 Sajjad Muhammad1,2,3*
Sajjad Muhammad1,2,3* Shafqat Rasul Chaudhry4
Shafqat Rasul Chaudhry4 Gergana Dobreva3
Gergana Dobreva3 Michael T. Lawton5
Michael T. Lawton5 Mika Niemelä2Daniel Hänggi1
Mika Niemelä2Daniel Hänggi1- 1Department of Neurosurgery, Faculty of Medicine, Heinrich-Heine-University, Düsseldorf, Germany
- 2Department of Neurosurgery, Helsinki University Hospital, University of Helsinki, Helsinki, Finland
- 3Department of Anatomy and Developmental Biology, Medical Faculty Mannheim and European Center for Angioscience (ECAS), University of Heidelberg, Mannheim, Germany
- 4Shifa College of Pharmaceutical Sciences, Shifa Tameer-e-Millat University, Islamabad, Pakistan
- 5Department of Neurosurgery, Barrow Brain and Spine, Barrow Neurological Institute, Phoenix, AZ, United States
Aneurysmal subarachnoid hemorrhage (aSAH) is a highly fatal and morbid type of hemorrhagic strokes. Intracranial aneurysms (ICAs) rupture cause subarachnoid hemorrhage. ICAs formation, growth and rupture involves cellular and molecular inflammation. Macrophages orchestrate inflammation in the wall of ICAs. Macrophages generally polarize either into classical inflammatory (M1) or alternatively-activated anti-inflammatory (M2)-phenotype. Macrophage infiltration and polarization toward M1-phenotype increases the risk of aneurysm rupture. Strategies that deplete, inhibit infiltration, ameliorate macrophage inflammation or polarize to M2-type protect against ICAs rupture. However, clinical translational data is still lacking. This review summarizes the contribution of macrophage led inflammation in the aneurysm wall and discuss pharmacological strategies to modulate the macrophageal response during ICAs formation and rupture.
Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) is a devastating subtype of hemorrhagic strokes and it accounts for 5% of all strokes. The worldwide incidence of aSAH is approximately 700000 person-years; the mortality of aSAH is approximately 40% despite appropriate surgical and medical care (1, 2). aSAH has a poor prognosis with significant lifelong morbidity and cognitive deficits for those who survive. Moreover, aSAH has a significant impact on society, as it often affects young people at the peak of their productive life (1, 2). This highly fatal and morbid type of intracranial hemorrhage is due to intracranial aneurysm (ICA) rupture in nearly 85% of SAH cases (3).
ICAs are weak ballooning, bulging, or abnormal dilatations that tend to form at arterial bifurcations due to chronic hemodynamic stress and inflammation (4). Intracranial aneurysms are usually found in 3% to 5% of the population and are slightly more prevalent among females (5). The risk factors for aneurysm development are arterial hypertension, smoking, chronic alcohol consumption, aging, female gender, and family history of aSAH in first-degree relatives (6). Some genetic disorders such as autosomal dominant polycystic kidney disease, Marfan syndrome, Ehlers-Danlos syndrome type IV, neurofibromatosis type 1, and fibromuscular dysplasia are associated with ICA formation (6). Moreover, single nucleotide gene polymorphisms (SNPs) in or near the genes CDKN2B-AS1, SOX17 transcription regulator gene, endothelin receptor gene, HDAC9, and the gene encoding elastin have been revealed in genome-wide association studies (GWAS). Linkage analysis suggests that these genes are strongly associated with intracranial aneurysms (5). An exome-wide association study identified a SNP of the collagen type XVIIα1 chain gene to be significantly associated with aSAH (7). Most ICAs are found incidentally and need preventive care to prevent enlargement and rupture. Prevention of growth and rupture is necessary, as the current treatment modalities, such as surgical clipping and endovascular modalities (coiling, with or without stent and flow diverter placement) are associated with some risks.
Patient factors (age, sex, comorbidities, family history, previous history of SAH, hypertension and smoking) and aneurysm characteristics (size, location, wall irregularity, presence of secondary pouches) are key factors that aid in deciding upon treatment for an unruptured ICA. It is challenging to predict exactly the rupture risk based on aneurysm characteristics and patient risk factors. It is thus, unclear which ICAs require active treatment.
A better understanding of the pathobiology of ICA is important to clarify when active treatment is needed and may facilitate development of pharmacological treatments with no or minimal risk.
Recent evidence from human and animal studies revealed that macrophage-mediated cellular and molecular inflammation is the key player in aneurysm formation and rupture. Here, we briefly review the current knowledge on the role of macrophages in aneurysm formation and their rupture.
Inflammation in Intracranial Aneurysms
The hallmarks of ICAs include endothelial cell dysfunction, smooth muscle cell phenotypic switch, matrix metalloproteinase secretion, and innate immune cell activation leading to vascular remodeling and vessel wall weakening (8–10). Histopathological analysis of aneurysm wall biopsies has revealed an upregulation of inflammatory mediators, disruption of lamina elastic interna, and thinning of media including mural cell death (11). Both cellular and molecular inflammation are crucial in aneurysm formation and rupture. Infiltration of inflammatory cells (especially macrophages) has been observed in the biopsies of ICAs, which shows a possible involvement of macrophages in aneurysm formation. NF-κB is a key transcription factor and is a major known regulator of important pro-inflammatory genes, including TNF, IL-1β, and COX-2. A genetic deletion of NF-κB has been shown to reduce ICA formation and growth (12). Moreover, pro-inflammatory genes regulated by NF-κB, including IL-1β (13), COX-2 (14), iNOS (15), and matrix metalloproteinase-9 (16) contribute to ICA formation. Furthermore, macrophage specific deletion of the prostaglandin E (PGE) receptor subtype 2 (EP2) (Ptger2), an upstream signaling receptor for NF-κB activation, significantly suppresses the development of ICAs in mice, indicating that prostaglandin E2-EP2-NF-κB signaling in macrophages plays a crucial role in ICA development (12). Intriguingly, macrophage-specific expression of a variant of IκBα, which abrogates the translocation of NF-κB, prevents ICA formation (17).
