 Mireia Boluda-Navarro1
Mireia Boluda-Navarro1 Mariam Ibáñez1,2,3,4,5
Mariam Ibáñez1,2,3,4,5 Alessandro Liquori1
Alessandro Liquori1 Clara Franco-Jarava6,7
Clara Franco-Jarava6,7 Mónica Martínez-Gallo6,7Héctor Rodríguez-Vega8
Mónica Martínez-Gallo6,7Héctor Rodríguez-Vega8 Jaijo Teresa9Carmen Carreras8Esperanza Such2,3Ángel Zúñiga9
Jaijo Teresa9Carmen Carreras8Esperanza Such2,3Ángel Zúñiga9 Roger Colobran6,7,10*†José Vicente Cervera2,9*†
Roger Colobran6,7,10*†José Vicente Cervera2,9*†- 1Accredited Research Group in Hematology and Hemotherapy, Instituto de Investigación Sanitaria La Fe, Valencia, Spain
- 2Department of Hematology, Hospital Universitario y Politécnico La Fe, Barcelona, Spain
- 3Centro de Investigación Biomédica en Red de Cáncer (CIBERONC), Madrid, Spain
- 4Department of Medicine, University of Valencia, Valencia, Spain
- 5Departamento de Ciencias Biomédicas, Facultad de Ciencias de la Salud, Universidad CEU Cardenal Herrera, Valencia, Spain
- 6Immunology Division, Hospital Universitari Vall d'Hebron (HUVH), Diagnostic Immunology, Vall d’Hebron Research Institute (VHIR), Barcelona, Spain
- 7Department of Cell Biology, Physiology and Immunology, Autonomous University of Barcelona (UAB), Barcelona, Spain
- 8Pediatric Hematology Unit, Hospital Universitario y Politécnico La Fe, Valencia, Spain
- 9Genetics Unit, Hospital Universitario y Politécnico La Fe, Valencia, Spain
- 10Department of Clinical and Molecular Genetics, Hospital Universitari Vall d’Hebron (HUVH), Barcelona, Spain
Chédiak-Higashi syndrome (CHS) is a rare autosomal recessive (AR) immune disorder that has usually been associated to missense, nonsense or indels mutations in the LYST gene. In this study, we describe for the first time the case of a CHS patient carrying a homozygous mutation in the LYST gene inherited as a result of a partial uniparental isodisomy (UPiD) of maternal origin. Sanger sequencing of the LYST cDNA and single nucleotide polymorphism (SNP)-arrays were performed to identify the causative mutation and to explain the molecular mechanism of inheritance, respectively. Partial-UPiD leads to a copy neutral loss of heterozygosity (CN-LOH) of the telomeric region of chromosome 1 (1q41q44), unmasking the potential effect of the mutation detected. The mutation (c.8380dupT) is an insertion located in exon 32 of the LYST gene resulting in a premature stop codon and leading to the loss of all the conserved domains at the C-terminal of the LYST protein. This would account for the severe phenotype observed. We also reviewed the only two previously reported cases of CHS as a result of a uniparental disomy. In this study, we show that the combination of different strategies, including the use of SNP-arrays, is pivotal to fine-tune the diagnosis of rare AR disorders, such as CHS. Moreover, this case highlights the relevance of uniparental disomy as a potential mechanism of CHS expression in non-consanguineous families.
Introduction
Uniparental disomy (UPD) is an unusual genetic mechanism in which homologous copies of a specific chromosome are inherited from only one parent (1). This event can occur as hetero- (UPhD) or iso-disomy (UPiD) depending on whether the two chromosomes transmitted are different or identical, respectively (2). Furthermore, UPiD can be partial (partial-UPiD) when only a fragment of a chromosome is affected.
UPD does not always turn out in disease, thus, the clinical outcome is directly related to the genetic content, the size of the affected chromosomal region, and the degree of mosaicism (3, 4). On this regard, abnormal phenotypes associated to UPD can result from the presence of imprinted genes on the chromosome involved by altering their regulation and dosage due to variations in epigenetic marks (5). UPD can also be implicated in autosomal recessive (AR) diseases by uncovering a recessive disease allele (6–9).
CHS (OMIM #214500) is a rare AR immune disorder characterized by immunodeficiency and severe hemophagocytic lymphohistiocytosis caused by altered functional cytotoxic lymphocytes, protein missorting in neutrophil and loss of natural killer (NK) cell function. CHS patients typically present oculocutaneous albinism, thin silvery white skin, a predisposition to bleeding, neurological dysfunction and infections, among others. In a morphological analysis, CHS is detected by the presence of large pathognomonic cytoplasmic lysosomal vesicles in granulocytic cells resulting from the dysregulated function of lysosomes (10). CHS immune dysfunction is linked to infections and/or lymphoproliferative disorders with a poor prognosis unless treated by allogeneic bone marrow transplantation (11).
CHS is caused by mutations in the lysosome trafficking regulator (LYST) gene, also known as CHS1, which is localized on chromosome 1q42.1-q42.2 (12). The LYST gene encompasses 53 exons and encodes for the LYST protein, which contains several highly conserved (BEACH, PH and WD40) domains at the C-terminal end. Even though the exact function of the LYST protein is still debated, the combined PH-BEACH motifs are thought to be implicated in different aspects of vesicular trafficking and, thus, likely playing a crucial role in regulating lysosome-related organelle size, fission and secretion (12–14). Both homozygous and compound heterozygous mutations in LYST gene have been previously described in CHS (10). Loss-of-function mutations are associated with the severe CHS childhood form leading to death if untreated. In contrast, missense mutations are observed in patients with a milder form of the disease, allowing survival into adulthood (11).
Here, we report the first case of CHS resulted from a partial-UPiD in a young patient harboring a novel mutation in the LYST gene. The mutation was present in homozygosis despite segregation analysis revealed the mother as the only carrier. Subsequent single nucleotide polymorphism (SNP)-arrays analysis allowed us to identify a segmental isodisomy of maternal origin as the cause of this uncommon genetic inheritance.
