 Sharon DB de Morais
Sharon DB de Morais Gunjan Kak1†
Gunjan Kak1† Joseph P. Menousek
Joseph P. Menousek Tammy Kielian
Tammy Kielian
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 February 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.625467
This article is part of the Research Topic Immune Response to Biofilms View all 10 articles
Bacterial infections in the central nervous system (CNS) can be life threatening and often impair neurological function. Biofilm infection is a complication following craniotomy, a neurosurgical procedure that involves the removal and replacement of a skull fragment (bone flap) to access the brain for surgical intervention. The incidence of infection following craniotomy ranges from 1% to 3% with approximately half caused by Staphylococcus aureus (S. aureus). These infections present a significant therapeutic challenge due to the antibiotic tolerance of biofilm and unique immune properties of the CNS. Previous studies have revealed a critical role for innate immune responses during S. aureus craniotomy infection. Experiments using knockout mouse models have highlighted the importance of the pattern recognition receptor Toll-like receptor 2 (TLR2) and its adaptor protein MyD88 for preventing S. aureus outgrowth during craniotomy biofilm infection. However, neither molecule affected bacterial burden in a mouse model of S. aureus brain abscess highlighting the distinctions between immune regulation of biofilm vs. planktonic infection in the CNS. Furthermore, the immune responses elicited during S. aureus craniotomy infection are distinct from biofilm infection in the periphery, emphasizing the critical role for niche-specific factors in dictating S. aureus biofilm-leukocyte crosstalk. In this review, we discuss the current knowledge concerning innate immunity to S. aureus craniotomy biofilm infection, compare this to S. aureus biofilm infection in the periphery, and discuss the importance of anatomical location in dictating how biofilm influences inflammatory responses and its impact on bacterial clearance.
Craniotomy and decompressive craniectomy are neurosurgical procedures where part of the skull (i.e. bone flap) is removed to access the brain (Figure 1). Craniotomy involves the temporary removal of the bone flap for procedures that include tumor resection, localization and resection of epileptogenic foci, and aneurysm clipping, where the bone is replaced intraoperatively (1). Decompressive craniectomy refers to the excision of the bone flap for an extended period following traumatic brain injury, ischemic stroke, or intracranial hemorrhage to treat intracranial hypertension (2). Upon removal, the bone flap is typically cryopreserved or implanted subcutaneously in the abdomen of the patient to preserve vascularization and replaced after cerebral edema has resolved (3, 4). However, complications can occur with prolonged absence of the bone flap including extracranial herniation, trephine syndrome, hydrocephalus, seizures, and neurological dysfunction (5, 6).
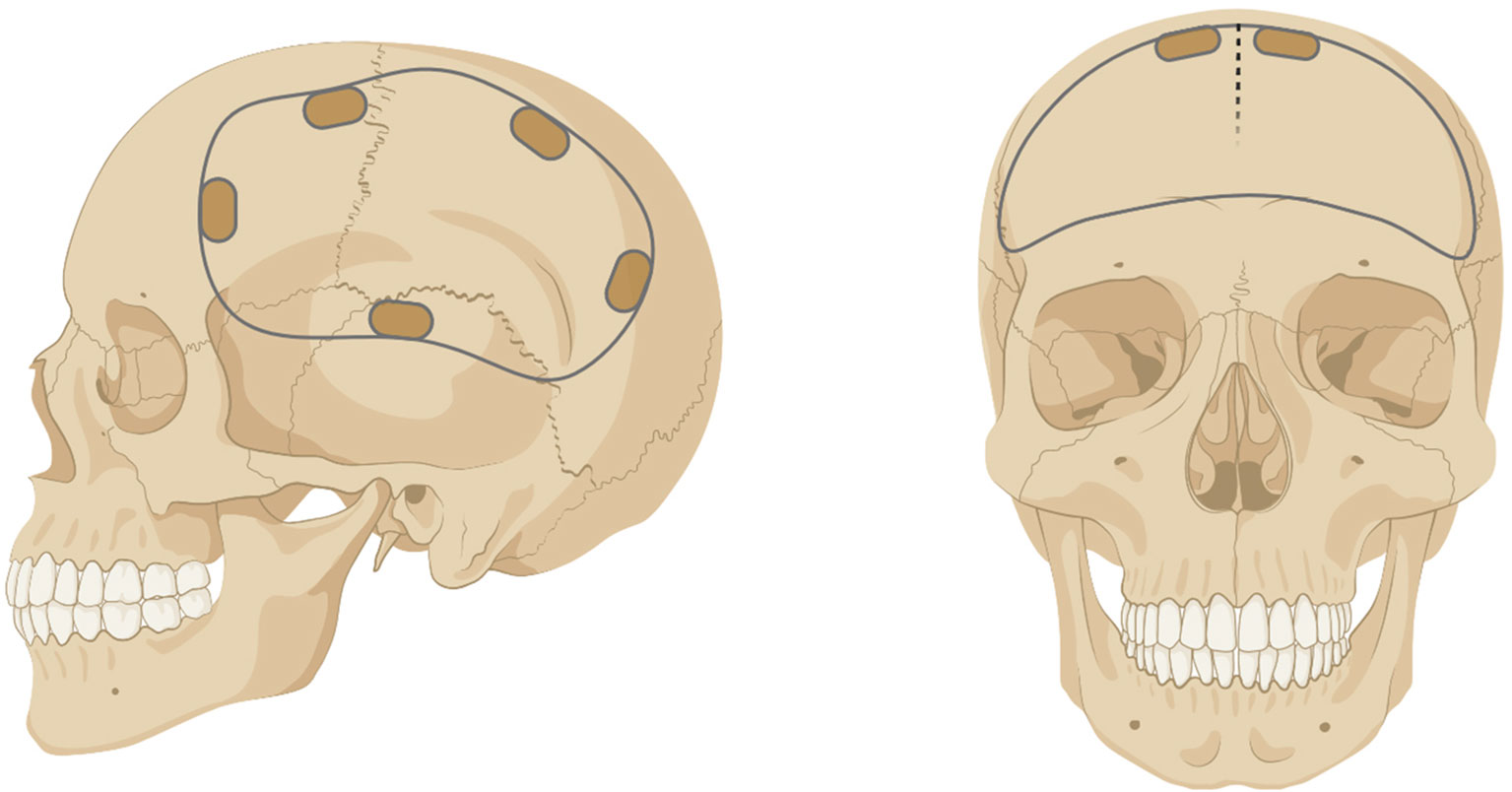
Figure 1 Example of a unilateral (left) or bilateral (right) craniotomy/craniectomy. Figure created with BioRender.
Despite peri- and post-operative prophylaxis, infectious complications occur in approximately 1% to 3% of craniotomy and craniectomy procedures (7, 8). These infections are associated with a high mortality rate and poor prognosis if not treated early (9–11). In terms of therapy, the decision of whether to salvage or discard the bone flap is left to the neurosurgeon and is often dictated by the length of time from the initial surgery to the presentation of clinical signs of infection. The first option is to salvage the infected bone flap with a combination of debridement and long-term antibiotic therapy. Alternatively, the bone flap can be discarded and, following extended antibiotic treatment, a cranioplasty is performed to correct the acquired skull defect with an autologous bone graft or prosthesis (12–15). Prior craniotomy infection increases the risk for re-infection, which may result from the outgrowth of residual bacteria that were not eliminated due to biofilm formation (see below).
Approximately one half of craniotomy/craniectomy infections are attributed to S. aureus (7, 16–18), a gram-positive pathogen that forms a biofilm on native bone (19). Infections can be caused by other bacteria and fungi, although these occur at a much lower rate (8, 17). Around 30% of the human population is colonized with S. aureus, typically in the nares and skin, and colonized individuals have an increased risk for invasive S. aureus infection (20). Although pre-surgical screening for S. aureus carrier status is routinely performed to decolonize carriers prior to orthopedic surgery (21), this approach has not been universally adopted in neurosurgery for patients that require a craniotomy/craniectomy.
S. aureus is a versatile pathogen, which is attributable to several features. First, the organism is prone to genetic adaptation, particularly the ability to acquire antibiotic resistance. An example is the mecA cassette that affords S. aureus resistance to the entire class of methicillin antibiotics (22). Second, S. aureus expresses an extensive repertoire of virulence factors that promote its pathogenesis and interfere with host immune recognition and bacterial clearance. These include cell surface attachment factors, capsular polysaccharides, enzymes, pore-forming toxins, superantigens, and numerous immune modulatory molecules (23–28). In addition to encoding a myriad of virulence factors, S. aureus can form biofilm that represents another virulence determinant (29). Biofilms are complex microbial communities surrounded by a matrix composed of extracellular DNA (eDNA), protein, and polysaccharide (29). The organization of bacteria within a biofilm creates microdomains with differential access to oxygen and nutrients leading to a sub-population of organisms that are less metabolically active, referred to as persisters (30, 31). Because most antibiotics target bacterial cell wall and protein synthesis, the metabolic dormancy of some biofilm-associated bacteria is responsible for the well-known antibiotic tolerance of biofilm. Compared to planktonic bacteria, the mechanisms responsible for S. aureus biofilm to evade immune-mediated clearance are only beginning to be understood. Work from our group and others has shown that S. aureus biofilm evades Toll-like receptor (TLR)-mediated recognition (32, 33), inhibits phagocytosis (33–37), and induces the recruitment of granulocytic-myeloid-derived suppressor cells (G-MDSCs) that inhibit monocyte/macrophage proinflammatory activity (38–40). Recent work has demonstrated that S. aureus metabolites (D- and L-lactate) play a key role in inducing epigenetic changes in G-MDSCs and macrophages to promote the production of the anti-inflammatory cytokine IL-10 and biofilm persistence (41).
Novel therapeutic approaches for S. aureus infection continued to be explored, since most antibiotics have poor efficacy against biofilm and a fine balance must be achieved with currently available antibiotics to reach an optimal minimum inhibitory concentration (MIC) during chronic administration while minimizing toxicity (31, 42). An effective vaccine against S. aureus has remained elusive (43, 44). This is likely explained by the fact that the organism can cause a wide range of infections with distinct attributes, and that it expresses numerous virulence factors that impair host immunity. The latter point has recently been shown to play an important role in a mouse model of S. aureus bacteremia where immunization with S. aureus toxoids reduced mortality, bacterial burden, and organ dysfunction (45). A better understanding of S. aureus colonization dynamics, how the organism interacts with different leukocyte populations, and influences of the local tissue milieu will be necessary to develop improved therapeutics for infections caused by S. aureus.
As mentioned above, S. aureus is a major cause of infectious complications following craniotomy (7, 9, 10); therefore, our laboratory developed a mouse model of S. aureus craniotomy infection to understand the immune mechanisms responsible for bacterial persistence (19). In the mouse model, a craniotomy is performed and the bone flap is colonized with S. aureus, which leads to biofilm formation on the bone and chronic infection in both the brain and subcutaneous galea that cannot be cleared with systemic antibiotics (46). Importantly, the mouse model shares several features with human craniotomy infection. This includes a conserved biofilm structure on the bone flap as revealed by scanning electron microscopy with similarities in extracellular matrix deposition, foci of bacterial aggregates on the bone flap, and complex tower-like structures (19). In addition, magnetic resonance imaging (MRI) revealed galeal inflammation with superficial cortical brain involvement (19), which is also an attribute of human infection and supports the translational relevance of the mouse model.
The CNS was once considered immune privileged based on the restrictive attributes of the blood brain barrier (BBB) (47–49). However, it is now clear that immune responses do occur in the CNS in a wide range of neurodegenerative and infectious diseases, and immune surveillance of the CNS takes place in the absence of pathology (50–52). Over the past decade, our laboratory has characterized the immune responses to S. aureus biofilm infection in both the CNS (craniotomy-associated infection) and periphery (prosthetic joint infection (PJI)) (19, 40, 41, 46, 53–56). Comparisons between these models clearly show that the immune responses elicited are distinct, which will be discussed later in this review, reflecting influences of the local tissue milieu. The impact of infection site and how this shapes the subsequent immune response has also been reported by other groups and emphasizes the need to understand niche-specific factors that influence S. aureus-immune crosstalk (57–61).
Even within a given infection, compartmentalization of immune responses can be observed. An example is the S. aureus craniotomy model where patterns of leukocyte recruitment and inflammatory mediator expression are distinct in the brain vs. subcutaneous galea despite both tissues being exposed to bacteria on the bone flap. For example, monocytes, innate lymphoid cells (NK and ɣδ T cells), and T cells are preferentially recruited to the brain, whereas G-MDSCs and neutrophils (PMNs) are the main leukocyte infiltrates in the galea (Figure 2) (19, 56, 62). The attributes of these cell types and their role during S. aureus infection will be described in more detail below. Likewise, the expression of chemokines, such as CCL2 (monocyte chemoattractant protein-1; MCP-1) and CXCL10 (interferon-inducible protein 10 kDa; IP-10) are higher in the brain (19, 56), which coincides with the enhanced recruitment of monocytes and lymphocyte populations. In the galea, chemokines responsible for PMN and G-MDSCs influx (CXCL2; macrophage inflammatory protein-2; MIP-2) are generally enriched (19, 56) in agreement with the preferential recruitment of these populations to this compartment.
