 Qi Jiang
Qi Jiang Guocan Yang
Guocan Yang Qi Liu
Qi Liu Shengjun Wang
Shengjun Wang Dawei Cui
Dawei Cui
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 01 April 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.626193
Rheumatoid arthritis (RA) is a systemic and heterogeneous autoimmune disease with symmetrical polyarthritis as its critical clinical manifestation. The basic cause of autoimmune diseases is the loss of tolerance to self or harmless antigens. The loss or functional deficiency of key immune cells, regulatory T (Treg) cells, has been confirmed in human autoimmune diseases. The pathogenesis of RA is complex, and the dysfunction of Tregs is one of the proposed mechanisms underlying the breakdown of self-tolerance leading to the progression of RA. Treg cells are a vital component of peripheral immune tolerance, and the transcription factor Foxp3 plays a major immunosuppressive role. Clinical treatment for RA mainly utilizes drugs to alleviate the progression of disease and relieve disease activity, and the ideal treatment strategy should be to re-induce self-tolerance before obvious tissue injury. Treg cells are one of the ideal options. This review will introduce the classification, mechanism of action, and characteristics of Treg cells in RA, which provides insights into clinical RA treatment.
Rheumatoid arthritis (RA) is a systemic inflammatory autoimmune disease. Musculoskeletal pain, joint swelling and stiffness are its common clinical symptoms, that seriously damage body function and reduce the quality of life of patients (1–3). Patients with RA are more likely to develop osteoporosis, infection, cardiovascular diseases, respiratory diseases, cancer and other diseases than the general population (2–4). More women than men are diagnosed with RA, and the proportion is approximately 3:1 (5). Early diagnosis, the emergence of new treatment methods, and the application of new effective treatment strategies significantly improve the long-term prognosis of the joints of patients with RA (3–6). The pathogenesis of RA is complex and includes synovial cell proliferation and fibrosis, vascular membrane formation, cartilage and bone erosion (2, 3). Naive CD4+ T cells can differentiate into different cells types under antigen presenting cell (APC) stimulation. An imbalance in the function and/or the number of these cells will lead to the abnormal cellular and humoral immunity (7, 8). Abnormal humoral immunity often leads to excessive activation of autoantigenic T and B cells, resulting in the abnormal production of antibodies, such as rheumatoid factor (RF) and anti-cycle citrullinated peptide (anti-CCP) antibodies, and the deposition of immune complexes in synovial tissue, resulting in persistent synovitis and joint destruction (4, 9, 10). Innate immune cells, including mast cells, dendritic cells (DCs), innate lymphocytes and adaptive immune cells, such as B cells, plasma cells, follicular regulatory T (TFR) cells and helper T (Th) cells mediate the systemic autoimmune inflammatory response (11). Abnormal activation of these cells may result in the excess production of pro-inflammatory cytokines such as IL-6, TNF and IL-17, which eventually lead to the destruction of bone tissue and cartilage (11–13). Th17 cells (T cell subsets characterized by secretion of IL-17) produce various pro-inflammatory cytokines to promote synovitis, while Treg cells inhibit inflammation and maintain immune tolerance (14–16).
Initially, Treg cells were divided into three categories according to their origin and differentiation: they are produced by immature T lymphocytes during thymus development, and with a phenotype is CD4+CD25+Foxp3+ T cells, which are called natural Treg (nTreg) cells that constitutively express CD25 and express the specific nuclear transcription factor Foxp3. Upon peripheral antigen stimulation or immunosuppressive factor induction, mature CD4+CD25- T cells are transformed into acquired Treg (iTreg) cells, including Tr1 and Th3 subsets; the former mainly secretes IL-10 and TGF-β, while the latter mainly produces TGF-β. In addition to regulatory CD4+ T cells, regulatory CD8+ T cells also exist in CD8+ T cells (17).
Some scholars also recommend distinguishing two primary Treg cell groups according to their origin. nTreg cells are called thymus-derived Treg (tTreg) cells, which originate from the thymus and have a relatively high self-affinity T cell receptor (TCR) (18). In the periphery, CD4+ effector cells begin to express Foxp3, under the influence of TCR signal transduction or other factors, such as TGF-β and IL-2. These cells are called pTreg cells and are most commonly found in peripheral barrier tissues and prevent local inflammation (19). Since naive CD4+Tconv cells and Treg cells have non-overlapping TCR sequences, the TCR libraries of tTreg cells and pTreg cells are also quite different (20). The TCR libraries of tTreg cells are biased toward self-recognition, while the TCRs of pTreg cells identify foreign antigens with high affinity (21).
Treg cells are distributed in the T cell region of lymphoid organs and in the B cell region TFR, controlling the production and maturation of antibodies (22). According to their location, these cells have been divided into peripheral lymphoid tissue Treg cells and non-lymphoid tissue-resident Treg cells, including central (cTreg) cells and effector (eTreg) cells (23). cTreg cells account for the majority of Treg cells in secondary lymphoid organs and express CCR7 and CD62L at high levels (24), while eTreg cells express surface markers, such as ICOS or CD44 (25). Non-lymphoid tissues where Tregs have been found include visceral adipose tissue (VAT), skin, the lamina propria of the colon, lung and skeletal muscle. Tissue-specific homing receptors such as GPR15 direct Treg cells to the colon, CCR4 promotes the migration of Treg cells to the skin to control tissue homeostasis (26–28). Significant differences between Treg cells in non-lymphoid tissues and lymphoid organs have been identified. The former has a tissue-specific phenotype and is functional (29). For example, VAT-Treg cells are functional specialized tissue resident cells that depend on the transcription factor PPAR-γ, limit inflammation of the skin, intestines and central nervous system and improve the sensitivity of adipose tissue to insulin (30). According to a recent study, significant differences in the transcriptional landscape, phenotype and chromatin accessibility of VAT-Treg cells exist between sexes (31). Tissue adaptation changes occur when Treg cells transfer from lymph nodes to barrier tissue (32). These dynamic adaptations lead to the co-expression and phenotypic acquisition of transcription factors associated with other pedigrees (for example, T-bet, GATA-3, IRF-4, BCL6 or STAT3) (33).
The specific inhibitory effect of Treg cells on T cells is related to the expression of these transcription factors. The ITIM domain protein (TIGIT) on the surface of Treg cells binds to CD155 on dendritic cells (DCs), resulting in an increase in IL-10 expression and a decrease in IL-12 expression in DCs, thus inhibiting the activation of effector T cells (34, 35). T-bet, a transcription factor associated with Th1 cells, is related to the expression of TIGIT. The T-bet+TIGIT+ Treg phenotype selectively inhibits the pro-inflammatory immune response mediated by Th1 and Th17 cells (36, 37). TheTh2-related transcription factor IRF-4 induces the expression of co-stimulatory molecules CTLA-4 and ICOS in Treg cells and cooperates with RBPJ and JUNB to limit the immune response mediated by Th2 cells (38–40). The expression of STAT3 (a typical Th17 transcription factor) in Treg cells is closely related to the Th17-mediated immune response, which increases the expression of the Ebi3, IL-10, and perforin-1 and granzyme B (41). Treg cells exert their inhibitory function through various mechanisms. Their mechanism of action is summarized below (Figure 1).
