 Deepak Salem
Deepak Salem Manoj Chelvanambi
Manoj Chelvanambi Walter J. Storkus
Walter J. Storkus Ronald J. Fecek
Ronald J. Fecek
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 04 March 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.629519
This article is part of the Research Topic Advances in Understanding the Role of Tertiary Lymphoid Structures in Cancer Immunotherapy View all 9 articles
Recent advances in immunotherapy have enabled rapid evolution of novel interventional approaches designed to reinvigorate and expand patient immune responses against cancer. An emerging approach in cancer immunology involves the conditional induction of tertiary lymphoid structures (TLS), which are non-encapsulated ectopic lymphoid structures forming at sites of chronic, pathologic inflammation. Cutaneous melanoma (CM), a highly-immunogenic form of solid cancer, continues to rise in both incidence and mortality rate, with recent reports supporting a positive correlation between the presence of TLS in melanoma and beneficial treatment outcomes amongst advanced-stage patients. In this context, TLS in CM are postulated to serve as dynamic centers for the initiation of robust anti-tumor responses within affected regions of active disease. Given their potential importance to patient outcome, significant effort has been recently devoted to gaining a better understanding of TLS neogenesis and the influence these lymphoid organs exert within the tumor microenvironment. Here, we briefly review TLS structure, function, and response to treatment in the setting of CM. To uncover potential tumor-intrinsic mechanisms that regulate TLS formation, we have taken the novel perspective of evaluating TLS induction in melanomas impacted by common driver mutations in BRAF, PTEN, NRAS, KIT, PRDM1, and MITF. Through analysis of The Cancer Genome Atlas (TCGA), we show expression of DNA repair proteins (DRPs) including BRCA1, PAXIP, ERCC1, ERCC2, ERCC3, MSH2, and PMS2 to be negatively correlated with expression of pro-TLS genes, suggesting DRP loss may favor TLS development in support of improved patient outcome and patient response to interventional immunotherapy.
Cutaneous melanoma (CM) is a deadly cancer that arises from molecular alterations in melanocytes, the pigment producing cells of the skin (1). Though CM accounts for <5% of all skin cancer cases, it accounts for the most deaths (2). Over the past 30 years, the incidence of CM has steadily increased and in 2020, there will be an estimated 100,350 new cases of CM with an estimated 6,850 deaths related to this disease (3, 4). The identification and presence of tumor infiltrating lymphocytes (TIL) in CM correlates with improved prognosis, supporting assertions that CM is an immunogenic cancer (5, 6). One reason for the strong immune system response to CM is believed to reflect it having one of the highest rates of tumor mutational burden (TMB) amongst solid cancers (7–9). The high TMB results in the generation of novel mutated neoantigens, which have not been subjected to central tolerance mechanisms shaping the repertoire of the adaptive immune system (9). Hence, tumors expressing high numbers of neoantigens tend to elicit more robust anti-tumor T cell responses in concert with a pro-inflammatory TME (10).
The presence of inflammation and particularly CD8+ effector T cells in CM may predict a favorable clinical response to immune checkpoint inhibitors (ICI), additionally reinforcing the important role the immune system plays in melanoma (11). Furthermore, because of their clinical efficacy, ICI such as ipilimumab (anti-cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) monoclonal antibody (mAb)), pembrolizumab, and nivolumab (both anti-programmed death-1 (PD-1) mAbs) when applied as monotherapies or combinational therapies have dominated recent immunotherapeutic clinical trial designs and are expected to continue to do so for many years to come (12–14). Nevertheless, most patients treated with these advanced immunotherapeutic agents exhibit intrinsic or acquired resistance to treatment (12, 15), emphasizing the need for an improved understanding of the dynamic interactions between the immune system and CM in the TME for the design of improved interventional approaches.
An exciting new paradigm in cancer immunology involves the role of TLS as regulators of disease progression and effective immunotherapy (16). TLS are ectopic lymphoid organs which are similar in function to secondary lymphoid organs (SLO), such as lymph nodes, but they form in areas of the body that do not normally house lymphoid tissue (17, 18). In this regard, SLO are considered “hard-wired” in forming at developmentally-programmed anatomic locations, while TLS evolve adaptively, forming in peripheral tissue sites impacted by chronic inflammation. Development of TLS in affected tissues is believed to be initiated by a complex crosstalk between recruited inflammatory leukocytes, local stromal cells, and a local source of sustained tissue insult (i.e., unresolved pathogens, infected cells, or mutated cells) (19).
TLS consist of organized aggregates of T cells, B cells, dendritic cells (DC), follicular dendritic cells (FDC), T follicular helper (Tfh) cells, specialized stromal fibroblasts, and high endothelial venules (HEV) (20, 21). When compared with SLO, TLS are non-encapsulated which is thought to facilitate rapid antigen presentation at the site of inflammation and, in the context of cancer, allows the locally primed immune system to quickly mediate surveillance of cognate tumor-associated (neo)antigens (18). Classic mature TLS exhibit compartmentalized zones for immune specialization, such as B cell follicles surrounding germinal centers (GC) containing rapidly proliferating/differentiating B cells (producing antibodies) and distinct T cell zones enriched in conventional DC-LAMP+ DC, akin to those found in SLO (22, 23). Non-classic TLS which are deficient in B cells/GC and composed largely of T cells and DC clustered around HEV have also been described in human cancers (24). Remarkably, both classic and non-classic TLS have been reported as prognostic biomarkers of improved clinical outcome amongst cancer patients (25). It is also important to note that TLS cellular composition is highly-variable over time, being impacted by local alterations in newly-recruited immune infiltrates and cytokine/chemokine profiles in the progressive/therapeutic TME (26–28).
Lymphangiogenesis is also strongly associated with TLS formation and the formation of HEVs that express peripheral node addressins (PNAd), a ligand for L-selectin, along with CCR7-ligand chemokine CCL21, characteristic of TLS formation vs. simple acute local inflammation (28, 29). Expression of CCL19, CCL21, ICAM-1, and MAdCAM on TLS-associated HEVs enables extravasation of circulating naïve CCR7+ T cells, as well as, CCR7+ DC into the TME (28). In a complementary fashion, expression of TLS-derived CXCL13 recruits naïve CXCR5+ B cells into the TME (18, 29). Once recruited, naïve B and T cells are exposed to cancer (neo)antigens presented by dendritic cell populations, allowing them to differentiate into antibody-producing plasma cells or effector T cells, respectively (30–33). Like conventional DC, memory B cells in classical TLS can serves as effective (neo)antigen-presenting cells to naïve and memory anti-tumor CD8+ T cells (32, 33).