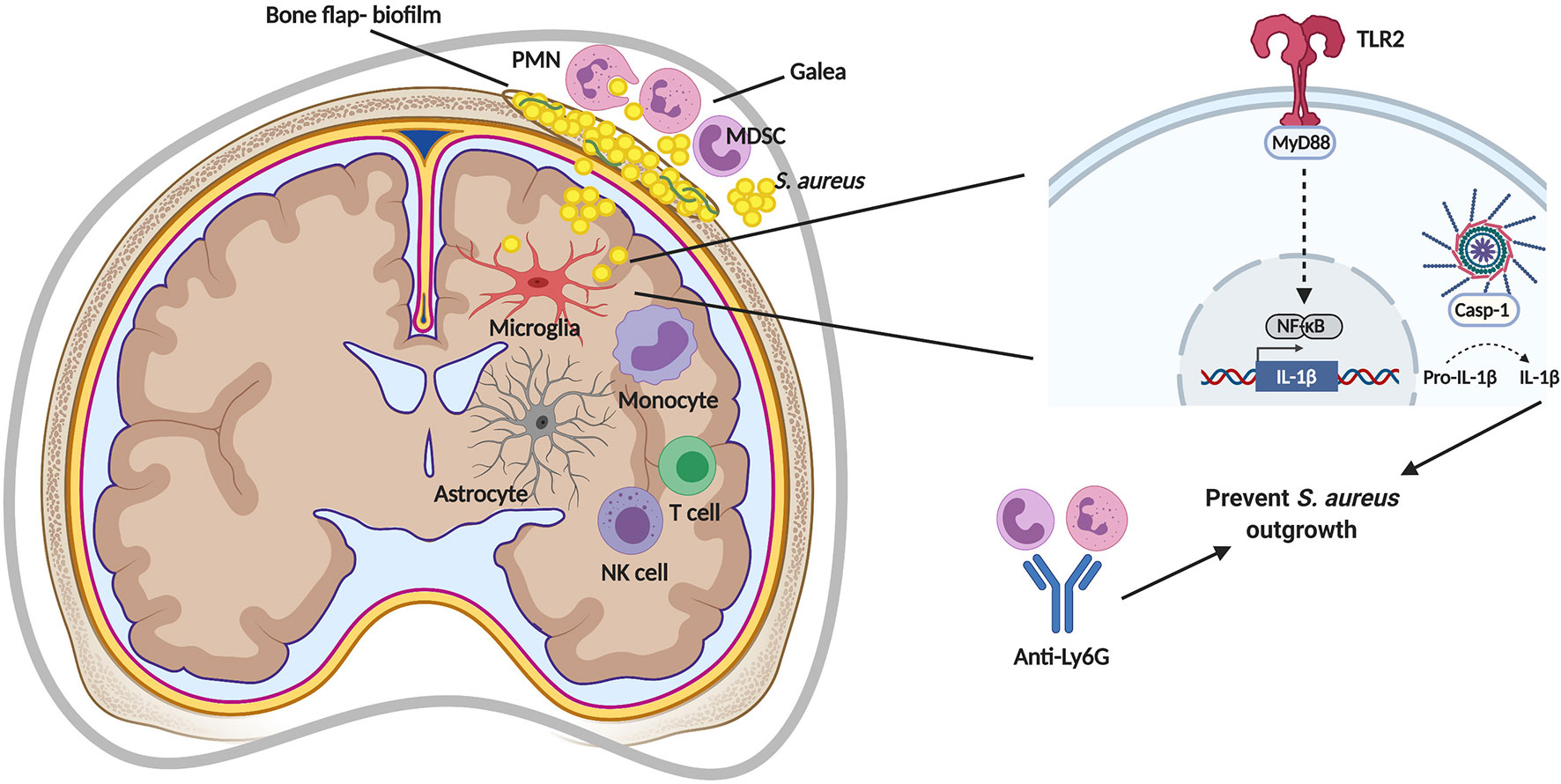
Figure 2 Immune responses during S. aureus craniotomy infection. S. aureus biofilm formation of the bone flap elicits a unique inflammatory response in the subcutaneous galea and brain. TLR2-mediated signaling via MyD88 induces pro-IL-1β production that is cleaved by a caspase-1 (casp-1)-containing inflammasome for secretion and to prevent S. aureus outgrowth. S. aureus containment is also mediated by neutrophils (PMNs), as shown by depletion using anti-Ly6G. MDSC, myeloid-derived suppressor cell; NF-κB, nuclear factor-kappa B; NK, natural killer. Figure created with BioRender.
Interactions between the immune system and CNS are not only important for controlling infection, but also for maintaining homeostatic functions including neurogenesis, behavior, and neuronal activity (63–66). Therefore, a delicate balance must be achieved to elicit sufficient inflammatory responses to clear infection without becoming overactive, which can lead to collateral tissue damage. Indeed, many bacterial infections in the CNS, including those caused by S. aureus, result in areas of tissue necrosis that vary according to infection severity. CNS biofilm infection represents an interesting dilemma since the chronicity of these infections is not characteristic of an overactive immune response, but instead one that is non-productive or anti-inflammatory. In this instance, CNS pathology may be mediated by products released from the biofilm, such as bacterial proteases, nucleases, or via the consumption of metabolites that are critical for CNS function (i.e. glucose). In the following section, we present an overview of the different immune populations associated with S. aureus craniotomy infection and their functional attributes.
Microglia comprise approximately 10% of brain parenchymal cells, which undergo slow proliferation throughout the lifespan of an organism to maintain their numbers (67, 68). Historically, there was much debate about the origin of microglia, where earlier dogma considered microglia to be bone marrow-derived; however, this has now been definitively disproved (69, 70). It is now well established that microglia arise from erythromyeloid precursors in the primitive yolk sac that migrate to the brain where they differentiate into microglia (71, 72). Microglia continuously survey the CNS parenchyma by expanding and retracting their processes to monitor the extracellular milieu, surrounding neurons and other glial cells, as well as to detect invading pathogens and CNS damage (73–75). Microglia play a key role in the phagocytosis of microbes, apoptotic cells, and protein aggregates, and produce a wide array of inflammatory mediators based on their diverse repertoire of pattern-recognition receptor (PRR) expression. Pertinent to S. aureus, microglia express 1) TLR2 and TLR9 that recognize bacterial lipoproteins and non-methylated CpG DNA motifs, respectively; 2) nucleotide-binding oligomerization domain-containing protein 2 (NOD2), an intracellular PRR that senses muramyl dipeptide, a component of peptidoglycan that is abundant in the cell wall of S. aureus and other gram-positive bacteria; and 3) CD14 (76–79). Following PRR activation, microglia produce a wide array of proinflammatory cytokines and chemokines (i.e. TNF-α, IL-6, IL-1β, IL-12, and CCL2) as well as reactive oxygen and nitrogen species (ROS, RNS). These mediators have pleotropic effects including promoting BBB permeability (TNF-α, IL-6, IL-1β), leukocyte recruitment (CCL2) and activation (TNF-α, IL-6, IL-1β, IL-12) and bactericidal activity (ROS/RNS), but they can also negatively impact neuronal function and survival if not tightly regulated (80). Therefore, the induction of anti-inflammatory mechanisms are critical to resolve inflammation and promote tissue repair, which are largely mediated by cytokines such as IL-10 and transforming growth factor-beta (TGF-β) (81–83). The chronicity of S. aureus craniotomy infection suggests a potential imbalance towards an anti-inflammatory state. This is supported by the presence of immune suppressive G-MDSCs as well as PMN and monocyte infiltrates that also possess anti-inflammatory activity as reflected by their ability to inhibit T cell activation (62).
Although as myeloid cells microglia and bone marrow-derived macrophages have distinct origins, they share many attributes including similarities in marker expression, cytokine production, and dependence on macrophage colony-stimulating factor-1 (CSF-1) for survival and proliferation (84–87). During CNS inflammatory conditions, it is not possible to discriminate between microglia and infiltrating macrophages in histological sections since activated microglia transform to an amoeboid morphology that is indistinguishable from macrophages. However, microglia can be discerned from infiltrating monocytes and macrophages by flow cytometry based on CD45 expression (macrophages are CD45high whereas microglia are CD45low/intermediate) (88). Furthermore, advances in next generation sequencing (NGS) and scRNA-seq have identified unique transcriptional profiles of resident microglia versus infiltrating macrophages (89–92), which has led to the identification of markers that are either uniquely (Tmem119, P2YR12, Hexb) or more highly expressed (CX3CR1) in microglia compared to macrophages to aid in their discrimination. Emerging studies from our laboratory have established the transcriptional heterogeneity of resident microglia and macrophage infiltrates in the brain during S. aureus craniotomy infection with the goal of identifying unique markers that will enable the purification of distinct microglial/macrophage clusters to understand their functional role and whether this shapes the chronicity of CNS biofilm infection (62).
Monocytes are bone marrow-derived and invade the inflamed CNS in response to injury or infection primarily via a CCR2-dependent pathway (93–95). Studies have implicated monocytes in the pathogenesis of several neurological diseases, including experimental autoimmune encephalomyelitis (EAE) an animal model of multiple sclerosis (MS), where preventing monocyte recruitment or monocyte depletion reduced disease severity (96, 97). To date, fewer studies have examined monocyte responses to S. aureus. TLR2 has been shown to regulate S. aureus intracellular survival in monocytes via a type I IFN pathway and induce IL-10 production to limit T cell responses (98, 99). Future studies are needed to assess the role of monocytes during craniotomy infection since they represent the predominant leukocyte infiltrate in the brain following S. aureus invasion (56).
Upon migrating into tissues, bone marrow-derived monocytes differentiate into macrophages. Macrophages are professional phagocytes that, along with microglia, play an important role in eliminating debris and apoptotic cells during inflammation in the brain parenchyma, which is critical for maintaining CNS homeostasis (100). There are three resident macrophage populations associated with the CNS, namely perivascular, meningeal, and choroid plexus macrophages. Perivascular and meningeal macrophages are derived from yolk sac progenitors, whereas choroid plexus macrophages originate from both yolk sac progenitors and the bone marrow (71). Each macrophage population possesses unique phenotypes with different capacities for self-renewal (71, 101), which is likely influenced by the local tissue microenvironment.
Macrophages are critical effector cells during infection, with planktonic S. aureus inducing robust proinflammatory cytokine and ROS/RNS production and bactericidal activity (102, 103). However, S. aureus expresses a number of virulence determinants to counteract macrophage effector mechanisms. This includes the production of molecules that interfere with TLR2-dependent recognition (104), such as lipase (Geh) (105), staphylococcal superantigen-like protein 3 (SSL3) (106), and molecular mimicry via blocking the Toll-interacting receptor (Tir) domain of TLR2 (107, 108). In addition, the paired-immunoglobulin-like receptor (PIR)-B contains an inhibitory immunoreceptor tyrosine-based inhibition motif (ITIM) that, upon binding S. aureus lipoteichoic acid, dampens proinflammatory cytokine production (109, 110). Biofilm formation by S. aureus also represents another virulence determinant to escape macrophage effector functions. S. aureus biofilm evades TLR2-mediated recognition, and macrophage invasion into biofilm is limited in vitro and in vivo. This biases cells towards an anti-inflammatory profile that prevents bacterial clearance (33). Macrophages are not capable of phagocytosing S. aureus biofilm (33), which leads to frustrated phagocytosis and cell death that is mediated, in part, through the action of toxins (α-toxin and leukocidin AB) (37). The adoptive transfer of proinflammatory macrophages in a mouse model of S. aureus catheter-associated infection was shown to transform the biofilm milieu into a proinflammatory state concomitant with reduced arginase-1 (Arg-1) expression, which decreased biofilm burden in a MyD88-dependent manner (34). Metabolic reprogramming of monocytes/macrophages to promote their proinflammatory activity was also capable of reducing biofilm burden in a mouse model of PJI (55) highlighting the importance of augmenting macrophage proinflammatory activity as a novel approach to target chronic biofilm infection. The impact of monocytes and macrophages in the pathogenesis of S. aureus craniotomy infection and how their metabolic status influences their inflammatory properties remains to be determined and represents an area of investigation in our laboratory.
PMNs are bone marrow-derived and are released into the circulation at a rate of 109/kg body weight per day (111). PMNs are the first leukocytes recruited to sites of bacterial infection by several chemoattractants including IL-8 (functional mouse homologs are CXCL1 and CXCL2), complement split products (C3a and C5a), and formylated peptides released from bacteria (f-Met-Leu-Phe). Upon extravasation, PMNs exert potent bactericidal activity through the action of antimicrobial peptides and granule enzymes, ROS production, neutrophil extracellular traps (NETs), and phagocytic activity (112, 113).
