 Kazuko Tajiri
Kazuko Tajiri Saori Yonebayashi
Saori Yonebayashi Siqi Li
Siqi Li Masaki Ieda
Masaki Ieda
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 22 February 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.624703
This article is part of the Research TopicTenascins: Key Players in Tissue Homeostasis and DefenseView all 24 articles
Accumulating evidence suggests that the breakdown of immune tolerance plays an important role in the development of myocarditis triggered by cardiotropic microbial infections. Genetic deletion of immune checkpoint molecules that are crucial for maintaining self-tolerance causes spontaneous myocarditis in mice, and cancer treatment with immune checkpoint inhibitors can induce myocarditis in humans. These results suggest that the loss of immune tolerance results in myocarditis. The tissue microenvironment influences the local immune dysregulation in autoimmunity. Recently, tenascin-C (TN-C) has been found to play a role as a local regulator of inflammation through various molecular mechanisms. TN-C is a nonstructural extracellular matrix glycoprotein expressed in the heart during early embryonic development, as well as during tissue injury or active tissue remodeling, in a spatiotemporally restricted manner. In a mouse model of autoimmune myocarditis, TN-C was detectable before inflammatory cell infiltration and myocytolysis became histologically evident; it was strongly expressed during active inflammation and disappeared with healing. TN-C activates dendritic cells to generate pathogenic autoreactive T cells and forms an important link between innate and acquired immunity.
Myocarditis is an inflammatory disease of the myocardium. It represents a public health challenge worldwide, as it is one of the leading causes of dilated cardiomyopathy, particularly in young, previously healthy individuals (1). Myocarditis can be triggered by a variety of infectious and non-infectious agents (2, 3), and the subsequent autoimmune response is thought to contribute to the disease progression to inflammatory cardiomyopathy (4, 5).
Tenascin-C (TN-C) is a non-structural extracellular matrix (ECM) expressed during embryonic development in the heart, but is not present in the normal adult heart (6). In tissue injury, inflammation, or active remodeling, TN-C is re-expressed in a spatiotemporally restricted manner (6). Recently, TN-C has gained attention as a local regulator of inflammation through various molecular mechanisms (7). Several animal studies have revealed that TN-C is involved in autoimmune disorders, including myocarditis, arthritis, glaucoma, and encephalomyelitis (8–11). In autoimmune myocarditis, TN-C activates dendritic cells (DCs) to generate pathogenic autoreactive T cells and forms an important link between innate and acquired immunity (9). In this mini review, we discuss the mechanistic insights into the development of myocarditis and its progression to inflammatory cardiomyopathy and focus on the role of TN-C in their pathology.
Myocarditis can occur in association with a wide spectrum of infectious agents (such as viruses, bacteria, and protozoans), systemic immune-mediated diseases, toxic substances, and drugs (such as immune checkpoint inhibitors) (2, 3, 12, 13). Viruses have been implicated as the leading trigger of myocarditis, with cardiotropic viruses (such as coxsackievirus B3 [CVB3] and adenoviruses), vasculotropic viruses (such as parvovirus B19), and lymphotropic viruses (such as human herpesvirus 6), which are common agents identified in the myocardium of patients with myocarditis/dilated cardiomyopathy (DCM) (14–16). However, the etiologic role of the viruses detected in myocarditis patients is not evident (17). For example, a high prevalence of parvovirus B19 has been observed in hearts both with (18) and without myocarditis (19). Thus, the causative or associative link between individual viral infections and the pathogenesis of myocarditis is still under investigation (17). In Latin America, infection by the protozoan parasite Trypanosoma cruzi is the most common cause of inflammatory heart disease (17).
Animal models of virus-induced myocarditis with CVB3 infection have been used to study how viruses trigger myocarditis. The pathogenesis of CVB3-induced myocarditis involves both viral cytotoxicity and subsequent host immune responses (2). Initially, CVB3 enters cardiomyocytes by binding to the coxsackievirus-adenovirus receptor, and causes direct cytotoxicity to the myocardium within three to four days post-infection (16). During the early stage of CVB3 infection, innate immune cells are activated through pattern recognition receptors, such as toll-like receptors (TLRs), and produce pro-inflammatory cytokines, such as interferons (IFNs), interleukin (IL)-1β, IL-6, IL-8, and tumor necrosis factor-α (16, 20). Subsequently, antigen-specific responses in adaptive immune cells are induced, eliminating the virus by up to 14 days post-infection (16, 20). However, even after viral clearance, a subset of individuals may develop chronic myocardial inflammation with virus-triggered uncontrolled immune response and the expansion of cardiac-autoreactive T cells, leading to inflammatory cardiomyopathy.
