 Tom O. J. Cockram
Tom O. J. Cockram Jacob M. Dundee
Jacob M. Dundee Alma S. Popescu
Alma S. Popescu Guy C. Brown
Guy C. Brown- Department of Biochemistry, University of Cambridge, Cambridge, United Kingdom
Mammalian phagocytes can phagocytose (i.e. eat) other mammalian cells in the body if they display certain signals, and this phagocytosis plays fundamental roles in development, cell turnover, tissue homeostasis and disease prevention. To phagocytose the correct cells, phagocytes must discriminate which cells to eat using a ‘phagocytic code’ - a set of over 50 known phagocytic signals determining whether a cell is eaten or not - comprising find-me signals, eat-me signals, don’t-eat-me signals and opsonins. Most opsonins require binding to eat-me signals – for example, the opsonins galectin-3, calreticulin and C1q bind asialoglycan eat-me signals on target cells - to induce phagocytosis. Some proteins act as ‘self-opsonins’, while others are ‘negative opsonins’ or ‘phagocyte suppressants’, inhibiting phagocytosis. We review known phagocytic signals here, both established and novel, and how they integrate to regulate phagocytosis of several mammalian targets - including excess cells in development, senescent and aged cells, infected cells, cancer cells, dead or dying cells, cell debris and neuronal synapses. Understanding the phagocytic code, and how it goes wrong, may enable novel therapies for multiple pathologies with too much or too little phagocytosis, such as: infectious disease, cancer, neurodegeneration, psychiatric disease, cardiovascular disease, ageing and auto-immune disease.
Introduction
Every second of a human life, about two million senescent red blood cells and one million apoptotic white blood cells are eliminated by phagocytosis, the cellular process of engulfing and degrading extracellular material (1). Countless more dead or dying host cells, pathogenic microbes, infected cells, cancer cells, excess synapses and cellular debris are similarly engulfed and digested. These phagocytic targets have to be accurately distinguished from around 30 trillion healthy cells. How is this distinction made, given that the phagocyte cannot look inside the cells it encounters, but can only read surface signals using its own surface receptors? The task may have seemed relatively simple when we only knew of two or three signals that regulated this phagocytosis, but we now know of over 50 such signals, some stimulating and some inhibiting phagocytosis. How is this information integrated to decide whether to eat or not to eat?
In this review, we outline the components of the phagocytic code i.e. established and novel find-me signals, eat-me signals, don’t eat-me signals and opsonins used to discriminate which mammalian cells (or sub-cellular material) to phagocytose. We have largely excluded signals mediating phagocytosis of pathogens, as this is a separate field well reviewed by others (2, 3), although many signals are known to regulate phagocytosis of both pathogens and mammalian cells. We describe how each eat-me signal, don’t-eat-me signal or opsonin interacts with receptors on phagocytes, as this is fundamental to operation of the phagocytic code. We then illustrate how these interactions determine the phagocytosis of particular targets, including: healthy cells, excess cells in development, senescent and aged cells, infected cells, cancer cells, dead or dying cells, cell debris and neuronal synapses. We outline how phagocytic signalling may go wrong in disease, and how this may inform novel therapies. Finally, we then offer some generalisations as to how the phagocytic code operates and integrates phagocytic signals.
Definitions. Terms used in the field of phagocytosis can be ambiguous, so it is important to clarify their definitions. Phagocytosis is a cellular process of engulfment and digestion of extracellular material > 0.5 microns in size, including other cells. We will call the cell doing the phagocytosis the phagocyte, and the cell to be phagocytosed the target cell. A find-me signal is a molecule released from a target cell to attract a phagocyte toward that cell. All find-me signals are chemotactic factors, but not all chemotactic factors are find-me signals, as many chemotactic factors are released by immune cells to attract other immune cells to remove pathogens and damage, but not to phagocytose the immune cells. An eat-me signal is a signal exposed on or released from a cell encouraging phagocytes to phagocytose that cell. A don’t-eat-me signal is a signal exposed on or released from a cell discouraging phagocytes to phagocytose that cell. An opsonin is a normally soluble, extracellular molecule, not derived from the phagocytosed cell, which, when bound to a cell, encourages phagocytes to phagocytose that cell. Opsonins can be confused with eat-me signals (as both stimulate phagocytosis of target cells), and indeed there is some overlap, but the fundamental distinction is that eat-me signals originate from the target cell, whereas opsonins do not. However, there are some eat-me signals released by target cells that can bind back onto the target cell to act as ‘self-opsonins’. The original opsonins were antibodies and complement proteins, but ‘opsonin’ now refers to any external molecule capable of bridging between target cells and phagocyte to stimulates phagocytosis of the target cell. The phagocytic code is the set of signals that determine whether a cell is phagocytosed by a phagocyte or not. A phagocyte is a cell capable of phagocytosing. The main professional phagocytes (cells specialised for phagocytosis) are: neutrophils, monocytes, dendritic cells and macrophages. Some tissues have specialised macrophages that enter the tissue prior to birth (e.g. microglia in the CNS); other macrophages differentiate from blood monocytes recruited into tissues during inflammation. A number of other cell types (such as fibroblasts) can act as non-professional phagocytes, capable of phagocytosing small, local targets, but with limited capacity to migrate, detect, engulf and digest targets. A target cell here means a cell potentially phagocytosed by a phagocyte. Targets include apoptotic and necrotic cells: necrotic cells have a ruptured cell membrane, whereas apoptotic cells have an intact cell membrane but exposed phosphatidylserine due to caspase activation. A phagocytic receptor is a receptor on a phagocyte that specifically regulates phagocytosis by responding to eat-me signals, don’t-eat-me signals or opsonins. A negative opsonin is a normally soluble, extracellular molecule, which, when bound to the target cell, discourages phagocytes from phagocytosing that cell. A phagocyte suppressant is a normally soluble, extracellular molecule, which, when bound to a phagocyte discourages it from phagocytosing targets. ‘Macrophage phagocytosis’ is ambiguous as it may refer to ‘phagocytosis of macrophages’ or ‘phagocytosis by macrophages’, and this can cause confusion. We will use the term to mean the latter, so in general ‘X phagocytosis’ will mean ‘phagocytosis by X’.
The Phagocytic Code
Find-Me Signals
Find-me signals are molecules released from a cell to attract phagocytes, resulting in phagocytosis of that cell, and include proteins, lipids and nucleotides (4). Table 1 includes known find-eat-me signals, whilst Figure 1 illustrates their binding receptors.
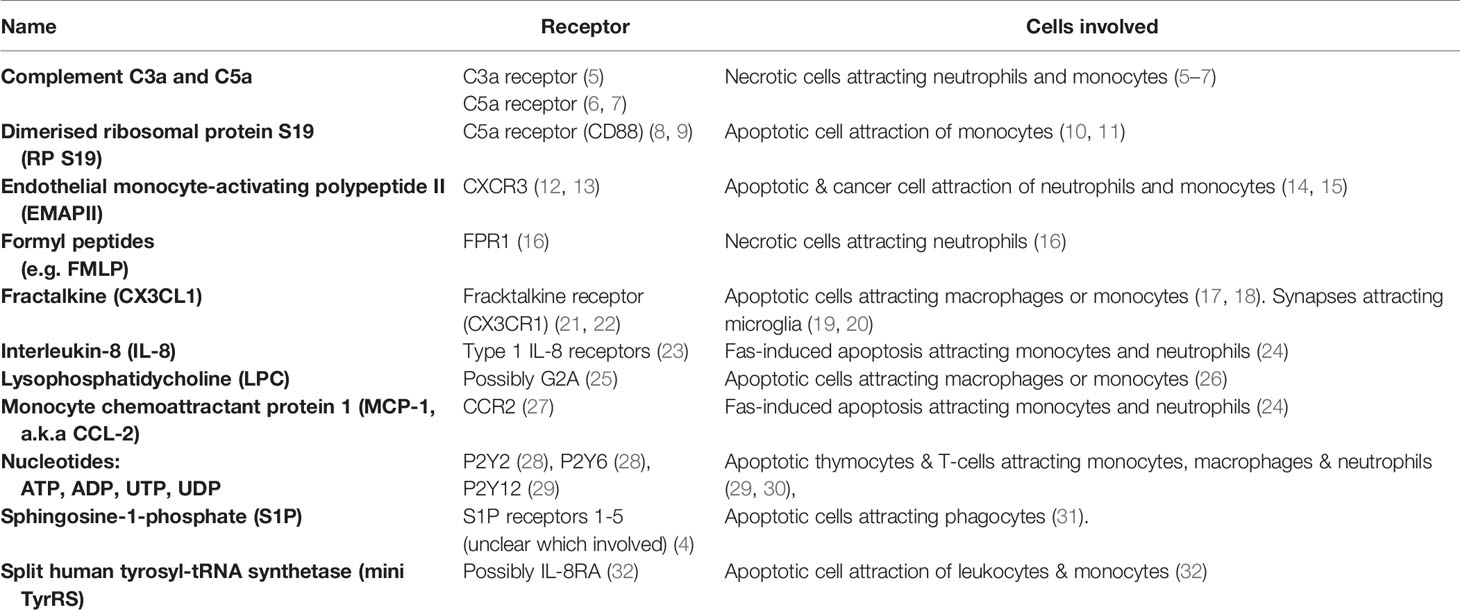
Table 1 Find-me signals.
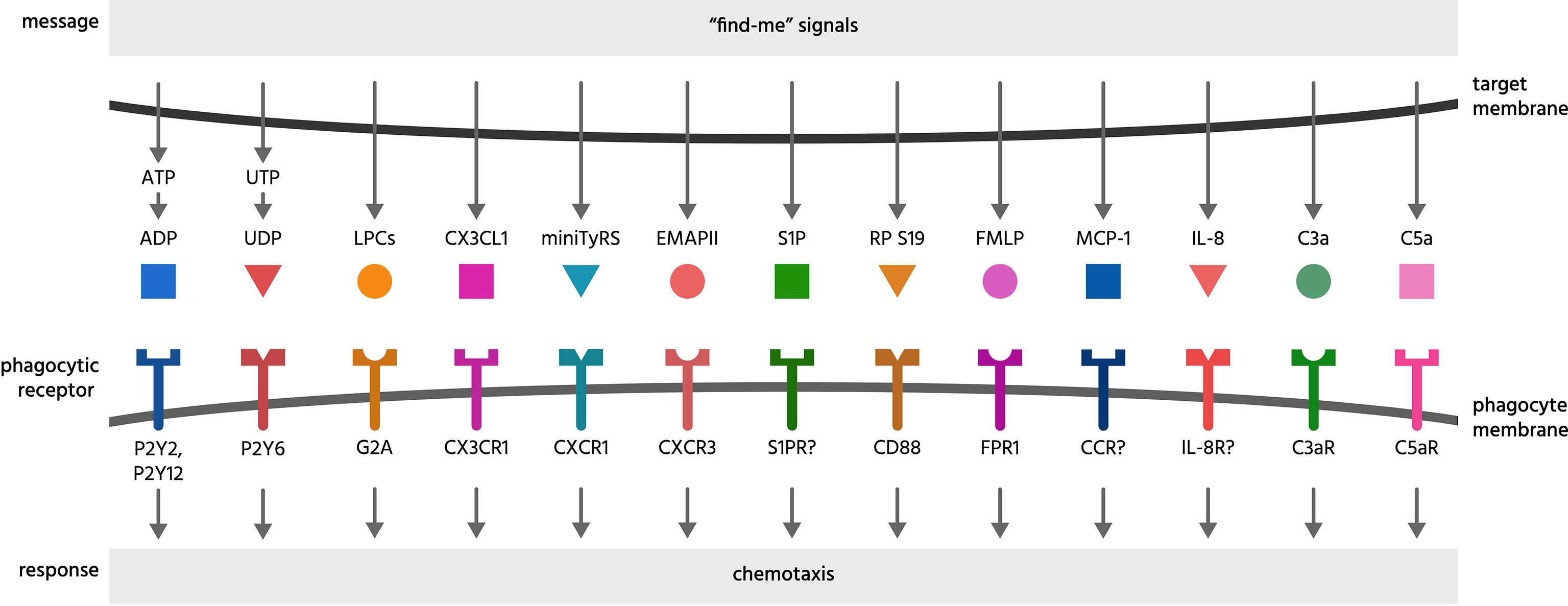
Figure 1 Find-me signals and their receptors. Find-me signals are molecules released from a cell to attract phagocytes, resulting in phagocytosis of that cell. The figure illustrates find-me signals released from mammalian target cells, and their putative receptors on phagocytes, mediating the recruitment of phagocytes to the target cells. ADP, adenosine-5’-diphosphate; ATP, adenosine-5’-triphosphate; C3a, complement component C3a; C5a, complement component C5a; C3aR, C3a receptor; C5aR, C5a receptor (CD88); CCR, chemokine receptor; CD88, cluster of differentiation 88 (a complement component C5a receptor); CX3CL1, chemokine C-X3-C ligand 1 (fractalkine); CX3CR1, C-X3-C chemokine receptor; CXCR1, C-X-C chemokine receptor type 1; CXCR3, C-X-C chemokine receptor type 3; EMAP II, endothelial monocyte-activating polypeptide II; FMLP, N-formylmethionyl-leucyl-phenylalanine; FPR1, formyl peptide receptor 1; G2A, G protein coupled receptor 132 (GPR132); IL-8, interleukin 8; IL-8R, interleukin 8 receptor; LPC, lysophosphatidylcholine; MCP-1, monocyte chemoattractant protein 1; miniTyRS, split tyrosyl tRNA synthetase; RP S19, (dimerised) ribosomal protein S19; S1P, sphingosine-1-phosphate; S1PR, sphingosine-1-phosphate receptor; P2Y, a family of purinergic receptors; UDP, uridine-5’-diphosphate; UTP, uridine-5’-triphosphate.
Complement components C3a and C5a are protein components of the complement system, generated from C3 and C5 respectively by proteolysis once the complement system is activated. Complement can be activated on necrotic cells particularly, generating C3a and C5a, which are both chemotactic for a variety of phagocytes (33). In muscle injury, C3a was found to be necessary to recruit monocytes (via the C3a receptor) to the necrotic tissue (5). In liver injury, C5a was necessary to recruit neutrophils (6). Both C3a and C5a are involved in recruitment of neutrophils and monocytes to arthritic joints (7).
Dimerised ribosomal protein S19 (RP S19) is a find-me signal for monocytes, first discovered in the context of rheumatoid arthritis (34). Later it was found to be released by apoptotic cells, and bind to the complement C5a receptor CD88 (8, 9). It is unclear how RP S19 is secreted by apoptotic cells, and may actually be released from apoptotic cells that have become necrotic (secondary necrosis).
Endothelial monocyte-activating polypeptide II (EMAP II) is a chemotactic cytokine released by apoptotic and cancer cells to recruit neutrophils and monocytes (14, 15). The main receptor is thought to be CXCR3 (12). It is unclear how EMAP II is released from cells, and may be released during necrosis.
Formyl peptides (including N-formylmethionyl-leucyl-phenylalanine, fMLP) are found in mitochondria, and are released from necrotic cells, inducing migration of neutrophils via formyl peptide receptor 1 (FPR1) (16).
Fractalkine (CX3CL1) is ubiquitously expressed as a membrane-anchored protein, but can be enzymatically cleaved to a soluble form by cells undergoing apoptosis, such as lymphocytes (17) and germinal B cells (18), to induce migration of macrophages or monocytes. Chemotactic responses to fractalkine act via the fractalkine receptor CX3CR1, expressed by macrophages, natural killer cells, T cells and circulating monocytes (21, 22). In the brain, soluble fractalkine is released by the metalloprotease ADAM10 on neurons, and may drive migration of microglia to developing neurites for synaptic pruning during circuitry development (19, 20, 35, 36).
Interleukin-8 (IL-8) and monocyte chemoattractant protein 1 (MCP-1) can be released from multiple cell types when apoptosis is induced by Fas, and chemoattract monocytes and neutrophils (24). It is not clear whether this is specific to Fas-induced apoptosis.
Lysophosphatidylcholine is a soluble lipid generated from membrane phosphatidylcholine by the action of phospholipase A2 (37). Chemotaxis may be induced via the receptor G2A (25), although lysophosphatidylcholine may also block this receptor in certain contexts (38). Lysophosphatidylcholine can be released from apoptotic cells following caspase-3 mediated activation of phospholipase A2 to attract monocytes and macrophages (26).
Nucleotides: adeonise-5’-triphosphate (ATP), adeonise-5’-diphosphate (ADP), uridine-5’-triphosphate (UTP) and uridine-5’-diphosphate (UDP) are released from a variety of cells undergoing apoptosis, including thymocytes and T-cells (39), and these extracellular nucleotides can promote migration of monocytes, macrophages and neutrophils to apoptotic cells in vitro and in vivo (29, 30). Nucleotide release from cells with intact membranes occurs via connexin or pannexin channels, and release from apoptotic cells can result from caspase-dependent cleavage of pannexins (40, 41). Phagocytes express a range of ionotropic P2X and metabotropic P2Y receptors for nucleotides that may mediate chemotaxis (42). In particular, the UTP and ATP-sensing P2Y2 receptor mediates macrophage chemotaxis towards apoptotic T cells and thymocytes (39). The ATP and ADP-sensing P2Y12 receptor mediates microglial migration and processes extension toward sites of brain damage in vivo (29). UDP induces migration of immature dendritic cells via the P2Y6 receptor (28). UDP may also chemoattract neutrophils, eosinophils and natural killer cells (43). Extracellular ectonucleotidases can degrade ATP/ADP and UTP/UDP to adenosine and uridine respectively and so may prevent these nucleotides from acting as find-me signals (44). For example, Thompson et al. showed that knockout of the ectonucleotidase CD73 increased lymphocyte migration to draining lymph nodes (45). Degradation of UDP by nucleotidases may also prevent it from acting as an eat-me signal (see ‘eat-me signals’ section below), and degradation of ATP/ADP generates adenosine, which may act as a don’t eat-me signal (see ‘don’t-eat-me’ section below).
Sphingosine-1-phosphate (S1P) is also released during apoptosis to induce phagocyte migration (31). S1P can activate five different S1P receptors S1P1-5, but which mediates chemotaxis is unclear (4).
Split tyrosyl tRNA synthetase (mini TyrRS) acts as a find-me signal for apoptotic cells once cleaved by the extracellular protease elastase (32). Cleavage produces two fragments, both acting as chemoattractants. The N-terminal fragment binds to the interleukin-8 type A receptor (32), although this has not been shown to mediate chemoattraction.
Eat-Me Signals
Eat-me signals are molecules exposed on or released from a target cell to directly induce phagocytosis by a phagocyte. Most eat-me signals (such as phosphatidylserine) are anchored in the target cell membrane, but some (such as calreticulin) are soluble proteins bound to the cell surface, and may be released and bind back onto the target cell. These overlap somewhat with opsonins and could be regarded as ‘self-opsonins’. Below we discuss all potential eat-me signals, but only those with reasonable evidence are listed in Table 2 and illustrated in Figure 2 with their binding partners.
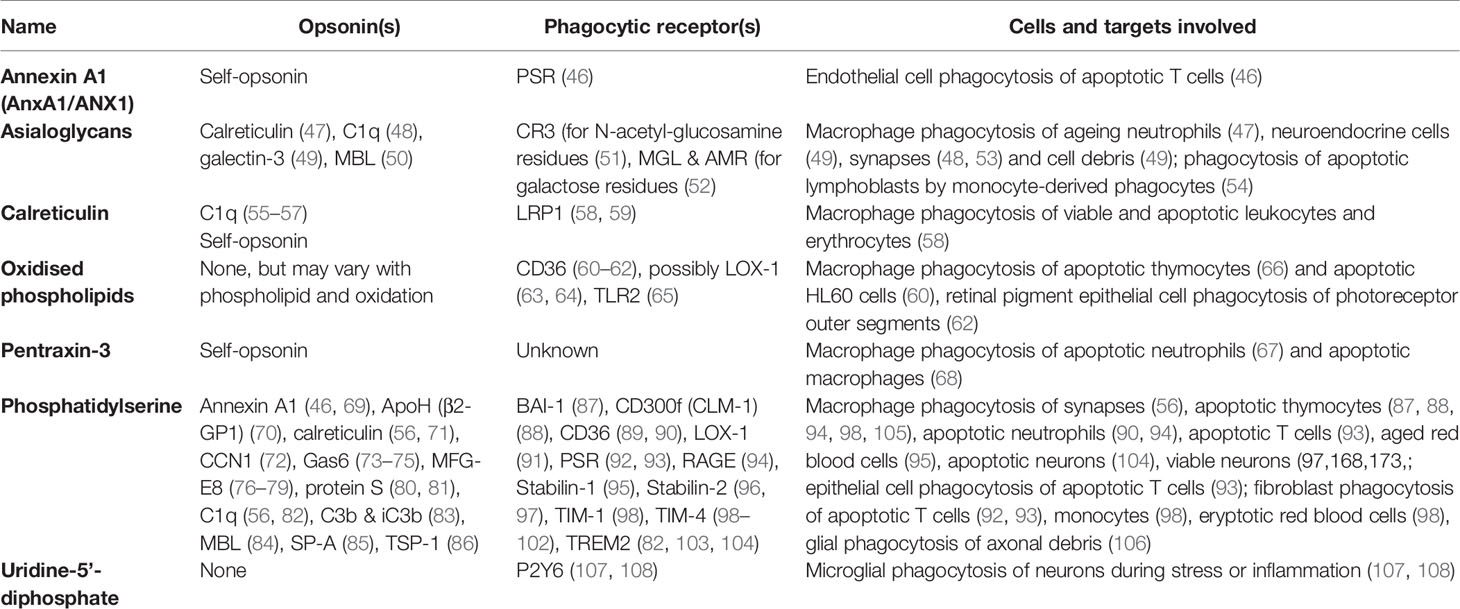
Table 2 Eat-me signals.
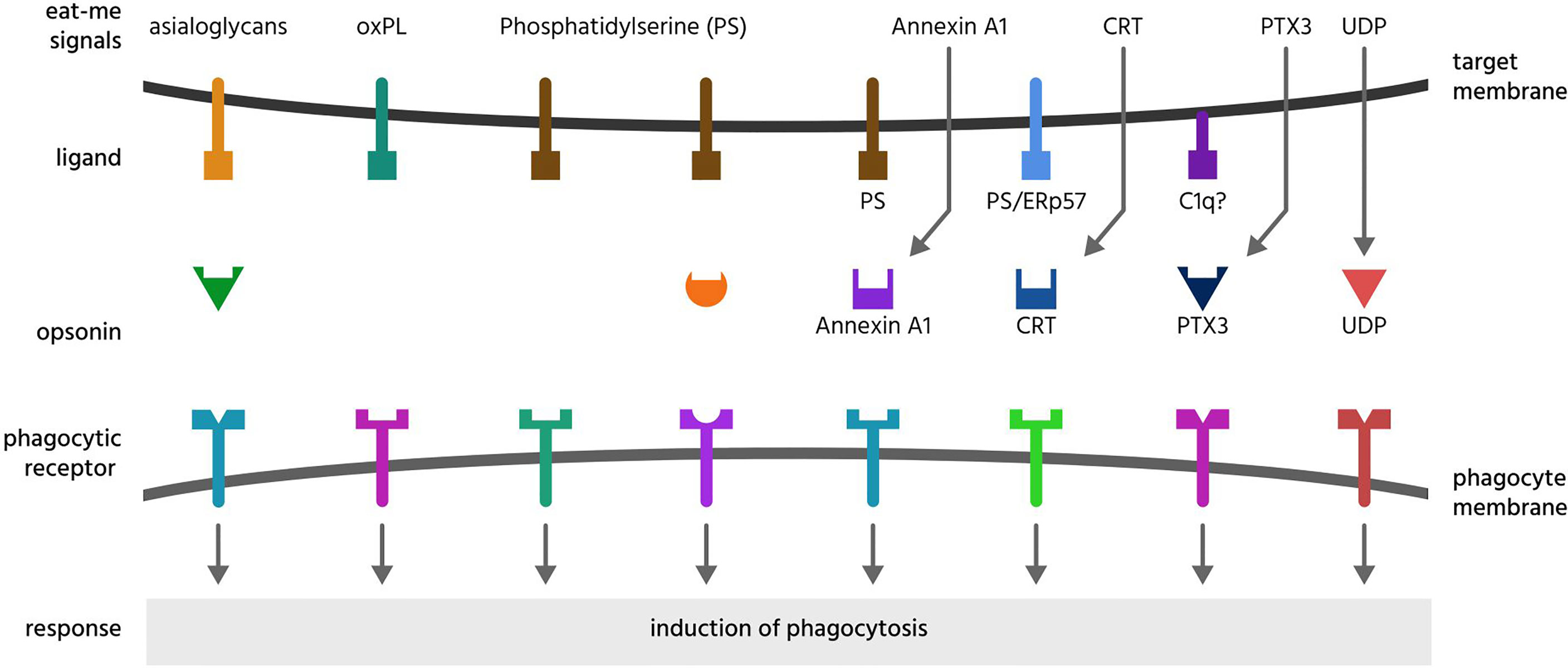
Figure 2 Eat-me signals. Eat-me signals are molecules exposed on or released from a target cell to directly induce phagocytosis of that cell by a phagocyte. The figure illustrates eat-me signals on or from mammalian target cells. Asialoglycans are desialylated glycoproteins or glycolipids, that can bind multiple opsonins, and induce phagocytosis via multiple receptors. Oxidised phospholipids (oxPL) can induce phagocytosis via receptors CD36 and LOX-1. Phosphatidylserine (PS) can induce phagocytosis either directly via multiple phagocytic receptors or indirectly via binding multiple opsonins. Annexin A1, CRT (calreticulin) and PTX3 (pentraxin 3) are soluble proteins released onto the surface of target cells, where they bind ligands: PS (phosphatidylserine), C1q (complement component C1q) or ERp57 (endoplasmic reticulum resident protein p57). UDP (uridine-5’-diphosphate) can induce phagocytosis via activating the P2Y6 receptor.
Annexin A1 (AnxA1) is a soluble protein known to facilitate a return to homeostasis following inflammation (109). AnxA1 can be externalised by apoptotic cells to induce phagocytosis of them by endothelial cells, possibly via the phosphatidylserine receptor (46). AnxA1 can bind phosphatidylserine, and colocalises with phosphatidylserine on apoptotic cells. Apoptotic lymphocytes may also expose AnxA1, inducing phagocytosis by macrophages (110). AnxA1 can also act as an opsonin by binding phosphatidylserine on target cells (see below). Thus, AnxA1 could be regarded as a self-opsonin.
Asialoglycans are glycans (molecules with sugar chains) that lack sialic acid, a monosaccharide normally at the end of glycan chains on glycoproteins and gangliosides (Figure 3). Desialylation (removal of sialic acid residues) of glycans can occur via neuraminidases (111), which can translocate to the cell surface during inflammation (112), thus generating surface asialoglycans. Such asialogylcans can then act as eat-me signals, for example for apoptotic cells (54), where the phagocytic signal of asialogylcans is relayed by binding opsonins. For example, the opsonin galectin-3 binds cell-surface galactose residues normally hidden by sialic acid, so galectin-3 binding to neuroendocrine cells was increased by desialylation, resulting in their phagocytosis by microglia (brain-resident macrophages) (49). Similarly, the binding of calreticulin to galactose-containing Galβ1→4GlcNAc residues exposed by desialylated neutrophils induced their phagocytosis by macrophages (47). Furthermore, the binding of the complement component C1q to neurites was greatly increased by desialylation, resulting in phagocytosis of C1q-opsonised neurites by microglia (48), possibly via C1q binding to exposed galactose (113). Exposed galactose residues on aged platelets induced their phagocytosis by liver macrophages via the macrophage galactose lectin (MGL) and Ashwell-Morell receptor (AMR), both of which bind exposed galactose residues directly (52). Desialylation exposes galactose residues, but removal of further sugar residues (e.g. by β-galactosidase and β-hexosaminidase) exposes N-acetylglucosamine and mannose residues, which are bound by mannose-binding lectin (MBL) (50). Exposed N-acetylglucosamine on aged platelets also directly activated phagocytosis via the lectin domain of the phagocytic complement receptor CR3 (51). Thus, asialoglycans act as eat-me signals in diverse tissues and cell-types (Figure 3).