S. aureus encodes an extensive repertoire of virulence factors to escape PMN killing. Molecules such as chemotaxis inhibitory protein of Staphylococcus aureus (CHIPS), formyl peptide receptor-like 1 (FPRL1), staphopain A, and staphylococcal superantigen-like proteins (SSLs) disrupt various aspects of PMN priming, activation, chemotaxis, and adhesion (23, 114, 115). Moreover, S. aureus secretes proteins that target complement and opsonophagocytosis (protein A), antioxidants that neutralize ROS (catalase, superoxide dismutase), and numerous toxins with pore-forming properties (phenol soluble modulins, leukocidins, α-toxin) all of which function to diminish PMN antibacterial activity (116–123). Individuals with mutations in NADPH oxidase (chronic granulomatous disease; CGD) are highly susceptible to severe and life-threatening S. aureus infections highlighting the critical role of PMNs in bacterial containment (124). Although PMNs are recognized for their beneficial roles during injury or infection (125), dysregulated activity has been implicated in tissue pathology originating from bystander damage via products released from activated PMNs (126–128).
The majority of PMNs during S. aureus craniotomy infection localize to the galea and bone flap, whereas PMN infiltrates are minimal in the brain. These patterns are similar to the profiles of G-MDSCs recruitment (56). Although earlier work in the S. aureus craniotomy infection model suggested an important role for PMNs in preventing bacterial outgrowth, this was with an anti-Gr-1 depletion strategy (19). This approach also targets Ly6C+ monocytes, since the Gr-1 antibody recognizes both Ly6G and Ly6C (129). The functional importance of PMNs during S. aureus craniotomy infection was recently demonstrated by our laboratory using a more selective targeting approach (i.e. Ly6G depletion). PMNs were critical for bacterial containment, although the chronicity of craniotomy infection indicates that PMNs are not capable of eliminating biofilm in the wild type setting (62).
Under physiological conditions, immature myeloid cells undergo maturation in the bone marrow, whereupon they are released and migrate to tissues to become effector macrophages, dendritic cells, or PMNs. During pathologic conditions such as cancer, infection, or chronic inflammation, proinflammatory mediators and/or endoplasmic reticulum (ER) stress drive immature myeloid cell expansion and their conversion into MDSCs (130–132). The growth factors G-CSF and GM-CSF are important for stimulating MDSC expansion with proinflammatory cytokines (IL-1β, TNF-α, IL-6) playing a key role in their activation (133–135). MDSCs exert potent immune-regulatory activity through several mechanisms, including suppressing macrophage and dendritic cell proinflammatory activity, promoting regulatory T cell (Treg) activation, and inhibiting CD4+ and CD8+ T cells. These effects are mediated by the action of several molecules including Arg-1, nitric oxide (NO), TGF-β, IL-10, cyclooxygenase-2 (COX-2), and ROS (133, 134, 136, 137). Through these mechanisms, MDSCs limit inflammation to perpetuate chronic infection by suppressing immune effectors that are important for disease resolution (138).
MDSCs consist of two groups referred to as granulocytic (G-MDSCs or PMN-MDSCs) and monocytic (M-MDSCs) that share phenotypic characteristics with PMNs and monocytes, respectively. Each MDSC subset utilizes distinct mechanisms to attenuate immune responses, where generally M-MDSCs suppress using NO (134, 139, 140), whereas G-MDSCs utilize ROS (141, 142). MDSCs have been best characterized in cancer; however, reports describing their importance during infection and chronic inflammation have emerged in recent years (143–146). Our group has been investigating MDSC-S. aureus biofilm crosstalk since 2014, and the role of MDSCs during S. aureus infection has been confirmed by other groups (38–41, 147–150). In response to peripheral S. aureus biofilm (i.e. PJI), G-MDSCs are critical for inhibiting monocyte/macrophage proinflammatory activity primarily through IL-10 production (38–41). IL-10 is induced by lactate released from S. aureus biofilm, which inhibits HDAC11 to induce epigenetic changes at the Il-10 promoter as well as other genes (41). G-MDSCs are also enriched in humans during PJI, and are expanded in the blood following orthopedic infection (151, 152). This suggests that they may play an important role in dictating infection persistence and/or susceptibility, respectively. MDSCs are the major leukocyte infiltrate in the galea and bone flap during S. aureus craniotomy infection, but are rare in the brain parenchyma (56). The transcriptional profiles of MDSCs during craniotomy infection identified them as G-MDSCs, which were shown to inhibit PMN S. aureus bactericidal activity (62). The effector molecules that are critical for G-MDSC suppressive activity in the context of S. aureus craniotomy infection remain to be identified.
Astrocytes are the most abundant cell type in the CNS parenchyma. They play a key role in maintaining neuronal homeostasis, BBB integrity, and can contribute to immune responses by the production of a wide array of chemokines that promote leukocyte recruitment to the CNS (153–155). S. aureus triggers TLR2 signaling in astrocytes and the secretion of NO, IL-1β, and TNF-α via NF-kB- and MAPK-dependent pathways (156). Other studies have shown that TLR activation induces astrocyte chemokine production (CCL2, CCL3, CCL5) and augments adhesion molecule expression (157, 158). In astrocytes, the intracellular pattern recognition receptor NOD2 was shown to activate NF-kB leading to IL-6, TNF-α, and co-stimulatory molecule expression, which amplified the anti-bacterial immune response (159). Based on their ability to influence immune responses via robust chemokine production, it is possible that astrocytes play an important role in leukocyte recruitment to the brain during S. aureus craniotomy infection. Of particular interest would be the production of monocyte, NK cell, and ɣδ T cell chemokines, since these cell types represent the most abundant leukocyte infiltrates in the brain (62). Studying this will require the use of transgenic mouse models where candidate chemokines are selectively depleted in astrocytes (i.e. Aldh1l1-Cre) (160), since it is not feasible to eliminate astrocytes due to their essential role in brain physiology. However, assigning a biological role to only one chemokine in the context of craniotomy infection might prove difficult based on the known redundancy in chemokine actions (161). An alternative approach would be to identify the chemokine receptors that are required for monocyte, NK cell, and ɣδ T cell recruitment into the brain and leverage this information to identify the responsible chemokines.
T cells participate in CNS immune surveillance and are important for normal learning and memory, behavior, and neurogenesis through IL-4 and IFN-ɣ production (162–164). It is important to note that these effects occur in the absence of CNS pathology when T cell numbers are low, since it is well recognized that increased T cell recruitment to the brain during diseases such as MS or normal aging is associated with adverse outcomes (164–166).
Interestingly, there are conflicting reports on the role of T cells during S. aureus biofilm infection in the periphery. In a model of tibial infection where titanium implants were pre-coated with S. aureus (high infectious inoculum), a beneficial role for Th2 and Treg cells in promoting biofilm clearance has been reported (167). In contrast, in a mouse model of S. aureus PJI with a low infectious inoculum, few T cells were observed, and tissues from PJI patients have fewer T cell infiltrates compared to individuals with aseptic trauma (39, 151). The reasons for this discrepancy are unclear, but they are likely influenced by differences in the infectious dose, background strain of mice, or site of implant infection. T cells are observed in the brain during S. aureus craniotomy infection, but are largely absent from the galea and bone flap (62). This pattern of recruitment suggests that T cells may play an important role in regulating the host response to craniotomy infection in the brain, but it remains to be determined whether this contributes to infection chronicity, or if T cells are a bystander population and do not significantly influence S. aureus biofilm persistence.
As discussed earlier, an intriguing aspect of S. aureus craniotomy infection is the generation of distinct immune responses within the CNS (brain) versus peripheral compartments (galea and bone flap; Figure 2). Although it might be expected that immune responses would differ in the brain compared to the periphery, prior studies in a mouse model of S. aureus brain abscess revealed that inflammatory changes in the brain were similar in nature to peripheral abscesses as described below. Therefore, the S. aureus craniotomy model can be leveraged to elucidate signals that orchestrate unique inflammatory events in the brain vs. periphery, which may lead to tailored therapies for each compartment. This would be particularly useful given the fact that CNS neurons cannot regenerate and, as such, eliciting efficient pathogen neutralization without excessive bystander damage that can accompany inflammation is paramount. During S. aureus craniotomy infection, PMN influx is significantly higher in the galea compared to the brain despite the presence of CXCL1 and CXCL2 in both compartments (19). This might be explained by the higher bacterial burden in the galea compared to the brain (typically 1-log), although both surfaces of the bone flap are colonized with S. aureus (19). Furthermore, the meninges that cover the surface of the brain are patrolled by resident meningeal macrophages that likely serve to limit S. aureus invasion into the brain. Interestingly, meningitis is not observed at the histological level in the S. aureus craniotomy infection model (19, 56), suggesting that any bacteria that detach from the ventral aspect of the bone flap are prevented from significant expansion in the subarachnoid space. The predominance of PMNs in the galea suggests that they are important for containing infection. This was supported by the finding that mice treated with a Gr-1 antibody became more moribund with increased bacterial burden (19). However, the Gr-1 antibody targets both Ly6G+ and Ly6C+ cells, meaning that not only were Ly6G+ PMNs (and G-MDSCs) depleted, but also Ly6C+ monocytes (168). A subsequent study from our group utilizing selective depletion of Ly6G+ cells has revealed that PMNs are critical for preventing S. aureus outgrowth during craniotomy infection, yet mice were not moribund unlike that observed following Gr-1 antibody treatment (62). By extension, this suggests that monocytes/macrophages also play a protective role during craniotomy infection, since they were not depleted with anti-Ly6G. An interesting observation is that although G-MDSCs are also targeted by anti-Ly6G, the removal of this suppressive population did not improve biofilm clearance. Instead, the opposite was observed, suggesting that PMNs are the main driver of biofilm containment in the craniotomy infection model (62).
Based on the chronicity of S. aureus craniotomy infection in the mouse model (at least 9 months, the latest time point examined to date), it might be assumed that there is minimal involvement of proinflammatory mechanisms (46). However, as alluded to above, there is some degree of proinflammatory tone during craniotomy biofilm infection because PMN/monocyte depletion with anti-Gr-1 results in rapid S. aureus outgrowth in the brain, galea, and on the bone flap (19). Another indication that proinflammatory cytokines are critical for bacterial containment has been through the examination of TLR signaling pathways. Our initial study examined MyD88, the adaptor molecule that facilitates signaling through all TLRs (except TLR3), IL-1R, and IL-18R, and is a critical factor in innate immune defense (169). MyD88 KO mice were extremely susceptible to S. aureus craniotomy infection with a significant reduction in PMN infiltrates and proinflammatory mediator production (CXCL1 and IL-1β) that resulted in increased bacterial burden in the brain, galea, and bone flap (19). These phenotypes combined with the enhanced morbidity of MyD88 KO mice were akin to the effects seen during anti-Gr-1 treatment where essentially all innate immune effectors were depleted (PMNs and monocytes) (19). The importance of TLRs and downstream effector mechanisms was further demonstrated by our recent work that revealed a crucial role for TLR2 and caspase-1 during S. aureus craniotomy infection, primarily via IL-1β action (56). Interleukin-1β is produced in an inactive pro-form that requires proteolytic cleavage by the inflammasome whose active moiety is caspase-1 (170). Inflammasome activation involves two signals; the first being delivered by a PRR, such as TLR2, which leads to the production of inflammasome subunits and pro-IL-1β. The second signal can be delivered by any number of stimuli depending on the type of NLR sensor (i.e. NLRP3, NLRC1, etc.) that results in inflammasome assembly and caspase-1 activation (170). Mice lacking either functional TLR2 or caspase-1 displayed increased bacterial burden in the brain, galea, and bone flap, which coincided with significant decreases in the production of several proinflammatory mediators including IL-1β (56). A critical role for IL-1β in bacterial containment was established by the fact that treatment of caspase-1 KO mice with IL-1β-containing microparticles returned the exaggerated bacterial burden in these animals to levels observed in WT mice (56). These findings revealed the essential role of the TLR2/caspase-1/IL-1β axis in bacterial containment during S. aureus craniotomy infection (Figure 2). The importance of TLR2 in preventing S. aureus outgrowth is intriguing given the number of S. aureus virulence factors that target this signaling pathway (28) as described earlier. One explanation is that these TLR2 evasion molecules have been described during planktonic growth, and it is unknown whether they are expressed during biofilm formation. In addition, it is clear that although TLR2-dependent pathways are capable of limiting S. aureus biofilm outgrowth, they are not sufficient to clear infection, since biofilm persists in the wild type setting. This was further demonstrated by the finding that exogenous IL-1β treatment was not able to reduce S. aureus burden in WT mice, revealing the recalcitrance of biofilm to proinflammatory signals (56). Interestingly, although TLR9 is an important sensor for staphylococcal DNA that is a major component of the S. aureus biofilm matrix (29), TLR9 had minimal impact on the course of craniotomy infection (56). This may result from the fact that TLR9 is an intracellular PRR that requires phagocytic uptake of bacteria or eDNA, and prior studies have demonstrated that S. aureus biofilm interferes with macrophage phagocytosis (33, 37). Collectively, these findings highlight the fact that anti-bacterial pathways are operative during craniotomy infection; however, this is difficult to appreciate in a wild type setting based on the chronicity of infection.
