 Lourdes Plaza-Rojas
Lourdes Plaza-Rojas José A. Guevara-Patiño
José A. Guevara-Patiño
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 26 February 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.624131
This article is part of the Research TopicImmunology of VitiligoView all 11 articles
Vitiligo is an acquired multifactorial disease that affects melanocytes and results in skin depigmentation. In this review, we examine the role of cells stress and self-reactive T cells responses. Given the canonical and non-canonical functions of NKG2D, such as authenticating stressed target and enhance TCR signaling, we examine how melanocyte stress leads to the expression of ligands that are recognized by the activating receptor NKG2D, and how its signaling results in the turning of T cells against self (melanocyte suicide by proxy). We also discuss how this initiation phase is followed by T cell perpetuation, as NKG2D signaling results in self-sustained long-lasting T cells, with improved cytolytic properties.
At the end of the eighteenth century, Paul Ehrlich introduced the concept of horror autotoxicus, which describes the body's innate aversion to immunological self-destruction. This model remains valid, as the immune system is entrusted with protecting the host against infections while avoiding autoimmunity. It is well-accepted that the killing of target cells by T cells occurs through recognition of the peptide-MHC Class I complex via the T cell receptor (TCR). In the thymus, T cells undergo positive and negative selection based on the TCR signal strength (1). T cells that recognize self-antigens with strong affinity undergo negative selection. In contrast, T cells that weakly respond to self-antigens undergo positive selection and populate the periphery as mature T cells. Thus, while self-reactive T cells are present in the periphery, their TCR interactions with peptide-MHC Class I complex are insufficient to mediate activation. Moreover, additional peripheral mechanisms are also in place to prevent their activation, namely regulatory T cells (Tregs) (2, 3).
Under certain pathophysiological conditions, T cells are responsible for several autoimmune disorders, including vitiligo. Vitiligo is an autoimmune disease characterized by the progressive loss of skin pigmentation as a result of melanocyte destruction. In this context, T cells are known to target and kill melanocytes, the pigment-producing cells in the skin. Vitiligo is the most common skin pigmentation disorder, affecting 0.1–2% of the population worldwide (4, 5), with no sex bias (6–9). While it can affect people at all ages, vitiligo appears more frequently before 20 years of age and its early onset (during childhood) is associated with hereditary disease (5, 7, 9–11). Vitiligo is characterized by white lesions on the skin, and the associated psychological and social effects make patients more prone to depression and low self-esteem (12). Regrettably, there are currently no FDA-approved treatments for vitiligo (13). Although great progress has been made throughout the past decades in elucidating the mechanisms that cause this disease, it remains incurable.
While T cells turn against melanocytes for reasons that remain largely unknown, recent studies suggest that CD8 T cells kill melanocytes through the recognition of stressed/damaged cells via Natural Killer Group 2D (NKG2D). NKG2D is a type II transmembrane receptor encoded by the killer cell lectin-like receptor subfamily K member 1 (Klrk1) gene (14, 15) expressed by CD8 T cells, NK cells, NKT cells, and a subset of γδ T cells, and by CD4 T cells in some pathologies (16, 17). Although NKG2D is expressed in both NK and CD8 T cells, the mechanisms of action differ. In NK cells, NKG2D signaling is sufficient to mediate direct killing of target cells (18). However, in CD8 T cells, NKG2D requires the concurrent activation of the TCR. In this case, NKG2D enhances TCR activation and thus, T cell function (18–20).
A number of elements are thought to contribute to the initiation of vitiligo, including genetic predisposition (21, 22), reduced proliferative capacity of melanocytes, increased oxidative stress in melanocytes, the subsequent expression of “danger” signals, reduced presence and function of Tregs, and increased activation of a self-reactive T cell repertoire (7, 23, 24). Moreover, there is evidence of an association between early inflammatory events (e.g., exposure to UV light, a bleaching agent, phenols, or trauma) and vitiligo (25, 26). We believe this is of significance, as the expression of NKG2D ligands (NKG2DL) is upregulated in response to cell stress (insult), rendering these cells visible to NKG2D-expressing immune cells (14, 18–20, 27). The ligands for NKG2D are composed of a variety of stress molecules, including the major histocompatibility complex (MHC) class-I-related chain (MIC) A/B and ULBP binding proteins 1–6 in humans (16) and members of the Rae-1 and H60a-c families in mice (28, 29). Importantly, NKG2DL expression on the cell surface is rigidly controlled. In contrast to physiological conditions in which few or no NKG2DL are expressed, their expression is induced under stress conditions, such as infection and transformation (28, 29). Of significance, proinflammatory signals such as IFN-α (30) and TLR4 and TLR7/8 signaling (28), as well as the ataxia telangiectasia mutated and Rad3-related (ATM/ATR) DNA damage response pathway also result in surface expression of these ligands (20, 27, 31, 32). Thus, the canonical function of NKG2D is to authenticate the stressed/damaged feature of the target cells. This cell authentication occurs in part by favoring the TCR signaling.