TLS have a prognostic, albeit dichotomous, value – in cancer, TLS are generally considered beneficial to the mounting of productive anti-tumor T and B cell responses, but in auto-immune disease such as rheumatoid arthritis, TLS potentiate the immune mediated attack on normal cells where they are considered detrimental (16–18). Furthermore, not all TLS are created operationally equal, and the composition of TLS-component immune subsets must be considered in interpreting impact on local disease pathology. Hence, the presence of regulatory immune cells, such as T regulatory cells (Treg) and myeloid-derived suppressor cells (MDSC) in TLS impacts immunobiologic output and disease severity (26). Although in the autoimmune setting, dominance of suppressor cell populations in TLS ameliorates disease severity and represents a preferred endpoint for treatment intervention (34) in cancer patients such immune deviation is associated with immune tolerance, tumor immune escape and disease progression (35, 36).
The generation of TLS shares many similarities with SLO generation, including the presence of complex interactions between mesenchymal lymphoid tissue stromal organizer (LTo) cells and hematopoietic lymphoid tissue inducer (LTi) cells (16, 37). LTβR+ (Lymphotoxin Beta receptor), PDPN+(podoplanin+), LTo cells associated with chronic or acute pathologically inflamed tissue express chemokines and adhesion molecules to attract hematopoietic cells locally (38, 39). Here, TLS neogenesis is initiated with the production of CXCL13, an important mediator of lymphoneogenesis (40). CXCL13, when secreted by activated DCs, stromal cells, and TGF-β stimulated CD8+ T cells, recruits and upregulates lymphotoxin (LTα1β2) on LTi cells (41–44). In CM, CXCL13 has been shown to be over-expressed in primary cells and metastases (45). In addition to CXCL13, IL-7 secretion from stromal cells is another important inducer of lymphotoxin expression on LTi cells (46). Lymphotoxin signaling between LTi cells, and LTβR+ LTo stromal cells is a critical step leading to the upregulation of pro-lymphangiogenic factors, such as VEGF-C, CXCL12, CXCL13, CCL19, and CCL21 (16, 38). The expression of CXCL13, CCL19, and CCL21 from TLS associated stromal cells following lymphotoxin signaling recruits naïve lymphocytes and synergizes with the upregulation of stromal cell/vascular adhesion molecules, facilitating TLS organization (37, 47, 48).
Secretion of these homeostatic chemokines from stromal cells, DCs, and other cells acting as functional LTo cells is important in TLS formation. Activated DCs have been investigated as strong secretors of pro-inflammatory cytokines, such as LTα and LIGHT (aka TNFSF14) (48). Furthermore, B cells in TLS-associated GC have also been reported to serve as strong producers of LIGHT, which is believed to sustain TLS durability in situ (49).
VEGF-C is another essential mediator of TLS development (17, 28) and a promoter of lymphangiogenesis (11). It is secreted from activated stromal cells and is a potent inducer of HEV formation (50). HEV formation following VEGF-C stimulation and further HEV-DC lymphotoxin signaling allows for the recruitment of lymphocytes from the circulating blood stream (51). VEGF-C secretion can be induced by IL-6, another pro-inflammatory cytokine secreted by activated DCs (52, 53).
The importance of stromal cells in TLS neogenesis is again highlighted by IL-17 signaling induced by Th17 cells (54, 55). Th17 cells, functioning as LTi cells, can secrete IL-17, and IL-22, promoting the stroma to induce expression of CXCL12 and CXCL13 (56, 57). B cells also have been shown to function as LTi cells, secreting IL-22, and regulating TLS formation (49). M1 macrophages, functioning as LTi cells, have also been shown to control TLS formation in colorectal carcinoma by secretion of the pro-inflammatory cytokine IL-36γ (58). M1 macrophages in rat models of chronic graft rejection additionally can produce high levels of LTα and TNF-α, thereby functioning as LTi cells (59). IL-13, another inflammatory cytokine present in the TME, has been implicated in stromal cell regulation, allowing for the development of PDPN+ immunofibroblasts that function as LTo for TLS development (60).
Additionally, LIGHT/TNFSF14, a T cell costimulatory molecule, can bind LTβR, and initiate lymphotoxin signaling in stromal cells resulting in lymphangiogenesis (61). High expression of LIGHT with expression of LTβR on target stromal cells in metastatic CM was found to associate with CCL21 expression from the stroma and significant T cell infiltration (62). Along with the lymphotoxin family (LTα1β2, LTα3, and LIGHT), TNF-α, another important cytokine associated with improved therapeutic response in CM (63), is also able to induce CCL21 expression (64). This is notable in CM, as tumor derived TNF-α was found to be secreted by murine B16 melanoma cells (65).
The presence of DC-Lamp+ DC, CD20+ B cells, and CD3+ T cells has been traditionally used to define TLS (16, 17). Additionally, transcriptional profiling for specific chemokine signatures has proven successful in discerning TLS in the TME. In CM, the presence of DC-Lamp+ DC and a 12-gene chemokine signature in the TME can effectively identify TLS+ tumors (16, 66–68).
The presence of high-levels of DC-LAMP+ and OX40+ lymphocytes in TLS in patients with stage Ia – IIIa CM has been associated with improved patient survival (66). In patients with metastatic melanoma, classic TLS with well-defined T cell and B cell/GC zones tend to be more commonly identified (69). The observation that CM metastases may have more developed TLS is interesting given that metastatic CM is genetically more complex than primary CM in exhibiting a higher TMB (7, 70). Interestingly, Posch et al. (71) reported that the TLS in colorectal carcinoma patients with microsatellite instability-high (MSI-H) tumor burden had higher numbers of “classical” TLS containing GC. Indeed, the higher TMB in metastatic CM has been associated with stronger inflammatory immune responses and patient responsiveness to ICI-based immunotherapy (72).
While the idea that advanced disease with higher TMB may be linked to TLS neogenesis, which is in turn associated with positive therapeutic response to anti-PD-1 and anti-CTLA-4 therapy (33, 73), is speculative, the concept of combining ICI with treatments promoting TLS neogenesis for improved clinical outcome is attractive and empirically testable. In advance of the development of such combination modality protocols, we note that there are currently two clinical trials with the induction of TLS in the TME as a clinical endpoint. One is assessing trustuzumab in the neoadjuvant setting for breast cancer patients (NCT03144947), with the other investigating the efficacy of nivolumab plus relatimab (anti-LAG3 mAb) in soft-tissue sarcoma (NCT04095208).
The identification of dominant driver mutations in melanoma initiated the development of successful targeted therapeutics against constitutively activated oncogenes (74, 75). Furthermore, BRAF, KIT, and NRAS mutational status predicts therapeutic response in CM patients (76).
The propensity for CM, a mutationally complex cancer, to form TLS is intriguing. Using the rich mutational data compiled in The Cancer Genome Atlas (TCGA), Thorsson et al. (77) classified CM into immune-oriented cohorts that expressed specific gene expression and immunologic responses. The immune expression biomarkers employed in this study included those predicted to promote TLS formation, including pro-angiogenic genes, high M1 macrophage ratio and high CD8+ TIL content (77). These pro-inflammatory immune features associated with notable driver mutations in CM may explain, at least in part, CM patient propensity to form TLS within the TME.