Transcriptomic analysis of ICAs revealed upregulation of pro-inflammatory cytokine genes associated with leukocyte infiltration (18–23). For instance, Nakaoka, Tajima (19) have shown an upregulation of genes related to inflammation, immune response and phagocytosis, whereas anti-inflammatory genes were downregulated. Similarly, upregulation of TNF-α and pro-apoptotic gene expression was shown along with suppressed IL-10 expression in ruptured ICAs. Moreover, SNPs in the IL-10 gene are associated with formation of ICAs (24, 25). Similar, transcriptomic and bioinformatic analyses of ruptured and unruptured ICAs have revealed enhanced expression and upregulation of inflammatory pathways such as TLR signaling, cytokine-cytokine receptor interaction, leukocyte trans-endothelial migration, NF-κB signaling, and many other inflammation-related gene ontology categories (18). Activation and involvement of the complement system has also been observed in ICAs and suggests that chronic inflammation underlies the pathogenesis of ICAs (10, 26). Further, shear stress due to disturbed blood flow at arterial branching points (which contributes to ICA development) upregulates inflammatory pathways such as NF-κB, promotes monocyte recruitment, and triggers sterile inflammation (27, 28). Inflammation in ICA walls is characterized by immune cell infiltration and altered composition of the immune cell populations such as natural killer cells, mast cells, lymphocytes, and importantly macrophages (29).
Monocytes/Macrophages in Intracranial Aneurysms
Monocytes/macrophages are among the main components of innate immunity and represent important members of the mononuclear phagocyte system comprised of myeloid-derived cells (30). Data from human and animal studies has revealed an increased infiltration of immune cells in the aneurysm wall (24, 25, 31, 32). Several lines of evidence have shown increased infiltration of T and B lymphocytes and macrophages along with increased pro-inflammatory molecular expression in clinical resections of ICAs (24, 31, 32).
Studies have clearly demonstrated that monocyte/macrophage infiltration in the wall of ruptured aneurysms is not only found after aneurysmal rupture, but contributes to aneurysm formation and rupture (33). Increased monocyte/macrophage marker CD68 expression has been observed in mice carrying negative mutations of PPARγ in smooth muscle cells of cerebral arteries along with CXCL1, MCP-1, TNF-α expression upregulation. These mice have an increased incidence of aneurysm formation and rupture (34). Aoki, Frò`sen (12) demonstrated that macrophage infiltration driven by MCP-1 and activation of NF-κB involving PGE2-PGEP2 (PGE receptor subtype 2) signaling in the macrophages of arterial wall leads to aneurysm formation, suggesting that inflammation is not only present after aneurysm rupture, but also drives aneurysm formation. As intracranial arteries lack vasa vasorum in the arterial wall, the macrophages may infiltrate through endothelial cell junctions. Sphingosine-1-phosphate (S1P) receptor type 1 signaling activation strengthens the endothelial barrier. Interestingly, activation of S1P receptor type 1 reduced the number of infiltrated macrophages and enlargement of ICAs (35).
Macrophage Polarization and Intracranial Aneurysms
Mills, Kincaid (36) described for the first time the M1/M2 paradigm, where M1 represents classically activated pro-inflammatory monocytes/macrophages, whereas M2 represents alternatively activated anti-inflammatory monocytes/macrophages. A very brief and simplistic overview of M1/M2 biology is represented in Figure 1. However, there is a considerable heterogeneity in macrophage phenotypes and several subtypes have been described such as M1, M2a, M2b, M2c, M2d, Mhem, Mox, M4 (37–40). This over simplistic representation of M1 as pro-inflammatory and M2 as anti-inflammatory macrophages is considered here to recognize different functional states of these polarized phenotypes to assign pro-inflammatory and anti-inflammatory role. Macrophage polarization has implications in aneurysm formation and rupture (Figure 2). Aortic aneurysms formation has been shown to be promoted by inflammatory M1 macrophages, whereas reparative M2 polarization prevents the formation, development and progression of aortic aneurysms (38, 41). It has been shown that GM-CSF contributes toward M1 polarization and M-CSF favors an M2 response (42). Intriguingly, GM-CSF has been shown to promote aortic aneurysm formation (43) and the levels of GM-CSF measured in plasma and lumen of the intracranial aneurysms have also shown a direct correlation with the size of intracranial aneurysms, highlighting a common inflammatory process upregulated by M1 macrophages underlie the development of both aortic and intracranial aneurysms (44). Consequently, immunohistochemical analysis of intracranial aneurysm dome resections have revealed that ruptured intracranial aneurysms from patients possess increased M1 (HLA-DR+) cells opposed to M2 (CD163+) cells (45). These findings suggest that a balance shift toward M2 may prevent aneurysm rupture. Previously, Froesen and colleagues demonstrated differences in CD68+ and CD163+ macrophages in human ruptured and unruptured ICAs (29). Intriguingly, CD68+ and CD163+ (hemoglobin-haptoglobin scavenger receptor) macrophages, mostly HLA-DR-, co-localize with glycophorin A (a component of the erythrocyte membrane) and infiltrate ICAs as a response to a luminal thrombus trapped and lysed erythrocytes, and may promote degenerative arterial wall remodeling (46). In a mouse model of ICAs, M1 (F4/80+ iNOS+) dominate over M2 (F4/80+ Arg1+) during aneurysm development (47). Interestingly, M1 dominance leading to aneurysm development is dependent on neutrophil infiltration, which when blocked led to an increased M2 polarization with reduced aneurysm formation (47). Shimada, Furukawa (48) employed different macrophage markers to assess the polarization of macrophages in ICAs. The authors employed CD68 as a macrophage marker and IL-12p40 and CD206 as M1 and M2 markers, respectively. They observed significant impairment in the M1/M2 ratio in ICAs associated with upregulation of M1-related gene expression (48).
