 Jiaojiao Tang
Jiaojiao Tang Zijun Yan
Zijun Yan Qiyu Feng
Qiyu Feng Lexing Yu
Lexing Yu Hongyang Wang
Hongyang Wang- 1Division of Life Sciences and Medicine, Cancer Research Center, The First Affiliated Hospital of University of Science and Technology of China, Hefei, China
- 2International Cooperation Laboratory on Signal Transduction, Ministry of Education Key Laboratory on Signaling Regulation and Targeting Therapy of Liver Cancer, Shanghai Key Laboratory of Hepato-Biliary Tumor Biology, Eastern Hepatobiliary Surgery Hospital, Shanghai, China
- 3Graduate Management Unit, Shanghai Changhai Hospital, Second Military Medical University, Shanghai, China
- 4National Center for Liver Cancer, Shanghai, China
Neutrophils are the largest population of circulating leukocytes and the first responder against invading pathogens or other danger signals. Sophisticated machineries help them play critical roles in immunity and inflammation, including phagocytosis, superoxide production, cytokine and chemokine production, degranulation, and formation of neutrophil extracellular traps (NETs). After maturation and release from the bone marrow, neutrophils migrate to inflamed tissues in response to many stimuli. Increasing evidences indicate that neutrophils are critically involved in the pathogenesis of liver diseases, including liver cancer, thus making them promising target for the treatment of liver diseases. Here, we would like to provide the latest finding about the role of neutrophils in liver diseases and discuss the potentiality of neutrophils as target for liver diseases.
Introduction
Neutrophils are the most abundant white blood cells in mammals, representing the first line of innate defense against invading pathogens or other foreign bodies. Moreover, they play significant roles in shaping adaptive immunity and function as coordinators of the overall immune and inflammatory responses. Sophisticated processes, including phagocytosis, reactive oxygen species (ROS) generation, degranulation, cytokines and chemokines production, and neutrophil extracellular traps (NETs) release are vital for the immunological functions of neutrophils (1). Neutrophil loss or deficiency due to diseases or side effects of therapy is usually associated with severe recurrent infection (2, 3). However, excess infiltration and/or activation of neutrophils in the tissue can cause chronic inflammation, limit tissue repair, and lead to loss of organ function (4). Previous studies have indicated that neutrophil-induced inflammation occurs during the pathogenesis of a range of chronic diseases and cancer. Therefore, neutrophils represent a promising therapeutic target for various diseases (1), and various targeting approaches, including targeting neutrophil development and production, interfering with neutrophil accumulation at the site of infection/inflammation and reversing the detrimental changes of neutrophil phenotype that occur during certain pathological conditions, as well as mitigating the harmful effects of NETs (1, 2) have emerged.
The role of neutrophils in the pathogenesis of liver diseases has garnered intense interest in recent years. Neutrophils routinely patrol the liver sinusoids and there are few resident neutrophils in the liver. Instead, they can be recruited into the liver rapidly during acute liver infection or injury and serve as the principal phagocyte type responsible for pathogen clearance. The infiltration of neutrophils is commonly seen in all types of liver diseases. However, the overwhelming activation of neutrophils can also induce liver damage. Therefore, neutrophils are considered to be double-edge swords during acute liver inflammation. The importance of neutrophils in the chronic liver diseases (CLD) has also been appreciated in recent years because they can communicate with other immune and non-immune cells within the liver. In the recent review, we would like to provide the emerging evidence for the relevance of neutrophils during various liver diseases, and discuss the potentiality of neutrophils as target for liver diseases. We also discuss how CLD affects granulopoiesis, neutrophil phenotype, and function.
Production of Neutrophils and Mediators of Their Function
Since they are among the shortest-lived cells in mammals, neutrophils rely on constant replenishment from the bone marrow through highly controlled granulopoiesis (~1011 neutrophils are produced from the human body every day) (5), and further increase under stress conditions (which is called “emergency granulopiesis”) (6). They originate from haematopoietic stem cells to common myeloid progenitor to lineage-committed progenitors that mature into neutrophils (3). Transcription factors, such as CCAAT/enhancer binding protein (C/EBPα), PU 1, and RUNX1 are necessary for neutrophil maturation during steady-state granulopoiesis, while C/EBPβ severs as a master regulator for emergency granulopoiesis (6, 7). Neutrophil production, maturation, release, and elimination are under tight control to maintain homeostatic stability and balance between antimicrobial and proinflammatory functions. A major regulator is granulocyte colony-stimulating factor (G-CSF), which promotes neutrophil development by engaging with G-CSF receptor and their release via downregulation of CXCR4 and upregulation of CXCR2 in neutrophils (1). Release mature neutrophils then migrate into inflamed tissues in response to various stimuli, such as chemokines (CXCL1, CXCL2) gradient.
The maturation of neutrophils is characterized by the condensation and mutilobular appearance of the nucleus, and the emergence of cell type-specific intracellular granules (7). During granulopoiesis, three types of granules are formed consecutively, namely, primary, secondary, and tertiary, as well as secretary vesicles of endocytic origin, all of which are prepackaged with antimicrobial and tissue-destructive factor, along with various neutrophil receptors. They are all readily available to be released to participate in the host response to inflammation or infection (1, 8). For instance, the azurophil (primary) granules are the reservoirs of myeloperoxidase (MPO), neutrophil elastase (NE), proteinase 3, (PR3) and most proteolytic and bactericidal proteins, and are considered to be the microbicidal compartment mobilized during phagocytosis. The specific (secondary) granules harbor antimicrobial lactoferrin, neutrophil gelatinase-associated lipocalin (LCN2), and chitinase-3-like protein-1. Gelatinase (tertiary) granules contain matrix metalloproteinase 9 (MMP9), collagenase (MMP8), and cathelicidin antimicrobial peptide. Secretory vesicles are rich in transmembrane receptors that integrate into the plasma membrane as exocytosis occurs (9).
Another weapon that helps neutrophils to capture pathogens is NETs, which are extracellular structures composed of chromatin coated with histones, proteases, and granular and cytosolic proteins. The formation of NETs is complicated and has been reviewed elsewhere (3, 10). NETs bind viruses, bacteria, fungi, and parasites, preventing their spread. They can also trap platelets and erythrocytes to initiate coagulation, and trap tumor cells to promote their spread (11). Thus, inappropriately formed or improperly degraded NETs can become pathogenic and are implicated in various non-infectious diseases such as CLD and cancer.
Neutrophils and Acute Liver Diseases
Neutrophils and Two-Thirds Partial Hepatectomy
The liver has a remarkable regenerative capacity with compensatory re-growth of the liver after liver damage, including physical resection or chemical injury (12, 13). Liver regeneration is a complicated and well-organized process involving multiple genes and signaling pathways that initiate or promote liver regeneration. Most knowledge on liver regeneration comes from the rodent model of two-thirds partial hepatectomy (PHx). In this model, two-thirds of the rodent liver is removed surgically, and mature quiescent hepatocytes of the remnant liver proliferate to restore the original liver mass and function (14). Inflammatory cells such as Kupffer cells (KCs), dendritic cells, and T cells control this process either through direct interactions with hepatocytes or indirectly by releasing inflammatory cytokines (15). However, studies regarding the role of neutrophils in liver regeneration are limited. Neutrophils promote liver regeneration by binding intracellular adhesion molecule (ICAM-1), triggering KC-dependent release of hepatocyte mitogens, interleukin (IL)-6 and tumor necrosis factor (TNF) α (16). This is demonstrated in neutropenic mice, which show delayed liver regeneration and reduced hepatic levels of TNFα and IL-6 (16). Furthermore, significant changes in neutrophil phenotype are observed in patients who undergo PHx. This has been proposed to be important in defense against gut-derived endotoxins following hepatic resection (17, 18).
Neutrophils and Drug/Chemical-Related Liver Injury
Drug/chemical-related liver injury [such as acetaminophen (APAP) and tetracarbon chloride (CCl4)] can result from chemical/drug-induced oxidant stress and tissue injury and/or by the local upregulation of inflammatory mediators (19), and is usually accompanied by a huge infiltration of neutrophils in the liver during the early phase (20). Danger-associated molecular patterns (DAMPs), such as HMGB1 and lipid peroxidation products from dying hepatocytes, and proinflammatory mediators such as IL-1β and TNFα released from KCs can guide neutrophils into damaged tissues, leading to a multistep process that involves ATP release, adhesion molecule upregulation, formation of a chemical gradient (CXCL1, CXCL2), formyl peptide signals, and finally clearance of necrotic debris (21, 22). Neutrophil invasion often aggravates the liver by the secretion of cytotoxic reactive oxygen and nitrogen species or proinflammatory cytokines such as IL-1β and TNF (22). In mouse models of acute and chronic liver injury, TLR2 and the S100A8–S100A9 signaling pathway act as key regulators of hepatic CXCL2 and TNF expression and subsequent neutrophil infiltration (23).