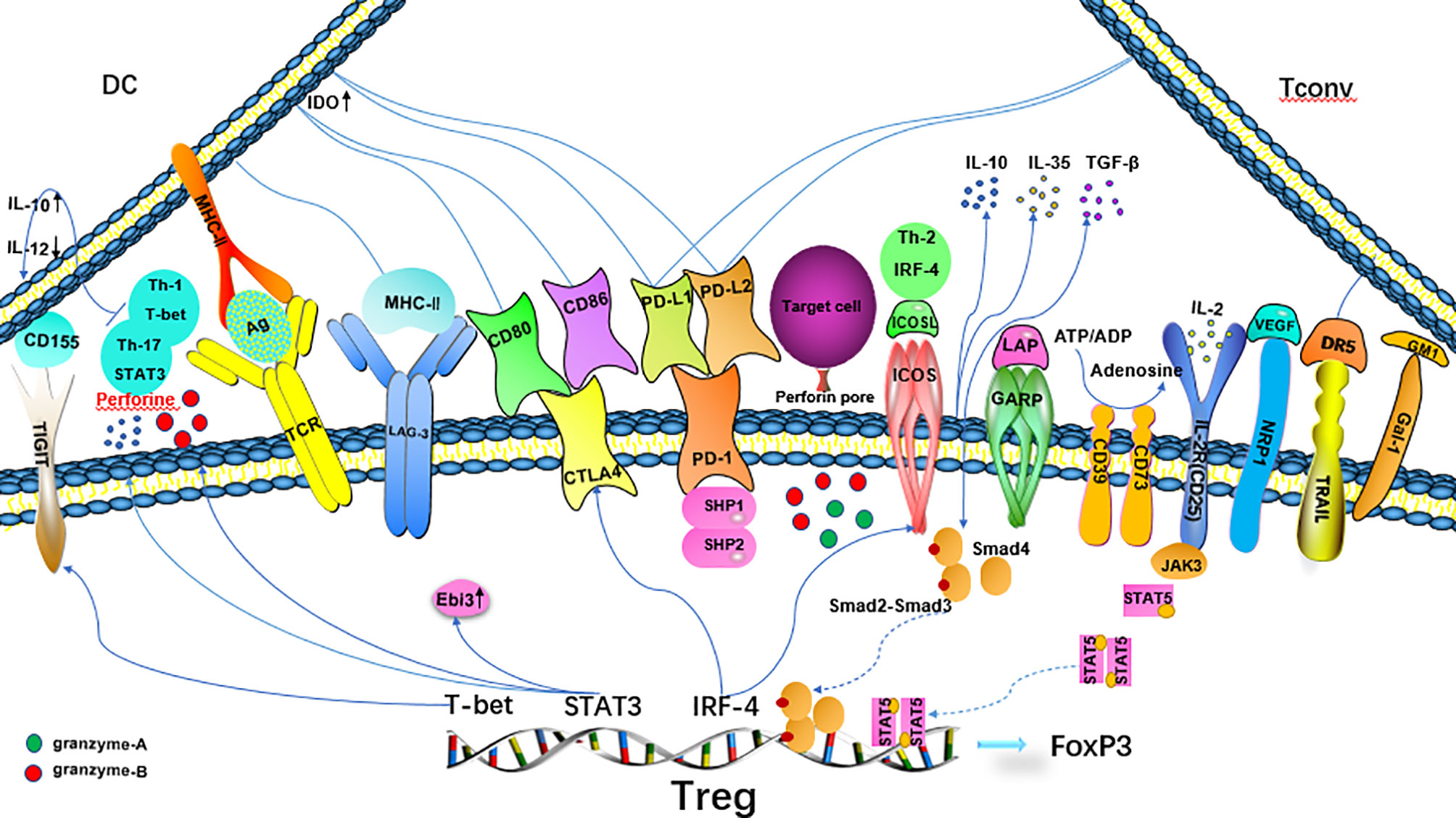
Figure 1 Mechanisms of Treg suppression. CTLA-4 binds to CD80/CD86 on DCs, inhibits the maturation and antigen presentation function of DCs and increases the expression of IDO in DCs, resulting in T effector incompetence. PD-1 binds to PD-L ligands on DCs and T cells to inhibit effector T cells, and synergistically enhance the transactivation of Smad3 by TGF-β.TIGIT increases IL-10 expression and decreases IL-12 expression in DCs by binding to CD155 on DCs and inhibits the activation of effector T cells. Tregs selectively inhibit the proinflammatory immune response mediated by Th1 and Th17 cells. The Th2-related transcription factor IRF-4 activates Treg expression through ICOS and CTLA-4, which restricts the Th2-mediated immune response. STAT3 increases the expression of the IL-10, Ebi3, and perforin-1 genes.
Treg cells share some surface markers with activated effector T cells, such as glucocorticoid-induced TNFR-related protein (GITR), cytotoxic T lymphocyte associated antigen-4 (CTLA-4), programmed death-1 (PD-1) and its ligand (PD-L1) (42). CTLA-4 binds to CD80 and CD86 on APCs (especially DCs), inhibits the antigen presentation and maturation function of APCs (43), and increases the expression of IDO in DCs, reducing the concentration of tryptophan necessary for effector T cell proliferation (44). PD-1 (CD279) binds to PD-L1 and PD-L2 ligands on DCs to inhibit effector T cells (45), and synergistically enhances the transactivation of Smad3 by TGF-β (46). LAG-3 binds to MHC-II, negatively regulates the function of T cells (47), and preferentially inhibits the response of T cells to the stable MHC complex (pMHC-II) (48). Neuropilin-1 (NRP1) is the receptor of vascular endothelial growth factor (VEGF). Its role differs in human and mouse, and the exact inhibitory mechanism remains to be confirmed (49). Galectin (Gal)-1 is a β-galactose-binding lectin that regulates the Treg/Th17 balance induced by DCs through the NF-κB/RelB-IL-27 pathway (50). The transmembrane protein GARP(LRRC32)/latency-related peptide (LAP) is related to the ability of Treg cells to activate TGF- β after stimulation by TCR (51). Down-regulation of GARP expression weakens the inhibitory function of Treg cells (52). TNF-related apoptosis-inducing ligand (TRAIL) is expressed when Treg cells are activated, while CD4+ effector cells express its ligand death receptor 5(DR5). The TRAIL/DR5 interaction activates caspase-8 to induce the apoptosis of effector lymphocytes (53, 54). CD25, also known as interleukin IL-2 receptor (IL-2R) is expressed at high levels on the surface of Treg cells. IL-2 is an important signal that induces cell proliferation in vivo. Treg cells compete with effector cells for IL-2 in the process of the immune response to prevent effector cells from acquiring a sufficient amount of IL-2 to proliferate (55). Treg cells exert their functions through soluble intermediates. The extracellular and/or pericellular accumulation of adenosine causes an immunosuppressive response (56). CD39/CD73 expressed on Treg cells degrade ATP into adenosine, and the increase in the adenosine concentration in the microenvironment will inhibit antigen presentation by DCs (57). The cellular lysis factors granzyme-A, granzyme-B and perforin (58, 59), anti-inflammatory cytokines IL-10, TGF-β, IL-35 and others also play a role in the immune regulation of Treg cells (60). TCR diversity was recently shown to be conducive to the expansion of Treg cells; if the specificity of TCR is the same but the affinity is different, the inhibition mechanism of Treg cells will be different (61). High-affinity receptor cells mainly express TCR-dependent mediators such as CTLA-4, GITR, IL-10 and TIGIT, in contrast, low-affinity receptor cells express more Ebi3,which is responsible for IL-35-mediated inhibition, indicating that affinity determines different inhibition mechanisms (20).
An increase in the number or enhancing the inhibitory function of Treg cells may be helpful in the treatment of autoimmune diseases while reducing the number of Treg cells or inhibiting their function enhances immunity toward tumors and chronic infectious pathogens. RA is characterized by long-term chronic synovitis, cartilage necrosis, and eventually joint destruction that lead to loss of function. Many studies have recently shown that Treg cells inhibit the autoimmune response. When the number and/or function of these cells are abnormal, related antigens and DR molecules cause immune cascade amplification, which leads to the rapid increase in the levels of various cytokines in the body, such as IL-2, and activates macrophages in the synovium of bones and joints to produce many inflammatory cytokines, such as IL-1, IL-6 and IL-8. These inflammatory reactions destroy articular cartilage and eventually lead to joint deformities, leading to the occurrence of RA. However, many studies have reported contradictory results. The number of Treg cells in the peripheral blood of patients with RA is increased (62, 63), unchanged (64–66)or decreased (67–71),and contradictory results were also reported for the functional characteristics of Treg cells from RA patients, namely enhancement or attenuation (72, 73)(Table 1).

Table 1 The frequency of Treg cells and associated cytokine levels in individuals with RA.
The explanation for the discrepancy is the persistent problems in the recognition of Treg cells. In most studies, the expression of Foxp3 is used to define Treg cells, but Foxp3 requires intracellular staining and the expression levels in Treg cells in the resting state and activated state are different (74). Tconv cells also express a low level of Foxp3 upon TCR stimulation (75, 76). CD25 is also a marker for activated Tconv, and Tconv also expresses low levels of CD127. Cell surface markers such as CD4+CD25+/high, CD127low/-, CD62 ligand, integrin Eα (CD103), GITR (TNFRSF18), CTLA-4 (CD152), CD45RO and neuropilin have been used as supplementary markers to identify Treg cells in clinical practice in addition to intracellular Foxp3 staining (77, 78). Among these markers, CD45RA and CD45RO are used to distinguish immature Treg cells (CD45RA+Foxp3low) from activated memory Treg (CD45RA-Foxp3high) cells (79). Currently, the CD3+CD4+CD25highCD127low phenotype is most commonly isolated from Treg population through flow cytometry or immunomagnetic bead separation. The determination of inhibitory activity and demethylation of Foxp3 CNS2 are considered to be the gold standard methods for Treg identification (80, 81), specially Treg-specific DNA hypomethylation, which distinguishes Treg cells from activated Tconv cells at the genetic level. Some scholars have performed a meta-analysis on the number and proportion of Treg cells in patients with RA. The conclusion is that the use of a more stringent method to define Treg cells will reveal decreased number of Treg cells in peripheral blood and increased number in synovial fluid (82). Do Treg cells in the synovial fluid function normally?