Notably, studies from our group have established in animal models that NKG2D signaling also leads to the development of long-lasting CD8 T cells with enhanced cytolytic function (33–35). NKG2DL upregulation and NKG2D signaling activation in effector CD8 T cells play a role in the onset or development of some autoimmune diseases, including vitiligo, rheumatoid arthritis, celiac disease, type 1 diabetes, alopecia areata, systemic lupus erythematosus (SLE), among others (36–41). Moreover, inhibiting NKG2D engagement can prevent inflammation and disease development in some models of type 1 diabetes (42), vitíligo (30), and other inflammatory diseases (16).
Here, we describe the relationship between multiple factors that can lead to vitiligo development with a main focus on the involvement of CD8 T cells and NKG2D signaling.
While multiple factors may induce the development of vitiligo, a dysfunction of the redox balance has been largely observed both systemically and in active lesions of vitiligo patients, causing an excessive accumulation of reactive oxygen species (ROS) and inducing cellular stress (43–45). Oxidative stress can arise when molecular oxygen (O2) is converted into oxygen radicals and the production of these species surpasses the anti-oxidant capacity of the cell, causing oxidation of cellular structures and DNA and potentially leading to cell death (46–49). A number of pathologies are associated with excessive ROS generation, which can also lead to inflammation, particularly in autoimmune diseases (50). Given the effects of ROS on inflammation as well as the active role of immune cells in the development of vitiligo, a relationship between oxidative stress and the immune system is apparent in the onset of the disease.
Melanocytes are derived from the neural crest and their main function is to produce the pigment melanin, which is responsible for skin and eye color (51). Keratinocytes, the most abundant cell type in the epidermis, regulate melanocytic functions, including proliferation, melanogenesis, and differentiation (52). Melanocytes and keratinocytes are arranged in epidermal melanin units, in which 1 melanocyte is surrounded by 36 keratinocytes. While its synthesis occurs in melanocytes, melanin is transported to the surrounding keratinocytes (53). This pigment is generated from the oxidation of tyrosine in a multistep reaction during which free radicals may arise (54). These reactions are highly regulated and carried on by a multienzyme complex unique to melanocytes, including the enzymes tyrosinase, TYRP1 and TYRP2 (55).
Exposure to UV light and its absorption by melanocytes causes photo-oxidation of melanin, generating superoxide radicals (54), which in turn induce melanin biosynthesis (56). Melanin creates a supranuclear cap to protect DNA and prevent or reduce its damage (57) and it has also been shown to act as an antioxidant and a free radical scavenger (58–60). However, not only does melanin biosynthesis consist of several oxidation reactions, it also requires energy production via mitochondrial respiration (61). Even though ROS generation has an important role in cell signaling, increased or altered mitochondrial activity may lead to excessive ROS production (62) that results in detrimental effects and apoptosis, as is the case in vitiliginous melanocytes. Although oxidative stress has been observed in both melanocytes and keratinocytes, the latter are less likely to undergo apoptosis. Instead, keratinocytes become senescent in response to UV irradiation (63, 64), and exposure to ROS induces the production of inflammatory cytokines by keratinocytes (65, 66).
Despite an increased O2 consumption rate, mitochondrial respiration and energy production is impaired, while ROS production is increased in vitiliginous melanocytes (67). Higher levels of ROS peroxynitrite and H2O2 are found in the skin of vitiligo patients, in concert with lower concentration of antioxidants and reducing enzymes (43, 68–70). High concentrations of H2O2 disrupt melanin synthesis by inhibiting tyrosinase and dihydropteridine reductase (71). Calreticulin, an endoplasmic reticulum (ER) protein that regulates Ca2+ homeostasis and signaling, is also modulated by H2O2, which increases calreticulin expression and translocation to the cell surface of melanocytes. This is associated with higher melanocyte apoptosis and production of pro-inflammatory cytokines IL-6 and TNF-α in vitiligo (72). As will be further discussed, pro-inflammatory cytokines have an important role in the vitiligo-associated immune response.
