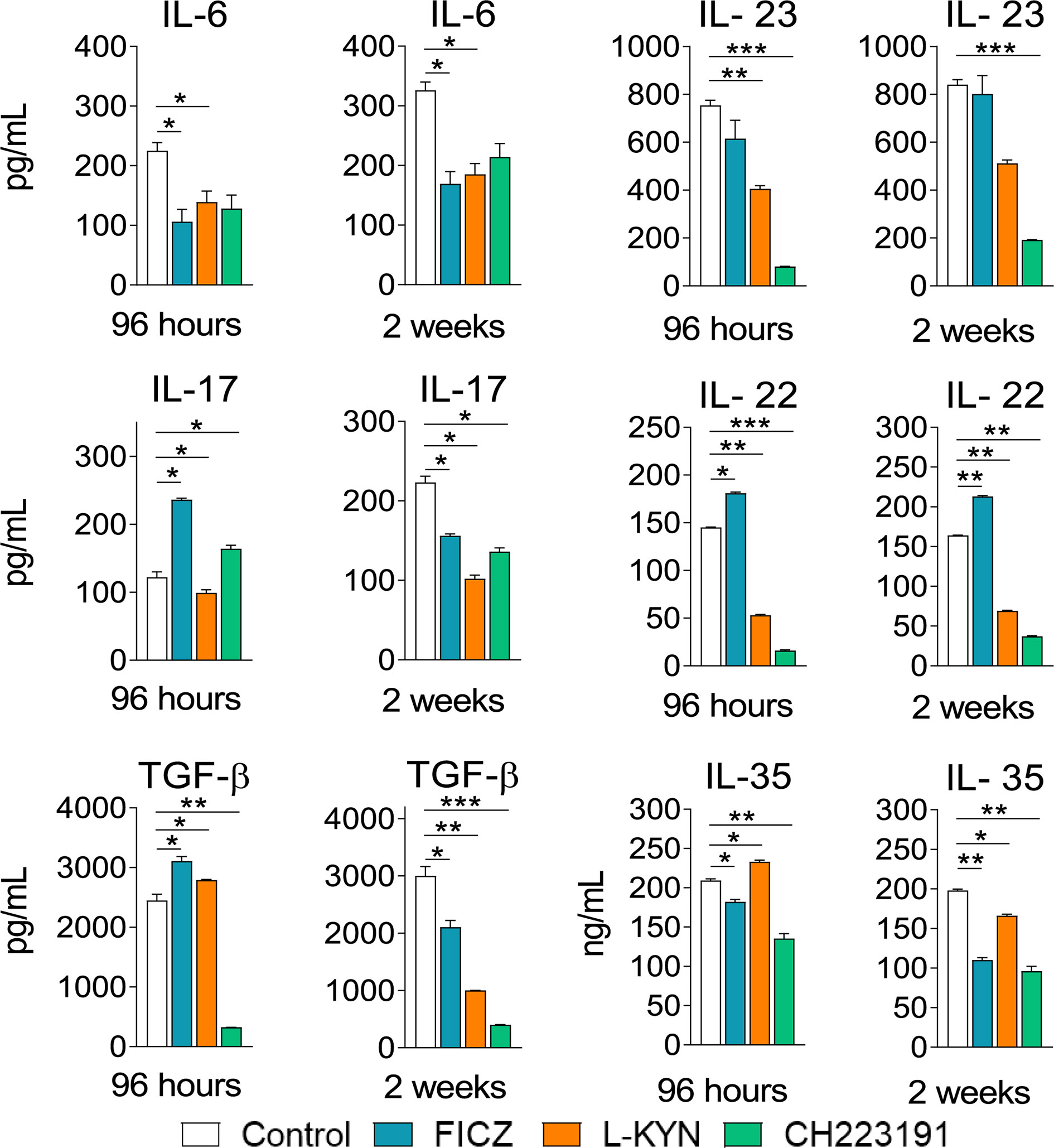
Figure 8 Treatment with AhR ligands alters the levels of pulmonary cytokines involved with Th17 and Treg cells development and activity. C57BL/6 mice (n = 5) were infected with 1 x 106 P. brasiliensis yeasts and treated with FICZ, L-Kyn, CH223191, as previously described. Control mice were treated with PBS. The animals were sacrificed 96 hours and 2 weeks after infection, their lungs removed, macerated and the supernatants analyzed for the presence of cytokines by ELISA. The experiment was repeated twice, and data are expressed as M ± SD. P values < 0.05 were considered significant (*p < 0.05; **p < 0.005 and ***p < 0.001).
Control and FICZ, L-Kyn, and CH223191 treated mice were sacrificed, their lungs removed, macerated, and the relative expression of mRNA for AhR, IDO, and master transcription factors for CD4+ T cell subsets differentiation were analyzed by RT-PCR. Similar results were obtained in both periods of infection (Figure 9). FICZ and L-Kyn agonists led to increased mRNA expression for IDO and AhR, while the antagonist CH223191 decreased their expression. FICZ reduced tbet but increased the expression of gata3 and rorc. L-Kyn treatment caused a robust increase in the foxp3 message while CH223191 did not significantly change the mRNA levels for all transcription factors assayed.