Neutrophils and Liver Ischemia-Reperfusion
Severe liver damage may occur in ischemia-reperfusion (IR) during liver transplantation or surgical liver tumor resection, when the blood supply is restored after a long period of ischemia. This hepatic inflammation is initiated by the ischemic period but occurs mainly during the reperfusion phase and is characterized by a large neutrophil recruitment to the liver (24). In liver IR (LIR), the acute inflammatory response has two consecutive stages: ROS exacerbates liver damage in the first 6 h of reperfusion, while neutrophil recruitment plays a major role in the next 18 h of reperfusion (25). The neutrophil-derived MMP9 can also promote the recruitment of neutrophils to the damaged site (24). The recruited neutrophils partially mediate damage through oxidative stress in a mitogen-activated protein kinase-activating protein kinase 2 (MK2)-dependent manner and the production of MPO (26, 27). And the neutrophils-derived MMP9 can promote recruitment and MPO activation of neutrophils (28), which form a positive loop to exacerbate the IR injury (IRI). COX-2 derived from hepatocytes reduces liver injury by decreasing endoplasmic reticulum stress, neutrophil infiltration, and oxidative stress, while escalating autophagy, and apoptosis (29). Similar to this mechanism, extracellular vesicles derived from human umbilical cord blood mesenchymal stem cells also moderate IRI by downregulating neutrophil respiratory burst and oxidative stress (30).
NE may have the ability to mediate adhesion and extravasation of neutrophils in IRI (31). In fact, the recruited neutrophils induce the production of macrophages monocyte chemoattractant protein-1 (MCP-1) through NE and oxygen-free radicals (32). MCP-1 upregulates the expression of ICAM-1 in endothelial cells and promotes the adhesion of neutrophils and endothelial cell damage (33). In addition to mediating endothelial cell damage and aggravating IRI, NE also aggravates IRI in other ways. In IRI, NE downregulate the expression of prostacyclin, which decreases the expression of downstream insulin-like growth factor 1, which has been reported to inhibit the expression of endothelial monocyte-activating polypeptide-II, a neutrophil chemotactic factor (34). Elevated NE, as the putative ligand of TLR4, causes the upregulation of TLR4 in macrophages and hepatocytes, which induces the inflammatory cascade responses in IR (35). NE inhibitor sivelestat treatment inhibits the infiltration and activation of neutrophils and apoptosis and reduces proinflammatory factors such as TNF-α and IL-6, and downregulates chemokines (36).
Net also mediates the inflammation, thrombotic diseases, cancer, and autoimmune diseases (37). LSECs/IL-33/ST2 axis (38), IL-17A (39), mast cell degranulation (40), and TIMP-1 (41) are the driving force of NET in LIR. NET has cytotoxic effects on hepatocytes in vivo and in vitro, and triggers a KC inflammation response by upregulating the inflammatory factors IL-1β, IL-6, TNF-α, and chemokines CXCL10 and MCP-1 (42). In addition, acrolein produced under chronic stress boosts oxidative burst and NET formation, which induces HepG2 nuclear and mitochondrial damage in IRI (43). Extravasated neutrophils cause hypochlorous acid (HOCl) to diffuse into hepatocytes and contribute to oxidative modification of proteins during the reperfusion phase (44). Neutrophils also damages hepatocytes by releasing proteases, TNF-α, TGF-β and leukotrienes (45). In turn, the histones and HMGB1, acting as DAMPs, from damaged hepatocytes also elicit NET formation by activating neutrophil TLR4- and TLR9-Myd88 signaling in LIR (42). The results indicate that a positive feedback loop is formed between NET formation and hepatocyte apoptosis, which mediates liver toxicity and organ injury. Therefore, targeting the associated mediator of neutrophils may be a useful way to improve the survival of patients after liver tranplantation or surgical liver tumor resection (Table 1). The other functions of neutrophils in liver IRI are shown in Tables 2, 3.
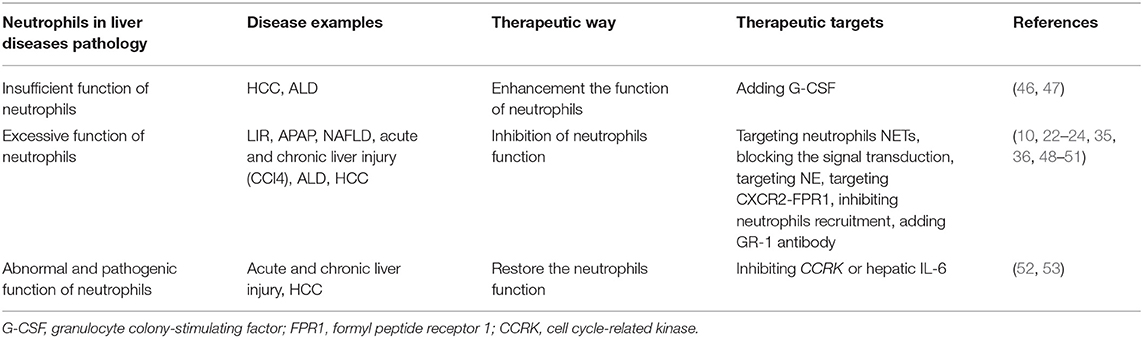
Table 1. Therapeutic targets of neutrophils in liver diseases.
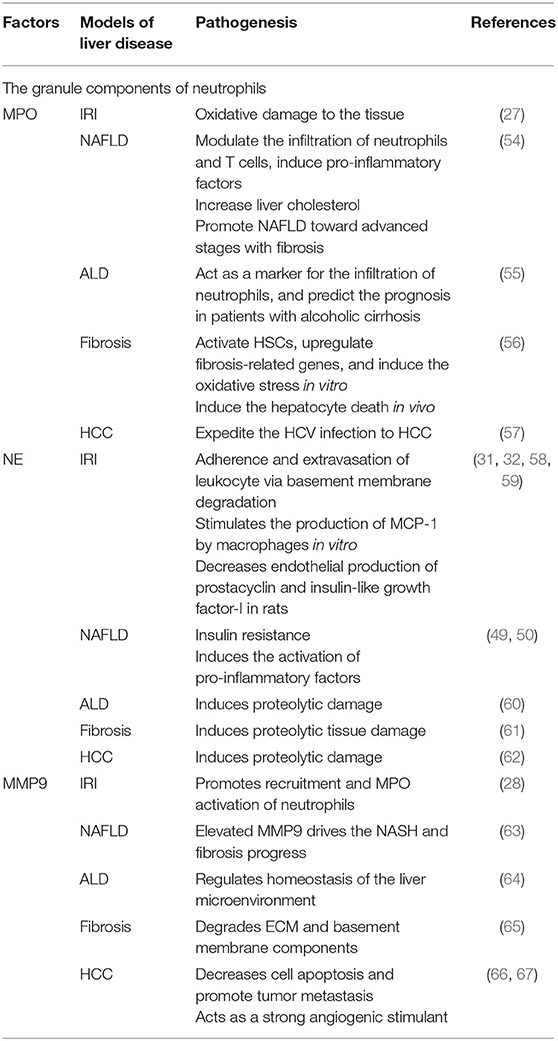
Table 2. The function of neutrophils granule component in liver disease.
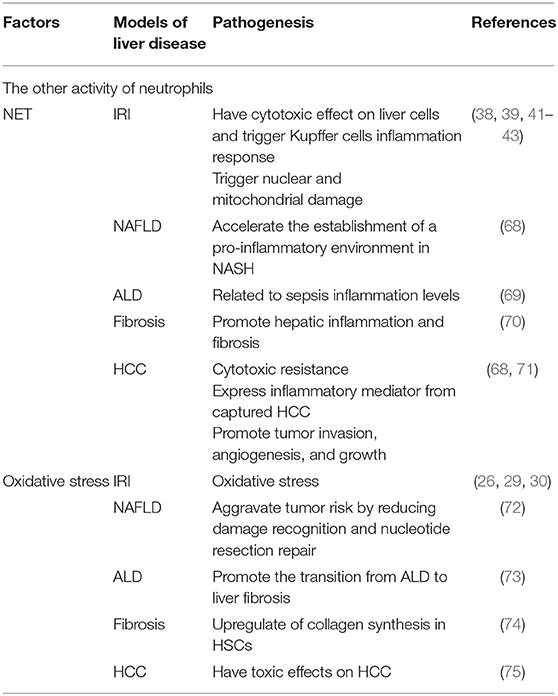
Table 3. The other activity of neutrophils in liver disease.
Neutrophils and Chronic Liver Inflammation
Liver Fibrosis
Liver fibrosis is the main consequence of chronic liver injury of any etiology and may progress to cirrhosis and liver cancer. Activation of hepatic stellate cells (HSCs) that transdifferentiate from vitamin A–storing pericyte-like cells to α-SMA-positive, collagen-producing myofibroblasts is now well-established as the central driver of fibrosis (76, 77).