Treg cells from patients with RA lack CTLA-4 expression in an inflammatory environment or show ineffective function due to the overexpression of IL-6 (83, 84). These Foxp3-cells are called “exTreg cells”, and a large number of exTreg cells with no inhibitory activity circulate in synovial fluid (85). However, these Treg cells show normal inhibitory activity in vitro, which proves that an essential disorder in these cells is not responsible for RA, rather it is caused by the inflammatory environment (67). Tregs isolated from peripheral blood might limit the proliferation of Teff cells but do not prevent the secretion of cytokines (86). Teff cells in an inflammatory environment are resistant to Treg-mediated inhibition (87). The sensitivity of CD4+CD25- T cells and APCs (most notably DCs) to Treg cell inhibition is also decreased (62, 88). In mice with collagen-induced arthritis (CIA), CD25loFoxp3+CD4+ T cells are transformed into Th17 cells (arthritic synovial fibroblasts promote this transformation). These cells, called exFoxp3Th17 cells, accumulate in inflammatory joints and show a stronger ability to induce osteoclast production than any other T cell subset (89). Another characteristic of RA is the anoxic microenvironment of synovial tissue, neovascularization and cell exudation lead to synovial oxygen deficiency (90). During hypoxia, immune-inflammatory cells make adaptive response and activate pro-inflammatory signal pathways, and hypoxia-inducible factor-1α (HIF-1α) pathway is activated under hypoxia condition (90, 91). HIF-1α is expressed at high levels in synovial fibroblasts and macrophages from individuals with RA (92). HIF-1α can not only induce RORγt transcription to promote Th17 differentiation at the mRNA level, but also cooperate with RORγt protein to regulate downstream Th17 related genes. HIF-1α can also ubiquitinate and proteasome degradation by binding to Foxp3, resulting in the decrease of Foxp3 gene transcriptional activity and down-regulation of Foxp3 expression (93). Therefore, HIF-1α may be a potential target for RA therapy (94, 95). In addition, synovial fibroblasts (SFSs) also induce T cell differentiation in a hypoxic environment, resulting in a decrease in the number of Treg cells and an increase in the number of Th17 cells (96). Synovial hypoxia also changes the metabolic environment, while hypoxia also stimulates osteoclast-mediated bone resorption and aggravates joint injury (97). In addition, knock-out of the PD-1 gene in mice will cause a delayed in the development of specific autoimmune diseases, indicating that PD-1 plays a role in maintaining immune tolerance in immune regulation (98). Li et al. showed that the expression of PD-1 on the surface of CD4+ T and CD8+ T cells and the level of soluble PD-1 (soluble PD-1, sPD-1) in serum were significantly decreased in patients with RA (99). According to recent studies, T cells and pathogenic PD-1+ B cells accumulate in RA joints, and the expression of CXCR3 and GM-CSF in PD-1+ B cells is higher than in PD-1- B cells (100).
Patients with autoimmune diseases often require lifelong immunotherapy, which is usually accompanied by serious adverse reactions and side effects. In recent years, the treatment of RA has gradually changed, and previous “step pyramid” treatment has been gradually replaced as guidelines have advocated the use of rheumatoid arthritis drugs such as disease-modifying anti-rheumatic drugs (DMARDs, methotrexate) at the early stage of the disease. For patients with a poor response to traditional DMARDs, biological DMARDs, such as TNF-α, CTLA-4 or small-molecule targeted DMARDs, such as the Janus kinase (JAK) inhibitor drugs facitinib and baracitinib, are recommended (4). Starting drug treatment in the early stage can effectively prevent the progression of the disease and reduce the rate of disease development. Because a large number of reports on the regenerative function of Treg cells have been published and the ideal treatment strategy is to induce self-tolerance before obvious tissue damage occurs, researchers have designed various strategies ways to increase the number of Treg cells and restore their function (101–107), by enhancing the function of Treg cells in vivo, including reducing the pro-inflammatory environment and enhancing the response of effector cells to inhibition (108–113) (Table 2). Specific Treg cell-specific targeted gene proliferation stimulators were used to promote the expansion of Treg cells, or Treg cells were induced and expanded in vitro following the addition of immune complexes, and then injected into patients (114). The adoptive transfer of Treg cells increased the survival of Scurfy mice and prevented autoimmune diseases, and the removal of Treg cells before the disease increases the incidence and severity of the disease (115). At the same time, the transfer of Treg cells can slow the disease process, confirming that these cells have the potential to treat autoimmune diseases (116, 117). Adoptive cell therapy (ACT) uses Treg cells isolated from blood based on the cell surface labeling of CD4+CD25+CD127- that are then expanded by treating them with anti-CD3, anti-CD28 and IL-2, followed by injection into the body (114). Expanded Treg cells have been used to treat a mouse autoimmune disease model before being used in the clinic. Early trials have been conducted in patients with type 1 diabetes and graft-versus-host disease after bone marrow transplantation showing a stable effect without serious adverse reactions (118–122). Models of CIA also showed inhibition, which significantly prevent the development of CIA (123). Importantly, when arthritis is inhibited in these models, not only are T and B cells inhibited by Treg cells but osteoclast-mediated bone destruction is also directly inhibited, preventing joint injury (124–127).

Table 2 The mechanisms by which the number or function of Treg cells is regulated.
Before the implementation of ACT, some key technical problems must be solved. Since Treg cells identify specific antigens, the first problem to be solved is the method used to isolate specific Treg cells in vitro. Both CD4+CD127low/- and CD4+CD127low/-CD25+ T cells have been used for Treg amplification. The expansion of CD4+CD127low/- cells requires the addition of rapamycin to maintain the purity of their lineage. CD4+CD127low/-CD25+ T cells, particularly the expansion of CD45RA+ subsets, produces a high yield of Treg cells that maintain high Foxp3 expression in the absence of rapamycin (128, 129). In the presence of anti-CD3/anti-CD28 and IL-2, this scheme can increase the number of cells up to thousands of times without losing the inhibitory activity of Treg cells (130). Since IL-2 is a key cytokine required for T cell activation and proliferation and nTregs express CD25 at high levels, they are highly sensitive to IL-2 stimulation. IL-2 (especially low-dose IL-2) preferentially amplifies Treg cells (131, 132). Although low-dose IL-2 directly increase the number of Treg cells in vivo, this effect is short-lived, once the treatment is stopped, the effect will be significantly reduced, and the effect of IL-2 itself on other effector cells must be considered.
The second problem is how to effectively expand antigen-specific Treg cells without losing their specificity or inhibitory function. Some studies have shown that amplified Treg cells tend to express IL-17, and CD4+CD25+Foxp3+ Tregs may be able to transform into pathogenic Th17 cells after repeated amplification (133–135). These studies show that the epigenetic stability of Tregs is unstable, and further studies have shown that the use of CD45RA+ as an additional marker for Treg isolation minimizes epigenetic instability due to amplification and avoids the increase in inflammation associated with Treg cell conversion into Th17 cells (135, 136).
The in vivo environment is complex, and in vitro cell therapy is inevitably time-consuming and expensive. Researchers have not clearly determined whether the expansion of Treg cells in vivo is better than in vitro expansion, and thus they have attempted to combine the two approaches, such as the application of autoantigen in an incomplete adjuvant (137), and the combination of tolDCs and Treg-induced peptide, which inactivate effector cells and promote the function of Treg cells (138).