Infection with the novel pathogen severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) may trigger myocarditis both directly and indirectly (14, 21). Several studies have suggested that the immune response triggered by the virus is the major cause of cardiomyocyte injury, rather than direct virus-mediated cytotoxicity (15, 22). In a recent series of hospitalized coronavirus disease 2019 (COVID-19) cases caused by SARS-CoV-2 infection, acute cardiac injury with serum troponin elevation occurred in 7% to 27% of patients, and elevated troponin levels were associated with increased mortality in patients with COVID-19 (23–25). However, elevated troponin can be caused not only by myocarditis but also by other heart diseases, such as ischemic heart disease, Takotsubo syndrome, or secondary cardiac injury due to systemic inflammation and hypoxemia due to respiratory dysfunction (14, 26). Recently, autopsies of 21 patients who died from COVID-19 identified multifocal lymphocytic myocarditis in three cases (14%) (26). Viral entry to cardiac cells using angiotensin converting enzyme 2 may directly induce myocarditis (27). However, the exact mechanism of SARS-CoV-2-induced myocarditis is currently unknown, and further investigations are required.
Myosin heavy chain α isoform (MyHC-α) represents a major cardiac autoantigen. MyHC-α immunization with immune adjuvants or the injection of MyHC-α-loaded DCs can induce autoimmune myocarditis in mice (28, 29). MyHC-α reactive T cells have been found in patients with myocarditis and, interestingly, in healthy subjects (30), suggesting that this may be due to impaired T cell tolerance mechanisms. In the thymus, most autoreactive T cells are eliminated through central immune tolerance or negative selection. In this process, the presentation of self-peptides by antigen-presenting medullary thymic epithelial cells is crucial for determining the fate of developing T cells. Importantly, unlike other cardiac antigens, MyHC-α is not expressed in thymic cells in either mice or humans. Therefore, a lack of central T cell tolerance to this protein allows MyHC-α-reactive T cells to escape negative selection and enter the peripheral circulation (30). MyHC-α-reactive T cells were markedly increased in myocarditis, and adoptive transfer of these cells induced myocarditis in the recipients, demonstrating the effector function of MyHC-α-reactive T cells (30). Although frequency is low, MyHC-α-reactive T cells are present in the periphery of healthy individuals (30), suggesting that peripheral immune tolerance is crucial to prevent these self-reactive T cells from inducing autoimmune myocarditis (5, 31, 32). The mechanism of peripheral immune tolerance is complicated, and immune checkpoints, including cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1)/PD ligand 1 (PD-L1), play important roles in maintaining peripheral tolerance to cardiac antigens (33). After viral infection, MyHC-α-specific CD4+ T cells expand, likely because of molecular mimicry (epitope cross-reactivity) or epitope spreading (self-antigen exposure from cardiomyocytes upon viral damage), and contribute to post-infectious myocarditis (34).
Until now, the role of gut bacteria in cardiac autoimmunity was unclear. However, recently, Gil-Cruz et al. (35) demonstrated that the commensal gut microbe Bacteroides thetaiotaomicron (B. theta) triggers a cross-immune response against the bacterial protein β-galactosidase and MyHC-α, causing inflammatory cardiomyopathy. B-galactosidase produced by B. theta has sequence homology to MyHC-α and induces proliferation and T helper 17 (Th17) polarization of MyHC-α-specific CD4+ T cells. Additionally, antibiotic therapy prevents lethal consequences. Patients with myocarditis have higher anti-B. theta antibody and circulating T cells from the patients show significantly higher IFN-γ production capacity against both MyHC-α and β-galactosidase than healthy subjects. These results suggest that targeting the microbiome could become a new therapeutic strategy.
In contrast to the well-defined cardiac antigen-specific T-cell responses, our understanding of the role of heart non-specific CD4+ T cells in myocarditis is limited. Recently, Zarak-Crnkovic et al. (36) demonstrated in a proof-of-concept study that heart non-specific effector T cells did not affect the severity of myocarditis, but protected the heart from adverse post-inflammatory fibrotic remodeling and cardiac dysfunction in the chronic stage. Moreover, bystander activation of effector T cells suppressed the myofibroblast phenotype of mouse and human cardiac fibroblasts (36), suggesting a dynamic and complex role of effector T cells and the interplay between T cells and fibroblasts in autoimmune myocarditis.