Infiltration of neutrophils is commonly observed in patients as well as in mice with alcohol or non-alcohol-induced steatohepatitis (78, 79). However, the role of neutrophils during liver fibrogenesis remains controversial. On one hand, increased expression of neutrophil (and mast cells)-derived IL-17 is a common signature of advanced liver fibrosis, which upregulates the expression of TGF-β receptor in HSCs and promotes liver fibrosis, and blocking IL-17/IL-22 alleviates liver fibrosis (80). IL-17A (secreted by Vγ2T or Th17 T cells) also promotes the recruitment of neutrophils into the liver and promotes liver fibrosis induced by Schistosoma japonicum infection (81) or bile duct ligation (BDL) (82). Mechanistically, neutrophils are shown to activate HSCs via the production of ROS and MPO (56, 74, 83). Activated HSCs produce GM-CSF and IL-15 to promote neutrophil survival (83), and cytokine-induced neutrophil chemoattractant to facilitate the recruitment of neutrophils (84), thus creating a positive feedback loop and exacerbating liver fibrosis. Moreover, neutrophils downregulate the butyrate receptor GPR43 and upregulate the secretion of TNF-α and IL-6, thereby promoting intestinal microbial translocation and exacerbating CCl4-induced liver fibrosis (85). Overexpression of HNP-1, a type of α-defensin, promotes the proliferation and activation of HSCs (86). And neutrophils induce proteolytic tissue damage by NE (61).
On the other hand, neutrophils have also been shown to contribute to collagen degradation during the resolution of fibrosis via their expression of MMPs (65). A recent report also demonstrated that neutrophils mediate the resolution of liver inflammation and fibrosis through microRNA (miR)-223 delivery to liver macrophages, favoring macrophage polarization toward a regenerative phenotype (87). Interestingly, another recent study by Yang et al. (88) also identified the beneficial effects of neutrophil-derived ROS on polarizing macrophages toward an alternative or reparative and anti-inflammatory phenotype in an APAP-induced liver injury model. In addition, the injection of autologous bone marrow-derived macrophages in mice during CCl4-mediated liver injury has been shown to lead to the recruitment of neutrophils into the liver, upregulation of MMPs, and anti-fibrotic effects (89).
What makes the thing complicated is that there are some reports showed that neutrophils were dispensable for establishing chronic inflammation and hepatic fibrosis. One report showed that neutrophils have minimal effects on BDL-induced liver fibrogenesis, as there is no significant difference in the production and deposition of collagen in the livers of anti-neutrophil antiserum treated mice or mice with neutrophil dysfunction due to transgenic expression of IL-8 (90). Another report showed that neutrophils are not essential to the hepatotoxin α-naphthylisothiocyanate-induced liver fibrosis, as there was comparable fibrosis between wild type and CXCR2 (the key receptor for neutrophil recruitment) konckout mice (91). While infiltration of neutrophils is a common feature of human liver diseases, defective neutrophil recruitment does not impact chronic liver fibrosis (23).
In conclusion, these data reveal an elaborate role of neutrophils during liver fibrosis, reflecting their adoptive ability to a phenotype tightly regulated by the integration of signals derived from the microenvironment.
Alcoholic Liver Disease
Alcoholic liver disease (ALD) is a spectrum of liver injury, ranging from hepatic steatosis to alcoholic hepatitis and cirrhosis (92), which is caused by excessive alcohol consumption. Chronic hepatocellular injury and death are intimately related to oxidative ethanol metabolism. A large number of neutrophils can be found in the liver of ALD patients, and markers of neutrophils (such as Ly6G, MPO, E-selectin) are upregulated (93). Moreover, neutrophils-derived MPO act as a marker for the infiltration of neutrophils, and predict the prognosis in patients with alcoholic cirrhosis (55). DAMPs, which are released following necrotic cell death, trigger macrophage and neutrophil activation, with senescence (via natural killer cells) and autophagy being the major regulators of liver inflammation (94). Factors that mediate hepatic infiltration of neutrophils include the CXCL1/CXCR2 axis (95), LCN2 (96), IL-33/ST2 (97), osteopontin (98), E-selectin (99), and activated type I natural killer T cells (100). Recruited neutrophils then release H2O2, NE (60), protease 3 (92), and proinflammatory factors (IL-8, TNF-α), or downregulate anti-inflammatory IL-10 to contribute to ALD (101). Therefore, neutrophils are a major contributor to the development of ALD, and targeting them may be a promising therapeutic strategy for ALD. Indeed, the blockade of inflammatory mediators involved in neutrophil infiltration or deletion of neutrophils ameliorates alcoholic liver injury in mouse models of early steatohepatitis (48).
However, excessive alcohol consumption frequently exerts negative effects on neutrophils, including granulopoiesis, and neutrophil release and function (46). Advanced ALD is also accompanied by granulicytopenia (102) and impairment of neutrophil function (103, 104). Infectious complications, including septic infections, occur in ~50% of ALD patients, which are the main cause of death in these individuals (104, 105). Therefore, neutrophil therapy in ALD patients requires special caution. Administration of G-CSF to increase neutrophil counts and improve their function, in adjunction to standard therapy, has been shown to substantially increase the survival of patients with either severe alcoholic hepatitis or alcoholic liver failure (46).
Non-alcoholic Fatty Liver Disease
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease, with a worldwide prevalence of 25% (106). It is an umbrella term that covers a continuum of liver conditions varying in severity of injury and resulting fibrosis: from hepatic steatosis alone (non-alcoholic fatty liver or NAFL) to a more serious condition with inflammation, hepatocyte damage, and pericellular fibrosis [non-alcoholic steatohepatitis [NASH]] (107). The presence of metabolic syndrome in an individual is the strongest risk factor for NAFLD. Its common pathologic drivers are the accumulation of toxic lipid species, which induce hepatocellular stress, injury, and death, leading to fibrogenesis and genomic instability that predispose individuals to cirrhosis and hepatocellular carcinoma (HCC) (107).
NASH is characterized by hepatic neutrophil infiltration (79). The ratio of NE to α1-antitrypsin (108), plasma concentration of PR3 and NE (109), neutrophil-to-lymphocyte ratio (110), serum levels of LCN2 (111), NETs (68), and MPO (112) significantly increase in patients with NAFLD. And elevated MMP9 from the neutrophils drives the NASH and fibrosis progress (63). Factors that mediate hepatic infiltration of neutrophils include activation of Th17 cells expressing IL-17A (113, 114), MPO (54), and gut-microbiome-derived DAMPs (115, 116).
Neutrophils then release a plethora of factors that play important roles in NAFLD. NE is an important regulator of insulin signaling, and depletion of NE results in enhanced insulin sensitivity, attenuated inflammation, and decreased liver damage in high-fat diet-fed mice (49, 50). Neutrophil PR3 also mediates insulin resistance in NAFLD (117). Neutrophil-derived serine proteases, namely NE, PR3, and cathepsin G, are important for the activation of pro-IL-1β/pro-IL-18, which are essential for NASH (118). The combined knockout of Caspase-1 and NE/PR3 genes in mice results in reduced inflammation and liver steatosis (118).
MPO, found in the primary granules of neutrophils, is released into both the phagolysosomes and the extracellular environment upon neutrophil activation. MPO catalyzes the formation of reactive oxygen intermediates, such as HOCl, a potent oxidant that interacts with superoxide anions to induce hepatocyte death (119, 120). HOCl can also damage DNA and inhibit DNA repair, thus leading to HCC development in NAFLD (72). MPO can also activate HSCs and promote fibrogenesis in methionine- and choline- deficient diet-induced NAFLD (56). In addition, MPO triggers the polarization of M2-type macrophages, which express high levels of TGF-β, MMP1, and MMP12, and promote fibrosis in NASH (49).
Neutrophil-derived NETs were found to promote the accumulation of macrophage in the liver, which then establish a favorable inflammatory microenvironment for HCC growth in an experimental NASH model (68). Interestingly, blocking NETs does not affect steatosis and free fatty acid accumulation but inhibits HCC development (68).
Neutrophils and HCC
HCC is a common result of chronic liver disease. Its pathogenesis varies, with the main cause being chronic viral infection or inflammatory environment caused by a large leukocyte infiltration. The multiplying neutrophils in the liver accelerate tumor angiogenesis, epithelial-mesenchymal transition, and growth by producing MMP-9 (66), NET, and hepatocyte growth factor, thereby exacerbating HCC and metastasis (121). Thus, the infiltrating neutrophils may be pro- or anti-tumorigenic depending on the complex tumor niche.
In the tumor microenvironment, under the action of TGF-β, tumor-associated neutrophils (TAN) are divided into N1- and N2-TAN. TGF-β in the tumor microenvironment induces tumor-promoting N2-TAN, and blocks TGF-β-induced tumor-inhibiting N1-TAN (122). N1-TAN inhibits tumor progression through tumor cytotoxicity, inflammation, and immunity response. On one hand, the secreted elastase can promote the degradation of vascular endothelial growth factor A (VEGF-A), basic fibroblast growth factor, and α-defensin and inhibit angiogenesis (123). On the other hand, the secretion of ICAM-1, CXCL10, and TNF-α promotes inflammation and inhibits tumor growth (124). However, N2-TAN is more common in the tumor microenvironment. The greater inflammation caused by infiltrated TAN and tumor-associated macrophage is the primary causative factor for the high morbidity of liver cancer (125). TAN is directly involved in HCC in vitro and vivo. TAN releases TGF-β and bone morphogenetic protein 2, which upregulate miR301b-3p, which in turn is crucial for the formation of caner stem cells in HCC, which are characterized by low levels of E-cadherin and high levels of vimentin and N-cadherin (126). These cancer stem cells form a positive feedback loop with TAN (126). The dual roles of TAN in HCC are summarized in Figure 1. In addition, there are a group of circulating neutrophils, which have been shown to be a poor prognostic factor for overall survival of patients with HCC. They promote the development of HCC through p53 and STAT3 signaling pathways (135).