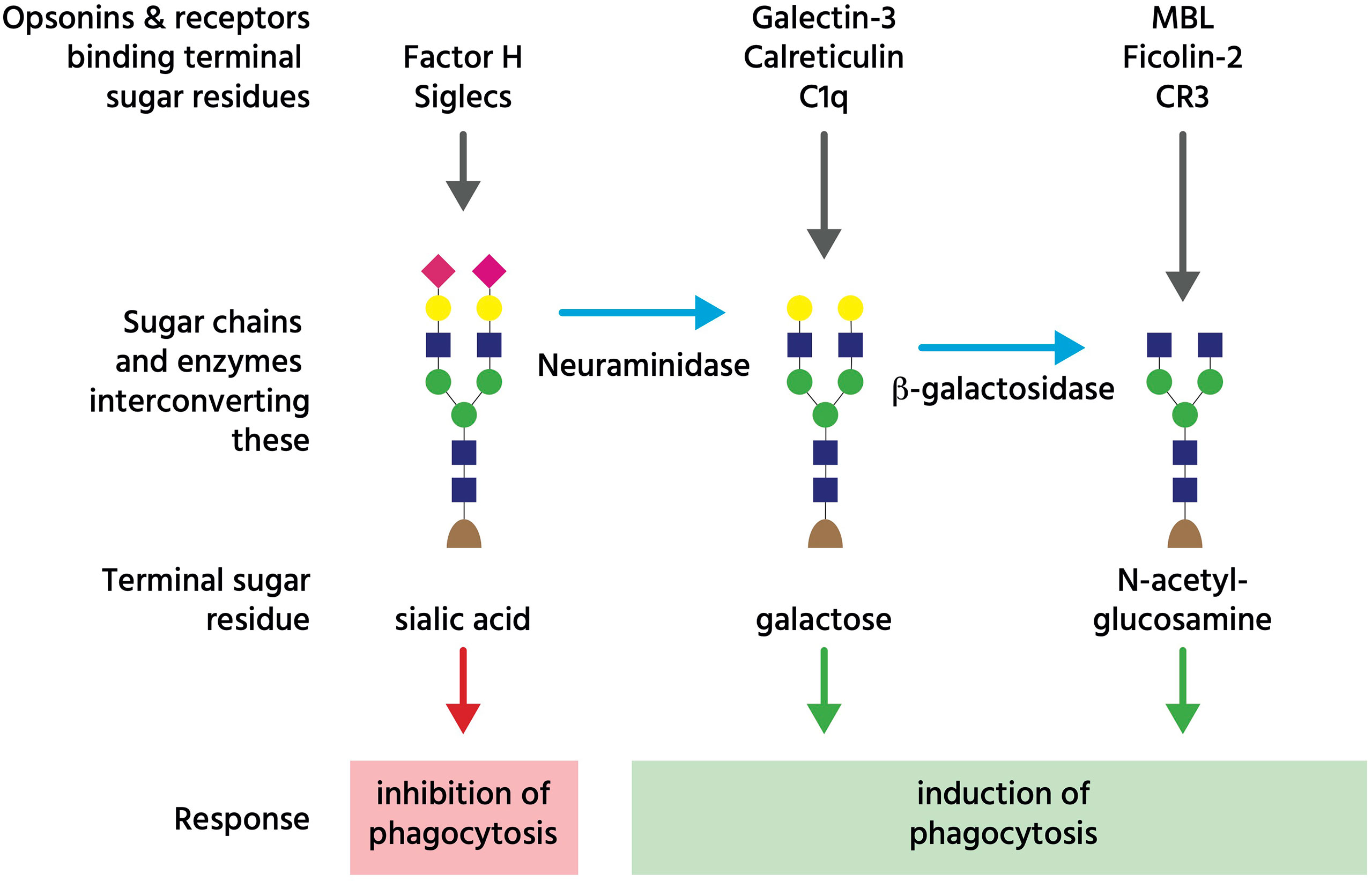
Figure 3 Glycans regulating phagocytosis. The coloured shapes illustrate sugar residues in a typical glycan chain on an N-glycosylated cell-surface glycoprotein, where the terminal (monosaccharide) sugar is normally sialic acid (pink diamond), which can bind either Factor H to inhibit complement, or Siglec receptors to inhibit phagocytosis. However, neuraminidases can remove that terminal sialic acid residue to reveal galactose residues (yellow circle), which bind opsonins galectin-3, calreticulin and C1q. Subsequently, beta-galactosidase can remove terminal galactose residues to reveal N-acetyl-glucosamine residues (blue square), which bind opsonins (and complement regulators) MBL (mannan binding lectin) and ficolin-2, as well as complement receptor 3 (CR3). This figure is a considerable simplification of actual sugar chains and their regulation of phagocytosis.
Calreticulin is a lectin (sugar-binding protein) that normally resides in the endoplasmic reticulum (ER), but can be translocated to the surface of the cell during apoptosis or ER stress (58, 114). Surface-exposed calreticulin acts as an eat-me signal to phagocytes via the LRP1 receptor, which mediates macrophage phagocytosis of healthy or apoptotic leukocytes, erythrocytes, and neurons (58, 114, 115). Calreticulin can bind directly to LRP1 (116), but it may also promote phagocytosis by binding the opsonin C1q (55), which can itself activate LRP1 to induce phagocytosis (117). Calreticulin can bind phosphatidylserine (56), and so may signal to phagocytes either alone or in association with exposed phosphatidylserine. Phagocytic signalling by apoptotic or ER-stressed cancer cells may also occur via translocation to the cell surface of calreticulin bound to ERp57 (114). However, as calreticulin is soluble and binding to ERp47 and phosphatidylserine is not covalent, calreticulin may also be released from cells. Indeed, calreticulin can be secreted as a soluble protein and act as an opsonin by binding asialoglycans on target cells (see ‘Asialoglycan’ section below), and potentially by binding phosphatidylserine or ERp57. Thus, calreticulin could be regarded as a ‘self-opsonin’ i.e. something released from and binding back onto a target cell to stimulate phagocytosis of the cell.
Deoxyribonucleic acid (DNA) - like histones, genomic DNA translocates from the nucleus to the cell-surface during necrosis and apoptosis, where it may act as an eat-me signal by binding opsonins. Xu et al. (118) found that the opsonin properdin strongly binds both double-stranded and single-stranded DNA, and associates with DNA on the surface of both necrotic and apoptotic cells. Jensen et al. (119) showed Ficolin-2 binds DNA, and induces phagocytosis of necrotic (but not apoptotic) T cells by monocytes. Additionally, C1q and C3 both directly bind DNA, and their recruitment at the surface of apoptotic cells is impaired by enzymatic degradation of DNA (120). Thus, DNA might act as an eat-me signal for cells exposing DNA, although it has not been shown that degrading cell-surface DNA inhibits phagocytosis of such cells. DNA does not itself integrate into the cell membrane, so may be better described as a self-opsonin or complement activator, although what DNA binds on the surface of dying cells is unclear.
Histones are normally located within the nucleus of all mammalian cells, but can appear on the cell surface during apoptosis (121). Here, they may act as eat-me signals by binding the opsonin apoJ (clusterin), thus facilitating phagocytic clearance of the apoptotic cell by macrophages (122). However, it is not yet known whether blocking histones at the surface of a target cell can inhibit phagocytosis.
Intercellular adhesion molecule 3 (ICAM-3, CD50) is constitutively expressed by leukocytes and mediates cell-cell adhesion by interacting with specific integrin receptors (61). ICAM-3 has been called an eat-me signal because antibodies blocking ICAM-3 on apoptotic neutrophils inhibit macrophage phagocytosis of the neutrophils in culture (123). ICAM-3 can directly bind the integrin receptor LFA-1 (lymphocyte function-associated antigen 1) (124), and knocking down or blocking LFA-1 in macrophages inhibited their phagocytosis of apoptotic neutrophils (123). Phagocytosis of ICAM-3-expressing apoptotic B cells, T cells and neutrophils may also involve the CD14 receptor on macrophages (125). As LFA-1 and CD14 are not phagocytic receptors, it seems likely that the role of ICAM-3 in phagocytosis is primarily adhesive, whereas phagocytosis per se is triggered by other signals (126). That ICAM-3 is not itself an eat-me signal is supported by the finding that ICAM-3 does not appear to change form and has somewhat lower expression during apoptosis (125). However, apoptotic leukocytes can release ICAM-3 as microparticles, which can chemoattract macrophages and may act as find-me signals (126).
Oxidized phospholipids (oxPL). Phospholipids can be oxidised by a variety of processes, and some oxidised phospholipids can act as eat-me signals. Monoclonal antibodies that target oxidation-specific epitopes on surface phospholipids have been shown to bind apoptotic cells and block their phagocytosis by macrophages, whilst the same antibodies failed to bind non-apoptotic cells (66). Interestingly, Greenberg et al. (60) found that incorporating oxidised (but not non-oxidised) phosphatidylserine into healthy cells was sufficient to induce their phagocytosis by macrophages. Thus, in certain contexts, phosphatidylserine may be insufficient to induce phagocytosis without oxidation. The scavenger receptor CD36 can directly bind oxPL (61), and mediate macrophage phagocytosis of apoptotic cells in culture (60). CD36 recognition of oxPL also mediated phagocytosis of photoreceptor outer segments by retinal pigment epithelial (62). The lectin-like OxLDL receptor 1 (LOX-1) can mediate phagocytosis of aged and apoptotic cells, possibly by binding oxidised low-density lipoproteins (LDLs) on these cells (63). Oxidized phosphatidylethanolamine exposed on the surface of cells undergoing ferroptosis (an iron-dependent form of programmed cell death) triggers macrophage phagocytosis of these cells via the macrophage toll-like receptor 2 (TLR2) (65).
Pentraxin-3 (PTX3) is a conserved member of the pentraxin family of acute phase proteins, and can translocate from intracellular granules to the surface of neutrophils during apoptosis, thereby promoting their phagocytosis by macrophages (67). PTX3 on the surface of apoptotic macrophages can induce phagocytosis of these apoptotic cells by non-apoptotic macrophages (68), although in this case it is unclear whether the surface PTX3 originated from inside or outside the apoptotic cell. PTX3 is a soluble protein that can also function as an opsonin (see below). Thus, PTX3 could be regarded as a self-opsonin for some apoptotic cells.
Phosphatidylserine is the most widely-documented eat-me signal. Phosphatidylserine constitutes approximately 10% of plasma membrane phospholipids, but in healthy cells is contained within the inner leaflet of the membrane via ATP-powered aminophospholipid translocases (127). During apoptosis, surface exposure of phosphatidylserine may increase over 100-fold within 1 or 2 hours (128) due to decreased translocase activity and increased activity of phospholipid scramblases (proteins that promote phosphatidylserine externalisation to the outer surface), where it acts as an eat-me signal. Blocking exposed phosphatidylserine on apoptotic cells by adding the phosphatidylserine-binding protein annexin V can fully prevent phagocytosis by macrophages (129).
There are several different phagocytic receptors for phosphatidylserine, which either bind to phosphatidylserine directly, or indirectly via opsonins. Non-opsonic receptors for phosphatidylserine include T-cell immunoglobulin and mucin (TIM) family receptors TIM-1, TIM-3 and TIM-4 (98, 99), brain angiogenesis inhibitor 1 (BAI-1) (87) and stabilin-2 (96) (Table 2). Opsonins binding phosphatidylserine (and their corresponding phagocytic receptors) include: Annexin A1 via formyl peptide receptor 2 (FPR2) or the phosphatidylserine receptor (PSR) (46, 69), apolipoprotein H via an unconfirmed receptor (70), calreticulin via lipoprotein receptor-related protein 1 (LRP1) (56, 71), milk fat globule-epidermal growth factor E8 (MFG-E8) via the integrin receptor αvβ3 (a vitronectin receptor, or VNR) (76), Cellular Communication Network Factor 1 (CCN1) via the integrin receptors αvβ3 and αvβ5 (72), and growth arrest-specific 6 (Gas6) & protein S via TAM receptors (Tyro, Axl and MerTK) (73–75, 80, 81) (Table 2).
Phosphatidylserine exposure was considered to be exclusively an apoptotic marker. However, transient phosphatidylserine exposure by live cells (not committed to death) has also been reported (130), and such exposure can be sufficient to induce death of these cells directly via ‘phagoptosis’ (death by phagocytosis) (131). Phosphatidylserine exposure on sub-cellular targets such as neuronal synapses (82) and axonal debris (106) has also been reported to facilitate their phagocytic clearance, which may be relevant in brain development and pathology.
Interestingly, phosphatidylserine can also negatively regulate phagocytosis by activating the inhibitory receptor CD300a on macrophages to inhibit phagocytosis (88). The function of this dual signalling is unclear, but would enable particular phagocytes to downregulate phagocytosis of phosphatidylserine-exposed cells when CD300a is upregulated.
Signalling lymphocytic activation molecule F7 (SLAMF7) is constitutively expressed on haematopoietic cells, and mediates macrophage phagocytosis of haematopoietic cells when CD47 (a don’t-eat-me signal) is blocked (132). This phagocytosis is apparently mediated by SLAM7 on target cells binding SLAM7 on macrophages, activating phagocytosis via CR3. Thus, SLAM7 could be regarded as a very specialised eat-me signal. However, as SLAM7 is constitutively expressed and does not appear to change as a result of CD47 blockade, it may be confusing to call it an eat-me signal.
Uridine-5’-diphosphate (UDP) is an exceptional eat-me signal, as unlike classical eat-me signals, it is a nucleotide released locally from target cells to induce phagocytosis by proximal phagocytes. UDP is released from dying or stressed neurons, and activates the phagocytic P2Y6 receptor expressed by microglia to induce phagocytosis (107, 108), so may be especially important in brain homeostasis. UDP may also be a find-me signal (see previous section), but it stimulates engulfment itself several fold (107). Indeed, UDP may be better thought of as an engulfment signal than as an eat-me signal.
Opsonins
Sometimes confused with eat-me signals, opsonins are normally soluble, extracellular proteins, which when bound to target cells induce phagocytes to phagocytose these cells (133, 134). To achieve this, opsonins must bind to both something on the target and to a phagocytic receptor on a phagocyte. Thus, opsonins are bridging proteins. As outlined above, opsonins differ fundamentally from eat-me signals in that they do not originate from the target cell. Opsonins of mammalian cells or sub-cellular targets are here reviewed, and summarised in Table 3 and Figures 4, 5.

Table 3 Opsonins for phagocytosis of mammalian cells/sub-cellular targets.
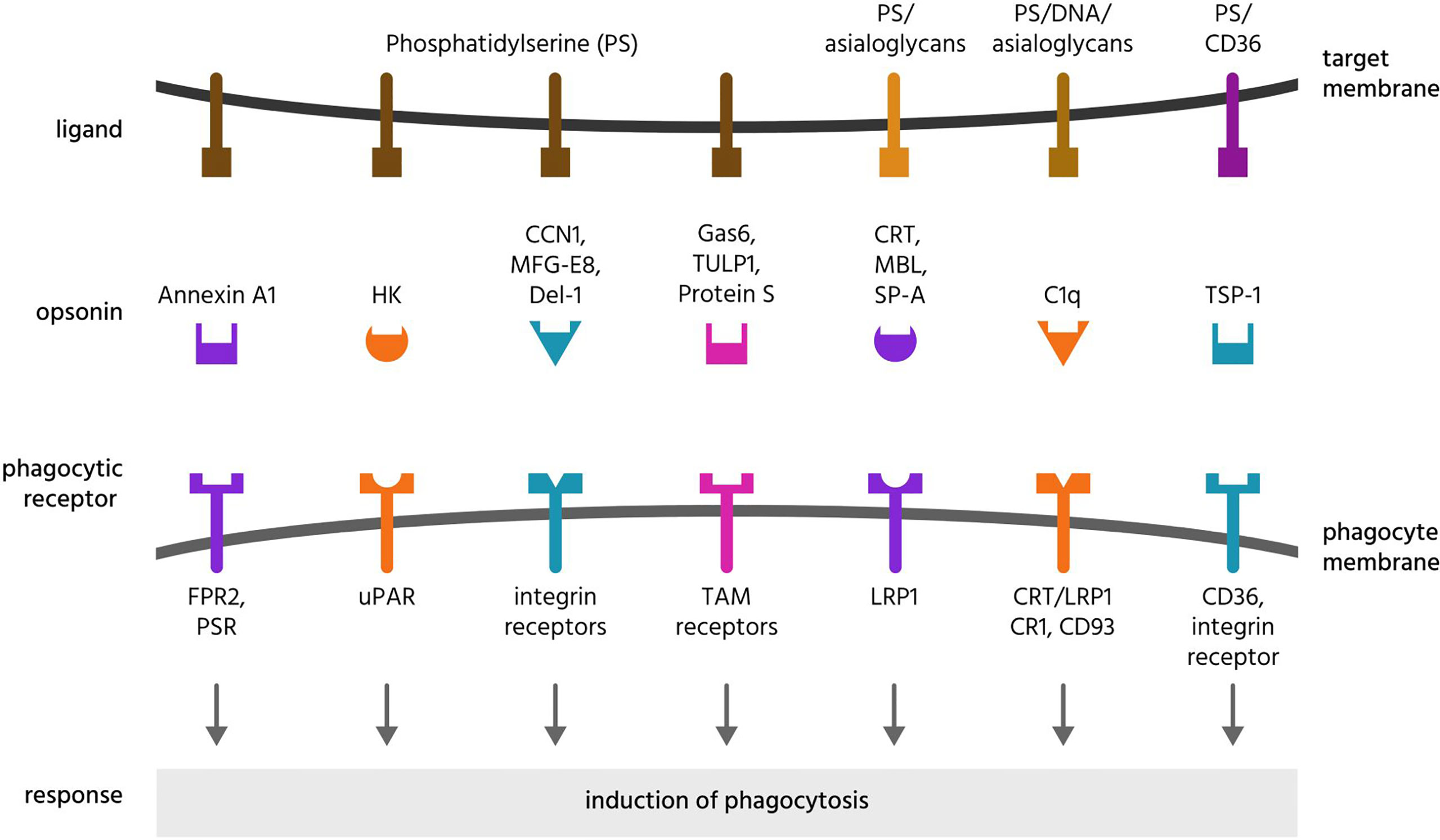
Figure 4 Opsonins and their ligands and receptors. Opsonins are normally soluble, extracellular proteins, which when bound to target cells induce phagocytes to phagocytose these cells. To achieve this, most opsonins bind eat-me signals on the target cell and phagocytic receptors on the phagocyte, and these are illustrated here for a variety of opsonins that bind phosphatidylserine or asialoglycans. CCN1, cellular communication network factor 1; CD, cluster of differentiation; CR1, complement receptor 1; CRT, calreticulin; Del-1, developmental endothelial locus 1; DNA, deoxyribonucleic acid; FPR2, formyl peptide receptor 2; HK, high molecular weight kininogen; LRP1, lipoprotein receptor-related protein 1; MBL, mannose-binding lectin; MFG-E8, milk fat globule-epidermal growth factor E8; PS, phosphatidylserine; PSR, phosphatidylserine receptor; SP-A, surfactant protein A; TAM receptors, Tyro, Axl and MerTK receptors; TSP-1, thrombospondin 1; TULP1, tubby-like protein 1; uPAR, urokinase plasminogen activator receptor.
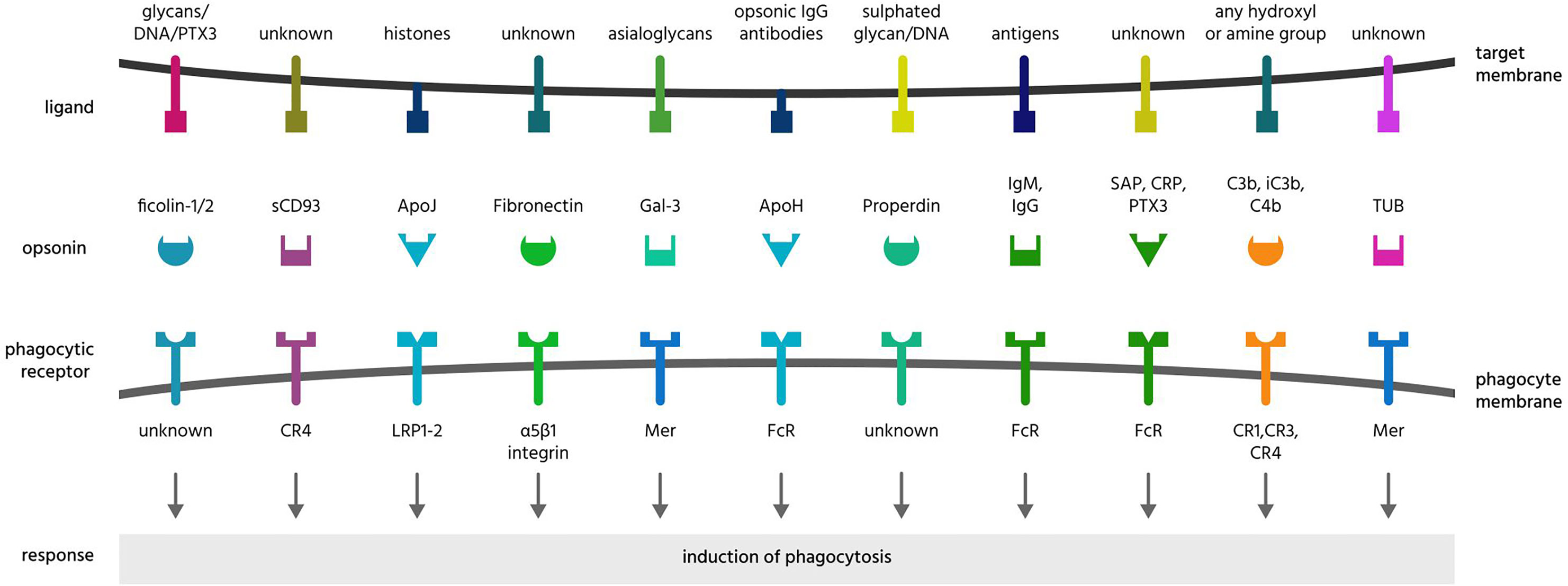
Figure 5 Further opsonins and their ligands and receptors. Apo, apolipoprotein; CR, complement receptor; CRP, C-reactive protein; DNA, deoxyribonucleic acid; Gal-3, galectin-3; Ig, immunoglobulin; LRP, lipoprotein receptor-related protein; PTX3, pentraxin 3; SAP, serum amyloid protein; sCD39, soluble cluster of differentiation 93; TUB, tubby.
Annexin A1 (AnxA1) can act as an eat-me signal (see above) in cells undergoing apoptosis, as intracellular AnxA1 translocates to the cell surface to induce phagocytosis by phagocytes (46). However, AnxA1 is a soluble protein, and soluble AnxA1 can opsonise various cells for phagocytosis: including apoptotic T cells and macrophages (136), apoptotic neurons (69), and apoptotic monocytes (135). AnxA1 binds phosphatidylserine (219), and co-localises with phosphatidylserine on the apoptotic cell surface, suggesting that AnxA1 opsonises by binding phosphatidylserine on cells. Phagocytic receptors for AnxA1 include the formyl peptide receptor 2 (FPR2) (69, 135) and the phosphatidylserine receptor (PSR) (46).
Antibodies opsonise cells by binding antigens on the target, and phagocytic Fcγ receptors on phagocytes (220). The major Fcγ receptors that promote phagocytosis of antibody-coated targets are FcγRI, FcγRIIA and FcγRIII (221). Some IgGs (immunoglobulin G) can preferentially bind apoptotic over non-apoptotic cells (144, 184). For example, antibodies to apolipoprotein H (a phosphatidylserine-binding opsonin) enhanced phagocytosis of phosphatidylserine-exposing apoptotic thymocytes (70). IgG has also been shown to opsonise apoptotic neutrophils for phagocytosis by macrophages (184), and apoptotic lymphoma cells for phagocytosis by dendritic cells (144). We have shown that antibodies targeting multiple antigens on live, malignant human B-cells enable their phagocytosis by macrophages (185). IgM antibodies binding to phosphatidylserine can also opsonise apoptotic T cells for phagocytosis by macrophages (187).
Apolipoproteins E, J (clusterin) and H. Apolipoproteins are a family of lipid-binding proteins, with multiple members now recognised as opsonins. ApoE opsonises neuroblastoma cells for phagocytosis by macrophages via the phagocytic receptor TREM2, to which ApoE directly binds (138). ApoE has also been shown to opsonise apoptotic thymocytes for phagocytosis by peripheral macrophages (140), and can enhance phagocytosis of neuronal synapses by astrocytes (141). ApoE may bind phosphatidylserine to mediate its opsonic effects (137), although such binding is unconfirmed. ApoE can also bind and activate the phagocytic receptor LRP1 (139), which could also relay the phagocytic signal of ApoE, but again this is unconfirmed. ApoJ (a.k.a. clusterin) has been reported to opsonise apoptotic cells (122) and cellular debris (142) by binding histones on cells and phagocytic receptors LRP1 and LRP2 (megalin) on phagocytes (142). ApoH (a.k.a. β2 glycoprotein 1) is recruited to the cell-surface of apoptotic cells (222), and can enhance phagocytosis of apoptotic thymocytes, lipid-symmetric red blood cells and platelet micro-vesicles by macrophages (70, 143). ApoH can associate with phosphatidylserine, which may be sufficient to mediate its opsonic function.
Calreticulin is an eat-me signal for cells undergoing apoptosis or ER-stress, inducing phagocytosis by activating the phagocytic LRP1 receptor (58, 59, 114, 115). However, calreticulin is a soluble protein that can be released from cells extracellularly, to bind the surface of cells and function as an opsonin. Calreticulin can bind phosphatidylserine (56), and Wijeyesakere et al. (71) showed that calreticulin directly bound phosphatidylserine exposed on apoptotic cells via its C-terminal acidic region, and enhanced phagocytosis of apoptotic fibroblasts by peritoneal macrophages, which was dependent on this phosphatidylserine-binding region. Feng et al. (47) found that calreticulin was actively secreted by macrophages, and bound to and opsonised co-incubated neutrophils, and blocking calreticulin on the neutrophils reduced their phagocytosis by macrophages. Calreticulin bound to the neutrophils via galactose (Galβ1➔4GlcNAc) residues of asialoglycans normally hidden by terminal sialic acid residues, so calreticulin acted as an opsonin for desialylated cells (47). Analogously, we found that calreticulin is released by activated microglia and binds to bacteria (via sugars), and induces microglial phagocytosis of the bacteria via LRP1 (133). To add to the complexity, calreticulin can also function as a phagocytic co-receptor - for example during the phagocytosis of apoptotic T cells by macrophages, via complex formation with macrophage-exposed LRP1 and surfactant proteins SP-A and SP-D (151) and/or C1q (148).
Cellular Communication Network factor 1 (CCN1, CYR61) is an extracellular matrix protein expressed in placental, skeletal, nervous and cardiovascular cells (223). Jun et al. (72) found that CCN1 opsonised apoptotic neutrophils for phagocytosis, by binding phosphatidylserine on the neutrophils and the integrins αvβ3 and αvβ5 on macrophages (72).
sCD93 (soluble CD93) is derived from the membrane protein CD93, and is released from myeloid cells in a soluble form detectable in human plasma (224). sCD93 can opsonise beads and apoptotic cells for phagocytosis by macrophages, via the phagocytic receptor CR4 (integrin αxβ2) (146), and CD93-/- mice exhibit impaired phagocytic clearance of apoptotic cells (225). What sCD93 binds to on apoptotic cells is not known, but is mediated by the lectin domain of sCD93, so may be glycans (146).
Collectins: Mannose-Binding Lectin (MBL), Surfactant Proteins A (SP-A) and Surfactant Protein D (SP-D). Collectins are a family of sugar-binding proteins, including MBL, SP-A and SP-D (191, 226). MBL can bind the surface of apoptotic cells along with C1q, which together forms a complex with calreticulin and LRP1 (CD91) present on the surface of local phagocytes to induce engulfment of the apoptotic cell (148). Apoptotic cells shown to be opsonised by MBL include erythrocytes (148), T cells (148) and adipocytes (192). Necrotic cells are also opsonised by MBL (191). MBL can bind phosphatidylserine (84) and asialoglycans (N‐acetyl‐glucosamine, mannose or fucose residues) (226), which may mediate binding to apoptotic cells. MBL can also bind MASP (mannan-binding lectin serine protease) to initiate the lectin-pathway of complement activation, thereby inducing opsonins C3b, iC3b and C4b (191). SP-A and SP-D also act as opsonins for macrophage phagocytosis of several apoptotic cell-types, mediated via calreticulin and LRP1 on the macrophage surface (151, 211). Binding of SP-A to apoptotic cells was via binding to exposed phosphatidylserine (85). Note, however, that SP-A and SP-D have also been reported to inhibit phagocytosis by activating SIRPα (227).