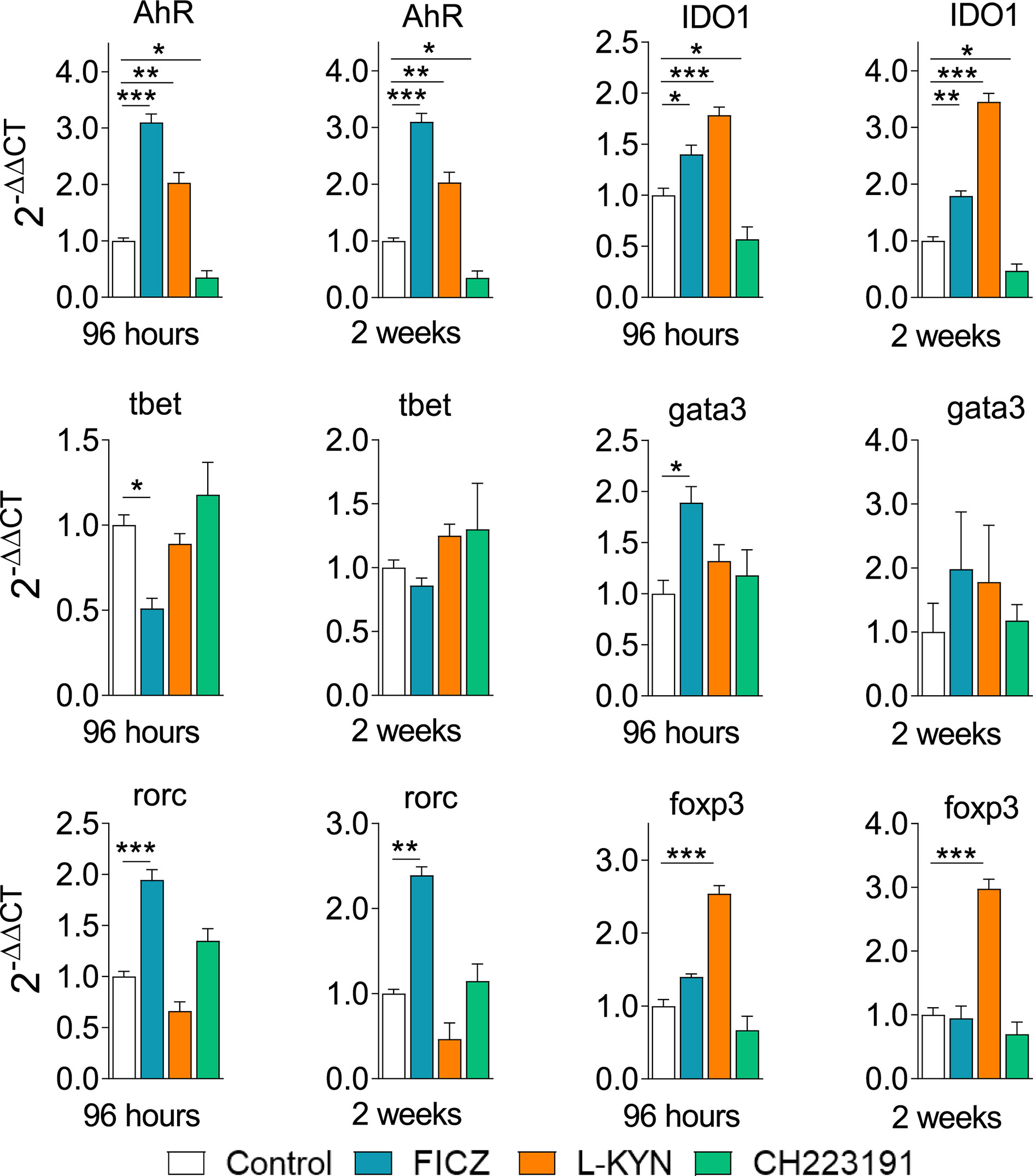
Figure 9 Treatment with AhR ligands alters the expression of mRNA for IDO, AhR, and transcription factors in the lungs of P. brasiliensis infected mice. C57BL/6 mice (n = 5) were infected with 1 x 106 P. brasiliensis yeasts and treated with FICZ, L-Kyn, CH223191 as previously described. Control mice were treated with PBS. The animals were sacrificed 96 hours and 2 weeks after infection, their lungs removed, macerated, and the RNA obtained was analyzed by RT-PCR as described in M&M. The experiment was repeated twice, and data are expressed as M ± SD. P values < 0.05 were considered significant (*p < 0.05; **p < 0.005 and ***p < 0.001).
Mice were treated as previously described. Two weeks after infection, isolated lung leukocytes were analyzed by flow cytometry regarding the presence of CD4+ T cell subsets. The gating strategies used to define CD4+ T cell subsets are shown in Figures 10A, B depicts the number of these cells present in the lungs of treated and untreated mice. The FICZ agonist significantly increased the migration of all cell T cell subpopulations (Th1, Th2, Th17, Th22, and Treg). Concomitant with the elevated expression of foxp3 mRNA, L-Kyn treatment caused a vigorous increase in Treg cells associated with Th1 and Th22 decrease. The CH223191 antagonist, on the other hand, reduced the migration of Th1, Th22, and Tregs but increased the presence of pulmonary Th17 lymphocytes. A general view of these results suggests that FICZ has a great inducing effect on the differentiation and migration of Th2, Th17, and Th22 subpopulations. L-Kyn has a great inducing effect on Treg cells, while the greatest effect of CH223191 is the expansion of Th17 lymphocytes associated with concomitant reduction of Treg cells.