There are many unknowns regarding the pathogenesis of S. aureus craniotomy infection. One critical point relates to identifying the mechanisms responsible for biofilm persistence despite antibiotic treatment. Second, we know little about the contributions of brain-resident cells, such as microglia and astrocytes, which are capable of influencing immune responses. In addition, we have recently identified a prominent influx of NK and γδ T cells in the brain during S. aureus craniotomy infection (62) and it will be interesting to examine the functional significance of each population in future studies. Finally, it will be critical to identify S. aureus virulence determinants that are important for promoting biofilm persistence, and to evaluate whether unique S. aureus transcriptional signatures are observed in bacteria recovered from the brain, galea, or the bone flap where the physical biofilm resides. These are all topics for future investigation.
Brain abscesses pose a challenging clinical problem, which can be associated with high mortality rates due to brain compression and neuronal death from associated edema (171). Pyogenic staphylococci and streptococci are among the most prevalent bacterial species associated with brain abscesses emanating from hematogenous spread (172). Abscess formation may also arise as a complication of neurosurgery or head trauma, and is commonly associated with S. aureus (8, 173). Anatomically, a brain abscess possesses a well-formed necrotic center containing bacteria and PMNs and is surrounded by a dense capsule composed of macrophages and myofibroblasts (174). Activated microglia and astrocytes are observed in the brain parenchyma surrounding the abscess margins along with extensive edema. Astrocytes have been shown to play an important role in regulating brain abscess pathology, since mice deficient for the astrocytic intermediate filament glial fibrillary acidic protein (GFAP) displayed increased bacterial burden, large lesion size, and diffuse leukocyte infiltration (175). These effects likely resulted from the inability to wall off the abscess, since GFAP is a cytoplasmic protein and its loss does not eliminate astrocytes in the brain parenchyma.
S. aureus craniotomy and brain abscess fundamentally differ based on their chronicity and degree of inflammation. In mouse models, S. aureus brain abscesses are shorter in duration typically resolving within 14–21 days (174); whereas during S. aureus craniotomy infection, bacteria are detectable on the infected bone flap, galea, and the brain for as long as 9 months (46) despite both models utilizing a similar infectious dose (i.e. 103 cfu vs. 104 cfu for craniotomy and brain abscess, respectively). Second, there are distinctions at the histological level. Craniotomy-associated infection is not typified by abscess formation in the brain parenchyma of WT animals (19, 56), which is obviously distinct from brain abscess where a solitary lesion is elicited (176, 177). Furthermore, brain abscesses are associated with significant edema (177, 178), whereas edema is not a prominent feature of craniotomy infection in the brain, although a purulent exudate forms in the galea (19, 56). Third, there are distinctions in the patterns of leukocyte recruitment. Craniotomy infections are typified by a more complex immune response owing to differential leukocyte recruitment across the brain, galea, and bone flap. The infected bone flap and galea are dominated by granulocytic infiltrates (i.e. PMNs and G-MDSCs) in regions that coincide with the highest bacterial burden (56). In contrast, the brain is typified by a monocytic infiltrate and an approximate 1-log reduction in bacteria compared to the bone flap and galea. Although brain abscesses are typically a solitary lesion, there is still some degree of specificity in leukocyte homing to particular niches. PMNs migrate primarily to the necrotic core, whereas macrophages are detected along the fibrotic abscess capsule (179).
When comparing the functional roles of TLR2 and MyD88 in both models, some similarities and distinctions are noted (Figure 3). First, differences between S. aureus craniotomy infection and brain abscesses can be seen at the level of TLR2 and MyD88 involvement in bacterial containment. For example, bacterial burden was similar in brain abscesses of MyD88 KO and WT mice (178); whereas, MyD88 was critical for preventing S. aureus outgrowth during craniotomy-associated infection (19). A similar finding was observed with respect to TLR2 where bacterial burden was exaggerated in TLR2 KO animals during craniotomy-associated infection (56), but was comparable in brain abscesses of WT and TLR2 KO mice (180). This is particularly interesting since proinflammatory mediator production was reduced in TLR2 KO animals in both models, revealing the involvement of TLR2-independent pathways in controlling bacterial burden during brain abscess formation.
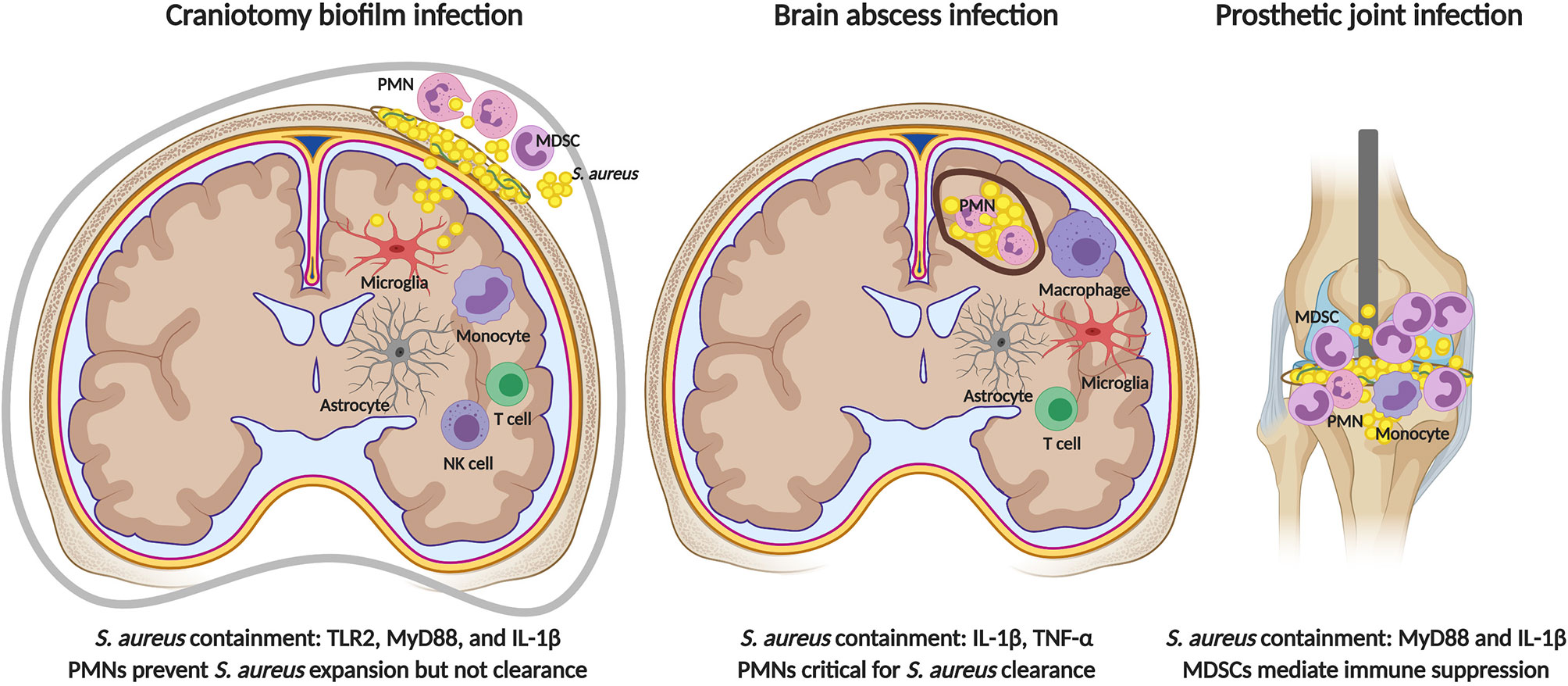
Figure 3 Comparisons in immune responses elicited by S. aureus in the CNS versus periphery. TLR2 signaling is critical for preventing bacterial outgrowth during S. aureus craniotomy infection (left), but is dispensable during brain abscess (center) and peripheral biofilm infection (right). MyD88-dependent signals are critical for restricting S. aureus growth during craniotomy and peripheral biofilm infection, and IL-1β production is key in all three models. PMNs are essential for brain abscess resolution and bacterial containment during craniotomy infection, but do not influence peripheral biofilm infection where infection chronicity is mediated by the inhibitory action of MDSCs on monocyte pro-inflammatory activity. Figure created with BioRender.
In terms of similarities between S. aureus craniotomy infection and brain abscess three examples are evident. First, neither model is dependent on TLR9, which could explained by the fact that TLR9 is an endosomal receptor that requires phagocytosis for pathogen-derived DNA to engage the receptor (181). S. aureus is known to inhibit opsonophagocytosis via protein A (SpA) production, and S. aureus biofilm attenuates macrophage phagocytic activity (33, 37, 182). Second, IL-1β plays an important role in S. aureus containment in both craniotomy infection and brain abscess although the pathways leading to IL-1β production differ (Figure 3) (56, 183, 184). During S. aureus craniotomy infection, caspase-1 was required for maximal IL-1β production; however, there was no role for the well-characterized inflammasome protein NLRP3 or its adaptor molecule ASC (56). Therefore, the NLR sensor that is required for inflammasome assembly and caspase-1 activation during craniotomy infection remains unknown. In contrast, IL-1β release during S. aureus brain abscess required the AIM2 inflammasome and ASC but, similar to craniotomy infection, was NLRP3-independent (184). These findings demonstrate the involvement of distinct inflammasome platforms for triggering IL-1β release during S. aureus biofilm-associated craniotomy infection versus brain abscess.
Finally, S. aureus craniotomy and brain abscess infection share a critical role for PMNs in bacterial containment (Figure 3). Mice lacking CXCR2, the receptor for the PMN chemokines CXCL1 and CXCL2, showed minimal PMN recruitment into the infected brain parenchyma and higher bacterial burden in the brain abscess model (185). Likewise, anti-Gr-1 treatment mimicked these findings with exaggerated bacterial burden and a failure to limit the extent of tissue damage during brain abscess development, although monocytes were also targeted with this antibody (185). Similar phenotypes were observed during S. aureus craniotomy infection, where anti-Gr-1 administration led to a significant outgrowth of bacteria in the brain, galea, and bone flap within 48 h post-infection concomitant with increased morbidity (19). Subsequent studies to refine cell depletion to only the PMN/G-MDSC populations with anti-Ly6G produced similar findings with exaggerated bacterial burden (62). However, unlike anti-Gr-1 depletion where effects were observed within 48 h the anti-Ly6G phenotype was delayed in comparison, becoming significant at day 7 post-infection, and no morbidity was observed (62). Collectively, these findings reveal the essential role of PMNs in S. aureus brain abscess resolution and although PMNs are important for preventing bacterial outgrowth during S. aureus craniotomy infection, the fact that animals tolerate PMN depletion suggests the involvement of other immune populations, the identity of which remains to be determined. Furthermore, it is important to emphasize that S. aureus craniotomy infection persists even when PMNs are present establishing their ineffectiveness at biofilm clearance in vivo. This may result, in part, by the ability of G-MDSCs to inhibit PMN killing of S. aureus (62).
S. aureus is a common etiologic agent of infections associated with prosthetic joints and other indwelling medical devices (186, 187). Over the years, our laboratory and others have identified numerous mechanisms used by S. aureus biofilm to evade host immune responses (186, 188, 189). One hallmark of S. aureus biofilm infection outside the CNS is the prevalence of leukocyte infiltrates that display anti-inflammatory properties. One example is MDSCs, immature myeloid cells that have the ability to suppress T cell activation and monocyte/macrophage proinflammatory activity (38). G-MDSCs are the major leukocyte infiltrate in a mouse model of S. aureus PJI in addition to patients with PJI (38, 40, 41, 151). Depletion of G-MDSCs and PMNs in the mouse S. aureus PJI model using anti-Ly6G transformed infiltrating monocytes to a proinflammatory state that led to a significant reduction in biofilm burden (38). In contrast, although G-MDSCs and PMNs are most abundant in the galea and bone flap during S. aureus craniotomy infection, Ly6G depletion of these cells resulted in bacterial outgrowth (62). Therefore, although both models are associated with G-MDSC infiltrates, these results suggest their differential involvement in dictating infection outcome (Figure 3). An alternative explanation could be differences in the abundance of PMNs in both infection models. PMN infiltrates are minimal in S. aureus PJI (which instead is dominated by G-MDSCs), whereas PMNs are more abundant in the galea and bone flap during S. aureus craniotomy infection. Therefore, the inability to contain S. aureus following Ly6G depletion in the craniotomy model may result from the loss of the larger PMN population that is a critical bactericidal effector.
Another distinction between PJI and craniotomy infection is the presence of innate and adaptive lymphoid populations in the latter. Both human PJI and the mouse model have few T cell infiltrates (39, 151) in agreement with the ability of the dominant G-MDSC population to inhibit T cell activation/proliferation (190). In contrast, S. aureus craniotomy infection is associated with significant NK and ɣδ T cell recruitment in the brain, with fewer T and B cells (62). These populations are largely absent from the galea and bone flap revealing a unique microenvironment in the brain that is responsible for the recruitment of these lymphoid populations.