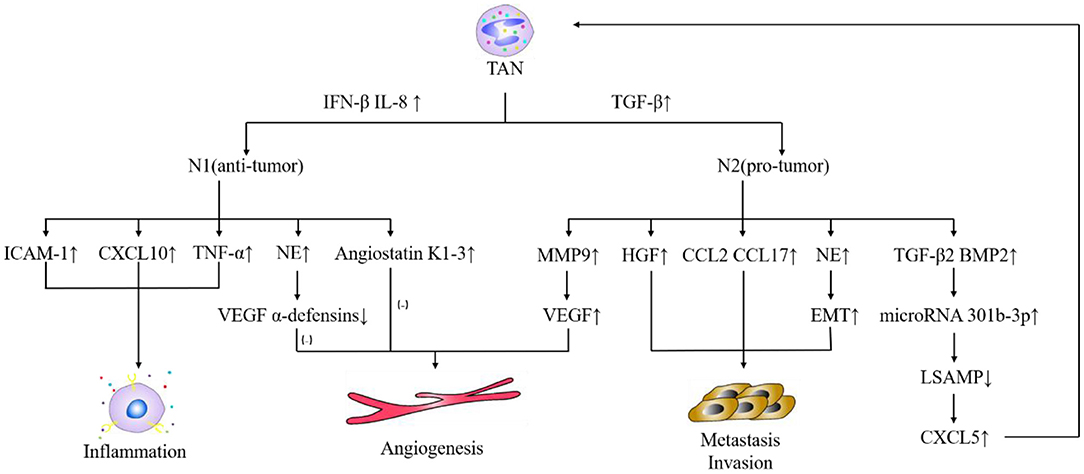
Figure 1. Dual roles of TAN in HCC. IFN-β, interferon-β; IL-8, interleukin-8; TGF-β, transforming growth factor-β; ICAM-1, intercellular cell adhesion molecule-1; CXCL, chemokine (C-X-C motif) ligand; TNF-α, tumor necrosis factor-α; NE, neutrophil elastase; VEGF, vascular endothelial growth factor; MMP9, matrix metalloproteinase 9; HGF, hepatocyte growth factor; CCL, chemokine (C-C motif) ligand; EMT, Epithelial-Mesenchymal Transitions; BMP2, bone morphogenetic protein 2; LSAMP, limbal gene expression membrane protein. TAN plays a critical role in pro-tumor and anti-tumor in HCC. Under the action of TGF-β (127), TAN differentiates into N2 phenotype, which promotes tumor development; while under the condition of IFN-β (128) and IL-8 (129), TAN differentiates into N1 phenotype, which limits tumor progression. N2 secretes MMP9, VEGF (128) to promote tumor angiogenesis; secretes HGF (130), CCL2, CCL17 (131), NE (132) to promote tumor metastasis and infiltration; secretes TGF-β2, BMP2 to promote the production of microRNA 301b-3p, inhibits the production of LSAMP, promotes the production of CXCL5. CXCL5 recruits more TAN (133). N1 secretes ICAM-1, CXCL10, TNF-α (124, 134) to promote inflammation. At this time, NE secreted by N1 will degrade VEGF α-defensins collaborating with Angiostatin K1-3 (123) secreted by N1 to inhibit angiogenesis and inhibit tumor progression.
NET formation from neutrophils is higher in patients with HCC (71), and the high expression of NET supports HCC through cytotoxic resistance and an elevated inflammatory response (68). The interaction between captured HCC and internalization NETs contributes to the acquisition of invasive potential of HCC via TLR4/9-COX2 signaling (71). More importantly, the hypoxic environment within the tumor exacerbates NET formation, which forms a positive feedback loop to aggravate liver injury (136). Although DNase/PAD4−/− mice do not exhibit changes in the progression of fatty liver, inhibiting the formation of NETs can successfully inhibit HCC growth (68). Recently, a clinical retrospective investigation revealed that higher pre-surgery NET levels are associated with shorter relapse-free survival/overall survival in patients with primary liver malignancies (137). This indicates that targeting NETs may be a potential therapeutic strategy against HCC.
Oxidative stress is critical for the development of HCC. Intracellular ROS and glutathione are elevated in neutrophils and others leukocytes in patients with early HCC (138). The neutrophils-derived oxidative stress partially initiates the HCC through MPO, which is expressed in neutrophils and KCs (139). MPO-derived HOCl damages DNA bases and impairs nucleotide excision repair, thus favoring somatic mutations and tumor progression (140). Activated neutrophils can also release cytochrome c via the production of ROS, which exerts anti-tumor effects against several carcinomas (141). The use of zinc oxide nanoparticles to mimic ROS from neutrophils or macrophages shows that ROS has toxic effects on HCC (75). The neutrophil-derived ROS against human HCC can be visualized at the cellular level (142).
Angiogenesis is critical for tumor progression, as blood provides oxygen and nutrients for cancer cells. The accumulation of neutrophils initiates the tumor angiogenic switch by releasing MMP9 in para-carcinoma from human HCC (51). MMP9 is produced by various types of cells, but human neutrophils can produce TIMP-free MMP9, which acts as a strong angiogenic stimulant (67). Elevated neutrophils can also upregulate the expression of VEGF and sinusoidal vasculature in HCC (51). Consistent with the above findings, neutrophils recruited in HCC and its products of IL-6 and IL-8 precipitate a proinflammatory microenvironment, which exacerbates the invasion of HCC in vitro and develops into angiogenesis and tumor metastasis in vivo (143). The inhibition of GR-1 with monoclonal antibodies has been shown to decrease tumor size and microvessel density in HCC-bearing mice (51).
In addition to affecting HCC itself, neutrophils affect the progression of HCC by acting on other immune cells. Cancer-associated fibroblasts in HCC enhance the level of programmed death-ligand 1 (PDL1)+ neutrophils via IL-6/STAT3, which is essential for the survival and functional activation of neutrophils (144). PD-L1+ neutrophils impair the function of T cells via PD-L1/programmed cell death protein 1 signaling (144). Neutrophils enhance the level of myeloid-derived suppressor cells, thereby inhibiting T cell function (145). Meanwhile, neutrophils also inhibit the interferon-γ production by peripheral blood mononuclear cells, which is associated with a low survival rate and high tumor burden (146), and can downregulate the IL-2-receptor-α and ICAM-1 receptor signaling, which in turn mediate cell-mediated immune resistance (147). This is indicated that targeting the neutrophils can improve the HCC by enhancing the activity of other immune cells.
Conclusion
Hepatic infiltration of neutrophils is a common feature of most types of liver diseases. While they mainly function against invading pathogens and remove debris and dead cells, they can also induce and aggravate hepatocyte injury and promote the progression of liver diseases by producing ROS, degranulation, inflammation mediators, and/or shaping immunity. Therefore, neutrophils represent a potential target for liver diseases. Targeting strategies should be disease-specific, either to enhance, inhibit or restore neutrophil function, and some strategies have been in clinical use or in different stages of clinical trials (1, 2). However, it is of note that many models use anti-Ly6G antibodies to deplete neutrophils in situ, and debris of these died neutrophils in the injured tissue may exacerbate immune cell activation and phenotype, thus promoting liver diseases (148). New models are needed to overcome these potential drawbacks. The phenotypic and functional heterogeneity of neutrophils has been recognized in recent years (149); however, heterogeneity of neutrophils is largely unknown in liver diseases. A recent report revealed that neutrophils enter the tissues and acquires remarkable heterogeneity at the chromatin, RNA, and receptor levels (150). Therefore, new techniques such as single-cell sequencing, Assay for Transposase-Accessible Chromatin-sequencing (ATAC-seq), or multispectral imaging may help to thoroughly characterize the heterogeneity of neutrophils during the development of liver diseases, and provide new therapeutic approaches for the treatment of liver diseases.