Recent clinical and pre-clinical reports have demonstrated that the loss of DNA repair protein expression is associated with increased emergence of neoantigens in a variety of tumor types and models (78–82). Germano et al. demonstrated accumulated neoantigens and improved immune surveillance in colorectal, breast, and pancreatic mouse cancer cell lines deficient in DNA mismatch repair (MMR-deficient). In fact, MMR-deficient tumors had increased infiltration of CD8+ and CD4+ T cells when compared to MMR-proficient tumors (78). Green et al. (82) similarly reported increased prevalence of CD8+ tumor-infiltrating lymphocytes in patient breast cancer tumors that expressed low levels of the XRCC1, ATM, and BRCA1 proteins involved with DNA damage response.
The tumor-associated inflammatory impact resulting from the loss of DNA repair and increased neoantigen expression presents an exploitable immunotherapeutic opportunity. Defects in DNA mismatch repair proteins may act as novel biomarkers for ICI efficacy, specifically anti-PD-1/PD-L1 immunotherapies (83). In a phase 2 clinical trial (NCT01876511), patients with progressive metastatic CRC with MMR-deficiency had increased immune-related objective response and progression-free survival when compared to patients with progressive metastatic CRC with MMR-proficiency after receiving pembrolizumab immunotherapy (84, 85). Additionally, in a mouse model of CRC, it was reported that anti-PD-l, and anti-CTLA-4 ICI significantly impaired the growth of MMR-deficient tumors compared to MMR-proficient tumors (78). Notably, increased levels of CD8+ TILs were found in the MMR-deficient tumors.
The presence of TLS in CM may provide a site for in situ neoantigen-specific CD8+ T cell clonal expansion, ultimately resulting in an effective antitumor adaptive immune response. Given that tumors with decreased DNA repair pathways may be pre-disposed for increased presence of CD8+ T cells, we hypothesize that the TMB of melanoma may be a predictor for TLS neogenesis. By analyzing the genome from two cohorts of cutaneous melanoma patients compiled in the TCGA using cBio Portal, we found that the expression of many DNA repair genes to be negatively correlated with expression of TLS-associated genes (86, 87) (Figure 1). We found a statistically significant inverse correlation between at least eight TLS-associated genes and DNA repair protein elements BRCA1, PAXIP, ERCC1, ERCC2, ERCC3, MSH2, and PMS2. These latter genes transcribe proteins involved in pathways of DNA repair systems including homologous recombination (BRCA1, PAXIP), nucleotide excision repair (ERCC1, ERCC2, ERCC3), and MMR (MSH2, PMS2). Although further in-depth studies are required, these preliminary data appear to support the potential utility of TMB as a predictor for TLS formation in CM.
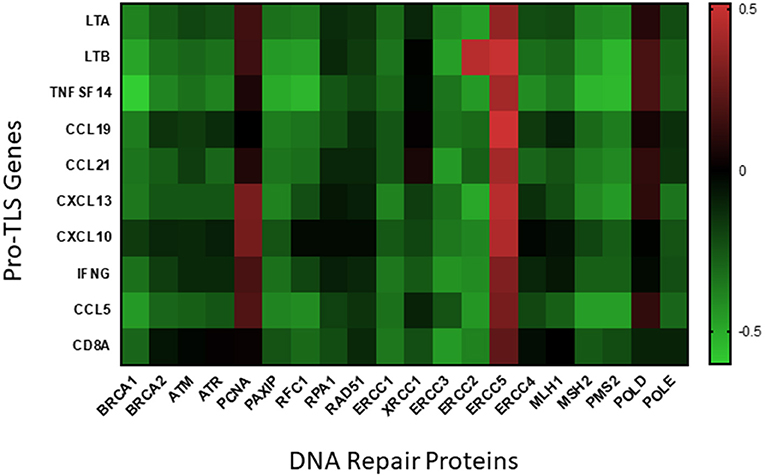
Figure 1. Correlation of DNA repair proteins with pro-TLS genes. TCGA data from two cohorts of cutaneous melanoma patients (SKCM_DFCI 2015 and SKCM_MSKCC 2014) was included to analyze if expression of DNA repair proteins correlated with the expression of inflammatory markers within the TME. TCGA data was uploaded onto cBio Portal, a published TCGA data analysis tool (86, 87), and correlative mRNA expression of the above inflammatory and DNA repair proteins visualized using linear regression models. Pearson's coefficients from each correlation is visualized as a heatmap (created using the Prism 8 software) above with positive correlations indicated in red and negative correlations indicated in green.
Cutaneous melanoma has significantly higher rates of somatic mutations than other cancers. The genes frequently mutated in CM resulting in melanoma driver mutations include BRAF, MITF, NRAS, KIT, PTEN, and PRDM1 (88). Extending these analyses to downstream driver mutations may uncover potential predictors for TLS formation in CM. Here, we briefly review molecular mechanisms downstream of driver mutations for their potential impact on TLS formation in CM (Figure 2).
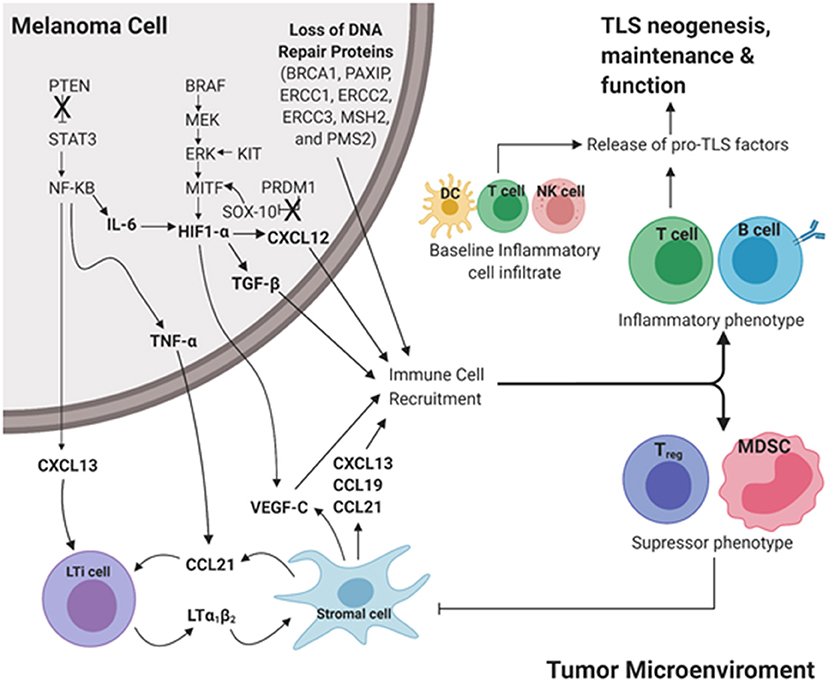
Figure 2. Proposed intrinsic molecular mechanisms that contribute to TLS development downstream of common driver mutations in cutaneous melanoma. Pro-inflammatory cues initiate TLS neogenesis in the peripheral tumor microenvironment (TME). In a melanoma tumor cell, loss of tumor suppressor genes PTEN and PRDM1 results in increased expression of STAT3 and SOX-10, respectively. Gain of function in the proto-oncogenes BRAF, KIT, MITF results in upregulation of MEK, ERK, and HIF-1α, respectively. Culmination of these mutations results in increased expression of pro-TLS cytokines (CCL21, CCL19, CXCL13, LTa1b2) and pro-inflammatory immune cell recruitment (T cell, B cell) contributing to the development, maintenance, and function of TLS in the TME (Inflammatory phenotype). The lack of proinflammatory cues in the TME contributes to the recruitment and maintenance of immunoregulation (Treg, MDSC) resulting in an immunosuppressive TME (Immunosuppressive phenotype). The ever-changing balance of pro-inflammatory vs. regulatory immune function in TLS likely dictates the anti- vs. pro-tumor influence TLS play in disease outcome. LTi cell, Lymphoid tissue inducer cell; MDSC, Myeloid derived suppressor cell; DC, Dendritic cell; NK cell, Natural killer cell. Created with BioRender.com.