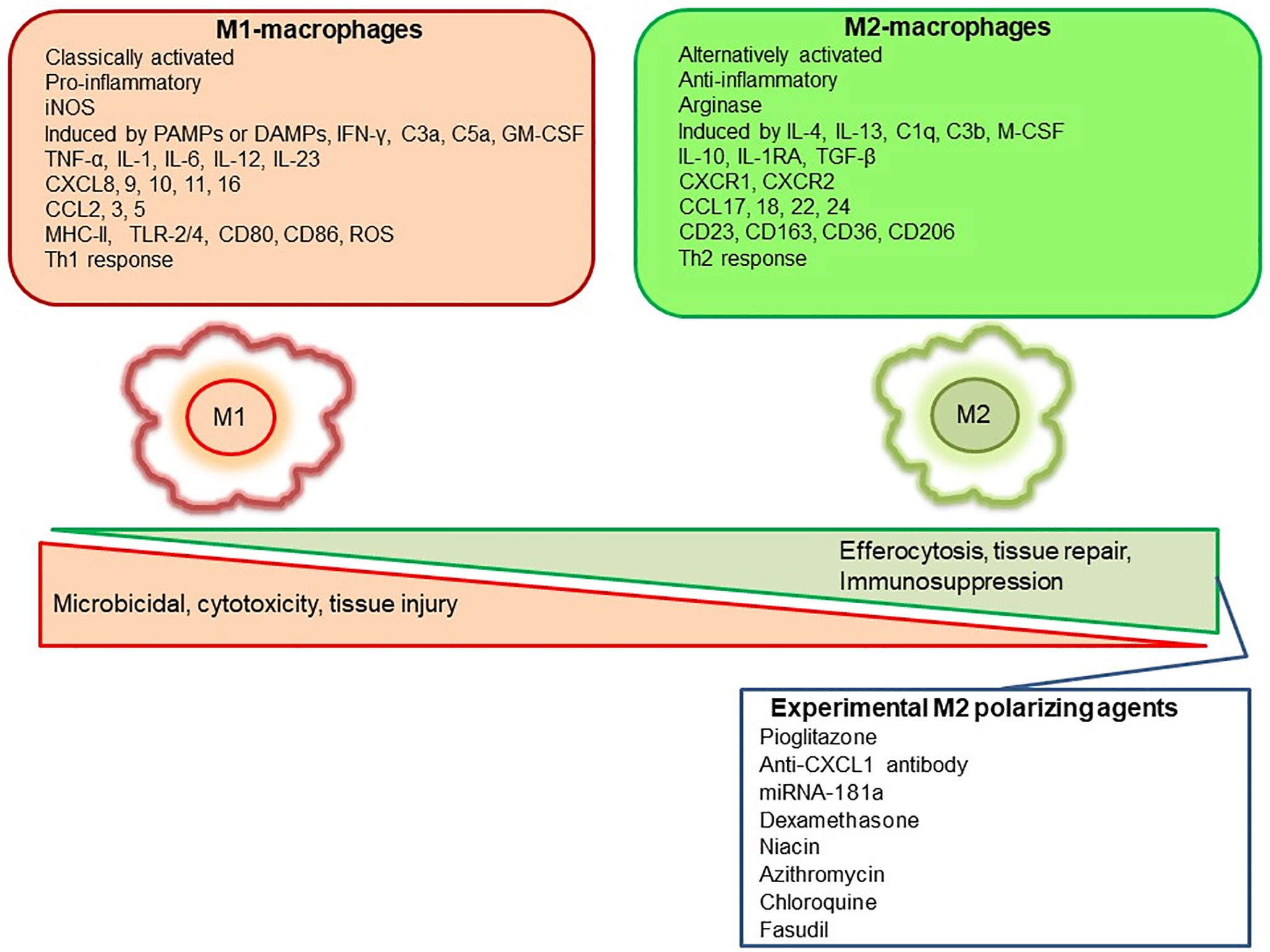
Figure 1 Representation of M1 and M2 distinguishing features and some of the pharmacological agents employed experimentally to polarize macrophages to M2-anti-inflammatory subtype.
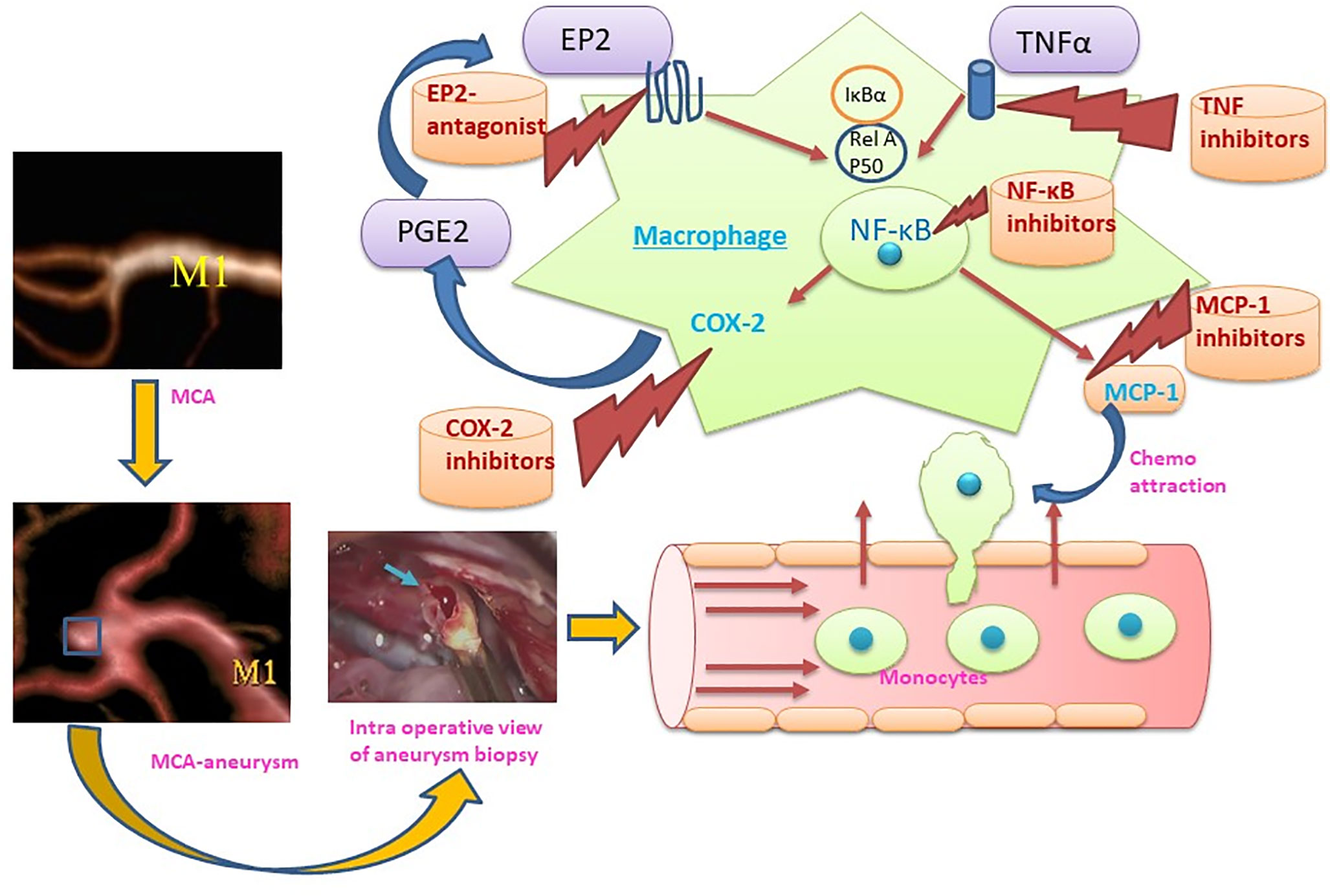
Figure 2 Representation of the M1 role in the formation and rupture of intracranial aneurysm and therapeutic strategies to modulate the macrophage mediated inflammation during aneurysm formation and rupture. (MCA, Middle cerebral artery: M1, M1 – Classically activated macrophages, MCP-1, Monocyte chemoattractant protein-1; COX-2, Cyclooxygenase-2, PGE2: Prostaglandin E2, EP2: Prostaglandin E2 receptor, NF-κB: nuclear factor-kappa B; TNFα, Tumor necrosis factor α).