In the heart, TN-C is transiently expressed at several important stages during embryonic development, but TN-C-deficient mice do not show a clear phenotype (37). TN-C is rarely expressed in normal adult hearts but is upregulated under pathological conditions with tissue injury, tissue repair/regeneration, and inflammation (38, 39), including myocarditis (40, 41), DCM (42), rheumatic heart disease (43), myocardial infarction (44, 45), hypertensive heart disease (46), and Kawasaki disease (47). Serum TN-C levels appear to be useful biomarkers for assessing disease activity and predicting disease prognosis. High serum TN-C levels are a significant independent predictor for cardiac events and have an incremental predictive power with brain natriuretic peptide (BNP) in both myocardial infarctions and DCM (48, 49). BNP is secreted from cardiomyocytes in the ventricles in response to stretching caused by increased wall tension and is broadly used as a marker for the diagnosis and treatment of heart failure (50, 51). On the other hand, fibroblasts are a major source of TN-C in the pathological heart (40, 52, 53). The combination of the two biomarkers may more accurately reflect the pathological condition of the entire heart than a single biomarker (54).
The expression of TN-C is detectable in the heart before inflammatory cell infiltration and myocytolysis become histologically apparent, persists during active inflammation, and is no longer present prior to mature collagen deposition in the healing phase in a mouse model of experimental autoimmune myocarditis (5). A major source of TN-C in the pathological heart consists of residential interstitial cells, primarily fibroblasts; however, precardiac mesodermal cells, a special population of cardiomyocytes in embryonic hearts, and several cell lines of cardiomyocytes also have the potential to produce TN-C (54). Its expression level reflects the activity of myocardial inflammation (40). We previously investigated the immunomodulatory effect of TN-C in experimental autoimmune myocarditis. TN-C-deficient mice were protected from severe myocarditis with lower Th17 cell infiltration to the heart compared to wild-type mice (9). Th17 cells are closely associated with autoimmunity, and IL-17-producing Th17 cells play a major role in the initiation and development of myocarditis (2, 55, 56). In human myocarditis/inflammatory cardiomyopathy, the Th17 immunophenotype is characterized by elevated Th17 levels with increases in the Th17-related cytokines IL-6, IL-1β, transforming growth factor-β1, IL-23, and granulocyte-macrophage colony-stimulating factor (GM-CSF) (57). Moreover, patients with severe heart failure have greater proportions of Th17 than those with low severity heart failure (57). IL-6 is a key cytokine that differentiates naïve CD4+ T cells into Th17 cells (58). The stimulation of DCs with exogenous TN-C produces high levels of IL-6 (9). Naive CD4+ T cells co-cultured with TN-C-stimulated DCs differentiate into Th17 cells, and the IL-6 blockade inhibits Th17 polarization (9). In addition, TN-C-stimulated DCs produce high levels of IL-1β and GM-CSF, which facilitate Th17 generation and maintenance (59, 60). Taken together, TN-C may promote Th17 expansion through its ability to induce Th17-inducing cytokine production from DCs and form an important link from innate to adaptive immunity.
DCs are antigen-presenting cells essential for priming T cell responses (61). Resting tolerogenic DCs that display cardiac myosin peptides in complex with class II major histocompatibility complex (MHC) are present in a healthy heart and are trafficked to the cardiac draining lymph nodes. In the lymph nodes, DCs present cardiac myosin peptides to naïve CD4+ T cells specific to those peptides, leading to deletion, anergy, or Treg induction (32). If a heart is damaged by tissue injury or inflammation, TN-C is produced by fibroblasts and stimulates myocardial DCs to migrate to the cardiac draining lymph nodes and activate cardiac myosin-specific T cells, which then differentiate into inflammatory effector T cells (Figure 1). In addition to presenting antigen-derived peptides on their MHCs with costimulatory molecules for naïve T cell activation and expansion, DCs release a cocktail of polarizing cytokines for the differentiation of CD4+ T cells into effector cells (30, 62). In autoimmune diseases, DCs play an important role in the regulation of autoreactive CD4+ T cells (30). A model of bone marrow-derived DC (BMDC)-induced autoimmune myocarditis is helpful for understanding how DCs activate autoreactive CD4+ T cells (28). In this model, the activation of TLRs on BMDCs loaded with a MyHC-α peptide is essential for the induction of autoimmune myocarditis (9, 28). TLR signaling triggers innate immunity upon stimulation with microbial products or endogenous danger signals (danger-associated molecular patterns [DAMPs]) (63). In sterile inflammation, DAMPs are released from either ECM (e.g., TN-C or biglycan) or from dying cells (e.g., histones, high mobility group box 1, heat-shock proteins, DNAs, or RNAs) and stimulate TLRs (64, 65). Popovic et al. reported that an endogenous TLR2/4 ligand biglycan enhanced the priming of autoreactive T cells and stimulated autoimmune perimyocarditis (66). We previously showed that TN-C provides DCs to induce myocarditis via TLR4 activation (9). The injection of MyHC-α-loaded BMDCs stimulated with TN-C induced myocarditis in the recipient mice (9). Upon stimulation with TN-C, DCs produced large amounts of proinflammatory cytokines, including the Th17-polarizing cytokines IL-1β, IL-6, and GM-CSF (9). Naïve CD4+ T cells co-cultured with TN-C-stimulated DCs differentiated into Th17 cells, but IL-6 blocking antibody inhibited Th17 polarization (9). Moreover, the blocking of TLR4 signaling reduced IL-6 secretion from DCs with less Th17 generation (9). TN-C-stimulated BMDCs from TLR4-deficient mice failed to induce myocarditis in the recipients, indicating that TN-C provides myocarditis inducibility to DCs, at least in part, via TLR4 interaction (Figure 1) (5, 9). However, this concept is based on limited experimental findings. Therefore, further studies are needed to fully determine the effect of TN-C on the onset and progression of myocarditis.
