 Samantha J. Paston
Samantha J. Paston Victoria A. Brentville
Victoria A. Brentville Peter Symonds
Peter Symonds Lindy G. Durrant
Lindy G. Durrant- 1Biodiscovery Institute, Scancell Limited, Nottingham, United Kingdom
- 2Biodiscovery Institute, University of Nottingham, Faculty of Medicine and Health Sciences, Nottingham, United Kingdom
Vaccination was first pioneered in the 18th century by Edward Jenner and eventually led to the development of the smallpox vaccine and subsequently the eradication of smallpox. The impact of vaccination to prevent infectious diseases has been outstanding with many infections being prevented and a significant decrease in mortality worldwide. Cancer vaccines aim to clear active disease instead of aiming to prevent disease, the only exception being the recently approved vaccine that prevents cancers caused by the Human Papillomavirus. The development of therapeutic cancer vaccines has been disappointing with many early cancer vaccines that showed promise in preclinical models often failing to translate into efficacy in the clinic. In this review we provide an overview of the current vaccine platforms, adjuvants and delivery systems that are currently being investigated or have been approved. With the advent of immune checkpoint inhibitors, we also review the potential of these to be used with cancer vaccines to improve efficacy and help to overcome the immune suppressive tumor microenvironment.
Introduction
The potential to develop a cancer vaccine has been extensively researched in both humans and animal models, the majority of these vaccines are therapeutic vaccines that aim to activate the immune system to recognize and kill established tumors. A prophylactic vaccine that prevents cancers caused by the Human Papillomavirus [Types 6, 11, 16, 18] has been approved and in the UK, children aged 12-13-years-old are routinely offered this vaccine. Developing a therapeutic cancer vaccine has been more problematic with many encouraging results in preclinical studies not translating into the clinic. To date the FDA has only approved one vaccine, sipuleucel-T, that is used to treat metastatic castration-resistant prostate cancer in a limited group of nearly asymptomatic patients (1). There are number of reasons for these failures such as the immune suppressive TME, lack of a robust T cell responses, sub optimal vaccine formulations, delivery, adjuvants and identifying the best patients to target. The ideal setting for a cancer vaccine to work is in patients following surgical resection, chemotherapy or radiotherapy, all of which stimulate an immune response themselves. Vaccination at this stage and in combination with a checkpoint inhibitor will provide the best setting to induce a potent anti-tumor immune response.
Over the last couple of decades, a better understanding of the tumor microenvironment (TME) and immune suppressive mechanisms have opened a number of new avenues that can be explored and has led to the next generation of new cancer therapies. The immune suppressive TME is a major obstacle to the success of any cancer vaccine with the description of immunologically “cold” tumors that on their own do not appear to be immunogenic with an absence of tumor infiltrating lymphocytes (TILs). In contrast “hot” tumors are immunogenic and have induced an immune response that has resulted in the infiltration of TILs but are not able to function due to the presence of various checkpoint molecules such as PD-1, CTLA-4, LAG-3, TIM-3 or the presence of immune suppressive cells such as regulatory T cells, myeloid-derived suppressor cells (MDSCs), M2 macrophages, regulatory natural killer (NK) T cells or cytokines such as transforming growth factor-beta [TGFβ], IL-10, and IL-13 (2, 3). The success of any cancer vaccine relies on overcoming the immune suppressive TME and converting “cold” tumors into “hot” tumors and therefore inducing a robust tumor specific immune response that can kill cancer cells.
Target Antigens
The choice of antigen to target in any cancer vaccine is extremely important to the efficacy of the vaccine in the clinic. The ideal antigen should be specifically expressed on cancer cells with no expression on normal cells, ideally the antigen should be necessary for cell survival and be highly immunogenic.
Tumor antigens fall into two broad categories, the tumor associated antigens (TAAs) and tumor-specific antigens (TSAs). Within each category a number of different types of tumor antigens have been described and are summarized in Table 1. The cancer germline antigens (also called cancer testis antigens) are the most studied group of cancer antigens, historically they were attractive antigens to target due to their expression only on germ cells of immune-privileged organs and high expression on tumor cells. The most common cancer germline antigens that are targeted include MAGE (4, 5), NY-ESO1 (6), GAGE (7, 8) and BAGE (9), both T cell and antibody responses to these antigen have been detected in patients.
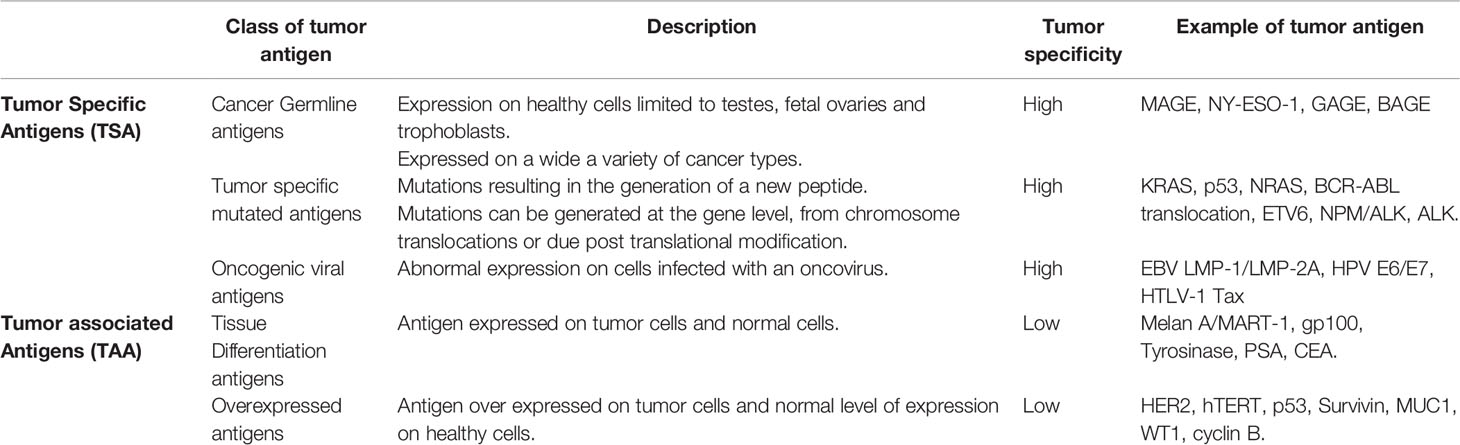
Table 1 Different types of tumor antigens.
The differentiation antigens are expressed on normal and tumor cells from the same tissue, targeting such antigens requires careful consideration to any potential toxicity to the normal tissue. Differentiation antigens include the melanoma antigens Melan A/MART-1 (10, 11), gp100 (12), tyrosinase (13), the prostate antigen prostate specific antigen (PSA) (14, 15) and the colon antigen carcinoembryonic antigen (CEA) (16, 17).
The overexpressed antigens are generally expressed at low levels on normal cells but are over expressed on tumor cells, there are many antigens that fall into this group including HER2 (18), hTERT (19, 20), p53 (21), survivin (22–25), MUC1 (26), WT1 (27), cyclin B (28, 29) and many more. Targeting over expressed antigens can be challenging, preclinical studies need to ensure that normal low-level expressing cells are not targeted by the vaccine induced immune response.
The oncogenic viral antigens are expressed on virus infected cells that have subsequently undergone malignant transformation. Oncogenic viral antigens have been targeted in both prophylactic vaccines such as HPV but also in therapeutic vaccines to treat existing malignancies. The most commonly targeted oncogenic viral antigens in this group include EBV LMP-1 and LMP-2A (30–32), HPV E6/E7 (33), HTLV-1 Tax (34).
The last group of cancer antigens are those antigens that are mutated, these mutations can be generated at the gene level or as a result of post translational modifications leading to the generation of a new peptide. In the last couple of years there has been a renewed effort in generating vaccines that target mutated antigens, in particular the neoantigens. There are very few mutated antigens described where the mutated peptide is shared across patients or cancer types, the most studied shared mutations are KRAS (35), NRAS (36), epitopes from BCR-ABL translocation (Chronic myeloid leukemia) (37, 38), ETV6 (acute myeloid leukemia) (39), NPM/ALK (anaplastic large cell lymphomas) (40, 41) and ALK (neuroblastoma) (42, 43). A number of groups are developing personalized vaccines that target neoantigens identified from the patient’s tumor, very few if any of these mutations are shared epitopes and therefore any generated vaccine is only specific to the individual. Neoantigens are immunogenic because they harbor mutations, they have escaped central tolerance and are recognized as “non self” by the adaptive immune system (44). Despite the higher immunogenicity of neoantigen’s only 1-2% of T cells recognize these antigens (45). The poor immunogenicity of many tumors means that designing an effective neoantigen tumor vaccine will need to overcome these challenges.
The post translational modified cancer antigens are another group of antigens, they are not subject to thymic deletion and are therefore attractive vaccine candidates. A number of different post-translational modifications have been described that generate tumor specific epitopes including glycopeptides (46), phosphopeptides (47, 48) and citrullinated peptides (49). Cancer cells often exhibit different phosphorylation patterns leading to the generation of phosphorylated antigens, these make attractive vaccine candidates (47, 48, 50). Phosphorylated epitopes can be naturally processed and presented on the cell surface in association with MHC class I molecules for recognition by CD8+ T cells (50–52). Unregulated signaling cascades in tumor cells often lead to an increase in protein phosphorylation within the cell which in turn leads to the generation of phosphopeptides (52). Phosphopeptides have been identified by mass spectrometry analysis of tumor biopsies and cancer cell lines (53). Engelhard et al. (53) identified two phosphorylated peptides derived from the insulin receptor substrate 2 (IRS2) protein and breast cancer anti-estrogen resistance 3 (BCAR3). The ISR2 protein is overexpressed in many cancer types and in vivo has been shown to enhance metastasis (54–56), BCAR3 is associated cellular migration and resistance to therapeutic anti-estrogens in breast cancer cells (57, 58). Phosphopeptides restricted by HLA-*02:01 were identified by mass spectrometry and included in a phase 1 clinical trial (NCT01846143) in patients with resected stage IIA–IV melanoma. All patients had treatment related adverse events, but none were grade 3-4, T cell responses were induced to the phosphorylated IRS2 (1097-1105) peptide in 5/12 patients and to the phosphorylated BCAR3 (126-134) peptide in 2/12 patients. This trial showed that phosphopeptides are safe and induced an immune response in some patients, however, with the advent of immune checkpoint inhibitors future studies will need to define and enhance the immune response induced to these peptides.
Our own research has focused on epitopes that are citrullinated in tumor cells. Citrullination is a post translation modification where positive charged arginine residues are converted into neutrally charged citrulline in a process catalyzed by the Ca2+ dependent peptidyl arginine deaminase (PADI) enzymes (59, 60) (Figure 1). This modification can impact the protein structure and induce changes that result in protein denaturation potentially altering the structure and the function of the protein (61, 62). We have detected T cell responses to citrullinated peptides in healthy donors (60) suggesting that the T cells recognizing them are positively and not negatively selected in the thymus. In healthy cells the PADI enzymes are maintained in an inactive state due to low concentrations of Ca2+ (34), in double membrane vesicles within viable cells the calcium concentrations can be high leading to the activation of the PADI enzymes. Citrullination can occur within autophagosomes as a result of autophagy, here high calcium levels activate PADI enzymes that then citrullinate engulfed proteins from the cytoplasm (36, 37), this process is induced in stressed cells (17) such as cancer cells. During stress induced autophagy and in the presence of inflammation citrullinated peptides can be presented on major histocompatibility complex (MHC) class II molecules for recognition by CD4+ T cells (63). During inflammation many cytokines are produced, the majority are proinflammatory that result in the upregulation of MHC class II expression that then activates CD4+ T cells (Figure 2).
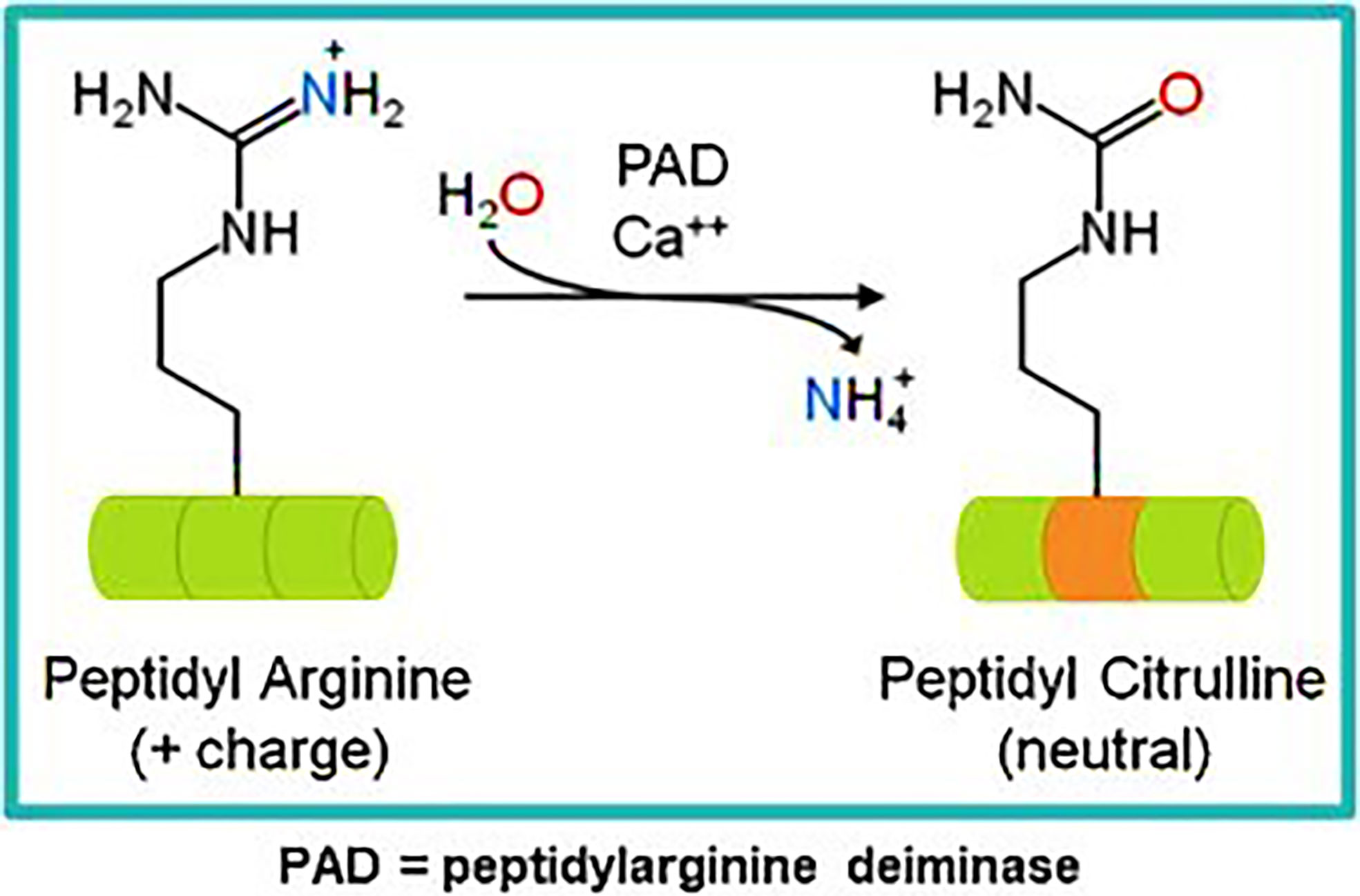
Figure 1 Schematic of the citrullination or deamidation of arginine.