Maintaining the stability and plasticity of Treg cells in vivo is one of the bottlenecks of ACT. Foxp3 is the main regulator of immunosuppression in Treg cells (139), and participates in the gene expression, function and survival of Treg cells. Its expression is regulated by transcriptional regulation, epigenetic regulation and post-translational regulation and is indispensable for the maintenance of immune self-tolerance (14, 140–142). The transcription factors NFAT, STAT5 and Foxo1 directly interact with the Foxp3 gene promoter to regulate the expression of Foxp3. The element of the conserved non-coding sequence (CNS) at the Foxp3 gene site regulates gene expression by recruiting transcription factors (143–145). The most noteworthy element is the methylation of CpG islets at the second intron enhancer site, also known as major TSDR(conserved non-coding sequence 2, CNS2), which is a region that is specifically demethylated in Treg cells. CNS2 demethylation stabilizes the expression of Foxp3 (146–148). Using CRISPR technology, recent studies have shown that ubiquitin-specific peptidase 22 (Usp22) is a positive regulator of stable Foxp3 expression, while ring finger protein 20 (Rnf20), an E3 ubiquitin ligase, is a negative regulator of Foxp3 (149). Phosphorylation, acetylation and ubiquitin are also considered factors regulating the stability of Foxp3 (145). Foxp3+nTreg cells are highly proliferative and highly stable. They may be able to recognize self-antigens or microbial antigens from symbiotic microorganisms (150–152).
In the past two years, researchers have performed numerous studies designed to improve the stability and optimize the function of Treg cells. For example, Chen et al. reported a more stable function for CD4 + CD126 low/- Foxp3+ cells than CD126highnTreg cells that remain stable even under inflammatory conditions (153). Human CD8+ regulatory T cells stimulated with rapamycin and TGF-β 1 also showed stable inhibitory ability in inflammatory environment (154). Park et al. found that daurinol, a natural arylnaphtholide isolated from the medicinal plant Haplophyllumdauricum, increases Treg cells stability by inducing DNA demethylation in the Foxp3 promoter region (155). PTPN2 promotes the stability of the Foxp3 mRNA in RORγt+ Treg cells, while the deletion or decreased expression of PTPN2 will promote the pathogenic transformation of Treg cells (156). TNFRII (TNF receptor type II) + Treg cells stably express Foxp3 through hypomethylation, and adoptive transfer of TNFRII+ Treg cells reduces the inflammatory response (157). The deletion of TNF receptor 2 causes Treg cells to show a Th17-like inflammatory phenotype (158). In addition, intracellular metabolic intermediates and environmental metabolites can also regulate the expression of Foxp3 in Treg cells (14). More interestingly, Treg cells have been modified to produce stable human Treg cells that target homing receptors, and these Treg cells migrate to specific sites or tissues to achieve more targeted immunosuppression and epigenetic stability under inflammatory conditions (159).
Genome editing technology is also used to enhance the stability of Treg cells. The expression of these genes is modified by CRISPR/Cas-9. Knockout of the PD-1 gene to modify T cells has been used in cancer therapy (160), which provides opportunities for the application of gene editing technology in Treg cells, such as knock-out of the genes that inhibit the function of Treg cells and up-regulation of the genes that can stabilize the expression of Foxp3. For example, knockout of the Stub1 gene increases the expression of Foxp3, and the up-regulation of CTLA-4, PD-1 and BACH2 will increase the stability of Treg cells (86). Using CRISPR technology, recent studies have shown that ubiquitin-specific peptidase 22 (Usp22) is a positive regulator of stable Foxp3 expression, while ring finger protein 20 (Rnf20), an E3 ubiquitin ligase, is a negative regulator of Foxp3 (149). In summary, CRISPR/Cas-9 technology is a new approach for RA therapy. The molecular mechanism of Foxp3 has been extensively studied. However, other studies have shown that the expression of Foxp3 alone is not sufficient to regulate gene expression in Treg cells (161–163). For example, an approximately 70% difference in the genomes of Foxp3+ nTreg cells and Foxp3-overexpressing Tconv cells has been identified, and the latter does not express some Treg signaling genes, such as Ikzf4 (Eos) and Ikzf2 (Helios) (163). However, the gene expression pattern of Foxp3-deleted Treg cells isolated from Foxp3 gene-deficient mice resembled Foxp3-intact normal nTreg cells (161, 164). The Treg-specific CpG demethylation (165) and histone modification also occurred before the expression of Foxp3 (166). Based on these findings, the cells that determine cell fate and differentiation develop long before Foxp3 expression. Therefore, the expression of Foxp3 alone does not represent the complete function of mature Treg cells. Foxp3 independent cell typing and immature tTreg cells differentiation indicate a Foxp3 independent genetic mechanism that controls the function and differentiation of early tTreg cells.
These Treg-specific genetic patterns are helpful for us understanding the function of Treg cells. The ideal strategy to stabilize the inhibitory function is to transform both initial and effector/memory Tconv cells into functionally stable Foxp3+ Treg cells. Foxp3 expression is induced in Tconv cells by treatments with targeting different signal transduction pathways, such as the TCR-DK8/19 pathway (167), AKT-mTOR pathway (168, 169), TGF-β-SMAD pathway (170). Continuous stimulation of Tconv cells by TCR might partially induce Treg-type DNA demethylation (171). Therefore, continuous TCR stimulation at the appropriate intensity likely induces the differentiation of developing T cells into functionally stable tTreg cells and the transformation from Tconv cells to Treg cells.
Treg cells use multiple molecular mechanisms to inhibit the adverse reactions of RA, and their application may significantly control the progression of RA. Treg cells as intelligent “drugs” are attracting researchers’ interest. The successful application of Treg cell therapy in autoimmune diseases and transplantation will encourage clinical application of this method in the treatment of other non-immune diseases, such as tissue repair and neurological disorders (172). Although the molecular characteristics of human and mouse Treg cells are very similar, they are not the same, and the assessment of animal models in vitro is limited to a certain pathway. However, the in vivo environment is complex, and thus methods to better transform and apply the results obtained in animal experiments to human are worth examining. The identification of Treg cells in vitro, in addition to the use of more stringent markers, the determination of whether the drugs used for the treatment of RA disease will affect the phenotype of Treg cells, and an assessment of whether the function of Treg cells improves after treatment in a specific or non-specific manner is also worthy of further discussion. In addition, the time and stage of disease development are related to the therapeutic effect of ACT, and this treatment will be more effective in the early stage of the disease (173). Given the ability of Treg cells to specifically detect antigens through the TCR, developing an ACT that acts directly on a specific site or detects an antigenic site may be an ideal approach, Chimeric antigen receptor (CAR) Treg cells specifically migrate to target sites and show more obvious antigen-specific inhibitory activity (174).
In addition, in this new era of gene and cell therapy development, technological advances such as gene therapy-induced by target specificity or methods for delivering one or more genes to treat RA (175), single-cell transcriptome sRNA-seq has been used to analyze the expression of hundreds of genes in a single cell (176). The development of ATAC-seq has facilitated analyses of the occupancy of transcription factors in specific cell types (177). CRISPR/Cas-9mediated technology quickly and effectively generates genetic interference to identify and regulate the proliferation and function of T cells (178). All of these findings provide opportunities for the future development of accurate medical strategies and promote the clinical application of these treatments.
QJ drafted the manuscript and designed the figures. GY and QL reviewed the manuscript structure and ideas. DC and SW conceived the topic and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81871709, 31711530025 and 81771759).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge the highly qualified native English speaking editors at American Journal Experts(AJE) for providing reputable English language editing service (Verification code: ECB3-D383-278C-6AB9-9A76) for our manuscript.
1. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet (2010) 376(9746):1094–108. doi: 10.1016/S0140-6736(10)60826-4
2. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (2016) 388(10055):2023–38. doi: 10.1016/S0140-6736(16)30173-8
3. Kumar LD, Karthik R, Gayathri N, Sivasudha T. Advancement in contemporary diagnostic and therapeutic approaches for rheumatoid arthritis. BioMed Pharmacother (2016) 79:52–61. doi: 10.1016/j.biopha.2016.02.001
4. Sparks JA. Rheumatoid Arthritis. Ann Intern Med (2019) 170(1):ITC1–ITC16. doi: 10.7326/AITC201901010
5. Cross M, Smith E, Hoy D, Carmona L, Wolfe F, Vos T, et al. The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis (2014) 73(7):1316–22. doi: 10.1136/annrheumdis-2013-204627
7. Wu R, Li N, Zhao X, Ding T, Xue H, Gao C, et al. Low-dose Interleukin-2: Biology and therapeutic prospects in rheumatoid arthritis. Autoimmun Rev (2020) 19(10):102645. doi: 10.1016/j.autrev.2020.102645
8. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature (2017) 542(7639):110–4. doi: 10.1038/nature20810
9. Derksen V, Huizinga TWJ, van der Woude D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin Immunopathol (2017) 39(4):437–46. doi: 10.1007/s00281-017-0627-z
10. Lee KH, Ahn BS, Cha D, Jang WW, Choi E, Park S, et al. Understanding the immunopathogenesis of autoimmune diseases by animal studies using gene modulation: A comprehensive review. Autoimmun Rev (2020) 19(3):102469. doi: 10.1016/j.autrev.2020.102469
11. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
12. Chen Z, Bozec A, Ramming A, Schett G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat Rev Rheumatol (2019) 15(1):9–17. doi: 10.1038/s41584-018-0109-2
13. Zhang Y, Li Y, Lv TT, Yin ZJ, Wang XB. Elevated circulating Th17 and follicular helper CD4(+) T cells in patients with rheumatoid arthritis. Apmis (2015) 123(8):659–66. doi: 10.1111/apm.12399
14. Shi H, Chi H. Metabolic Control of Treg Cell Stability, Plasticity, and Tissue-Specific Heterogeneity. Front Immunol (2019) 10:2716. doi: 10.3389/fimmu.2019.02716
15. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol (2012) 30:733–58. doi: 10.1146/annurev-immunol-020711-075043
16. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev (2014) 13(6):668–77. doi: 10.1016/j.autrev.2013.12.004
17. Carrier Y, Yuan J, Kuchroo VK, Weiner HL. Th3 cells in peripheral tolerance. II. TGF-beta-transgenic Th3 cells rescue IL-2-deficient mice from autoimmunity. J Immunol (2007) 178(1):172–8. doi: 10.4049/jimmunol.178.1.172
18. Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature (2012) 482:395–9:7385. doi: 10.1038/nature10772
19. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14(4):307–8. doi: 10.1038/ni.2554
20. Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
21. Wyss L, Stadinski BD, King CG, Schallenberg S, McCarthy NI, Lee JY, et al. Affinity for self antigen selects Treg cells with distinct functional properties. Nat Immunol (2016) 17(9):1093–101. doi: 10.1038/ni.3522
22. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17(8):975–82. doi: 10.1038/nm.2425
23. Smigiel KS, Srivastava S, Stolley JM, Campbell DJ. Regulatory T-cell homeostasis: steady-state maintenance and modulation during inflammation. Immunol Rev (2014) 259(1):40–59. doi: 10.1111/imr.12170
24. Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol (2014) 14(3):154–65. doi: 10.1038/nri3605
25. Dias S, D’Amico A, Cretney E, Liao Y, Tellier J, Bruggeman C, et al. Effector Regulatory T Cell Differentiation and Immune Homeostasis Depend on the Transcription Factor Myb. Immunity (2017) 46(1):78–91. doi: 10.1016/j.immuni.2016.12.017
26. Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol (2016) 34:609–33. doi: 10.1146/annurev-immunol-032712-095948
27. Feuerer M, Hill JA, Mathis D, Benoist C. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nat Immunol (2009) 10(7):689–95. doi: 10.1038/ni.1760
28. Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz-Wagenblatt A, Träger U, et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat Immunol (2017) 18(10):1160–72. doi: 10.1038/ni.3799
29. Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol (2013) 14(10):1007–13. doi: 10.1038/ni.2683
30. Lu J, Meng H, Zhang A, Yang J, Zhang X. Phenotype and function of tissue-resident unconventional Foxp3-expressing CD4(+) regulatory T cells. Cell Immunol (2015) 297(1):53–9. doi: 10.1016/j.cellimm.2015.06.005
31. Vasanthakumar A, Chisanga D, Blume J, Gloury R, Britt K, Henstridge DC, et al. Sex-specific adipose tissue imprinting of regulatory T cells. Nature (2020) 579(7800):581–5. doi: 10.1038/s41586-020-2040-3
32. Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, et al. Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity (2019) 50(2):493–504.e7. doi: 10.1016/j.immuni.2019.01.001
33. Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol (2011) 11(2):119–30. doi: 10.1038/nri2916
34. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
35. Kučan Brlić P, Lenac Roviš T, Cinamon G, Tsukerman P, Mandelboim O, Jonjić S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell Mol Immunol (2019) 16(1):40–52. doi: 10.1038/s41423-018-0168-y
36. Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature (2017) 546(7658):421–5. doi: 10.1038/nature22360
37. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40(4):569–81. doi: 10.1016/j.immuni.2014.02.012
38. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature (2009) 458(7236):351–6. doi: 10.1038/nature07674
39. Delacher M, Schmidl C, Herzig Y, Breloer M, Hartmann W, Brunk F, et al. Rbpj expression in regulatory T cells is critical for restraining T(H)2 responses. Nat Commun (2019) 10:1621. doi: 10.1038/s41467-019-09276-w
40. Koizumi SI, Sasaki D, Hsieh TH, Taira N, Arakaki N, Yamasaki S, et al. JunB regulates homeostasis and suppressive functions of effector regulatory T cells. Nat Commun (2018) 9:5344. doi: 10.1038/s41467-018-07735-4
41. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science (2009) 326(5955):986–91. doi: 10.1126/science.1172702
42. Rosenblum MD, Way SS, Abbas AK. Regulatory T cell memory. Nat Rev Immunol (2016) 16(2):90–101. doi: 10.1038/nri.2015.1
43. Paust S, Lu L, McCarty N, Cantor H. Engagement of B7 on effector T cells by regulatory T cells prevents autoimmune disease. Proc Natl Acad Sci U.S.A. (2004) 101(28):10398–403. doi: 10.1073/pnas.0403342101
44. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol (2003) 4(12):1206–12. doi: 10.1038/ni1003
45. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi: 10.1038/nri3405
46. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206(13):3015–29. doi: 10.1084/jem.20090847
47. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells,” (in eng). Nat Med (2013) 19(6):739–46. doi: 10.1038/nm.3179
48. Maruhashi T, Okazaki IM, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG-3 inhibits the activation of CD4(+) T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat Immunol (2018) 19(12):1415–26. doi: 10.1038/s41590-018-0217-9
49. Chaudhary B, Khaled YS, Ammori BJ, Elkord E. Neuropilin 1: function and therapeutic potential in cancer. Cancer Immunol Immunother (2014) 63(2):81–99. doi: 10.1007/s00262-013-1500-0
50. Lu H, Dai X, Li X, Sun Y, Gao Y, Zhang C. Gal-1 regulates dendritic cells-induced Treg/Th17 balance though NF-κB/RelB-IL-27 pathway. Ann Transl Med (2019) 7(22):628. doi: 10.21037/atm.2019.11.02
51. Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur J Immunol (2009) 39(12):3315–22. doi: 10.1002/eji.200939684
52. Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U.S.A. (2009) 106(32):13445–50. doi: 10.1073/pnas.0901944106
53. Ren X, Ye F, Jiang Z, Chu Y, Xiong S, Wang Y. Involvement of cellular death in TRAIL/DR5-dependent suppression induced by CD4(+)CD25(+) regulatory T cells. Cell Death Differ (2007) 14(12):2076–84. doi: 10.1038/sj.cdd.4402220