Author Contributions
JT drafted the manuscript. ZY and QF reviewed the manuscript structure and ideas. LY evaluated and reviewed manuscript structure, ideas, and science. HW conceived the topic and revised the manuscript. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by National Key R&D Program of China (2018YFA0107500), National Natural Science Foundation of China (81472592, 81773077, 81830054, 91859205, and 81988101), Shanghai Municipal Education Commission Project (201901070007E00065), and Shanghai Pujiang Program (18PJD060).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Németh T, Sperandio M, Mócsai A. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov. (2020) 19:253–75. doi: 10.1038/s41573-019-0054-z
2. Soehnlein O, Steffens S, Hidalgo A, Weber C. Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol. (2017) 17:248–61. doi: 10.1038/nri.2017.10
3. Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, et al. Neutrophils: new insights and open questions. Sci Immunol. (2018) 3:eaat4579. doi: 10.1126/sciimmunol.aat4579
4. de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. (2016) 16:378–91. doi: 10.1038/nri.2016.49
5. Dancey JT, Deubelbeiss KA, Harker LA, Finch CA. Neutrophil kinetics in man. J Clin Invest. (1976) 58:705–15. doi: 10.1172/JCI108517
6. Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. (2014) 14:302–14. doi: 10.1038/nri3660
7. Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. (2016) 273:11–28. doi: 10.1111/imr.12440
8. Gullberg U, Andersson E, Garwicz D, Lindmark A, Olsson I. Biosynthesis, processing and sorting of neutrophil proteins: insight into neutrophil granule development. Eur J Haematol. (1997) 58:137–53. doi: 10.1111/j.1600-0609.1997.tb00940.x
9. Borregaard N, Sørensen OE, Theilgaard-Mönch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol. (2007) 28:340–5. doi: 10.1016/j.it.2007.06.002
10. Honda M, Kubes P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol. (2018) 15:206–21. doi: 10.1038/nrgastro.2017.183
11. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. (2020) 583:133–8. doi: 10.1038/s41586-020-2394-6
12. Michalopoulos GK. Liver regeneration. J Cell Physiol. (2007) 213:286–300. doi: 10.1002/jcp.21172
13. Michalopoulos GK. Liver regeneration after partial hepatectomy: critical analysis of mechanistic dilemmas. Am J Pathol. (2010) 176:2–13. doi: 10.2353/ajpath.2010.090675
14. Michalopoulos GK, Bhushan B. Liver regeneration: biological and pathological mechanisms and implications. Nat Rev Gastroenterol Hepatol. (2021) 18:40–55. doi: 10.1038/s41575-020-0342-4
15. Li N, Hua J. Immune cells in liver regeneration. Oncotarget. (2017) 8:3628–39. doi: 10.18632/oncotarget.12275
16. Selzner N, Selzner M, Odermatt B, Tian Y, Van Rooijen N, Clavien PA. ICAM-1 triggers liver regeneration through leukocyte recruitment and Kupffer cell-dependent release of TNF-alpha/IL-6 in mice. Gastroenterology. (2003) 124:692–700. doi: 10.1053/gast.2003.50098
17. Holzer K, Hofmann D, Oppermann E, Zeuzem S, Mönch C, Henrich D, et al. Neutrophil phenotype and function in partial hepatectomy in man. Langenbeck's Arch Surg. (2010) 395:643–53. doi: 10.1007/s00423-009-0557-x
18. Xu CP, Liu J, Liu JC, Han DW, Zhang Y, Zhao YC. Dynamic changes and mechanism of intestinal endotoxemia in partially hepatectomized rats. World J Gastroenterol. (2007) 13:3592–7. doi: 10.3748/wjg.v13.i26.3592
19. Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol. (2007) 35:757–66. doi: 10.1080/01926230701584163
20. Graubardt N, Vugman M, Mouhadeb O, Caliari G, Pasmanik-Chor M, Reuveni D, et al. Ly6C(hi) monocytes and their macrophage descendants regulate neutrophil function and clearance in acetaminophen-induced liver injury. Front Immunol. (2017) 8:626. doi: 10.3389/fimmu.2017.00626
21. Markose D, Kirkland P, Ramachandran P, Henderson NC. Immune cell regulation of liver regeneration and repair. J Immunol Regener Med. (2018) 2:1–10. doi: 10.1016/j.regen.2018.03.003
22. Heymann F, Tacke F. Immunology in the liver — from homeostasis to disease. Nat Rev Gastroenterol Hepatol. (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
23. Moles A, Murphy L, Wilson CL, Chakraborty JB, Fox C, Park EJ, et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol. (2014) 60:782–91. doi: 10.1016/j.jhep.2013.12.005
24. Oliveira THC, Marques PE, Proost P, Teixeira MMM. Neutrophils: a cornerstone of liver ischemia and reperfusion injury. Lab Invest. (2018) 98:51–62. doi: 10.1038/labinvest.2017.90
25. Xu R, Huang H, Zhang Z, Wang FS. The role of neutrophils in the development of liver diseases. Cell Mol Immunol. (2014) 11:224–31. doi: 10.1038/cmi.2014.2
26. Sun L, Wu Q, Nie Y, Cheng N, Wang R, Wang G, et al. A role for MK2 in enhancing neutrophil-derived ROS production and aggravating liver ischemia/reperfusion injury. Front Immunol. (2018) 9:2610. doi: 10.3389/fimmu.2018.02610
27. Shao B, Oda MN, Oram JF, Heinecke JW. Myeloperoxidase: an oxidative pathway for generating dysfunctional high-density lipoprotein. Chemical research in toxicology. (2010) 23:447–54. doi: 10.1021/tx9003775
28. Moore C, Shen XD, Gao F, Busuttil RW, Coito AJ. Fibronectin-alpha4beta1 integrin interactions regulate metalloproteinase-9 expression in steatotic liver ischemia and reperfusion injury. Am J Pathol. (2007) 170:567–77. doi: 10.2353/ajpath.2007.060456
29. Motiño O, Francés DE, Casanova N, Fuertes-Agudo M, Cucarella C, Flores JM, et al. Protective role of hepatocyte cyclooxygenase-2 expression against liver ischemia-reperfusion injury in mice. Hepatology. (2019) 70:650–65. doi: 10.1002/hep.30241
30. Yao J, Zheng J, Cai J, Zeng K, Zhou C, Zhang J, et al. Extracellular vesicles derived from human umbilical cord mesenchymal stem cells alleviate rat hepatic ischemia-reperfusion injury by suppressing oxidative stress and neutrophil inflammatory response. FASEB J. (2019) 33:1695–710. doi: 10.1096/fj.201800131RR
31. Zimmerman BJ, Granger DN. Reperfusion-induced leukocyte infiltration: role of elastase. Am J Physiol. (1990) 259:H390–4. doi: 10.1152/ajpheart.1990.259.2.H390
32. Yamaguchi Y, Matsumura F, Liang J, Okabe K, Ohshiro H, Ishihara K, et al. Neutrophil elastase and oxygen radicals enhance monocyte chemoattractant protein- expression after ischemia/reperfusion in rat liver. Transplantation. (1999) 68:1459–68. doi: 10.1097/00007890-199911270-00005
33. Yamaguchi Y, Matsumura F, Takeya M, Ichiguchi O, Kuratsu JI, Horiuchi T, et al. Monocyte chemoattractant protein-1 enhances expression of intercellular adhesion molecule-1 following ischemia-reperfusion of the liver in rats. Hepatology. (1998) 27:727–34. doi: 10.1002/hep.510270314
34. Zhao J, Harada N, Sobue K, Katsuya H, Okajima K. Insulin-like growth factor-I reduces stress-induced gastric mucosal injury by inhibiting neutrophil activation in mice. Growth Horm IGF Res. (2009) 19:136–45. doi: 10.1016/j.ghir.2008.08.003
35. Uchida Y, Freitas MC, Zhao D, Busuttil RW, Kupiec-Weglinski JW. The inhibition of neutrophil elastase ameliorates mouse liver damage due to ischemia and reperfusion. Liver Transpl. (2009) 15:939–47. doi: 10.1002/lt.21770
36. Uchida Y, Freitas MC, Zhao D, Busuttil RW, Kupiec-Weglinski JW. The protective function of neutrophil elastase inhibitor in liver ischemia/reperfusion injury. Transplantation. (2010) 89:1050–6. doi: 10.1097/TP.0b013e3181d45a98
37. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
38. Yazdani HO, Chen HW, Tohme S, Tai S, van der Windt DJ, Loughran P, et al. IL-33 exacerbates liver sterile inflammation by amplifying neutrophil extracellular trap formation. J Hepatol. doi: 10.1016/j.jhep.2017.09.010. [Epub ahead of print].
39. Tohme S, Yazdani HO, Sud V, Loughran P, Huang H, Zamora R, et al. Computational analysis supports IL-17A as a central driver of neutrophil extracellular trap-mediated injury in liver ischemia reperfusion. J Immunol. (2019) 202:268–77. doi: 10.4049/jimmunol.1800454
40. He Z, Li Y, Ma S, Yang M, Ma Y, Ma C, et al. Degranulation of gastrointestinal mast cells contributes to hepatic ischemia-reperfusion injury in mice. Clin Sci. (2018) 132:2241–59. doi: 10.1042/CS20180662
41. Duarte S, Matian P, Ma S, Busuttil RW, Coito AJ. Adeno-associated virus-mediated gene transfer of tissue inhibitor of metalloproteinases-1 impairs neutrophil extracellular trap formation and ameliorates hepatic ischemia and reperfusion injury. Am J Pathol. (2018) 188:1820–32. doi: 10.1016/j.ajpath.2018.05.002
42. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. (2015) 62:600–14. doi: 10.1002/hep.27841
43. Arumugam S, Girish Subbiah K, Kemparaju K, Thirunavukkarasu C. Neutrophil extracellular traps in acrolein promoted hepatic ischemia reperfusion injury: therapeutic potential of NOX2 and p38MAPK inhibitors. J Cell Physiol. (2018) 233:3244–61. doi: 10.1002/jcp.26167
44. Hasegawa T, Malle E, Farhood A, Jaeschke H. Generation of hypochlorite-modified proteins by neutrophils during ischemia-reperfusion injury in rat liver: attenuation by ischemic preconditioning. Am J Physiol Gastrointest Liver Physiol. (2005) 289:G760–7. doi: 10.1152/ajpgi.00141.2005
45. Helewski KJ, Kowalczyk-Ziomek GI, Konecki J. [Neutrophils' contribution to ischaemia and reperfusion injury in liver]. Wiad Lek. (2007) 60:47–52.