A final difference between S. aureus biofilm infection in the CNS vs. periphery is demonstrated by the role of TLR2 in disease. As mentioned earlier, TLR2 is critical for bacterial containment during S. aureus craniotomy infection (56), whereas the receptor is dispensable during peripheral biofilm infection (Figure 3) (33). Despite the differential involvement of TLR2, the TLR/IL-1R adaptor MyD88 is plays an equally important role in preventing S. aureus outgrowth during both CNS and peripheral biofilm infection (19, 34, 191). This phenotype is likely driven by IL-1β given that the loss of IL-1β production or signaling results in increased bacterial outgrowth in both CNS and peripheral models of S. aureus biofilm infection (32, 56, 192).
Despite extensive precautionary measures, post-operative complications following craniotomy continue to occur with S. aureus responsible for approximately one-half of these infections (18). Multiple surgeries, prolonged hospital stays, and significant mortality confound the complications arising from craniotomy infections (193). Current treatment strategies include the ex vivo submergence of the infected bone flap in an antiseptic solution and aggressive debridement prior to re-insertion; however, these have not yet been adopted as standard-of-care practices (194, 195). Surgical drainage in combination with a prolonged antibiotic regimen can often effectively control infection (196), which is largely dictated by the interval between surgery and presentation of clinical signs of infection. In some instances, the bone flap cannot be salvaged and a cranioplasty is performed using a bone graft or alloplastic prosthesis. In either case, patients are subjected to an extended antibiotic regimen lasting for months. Because a second surgery is often required for treatment, and the potential for infection recurrence, devising novel therapeutic approaches may significantly improve the outcome of craniotomy infection without the need for more radical interventions.
Previously, our laboratory demonstrated the efficacy of administering proinflammatory macrophages to promote biofilm clearance in vivo (34). We leveraged this observation to evaluate the efficacy of a 3D bioprinted bone scaffold that incorporated viable macrophages and an antibiotic cocktail as a localized delivery system for the treatment of S. aureus craniotomy infection (46). The rationale for including viable macrophages was that they might facilitate biofilm dispersal making bacteria more susceptible to antibiotic action. The 3D bioprinted scaffold was capable of reducing established biofilm infection, since scaffold implantation at day 7 post-infection led to a significant reduction in bacterial burden and reduced BBB damage that is associated with chronic S. aureus infection (46). Interestingly, although macrophage incorporation into 3D scaffolds was effective at diminishing an established biofilm, this was not the case in a prophylactic paradigm; therefore, subsequent studies focused on increasing the antibiotic dose in the scaffold. This approach mitigated bacterial burden to below the limit of detection for 2 weeks; however, S. aureus outgrowth was observed after this period due to the loss of antibiotic from the scaffold. Therefore, second generation 3D scaffolds are currently being developed by our group that incorporate additional bioactive moieties designed to negate the outgrowth of residual bacteria after the antibiotic has exited the scaffold. Other strategies that could be leveraged to enhance therapeutic efficacy include the use of tagged nanoparticles to target a specific immune population to enhance its microbicidal activity. Furthermore, the use of systemic antibiotics once the primary biofilm burden has been reduced by 3D bioprinted scaffolds will be critical to clear residual bacteria, which was supported by our recent study (46).
Other therapeutic approaches have utilized nanoparticle-based delivery systems to augment immune cell function (197, 198). We recently employed a similar strategy to deliver IL-1β-containing microparticles to attenuate bacterial burden in caspase-1 KO mice during S. aureus craniotomy infection (56). It will be interesting to see how these rapidly evolving therapeutics can modulate infection in mouse biofilm models for potential translation to the clinic.
Several studies have highlighted niche-specific differences in the composition of leukocyte infiltrates and their ensuing inflammatory responses during infection (199, 200). One example is a mouse model of visceral leishmaniasis, where parasites are cleared within 2 months following intravenous injection in the liver but are present in the spleen and bone marrow throughout the life of the animal (201). This is thought to result from alterations in adaptive immunity and macrophage function in each of these locations. Tissue-specific cues are exemplified when comparing the immune responses elicited by S. aureus biofilm in the periphery that, in general, are characterized by an anti-inflammatory phenotype whereas more proinflammatory responses are elicited during CNS biofilm infection. Deciphering the signals emanating from different tissues will be crucial for understanding the pathogenesis of biofilm infection and for developing selective treatment strategies to avoid adverse side effects. The use of conditional KO mice will be an extremely important tool to understand the role of immune mediators in leukocyte populations enriched in a given tissue niche. This is particularly relevant in the case of S. aureus craniotomy-associated infection that is more complex in terms of distinct leukocyte subsets across various tissue domains.
It is intriguing how S. aureus can elicit markedly different immune responses depending upon the site of infection. As discussed, S. aureus craniotomy infection displays a compartmentalized immune response within affected CNS regions, and recent RNA-seq studies have begun to decipher the pathways that program a given immune population in its unique niche (62). Imaging modalities such as intravital microscopy would provide an unprecedented window into S. aureus-leukocyte interactions and migratory patterns associated with CNS resident vs. invading immune cells in real time. Identifying the factors that modulate changes in transcriptional networks, the nature of host-pathogen interactions, and patterns of leukocyte migration would provide a better understanding of S. aureus-leukocyte crosstalk and ultimately pave the way for developing tailored therapeutic strategies to mitigate S. aureus biofilm infections within the CNS or periphery.
SD and GK contributed equally to writing the manuscript draft with clinical information provided by JM. The initial editing was performed by TK and all authors contributed to subsequent editing. All authors contributed to the article and approved the submitted version.
The Kielian laboratory is supported by NIH grants R01 NS107369 and 3P01AI083211 (Project 4 to TK).
The authors apologize to those whose work could not be cited due to space limitations.
A patent has been filed with the US Patent and Trademark Office covering the application of 3D bioprinted scaffolds for the treatment of craniotomy-associated infections that is discussed in this review (PCT/US2020/021440; TK).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
3D: three-dimensional
Arg-1: arginase-1
AIM2: absent in melanoma 2
ASC: apoptosis-associated speck-like protein containing a carboxy-terminal CARD
BBB: blood-brain barrier
CCL2: monocyte chemoattractant protein-1
CNS: central nervous system
CSF-1: macrophage colony stimulating factor-1
CXCL1: keratinocyte chemoattractant
CXCL2: macrophage inflammatory protein-2
CXCL10: interferon-inducible protein 10 kDa
CXCR2: C-X-C receptor 2
EAE: experimental autoimmune encephalomyelitis
eDNA: extracellular DNA
ER: endoplasmic reticulum
G-CSF: granulocyte colony-stimulating factor
GFAP: glial fibrillary acidic protein
G-MDSC: granulocyte myeloid-derived suppressor cell
γδ T cell: gamma-delta
GM-CSF: granulocyte-macrophage colony-stimulating factor
IFN: interferon
IL: interleukin
KO: knockout
MAPK: mitogen-activated protein kinase
MDSC: myeloid-derived suppressor cell
M-MDSC: monocyte myeloid-derived suppressor cell
MyD88: myeloid differentiation factor 88
NET: neutrophil extracellular trap
NGS: next generation sequencing
NF-κB: nuclear factor-kappa B
NK cell: natural killer cell
NLR: NOD-like receptor
NO: nitric oxide
NOD2: nucleotide-binding oligomerization domain-containing protein 2
PJI: prosthetic joint infection
PMN: neutrophil
PRR: pattern recognition receptor
RNI: reactive nitrogen intermediate
ROS: reactive oxygen species
S. aureus: Staphylococcus aureus
scRNA-seq: single cell RNA-sequencing
Spa: protein A
TGF-β: transforming growth factor-beta
TLR: Toll-like receptor
TNF-α: tumor necrosis factor-alpha
WT: wild type
2. Pinto VL, Tadi P, Adeyinka A. Increased Intracranial Pressure. Treasure Island (FL): StatPearls (2020).
3. Rosinski CL, Chaker AN, Zakrzewski J, Geever B, Patel S, Chiu RG, et al. Autologous Bone Cranioplasty: A Retrospective Comparative Analysis of Frozen and Subcutaneous Bone Flap Storage Methods. World Neurosurg (2019) 131:e312–e20. doi: 10.1016/j.wneu.2019.07.139
4. Corliss B, Gooldy T, Vaziri S, Kubilis P, Murad G, Fargen K. Complications After In Vivo and Ex Vivo Autologous Bone Flap Storage for Cranioplasty: A Comparative Analysis of the Literature. World Neurosurg (2016) 96:510–5. doi: 10.1016/j.wneu.2016.09.025
5. Honeybul S, Ho KM. Long-term complications of decompressive craniectomy for head injury. J Neurotrauma (2011) 28(6):929–35. doi: 10.1089/neu.2010.1612
6. Stiver SI. Complications of decompressive craniectomy for traumatic brain injury. Neurosurg Focus (2009) 26(6):E7. doi: 10.3171/2009.4.FOCUS0965
7. Chughtai KA, Nemer OP, Kessler AT, Bhatt AA. Post-operative complications of craniotomy and craniectomy. Emerg Radiol (2019) 26(1):99–107. doi: 10.1007/s10140-018-1647-2
8. Dashti SR, Baharvahdat H, Spetzler RF, Sauvageau E, Chang SW, Stiefel MF, et al. Operative intracranial infection following craniotomy. Neurosurg Focus (2008) 24(6):E10. doi: 10.3171/FOC/2008/24/6/E10
9. Wang LY, Cao XH, Shi LK, Ma ZZ, Wang Y, Liu Y. Risk factors for intracranial infection after craniotomy: A case-control study. Brain Behav (2020) 10(7):e01658. doi: 10.1002/brb3.1658
10. Shi ZH, Xu M, Wang YZ, Luo XY, Chen GQ, Wang X, et al. Post-craniotomy intracranial infection in patients with brain tumors: a retrospective analysis of 5723 consecutive patients. Br J Neurosurg (2017) 31(1):5–9. doi: 10.1080/02688697.2016.1253827
11. Fang C, Zhu T, Zhang P, Xia L, Sun C. Risk factors of neurosurgical site infection after craniotomy: A systematic review and meta-analysis. Am J Infect Control (2017) 45(11):e123–e34. doi: 10.1016/j.ajic.2017.06.009
12. Baumeister S, Peek A, Friedman A, Levin LS, Marcus JR. Management of postneurosurgical bone flap loss caused by infection. Plast Reconstr Surg (2008) 122(6):195e–208e. doi: 10.1097/PRS.0b013e3181858eee
13. Le C, Guppy KH, Axelrod YV, Hawk MW, Silverthorn J, Inacio MC, et al. Lower complication rates for cranioplasty with peri-operative bundle. Clin Neurol Neurosurg (2014) 120:41–4. doi: 10.1016/j.clineuro.2014.02.009
14. Gooch MR, Gin GE, Kenning TJ, German JW. Complications of cranioplasty following decompressive craniectomy: analysis of 62 cases. Neurosurg Focus (2009) 26(6):E9. doi: 10.3171/2009.3.FOCUS0962
15. Talwar AA, Bhat DK, Heiman AJ, Ricci JA. Outcomes of Immediate Titanium Cranioplasty Following Post-Craniotomy Infection. J Craniofac Surg (2020) 31(5):1404–7. doi: 10.1097/SCS.0000000000006488