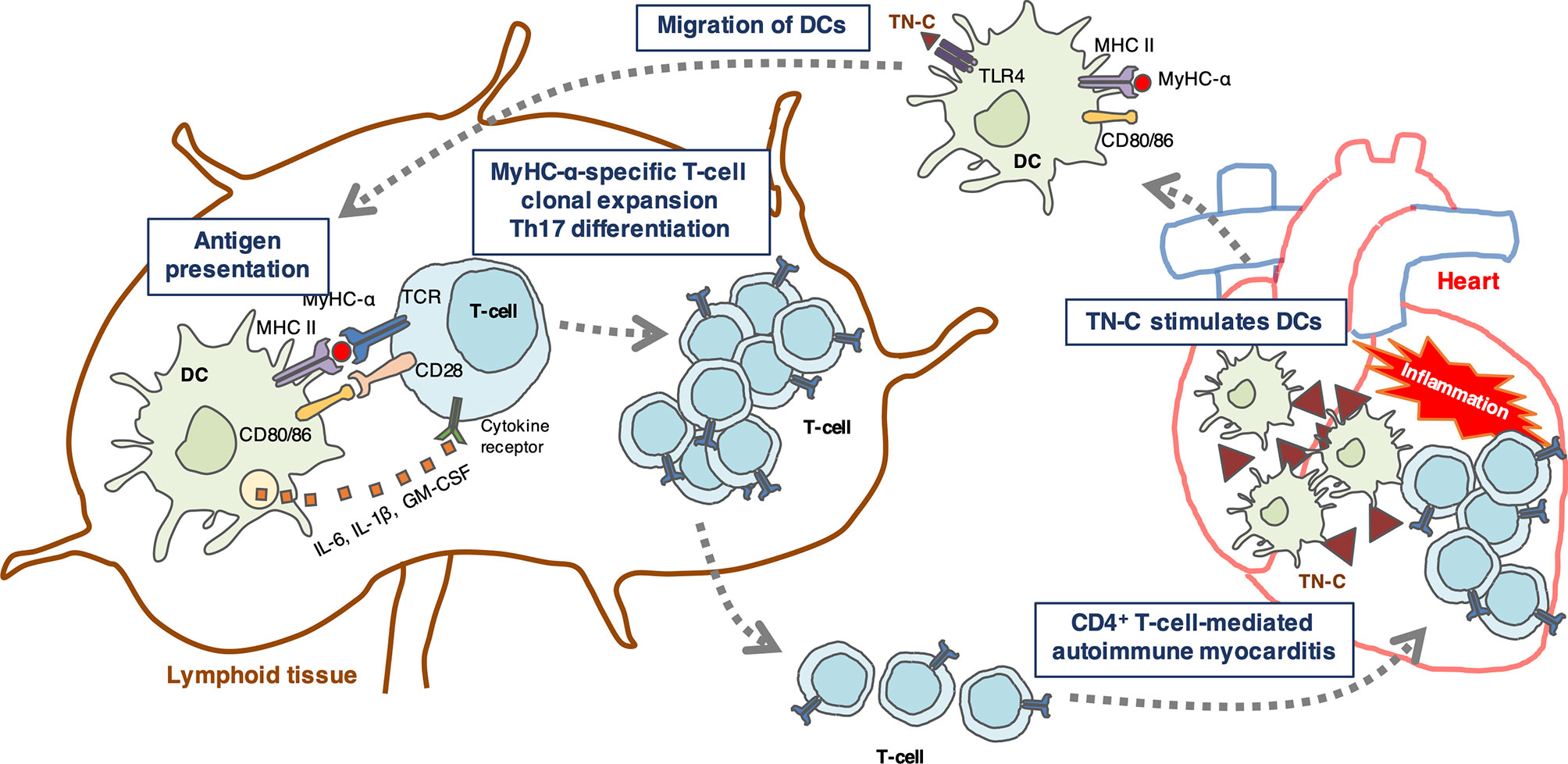
Figure 1 Schematic illustration showing a hypothetical mechanism by which tenascin-C (TN-C)-stimulated dendritic cells (DCs) induce Th17 differentiation. TN-C is upregulated in the heart under pathological conditions such as tissue injury and inflammation, and stimulates myocardial dendritic cells (DCs) via toll-like receptor 4 (TLR4) activation in the heart. Activated DCs migrate to the cardiac draining lymph nodes where they activate cardiac myosin-specific T cells. DCs produce Th17-polarizing cytokines (IL-6, IL-1, and GM-CSF) that contribute to the generation of Th17 cells. In turn, CD4+ T cells migrate back to the heart and cause autoimmune myocarditis.