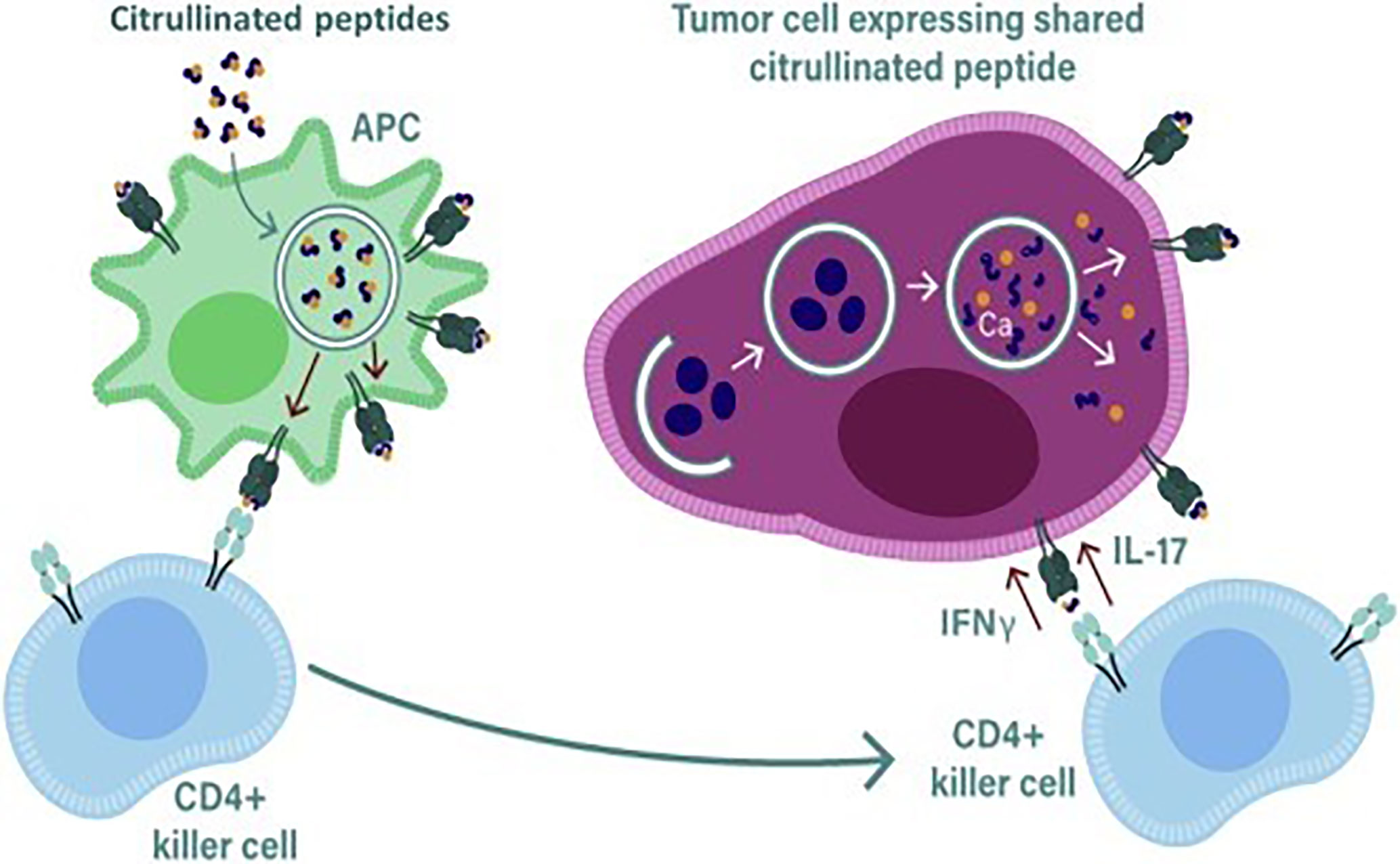
Figure 2 During stress induced autophagy and in the presence of inflammation citrullinated peptides can be presented on major histocompatibility complex (MHC) class II molecules for recognition by CD4+ T cells. During inflammation many cytokines are produced, the majority are proinflammatory that result in the upregulation of MHC class II expression that then activates CD4+ T cells. Primed killer CD4 T cells enter the tumor and are reactivated by APCs presenting citrullinated peptides from tumors allowing recognition and lysis by the killer CD4 T cells.
A number of studies performed in autoimmune patients have demonstrated that CD4+ T cell responses can be detected to citrullinated proteins such as the intermediate filament protein vimentin and the glycolytic enzyme enolase (64–68). In ovarian cancer patients we have demonstrated the presence of CD4+ T cell responses to citrullinated peptides derived from α-enolase and vimentin (60). The constitutive expression of MHC class II is mainly restricted to APCs such as DCs, B cells and macrophages but other cells such thymic epithelia cells and activated T cells can also express MHC class II (69). The expression of MHC class II on most other cells can be induced by interferon gamma (IFNγ) present in the local vicinity. The expression of MHC class II is controlled by the Class II Major Histocompatibility complex transactivator (CIITA) which is regulated by four different promoters, promoter I is active in myeloid cells, promoter III in lymphocytes and promoter IV is necessary for responsiveness to IFNγ (70), the function of promoter II is unknown, transcripts from this promoter are rare and therefore its function has not been pursued. Most tumors in the presence of inflammation and IFNγ will express MHC class II, if citrullinated peptides are then generated in response to stress or autophagy these can then be loaded onto MHC class II for presentation on the surface of tumor cells.
We have focused on citrullinated vimentin and α-enolase as attractive cancer vaccine targets. Vimentin is an intermediate filament protein that is known to be citrullinated and overexpressed in a wide range of cancers (71–76), particularly during EMT (77). The glycolytic enzyme α-Enolase (ENO1) catalyzes the final step in glycolysis (78). Many tumors switch to generating their energy via glycolysis in a process termed the “Warburg effect” and therefore overexpress ENO1, a wide range of tumors overexpress ENO1 (79–82). Due to its ubiquitous expression, ENO1 is often degraded during autophagy; previous studies have also shown that ENO1 can be citrullinated (65, 83). We have shown that these citrullinated peptides are recognized and presented to CD4+ T cells by both MHC class II HLA-DR4 and HLA-DP4 molecules (49, 84, 85). HLA transgenic mice vaccinated with citrullinated vimentin and α-enolase peptides linked to an adjuvant (Modi-1 vaccine) can stimulate CD4+ T cells (49, 64, 86) and generate potent anti-tumor responses resulting in tumor regression and eradication with no associated toxicity (49, 87). We have also shown that healthy donors have a repertoire of T cells that can be detected following stimulation with the citrullinated vimentin and α-enolase peptides showing that citrullinated peptides can be presented in the thymus allowing positive selection and resulting in specific T cell repertoires capable of recognizing these peptides (87). Our preclinical data shows that citrullinated vimentin and α-enolase are promising candidate vaccine targets and as a vaccine have generated impressive anti-tumor responses in preclinical murine models.
Adjuvants
Antigens alone in a vaccine are poor inducers of the adaptive immune response. In the absence of an adjuvant antigens targeted to immature DCs in the absence of inflammation or any microbial stimulation induce tolerance instead of a potent immune response (88). Adjuvants need to attract immune cells to the site of injection while also promoting cell mediated trafficking of antigen to draining lymph nodes and triggering the activation of APCs.
Current Vaccine Adjuvants
The water-in-oil emulsions such as Montanide ISA 720 and Montanide ISA-51 have been widely adopted as adjuvants, they form a depot at the injection site, this results in the trapping of the soluble antigens preventing their rapid trafficking to local lymph nodes, this induces inflammation and the gradual release of the antigen. In a clinical trial Montanide ISA-51 was shown to induce both CD4+ and CD8+ T cell responses in patients vaccinated with long peptides of the oncoproteins E6 and E7 (89).
New vaccine adjuvants have been developed that target specific components of the immune system to generate a more robust and longer lasting immune response. Newer adjuvants that consist of Pathogen-associated molecular pattern molecules (PAMPs) are now being used, these provide a danger signal that is recognized by pattern recognition receptors (PRRs) inducing an immune response. Innate cells express PPRs, these receptors include the Toll-like receptors (TLRs), nucleotide binding oligomerization domain like receptors and the mannose receptor. TLR agonists are increasingly being used as a vaccine adjuvant, they mimic microbial stimulation and have been shown to increase vaccine efficacy (90) particularly for cancers (91). Lymph node targeted TLR agonists have shown a direct relationship between the magnitude of CD8+ T cell responses and the amount of TLR agonist accumulated in draining lymph nodes, demonstrating the importance of providing sufficient inflammatory signals during immunization (92). A number of TLR agonists are currently in trial as adjuvants for cancer vaccines, one of the most commonly used TLR agonist is polyinosinic–polycytidylic acid with polylysine and carboxymethylcellulose (Poly-ICLC) a TLR3 agonist (93), others include monophosphoryl lipid A (MPLA) a TLR4 agonist (94, 95), imiquimod a TLR7 agonist (96, 97), resiquimod a TLR7 and TLR8 agonist (98, 99), CpG oligodeoxynucleotide (CpG ODN) a TLR9 agonist (90, 100, 101).
New Emerging Vaccine Adjuvants
Other newer adjuvants are also being investigated to increase the efficacy of a cancer vaccine, these include the CD40 agonists, these directly target the antigen to the early endosomes of DCs and mediate cross presentation. Although CD40 agonist antibodies have not been extensively studied in clinical trials as a vaccine adjuvant, they have been studied independently as monotherapy (102). A number of preclinical mouse models have shown that CD40 agonists can be used in combination with TLR agonists in a vaccination strategy (103, 104), whether this translates into clinical efficacy is still to be determined.
Another class of potential adjuvants is the Stimulator of interferon genes protein (STING) agonists. STING is a transmembrane protein located in the endoplasmic reticulum (105), its activation triggers a type I interferon response in response to intracellular DNA (106). STING agonists include synthetic cyclic dinucleotide derivatives and cyclic di-guanosine monophosphate, these have all shown anti-tumor activity in mice (107, 108). STING expression is highest on T cells and STING activation can lead to T cell apoptosis, such effects are not seen with macrophages and DCs (109). To use a STING agonist in a cancer vaccine it would need to be combined with an adjuvant or delivery system that targets only myeloid cells in vivo (110) preventing T cell apoptosis. STING agonists do induce some systemic toxicity and to overcome this intratumoral injection is the preferred route of administration. In addition, preclinical studies of STING agonists have been complicated by their differential binding properties in murine and human cells (111). The potential toxicity of STING agonists and lack of specific targeting could limit their use as adjuvants in a cancer vaccine.
In addition to using pathogen derived molecules as adjuvants a number of cytokines have also been shown to act as an adjuvant. Immunostimulatory cytokines such as IL-2 (112, 113), IFN (114), IL-12 (115, 116) and granulocyte-macrophage colony stimulating factor (GM-CSF) (117–119) have all been investigated, although recent studies have focused mainly on their application in cellular based therapies and vaccines. GM-CSF is the most studied immunostimulatory factor and has been included in numerous cancer vaccine trials (120). In preclinical studies GM-CSF looked a very promising candidate, it helps recruit DCs to the injection site, it can promote the maturation of DCs and antigen presentation resulting in an enhanced adaptive immune response (117). However, in clinical trials GM-CSF has generated disappointing results with only a few trials having shown a clinical benefit, the results across the majority of trials have been inconsistent. Preclinical studies indicated that GM-CSF could expand MDSCs resulting in the suppression of cell mediated anti-tumor responses (118). The effect of GM-CSF was also observed in clinical trials where a low dose of GM-CSF induced the expansion of CD14 positive, HLA-DR low/negative myeloid cells. In another study GM-CSF was used with incomplete Freund’s adjuvant and resulted in a low T cell response when compared to vaccine adjuvant without GM-CSF (119). Despite these results a number of clinical trials are currently underway using GM-CSF as an adjuvant component.
We have previously described our Modi-1 peptide vaccine (60), this vaccine comprises of two citrullinated vimentin peptides, as well as a citrullinated peptide from α-enolase, each peptide is conjugated to the TLR1/2 ligand adjuvant AMPLIVANT® (ISA Pharmaceuticals BV, Leiden, the Netherlands). In preclinical studies we have shown that by combining a peptide vaccine with a TLR ligand adjuvant promotes a Th1 response that is capable of inducing a potent anti-tumor response in tumor bearing mice (60). The CD4+ but not CD8+ T cells were essential for the generation of the anti-tumor response, depleting CD4+ T cells abrogated this response and a corresponding increase in CD4+ tumor-infiltrating lymphocytes (TILs) was associated with tumor regression (60). A comparison of different Toll-like receptor (TLR)-stimulating adjuvants showed that Modi-1 induced strong Th1 responses when combined with GM-CSF, TLR9/TLR4, TLR9, TLR3, TLR1/2 and TLR7 agonists. The strongest response was observed with TLR1/2 AMPLIVANT® adjuvant. The AMPLIVANT® adjuvant is already being used in an ongoing study evaluating two HPV-16 peptides in patients with head and neck squamous cell carcinoma (NCT02821494). These results highlight the importance of screening a range of adjuvants and doses to find the optimal adjuvant and dose to induce a potent immune response. The Modi-1 vaccine will enter a Phase 1/2 clinical trial in 2021.
Delivery Systems
Electroporation and Gene Gun Vaccine Delivery
There have been significant improvements in optimizing vaccine administration routes to overcome poor cellular uptake. Also improvements with delivery and plasmid design have improved the efficacy of DNA vaccines in both pre-clinical and clinical studies (121). One strategy for improving the uptake of plasmid DNA into antigen presenting cells (APCs) is by using electroporation. Electroporation delivers small electrical pulses that causes transient pores to form in the cell membrane. During the period of membrane destabilization plasmid DNA present in the extracellular environment around the target cell gains access to the intracellular compartment (122). Following the transfer of DNA into the cell the membrane then reseals. The transient increase in the permeability of the target cell membrane enhances the uptake of plasmid DNA (123). Electroporation increases DNA uptake by over a 1000-fold and has an adjuvant effect due to local tissue damage and the resulting stimulation of proinflammatory cytokine in the local vicinity (124, 125). A number of DNA vaccines currently in clinical trials are using electroporation to delivery DNA plasmids, the ability of these plasmids to induce an immune response has been demonstrated in prostate cancer and melanoma (126). Another similar strategy is using a gene gun to deliver plasmid DNA that is coated with a heavy metal, typically gold particle are used, APCs at the injection site are bombarded with plasmid coated particles. The gene gun strategy reduces the amount of DNA required by 100-1,000 (127); some promising preclinical data has led to phase 1 and 2 clinical trials in head and neck squamous cell carcinoma and cervical cancer (128). Electroporation is also being used to deliver plasmid DNA in the infectious disease field, there are a number of COVID-19 DNA vaccines currently in in clinical trial (WHO landscape report Dec 2020) that use DNA plasmids encoding the Spike antigen and using electroporation as a delivery system (NCT04445389, NCT04447781, NCT04642638, NCT04627675). The main disadvantages of a DNA vaccine is when electroporation is used as a delivery system, electroporation can cause considerable pain and anxiety on administration and not suitable for mass vaccination programs, alternative delivery systems are currently being pursued.
Nanoparticle Vaccine Delivery Systems
Nanoparticle based drug delivery platforms offer an alternative vehicle for delivering drugs that have previously suffered from pharmacokinetic limitations including poor bioavailability, a short half-life or poor solubility. A variety of nanoparticles have been explored as delivery systems or as adjuvants, such as polymeric nanoparticles, liposomes, micelles, carbon nanotubes, mesoporous silica nanoparticles, gold nanoparticles and virus nanoparticles, that can all be used alone or in combination (129). Liposomes are a popular nanoparticle vaccine delivery system, they are versatile and can be constructed with a variety of different properties by changing the lipid composition, charge, size and surface properties (130–132). Nanoparticle based drug delivery platforms use well-known lipid carriers to deliver biotherapeutic encoding tumor antigens directly into APCs such as dendritic cells. The targeting of APCs in the lymphoid compartments is accomplished by using well-known lipid carriers such as DOTMA, DOTAP, DOPE and cholesterol and by adjusting negative net charge of the nanoparticles to provide optimal drug delivery. Cationic liposomes are mainly composed of the lipids DOTMA and DOPE that form colloidally stable nanoparticles of reproducible particle size (200–400 nm) with an excess of positive charge preventing excessive aggregation (133). Liposomes can increase the immunogenicity of target antigens for cancer vaccines and have been used to deliver RNA, DNA and antigens. Hydrophilic and lipophilic antigens can be loaded into liposomes, the hydrophilic antigens are trapped in the aqueous inner space and the lipophilic components are inserted into the lipid bilayer by adsorption or chemical attachment. Liposomes have been utilized to improve lymph node trafficking of small molecule adjuvants to the lymph node (134)
BioNTech have developed a lipid-based nanoparticle formulation called Lipoplex. Lipoplex has been used to provide efficient targeting of RNA to dendritic cells. The optimized Lipoplex:RNA formulation uses a charge ratio of 1.3:2, this was found to effectively target RNA to the spleen, form monodisperse and stable particles and was fully resistant to degradation by mouse serum at 37°C (135). In addition to targeting APCs, liposomes can also protect the RNA to be delivered from extracellular ribonucleases and mediates efficient uptake and expression by DCs and macrophages located in various lymphoid compartments. Lipoplex complexed with RNA encoding tumor antigens has also been shown to induce strong effector and memory T cells responses and mediate IFNα dependent rejection of progressive tumors (135). Vaccines using Lipoplex complexed with RNA induce and mobilize both the adaptive and innate immune responses mimicking an antiviral response.
Sahin et al. (136) recently conducted at phase 1 trial (NCT02410733) in melanoma patients who received melanoma FixVac (BNT111) an intravenously administered liposomal RNA (RNA-LPX) vaccine that targets four tumor associated antigens (NY-ESO-1, MAGE-A3, tyrosinase and TPTE) (135). Interim analysis (136) has shown that melanoma FixVac when used alone or in combination with the checkpoint inhibitor PD-1 mediates durable responses in patients with unresectable melanoma and with prior experience with a checkpoint inhibitor. FixVac induced clinical responses and potent CD4+ and CD8+ T cell responses could be detected with the cytotoxic T cell responses in some patients reaching the same level as reported for patients on T cell therapy trials. The completion date for this trial is estimated for December 2021, so far 119 patients (as of August 2020) have been enrolled.
Self-Assembling Peptides
Self-assembling peptides can also be used as a delivery system to deliver antigens to target cells. Self-assembling peptides can spontaneously form into ordered structures in response to changes in pH, solvent, co-assembling molecules, temperature and ionic strength (137, 138). They can have a diverse range of properties and can be manufactured to form nanomicelles, nanovesicles, nanofibers, nanotubes, nanoribbons and hydrogels (139). Self-assembling peptide deliver systems have a number of advantages over liposomes or nanoparticles including high drug loading, low drug leakage, biodegradability and are highly permeable to target cell membranes. The particle size is important for vaccine delivery and can impact the efficiency of uptake by APCs, with smaller particles (20-200 nm) being more immunogenic but there is no optimal size and this should be optimized for each vaccine candidate (140–142). The smaller particle size is thought to improve uptake into DCs and also the lymphatic system, in addition to size the shape, stability and ability to display multiple antigen can also improve immunogenicity. Self-assembling peptides can be designed to provide vaccines with the desired properties to enable efficient delivery to the target cell.