Microphthalmia-associated transcription factor (MITF) encodes an important transcription factor for early melanocyte development, and when mutated, functions as a constitutively active oncoprotein to promote tumorigenesis (89). MITF is downstream of the MAPK signaling pathway and also is able to be activated by other transcription factors that can be mutated in CM, such as SRY-box 10 (SOX-10) and cyclic adenosine monophosphate response element-binding protein (CREB) (90).
MITF has been shown to have roles in anti-tumor immunity. MITF knockdown in B16.F10 melanoma cells results in decreased expression of (TLS-promoting) CCL21 and CXCL10 chemokine levels, in association with reduced immune infiltration/inflammation in the TME and enhanced tumor progression (91). MITF is additionally able to upregulate HIF-1α expression by upstream promoter binding of the HIF-1α gene (92, 93). HIF-1α is a potent inducer of VEGF-C, TGF-β, and CXCL12 (93–95) which are known to promote HEV formation and to stimulate LTi cells, both supportive of TLS formation (16, 43, 94).
The proto-oncogene BRAF codes for a serine/threonine protein kinase involved in the MAPK pathway (96). BRAF mutations are present in 66% of malignant melanoma, with the V600E mutant being the most prevalent (97). The use of FDA-approved targeted BRAFV600E inhibitors (BRAFi) such as dabrafenib and vemurafenib revealed the immunomodulating role of oncogenic BRAF in the TME. BRAFi promote CD4+ and CD8+ T cell infiltration into human metastatic melanoma in addition to increasing expression of melanocyte differentiation antigens by melanoma cells, leading to enhanced recognition by antigen-specific T cells (98–100). Furthermore, BRAFV600E regulates IL-1α/β transcription in melanocytes leading to upregulation of immunosuppressive genes such as PD-1 ligands and COX-2 in tumor associated fibroblasts (101). BRAF inhibition as well as knockdown of BRAFV600E in multiple patient-derived melanoma cell lines blocked IL-1α production. Although no direct links between BRAFV600E and the production of TLS-associated factors (CCL19, CCL21, CXCL13, LTα/β, or LIGHT) have been reported it stands to reason that BRAFV600E may promote/support TLS formation through indirect mechanisms and/or pathways.
BRAF V600E has been shown to regulate MITF through MEK and ERK mediated phosphorylation of MITF (90). This phosphorylation by ERK can increase MITF via BRN2 and simultaneously down-regulate it by targeting MITF for degradation (102). BRAF mutations in CM have also been associated with overexpression of the scaffold protein, Grb-2-associated binder 2 (GAB2). GAB2 is a stimulator of HIF-1α, stimulating VEGF-C, and CXCL12, thus inducing HEV formation (103). Interestingly, a retrospective cohort study of patients with stage II or III non-metastatic colorectal adenocarcinoma, reported that tumors with MSI-H and/or BRAF V600E mutation had higher numbers of TLS (defined as CXCL13+CD21+CD23+) (71). Furthermore, the authors observed a significant two-way association between BRAF V600E mutation and MSI-H status.
NRAS was the first oncogene to be identified in melanoma (104). Mutations of the NRAS GTPase account for 20% of all melanomas and are the second most common driver mutation (105). NRAS G12D positive CM is associated with an overexpression of GAB2, leading to increased HIF-1α, CXCL12, and VEGF-C allowing for TLS development (103).
KIT (KIT proto-oncogene receptor tyrosine kinase) mutations are found in about 2% of all CM cases (106). Small molecule tyrosine kinase inhibitors (TKIs) have been developed to compete with the ATP-binding site of oncogenic tyrosine kinases. Constitutively active oncogenic KIT is involved in MITF regulation through downstream phosphorylation of ERK, leading to MITF activation (90, 107). Increased MITF expression is subsequently able to upregulate pro-TLS cytokines through HIF-1α (92). Similarly, to BRAF and NRAS mutations, the upregulation of these downstream effectors from mutated KIT can support TLS formation.
Phosphatase and tensin homolog (PTEN) is a commonly mutated tumor-suppressor gene in invasive and metastatic CM. PTEN encodes a phosphatidyl-inositol-3,4,5-triphosphate 3-phosphatase protein that is a key regulator of the PI3k signaling pathway and has a role in maintaining anti-tumor immunity (90, 108). In adipose cells, PTEN mutations resulting in loss of function were found to be associated with increased levels of STAT3 (109, 110). Higher levels of STAT3 can activate NF-κB and results in TNF-α upregulation, leading to increased CCL21 expression in support of TLS neogenesis (64, 109).
Increased STAT3 following PTEN loss of function in CM was also found to increase IL-6 levels via NF-κB (111). The increased IL-6 levels can then upregulate VEGF-C, leading to HEV formation and pro-TLS growth (52, 112, 113). Loss of PTEN in a mouse model of prostate cancer lead to NF-κB mediated transcriptional activation of the CXCL13 gene (114). CXCL13 is pivotal in inducing LTi cells, thereby facilitating classical TLS formation (16).