The etiology and pathogenesis of myocarditis are not yet fully understood. TN-C may be a key extracellular modulator that controls immune responses in myocarditis and inflammatory cardiomyopathy. To date, no attempt has been made to suppress the function of TN-C during myocarditis; however, it has been reported that the administration of an antibody against a domain of TN-C to a rheumatoid arthritis model ameliorated disease severity. Thus, blocking TN-C-dependent inflammatory signals may be a potential novel therapeutic strategy for treating autoimmune myocarditis. Studies involving various animal models have provided a plethora of information, but there remains a gap in knowledge regarding how myocarditis in animal models may differ from that in humans. The prevalence of myocarditis will increase together with the expanding use of immune checkpoint inhibitors and the progression of the COVID-19 pandemic (67). As a result, sophisticated technologies, computational models, and insights are needed.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication. All authors contributed to the article and approved the submitted version.
This study was supported in part by a Grant-in-Aid for Scientific Research (Japan Society for the Promotion of Science KAKENHI grant number 20K08396) to KT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Editage (www.editage.jp) for English language editing.
1. Corsten MF, Schroen B, Heymans S. Inflammation in viral myocarditis: Friend or foe? Trends Mol Med (2012) 18:426–37. doi: 10.1016/j.molmed.2012.05.005
2. Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol (2008) 99:95–114. doi: 10.1016/S0065-2776(08)00604-4
3. Kindermann I, Barth C, Mahfoud F, Ukena C, Lenski M, Yilmaz A, et al. Update on myocarditis. J Am Coll Cardiol (2012) 59:779–92. doi: 10.1016/j.jacc.2011.09.074
4. Leuschner F, Katus HA, Kaya Z. Autoimmune myocarditis: past, present and future. J Autoimmun (2009) 33:282–9. doi: 10.1016/j.jaut.2009.07.009
5. Tajiri K, Yasutomi Y, Aonuma K. Recent advances in the management of autoimmune myocarditis: insights from animal studies. Curr Pharm Des (2016) 22:427–39. doi: 10.2174/1381612822666151222160702
6. Imanaka-Yoshida K, Yoshida T, Miyagawa-Tomita S. Tenascin-C in development and disease of blood vessels. Anat Rec (2014) 297:1747–57. doi: 10.1002/ar.22985
7. Marzeda AM, Midwood KS. Internal Affairs: Tenascin-C as a Clinically Relevant, Endogenous Driver of Innate Immunity. J Histochem Cytochem (2018) 66:289–304. doi: 10.1369/0022155418757443
8. Kanayama M, Morimoto J, Matsui Y, Ikesue M, Danzaki K, Kurotaki D, et al. α9β1 integrin-mediated signaling serves as an intrinsic regulator of pathogenic Th17 cell generation. J Immunol (2011) 187:5851–64. doi: 10.4049/jimmunol.1101524
9. Machino-Ohtsuka T, Tajiri K, Kimura T, Sakai S, Sato A, Yoshida T, et al. Tenascin-C aggravates autoimmune myocarditis via dendritic cell activation and Th17 cell differentiation. J Am Heart Assoc (2014) 3:e001052. doi: 10.1161/JAHA.114.001052
10. Momčilović M, Stamenković V, Jovanović M, Andjus PR, Jakovčevski I, Schachner M, et al. Tenascin-C deficiency protects mice from experimental autoimmune encephalomyelitis. J Neuroimmunol (2017) 302:1–6. doi: 10.1016/j.jneuroim.2016.12.001
11. Reinehr S, Reinhard J, Wiemann S, Stute G, Kuehn S, Woestmann J, et al. Early remodelling of the extracellular matrix proteins tenascin-C and phosphacan in retina and optic nerve of an experimental autoimmune glaucoma model. J Cell Mol Med (2016) 20:2122–37. doi: 10.1111/jcmm.12909
12. Tajiri K, Ieda M. Cardiac Complications in Immune Checkpoint Inhibition Therapy. Front Cardiovasc Med (2019) 6:3. doi: 10.3389/fcvm.2019.00003
13. Tajiri K, Aonuma K, Sekine I. Immune checkpoint inhibitor-related myocarditis. Jpn J Clin Oncol (2018) 48:7–12. doi: 10.1093/jjco/hyx154