Case Presentation
Here we present a 3-year-old girl, born from non-consanguineous parents after a normal pregnancy and cesarean delivery, hospitalized after 2 days of high fever, productive cough, clear nasal secretions and lack of appetite. One week before, she had suffered a non-febrile respiratory infection treated with azithromycin. She presented incomplete ocular albinism characterized by absence of pigmentation in the peripheral retina but had no history of serious recurrent infections or excessive bleeding. There was no family history of albinism or hypopigmentation, immunological or hematological alterations, or of deaths at an early age.
Clinical, Laboratory and Genetic Results
During her evaluation, pancytopenia and oculocutaneous albinism were identified whilst chest x-rays (CXR) did not show significant alterations. Infectious diseases were ruled out by serological screening and direct observation in blood smears. Analysis for respiratory viruses was positive for adenovirus and respiratory syncytial virus (RSV). She showed normal psychomotor development and her vaccination schedule was up to date. Morphological studies were performed both on peripheral blood (PB) and bone marrow (BM) samples. Results for PB cells were consistent with CHS alterations. Hypercellular BM with increased levels of the granulocytic series and lower erythroid and lymphocytes series was also observed. In PB as in BM, purple inclusions were detected in both lymphocytes and all stages of the granulocytic series. Intense vacuolization, sometimes with a purple inclusion, was also seen in all cell types (Figures 1A–D). BM’s flow cytometry showed a normal immunophenotype, with an increase of immature myeloid cells (Supplementary Table 1). NK cells showed defective granule exocytosis with low expression of CD107a on stimulated NK cells (Figure 1E) and a slightly reduced cytotoxic capacity (Figure 1F) evaluated by degranulation assay.
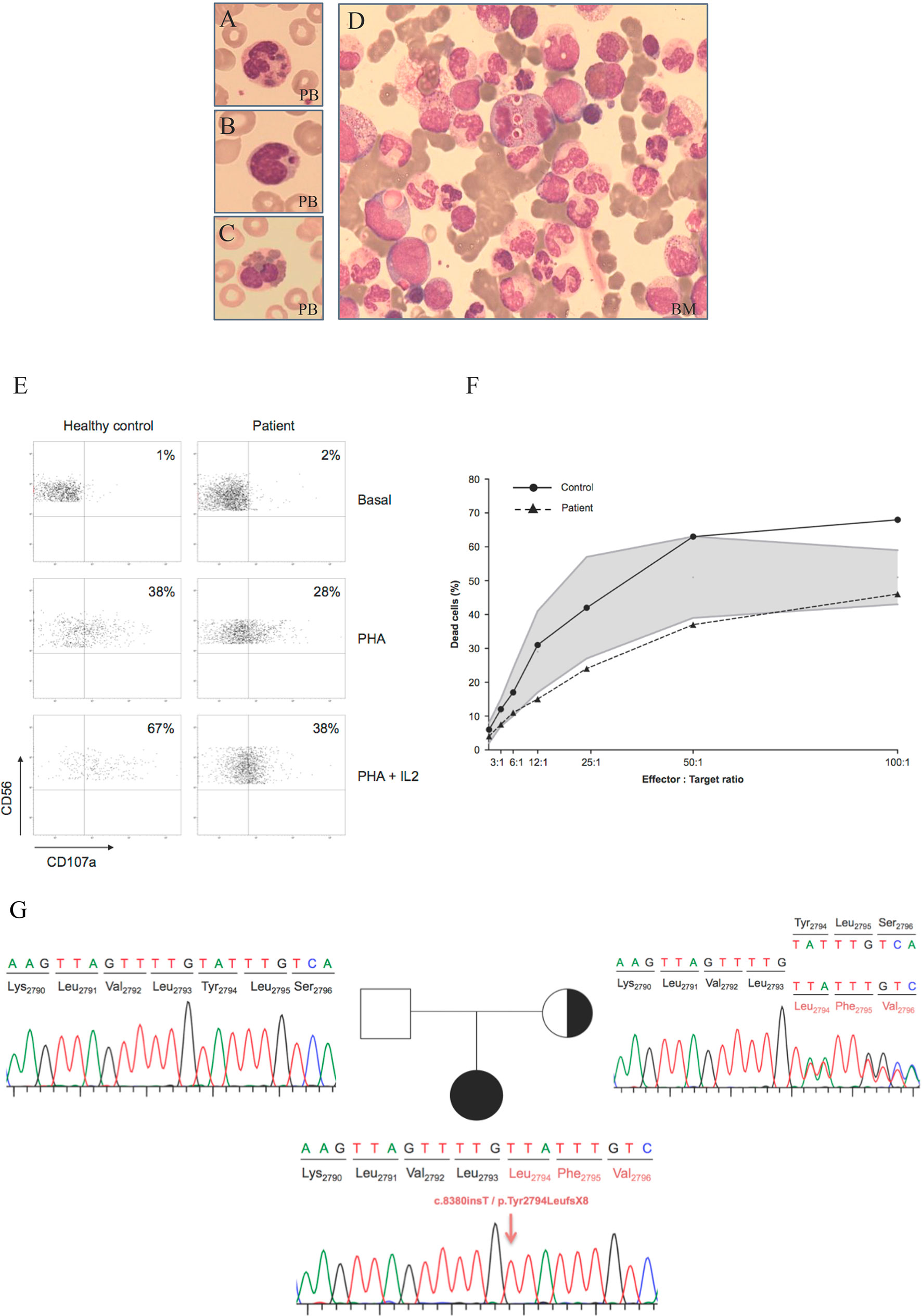
Figure 1 Morphological, cellular and molecular findings in the CHS patient. (A–C) Wright stains of peripheral smear showing a neutrophil, lymphocyte and eosinophil with giant intracytoplasmic granules. (D) Bone marrow aspirate. Intense vacuolization and purple inclusions are also observed in immature cells of the myeloid linage. PB: peripheral blood; BM: bone marrow. (E) Degranulation assay was decreased compared to the healthy control both with PHA and PHA + IL2. PBMCs were incubated with or without (resting cells) PHA either alone or with IL-2 for 4 hr at 37°C. Thereafter, cells were stained with fluorochrome-conjugated anti-CD56, and anti-CD107a mAbs. CD107a surface expression was gated on CD56 cells. (F) A flow cytometry-based NK cytotoxicity assay was performed by measuring cytotoxicity against K562 cell line in a 4-hour staining with propidium iodide. The shaded area indicates normal ranges of cytotoxicity tested in healthy individuals. The patient showed a cytotoxic function on the lower range of normality. (G) Family tree and sequence analysis of the patient and his parents. Sequence patterns showing the patient’s mutation c.8380dupT in homozygosis, the normal sequence of the non-carrier father and the mother’s sequence showing the mutation in heterozygosis.