Complement proteins C1q, C3b and C4b and cleavage products iC3b, C3c, C3d and C3dg can opsonise targets via complement receptors CR1, CR2, CR3, CR4 and CRIg (83, 158, 160) expressed by several phagocytes, notably neutrophil monocytes and macrophages (228, 229). C1q can also induce phagocytosis via LRP1 (55, 117) or the calreticulin/LRP1 receptor complex (148). C1q, C3b, iC3b and C4b have together been implicated in the opsonisation of a wide range of apoptotic cells, as well as necrotic cells (Table 3). C1q and C3b can each bind phosphatidylserine (83, 147), which may mediate their opsonisation of phosphatidylserine-exposing apoptotic cells. However, C1q may also opsonise by binding calreticulin (151), a known C1q receptor and established eat-me signal present on the surface of apoptotic cells. C1q can also bind DNA (230), initiating complement activation, and nucleic acid exposure by apoptotic cells increases detectable C1q and C3b on the apoptotic cell surface (120). Complement can also opsonise neuronal synapses for phagocytosis by microglia (155). Desialylation of neurons caused C1q to bind to neurons, resulting in CR3-mediated microglial phagocytosis of neurites (presumably by inducing C3b deposition) (48). It is unclear why C1q binds more to desialylated neurons, though this may occur via binding exposed galactose and N-acetyl-glucosamine residues (113). Desialylation of pentraxin-3 has also been shown to induce C1q binding and complement activation (231). Complement opsonisation of myelin has also been reported (159). C1q binding to a cell can induce the complement cascade resulting in local deposition of C4b and C3b on the cell. C3b and C4b covalently attach to cell surface hydroxyl groups, typically on sugars, opsonising such surfaces for phagocytosis (157, 162). Cell surface sialylation inhibits C3b production and stability via recruiting the complement inhibitor Factor H (232). Thus, the complement system of proteins contains multiple opsonins with complex regulation of phagocytosis.
Developmental endothelial locus-1 (Del-1) is a cell-secreted glycoprotein, recently shown to opsonise apoptotic neutrophils for phagocytosis by macrophages (171). Del-1 can bind directly to phosphatidylserine via it’s C-terminal domain, and Del-1-mediated phagocytosis required the αvβ3 integrin receptor (171).
Ficolins are group of pattern recognition proteins known to act as opsonins for pathogens via binding surface sugar residues such as N-acetyl-glucosamine, but may also opsonise necrotic host cells via binding DNA (119) or pentraxins (176).
Fibronectin is a large (440 kDa) soluble glycoprotein found at high levels in plasma (233), and early reports indicated that fibronectin could enhance monocyte phagocytosis of erythrocytes (172), possibly via Fc receptors expressed by the monocyte. Subsequently, fibronectin was reported to bind and opsonise apoptotic Jurkat T cells and activate macrophage phagocytosis of such cells via also binding and activating the α5β1 integrin on the macrophages (174).
Galectin-3 is a β-galactoside-binding protein expressed in myeloid cells including macrophages, monocytes, dendritic cells and neutrophils (234). Galectin-3 is released from inflamed macrophages, and can bind to exposed galactose residues on cells to opsonise these cells (49). Galectin can enhance the uptake of apoptotic neutrophils by monocyte-derived macrophages, blocked by lactose, which competes for sugar binding (178). Whilst it usually exists as a monomer, binding to galactose residues induces oligomerisation, which may facilitate the bridging between phagocytes and target cells during phagocytosis (235). Galectin-3 can directly bind the phagocytic receptor MerTK, and stimulate macrophage phagocytosis of apoptotic T cells (177). We found that galectin-3 binds to desialylated cells and induces microglial phagocytosis of cellular debris and neuroendocrine cells, inhibited by blocking MerTK (49). More recently, we have shown that galectin-3 can opsonise gram-negative bacterial E. coli for phagocytosis by microglia via MerTK (133). Thus, galectin-3 can opsonise both host and foreign targets for phagocytic elimination in mammalian systems.
Growth arrest-specific protein 6 (Gas6) is an extracellular protein ligand for the phagocytic receptor MerTK (as well as for other TAM members Tyro3 and Axl) (75, 179, 180). Gas6 binds phosphatidylserine, which mediates the phagocytosis of apoptotic thymocytes by macrophages (74), and Gas6-dependent phagocytosis is abolished by genetic deletion of MerTK (180). Gas6 has further been described to induce phagocytosis of apoptotic cells by microglia (181), and also of photoreceptor outer segments by retinal pigment epithelial cells (182).
High-molecular weight kininogen (HK) is a serum protein, increased by inflammation, found to opsonise apoptotic cells by binding phosphatidylserine on these cells and activating the urokinase plasminogen activator receptor (uPAR) on macrophages (183).
Milk fat globule-epidermal growth factor 8 (MFG-E8) is an extracellular protein ligand for the integrin receptors αvβ3 and αvβ5 (236). MFG-E8 is secreted from dendritic cells (237) and macrophages (197), and can opsonise apoptotic cells for phagocytic removal (236). Soluble MFG-E8 binds phosphatidylserine exposed on the target cell (77) and either of the integrin receptors αvβ3 (76) or αvβ5 (193) on the phagocyte, thus inducing phagocytosis. MFG-E8 can promote phagocytosis of defective red blood cells (sickle cells) (196), apoptotic lymphocytes (195) and live neurons (198) by macrophages, photoreceptor cells or cell segments by retinal pigment epithelia (193), and apoptotic thymocytes by fibroblasts expressing αvβ3 (76).
Pentraxins: C-reactive protein, serum amyloid P and PTX3. Pentraxins (PTX) are extracellular pentameric proteins that function as pattern recognition molecules (238). Three pentraxins - C-reactive protein (CRP, a.k.a PTX1), serum amyloid P (SAP, a.k.a PTX2) and PTX3 opsonise mammalian and microbial cells for phagocytosis. CRP binds and opsonises apoptotic T cells for phagocytosis by macrophages (169, 170), and also erythrocytes for phagocytosis by peripheral blood mononuclear cells (PBMCs) (168). In both cases, opsonisation was mediated by phagocytic Fc receptors. SAP also binds and opsonises apoptotic cells, including T cells and neutrophils (170), for phagocytosis by macrophages, again mediated by phagocytic Fc receptors. As noted previously, PTX3 has been described as an eat-me signal. However, Ma et al. (176) showed that exogenous PTX3 associates with apoptotic or necrotic (but not viable) T cells, where it complexes with ficolin-1 to enhance phagocytosis of these cells by macrophages. Lech et al. (239) found that murine macrophages lacking PTX-3 had a reduced ability to phagocytose apoptotic cells with normal PTX-3 levels, confirming the importance of non-target-cell PTX-3 for phagocytosis. It is unclear to what CRP, SAP and PTX-3 bind to on target cells, although each can bind to C1q (167, 205), which might mediate the binding to apoptotic cells (169).
Properdin is a soluble protein released by leukocytes, and can modulate inflammation by stimulating the alternative pathway of complement activation (240). Properdin can bind exposed DNA and induce deposition of the opsonin iC3b on apoptotic cells, indirectly stimulating phagocytosis via complement (118). However, properdin can opsonise independently of complement by binding sulphated glycosaminoglycans exposed on apoptotic and malignant T cells, inducing their phagocytosis by macrophages or dendritic cells via unknown receptors (199).
Protein S (Pros1) is a serum protein that can bind phosphatidylserine (81) and the phagocytic receptor MerTK (241). McColl et al. (200) found that the phagocytosis of apoptotic neutrophils by dexamethasone-treated macrophages involved opsonisation by protein S signalling via MerTK. Similarly, phagocytosis of photoreceptor cells by retinal pigment epithelia was mediated by Pros1 and MerTK (75). Protein S binding to phosphatidylserine opsonises apoptotic lymphocytes for phagocytosis by macrophages (80). Opsonisation by protein S has also been demonstrated in macrophage phagocytosis of apoptotic T cells (202) and apoptotic neutrophils (203). Protein S can also activate the phagocytic receptor Tyro3 (75), but it is unclear where this might regulate phagocytosis.
Thrombospondin (TSP-1) is an extracellular matrix protein expressed by endothelial cells, monocytes and macrophages (242). Extracellular secretion of TSP-1 by cells increases during apoptosis (86), and extracellular TSP-1 has been shown to opsonise apoptotic neutrophils (213), eosinophils (214) and fibroblasts (86) for phagocytosis by macrophages. TSP-1 can bind phosphatidylserine (212), which may mediate opsonisation by TSP-1. However, Moodley et al. (86) found that TSP-1 acted as a bridging protein between CD36 on apoptotic fibroblasts and CD36 on macrophages, and induced phagocytosis independent of phosphatidylserine. TSP-1 also indirectly associates with αvβ3 via the integrin-associated protein (IAP) (243), which may also mediate TSP-1 opsonisation in contexts of efferocytosis.
Tubby (TUB) and tubby-like protein 1 (TULP1) - two structurally related members of the tubby protein family – are expressed in the brain (244) and reside intracellularly, but can also be secreted by cells (245). Extracellular TUB and TULP1 enhance the phagocytosis of photoreceptor outer segments by retinal pigment epithelia by activating the phagocytic receptor MerTK (215, 216). Moreover, TULP1 enhanced the microglial phagocytosis of apoptotic (but not healthy) T cells, and also of neuroblastoma-derived membrane vesicles, both mediated via MerTK (218). TULP1 can also interact with the other TAM receptors Tyro3 and Axl (215). It is unclear to what TUB binds to on target cells, although TULP1 can bind phosphatidylserine (217), which likely mediates its opsonic effect.
Don’t-Eat-Me Signals
The vast majority of cells in the body express don’t-eat-me signals in order to prevent themselves being eaten by phagocytes. Don’t-eat-me signals are signals on or from target cells that inhibit the phagocytosis of these cell. Table 4 lists known don’t-eat-me signals, whilst Figure 6 illustrates don’t-eat-me signals and their binding partners.
Table 4 Don’t-eat-me signals.
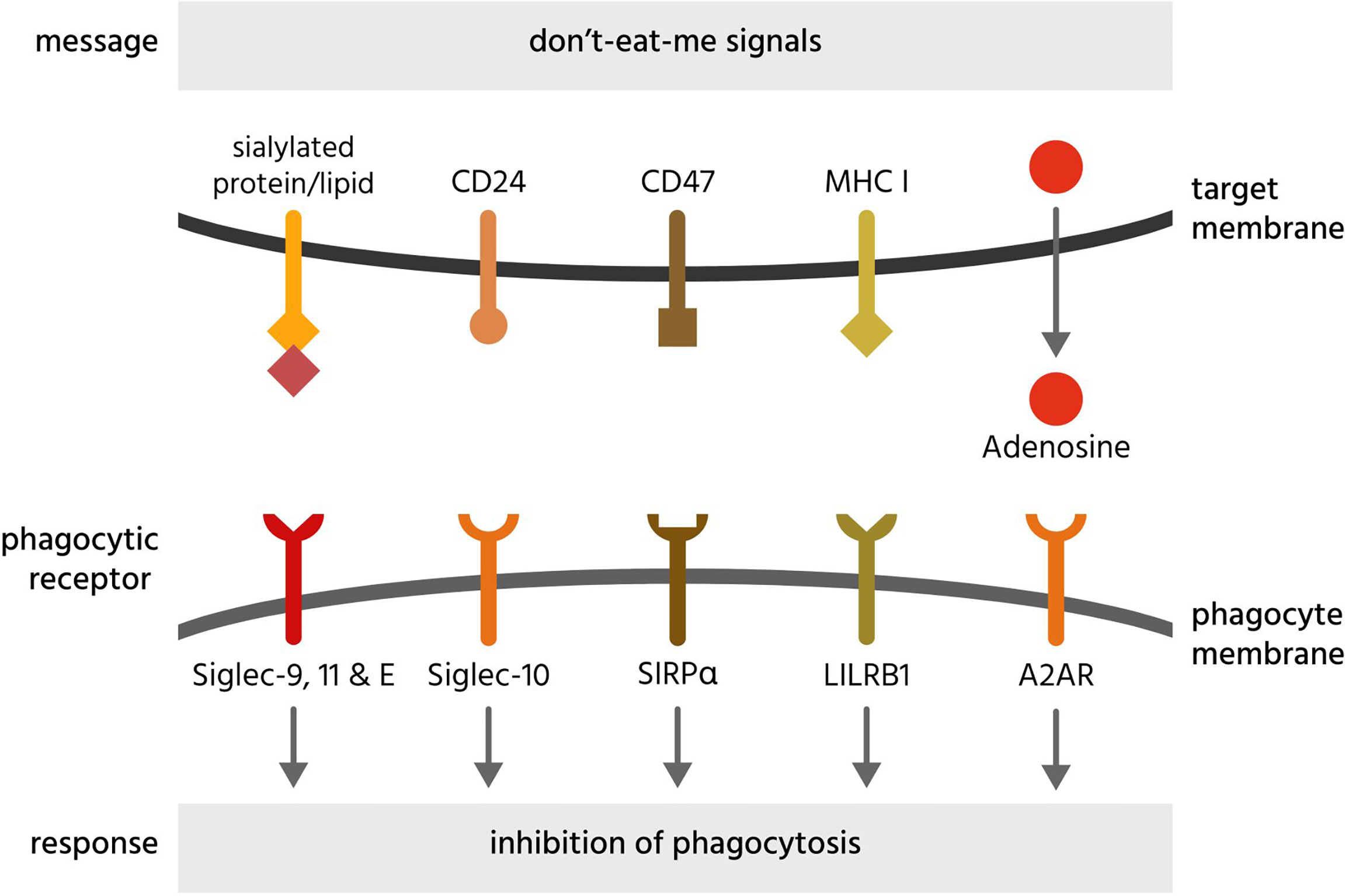
Figure 6 Don’t-eat-me signals and their receptors. Don’t-eat-me signals are molecules exposed on or released from a target cell to directly inhibit phagocytosis of that cell by a phagocyte. The figure illustrates eat-me signals on or from mammalian target cells. A2AR, adenosine 2A receptors; CD, cluster of differentiation; LILRB1, leukocyte immunoglobulin-like receptor subfamily B member 1; MHC I, major histocompatibility complex 1; Siglec, sialic acid-binding immunoglobulin-type lectin; SIRPα, signal regulatory protein α.
Adenosine can be released by stressed or dying cells and inhibits phagocytosis of such cells via adenosine 2A receptors (A2AR) on phagocytes (246). Antibody-induced phagocytosis of cancer cells was inhibited by adenosine and enhanced by knockout or inhibition of A2AR receptors in vivo (246).
CD24 is a sialylated glycoprotein present on the surface of B cells and neutrophils, inhibiting the phagocytosis of such cells by binding and activating Siglec-10 on phagocytes (247). CD24 is upregulated on ovarian or breast cancer cells, and Barkal et al. (247) found that blocking both CD47 and CD24 synergistically enhanced phagocytosis, indicating redundancy of these two membrane-bound don’t-eat-me signals. Siglec-10 binding to CD24 is partly mediated by sialic acid residues on CD24 but also some amino acid residues (247), indicating a level of specificity towards CD24.
CD31 (PECAM-1) is expressed on the surface of leukocytes, macrophages and endothelial cells, and was reported to promote the detachment of leukocytes from macrophages under flow (259). During apoptosis, CD31 can change conformation to promote attachment and enable phagocytosis (259), possibly by stimulating the α5β1 integrin receptor (174). However, this role of CD31 has not confirmed, and the reported mechanism does not conform to a traditional don’t-eat-me signal, but rather a detachment signal under flow.
CD47 is a transmembrane receptor expressed by virtually all cells, and inhibits phagocytosis of cells by binding and activating the transmembrane receptor SIRPα on phagocytes (249, 250). Loss of CD47 expression induces phagocytosis of apoptotic cells (249) and senescent erythrocytes (250). Expression of CD47 appears sufficient to inhibit phagocytosis mediated by antibodies and complement (251). Loss of CD47 or antibody blockade is sufficient to induce phagocytosis of some cells (erythrocytes and cancer cells) (252, 253), but not other cells (260), suggesting either that other don’t-eat-me signals can replace CD47 in particular cells, or that CD47 blockade is sufficient to induce phagocytosis only in cells exposing eat-me signals. CD47 can be cleaved by metalloproteases on apoptotic cells, to remove this don’t-eat-me signals but also release the soluble ectodomain of CD47 (sCD47), which can antagonise SIRPα on phagocytes to stimulate phagocytosis. Thus, sCD47 may potentially act as a soluble eat-me signal (261).
CD200 (OX-2 membrane glycoprotein) is a transmembrane protein that is typically expressed on the surface of hematopoietic-derived cells, as well as B-cells, activated T cells, endothelial cells and neuronal cells (262). Expression of CD200 on endothelial cells was found to inhibit phagocytosis of those cells by macrophages, possibly by engaging the CD200 receptor, CD200R (263). However, CD200 inhibits inflammation via CD200R (262), so it is possible that the inhibition of phagocytosis is indirect via inhibiting inflammation.
MHC class I (major histocompatibility complex class I; MHC1) is a transmembrane protein complex present on the surface of all host cells (except erythrocytes) and is a dimer of a variable α-subunit and an invariant β2-microglobulin. This β2-microglobulin subunit of MHC1 inhibits phagocytosis by binding and activating LILRB1 (leukocyte immunoglobulin-like receptor subfamily B member 1) on macrophages (254). Because MHC1 is also a core component of adaptive immunity, it is downregulated on many cancer cells and virally-infected cells, potentially making them susceptible to phagocytosis by macrophages (254).
Programmed death-ligand 1 (PD-L1) is transmembrane protein, expressed on immune cells, epithelial cells, and vascular endothelial cells, suppressing adaptive immunity by activating the inhibitory receptor programmed cell death protein 1 (PD-1) on T cells. Recently, it was reported that PD-L1 on tumour cells acted as a don’t-eat-me signal to inhibit their phagocytosis by tumour associated macrophages by activating PD-1 on the macrophages (264). However, this paper showed little evidence that PD-L1 directly inhibited phagocytosis, rather than inhibiting inflammatory activation of the macrophages, so PD-L1 status as a don’t-eat-me signal requires verification.
Sialic acid (N-acetylneuraminic acid) residues on the surface of virtually all cells inhibits phagocytosis of such cells by engaging and activating sialic acid-binding immunoglobulin-type lectin receptors (Siglec-9, -10, -11 and -E) on phagocytes (265). Sialic acid residues terminate the sugar chains of most glycoproteins and glycolipids on the cell surface, but these residues can be removed by sialidases (266). Loss of sialic acid residues (known as desialylation) can induce phagocytosis of apoptotic cells (54) and senescent cells (258). Cancer cells are hypersialylated and may overexpress highly sialylated mucins to prevent themselves being phagocytosed (267).
Negative Opsonins and Phagocyte Suppressants
Some extracellular proteins, not derived from the target cell, can inhibit phagocytosis, and we refer to these as either ‘negative opsonins’ (‘nopsonins’ for short) if they bind the target cell, and ‘phagocyte suppressants’ if they bind the phagocyte. Nopsonins and phagocyte suppressants are summarised in Table 5 and depicted in Figure 7. Note, however, it has rarely been demonstrated that these proteins can inhibit phagocytosis physiologically in vivo. Why might there be negative regulators of phagocytosis? Opsonins are probably expressed to amplify phagocytosis of eat-me signalling cells in particular conditions, such as inflammation, or alternatively to direct phagocytosis via particular phagocytes or phagocytic receptors, which determines what happens to the target cell. So, reasons for having negative regulators of phagocytosis might include: to suppress (or set a threshold for) phagocytosis of eat-me signalling cells in other conditions, such as in the healthy adult, or alternatively to suppress phagocytosis via particular phagocytes or particular receptors, thereby directing the fate of target cells.
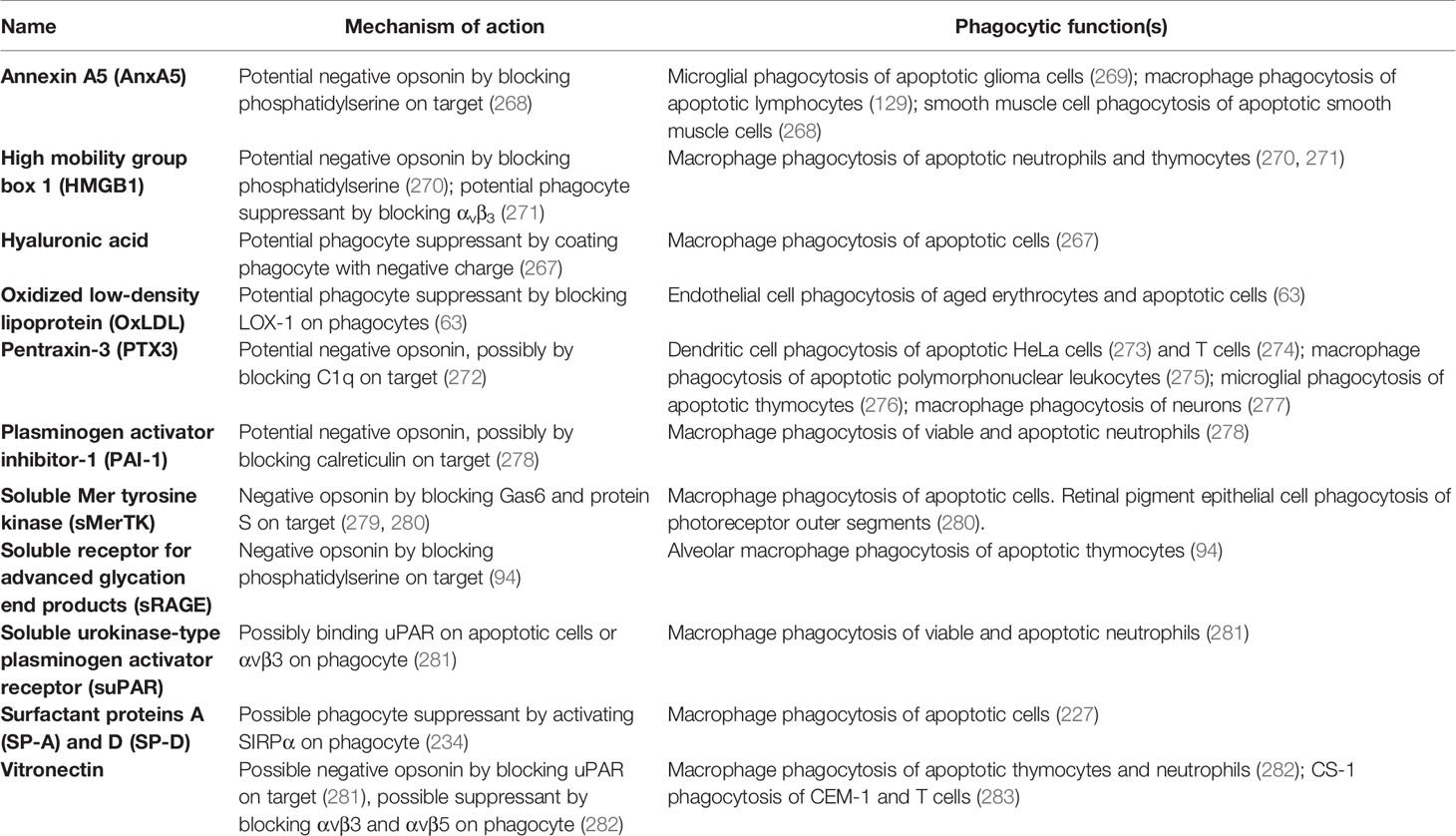
Table 5 Negative opsonins and phagocyte suppressants.
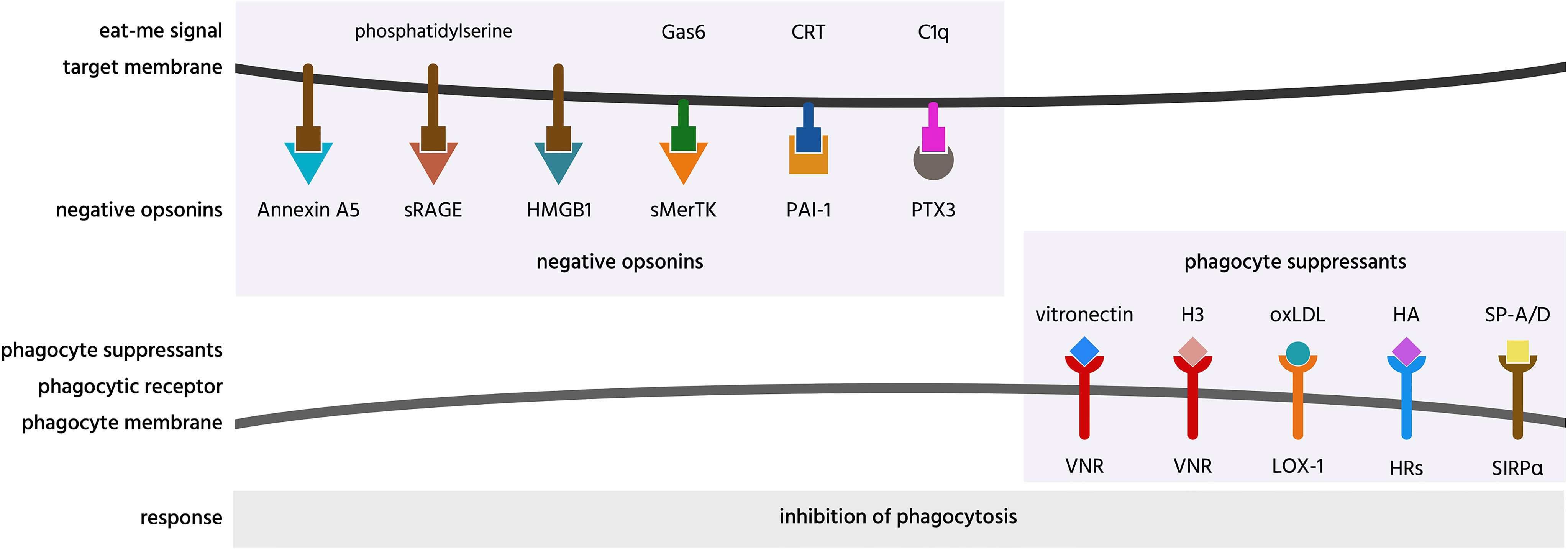
Figure 7 Negative opsonins and phagocyte suppressants, and their potential mechanisms of action. Negative opsonins are extracellular proteins that, when bound to a target cell, inhibit phagocytosis of that cell by phagocytes; a phagocyte suppressant is a normally soluble, extracellular molecule, which, when bound to a phagocyte discourages it from phagocytosing targets. CRT, calreticulin; H3, histone H3; HA, hyaluronic acid; HMGB1, high mobility group box 1 protein; HRs, hyaluronic acid receptors; LOX-1, lectin-like OxLDL receptor 1; oxLDL, oxidised low-density lipoprotein; PAI-1, plasminogen activator inhibitor-1; PTX3, pentraxin 3; SIRPα, signal regulatory protein α; sMerTK, soluble Mer tyrosine kinase; sRAGE, soluble receptor for advanced glycation end products; SP, surfactant protein; VNR, vitronectin receptors.
Annexin A5 (AnxA5) is normally an intracellular protein, but is present in serum, and can inhibit phagocytosis of apoptotic cells by binding and blocking phosphatidylserine exposed on the apoptotic cell surface (129, 268, 269, 284). Thus, annexin A5 is a potential negative opsonin.
High mobility group box 1 protein (HMGB1) is normally located in the nucleus, but can be released extracellularly during inflammation, and elevated levels are present in blood plasma in inflammatory states (270). HMGB1 can inhibit phagocytosis of apoptotic neutrophils by binding and blocking phosphatidylserine on neutrophils (270), and/or binding and blocking the phagocytic receptor αvβ3 on phagocytes (72, 271). Thus, HMGB1 is potentially both a negative opsonin and a phagocyte suppressant.
Histones are normally located in the nucleus, but can be released onto the surface of the cell to act as eat-me signals (see above), or released extracellularly during inflammation (285). Histone H3 was reported to inhibit macrophage phagocytosis of apoptotic neutrophils and thymocytes, by blocking the phagocytic receptors MerTK and the integrin receptor αvβ5 (286). However, this has not been verified at physiologically relevant levels. Thus, histone H3 is a potential phagocyte suppressant.