54. Bodmer JL, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P, et al. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol (2000) 2(4):241–3. doi: 10.1038/35008667
55. von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol (2005) 6(4):338–44. doi: 10.1038/ni1180
56. Sitkovsky MV, Ohta A. The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol (2005) 26(6):299–304. doi: 10.1016/j.it.2005.04.004
57. Ernst PB, Garrison JC, Thompson LF. Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J Immunol (2010) 185(4):1993–8. doi: 10.4049/jimmunol.1000108
58. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity (2004) 21(4):589–601. doi: 10.1016/j.immuni.2004.09.002
59. Gondek DC, Devries V, Nowak EC, Lu LF, Bennett KA, Scott ZA, et al. Transplantation survival is maintained by granzyme B+ regulatory cells and adaptive regulatory T cells. J Immunol (2008) 181(7):4752–60. doi: 10.4049/jimmunol.181.7.4752
60. Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25- cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol (2004) 172(9):5213–21. doi: 10.4049/jimmunol.172.9.5213
61. Föhse L, Suffner J, Suhre K, Wahl B, Lindner C, Lee CW, et al. High TCR diversity ensures optimal function and homeostasis of Foxp3+ regulatory T cells. Eur J Immunol (2011) 41(11):3101–13. doi: 10.1002/eji.201141986
62. van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum (2004) 50(9):2775–85. doi: 10.1002/art.20499
63. Han GM, O’Neil-Andersen NJ, Zurier RB, Lawrence DA. CD4+CD25high T cell numbers are enriched in the peripheral blood of patients with rheumatoid arthritis. Cell Immunol (2008) 253(1-2):92–101. doi: 10.1016/j.cellimm.2008.05.007
64. Cao D, Malmström V, Baecher-Allan C, Hafler D, Klareskog L, Trollmo C. Isolation and functional characterization of regulatory CD25brightCD4+ T cells from the target organ of patients with rheumatoid arthritis. Eur J Immunol (2003) 33(1):215–23. doi: 10.1002/immu.200390024
65. Möttönen M, Heikkinen J, Mustonen L, Isomäki P, Luukkainen R, Lassila O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol (2005) 140(2):360–7. doi: 10.1111/j.1365-2249.2005.02754.x
66. Liu MF, Wang CR, Fung LL, Lin LH, Tsai CN. The presence of cytokine-suppressive CD4+CD25+ T cells in the peripheral blood and synovial fluid of patients with rheumatoid arthritis. Scand J Immunol (2005) 62(3):312–7. doi: 10.1111/j.1365-3083.2005.01656.x
67. Cao D, van Vollenhoven R, Klareskog L, Trollmo C, Malmström V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res Ther (2004) 6(4):R335–46. doi: 10.1186/ar1192
68. Niu Q, Cai B, Huang ZC, Shi YY, Wang LL. Disturbed Th17/Treg balance in patients with rheumatoid arthritis. Rheumatol Int (2012) 32(9):2731–6. doi: 10.1007/s00296-011-1984-x
69. Kawashiri SY, Kawakami A, Okada A, Koga T, Tamai M, Yamasaki S, et al. CD4+CD25(high)CD127(low/-) Treg cell frequency from peripheral blood correlates with disease activity in patients with rheumatoid arthritis. J Rheumatol (2011) 38(12):2517–21. doi: 10.3899/jrheum.110283
70. Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J, et al. Brief report: inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum (2012) 64(8):2499–503. doi: 10.1002/art.34477
71. Lina C, Conghua W, Nan L, Ping Z. Combined treatment of etanercept and MTX reverses Th1/Th2, Th17/Treg imbalance in patients with rheumatoid arthritis. J Clin Immunol (2011) 31(4):596–605. doi: 10.1007/s10875-011-9542-6
72. Walter GJ, Evans HG, Menon B, Gullick NJ, Kirkham BW, Cope AP, et al. Interaction with activated monocytes enhances cytokine expression and suppressive activity of human CD4+CD45ro+CD25+CD127(low) regulatory T cells. Arthritis Rheum (2013) 65(3):627–38. doi: 10.1002/art.37832
73. Sun H, Gao W, Pan W, Zhang Q, Wang G, Feng D, et al. Tim3(+) Foxp3 (+) Treg Cells Are Potent Inhibitors of Effector T Cells and Are Suppressed in Rheumatoid Arthritis. Inflammation (2017) 40(4):1342–50. doi: 10.1007/s10753-017-0577-6
74. Miyara M, Sakaguchi S. Human FoxP3(+)CD4(+) regulatory T cells: their knowns and unknowns. Immunol Cell Biol (2011) 89(3):346–51. doi: 10.1038/icb.2010.137
75. Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol (2007) 4:345–54. doi: 10.1093/intimm/dxm014
76. Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol (2007) 37(1):129–38. doi: 10.1002/eji.200636435
77. Holm TL, Nielsen J, Claesson MH. CD4+CD25+ regulatory T cells: I. Phenotype and physiology. Apmis (2004) 112(10):629–41. doi: 10.1111/j.1600-0463.2004.apm1121001.x
78. Schmetterer KG, Neunkirchner A, Pickl WF. Naturally occurring regulatory T cells: markers, mechanisms, and manipulation. FASEB J (2012) 26(6):2253–76. doi: 10.1096/fj.11-193672
79. Machura E, Mazur B, Pieniazek W, Karczewska K. Expression of naive/memory (CD45RA/CD45RO) markers by peripheral blood CD4+ and CD8 + T cells in children with asthma. Arch Immunol Ther Exp (Warsz) (2008) 56(1):55–62. doi: 10.1007/s00005-008-0005-6
80. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
81. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10(7):490–500. doi: 10.1038/nri2785
82. Morita T, Shima Y, Wing JB, Sakaguchi S, Ogata A, Kumanogoh A. The Proportion of Regulatory T Cells in Patients with Rheumatoid Arthritis: A Meta-Analysis. PloS One (2016) 11(9):e0162306. doi: 10.1371/journal.pone.0162306
83. Flores-Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci USA (2008) 105(49):19396–401. doi: 10.1073/pnas.0806855105
84. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24(2):179–89. doi: 10.1016/j.immuni.2006.01.001
85. Li S, Yin H, Zhang K, Wang T, Yang Y, Liu X, et al. Effector T helper cell populations are elevated in the bone marrow of rheumatoid arthritis patients and correlate with disease severity. Sci Rep (2017) 7:4776. doi: 10.1038/s41598-017-05014-8
86. Safari F, Farajnia S, Arya M, Zarredar H, Nasrolahi A. CRISPR and personalized Treg therapy: new insights into the treatment of rheumatoid arthritis. Immunopharmacol Immunotoxicol (2018) 40(3):201–11. doi: 10.1080/08923973.2018.1437625
87. Rossetti M, Spreafico R, Consolaro A, Leong JY, Chua C, Massa M, et al. TCR repertoire sequencing identifies synovial Treg cell clonotypes in the bloodstream during active inflammation in human arthritis. Ann Rheum Dis (2017) 76(2):435–41. doi: 10.1136/annrheumdis-2015-208992
88. de Kleer IM, Wedderburn LR, Taams LS, Patel A, Varsani H, Klein M, et al. CD4+CD25bright regulatory T cells actively regulate inflammation in the joints of patients with the remitting form of juvenile idiopathic arthritis. J Immunol (2004) 172(10):6435–43. doi: 10.4049/jimmunol.172.10.6435
89. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20(1):62–8. doi: 10.1038/nm.3432
90. Fearon U, Canavan M, Biniecka M, Veale DJ. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat Rev Rheumatol (2016) 12(7):385–97. doi: 10.1038/nrrheum.2016.69
91. Wu J, Cui H, Zhu Z, Wang L, Li H, Wang D. Effect of HIF1alpha on Foxp3 expression in CD4+ CD25- T lymphocytes. Microbiol Immunol (2014) 58(7):409–15. doi: 10.1111/1348-0421.12168
92. Yang ZC, Liu Y. Hypoxia-Inducible Factor-1α and Autoimmune Lupus, Arthritis. Inflammation (2016) 39(3):1268–73. doi: 10.1007/s10753-016-0337-z
93. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
94. Hu Y, Zhang T, Chen J, Cheng W, Chen J, Zheng Z, et al. Downregulation of Hypoxia-Inducible Factor-1α by RNA Interference Alleviates the Development of Collagen-Induced Arthritis in Rats. Mol Ther Nucleic Acids (2020) 19:1330–42. doi: 10.1016/j.omtn.2020.01.014
95. Guo X, Chen G. Hypoxia-Inducible Factor Is Critical for Pathogenesis and Regulation of Immune Cell Functions in Rheumatoid Arthritis. Front Immunol (2020) 11:1668. doi: 10.3389/fimmu.2020.01668
96. Ding Y, Wang L, Wu H, Zhao Q, Wu S. Exosomes derived from synovial fibroblasts under hypoxia aggravate rheumatoid arthritis by regulating Treg/Th17 balance. Exp Biol Med (Maywood) (2020) 245(14):1535370220934736. doi: 10.1177/1535370220934736