Macrophage Modulation as a Treatment Strategy for Intracranial Aneurysms
Recent case-control studies have shown that the use of statins and non-steroidal anti-inflammatory drugs (NSAIDs) is inversely associated with SAH by affecting rupture of ICAs (49–52), supporting the notion that rupture of ICAs can be prevented by pharmacological therapy.
Aspirin protects against ICA rupture through modulation of inflammatory pathways (COX-2 and microsomal PGE2 synthase-1 inhibition) and macrophage burden in ICAs (53–55). A prospective cohort study revealed that usage of atorvastatin in secondary prevention for ischemic stroke increases the incidence of hemorrhagic stroke (hazard ratio 1.66) (29). Therefore, statin usage to prevent aneurysm rupture should be approached with caution. Due to hemorrhagic diathesis by the antiplatelet effect, the use of NSAIDs as a pre-emptive medication to prevent SAH also requires caution. Thus, drugs with highly specific targets with minimal or no side effects should be explored for long-term prophylaxis.
Given the fact that macrophages are key players in orchestrating the inflammatory response during ICA formation and rupture, they may represent vital therapeutic targets to modulate and inhibit inflammation (48). Multiple targets at the level of macrophages and macrophage-mediated inflammation have been explored recently (Figure 2). Inhibition of a key chemoattractant molecule, monocyte chemotactic protein-1 (MCP-1) and depletion of macrophages are associated with reduced ICAs in animal models (8, 33) demonstrating that reducing macrophage burden with both strategies effectively prevented aneurysm formation and rupture. In contrast, CXCL1 (neutrophil chemoattractant) blockade reduced neutrophil infiltration and prevented aneurysm formation without modifying the macrophage burden in a mouse model of ICAs (47), which suggests that other molecular and cellular mechanisms may be involved. Interestingly, inhibition of neutrophil infiltration with CXCL1 inhibition is associated with a shift toward M2 macrophages from a M1 phenotype (47). Activation of PPARγ by Pioglitazone has also been shown to effectively reduce the rupture of ICAs through a reduction in infiltrating macrophages and the M1/M2 ratio (48). Clodronate liposome-mediated depletion of macrophages also reduced the rupture of ICAs similar to that shown with pioglitazone, which was associated with a decrease in M1-phenotype related gene expression (48).
In addition to cellular targets, molecular targets have also been successful in experimental models. Anagliptin, a dipeptidyl peptidase-4 inhibitor, suppresses ICA growth through inhibition of macrophage infiltration and activation via ERK-5-mediated suppression of NF-κB (56). Moreover, Eplerenone, a mineralocorticoid receptor blocker, has been shown to reduce ICA formation, in part via reduction in MCP-1, MMP-9 expression and CD68+ macrophage infiltration in a rat model of ICAs (57). A pilot clinical study showed the beneficial effects of Eplerenone in preventing growth and rupture of ICAs (58). Employment of NF-κB p50 decoy oligodeoxynucleotide (ODN) has been shown to downregulate the expression of macrophage related inflammatory genes and reduced macrophage infiltration with a decline in ICAs growth in a rat model (59). Nifedipine, the drug known to be associated with better outcomes after aSAH, has been shown to prevent the enlargement and degenerative ICAs wall changes through reduced macrophage infiltration, MCP-1, and MMP-2 expression probably by modulating the DNA binding capacity of NF-κB (60).
Macrophage polarization as a therapeutic venture has been studied across various disease models. For instance, tumor associated macrophages (TAMs) represent primarily M2 like macrophages promoting tumors and their polarization toward M1 phenotype through the application of various agents such as CSF-1R inhibitor BLZ945, anti-CSF-1 mAb, Zoledronic acid, Histidine-rich glycoprotein, Hydrazinocurcumin, vadimezan (5,6-dimethylxanthenone-4-acetic acid; DMXAA), flavone glycoside Baicalin, IL10R mAb, CD40 mAb, corosolic acid, N-(2-hydroxy acetophenone) glycinate (CuNG), imiquimod, etc. have been investigated as potential tumoricidal drugs (61, 62). Interestingly, certain pieces of evidence support the antagonism of M-CSF as a beneficial tumor therapy leading to M1 polarization of macrophages from M2 tumor associated macrophage phenotype (42). Similarly, in rheumatoid arthritis with a predominance of M1 response, an opposite approach polarizing macrophages from M1 to M2 type has been shown to reduce inflammation and disease severity (63). Several M1 to M2 polarizing agents such as gene therapy by using IL-10 DNA plasmid incorporated in nanoparticles carrying tuftsin protein to target synovial tissue macrophages, Withaferin-A incorporated in manosylated liposomes, paeoniflorin-6′-O-benzene sulfonate (CP-25), sSiglec-9, fucose/galactose analog 2-D-gal and JWH133 have been shown to reduce disease severity in experimental arthritis models and polarize macrophages to anti-inflammatory M2 type (63). Similar, approaches to modulate macrophage polarization from M1 to M2 may prevent the rupture of ICAs. For instance, molecular genetic approaches leading to polarization toward M2 macrophages using miRNAs such as miRNA-181a (64) could be of great potential. Aptamer based enrichment of M2 polarized macrophages in ICAs may also be developed (65). Epigenetic control of macrophage polarization could also be exploited to abrogate the chronic inflammation leading to ICAs formation (66). Egress of macrophages may also be promoted to decrease macrophage burden (67). Berberine, an alkaloid from Coptis chinensis, has been shown to inhibit macrophage activation and infiltration in ICAs by modulation of the phospho-focal adhesion kinase (pFAK)/Grp78/unfolded protein response signaling pathway and reduced the elaboration of inflammatory factors from macrophages such as MCP-1, IL-1β, IL-6, TNF-α, and MMPs (68). Intriguingly, cutaneous non-invasive vagus nerve stimulation has been shown to reduce aneurysm rupture rates and improve outcomes after aneurysm rupture, and may implicate reduced MMP-9 expression as a potential mechanism of action (69). Suppression of MMP-9 expression in macrophages and polarization of macrophages/microglia to the M2 phenotype has already been shown to stem from vagus nerve stimulation-mediated modulation of inflammatory pathways (69, 70). Taken together, there are multiple strategies at the level of inflammation and macrophage modulation that have translational potential in human disease. However, the heterogeneity and the complexity of macrophageal response should be cautiously considered in future studies aiming at characterizing the role of these main sentinel cells of ICAs inflammation (39, 40). A recent study revealed that during Ang-II induced inflammation of the aorta, primarily adventitial macrophage population expanded due to the infiltration of the bone marrow derived macrophages, whereas the residential embryonic macrophages do show local proliferation, but retain their homeostatic roles (40). Similar, studies utilizing fate mapping, mass cytometry, single cell transcriptomics and proteomics may be required to unveil the complexity and heterogeneity of ICAs macrophages, which may be helpful to design better therapeutic strategies. A summary of various clinical and preclinical observations and interventions aiming to prevent macrophage mediated inflammation in ICAs is represented in Table 1. Clinical trials are needed to confirm the efficacy observed in animal studies.