PRDM1 (PR domain zinc finger protein 1), also known as BLIMP-1, is a tumor-suppressor gene that is an important regulator of neural crest cell (NCC) development and is frequently lost in metastatic melanoma (115). Loss of function in PRDM1 in a zebrafish model of melanoma has been found to result in faster melanoma tumorigenesis and a more aggressive cancer (115). In this same zebrafish model, loss of PRDM1 resulted in increased expression of SRY-box 10 (SOX10), an important nuclear transcription factor involved in NCC progenitor differentiation to melanocytes (115, 116). The increased SOX10 can upregulate MITF expression, resulting in pro-TLS cytokine expression (117).
The mutational genotype of melanoma plays an undeniable role in constitutive tumor immunogenicity and the promotion of intrinsic or therapy-induced anti-tumor immunity. The association of driver mutations with sustained inflammation in TME contributes to fertile soil for the development of TLS in association with superior prognosis and response to interventional immunotherapy. In this regard, we propose that CM, known to exhibit a consensus high TMB, may be predisposed to develop TLS formation, thereby contributing to the classification of CM as an immunogenic disease. Further experimental evidence needs to be conducted to assess the specific role of each driver gene in the regulation of TLS induction and the constitutive/on-treatment operational immune functionality of these lymphoid organs as it relates to patient outcomes and the development of refined treatment designs centered on therapeutic TLS development.
DS and RF contributed intellectually, performed literature searches, and prepared/revised the manuscript and figures. MC contributed intellectually, performed data analyses, and prepared figures. WS contributed intellectually, performed data analyses, and revised manuscript and figures. All authors contributed to the article and approved the submitted version.
This work was supported by internal funding at the Lake Erie College of Osteopathic Medicine.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. (2007) 445:851–7. doi: 10.1038/nature05661
2. Linos E, Swetter SM, Cockburn MG, Colditz GA, Clarke CA. Increasing burden of melanoma in the United States. J Invest Dermatol. (2009) 129:1666–74. doi: 10.1038/jid.2008.423
3. Ali Z, Yousaf N, Larkin J. Melanoma epidemiology, biology, and prognosis. Eur J Cancer. (2013) 11:81–91. doi: 10.1016/j.ejcsup.2013.07.012
4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. (2020) 70:7–30. doi: 10.3322/caac.21590
5. Domingues B, Lopes J, Soares P, Populo H. Melanoma treatment in review. ImmunoTargets Ther. (2018) 7:35–49. doi: 10.2147/ITT.S134842
6. Passarelli A, Mannavola F, Stucci LS, Tucci M, Silvestris F. Immune system and melanoma biology: a balance between immunosurveillance and immune escape. Oncotarget. (2017) 8:106132–42. doi: 10.18632/oncotarget.22190
7. Galuppini F, Dal Pozzo CA, Deckert J, Loupakis F, Fassan M, Baffa R. Tumor mutation burden: from comprehensive mutational screening to the clinic. Cancer Cell Int. (2019) 19:209. doi: 10.1186/s12935-019-0929-4
8. Steuer CE, Ramalingam SS. Tumor mutation burden: leading immunotherapy to the era of precision medicine? J Clin Oncol. (2018) 36:631–2. doi: 10.1200/JCO.2017.76.8770
9. Castle JC, Uduman M, Pabla S, Stein RB, Buell JS. Mutation-derived neoantigens for cancer immunotherapy. Front Immunol. (2019) 10:1856. doi: 10.3389/fimmu.2019.01856
10. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
11. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. doi: 10.1038/nature13954
12. Callahan MK, Kluger H, Postow MA, Segal NH, Lesokhin A, Atkins MB, et al. Nivolumab plus ipilimumab in patients with advanced melanoma: updated survival, response, and safety data in a phase i dose-escalation study. J Clin Oncol. (2018) 36:391–8. doi: 10.1200/JCO.2017.72.2850
13. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
14. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2017) 377:1345–56. doi: 10.1056/NEJMoa1709684
15. Imbert C, Montfort A, Fraisse M, Marcheteau E, Gilhodes J, Martin E, et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat Commun. (2020) 11:1–14. doi: 10.1038/s41467-019-14218-7
16. Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. (2019) 19:307–25. doi: 10.1038/s41568-019-0144-6
17. Sautès-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol. (2016) 7:407. doi: 10.3389/fimmu.2016.00407
18. Pipi E, Nayar S, Gardner DH, Colafrancesco S, Smith C, Barone F. Tertiary lymphoid structures: autoimmunity goes local. Front Immunol. (2018) 9:1952. doi: 10.3389/fimmu.2018.01952
19. Barone F, Gardner DH, Nayar S, Steinthal N, Buckley CD, Luther SA. Stromal fibroblasts in tertiary lymphoid structures: a novel target in chronic inflammation. Front Immunol. (2016) 7:477. doi: 10.3389/fimmu.2016.00477
20. Yamakoshi Y, Tanaka H, Sakimura C, Deguchi S, Mori T, Tamura T, et al. Immunological potential of tertiary lymphoid structures surrounding the primary tumor in gastric cancer. Int J Oncol. (2020) 57:171–82. doi: 10.3892/ijo.2020.5042
21. Sakimura C, Tanaka H, Okuno T, Hiramatsu S, Muguruma K, Hirakawa K, et al. B cells in tertiary lymphoid structures are associated with favorable prognosis in gastric cancer. J Surg Res. (2017) 215:74–82. doi: 10.1016/j.jss.2017.03.033
22. Jones GW, Hill DG, Jones SA. Understanding immune cells in tertiary lymphoid organ development: it is all starting to come together. Front Immunol. (2016) 7:1. doi: 10.3389/fimmu.2016.00401
23. Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol. (2012) 33:297–305. doi: 10.1016/j.it.2012.04.006
24. Weinstein AM, Storkus WJ. Biosynthesis and functional significance of peripheral node addressin in cancer-associated TLO. Front Immunol. (2016) 7:301. doi: 10.3389/fimmu.2016.00301
25. Wirsing AM, Rikardsen OG, Steigen SE, Uhlin-Hansen L, Hadler-Olsen E. Characterisation and prognostic value of tertiary lymphoid structures in oral squamous cell carcinoma. BMC Clin Pathol. (2014) 14:38. doi: 10.1186/1472-6890-14-38