A delivery system based on modified cell penetrating peptide (CPP) based gene vectors, the Glycosaminoglycan (GAG)-binding enhanced transduction (GET) delivery system has been used to enhance delivery of nucleic acids for lung gene therapy and bone regeneration in vivo (143, 144). The GET peptides (143–150) are multi-domain sequences comprising of a heparan sulphate (HS) cell targeting sequence fused to a CPP for improved membrane association and synergistically enhanced intracellular delivery of therapeutic cargoes (145). GET peptides can deliver self-reporting cargo (monomeric red fluorescent protein; mRFP) into difficult to transduce cell types including mesenchymal stem cells (MSCs), human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs). Delivery involves a heparin sulphate (HS) cell targeting system fused to a CPP and an endosomal escape peptide and a system which stabilizes particles to prevent aggregation and promote diffusion and cell uptake by PEGylation. The tripeptide complexes the DNA into nanoparticles and can be delivered by simple intramuscular injection. This tripeptide formulation has achieved exceptional results in DNA delivery applications in particular in lung, brain, and has huge potential in vaccine delivery.
Genetic Vaccines
DNA Vaccines
DNA vaccines have a number of advantages, they are simple to design, relatively low production costs, good stability (stable at +2-8°C) and solubility, can be rapidly modified, it is a versatile platform that can have many applications including in infectious diseases and oncology. DNA vaccines were first shown to be immunogenic in the 1990s (151–153), they are an attractive cancer vaccine platform (154) and this led to a flood in preclinical and clinical trials. Plasmid DNA vaccines can be designed to act as both an antigen and adjuvant (155), unmethylated DNA containing cytosine-guanine rich regions can act as an adjuvant stimulating an immune response (156). DNA vectors have a negatively charged backbone and have a low molecular weight, therefore naked DNA often suffers from poor cellular uptake resulting in poor antigen production (157–159). With a significant improvement in our knowledge of cancer immunology particular the TME and immune suppression the reasons for the failures of early DNA vaccines are now better understood.
In addition to improving the delivery of plasmid DNA the vector itself can also be modified to specifically target epitopes directly to APCs. In preclinical models we have previously demonstrated that a DNA plasmid encoding T cell epitopes within the complementarity determining regions of a human IgG1 antibody (ImmunoBody®) (160), when administered with electroporation (EP) stimulates high avidity T cell responses (161). ImmunoBody® works by the direct uptake of the DNA into APCs, it is then transcribed, translated and processed, with epitopes being presented on the cell surface in combination with MHC. ImmunoBody® can also be taken up by both antigen presenting cells and non-antigen presenting cells and the transcribed antibody protein secreted. The secreted antibody is internalized via the high affinity FcγR1 receptor (CD64) on antigen presenting cells, it is then processed, and epitopes cross presented on MHC class I. The combination of direct and cross presentation induces T cells with sufficiently high avidity to eradicate established tumors in preclinical models (160). Immunization with ImmunoBody® DNA vectors induces high frequency and avidity of T cell responses that are superior when compared to those induced by immunization with DNA encoding full-length antigen or when using peptides or peptide loaded onto dendritic cells (161–164). The first ImmunoBody® in the SCIB series, SCIB1, targets four epitopes from the melanoma-associated antigens TRP2 and gp100, our second vaccine, SCIB2, incorporates several epitopes derived from the NY-ESO-1 cancer testis antigen. A first-in-human study performed by Patel et al. used SCIB1 ImmunoBody® that incorporated HLA-A*02:01 restricted epitopes from gp100 and TRP-2 in addition to HLA-DR*04:01 and HLA-DR7/DR53/DQ6 restricted epitopes from gp100. In a cohort of 15 melanoma patients SCIB1 was shown to be safe (165). In this trial 7/15 patients had stable disease, 5/20 fully resected patients experienced disease recurrence and 1 patient had measurable disease, all patients were still alive at the last observation time of 37 months. A phase 2 study in melanoma patients receiving pembrolizumab is now recruiting (ClinicalTrials.gov Identifier: NCT04079166).
RNA Vaccines
The RNA vaccine platform has the advantage that RNA does not integrate into the host cell genome and thus avoids potential safety concerns, it is also quick to manufacture and can encode multiple epitopes. RNA is single stranded and therefore has a built-in adjuvant function through TLR7 and TLR8 stimulation. However, RNA is very susceptible to cellular degradation, to overcome this in clinical trials it has either been injected directly into inguinal lymph nodes or delivered using a nanoparticle delivery system that protects the RNA. RNA is particularly susceptible to degradation by RNases, to improve transfection and avoid degradation many groups have used delivery systems such as nanoparticles and liposomes (135, 166–170). RNA has an advantage over DNA in that it only needs to be delivered to the cytoplasm for translational into protein unlike DNA that needs to enter the nucleus for transcription.
The first clinical trials using RNA was performed by Weide et al. (171, 172) in patients with metastatic melanoma. In a phase 1 trial in 15 melanoma patients (171) the intradermal administration of naked mRNA was shown to be safe. In a phase 1/2 trial (172) 21 patients received i.d. injections of protamine stabilized mRNA coding for Melan-A, Tyrosinase, gp100, Mage-A1, Mage-A3 and survivin; GM-CSF was used as an adjuvant and half the patients also had keyhole limpet hemocyanin added to the vaccine. The number of clinical responses to the vaccine in this trial was low with only 1 promising clinical response observed in a patient with measurable disease. In a phase 1/2 trial performed by Rittig et al. in 30 patients with stage IV renal cell cancer (173), naked mRNA coding for TAA’s was administered intradermally. This trial demonstrated that vaccination was safe and well tolerated and induced clinical responses in 16 patients; this trial also demonstrated that vaccination induced CD4 and CD8 T cell responses as determined by IFNγ ELISpot and Cr-release assays. The results from these trials demonstrated that vaccination with RNA was feasible and safe. With improvements to trial design, frequency, and route of administration the RNA vaccine platform is progressing through clinical trials in the fields of cancer and infectious diseases. For COVID-19 two RNA vaccines (Tozinameran from Pfizer–BioNTech and mRNA-1273 from Moderna) have now been approved by national health regulators.
In a study performed by Sahin et al. the use of an RNA vaccine encoding neoantigens was explored (174) in melanoma patients. In this study neoantigens were identified by comparative exome analysis in tumors from thirteen patients with stage III and IV melanoma. Mutations were selected for incorporation into the vaccine based firstly on the predicted binding score for HLA class II and secondly based on the predicted binding score for HLA class I. For each patient two synthetic RNAs were synthesized incorporating the identified mutations. The RNA vaccine was produced within 68 days (range 49 to 102 days), following analytical testing they were released within 103 days (range 89 to 160 days). RNA vaccines work in a similar way to the long peptide vaccines, the RNA is translated into protein which is then processed into long peptides by APCs, these peptides are then loaded onto MHC class I or class II molecules and presented on the cell surface to prime and activate T cells. This study demonstrated the clinical feasibility and safety of RNA neo-epitope vaccines. In this study 8/13 patients had no tumors develop during the monitoring period and neoantigen specific T cells could be detected in the peripheral blood of these patients. The use of many neoantigen epitopes in a vaccine reduces the risk of single antigen loss variants (175), however, in this study the outgrowth of B2M deficient tumor cells in one patient demonstrates the complexity of the TME and the selective pressures that drive resistance to therapy.
Viral Vector Vaccines
Viral vectors have been used in both the gene therapy and vaccine fields. Viral vectors have the advantage of being recognized as foreign by the immune system, inducing potent innate and adaptive immune responses resulting in the induction of strong and durable immune responses. Viral vectors enable the presentation of intracellular antigens incorporated into the vector such as cancer antigens, viral antigens, or a specific gene for gene therapy.
The most commonly used viral vectors are derived from adenoviruses, poxviruses and alpha viruses. The majority of viral vectors are replication defective or attenuated versions, these are preferred from a safety point of view. Viral vectors have a very good safety record with many approved in the infectious disease field such a recently approved Ebola vaccine and COVID-19 vaccines that use adenovirus virus vectors. A disadvantage of the viral vectors is their ability to also induce immune responses that also neutralizes the vector preventing further repeat immunizations. Pre-existing immunity to measles and adenovirus can be problematic limiting the effectiveness and ability to boost responses when adenovirus or measles virus vectors are used. A prime boost vaccine regime is commonly used, and a number of different strategies have been used to overcome the problem with pre-exiting immunity. Strategies using a non-human specific virus such as the replication-defective chimpanzee adenovirus (ChAd68 serotype), or using different vectors derived from different viruses for the prime and boost immunization or using different vaccine platforms for the prime and boost immunizations can all avoid problems associated with pre-existing immunity to the virus vector. A common combination is the use of a DNA prime and a viral vector boost. Another commonly used combination is the Modified Vaccinia virus Ankara (MVA) and Adenovirus (Ad) vectors, both vectors induce potent immune responses that when used in combination in a prime boost regime these responses are further enhanced (176, 177). These strategies have all been used successfully in the infectious disease field, particularly more recently to target SARS-CoV-2, where 40 viral vector vaccines are currently being assessed in preclinical studies, an additional 19 vaccines are currently in clinical trials and another 4 vaccines have already received approval from regulatory authorities (178).
A number of different viral vectors have been used for cancer vaccines (179); with some having progressed into clinical trials. In clinical trials the efficacy of cancer vaccines using viral vectors have not delivered the same results as those generated in the infectious disease field. The immunosuppressive TME and selection of the best cancer antigen to target is problematic and impacts all cancer vaccine platforms. To overcome central tolerance and the immune suppressive TME a cancer vaccine would need multiple boosts in order to induce and sustain a potent immune response, however, this can be problematic due to anti-vector immunity. In preclinical and clinical studies, a prime-boost approach using a recombinant vaccinia vector and a recombinant avipox virus have been successfully used, and multiple boosts using recombinant avipox such as fowlpox is possible. Preclinical and clinical studies have shown that multiple booster vaccinations using a fowlpox does not induce host anti-vector immune responses (180, 181). Both viruses have been shown to be safe, vaccinia was used in the smallpox vaccine that has been delivered to over 1 billion people worldwide. Avipox is an avian virus that is unable to replicate in mammals. Both vectors do not integrate into DNA and successfully infect APCs thus stimulate potent immune responses.
The TRICOM vaccine platform uses the recombinant vaccinia virus (rV-) for the prime and recombinant avipox (fowlpox, rF-) for multiple booster vaccinations. Each vector contains one or more TAAs and transgenes for the costimulatory molecules CD80, ICAM1 and LFA-3. In a phase 2 clinical trial, 125 men with metastatic castration-resistant prostate cancer received a vaccinia virus encoding PSA in combination with GM-CSF followed by six subsequent boosts using a fowlpox virus encoding PSA (PROSTVAC-VF) (182). The results from this phase 2 trial was encouraging with a 10-month improvement in overall survival compared to the empty vector control group (183). Unfortunately, these results were not seen in a large phase 3 study and the study was subsequently stopped (184). It is likely that despite the activation of specific T cells they were either not potent or unable to overcome the immunosuppressive TME (184). Trials are now ongoing to see if combining PROSTVAC-VF/TRICOM with check point inhibitors can improve clinical responses (NCT02933255, NCT04020094, NCT03532217, and NCT03315871).
Peptide Vaccines
The number of peptide vaccines being explored has increased due to the discovery of neoantigens. Targeting neoantigens is a personalized therapy and the rapid synthesis of peptides makes the peptides vaccines an attractive platform. Almost half of the clinical trials currently recruiting (as of August 2020) that target neoantigens are using peptide vaccines, with the RNA and DNA vaccine platforms also represented (Figure 3). Following administration the peptides included in a vaccine need to be presented on antigen presenting cells (APCs) in order to trigger an adaptive immune response. To efficiently prime an immune response the coadministration of an adjuvant is required to activate the immune system to kill tumor cells expressing the peptide (185–187). Tumor antigens need to be processed and the resulting peptides presented on the cell surface in association with MHC class I or class II molecules. Cancer specific T cells in the TME need to recognize the relevant peptides and kill tumor cells expressing them. The key to the success of a peptide vaccine relies on the correct choice of peptides to include and the best adjuvant to use to generate a local immune response and promote antigen trafficking to local draining lymph nodes. Bioinformatic applications and algorithm prediction program are commonly used to define peptides capable of binding MHC I or MHC class II molecules. Identification of peptides bound to the MHC molecules on the cell surface can be achieved via mass spectrometry (MS) analysis. Combining data from MS analysis, epitope predicting algorithms and gene expression data help to predict the best candidate peptides to include in a vaccine.
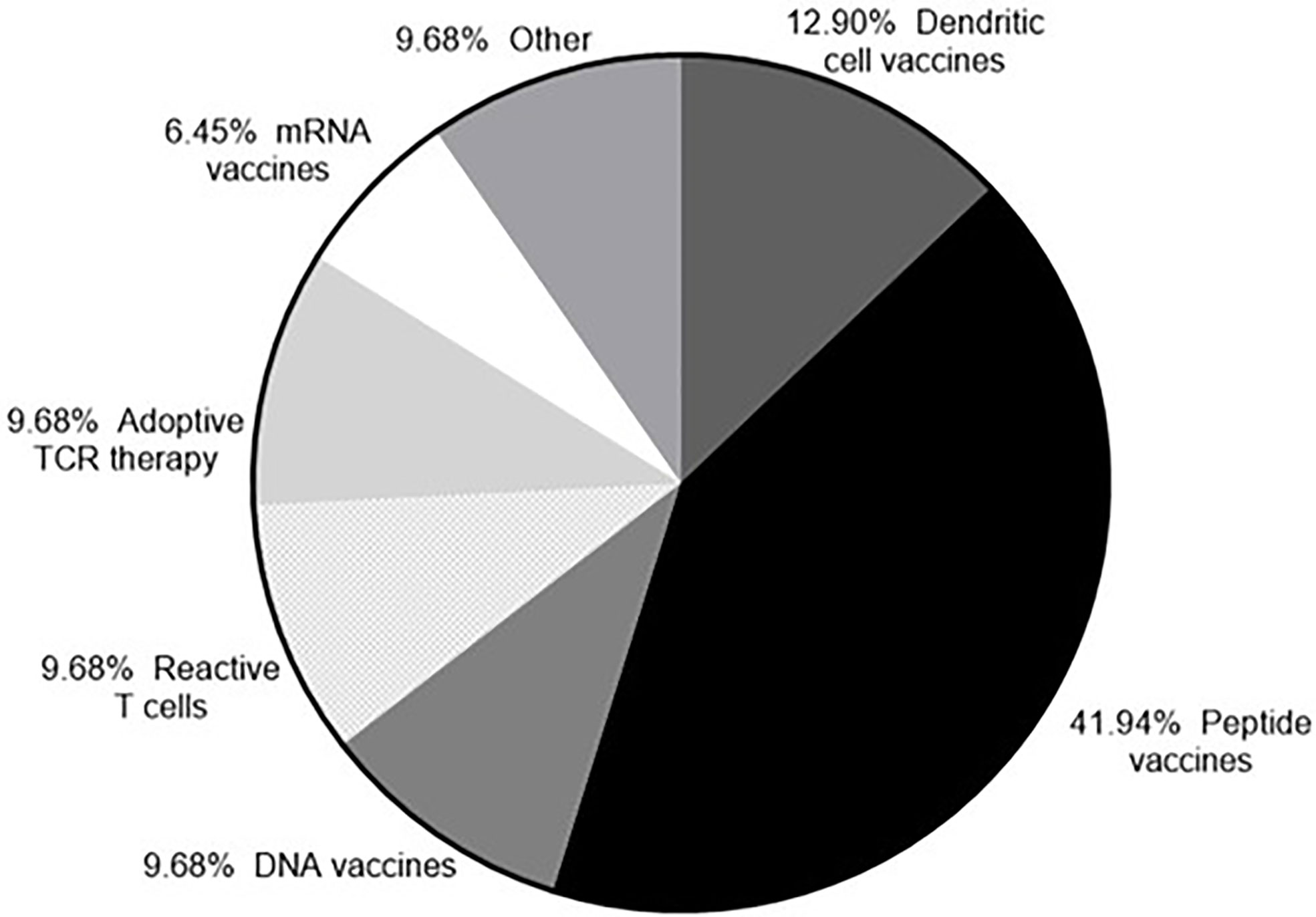
Figure 3 Neoantigens currently in clinical trial. According to clinicaltrials.gov (as of 23rd September 2020) there are currently 33 clinical trials recruiting that target neoantigens.
A vaccine needs to stimulate both CD4+ and CD8+ specific T cells. The majority of peptide vaccines use longer peptides typically 20-30mers, these are likely to contain nested CD8+ T cell epitopes in addition to longer CD4 T cell epitopes and therefore are able to stimulate both CD4+ and CD8+ T cells. In addition, multi-peptide vaccines are often used, incorporating many peptides meaning that many antigens can be targeted, increasing the chances of overcoming any antigen loss on the tumor cells. A number of peptide vaccines targeting neoantigens have been developed by a number of groups, this personalized therapy can target a patient’s individual tumor. Ott et al. (188) used whole-exome sequencing (WES) and RNA-sequencing (RNA-Seq) to identify neoantigens from six stage III/IV melanoma patients. Using NetMHCpan (v2.4) a list of peptides that bind to MHC class I was generated, synthesized peptides were between 15 to 30 amino acids in length and thus capable of stimulating CD4+ and CD8+ T cells. Patients were immunized with 30 peptides given in combination with poly-ICLC. This vaccine-induced polyfunctional CD4+ and CD8+ T cells targeting 58/97 and 15/97 neoantigens respectively across the patients. At 25 months post vaccination 4 patients had no recurrence while 2 patients had progressive disease that was successfully treated with anti-PD-1 therapy.