Differential diagnosis by cytogenetic analysis, including conventional karyotype by C-banding and FISH revealed a normal female karyotype (46,XX). Due to the clinical phenotype that included albinism, together with the defective degranulation assay and the presence of cytoplasmic lysosomal vesicles in granulocytic cells, we amplified by PCR the entire patient’s cDNA sequence of LYST gene (53 exons) in 5 overlapping fragments (Supplementary Table 2, Supplementary Figure 1A). PCR products were then sequenced by Sanger. The genotype analysis of the patient revealed a homozygous frameshift c.8380dupT (NM_000081.3) variant in the exon 32 (Figure 1G) that would lead to a premature stop codon (PTC) (NP_000072.2: p.Tyr2794Leufs*8). The presence of this variant was then confirmed by the targeted sequencing of the corresponding genomic region (gDNA). The LYST c.8380dupT variant was not reported in the literature or in the main population databases (e.g. dbSNP, ExAC and gnomAD). The effect of the variant on the encoded protein was assessed by identifying and comparing conserved domains in both the reference and the resulting mutated LYST protein with the NCBI Conserved Domain Search Tool (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) (Figure 2). This tool revealed that the variant identified might remove all conserved domains responsible for the protein function (PH, BEACH and WD40). Given the analysis performed on the variant effect and the patient phenotype, we have classified this variant as pathogenic (PVS1 according to the American College of Medical Genetics and Genomics recommendation) (16). Written informed consent from the parents was provided for all genetic studies reported here.
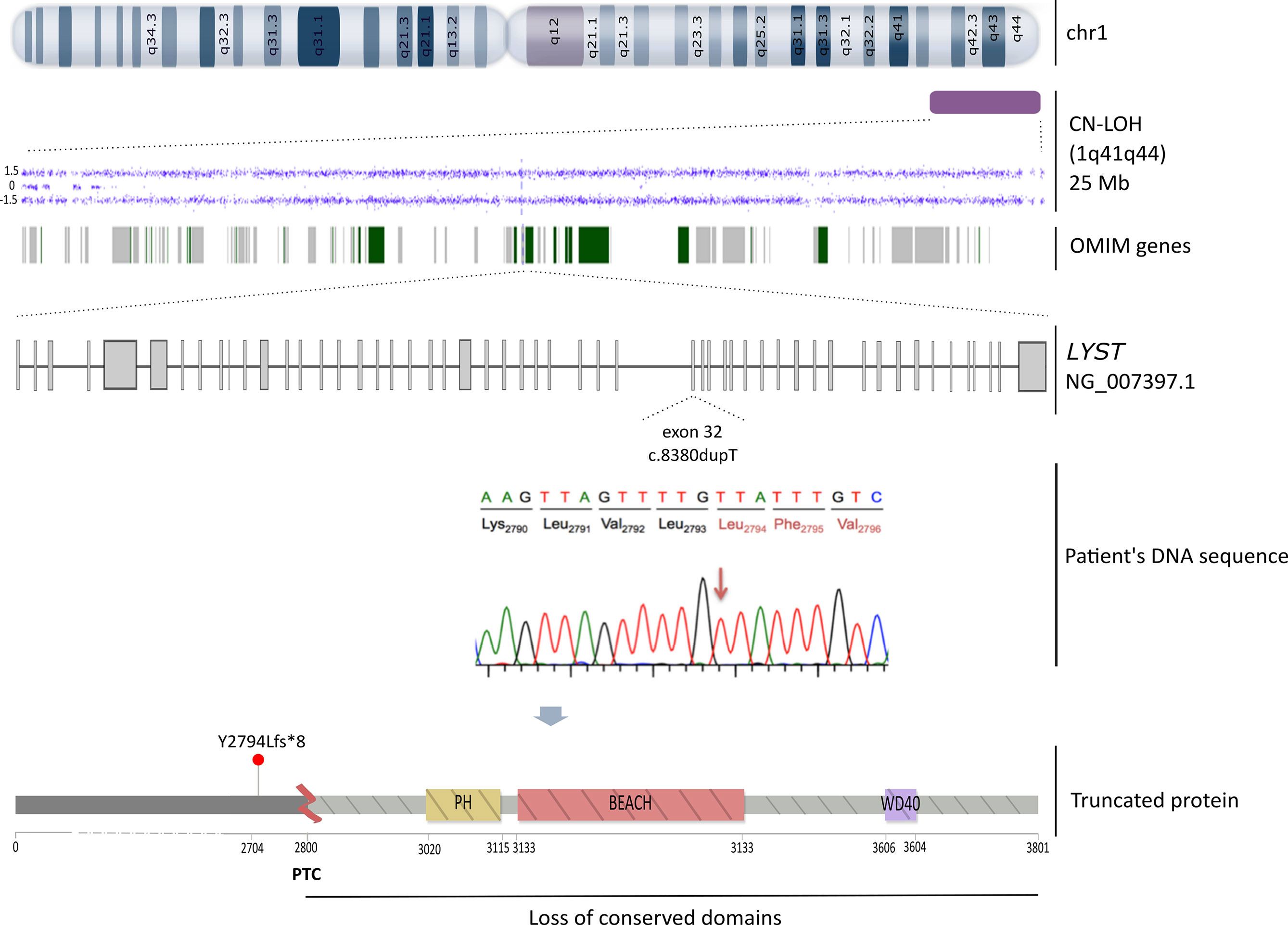
Figure 2 Schematic representation of the triggering molecular mechanism of CHS in our patient. From top to bottom: the CN-LOH detected by SNP-array (Cytoscan HD, Affymetrix) in chromosome 1 (1q41q44) is delineated as a purple rectangle, the dots depict the altered markers in the array and the green and grey bands represent the 93 OMIM genes affected, including the LYST gene. Below, the LYST gene (NG_007397.1) that encodes for the entire functional protein is depicted. The c.8380dupT (NM_000081.3) frameshift mutation is localized in the exon 32. A lollipop-like (15) image reflects the resulting truncated protein owing to a PTC at 2,800 amino acid that would remove all conserved domains (PH, BEACH and WD40).