High ROS can also induce ER oxidation; in particular, H2O2 can interfere with the ion channel TRPM2, resulting in higher Ca2+ influx into the cell and the mitochondria (73). Increased mitochondrial Ca2+ concentration ([Ca2+]), in addition to augmented ROS production, induces a reduction of the mitochondrial membrane potential (ΔΨm) in melanocytes and the circulating mononuclear cells of vitiligo patients (44, 72). While melanin is known to chelate Ca2+ and control intracellular [Ca2+], protecting the cell from DNA damage caused by ROS or UV rays, eventually, these alterations may lead to cytochrome c release and apoptosis (74, 75). In the ER, ROS induces accumulation of dysfunctional and unfolded proteins, triggering the unfolded protein response (UPR) within vitiliginous melanocytes, potentially leading to cell death (76). Moreover, aberrant self-proteins, perceived as foreign antigens, can elicit immune responses (77). Therefore, oxidative stress in vitiligo skin is likely to precede the autoimmune response. In particular, NKG2DL expression can be triggered by ROS in multiple cell types and in cancer (78–83). NKG2D-mediated activation of CD8 T cells may thus play a significant role in the development of vitiligo, as discussed below.
NKG2D is an activating receptor that is expressed by many cytotoxic lymphocytes and binds to a variety of ligands (i.e., MICA and Rae-1 in human and mouse, respectively) expressed on stressed cells. Studies including ours have shown that the function of these ligands is to enable stressed cell recognition and destruction in a suicide by proxy mechanism, facilitated by immune cells expressing NKG2D (34, 84, 85). Interestingly, increased expression of NKG2DL has been shown in response to multiple insults, including oxidative cell stress (86, 87). Moreover, studies have shown an association between CD8 T cells expressing NKG2D and various autoimmune diseases, including skin conditions such as alopecia areata (88–90), vitiligo, pancreatitis, diabetes (91), and celiac disease (39, 92). For example, in type 1 diabetes, NKG2D engagement is necessary for the development of the disease (42) and destruction of β-cells in the pancreas by CD8 T cells has been compared to melanocyte death in skin patches of vitiligo patients (93, 94).
In a mouse model of vitiligo, we show that engagement of NKG2D results in exacerbation of CD8 T cell-mediated vitiligo (34). Consistent with this, melanocyte-reactive T cells are further activated and drawn to stressed melanocytes that express NKG2DL (19). Moreover, NKG2D-expressing T cells are enriched in vitiligo patients (30). Thus, understanding the mechanisms that regulate NKG2DL expression on stressed melanocytes can lead to development of therapeutic approaches targeting the interactions of NKG2D+ self-reactive CD8 T cells with melanocytes in vitiligo.
The expression of NKG2DL is upregulated in response to cell stress, rendering stressed or altered cells visible to NKG2D-expressing CD8 T cells. Studies have shown that stress signals induce the expression of NKG2DL on most cell types, including melanocytes (30). Increased expression of such signals in healthy tissue may induce the destruction and inflammation associated with autoimmune diseases (36, 37, 39, 41, 42, 88, 95–97). In fact, melanocyte-reactive T cells, normally suppressed by Tregs, have been found in circulating blood of individuals that do not exhibit vitiligo (98). While such self-reactive cytolytic T cells can be exploited for anti-tumor therapy, loss of functional Treg cells can result in the onset of vitiligo (98, 99).