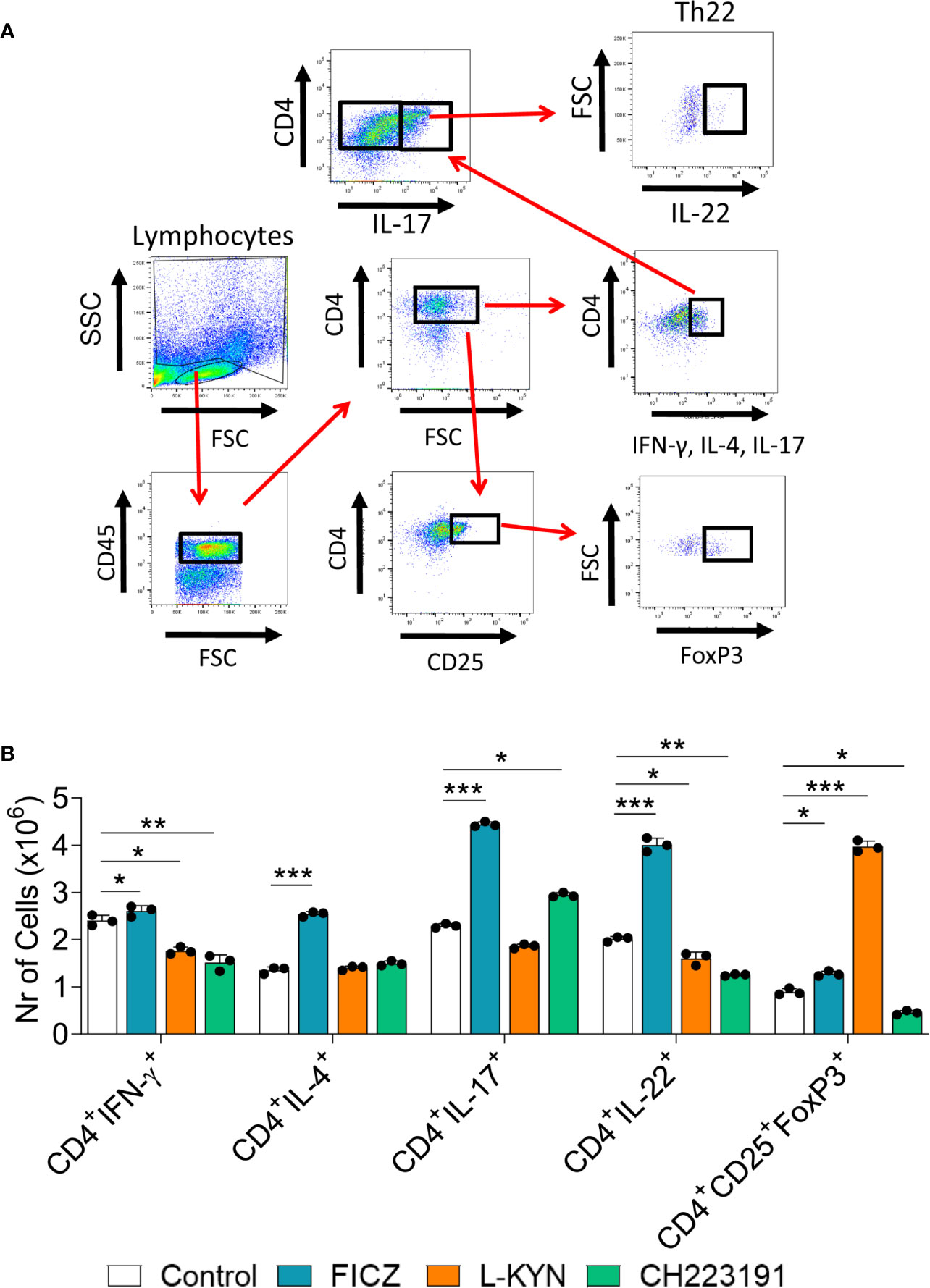
Figure 10 Treatment with AhR ligands alters the differentiation and migration of CD4+ T cell subsets to the lungs of P. brasiliensis infected mice. C57BL/6 mice were infected with 1 x 106 P. brasiliensis yeasts and treated on alternate days with FICZ, L-Kyn, or CH223191 at doses of 200, 800, or 400 μg/animal, respectively. Control mice were treated with PBS. The animals were sacrificed 2 weeks after infection, their lungs removed, macerated and the CD4+ T lymphocytes evaluated by flow cytometry for intracellular expression of Th signature cytokines (IFN-γ, IL-4, IL-17, IL-22) and the Treg phenotype (CD4+CD25+Foxp3+). (A) Gate strategy to define T cell subsets. (B) Number of Th1, Th2, Th17, Th22, and Treg cells present in the lungs of control and AhR ligands treated mice. The experiment was repeated twice, and data are expressed as M ± SD. P values < 0.05 were considered significant (*p < 0.05; **p < 0.005 and ***p < 0.001).
Traditionally considered a mediator in the toxic response to dioxin, AhR was later described as an important regulator of the immune response, including the immunity against infectious agents (13). In addition to inducing detoxifying enzymes, AhR modulates the differentiation and activity of innate and adaptive immune cells (10, 11, 13, 44), profoundly influencing the outcome of infectious and inflammatory processes (13, 45).