To determine the inheritance pattern, segregation analysis was performed revealing only the mother as the single carrier of this variant, whereas the father was wild-type (Figure 1C). Bearing in mind the non-consanguinity of the parents, paternity tests to confirm family relationships were carried out. Given that CHS is an AR disorder, we conducted a SNP-array (Cytoscan HD, Affymetrix) according to manufacturer’s protocol (Affymetrix Santa Clara, C.A., U.S.A.). The chromosomal region containing the LYST gene was assessed for copy number variations (CNVs) and CN-LOH using Chromosome Analysis Suite software (Affymetrix) for both the patient and her parents. Filters applied for the detection of CNV were ≥ 20 consecutive markers in a region of at least 50kb, and for regions of CN-LOH, ≥ 100 markers in at least 5,000kb. Whilst no CNVs were observed in the parents’ DNA, a CN-LOH on the telomeric region of the long arm of chromosome 1 encompassing 25Mb (1q41q44) was detected in the child, accounting for the candidate variant in homozygosis in the LYST gene (Figure 2). A time-line summary of genetic investigations is represented in Supplementary Figure 2.
In addition, UPiD can alter the dosage of imprinted genes contributing to the phenotype of the disease (5, 17). Therefore, although the truncated LYST protein derived from the c.8380dupT variant would explain the severe phenotype in our patient, the possibility of an altered imprinted gene dosage as a consequence of the partial UPiD was also examined. We consulted the “Geneimprint” portal (http://www.geneimprint.com/site/genes-by-species) and found that OBSCN and OR11L1 genes, in the region affected by the partial-UPiD, were predicted as paternally imprinted. However, the alteration of such genes cannot be related to the CHS patient phenotype.
The patient experienced a spontaneous recovery from the cytopenias. However, she was repeatedly admitted to the hospital due to frequent episodes of high fever, general condition impairment, and splenomegaly varying between 5 and 15 cm. Recurrence of exacerbated cytopenias required a transfusion of red blood cells in one occasion and G-CSF in two episodes. Although the patient did not meet the criteria for massive hemophagocytic lymphohistiocytosis (HLH) established by the 2004 Hystiocyte Society guidelines the last time she was hospitalized (Table 1), given the progression of the condition and clinical deterioration, she was treated with Dexamethasone and Etoposide according to the HLH protocol - 94/2004 (18, 19) to which she showed good response. Once remission was achieved, the patient received a stem cell transplant (HCT) from an unrelated HLA 10/10 compatible donor at 51/2 years after following a conditioning regimen with busulfan, fludarabine and ATG. After 2 years since the transplant, the patient maintains complete donor chimerism, showing a normal physical and neurological development, and has not required subsequent admissions into the hospital.
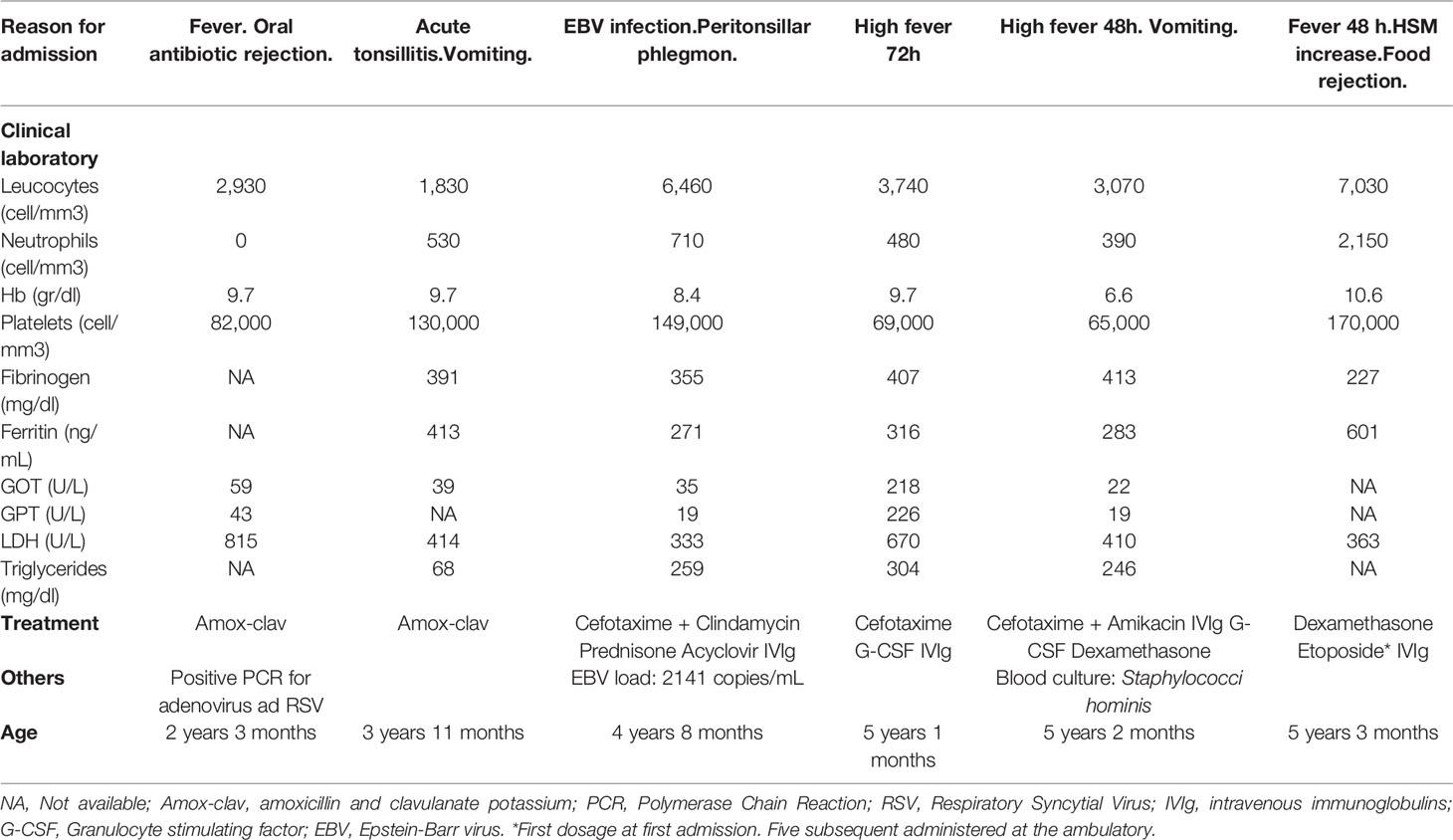
Table 1 Patient’s clinical and treatment progression from the first admission to date.