46. Shi X, DeLucia AL, Bao J, Zhang P. Alcohol abuse and disorder of granulopoiesis. Pharmacol Ther. (2019) 198:206–19. doi: 10.1016/j.pharmthera.2019.03.001
47. Himoto T, Watanabe S, Nishioka M, Maeba T, Tanaka S, Saito M. Combination immunotherapy with OK-432, recombinant granulocyte-colony-stimulating factor and recombinant interleukin-2 for human hepatocellular carcinoma. Cancer Immunol Immunother. (1996) 42:127–31. doi: 10.1007/s002620050262
48. Chang B, Xu MJ, Zhou Z, Cai Y, Li M, Wang W, et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: an important role for CXCL1. Hepatology. (2015) 62:1070–85. doi: 10.1002/hep.27921
49. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. (2012) 18:1407–12. doi: 10.1038/nm.2885
50. Zang SF, Ma XJ, Wang L, Zhu GL, Yang WJ, Liu YL, et al. [Sivelestat alleviates nonalcoholic steatohepatitis in mice through inhibiting activation of Kupffer cells]. Zhonghua Gan Zang Bing Za Zhi. (2017) 25:371–6. doi: 10.3760/cma.j.issn.1007-3418.2017.05.012
51. Kuang D-M, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, et al. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol. (2011) 54:948–55. doi: 10.1016/j.jhep.2010.08.041
52. Hammerich L, Tacke F. Emerging roles of myeloid derived suppressor cells in hepatic inflammation and fibrosis. World J Gastrointest Pathophysiol. (2015) 6:43–50. doi: 10.4291/wjgp.v6.i3.43
53. Zhou J, Liu M, Sun H, Feng Y, Xu L, Chan AWH, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut. (2018) 67:931–44. doi: 10.1136/gutjnl-2017-314032
54. Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri-Sverdlov R, et al. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS ONE. (2012) 7:e52411. doi: 10.1371/journal.pone.0052411
55. Zhang Z, Xie G, Liang L, Liu H, Pan J, Cheng H, et al. RIPK3-mediated necroptosis and neutrophil infiltration are associated with poor prognosis in patients with alcoholic cirrhosis. J Immunol Res. (2018) 2018:1509851. doi: 10.1155/2018/1509851
56. Pulli B, Ali M, Iwamoto Y, Zeller MW, Schob S, Linnoila JJ, et al. Myeloperoxidase-hepatocyte-stellate cell cross talk promotes hepatocyte injury and fibrosis in experimental nonalcoholic steatohepatitis. Antioxid Redox Signal. (2015) 23:1255–69. doi: 10.1089/ars.2014.6108
57. Abdel-Hamid M, Nada OH, Ellakwa DES, Ahmed LK. Role of myeloperoxidase in hepatitis C virus related hepatocellular carcinoma. Meta Gene. (2018) 18:1–8. doi: 10.1016/j.mgene.2018.07.008
58. Okajima K, Harada N, Uchiba M, Mori M. Neutrophil elastase contributes to the development of ischemia-reperfusion-induced liver injury by decreasing endothelial production of prostacyclin in rats. Am J Physiol Gastrointest Liver Physiol. (2004) 287:G1116–23. doi: 10.1152/ajpgi.00061.2004
59. Kawai M, Harada N, Takeyama H, Okajima K. Neutrophil elastase contributes to the development of ischemia/reperfusion-induced liver injury by decreasing the production of insulin-like growth factor-I in rats. Transl Res. (2010) 155:294–304. doi: 10.1016/j.trsl.2010.02.003
60. Yoosefian M, Mirhaji E, Moghani MZ, Ebrahimpour Y, Fouladi M. The effect of water/ethanol solvent mixtures on interactions of an antibody selective for wild-type alpha-1-antitrypsin in complex with its antigen. J Mol Liq. (2020) 312:113437. doi: 10.1016/j.molliq.2020.113437
61. Dabbagh K, Laurent GJ, Shock A, Leoni P, Papakrivopoulou J, Chambers RC. Alpha-1-antitrypsin stimulates fibroblast proliferation and procollagen production and activates classical MAP kinase signalling pathways. J Cell Physiol. (2001) 186:73–81. doi: 10.1002/1097-4652(200101)186:1<73::AID-JCP1002>3.0.CO;2-Q
62. Ghouse R, Chu A, Wang Y, Perlmutter DH. Mysteries of α1-antitrypsin deficiency: emerging therapeutic strategies for a challenging disease. Dis Model Mech. (2014) 7:411–9. doi: 10.1242/dmm.014092
63. D'Amico F, Consolo M, Amoroso A, Skarmoutsou E, Mauceri B, Stivala F, et al. Liver immunolocalization and plasma levels of MMP-9 in non-alcoholic steatohepatitis (NASH) and hepatitis C infection. Acta Histochem. (2010) 112:474–81. doi: 10.1016/j.acthis.2009.05.005
64. Chen JM, Tian SX, Xing LJ, Zheng PY, Ji G. Role of matrix metalloproteinases and tissue inhibitor of metalloproteinases type-1 in the development of alcoholic liver fibrosis in rats. World Chin J Digestol. (2013) 21:1595–601. doi: 10.11569/wcjd.v21.i17.1595
65. Harty MW, Muratore CS, Papa EF, Gart MS, Ramm GA, Gregory SH, et al. Neutrophil depletion blocks early collagen degradation in repairing cholestatic rat livers. Am J Pathol. (2010) 176:1271–81. doi: 10.2353/ajpath.2010.090527
66. Li XF, Chen DP, Ouyang FZ, Chen MM, Wu Y, Kuang DM, et al. Increased autophagy sustains the survival and pro-tumourigenic effects of neutrophils in human hepatocellular carcinoma. J Hepatol. (2015) 62:131–9. doi: 10.1016/j.jhep.2014.08.023
67. Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci USA. (2007) 104:20262–7. doi: 10.1073/pnas.0706438104
68. van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology. (2018) 68:1347–60. doi: 10.1002/hep.29914
69. Bukong TN, Cho Y, Iracheta-Vellve A, Saha B, Lowe P, Adejumo A, et al. Abnormal neutrophil traps and impaired efferocytosis contribute to liver injury and sepsis severity after binge alcohol use. J Hepatol. (2018) 69:1145–54. doi: 10.1016/j.jhep.2018.07.005
70. Zhao X, Yang L, Chang N, Hou L, Zhou X, Yang L, et al. Neutrophils undergo switch of apoptosis to NETosis during murine fatty liver injury via S1P receptor 2 signaling. Cell Death Dis. (2020) 11:379. doi: 10.1038/s41419-020-2582-1
71. Yang LY, Luo Q, Lu L, Zhu WW, Sun HT, Wei R, et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J Hematol Oncol. (2020) 13:3. doi: 10.1186/s13045-019-0836-0
72. Schults MA, Nagle PW, Rensen SS, Godschalk RW, Munnia A, Peluso M, et al. Decreased nucleotide excision repair in steatotic livers associates with myeloperoxidase-immunoreactivity. Mutat Res. (2012) 736:75–81. doi: 10.1016/j.mrfmmm.2011.11.001
73. Nieto N, Greenwel P, Friedman SL, Zhang F, Dannenberg AJ, Cederbaum AI. Ethanol and arachidonic acid increase alpha 2(I) collagen expression in rat hepatic stellate cells overexpressing cytochrome P450 2E1. Role of H2O2 and cyclooxygenase-2. J Biol Chem. (2000) 275:20136–45. doi: 10.1074/jbc.M001422200
74. Casini A, Ceni E, Salzano R, Biondi P, Parola M, Galli A, et al. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: role of nitric oxide. Hepatology. (1997) 25:361–7. doi: 10.1002/hep.510250218
75. Kim AR, Ahmed FR, Jung GY, Cho SW, Kim DI, Um SH. Hepatocyte cytotoxicity evaluation with zinc oxide nanoparticles. J Biomed Nanotechnol. (2013) 9:926–9. doi: 10.1166/jbn.2013.1495
76. Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. (2011) 6:425–56. doi: 10.1146/annurev-pathol-011110-130246
77. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
78. Das S, Maras JS, Hussain MS, Sharma S, David P, Sukriti S, et al. Hyperoxidized albumin modulates neutrophils to induce oxidative stress and inflammation in severe alcoholic hepatitis. Hepatology. (2017) 65:631–46. doi: 10.1002/hep.28897
79. Gao B, Tsukamoto H. Inflammation in alcoholic and nonalcoholic fatty liver disease: friend or foe? Gastroenterology. (2016) 150:1704–9. doi: 10.1053/j.gastro.2016.01.025
80. Fabre T, Molina MF, Soucy G, Goulet JP, Willems B, Villeneuve JP, et al. Type 3 cytokines IL-17A and IL-22 drive TGF-beta-dependent liver fibrosis. Sci Immunol. (2018) 3:eaar7754. doi: 10.1126/sciimmunol.aar7754