97. Knowles HJ, Athanasou NA. Acute hypoxia and osteoclast activity: a balance between enhanced resorption and increased apoptosis. J Pathol (2009) 218(2):256–64. doi: 10.1002/path.2534
98. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science (2001) 291(5502):319–22. doi: 10.1126/science.291.5502.319
99. Li S, Liao W, Chen M, Shan S, Song Y, Zhang S, et al. Expression of programmed death-1 (PD-1) on CD4+ and CD8+ T cells in rheumatoid arthritis. Inflammation (2014) 37(1):116–21. doi: 10.1007/s10753-013-9718-8
100. Floudas A, Neto N, Marzaioli V, Murray K, Moran B, Monaghan MG, et al. Pathogenic, glycolytic PD-1+ B cells accumulate in the hypoxic RA joint. JCI Insight (2020) 5(21):e139032. doi: 10.1172/jci.insight.139032
101. Gonzalez-Rey E, Delgado M. Vasoactive intestinal peptide and regulatory T-cell induction: a new mechanism and therapeutic potential for immune homeostasis. Trends Mol Med (2007) 13(6):241–51. doi: 10.1016/j.molmed.2007.04.003
102. Lu YC, Chuang CH, Chuang KH, Chen IJ, Huang BC, Lee WH, et al. Specific activation of pro-Infliximab enhances selectivity and safety of rheumatoid arthritis therapy. PloS Biol (2019) 17(6):e3000286. doi: 10.1371/journal.pbio.3000286
103. Ko HJ, Cho ML, Lee SY, Oh HJ, Heo YJ, Moon YM, et al. CTLA4-Ig modifies dendritic cells from mice with collagen-induced arthritis to increase the CD4+CD25+Foxp3+ regulatory T cell population. J Autoimmun (2010) 34(2):111–20. doi: 10.1016/j.jaut.2009.07.006
104. Razmara M, Hilliard B, Ziarani AK, Chen YH, Tykocinski ML. CTLA-4 x Ig converts naive CD4+CD25- T cells into CD4+CD25+ regulatory T cells. Int Immunol (2008) 20(4):471–83. doi: 10.1093/intimm/dxn007
105. Zhou X, Kong N, Zou H, Brand D, Li X, Liu Z, et al. Therapeutic potential of TGF-β-induced CD4(+) Foxp3(+) regulatory T cells in autoimmune diseases. Autoimmunity (2011) 44(1):43–50. doi: 10.3109/08916931003782163
106. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in T(reg) cell function. Nat Immunol (2016) 17(11):1322–33. doi: 10.1038/ni.3540
107. Kisiel B, Kruszewski R, Juszkiewicz A, Raczkiewicz A, Bachta A, Tłustochowicz M, et al. Methotrexate, Cyclosporine A, and Biologics Protect against Atherosclerosis in Rheumatoid Arthritis. J Immunol Res (2015) 2015:759610. doi: 10.1155/2015/759610
108. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol (2006) 177(12):8338–47. doi: 10.4049/jimmunol.177.12.8338
109. Scott LJ. Tocilizumab: A Review in Rheumatoid Arthritis. Drugs (2017) 77(17):1865–79. doi: 10.1007/s40265-017-0829-7
110. Biggioggero M, Crotti C, Becciolini A, Favalli EG. Tocilizumab in the treatment of rheumatoid arthritis: an evidence-based review and patient selection. Drug Des Devel Ther (2019) 13:57–70. doi: 10.2147/DDDT.S150580
111. Jones G, Sebba A, Gu J, Lowenstein MB, Calvo A, Gomez-Reino JJ, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis (2010) 69(1):88–96. doi: 10.1136/ard.2008.105197
112. Zhang J, Hu X, Dong X, Chen W, Zhang L, Chang Y, et al. Regulation of T Cell Activities in Rheumatoid Arthritis by the Novel Fusion Protein IgD-Fc-Ig. Front Immunol (2020) 11:755. doi: 10.3389/fimmu.2020.00755
113. Yoo J, Shah F, Velangi S, Stewart G, Scarisbrick JS. Secukinumab for treatment of psoriasis: does secukinumab precipitate or promote the presentation of cutaneous T-cell lymphoma? Clin Exp Dermatol (2019) 44(4):414–7. doi: 10.1111/ced.13777
114. Mosanya CH, Isaacs JD. Tolerising cellular therapies: what is their promise for autoimmune disease? Ann Rheum Dis (2019) 78(3):297–310. doi: 10.1136/annrheumdis-2018-214024
115. Huter EN, Punkosdy GA, Glass DD, Cheng LI, Ward JM, Shevach EM. TGF-beta-induced Foxp3+ regulatory T cells rescue scurfy mice. Eur J Immunol (2008) 38(7):1814–21. doi: 10.1002/eji.200838346
116. Morgan ME, Sutmuller RP, Witteveen HJ, van Duivenvoorde LM, Zanelli E, Melief CJ, et al. CD25+ cell depletion hastens the onset of severe disease in collagen-induced arthritis. Arthritis Rheum (2003) 48(5):1452–60. doi: 10.1002/art.11063
117. Kelchtermans H, De Klerck B, Mitera T, Van Balen M, Bullens D, Billiau A, et al. Defective CD4+CD25+ regulatory T cell functioning in collagen-induced arthritis: an important factor in pathogenesis, counter-regulated by endogenous IFN-gamma. Arthritis Res Ther (2005) 7(2):R402–15. doi: 10.1186/ar1500
118. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199(11):1455–65. doi: 10.1084/jem.20040139
119. Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med (2004) 199(11):1467–77. doi: 10.1084/jem.20040180
120. Masteller EL, Warner MR, Tang Q, Tarbell KV, McDevitt H, Bluestone JA. Expansion of functional endogenous antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J Immunol (2005) 175(5):3053–9. doi: 10.4049/jimmunol.175.5.3053
121. Yamazaki S, Inaba K, Tarbell KV, Steinman RM. Dendritic cells expand antigen-specific Foxp3+ CD25+ CD4+ regulatory T cells including suppressors of alloreactivity. Immunol Rev (2006) 212:314–29. doi: 10.1111/j.0105-2896.2006.00422.x
122. Tarbell KV, Yamazaki S, Steinman RM. The interactions of dendritic cells with antigen-specific, regulatory T cells that suppress autoimmunity. Semin Immunol (2006) 18(2):93–102. doi: 10.1016/j.smim.2006.01.009
123. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117(3):1061–70. doi: 10.1182/blood-2010-07-293795
124. Mizrahi M, Ilan Y. The gut mucosa as a site for induction of regulatory T-cells. Curr Pharm Des (2009) 15(11):1191–202. doi: 10.2174/138161209787846784
125. Singer BD, King LS, D'Alessio FR. Regulatory T cells as immunotherapy. Front Immunol (2014) 5:46. doi: 10.3389/fimmu.2014.00046
126. Morgan ME, Flierman R, van Duivenvoorde LM, Witteveen HJ, van Ewijk W, van Laar JM, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum (2005) 52(7):2212–21. doi: 10.1002/art.21195
127. Zaiss MM, Axmann R, Zwerina J, Polzer K, Gückel E, Skapenko A, et al. Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum (2007) 56(12):4104–12. doi: 10.1002/art.23138
128. Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, et al. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes (2009) 58(3):652–62. doi: 10.2337/db08-1168
129. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
130. Zavvar M, Abdolmaleki M, Farajifard H, Noorbakhsh F, Azadmanesh K, Vojgani M, et al. Collagen II-primed Foxp3 Transduced T Cells Ameliorate Collagen-induced Arthritis in Rats: The Effect of Antigenic Priming on T Regulatory Cell Function. Iran J Allergy Asthma Immunol (2018) 17(4):361–71. doi: 10.18502/ijaai.v17i4.95
131. Lemoine FM, Cherai M, Giverne C, Dimitri D, Rosenzwajg M, Trebeden-Negre H, et al. Massive expansion of regulatory T-cells following interleukin 2 treatment during a phase I-II dendritic cell-based immunotherapy of metastatic renal cancer. Int J Oncol (2009) 35(3):569–81. doi: 10.3892/ijo_00000368
132. Bjoern J, Brimnes MK, Andersen MH, Thor Straten P, Svane IM. Changes in peripheral blood level of regulatory T cells in patients with malignant melanoma during treatment with dendritic cell vaccination and low-dose IL-2. Scand J Immunol (2011) 73(3):222–33. doi: 10.1111/j.1365-3083.2010.02494.x
133. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol (2009) 10(9):1000–7. doi: 10.1038/ni.1774
134. Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol (2009) 3:281–5. doi: 10.1016/j.coi.2009.05.007
135. Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, Wieczorek G, et al. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol (2009) 39(4):1088–97. doi: 10.1002/eji.200838904
136. Schmidl C, Hansmann L, Andreesen R, Edinger M, Hoffmann P, Rehli M. Epigenetic reprogramming of the RORC locus during in vitro expansion is a distinctive feature of human memory but not naïve Treg. Eur J Immunol (2011) 41(5):1491–8. doi: 10.1002/eji.201041067