Discussion
This study reports the first case of CHS as a result of partial-UPiD. The patient harbored a novel pathogenic frameshift variant (NM_000081.3:c.8380dupT) in homozygosis in the LYST gene. To date, 72 mutations in this gene have been associated with CHS (20), and UPD has been reported as the causative mechanism only in three CHS cases (including the patient described here) (17, 21). A comparison of the three cases of UPD in CHS is shown in Table 2. In addition, 11 cases of primary immunodeficiencies caused by UPD have been reported to date (9) (considering this study) but only SCID (22) and LCK (23) in chromosome 1. Taken together, these data highlight the rarity of this event.
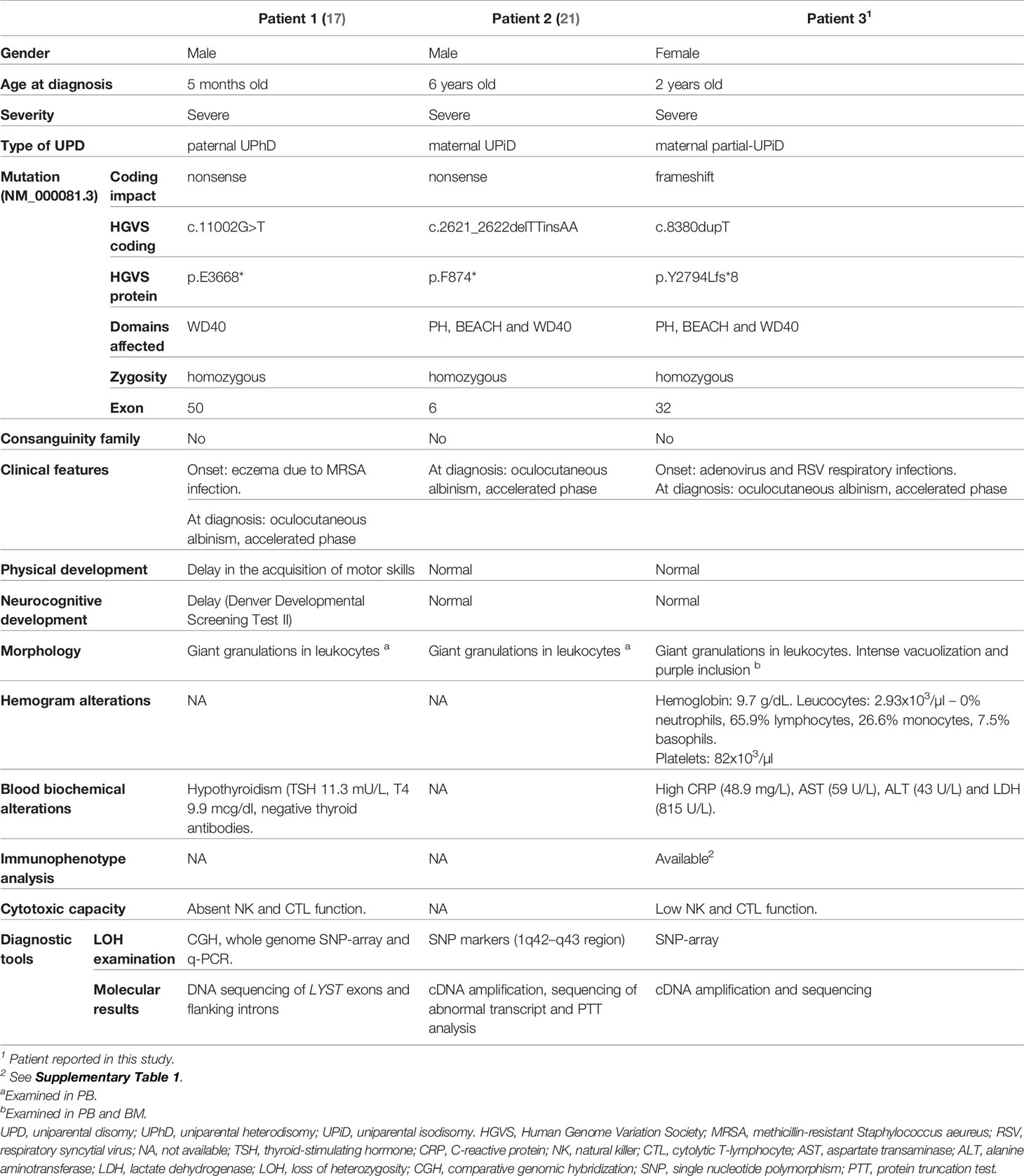
Table 2 Summary of reported cases of UPD in CHS with clinical and diagnostic tools detailed.