In pulmonary PCM, our group showed that P. brasiliensis infection induces and activates the enzyme IDO that causes TRP deprivation and L-Kyn production. TRP shortage leads to reduced fungal growth and diminished infection of DCs and macrophages. The enhanced L-Kyn synthesis, via AhR activation, increases the expansion of Treg cells that control excessive Th17-mediated tissue pathology (34–37, 46). Our studies also demonstrated an important interconnection between IDO and AhR expression, where a balanced activation of these mediators proved to be fundamental for the control of fungal immunity and disease tolerance (34–37, 46).
Since its description, the immunomodulatory effect of AhR has been associated with the control of both, exacerbated and deficient immune responses. These opposed effects result from the diverse interactions between AhR and its ligands and environmental cytokines, among other factors (10, 11, 47–49). Thus, the AhR activation can enhance pro-inflammatory responses, usually mediated by the Th1/Th17 subpopulations, or anti-inflammatory or suppressive immunity mediated by Treg and Tr1 cells (10, 11, 14). Environmental cytokines at the time of cell differentiation are fundamental to this process, and each pathogen and each specific disease induces complex patterns of mediators that are greatly influenced by the host’s genetic pattern, as well as more general environmental and metabolic factors (13, 14, 45). Indeed, comparing the effect of four different AhR agonists in the immunity and severity of a viral infection, Boule et al. (45) elegantly demonstrated that ligand metabolism and binding affinity, but not the chemical source, determines their immunological effects. Despite this great variability, some studies have shown that AhR agonists that are more difficult to be metabolized (eg: TCDD) induce increased expression of Treg (or Tr1) cells, while others, such as FICZ, induce greater polarization of T cells to the Th17 phenotype that synthesizes IL-17 and are also competent IL-22 producers (41, 50, 51). Th22 subpopulations, on the other hand, are most dependent on AhR expression (52, 53).
Our previous findings demonstrating the important role of the IDO/AhR/Treg/Th17 axis in the control of pulmonary PCM led us to comparatively investigate the immunomodulatory effect of two different agonists and one antagonist of AhR signaling. Thus, different groups of infected mice were treated with FICZ, a high-affinity agonist, L-Kyn, a low-affinity agonist, and CH223191 a low-affinity antagonist of AhR (49). The findings here reported demonstrate the peculiar effects of each of the AhR ligands studied in the control of pulmonary PCM. Some similar effects were shared by the FICZ and L-Kyn agonists, such as reduced fungal loads, decreased number of pulmonary CD11c+ myeloid cells expressing activation markers (IA, CD40, CD80, CD86), and increased expression of AhR and IDO. In contrast, these effects were opposed when the animals were treated with the AhR antagonist CH22319: there was an increase in pulmonary fungal loads and the number of CD11c+ leukocytes expressing activation markers, besides a drastic reduction in the expression of AhR and IDO. In the adaptive immune response, however, each of these ligands induced a characteristic profile. The large expansion of myeloid, lymphoid, and plasmacytoid DCs induced by FICZ increased all CD4+ T cell subpopulations (Th1, Th2, Th17, Th22, and Treg), but with the predominance of the Th17 and Th22 subsets. L-Kyn, which preferentially activated plasmacytoid DCs, possibly tolerogenic (35, 46, 54, 55), reduced the expansion of Th1 and Th22 cells but caused a great expansion of Treg cells differentiation. CH223191, the AhR antagonist, induced the preferential expansion of myeloid DCs, reduced the number of Th1, Th22, and Treg lymphocytes, but caused a significant increase in Th17 cells. This compound also induced a great decrease in the synthesis of pulmonary cytokines, with emphasis on the reduction of the various cytokines associated with the suppressive function of Tregs (IL-10, IL-35, and TGF-β). ILC3 was the most ILC subpopulation affected by AhR ligands treatment. While treatment with FICZ promoted a large increase in ILC3, the CH223191 antagonist drastically reduced this cell population. L-Kyn increased the number of ILC1 in the lungs but, similarly to CH223191, reduced the expansion of ILC3.
Treatment with FICZ augmented the number of CD11c+ myeloid cells expressing some intracellular pro- and anti-inflammatory cytokines that appear to have influenced the differentiation of all CD4+ T cell subsets here described. Interestingly, FICZ treatment caused a significant reduction in almost all secreted pulmonary cytokines except for IL-1β, TGF-β, IL-17, and IL-22, all cytokines involved in Th17 expansion and activity, and a prominent Th cell expanded by this treatment. Besides, the characterization of lung mRNA demonstrated constant increases in ahr, rorc, il-22, and il-17, indicating the tendency of this AhR agonist to induce prevalent Th17 and Th22 responses. This finding is in agreement with a pioneering publication by Quintana et al. (11) demonstrating that FICZ induces the production of transcription factors and cytokines that coordinate the preferential differentiation of T cells to the Th17 profile. The increased differentiation of Th22 is also in agreement with the IL-22 dependence of AhR expression (56). This AhR agonist also induced a large increase in ILC3 lymphocytes, IL-17, and IL-22 producers and highly dependent on the transcription factors RORc and AhR [12, 57).