26. Weinstein AM, Storkus WJ. Therapeutic lymphoid organogenesis in the tumor microenvironment. Adv Cancer Res. (2015) 128:197–233. doi: 10.1016/bs.acr.2015.04.003
27. Astorri E, Bombardieri M, Gabba S, Peakman M, Pozzilli P, Pitzalis C. Evolution of ectopic lymphoid neogenesis and in situ autoantibody production in autoimmune nonobese diabetic mice: cellular and molecular characterization of tertiary lymphoid structures in pancreatic islets. J Immunol. (2010) 185:3359–68. doi: 10.4049/jimmunol.1001836
28. Ruddle NH. High endothelial venules and lymphatic vessels in tertiary lymphoid organs: characteristics, functions, and regulation. Front Immunol. (2016) 7:491. doi: 10.3389/fimmu.2016.00491
29. Engelhard VH, Rodriguez AB, Mauldin IS, Woods AN, Peske JD, Slingluff CL. Immune cell infiltration and tertiary lymphoid structures as determinants of antitumor immunity. J Immunol. (2018) 200:432–42. doi: 10.4049/jimmunol.1701269
30. Alsughayyir J, Pettigrew GJ, Motallebzadeh R. Spoiling for a fight: B lymphocytes as initiator and effector populations within tertiary lymphoid organs in autoimmunity and transplantation. Front Immunol. (2017) 8:1639. doi: 10.3389/fimmu.2017.01639
31. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer, and autoimmunity. Nat Rev Immunol. (2014) 14:447–62. doi: 10.1038/nri3700
32. Petitprez F, de Reyniès A, Keung EZ, Chen TWW, Sun CM, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. (2020) 577:556–60. doi: 10.1038/s41586-019-1906-8
33. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. (2020) 577:549–55. doi: 10.1038/s41586-019-1922-8
34. Gräbner R, Lötzer K, Döpping S, Hildner M, Radke D, Beer M, et al. Lymphotoxin β receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE -/- mice. J Exp Med. (2009) 206:233–48. doi: 10.1084/jem.20080752
35. Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, et al. Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity. (2015) 43:579–90. doi: 10.1016/j.immuni.2015.08.006
36. Zlotnik A, Burkhardt AM, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol. (2011) 11:597–606. doi: 10.1038/nri3049
37. Zhu G, Nemoto S, Mailloux AW, Perez-Villarroel P, Nakagawa R, Falahat R, et al. Induction of tertiary lymphoid structures with antitumor function by a lymph node-derived stromal cell line. Front Immunol. (2018) 9:1609. doi: 10.3389/fimmu.2018.01609
38. Colbeck EJ, Ager A, Gallimore A, Jones GW. Tertiary lymphoid structures in cancer: drivers of antitumor immunity, immunosuppression, or Bystander Sentinels in disease? Front Immunol. (2017) 8:1830. doi: 10.3389/fimmu.2017.01830
39. Van De Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. (2010) 10:664–74. doi: 10.1038/nri2832
40. Van De Pavert SA, Olivier BJ, Goverse G, Vondenhoff MF, Greuter M, Beke P, et al. Chemokine cxcl13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol. (2009) 10:1193–9. doi: 10.1038/ni.1789
41. Cupedo T, Jansen W, Kraal G, Mebius RE. Induction of secondary and tertiary lymphoid structures in the skin. Immunity. (2004) 21:655–67. doi: 10.1016/j.immuni.2004.09.006
42. Ansel KM, Ngo VN, Hyman PL, Luther SA, Förster R, Sedgwlck JD, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. (2000) 406:309–14. doi: 10.1038/35018581
43. Workel HH, Lubbers JM, Arnold R, Prins TM, van der Vlies P, de Lange K, et al. A transcriptionally distinct CXCL13þCD103þCD8þ T-cell population is associated with B-cell recruitment and neoantigen load in human cancer. Cancer Immunol Res. (2019) 7:784–96. doi: 10.1158/2326-6066.CIR-18-0517
44. Buckley CD, Barone F, Nayar S, Bénézech C, Caamaño J. Stromal cells in chronic inflammation and tertiary lymphoid organ formation. Annu Rev Immunol. (2015) 33:715–45. doi: 10.1146/annurev-immunol-032713-120252
45. Liu W, Peng Y, Tobin DJ. A new 12-gene diagnostic biomarker signature of melanoma revealed by integrated microarray analysis. PeerJ. (2013) 2013:e49. doi: 10.7717/peerj.49
46. Onder L, Narang P, Scandella E, Chai Q, Iolyeva M, Hoorweg K, et al. IL-7-producing stromal cells are critical for lymph node remodeling. Blood. (2012) 120:4675–83. doi: 10.1182/blood-2012-03-416859
47. Genovese L, Brendolan A. Lymphoid tissue mesenchymal stromal cells in development and tissue remodeling. Stem Cells Int. (2016) 2016:8419104. doi: 10.1155/2016/8419104
48. Tang H, Zhu M, Qiao J, Fu YX. Lymphotoxin signalling in tertiary lymphoid structures and immunotherapy. Cell Mol Immunol. (2017) 14:809–18. doi: 10.1038/cmi.2017.13
49. Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORγt and LTi cells. J Exp Med. (2011) 208:125–34. doi: 10.1084/jem.20100052
50. Furtado GC, Marinkovic T, Martin AP, Garin A, Hoch B, Hubner W, et al. Lymphotoxin β receptor signaling is required for inflammatory lymphangiogenesis in the thyroid. Proc Natl Acad Sci U S A. (2007) 104:5026–31. doi: 10.1073/pnas.0606697104
51. Moussion C, Girard JP. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature. (2011) 479:542–6. doi: 10.1038/nature10540
52. Huang Y-H, Yang H-Y, Huang S-W, Ou G, Hsu Y-F, Hsu M-J. Interleukin-6 induces vascular endothelial growth factor-C expression via Src-FAK-STAT3 signaling in lymphatic endothelial cells. PLoS ONE. (2016) 11:e0158839. doi: 10.1371/journal.pone.0158839
53. Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic T H 17 cells. Nat Immunol. (2017) 18:74–85. doi: 10.1038/ni.3632
54. Deteix C, Attuil-Audenis V, Duthey A, Patey N, McGregor B, Dubois V, et al. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol. (2010) 184:5344–51. doi: 10.4049/jimmunol.0902999
55. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. (2011) 35:986–96. doi: 10.1016/j.immuni.2011.10.015
56. Barone F, Nayara S, Camposa J, Cloakea T, Withers DR, Toellner KM, et al. IL-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs. Proc Natl Acad Sci U S A. (2015) 112:11024–9. doi: 10.1073/pnas.1503315112
57. Fleige H, Ravens S, Moschovakis GL, Bölter J, Willenzon S, Sutter G, et al. IL-17-induced CXCL12 recruits B cells and induces follicle formation in BALT in the absence of differentiated FDCs. J Exp Med. (2014) 211:643–51. doi: 10.1084/jem.20131737