14. Tschöpe C, Ammirati E, Bozkurt B, Caforio ALP, Cooper LT, Felix SB, et al. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol (2020) 1–25. doi: 10.1038/s41569-020-00435-x
15. Ammirati E, Veronese G, Bottiroli M, Wang DW, Cipriani M, Garascia A, et al. Update on acute myocarditis. Trends Cardiovasc Med (2020). doi: 10.1016/j.tcm.2020.05.008
16. Lasrado N, Reddy J. An overview of the immune mechanisms of viral myocarditis. Rev Med Virol (2020) 6:1–14. doi: 10.1002/rmv.2131
17. Błyszczuk P. Myocarditis in Humans and in Experimental Animal Models. Front Cardiovasc Med (2019) 6:64. doi: 10.3389/fcvm.2019.00064
18. Mahrholdt H, Wagner A, Deluigi CC, Kispert E, Hager S, Meinhardt G, et al. Presentation, patterns of myocardial damage, and clinical course of viral myocarditis. Circulation (2006) 114:1581–90. doi: 10.1161/CIRCULATIONAHA.105.606509
19. Schenk T, Enders M, Pollak S, Hahn R, Huzly D. High prevalence of human parvovirus B19 DNA in myocardial autopsy samples from subjects without myocarditis or dilative cardiomyopathy. J Clin Microbiol (2009) 47:106–10. doi: 10.1128/JCM.01672-08
20. Maisch B. Cardio-Immunology of Myocarditis: Focus on Immune Mechanisms and Treatment Options. Front Cardiovasc Med (2019) 6:48. doi: 10.3389/fcvm.2019.00048
21. Sawalha K, Abozenah M, Kadado AJ, Battisha A, Al-Akchar M, Salerno C, et al. Systematic review of COVID-19 related myocarditis: Insights on management and outcome. Cardiovasc Revasc Med (2020). doi: 10.1016/j.carrev.2020.08.028
22. Wang J, Han B. Dysregulated CD4+ T Cells and microRNAs in Myocarditis. Front Immunol (2020) 11:539. doi: 10.3389/fimmu.2020.00539
23. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol (2020) 5:811–8. doi: 10.1001/jamacardio.2020.1017
24. Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of Cardiac Injury with Mortality in Hospitalized Patients with COVID-19 in Wuhan, China. JAMA Cardiol (2020) 5:802–10. doi: 10.1001/jamacardio.2020.0950
25. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
26. Basso C, Leone O, Rizzo S, De Gaspari M, van der Wal AC, Aubry M-C, et al. Pathological features of COVID-19-associated myocardial injury: a multicentre cardiovascular pathology study. Eur Heart J (2020) 41:3827–35. doi: 10.1093/eurheartj/ehaa664
27. Siripanthong B, Nazarian S, Muser D, Deo R, Santangeli P, Khanji MY, et al. Recognizing COVID-19–related myocarditis: The possible pathophysiology and proposed guideline for diagnosis and management. Hear Rhythm (2020) 17:1463–71. doi: 10.1016/j.hrthm.2020.05.001
28. Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med (2003) 9:1484–90. doi: 10.1038/nm960
29. Tajiri K, Imanaka-Yoshida K, Matsubara A, Tsujimura Y, Hiroe M, Naka T, et al. Suppressor of Cytokine Signaling 1 DNA Administration Inhibits Inflammatory and Pathogenic Responses in Autoimmune Myocarditis. J Immunol (2012) 189:2043–53. doi: 10.4049/jimmunol.1103610
30. Lv H, Havari E, Pinto S, Gottumukkala RVSRK, Cornivelli L, Raddassi K, et al. Impaired thymic tolerance to α-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest (2011) 121:1561–73. doi: 10.1172/JCI44583
31. Lv H, Lipes MA. Role of impaired central tolerance to α-myosin in inflammatory heart disease. Trends Cardiovasc Med (2012) 22:113–7. doi: 10.1016/j.tcm.2012.07.005
32. Lichtman AH. The heart of the matter: protection of the myocardium from T cells. J Autoimmun (2013) 45:90–6. doi: 10.1016/j.jaut.2013.05.004
33. Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol (2010) 11:21–7. doi: 10.1038/ni.1817
34. Gangaplara A, Massilamany C, Brown DM, Delhon G, Pattnaik AK, Chapman N, et al. Coxsackievirus B3 infection leads to the generation of cardiac myosin heavy chain-α-reactive CD4 T cells in A/J mice. Clin Immunol (2012) 144:237–49. doi: 10.1016/j.clim.2012.07.003