16. McClelland S,3, Hall WA. Postoperative central nervous system infection: incidence and associated factors in 2111 neurosurgical procedures. Clin Infect Dis (2007) 45(1):55–9. doi: 10.1086/518580
17. Chen Y, Zhang L, Qin T, Wang Z, Li Y, Gu B. Evaluation of neurosurgical implant infection rates and associated pathogens: evidence from 1118 postoperative infections. Neurosurg Focus (2019) 47(2):E6. doi: 10.3171/2019.5.FOCUS18582
18. Chiang HY, Steelman VM, Pottinger JM, Schlueter AJ, Diekema DJ, Greenlee JD, et al. Clinical significance of positive cranial bone flap cultures and associated risk of surgical site infection after craniotomies or craniectomies. J Neurosurg (2011) 114(6):1746–54. doi: 10.3171/2011.1.JNS10782
19. Cheatle J, Aldrich A, Thorell WE, Boska MD, Kielian T. Compartmentalization of immune responses during Staphylococcus aureus cranial bone flap infection. Am J Pathol (2013) 183(2):450–8. doi: 10.1016/j.ajpath.2013.04.031
20. Turner NA, Sharma-Kuinkel BK, Maskarinec SA, Eichenberger EM, Shah PP, Carugati M, et al. Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat Rev Microbiol (2019) 17(4):203–18. doi: 10.1038/s41579-018-0147-4
21. Romero-Palacios A, Petruccelli D, Main C, Winemaker M, de Beer J, Mertz D. Screening for and decolonization of Staphylococcus aureus carriers before total joint replacement is associated with lower S aureus prosthetic joint infection rates. Am J Infect Control (2020) 48(5):534–7. doi: 10.1016/j.ajic.2019.09.022
22. Harkins CP, Pichon B, Doumith M, Parkhill J, Westh H, Tomasz A, et al. Methicillin-resistant Staphylococcus aureus emerged long before the introduction of methicillin into clinical practice. Genome Biol (2017) 18(1):130. doi: 10.1186/s13059-017-1252-9
23. Guerra FE, Borgogna TR, Patel DM, Sward EW, Voyich JM. Epic Immune Battles of History: Neutrophils vs. Staphylococcus aureus. Front Cell Infect Microbiol (2017) 7:286. doi: 10.3389/fcimb.2017.00286
24. de Vor L, Rooijakkers SHM, van Strijp JAG. Staphylococci evade the innate immune response by disarming neutrophils and forming biofilms. FEBS Lett (2020) 594(16):2556–69. doi: 10.1002/1873-3468.13767
25. Watkins KE, Unnikrishnan M. Evasion of host defenses by intracellular Staphylococcus aureus. Adv Appl Microbiol (2020) 112:105–41. doi: 10.1016/bs.aambs.2020.05.001
26. Muthukrishnan G, Masters EA, Daiss JL, Schwarz EM. Mechanisms of Immune Evasion and Bone Tissue Colonization That Make Staphylococcus aureus the Primary Pathogen in Osteomyelitis. Curr Osteoporos Rep (2019) 17(6):395–404. doi: 10.1007/s11914-019-00548-4
27. Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol (2015) 13(9):529–43. doi: 10.1038/nrmicro3521
28. Askarian F, Wagner T, Johannessen M, Nizet V. Staphylococcus aureus modulation of innate immune responses through Toll-like (TLR), (NOD)-like (NLR) and C-type lectin (CLR) receptors. FEMS Microbiol Rev (2018) 42(5):656–71. doi: 10.1093/femsre/fuy025
29. Schilcher K, Horswill AR. Staphylococcal Biofilm Development: Structure, Regulation, and Treatment Strategies. Microbiol Mol Biol Rev (2020) 84(3):e00026–19. doi: 10.1128/MMBR.00026-19
30. Conlon BP, Rowe SE, Lewis K. Persister cells in biofilm associated infections. Adv Exp Med Biol (2015) 831:1–9. doi: 10.1007/978-3-319-09782-4_1
31. Lewis K. The Science of Antibiotic Discovery. Cell (2020) 181(1):29–45. doi: 10.1016/j.cell.2020.02.056
32. Bernthal NM, Pribaz JR, Stavrakis AI, Billi F, Cho JS, Ramos RI, et al. Protective role of IL-1β against post-arthroplasty Staphylococcus aureus infection. J Orthop Res (2011) 29(10):1621–6. doi: 10.1002/jor.21414
33. Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH, et al. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol (2011) 186(11):6585–96. doi: 10.4049/jimmunol.1002794
34. Hanke ML, Heim CE, Angle A, Sanderson SD, Kielian T. Targeting macrophage activation for the prevention and treatment of Staphylococcus aureus biofilm infections. J Immunol (2013) 190(5):2159–68. doi: 10.4049/jimmunol.1202348
35. Alhede M, Lorenz M, Fritz BG, Jensen P, Ring HC, Bay L, et al. Bacterial aggregate size determines phagocytosis efficiency of polymorphonuclear leukocytes. Med Microbiol Immunol (2020) 209:669–80. doi: 10.1007/s00430-020-00691-1
36. Bhattacharya M, Berends ETM, Zheng X, Hill PJ, Chan R, Torres VJ, et al. Leukocidins and the Nuclease Nuc Prevent Neutrophil-Mediated Killing of Staphylococcus aureus Biofilms. Infect Immun (2020) 88(10):e00372-20. doi: 10.1128/IAI.00372-20
37. Scherr TD, Hanke ML, Huang O, James DB, Horswill AR, Bayles KW, et al. Staphylococcus aureus Biofilms Induce Macrophage Dysfunction Through Leukocidin AB and Alpha-Toxin. mBio (2015) 6(4):e01021-15. doi: 10.1128/mBio.01021-15
38. Heim CE, Vidlak D, Scherr TD, Kozel JA, Holzapfel M, Muirhead DE, et al. Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J Immunol (2014) 192(8):3778–92. doi: 10.4049/jimmunol.1303408
39. Heim CE, Vidlak D, Scherr TD, Hartman CW, Garvin KL, Kielian T. IL-12 promotes myeloid-derived suppressor cell recruitment and bacterial persistence during Staphylococcus aureus orthopedic implant infection. J Immunol (2015) 194(8):3861–72. doi: 10.4049/jimmunol.1402689
40. Heim CE, Vidlak D, Kielian T. Interleukin-10 production by myeloid-derived suppressor cells contributes to bacterial persistence during Staphylococcus aureus orthopedic biofilm infection. J Leukoc Biol (2015) 98(6):1003–13. doi: 10.1189/jlb.4VMA0315-125RR
41. Heim CE, Bosch ME, Yamada KJ, Aldrich AL, Chaudhari SS, Klinkebiel D, et al. Lactate production by Staphylococcus aureus biofilm inhibits HDAC11 to reprogramme the host immune response during persistent infection. Nat Microbiol (2020) 5(10):1271–84. doi: 10.1038/s41564-020-0756-3
42. Balaban NQ, Helaine S, Lewis K, Ackermann M, Aldridge B, Andersson DI, et al. Definitions and guidelines for research on antibiotic persistence. Nat Rev Microbiol (2019) 17(7):441–8. doi: 10.1038/s41579-019-0196-3
43. Miller LS, Fowler VG, Shukla SK, Rose WE, Proctor RA. Development of a vaccine against Staphylococcus aureus invasive infections: Evidence based on human immunity, genetics and bacterial evasion mechanisms. FEMS Microbiol Rev (2020) 44(1):123–53. doi: 10.1093/femsre/fuz030
44. O’Brien EC, McLoughlin RM. Considering the ‘Alternatives’ for Next-Generation Anti-Staphylococcus aureus Vaccine Development. Trends Mol Med (2019) 25(3):171–84. doi: 10.1016/j.molmed.2018.12.010
45. Tam K, Lacey KA, Devlin JC, Coffre M, Sommerfield A, Chan R, et al. Targeting leukocidin-mediated immune evasion protects mice from Staphylococcus aureus bacteremia. J Exp Med (2020) 217(9):e20190541. doi: 10.1084/jem.20190541
46. Aldrich A, Kuss MA, Duan B, Kielian T. 3D Bioprinted Scaffolds Containing Viable Macrophages and Antibiotics Promote Clearance of Staphylococcus aureus Craniotomy-Associated Biofilm Infection. ACS Appl Mater Interf (2019) 11(13):12298–307. doi: 10.1021/acsami.9b00264
47. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol (2012) 12(9):623–35. doi: 10.1038/nri3265
48. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis (2010) 37(1):13–25. doi: 10.1016/j.nbd.2009.07.030
49. Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol (2009) 31(4):497–511. doi: 10.1007/s00281-009-0177-0
50. Runtsch MC, Ferrara G, Angiari S. Metabolic determinants of leukocyte pathogenicity in neurological diseases. J Neurochem (2020) 00:1–23. doi: 10.1111/jnc.15169
51. Klein RS, Garber C, Funk KE, Salimi H, Soung A, Kanmogne M, et al. Neuroinflammation During RNA Viral Infections. Annu Rev Immunol (2019) 37:73–95. doi: 10.1146/annurev-immunol-042718-041417
52. Norris GT, Kipnis J. Immune cells and CNS physiology: Microglia and beyond. J Exp Med (2019) 216(1):60–70. doi: 10.1084/jem.20180199
53. Heim CE, West SC, Ali H, Kielian T. Heterogeneity of Ly6G(+) Ly6C(+) Myeloid-Derived Suppressor Cell Infiltrates during Staphylococcus aureus Biofilm Infection. Infect Immun (2018) 86(12):e00684-18. doi: 10.1128/IAI.00684-18
54. Bosch ME, Bertrand BP, Heim CE, Alqarzaee AA, Chaudhari SS, Aldrich AL, et al. Staphylococcus aureus ATP Synthase Promotes Biofilm Persistence by Influencing Innate Immunity. mBio (2020) 11(5):e01581-20. doi: 10.1128/mBio.01581-20
55. Yamada KJ, Heim CE, Xi X, Attri KS, Wang D, Zhang W, et al. Monocyte metabolic reprogramming promotes pro-inflammatory activity and Staphylococcus aureus biofilm clearance. PloS Pathog (2020) 16(3):e1008354. doi: 10.1371/journal.ppat.1008354
56. Aldrich AL, Heim CE, Shi W, Fallet RW, Duan B, Kielian T. TLR2 and caspase-1 signaling are critical for bacterial containment but not clearance during craniotomy-associated biofilm infection. J Neuroinflamm (2020) 17(1):114. doi: 10.1186/s12974-020-01793-6
57. Baral P, Umans BD, Li L, Wallrapp A, Bist M, Kirschbaum T, et al. Nociceptor sensory neurons suppress neutrophil and γδ T cell responses in bacterial lung infections and lethal pneumonia. Nat Med (2018) 24(4):417–26. doi: 10.1038/nm.4501
58. Rowe SE, Wagner NJ, Li L, Beam JE, Wilkinson AD, Radlinski LC, et al. Reactive oxygen species induce antibiotic tolerance during systemic Staphylococcus aureus infection. Nat Microbiol (2020) 5(2):282–90. doi: 10.1038/s41564-019-0627-y
59. Toledo AG, Golden G, Campos AR, Cuello H, Sorrentino J, Lewis N, et al. Proteomic atlas of organ vasculopathies triggered by Staphylococcus aureus sepsis. Nat Commun (2019) 10(1):4656. doi: 10.1038/s41467-019-12672-x
60. Jenkins A, Diep BA, Mai TT, Vo NH, Warrener P, Suzich J, et al. Differential expression and roles of Staphylococcus aureus virulence determinants during colonization and disease. mBio (2015) 6(1):e02272–14. doi: 10.1128/mBio.02272-14
61. Kalinka J, Hachmeister M, Geraci J, Sordelli D, Hansen U, Niemann S, et al. Staphylococcus aureus isolates from chronic osteomyelitis are characterized by high host cell invasion and intracellular adaptation, but still induce inflammation. Int J Med Microbiol (2014) 304(8):1038–49. doi: 10.1016/j.ijmm.2014.07.013
62. Aldrich A, Horn CM, Heim CE, Korshoj LE, Kielian T. Transcriptional diversity and niche-specific distribution of leukocyte populations during Staphylococcus aureus craniotomy-associated biofilm infection. J Immunol (2021) 206:751–65. doi: 10.4049/jimmunol.2001042
63. Kipnis J, Gadani S, Derecki NC. Pro-cognitive properties of T cells. Nat Rev Immunol (2012) 12(9):663–9. doi: 10.1038/nri3280
64. Veiga-Fernandes H, Artis D. Neuronal-immune system cross-talk in homeostasis. Science (2018) 359(6383):1465–6. doi: 10.1126/science.aap9598
65. Kierdorf K, Masuda T, Jordão MJC, Prinz M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci (2019) 20(9):547–62. doi: 10.1038/s41583-019-0201-x
66. Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol Cell Biol (2020) 98(1):28–41. doi: 10.1111/imcb.12301
67. Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci (2018) 21(4):530–40. doi: 10.1038/s41593-018-0090-8
68. Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, et al. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci (2016) 19(1):94–101. doi: 10.1038/nn.4189
69. Ginhoux F, Prinz M. Origin of microglia: current concepts and past controversies. Cold Spring Harb Perspect Biol (2015) 7(8):a020537. doi: 10.1101/cshperspect.a020537
70. Prinz M, Jung S, Priller J. Microglia Biology: One Century of Evolving Concepts. Cell (2019) 179(2):292–311. doi: 10.1016/j.cell.2019.08.053
71. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol (2018) 18(4):225–42. doi: 10.1038/nri.2017.125
72. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol (2014) 14(6):392–404. doi: 10.1038/nri3671
73. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (2005) 308(5726):1314–8. doi: 10.1126/science.1110647
74. Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci (2009) 29(13):3974–80. doi: 10.1523/JNEUROSCI.4363-08.2009
75. Tremblay M, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PloS Biol (2010) 8(11):e1000527. doi: 10.1371/journal.pbio.1000527
76. Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia (2005) 49(4):567–76. doi: 10.1002/glia.20144
77. Esen N, Kielian T. Recognition of Staphylococcus aureus-derived peptidoglycan (PGN) but not intact bacteria is mediated by CD14 in microglia. J Neuroimmunol (2005) 170(1-2):93–104. doi: 10.1016/j.jneuroim.2005.09.003
78. Liu X, Chauhan VS, Marriott I. NOD2 contributes to the inflammatory responses of primary murine microglia and astrocytes to Staphylococcus aureus. Neurosci Lett (2010) 474(2):93–8. doi: 10.1016/j.neulet.2010.03.013
79. Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol (2004) 173(6):3916–24. doi: 10.4049/jimmunol.173.6.3916
80. Stenzel W, Soltek S, Sanchez-Ruiz M, Akira S, Miletic H, Schluter D, et al. Both TLR2 and TLR4 are required for the effective immune response in Staphylococcus aureus-induced experimental murine brain abscess. Am J Pathol (2008) 172(1):132–45. doi: 10.2353/ajpath.2008.070567
81. Burmeister AR, Marriott I. The Interleukin-10 Family of Cytokines and Their Role in the CNS. Front Cell Neurosci (2018) 12:458. doi: 10.3389/fncel.2018.00458
82. Sochocka M, Diniz BS, Leszek J. Inflammatory Response in the CNS: Friend or Foe? Mol Neurobiol (2017) 54(10):8071–89. doi: 10.1007/s12035-016-0297-1
83. Chitnis T, Weiner HL. CNS inflammation and neurodegeneration. J Clin Invest (2017) 127(10):3577–87. doi: 10.1172/JCI90609
84. Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol (2017) 18(4):385–92. doi: 10.1038/ni.3703
85. Otero K, Turnbull IR, Poliani PL, Vermi W, Cerutti E, Aoshi T, et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol (2009) 10(7):734–43. doi: 10.1038/ni.1744
86. Lei F, Cui N, Zhou C, Chodosh J, Vavvas DG, Paschalis EI. CSF1R inhibition by a small-molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proc Natl Acad Sci U S A (2020) 117(38):23336–8. doi: 10.1073/pnas.1922788117
87. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron (2014) 82(2):380–97. doi: 10.1016/j.neuron.2014.02.040
88. Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol (1995) 154(9):4309–21. doi: 10.1038/ni.2419
89. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol (2012) 13(11):1118–28. doi: 10.1038/ni.2419
90. Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol (2016) 17(7):797–805. doi: 10.1038/ni.3423
91. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci (2013) 16(12):1896–905. doi: 10.1038/nn.3554
92. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity (2018) 48(2):380–95.e6. doi: 10.1016/j.immuni.2018.01.011
93. Prinz M, Priller J. Tickets to the brain: role of CCR2 and CX3CR1 in myeloid cell entry in the CNS. J Neuroimmunol (2010) 224(1-2):80–4. doi: 10.1016/j.jneuroim.2010.05.015
94. Lim JK, Obara CJ, Rivollier A, Pletnev AG, Kelsall BL, Murphy PM. Chemokine receptor Ccr2 is critical for monocyte accumulation and survival in West Nile virus encephalitis. J Immunol (2011) 186(1):471–8. doi: 10.4049/jimmunol.1003003
95. Chen BP, Kuziel WA, Lane TE. Lack of CCR2 results in increased mortality and impaired leukocyte activation and trafficking following infection of the central nervous system with a neurotropic coronavirus. J Immunol (2001) 167(8):4585–92. doi: 10.4049/jimmunol.167.8.4585
96. Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med (2000) 192(6):899–905. doi: 10.1084/jem.192.6.899
97. Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med (2000) 192(7):1075–80. doi: 10.1084/jem.192.7.1075
98. Musilova J, Mulcahy ME, Kuijk MM, McLoughlin RM, Bowie AG. Toll-like receptor 2-dependent endosomal signaling by Staphylococcus aureus in monocytes induces type I interferon and promotes intracellular survival. J Biol Chem (2019) 294(45):17031–42. doi: 10.1074/jbc.RA119.009302
99. Frodermann V, Chau TA, Sayedyahossein S, Toth JM, Heinrichs DE, Madrenas J. A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus. J Infect Dis (2011) 204(2):253–62. doi: 10.1093/infdis/jir276
100. Okabe Y, Medzhitov R. Tissue biology perspective on macrophages. Nat Immunol (2016) 17(1):9–17. doi: 10.1038/ni.3320
101. Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci (2019) 22(6):1021–35. doi: 10.1038/s41593-019-0393-4
102. Grayczyk JP, Alonzo F. Staphylococcus aureus Lipoic Acid Synthesis Limits Macrophage Reactive Oxygen and Nitrogen Species Production To Promote Survival during Infection. Infect Immun (2019) 87(10):e00344-19. doi: 10.1128/IAI.00344-19
103. Flannagan RS, Heit B, Heinrichs DE. Antimicrobial Mechanisms of Macrophages and the Immune Evasion Strategies of Staphylococcus aureus. Pathogens (2015) 4(4):826–68. doi: 10.3390/pathogens4040826
104. Peres AG, Stegen C, Li J, Xu AQ, Levast B, Surette MG, et al. Uncoupling of pro- and anti-inflammatory properties of Staphylococcus aureus. Infect Immun (2015) 83(4):1587–97. doi: 10.1128/IAI.02832-14
105. Chen X, Alonzo F,3. Bacterial lipolysis of immune-activating ligands promotes evasion of innate defenses. Proc Natl Acad Sci U S A (2019) 116(9):3764–73. doi: 10.1073/pnas.1817248116
106. Koymans KJ, Feitsma LJ, Brondijk TH, Aerts PC, Lukkien E, Lössl P, et al. Structural basis for inhibition of TLR2 by staphylococcal superantigen-like protein 3 (SSL3). Proc Natl Acad Sci U S A (2015) 112(35):11018–23. doi: 10.1073/pnas.1502026112
107. Askarian F, van Sorge NM, Sangvik M, Beasley FC, Henriksen JR, Sollid JU, et al. A Staphylococcus aureus TIR domain protein virulence factor blocks TLR2-mediated NF-κB signaling. J Innate Immun (2014) 6(4):485–98. doi: 10.1159/000357618
108. Patot S, Imbert PR, Baude J, Martins Simões P, Campergue JB, Louche A, et al. The TIR Homologue Lies near Resistance Genes in Staphylococcus aureus, Coupling Modulation of Virulence and Antimicrobial Susceptibility. PloS Pathog (2017) 13(1):e1006092. doi: 10.1371/journal.ppat.1006092
109. Nakayama M, Kurokawa K, Nakamura K, Lee BL, Sekimizu K, Kubagawa H, et al. Inhibitory receptor paired Ig-like receptor B is exploited by Staphylococcus aureus for virulence. J Immunol (2012) 189(12):5903–11. doi: 10.4049/jimmunol.1201940
110. Nakayama M, Underhill DM, Petersen TW, Li B, Kitamura T, Takai T, et al. Paired Ig-like receptors bind to bacteria and shape TLR-mediated cytokine production. J Immunol (2007) 178(7):4250–9. doi: 10.4049/jimmunol.178.7.4250
111. Rankin SM. The bone marrow: a site of neutrophil clearance. J Leukoc Biol (2010) 88(2):241–51. doi: 10.1189/jlb.0210112
112. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
113. Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol (2014) 15(7):602–11. doi: 10.1038/ni.2921
114. de Jong NWM, van Kessel KPM, van Strijp JAG. Immune Evasion by Staphylococcus aureus. Microbiol Spectr (2019) 7(2):GPP3-0061-2019. doi: 10.1128/microbiolspec.GPP3-0061-2019
115. Kobayashi SD, Malachowa N, DeLeo FR. Neutrophils and Bacterial Immune Evasion. J Innate Immun (2018) 10(5-6):432–41. doi: 10.1159/000487756
116. Yatime L, Merle NS, Hansen AG, Friis NA, Østergaard JA, Bjerre M, et al. A Single-Domain Antibody Targeting Complement Component C5 Acts as a Selective Inhibitor of the Terminal Pathway of the Complement System and Thus Functionally Mimicks the C-Terminal Domain of the Staphylococcus aureus SSL7 Protein. Front Immunol (2018) 9:2822. doi: 10.3389/fimmu.2018.02822
117. Amdahl H, Jongerius I, Meri T, Pasanen T, Hyvärinen S, Haapasalo K, et al. Staphylococcal Ecb protein and host complement regulator factor H enhance functions of each other in bacterial immune evasion. J Immunol (2013) 191(4):1775–84. doi: 10.4049/jimmunol.1300638
118. DuMont AL, Yoong P, Day CJ, Alonzo F,3, McDonald WH, Jennings MP, et al. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc Natl Acad Sci U S A (2013) 110(26):10794–9. doi: 10.1073/pnas.1305121110
119. Kobayashi SD, DeLeo FR. Staphylococcus aureus protein A promotes immune suppression. mBio (2013) 4(5):e00764–13. doi: 10.1128/mBio.00764-13
120. Gonzalez CD, Ledo C, Giai C, Garófalo A, Gómez MI. The Sbi Protein Contributes to Staphylococcus aureus Inflammatory Response during Systemic Infection. PloS One (2015) 10(6):e0131879. doi: 10.1371/journal.pone.0131879
121. Niemann S, Bertling A, Brodde MF, Fender AC, Van de Vyver H, Hussain M, et al. Panton-Valentine Leukocidin associated with S. aureus osteomyelitis activates platelets via neutrophil secretion products. Sci Rep (2018) 8(1):2185. doi: 10.1038/s41598-018-20582-z
122. Ma J, Gulbins E, Edwards MJ, Caldwell CC, Fraunholz M, Becker KA. Staphylococcus aureus α-Toxin Induces Inflammatory Cytokines via Lysosomal Acid Sphingomyelinase and Ceramides. Cell Physiol Biochem (2017) 43(6):2170–84. doi: 10.1159/000484296
123. Cheung GY, Joo HS, Chatterjee SS, Otto M. Phenol-soluble modulins–critical determinants of staphylococcal virulence. FEMS Microbiol Rev (2014) 38(4):698–719. doi: 10.1111/1574-6976.12057
124. Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med (2000) 343(23):1703–14. doi: 10.1056/NEJM200012073432307
125. Phillipson M, Kubes P. The Healing Power of Neutrophils. Trends Immunol (2019) 40(7):635–47. doi: 10.1016/j.it.2019.05.001
126. Fattori V, Amaral FA, Verri WA Jr. Neutrophils and arthritis: Role in disease and pharmacological perspectives. Pharmacol Res (2016) 112:84–98. doi: 10.1016/j.phrs.2016.01.027
127. Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol (2012) 5(4):354–66. doi: 10.1038/mi.2012.24
128. Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol (2013) 48(5):531–9. doi: 10.1165/rcmb.2012-0492TR
129. Bao Y, Cao X. Revisiting the protective and pathogenic roles of neutrophils: Ly-6G is key! Eur J Immunol (2011) 41(9):2535–8. doi: 10.1002/eji.201141979
130. Lee BR, Chang SY, Hong EH, Kwon BE, Kim HM, Kim YJ, et al. Elevated endoplasmic reticulum stress reinforced immunosuppression in the tumor microenvironment via myeloid-derived suppressor cells. Oncotarget (2014) 5(23):12331–45. doi: 10.18632/oncotarget.2589
131. Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest (2014) 124(6):2626–39. doi: 10.1172/JCI74056
132. Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, et al. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity (2014) 41(3):389–401. doi: 10.1016/j.immuni.2014.08.015
133. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest (2015) 125(9):3356–64. doi: 10.1172/JCI80005
134. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
135. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity (2010) 32(6):790–802. doi: 10.1016/j.immuni.2010.05.010
136. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202(7):931–9. doi: 10.1084/jem.20050715
137. Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res (2017) 5(1):3–8. doi: 10.1158/2326-6066.CIR-16-0297
138. Medina E, Hartl D. Myeloid-Derived Suppressor Cells in Infection: A General Overview. J Innate Immun (2018) 10(5-6):407–13. doi: 10.1159/000489830
139. Ribechini E, Eckert I, Beilhack A, Du Plessis N, Walzl G, Schleicher U, et al. Heat-killed Mycobacterium tuberculosis prime-boost vaccination induces myeloid-derived suppressor cells with spleen dendritic cell-killing capability. JCI Insight (2019) 5:e128664. doi: 10.1172/jci.insight.128664
140. Tam JW, Kullas AL, Mena P, Bliska JB, van der Velden AW. CD11b+ Ly6Chi Ly6G- immature myeloid cells recruited in response to Salmonella enterica serovar Typhimurium infection exhibit protective and immunosuppressive properties. Infect Immun (2014) 82(6):2606–14. doi: 10.1128/IAI.01590-13
141. Aarts CEM, Hiemstra IH, Beguin EP, Hoogendijk AJ, Bouchmal S, van Houdt M, et al. Activated neutrophils exert myeloid-derived suppressor cell activity damaging T cells beyond repair. Blood Adv (2019) 3(22):3562–74. doi: 10.1182/bloodadvances.2019031609
142. Ohl K, Tenbrock K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front Immunol (2018) 9:2499. doi: 10.3389/fimmu.2018.02499
143. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