To assess the pathogenicity of the c.8380dupT variant and its association to the clinical phenotype of the patient, an in silico analysis was performed, suggesting that the LYST protein might be affected. The frameshift produced by the c.8380dupT leads to a PTC in exon 32 resulting in the loss of PH, BEACH and WD40 highly conserved domains at the C-terminal end of the protein. Both WD40 and PH domains are involved in targeting proteins to appropriate subcellular compartments; PH domain also participates in the protein-protein interaction with a binding partner. Therefore, the LYST protein would be truncated and non-functional (13). PTC could also initiate an mRNA degradation by the nonsense mediated decay (NMD), preventing its translation into the protein (24). However, the patient’s LYST RNA was successfully retro-transcribed into cDNA for molecular studies, suggesting that this transcript would escape the NMD surveillance mechanism. Additionally, we also amplified and sequenced the mother’s LYST cDNA region including the c.8380dupT mutation. The similar intensity of both alleles in the mother’s Sanger sequencing electropherogram rules out a significant degradation of the mutated allele (Supplementary Figure 1B). Therefore, all these data point out that the LYST c.8380dupT variant is pathogenic and would explain the severe CHS phenotype and childhood onset observed in our patient. In addition, although the genotype-phenotype correlation in CHS cases remains controversial, several studies have reported patients harboring frameshift, nonsense and splice-site mutations resulting in an absent LYST protein and similar clinical outcomes to that of our patient (10, 11). In contrast, the milder adult form of CHS is often due to missense mutation encoding for a partially functioning protein. However, Sánchez-Guiu et al. (25) demonstrated different outcomes due to a direct consequence of changes in protein structure and, especially, in the case of missense mutations, in electrostatic surface potential (25). Therefore, identifying the real molecular effect of the mutation is crucial to solve genotype-phenotype relationship.
Nevertheless, the detection of the c.8380dupT variant in homozygosity, the non-consanguinity of the parents and the results of segregation studies, showing the mother as the only heterozygous carrier, led us to consider the presence of other underlying genetic events, such as chromosomal deletion or loss of heterozygosity. Consequently, to determine the causative molecular mechanism of the disease, a SNP-array was performed. This analysis revealed that the patient harbored a CN-LOH affecting the 1q42.12 – 1q44 region, which suggests a partial-UPiD of maternal origin. In CHS, UPiD owing to CN-LOH has been previously identified (21), however, to our knowledge, this is the first CHS case reported as a consequence of partial-UPiD (17, 21). The two previously reported CHS patients have a severe phenotype at early stages caused by nonsense mutations. Manoli et al. (17) reported a paternal UPhD proband with additional developmental delays despite carrying the most distal mutation among the three cases (Table 2). The authors argued that this could be the result of additional non-detectable effects linked to a deregulation in genomic imprinting of chromosome 1 as previously suggested (26). Nonetheless, several AR disorders, including the other CHS case of maternal UPiD, have been purely described as a consequence of UPD of chromosome 1 without any atypical phenotype, suggesting the lack of imprinted genes on either the paternal or maternal alleles (21, 27). In our case, although partial-UPiD encompassed 93 OMIM genes besides LYST, of which OBSCN and OR11L1 (http://www.geneimprint.com/site/genes-by-species) were predicted to be paternal imprinted genes, only OBSCN has been related to an AR disorder (i.e. fibromuscular dysplasia and Limb-Girdle muscular dystrophy) (28). Since our patient exhibited no other uncommon conditions other than classic CHS, we concluded that the alteration of such genes is not related to the patient phenotype.
Although there is no clear explanation for the mechanism that leads to partial-UPiD observed in our patient, this could be produced by an early mitotic division error resulting in the loss of the paternal chromosome 1 segment, as indicated in previous reports (3, 29). In other AR diseases (30–32), partial-UPiD has been associated with mosaicism as a very rare event (33). However, the SNP-arrays technology analyses may have disregarded the presence of mosaicism in our patient, especially if we consider that the sensitivity of this technique is 10%, as well as the premature alteration in mitotic division (34).
On the other hand, CHS is also associated with the development of HLH, which affects more than 75% of CHS patients within their first decade and it is fatal unless treated (35). HLH is a main characteristic of the ‘accelerated phase’ of the disease, hence, being particularly useful when determining the need for a HCT. A decrease in NK-cell and cytotoxic T lymphocytes capacity provides a diagnostic criteria and predictor of the severity and transformation to HLH (36). In addition to function impairment in NK cells, our patient displayed a clinical deterioration with consecutives hospitalization matching HLH-94/2004 guidelines. Therefore, since transplantation appears to be most successful if performed prior to the onset of HLH or during remission, we decided to perform HCT once remission was achieved. Although a HCT does not prevent the possible neurologic detriment, our patient remains healthy after two years from the HCT, showing a normal immunologic, hematologic and neurological development.