35. Gil-Cruz C, Perez-Shibayama C, de Martin A, Ronchi F, van der Borght K, Niederer R, et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Sci (80- ) (2019) 366:881–6. doi: 10.1126/science.aav3487
36. Zarak-Crnkovic M, Kania G, Jaźwa-Kusior A, Czepiel M, Wijnen WJ, Czyż J, et al. Heart non-specific effector CD4+ T cells protect from postinflammatory fibrosis and cardiac dysfunction in experimental autoimmune myocarditis. Basic Res Cardiol (2020) 115:6. doi: 10.1007/s00395-019-0766-6
37. Saga Y, Yagi T, Ikawa Y, Sakakura T, Aizawa S. Mice develop normally without tenascin. Genes Dev (1992) 6:1821–31. doi: 10.1101/gad.6.10.1821
38. Midwood KS, Hussenet T, Langlois B, Orend G. Advances in tenascin-C biology. Cell Mol Life Sci (2011) 68:3175–99. doi: 10.1007/s00018-011-0783-6
39. Imanaka-Yoshida K. Tenascin-C in cardiovascular tissue remodeling: from development to inflammation and repair. Circ J (2012) 76:2513–20. doi: 10.1253/circj.CJ-12-1033
40. Imanaka-Yoshida K, Hiroe M, Yasutomi Y, Toyozaki T, Tsuchiya T, Noda N, et al. Tenascin-C is a useful marker for disease activity in myocarditis. J Pathol (2002) 197:388–94. doi: 10.1002/path.1131
41. Sato M, Toyozaki T, Odaka K, Uehara T, Arano Y, Hasegawa H, et al. Detection of experimental autoimmune myocarditis in rats by 111In monoclonal antibody specific for tenascin-C. Circulation (2002) 106:1397–402. doi: 10.1161/01.CIR.0000027823.07104.86
42. Tsukada B, Terasaki F, Shimomura H, Otsuka K, Otsuka K, Katashima T, et al. High prevalence of chronic myocarditis in dilated cardiomyopathy referred for left ventriculoplasty: expression of tenascin C as a possible marker for inflammation. Hum Pathol (2009) 40:1015–22. doi: 10.1016/j.humpath.2008.12.017
43. Shiba M, Sugano Y, Ikeda Y, Okada H, Nagai T, Ishibashi-Ueda H, et al. Presence of increased inflammatory infiltrates accompanied by activated dendritic cells in the left atrium in rheumatic heart disease. PloS One (2018) 13:e0203756. doi: 10.1371/journal.pone.0203756
44. Sato A, Aonuma K, Imanaka-Yoshida K, Yoshida T, Isobe M, Kawase D, et al. Serum tenascin-C might be a novel predictor of left ventricular remodeling and prognosis after acute myocardial infarction. J Am Coll Cardiol (2006) 47:2319–25. doi: 10.1016/j.jacc.2006.03.033
45. Kimura T, Tajiri K, Sato A, Sakai S, Wang Z, Yoshida T, et al. Tenascin-C accelerates adverse ventricular remodelling after myocardial infarction by modulating macrophage polarization. Cardiovasc Res (2019) 115:614–24. doi: 10.1093/cvr/cvy244
46. Nishioka T, Suzuki M, Onishi K, Takakura N, Inada H, Yoshida T, et al. Eplerenone attenuates myocardial fibrosis in the angiotensin II-induced hypertensive mouse: involvement of tenascin-C induced by aldosterone-mediated inflammation. J Cardiovasc Pharmacol (2007) 49:261–8. doi: 10.1097/FJC.0b013e318033dfd4
47. Yokouchi Y, Oharaseki T, Enomoto Y, Sato W, Imanaka-Yoshida K, Takahashi K. Expression of tenascin C in cardiovascular lesions of Kawasaki disease. Cardiovasc Pathol (2019) 38:25–30. doi: 10.1016/j.carpath.2018.10.005
48. Sato A, Hiroe M, Akiyama D, Hikita H, Nozato T, Hoshi T, et al. Prognostic value of serum tenascin-C levels on long-term outcome after acute myocardial infarction. J Card Fail (2012) 18:480–6. doi: 10.1016/j.cardfail.2012.02.009
49. Fujimoto N, Onishi K, Sato A, Terasaki F, Tsukada B, Nozato T, et al. Incremental prognostic values of serum tenascin-C levels with blood B-type natriuretic peptide testing at discharge in patients with dilated cardiomyopathy and decompensated heart failure. J Card Fail (2009) 15:898–905. doi: 10.1016/j.cardfail.2009.06.443
50. Maisel AS, Krishnaswamy P, Nowak RM, McCord J, Hollander JE, Duc P, et al. Rapid Measurement of B-Type Natriuretic Peptide in the Emergency Diagnosis of Heart Failure. N Engl J Med (2002) 347:161–7. doi: 10.1056/NEJMoa020233