A number of early peptide-based cancer vaccine trials primarily focused on short peptides from tumor associated antigens, and did not include any delivery system or longer peptide formulations (189). The majority of these trials failed when reaching phase 3 trials, a number of additional reasons for these failures include adjuvant selection, timing of vaccination and peptide formulation (120). Peptide vaccines do generate an immune response but as a monotherapy they can struggle to show efficacy in the clinic. Kimura et al. immunized 39 patients with premalignant colon adenomas with a MUC1 peptide vaccine, they showed the peptides were immunogenic however in 22 patients there was a lack of a response which correlated with a high number of myeloid derived suppressor cells in the TME pre vaccination (190). In another two trials in melanoma and ovarian cancer patients (191, 192), a mixture of peptides were used to immunize patients, vaccination induced an immune response which was associated with some favorable outcomes. The majority of peptide vaccines did not generate an immune response that was robust enough to see any significant clinical benefit (189). These early studies highlighted the need to further optimize peptide vaccines, either by better targeting, better adjuvants or used in combination to overcome the immunosuppressive TME.
Peptide-Adjuvant Conjugate Vaccines
The administration of free adjuvant with antigens in a cancer vaccine can result in their dissociation following injection and subsequently do not enter the lymphatic system. Adjuvants can be rapidly degraded (193) reducing the amount that reaches the target cells resulting in suboptimal antigen priming and immune response. Free adjuvant in the circulation can also induce autoimmunity (194) or toxicity. The co-delivery of adjuvant with antigens is required to induce a potent immune response while avoiding any autoimmunity or toxicity, as already described a delivery system can be used to deliver the adjuvant and antigen, alternatively the adjuvant and antigen can be linked to improve targeting.
The direct conjugation of a peptide to an adjuvant is gaining increasing attention, particularly from groups targeting neoantigens. Previous reports have described the direct conjugation of peptides to TLR ligands can enhance the immune response by directly targeting the peptide and adjuvant to the same APC (195–199). Peptides can be linked to hydrophobic carriers such as lipids (92), fatty acids (200) and TLR agonists (196, 201, 202) for more efficient delivery to APCs and subsequently lymph nodes. A recent preclinical study performed by Lynn et al. (203) used a peptide platform based on charge modified TLR-7/8a peptide conjugates, these conjugates self-assemble into nanoparticles of uniform size which is independent of the peptide antigen composition. This platform is used to conjugate identified neoantigen peptides, these peptides would possess a variety of properties, such a platform would be able to incorporate peptides with a wide range of characteristics. Neoantigen peptides were predicted (179 peptides) from three murine tumor models, vaccination of mice induced a CD8+ T cell response with approximately 50% of the peptides being recognized, this led to enhanced tumor clearance.
We have directly conjugated citrullinated peptides from vimentin and α-enolase (Vim28-49cit, Vim415-433cit and Eno241-260cit) to the TLR1/2 ligand AMPLIVANT®. The direct linkage of a TLR agonist to a peptide can enhances the immunogenicity of the vaccine (196–199). In HLA-DR4 and DP4 transgenic mice vaccination with the conjugated peptides induced a high frequency of specific T cells. The direct conjugation of the Vim28cit, Vim415cit and Eno241cit peptides to the TLR1/2 ligand Amplivant® reduced the peptide-equivalent dose required to induce immune responses by at least 1 log without the loss anti-tumor responses (60). These results demonstrate that the linkage of a TLR ligand to a peptide enhances the immune response and supports the development and application of these peptide/TLR ligand linked conjugates in a clinical setting.
Peptide vaccines have many advantages, they can be chemically synthesized, manufactured at large scale and cost effective (204) when compared to other cancer therapies. In pre-clinical and clinical studies they have been shown to be safe and well tolerated (204, 205). Peptide vaccines should include peptides that target multiple antigens to generate a polyclonal antigen T cell response (206–208). Like DNA and RNA vaccines the use of a delivery system can help improve the targeting and stability of peptides used in a vaccine which reduces any potential off target effects (209–212). The production of conjugated peptide vaccines can be problematic for some peptides, particularly hydrophobic peptides that tend to form aggregates that complicate manufacturing and when injected they form injection site depots leading suboptimal immune responses (213). The conjugation of peptides with adjuvants improves the delivery of both components to APCs which may be needed to induce T cell priming (203, 214). Peptide conjugates used in combination with other therapies have a huge potential. A peptide conjugate vaccine can specifically target APCs and that has the advantage of being dose sparing, in combination with a checkpoint inhibitor to relieve immunosuppression in the TME this would provide the best opportunity for these vaccines to work in the clinic.
Vaccines in Combination With Other Therapies
Immune checkpoint inhibitors constitute an important breakthrough positively influencing treatment outcomes in cancer patients. Cancer vaccines have the potential to induce potent immune responses but are hampered as tumor cells possess a variety of immune evasions mechanisms that interfere with the function of T cells (215–220). Upon activation, T cells migrate and accumulate in the TME where they can induce tumor cell killing, however, tumors have evolved multiple mechanisms that can dampen or inhibit T cell mediated killing. Tumor cells can alter the antigen processing machinery, secrete immunosuppressive factors that kill the T cells or activate pathways that induce tolerance rendering any tumor therapy ineffective (221). The identification of the key regulators of the immune response has led to the generation of new therapies that have the potential to reverse some of the immune suppression in the TME. Immune checkpoint inhibitors are cell surface receptors that regulate the immune response, they enable self-tolerance while preventing over activation of the immune system resulting in autoimmune disease (222). In the TME the expression of checkpoint receptors suppresses T cell activation and thus provides the tumor with a growth advantage (223). The cytotoxic T lymphocyte protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) are the best characterised checkpoint receptors in the immunotherapy field, others have been described that are emerging from preclinical studies and entering the clinic.
The checkpoint receptor, PD-1, is expressed on activated T cells and overexpressed on exhausted T cells (90). There are two PD-1 ligands, PD-L1 and PD-L2, PD-L1 is expressed on many cells including immune cells, epithelial cells, endothelial cells and tumor cells (88, 91). PD-L2 is expressed on professional antigen presenting cells including DCs and macrophages (224). The binding of PD-1 on T cells to PD-L1 expressed on a tumor cell or PD-L2 expressed on an APC leads to TCR downregulation, resulting in lower secretion of TNF-α, IFN-γ, and IL-2 (92). The expression of CTLA-4 is induced upon T cell activation and competes with the costimulatory molecule CD28 for its co-stimulatory ligands. CTLA-4 suppresses the early activation of naïve and memory T cells by competing with CD28 (88–90), PD-1 inhibits T cell function at a later activation stage by down regulating TCR expression. Monoclonal antibodies that block CTLA-4 (223), PD-1 (225) or PD-L1 (226) pathways remove the inhibition of T cell function (227) and have made significant clinical impacts. Antibodies that specifically block the CTLA-4 or PD-1/PD-L1 pathway have the potential to remove T cell immune suppression enabling the successful recognition and killing of tumor cells.
Checkpoint blockade has shown promising results in clinical trials and have gained approval for an increasing number of cancers including melanoma, renal-cell carcinoma (RCC), advanced non-small-cell lung cancer (NSCLC), classic Hodgkin’s lymphoma (HL), bladder carcinoma, Merkel cell carcinoma, head and neck cancer, and more recently, solid tumors with mismatch repair-deficiency. PD-1 and CTLA-4 inhibit T cell responses at different stages and by different mechanisms, it is therefore tempting to block both pathways in order to overcome immune suppression. Clinical trials in melanoma patients that have combined PD-1 and CTLA-4 blockade have shown improved clinical responses, however, these have come at a cost with an increase in toxicities being reported (228–230). The combination of anti CTLA-4 and anti PD-1 is now approved as the first line therapy for advanced melanoma patients; however, the toxicities have limited the use of this combination, trials are ongoing to vary the dose and interval of dosing to reduce toxicity.
Many cancer vaccines currently in clinical trials are combined with a checkpoint inhibitor such as CTLA-4, PD-1 or PD-L1 inhibitors, which are offered as standard treatment for an increasing number of cancers. A couple of comparative studies have shown that the combination of a tumor vaccine with a checkpoint inhibitor is more effective than monotherapy (231, 232). Less than 50% of patients respond to checkpoint inhibitors (233–235), there are several other factors that can lead to immune suppression in the TME, such as the action of T-regulatory cells, myeloid-derived suppressor cells (MDSCs), tumor associated macrophages and immunosuppressive DCs (236). For a vaccine to show efficacy in the clinic it is likely a combination with another form of therapy is required to improve tumor specific T cell function in the TME.
In preclinical murine models using the ImmunoBody® vaccine SCIB2 a synergistic effect was observed when given in combination with anti-PD-1 (Figure 4) (237). The synergistic effect was also observed with SCIB1 when given with anti-PD1 (163, 237). These results demonstrate that a cancer vaccine on its own will not achieve the expected results in the clinic without combining with other therapies aimed at modifying the TME by reducing immune suppression and also improving T cell trafficking into the tumor.
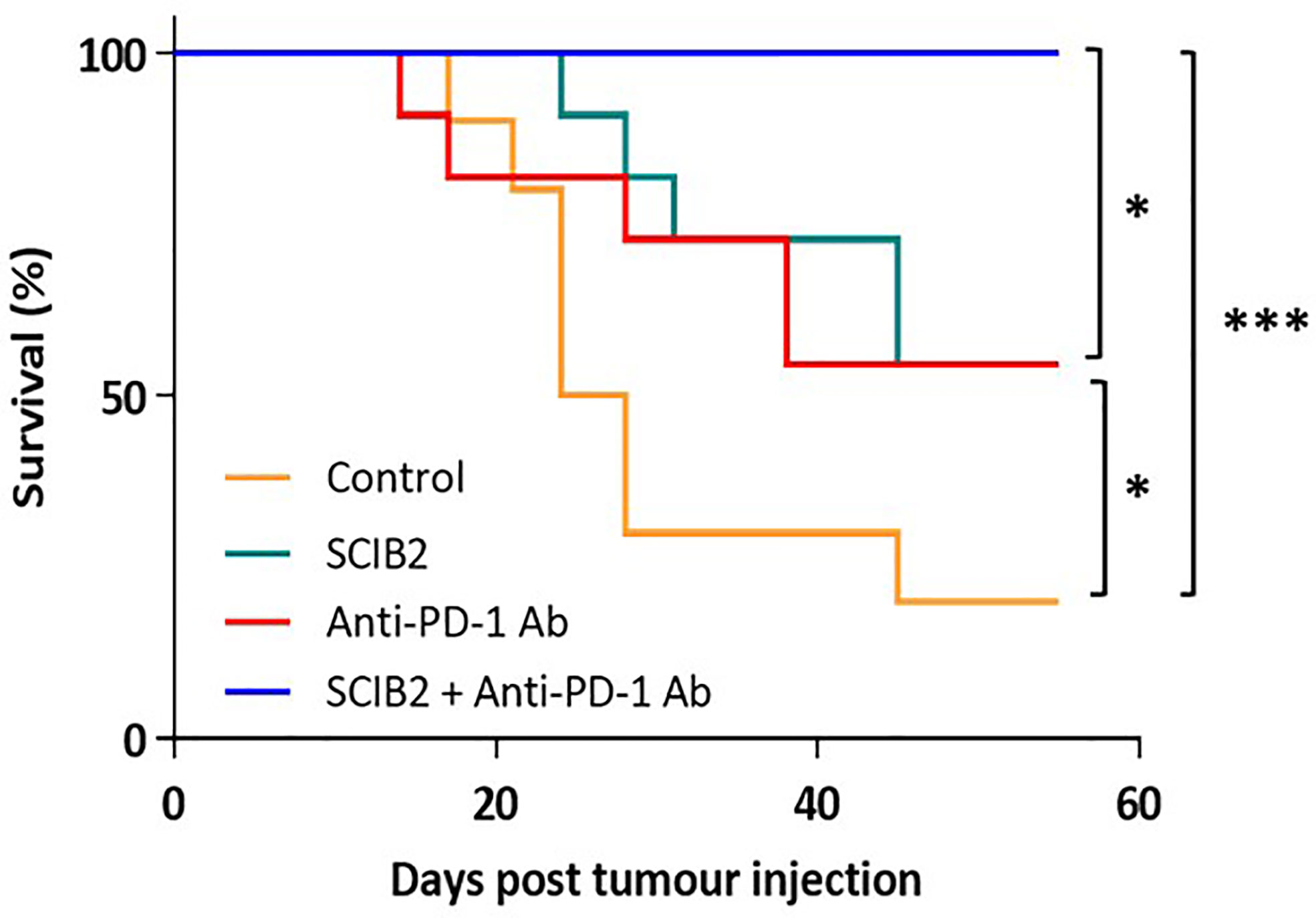
Figure 4 Survival of HHDII mice challenged with 5 x104 tumor cells and immunized with SCIB2 and anti-PD-1 antibody alone or in combination. Control vs SCIB2 (*p = 0.037); Control vs anti-PD-1 antibody (p = 0.111); Control vs SCIB2 and anti-PD-1 antibody (***p = 0.0003); SCIB2 vs SCIB2 and anti-PD-1 (* = 0.0177); anti-PD-1 antibody vs SCIB2 and anti-PD-1 (*P = 0.0177). Lack of survival was defined as tumor size > 528 mm3. Each curve represents at least 10 mice per group.
The first line treatment for the majority of cancer indications is radiotherapy or chemotherapy. These traditional treatments are not targeted therapies but the damage they cause to tumors results in the release of more antigens from the tumor cells. Damage to tissue surrounding a tumor can also induce the recruitment of T cells into the vicinity, this is particularly valuable when the tumor mutational burden (TMB) is low (238). A number of clinical trials are using either radiotherapy or chemotherapy to enhance the immune response to a vaccine. Radiotherapy can enhance the recruitment of T cells into tumor tissue and increase the intensity of specific anti-tumor immune responses (239). Other studies have shown that some chemotherapeutic drugs can enhance the antitumor activity of tumor vaccines (140, 141) and adoptively transferred T cells (137, 138). A vaccine combined with immune checkpoint inhibitors or traditional treatments can induce stronger anti-tumor responses (188), as such the majority of tumor vaccines in clinical trials are in combination with other therapies. The majority of cancer patients will be offered first line standard of care treatment prior to being offered alternative therapies or participation in a clinical trial.
Tumor Microenvironment and Tumor Induced Immune Suppression
Tumors evolve and change, they are heterogenous and genetically unstable. Tumors generate many mutations overtime, some are cloned, altered or lost in the tumor genome. Advances in sequencing technology now allows the analysis of a resected tumor or biopsy, the data gathered is a snapshot of a single tumor or part of a tumor at a specific time and does not provide information regarding the overall heterogeneity in the tumor (240, 241). This is a particular problem with the development of a personalized vaccine targeting a neoantigen, the information gained via biopsy or resection may represent a mutated tumor subclone or the neoantigen may be not expressed in the whole tumor or metastatic tumors compromising the effectiveness of the vaccine (242). The ideal mutations to target are driver mutations, these are critical for the growth of the tumor and are usually expressed in every tumor cell. However, the number of driver mutations can be low, for example in melanoma only 8% neoantigens are driver mutations (243). The degree of tumor heterogeneity will vary between patients, indications, and tumors. Improving our understanding of tumor heterogeneity will help identify the best epitopes to include in a cancer vaccine and targeting more than one antigen with help overcome tumor heterogeneity. The cancer vaccines targeting neoantigens address this heterogeneity by targeting more than one neoantigen and also addressing potential antigen loss.
Another factor that significantly contributes to the success immunotherapy is the TMB, studies by Rooney et al. demonstrated that the TMB correlated with immune responses (244). Tumors with high TMB, such as melanoma and NSCLC have a higher response rate to immunotherapy compared to tumors with a low TMB, however, this is not the case with all tumors. Pediatric tumors generally have fewer somatic mutations, a study performed by Zamora et al. showed that tumors from children with acute lymphoblastic leukemia had a low TMB but they could still induce a strong anti-tumor response (245). A number of clinical trials are underway for cancer indications that have an unmet need, poor survival and also have a low TMB e.g. glioblastoma and pancreatic cancer. Hopefully the results from these trials will help with our understanding of how to target tumors with a low TMB.
The TME consists of many different cell types including immune and stromal cells, vasculature, extracellular matrix and a variety of cytokines and chemokines. The extracellular matrix is made up of cells from endothelial, mesenchymal and haematopoietic origins. Changes in the TME impact the trafficking of TILs and efficacy of cancer vaccines that have induced specific T cells but are unable to traffic into the tumor.