Hyaluronic acid (HA, hyaluronan) is a glycosaminoglycan component of extracellular matrix that can bind some cells including macrophages coating them in a negative charge that can inhibit macrophage phagocytosis of other cells (267). Thus, OxLDL is a potential phagocyte suppressant.
Oxidized low-density lipoprotein (OxLDL) is LDL with oxidised lipids, and is found in serum, and may contribute to atherosclerosis. OxLDL is reported to inhibit endothelial cell phagocytosis of aged and apoptotic cells via blocking LOX-1 on endothelial cells (63). Thus, OxLDL may be a phagocyte suppressant.
Pentraxin 3 (PTX3) is a soluble protein found in plasma and released by inflammatory-activated cells (287). PTX3 strongly binds to complement factor C1q and enhances C1-mediated deposition of C3 on cells, thereby enhancing phagocytosis (272). As mentioned previously, PTX3 can act as an opsonin for apoptotic cells (67, 68), but it is unclear whether this is in fact indirect via binding to C1q. PTX3 has also been found to inhibit phagocytosis of apoptotic cells in certain contexts (273–277), but again the mechanisms are unclear, possibly again by binding C1q (272). Thus, PTX3 may also act as a negative opsonin.
Plasminogen activator inhibitor-1 (PAI-1) is a serum protein reported to inhibit phagocytosis of viable and apoptotic neutrophils by macrophages, possibly by interfering with calreticulin on the surface of neutrophils (278). Macrophages showed increased phagocytosis of neutrophils from PAI-1 knock-out mice, which was later reversed in the presence of exogenous PAI-1 (278). PAI-1 also inhibited microglial phagocytosis, apparently via vitronectin and Toll-like receptors (288). Thus, PAI-1 is a potential negative opsonin and phagocyte suppressant.
Soluble intercellular adhesion molecule 5 (sICAM-5) is the soluble ectodomain of the transmembrane protein ICAM-5. Activated neurons can release sICAM-5, which binds integrins on microglia to inhibit microglial adhesion, activation and phagocytosis (289). However, supraphysiological levels of soluble ICAM-5 were used to inhibit microglial phagocytosis of beads, and this action might be via acting on microglial activation or adhesion, rather than on phagocytosis per se. As sICAM-5 is present in plasma it might act as a physiological phagocyte suppressant, but this has not investigated.
Soluble Mer tyrosine kinase (sMerTK) is the soluble ectodomain of the phagocytic receptor MerTK. sMerTK is cleaved from MerTK by metalloproteases, and is present in plasma (261). sMerTK is released by activated macrophages and inhibits macrophage phagocytosis of apoptotic cells by binding opsonins ProteinS or Gas6, preventing their binding to full length MerTK (279). Phagocytosis of photoreceptor outer segments by retinal pigment epithelial (RPE) induces release of sMerTK that inhibits further phagocytosis by binding the opsonins (280). Increasing sMerTK release further inhibited phagocytosis, while inhibiting sMerTK release stimulated phagocytosis (280), indicating that sMerTK is a negative opsonin.
Soluble receptor for advanced glycation end products (sRAGE) is produced by the proteolytic cleavage of the receptor RAGE or by alternative splicing of RAGE mRNA (290). RAGE was shown to act as a macrophage receptor for phosphatidylserine on apoptotic cells, while sRAGE was shown to bind to phosphatidylserine on apoptotic cells, and thereby inhibit phagocytosis of such cells (94) by acting as a negative opsonin. Whether it can do this physiologically is unknown, but sRAGE levels in serum change with inflammation and disease states (290).
Soluble urokinase-type plasminogen activator receptor (suPAR) is present in plasma and released from membrane-bound uPAR following cleavage by phospholipase C or D (291). Park et al. (281) reported increased macrophage phagocytosis of neutrophils if uPAR were knocked out in either the macrophages or the neutrophils, and in both cases addition of suPAR inhibited phagocytosis, but the mechanisms are unclear.
Surfactant protein A (SP-A) and surfactant protein D (SP-D) are collectins and extracellular proteins that can act as opsonins (see ‘Opsonins’ section). However, SP-A and SP-D have also been reported to inhibit phagocytosis (as a phagocyte suppressant) by activating SIRPα on macrophages (227). The authors speculated that these opposite effects are determined by whether SP-A and SP-D bind to phagocytes via the N-terminal that engages the calreticulin/CD91 receptor or the C-terminal that engages SIRPα.
Vitronectin is a protein found on the surface of cells and in a soluble form in the extracellular matrix (248). Soluble vitronectin was reported to inhibit macrophage phagocytosis of apoptotic cells in vitro and in vivo, both by blocking uPAR on apoptotic cells and by blocking the vitronectin receptor on macrophages (282). Thus, vitronectin may be both a negative opsonin and a phagocyte suppressant.
The Phagocytic Code for Particular Phagocytic Targets
Having introduced the components of the phagocytic code, we will now outline how they operate to regulate the phagocytosis of particular targets below.
Healthy Cells
As far as we know, healthy self-cells are not normally phagocytosed, unless they are excess to requirements or somehow senescent. Thus, the vast majority of host cells are not phagocytosed, preventing loss of healthy cells, but how is this achieved? We only know the answer to this in specific contexts, so much of what follows is speculative. Firstly, most healthy cells do not expose eat-me signals, such as phosphatidylserine (292). However, activated cells (with increased cytosolic calcium) can reversibly expose phosphatidylserine (293), and the eat-me signal calreticulin is constitutively exposed on neutrophils (294). Thus, there is probably a need for healthy cells to express don’t-eat-me signals, and indeed most healthy host cells express CD47 and MHC1 and are sialylated, inhibiting their clearance by phagocytosis (235, 295, 296). In healthy tissues, opsonin levels are relatively low (because most opsonins are induced by inflammation), and in both healthy and inflamed tissues, opsonins do not normally bind to healthy cells. Thus, healthy, self-cells can avoid phagocytosis in principle by: expressing don’t-eat-me signals, not releasing find-me signals, not exposing eat-me signals, and not binding opsonins. However, we don’t actually know the extent to which healthy cells are phagocytosed as part of physiological turnover of tissues – and it would be useful to know this. Parts of healthy cells are phagocytosed, for example, synapses (see below) and it has been suggested that healthy microglia phagocytose their own processes (297), but we don’t know the extent of cellular self-eating generally. Healthy self-cells may be aberrantly phagocytosed in some pathologies associated with inflammation (see below). For example, activation of macrophages with CpG DNA, interferon-γ, and anti-interleukin-10 receptor antibody results in macrophage phagocytosis of live T cells, B cells and myeloid cells via ICAM-1 or VCAM-1-mediated adhesion (298).
Excess Cells During Development and After Inflammation
A variety of cells, especially during development, become redundant and can be considered ‘excess’ cells, needing removal by phagocytosis. In some cases, the phagocytosis is induced by the cell undergoing apoptosis, but in other cases the excess cells are phagocytosed when alive, which raises the question of what signals mediate this phagocytosis of apparently healthy cells. For example, excess neurons in the retina are phagocytosed alive by microglia during development apparently via tagging new-born neurons with C1q and microglial phagocytosis of these via the CR3 receptor (299).
Many myeloid cells expand during infection and become redundant and potentially damaging post-infection, so are preferentially phagocytosed, usually as a result of phosphatidylserine exposure on live cells. For example, macrophages can induce phosphatidylserine exposure on live neutrophils, which enables the macrophages to phagocytose these neutrophils if the macrophages also release MGF-E8 to bind the phosphatidylserine and vitronectin receptor (300). Inflammatory-activated neutrophils expose phosphatidylserine and oxidise this to lysophosphatidylserine, which induces phagocytosis of such neutrophils by macrophages (301). However, activated neutrophils also desialylate their surface (302), and macrophages can release calreticulin that binds to desialylated neutrophils to induce macrophage phagocytosis of such neutrophils (47). Antigen recognition by live CD8+ T cells induces phosphatidylserine exposure on these cells (303), and the phosphatidylserine receptor TIM-4 has been shown to mediate phagocytosis of antigen-specific T cells post-infection (304).
Apoptotic Cells
Apoptosis is cell death mediated by Bcl-2 homologous proteins and/or caspase activation, resulting in phosphatidylserine exposure on an intact plasma membrane, which normally suppresses inflammation (305, 306). Phosphatidylserine on apoptotic cells is then bound by opsonins MFG-E8 or Gas6, which then bind and activate the phagocytic vitronectin receptors and MerTK, respectively (Table 3). Other phagocytic receptors bind phosphatidylserine directly, such as Tim4, which acts in conjunction with MerTK to induce phagocytosis of apoptotic cells (Table 3). The full range of phagocytic receptors binding to phosphatidylserine on apoptotic cells includes BAI-1, CD300f, CD36, LOX-1, PSR, RAGE, Stablin-1 & -2, TIM-1 & -4 and TREM2 (Table 2). The full range of opsonins binding to phosphatidylserine on apoptotic cells includes Annexin A1, β2-GP1, calreticulin, CCN1, Gas6, MFG-E8, protein S, C1q, C3b & iC3b, MBL, SP-A, TSP-1 (Tables 2 and 3). Other eat-me signals mediating phagocytosis of apoptotic cells include: calreticulin, oxidised phospholipids, DNA and pentraxin-3 (Table 2).
Find-me signals released from apoptotic cells includes proteins (chemokines): fractalkine, monocyte chemoattractant protein 1 (MCP-1), interleukin-8 (IL-8), S19 ribosomal protein dimer (RP S19), endothelial monocyte-activating polypeptide II (EMAPII) and split human tyrosyl-tRNA synthetase (mini TyrRS), lipids: lysophosphatidycholine and sphingosine-1-phosphate (S1P), and nucleotides: ATP, ADP, UTP, UDP (Table 1). Don’t-eat-me signals and negative opsonins found to block phagocytosis of apoptotic cells include: CD47, sialic acid, annexin A5, histone H3, HMGB1, PAI-1, PTX3, sRAGE, suPAR and vitronectin (Tables 4 and 5). CD31 was reported to act as a don’t-eat-me (or detachment) signal, but switch to an eat-me (or attachment) signal as a result of a conformational change induced by apoptosis (259). Analogously, apoptosis can induce desialylation of the apoptotic cell membrane, thereby removing a don’t-eat-me signal (sialic acid residues) and inducing an eat-me signal (asialoglycans) (111). Apoptosis can also expose the eat-me signal calreticulin, while reducing exposure of the don’t-eat-me signal CD47 (58).
Necrotic Cells and Cellular Debris
Necrotic cells are defined by rupture of the plasma membrane, causing leakage of cellular content that induces inflammation, and if these necrotic cells are not removed rapidly, break up into cellular debris. Phosphatidylserine exposure occurs on all dead and dying cells and debris, but earlier on apoptotic cells, as they have specific mechanisms to induce this. Phosphatidylserine exposure on necrotic cells normally mediates phagocytosis of these cells (307), however, other phagocytic signals from necrotic cells differ from those coming from apoptotic cells, reviewed in (33) and (308). Find-me signals from necrotic cells include formyl-peptides, nucleotides, complement C3a and C5a (Table 1). Opsonins shown to mediate phagocytosis of necrotic cells and debris, include complement C1q, C3b and C4b, antibodies IgG and IgM, MBL, pentraxins CRP, SAP and PTX3, AnxA1 and TSP1 (Table 3). Necrosis can occur by different mechanisms, including necroptosis, pyroptosis and ferroptosis, but it is unclear whether phagocytosis of these cells occurs via different signals.
The biological remnants of cell death are collectively referred to as cellular debris, and their efficient clearance by phagocytes is thought to be important to avoid chronic inflammation and autoimmunity (33). Phosphatidylserine is exposed on cell debris and is probably the main eat-me signal. However, other myelin lipids such as sulfatide and sphingomyelin may contribute to TREM2-mediated phagocytosis of myelin debris by microglia (309). CD47 on myelin debris can act as a don’t-eat-me signal to block its uptake (310). Schwann cells, which myelinate axons in the peripheral nervous system, upregulate phagocytic receptors Axl and MerTK after nerve damage to clear the resulting myelin debris, possibly via phosphatidylserine exposure on the debris (311). Phagocytes can also clear myelin debris via the complement receptor CR3 (312) or the scavenger-receptor-AI/II (159). Phagocytic clearance of retinal debris occurs via αVβ3 and phosphatidylserine receptor (PSR) binding to phosphatidylserine on the debris (313). However, phagocytic uptake of debris from lysed cells by non-professional phagocytes, including fibroblasts and epithelial cells, occurred via the opsonin ApoJ (clusterin) binding histones debris and activating uptake through the phagocytic receptors megalin and LRP (142). Galectin-3 can also act as an opsonin for desialylated neuronal debris, facilitating uptake into microglia via the MerTK receptor (49). Microglia may be recruited to debris via fractalkine and the fractalkine receptor CX3CR1 (314).
Senescent and Ageing Cells
Cellular senescence refers to irreversible loss of proliferation or other cellular function with age or stress. Senescent cells accumulate in aged organisms, and may contribute to loss of function with age. However, some cells, such as erythrocytes, senesce rapidly as part of physiological turnover. CD47 undergoes a conformation change on senescent erythrocytes, coupled with reduced CD47 expression, promoting the phagocytosis of erythrocytes by macrophages (250, 315). Senescent erythrocytes also expose phosphatidylserine and desialylated membrane glycoproteins - both of which can enhance phagocytosis (316). Neutrophils senescence is even more rapid (24 hours), and is associated with phosphatidylserine exposure, opsonisation by MFG-E8 and annexin A1, and phagocytosis by activated macrophages in the bone marrow (300). Phagocytosis of senescent neutrophils in the spleen depends on phosphatidylserine-binding opsonin Gas6 and its phagocytic receptor MerTK (317). Calreticulin is exposed by aging neutrophils, promoting phagocytosis via the phagocytic receptor LRP1 (47, 71). Senescent platelets appear to be phagocytosed as a result of desialylation exposing galactose residues (52). Cells in the post-partem uterus senesce and are removed by macrophages, but the signals driving this are unknown (318). Similarly, macrophages clear senescent cancer cells by unknown phagocytic signals (319).
In the aged brain of mice, there appears to be excessive microglial phagocytosis of synapses and neurons. This appears to be partly mediated by complement, as C3 knockout mice lost less synapses and neurons in the hippocampus, and had improved learning in memory (320). TREM2 knockout mice also had reduced neuronal loss in the hippocampus and substantia nigra (321), suggesting that excessive microglial phagocytosis of neurons may contribute to aging-induced neuronal loss.
Cancer Cells
Cancer cells have increased expression of both ‘eat-me’ signals and ‘don’t-eat-me’ signals, though expression varies between cancer cell-types and changes with tumour progression (322). Many human cancer cells have the eat-me signal calreticulin on the surface (possibly due to ER stress, or possibly a tumour suppressor mechanism), but overexpress the don’t-eat-me signal CD47 to prevent host phagocytes from phagocytosing the cancer cells (323). Thus, function blocking antibodies and peptides to CD47 or its receptor SIRPα induce macrophage phagocytosis of live cancer cells in culture and in vivo, and are being developed as a potential treatment for multiple cancers (322, 324). However, some tumour cells overexpress stanniocalcin 1 (STC1), which obstructs calreticulin exposure on the cell surface, reducing phagocytosis of the cancer cells (325). Macrophages may also recognise and clear haematopoietic cancer cells via SLAMF7 heterodimerising between cancer cells and macrophage, and activating CR3 on the macrophage, resulting in phagocytic clearance of the cancer cell (132). MHC-I expression on cancer cells may act as another ‘don’t-eat-me’ signal when bound by the receptor LILRB1 on phagocytes, as disruption of this interaction increases clearance of tumour cells (254). Antibodies bound to cancer cell antigens can induce phagocytosis of the live cancer cell via Fc receptors on phagocytes (185). Cancer cells often have increased levels of cell surface sialylation (326), which potentially inhibits phagocytosis of these cells. This inhibition may be partly mediated by sialylation of CD24 on cancer cells, which inhibits macrophage phagocytosis via activating Siglec-10 on macrophages (247).
Infected Cells
Live mammalian cells infected with virus, bacteria or other intracellular pathogens can release signals inducing phagocytosis of the infected cell by phagocytes, thereby limiting infection of the host organism (327). For example, E. coli-infection (which presumably involves infected host-cells) induced nucleotides ATP and UDP release from host cells, which stimulated macrophage phagocytosis and reduced bacterial loads in vivo (328, 329), and similarly for live host infected with vesicular stomatitis virus (330). Chemokines that attract phagocytes are released by host cells infected with influenza (331), hepatitis C virus (332), and the bacterium O. tsutsugamushi (333). Live HIV-infected cells were shown to externalise phosphatidylserine, which induced macrophages to phagocytose these cells, via MerTK, Gas6 and Protein S (334). Infection of mouse brain with adenovirus caused phosphatidylserine exposure on live brain cells, with subsequent phagocytosis by microglia of the infected cells mediated by MerTK (335). Similarly, human cells infected by Chlamydia rapidly and reversibly exposed phosphatidylserine, induced macrophages to phagocytose the live, infected cells (336). Mycobacterium tuberculosis and cytomegalovirus infection caused calreticulin exposure on infected cells (337, 338).
Red blood cells infected with parasitic Plasmodium reduced CD47 levels to induce their phagocytic removal (339). Influenza infection induces desialylation of infected cells, which increases phagocytosis of the infected cells (340). Infected cells rapidly cause alternative complement pathway activation at their surface, which enables binding to phagocytes (341). West Nile virus infection of neurons induced complement tagging of the neurons, and complement-mediated phagocytosis of the live, infected neurons or synapses by microglia in culture and in vivo (342).
Synapses
The phagocytosis of synapses is required for brain development and synaptic plasticity, where less active synaptic connections are phagocytosed by neighbouring microglia and astrocytes (155, 343). Synapses can facilitate phagocytosis by the release of the find-me signal fractalkine to recruit microglia through CX3CR1 binding, so CX3CR1 knock out mice have both reduced synaptic pruning and impaired synapse maturation (35). ATP also acts as a find-me signal leading to the migration of microglia to stressed or damaged neurons or synapses via P2Y12 receptor binding (29), and this may contribute to microglial phagocytosis of synapses during experience-dependent plasticity (344). Active synapses express CD47 to inhibit their phagocytosis, and knock-out of CD47 in neurons or its receptor SIRPα in microglia increases phagocytosis of active synapses by microglia (345). To direct phagocytosis of inactive synapses, complement opsonins C1q and iC3b have been reported to mark synapses for pruning, so knockout of C1q, C3 or CR3 prevents synaptic pruning during development or ageing (346). Candidate eat-me signals for complement-mediated opsonisation of synapses include asialoglycans (53) and phosphatidylserine (82).
Astrocytes also contribute to pruning via the phagocytic receptors Megf10 and MerTK, all being able to bind to, either directly or indirectly, to C1q (343). The opsonin ApoE can also affect astrocyte phagocytosis of synapses, depending on the isoform (141). The microglial phagocytic receptor TREM2 mediates synaptic pruning, as TREM knockout mice have reduced microglial internalisation of synapses and increased synaptic density (347). Excessive microglial phagocytosis of synapses may contribute to neurodegeneration (348, 349), emphasising the clinical importance for better understanding the phagocytic code regulating synapse removal.
Consequences of Phagocytosis via Different Signals: Reprogramming, Killing, Inflammation and/or Antigen Presentation
What are the consequences for the phagocyte following phagocytosis mediated by different phagocytic signals? The phagocyte needs to decide what to do with what has been phagocytosed, and this decision is influenced by the signals mediating the phagocytosis. An immediate question the phagocyte needs to address is whether to try to kill what is in the phagosome - if it has a live pathogen, then it needs to kill it rapidly, although doing so may also damage the phagocyte and surrounding host cells. Phagocytosis mediated by CR3, Fc receptors and TREM2 activates the NADPH oxidase to generate superoxide, hydrogen peroxide and derivative species in the phagosome, which kill enclosed cells and activate proteases (350). The hydrogen peroxide produced can also activate the phagocyte, promoting inflammation (351). If the phagosome contains a pathogen, it may activate pattern recognition receptors present. However, phagocytosis mediated by phosphatidylserine and phosphatidylserine receptors normally inhibits inflammatory activation of the phagocyte and antigen presentation by the phagocyte (352). For example, activation of TAM receptors, such as MerTK, is anti-inflammatory by multiple mechanisms (353), whereas phagocytosis mediated by calreticulin can promote antigen presentation by the phagocyte (354). Phagocytosis of non-apoptotic, live self-cells results in much slower digestion of the phagocytosed cells (298), possibly because lysosomal enzymes can’t get into the live cell. Phagocytosis of cells can also reprogram phagocytes via changes in epigenetics, transcription, expression and release of multiple factors (355–357). Most targets are phagocytosed via the eat-me signal phosphatidylserine, but the particular opsonin bridging phosphatidylserine to particular phagocytic receptors determines the phagocytes response. For example, Del-1-mediated phagocytosis of phosphatidylserine-exposed neutrophils in inflamed tissue reprogrammes the phagocytosing macrophages into pro-resolution phenotype, for example, secreting anti-inflammatory TGFβ (171).
Pathologies Involving Phagocytosis and Their Potential Treatment
In various pathologies, phagocytosis plays beneficial roles. During pathogenic infection, phagocytosis of the infectious agent (and of infected cells) via innate and adaptive immunity is beneficial, and insufficient phagocytosis can enable disease (3). Thus, upregulating phagocytosis, or opsonising inactivated pathogens with immunogenic opsonins [e.g. calreticulin (133)] that promote both phagocytosis and antigen presentation by dendritic cells, may be therapeutically beneficial. The autoimmune disease lupus may be caused by insufficient phagocytosis of apoptotic cells, resulting in secondary necrosis that promotes presentation of self-antigens and thus autoimmunity (358). In multiple sclerosis, myelin degeneration causes an accumulation of extracellular myelin, and in models of the disease, microglial phagocytosis of myelin debris promotes myelin regeneration through activation of oligodendrocyte precursor cells (359). Following ischemic stroke, microglial phagocytosis can be beneficial in clearing debris and restoring tissue homeostasis (360). In cancer, insufficient phagocytosis of cancer cells due their overexpression of CD47, and animal models benefit from increasing phagocytosis of cancer cells using function blocking antibodies to CD47 or its receptor SIRPα (361). In atherosclerosis, defective phagocytosis of apoptotic cells within atherosclerotic lesions results in inflammation and formation of a necrotic core, which can then drive acute cardiovascular injury (362). Chédiak–Higashi syndrome is also characterised by insufficient phagocytosis (363). For all these cases where phagocytosis helps combats disease, augmenting phagocytosis may be therapeutically beneficial – providing this augmentation does not promote excessive phagocytosis of healthy cell targets, or otherwise promote inflammation, which might itself cause disease.
In other pathologies, phagocytosis plays detrimental roles. In Alzheimer’s disease, excessive phagocytosis of synapses and neurons by microglia may contribute to disease progression, and blocking phagocytosis can ameliorate animal models of the disease (348, 349). Excessive phagocytosis of neurons and/or synapses within the brain has been linked to several other neuropathologies, including Parkinson’s disease, frontotemporal dementia (364), schizophrenia (166), glaucoma (365) and disease resulting from West Nile Virus infection (342). In such cases, inhibiting (rather than augmenting) phagocytosis may be therapeutically beneficial. However, therapies inhibiting detrimental phagocytosis should be designed with the knowledge that inhibiting phagocytosis can cause pathology per se (and vice versa for therapies augmenting beneficial phagocytosis).
Other diseases where phagocytosis has been suggested to be dysfunctional include: hemophagocytic lymphohistiocytosis, arthritis, osteoporosis, cystic fibrosis, chronic obstructive pulmonary disease, autoimmune hepatitis, fatty liver disease, primary biliary cholangitis, type 1 diabetes and wound healing (308). For such pathologies, a full understanding of the phagocytic code is important.
Open Questions on the Logic of the Phagocytic Code
When a phagocyte encounters another cell, it may receive several different phagocytic signals encouraging it to phagocytose or not to phagocytose the cell. How does the phagocyte integrate this information and make the crucial decision to eat or not to eat? And is there any simplifying logic to the mass of phagocytic signals? The short answer is that we do not know yet, but below we speculate on some open questions (summarised in Figure 8), and make a few tentative generalisations.
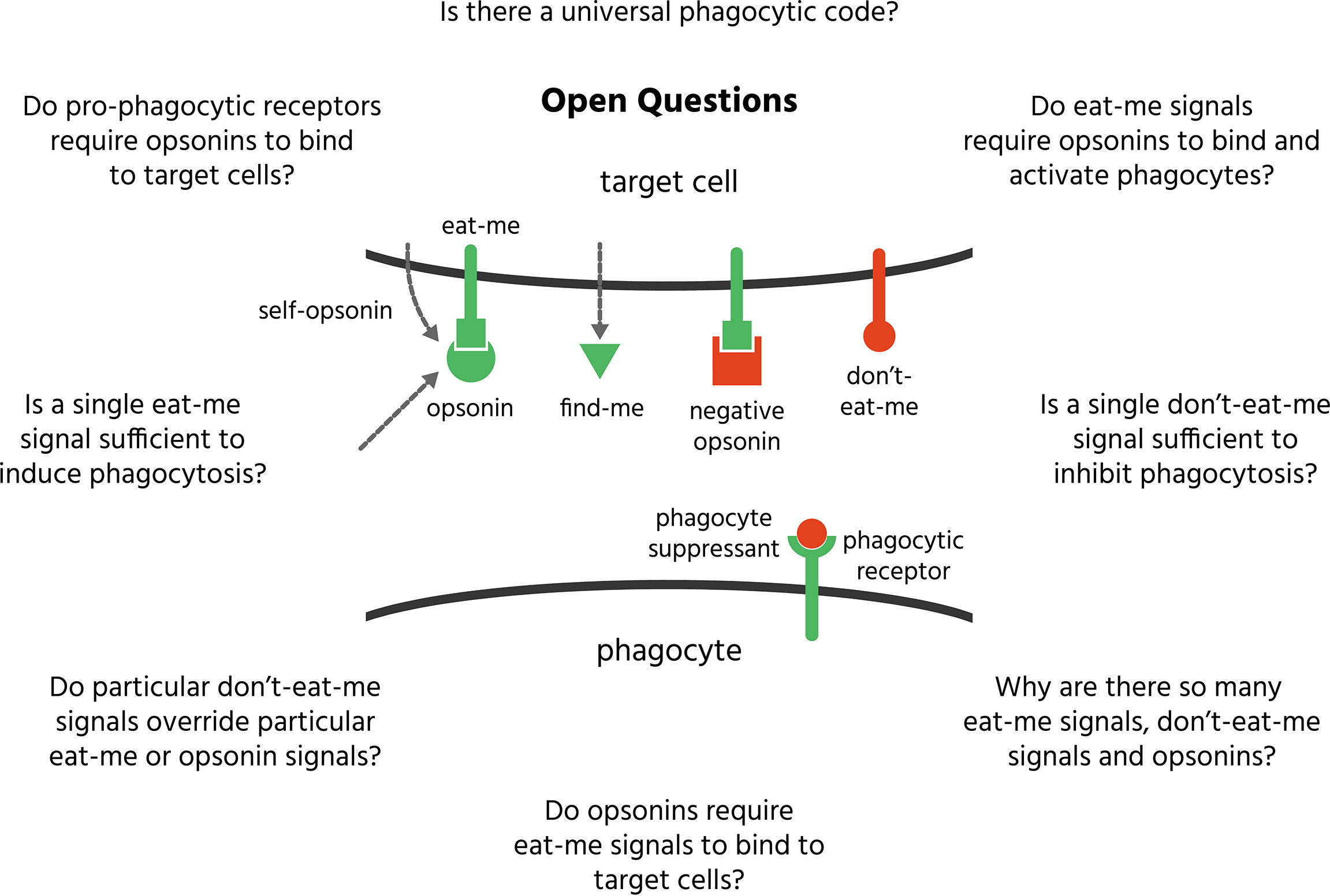
Figure 8 Open questions on the phagocytic code. Multiple signals present on the target cell surface stimulate or inhibit phagocytosis of these targets by phagocytes. How these signals combine to determine whether the target is phagocytosed or not is known as the phagocytic code. However, there are multiple open questions as to how this code works, some of which are listed here and discussed in the text.
Is a Single Eat-Me Signal Sufficient to Induce Phagocytosis?
Given the range of eat-me signals, is any one sufficient to trigger phagocytosis, or are particular combinations required?