58. Weinstein AM, Giraldo NA, Petitprez F, Julie C, Lacroix L, Peschaud F, et al. Association of IL-36γ with tertiary lymphoid structures and inflammatory immune infiltrates in human colorectal cancer. Cancer Immunol Immunother. (2019) 68:109–20. doi: 10.1007/s00262-018-2259-0
59. Thaunat O, Field AC, Dai J, Louedec L, Patey N, Bloch MF, et al. Lymphoid neogenesis in chronic rejection: evidence for a local humoral alloimmune response. Proc Natl Acad Sci U S A. (2005) 102:14723–8. doi: 10.1073/pnas.0507223102
60. Nayar S, Campos J, Smith CG, Iannizzotto V, Gardner DH, Mourcin F, et al. Immunofibroblasts are pivotal drivers of tertiary lymphoid structure formation and local pathology. Proc Natl Acad Sci U S A. (2019) 116:13490–7. doi: 10.1073/pnas.1905301116
61. Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin α are ligands for herpesvirus entry mediator. Immunity. (1998) 8:21–30. doi: 10.1016/S1074-7613(00)80455-0
62. Mortarini R, Scarito A, Nonaka D, Zanon M, Bersani I, Montaldi E, et al. Constitutive expression and costimulatory function of LIGHT/TNFSF14 on human melanoma cells and melanoma-derived microvesicles. Cancer Res. (2005) 65:3428–36. doi: 10.1158/0008-5472.CAN-04-3239
63. Fraker DL, Alexander HR, Andrich M, Rosenberg SA. Treatment of patients with melanoma of the extremity using hyperthermic isolated limb perfusion with melphalan, tumor necrosis factor, and interferon gamma: results of a tumor necrosis factor dose-escalation study. J Clin Oncol. (1996) 14:479–89. doi: 10.1200/JCO.1996.14.2.479
64. Wiede F, Vana K, Sedger LM, Lechner A, Körner H. TNF-dependent overexpression of CCL21 is an underlying cause of progressive lymphoaccumulation in generalized lymphoproliferative disorder. Eur J Immunol. (2007) 37:351–7. doi: 10.1002/eji.200636218
65. Li B, Vincent A, Cates J, Brantley-Sieders DM, Polk DB, Young PP. Low levels of tumor necrosis factor α increase tumor growth by inducing an endothelial phenotype of monocytes recruited to the tumor site. Cancer Res. (2009) 69:338–48. doi: 10.1158/0008-5472.CAN-08-1565
66. Ladányi A, Kiss J, Somlai B, Gilde K, Fejos Z, Mohos A, et al. Density of DC-LAMP + mature dendritic cells in combination with activated T lymphocytes infiltrating primary cutaneous melanoma is a strong independent prognostic factor. Cancer Immunol Immunother. (2007) 56:1459–69. doi: 10.1007/s00262-007-0286-3
67. Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, et al. 12-chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep. (2012) 2:765. doi: 10.1038/srep00765
68. Zhu G, Falahat R, Wang K, Mailloux A, Artzi N, Mulé JJ. Tumor-associated tertiary lymphoid structures: gene-expression profiling and their bioengineering. Front Immunol. (2017) 8:767. doi: 10.3389/fimmu.2017.00767
69. Cipponi A, Mercier M, Seremet T, Baurain JF, Theáte I, Van Den Oord J, et al. Neogenesis of lymphoid structures and antibody responses occur in human melanoma metastases. Cancer Res. (2012) 72:3997–4007. doi: 10.1158/0008-5472.CAN-12-1377
70. Birkeland E, Zhang S, Poduval D, Geisler J, Nakken S, Vodak D, et al. Patterns of genomic evolution in advanced melanoma. Nat Commun. (2018) 9:2665. doi: 10.1038/s41467-018-05063-1
71. Posch F, Silina K, Leibl S, Mündlein A, Moch H, Siebenhüner A, et al. Maturation of tertiary lymphoid structures and recurrence of stage II and III colorectal cancer. Oncoimmunology. (2018) 7:e1378844. doi: 10.1080/2162402X.2017.1378844
72. Kim JY, Kronbichler A, Eisenhut M, Hong SH, van der Vliet HJ, Kang J, et al. Tumor mutational burden and efficacy of immune checkpoint inhibitors: a systematic review and meta-analysis. Cancers. (2019) 11:11. doi: 10.3390/cancers11111798
73. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. (2020) 577:561–5. doi: 10.1038/s41586-019-1914-8
74. Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. (2015) 372:30–9. doi: 10.1056/NEJMoa1412690
75. Dobry AS, Zogg CK, Hodi FS, Smith TR, Ott PA, Iorgulescu JB. Management of metastatic melanoma: improved survival in a national cohort following the approvals of checkpoint blockade immunotherapies and targeted therapies. Cancer Immunol Immunother. (2018) 67:1833–44. doi: 10.1007/s00262-018-2241-x
76. Flaherty KT, Hodi FS, Fisher DE. From genes to drugs: targeted strategies for melanoma. Nat Rev Cancer. (2012) 12:349–61. doi: 10.1038/nrc3218
77. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity. (2018) 48:812-30.e14. doi: 10.1016/j.immuni.2018.03.023
78. Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. (2017) 552:1–5. doi: 10.1038/nature24673
79. Chae YK, Anker JF, Oh MS, Bais P, Namburi S, Agte S, et al. Mutations in DNA repair genes are associated with increased neoantigen burden and a distinct immunophenotype in lung squamous cell carcinoma. Sci Rep. (2019) 9:3235. doi: 10.1038/s41598-019-39594-4
80. Koi M, Tseng-Rogenski SS, Carethers JM. Inflammation-associated microsatellite alterations: mechanisms and significance in the prognosis of patients with colorectal cancer. World J Gastrointest Oncol. (2018) 10:1–14. doi: 10.4251/wjgo.v10.i1.1
81. Germano G, Amirouchene-Angelozzi N, Rospo G, Bardelli A. The clinical impact of the genomic landscape of mismatch repair–deficient cancers. Cancer Discov. (2018) 8:1518–28. doi: 10.1158/2159-8290.CD-18-0150
82. Green AR, Aleskandarany MA, Ali R, Hodgson EG, Atabani S, De Souza K, et al. Clinical impact of tumor DNA repair expression and T-cell infiltration in breast cancers. Cancer Immunol Res. (2017) 5:292–9. doi: 10.1158/2326-6066.CIR-16-0195
83. Zhao D, Klempner SJ, Chao J. Progress and challenges in HER2-positive gastroesophageal adenocarcinoma. J Hematol Oncol. (2019) 12:50. doi: 10.1186/s13045-019-0737-2
84. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. (2015) 372:2509–20. doi: 10.1200/jco.2015.33.15_suppl.lba100
85. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. (2017) 357:409–13. doi: 10.1126/science.aan6733
86. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
87. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. (2013) 6:pl1. doi: 10.1126/scisignal.2004088