In summary, we report the first CHS case with a pathogenic variant in homozygosis as a consequence of partial-UPiD. Risk of AR conditions, such as CHS, are easily estimated with siblings having a 25% risk of developing the disease. However, the prevalence of germline UPD is harder to quantify with frequencies from 0.03 – 0.3% (37–40), being partial-UPiD significantly lower (40). Despite the low risk of recurrence, having detected the heterozygosis c.8380dupT variant in the mother of this child and defined the inheritance mechanism, will allow a more accurate genetic counseling for future pregnancies and other family members at risk. Taken together, in non-consanguineous families, UPD should be considered as a potential mechanism of CHS when a homozygous mutation is identified. This case also underlines the pivotal role of SNP-array as a diagnostic approach to assess the molecular mechanism responsible of AR disorders, such as CHS.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité de Ética de la Investigación con medicamentos del Hospital Universitario y Politécnico la Fe. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
MB-N analyzed molecular studies and wrote the paper. MI and AL supervised experimental findings. CF-J and MM-G performed and analyzed the functional assays research and data analysis. HR-V, JT and CC were involved in the management of the patient. ES performed cytogenetic analysis. ÁZ supervised clinical and experimental findings. RC performed and analyzed the molecular studies. JC and RC were responsible for the conception of the study and final approval of the draft. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by research funding from FEDER funds (CIBERONC, CB16/12/00284), Instituto de Salud Carlos III grants PI16/01113, PI17/00660, PI18/1472, PI19/00812 cofinanced by the European Regional Development Fund (ERDF); as well as from the “Conselleria de Educación, Cultura y Deporte” GV/2019/084. MB-N and AL are recipients of a fellowship from the “Asociación Española Contra El Cáncer” and the “Fundación Española de Hematología y Hemoterapia”, respectively.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank “Biobank La Fe” for their useful help in sample processing and supply.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.625591/full#supplementary-material
References
1. Engel E. A new genetic concept: Uniparental disomy and its potential effect, isodisomy. Am J Med Genet (1980) 6:137–43. doi: 10.1002/ajmg.1320060207
2. Makishima H, Maciejewski JP. Pathogenesis and consequences of uniparental disomy in cancer. Clin Cancer Res (2011) 17:3913–23. doi: 10.1158/1078-0432.CCR-10-2900
3. Lapunzina P, Monk D. The consequences of uniparental disomy and copy number neutral loss-of-heterozygosity during human development and cancer. Biol Cell (2011) 103:303–17. doi: 10.1042/bc20110013
4. Gaudio Dd, Shinawi M, Astbury C, MK T, KL D, Raca G. Diagnostic testing for uniparental disomy: a points to consider statement from the American College of Medical Genetics and Genomics (ACMG). Genet Med (2020) 2020:1–9. doi: 10.1038/s41436-020-0782-9
5. Monk D, Mackay DJG, Eggermann T, Maher ER, Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet (2019) 20:235–48. doi: 10.1038/s41576-018-0092-0
6. Gelb BD, Willner JP, Dunn TM, Kardon NB, Verloes A, Poncin J, et al. Paternal uniparental disomy for chromosome 1 revealed by molecular analysis of a patient with pycnodysostosis. Am J Hum Genet (1998) 62:848–54. doi: 10.1086/301795
7. Travaglini L, Aiello C, Alesi V, Loddo S, Novelli A, Tozzi G, et al. Uniparental disomy of chromosome 1 unmasks recessive mutations of PPT1 in a boy with neuronal ceroid lipofuscinosis type 1. Brain Dev (2017) 39:182–3. doi: 10.1016/j.braindev.2016.08.010
8. Mannurita SC, Vignoli M, Bianchi L, Kondi A, Gerloni V, Breda L, et al. CACP syndrome: Identification of five novel mutations and of the first case of UPD in the largest European cohort. Eur J Hum Genet (2014) 22:197–201. doi: 10.1038/ejhg.2013.123
9. Soler-Palacín P, Garcia-Prat M, Martín-Nalda A, Franco-Jarava C, Rivière JG, Plaja A, et al. LRBA deficiency in a patient with a novel homozygous mutation due to chromosome 4 segmental uniparental isodisomy. Front Immunol (2018) 9:2397. doi: 10.3389/fimmu.2018.02397
10. Zarzour W, Kleta R, Frangoul H, Suwannarat P, Jeong A, Kim SY, et al. Two novel CHS1 (LYST) mutations: Clinical correlations in an infant with Chediak–Higashi syndrome. Mol Genet Metab (2005) 85:125–32. doi: 10.1016/J.YMGME.2005.02.011
11. Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ, et al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet (2002) 108:16–22. doi: 10.1002/ajmg.10184
12. Nagle DL, Karim MA, Woolf EA, Holmgren L, Bork P, Misumi DJ, et al. Identification and mutation analysis of the complete gene for Chediak–Higashi syndrome. Nat Genet (1996) 14:307–11. doi: 10.1038/ng1196-307
13. Cullinane AR, Schäffer AA, Huizing M. The BEACH Is Hot: A LYST of Emerging Roles for BEACH-Domain Containing Proteins in Human Disease. Traffic (2013) 14:749–66. doi: 10.1111/tra.12069
14. Durchfort N, Verhoef S, Vaughn MB, Shrestha R, Adam D, Kaplan J, et al. The Enlarged Lysosomes in beigej Cells Result From Decreased Lysosome Fission and Not Increased Lysosome Fusion. Traffic (2012) 13:108–19. doi: 10.1111/j.1600-0854.2011.01300.x
15. Jay JJ, Brouwer C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PloS One (2016) 11:e0160519. doi: 10.1371/journal.pone.0160519
16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
17. Manoli I, Golas G, Westbroek W, Vilboux T, Markello TC, Introne W, et al. Chediak-Higashi syndrome with early developmental delay resulting from paternal heterodisomy of chromosome. Am J Med Genet Part A (2010) 152:1474–83. doi: 10.1002/ajmg.a.33389