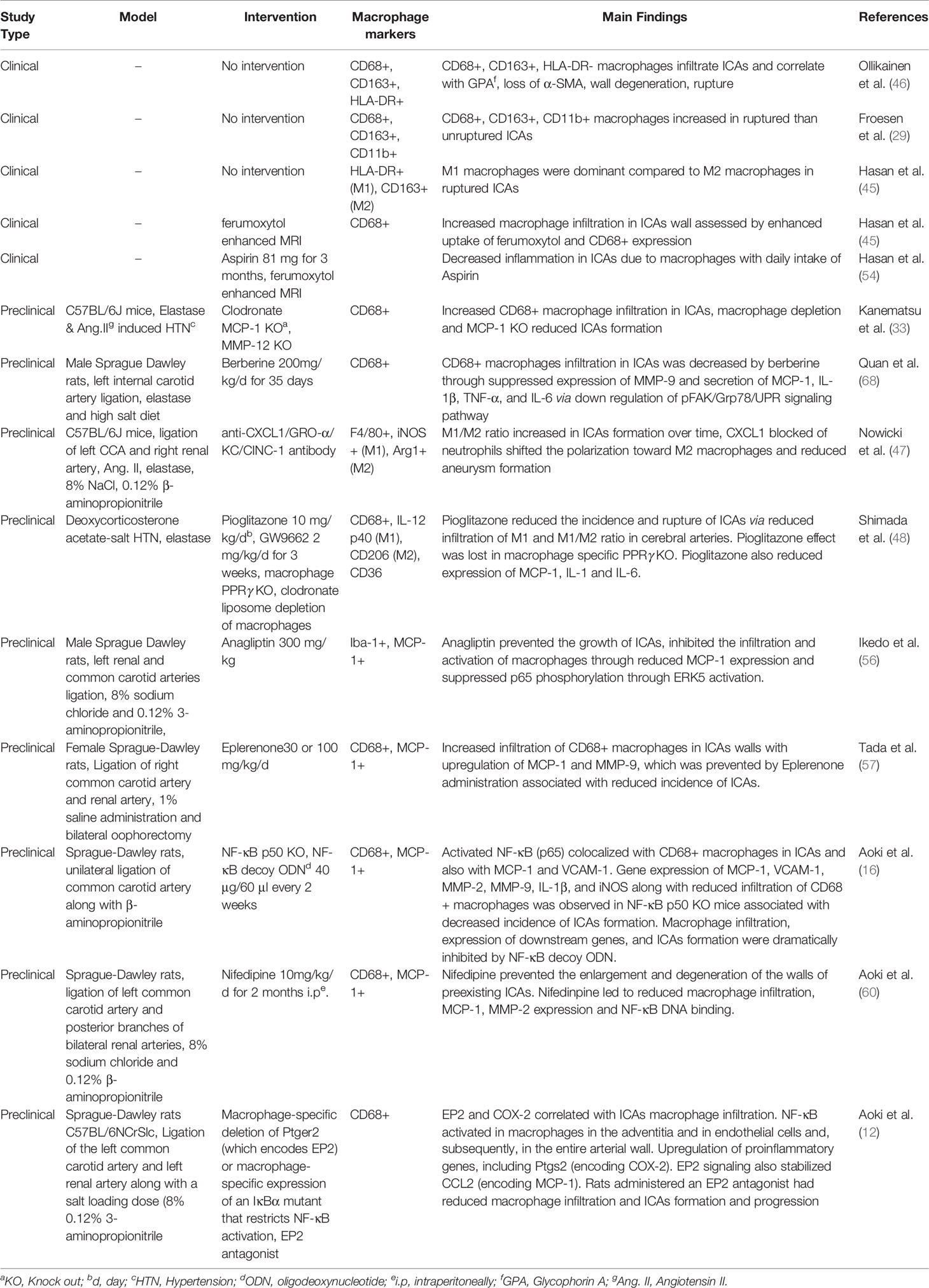
Table 1 A brief summary of macrophage modulation studies for prevention of intracranial aneurysms (ICAs) formation and rupture.
Conclusion
Inflammation and macrophages represent the cornerstones of ICAs development and rupture. Macrophage modulation seems to represent an important therapeutic target and may lead to treatments against ICAs growth and rupture.