The analysis of mRNA present in the lungs of L-Kyn treated mice confirmed a large increase in the message for AhR and IDO in the two post-infection periods analyzed. Analogous to FICZ, treatment with L-Kyn increased the numbers of CD11c+ myeloid cells expressing pro- and anti-inflammatory cytokines. However, a large expansion of plasmacytoid DCs, which have a tolerogenic profile in pulmonary PCM (35, 46), was detected in L-Kyn treated mice. Indeed, L-Kyn treatment induced a robust increase in pulmonary Treg (CD4+CD25+Foxp3+) cells, and this finding was associated with the large expression of mRNA for Foxp3 in the two periods of infection studied. The profile of soluble pulmonary cytokines in L-Kyn treated mice showed a consistent reduction in almost all cytokines assayed, except for TGF-β and IL-35 that could be associated with the increased Treg cells expansion here described. Still, a reduction in Th22 cells observed at the second week post-infection, and this finding was accompanied by a reduction in IL-22 in the lung supernatants and in mRNA for IL-22 of L-Kyn treated mice at the two periods of disease assayed. Since the synthesis of IL-22 is highly dependent on AhR (56), and this transcription factor appeared at high levels as protein in myeloid cells, and as mRNA in total lung cells, we can suppose that the reduction in Th22 lymphocytes mediated by L-Kyn treatment could have been influenced by the concomitant activity of other transcription factors or the increased expansion of Treg cells. In this respect, it is also worth mentioning the great reduction of ILC3, which depends on the RORc transcription factor for IL-17 synthesis, but on AhR for IL-22 production (12, 57). The decrease in ILC3 was consistently accompanied by a reduction in IL-17 and IL-22 in the lung supernatants, but not in the expression of AhR. However, to better analyze the effect of AhR on ILCs, we should have phenotypically characterized the simultaneous synthesis of IL-17 and IL-22 by ILC3 as well as the NCR-IL-22 subpopulation whose IL-22 synthesis is AhR-dependent (57). In summary, treatment with L-Kyn appears to have exerted a predominant anti-inflammatory effect on PCM, mainly due to the early increased expression of AhR and IDO that controlled the fungal load and established a low expression of activation molecules by myeloid cells, a predominant expansion of plasmacytoid DCs and an increased differentiation of Foxp3+ Treg cells.
The AhR antagonist CH223191, as expected, caused opposed effects to the studied agonists, in particular FICZ. CH223191 treatment reduced the number of IDO+ CD11c+ cells and increased the number of viable P. brasiliensis yeasts present in the lungs at both time points studied. This agrees with our previous reports demonstrating the increased P. brasiliensis growth when IDO is metabolically inhibited or genetically ablated, possibly by the increased TRP availability for fungal metabolism (34–37), The reduction in IDO was concomitant with that of AhR, the two components that are mutually controlled (14). Contrasting the treatments with FICZ and L-Kyn, CH223191 caused a large increase in the number of CD11c+ myeloid cells expressing activation molecules, intracellular TNF-α and a preferential expansion of myeloid DCs. It was also observed a reduction in Th1, Th22, and Treg cells besides an increase in Th17 cells. The reduction in Treg cells was concomitant with decreased levels of several cytokines (IL-10, TGF-β, and IL-35) associated with their anti-inflammatory function. The reduction of Th1 lymphocytes was concomitant with a low presence of IFN-γ in pulmonary cell supernatants, while the increase in Th17 lymphocytes occurred with an increase in IL-17 only at 96 hours after infection. Contrasting the AhR agonists, CH223191 reduced, as expected, the mRNA expression for IDO and AhR. This reduced AhR expression was associated with decreased numbers of AhR-dependent ILC3 since we did not notice a reduction in RORc expression. As a whole, treatment with an AhR antagonist reproduced the main findings that we observed in P. brasiliensis infected AhR-/- mice (37), increased fungal loads, and Th17 immunity not adequately controlled by insufficient Treg cells expansion.
The lung is a barrier organ that expresses AhR at high levels (58). This transcription factor is expressed by epithelial and immune cells, is involved in mucous secretion (59) and the balanced immune response in the lung (49). In accordance, our studies have demonstrated the important participation of AhR in the control of disease severity and immune response in pulmonary PCM (34–37, 46). The present study allowed us to demonstrate that this fungal infection can be modulated by AhR ligands in opposed directions as previously demonstrated in other experimental models (10, 11, 45), suggesting their therapeutic use in different forms of the disease. FICZ, due to its fungicidal effect and prominent pro-inflammatory activity that mediates the increased expansion of all T cell subsets but prevalent Th17 differentiation, could be used as adjunct therapy of severe human PCM characterized by T cell anergy and high fungal loads (25). L-Kyn, which favors the expansion of Treg cells but reduces fungal loads, could be used as an immunomodulator in those situations of severe tissue damage associated with hyperreactivity of the immune system, which occasionally occurs in the human PCM (60). The use of the AhR antagonist, which leads to excessive fungal growth and increased Th17 immunity should be therapeutically discarded since its effect mimics those observed with AhR-/- mice, where the uncontrolled fungal growth associated with unrestrained pro-inflammatory reactions lead to extremely severe disease. Finally, our data encourage further studies on the immunomodulation of PCM by AhR agonists and open the perspective of their use in future immunotherapeutic procedures.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Committee on Animal Experiments of the Institute of Biomedical Sciences of the University of São Paulo (Proc.180/11/CEEA).