144. Dorhoi A, Glaría E, Garcia-Tellez T, Nieuwenhuizen NE, Zelinskyy G, Favier B, et al. MDSCs in infectious diseases: regulation, roles, and readjustment. Cancer Immunol Immunother (2019) 68(4):673–85. doi: 10.1007/s00262-018-2277-y
145. Schrijver IT, Théroude C, Roger T. Myeloid-Derived Suppressor Cells in Sepsis. Front Immunol (2019) 10:327. doi: 10.3389/fimmu.2019.00327
146. Hu C, Pang B, Lin G, Zhen Y, Yi H. Energy metabolism manipulates the fate and function of tumour myeloid-derived suppressor cells. Br J Cancer (2020) 122(1):23–9. doi: 10.1038/s41416-019-0644-x
147. Stoll H, Ost M, Singh A, Mehling R, Neri D, Schafer I, et al. Staphylococcal Enterotoxins Dose-Dependently Modulate the Generation of Myeloid-Derived Suppressor Cells. Front Cell Infect Microbiol (2018) 8:321. doi: 10.3389/fcimb.2018.00321
148. Ledo C, Gonzalez CD, Poncini CV, Mollerach M, Gomez MI. TNFR1 Signaling Contributes to T Cell Anergy During Staphylococcus aureus Sepsis. Front Cell Infect Microbiol (2018) 8:259. doi: 10.3389/fcimb.2018.00259
149. Tebartz C, Horst SA, Sparwasser T, Huehn J, Beineke A, Peters G, et al. A major role for myeloid-derived suppressor cells and a minor role for regulatory T cells in immunosuppression during Staphylococcus aureus infection. J Immunol (2015) 194(3):1100–11. doi: 10.4049/jimmunol.1400196
150. Skabytska Y, Wolbing F, Gunther C, Koberle M, Kaesler S, Chen KM, et al. Cutaneous innate immune sensing of Toll-like receptor 2-6 ligands suppresses T cell immunity by inducing myeloid-derived suppressor cells. Immunity (2014) 41(5):762–75. doi: 10.1016/j.immuni.2014.10.009
151. Heim CE, Vidlak D, Odvody J, Hartman CW, Garvin KL, Kielian T. Human prosthetic joint infections are associated with myeloid-derived suppressor cells (MDSCs): Implications for infection persistence. J Orthop Res (2018) 36(6):1605–13. doi: 10.1002/jor.23806
152. Heim CE, Yamada KJ, Fallet R, Odvody J, Schwarz DM, Lyden ER, et al. Orthopaedic Surgery Elicits a Systemic Anti-Inflammatory Signature. J Clin Med (2020) 9(7):2123. doi: 10.3390/jcm9072123
153. Allen NJ, Lyons DA. Glia as architects of central nervous system formation and function. Science (2018) 362(6411):181–5. doi: 10.1126/science.aat0473
154. Colombo E, Farina C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol (2016) 37(9):608–20. doi: 10.1016/j.it.2016.06.006
155. Sofroniew MV. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol (2020) 41(9):758–70. doi: 10.1016/j.it.2020.07.004
156. Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem (2004) 88(3):746–58. doi: 10.1046/j.1471-4159.2003.02202.x
157. Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia (2005) 49(3):360–74. doi: 10.1002/glia.20117
158. Li L, Acioglu C, Heary RF, Elkabes S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav Immun (2020) 91:740–55. doi: 10.1016/j.bbi.2020.10.007
159. Sterka D Jr, Rati DM, Marriott I. Functional expression of NOD2, a novel pattern recognition receptor for bacterial motifs, in primary murine astrocytes. Glia (2006) 53(3):322–30. doi: 10.1002/glia.20286
160. Srinivasan R, Lu TY, Chai H, Xu J, Huang BS, Golshani P, et al. New Transgenic Mouse Lines for Selectively Targeting Astrocytes and Studying Calcium Signals in Astrocyte Processes In Situ and In Vivo. Neuron (2016) 92(6):1181–95. doi: 10.1016/j.neuron.2016.11.030
161. Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol (2000) 18:217–42. doi: 10.1146/annurev.immunol.18.1.217
162. Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR, et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med (2010) 207(5):1067–80. doi: 10.1084/jem.20091419
163. Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature (2016) 535(7612):425–9. doi: 10.1038/nature18626
164. Mundt S, Mrdjen D, Utz SG, Greter M, Schreiner B, Becher B. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci Immunol (2019) 4(31):eaau8380. doi: 10.1126/sciimmunol.aau8380
165. Dulken BW, Buckley MT, Navarro Negredo P, Saligrama N, Cayrol R, Leeman DS, et al. Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature (2019) 571(7764):205–10. doi: 10.1038/s41586-019-1362-5
166. Hauser SL, Oksenberg JR. The neurobiology of multiple sclerosis: genes, inflammation, and neurodegeneration. Neuron (2006) 52(1):61–76. doi: 10.1016/j.neuron.2006.09.011
167. Prabhakara R, Harro JM, Leid JG, Keegan AD, Prior ML, Shirtliff ME. Suppression of the inflammatory immune response prevents the development of chronic biofilm infection due to methicillin-resistant Staphylococcus aureus. Infect Immun (2011) 79(12):5010–8. doi: 10.1128/IAI.05571-11
168. Boivin G, Faget J, Ancey P-B, Gkasti A, Mussard J, Engblom C, et al. Durable and controlled depletion of neutrophils in mice. Nat Commun (2020) 11(1):2762. doi: 10.1038/s41467-020-16596-9
169. Deguine J, Barton GM. MyD88: a central player in innate immune signaling. F1000Prime Rep (2014) 6:97–. doi: 10.12703/P6-97
170. Christgen S, Place DE, Kanneganti TD. Toward targeting inflammasomes: insights into their regulation and activation. Cell Res (2020) 30(4):315–27. doi: 10.1038/s41422-020-0295-8
171. Brouwer MC, Tunkel AR, McKhann GM, van de Beek D. Brain Abscess. New Engl J Med (2014) 371(5):447–56. doi: 10.1056/NEJMra1301635
172. Helweg-Larsen J, Astradsson A, Richhall H, Erdal J, Laursen A, Brennum J. Pyogenic brain abscess, a 15 year survey. BMC Infect Dis (2012) 12:332. doi: 10.1186/1471-2334-12-332
173. Yang KY, Chang WN, Ho JT, Wang HC, Lu CH. Postneurosurgical nosocomial bacterial brain abscess in adults. Infection (2006) 34(5):247–51. doi: 10.1007/s15010-006-5607-5
174. Kielian T. Immunopathogenesis of brain abscess. J Neuroinflamm (2004) 1(1):16. doi: 10.1186/1742-2094-1-16
175. Stenzel W, Soltek S, Schlüter D, Deckert M. The intermediate filament GFAP is important for the control of experimental murine Staphylococcus aureus-induced brain abscess and Toxoplasma encephalitis. J Neuropathol Exp Neurol (2004) 63(6):631–40. doi: 10.1093/jnen/63.6.631
176. Karpuk N, Burkovetskaya M, Fritz T, Angle A, Kielian T. Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J Neurosci (2011) 31(2):414–25. doi: 10.1523/JNEUROSCI.5247-10.2011
177. Kielian T, Syed MM, Liu S, Phulwani NK, Phillips N, Wagoner G, et al. The synthetic peroxisome proliferator-activated receptor-gamma agonist ciglitazone attenuates neuroinflammation and accelerates encapsulation in bacterial brain abscesses. J Immunol (2008) 180(7):5004–16. doi: 10.4049/jimmunol.180.7.5004
178. Kielian T, Phulwani NK, Esen N, Syed MM, Haney AC, McCastlain K, et al. MyD88-dependent signals are essential for the host immune response in experimental brain abscess. J Immunol (2007) 178(7):4528–37. doi: 10.4049/jimmunol.178.7.4528
179. Baldwin AC, Kielian T. Persistent immune activation associated with a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuroimmunol (2004) 151(1-2):24–32. doi: 10.1016/j.jneuroim.2004.02.002
180. Kielian T, Haney A, Mayes PM, Garg S, Esen N. Toll-like receptor 2 modulates the proinflammatory milieu in Staphylococcus aureus-induced brain abscess. Infect Immun (2005) 73(11):7428–35. doi: 10.1128/IAI.73.11.7428-7435.2005
181. Fitzgerald KA, Kagan JC. Toll-like Receptors and the Control of Immunity. Cell (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
182. Higgins J, Loughman A, Van Kessel KPM, Van Strijp JAG, Foster TJ. Clumping factor A of Staphylococcus aureus inhibits phagocytosis by human polymorphonuclear leucocytes. FEMS Microbiol Lett (2006) 258(2):290–6. doi: 10.1111/j.1574-6968.2006.00229.x
183. Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-alpha play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuropathol Exp Neurol (2004) 63(4):381–96. doi: 10.1093/jnen/63.4.381
184. Hanamsagar R, Aldrich A, Kielian T. Critical role for the AIM2 inflammasome during acute CNS bacterial infection. J Neurochem (2014) 129(4):704–11. doi: 10.1111/jnc.12669
185. Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J Immunol (2001) 166(7):4634–43. doi: 10.4049/jimmunol.166.7.4634
186. Arciola CR, Campoccia D, Montanaro L. Implant infections: adhesion, biofilm formation and immune evasion. Nat Rev Microbiol (2018) 16(7):397–409. doi: 10.1038/s41579-018-0019-y
187. Ricciardi BF, Muthukrishnan G, Masters E, Ninomiya M, Lee CC, Schwarz EM. Staphylococcus aureus Evasion of Host Immunity in the Setting of Prosthetic Joint Infection: Biofilm and Beyond. Curr Rev Musculoskelet Med (2018) 11(3):389–400. doi: 10.1007/s12178-018-9501-4
188. Bhattacharya M, Berends ETM, Chan R, Schwab E, Roy S, Sen CK, et al. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc Natl Acad Sci U S A (2018) 115(28):7416–21. doi: 10.1073/pnas.1721949115
189. Scherr TD, Heim CE, Morrison JM, Kielian T. Hiding in Plain Sight: Interplay between Staphylococcal Biofilms and Host Immunity. Front Immunol (2014) 5:37. doi: 10.3389/fimmu.2014.00037
190. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res (2010) 70(1):68–77. doi: 10.1158/0008-5472.CAN-09-2587
191. Hanke ML, Angle A, Kielian T. MyD88-dependent signaling influences fibrosis and alternative macrophage activation during Staphylococcus aureus biofilm infection. PloS One (2012) 7(8):e42476. doi: 10.1371/journal.pone.0042476
192. Wang Y, Ashbaugh AG, Dikeman DA, Zhang J, Ackerman NE, Kim SE, et al. Interleukin-1β and tumor necrosis factor are essential in controlling an experimental orthopedic implant-associated infection. J Orthop Res (2020) 38(8):1800–9. doi: 10.1002/jor.24608
193. O’Keeffe AB, Lawrence T, Bojanic S. Oxford craniotomy infections database: A cost analysis of craniotomy infection. Br J Neurosurg (2012) 26(2):265–9. doi: 10.3109/02688697.2011.626878
194. Wallace DJ, McGinity MJ, Floyd JR. 2nd. Bone flap salvage in acute surgical site infection after craniotomy for tumor resection. Neurosurg Rev (2018) 41(4):1071–7. doi: 10.1007/s10143-018-0955-z
195. Widdel L, Winston KR. Pus and free bone flaps. J Neurosurg Pediatr (2009) 4(4):378–82. doi: 10.3171/2009.5.PEDS0963
196. Kural C, Kırmızıgoz S, Ezgu MC, Bedir O, Kutlay M, Izci Y. Intracranial infections: lessons learned from 52 surgically treated cases. Neurosurg Focus (2019) 47: (2):E10. doi: 10.3171/2019.5.FOCUS19238
197. Gendelman HE, Anantharam V, Bronich T, Ghaisas S, Jin H, Kanthasamy AG, et al. Nanoneuromedicines for degenerative, inflammatory, and infectious nervous system diseases. Nanomedicine (2015) 11(3):751–67. doi: 10.1016/j.nano.2014.12.014
198. Thamphiwatana S, Angsantikul P, Escajadillo T, Zhang Q, Olson J, Luk BT, et al. Macrophage-like nanoparticles concurrently absorbing endotoxins and proinflammatory cytokines for sepsis management. Proc Natl Acad Sci (2017) 114(43):11488–93. doi: 10.1073/pnas.1714267114
199. Lionakis MS, Lim JK, Lee C-CR, Murphy PM. Organ-specific innate immune responses in a mouse model of invasive candidiasis. J Innate Immunity (2011) 3(2):180–99. doi: 10.1159/000321157
200. Hu W, Pasare C. Location, location, location: tissue-specific regulation of immune responses. J Leukoc Biol (2013) 94(3):409–21. doi: 10.1189/jlb.0413207
Keywords: biofilm, Staphylococcus aureus, craniotomy, myeloid-derived suppressor cell, neutrophil, macrophage, microglia
Citation: de Morais SDB, Kak G, Menousek JP and Kielian T (2021) Immunopathogenesis of Craniotomy Infection and Niche-Specific Immune Responses to Biofilm. Front. Immunol. 12:625467. doi: 10.3389/fimmu.2021.625467
Received: 03 November 2020; Accepted: 05 January 2021;
Published: 23 February 2021.
Edited by:
Luisa Martinez-Pomares, University of Nottingham, United KingdomReviewed by:
Mirian Nacagami Sotto, University of São Paulo, BrazilCopyright © 2021 de Morais, Kak, Menousek and Kielian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tammy Kielian, dGtpZWxpYW5AdW5tYy5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.