Tumors have a number of mechanisms that have evolved to suppress anti-tumor immune responses. Apart from checkpoint mediators such as PD-1 and CTLA-4 a number of cell types have been identified that contribute to immune tolerance and evasion in the TME, Myeloid-derived suppressor cells (MDSCs), T regs, tumor associated macrophages (TAMs) and cancer associated fibroblasts all contribute to this immune suppression (246). MDSCs are a type of regulatory cell that are found within the TME (246, 247), they produce nitric oxide, cytokines and reactive oxygen species that can suppress T cells. MDSCs play a critical role in tumor invasion, metastasis and angiogenesis (248, 249). The presence of MDSCs in the TME correlates with poor overall survival and progression free survival (250). In a murine model of rhabdomyosarcoma, the trafficking of MDSCs was inhibited and subsequently an enhanced response to anti-PD-1 therapy was observed (251). In addition to MDSCs the presence of Tregs in the TME in many cancer types is also associated with a poor prognosis (252). Tregs are critical for the maintenance of cell tolerance, they suppress T cell responses by binding IL-2 therefore limiting the amount of free IL-2 available to drive T cell proliferation and activation (253). Tregs express CTLA-4 and can produce immunosuppressive cytokines that further contribute to the immune suppressive TME (252). The TAMs, in particular the M2 macrophages are another cell type that can contribute to the immune suppressive TME (254). The M2 macrophages can promote tumor growth by stimulating tumor cell motility, angiogenesis, and immune evasion (255). Murine studies have demonstrated that the depletion of macrophages reduces tumor growth and also by inhibiting the myeloid growth factor signaling pathway in macrophages overcome resistance to checkpoint inhibitors in a pancreatic cancer murine model (256, 257). The depletion or inhibition of MDSCs, Tregs or TAMs all have the potential to improve anti-tumor responses induced by vaccination or a cellular therapy, however, the impact of depleting these cells in the periphery as well as the tumor increases the potential to induce autoimmunity.
Within the TME the most abundant stromal cells are the cancer associated fibroblasts (CAF), these have been shown to play a role in tumorigenesis, angiogenesis, metastasis, drug resistance, immunosuppression, extracellular matrix (ECM), remodeling and maintenance of cancer stemness (258–264). Different subtypes of CAFs exist each capable of secreting a number of cytokines and chemokines such as TGF-β, IL-6, IL-8, IL-13, CXCL12, CXCL14, and VEGF that inhibit anti-tumor immune responses. Some CAFs also express PD-L1/PD-L2 or produce metabolites or enzymes such as indoleamine-2,3-dioxygenase (IDO), arginase (Arg), adenosine, and tryptase that recruit Tregs, mast cells and TAMs. CAFs can also contribute to the integrity of tumors, they can synthesize components that make up the ECM such as collagen, fibronectin, matrix metalloproteinases and can contribute to ECM stiffness and thus prevent T cell infiltration. The role of CAFs in cancer progression makes them a promising target for cancer therapy. There have been a few studies looking at anti-CAF based therapies, however in a murine CAR T cell-based study targeting the fibroblast marker FAP, toxicity was observed due to expression of FAP on other tissues (265). Other studies are looking at depleting CAFs, blocking their function or altering their function.
Cytokines and chemokines within the TME can also induce an immune suppressive TME and reduce T cells responses. One of the most studied cytokines is transforming growth factor beta (TGF-β). TGF-β signaling has a massive impact in the TME where it can influence cell growth, differentiation and apoptosis while also inhibiting T cells responses and upregulating Tregs (266). In patients with colon cancer TGF-β tends to suppress cell growth but in advanced stages of the disease the presence of cells expressing members of TGF-β superfamily tend to have a poor prognosis. In murine models the inhibition of TGF-β reverses its immunosuppressive effects and improves the activity of T cells also rendering tumors suspectable to treatment with checkpoint blockade (267, 268). In addition to cytokines in the TME, chemokines such as CXCR2 and CXCR4 that bind to MDSCs and Tregs respectively contribute to tumor immune evasion. In murine models the inhibition of CXCR2 and CXCR4 in combination with anti-PD-1 reversed immune evasion (251, 269). Targeting cytokines and chemokines in TME could be a good cancer immunotherapy strategy that will help change the immune suppressive environment by preventing the recruitment and activation of Tregs, TAMs and MDSCs.
Cancer cells can also lose surface antigens as an immune evasion mechanism following natural or therapy induced selective pressure. Antigen loss has been observed for CD19 in acute lymphocytic leukemia and CD20 in chronic lymphocytic leukemia. Antigen loss is a common reason for resistance to therapy and subsequent relapse. To address the problem with potential antigen loss we have targeted two antigens in our Modi-1 and SCIB1 vaccines, with SCIB1 also inducing high avidity T cells that are capable of responding to a lower number of MHC:peptide complexes on tumor cells. In addition to antigen loss, tumors can also decrease MHC class I expression rendering the immune response powerless. The downregulation of MHC class I has been observed in both human and murine tumors (270–272). The majority of early primary tumors express MHC class I but this profile often changes as the tumor progresses and escape immune surveillance (273). The percentage of cancers that have HLA class I loss, total loss, haplotype loss or allelic loss can range from 65-90% (274, 275). We have addressed MHC class I loss by incorporating both MHC class I and class II peptides in our SCIB1 vaccine, our Modi-1 vaccine only includes MHC class II restricted peptides.
Conclusions
A large number of cancer vaccines have failed to show clinical efficacy, this can be due to the tumor’s own mechanisms of immune evasion and escape that have evolved including antigen loss, MHC loss, the presence of immune suppressive cells or soluble factors in the TME and lack of a robust anti-tumor immune response (276–279) and also due to the inability of the cancer vaccines to induce sufficiently high avidity T cell responses to efficiently destroy tumors. Early cancer vaccines were primarily focused on stimulating CD8+ T cell responses against tumor associated antigens often using short minimal epitope sequences, T cells recognizing these antigens are highly tolerized and subsequently these vaccines fail in the clinic. The incorporation of CD4+ T cell epitopes into peptides, RNA or DNA vaccine platforms is essential to induce specific CD4+ and CD8+ T cell responses. Targeting non self or mutated tumor antigens will induce high avidity T cells responses when delivered with optimal adjuvant and delivery systems, as such the neoantigens and post translational modified antigens currently show the most promise.
Generating a neoantigen vaccine can be costly both in terms of time and money, the peptide vaccines are personalized and require a significant amount of bioinformatics input to generate the best candidate neoepitopes. The Modi-1 vaccine is not a personalized therapy and is broadly applicable to many patients and cancer types, it is cheaper to manufacture when compared to other platforms and in addition has no time delay constraints that is associated with the production of neoepitope vaccines.
The vaccine platform and the delivery systems used have undergone a huge number of improvements over the last decade. Many new adjuvants have emerged or are currently being investigated in order to improve the immune response at the injection site while also increasing antigen trafficking to the lymph nodes. The majority of cancer vaccines are currently using TLR agonists as adjuvants, these have also been conjugated to peptides or included in nanoparticles to improve targeting. Other adjuvants such as STING, CD40 agonist and GM-CSF are currently being investigated in clinical trials. The correct selection of an adjuvant is key to the ability of the vaccine to induce a robust immune response. We have previously screened a number of different potential adjuvants to use in our Modi-1 vaccine, this included CpG (TLR9), MPLA (TLR4), CpG/MPLA (TLR9/TLR4), GM-CSF, imiquimod (TLR7), Poly I:C (TLR3) or TLR1/2 (AMPLIVANT®). Preclinical studies have shown that when AMPLIVANT® is given in combination with the Modi-1 peptides it induced the strongest anti-tumor response (60). This highlights the importance of determining the best delivery and targeting approach for a vaccine that generates the strongest immune response while reducing any possible toxicity.
With our ImmunoBody® platform we have modified a DNA vector by engineering T cell epitopes into the IgG1 CDR regions (163), the Fc region of the antibody targets the high affinity Fc receptor CD64 that is expressed on activated APCs. The SCIB DNA vaccines allow both direct- and cross-presentation of epitopes by targeting dendritic cells, and are able to generate high avidity CD8+ T cells that efficiently eradicate tumors.. Vaccination with SCIB1 induces high frequency and high avidity specific T cells (165).
Improvement with vaccine delivery systems has led to the generation of nanoparticles, self-assembling peptides, and needle free delivery systems. Electroporation was used to administer SCIB1 in our phase 1 clinical trial, and has been used to deliver other cancer and infectious disease vaccines. However, the pain on administration using electroporation and the requirement for specialized vaccine delivery instrument have limited their use, these newer delivery systems provide better alternatives. Liposomes are increasingly being used as delivery system, they are versatile, incorporating small drug candidates or antigens in the form of RNA or peptides, and have a good safety profile. Liposomes do require optimization in order to determine the optimal charge/size of the particle to incorporate their cargo and deliver it across the cell membrane. The targeting of cancer antigens to improve the local immune response and trafficking to lymph nodes can be achieved through either the linking of peptide to adjuvant, incorporating the antigen into nanoparticles or by modifying genetic vectors eg ImmunoBody® platform.
There are a number of challenges to address in the development of a successful cancer vaccine, we have tried to address a number of these challenges with our SCIB1, SCIB2 and Modi-1 vaccines. The SCIB1 vaccine is currently in phase 2 trials in combination with anti-PD-1 therapy, and the Modi-1 vaccine will be entering clinical trials in 2021. The success of any cancer vaccine does not rely only on the ability of the vaccine to induce a robust immune response but also on the modification of the immune suppressive TME to enable the successful trafficking of T cells and the ability of these T cells to recognize and kill tumor cells. The tumor size, TMB and previous treatments will all influence the success of a cancer vaccine and this will vary among patients and cancer types. With huge improvements in the cancer vaccine field and a better knowledge of the TME with time cancer vaccines will start to show good clinical efficacy.
Author Contributions
SP wrote the review article. VB and PS contributed and analyzed the data. LD proof read the review and conceived the ideas and work around the Modi-1 and Immunobody vaccine platforms. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by Scancell Ltd.
Conflict of Interest
LD is CSO and shareholder in Scancell Ltd. SP, VB, and PS are employees of Scancell Ltd.
The authors declare that this study was funded by Scancell Ltd. The funder was involved in the study design, collection, analysis, interpretation of data, the writing of this article and the decision to submit it for publication.
References
1. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med (2010) 363(5):411–22. doi: 10.1056/NEJMoa1001294
2. Berzofsky JA, Terabe M, Wood LV. Strategies to use immune modulators in therapeutic vaccines against cancer. Semin Oncol (2012) 39(3):348–57. doi: 10.1053/j.seminoncol.2012.02.002
3. Parchment RE, Voth AR, Doroshow JH, Berzofsky JA. Immuno-pharmacodynamics for evaluating mechanism of action and developing immunotherapy combinations. Semin Oncol (2016) 43(4):501–13. doi: 10.1053/j.seminoncol.2016.06.008
4. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science (1991) 254(5038):1643–7. doi: 10.1126/science.1840703
5. De Plaen E, Arden K, Traversari C, Gaforio JJ, Szikora JP, De Smet C, et al. Structure, chromosomal localization, and expression of 12 genes of the MAGE family. Immunogenetics (1994) 40(5):360–9. doi: 10.1007/BF01246677
6. Chen YT, Scanlan MJ, Sahin U, Tureci O, Gure AO, Tsang S, et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci U S A (1997) 94(5):1914–8. doi: 10.1073/pnas.94.5.1914
7. De Backer O, Arden KC, Boretti M, Vantomme V, De Smet C, Czekay S, et al. Characterization of the GAGE genes that are expressed in various human cancers and in normal testis. Cancer Res (1999) 59(13):3157–65.
8. Van den Eynde B, Peeters O, De Backer O, Gaugler B, Lucas S, Boon T. A new family of genes coding for an antigen recognized by autologous cytolytic T lymphocytes on a human melanoma. J Exp Med (1995) 182(3):689–98. doi: 10.1084/jem.182.3.689
9. Boel P, Wildmann C, Sensi ML, Brasseur R, Renauld JC, Coulie P, et al. BAGE: a new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity (1995) 2(2):167–75. doi: 10.1016/S1074-7613(95)80053-0
10. Coulie PG, Brichard V, Van Pel A, Wolfel T, Schneider J, Traversari C, et al. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med (1994) 180(1):35–42. doi: 10.1084/jem.180.1.35
11. Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci U S A (1994) 91(9):3515–9. doi: 10.1073/pnas.91.9.3515
12. Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci U S A (1994) 91(14):6458–62. doi: 10.1073/pnas.91.14.6458
13. Brichard V, Van Pel A, Wolfel T, Wolfel C, De Plaen E, Lethe B, et al. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med (1993) 178(2):489–95. doi: 10.1084/jem.178.2.489
14. Watt KW, Lee PJ, M’Timkulu T, Chan WP, Loor R. Human prostate-specific antigen: structural and functional similarity with serine proteases. Proc Natl Acad Sci U S A (1986) 83(10):3166–70. doi: 10.1073/pnas.83.10.3166
15. Lilja H. A kallikrein-like serine protease in prostatic fluid cleaves the predominant seminal vesicle protein. J Clin Invest (1985) 76(5):1899–903. doi: 10.1172/JCI112185
16. Ilantzis C, DeMarte L, Screaton RA, Stanners CP. Deregulated expression of the human tumor marker CEA and CEA family member CEACAM6 disrupts tissue architecture and blocks colonocyte differentiation. Neoplasia (2002) 4(2):151–63. doi: 10.1038/sj.neo.7900201
17. Bajenova O, Chaika N, Tolkunova E, Davydov-Sinitsyn A, Gapon S, Thomas P, et al. Carcinoembryonic antigen promotes colorectal cancer progression by targeting adherens junction complexes. Exp Cell Res (2014) 324(2):115–23. doi: 10.1016/j.yexcr.2014.04.007
18. Yarden Y. Biology of HER2 and its importance in breast cancer. Oncology (2001) 61(Suppl 2):1–13. doi: 10.1159/000055396
19. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science (2013) 339(6122):957–9. doi: 10.1126/science.1229259
20. Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell (1997) 90(4):785–95. doi: 10.1016/S0092-8674(00)80538-3
21. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell (1997) 88(3):323–31. doi: 10.1016/S0092-8674(00)81871-1
22. Andersen MH, Svane IM, Becker JC, Straten PT. The universal character of the tumor-associated antigen survivin. Clin Cancer Res (2007) 13(20):5991–4. doi: 10.1158/1078-0432.CCR-07-0686
23. Andersen MH, thor SP. Survivin–a universal tumor antigen. Histol Histopathol (2002) 17(2):669–75. doi: 10.14670/HH-17.669
24. Adida C, Haioun C, Gaulard P, Lepage E, Morel P, Briere J, et al. Prognostic significance of survivin expression in diffuse large B-cell lymphomas. Blood (2000) 96(5):1921–5. doi: 10.1182/blood.V96.5.1921
25. Schmidt SM, Schag K, Muller MR, Weck MM, Appel S, Kanz L, et al. Survivin is a shared tumor-associated antigen expressed in a broad variety of malignancies and recognized by specific cytotoxic T cells. Blood (2003) 102(2):571–6. doi: 10.1182/blood-2002-08-2554
26. Lau SK, Weiss LM, Chu PG. Differential expression of MUC1, MUC2, and MUC5AC in carcinomas of various sites: an immunohistochemical study. Am J Clin Pathol (2004) 122(1):61–9. doi: 10.1309/9R6673QEC06D86Y4
27. Miwa H, Beran M, Saunders GF. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia (1992) 6(5):405–9.
28. Koziol JA, Zhang JY, Casiano CA, Peng XX, Shi FD, Feng AC, et al. Recursive partitioning as an approach to selection of immune markers for tumor diagnosis. Clin Cancer Res (2003) 9(14):5120–6.
29. Suzuki H, Graziano DF, McKolanis J, Finn OJ. T cell-dependent antibody responses against aberrantly expressed cyclin B1 protein in patients with cancer and premalignant disease. Clin Cancer Res (2005) 11(4):1521–6. doi: 10.1158/1078-0432.CCR-04-0538
30. Epstein MA, Achong BG, Barr YM. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet (1964) 1(7335):702–3. doi: 10.1016/S0140-6736(64)91524-7
31. Hjalgrim H, Askling J, Rostgaard K, Hamilton-Dutoit S, Frisch M, Zhang JS, et al. Characteristics of Hodgkin’s lymphoma after infectious mononucleosis. N Engl J Med (2003) 349(14):1324–32. doi: 10.1056/NEJMoa023141
32. zur Hausen H, Schulte-Holthausen H, Klein G, Henle W, Henle G, Clifford P, et al. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature (1970) 228(5276):1056–8. doi: 10.1038/2281056a0
33. Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med (2003) 348(6):518–27. doi: 10.1056/NEJMoa021641
34. Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood (1977) 50(3):481–92. doi: 10.1182/blood.V50.3.481.481
35. Chu NJ, Armstrong TD, Jaffee EM. Nonviral oncogenic antigens and the inflammatory signals driving early cancer development as targets for cancer immunoprevention. Clin Cancer Res (2015) 21(7):1549–57. doi: 10.1158/1078-0432.CCR-14-1186
36. Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res (2003) 9(17):6483–8.
37. Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst (1960) 25:85–109.
38. Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature (1973) 243(5405):290–3. doi: 10.1038/243290a0
39. Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell (1994) 77(2):307–16. doi: 10.1016/0092-8674(94)90322-0
40. Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science (1994) 263(5151):1281–4. doi: 10.1126/science.8122112
41. Shiota M, Fujimoto J, Semba T, Satoh H, Yamamoto T, Mori S. Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene (1994) 9(6):1567–74.
42. Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell (2014) 26(5):682–94. doi: 10.1016/j.ccell.2014.09.019
43. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature (2008) 455(7215):967–70. doi: 10.1038/nature07398
44. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature (2014) 515(7528):572–6. doi: 10.1038/nature14001
45. Karpanen T, Olweus J. The Potential of Donor T-Cell Repertoires in Neoantigen-Targeted Cancer Immunotherapy. Front Immunol (2017) 8:1718. doi: 10.3389/fimmu.2017.01718
46. Ryan SO, Vlad AM, Islam K, Gariepy J, Finn OJ. Tumor-associated MUC1 glycopeptide epitopes are not subject to self-tolerance and improve responses to MUC1 peptide epitopes in MUC1 transgenic mice. Biol Chem (2009) 390(7):611–8. doi: 10.1515/BC.2009.070
47. Depontieu FR, Qian J, Zarling AL, McMiller TL, Salay TM, Norris A, et al. Identification of tumor-associated, MHC class II-restricted phosphopeptides as targets for immunotherapy. Proc Natl Acad Sci U S A (2009) 106(29):12073–8. doi: 10.1073/pnas.0903852106
48. Cobbold M, De La Pena H, Norris A, Polefrone JM, Qian J, English AM, et al. MHC class I-associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci Transl Med (2013) 5(203):203ra125. doi: 10.1126/scitranslmed.3006061
49. Brentville VA, Metheringham RL, Gunn B, Symonds P, Daniels I, Gijon M, et al. Citrullinated Vimentin Presented on MHC-II in Tumor Cells Is a Target for CD4+ T-Cell-Mediated Antitumor Immunity. Cancer Res (2016) 76(3):548–60. doi: 10.1158/0008-5472.CAN-15-1085
50. Zarling AL, Obeng RC, Desch AN, Pinczewski J, Cummings KL, Deacon DH, et al. MHC-restricted phosphopeptides from insulin receptor substrate-2 and CDC25b offer broad-based immunotherapeutic agents for cancer. Cancer Res (2014) 74(23):6784–95. doi: 10.1158/0008-5472.CAN-14-0043
51. Zarling AL, Ficarro SB, White FM, Shabanowitz J, Hunt DF, Engelhard VH. Phosphorylated peptides are naturally processed and presented by major histocompatibility complex class I molecules in vivo. J Exp Med (2000) 192(12):1755–62. doi: 10.1084/jem.192.12.1755
52. Zarling AL, Polefrone JM, Evans AM, Mikesh LM, Shabanowitz J, Lewis ST, et al. Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc Natl Acad Sci U S A (2006) 103(40):14889–94. doi: 10.1073/pnas.0604045103
53. Engelhard VH, Obeng RC, Cummings KL, Petroni GR, Ambakhutwala AL, Chianese-Bullock KA, et al. MHC-restricted phosphopeptide antigens: preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J Immunother Cancer (2020) 8(1). doi: 10.1136/jitc-2019-000262
54. Dearth RK, Cui X, Kim HJ, Hadsell DL, Lee AV. Oncogenic transformation by the signaling adaptor proteins insulin receptor substrate (IRS)-1 and IRS-2. Cell Cycle (2007) 6(6):705–13. doi: 10.4161/cc.6.6.4035
55. Dearth RK, Cui X, Kim HJ, Kuiatse I, Lawrence NA, Zhang X, et al. Mammary tumorigenesis and metastasis caused by overexpression of insulin receptor substrate 1 (IRS-1) or IRS-2. Mol Cell Biol (2006) 26(24):9302–14. doi: 10.1128/MCB.00260-06
56. Gorgisen G, Gulacar IM, Ozes ON. The role of insulin receptor substrate (IRS) proteins in oncogenic transformation. Cell Mol Biol (Noisy-le-grand) (2017) 63(1):1–5. doi: 10.14715/cmb/2017.63.1.1
57. Riggins RB, Quilliam LA, Bouton AH. Synergistic promotion of c-Src activation and cell migration by Cas and AND-34/BCAR3. J Biol Chem (2003) 278(30):28264–73. doi: 10.1074/jbc.M303535200
58. Schrecengost RS, Riggins RB, Thomas KS, Guerrero MS, Bouton AH. Breast cancer antiestrogen resistance-3 expression regulates breast cancer cell migration through promotion of p130Cas membrane localization and membrane ruffling. Cancer Res (2007) 67(13):6174–82. doi: 10.1158/0008-5472.CAN-06-3455
59. Cantarino N, Musulen E, Valero V, Peinado MA, Perucho M, Moreno V, et al. Downregulation of the Deiminase PADI2 Is an Early Event in Colorectal Carcinogenesis and Indicates Poor Prognosis. Mol Cancer Res (2016) 14(9):841–8. doi: 10.1158/1541-7786.MCR-16-0034
60. Brentville VA, Metheringham RL, Daniels I, Atabani S, Symonds P, Cook KW, et al. Combination vaccine based on citrullinated vimentin and enolase peptides induces potent CD4-mediated anti-tumor responses. J Immunother Cancer (2020) 8(1). doi: 10.1136/jitc-2020-000560
61. Witalison EE, Thompson PR, Hofseth LJ. Protein Arginine Deiminases and Associated Citrullination: Physiological Functions and Diseases Associated with Dysregulation. Curr Drug Targets (2015) 16(7):700–10. doi: 10.2174/1389450116666150202160954
62. Alghamdi M, Alasmari D, Assiri A, Mattar E, Aljaddawi AA, Alattas SG, et al. An Overview of the Intrinsic Role of Citrullination in Autoimmune Disorders. J Immunol Res (2019) 2019:7592851. doi: 10.1155/2019/7592851
63. Ireland JM, Unanue ER. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med (2011) 208(13):2625–32. doi: 10.1084/jem.20110640
64. Feitsma AL, van der Voort EI, Franken KL, el Bannoudi H, Elferink BG, Drijfhout JW, et al. Identification of citrullinated vimentin peptides as T cell epitopes in HLA-DR4-positive patients with rheumatoid arthritis. Arthritis Rheum (2010) 62(1):117–25. doi: 10.1002/art.25059
65. Gerstner C, Dubnovitsky A, Sandin C, Kozhukh G, Uchtenhagen H, James EA, et al. Functional and Structural Characterization of a Novel HLA-DRB1*04:01-Restricted alpha-Enolase T Cell Epitope in Rheumatoid Arthritis. Front Immunol (2016) 7:494. doi: 10.3389/fimmu.2016.00494
66. Gerstner C, Dubnovitsky A, Sandin C, Kozhukh G, Uchtenhagen H, James EA, et al. Corrigendum: Functional and Structural Characterization of a Novel HLA-DRB1*04:01-Restricted alpha-Enolase T Cell Epitope in Rheumatoid Arthritis. Front Immunol (2017) 8:1236. doi: 10.3389/fimmu.2017.01236
67. James EA, Rieck M, Pieper J, Gebe JA, Yue BB, Tatum M, et al. Citrulline-specific Th1 cells are increased in rheumatoid arthritis and their frequency is influenced by disease duration and therapy. Arthritis Rheumatol (2014) 66(7):1712–22. doi: 10.1002/art.38637
68. Snir O, Rieck M, Gebe JA, Yue BB, Rawlings CA, Nepom G, et al. Identification and functional characterization of T cells reactive to citrullinated vimentin in HLA-DRB1*0401-positive humanized mice and rheumatoid arthritis patients. Arthritis Rheum (2011) 63(10):2873–83. doi: 10.1002/art.30445
69. Seliger B, Kloor M, Ferrone S. HLA class II antigen-processing pathway in tumors: Molecular defects and clinical relevance. Oncoimmunology (2017) 6(2):e1171447. doi: 10.1080/2162402X.2016.1171447
70. Lhuillier C, Rudqvist NP, Elemento O, Formenti SC, Demaria S. Radiation therapy and anti-tumor immunity: exposing immunogenic mutations to the immune system. Genome Med (2019) 11(1):40. doi: 10.1186/s13073-019-0653-7
71. Coppola D, Fu L, Nicosia SV, Kounelis S, Jones M. Prognostic significance of p53, bcl-2, vimentin, and S100 protein-positive Langerhans cells in endometrial carcinoma. Hum Pathol (1998) 29(5):455–62. doi: 10.1016/S0046-8177(98)90060-0
72. Fuyuhiro Y, Yashiro M, Noda S, Kashiwagi S, Matsuoka J, Doi Y, et al. Clinical significance of vimentin-positive gastric cancer cells. Anticancer Res (2010) 30(12):5239–43.
73. Gilles C, Polette M, Piette J, Delvigne AC, Thompson EW, Foidart JM, et al. Vimentin expression in cervical carcinomas: association with invasive and migratory potential. J Pathol (1996) 180(2):175–80. doi: 10.1002/(SICI)1096-9896(199610)180:2<175::AID-PATH630>3.0.CO;2-G
74. Gustmann C, Altmannsberger M, Osborn M, Griesser H, Feller AC. Cytokeratin expression and vimentin content in large cell anaplastic lymphomas and other non-Hodgkin’s lymphomas. Am J Pathol (1991) 138(6):1413–22.
75. Williams AA, Higgins JP, Zhao H, Ljunberg B, Brooks JD. CD 9 and vimentin distinguish clear cell from chromophobe renal cell carcinoma. BMC Clin Pathol (2009) 9:9. doi: 10.1186/1472-6890-9-9
76. Yamamoto Y, Izumi K, Otsuka H. An immunohistochemical study of epithelial membrane antigen, cytokeratin, and vimentin in papillary thyroid carcinoma. Recognition of lethal and favorable prognostic types. Cancer (1992) 70(9):2326–33. doi: 10.1002/1097-0142(19921101)70:9<2326::AID-CNCR2820700919>3.0.CO;2-D
77. Palena C, Fernando RI, Litzinger MT, Hamilton DH, Huang B, Schlom J. Strategies to target molecules that control the acquisition of a mesenchymal-like phenotype by carcinoma cells. Exp Biol Med (Maywood) (2011) 236(5):537–45. doi: 10.1258/ebm.2011.010367
78. Miles LA, Dahlberg CM, Plescia J, Felez J, Kato K, Plow EF. Role of cell-surface lysines in plasminogen binding to cells: identification of alpha-enolase as a candidate plasminogen receptor. Biochemistry (1991) 30(6):1682–91. doi: 10.1021/bi00220a034
79. Cappello P, Tomaino B, Chiarle R, Ceruti P, Novarino A, Castagnoli C, et al. An integrated humoral and cellular response is elicited in pancreatic cancer by alpha-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int J Cancer (2009) 125(3):639–48. doi: 10.1002/ijc.24355
80. Fu QF, Liu Y, Fan Y, Hua SN, Qu HY, Dong SW, et al. Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. J Hematol Oncol (2015) 8:22. doi: 10.1186/s13045-015-0117-5
81. Principe M, Ceruti P, Shih NY, Chattaragada MS, Rolla S, Conti L, et al. Targeting of surface alpha-enolase inhibits the invasiveness of pancreatic cancer cells. Oncotarget (2015) 6(13):11098–113. doi: 10.18632/oncotarget.3572
82. Zhao M, Fang W, Wang Y, Guo S, Shu L, Wang L, et al. Enolase-1 is a therapeutic target in endometrial carcinoma. Oncotarget (2015) 6(17):15610–27. doi: 10.18632/oncotarget.3639
83. Lundberg K, Kinloch A, Fisher BA, Wegner N, Wait R, Charles P, et al. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum (2008) 58(10):3009–19. doi: 10.1002/art.23936
84. Brentville VA, Symonds P, Cook KW, Daniels I, Pitt T, Gijon M, et al. T cell repertoire to citrullinated self-peptides in healthy humans is not confined to the HLA-DR SE alleles; Targeting of citrullinated self-peptides presented by HLA-DP4 for tumour therapy. Oncoimmunology (2019) 8(5):e1576490. doi: 10.1080/2162402X.2019.1576490
85. Cook K, Daniels I, Symonds P, Pitt T, Gijon M, Xue W, et al. Citrullinated alpha-enolase is an effective target for anti-cancer immunity. Oncoimmunology (2018) 7(2):e1390642. doi: 10.1080/2162402X.2017.1390642
86. Durrant LG, Metheringham RL, Brentville VA. Autophagy, citrullination and cancer. Autophagy (2016) 12(6):1055–6. doi: 10.1080/15548627.2016.1166326
87. Brentville VA, Vankemmelbeke M, Metheringham RL, Durrant LG. Post-translational modifications such as citrullination are excellent targets for cancer therapy. Semin Immunol (2020) 47:101393. doi: 10.1016/j.smim.2020.101393
88. Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell (2001) 106(3):255–8. doi: 10.1016/S0092-8674(01)00449-4
89. Kenter GG, Welters MJ, Valentijn AR, Lowik MJ, Berends-van der Meer DM, Vloon AP, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med (2009) 361(19):1838–47. doi: 10.1056/NEJMoa0810097
90. Maisonneuve C, Bertholet S, Philpott DJ, De Gregorio E. Unleashing the potential of NOD- and Toll-like agonists as vaccine adjuvants. Proc Natl Acad Sci U S A (2014) 111(34):12294–9. doi: 10.1073/pnas.1400478111
91. Yang Y, Huang CT, Huang X, Pardoll DM. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat Immunol (2004) 5(5):508–15. doi: 10.1038/ni1059
92. Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature (2014) 507(7493):519–22. doi: 10.1038/nature12978
93. Ammi R, De Waele J, Willemen Y, Van Brussel I, Schrijvers DM, Lion E, et al. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol Ther (2015) 146:120–31. doi: 10.1016/j.pharmthera.2014.09.010
94. Vosika GJ, Barr C, Gilbertson D. Phase-I study of intravenous modified lipid A. Cancer Immunol Immunother (1984) 18(2):107–12. doi: 10.1007/BF00205743
95. Johnson AG, Tomai M, Solem L, Beck L, Ribi E. Characterization of a nontoxic monophosphoryl lipid A. Rev Infect Dis (1987) 9(Suppl 5):S512–6. doi: 10.1093/clinids/9.Supplement_5.S512
96. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (2004) 303(5663):1529–31. doi: 10.1126/science.1093616
97. Lee J, Chuang TH, Redecke V, She L, Pitha PM, Carson DA, et al. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc Natl Acad Sci U S A (2003) 100(11):6646–51. doi: 10.1073/pnas.0631696100
98. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science (2004) 303(5663):1526–9. doi: 10.1126/science.1093620
99. Jurk M, Kritzler A, Schulte B, Tluk S, Schetter C, Krieg AM, et al. Modulating responsiveness of human TLR7 and 8 to small molecule ligands with T-rich phosphorothiate oligodeoxynucleotides. Eur J Immunol (2006) 36(7):1815–26. doi: 10.1002/eji.200535806
100. Krug A, Rothenfusser S, Hornung V, Jahrsdorfer B, Blackwell S, Ballas ZK, et al. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur J Immunol (2001) 31(7):2154–63. doi: 10.1002/1521-4141(200107)31:7<2154::AID-IMMU2154>3.0.CO;2-U
101. Kranzer K, Bauer M, Lipford GB, Heeg K, Wagner H, Lang R. CpG-oligodeoxynucleotides enhance T-cell receptor-triggered interferon-gamma production and up-regulation of CD69 via induction of antigen-presenting cell-derived interferon type I and interleukin-12. Immunology (2000) 99(2):170–8. doi: 10.1046/j.1365-2567.2000.00964.x
102. Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res (2013) 19(5):1035–43. doi: 10.1158/1078-0432.CCR-12-2064
103. McWilliams JA, Sanchez PJ, Haluszczak C, Gapin L, Kedl RM. Multiple innate signaling pathways cooperate with CD40 to induce potent, CD70-dependent cellular immunity. Vaccine (2010) 28(6):1468–76. doi: 10.1016/j.vaccine.2009.11.071
104. Nimanong S, Ostroumov D, Wingerath J, Knocke S, Woller N, Gurlevik E, et al. CD40 Signaling Drives Potent Cellular Immune Responses in Heterologous Cancer Vaccinations. Cancer Res (2017) 77(8):1918–26. doi: 10.1158/0008-5472.CAN-16-2089
105. Dubensky TW Jr, Kanne DB, Leong ML. Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Ther Adv Vaccines (2013) 1(4):131–43. doi: 10.1177/2051013613501988
106. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455(7213):674–8. doi: 10.1038/nature07317
107. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep (2015) 11(7):1018–30. doi: 10.1016/j.celrep.2015.04.031
108. Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, et al. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res (2014) 2(9):901–10. doi: 10.1158/2326-6066.CIR-13-0123
109. Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, et al. Signalling strength determines proapoptotic functions of STING. Nat Commun (2017) 8(1):427. doi: 10.1038/s41467-017-00573-w
110. Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, et al. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest (2015) 125(6):2532–46. doi: 10.1172/JCI79915
111. Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol (2013) 190(10):5216–25. doi: 10.4049/jimmunol.1300097
112. Kim H, Kwon B, Sin JI. Combined stimulation of IL-2 and 4-1BB receptors augments the antitumor activity of E7 DNA vaccines by increasing Ag-specific CTL responses. PLoS One (2013) 8(12):e83765. doi: 10.1371/journal.pone.0083765
113. Pavlenko M, Roos AK, Lundqvist A, Palmborg A, Miller AM, Ozenci V, et al. A phase I trial of DNA vaccination with a plasmid expressing prostate-specific antigen in patients with hormone-refractory prostate cancer. Br J Cancer (2004) 91(4):688–94. doi: 10.1038/sj.bjc.6602019
114. Sikora AG, Jaffarzad N, Hailemichael Y, Gelbard A, Stonier SW, Schluns KS, et al. IFN-alpha enhances peptide vaccine-induced CD8+ T cell numbers, effector function, and antitumor activity. J Immunol (2009) 182(12):7398–407. doi: 10.4049/jimmunol.0802982
115. Gajewski TF, Fallarino F, Ashikari A, Sherman M. Immunization of HLA-A2+ melanoma patients with MAGE-3 or MelanA peptide-pulsed autologous peripheral blood mononuclear cells plus recombinant human interleukin 12. Clin Cancer Res (2001) 7(3 Suppl):895s–901s.