EA and VC conceived and planned experiments. EA, NP, CF, NG, and TC carried out the experiments. EA, FL, and VC contributed to the interpretation of the results. EA, FL, and VC wrote the paper. EA, NP, and FL prepared the figures. VC supervised the project and provided financial support. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP- grant to VC 2011/51258-2 and 2016/23189-0; fellowship to EA 2014/18668-2; grant to FL 2018/14762-3; fellowship to NP 2019-09278-8), and Conselho Nacional de Pesquisa (CNPq).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.630938/full#supplementary-material
1. Mandal PK. Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol B (2005) 175:221–30. doi: 10.1007/s00360-005-0483-3
2. Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol (2003) 43:309–34. doi: 10.1146/annurev.pharmtox.43.100901.135828
3. Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol (2014) 32:403–32. doi: 10.1146/annurev-immunol-032713-120245
4. Schrenk D. Impact of dioxin-type induction of drug-metabolizing enzymes on the metabolism of endo- and xenobiotics. Biochem Pharmacol (1998) 55:1155–62. doi: 10.1016/s0006-2952(97)00591-1
5. Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol (2008) 21:102. doi: 10.1021/tx7001965
6. DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, et al. L-Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol Sci (2010) 115:89–97. doi: 10.1093/toxsci/kfq024
7. Gutiérrez-Vázquez C, Quintana FJ. Regulation of immune response by the aryl hydrocarbon receptor. Immunity (2017) 48:19–33. doi: 10.1016/j.immuni.2017.12.012
8. Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, et al. The aryl hydrocarbon receptor interacts with cMaf to promote differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol (2010) 11:854–61. doi: 10.1038/ni.1912
9. Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci USA (2008) 105:9721. doi: 10.1073/pnas.0804231105
10. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links Th17-cell-mediated autoimmunity to environmental toxins. Nature (2008) 453:106–9. doi: 10.1038/nature06881
11. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(h)17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453:65. doi: 10.1038/nature06880
12. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science (2011) 334:1561–5. doi: 10.1126/science.1214914
13. Lawrence BP, Vorderstrasse BA. New insights into the aryl hydrocarbon receptor as a modulator of host responses to infection. Semin Immunopathol (2013) 35:615–26. doi: 10.1007/s00281-013-0395-3
14. Nguyen NT, Hanieh H, Nakahama T, Kishimoto T. The roles of aryl hydrocarbon receptor in immune responses. Int Immunol (2013) 25:335–43. doi: 10.1093/intimm/dxt011
15. Safe S, Cheng Y, Un-Ho J. The aryl hydrocarbon receptor (AhR) as a drug target for cancer chemotherapy. Curr Opin Toxicol (2017) 2:24–9. doi: 10.1016/j.cotox.2017.01.012
16. Loures FV, Pina A, Felonato M, Araújo EF, Leite KRM, Calich VLG. TLR4 signaling leads to a more severe fungal infection associated with enhanced proinflammatory immunity and impaired expansion of regulatory T cells. Infect Immun (2010) 78:1078–88. doi: 10.1128/IAI.01198-09
17. Loures FV, Pina A, Felonato M, Feriotti C, de Araújo EF, Calich VLG. MyD88 signaling is required for efficient innate and adaptive immune responses to Paracoccidioides brasiliensis infection. Infect Immun (2011) 79:2470–80. doi: 10.1128/IAI.00375-10
18. Loures FV, de Araújo EF, Feriotti C, Bazan SB, Costa TA, Brown GD, et al. Dectin-1 Induces M1 Macrophages and Prominent Expansion of CD8+IL-17+ Cells in a Pulmonary Model of Fungal Infection. J Infect Dis (2014) 210:762–73. doi: 10.1093/infdis/jiu136
19. Loures FV, de Araújo EF, Feriotti C, Bazan SB, Calich VLG. TLR-4 Cooperates with Dectin-1 and Mannose Receptor to Expand Th17 and Tc17 Cells Induced by Paracoccidioides brasiliensis Stimulated Dendritic Cells. Front Microbiol (2015) 6:261. doi: 10.3389/fmicb.2015.00261
20. Feriotti C, Loures FV, de Araújo EF, Costa TA, Calich VLG. Mannosyl-Recognizing Receptors Induce an M1-Like Phenotype in Macrophages of Susceptible Mice whereas an M2-Like Phenotype in Resistant Mice to a Fungal Infection. PloS One (2013) 8:e54845. doi: 10.371/journal.pone.0054845
21. Feriotti C, Bazan SB, Loures FV, deAraújo EF, Costa TA, Calich VLG. Expression of dectin1 and enhanced activation of NALP3 inflammasome are associated with resistance to paracoccidioidomycosis. Front Microbiol (2015) 6:913. doi: 10.3389/fmicb.2015.00913
22. Feriotti C, de Araújo EF, Loures FV, Costa TA, Galdino NAL, Zamboni DA, et al. NOD-Like Receptor P3 inflammasome controls protective Th1/Th17 immunity against pulmonary paracoccidioidomycosis. Front Immunol (2017) 8:786. doi: 10.3389/fimmu.2017.00786