51. Omar HR, Guglin M. Longitudinal BNP follow-up as a marker of treatment response in acute heart failure: Relationship with objective markers of decongestion. Int J Cardiol (2016) 221:167–70. doi: 10.1016/j.ijcard.2016.06.174
52. Morimoto S-I, Imanaka-Yoshida K, Hiramitsu S, Kato S, Ohtsuki M, Uemura A, et al. Diagnostic utility of tenascin-C for evaluation of the activity of human acute myocarditis. J Pathol (2005) 205:460–7. doi: 10.1002/path.1730
53. Imanaka-Yoshida K, Hiroe M, Nishikawa T, Ishiyama S, Shimojo T, Ohta Y, et al. Tenascin-C modulates adhesion of cardiomyocytes to extracellular matrix during tissue remodeling after myocardial infarction. Lab Invest (2001) 81:1015–24. doi: 10.1038/labinvest.3780313
54. Imanaka-Yoshida K, Tawara I, Yoshida T. Tenascin-C in cardiac disease: A sophisticated controller of inflammation, repair, and fibrosis. Am J Physiol Physiol (2020) 319:C781–96. doi: 10.1152/ajpcell.00353.2020
55. Valaperti A, Marty RR, Kania G, Germano D, Mauermann N, Dirnhofer S, et al. CD11b+ monocytes abrogate Th17 CD4+ T cell-mediated experimental autoimmune myocarditis. J Immunol (2008) 180:2686–95. doi: 10.4049/jimmunol.180.4.2686
56. Sonderegger I, Röhn TA, Kurrer MO, Iezzi G, Zou Y, Kastelein RA, et al. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur J Immunol (2006) 36:2849–56. doi: 10.1002/eji.200636484
57. Myers JM, Cooper LT, Kem DC, Stavrakis S, Kosanke SD, Shevach EM, et al. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight (2016) 1:e85851. doi: 10.1172/jci.insight.85851
58. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi: 10.1038/nature04753
59. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity (2009) 30:576–87. doi: 10.1016/j.immuni.2009.02.007
60. Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med (2008) 205:2281–94. doi: 10.1084/jem.20071119
61. Van Der Borght K, Scott CL, Martens L, Sichien D, Van Isterdael G, Nindl V, et al. Myocarditis elicits dendritic cell and monocyte infiltration in the heart and self-antigen presentation by conventional type 2 dendritic cells. Front Immunol (2018) 9:2714. doi: 10.3389/fimmu.2018.02714
62. Hilligan KL, Ronchese F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell Mol Immunol (2020) 17:587–99. doi: 10.1038/s41423-020-0465-0
63. Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett (2003) 85:85–95. doi: 10.1016/S0165-2478(02)00228-6
64. Frevert CW, Felgenhauer J, Wygrecka M, Nastase MV, Schaefer L. Danger-Associated Molecular Patterns Derived From the Extracellular Matrix Provide Temporal Control of Innate Immunity. J Histochem Cytochem (2018) 66:213–27. doi: 10.1369/0022155417740880
65. Zuurbier CJ, Abbate A, Cabrera-Fuentes HA, Cohen MV, Collino M, De Kleijn DPV, et al. Innate immunity as a target for acute cardioprotection. Cardiovasc Res (2019) 115:1131–42. doi: 10.1093/cvr/cvy304
66. Popovic ZV, Wang S, Papatriantafyllou M, Kaya Z, Porubsky S, Meisner M, et al. The proteoglycan biglycan enhances antigen-specific T cell activation potentially via MyD88 and TRIF pathways and triggers autoimmune perimyocarditis. J Immunol (2011) 187:6217–26. doi: 10.4049/jimmunol.1003478
Keywords: tenascin-C, myocarditis, inflammatory cardiomyopathy, autoimmunity, extracellular matrix
Citation: Tajiri K, Yonebayashi S, Li S and Ieda M (2021) Immunomodulatory Role of Tenascin-C in Myocarditis and Inflammatory Cardiomyopathy. Front. Immunol. 12:624703. doi: 10.3389/fimmu.2021.624703
Received: 01 November 2020; Accepted: 06 January 2021;
Published: 22 February 2021.
Edited by:
Gertraud Orend, INSERM Immuno Rhumatologie Moléculaire (IRM), FranceReviewed by:
Przemyslaw Blyszczuk, Jagiellonian University Medical College, PolandCopyright © 2021 Tajiri, Yonebayashi, Li and Ieda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuko Tajiri, a3RhamlyaUBtZC50c3VrdWJhLmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.