116. Lee P, Wang F, Kuniyoshi J, Rubio V, Stuges T, Groshen S, et al. Effects of interleukin-12 on the immune response to a multipeptide vaccine for resected metastatic melanoma. J Clin Oncol (2001) 19(18):3836–47. doi: 10.1200/JCO.2001.19.18.3836
117. Dranoff G. GM-CSF-based cancer vaccines. Immunol Rev (2002) 188:147–54. doi: 10.1034/j.1600-065X.2002.18813.x
118. Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res (2004) 64(17):6337–43. doi: 10.1158/0008-5472.CAN-04-0757
119. Slingluff CL Jr, Petroni GR, Olson WC, Smolkin ME, Ross MI, Haas NB, et al. Effect of granulocyte/macrophage colony-stimulating factor on circulating CD8+ and CD4+ T-cell responses to a multipeptide melanoma vaccine: outcome of a multicenter randomized trial. Clin Cancer Res (2009) 15(22):7036–44. doi: 10.1158/1078-0432.CCR-09-1544
120. Wong KK, Li WA, Mooney DJ, Dranoff G. Advances in Therapeutic Cancer Vaccines. Adv Immunol (2016) 130:191–249. doi: 10.1016/bs.ai.2015.12.001
121. Fioretti D, Iurescia S, Fazio VM, Rinaldi M. DNA vaccines: developing new strategies against cancer. J BioMed Biotechnol (2010) 2010:174378. doi: 10.1155/2010/174378
122. Becker SM, Kuznetsov AV. Local temperature rises influence in vivo electroporation pore development: a numerical stratum corneum lipid phase transition model. J Biomech Eng (2007) 129(5):712–21. doi: 10.1115/1.2768380
123. Roos AK, Eriksson F, Timmons JA, Gerhardt J, Nyman U, Gudmundsdotter L, et al. Skin electroporation: effects on transgene expression, DNA persistence and local tissue environment. PLoS One (2009) 4(9):e7226. doi: 10.1371/journal.pone.0007226
124. Chiarella P, Massi E, De Robertis M, Sibilio A, Parrella P, Fazio VM, et al. Electroporation of skeletal muscle induces danger signal release and antigen-presenting cell recruitment independently of DNA vaccine administration. Expert Opin Biol Ther (2008) 8(11):1645–57. doi: 10.1517/14712598.8.11.1645
125. van Drunen Littel-van den Hurk S, Hannaman D. Electroporation for DNA immunization: clinical application. Expert Rev Vaccines (2010) 9(5):503–17. doi: 10.1586/erv.10.42
126. Tiptiri-Kourpeti A, Spyridopoulou K, Pappa A, Chlichlia K. DNA vaccines to attack cancer: Strategies for improving immunogenicity and efficacy. Pharmacol Ther (2016) 165:32–49. doi: 10.1016/j.pharmthera.2016.05.004
127. Nguyen-Hoai T, Pezzutto A, Westermann J. Gene Gun Her2/neu DNA Vaccination: Evaluation of Vaccine Efficacy in a Syngeneic Her2/neu Mouse Tumor Model. Methods Mol Biol (2015) 1317:17–37. doi: 10.1007/978-1-4939-2727-2_2
128. Trimble C, Lin CT, Hung CF, Pai S, Juang J, He L, et al. Comparison of the CD8+ T cell responses and antitumor effects generated by DNA vaccine administered through gene gun, biojector, and syringe. Vaccine (2003) 21(25-26):4036–42. doi: 10.1016/S0264-410X(03)00275-5
129. Wen R, Banik B, Pathak RK, Kumar A, Kolishetti N, Dhar S. Nanotechnology inspired tools for mitochondrial dysfunction related diseases. Adv Drug Delivery Rev (2016) 99(Pt A):52–69. doi: 10.1016/j.addr.2015.12.024
130. Cao J, Wang R, Gao N, Li M, Tian X, Yang W, et al. A7RC peptide modified paclitaxel liposomes dually target breast cancer. Biomater Sci (2015) 3(12):1545–54. doi: 10.1039/C5BM00161G
131. Li MH, Yu H, Wang TF, Chang ND, Zhang JQ, Du D, et al. Tamoxifen embedded in lipid bilayer improves the oncotarget of liposomal daunorubicin in vivo. J Mater Chem B (2014) 2(12):1619–25. doi: 10.1039/c3tb21423k
132. Nguyen TX, Huang L, Gauthier M, Yang G, Wang Q. Recent advances in liposome surface modification for oral drug delivery. Nanomed (Lond) (2016) 11(9):1169–85. doi: 10.2217/nnm.16.9
133. Pires P, Simoes S, Nir S, Gaspar R, Duzgunes N, Pedroso de Lima MC. Interaction of cationic liposomes and their DNA complexes with monocytic leukemia cells. Biochim Biophys Acta (1999) 1418(1):71–84. doi: 10.1016/S0005-2736(99)00023-1
134. Detienne S, Welsby I, Collignon C, Wouters S, Coccia M, Delhaye S, et al. Central Role of CD169(+) Lymph Node Resident Macrophages in the Adjuvanticity of the QS-21 Component of AS01. Sci Rep (2016) 6:39475. doi: 10.1038/srep39475
135. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature (2016) 534(7607):396–401. doi: 10.1038/nature18300
136. Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature (2020) 585(7823):107–12. doi: 10.1038/s41586-020-2537-9
137. Cui H, Webber MJ, Stupp SI. Self-assembly of peptide amphiphiles: from molecules to nanostructures to biomaterials. Biopolymers (2010) 94(1):1–18. doi: 10.1002/bip.21328
138. Mandal D, Nasrolahi Shirazi A, Parang K. Self-assembly of peptides to nanostructures. Org Biomol Chem (2014) 12(22):3544–61. doi: 10.1039/C4OB00447G
139. Rudra JS, Tian YF, Jung JP, Collier JH. A self-assembling peptide acting as an immune adjuvant. Proc Natl Acad Sci U S A (2010) 107(2):622–7. doi: 10.1073/pnas.0912124107
140. Foged C. Subunit vaccines of the future: the need for safe, customized and optimized particulate delivery systems. Ther Deliv (2011) 2(8):1057–77. doi: 10.4155/tde.11.68
141. Xiang SD, Scholzen A, Minigo G, David C, Apostolopoulos V, Mottram PL, et al. Pathogen recognition and development of particulate vaccines: does size matter? Methods (2006) 40(1):1–9. doi: 10.1016/j.ymeth.2006.05.016
142. Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nat Mater (2013) 12(11):978–90. doi: 10.1038/nmat3775
143. Osman G, Rodriguez J, Chan SY, Chisholm J, Duncan G, Kim N, et al. PEGylated enhanced cell penetrating peptide nanoparticles for lung gene therapy. J Control Release (2018) 285:35–45. doi: 10.1016/j.jconrel.2018.07.001
144. Raftery RM, Walsh DP, Blokpoel Ferreras L, Mencia Castano I, Chen G, LeMoine M, et al. Highly versatile cell-penetrating peptide loaded scaffold for efficient and localised gene delivery to multiple cell types: From development to application in tissue engineering. Biomaterials (2019) 216:119277. doi: 10.1016/j.biomaterials.2019.119277
145. Dixon JE, Osman G, Morris GE, Markides H, Rotherham M, Bayoussef Z, et al. Highly efficient delivery of functional cargoes by the synergistic effect of GAG binding motifs and cell-penetrating peptides. Proc Natl Acad Sci U S A (2016) 113(3):E291–9. doi: 10.1073/pnas.1518634113
146. Eltaher HM, Yang J, Shakesheff KM, Dixon JE. Highly efficient intracellular transduction in three-dimensional gradients for programming cell fate. Acta Biomater (2016) 41:181–92. doi: 10.1016/j.actbio.2016.06.004
147. Abu-Awwad HAM, Thiagarajan L, Dixon JE. Controlled release of GAG-binding enhanced transduction (GET) peptides for sustained and highly efficient intracellular delivery. Acta Biomater (2017) 57:225–37. doi: 10.1016/j.actbio.2017.04.028
148. Thiagarajan L, Abu-Awwad HAM, Dixon JE. Osteogenic Programming of Human Mesenchymal Stem Cells with Highly Efficient Intracellular Delivery of RUNX2. Stem Cells Transl Med (2017) 6(12):2146–59. doi: 10.1002/sctm.17-0137
149. Markides H, Newell KJ, Rudorf H, Ferreras LB, Dixon JE, Morris RH, et al. Ex vivo MRI cell tracking of autologous mesenchymal stromal cells in an ovine osteochondral defect model. Stem Cell Res Ther (2019) 10(1):25. doi: 10.1186/s13287-018-1123-7
150. Spiliotopoulos A, Blokpoel Ferreras L, Densham RM, Caulton SG, Maddison BC, Morris JR, et al. Discovery of peptide ligands targeting a specific ubiquitin-like domain-binding site in the deubiquitinase USP11. J Biol Chem (2019) 294(2):424–36. doi: 10.1074/jbc.RA118.004469
151. Tang DC, DeVit M, Johnston SA. Genetic immunization is a simple method for eliciting an immune response. Nature (1992) 356(6365):152–4. doi: 10.1038/356152a0
152. Ulmer JB, Donnelly JJ, Parker SE, Rhodes GH, Felgner PL, Dwarki VJ, et al. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science (1993) 259(5102):1745–9. doi: 10.1126/science.8456302
153. Wang B, Ugen KE, Srikantan V, Agadjanyan MG, Dang K, Refaeli Y, et al. Gene inoculation generates immune responses against human immunodeficiency virus type 1. Proc Natl Acad Sci U S A (1993) 90(9):4156–60. doi: 10.1073/pnas.90.9.4156
154. Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest (2015) 125(9):3401–12. doi: 10.1172/JCI80009
155. Kojima Y, Xin KQ, Ooki T, Hamajima K, Oikawa T, Shinoda K, et al. Adjuvant effect of multi-CpG motifs on an HIV-1 DNA vaccine. Vaccine (2002) 20(23-24):2857–65. doi: 10.1016/S0264-410X(02)00238-4
156. Klinman DM, Yamshchikov G, Ishigatsubo Y. Contribution of CpG motifs to the immunogenicity of DNA vaccines. J Immunol (1997) 158(8):3635–9.
157. Lee SH, Danishmalik SN, Sin JI. DNA vaccines, electroporation and their applications in cancer treatment. Hum Vaccin Immunother (2015) 11(8):1889–900. doi: 10.1080/21645515.2015.1035502
158. Yang B, Jeang J, Yang A, Wu TC, Hung CF. DNA vaccine for cancer immunotherapy. Hum Vaccin Immunother (2014) 10(11):3153–64. doi: 10.4161/21645515.2014.980686
159. Kuang H, Ku SH, Kokkoli E. The design of peptide-amphiphiles as functional ligands for liposomal anticancer drug and gene delivery. Adv Drug Deliv Rev (2017) 110-111:80–101. doi: 10.1016/j.addr.2016.08.005
160. Metheringham RL, Pudney VA, Gunn B, Towey M, Spendlove I, Durrant LG. Antibodies designed as effective cancer vaccines. MAbs (2009) 1(1):71–85. doi: 10.4161/mabs.1.1.7492
161. Pudney VA, Metheringham RL, Gunn B, Spendlove I, Ramage JM, Durrant LG. DNA vaccination with T-cell epitopes encoded within Ab molecules induces high-avidity anti-tumor CD8+ T cells. Eur J Immunol (2010) 40(3):899–910. doi: 10.1002/eji.200939857
162. Brentville VA, Metheringham RL, Gunn B, Durrant LG. High avidity cytotoxic T lymphocytes can be selected into the memory pool but they are exquisitely sensitive to functional impairment. PLoS One (2012) 7(7):e41112. doi: 10.1371/journal.pone.0041112
163. Xue W, Brentville VA, Symonds P, Cook KW, Yagita H, Metheringham RL, et al. SCIB1, a huIgG1 antibody DNA vaccination, combined with PD-1 blockade induced efficient therapy of poorly immunogenic tumors. Oncotarget (2016) 7(50):83088–100. doi: 10.18632/oncotarget.13070
164. Saif JM, Vadakekolathu J, Rane SS, McDonald D, Ahmad M, Mathieu M, et al. Novel prostate acid phosphatase-based peptide vaccination strategy induces antigen-specific T-cell responses and limits tumour growth in mice. Eur J Immunol (2014) 44(4):994–1004. doi: 10.1002/eji.201343863
165. Patel PM, Ottensmeier CH, Mulatero C, Lorigan P, Plummer R, Pandha H, et al. Targeting gp100 and TRP-2 with a DNA vaccine: Incorporating T cell epitopes with a human IgG1 antibody induces potent T cell responses that are associated with favourable clinical outcome in a phase I/II trial. Oncoimmunology (2018) 7(6):e1433516. doi: 10.1080/2162402X.2018.1433516
166. Lu D, Benjamin R, Kim M, Conry RM, Curiel DT. Optimization of methods to achieve mRNA-mediated transfection of tumor cells in vitro and in vivo employing cationic liposome vectors. Cancer Gene Ther (1994) 1(4):245–52.
167. Wasungu L, Hoekstra D. Cationic lipids, lipoplexes and intracellular delivery of genes. J Control Release (2006) 116(2):255–64. doi: 10.1016/j.jconrel.2006.06.024
168. Little SR, Lynn DM, Ge Q, Anderson DG, Puram SV, Chen J, et al. Poly-beta amino ester-containing microparticles enhance the activity of nonviral genetic vaccines. Proc Natl Acad Sci U S A (2004) 101(26):9534–9. doi: 10.1073/pnas.0403549101
169. Phua KK, Leong KW, Nair SK. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J Control Release (2013) 166(3):227–33. doi: 10.1016/j.jconrel.2012.12.029
170. Su X, Fricke J, Kavanagh DG, Irvine DJ. In vitro and in vivo mRNA delivery using lipid-enveloped pH-responsive polymer nanoparticles. Mol Pharm (2011) 8(3):774–87. doi: 10.1021/mp100390w
171. Weide B, Carralot JP, Reese A, Scheel B, Eigentler TK, Hoerr I, et al. Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J Immunother (2008) 31(2):180–8. doi: 10.1097/CJI.0b013e31815ce501
172. Weide B, Pascolo S, Scheel B, Derhovanessian E, Pflugfelder A, Eigentler TK, et al. Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. J Immunother (2009) 32(5):498–507. doi: 10.1097/CJI.0b013e3181a00068
173. Rittig SM, Haentschel M, Weimer KJ, Heine A, Muller MR, Brugger W, et al. Intradermal vaccinations with RNA coding for TAA generate CD8+ and CD4+ immune responses and induce clinical benefit in vaccinated patients. Mol Ther (2011) 19(5):990–9. doi: 10.1038/mt.2010.289
174. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature (2017) 547(7662):222–6. doi: 10.1038/nature23003
175. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature (2012) 482(7385):400–4. doi: 10.1038/nature10755
176. Wang HB, Kondo A, Yoshida A, Yoshizaki S, Abe S, Bao LL, et al. Partial protection against SIV challenge by vaccination of adenovirus and MVA vectors in rhesus monkeys. Gene Ther (2010) 17(1):4–13. doi: 10.1038/gt.2009.122
177. Bliss CM, Bowyer G, Anagnostou NA, Havelock T, Snudden CM, Davies H, et al. Assessment of novel vaccination regimens using viral vectored liver stage malaria vaccines encoding ME-TRAP. Sci Rep (2018) 8(1):3390. doi: 10.1038/s41598-018-21630-4
178. WHO. W.H.o., The COVID-19 candidate vaccine landscape and tracker. (2021). Available at: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines.