23. Preite NW, Feriotti C, Souza de Lima D, Silva BB, Condino-Neto A, Pontillo A, et al. The Syk-coupled C-type lectin receptors dectin-2 and dectin-3 are involved in Paracoccidioides brasiliensis recognition by human plasmacytoid dendritic cells. Front Immunol (2018) 9:464. doi: 10.3389/fimmu.2018.00464
24. Felonato M, Pina A, de Araujo EF, Loures FV, Bazan SB, Feriotti C, et al. Anti-CD25 treatment depletes Treg cells and decreases disease severity in susceptible and resistant mice infected with Paracoccidioides brasiliensis. PloS One (2012) 7:e51071. doi: 10.1371/journal.pone.0051071
25. de Castro LF, Ferreira MC, da Silva RM, Blotta MH, Longhi LN, Mamoni RL. Characterization of the immune response in human paracoccidioidomycosis. J Infect (2013) 67:470–85. doi: 10.1016/j.jinf.2013.07.019
26. Cavassani KA, Campanelli AP, Moreira AP, Vancim JO, Vitali LH, Mamede RC, et al. Systemic and local characterization of regulatory T cells in a chronic fungal infection in humans. J Immunol (2006) 177:5811–8. doi: 10.4049/jimmunol.177.9.5811
27. Ferreira MC, Oliveira RTD, Silva RM, Blotta MHSL, Mamoni RL. Involvement of regulatory T Cells in the immunosuppression characteristic of patients with paracoccidioidomycosis. Infect Immun (2010) 78:4392–401. doi: 10.1128/IAI.00487-10
28. Calich VLG, Mamoni RL, Loures FV. Regulatory T cells in paracoccidioidomycosis. Virulence (2019) 10:810–21. doi: 10.1080/21505594.2018.1483674
29. Bazan SB, Costa TA, de Araújo EF, Feriotti C, Loures FV, Pretel FD, et al. Loss- and gain-of-function approaches indicate a dual role exerted by regulatory T cells in pulmonary paracoccidioidomycosis. PloS Negl Trop Dis (2015) 9:e0004189. doi: 10.1371/journal.pntd.0004189
30. De Luca A, Montagnoli C, Zelante T, Bonifazi P, Bozza S, Moretti S, et al. Functional yet balanced reactivity to Candida albicans requires TRIF, MyD88, and IDO-dependent inhibition of Rorc. J Immunol (2007) 179:5999–6008. doi: 10.4049/jimmunol.179.9.5999
31. Romani L, Zelante T, De Luca A, Fallarino F, Puccetti P. IL-17 and therapeutic L-Kynurenines in pathogenic inflammation to fungi. J Immunol (2008) 180:5157–62. doi: 10.4049/jimmunol.180.8.5157
32. De Luca A, Zelante T, D’Angelo C, Zagarella S, Fallarino F, Spreca A, et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol (2010) 3:361–73. doi: 10.1038/mi.2010.22
33. Veldhoen M, Hirota K, Christensen J, O’Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 cells. J Exp Med (2009) 206:43–9. doi: 10.1084/jem.20081438
34. Araújo EF, Loures FV, Bazan SB, Feriotti C, Pina A, Schanoski AS, et al. Indoleamine 2,3-Dioxygenase controls fungal loads and immunity in paracoccidioidomicosis but is more important to susceptible than resistant hosts. PloS Negl Trop Dis (2014) 8:e3330. doi: 10.1371/journal.pntd.0003330
35. Araújo EF, Loures FV, Feriotti C, Costa T, Vacca C, Puccetti P, et al. Disease tolerance mediated by phosphorylated indoleamine-2,3 dioxygenase confers resistance to a primary fungal pathogen. Front Immunol (2017) 8:1522. doi: 10.3389/fimmu.2017.01522
36. Araújo EF, Costa T, Preite NW, Loures FV, Calich VLG. The IDO-AhR-Treg axis controls Th17/Th22 immunity in a pulmonary model of fungal infection. Front Immunol (2018) 8:880. doi: 10.3389/fimmu.2017.00880
37. Araújo EF, Preite NW, Veldhoen M, Loures FV, Calich VLG. Pulmonary paracoccidioidomycosis in AhR deficient hosts is severe and associated with defective Treg and Th22 responses. Sci Rep (2020) 10:11312. doi: 10.1038/s41598-020-68322-6
38. Duarte JH, Di Meglio P, Hirota K, Ahlfors H, Stockinger B. Differential Influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PloS One (2013) 8:e79819. doi: 10.1371/journal.pone.0079819
39. Parks AJ, Pollastri MP, Hahn ME, Stanford EA, Novikov O, Franks DG, et al. In silico identification of an aryl hydrocarbon receptor antagonist with biological activity in vitro and in vivo. Mol Pharmacol (2014) 86:593–608. doi: 10.1124/mol.114.093369
40. Kim SH, Henry EC, Kim DK, Kim YH, Shin KJ, Han MS, et al. Novel compound 2-methyl-2H-pyrazole-3-carboxylic acid (2-methyl-4-o-tolylazo-phenyl)-amide (CH-223191) prevents 2,3,7,8-TCDD-induced toxicity by antagonizing the aryl hydrocarbon receptor. Mol Pharmacol (2006) 69:1871–78. doi: 10.1124/mol.105.021832
41. De Luca A, Carvalho A, Cunha C, Iannitti RG, Pitzurra L, Giovannini G, et al. IL-22 and IDO1 affect immunity and tolerance to murine and human vaginal candidiasis. PloS Pathog (2013) 9:e1003486. doi: 10.1371/journal.ppat.1003486
42. Galdino NAL, Loures FV, de Araújo EF, da Costa TA, Preite NW, Calich VLG. Depletion of regulatory T cells in ongoing paracoccidioidomycosis rescues protective Th1/Th17immunity and prevents fatal disease outcomes. Sci Rep (2018) 8:16544. doi: 10.1038/s41598-018-35037-8
43. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13:145–9. doi: 10.1038/nri3365
44. Mulero-Navarro S, Fernandez-Salguero PM. New trends in aryl hydrocarbon receptor biology. Front Cell Dev Biol (2016) 4:45. doi: 10.3389/fcell.2016.00045
45. Boule LA, Burke CG, Jin GB, Laurence BP. Aryl hydrocarbon receptor signaling modulates antiviral immune responses: ligand metabolism rather than chemical source is the stronger predictor of outcome. Sci Rep (2018) 8:1286. doi: 10.1038/s41598-018-20197-4
46. Araújo EF, Medeiros DH, Galdino NA, Condino-Neto A, Calich VL, Loures FV. Tolerogenic plasmacytoid dendritic cells control Paracoccidioides brasiliensis infection by inducing regulatory T cells in an IDO-dependent manner. PloS Pathog (2016) 12:e1006115. doi: 10.1371/journal.ppat.1006115
47. Wheeler JL, Martin KC, Resseguie E, Lawrence BP. Differential consequences of two distinct AhR ligands on innate and adaptive immune responses to influenza A virus. Toxicol Sci (2014) 137:324–34. doi: 10.1093/toxsci/kft255
48. Benson J, Shepherd DM. Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn’s disease. Toxicol Sci (2011) 120:68–78. doi: 10.1093/toxsci/kfq360
49. Esser C, Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol Rev (2015) 67:259–79. doi: 10.1124/pr.114.009001
50. Monteleone I, Rizzo A, Sarra M, Sica G, Biancone L, McDonald TT, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production an inhibit inflammation in the gastrointestinal tract. Gastroenterology (2011) 141:237–48. doi: 10.1053/j.gastro.2011.04.007
51. Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity (2008) 29:958–70. doi: 10.1016/j.immuni.2008.11.001
52. Wang J, Wang P, Tian H, Tian F, Zhang Y, Zhang L, et al. Aryl hydrocarbon receptor/IL-22/Stat3 signaling pathway is involved in the modulation of intestinal mucosa antimicrobial molecules by commensal microbiota in mice. Innate Immun (2018) 24:297–306. doi: 10.1177/1753425918785016
53. Yeste A, Mascanfroni ID, Nadeau M, Burns EJ, Tukpah AM, Santiago A, et al. IL-21 induces IL-22 production in CD4+ T cells. Nat Commun (2014) 5:3753. doi: 10.1038/ncomms4753
54. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. Tryptophan catabolism generates autoimmune-preventive regulatory T cells. Transpl Immunol (2006) 17:58–60. doi: 10.1016/j.trim.2006.09.017
55. Fallarino F, Grohmann U, Puccetti P. Indoleamine 2,3-dioxygenase: from catalyst to signaling function. Eur J Immunol (2012) 42:1932–7. doi: 10.1002/eji.201242572
56. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity (2008) 28:29–39. doi: 10.1016/j.immuni.2007.11.016
57. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol (2016) 17:758–64. doi: 10.1038/ni.3482
58. Frericks M, Meissner M, Esser C. Microarray analysis of the AHR system: tissue-specific flexibility in signal and target genes. Toxicol Appl Pharmacol (2007) 220:320–32. doi: 10.1016/j.taap.2007.01.014
59. Chiba T, Uchi H, Tsuji G, Gondo H, Moroi Y, Furue M, et al. Aryl hydrocarbon receptor (AhR) activation in airway epithelial cells induces MUC5AC via reactive oxygen species (ROS) production. Pulm Pharmacol Ther (2011) 24:133–40. doi: 10.1016/j.pupt.2010.08.002
60. Gryschek RCB, Pereira RM, Kono A, Patzina RA, Tresoldi AT, Shikanai-Yasuda MA, et al. Paradoxical reaction to treatment in 2 patients with severe acute paracoccidioidomycosis: a previously unreported complication and its management with corticosteroids. Clin Infect Dis (2010) 50:e56–8. doi: 10.1086/652290
Keywords: innate lymphoid cells (ILCs), L-kynurenine, FICZ, T cell subsets, paracoccidiodomycosis, IDO - Indoleamine 2 3-dioxygenase, AhR (Aryl hydrocarbon Receptor)
Citation: de Araújo EF, Loures FV, Preite NW, Feriotti C, Galdino NAL, Costa TA and Calich VLG (2021) AhR Ligands Modulate the Differentiation of Innate Lymphoid Cells and T Helper Cell Subsets That Control the Severity of a Pulmonary Fungal Infection. Front. Immunol. 12:630938. doi: 10.3389/fimmu.2021.630938
Received: 18 November 2020; Accepted: 30 March 2021;
Published: 16 April 2021.
Edited by:
Gilles J. Guillemin, Macquarie University, AustraliaCopyright © 2021 de Araújo, Loures, Preite, Feriotti, Galdino, Costa and Calich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vera L. G. Calich, dmxjYWxpY2hAaWNiLnVzcC5icg==
†Present address: Flávio V. Loures, Institute of Sciences and Technology, Federal University of São Paulo, São José dos Campos, Brazil Nycolas W. Preite, Institute of Sciences and Technology, Federal University of São Paulo, São José dos Campos, Brazil Nayane AL Galdino, Cancer Hospital of São Paulo, São Paulo, Brazil
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.