NKG2DL expression on melanocytes may also drive the onset of the disease. This was demonstrated in a mouse model which revealed that NKG2DL expression on target cells is sufficient to induce vitiligo (34). Accordingly, melanocyte-reactive T cells also have a higher NKG2D expression. In particular, a subset of tissue-resident memory CD8 T (TRM) cells populating lesion areas in the skin of vitiligo patients was shown to have increased NKG2D levels and to be responsible for the increased production of IFN-γ and TNF-α (30). This TRM cell activation was mediated not only by IL-15, a key cytokine in memory formation, but also by skin dendritic cells (DCs) expressing human NKG2DL MICA/B (30). Engagement of NKG2D on CD8 T cells in combination with IL-15 stimulation can trigger the killing of target cells, a feature of autoimmune diseases (39, 84, 95, 100–102). While NKG2D plays an important role in the effector phase of T cells, we have shown that engagement of this receptor also has long-term effects, including the promotion of memory formation (35). In the mentioned study, we show that signaling through NKG2D mediates a process that we termed memory certification. We found that temporary blockade of NKG2D signaling during the effector phase resulted in the formation of highly defective memory CD8 T cells characterized by altered expression of the ribosomal protein S6 and epigenetic modifiers, suggesting modifications in the T cell translational machinery and epigenetic programming. Based on these data, we concluded that NKG2D signaling during this initial effector phase, poises NKG2D engaged cells with a certification or molecular accreditation that results in their optimal development as memory T cells. This process of certification guarantees that the memory compartment is populated with CD8 T cells that have demonstrated their ability to kill the correct targets through a two-step process that utilizes the TCR and NKG2D signaling. Interestingly, in this system, NKG2D-certtified memory T cells will be able to kill melanocytes independently of NKG2D, thus resulting in the destruction of healthy melanocytes. We postulate that melanocyte-reactive CD8 T cells that receive NKG2D signaling during the killing phase will also persist as memory cells and potentially enrich the skin TRM population in vitiligo patients.
Heat shock protein are stress-inducible chaperones that protect cells from undergoing apoptosis through several mechanisms, including the binding and renaturing or degradation of misfolded proteins during the stress response (103). In particular, Hsp70 expression in tumor cells has been shown to elicit both innate and adaptive immune responses (104–108). In vitiligo, stressed melanocytes increase the expression of Hsp70, which binds and transports potentially immunogenic antigens to the MHC complex, allowing for their presentation on the cell surface to cytotoxic T cells (109). Moreover, free Hsp70 also triggers an immune response by interacting with DC surface receptors (107, 110, 111), inducing the expression of human NKG2DL MICA (107). This acts as a danger signal, rendering stressed cells visible to both NK and CD8 T cells expressing NKG2D. Hsp70 expression can be induced not only by physical and chemical stress, but also in physiological conditions, such as under high ROS production, as seen in vitiligo and other pathologies (110, 112, 113). Expression of both inducible HSP70 and NKG2DL is controlled by the transcriptional activator heath shock factor 1 (HSF1) (83). In response to cell stress, HSF1 is released in the cytoplasm. Following nuclear translocation, HSF1 binds to the promoter regions of both HSP70i and NKG2DL, initiating their transcription. These observations raise a novel paradox: while the canonical function of intracellular HSP70i is cytoprotectant, expression of NKG2DL results in cell suicide by proxy. While it is not known how melanocytes survive or render themselves visible for killing after cell stress, we believe that focusing on answering this question may in turn help identify actionable therapies in vitiligo.
Environmental insults can trigger inflammation and result in the onset of vitiligo, especially in adulthood (25, 26). For example, monobenzone, a skin bleaching agent is used to complete skin depigmentation in vitiligo patients. Monobenzone induces ROS generation and oxidative stress in the skin (114), further contributing to the autoimmune response in vitiligo. In addition, monobenzone is converted into a reactive quinone by tyrosinase (114); these quinone products bind covalently to proteins in melanocytes, forming aberrant antigens targeted by T cells (115). Therefore, monobenzone induces a systemic T cell response, effectively targeting all melanocytes in the skin.
Cytokines have a crucial role in regulating immune responses. High levels of cytokines are often found in vitiligo patients, including cytokines corresponding to Th1, Th17, and innate cell responses (116). Interestingly, keratinocytes in vitiligo lesions aberrantly produce IL-1, IL-6, and TNF-α, which inhibit melanocyte function (65, 66) and elicit an inflammatory response.
The production of pro-inflammatory cytokines allows for enhanced T cell recruitment and activation, resulting in increased presence of activated CD8 T cells in vitiligo lesions (13, 117–122). Produced by multiple immune cell types, IFN-γ is associated with vitiligo. IFN-γ promotes the production of other cytokines and chemokines, and the recruitment of T cells as well as other types of immune cells (123). Studies show that IFN-γ directly induces melanocyte apoptosis (124, 125) and its signaling enhances CD8 T cell function and expansion (126). Notably, we and others have demonstrated that NKG2D signaling upregulates IFN-γ production (18, 33). IFN-γ also promotes the expression of MHC Class I and therefore antigen presentation for CD8 T cells. IFN-γ is also responsible for stimulating the production of CXCL9 and CXCL10 in keratinocytes, which engages the chemokine receptor CXCR3 on melanocytes and triggers apoptosis, further contributing to the disease (127).