18. Bergsten E, Horne AC, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: Long-Term results of the cooperative HLH-2004 study. Blood (2017) 130:2728–38. doi: 10.1182/blood-2017-06-788349
19. Henter JI, Samuelsson-Horne AC, Aricò M, Maarten Egeler R, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood (2002) 100:2367–73. doi: 10.1182/blood-2002-01-0172
20. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res (2018) 46:D1062–7. doi: 10.1093/nar/gkx1153
21. Dufourcq-Lagelouse R, Lambert N, Duval M, Viot G, Vilmer E, Fischer A, et al. Chediak-Higashi syndrome associated with maternal uniparental isodisomy of chromosome 1. Eur J Hum Genet (1999) 7:633–7. doi: 10.1038/sj.ejhg.5200355
22. Roberts JL, Buckley RH, Luoc B, Pei J, Lapidus A, Peri S, et al. CD45-deficient severe combined immunodeficiency caused by uniparental disomy. Proc Natl Acad Sci USA (2012) 109:10456–61. doi: 10.1073/pnas.1202249109
23. Hauck F, Randriamampita C, Martin E, Gerart S, Lambert N, Lim A, et al. Primary T-cell immunodeficiency with immunodysregulation caused by autosomal recessive LCK deficiency. J Allergy Clin Immunol (2012) 130(5):1144–52. doi: 10.1016/j.jaci.2012.07.029
24. Gudipati RK, Xu Z, Lebreton A, Séraphin B, Steinmetz LM, Jacquier A, et al. Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol Cell (2012) 48:409–21. doi: 10.1016/j.molcel.2012.08.018
25. Sánchez-Guiu I, Antón AI, García-Barberá N, Navarro-Fernández J, Martínez C, Fuster JL, et al. Chediak-Higashi syndrome: description of two novel homozygous missense mutations causing divergent clinical phenotype. Eur J Haematol (2014) 92:49–58. doi: 10.1111/ejh.12203
26. Wassink TH, Losh M, Frantz RS, Vieland VJ, Goedken R, Piven J, et al. A case of autism and uniparental disomy of chromosome 1. Hum Genet (2005) 117:200–6. doi: 10.1007/s00439-005-1257-4
27. Turner CLS, Bunyan DJ, Thomas NS, Mackay DJG, Jones HP, Waterham HR, et al. Zellweger syndrome resulting from maternal isodisomy of chromosome 1. Am J Med Genet Part A (2007) 143A:2172–7. doi: 10.1002/ajmg.a.31912
28. GeneCards. Human Genes | Gene Database | Gene Search . Available at: https://www.genecards.org/ (Accessed March 4, 2021).
29. Niida Y, Ozaki M, Shimizu M, Ueno K, Tanaka T. Classification of uniparental isodisomy patterns that cause autosomal recessive disorders: Proposed mechanisms of different proportions and parental origin in each pattern. Cytogenet Genome Res (2018) 154:137–46. doi: 10.1159/000488572
30. Keren B, Chantot-Bastaraud S, Brioude F, Mach C, Fonteneau E, Azzi S, et al. SNP arrays in Beckwith–Wiedemann syndrome: An improved diagnostic strategy. Eur J Med Genet (2013) 56:546–50. doi: 10.1016/J.EJMG.2013.06.005
31. Henry I, Puech A, Riesewijk A, Ahnine L, Mannens M, Beldjord C, et al. Somatic Mosaicism for Partial Paternal Isodisomy in Wiedemann-Beckwith Syndrome: A Post-Fertilization Event. Eur J Hum Genet (1993) 1:19–29. doi: 10.1159/000472384
32. Su J, Wang J, Fan X, Fu C, Zhang S, Zhang Y, et al. Mosaic UPD(7q)mat in a patient with silver Russell syndrome. Mol Cytogenet (2017) 10:36. doi: 10.1186/s13039-017-0337-1
33. Eggermann T, Soellner L, Buiting K, Kotzot D. Mosaicism and uniparental disomy in prenatal diagnosis. Trends Mol Med (2015) 21:77–87. doi: 10.1016/j.molmed.2014.11.010
34. Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, et al. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet (2010) 19:1263–75. doi: 10.1093/hmg/ddq003
35. Aricò M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia (1996) 10:197–203.
36. Jessen B, Maul-Pavicic A, Ufheil H, Vraetz T, Enders A, Lehmberg K, et al. Subtle differences in CTL cytotoxicity determine susceptibility to hemophagocytic lymphohistiocytosis in mice and humans with Chediak-Higashi syndrome. Blood (2011) 118:4620–9. doi: 10.1182/blood-2011-05-356113
37. Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays (2000) 22:452–9. doi: 10.1002/(SICI)1521-1878(200005)22:5<452::AID-BIES7>3.0.CO;2-K
38. Nakka P, Pattillo Smith S, O'Donnell-Luria AH, McManus KF, Agee M, Auton A, et al. Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population. Am J Hum Genet (2019) 105:921–32. doi: 10.1016/j.ajhg.2019.09.016
39. Yauy K, de Leeuw N, Yntema HG, Pfundt R, Gilissen C. Accurate detection of clinically relevant uniparental disomy from exome sequencing data. Genet Med (2020) 22:803–8. doi: 10.1038/s41436-019-0704-x
Keywords: Chédiak-Higashi syndrome, CHS, primary immunodeficiency, hemophagocytic lymphohistiocytosis, LYST, SNP-array, uniparental disomy, loss of heterozygosity
Citation: Boluda-Navarro M, Ibáñez M, Liquori A, Franco-Jarava C, Martínez-Gallo M, Rodríguez-Vega H, Teresa J, Carreras C, Such E, Zúñiga Á, Colobran R and Cervera JV (2021) Case Report: Partial Uniparental Disomy Unmasks a Novel Recessive Mutation in the LYST Gene in a Patient With a Severe Phenotype of Chédiak-Higashi Syndrome. Front. Immunol. 12:625591. doi: 10.3389/fimmu.2021.625591
Received: 03 November 2020; Accepted: 16 March 2021;
Published: 31 March 2021.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Raz Somech, Sheba Medical Center, IsraelEkaterini Simoes Goudouris, Federal University of Rio de Janeiro, Brazil
Khan Saadullah, Kohat University of Science and Technology, Pakistan
Copyright © 2021 Boluda-Navarro, Ibáñez, Liquori, Franco-Jarava, Martínez-Gallo, Rodríguez-Vega, Teresa, Carreras, Such, Zúñiga, Colobran and Cervera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger Colobran, cmNvbG9icmFuQHZoZWJyb24ubmV0; José Vicente Cervera, Y2VydmVyYV9qb3NAZ3ZhLmVz
†These authors have contributed equally to this work