Author Contributions
SM conceived the idea, contributed to the initial manuscript draft, and reviewed and edited it. SC significantly contributed to the initial manuscript draft. GD, ML, MN, and DH critically reviewed the final draft of the manuscript for the intellectual content. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hackenberg Katharina AM, Hänggi D, Etminan N. Unruptured Intracranial Aneurysms. Stroke (2018) 49(9):2268–75. doi: 10.1161/STROKEAHA.118.021030
2. Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol (2014) 10(1):44–58. doi: 10.1038/nrneurol.2013.246
3. Macdonald RL, Schweizer TA. Spontaneous subarachnoid haemorrhage. Lancet (2017) 389(10069):655–66. doi: 10.1016/S0140-6736(16)30668-7
4. Lawton MT, Vates GE. Subarachnoid Hemorrhage. New Engl J Med (2017) 377(3):257–66. doi: 10.1056/NEJMcp1605827
5. Etminan N, Rinkel GJ. Unruptured intracranial aneurysms: development, rupture and preventive management. Nat Rev Neurol (2016) 12(12):699–713. doi: 10.1038/nrneurol.2016.150
6. D’Souza S. Aneurysmal Subarachnoid Hemorrhage. J Neurosurg Anesthesiol (2015) 27(3):222–40. doi: 10.1097/ANA.0000000000000130
7. Yamada Y, Sakuma J, Takeuchi I, Yasukochi Y, Kato K, Oguri M, et al. Identification of six polymorphisms as novel susceptibility loci for ischemic or hemorrhagic stroke by exome-wide association studies. Int J Mol Med (2017) 39(6):1477–91. doi: 10.3892/ijmm.2017.2972
8. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Egashira K, Hashimoto N. Impact of monocyte chemoattractant protein-1 deficiency on cerebral aneurysm formation. Stroke (2009) 40(3):942–51. doi: 10.1161/STROKEAHA.108.532556
9. Strong MJ, Amenta PS, Dumont AS, Medel R. The role of leukocytes in the formation and rupture of intracranial aneurysms. Neuroimmunol Neuroinflamm (2015) 2:107–14. doi: 10.4103/2347-8659.153972
10. Tulamo R, Frosen J, Junnikkala S, Paetau A, Kangasniemi M, Pelaez J, et al. Complement system becomes activated by the classical pathway in intracranial aneurysm walls. Lab Invest (2010) 90(2):168–79. doi: 10.1038/labinvest.2009.133
11. Frosen J, Tulamo R, Paetau A, Laaksamo E, Korja M, Laakso A, et al. Saccular intracranial aneurysm: pathology and mechanisms. Acta Neuropathol (2012) 123(6):773–86. doi: 10.1007/s00401-011-0939-3
12. Aoki T, Frò`sen J, Fukuda M, Bando K, Shioi G, Tsuji K, et al. Prostaglandin E2–EP2–NF-κB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci Signaling (2017) 10(465):eaah6037. doi: 10.1126/scisignal.aah6037
13. Moriwaki T, Takagi Y, Sadamasa N, Aoki T, Nozaki K, Hashimoto N. Impaired progression of cerebral aneurysms in interleukin-1beta-deficient mice. Stroke (2006) 37(3):900–5. doi: 10.1161/01.STR.0000204028.39783.d9
14. Aoki T, Nishimura M, Matsuoka T, Yamamoto K, Furuyashiki T, Kataoka H, et al. PGE2-EP2 signalling in endothelium is activated by haemodynamic stress and induces cerebral aneurysm through an amplifying loop via NF-κB. Br J Pharmacol (2011) 163(6):1237–49. doi: 10.1111/j.1476-5381.2011.01358.x
15. Sadamasa N, Nozaki K, Hashimoto N. Disruption of gene for inducible nitric oxide synthase reduces progression of cerebral aneurysms. Stroke (2003) 34(12):2980–4. doi: 10.1161/01.str.0000102556.55600.3b
16. Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke (2007) 38(1):162–9. doi: 10.1161/01.STR.0000252129.18605.c8
17. Shimizu K, Kushamae M, Mizutani T, Aoki T. Intracranial Aneurysm as a Macrophage-mediated Inflammatory Disease. Neurol Med-Chirurg (2019) 59(4):126–32. doi: 10.2176/nmc.st.2018-0326
18. Kurki MI, Hakkinen SK, Frosen J, Tulamo R, von und zu Fraunberg M, Wong G, et al. Upregulated signaling pathways in ruptured human saccular intracranial aneurysm wall: an emerging regulative role of Toll-like receptor signaling and nuclear factor-kappaB, hypoxia-inducible factor-1A, and ETS transcription factors. Neurosurgery (2011) 68(6):1667–75; discussion 75-6. doi: 10.1227/NEU.0b013e318210f001
19. Nakaoka H, Tajima A, Yoneyama T, Hosomichi K, Kasuya H, Mizutani T, et al. Gene expression profiling reveals distinct molecular signatures associated with the rupture of intracranial aneurysm. Stroke (2014) 45(8):2239–45. doi: 10.1161/strokeaha.114.005851
20. Pera J, Korostynski M, Krzyszkowski T, Czopek J, Slowik A, Dziedzic T, et al. Gene expression profiles in human ruptured and unruptured intracranial aneurysms: what is the role of inflammation? Stroke (2010) 41(2):224–31. doi: 10.1161/strokeaha.109.562009
21. Shi C, Awad IA, Jafari N, Lin S, Du P, Hage ZA, et al. Genomics of human intracranial aneurysm wall. Stroke (2009) 40(4):1252–61. doi: 10.1161/strokeaha.108.532036
22. Weinsheimer S, Lenk GM, van der Voet M, Land S, Ronkainen A, Alafuzoff I, et al. Integration of expression profiles and genetic mapping data to identify candidate genes in intracranial aneurysm. Physiol Genomics (2007) 32(1):45–57. doi: 10.1152/physiolgenomics.00015.2007
23. Krischek B, Kasuya H, Tajima A, Akagawa H, Sasaki T, Yoneyama T, et al. Network-based gene expression analysis of intracranial aneurysm tissue reveals role of antigen presenting cells. Neuroscience (2008) 154(4):1398–407. doi: 10.1016/j.neuroscience.2008.04.049
24. Jayaraman T, Berenstein V, Li X, Mayer J, Silane M, Shin YS, et al. Tumor Necrosis Factor α is a Key Modulator of Inflammation in Cerebral Aneurysms. Neurosurgery (2005) 57(3):558–64. doi: 10.1227/01.NEU.0000170439.89041.D6