Phosphatidylserine exposure is sufficient to induce phagocytosis of some cells but not others (366). Phosphatidylserine exposure might be insufficient in certain cases due to the presence of don’t-eat-me signals, or the absence of phosphatidylserine-binding opsonins or other co-stimulatory signals. Alternatively, the quantity of phosphatidylserine exposure could be key, i.e. high levels of phosphatidylserine exposure may induce phagocytosis regardless of other don’t-eat-me signals/opsonins present, while low levels may be insufficient without opsonins, additional eat-me signals, or loss of don’t-eat-me signals.
There is some indication that both calreticulin and phosphatidylserine can be required for phagocytosis in some cells and conditions. Gardai et al. (58) found that calreticulin knockout prevented phagocytosis of phosphatidylserine-exposing apoptotic cells, and addition of exogenous calreticulin restored phagocytosis, but this phagocytosis remained phosphatidylserine-dependent. Similarly, we found that microglial phagocytosis of stressed neurons in culture required both calreticulin and phosphatidylserine: calreticulin was constitutively expressed at a low level on cerebellar neurons, and stress induced phosphatidylserine exposure, but blocking either calreticulin or phosphatidylserine prevented phagocytosis (59). The requirement for phosphatidylserine could be overcome by adding high levels of calreticulin, and microglial activation with lipopolysaccharide (LPS) was required for phagocytosis of neurons to occur. Blocking CD47 with anti-CD47 antibodies was not sufficient to induce phagocytosis with these cells, but Gardai et al. (58) found that blocking CD47 was sufficient to induce phagocytosis of viable cells, but this phagocytosis required surface calreticulin. They found that calreticulin was present at low levels on a range of cells, but apoptosis induced calreticulin clumping on the cell surface, together with phosphatidylserine, and away from CD47. Thus, it is possible that clumping of eat-me signals together, and away from don’t-eat-me signals, promotes phagocytosis. However, this hypothesis has not been causally tested.
Is desialylation of cells sufficient to induce phagocytosis? It was reported that desialylation of apoptotic lymphocytes or non-apoptotic lymphoblasts increased their engulfment, whereas desialylation of healthy, resting lymphocytes did not increase their engulfment by monocyte-derived macrophages (54). Thus, desialylation is sufficient to induce phagocytosis of some cells but not others, and can change with conditions. Resting lymphocytes may lack an eat-me signal that is present on apoptotic lymphocytes (e.g. phosphatidylserine) or lymphoblasts (e.g. calreticulin), and the asialoglycans generated by desialylation may be insufficient to induce phagocytosis without asialoglycan opsonins (e.g. calreticulin, C1q, Gal-3 and MBL) being present.
Is a Single Don’t-Eat-Me Signal Sufficient to Inhibit Phagocytosis?
Is one don’t-eat-me signal sufficient to prevent phagocytosis, making the others redundant? Or is removing one don’t-eat-me signal sufficient to induce phagocytosis? Barkal et al. (254) found that CD47-blocking antibodies induced phagocytosis of many cancer cell lines, while knockout of MHC-I had only a modest effect, however, cell lines resistant to anti-CD47 had high MHC-I expression, and MHC-I knockout substantially increased phagocytosis in these cell line. Similarly, Barkal et al. (247) found that CD24 was expressed in ovarian and breast cancer cells, together with CD47, such that anti-CD24 and anti-CD47 antibodies synergistically induced phagocytosis of these cells by macrophages. Thus, for viable cancer cells, the signals appear to be partially redundant, so that removing more than one don’t-eat-me signal stimulates phagocytosis at least additively, but varying with cell type, depending on their expression of these signals. This emphasises that therapeutic strategies targeting phagocytosis need to consider whether the target is redundant.
Do Particular Don’t-Eat-Me Signals Override Particular Eat-Me or Opsonin Signals?
There is insufficient evidence to answer this important question for particular eat-me, opsonins and don’t-eat-me signals. However, in order for a specific don’t-eat-me signal to override a specific eat-me signal, the downstream signalling has to interact somewhere before the phagocytic machinery, and the inhibitory signalling must be stronger (and vice versa to induce phagocytosis). An important subset of phagocytic receptors (Fcγ receptors, CR3, TREM2, Dectin-1, CLEC2, MEFG10, Jedi-1) for eat-me signals and opsonins induce phagocytosis via ITAM domains and the tyrosine kinase Syk; while most receptors for don’t-eat-me signals (Siglec receptors for sialoglycans and CD24; LILRB1 for MHC-1; SIRPα for CD47) inhibit phagocytosis via ITIM domains and tyrosine phosphatases that reverse Syk signalling (367). Thus, don’t-eat-me signals that antagonise Syk signalling may potentially block eat-me and opsonin signalling mediated by the Syk pathway, but not eat-me and opsonin signalling not mediated by Syk, such as that mediated via MerTK or αvβ3. This is consistent with the finding that CD47/SIRPα signalling can inhibit phagocytosis mediated by Fcγ receptors and CR3 (282).
Do Opsonins Require Eat-Me Signals to Bind to Target Cells?
Many opsonins require eat-me signals on the surface of the cell to bind to, but do all of them? There is a rational for opsonin binding to mammalian cells requiring an eat-me signal, because otherwise: how could opsonin binding be specific to target cells rather than healthy cells? The target cell must be displaying something different from healthy cells in order for the opsonin to specifically bind to it, and whatever that difference is, it can be called an eat-me signal. A potential exception is C3b, which appears to bind randomly to cells. However, the specificity of C3b regarding phagocytic regulation arises from: i) the C3 convertase that generates C3b from C3 is specifically located on the target cell by the classical, alternate (C3b) or lectin (MBL) pathway by specific binding to the cell of antibodies and/or C1q, C3b or MBL respectively, and ii) complement factors that specifically bind to target cells and regulate C3b production or disposal, e.g. factor H specifically binds sialylated surfaces and blocks C3b production and induces C3b conversion to iC3b (368). Thus, C3b does not itself bind to an eat-me signal, but it does require an eat-me signal to generate it on the target cell surface and the absence of a don’t-eat-me signal to survive there. Antibody binding to cells does not require a conventional eat-me signal, however, it does require a new epitope, which fits the criteria for an eat-me signal. All other opsonins for mammalian cells either have an identified eat-me signal that they bind to or are presumed to have an as yet unidentified eat-me signal (Table 3).
Do Eat-Me Signals Require Opsonins to Bind and Activate Phagocytes?
Some eat-me signals require opsonins to induce phagocytosis, whilst others don’t, and some eat-me signals can induce phagocytosis in both opsonin-dependent and independent manners (see Tables 2 and 3). For example, phosphatidylserine can bind some phagocytic receptors directly, but also can activate phagocytosis indirectly via many different opsonins. Similarly, asialoglycans can activate some receptors directly, but is generally recognised via multiple opsonins. Calreticulin acts as a self-opsonin (i.e. released from and binding back onto the target cell) that may activate LRP1 directly, but also induces phagocytosis via binding the opsonin C1q. Annexin A1 and pentraxin-3 also may act as self-opsonin that activate phagocytic receptors directly.
Do Pro-Phagocytic Receptors Require Opsonins to Bind to Target Cells?
Most pro-phagocytic receptors on phagocytes bind to target cells via opsonins bound to eat-me signals on the target cell (Tables 2 and 3). Exceptions are some of the phosphatidylserine receptors, TIM-4, BAI1, PSR and TREM2, which can bind to phosphatidylserine directly (Table 2). Note that many opsonins are upregulated in inflammatory states to increase phagocytosis of particular targets.
If most eat-me signals require an opsonin, and most opsonins require an eat-me signal, then an eat-me signal or opsonin alone may not be sufficient to induce phagocytosis. Experimentally, it may not make sense to test whether an eat-me signal or opsonin alone induces phagocytosis. Alternatively, opsonins may be there to amplify the eat-me signal by making it visible or readable by phagocytes, or different opsonins for the same eat-me signal (e.g. phosphatidylserine) may convey different signals to the phagocyte or activate particular types of phagocyte. Opsonins might also potentially act as hub integrating signals by binding several different signals, other opsonins and/or phagocytic receptors to coordinate a response.
Why Are There so Many Phagocytic Signals?
We now know of over 50 mammalian phagocytic signals. Why so many? Potential explanations for multiple eat-me signals include: i) different eat-me signals can induce different downstream events within the phagocyte once the target cell is phagocytosed, ii) different eat-me signals can induce different phagocytes to phagocytose the cell, and iii) cells are phagocytosed for many different reasons (apoptosis, infection, senescence, etc) that may trigger different eat-me signals to be exposed.
Why are there so many opsonins? Most opsonins work by binding an eat-me signal on the target and a phagocytic receptor on the phagocyte. Thus, because there are several different eat-me signals and phagocytic receptors, multiple opsonins are required to couple them together. Because different eat-me signals can represent different cellular states, and different phagocytic receptors trigger different events within the phagocyte and different fates for the target cells, consequently, different opsonins have somewhat different functions.
Is There Is a Universal Phagocytic Code?
It is clear that there are somewhat different phagocytic codes for different targets, different phagocytes and different conditions. Unsurprisingly, the phagocytic signals used to remove different phagocytic targets differ, because these target cells are undergoing different cellular processes to identify them as in need of removal. These different targets may also require different treatment by phagocytes, requiring different signals on the target cell (see section on ‘Consequences of Phagocytosis Via Different Signals’). Different phagocytes may use different phagocytic codes simply as a result of expressing different phagocytic receptors detecting different signals. A rationale for this would be that different phagocytes specialise in the phagocytosis of different targets – for example, neutrophils specialise in phagocytosis of bacteria and therefore express relevant phagocytic receptors to detect such targets. The phagocytic code can change somewhat in different conditions as a result of phagocytes changing the phagocytic receptors they express. For example, during inflammation, different cytokines and inflammatory stimuli can drive the expression of different phagocytic receptors and opsonins (367). And the phagocytosis of targets can itself drive the expression of different phagocytic receptors (369).
If different phagocytes express different receptors (for find-me, eat-me and don’t-eat-me signals and opsonins), and in different conditions, then it follows that all phagocytic signals are conditional – not only on which phagocytic receptors are expressed on the particular phagocyte in a given circumstance, but also on expression levels and activation states of those receptors and their downstream signalling components. However, changes in the phagocytic code for different targets, phagocytes and conditions are limited, and there are commonalities - such as phosphatidylserine and asialoglycans being eat-me signals for most targets.
Conclusion
It should be clear from the above that we are only just beginning to understand the phagocytic code, its consequences, how it goes wrong in disease and how to manipulate it for treatment. However, seeing the interaction between phagocytes and targets in terms of the phagocytic code helps frame new questions, that will in turn help elucidate this important interaction.
Author Contributions
Text and tables contained within this review were written and reviewed by TC, JD, AP and GB. Figures were designed by AP, GB, JD and TC, and created by AP. All authors contributed to the article and approved the submitted version.
Funding
TC received funding from the Biotechnology and Biological Sciences Research Council UK. JD received funding from Eli Lilly and the Biotechnology and Biological Sciences Research Council UK. GB received funding from and from the Medical Research Council UK (No. MR/L010593). This project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115976. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA. The authors declare that they have no competing interests.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
GLOSSARY

References
1. Bratosin D, Mazurier J, Tissier JP, Estaquier J, Huart JJ, Ameisen JC, et al. Cellular and Molecular Mechanisms of Senescent Erythrocyte Phagocytosis by Macrophages. A Rev Biochim (1998) 80(2):173–95. doi: 10.1016/S0300-9084(98)80024-2
2. Rosales C, Uribe-Querol E. Phagocytosis: A Fundamental Process in Immunity. BioMed Res Int (2017) 2017:9042851. doi: 10.1155/2017/9042851
3. Uribe-Querol E, Rosales C. Control of Phagocytosis by Microbial Pathogens. Front Immunol (2017) 8:1368. doi: 10.3389/fimmu.2017.01368
4. Medina CB, Ravichandran KS. Do Not Let Death do Us Part: “Find-Me” Signals in Communication Between Dying Cells and the Phagocytes. Cell Death Differ (2016) 23(6):979–89. doi: 10.1038/cdd.2016.13
5. Zhang C, Wang C, Li Y, Miwa T, Liu C, Cui W, et al. Complement C3a Signaling Facilitates Skeletal Muscle Regeneration by Regulating Monocyte Function and Trafficking. Nat Commun (2017) 8(1):1–12. doi: 10.1038/s41467-017-01526-z
6. Marshall KM, He S, Zhong Z, Atkinson C, Tomlinson S. Dissecting the Complement Pathway in Hepatic Injury and Regeneration With a Novel Protective Strategy. J Exp Med (2014) 211(9):1793–805. doi: 10.1084/jem.20131902
7. Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, et al. Alternative Complement Pathway Activation Is Essential for Inflammation and Joint Destruction in the Passive Transfer Model of Collagen-Induced Arthritis. J Immunol (2006) 177(3):1904–12. doi: 10.4049/jimmunol.177.3.1904
8. Nishimura T, Horino K, Nishiura H, Shibuya Y, Hiraoka T, Tanase S, et al. Apoptotic Cells of an Epithelial Cell Line, AsPC-1, Release Monocyte Chemotactic S19 Ribosomal Protein Dimer. J Biochem (2001) 129(3):445–54. doi: 10.1093/oxfordjournals.jbchem.a002876
9. Shibuya Y, Shiokawa M, Nishiura H, Nishimura T, Nishino N, Okabe H, et al. Identification of Receptor-Binding Sites of Monocyte Chemotactic S19 Ribosomal Protein Dimer. Am J Pathol (2001) 159(6):2293–301. doi: 10.1016/S0002-9440(10)63079-9
10. Yamamoto T. Roles of the Ribosomal Protein S19 Dimer and the C5a Receptor in Pathophysiological Functions of Phagocytic Leukocytes. Pathol Int (2007) 57(1):1–11. doi: 10.1111/j.1440-1827.2007.02049.x
11. Umeda Y, Shibuya Y, Semba U, Tokita K, Nishino N, Yamamoto T. Guinea Pig S19 Ribosomal Protein as Precursor of C5a Receptor-Directed Monocyte-Selective Leukocyte Chemotactic Factor. Inflammation Res (2004) 53(11):623–30. doi: 10.1007/s00011-004-1302-0
12. Hou Y, Plett PA, Ingram DA, Rajashekhar G, Orschell CM, Yoder MC, et al. Endothelial-Monocyte-Activating Polypeptide II Induces Migration of Endothelial Progenitor Cells Via the Chemokine Receptor CXCR3. Exp Hematol (2006) 34(8):1125–32. doi: 10.1016/j.exphem.2006.05.021
13. Traves SL, Tudhope SJ, Fenwick PS, Belchamber KBR, Russell REK, Barnes PJ, et al. Enhanced Monocyte Migration to CXCR3 and CCR5 Chemokines in COPD Claudia Costa1. Eur Respir J (2016) 47(4):1093–102. doi: 10.1183/13993003.01642-2015
14. Kao J, Ryan J, Brett G, Chen J, Shen H, Fan YG, et al. Endothelial Monocyte-Activating Polypeptide II. A Novel Tumor-Derived Polypeptide That Activates Host-Response Mechanisms. J Biol Chem (1992) 267(28):20239–47. doi: 10.1016/S0021-9258(19)88692-1
15. Knies UE, Behrensdorf HA, Mitchell CA, Deutsch U, Risau W, Drexler HCA, et al. Regulation of Endothelial Monocyte-Activating Polypeptide II Release by Apoptosis. Proc Natl Acad Sci USA (1998) 95(21):12322–7. doi: 10.1073/pnas.95.21.12322
16. Raoof M, Zhang Q, Itagaki K, Hauser CJ. Mitochondrial Peptides are Potent Immune Activators That Activate Human Neutrophils Via FPR-1. J Trauma - Inj Infect Crit Care (2010) 68(6):1328–32. doi: 10.1097/TA.0b013e3181dcd28d
17. Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, et al. CX3CL1/Fractalkine Is Released From Apoptotic Lymphocytes to Stimulate Macrophage Chemotaxis. Blood (2008) 112(13):5026–36. doi: 10.1182/blood-2008-06-162404
18. Segundo C, Medina F, Rodríguez C, Martínez-Palencia R, Leyva-Cobián F, Brieva JA. Surface Molecule Loss and Bleb Formation by Human Germinal Center B Cells Undergoing Apoptosis: Role of Apoptotic Blebs in Monocyte Chemotaxis. Blood (1999) 94(3):1012–20. doi: 10.1182/blood.V94.3.1012.415k05_1012_1020
19. Paolicelli RC, Bisht K, Tremblay M-È. Fractalkine Regulation of Microglial Physiology and Consequences on the Brain and Behavior. Front Cell Neurosci (2014) 8:129. doi: 10.3389/fncel.2014.00129
20. Gunner G, Cheadle L, Johnson KM, Ayata P, Badimon A, Mondo E, et al. Sensory Lesioning Induces Microglial Synapse Elimination Via ADAM10 and Fractalkine Signaling. Nat Neurosci (2019) 22(7):1075–88. doi: 10.1038/s41593-019-0419-y
21. Panek CA, Ramos MV, Mejias MP, Abrey-Recalde MJ, Fernandez-Brando RJ, Gori MS, et al. Differential Expression of the Fractalkine Chemokine Receptor (CX3CR1) in Human Monocytes During Differentiation. Cell Mol Immunol (2015) 12(6):669–80. doi: 10.1038/cmi.2014.116
22. Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and Fractalkine Receptor (CX3CR1) Distribution in Spinal Cord and Dorsal Root Ganglia Under Basal and Neuropathic Pain Conditions. Eur J Neurosci (2004) 20(5):1150–60. doi: 10.1111/j.1460-9568.2004.03593.x
23. Hammond ME, Lapointe GR, Feucht PH, Hilt S, Gallegos CA, Gordon CA, et al. IL-8 Induces Neutrophil Chemotaxis Predominantly Via Type I IL-8 Receptors. J Immunol (1995) 155(3):1428–33.
24. Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, Tynan GA, et al. Fas/Cd95-Induced Chemokines can Serve as “ Find-Me” Signals for Apoptotic Cells. Mol Cell (2013) 49(6):1034–48. doi: 10.1016/j.molcel.2013.01.025
25. Peter C, Waibel M, Radu CG, Yang LV, Witte ON, Schulze-Osthoff K, et al. Migration to Apoptotic “Find-Me” Signals Is Mediated Via the Phagocyte Receptor G2a. J Biol Chem (2008) 283(9):5296–305. doi: 10.1074/jbc.M706586200
26. Lauber K, Bohn E, Kröber SM, Xiao Y, Blumenthal SG, Lindemann RK, et al. Apoptotic Cells Induce Migration of Phagocytes Via Caspase-3-Mediated Release of a Lipid Attraction Signal. Cell (2003) 113(6):717–30. doi: 10.1016/S0092-8674(03)00422-7
27. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J Interferon Cytokine Res Mary Ann Liebert Inc (2009) 29:313–25. doi: 10.1089/jir.2008.0027
28. Idzko M, Panther E, Sorichter S, Herouy Y, Berod L, Geissler M, et al. Characterization of the Biological Activities of Uridine Diphosphate in Human Dendritic Cells: Influence on Chemotaxis and CXCL8 Release. J Cell Physiol (2004) 201(2):286–93. doi: 10.1002/jcp.20070
29. Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, et al. The P2Y12 Receptor Regulates Microglial Activation by Extracellular Nucleotides. Nat Neurosci (2006) 9(12):1512–9. doi: 10.1038/nn1805
30. Lu Z, Elliott MR, Chen Y, Walsh JT, Klibanov AL, Ravichandran KS, et al. Phagocytic Activity of Neuronal Progenitors Regulates Adult Neurogenesis. Nat Cell Biol (2011) 13(9):1076–83. doi: 10.1038/ncb2299
31. Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Sphingosine Kinases, Sphingosine 1-Phosphate, Apoptosis and Diseases. Biochim Biophys Acta - Biomembr (2006) 1758(12):2016–26. doi: 10.1016/j.bbamem.2006.08.007
32. Wakasugi K, Schimmel P. Two Distinct Cytokines Released From a Human aminoacyl-tRNA Synthetase. Science (1999) 284(5411):147–51. doi: 10.1126/science.284.5411.147
33. Westman J, Grinstein S, Marques PE. Phagocytosis of Necrotic Debris At Sites of Injury and Inflammation. Front Immunol (2020) 10:3030. doi: 10.3389/fimmu.2019.03030
34. Nishiura H, Shibuya Y, Matsubara S, Tanase S, Kambara T, Yamamoto T. Monocyte Chemotactic Factor in Rheumatoid Arthritis Synovial Tissue. J Biol Chem (1996) 271(2):878–82. doi: 10.1074/jbc.271.2.878
35. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science (2011) 333(6048):1456–8. doi: 10.1126/science.1202529
36. Hoshiko M, Arnoux I, Avignone E, Yamamoto N, Audinat E. Deficiency of the Microglial Receptor CX3CR1 Impairs Postnatal Functional Development of Thalamocortical Synapses in the Barrel Cortex. J Neurosci (2012) 32(43):15106–11. doi: 10.1523/JNEUROSCI.1167-12.2012
37. Law S-H, Chan M-L, Marathe GK, Parveen F, Chen C-H, Ke L-Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int J Mol Sci (2019) 20(5):1149. doi: 10.3390/ijms20051149
38. Murakami N, Yokomizo T, Okuno T, Shimizu T. G2a Is a Proton-Sensing G-Protein-Coupled Receptor Antagonized by Lysophosphatidylcholine. J Biol Chem (2004) 279(41):42484–91. doi: 10.1074/jbc.M406561200
39. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides Released by Apoptotic Cells Act as a Find-Me Signal to Promote Phagocytic Clearance. Nature (2009) 461(7261):282–6. doi: 10.1038/nature08296
40. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 Channels Mediate “Find-Me” Signal Release and Membrane Permeability During Apoptosis. Nature (2010) 467(7317):863–7. doi: 10.1038/nature09413
41. Sandilos JK, Chiu Y-H, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, et al. Pannexin 1, an ATP Release Channel, Is Activated by Caspase Cleavage of Its Pore-Associated C-Terminal Autoinhibitory Region. J Biol Chem (2012) 287(14):11303–11. doi: 10.1074/jbc.M111.323378
42. Idzko M, Ferrari D, Eltzschig HK. Nucleotide Signalling During Inflammation. Nature (2014) 509(7500):310–7. doi: 10.1038/nature13085
43. Myrtek D, Idzko M. Chemotactic Activity of Extracellular Nucleotideson Human Immune Cells. Purinergic Signal (2007) 3(1–2):5–11. doi: 10.1007/s11302-006-9032-0
44. Giuliani AL, Sarti AC, Di Virgilio F. Ectonucleotidases in Acute and Chronic Inflammation [Internet]. Front Pharmacol Front Media SA (2021) 11:2419. doi: 10.3389/fphar.2020.619458
45. Thompson LF, Takedachi M, Ebisuno Y, Tanaka T, Miyasaka M, Mills JH, et al. Regulation of Leukocyte Migration Across Endothelial Barriers by ecto-5′-Nucleotidase-Generated Adenosine. Nucleosides Nucleotides Nucleic Acids (2008) 27(6):755–60. doi: 10.1080/15257770802145678
46. Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, et al. Annexin I is an Endogenous Ligand That Mediates Apoptotic Cell Engulfment. Dev Cell (2003) 4(4):587–98. doi: 10.1016/S1534-5807(03)00090-X
47. Feng M, Marjon KD, Zhu F, Weissman-Tsukamoto R, Levett A, Sullivan K, et al. Programmed Cell Removal by Calreticulin in Tissue Homeostasis and Cancer. Nat Commun (2018) 9(1):3194. doi: 10.1038/s41467-018-05211-7
48. Linnartz B, Kopatz J, Tenner a J, Neumann H. Sialic Acid on the Neuronal Glycocalyx Prevents Complement C1 Binding and Complement Receptor-3-Mediated Removal by Microglia. J Neurosci (2012) 32(3):946–52. doi: 10.1523/JNEUROSCI.3830-11.2012%5Cn
49. Nomura K, Vilalta A, Allendorf DH, Hornik TC, Brown GC. Activated Microglia Desialylate and Phagocytose Cells Via Neuraminidase, Galectin-3, and Mer Tyrosine Kinase. J Immunol (2017) 198(12):4792–801. doi: 10.4049/jimmunol.1502532
50. Saevarsdottir S, Vikingsdottir T, Valdimarsson H. The Potential Role of Mannan-Binding Lectin in the Clearance of Self-Components Including Immune Complexes. Scand J Immunol (2004) 60(1–2):23–9. doi: 10.1111/j.0300-9475.2004.01437.x
51. Hoffmeister KM, Josefsson EC, Isaac NA, Clausen H, Hartwig JH, Stossel TP. Glycosylation Restores Survival of Chilled Blood Platelets. Science (2003) 301(5639):1531–4. doi: 10.1126/science.1085322
52. Deppermann C, Kratofil RM, Peiseler M, David BA, Zindel J, Silva Castanheira FVE, et al. Macrophage Galactose Lectin Is Critical for Kupffer Cells to Clear Aged Platelets. J Exp Med (2020) 217(4). doi: 10.1084/jem.20190723
53. Linnartz-Gerlach B, Schuy C, Shahraz A, Tenner AJ, Neumann H. Sialylation of Neurites Inhibits Complement-Mediated Macrophage Removal in a Human Macrophage-Neuron Co-Culture System. Glia (2016) 64(1):35–47. doi: 10.1002/glia.22901
54. Meesmann HM, Fehr EM, Kierschke S, Herrmann M, Bilyy R, Heyder P, et al. Decrease of Sialic Acid Residues as an Eat-Mesignal on the Surface of Apoptotic Lymphocytes. J Cell Sci (2010) 123(19):3347–56. doi: 10.1242/jcs.066696
55. Verneret M, Tacnet-Delorme P, Osman R, Awad R, Grichine A, Kleman JP, et al. Relative Contribution of C1q and Apoptotic Cell-Surface Calreticulin to Macrophage Phagocytosis. J Innate Immun (2014) 6(4):426–34. doi: 10.1159/000358834
56. Païdassi H, Tacnet-Delorme P, Verneret M, Gaboriaud C, Houen G, Duus K, et al. Investigations on the C1q-Calreticulin-Phosphatidylserine Interactions Yield New Insights Into Apoptotic Cell Recognition. J Mol Biol (2011) 408(2):277–90. doi: 10.1016/j.jmb.2011.02.029
57. Steinø A, Jørgensen CS, Laursen I, Houen G. Interaction of C1q With the Receptor Calreticulin Requires a Conformational Change in C1q. Scand J Immunol (2004) 59(5):485–95. doi: 10.1111/j.0300-9475.2004.01425.x
58. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells Through Trans-Activation of LRP on the Phagocyte. Cell (2005) 123(2):321–34. doi: 10.1016/j.cell.2005.08.032
59. Fricker M, Oliva-Martín MJ, Brown GC. Primary Phagocytosis of Viable Neurons by Microglia Activated With LPS or Aβ Is Dependent on Calreticulin/LRP Phagocytic Signalling. J Neuroinflamm (2012) 9(1):196. doi: 10.1186/1742-2094-9-196
60. Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 Interactions Play an Essential Role in Macrophage-Dependent Phagocytosis of Apoptotic Cells. J Exp Med (2006) 203(12):2613–25. doi: 10.1084/jem.20060370
61. Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, et al. Identification of a Novel Family of Oxidized Phospholipids That Serve as Ligands for the Macrophage Scavenger Receptor CD36. J Biol Chem (2002) 277(41):38503–16. doi: 10.1074/jbc.M203318200
62. Sun M, Finnemann SC, Febbraio M, Shan L, Annangudi SP, Podrez EA, et al. Light-Induced Oxidation of Photoreceptor Outer Segment Phospholipids Generates Ligands for CD36-Mediated Phagocytosis by Retinal Pigment Epithelium: A Potential Mechanism for Modulating Outer Segment Phagocytosis Under Oxidant Stress Conditions. J Biol Chem (2006) 281(7):4222–30. doi: 10.1074/jbc.M509769200
63. Mitra S, Goyal T, Mehta JL, Oxidized LDL. LOX-1 and Atherosclerosis. Cardiovasc Drugs Ther (2011) 25(5):419–29. doi: 10.1007/s10557-011-6341-5
64. Oka K, Sawamura T, Kikuta KI, Itokawa S, Kume N, Kita T, et al. Lectin-Like Oxidized Low-Density Lipoprotein Receptor 1 Mediates Phagocytosis of Aged/Apoptotic Cells in Endothelial Cells. Proc Natl Acad Sci USA (1998) 95(16):9535–40. doi: 10.1073/pnas.95.16.9535
65. Luo X, Gong HB, Gao HY, Wu YP, Sun WY, Li ZQ, et al. Oxygenated Phosphatidylethanolamine Navigates Phagocytosis of Ferroptotic Cells by Interacting With TLR2. Cell Death Differ (2021). doi: 10.1038/s41418-020-00719-2
66. Chang MIK, Bergmark C, Laurila A, Hörkkö S, Han KH, Friedman P, et al. Monoclonal Antibodies Against Oxidized Low-Density Lipoprotein Bind to Apoptotic Cells and Inhibit Their Phagocytosis by Elicited Macrophages: Evidence That Oxidation-Specific Epitopes Mediate Macrophage Recognition. Proc Natl Acad Sci USA (1999) 96(11):6353–8. doi: 10.1073/pnas.96.11.6353
67. Jaillon S, Jeannin P, Hamon Y, Frémaux I, Doni A, Bottazzi B, et al. Endogenous PTX3 Translocates At the Membrane of Late Apoptotic Human Neutrophils and Is Involved in Their Engulfment by Macrophages. Cell Death Differ (2009) 16(3):465–74. doi: 10.1038/cdd.2008.173
68. Guo T, Ke L, Qi B, Wan J, Ge J, Bai L, et al. Ptx3 Is Located At the Membrane of Late Apoptotic Macrophages and Mediates the Phagocytosis of Macrophages. J Clin Immunol (2012) 32(2):330–9. doi: 10.1007/s10875-011-9615-6
69. McArthur S, Cristante E, Paterno M, Christian H, Roncaroli F, Gillies GE, et al. Annexin A1: A Central Player in the Anti-Inflammatory and Neuroprotective Role of Microglia. J Immunol (2010) 185(10):6317–28. doi: 10.4049/jimmunol.1001095
70. Balasubramanian K, Chandra J, Schroit AJ. Immune Clearance of Phosphatidylserine-Expressing Cells by Phagocytes: The Role of β2-Glycoprotein I in Macrophage Recognition. J Biol Chem (1997) 272(49):31113–7. doi: 10.1074/jbc.272.49.31113
71. Wijeyesakere SJ, Bedi SK, Huynh D, Raghavan M. The C-Terminal Acidic Region of Calreticulin Mediates Phosphatidylserine Binding and Apoptotic Cell Phagocytosis. J Immunol (2016) 196(9):3896–909. doi: 10.4049/jimmunol.1502122
72. Jun J, KH K, Lau LF. The Matricellular Protein CCN1 Mediates Neutrophil Efferocytosis in Cutaneous Wound Healing. Nat Commun (2015) 6(1):1–14. doi: 10.1038/ncomms8386
73. Nakano T, Ishimoto Y, Kishino J, Umeda M, Inoue K, Nagata K, et al. Cell Adhesion to Phosphatidylserine Mediated by a Product of Growth Arrest-Specific Gene 6. J Biol Chem (1997) 272(47):29411–4. doi: 10.1074/jbc.272.47.29411
74. Ishimoto Y, Ohashi K, Mizuno K, Nakano T. Promotion of the Uptake of PS Liposomes and Apoptotic Cells by a Product of Growth Arrest-Specific Gene, Gas6. J Biochem (2000) 127(3):411–7. doi: 10.1093/oxfordjournals.jbchem.a022622
75. Lew ED, Oh J, Burrola PG, Lax I, Zagórska A, Través PG, et al. Differential TAM Receptor-Ligand-Phospholipid Interactions Delimit Differential TAM Bioactivities. Elife (2014) 3:e03385. doi: 10.7554/eLife.03385
76. Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a Factor That Links Apoptotic Cells to Phagocytes. Nature (2002) 417(6885):182–7. doi: 10.1038/417182a
77. Shi J, Heegaard CW, Rasmussen JT, Gilbert GE. Lactadherin Binds Selectively to Membranes Containing Phosphatidyl-L-Serine and Increased Curvature. Biochim Biophys Acta - Biomembr (2004) 1667(1):82–90. doi: 10.1016/j.bbamem.2004.09.006
78. Andersen MH, Graversen H, Fedosov SN, Petersen TE, Rasmussen JT. Functional Analyses of Two Cellular Binding Domains of Bovine Lactadherin †. Biochemistry (2000) 39(20):6200–6. doi: 10.1021/bi992221r
79. Yamaguchi H, Takagi J, Miyamae T, Yokota S, Fujimoto T, Nakamura S, et al. Milk Fat Globule EGF Factor 8 in the Serum of Human Patients of Systemic Lupus Erythematosus. J Leukoc Biol (2008) 83(5):1300–7. doi: 10.1189/jlb.1107730
80. Anderson HA, Maylock CA, Williams JA, Paweletz CP, Shu H, Shacter E. Serum-Derived Protein S Binds to Phosphatidylserine and Stimulates the Phagocytosis of Apoptotic Cells. Nat Immunol (2003) 4(1):87–91. doi: 10.1038/ni871
81. Barth ND, Marwick JA, Heeb MJ, Gale AJ, Rossi AG, Dransfield I. Augmentation of Human Monocyte Responses to Lipopolysaccharide by the Protein S and Mer/Tyro3 Receptor Tyrosine Kinase Axis. J Immunol (2018) 201(9):2602–11. doi: 10.4049/jimmunol.1800249
82. Scott-Hewitt NJ, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, et al. Local Externalization of Phosphatidylserine Mediates Developmental Synaptic Pruning by Microglia. bioRxiv (2020) 2020.04.24.059584.
83. Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-Dependent Clearance of Apoptotic Cells by Human Macrophages. J Exp Med (1998) 188(12):2313–20. doi: 10.1084/jem.188.12.2313
84. Kilpatrick DC. Phospholipid-Binding Activity of Human Mannan-Binding Lectin. Immunol Lett (1998) 61(2–3):191–5. doi: 10.1016/S0165-2478(98)00031-5
85. Jäkel A, Reid KBM, Clark H. Surfactant Protein A (Sp-A) Binds to Phosphatidylserine and Competes With Annexin V Binding on Late Apoptotic Cells. Protein Cell (2010) 1(2):188–97. doi: 10.1007/s13238-010-0024-z
86. Moodley Y, Rigby P, Bundell C, Bunt S, Hayashi H, Misso N, et al. Macrophage Recognition and Phagocytosis of Apoptotic Fibroblasts Is Critically Dependent on Fibroblast-Derived Thrombospondin 1 and CD36. Am J Pathol (2003) 162(3):771–9. doi: 10.1016/S0002-9440(10)63874-6
87. Park D, Tosello-Trampont A-C, Elliott MR, Lu M, Haney LB, Ma Z, et al. BAI1 Is an Engulfment Receptor for Apoptotic Cells Upstream of the ELMO/Dock180/Rac Module. Nature (2007) 450(7168):430–4. doi: 10.1038/nature06329
88. Simhadri VR, Andersen JF, Calvo E, Choi SC, Coligan JE, Borrego F. Human CD300a Binds to Phosphatidylethanolamine and Phosphatidylserine, and Modulates the Phagocytosis of Dead Cells. Blood (2012) 119(12):2799–809. doi: 10.1182/blood-2011-08-372425
89. Rigotti A, Acton SL, Krieger M. The Class B Scavenger Receptors SR-BI and CD36 Are Receptors for Anionic Phospholipids. J Biol Chem (1995) 270(27):16221–4. doi: 10.1074/jbc.270.27.16221
90. Cd36 Is Required for Phagocytosis of Apoptotic Cells by Human Macrophages That Use Either a Phosphatidylserine Receptor or the Vitronectin Receptor (Alpha V Beta 3) - Pubmed. Available at: https://pubmed.ncbi.nlm.nih.gov/9834113/.
91. Murphy JE, Tacon D, Tedbury PR, Hadden JM, Knowling S, Sawamura T, et al. LOX-1 Scavenger Receptor Mediates Calcium-Dependent Recognition of Phosphatidylserine and Apoptotic Cells. Biochem J (2006) 393(1):107–15. doi: 10.1042/BJ20051166
92. Hoffmann PR, DeCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, et al. Phosphatidylserine (PS) Induces PS Receptor-Mediated Macropinocytosis and Promotes Clearance of Apoptotic Cells. J Cell Biol (2001) 155(4):649–59. doi: 10.1083/jcb.200108080
93. Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RAB, Henson PM. A Receptor for Phosphatidylserine-Specific Clearance of Apoptotic Cells. Nature (2000) 405(6782):85–90. doi: 10.1038/35011084
94. He M, Kubo H, Morimoto K, Fujino N, Suzuki T, Takahasi T, et al. Receptor for Advanced Glycation End Products Binds to Phosphatidylserine and Assists in the Clearance of Apoptotic Cells. EMBO Rep (2011) 12(4):358–64. doi: 10.1038/embor.2011.28
95. Park SY, Jung MY, Lee SJ, Kang KB, Gratchev A, Riabov V, et al. Stabilin-1 Mediates Phosphatidylserine-Dependent Clearance of Cell Cropses in Alternatively Activated Macrophages. J Cell Sci (2009) 122(18):3365–73. doi: 10.1242/jcs.049569
96. Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, et al. Rapid Cell Corpse Clearance by Stabilin-2, a Membrane Phosphatidylserine Receptor. Cell Death Differ (2008) 15(1):192–201. doi: 10.1038/sj.cdd.4402242
97. Park S-Y, Kim S-Y, Jung M-Y, Bae D-J, Kim I-S. Epidermal Growth Factor-Like Domain Repeat of Stabilin-2 Recognizes Phosphatidylserine During Cell Corpse Clearance. Mol Cell Biol (2008) 28(17):5288–98. doi: 10.1128/MCB.01993-07
98. Kobayashi N, Karisola P, Peña-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 Glycoproteins Bind Phosphatidylserine and Mediate Uptake of Apoptotic Cells. Immunity (2007) 27(6):927–40. doi: 10.1016/j.immuni.2007.11.011
99. Bu X, DeKruyff R, Umetsu D, Freeman G. Tim-3- and TIM-4-Mediated Phagocytosis of Apoptotic Cells Induces Cytokine Production in Peritoneal Macrophages. J Immunol (2011) 186(1 Supplement):100.55.
100. Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a Phosphatidylserine Receptor. Nature (2007) 450(7168):435–9. doi: 10.1038/nature06307
101. Miyanishi M, Segawa K, Nagata S. Synergistic Effect of Tim4 and MFG-E8 Null Mutations on the Development of Autoimmunity. Int Immunol (2012) 24(9):551–9. doi: 10.1093/intimm/dxs064
102. Fond AM, Lee CS, Schulman IG, Kiss RS, Ravichandran KS. Apoptotic Cells Trigger a Membrane-Initiated Pathway to Increase ABCA1. J Clin Invest (2015) 125(7):2748–58. doi: 10.1172/JCI80300
103. Sudom A, Talreja S, Danao J, Bragg E, Kegel R, Min X, et al. Molecular Basis for the Loss-of-Function Effects of the Alzheimer’s Disease–Associated R47H Variant of the Immune Receptor TREM2. J Biol Chem (2018) 293(32):12634–46. doi: 10.1074/jbc.RA118.002352
104. Takahashi K, Rochford CDP, Neumann H. Clearance of Apoptotic Neurons Without Inflammation by Microglial Triggering Receptor Expressed on Myeloid Cells-2. J Exp Med (2005) 201(4):647–57. doi: 10.1084/jem.20041611
105. Tian L, Choi SC, Murakami Y, Allen J, Morse HC, Qi CF, et al. P85α Recruitment by the CD300f Phosphatidylserine Receptor Mediates Apoptotic Cell Clearance Required for Autoimmunity Suppression. Nat Commun (2014) 5:3146. doi: 10.1038/ncomms4146
106. Shacham-Silverberg V, Sar Shalom H, Goldner R, Golan-Vaishenker Y, Gurwicz N, Gokhman I, et al. Phosphatidylserine Is a Marker for Axonal Debris Engulfment But Its Exposure can be Decoupled From Degeneration. Cell Death Dis (2018) 9(11):1–15. doi: 10.1038/s41419-018-1155-z
107. Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, et al. UDP Acting At P2Y6 Receptors is a Mediator of Microglial Phagocytosis. Nature (2007) 446(7139):1091–5. doi: 10.1038/nature05704
108. Neher JJ, Neniskyte U, Hornik T, Brown GC. Inhibition of UDP/P2Y6 Purinergic Signaling Prevents Phagocytosis of Viable Neurons by Activated Microglia In Vitro and In Vivo. Glia (2014) 62(9):1463–75. doi: 10.1002/glia.22693
109. Sugimoto MA, Vago JP, Teixeira MM, Sousa LP. Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J Immunol Res (2016) 2016:1469780. doi: 10.1155/2016/8239258
110. Fan X, Krahling S, Smith D, Williamson P, Schlegel RA. Macrophage Surface Expression of Annexins I and II in the Phagocytosis of Apoptotic Lymphocytes. Mol Biol Cell (2004) 15(6):2863–72. doi: 10.1091/mbc.e03-09-0670
111. Zhang X-H, Wang Q-M, Zhang J-M, Feng F-E, Wang F-R, Chen H, et al. Desialylation is Associated With Apoptosis and Phagocytosis of Platelets in Patients With Prolonged Isolated Thrombocytopenia After Allo-HSCT. J Hematol Oncol (2015) 8:116. doi: 10.1186/s13045-015-0216-3
112. Lübbers J, Rodríguez E, van Kooyk Y. Modulation of Immune Tolerance Via Siglec-Sialic Acid Interactions. Front Immunol (2018) 9:2807. doi: 10.3389/fimmu.2018.02807
113. Païdassi H, Tacnet-Delorme P, Lunardi T, Arlaud GJ, Thielens NM, Frachet P. The Lectin-Like Activity of Human C1q and Its Implication in DNA and Apoptotic Cell Recognition. FEBS Lett (2008) 582(20):3111–6. doi: 10.1016/j.febslet.2008.08.001
114. Martins I, Kepp O, Galluzzi L, Senovilla L, Schlemmer F, Adjemian S, et al. Surface-Exposed Calreticulin in the Interaction Between Dying Cells and Phagocytes. Ann N Y Acad Sci (2010) 1209(1):77–82. doi: 10.1111/j.1749-6632.2010.05740.x
115. Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, et al. A Novel Pathway Combining Calreticulin Exposure and ATP Secretion in Immunogenic Cancer Cell Death. EMBO J (2012) 31(5):1062–79. doi: 10.1038/emboj.2011.497
116. Orr AW, Pedraza CE, Pallero MA, Elzie CA, Goicoechea S, Strickland DK, et al. Low Density Lipoprotein Receptor-Related Protein Is a Calreticulin Coreceptor That Signals Focal Adhesion Disassembly. J Cell Biol (2003) 161(6):1179–89. doi: 10.1083/jcb.200302069
117. Duus K, Hansen EW, Tacnet P, Frachet P, Arlaud GJ, Thielens NM, et al. Direct Interaction Between CD91 and C1q. FEBS J (2010) 277(17):3526–37. doi: 10.1111/j.1742-4658.2010.07762.x
118. Wei Xu MR, van Kooten Boer C, Schlagwein N, Mutsaers C, Berger SP, Trouw LA, et al. Properdin Binds to Late Apoptotic and Necrotic Cells Independently of C3b and Regulates Alternative Pathway Complement Activation. J Immunol Ref (2008) 180:7613–21. doi: 10.4049/jimmunol.180.11.7613
119. Jensen ML, Honoré C, Hummelshøj T, Hansen BE, Madsen HO, Garred P. Ficolin-2 Recognizes DNA and Participates in the Clearance of Dying Host Cells. Mol Immunol (2007) 44(5):856–65. doi: 10.1016/j.molimm.2006.04.002
120. Elward K, Griffiths M, Mizuno M, Harris CL, Neal JW, Paul Morgan B, et al. Cd46 Plays a Key Role in Tailoring Innate Immune Recognition of Apoptotic and Necrotic Cells. J Biol Chem (2005) 280(43):36342–54. doi: 10.1074/jbc.M506579200
121. Radic M, Marion T, Monestier M. Nucleosomes Are Exposed At the Cell Surface in Apoptosis. J Immunol (2004) 172(11):6692–700. doi: 10.4049/jimmunol.172.11.6692
122. Cunin P, Beauvillain C, Miot C, Augusto JF, Preisser L, Blanchard S, et al. Clusterin Facilitates Apoptotic Cell Clearance and Prevents Apoptotic Cell-Induced Autoimmune Responses. Cell Death Dis (2016) 7(5):e2215. doi: 10.1038/cddis.2016.113
123. Kristóf E, Zahuczky G, Katona K, Doró Z, Nagy É, Fésüs L. Novel Role of ICAM3 and LFA-1 in the Clearance of Apoptotic Neutrophils by Human Macrophages. Apoptosis (2013) 18(10):1235–51. doi: 10.1007/s10495-013-0873-z
124. Fawcett J, Holness CLL, Needham LA, Turley H, Gattert KC, Mason DY, et al. Molecular Cloning of ICAM-3, A Third Ligand for LFA-1, Constitutively Expressed on Resting Leukocytes. Nature (1992) 360(6403):481–4. doi: 10.1038/360481a0
125. Moffatt OD, Devitt A, Simmons DL, Gregory CD. Macrophage Recognition of ICAM-3 on Apoptotic Leukocytes. J Immunol (1999) 162(11):6800–10.
126. Torr EE, Gardner DH, Thomas L, Goodall DM, Bielemeier A, Willetts R, et al. Apoptotic Cell-Derived ICAM-3 Promotes Both Macrophage Chemoattraction to and Tethering of Apoptotic Cells. Cell Death Differ (2012) 19(4):671–9. doi: 10.1038/cdd.2011.167
127. Verhoven B, Schlegel RA, Williamson P. Mechanisms of Phosphatidylserine Exposure, a Phagocyte Recognition Signal, on Apoptotic T Lymphocytes. J Exp Med (1995) 182(5):1597–601. doi: 10.1084/jem.182.5.1597
128. Borisenko GG, Matsura T, Liu S-X, Tyurin VA, Jianfei J, Serinkan FB, et al. Macrophage Recognition of Externalized Phosphatidylserine and Phagocytosis of Apoptotic Jurkat Cells–Existence of a Threshold. Arch Biochem Biophys (2003) 413(1):41–52. doi: 10.1016/S0003-9861(03)00083-3
129. Krahling S, Callahan MK, Williamson P, Schlegel RA. Exposure of Phosphatidylserine Is a General Feature in the Phagocytosis of Apoptotic Lymphocytes by Macrophages. Cell Death Differ (1999) 6(2):183–9. doi: 10.1038/sj.cdd.4400473
130. Neher JJ, Neniskyte U, Zhao J-W, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of Microglial Phagocytosis Is Sufficient To Prevent Inflammatory Neuronal Death. J Immunol (2011) 186(8):4973–83. doi: 10.4049/jimmunol.1003600
131. Brown GC, Neher JJ. Eaten Alive! Cell Death by Primary Phagocytosis: “Phagoptosis”. Trends Biochem Sci (2012) 37(8):325–32. doi: 10.1016/j.tibs.2012.05.002
132. Chen J, Zhong MC, Guo H, Davidson D, Mishel S, Lu Y, et al. SLAMF7 Is Critical for Phagocytosis of Haematopoietic Tumour Cells Via Mac-1 Integrin. Nature (2017) 544(7651):493–7. doi: 10.1038/nature22076
133. Cockram TOJ, Puigdellívol M, Brown GC. Calreticulin and Galectin-3 Opsonise Bacteria for Phagocytosis by Microglia. Front Immunol (2019) 10:2647–58. doi: 10.3389/fimmu.2019.02647
134. Hart SP, Smith JR, Dransfield I. Phagocytosis of Opsonized Apoptotic Cells: Roles for “Old-Fashioned” Receptors for Antibody and Complement. Clin Exp Immunol (2004) 135(2):181–5. doi: 10.1111/j.1365-2249.2003.02330.x
135. Maderna P, Yona S, Perretti M, Godson C. Modulation of Phagocytosis of Apoptotic Neutrophils by Supernatant From Dexamethasone-Treated Macrophages and Annexin-Derived Peptide Ac 2–26. J Immunol (2005) 174(6):3727–33. doi: 10.4049/jimmunol.174.6.3727
136. Blume KE, Soeroes S, Waibel M, Keppeler H, Wesselborg S, Herrmann M, et al. Cell Surface Externalization of Annexin A1 as a Failsafe Mechanism Preventing Inflammatory Responses During Secondary Necrosis. J Immunol (2009) 183(12):8138–47. doi: 10.4049/jimmunol.0902250
137. Lee G, Pollard HB, Arispe N. Annexin 5 and Apolipoprotein E2 Protect Against Alzheimer’s Amyloid-β-Peptide Cytotoxicity by Competitive Inhibition At a Common Phosphatidylserine Interaction Site. Peptides (2002) 23(7):1249–63. doi: 10.1016/S0196-9781(02)00060-8
138. Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J Biol Chem (2015) 290(43):26043–50. doi: 10.1074/jbc.M115.679043
139. Fagan AM, Bu G, Sun Y, Daugherty A, Holtzman DM. Apolipoprotein E-Containing High Density Lipoprotein Promotes Neurite Outgrowth and Is a Ligand for the Low Density Lipoprotein Receptor-Related Protein. J Biol Chem (1996) 271(47):30121–5. doi: 10.1074/jbc.271.47.30121
140. Grainger DJ, Reckless J, McKilligin E. Apolipoprotein E Modulates Clearance of Apoptotic Bodies In Vitro and In Vivo, Resulting in a Systemic Proinflammatory State in Apolipoprotein E-Deficient Mice. J Immunol (2004) 173(10):6366–75. doi: 10.4049/jimmunol.173.10.6366
141. Chung W-S, Verghese PB, Chakraborty C, Joung J, Hyman BT, Ulrich JD, et al. Novel Allele-Dependent Role for APOE in Controlling the Rate of Synapse Pruning by Astrocytes. Proc Natl Acad Sci (2016) 113(36):10186–91. doi: 10.1073/pnas.1609896113
142. Bartl MM, Luckenbach T, Bergner O, Ullrich O, Koch-Brandt C. Multiple Receptors Mediate apoJ-Dependent Clearance of Cellular Debris Into Nonprofessional Phagocytes. Exp Cell Res (2001) 271(1):130–41. doi: 10.1006/excr.2001.5358
143. Abdel-Monem H, Dasgupta SK, Le A, Prakasam A, Thiagarajan P. Phagocytosis of Platelet Microvesicles and β2- Glycoprotein I. Thromb Haemost (2010) 104(2):335–41. doi: 10.1160/TH09-12-0849
144. Rovere P, Manfredi AA, Vallinoto C, Zimmermann VS, Fascio U, Balestrieri G, et al. Dendritic Cells Preferentially Internalize Apoptotic Cells Opsonized by Anti-β2-Glycoprotein I Antibodies. J Autoimmun (1998) 11(5):403–11. doi: 10.1006/jaut.1998.0224
145. Steinberger P, Szekeres A, Wille S, Stöckl J, Selenko N, Prager E, et al. Identification of Human CD93 as the Phagocytic C1q Receptor (C1qRp) by Expression Cloning. J Leukoc Biol (2002) 71(1):133–40. doi: 10.1189/jlb.71.1.133
146. Blackburn JWD, Lau DHC, Liu EY, Ellins J, Vrieze AM, Pawlak EN, et al. Soluble CD93 Is an Apoptotic Cell Opsonin Recognized by α X β 2. Eur J Immunol (2019) 49(4):600–10. doi: 10.1002/eji.201847801
147. Païdassi H, Tacnet-Delorme P, Garlatti V, Darnault C, Ghebrehiwet B, Gaboriaud C, et al. C1q Binds Phosphatidylserine and Likely Acts as a Multiligand-Bridging Molecule in Apoptotic Cell Recognition. J Immunol (2008) 180(4):2329–38. doi: 10.4049/jimmunol.180.4.2329
148. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and Mannose Binding Lectin Engagement of Cell Surface Calreticulin and Cd91 Initiates Macropinocytosis and Uptake of Apoptotic Cells. J Exp Med (2001) 194(6):781–96. doi: 10.1084/jem.194.6.781
149. Klickstein LB, Barbashov SF, Liu T, Jack RM, Nicholson-Weller A. Complement Receptor Type 1 (CR1, CD35) Is a Receptor for C1q. Immun (1997) 7(3):345–55. doi: 10.1016/S1074-7613(00)80356-8
150. Fraser DA, Laust AK, Nelson EL, Tenner AJ. C1q Differentially Modulates Phagocytosis and Cytokine Responses During Ingestion of Apoptotic Cells by Human Monocytes, Macrophages, and Dendritic Cells. J Immunol (2009) 183(10):6175–85. doi: 10.4049/jimmunol.0902232
151. Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, et al. Role of Surfactant Proteins A, D, and C1q in the Clearance of Apoptotic Cells In Vivo and In Vitro: Calreticulin and CD91 as a Common Collectin Receptor Complex. J Immunol (2002) 169(7):3978–86. doi: 10.4049/jimmunol.169.7.3978
152. Quartier P, Potter PK, Ehrenstein MR, Walport MJ, Botto M. Predominant Role of IgM-Dependent Activation of the Classical Pathway in the Clearance of Dying Cells by Murine Bone Marrow-Derived Macrophages In Vitro. Eur J Immunol (2005) 35(1):252–60. doi: 10.1002/eji.200425497
153. Fraser DA, Pisalyaput K, Tenner AJ. C1q Enhances Microglial Clearance of Apoptotic Neurons and Neuronal Blebs, and Modulates Subsequent Inflammatory Cytokine Production. J Neurochem (2010) 112(3):733–43. doi: 10.1111/j.1471-4159.2009.06494.x
154. Böttcher A, Gaipl US, Fürnrohr BG, Herrmann M, Girkontaite I, Kalden JR, et al. Involvement of Phosphatidylserine, αvβ3, CD14, CD36, and Complement C1q in the Phagocytosis of Primary Necrotic Lymphocytes by Macrophages. Arthritis Rheum (2006) 54(3):927–38. doi: 10.1002/art.21660
155. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron (2012) 74(4):691–705. doi: 10.1016/j.neuron.2012.03.026
156. Bialas AR, Stevens B. Tgf-β Signaling Regulates Neuronal C1q Expression and Developmental Synaptic Refinement. Nat Neurosci (2013) 16(12):1773–82. doi: 10.1038/nn.3560
157. Pangburn MK, Muller-Eberhard HJ. Relation of a Putative Thioester Bond in C3 to Activation of the Alternative Pathway and the Binding of C3b to Biological Targets of Complement. J Exp Med (1980) 152(4):1102–14. doi: 10.1084/jem.152.4.1102
158. Takizawa F, Tsuji S, Nagasawa S. Enhancement of Macrophage Phagocytosis Upon iC3b Deposition on Apoptotic Cells. FEBS Lett (1996) 397(2–3):269–72. doi: 10.1016/S0014-5793(96)01197-0
159. Reichert F, Rotshenker S. Complement-Receptor-3 and scavenger-receptor-AI/II Mediated Myelin Phagocytosis in Microglia and Macrophages. Neurobiol Dis (2003) 12(1):65–72. doi: 10.1016/S0969-9961(02)00008-6
160. Gaither TA, Vargas I, Inada S, Frank MM. The Complement Fragment C3d Facilitates Phagocytosis by Monocytes [Published Erratum Appears in Immunology 1988 Mar;63(3):559]. Immunology (1987) 62(0019-2805 SB-M SB-X):405–11.