81. Zheng L, Hu Y, Wang Y, Huang X, Xu Y, Shen Y, et al. Recruitment of neutrophils mediated by vgamma2 gammadelta T cells deteriorates liver fibrosis induced by schistosoma japonicum infection in C57BL/6 mice. Infect Immun. (2017) 85:e01020–16. doi: 10.1128/IAI.01020-16
82. O'Brien KM, Allen KM, Rockwell CE, Towery K, Luyendyk JP, Copple BL. IL-17A synergistically enhances bile acid–induced inflammation during obstructive cholestasis. Am J Pathol. (2013) 183:1498–507. doi: 10.1016/j.ajpath.2013.07.019
83. Zhou Z, Xu MJ, Cai Y, Wang W, Jiang JX, Varga ZV, et al. Neutrophil-hepatic stellate cell interactions promote fibrosis in experimental steatohepatitis. Cell Mol Gastroenterol Hepatol. (2018) 5:399–413. doi: 10.1016/j.jcmgh.2018.01.003
84. Maher JJ, Lozier JS, Scott MK. Rat hepatic stellate cells produce cytokine-induced neutrophil chemoattractant in culture and in vivo. Am J Physiol. (1998) 275:G847–53. doi: 10.1152/ajpgi.1998.275.4.G847
85. Gomez-Hurtado I, Santacruz A, Peiro G, Zapater P, Gutierrez A, Perez-Mateo M, et al. Gut microbiota dysbiosis is associated with inflammation and bacterial translocation in mice with CCl4-induced fibrosis. PLoS ONE. (2011) 6:e23037. doi: 10.1371/journal.pone.0023037
86. Ibusuki R, Uto H, Arima S, Mawatari S, Setoguchi Y, Iwashita Y, et al. Transgenic expression of human neutrophil peptide-1 enhances hepatic fibrosis in mice fed a choline-deficient, L-amino acid-defined diet. Liver Int. (2013) 33:1549–56. doi: 10.1111/liv.12203
87. Calvente CJ, Tameda M, Johnson CD, del Pilar H, Lin YC, Adronikou N, et al. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J Clin Invest. (2019) 129:4091–109. doi: 10.1172/JCI122258
88. Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat Commun. (2019) 10:1076. doi: 10.1038/s41467-019-09046-8
89. Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon-Walker TT, Hartland S, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology. (2011) 53:2003–15. doi: 10.1002/hep.24315
90. Saito JM, Bostick MK, Campe CB, Xu J, Maher JJ. Infiltrating neutrophils in bile duct-ligated livers do not promote hepatic fibrosis. Hepatol Res. (2003) 25:180–91. doi: 10.1016/S1386-6346(02)00247-4
91. Xu J, Lee G, Wang H, Vierling JM, Maher JJ. Limited role for CXC chemokines in the pathogenesis of alpha-naphthylisothiocyanate-induced liver injury. Am J Physiol Gastrointest Liver Physiol. (2004) 287:G734–41. doi: 10.1152/ajpgi.00300.2003
92. Magdaleno F, Blajszczak CC, Nieto N. Key events participating in the pathogenesis of alcoholic liver disease. Biomolecules. (2017) 7:9. doi: 10.3390/biom7010009
93. Ambade A, Satishchandran A, Gyongyosi B, Lowe P, Szabo G. Adult mouse model of early hepatocellular carcinoma promoted by alcoholic liver disease. World J Gastroenterol. (2016) 22:4091–108. doi: 10.3748/wjg.v22.i16.4091
94. Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. (2014) 147:765–83.e4. doi: 10.1053/j.gastro.2014.07.018
95. Roh YS, Zhang B, Loomba R, Seki E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am J Physiol Gastrointest Liver Physiol. (2015) 309:G30–41. doi: 10.1152/ajpgi.00031.2015
96. Wieser V, Tymoszuk P, Adolph TE, Grander C, Grabherr F, Enrich B, et al. Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J Hepatol. (2016) 64:872–80. doi: 10.1016/j.jhep.2015.11.037
97. Artru F, Bou Saleh M, Maggiotto F, Lassailly G, Ningarhari M, Demaret J, et al. IL-33/ST2 pathway regulates neutrophil migration and predicts outcome in patients with severe alcoholic hepatitis. J Hepatol. (2020) 72:1052–61. doi: 10.1016/j.jhep.2019.12.017
98. Banerjee A, Apte UM, Smith R, Ramaiah SK. Higher neutrophil infiltration mediated by osteopontin is a likely contributing factor to the increased susceptibility of females to alcoholic liver disease. J Pathol. (2006) 208:473–85. doi: 10.1002/path.1917
99. Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. (2013) 58:1814–23. doi: 10.1002/hep.26419
100. Maricic I, Sheng H, Marrero I, Seki E, Kisseleva T, Chaturvedi S, et al. Inhibition of type I natural killer T cells by retinoids or following sulfatide-mediated activation of type II natural killer T cells attenuates alcoholic liver disease in mice. Hepatology. (2015) 61:1357–69. doi: 10.1002/hep.27632
101. Taieb J, Mathurin P, Elbim C, Cluzel P, Arce-Vicioso M, Bernard B, et al. Blood neutrophil functions and cytokine release in severe alcoholic hepatitis: effect of corticosteroids. J Hepatol. (2000) 32:579–86. doi: 10.1016/S0168-8278(00)80219-6
102. Jain D, Aggarwal HK, Rao A, Dahiya S, Singla S. Hematological spectrum in patients with alcoholic liver cirrhosis: a model of end-stage liver disease score based approach. Int J Adv Med. (2017) 3:7. doi: 10.18203/2349-3933.ijam20160494
103. Rolas L, Makhezer N, Hadjoudj S, El-Benna J, Djerdjouri B, Elkrief L, et al. Inhibition of mammalian target of rapamycin aggravates the respiratory burst defect of neutrophils from decompensated patients with cirrhosis. Hepatology. (2013) 57:1163–71. doi: 10.1002/hep.26109
104. Karakike E, Moreno C, Gustot T. Infections in severe alcoholic hepatitis. Ann Gastroenterol. (2017) 30:152–60. doi: 10.20524/aog.2016.0101
105. Bruns T, Zimmermann HW, Stallmach A. Risk factors and outcome of bacterial infections in cirrhosis. World J Gastroenterol. (2014) 20:2542–54. doi: 10.3748/wjg.v20.i10.2542
106. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. (2016) 64:73–84. doi: 10.1002/hep.28431
107. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
108. Zang S, Ma X, Zhuang Z, Liu J, Bian D, Xun Y, et al. Increased ratio of neutrophil elastase to α1-antitrypsin is closely associated with liver inflammation in patients with nonalcoholic steatohepatitis. Clin Exp Pharmacol Physiol. (2016) 43:13–21. doi: 10.1111/1440-1681.12499
109. Mirea AM, Toonen EJM, van den Munckhof I, Munsterman ID, Tjwa E, Jaeger M, et al. Increased proteinase 3 and neutrophil elastase plasma concentrations are associated with non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes. Mol Med. (2019) 25:16. doi: 10.1186/s10020-019-0084-3
110. Khoury T, Mari A, Nseir W, Kadah A, Sbeit W, Mahamid M. Neutrophil-to-lymphocyte ratio is independently associated with inflammatory activity and fibrosis grade in nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. (2019) 31:1110–5. doi: 10.1097/MEG.0000000000001393
111. Ye D, Yang K, Zang S, Lin Z, Chau HT, Wang Y, et al. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J Hepatol. (2016) 65:988–97. doi: 10.1016/j.jhep.2016.05.041
112. Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol. (2009) 175:1473–82. doi: 10.2353/ajpath.2009.080999
113. Paquissi FC. Immune imbalances in non-alcoholic fatty liver disease: from general biomarkers and neutrophils to interleukin-17 axis activation and new therapeutic targets. Front Immunol. (2016) 7:490. doi: 10.3389/fimmu.2016.00490
114. Gomes AL, Teijeiro A, Buren S, Tummala KS, Yilmaz M, Waisman A, et al. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell. (2016) 30:161–75. doi: 10.1016/j.ccell.2016.05.020
115. Mridha AR, Haczeyni F, Yeh MM, Haigh WG, Ioannou GN, Barn V, et al. TLR9 is up-regulated in human and murine NASH: pivotal role in inflammatory recruitment and cell survival. Clin Sci. (2017) 131:2145–59. doi: 10.1042/CS20160838
116. Schneider KM, Mohs A, Kilic K, Candels LS, Elfers C, Bennek E, et al. Intestinal microbiota protects against MCD diet-induced steatohepatitis. Int J Mol Sci. (2019) 20:308. doi: 10.3390/ijms20020308
117. Toonen EJ, Mirea AM, Tack CJ, Stienstra R, Ballak DB, van Diepen JA, et al. Activation of proteinase 3 contributes to non-alcoholic fatty liver disease (NAFLD) and insulin resistance. Mol Med. (2016) 22:202–14. doi: 10.2119/molmed.2016.00033