25. Sathyan S, Koshy LV, Srinivas L, Easwer HV, Premkumar S, Nair S, et al. Pathogenesis of intracranial aneurysm is mediated by proinflammatory cytokine TNFA and IFNG and through stochastic regulation of IL10 and TGFB1 by comorbid factors. J Neuroinflamm (2015) 12(1):1–10. doi: 10.1186/s12974-015-0354-0
26. Tulamo R, Frosen J, Junnikkala S, Paetau A, Pitkaniemi J, Kangasniemi M, et al. Complement activation associates with saccular cerebral artery aneurysm wall degeneration and rupture. Neurosurgery (2006) 59(5):1069–76; discussion 76-7. Epub 2006/10/04. doi: 10.1227/01.NEU.0000245598.84698.26
27. Baeriswyl DC, Prionisti I, Peach T, Tsolkas G, Chooi KY, Vardakis J, et al. Disturbed flow induces a sustained, stochastic NF-κB activation which may support intracranial aneurysm growth in vivo. Sci Rep (2019) 9(1):4738. doi: 10.1038/s41598-019-40959-y
28. Cuhlmann S, Van der Heiden K, Saliba D, Tremoleda JL, Khalil M, Zakkar M, et al. Disturbed blood flow induces RelA expression via c-Jun N-terminal kinase 1: a novel mode of NF-kappaB regulation that promotes arterial inflammation. Circ Res (2011) 108(8):950–9. doi: 10.1161/circresaha.110.233841
29. Frosen J, Piippo A, Paetau A, Kangasniemi M, Niemela M, Hernesniemi J, et al. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke (2004) 35(10):2287–93. doi: 10.1161/01.STR.0000140636.30204.da
30. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol (2017) 17(6):349–62. doi: 10.1038/nri.2017.28
31. Kataoka K, Taneda M, Asai T, Kinoshita A, Ito M, Kuroda R. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke (1999) 30(7):1396–401. doi: 10.1161/01.str.30.7.1396
32. Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms. Neurosurgery (1999) 45(5):1137–46; discussion 46-7. doi: 10.1097/00006123-199911000-00024
33. Kanematsu Y, Kanematsu M, Kurihara C, Tada Y, Tsou TL, van Rooijen N, et al. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke (2011) 42(1):173–8. doi: 10.1161/strokeaha.110.590976
34. Hasan DM, Starke RM, Gu H, Wilson K, Chu Y, Chalouhi N, et al. Smooth Muscle Peroxisome Proliferator-Activated Receptor gamma Plays a Critical Role in Formation and Rupture of Cerebral Aneurysms in Mice In Vivo. Hypertens (Dallas Tex 1979) (2015) 66(1):211–20. doi: 10.1161/hypertensionaha.115.05332
35. Yamamoto R, Aoki T, Koseki H, Fukuda M, Hirose J, Tsuji K, et al. A sphingosine-1-phosphate receptor type 1 agonist, ASP4058, suppresses intracranial aneurysm through promoting endothelial integrity and blocking macrophage transmigration. Br J Pharmacol (2017) 174(13):2085–101. doi: 10.1111/bph.13820
36. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2000) 164(12):6166–73. doi: 10.4049/jimmunol.164.12.6166
37. Boyle JJ, Johns M, Kampfer T, Nguyen AT, Game L, Schaer DJ, et al. Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ Res (2012) 110(1):20–33. doi: 10.1161/circresaha.111.247577
38. Cheng Z, Zhou Y-Z, Wu Y, Wu Q-Y, Liao X-B, Fu X-M, et al. Diverse roles of macrophage polarization in aortic aneurysm: destruction and repair. J Trans Med (2018) 16(1):354. doi: 10.1186/s12967-018-1731-0
39. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
40. Weinberger T, Esfandyari D, Messerer D, Percin G, Schleifer C, Thaler R, et al. Ontogeny of arterial macrophages defines their functions in homeostasis and inflammation. Nat Commun (2020) 11(1):4549. doi: 10.1038/s41467-020-18287-x
41. Li H, Bai S, Ao Q, Wang X, Tian X, Li X, et al. Modulation of Immune-Inflammatory Responses in Abdominal Aortic Aneurysm: Emerging Molecular Targets. J Immunol Res (2018) 2018:7213760. doi: 10.1155/2018/7213760
42. Hamilton TA, Zhao C, Pavicic PG, Datta S. Myeloid Colony-Stimulating Factors as Regulators of Macrophage Polarization. Front Immunol (2014) 5:554. doi: 10.3389/fimmu.2014.00554
43. Ye P, Chen W, Wu J, Huang X, Li J, Wang S, et al. GM-CSF contributes to aortic aneurysms resulting from SMAD3 deficiency. J Clin Invest (2013) 123(5):2317–31. doi: 10.1172/JCI67356
44. Chalouhi N, Theofanis T, Starke RM, Zanaty M, Jabbour P, Dooley SA, et al. Potential role of granulocyte-monocyte colony-stimulating factor in the progression of intracranial aneurysms. DNA Cell Biol (2015) 34(1):78–81. doi: 10.1089/dna.2014.2618
45. Hasan D, Chalouhi N, Jabbour P, Hashimoto T. Macrophage imbalance (M1 vs. M2) and upregulation of mast cells in wall of ruptured human cerebral aneurysms: preliminary results. J Neuroinflamm (2012) 9(1):1–7. doi: 10.1186/1742-2094-9-222
46. Ollikainen E, Tulamo R, Kaitainen S, Honkanen P, Lehti S, Liimatainen T, et al. Macrophage Infiltration in the Saccular Intracranial Aneurysm Wall as a Response to Locally Lysed Erythrocytes That Promote Degeneration. J Neuropathol Exp Neurol (2018) 77(10):890–903. doi: 10.1093/jnen/nly068
47. Nowicki KW, Hosaka K, Walch FJ, Scott EW, Hoh BL. M1 macrophages are required for murine cerebral aneurysm formation. J Neurointervent Surg (2018) 10(1):93–7. doi: 10.1136/neurintsurg-2016-012911
48. Shimada K, Furukawa H, Wada K, Korai M, Wei Y, Tada Y, et al. Protective Role of Peroxisome Proliferator-Activated Receptor-gamma in the Development of Intracranial Aneurysm Rupture. Stroke (2015) 46(6):1664–72. doi: 10.1161/strokeaha.114.007722
49. Hasan DM, Mahaney KB, Brown RD Jr, Meissner I, Piepgras DG, Huston J, et al. Aspirin as a promising agent for decreasing incidence of cerebral aneurysm rupture. Stroke (2011) 42(11):3156–62. doi: 10.1161/STROKEAHA.111.619411