161. Ehlenberger AG, Nussenzweig V. The Role of Membrane Receptors for C3b and C3d in Phagocytosis*. J Exp Med (1977) 145(2):357–71. doi: 10.1084/jem.145.2.357
162. Sepp A, Dodds AW, Anderson MJ, Duncan Campbell R, Willis AC, Alex Law SK. Covalent Binding Properties of the Human Complement Protein C4 and Hydrolysis Rate of the Internal Thioester Upon Activation. Protein Sci (1993) 2(5):706–16. doi: 10.1002/pro.5560020502
163. Wilson JG, Andriopoulos NA, Fearon DT. CR1 and the Cell Membrane Proteins That Bind C3 and C4 - A Basic and Clinical Review. Immunol Res (1987) 6(3):192–209. doi: 10.1007/BF02918091
164. Bijl M, Reefman E, Horst G, Limburg PC, Kallenberg CGM. Reduced Uptake of Apoptotic Cells by Macrophages in Systemic Lupus Erythematosus: Correlates With Decreased Serum Levels of Complement. Ann Rheum Dis (2006) 65(1):57–63. doi: 10.1136/ard.2005.035733
165. Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, et al. A Hierarchical Role for Classical Pathway Complement Proteins in the Clearance of Apoptotic Cells In Vivo. J Exp Med (2000) 192(3):359–66. doi: 10.1084/jem.192.3.359
166. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia Risk From Complex Variation of Complement Component 4. Nature (2016) 530(7589):177–83. doi: 10.1038/nature16549
167. Roumenina LT, Ruseva MM, Zlatarova A, Ghai R, Kolev M, Olova N, et al. Interaction of C1q With IgG1, C-Reactive Protein and Pentraxin 3: Mutational Studies Using Recombinant Globular Head Modules of Human C1q a, B, and C Chains. Biochem (2006) 45(13):4093–104. doi: 10.1021/bi052646f
168. Bodman-Smith KB, Melendez AJ, Campbell I, Harrison PT, Allen JM, Raynes JG. C-Reactive Protein-Mediated Phagocytosis and Phospholipase D Signalling Through the High-Affinity Receptor for Immunoglobulin G (Fcγri). Immunology (2002) 107(2):252–60. doi: 10.1046/j.1365-2567.2002.01481.x
169. Gershov D, Kim S, Brot N, Elkon KB. C-Reactive Protein Binds to Apoptotic Cells, Protects the Cells From Assembly of the Terminal Complement Components, and Sustains an Antiinflammatory Innate Immune Response: Implications for Systemic Autoimmunity. J Exp Med (2000) 192(9):1353–64. doi: 10.1084/jem.192.9.1353
170. Mold C, Baca R, Du Clos TW. Serum Amyloid P Component and C-Reactive Protein Opsonize Apoptotic Cells for Phagocytosis Through Fcγ Receptors. J Autoimmun (2002) 19(3):147–54. doi: 10.1006/jaut.2002.0615
171. Kourtzelis I, Li X, Mitroulis I, Grosser D, Kajikawa T, Wang B, et al. DEL-1 Promotes Macrophage Efferocytosis and Clearance of Inflammation. Nat Immunol (2019) 20(1):40–9. doi: 10.1038/s41590-018-0249-1
172. Human Fibronectin Is an Opsonin for IgG Antibody-Coated Sheep Erythrocytes - Pubmed. Available at: https://pubmed.ncbi.nlm.nih.gov/6699476/.
173. Shimaoka T, Kume N, Minami M, Hayashida K, Sawamura T, Kita T, et al. Lectin-Like Oxidized Low Density Lipoprotein Receptor-1 (LOX-1) Supports Cell Adhesion to Fibronectin. FEBS Lett (2001) 504(1–2):65–8. doi: 10.1016/S0014-5793(01)02774-0
174. Vernon-Wilson EF. CD31 Promotes 1 Integrin-Dependent Engulfment of Apoptotic Jurkat T Lymphocytes Opsonized for Phagocytosis by Fibronectin. J Leukoc Biol (2006) 79(6):1260–7. doi: 10.1189/jlb.1005571
175. Czop JK, Kadisho JL, Frank Austen K. Augmentation of Human Monocyte Opsonin-Independent Phagocytosis by Fragments of Human Plasma Fibronectin (Particulate Activators/Alternative Complement Pathway/Fragmented Cold-Insoluble Globulin). Proc Natl Acad Sci USA (1981) 78(6):3649–53. doi: 10.1073/pnas.78.6.3649
176. Ma YJ, Doni A, Romani L, Jürgensen HJ, Behrendt N, Mantovani A, et al. Ficolin-1–PTX3 Complex Formation Promotes Clearance of Altered Self-Cells and Modulates Il-8 Production. J Immunol (2013) 191(3):1324–33. doi: 10.4049/jimmunol.1300382
177. Caberoy NB, Alvarado G, Bigcas J-L, Li W. Galectin-3 Is a New MerTK-specific Eat-Me Signal. J Cell Physiol (2012) 227(2):401–7. doi: 10.1002/jcp.22955
178. Karlsson A, Christenson K, Matlak M, Bjorstad A, Brown KL, Telemo E, et al. Galectin-3 Functions as an Opsonin and Enhances the Macrophage Clearance of Apoptotic Neutrophils. Glycobiology (2008) 19(1):16–20. doi: 10.1093/glycob/cwn104
179. Nagata K, Ohashi K, Nakano T, Arita H, Zong C, Hanafusa H, et al. Identification of the Product of Growth Arrest-Specific Gene 6 as a Common Ligand for Axl, Sky, and Mer Receptor Tyrosine Kinases. J Biol Chem (1996) 271(47):30022–7. doi: 10.1074/jbc.271.47.30022
180. Zagórska A, Través PG, Lew ED, Dransfield I, Lemke G. Diversification of TAM Receptor Tyrosine Kinase Function. Nat Immunol (2014) 15(10):920–8. doi: 10.1038/ni.2986
181. Grommes C, Lee CYD, Wilkinson BL, Jiang Q, Koenigsknecht-Talboo JL, Varnum B, et al. Regulation of Microglial Phagocytosis and Inflammatory Gene Expression by Gas6 Acting on the Axl/Mer Family of Tyrosine Kinases. J Neuroimmun Pharmacol (2008) 3(2):130–40. doi: 10.1007/s11481-007-9090-2
182. Hall MO, Prieto AL, Obin MS, Abrams TA, Burgess BL, Heeb MJ, et al. Outer Segment Phagocytosis by Cultured Retinal Pigment Epithelial Cells Requires Gas6. Exp Eye Res (2001) 73(4):509–20. doi: 10.1006/exer.2001.1062
183. Yang A, Dai J, Xie Z, Colman RW, Wu Q, Birge RB, et al. High Molecular Weight Kininogen Binds Phosphatidylserine and Opsonizes Urokinase Plasminogen Activator Receptor–Mediated Efferocytosis. J Immunol (2014) 192(9):4398–408. doi: 10.4049/jimmunol.1302590
184. Hart SP, Alexander KM, Dransfield I. Immune Complexes Bind Preferentially to Fc Gamma RIIA (CD32) on Apoptotic Neutrophils, Leading to Augmented Phagocytosis by Macrophages and Release of Proinflammatory Cytokines. J Immunol (2004) 172(3):1882–7. doi: 10.4049/jimmunol.172.3.1882
185. Métayer LE, Vilalta A, Amos Burke GA, Brown GC. Anti-CD47 Antibodies Induce Phagocytosis of Live, Malignant B Cells by Macrophages Via the Fc Domain, Resulting in Cell Death by Phagoptosis. Oncotarget (2017) 8(37):60892–903. doi: 10.18632/oncotarget.18492
186. Kim SJ, Gershov D, Ma X, Brot N, Elkon KB. I-PLA 2 Activation During Apoptosis Promotes the Exposure of Membrane Lysophosphatidylcholine Leading to Binding by Natural Immunoglobulin M Antibodies and Complement Activation. J Exp Med (2002) 196(5):655–65. doi: 10.1084/jem.20020542
187. Kim J. Identification of a Human Monoclonal Natural IgM Antibody That Recognizes Early Apoptotic Cells and Promotes Phagocytosis. Hybridoma (2010) 29(4):275–81. doi: 10.1089/hyb.2010.0012
188. Kubagawa H, Honjo K, Ohkura N, Sakaguchi S, Radbruch A, Melchers F, et al. Functional Roles of the IgM Fc Receptor in the Immune System. Front Immunol Front Media SA (2019) 10:945. doi: 10.3389/fimmu.2019.00945
189. Uher F, Dobronyi I, Gergel J. IgM-Fc Receptor-Mediated Phagocytosis of Rat Macrophages. Immunol (1981) 42(3):419–25.
190. Malhotra R, Thiel S, Reid KB, Sim RB. Human Leukocyte C1q Receptor Binds Other Soluble Proteins With Collagen Domains. J Exp Med (1990) 172(3):955–9. doi: 10.1084/jem.172.3.955
191. Nauta AJ, Raashou-Jensen N, Roos A, Daha MR, Madsen HO, Borrias-Essers MC, et al. Mannose-Binding Lectin Engagement With Late Apoptotic and Necrotic Cells. Eur J Immunol (2003) 33(10):2853–63. doi: 10.1002/eji.200323888
192. Stienstra R, Dijk W, Van Beek L, Jansen H, Heemskerk M, Houtkooper RH, et al. Mannose-Binding Lectin Is Required for the Effective Clearance of Apoptotic Cells by Adipose Tissue Macrophages During Obesity. Diabetes (2014) 63(12):4143–53. doi: 10.2337/db14-0256
193. Finnemann SC, Bonilha VL, Marmorstein AD, Rodriguez-Boulan E. Essential Role for MFG-E8 as Ligand for αvβ5 Integrin in Diurnal Retinal Phagocytosis. Proc Natl Acad Sci USA (2007) 104(29):12005–10. doi: 10.1073/pnas.0704756104
194. Miksa M, Amin D, Wu R, Dong W, Ravikumar TS, Wang P. Fractalkine-Induced MFG-E8 Leads to Enhanced Apoptotic Cell Clearance by Macrophages. Mol Med (2007) 13(11–12):553–60. doi: 10.2119/2007-00019.Miksa
195. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune Disease and Impaired Uptake of Apoptotic Cells in MFG-E8-Deficient Mice. Science (2004) 304(5674):1147–50. doi: 10.1126/science.1094359
196. Dasgupta SK, Thiagarajan P. The Role of Lactadherin in the Phagocytosis of Phosphatidylserine-Expressing Sickle Red Blood Cells by Macrophages. Haematol (2005) 90(9):1267–8.
197. Neniskyte U, Brown GC. Lactadherin/MFG-E8 Is Essential for Microglia-Mediated Neuronal Loss and Phagoptosis Induced by Amyloid β. J Neurochem (2013) 126(3):312–7. doi: 10.1111/jnc.12288
198. Fricker M, Neher JJ, Zhao J-W, Thery C, Tolkovsky AM, Brown GC. Mfg-E8 Mediates Primary Phagocytosis of Viable Neurons During Neuroinflammation. J Neurosci (2012) 32(8):2657–66. doi: 10.1523/JNEUROSCI.4837-11.2012
199. Kemper C, Mitchell LM, Zhang L, Hourcade DE. The Complement Protein Properdin Binds Apoptotic T Cells and Promotes Complement Activation and Phagocytosis. Proc Natl Acad Sci USA (2008) 105(26):9023–8. doi: 10.1073/pnas.0801015105
200. McColl A, Bournazos S, Franz S, Perretti M, Morgan BP, Haslett C, et al. Glucocorticoids Induce Protein S-Dependent Phagocytosis of Apoptotic Neutrophils by Human Macrophages. J Immunol (2009) 183(3):2167–75. doi: 10.4049/jimmunol.0803503
201. Nandrot EF. Opposite Roles of MerTK Ligands Gas6 and Protein S During Retinal Phagocytosis. In: Advances in Experimental Medicine and Biology. New York LLC: Springer (2018). p. 577–83. Available at: https://pubmed.ncbi.nlm.nih.gov/29721990/.
202. Kask L, Trouw LA, Dahlbä ck B, Blom AM. The C4b-Binding Protein-Protein S Complex Inhibits the Phagocytosis of Apoptotic Cells. J Biol Chem (2004) 279(23):23869–73. doi: 10.1074/jbc.C400159200
203. Lumbroso D, Soboh S, Maimon A, Schif-Zuck S, Ariel A, Burstyn-Cohen T. Macrophage-Derived Protein S Facilitates Apoptotic Polymorphonuclear Cell Clearance by Resolution Phase Macrophages and Supports Their Reprogramming. Front Immunol (2018) 9:358. doi: 10.3389/fimmu.2018.00358
204. Christner RB, Mortensen RF. Binding of Human Serum Amyloid P-component to Phosphocholine. Arch Biochem Biophys (1994) 314(2):337–43. doi: 10.1006/abbi.1994.1451
205. Ying SC, Gewurz AT, Jiang H, Gewurz H. Human Serum Amyloid P Component Oligomers Bind and Activate the Classical Complement Pathway Via Residues 14-26 and 76-92 of the A Chain Collagen-Like Region of C1q. J Immunol (1993) 150(1):169–76.
206. Bharadwaj D, Mold C, Markham E, Du Clos TW. Serum Amyloid P Component Binds to Fcγ Receptors and Opsonizes Particles for Phagocytosis. J Immunol (2001) 166(11):6735–41. doi: 10.4049/jimmunol.166.11.6735
207. Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD. Structural Recognition and Functional Activation of Fcγr by Innate Pentraxins. Nature (2008) 456(7224):989–92. doi: 10.1038/nature07468
208. Lu J, Marjon KD, Mold C, Marnell L, Du Clos TW, Sun P. Pentraxins and IgA Share a Binding Hot-Spot on Fcαri. Protein Sci (2014) 23(4):378–86. doi: 10.1002/pro.2419
209. Kuroki Y, Akino T. Pulmonary Surfactant Protein A (Sp-A) Specifically Binds Dipalmitoylphosphatidylcholine. J Biol Chem (1991) 266(5):3068–73. doi: 10.1016/S0021-9258(18)49955-3
210. Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, et al. By Binding Sirpα or Calreticulin/CD91, Lung Collectins Act as Dual Function Surveillance Molecules to Suppress or Enhance Inflammation. Cell (2003) 115(1):13–23. doi: 10.1016/S0092-8674(03)00758-X
211. Schagat TL, Wofford JA, Wright JR. Surfactant Protein A Enhances Alveolar Macrophage Phagocytosis of Apoptotic Neutrophils. J Immunol (2001) 166(4):2727–33. doi: 10.4049/jimmunol.166.4.2727
212. Manodori AB, Barabino GA, Lubin BH, Kuypers FA. Adherence of Phosphatidylserine-Exposing Erythrocytes to Endothelial Matrix Thrombospondin. Blood (2000) 95(4):1293–300. doi: 10.1182/blood.V95.4.1293.004k42_1293_1300
213. Savill J, Hogg N, Ren Y, Haslett C. Thrombospondin Cooperates With CD36 and the Vitronectin Receptor in Macrophage Recognition of Neutrophils Undergoing Apoptosis. J Clin Invest (1992) 90(4):1513–22. doi: 10.1172/JCI116019
214. Stern M, Savill J, Haslett C. Human Monocyte-Derived Macrophage Phagocytosis of Senescent Eosinophils Undergoing Apoptosis: Mediation by α(V)β3/CD36/Thrombospondin Recognition Mechanism and Lack of Phlogistic Response. Am J Pathol (1996) 149(3):911–21.
215. Caberoy NB, Zhou Y, Li W. Tubby and Tubby-Like Protein 1 Are New MerTK Ligands for Phagocytosis. EMBO J (2010) 29(23):3898–910. doi: 10.1038/emboj.2010.265
216. Caberoy NB, Maiguel D, Kim Y, Li W. Identification of Tubby and Tubby-Like Protein 1 as Eat-Me Signals by Phage Display. Exp Cell Res (2010) 316(2):245–57. doi: 10.1016/j.yexcr.2009.10.008
217. Xi Q, Pauer GJT, Marmorstein AD, Crabb JW, Hagstrom SA. Tubby-Like Protein 1 (TULP1) Interacts With F-actin in Photoreceptor Cells. Investig Ophthalmol Vis Sci (2005) 46(12):4754–61. doi: 10.1167/iovs.05-0693
218. Caberoy NB, Alvarado G, Li W. Tubby Regulates Microglial Phagocytosis Through Mertk. J Neuroimmunol (2012) 252(1–2):40–8. doi: 10.1016/j.jneuroim.2012.07.009
219. Lizarbe MA, Barrasa JI, Olmo N, Gavilanes F, Turnay J. Annexin-Phospholipid Interactions. Functional Implications [Internet]. Int J Mol Sci Multidiscip Digital Publ Inst (MDPI) (2013) 14:2652–83. doi: 10.3390/ijms14022652
220. Janeway CJ, Travers P, Walport M, Al E. The Destruction of Antibody-Coated Pathogens Via Fc Receptors. Immunobiol Immune Syst Heal Dis (2001), 1–6.
221. Aderem A, Underhill DM. Mechanisms of Phagocytosis in Macrophages. Annu Rev Immunol (1999) 17:593–623. doi: 10.1146/annurev.immunol.17.1.593
222. Balasubramanian K, Maiti SN, Schroit AJ. Recruitment of Beta-2-Glycoprotein 1 to Cell Surfaces in Extrinsic and Intrinsic Apoptosis. Apoptosis (2005) 10(2):439–46. doi: 10.1007/s10495-005-0817-3
223. Yang R, Chen Y, Chen D. Biological Functions and Role of CCN1/Cyr61 in Embryogenesis and Tumorigenesis in the Female Reproductive System (Review). Mol Med Rep Spandidos Publ (2018) 17:3–10. doi: 10.3892/mmr.2017.7880
224. Bohlson SS, Silva R, Fonseca MI, Tenner AJ. Cd93 Is Rapidly Shed From the Surface of Human Myeloid Cells and the Soluble Form Is Detected in Human Plasma. J Immunol (2005) 175(2):1239–47. doi: 10.4049/jimmunol.175.2.1239
225. Norsworthy PJ, Fossati-Jimack L, Cortes-Hernandez J, Taylor PR, Bygrave AE, Thompson RD, et al. Murine CD93 (C1qrp) Contributes to the Removal of Apoptotic Cells In Vivo But Is Not Required for C1q-Mediated Enhancement of Phagocytosis. J Immunol (2004) 172(6):3406–14. doi: 10.4049/jimmunol.172.6.3406
226. Lu J, Teh C, Kishore U, Reid KB. Collectins and Ficolins: Sugar Pattern Recognition Molecules of the Mammalian Innate Immune System. Biochim Biophys Acta - Gen Subj (2002) 1572(2–3):387–400. doi: 10.1016/S0304-4165(02)00320-3
227. Janssen WJ, McPhillips KA, Dickinson MG, Linderman DJ, Morimoto K, Xiao YQ, et al. Surfactant Proteins A and D Suppress Alveolar Macrophage Phagocytosis Via Interaction With Sirpα. Am J Respir Crit Care Med (2008) 178(2):158–67. doi: 10.1164/rccm.200711-1661OC
228. Martin U, Bock D, Arseniev L, Tornetta MA, Ames RS, Bautsch W, et al. The Human C3a Receptor Is Expressed on Neutrophils and Monocytes, But Not on B or T Lymphocytes. J Exp Med (1997) 186(2):199–207. doi: 10.1084/jem.186.2.199
229. Zwirner J, Götze O, Moser A, Sieber A, Begemann G, Kapp A, et al. Blood- and Skin-Derived Monocytes/Macrophages Respond to C3a But Not to C3a(desArg) With a Transient Release of Calcium Via a Pertussis Toxin-Sensitive Signal Transduction Pathway. Eur J Immunol (1997) 27(9):2317–22. doi: 10.1002/eji.1830270928
230. Jiang H, Cooper B, Robey FA, Gewurz H. DNA Binds and Activates Complement Via Residues 14-26 of the Human C1q A Chain. J Biol Chem (1992) 267(35):25597–601. doi: 10.1016/S0021-9258(19)74082-4
231. Inforzato A, Peri G, Doni A, Garlanda C, Mantovani A, Bastone A, et al. Structure and Function of the Long Pentraxin PTX3 Glycosidic Moiety: Fine-tuning of the Interaction With C1q and Complement Activation. Biochem (2006) 45(38):11540–51. doi: 10.1021/bi0607453
232. Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrín D, Stehle T. Structural Basis for Sialic Acid-Mediated Self-Recognition by Complement Factor H. Nat Chem Biol (2015) 11(1):77–82. doi: 10.1038/nchembio.1696
233. Mccafferty MH, Lepow M, Saba TM, Cho E, Meuwissen H, White J, et al. Normal Fibronectin Levels as a Function of Age in the Pediatric Population. Pediatr Res (1983) 17(6):482–5. doi: 10.1203/00006450-198306000-00012
234. Díaz-Alvarez L, Ortega E. The Many Roles of Galectin-3, A Multifaceted Molecule, in Innate Immune Responses Against Pathogens. Mediators Inflammation (2017) 2017:1–10. doi: 10.1155/2017/9247574
235. Puigdellívol M, Allendorf DH, Brown GC. Sialylation and Galectin-3 in Microglia-Mediated Neuroinflammation and Neurodegeneration. Front Cell Neurosci Front Media SA (2020) 14:162. doi: 10.3389/fncel.2020.00162
236. Yi Y-S. Functional Role of Milk Fat Globule-Epidermal Growth Factor VIII in Macrophage-Mediated Inflammatory Responses and Inflammatory/Autoimmune Diseases. Mediators Inflammation (2016) 2016:5628486. doi: 10.1155/2016/5628486
237. Abe T, Shin J, Hosur K, Udey MC, Chavakis T, Hajishengallis G. Regulation of Osteoclast Homeostasis and Inflammatory Bone Loss by MFG-E8. J Immunol (2014) 193(3):1383–91. doi: 10.4049/jimmunol.1400970
238. Du Clos TW. Pentraxins: Structure, Function, and Role in Inflammation. ISRN Inflammation (2013) 2013:379040. doi: 10.1155/2013/379040
239. Lech M, Römmele C, Kulkarni OP, Susanti HE, Migliorini A, Garlanda C, et al. Lack of the Long Pentraxin PTX3 Promotes Autoimmune Lung Disease But Not Glomerulonephritis in Murine Systemic Lupus Erythematosus. PloS One (2011) 6(5). doi: 10.1371/journal.pone.0020118
240. Blatt AZ, Pathan S, Ferreira VP. Properdin: A Tightly Regulated Critical Inflammatory Modulator. Immunol Rev Blackwell Publ Ltd (2016) 274:172–90. doi: 10.1111/imr.12466
241. Dahlbäck B. Vitamin K-Dependent Protein S: Beyond the Protein C Pathway. Semin Thromb Hemost (2018) 44(2):176–84. doi: 10.1055/s-0037-1604092
242. Lopez-Dee Z, Pidcock K, Gutierrez LS. Thrombospondin-1: Multiple Paths to Inflammation. Mediators Inflammation (2011) 2011:296069. doi: 10.1155/2011/296069
243. Gao AG, Lindberg FP, Dimitry JM, Brown EJ, Frazier WA. Thrombospondin Modulates α(V)β3 Function Through Integrin-Associated Protein. J Cell Biol (1996) 135(2):533–44. doi: 10.1083/jcb.135.2.533
244. Carroll K, Gomez C, Shapiro L. Tubby Proteins: The Plot Thickens. Nat Rev Mol Cell Biol Nat Publ Group (2004) 5:55–63. doi: 10.1038/nrm1278
245. Caberoy NB, Li W. Unconventional Secretion of Tubby and Tubby-Like Protein 1. FEBS Lett (2009) 583(18):3057–62. doi: 10.1016/j.febslet.2009.08.015
246. Nakamura K, Casey M, Oey H, Vari F, Stagg J, Gandhi MK, et al. Targeting an Adenosine-Mediated “Don’t Eat Me Signal” Augments Anti-Lymphoma Immunity by Anti-CD20 Monoclonal Antibody. Leukemia (2020) 34(10):2708–21. doi: 10.1038/s41375-020-0811-3
247. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 Signalling Through Macrophage Siglec-10 Is a Target for Cancer Immunotherapy. Nature (2019) 572(7769):392–6. doi: 10.1038/s41586-019-1456-0
248. Boyd NAM, Bradwell AR, Thompson RA. Quantitation of Vitronectin in Serum: Evaluation of Its Usefulness in Routine Clinical Practice. J Clin Pathol (1993) 46(11):1042–5. doi: 10.1136/jcp.46.11.1042
249. Lv Z, Bian Z, Shi L, Niu S, Ha B, Tremblay A, et al. Loss of Cell Surface CD47 Clustering Formation and Binding Avidity to Sirpα Facilitate Apoptotic Cell Clearance by Macrophages. J Immunol (2015) 195(2):661–71. doi: 10.4049/jimmunol.1401719
250. Burger P, Hilarius-Stokman P, De Korte D, Van Den Berg TK, Van Bruggen R. CD47 Functions as a Molecular Switch for Erythrocyte Phagocytosis. Blood (2012) 119(23):5512–21. doi: 10.1182/blood-2011-10-386805
251. Oldenborg PA, Gresham HD, Lindberg FP. CD47-Signal Regulatory Protein Alpha (Sirpalpha) Regulates Fcgamma and Complement Receptor-Mediated Phagocytosis. J Exp Med (2001) 193(7):855–62. doi: 10.1084/jem.193.7.855
252. Fossati-Jimack L, Azeredo Da Silveira S, Moll T, Kina T, Kuypers FA, Oldenborg PA, et al. Selective Increase of Autoimmune Epitope Expression on Aged Erythrocytes in Mice: Implications in Anti-Erythrocyte Autoimmune Responses. J Autoimmun (2002) 18(1):17–25. doi: 10.1006/jaut.2001.0563
253. Gu S, Ni T, Wang J, Liu Y, Fan Q, Wang Y, et al. Cd47 Blockade Inhibits Tumor Progression Through Promoting Phagocytosis of Tumor Cells by M2 Polarized Macrophages in Endometrial Cancer. J Immunol Res (2018) 2018:6156757. doi: 10.1155/2018/6156757
254. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC Class I by the Inhibitory Receptor LILRB1 Suppresses Macrophages and Is a Target of Cancer Immunotherapy. Nat Immunol (2018) 19(1):76–84. doi: 10.1038/s41590-017-0004-z
255. Carlin AF, Uchiyama S, Chang YC, Lewis AL, Nizet V, Varki A. Molecular Mimicry of Host Sialylated Glycans Allows a Bacterial Pathogen to Engage Neutrophil Siglec-9 and Dampen the Innate Immune Response. Blood (2009) 113(14):3333–6. doi: 10.1182/blood-2008-11-187302
256. Wang Y, Neumann H. Alleviation of Neurotoxicity by Microglial Human Siglec-11. J Neurosci (2010) 30(9):3482–8. doi: 10.1523/JNEUROSCI.3940-09.2010
257. Claude J, Linnartz-Gerlach B, Kudin AP, Kunz WS, Neumann H. Microglial CD33-related Siglec-E Inhibits Neurotoxicity by Preventing the Phagocytosis-Associated Oxidative Burst. J Neurosci (2013) 33(46):18270–6. doi: 10.1523/JNEUROSCI.2211-13.2013
258. Ensinck A, Biondi CS, Marini A, García Borrás S, Racca LL, Cotorruelo CM, et al. Effect of Membrane-Bound IgG and Desialysation in the Interaction of Monocytes With Senescent Erythrocytes. Clin Exp Med (2006) 6(3):138–42. doi: 10.1007/s10238-006-0110-y
259. Brown S, Heinisch I, Ross E, Shaw K, Buckley CO, Savill J. Apoptosis Disables CD31-mediated Cell Detachment From Phagocytes Promoting Binding and Engulfment. Nature (2002) 418(6894):200–3. doi: 10.1038/nature00811
260. Wang H, Madariaga ML, Wang S, Van Rooijen N, Oldenborg PA, Yang YG. Lack of CD47 on Nonhematopoietic Cells Induces Split Macrophage Tolerance to CD47null Cells. Proc Natl Acad Sci USA (2007) 104(34):13744–9. doi: 10.1073/pnas.0702881104
261. Tajbakhsh A, Gheibi Hayat SM, Butler AE, Sahebkar A. Effect of Soluble Cleavage Products of Important Receptors/Ligands on Efferocytosis: Their Role in Inflammatory, Autoimmune and Cardiovascular Disease. Ageing Res Rev Elsevier Ireland Ltd (2019) 50:43–57. doi: 10.1016/j.arr.2019.01.007
262. Barclay AN, Wright GJ, Brooke G, Brown MH. CD200 and Membrane Protein Interactions in the Control of Myeloid Cells. Trends Immunol (2002) 23(6):285–90. doi: 10.1016/S1471-4906(02)02223-8
263. Sakai R, Maeda A, Choi TV, Lo PC, Jiaravuthisan P, Shabri AM, et al. Human CD200 Suppresses Macrophage-Mediated Xenogeneic Cytotoxicity and Phagocytosis. Surg Today (2018) 48(1):119–26. doi: 10.1007/s00595-017-1546-2
264. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature (2017) 545(7655):495–9. doi: 10.1038/nature22396
265. Macauley MS, Crocker PR, Paulson JC. Siglec-Mediated Regulation of Immune Cell Function in Disease. Nat Rev Immunol (2014) 14(10):653–66. doi: 10.1038/nri3737
266. Cabral MG, Silva Z, Ligeiro D, Seixas E, Crespo H, Carrascal MA, et al. The Phagocytic Capacity and Immunological Potency of Human Dendritic Cells Is Improved by α2,6-Sialic Acid Deficiency. Immunology (2013) 138(3):235–45. doi: 10.1111/imm.12025
267. Imbert PRC, Saric A, Pedram K, Bertozzi CR, Grinstein S, Freeman SA. An Acquired and Endogenous Glycocalyx Forms a Bidirectional “Don’t Eat” and “Don’t Eat Me” Barrier to Phagocytosis. Curr Biol (2021) 31(1):77–89.e5. doi: 10.1016/j.cub.2020.09.082
268. Bennett MR, Gibson DF, Schwartz SM, Tait JF. Binding and Phagocytosis of Apoptotic Vascular Smooth Muscle Cells Is Mediated in Part by Exposure of Phosphatidylserine. Circ Res (1995) 77(6):1136–42. doi: 10.1161/01.RES.77.6.1136
269. Chang GH-F, Barbaro NM, Pieper RO. Phosphatidylserine-Dependent Phagocytosis of Apoptotic Glioma Cells by Normal Human Microglia, Astrocytes, and Glioma Cells. Neuro Oncol (2000) 2(3):174–83. doi: 10.1215/15228517-2-3-174
270. Liu G, Wang J, Park Y-J, Tsuruta Y, Lorne EF, Zhao X, et al. High Mobility Group Protein-1 Inhibits Phagocytosis of Apoptotic Neutrophils Through Binding to Phosphatidylserine. J Immunol (2008) 181(6):4240–6. doi: 10.4049/jimmunol.181.6.4240
271. Friggeri A, Yang Y, Banerjee S, Park YJ, Liu G, Abraham E. HMGB1 Inhibits Macrophage Activity in Efferocytosis Through Binding to the αvβ3-Integrin. Am J Physiol - Cell Physiol (2010) 299(6):1267–76. doi: 10.1152/ajpcell.00152.2010
272. Nauta AJ, Bottazzi B, Mantovani A, Salvatori G, Kishore U, Schwaeble WJ, et al. Biochemical and Functional Characterization of the Interaction Between Pentraxin 3 and C1q. Eur J Immunol (2003) 33(2):465–73. doi: 10.1002/immu.200310022
273. Baruah P, Dumitriu IE, Peri G, Russo V, Mantovani A, Manfredi AA, et al. The Tissue Pentraxin PTX3 Limits C1q-Mediated Complement Activation and Phagocytosis of Apoptotic Cells by Dendritic Cells. J Leukoc Biol (2006) 80(1):87–95. doi: 10.1189/jlb.0805445
274. Rovere P, Peri G, Fazzini F, Bottazzi B, Doni A, Bondanza A, et al. The Long Pentraxin PTX3 Binds to Apoptotic Cells and Regulates Their Clearance by Antigen-Presenting Dentritic Cells. Blood (2000) 96(13):4300–6. doi: 10.1182/blood.V96.13.4300
275. Van Rossum AP, Fazzini F, Limburg PC, Manfredi AA, Rovere-Querini P, Mantovani A, et al. The Prototypic Tissue Pentraxin PTX3, in Contrast to the Short Pentraxin Serum Amyloid P, Inhibits Phagocytosis of Late Apoptotic Neutrophils by Macrophages. Arthritis Rheum (2004) 50(8):2667–74. doi: 10.1002/art.20370
276. Jeon H, Lee S, Lee WH, Suk K. Analysis of Glial Secretome: The Long Pentraxin PTX3 Modulates Phagocytic Activity of Microglia. J Neuroimmunol (2010) 229(1–2):63–72. doi: 10.1016/j.jneuroim.2010.07.001
277. Ko CY, Chang LH, Lee YC, Sterneck E, Cheng CP, Chen SH, et al. CCAAT/Enhancer Binding Protein Delta (CEBPD) Elevating PTX3 Expression Inhibits Macrophage-Mediated Phagocytosis of Dying Neuron Cells. Neurobiol Aging (2012) 33(2):422.e11–25.