179. Larocca C, Schlom J. Viral vector-based therapeutic cancer vaccines. Cancer J (2011) 17(5):359–71. doi: 10.1097/PPO.0b013e3182325e63
180. Marshall JL, Gulley JL, Arlen PM, Beetham PK, Tsang KY, Slack R, et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol (2005) 23(4):720–31. doi: 10.1200/JCO.2005.10.206
181. Capone S, Reyes-Sandoval A, Naddeo M, Siani L, Ammendola V, Rollier CS, et al. Immune responses against a liver-stage malaria antigen induced by simian adenoviral vector AdCh63 and MVA prime-boost immunisation in non-human primates. Vaccine (2010) 29(2):256–65. doi: 10.1016/j.vaccine.2010.10.041
182. DiPaola RS, Plante M, Kaufman H, Petrylak DP, Israeli R, Lattime E, et al. A phase I trial of pox PSA vaccines (PROSTVAC-VF) with B7-1, ICAM-1, and LFA-3 co-stimulatory molecules (TRICOM) in patients with prostate cancer. J Transl Med (2006) 4:1. doi: 10.1186/1479-5876-4-1
183. Kantoff PW, Gulley JL, Pico-Navarro C. Revised Overall Survival Analysis of a Phase II, Randomized, Double-Blind, Controlled Study of PROSTVAC in Men With Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol (2017) 35(1):124–5. doi: 10.1200/JCO.2016.69.7748
184. Gulley JL, Borre M, Vogelzang NJ, Ng S, Agarwal N, Parker CC, et al. Phase III Trial of PROSTVAC in Asymptomatic or Minimally Symptomatic Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol (2019) 37(13):1051–61. doi: 10.1200/JCO.18.02031
186. Zahm CD, Colluru VT, McNeel DG. Vaccination with High-Affinity Epitopes Impairs Antitumor Efficacy by Increasing PD-1 Expression on CD8(+) T Cells. Cancer Immunol Res (2017) 5(8):630–41. doi: 10.1158/2326-6066.CIR-16-0374
187. Mahdavi M, Moreau V, Kheirollahi M. Identification of B and T cell epitope based peptide vaccine from IGF-1 receptor in breast cancer. J Mol Graph Model (2017) 75:316–21. doi: 10.1016/j.jmgm.2017.06.004
188. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature (2017) 547(7662):217–21. doi: 10.1038/nature22991
189. Pol J, Bloy N, Buque A, Eggermont A, Cremer I, Sautes-Fridman C, et al. Trial Watch: Peptide-based anticancer vaccines. Oncoimmunology (2015) 4(4):e974411. doi: 10.4161/2162402X.2014.974411
190. Kimura T, McKolanis JR, Dzubinski LA, Islam K, Potter DM, Salazar AM, et al. MUC1 vaccine for individuals with advanced adenoma of the colon: a cancer immunoprevention feasibility study. Cancer Prev Res (Phila) (2013) 6(1):18–26. doi: 10.1158/1940-6207.CAPR-12-0275
191. Morse MA, Secord AA, Blackwell K, Hobeika AC, Sinnathamby G, Osada T, et al. MHC class I-presented tumor antigens identified in ovarian cancer by immunoproteomic analysis are targets for T-cell responses against breast and ovarian cancer. Clin Cancer Res (2011) 17(10):3408–19. doi: 10.1158/1078-0432.CCR-10-2614
192. Reed CM, Cresce ND, Mauldin IS, Slingluff CL Jr, Olson WC. Vaccination with Melanoma Helper Peptides Induces Antibody Responses Associated with Improved Overall Survival. Clin Cancer Res (2015) 21(17):3879–87. doi: 10.1158/1078-0432.CCR-15-0233
193. Sands H, Gorey-Feret LJ, Cocuzza AJ, Hobbs FW, Chidester D, Trainor GL. Biodistribution and metabolism of internally 3H-labeled oligonucleotides. I. Comparison of a phosphodiester and a phosphorothioate. Mol Pharmacol (1994) 45(5):932–43.
194. Kreutz M, Giquel B, Hu Q, Abuknesha R, Uematsu S, Akira S, et al. Antibody-antigen-adjuvant conjugates enable co-delivery of antigen and adjuvant to dendritic cells in cis but only have partial targeting specificity. PLoS One (2012) 7(7):e40208. doi: 10.1371/journal.pone.0040208
195. Zom GG, Filippov DV, van der Marel GA, Overkleeft HS, Melief CJ, Ossendorp F. Two in one: improving synthetic long peptide vaccines by combining antigen and adjuvant in one molecule. Oncoimmunology (2014) 3(7):e947892. doi: 10.4161/21624011.2014.947892
196. Zom GG, Khan S, Britten CM, Sommandas V, Camps MG, Loof NM, et al. Efficient induction of antitumor immunity by synthetic toll-like receptor ligand-peptide conjugates. Cancer Immunol Res (2014) 2(8):756–64. doi: 10.1158/2326-6066.CIR-13-0223
197. Zom GG, Welters MJ, Loof NM, Goedemans R, Lougheed S, Valentijn RR, et al. TLR2 ligand-synthetic long peptide conjugates effectively stimulate tumor-draining lymph node T cells of cervical cancer patients. Oncotarget (2016) 7(41):67087–100. doi: 10.18632/oncotarget.11512
198. Heit A, Schmitz F, O’Keeffe M, Staib C, Busch DH, Wagner H, et al. Protective CD8 T cell immunity triggered by CpG-protein conjugates competes with the efficacy of live vaccines. J Immunol (2005) 174(7):4373–80. doi: 10.4049/jimmunol.174.7.4373
199. Wille-Reece U, Wu CY, Flynn BJ, Kedl RM, Seder RA. Immunization with HIV-1 Gag protein conjugated to a TLR7/8 agonist results in the generation of HIV-1 Gag-specific Th1 and CD8+ T cell responses. J Immunol (2005) 174(12):7676–83. doi: 10.4049/jimmunol.174.12.7676
200. Cho HI, Barrios K, Lee YR, Linowski AK, Celis E. BiVax: a peptide/poly-IC subunit vaccine that mimics an acute infection elicits vast and effective anti-tumor CD8 T-cell responses. Cancer Immunol Immunother (2013) 62(4):787–99. doi: 10.1007/s00262-012-1382-6
201. Ignacio BJ, Albin TJ, Esser-Kahn AP, Verdoes M. Toll-like Receptor Agonist Conjugation: A Chemical Perspective. Bioconjug Chem (2018) 29(3):587–603. doi: 10.1021/acs.bioconjchem.7b00808
202. Lu BL, Williams GM, Verdon DJ, Dunbar PR, Brimble MA. Synthesis and Evaluation of Novel TLR2 Agonists as Potential Adjuvants for Cancer Vaccines. J Med Chem (2020) 63(5):2282–91. doi: 10.1021/acs.jmedchem.9b01044
203. Lynn GM, Sedlik C, Baharom F, Zhu Y, Ramirez-Valdez RA, Coble VL, et al. Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat Biotechnol (2020) 38(3):320–32. doi: 10.1038/s41587-019-0390-x
204. Li W, Joshi MD, Singhania S, Ramsey KH, Murthy AK. Peptide Vaccine: Progress and Challenges. Vaccines (Basel) (2014) 2(3):515–36. doi: 10.3390/vaccines2030515
205. Nascimento IP, Leite LC. Recombinant vaccines and the development of new vaccine strategies. Braz J Med Biol Res (2012) 45(12):1102–11. doi: 10.1590/S0100-879X2012007500142
206. Slingluff CL Jr. The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination? Cancer J (2011) 17(5):343–50. doi: 10.1097/PPO.0b013e318233e5b2
207. Chianese-Bullock KA, Lewis ST, Sherman NE, Shannon JD, Slingluff CL Jr. Multi-peptide vaccines vialed as peptide mixtures can be stable reagents for use in peptide-based immune therapies. Vaccine (2009) 27(11):1764–70. doi: 10.1016/j.vaccine.2009.01.018
208. Li AW, Sobral MC, Badrinath S, Choi Y, Graveline A, Stafford AG, et al. A facile approach to enhance antigen response for personalized cancer vaccination. Nat Mater (2018) 17(6):528–34. doi: 10.1038/s41563-018-0028-2
209. Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater (2017) 16(4):489–96. doi: 10.1038/nmat4822
210. Tan ML, Choong PF, Dass CR. Recent developments in liposomes, microparticles and nanoparticles for protein and peptide drug delivery. Peptides (2010) 31(1):184–93. doi: 10.1016/j.peptides.2009.10.002
211. Zhai Y, Su J, Ran W, Zhang P, Yin Q, Zhang Z, et al. Preparation and Application of Cell Membrane-Camouflaged Nanoparticles for Cancer Therapy. Theranostics (2017) 7(10):2575–92. doi: 10.7150/thno.20118
212. Zhang R, Leeper CN, Wang X, White TA, Ulery BD. Immunomodulatory vasoactive intestinal peptide amphiphile micelles. Biomater Sci (2018) 6(7):1717–22. doi: 10.1039/C8BM00466H
213. Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med (2013) 19(4):465–72. doi: 10.1038/nm.3105
214. Wille-Reece U, Flynn BJ, Lore K, Koup RA, Kedl RM, Mattapallil JJ, et al. HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates. Proc Natl Acad Sci U S A (2005) 102(42):15190–4. doi: 10.1073/pnas.0507484102
215. Jin K, Wang S, Zhang Y, Xia M, Mo Y, Li X, et al. Long non-coding RNA PVT1 interacts with MYC and its downstream molecules to synergistically promote tumorigenesis. Cell Mol Life Sci (2019) 76(21):4275–89. doi: 10.1007/s00018-019-03222-1
216. Tang Y, He Y, Zhang P, Wang J, Fan C, Yang L, et al. LncRNAs regulate the cytoskeleton and related Rho/ROCK signaling in cancer metastasis. Mol Cancer (2018) 17(1):77. doi: 10.1186/s12943-018-0825-x
217. Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, et al. Role of tumor microenvironment in tumorigenesis. J Cancer (2017) 8(5):761–73. doi: 10.7150/jca.17648
218. Wei F, Wu Y, Tang L, He Y, Shi L, Xiong F, et al. BPIFB1 (LPLUNC1) inhibits migration and invasion of nasopharyngeal carcinoma by interacting with VTN and VIM. Br J Cancer (2018) 118(2):233–47. doi: 10.1038/bjc.2017.385
219. Wu Y, Wei F, Tang L, Liao Q, Wang H, Shi L, et al. Herpesvirus acts with the cytoskeleton and promotes cancer progression. J Cancer (2019) 10(10):2185–93. doi: 10.7150/jca.30222
220. Xia M, Zhang Y, Jin K, Lu Z, Zeng Z, Xiong W. Communication between mitochondria and other organelles: a brand-new perspective on mitochondria in cancer. Cell Biosci (2019) 9:27. doi: 10.1186/s13578-019-0289-8
221. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348(6230):74–80. doi: 10.1126/science.aaa6204
222. Jung K, Choi I. Emerging Co-signaling Networks in T Cell Immune Regulation. Immune Netw (2013) 13(5):184–93. doi: 10.4110/in.2013.13.5.184
223. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
224. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol (2007) 8(3):239–45. doi: 10.1038/ni1443
225. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
226. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
227. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
228. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol (2015) 194(3):950–9. doi: 10.4049/jimmunol.1401686
229. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515(7528):577–81. doi: 10.1038/nature13988
230. Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res (2019) 38(1):255. doi: 10.1186/s13046-019-1259-z
231. Ali OA, Lewin SA, Dranoff G, Mooney DJ. Vaccines Combined with Immune Checkpoint Antibodies Promote Cytotoxic T-cell Activity and Tumor Eradication. Cancer Immunol Res (2016) 4(2):95–100. doi: 10.1158/2326-6066.CIR-14-0126
232. Karyampudi L, Lamichhane P, Scheid AD, Kalli KR, Shreeder B, Krempski JW, et al. Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer Res (2014) 74(11):2974–85. doi: 10.1158/0008-5472.CAN-13-2564
233. Rotte A, Jin JY, Lemaire V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol (2018) 29(1):71–83. doi: 10.1093/annonc/mdx686
234. Tarhini A. Immune-mediated adverse events associated with ipilimumab ctla-4 blockade therapy: the underlying mechanisms and clinical management. Scientifica (Cairo) (2013) 2013:857519. doi: 10.1155/2013/857519
235. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer (2016) 54:139–48. doi: 10.1016/j.ejca.2015.11.016
236. Bakdash G, Buschow SI, Gorris MA, Halilovic A, Hato SV, Skold AE, et al. Expansion of a BDCA1+CD14+ Myeloid Cell Population in Melanoma Patients May Attenuate the Efficacy of Dendritic Cell Vaccines. Cancer Res (2016) 76(15):4332–46. doi: 10.1158/0008-5472.CAN-15-1695
237. Xue W, Metheringham RL, Brentville VA, Gunn B, Symonds P, Yagita H, et al. SCIB2, an antibody DNA vaccine encoding NY-ESO-1 epitopes, induces potent antitumor immunity which is further enhanced by checkpoint blockade. Oncoimmunology (2016) 5(6):e1169353. doi: 10.1080/2162402X.2016.1169353
238. Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med (2007) 204(1):49–55. doi: 10.1084/jem.20062056
239. Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol (2005) 174(12):7516–23. doi: 10.4049/jimmunol.174.12.7516
240. de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science (2014) 346(6206):251–6. doi: 10.1126/science.1253462
241. Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet (2014) 46(3):225–33. doi: 10.1038/ng.2891
242. McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med (2015) 7(283):283ra54. doi: 10.1126/scitranslmed.aaa1408
243. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348(6230):69–74. doi: 10.1126/science.aaa4971
244. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell (2015) 160(1-2):48–61. doi: 10.1016/j.cell.2014.12.033
245. Zamora AE, Crawford JC, Allen EK, Guo XJ, Bakke J, Carter RA, et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8(+) T cell responses. Sci Transl Med (2019) 11(498). doi: 10.1126/scitranslmed.aat8549
246. Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol (2013) 91(8):493–502. doi: 10.1038/icb.2013.29
247. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
248. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell (2004) 6(4):409–21. doi: 10.1016/j.ccr.2004.08.031
249. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell (2008) 13(1):23–35. doi: 10.1016/j.ccr.2007.12.004
250. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother (2014) 63(3):247–57. doi: 10.1007/s00262-013-1508-5
251. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med (2014) 6(237):237ra67. doi: 10.1126/scitranslmed.3007974
252. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep (2015) 5:15179. doi: 10.1038/srep15179
253. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27(1):109–18. doi: 10.1038/cr.2016.151
254. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210(9):1695–710. doi: 10.1084/jem.20130579
255. Du X, Tang F, Liu M, Su J, Zhang Y, Wu W, et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res (2018) 28(4):416–32. doi: 10.1038/s41422-018-0011-0
256. Dominguez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, et al. Selective Targeting of Myeloid-Derived Suppressor Cells in Cancer Patients Using DS-8273a, an Agonistic TRAIL-R2 Antibody. Clin Cancer Res (2017) 23(12):2942–50. doi: 10.1158/1078-0432.CCR-16-1784
257. Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell (2011) 19(6):715–27. doi: 10.1016/j.ccr.2011.04.016
258. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer (2016) 16(9):582–98. doi: 10.1038/nrc.2016.73
259. Bartoschek M, Oskolkov N, Bocci M, Lovrot J, Larsson C, Sommarin M, et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun (2018) 9(1):5150. doi: 10.1038/s41467-018-07582-3
260. Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res (2011) 1(4):482–97.
261. Madar S, Goldstein I, Rotter V. ‘Cancer associated fibroblasts’–more than meets the eye. Trends Mol Med (2013) 19(8):447–53. doi: 10.1016/j.molmed.2013.05.004
262. Valcz G, Sipos F, Tulassay Z, Molnar B, Yagi Y. Importance of carcinoma-associated fibroblast-derived proteins in clinical oncology. J Clin Pathol (2014) 67(12):1026–31. doi: 10.1136/jclinpath-2014-202561
263. Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med (2014) 211(8):1503–23. doi: 10.1084/jem.20140692
264. Paraiso KH, Smalley KS. Fibroblast-mediated drug resistance in cancer. Biochem Pharmacol (2013) 85(8):1033–41. doi: 10.1016/j.bcp.2013.01.018
265. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med (2013) 210(6):1125–35. doi: 10.1084/jem.20130110
266. Najafi M, Farhood B, Mortezaee K. Contribution of regulatory T cells to cancer: A review. J Cell Physiol (2019) 234(6):7983–93. doi: 10.1002/jcp.27553
267. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
268. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
269. Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology (2015) 61(5):1591–602. doi: 10.1002/hep.27665
270. Garrido F, Cabrera T, Concha A, Glew S, Ruiz-Cabello F, Stern PL. Natural history of HLA expression during tumour development. Immunol Today (1993) 14(10):491–9. doi: 10.1016/0167-5699(93)90264-L
271. Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol (2000) 74:181–273. doi: 10.1016/S0065-2776(08)60911-6
272. Seliger B, Cabrera T, Garrido F, Ferrone S. HLA class I antigen abnormalities and immune escape by malignant cells. Semin Cancer Biol (2002) 12(1):3–13. doi: 10.1006/scbi.2001.0404
273. Garrido F, Algarra I. MHC antigens and tumor escape from immune surveillance. Adv Cancer Res (2001) 83:117–58. doi: 10.1016/S0065-230X(01)83005-0
274. Garrido F, Ruiz-Cabello F, Cabrera T, Perez-Villar JJ, Lopez-Botet M, Duggan-Keen M, et al. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol Today (1997) 18(2):89–95. doi: 10.1016/S0167-5699(96)10075-X
275. Koopman LA, Corver WE, van der Slik AR, Giphart MJ, Fleuren GJ. Multiple genetic alterations cause frequent and heterogeneous human histocompatibility leukocyte antigen class I loss in cervical cancer. J Exp Med (2000) 191(6):961–76. doi: 10.1084/jem.191.6.961
276. Garrido F. MHC/HLA Class I Loss in Cancer Cells. Adv Exp Med Biol (2019) 1151:15–78. doi: 10.1007/978-3-030-17864-2_2
277. Ignatov T, Gorbunow F, Eggemann H, Ortmann O, Ignatov A. Loss of HER2 after HER2-targeted treatment. Breast Cancer Res Treat (2019) 175(2):401–8. doi: 10.1007/s10549-019-05173-4
278. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol (2010) 28(31):4722–9. doi: 10.1200/JCO.2010.28.6963
Keywords: adjuvant, peptide vaccine, DNA vaccine, cancer, vaccine
Citation: Paston SJ, Brentville VA, Symonds P and Durrant LG (2021) Cancer Vaccines, Adjuvants, and Delivery Systems. Front. Immunol. 12:627932. doi: 10.3389/fimmu.2021.627932
Received: 10 November 2020; Accepted: 12 March 2021;
Published: 30 March 2021.
Edited by:
Pål Johansen, University of Zurich, SwitzerlandReviewed by:
Kwong Tsang, Precision Biologics, Inc., United StatesSteve Pascolo, University of Zurich, Switzerland
Copyright © 2021 Paston, Brentville, Symonds and Durrant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lindy G. Durrant, bGluZHkuZHVycmFudEBub3R0aW5naGFtLmFjLnVr