88. Zhang T, Dutton-Regester K, Brown KM, Hayward NK. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. (2016) 29:266–83. doi: 10.1111/pcmr.12459
89. Wellbrock C, Arozarena I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res. (2015) 28:390–406. doi: 10.1111/pcmr.12370
90. Reddy BY, Miller DM, Tsao H. Somatic driver mutations in melanoma. Cancer. (2017) 123:2104–17. doi: 10.1002/cncr.30593
91. Wiedemann GM, Aithal C, Kraechan A, Heise C, Cadilha BL, Zhang J, et al. Microphthalmia-associated transcription factor (MITF) regulates immune cell migration into melanoma. Transl Oncol. (2019) 12:350–60. doi: 10.1016/j.tranon.2018.10.014
92. Buscà R, Berra E, Gaggioli C, Khaled M, Bille K, Marchetti B, et al. Hypoxia-inducible factor 1α is a new target of microphthalmia- associated transcription factor (MITF) in melanoma cells. J Cell Biol. (2005) 170:49–59. doi: 10.1083/jcb.200501067
93. Zbytek B, Peacock DL, Seagroves TN, Slominski A. Putative role of hif transcriptional activity in melanocytes and melanoma biology. Dermatoendocrinol. (2013) 5:239–51. doi: 10.4161/derm.22678
94. Schoppmann SF, Fenzl A, Schindl M, Bachleitner-Hofmann T, Nagy K, Gnant M, et al. Hypoxia inducible factor-1α correlates with VEGF-C expression and lymphangiogenesis in breast cancer. Breast Cancer Res Treat. (2006) 99:135–41. doi: 10.1007/s10549-006-9190-3
95. Triner D, Shah YM. Hypoxia-inducible factors: a central link between inflammation and cancer. J Clin Invest. (2016) 126:3689–98. doi: 10.1172/JCI84430
96. Hawryluk EB, Tsao H. Melanoma: clinical features and genomic insights. Cold Spring Harb Perspect Med. (2014) 4:a015388. doi: 10.1101/cshperspect.a015388
97. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. (2002) 417:949–54. doi: 10.1038/nature00766
98. Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. (2012) 18:1386–94. doi: 10.1158/1078-0432.CCR-11-2479
99. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. (2013) 19:1225–31. doi: 10.1158/1078-0432.CCR-12-1630
100. Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CNJ, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. (2010) 70:5213–9. doi: 10.1158/0008-5472.CAN-10-0118
101. Khalili JS, Liu S, Rodríguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. (2012) 18:5329–40. doi: 10.1158/1078-0432.CCR-12-1632
102. Haq R, Yokoyama S, Hawryluk EB, Jönsson GB, Frederick DT, McHenry K, et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc Natl Acad Sci U S A. (2013) 110:4321–6. doi: 10.1073/pnas.1205575110
103. Yang Y, Wu J, Demir A, Castillo-Martin M, Melamed RD, Zhang G, et al. GAB2 induces tumor angiogenesis in NRAS-driven melanoma. Oncogene. (2013) 32:3627–37. doi: 10.1038/onc.2012.367
104. Albino AP, Strange RL, Oliff AI, Furth ME, Old LJ. Transforming ras genes from human melanoma: a manifestation of tumour heterogeneity? Nature. (1984) 308:69–72. doi: 10.1038/308069a0
105. Manzano JL, Layos L, Bugés C, De los Llanos Gil M, Vila L, Martínez-Balibrea E, et al. Resistant mechanisms to BRAF inhibitors in melanoma. Ann Transl Med. (2016) 4:1–9. doi: 10.21037/atm.2016.06.07
106. Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. (2008) 14:6821–8. doi: 10.1158/1078-0432.CCR-08-0575
107. Hemesath TJ, Price ER, Takemoto C, Badalian T, Fisher DE. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature. (1998) 391:298–301. doi: 10.1038/34681
108. Brandmaier A, Hou SQ, Demaria S, Formenti SC, Shen WH. PTEN at the interface of immune tolerance and tumor suppression. Front Biol. (2017) 12:163–74. doi: 10.1007/s11515-017-1443-5
109. Hoareau L, Bencharif K, Rondeau P, Murumalla R, Ravanan P, Tallet F, et al. Signaling pathways involved in LPS induced TNFalpha production in human adipocytes. J Inflamm. (2010) 7:1. doi: 10.1186/1476-9255-7-1
110. Hill DG, Yu L, Gao H, Balic JJ, West A, Oshima H, et al. Hyperactive gp130/STAT3-driven gastric tumourigenesis promotes submucosal tertiary lymphoid structure development. Int J Cancer. (2018) 143:167–78. doi: 10.1002/ijc.31298
111. Piro G, Carbone C, Carbognin L, Pilotto S, Ciccarese C, Iacovelli R, et al. Revising PTEN in the era of immunotherapy: new perspectives for an old story. Cancers. (2019) 11:1525. doi: 10.3390/cancers11101525
112. Jin C, Wang A, Chen J, Liu X, Wang G. Relationship between expression and prognostic ability of PTEN, STAT3 and VEGF-C in colorectal cancer. Exp Ther Med. (2012) 4:633–9. doi: 10.3892/etm.2012.651
113. Klatte T, Seligson DB, LaRochelle J, Shuch B, Said JW, Riggs SB, et al. Molecular signatures of localized clear cell renal cell carcinoma to predict disease-free survival after nephrectomy. Cancer Epidemiol Biomarkers Prev. (2009) 18:894–900. doi: 10.1158/1055-9965.EPI-08-0786
114. Garg R, Blando JM, Perez CJ, Abba MC, Benavides F, Kazanietz MG. Protein kinase C epsilon cooperates with PTEN loss for prostate tumorigenesis through the CXCL13-CXCR5 pathway. Cell Rep. (2017) 19:375–88. doi: 10.1016/j.celrep.2017.03.042
115. Iwanaga R, Truong BT, Hsu JY, Lambert KA, Vyas R, Orlicky D, et al. Loss of prdm1a accelerates melanoma onset and progression. Mol Carcinog. (2020) 59:1052–63. doi: 10.1101/2019.12.20.884767
116. Willis BC, Johnson G, Wang J, Cohen C. SOX10: a useful marker for identifying metastatic melanoma in sentinel lymph nodes. Appl Immunohistochem Mol Morphol. (2015) 23:109–12. doi: 10.1097/PAI.0000000000000097
Keywords: tertiary lymphoid structures, cutaneous melanoma, tumor mutational burden, driver mutations, DNA repair proteins
Citation: Salem D, Chelvanambi M, Storkus WJ and Fecek RJ (2021) Cutaneous Melanoma: Mutational Status and Potential Links to Tertiary Lymphoid Structure Formation. Front. Immunol. 12:629519. doi: 10.3389/fimmu.2021.629519
Received: 15 November 2020; Accepted: 11 February 2021;
Published: 04 March 2021.
Edited by:
Peng Qu, National Institutes of Health (NIH), United StatesReviewed by:
Edoardo Migliori, Columbia University Irving Medical Center, United StatesCopyright © 2021 Salem, Chelvanambi, Storkus and Fecek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronald J. Fecek, cmZlY2VrQGxlY29tLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.