278. Park Y-J, Liu G, Lorne EF, Zhao X, Wang J, Tsuruta Y, et al. PAI-1 Inhibits Neutrophil Efferocytosis. Proc Natl Acad Sci USA (2008) 105(33):11784–9. doi: 10.1073/pnas.0801394105
279. Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, et al. A Soluble Form of the Mer Receptor Tyrosine Kinase Inhibits Macrophage Clearance of Apoptotic Cells and Platelet Aggregation. Blood (2007) 109(3):1026–33. doi: 10.1182/blood-2006-05-021634
280. Law AL, Parinot C, Chatagnon J, Gravez B, Sahel JA, Bhattacharya SS, et al. Cleavage of Mer Tyrosine Kinase (MerTK) From the Cell Surface Contributes to the Regulation of Retinal Phagocytosis. J Biol Chem (2015) 290(8):4941–52. doi: 10.1074/jbc.M114.628297
281. Park YJ, Liu G, Tsuruta Y, Lorne E, Abraham E. Participation of the Urokinase Receptor in Neutrophil Efferocytosis. Blood (2009) 114(4):860–70. doi: 10.1182/blood-2008-12-193524
282. Bae H-B, Tadie J-M, Jiang S, Park DW, Bell CP, Thompson LC, et al. Vitronectin Inhibits Efferocytosis Through Interactions With Apoptotic Cells as Well as With Macrophages. J Immunol (2013) 190(5):2273–81. doi: 10.4049/jimmunol.1200625
283. D’mello V, Singh S, Wu Y, Birge RB. The Urokinase Plasminogen Activator Receptor Promotes Efferocytosis of Apopotic Cells. J Biol Chem (2009) 284(25):17030–8. doi: 10.1074/jbc.M109.010066
284. Kenis H, Van Genderen H, Deckers NM, Lux PAG, Hofstra L, Narula J, et al. Annexin A5 Inhibits Engulfment Through Internalization of PS-expressing Cell Membrane Patches. Exp Cell Res (2006) 312(6):719–26. doi: 10.1016/j.yexcr.2005.11.023
285. Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular Histones are Mediators of Death Through TLR2 and TLR4 in Mouse Fatal Liver Injury. J Immunol (2011) 187(5):2626–31. doi: 10.4049/jimmunol.1003930
286. Friggeri A, Banerjee S, Xie N, Cui H, de Freitas A, Zerfaoui M, et al. Extracellular Histones Inhibit Efferocytosis. Mol Med (2012) 18(5):825–33. doi: 10.2119/molmed.2012.00005
287. Imamura M, Kawasaki T, Savchenko AS, Ohashi R, Jiang S, Miyamoto K, et al. Lipopolysaccharide Induced Expression of Pentraxin 3 in Human Neutrophils and Monocyte-Derived Macrophages. Cell Immunol (2007) 248(2):86–94. doi: 10.1016/j.cellimm.2007.09.003
288. Jeon H, Kim J-H, Kim J-H, Lee W-H, Lee M-S, Suk K. Plasminogen Activator Inhibitor Type 1 Regulates Microglial Motility and Phagocytic Activity. J Neuroinflamm (2012) 9(1):637. doi: 10.1186/1742-2094-9-149
289. Paetau S, Rolova T, Ning L, Gahmberg CG. Neuronal ICAM-5 Inhibits Microglia Adhesion and Phagocytosis and Promotes an Anti-Inflammatory Response in LPS Stimulated Microglia. Front Mol Neurosci (2017) 10:1–12. doi: 10.3389/fnmol.2017.00431
290. Santilli F, Vazzana N, Bucciarelli L, Davi G. Soluble Forms of RAGE in Human Diseases: Clinical and Therapeutical Implications. Curr Med Chem (2009) 16(8):940–52. doi: 10.2174/092986709787581888
291. Ragno P. The Urokinase Receptor: A Ligand or a Receptor? Story of a Sociable Molecule. Cell Mol Life Sci (2006) 63(9):1028–37. doi: 10.1007/s00018-005-5428-1
292. Leventis PA, Grinstein S. The Distribution and Function of Phosphatidylserine in Cellular Membranes. Annu Rev Biophys Annu Rev Biophys (2010) 39:407–27. doi: 10.1146/annurev.biophys.093008.131234
293. Shin HW, Takatsu H. Phosphatidylserine Exposure in Living Cells. Crit Rev Biochem Mol Biol Taylor Francis Ltd (2020) 55:166–78. doi: 10.1080/10409238.2020.1758624
294. Ghiran I, Klickstein LB, Nicholson-Weller A. Calreticulin is At the Surface of Circulating Neutrophils and Uses CD59 as an Adaptor Molecule. J Biol Chem (2003) 278(23):21024–31. doi: 10.1074/jbc.M302306200
295. Flannagan RS, Jaumouillé V, Grinstein S. The Cell Biology of Phagocytosis. Annu Rev Pathol Mech Dis (2012) 7(1):61–98. doi: 10.1146/annurev-pathol-011811-132445
296. Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, Noé F, et al. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front Immunol Front Res Found (2017) 8:1. doi: 10.3389/fimmu.2017.00292
297. Bolasco G, Weinhard L, Boissonnet T, Neujahr R, Gross CT. Three-Dimensional Nanostructure of an Intact Microglia Cell. Front Neuroanat (2018) 12:105. doi: 10.3389/fnana.2018.00105
298. Ishidome T, Yoshida T, Hanayama R. Induction of Live Cell Phagocytosis by a Specific Combination of Inflammatory Stimuli. EBioMedicine (2017) 22:89–99. doi: 10.1016/j.ebiom.2017.07.011
299. Anderson SR, Zhang J, Steele MR, Romero CO, Kautzman AG, Schafer DP, et al. Complement Targets Newborn Retinal Ganglion Cells for Phagocytic Elimination by Microglia. J Neurosci (2019) 39(11):2025–40. doi: 10.1523/JNEUROSCI.1854-18.2018
300. Jitkaew S, Witasp E, Zhang S, Kagan VE, Fadeel B. Induction of Caspase- and Reactive Oxygen Species-Independent Phosphatidylserine Externalization in Primary Human Neutrophils: Role in Macrophage Recognition and Engulfment. J Leukoc Biol (2009) 85(3):427–37. doi: 10.1189/jlb.0408232
301. Frasch SC, Berry KZ, Fernandez-Boyanapalli R, Jin HS, Leslie C, Henson PM, et al. NADPH Oxidase-Dependent Generation of Lysophosphatidylserine Enhances Clearance of Activated and Dying Neutrophils Via G2A. J Biol Chem (2008) 283(48):33736–49. doi: 10.1074/jbc.M807047200
302. Cross AS, Sakarya S, Rifat S, Held TK, Drysdale BE, Grange PA, et al. Recruitment of Murine Neutrophils In Vivo Through Endogenous Sialidase Activity. J Biol Chem (2003) 278(6):4112–20. doi: 10.1074/jbc.M207591200
303. Fischer K, Voelkl S, Berger J, Andreesen R, Pomorski T, Mackensen A. Antigen Recognition Induces Phosphatidylserine Exposure on the Cell Surface of Human CD8+ T Cells. Blood (2006) 108(13):4094–101. doi: 10.1182/blood-2006-03-011742
304. Albacker LA, Karisola P, Chang Y-J, Umetsu SE, Zhou M, Akbari O, et al. Tim-4, A Receptor for Phosphatidylserine, Controls Adaptive Immunity by Regulating the Removal of Antigen-Specific T Cells. J Immunol (2010) 185(11):6839–49. doi: 10.4049/jimmunol.1001360
305. Arandjelovic S, Ravichandran KS. Phagocytosis of Apoptotic Cells in Homeostasis. Nat Immunol (2015) 16(9):907–17. doi: 10.1038/ni.3253
306. Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-Mediated Cleavage of Phospholipid Flippase for Apoptotic Phosphatidylserine Exposure. Science (2014) 344(6188):1164–8. doi: 10.1126/science.1252809
307. Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA, et al. Phosphatidylserine Externalization, “Necroptotic Bodies” Release, and Phagocytosis During Necroptosis. PloS Biol (2017) 15(6):1–23. doi: 10.1371/journal.pbio.2002711
308. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The Clearance of Dead Cells by Efferocytosis. Nat Rev Mol Cell Biol Nat Res (2020) 21:398–414. doi: 10.1038/s41580-020-0232-1
309. Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, et al. TREM2 Sustains Microglial Expansion During Aging and Response to Demyelination. J Clin Invest (2015) 125(5):2161–70. doi: 10.1172/JCI77983
310. Gitik M, Liraz-Zaltsman S, Oldenborg P-A, Reichert F, Rotshenker S. Myelin Down-Regulates Myelin Phagocytosis by Microglia and Macrophages Through Interactions Between CD47 on Myelin and Sirpα (Signal Regulatory Protein-α) on Phagocytes. J Neuroinflamm (2011) 8:24. doi: 10.1186/1742-2094-8-24
311. Lutz AB, Chung WS, Sloan SA, Carson GA, Zhou L, Lovelett E, et al. Schwann Cells Use TAM Receptor-Mediated Phagocytosis in Addition to Autophagy to Clear Myelin in a Mouse Model of Nerve Injury. Proc Natl Acad Sci USA (2017) 114(38):E8072–80. doi: 10.1073/pnas.1710566114
312. Brück W, Friede RL. Anti-Macrophage CR3 Antibody Blocks Myelin Phagocytosis by Macrophages In Vitro. Acta Neuropathol (1990) 80(4):415–8. doi: 10.1007/BF00307696
313. Hisatomi T, Sakamoto T, Sonoda KH, Tsutsumi C, Qiao H, Enaida H, et al. Clearance of Apoptotic Photoreceptors: Elimination of Apoptotic Debris Into the Subretinal Space and Macrophage-Mediated Phagocytosis Via Phosphatidylserine Receptor and Integrin αvβ3. Am J Pathol (2003) 162(6):1869–79. doi: 10.1016/j.ajo.2003.09.027
314. Lampron A, Larochelle A, Laflamme N, Préfontaine P, Plante MM, Sánchez MG, et al. Inefficient Clearance of Myelin Debris by Microglia Impairs Remyelinating Processes. J Exp Med (2015) 212(4):481–95. doi: 10.1084/jem.20141656
315. Khandelwal S, Van Rooijen N, Saxena RK. Reduced Expression of CD47 During Murine Red Blood Cell (RBC) Senescence and its Role in RBC Clearance From the Circulation. Transfusion (2007) 47(9):1725–32. doi: 10.1111/j.1537-2995.2007.01348.x
316. Lutz HU, Bogdanova A. Mechanisms Tagging Senescent Red Blood Cells for Clearance in Healthy Humans. Front Physiol (2013) 4:1–15. doi: 10.3389/fphys.2013.00387
317. Hong C, Kidani Y, A-Gonzalez N, Phung T, Ito A, Rong X, et al. Coordinate Regulation of Neutrophil Homeostasis by Liver X Receptors in Mice. J Clin Invest (2012) 122(1):337–47. doi: 10.1172/JCI58393
318. Egashira M, Hirota Y, Shimizu-Hirota R, Saito-Fujita T, Haraguchi H, Matsumoto L, et al. F4/80+ Macrophages Contribute to Clearance of Senescent Cells in the Mouse Postpartum Uterus. Endocrinol (2017) 158(7):2344–53. doi: 10.1210/en.2016-1886
319. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and Tumour Clearance is Triggered by p53 Restoration in Murine Liver Carcinomas. Nature (2007) 445(7128):656–60. doi: 10.1038/nature05529
320. Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J Neurosci (2015) 35(38):13029–42. doi: 10.1523/JNEUROSCI.1698-15.2015
321. Linnartz-Gerlach B, Bodea LG, Klaus C, Ginolhac A, Halder R, Sinkkonen L, et al. TREM2 Triggers Microglial Density and Age-Related Neuronal Loss. Glia (2019) 67(3):539–50. doi: 10.1002/glia.23563
322. Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis Checkpoints as New Targets for Cancer Immunotherapy. Nat Rev Cancer Nat Publ Group (2019) 19:568–86. doi: 10.1038/s41568-019-0183-z
323. Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, et al. Calreticulin Is the Dominant Pro-Phagocytic Signal on Multiple Human Cancers and Is Counterbalanced by CD47. Sci Transl Med (2010) 2(63):63–94. doi: 10.1126/scitranslmed.3001375
324. Wang X, Wang Y, Hu J, Xu H. An Antitumor Peptide RS17-targeted CD47, Design, Synthesis, and Antitumor Activity. Cancer Med (2021) 10(6):2125–36. doi: 10.1002/cam4.3768
325. Lin H, Kryczek I, Li S, Green MD, Ali A, Hamasha R, et al. Stanniocalcin 1 Is a Phagocytosis Checkpoint Driving Tumor Immune Resistance. Cancer Cell (2021) 39(4):480–93.e6. doi: 10.1016/j.ccell.2020.12.023
326. Rodrigues E, Macauley MS. Hypersialylation in Cancer: Modulation of Inflammation and Therapeutic Opportunities. Cancers MDPI AG (2018) 10(6):207. doi: 10.3390/cancers10060207
327. Birkle T, Brown GC. I’m Infected, Eat Me! Innate Immunity Mediated by Live, Infected Cells Signaling To be Phagocytosed. Infect Immun (2021) 89(5). doi: 10.1128/IAI.00476-20
328. Qin J, Zhang G, Zhang X, Tan B, Lv Z, Liu M, et al. TLR-Activated Gap Junction Channels Protect Mice Against Bacterial Infection Through Extracellular Udp Release. J Immunol (2016) 196(4):1790–8. doi: 10.4049/jimmunol.1501629
329. Ren H, Teng Y, Tan B, Zhang X, Jiang W, Liu M, et al. Toll-Like Receptor-Triggered Calcium Mobilization Protects Mice Against Bacterial Infection Through Extracellular ATP Release. Infect Immun (2014) 82(12):5076–85. doi: 10.1128/IAI.02546-14
330. Li R, Tan B, Yan Y, Ma X, Zhang N, Zhang Z, et al. Extracellular UDP and P2Y 6 Function as a Danger Signal To Protect Mice From Vesicular Stomatitis Virus Infection Through an Increase in IFN-β Production. J Immunol (2014) 193(9):4515–26. doi: 10.4049/jimmunol.1301930
331. Hofmann P, Sprenger H, Kaufmann A, Bender A, Hasse C, Nain M, et al. Susceptibility of Mononuclear Phagocytes to Influenza A Virus Infection and Possible Role in the Antiviral Response. J Leukoc Biol (1997) 61(4):408–14. doi: 10.1002/jlb.61.4.408
332. Liu CJ, Chuang WL, Sheen IS, Wang HY, Chen CY, Tseng KC, et al. Efficacy of Ledipasvir and Sofosbuvir Treatment of HCV Infection in Patients Coinfected With Hbv. Gastroenterology (2018) 154(4):989–97. doi: 10.1053/j.gastro.2017.11.011
333. Cho NH, Seong SY, Huh MS, Han TH, Koh YS, Choi MS, et al. Expression of Chemokine Genes in Murine Macrophages Infected With Orientia Tsutsugamushi. Infect Immun (2000) 68(2):594–602. doi: 10.1128/IAI.68.2.594-602.2000
334. Chua BA, Ngo JA, Situ K, Ramirez CM, Nakano H, Morizono K. Protein S and Gas6 Induce Efferocytosis of HIV-1-Infected Cells. Virology (2018) 515:176–90. doi: 10.1016/j.virol.2017.12.025
335. Tufail Y, Cook D, Fourgeaud L, Powers CJ, Merten K, Clark CL, et al. Phosphatidylserine Exposure Controls Viral Innate Immune Responses by Microglia. Neuron (2017) 93(3):574–86.e8. doi: 10.1016/j.neuron.2016.12.021
336. Goth SR, Stephens RS. Rapid, Transient Phosphatidylserine Externalization Induced in Host Cells by Infection With Chlamydia Spp. Infect Immun (2001) 69(2):1109–19. doi: 10.1128/IAI.69.2.1109-1119.2001
337. Jo SH, Choi JA, Lim YJ, Lee J, Cho SN, Oh SM, et al. Calreticulin Modulates the Intracellular Survival of Mycobacteria by Regulating ER-Stress-Mediated Apoptosis. Oncotarget (2017) 8(35):58686–98. doi: 10.18632/oncotarget.17419
338. Zhu J, Newkirk MM. Viral Induction of the Human Autoantigen Calreticulin. Clin Investig Med (1994) 17(3):196–205.
339. Ayi K, Lu Z, Serghides L, Ho JM, Finney C, Wang JCY, et al. Cd47-Sirpα Interactions Regulate Macrophage Uptake of Plasmodium Falciparum-Infected Erythrocytes and Clearance of Malaria In Vivo. Infect Immun (2016) 84(7):2002–11. doi: 10.1128/IAI.01426-15
340. Watanabe Y, Shiratsuchi A, Shimizu K, Takizawa T, Nakanishi Y. Role of Phosphatidylserine Exposure and Sugar Chain Desialylation At the Surface of Influenza Virus-Infected Cells in Efficient Phagocytosis by Macrophages. J Biol Chem (2002) 277(20):18222–8. doi: 10.1074/jbc.M201074200
341. van Strijp JA, van Kessel KP, Miltenburg LA, Fluit AC, Verhoef J. Attachment of Human Polymorphonuclear Leukocytes to Herpes Simplex Virus-Infected Fibroblasts Mediated by Antibody-Independent Complement Activation. J Virol (1988) 62(3):847–50. doi: 10.1128/JVI.62.3.847-850.1988
342. Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A Complement–Microglial Axis Drives Synapse Loss During Virus-Induced Memory Impairment. Nature (2016) 534(7608):538–43. doi: 10.1038/nature18283
343. Chung W-S, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes Mediate Synapse Elimination Through MEGF10 and MERTK Pathways. Nature (2013) 504(7480):394–400. doi: 10.1038/nature12776
344. Sipe GO, Lowery RL, Tremblay M, Kelly EA, Lamantia CE, Majewska AK. Microglial P2Y12 Is Necessary for Synaptic Plasticity in Mouse Visual Cortex. Nat Commun (2016) 7:10905. doi: 10.1038/ncomms10905
345. Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, et al. Cd47 Protects Synapses From Excess Microglia-Mediated Pruning During Development. Neuron (2018) 100(1):120–34.e6. doi: 10.1016/j.neuron.2018.09.017
346. Lee E, Chung WS. Glial Control of Synapse Number in Healthy and Diseased Brain. Front Cell Neurosci (2019) 13:1–8. doi: 10.3389/fncel.2019.00042
347. Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity (2018) 48(5):979–991.e8. doi: 10.1016/j.immuni.2018.04.016
348. Vilalta A, Brown GC. Neurophagy, the Phagocytosis of Live Neurons and Synapses by Glia, Contributes to Brain Development and Disease. FEBS J (2017) 285(19):3566–75. doi: 10.1111/febs.14323
349. Butler CA, Popescu AS, Kitchener EJA, Allendorf DH, Puigdellívol M, Brown GC. Microglial Phagocytosis of Neurons in Neurodegeneration, and Its Regulation. J Neurochem (2021). doi: 10.1111/jnc.15327
350. Charles JF, Humphrey MB, Zhao X, Quarles E, Nakamura MC, Aderem A, et al. The Innate Immune Response to Salmonella Enterica Serovar Typhimurium by Macrophages Is Dependent on TREM2-DAP. Infect Immun (2008) 76(6):2439–47. doi: 10.1128/IAI.00115-08
351. Jekabsone A, Mander PK, Tickler A, Sharpe M, Brown GC. Fibrillar Beta-Amyloid Peptide Aβ1-40 Activates Microglial Proliferation Via Stimulating TNF-α Release and H2O2 Derived From NADPH Oxidase: A Cell Culture Study. J Neuroinflamm (2006), 3. doi: 10.1186/1742-2094-3-24
352. Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, et al. Phosphatidylserine is a Global Immunosuppressive Signal in Efferocytosis, Infectious Disease, and Cancer. Cell Death Diff Nat Publ Group (2016) 23:962–78. doi: 10.1038/cdd.2016.11
353. Cai B, Kasikara C, Doran AC, Ramakrishnan R, Birge RB, Tabas I. MerTK Signaling in Macrophages Promotes the Synthesis of Inflammation Resolution Mediators by Suppressing CaMKII Activity. Sci Signal (2018) 11(549):eaar3721. doi: 10.1126/scisignal.aar3721
354. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat Med (2007) 13(1):54–61. doi: 10.1038/nm1523
355. Diaz-Aparicio I, Paris I, Sierra-Torre V, Plaza-Zabala A, Rodríguez-Iglesias N, Márquez-Ropero M, et al. Microglia Actively Remodel Adult Hippocampal Neurogenesis Through the Phagocytosis Secretome. J Neurosci (2020) 40(7):1453–82. doi: 10.1523/JNEUROSCI.0993-19.2019
356. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity (2017) 47(3):566–81.e9. doi: 10.1016/j.immuni.2017.08.008
357. Ayata P, Badimon A, Strasburger HJ, Duff MK, Montgomery SE, Loh YHE, et al. Epigenetic Regulation of Brain Region-Specific Microglia Clearance Activity. Nat Neurosci (2018) 21(8):1049–60. doi: 10.1038/s41593-018-0192-3
358. Tsokos GC, Lo MS, Reis PC, Sullivan KE. New Insights Into the Immunopathogenesis of Systemic Lupus Erythematosus. Nat Rev Rheumatol Nat Publ Group (2016) 12:716–30. doi: 10.1038/nrrheum.2016.186
359. Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K, et al. Identification of a Microglia Phenotype Supportive of Remyelination. Glia (2012) 60(2):306–21. doi: 10.1002/glia.21266
360. Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. Triggering Receptor Expressed on Myeloid Cells 2 (Trem2) Deficiency Attenuates Phagocytic Activities of Microglia and Exacerbates Ischemic Damage in Experimental Stroke. J Neurosci (2015) 35(8):3384. doi: 10.1523/JNEUROSCI.2620-14.2015
361. Smolle MA, Pichler M. Inflammation, Phagocytosis and Cancer: Another Step in the CD47 Act. J Thorac Dis (2017) 9(8):2279–82. doi: 10.21037/jtd.2017.07.38
362. Yurdagul A, Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front Cardiovasc Med Front Media SA (2018) 4:1. doi: 10.3389/fcvm.2017.00086
363. White CJ, Gallin JI. Phagocyte Defects. Clin Immunol Immunopathol (1986) 40(1):50–61. doi: 10.1016/0090-1229(86)90068-1
364. Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang H-Y, et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia Via Complement Activation. Cell (2016) 165(4):921–35. doi: 10.1016/j.cell.2016.04.001
365. Williams PA, Tribble JR, Pepper KW, Cross SD, Morgan BP, Morgan JE, et al. Inhibition of the Classical Pathway of the Complement Cascade Prevents Early Dendritic and Synaptic Degeneration in Glaucoma. Mol Neurodegener (2016) 11(1):26. doi: 10.1186/s13024-016-0091-6
366. Segawa K, Suzuki J, Nagata S. Constitutive Exposure of Phosphatidylserine on Viable Cells. Proc Natl Acad Sci USA (2011) 108(48):19246–51. doi: 10.1073/pnas.1114799108
367. Freeman SA, Grinstein S. Phagocytosis: Receptors, Signal Integration, and the Cytoskeleton. Immunol Rev (2014) 262(1):193–215. doi: 10.1111/imr.12212
368. Parente R, Clark SJ, Inforzato A, Day AJ. Complement Factor H in Host Defense and Immune Evasion. Cell Mol Life Sci Birkhauser Verlag AG (2017) 74:1605–24. doi: 10.1007/s00018-016-2418-4
Keywords: phagocytosis, cell, signal, opsonin, immunity, cancer, neurodegeneration, signalling
Citation: Cockram TOJ, Dundee JM, Popescu AS and Brown GC (2021) The Phagocytic Code Regulating Phagocytosis of Mammalian Cells. Front. Immunol. 12:629979. doi: 10.3389/fimmu.2021.629979
Received: 16 November 2020; Accepted: 18 May 2021;
Published: 09 June 2021.
Edited by:
Alexandre Corthay, Oslo University Hospital, NorwayReviewed by:
Christine Gaboriaud, UMR5075 Institut de Biologie Structurale (IBS), FranceAmanda Sierra, Achucarro Basque Center for Neuroscience, Spain
Vitaly Balan, Refuge Biotechnologies Inc., United States
Copyright © 2021 Cockram, Dundee, Popescu and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guy C. Brown, Z2NiM0BjYW0uYWMudWs=