TNF-α is also produced at high levels in vitiligo (128). While its role as an apoptotic mediator has been shown in multiple cell types (129), in vitiligo, TNF-α inhibits melanogenesis by activating the transcription factor NF-κB (130). In addition, it induces the expression of ICAM-1 on melanocytes (131), allowing for the attachment of T cells and triggering cytotoxicity (132). Moreover, TNF-α inhibits tyrosinase and Trp1 activity, both essential for melanin synthesis (61). ROS production is also increased by TNF-α, further promoting stress signals in vitiligo melanocytes.
While keratinocytes can contribute to the production of inflammatory factors, melanocyte-reactive CD8 T cells are sufficient to initiate the development of vitiligo, as it has been shown that perilesional T cells from vitiligo patients can recognize and kill melanocytes in healthy skin (122). Activated self-reactive T cells remain in the skin and differentiate into TRM cells. This differentiation is mediated by IL-15 derived from keratinocytes (133). Blockade of IL-15Rβ (CD122) reduces IFN-γ production and can eliminate skin TRM and reverse vitiligo (133).
Another indicator of an existing melanocyte-reactive T cell population is the development of vitiligo in melanoma patients who receive immunotherapy (134–140). In patients without the right stimuli, these T cells do not mount a response, but can potentially be activated for the treatment of cancer. This observation also indicates the possibility to treat melanoma with agents known to trigger vitiligo, such as skin bleaching agents (114, 115).
In addition to a high production of pro-inflammatory cytokines, there is a corresponding reduction in suppressive cytokines in the skin of vitiligo patients. Tregs maintain immune tolerance by controlling self-reactive cytotoxic T cells. In healthy adults, Tregs constitute about half of the CD4 T cell population in the skin (141). In vitiligo, however, a much lower percentage of Tregs is often found surrounding lesions (23), which has been associated with reduced levels of Treg growth and differentiation factors TGF-β and IL-10 (142, 143). In particular, low TGF-β concentrations in both serum and skin have been found to correlate with skin depigmentation in vitiligo (142, 144). In addition, there is an association between reduced expression of CCL22, a skin-homing chemokine, and Treg infiltration in vitiligo skin, suggesting impaired migration of Tregs to the skin in vitiligo patients (24). These events can thus result in the alteration of the Treg/cytotoxic T cell ratio, impaired Treg differentiation, and increased inflammation in vitiligo.
In summation, the imbalance of pro-inflammatory and suppressive cytokines further contributes to disease progression. The interplay of these messengers, the immune system, and the affected skin cells is being actively investigated to identify novel therapeutic avenues.
Genetic predisposition is heavily linked to an overreactive immune system and patients often exhibit more than one autoimmune disease. While the inflammation and stress observed in vitiligo patients can have a variety of causes, including environmental, there is also a high risk of vitiligo heritability, with about 8% of patients having at least one affected relative (7). Gene analysis studies have revealed a higher expression of Class II MHC haplotypes HLA-DR and HLA-DQ in Caucasian patients resulting from a polymorphism in a super-enhancer within the Class II region (6, 145). Moreover, there is a correlation in the expression of such MHC haplotypes and a higher production of pro-inflammatory cytokines IFN-γ and IL-1β (6). In addition, mutations affecting other loci implicated in immune regulation have been associated with vitiligo and other autoimmune diseases. These genes include those encoding IL-2Rα (146), the TCR signaling regulator UBASH3A (146), and CCL20 receptor CCR6 (147), among others (21). While these reports indicate that there is an association between immune regulation and the onset of autoimmunity and vitiligo, there is also evidence that some polymorphisms lead to reactivity specifically against melanocytes. These include peptides derived from tyrosinase, the transmembrane protein OCA2, and melanocortin 1 receptor (MC1R), all of which are naturally processed and presented as antigens by Class I MHC molecules (5, 148, 149). This, in combination with higher MHC expression, increases the potential for the presentation of melanocyte-specific antigens and for self-reactive T cells targeting these cells.
Reduced function and number of the tolerogenic Tregs has also been associated with the development of vitiligo. Genetic polymorphisms in Foxp3, a transcription factor critical for Treg differentiation, and transcription factor FoxP1 (147), also critical for Treg homeostasis (150, 151), have been associated with Treg dysfunction in vitiligo (23, 152–154). Similarly, variants in the genes encoding TGF-β receptor II (155) and IL-10 (156) may affect Treg-mediated immune suppression in vitiligo. As mentioned, both TGF-β and IL-10 are cytokines essential for Treg differentiation and development (157–159).