50. Can A, Castro VM, Dligach D, Finan S, Yu S, Gainer V, et al. Lipid-Lowering Agents and High HDL (High-Density Lipoprotein) Are Inversely Associated With Intracranial Aneurysm Rupture. Stroke (2018) 49(5):1148–54. doi: 10.1161/strokeaha.117.019972
51. Yoshimura Y, Murakami Y, Saitoh M, Yokoi T, Aoki T, Miura K, et al. Statin Use and Risk of Cerebral Aneurysm Rupture: A Hospital-based Case–control Study in Japan. J Stroke Cerebrovasc Dis (2014) 23(2):343–8. doi: 10.1016/j.jstrokecerebrovasdis.2013.04.022
52. Can A, Rudy RF, Castro VM, Yu S, Dligach D, Finan S, et al. Association between aspirin dose and subarachnoid hemorrhage from saccular aneurysms: A case-control study. Neurology (2018) 91(12):e1175–81. doi: 10.1212/wnl.0000000000006200
53. Chalouhi N, Atallah E, Jabbour P, Patel PD, Starke RM, Hasan D. Aspirin for the Prevention of Intracranial Aneurysm Rupture. Neurosurgery (2017) 64(CN_suppl_1):114–8. doi: 10.1093/neuros/nyx299
54. Hasan DM, Chalouhi N, Jabbour P, Dumont AS, Kung DK, Magnotta VA, et al. Evidence that acetylsalicylic acid attenuates inflammation in the walls of human cerebral aneurysms: preliminary results. J Am Heart Assoc (2013) 2(1):e000019. doi: 10.1161/jaha.112.000019
55. Hasan DM, Chalouhi N, Jabbour P, Magnotta VA, Kung DK, Young WL. Imaging aspirin effect on macrophages in the wall of human cerebral aneurysms using ferumoxytol-enhanced MRI: preliminary results. J Neuroradiol J Neuroradiol (2013) 40(3):187–91. doi: 10.1016/j.neurad.2012.09.002
56. Ikedo T, Minami M, Kataoka H, Hayashi K, Nagata M, Fujikawa R, et al. Dipeptidyl Peptidase-4 Inhibitor Anagliptin Prevents Intracranial Aneurysm Growth by Suppressing Macrophage Infiltration and Activation. J Am Heart Assoc (2017) 6(6):e004777. doi: 10.1161/jaha.116.004777
57. Tada Y, Kitazato KT, Tamura T, Yagi K, Shimada K, Kinouchi T, et al. Role of Mineralocorticoid Receptor on Experimental Cerebral Aneurysms in Rats. Hypertens (Dallas Tex 1979) (2009) 54(3):552–7. doi: 10.1161/HYPERTENSIONAHA.109.134130
58. Nagahiro S, Tada Y, Satomi J, Kinouchi T, Kuwayama K, Yagi K, et al. Treatment of Unruptured Cerebral Aneurysms with the Mineralocorticoid Receptor Blocker Eplerenone—Pilot Study. J Stroke Cerebrovasc Dis (2018) 27(8):2134–40. doi: 10.1016/j.jstrokecerebrovasdis.2018.03.008
59. Aoki T, Kataoka H, Shimamura M, Nakagami H, Wakayama K, Moriwaki T, et al. NF-κB Is a Key Mediator of Cerebral Aneurysm Formation. Circulation (2007) 116(24):2830–40. doi: 10.1161/CIRCULATIONAHA.107.728303
60. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Nifedipine inhibits the progression of an experimentally induced cerebral aneurysm in rats with associated down-regulation of NF-kappa B transcriptional activity. Curr Neurovasc Res (2008) 5(1):37–45. doi: 10.2174/156720208783565663
61. Zheng X, Turkowski K, Mora J, Brüne B, Seeger W, Weigert A, et al. Redirecting TAMs to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget (2017) 8(29):48436–52. doi: 10.18632/oncotarget.17061
62. Genard G, Lucas S, Michiels C. Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies. Front Immunol (2017) 8:828. doi: 10.3389/fimmu.2017.00828
63. Tardito S, Martinelli G, Soldano S, Paolino S, Pacini G, Patane M, et al. Macrophage M1/M2 polarization and rheumatoid arthritis: A systematic review. Autoimmun Rev (2019) 18(11):102397. doi: 10.1016/j.autrev.2019.102397
64. Bi J, Zeng X, Zhao L, Wei Q, Yu L, Wang X, et al. miR-181a Induces Macrophage Polarized to M2 Phenotype and Promotes M2 Macrophage-mediated Tumor Cell Metastasis by Targeting KLF6 and C/EBPalpha. Mol Ther Nucleic Acids (2016) 5(9):e368. doi: 10.1038/mtna.2016.71
65. Enam SF, Bellamkonda RV. FKN-aptamer functionalized hydrogels for local enrichment of M2 macrophages after traumatic brain injury. Front Bioeng Biotechnol. (2016) 4. doi: 10.3389/conf.FBIOE.2016.01.00132
66. de Groot AE, Pienta KJ. Epigenetic control of macrophage polarization: implications for targeting tumor-associated macrophages. Oncotarget (2018) 9(29):20908–27. doi: 10.18632/oncotarget.24556
67. Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis. Circ Res (2014) 114(11):1757–71. doi: 10.1161/circresaha.114.301174
68. Quan K, Li S, Wang D, Shi Y, Yang Z, Song J, et al. Berberine Attenuates Macrophages Infiltration in Intracranial Aneurysms Potentially Through FAK/Grp78/UPR Axis. Front Pharmacol (2018) 9:565. doi: 10.3389/fphar.2018.00565
69. Suzuki T, Takizawa T, Kamio Y, Qin T, Hashimoto T, Fujii Y, et al. Noninvasive Vagus Nerve Stimulation Prevents Ruptures and Improves Outcomes in a Model of Intracranial Aneurysm in Mice. Stroke (2019) 50(5):Strokeaha118023928. doi: 10.1161/strokeaha.118.023928
Keywords: intracranial aneurysms, monocytes, macrophages, inflammation, subarachnoid hemorrhage, stroke, macrophage polarization
Citation: Muhammad S, Chaudhry SR, Dobreva G, Lawton MT, Niemelä M and Hänggi D (2021) Vascular Macrophages as Therapeutic Targets to Treat Intracranial Aneurysms. Front. Immunol. 12:630381. doi: 10.3389/fimmu.2021.630381
Received: 17 November 2020; Accepted: 11 January 2021;
Published: 08 March 2021.
Edited by:
Alexander Steinkasserer, University Hospital Erlangen, GermanyReviewed by:
Angel L. Corbi, Consejo Superior de Investigaciones Científicas (CSIC), SpainOsamu Takeuchi, Kyoto University, Japan
Copyright © 2021 Muhammad, Chaudhry, Dobreva, Lawton, Niemelä and Hänggi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sajjad Muhammad, c2FqamFkLm11aGFtbWFkQG1lZC51bmktZHVlc3NlbGRvcmYuZGU=