137. Orban T, Farkas K, Jalahej H, Kis J, Treszl A, Falk B, et al. Autoantigen-specific regulatory T cells induced in patients with type 1 diabetes mellitus by insulin B-chain immunotherapy. J Autoimmun (2010) 34(4):408–15. doi: 10.1016/j.jaut.2009.10.005
138. van Eden W. “Vaccination against autoimmune diseases moves closer to the clinic. Hum Vaccin Immunother (2020) 16(2):228–32. doi: 10.1080/21645515.2019.1593085
139. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
140. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
141. Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol (2004) 22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122
142. Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev (2011) 241(1):260–8. doi: 10.1111/j.1600-065X.2011.01018.x
143. Okada M, Hibino S, Someya K, Yoshmura A. Regulation of regulatory T cells: epigenetics and plasticity. Adv Immunol (2014) 124:249–73. doi: 10.1016/B978-0-12-800147-9.00008-X
144. Li X, Zheng Y. Regulatory T cell identity: formation and maintenance. Trends Immunol (2015) 36(6):344–53. doi: 10.1016/j.it.2015.04.006
145. Barbi J, Pardoll D, Pan F. Treg functional stability and its responsiveness to the microenvironment. Immunol Rev (2014) 259(1):115–39. doi: 10.1111/imr.12172
146. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell (2014) 158(4):749–63. doi: 10.1016/j.cell.2014.07.031
147. Kitagawa Y, Ohkura N, Sakaguchi S. Molecular determinants of regulatory T cell development: the essential roles of epigenetic changes. Front Immunol (2013) 4:106. doi: 10.3389/fimmu.2013.00106
148. Sekiya T, Nakatsukasa H, Lu Q, Yoshimura A. Roles of transcription factors and epigenetic modifications in differentiation and maintenance of regulatory T cells. Microbes Infect (2016) 18(6):378–86. doi: 10.1016/j.micinf.2016.02.004
149. Cortez JT, Montauti E, Shifrut E, Gatchalian J, Zhang Y, Shaked O, et al. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature (2020) 582(7812):416–20. doi: 10.1038/s41586-020-2246-4
150. Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol (2009) 9(2):83–9. doi: 10.1038/nri2474
151. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol (2008) 9(2):194–202. doi: 10.1038/ni1549
152. Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood (2009) 114(18):3727–35. doi: 10.1182/blood-2009-05-219584
153. Chen Y, Xu Z, Liang R, Wang J, Xu A, Na N, et al. CD4(+)CD126(low/-) Foxp3(+) Cell Population Represents a Superior Subset of Regulatory T Cells in Treating Autoimmune Diseases. Mol Ther (2020) 28(11):2406–16. doi: 10.1016/j.ymthe.2020.07.020
154. Sun J, Yang Y, Huo X, Zhu B, Li Z, Jiang X, et al. Efficient Therapeutic Function and Mechanisms of Human Polyclonal CD8(+)CD103(+)Foxp3(+) Regulatory T Cells on Collagen-Induced Arthritis in Mice. J Immunol Res (2019) 2019:8575407. doi: 10.1155/2019/8575407
155. Park MJ, Moon SJ, Lee EJ, Kim EK, Baek JA, Kim SY, et al. Daurinol Attenuates Autoimmune Arthritis via Stabilization of Nrp1-PTEN-Foxp3 Signaling in Regulatory T Cells. Front Immunol (2019) 10:1526. doi: 10.3389/fimmu.2019.01526
156. Svensson MN, Doody KM, Schmiedel BJ, Bhattacharyya S, Panwar B, Wiede F, et al. Reduced expression of phosphatase PTPN2 promotes pathogenic conversion of Tregs in autoimmunity. J Clin Invest (2019) 129(3):1193–210. doi: 10.1172/JCI123267
157. Santinon F, Batignes M, Mebrek ML, Biton J, Clavel G, Hervé R, et al. Involvement of Tumor Necrosis Factor Receptor Type II in FoxP3 Stability and as a Marker of Treg Cells Specifically Expanded by Anti-Tumor Necrosis Factor Treatments in Rheumatoid Arthritis. Arthritis Rheumatol (2020) 72(4):576–87. doi: 10.1002/art.41134
158. Tseng WY, Huang YS, Clanchy F, McNamee K, Perocheau D, Ogbechi J, et al. TNF receptor 2 signaling prevents DNA methylation at the Foxp3 promoter and prevents pathogenic conversion of regulatory T cells. Proc Natl Acad Sci U.S.A. (2019) 116(43):21666–72. doi: 10.1073/pnas.1909687116
159. Hoeppli RE, MacDonald KN, Leclair P, Fung VCW, Mojibian M, Gillies J, et al. Tailoring the homing capacity of human Tregs for directed migration to sites of Th1-inflammation or intestinal regions. Am J Transplant (2019) 19(1):62–76. doi: 10.1111/ajt.14936
160. Su S, Hu B, Shao J, Shen B, Du J, Du Y, et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep (2016) 6:20070. doi: 10.1038/srep20070
161. Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity (2007) 27(5):786–800. doi: 10.1016/j.immuni.2007.09.010
162. Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, et al. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol (2007) 8(4):359–68. doi: 10.1038/ni1445
163. Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, et al. “Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol (2006) 18(8):1197–209. doi: 10.1093/intimm/dxl060
164. Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature (2007) 445(7130):936–40. doi: 10.1038/nature05563
165. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol (2017) 18(2):173–83. doi: 10.1038/ni.3646
166. Ohkura N, Sakaguchi S. Transcriptional and epigenetic basis of Treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res (2020) 30(6):465–74. doi: 10.1038/s41422-020-0324-7
167. Akamatsu M, Mikami N, Ohkura N, Kawakami R, Kitagawa Y, Sugimoto A, et al. Conversion of antigen-specific effector/memory T cells into Foxp3-expressing T(reg) cells by inhibition of CDK8/19. Sci Immunol (2019) 4(40):eaaw2707. doi: 10.1126/sciimmunol.aaw2707
168. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA (2008) 105(22):7797–802. doi: 10.1073/pnas.0800928105
169. Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med (2008) 205(3):565–74. doi: 10.1084/jem.20071477
170. Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol (2016) 37(11):803–11. doi: 10.1016/j.it.2016.08.012
171. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity (2012) 37(5):785–99. doi: 10.1016/j.immuni.2012.09.010
172. Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol (2020) 20(3):158–72. doi: 10.1038/s41577-019-0232-6
173. Felcenloben I, Piasecki T, Miller J, Rossowska J, Bańcyr E, Atamaniuk W, et al. Adoptively transferred Tregs accumulate in a site-specific manner and ameliorate signs of less advanced collagen-induced arthritis progress in rats. Immunotherapy (2015) 7(3):215–28. doi: 10.2217/imt.14.121
174. Zhang Q, Lu W, Liang CL, Chen Y, Liu H, Qiu F, et al. Chimeric Antigen Receptor (CAR) Treg: A Promising Approach to Inducing Immunological Tolerance. Front Immunol (2018) 9:2359. doi: 10.3389/fimmu.2018.02359
175. Zavvar M, Assadiasl S, Soleimanifar N, Pakdel FD, Abdolmohammadi K, Fatahi Y, et al. Gene therapy in rheumatoid arthritis: Strategies to select therapeutic genes. J Cell Physiol (2019) 234(10):16913–24. doi: 10.1002/jcp.28392
176. Scheinecker C, Göschl L, Bonelli M. Treg cells in health and autoimmune diseases: New insights from single cell analysis. J Autoimmun (2020) 110:102376. doi: 10.1016/j.jaut.2019.102376
177. Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen TM, Xu B, et al. Metabolic heterogeneity underlies reciprocal fates of T(H)17 cell stemness and plasticity. Nature (2019) 565(7737):101–5. doi: 10.1038/s41586-018-0806-7
Keywords: Treg cells, rheumatoid arthritis, autoimmune diseases, immune tolerance, transcription factor Foxp3
Citation: Jiang Q, Yang G, Liu Q, Wang S and Cui D (2021) Function and Role of Regulatory T Cells in Rheumatoid Arthritis. Front. Immunol. 12:626193. doi: 10.3389/fimmu.2021.626193
Received: 05 November 2020; Accepted: 15 March 2021;
Published: 01 April 2021.
Edited by:
Zhiguang Zhou, Central South University, ChinaReviewed by:
Fan Pan, Chinese Academy of Sciences (CAS), ChinaCopyright © 2021 Jiang, Yang, Liu, Wang and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dawei Cui, ZGF3ZWljdWlAemp1LmVkdS5jbg==; Shengjun Wang, c2p3anNAdWpzLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.