Additionally, transcriptional analyses have demonstrated altered expression of factors involved in proliferation, apoptosis and regeneration of melanocytes (160). This is supported by the observation that vitiliginous melanocytes display poor proliferative capacity and cannot be sub-cultured in vitro (161, 162).
Aberrant expression of miRNAs has also been observed in vitiligo (163–165). In particular, miRNA-211 is highly expressed in healthy melanocytes, but its expression is severely reduced in non-pigmented melanoma and in the vitiligo cell line PIG3V (22). miRNA-211 target genes have functions involved in cell respiration and metabolism, including mitochondrial complexes I, II, and IV. In consequence, reduced miRNA-211 expression in vitiligo is associated with increased O2 consumption and may partially explain the oxidative stress observed in patients (22, 166).
Vitiligo is a complex disease with multiple causal factors, including genetic predisposition, enhanced self-antigen presentation, and increased pro-inflammatory signals. Inflammation is known to induce DNA and cell damage and increased ROS production, triggering the expression of NKG2DL on melanocytes. It is well-accepted that the interaction between the autoantigen:MHC-I complex and the self-reactive TCR on CD8 T cells is weak. However, under stress and inflammation, the recognition of both autoantigen and NKG2DL by NKG2D-expressing T cells causes the reduction of the T cell activating threshold (Figure 1). In this scenario, the expression of NKG2D ligands on stressed melanocytes is sensed by the immune system as an instruction for destruction (suicide by proxy). This initiation phase is followed by the perpetuation phase: once T cells have killed stressed melanocytes via engagement of both the TCR and NKG2D, these T cells are endowed with a transcriptional program that enables them to become long-lasting, with enhanced cytolytic properties as well as independence from immunological help. Evidence suggests that these NKG2D-engaged T cells are then responsible for the killing of healthy melanocytes in an NKG2D-independent mechanism, thus perpetuating the disease.
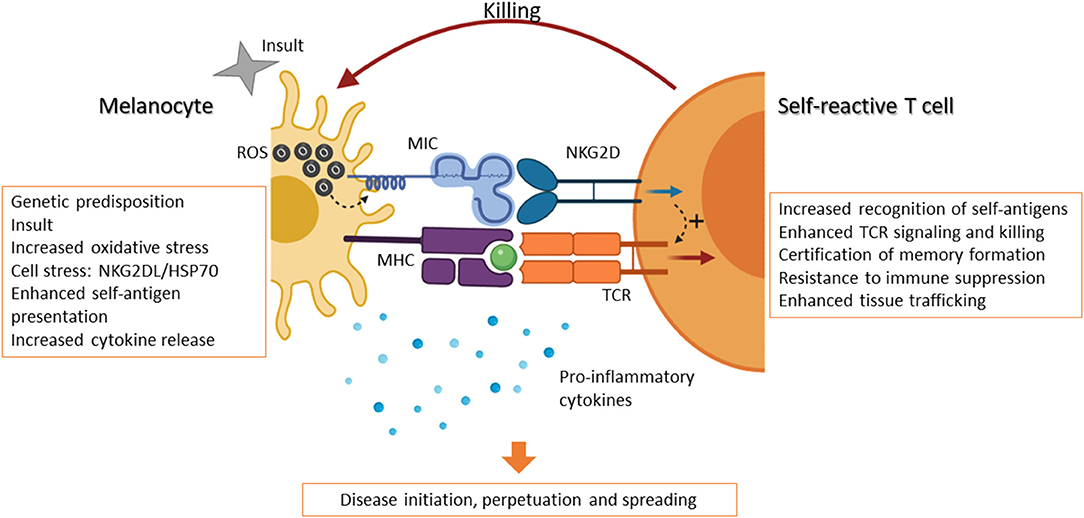
Figure 1. NKG2D-expressing T cells initiate and perpetuate vitiligo. The occurrence of vitiligo can be associated with genetic predisposition and hereditary disease, which in turned is linked to an overreactive immune system. Expression of certain MHC variants, for example, can lead to increased self-antigen presentation. However, other contributing factors can trigger the onset of the disease. An inflammatory event, or insult, may lead to increased ROS production and oxidative stress in melanocytes and their surroundings. In response to this, the production of cytokines by immune cells and keratinocytes can further induce inflammation and an immune response. Melanocytes in turn express “danger” signals, including NKG2D ligands (e.g., MICA/B in humans) and chaperone proteins, such as hsp70. Self-reactive cytolytic T cells are thus able to recognize melanocytes through the TCR and become activated via the engagement of NKG2D, whose signaling further enhances the downstream effects of TCR signaling, including cytotoxicity. Moreover, NKG2D activation certifies these self-reactive T cells to become memory cells and renders them more resistant to immune suppression. These certified T cells have enhanced local trafficking and are able to continue killing melanocytes. These events, in combination with reduced suppression by dysfunctional Treg cells, result in the onset, perpetuation, and spreading of vitiligo. Created with BioRender.com.