118. Mirea AM, Stienstra R, Kanneganti TD, Tack CJ, Chavakis T, Toonen EJM, et al. Mice deficient in the IL-1β activation genes prtn3, elane, and casp1 are protected against the development of obesity-induced NAFLD. Inflammation. (2020) 43:1054–64. doi: 10.1007/s10753-020-01190-4
119. Aratani Y. Myeloperoxidase: its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys. (2018) 640:47–52. doi: 10.1016/j.abb.2018.01.004
120. Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood. (2011) 117:953–9. doi: 10.1182/blood-2010-06-290171
121. He M, Peng A, Huang XZ, Shi DC, Wang JC, Zhao Q, et al. Peritumoral stromal neutrophils are essential for c-Met-elicited metastasis in human hepatocellular carcinoma. Oncoimmunology. (2016) 5:e1219828. doi: 10.1080/2162402X.2016.1219828
122. Benson DD, Meng X, Fullerton DA, Moore EE, Lee JH, Ao L, et al. Activation state of stromal inflammatory cells in murine metastatic pancreatic adenocarcinoma. Am J Physiol Regul Integr Comp Physiol. (2012) 302:R1067–75. doi: 10.1152/ajpregu.00320.2011
123. Scapini P, Nesi L, Morini M, Tanghetti E, Belleri M, Noonan D, et al. Generation of biologically active angiostatin kringle 1-3 by activated human neutrophils. J Immunol. (2002) 168:5798–804. doi: 10.4049/jimmunol.168.11.5798
124. Ohms M, Möller S, Laskay T. An attempt to polarize human neutrophils toward N1 and N2 phenotypes in vitro. Front Immunol. (2020) 11:532. doi: 10.3389/fimmu.2020.00532
125. Yan C, Yang Q, Gong Z. Tumor-associated neutrophils and macrophages promote gender disparity in hepatocellular carcinoma in zebrafish. Cancer Res. (2017) 77:1395–407. doi: 10.1158/0008-5472.CAN-16-2200
126. Zhou SL, Yin D, Hu ZQ, Luo CB, Zhou ZJ, Xin HY, et al. A positive feedback loop between cancer stem-like cells and tumor-associated neutrophils controls hepatocellular carcinoma progression. Hepatology. (2019) 70:1214–30. doi: 10.1002/hep.30630
127. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
128. Pylaeva E, Lang S, Jablonska J. The essential role of type i interferons in differentiation and activation of tumor-associated neutrophils. Front Immunol. (2016) 7:629. doi: 10.3389/fimmu.2016.00629
129. Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. (2004) 6:447–58. doi: 10.1016/j.ccr.2004.09.028
130. Mizuno R, Kawada K, Itatani Y, Ogawa R, Kiyasu Y, Sakai Y. The role of tumor-associated neutrophils in colorectal cancer. Int J Mol Sci. (2019) 20:529. doi: 10.3390/ijms20030529
131. Xiang DM, Sun W, Ning BF, Zhou TF, Li XF, Zhong W, et al. The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis. Gut. (2018) 67:1704–15. doi: 10.1136/gutjnl-2016-313392
132. Grosse-Steffen T, Giese T, Giese N, Longerich T, Schirmacher P, Hänsch GM, et al. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: the role of neutrophils and neutrophil-derived elastase. Clin Dev Immunol. (2012) 2012:720768. doi: 10.1155/2012/720768
133. Zhao L, Hu H, Gustafsson JÅ, Zhou S. Nuclear receptors in cancer inflammation and immunity. Trends Immunol. (2020) 41:172–85. doi: 10.1016/j.it.2019.12.006
134. Shaul ME, Fridlender ZG. Neutrophils as active regulators of the immune system in the tumor microenvironment. J Leukoc Biol. (2017) 102:343–9. doi: 10.1189/jlb.5MR1216-508R
135. Wang Y, Yao R, Zhang D, Chen R, Ren Z, Zhang L. Circulating neutrophils predict poor survival for HCC and promote HCC progression through p53 and STAT3 signaling pathway. J Cancer. (2020) 11:3736–44. doi: 10.7150/jca.42953
136. Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. (2016) 76:1367–80. doi: 10.1158/0008-5472.CAN-15-1591
137. Kaltenmeier CT, Yazdani H, van der Windt D, Molinari M, Geller D, Tsung A, et al. Neutrophil extracellular traps as a novel biomarker to predict recurrence-free and overall survival in patients with primary hepatic malignancies. HPB. (2020) 23:309–20. doi: 10.1016/j.hpb.2020.06.012
138. Lluch P, Segarra G, Tosca J, Navarro L, Navarrete-Navarro J, Herrera G, et al. Oxidative and nitrosative pattern in circulating leukocytes of very early/early hepatocellular carcinoma patients. Anticancer Res. (2020) 40:6853–61. doi: 10.21873/anticanres.14707
139. Nahon P, Sutton A, Rufat P, Ziol M, Akouche H, Laguillier C, et al. Myeloperoxidase and superoxide dismutase 2 polymorphisms comodulate the risk of hepatocellular carcinoma and death in alcoholic cirrhosis. Hepatology. (2009) 50:1484–93. doi: 10.1002/hep.23187
140. Weitzman SA, Weitberg AB, Clark EP, Stossel TP. Phagocytes as carcinogens: malignant transformation produced by human neutrophils. Science. (1985) 227:1231–3. doi: 10.1126/science.3975611
141. Dissemond J, Weimann TK, Schneider LA, Schneeberger A, Scharffetter-Kochanek K, Goos M, et al. Activated neutrophils exert antitumor activity against human melanoma cells: reactive oxygen species-induced mechanisms and their modulation by granulocyte-macrophage-colony-stimulating factor. J Invest Dermatol. (2003) 121:936–8. doi: 10.1046/j.1523-1747.2003.12475.x
142. Saito H, Fukumura D, Kurose I, Suematsu M, Tada S, Kagawa T, et al. Visualization of oxidative processes at the cellular level during neutrophil-mediated cytotoxicity against a human hepatoma cell line, HCC-M. Int J Cancer. (1992) 51:124–9. doi: 10.1002/ijc.2910510122
143. Gao Q, Zhao YJ, Wang XY, Qiu SJ, Shi YH, Sun J, et al. CXCR6 upregulation contributes to a proinflammatory tumor microenvironment that drives metastasis and poor patient outcomes in hepatocellular carcinoma. Cancer Res. (2012) 72:3546–56. doi: 10.1158/0008-5472.CAN-11-4032
144. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. (2018) 9:422. doi: 10.1038/s41419-018-0458-4
145. Li X, Xing YF, Lei AH, Xiao Q, Lin ZH, Hong YF, et al. Neutrophil count is associated with myeloid derived suppressor cell level and presents prognostic value of for hepatocellular carcinoma patients. Oncotarget. (2017) 8:24380–8. doi: 10.18632/oncotarget.15456
146. Tsuda Y, Fukui H, Asai A, Fukunishi S, Miyaji K, Fujiwara S, et al. An immunosuppressive subtype of neutrophils identified in patients with hepatocellular carcinoma. J Clin Biochem Nutr. (2012) 51:204–12. doi: 10.3164/jcbn.12-32
147. Thieringer FR, Maass T, Anthon B, Meyer E, Schirmacher P, Longerich T, et al. Liver-specific overexpression of matrix metalloproteinase 9 (MMP-9) in transgenic mice accelerates development of hepatocellular carcinoma. Mol Carcinog. (2012) 51:439–48. doi: 10.1002/mc.20809
148. Guillot A, Tacke F. The unexpected role of neutrophils for resolving liver inflammation by transmitting microRNA-223 to macrophages. Hepatology. (2020) 71:749–51. doi: 10.1002/hep.30925
149. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. (2019) 19:255–65. doi: 10.1038/s41577-019-0141-8
Keywords: neutrophils, liver regeneration, alcoholic liver disease, non-aloholic liver disease, hepatocellular carcinoma
Citation: Tang J, Yan Z, Feng Q, Yu L and Wang H (2021) The Roles of Neutrophils in the Pathogenesis of Liver Diseases. Front. Immunol. 12:625472. doi: 10.3389/fimmu.2021.625472
Received: 03 November 2020; Accepted: 25 January 2021;
Published: 08 March 2021.
Edited by:
Markus H. Hoffmann, University of Erlangen Nuremberg, GermanyReviewed by:
Irina Miralda, Seattle Children's Research Institute, United StatesYi Zhao, Sichuan University, China
Copyright © 2021 Tang, Yan, Feng, Yu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyang Wang, aHl3YW5na0B2aXAuc2luYS5jb20=; Lexing Yu, eXVseDAwMUAxNjMuY29t