JG-P and LP-R prepared and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the NIH R01CA188395, R01CA250514, and the generous gift from the Volo Family Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Daniels MA, Teixeiro E, Gill J, Hausmann B, Roubaty D, Holmberg K, et al. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature. (2006) 444:724–9. doi: 10.1038/nature05269
2. Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4 + CD25 high regulatory cells in human peripheral blood. J Immunol. (2001) 167:1245–53. doi: 10.4049/jimmunol.167.3.1245
3. Barthlott T, Moncrieffe H, Veldhoen M, Atkins CJ, Christensen J, O'Garra A, et al. CD25+CD4+ T cells compete with naive CD4+ T cells for IL-2 and exploit it for the induction of IL-10 production. Int Immunol. (2005) 17:279–88. doi: 10.1093/intimm/dxh207
4. Zhang Y, Cai Y, Shi M, Jiang S, Cui S, Wu Y, et al. The prevalence of vitiligo: a meta-analysis. PLoS ONE. (2016) 11:163806. doi: 10.1371/journal.pone.0163806
5. Jin Y, Birlea SA, Fain PR, Ferrara TM, Ben S, Riccardi SL, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. (2012) 44:676–80. doi: 10.1038/ng.2272
6. Cavalli G, Hayashi M, Jin Y, Yorgov D, Santorico SA, Holcomb C, et al. MHC class II super-enhancer increases surface expression of HLA-DR and HLA-DQ and affects cytokine production in autoimmune vitiligo. Proc Natl Acad Sci USA. (2016) 113:1363–8. doi: 10.1073/pnas.1523482113
7. Alkhateeb A, Fain PR, Thody A, Bennett DC, Spritz RA. Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families. Pigment Cell Res. (2003) 16:208–14. doi: 10.1034/j.1600-0749.2003.00032.x
8. Bergqvist C, Ezzedine K. Vitiligo: a review. Dermatology. (2020) 236:571–92. doi: 10.1159/000506103
9. Ezzedine K, Eleftheriadou V, Whitton M, Van Geel N. Vitiligo. Lancet. (2015) 386:74–84. doi: 10.1016/S0140-6736(14)60763-7
10. Skov J, Eriksson D, Kuja-Halkola R, Höijer J, Gudbjörnsdottir S, Svensson AM, et al. Co-aggregation and heritability of organ-specific autoimmunity: a population-based twin study. Eur J Endocrinol. (2020) 182:473. doi: 10.1530/EJE-20-0049
11. Handa S, Kaur I. Vitiligo: clinical findings in 1436 patients. J Dermatol. (1999) 26:653–7. doi: 10.1111/j.1346-8138.1999.tb02067.x
12. Ezzedine K, Lim HW, Suzuki T, Katayama I, Hamzavi I, Lan CCE, et al. Revised classification/nomenclature of vitiligo and related issues: the Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. (2012) 25:E1–13. doi: 10.1111/j.1755-148X.2012.00997.x
13. Ezzedine K, Sheth V, Rodrigues M, Eleftheriadou V, Harris JE, Hamzavi IH, et al. Vitiligo is not a cosmetic disease. J Am Acad Dermatol. (2015) 73:883–5. doi: 10.1016/j.jaad.2015.07.039
14. Houchins JP, Yabe T, McSherry C, Bach FH. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J Exp Med. (1991) 173:1017–20. doi: 10.1084/jem.173.4.1017
15. Brown MG, Fulmek S, Matsumoto K, Robert C, Lyons PA, Levy ER, et al. A 2-Mb YAC contig and physical map of the natural killer gene complex on mouse chromosome 6. Genomics. (1997) 42:16–25. doi: 10.1006/geno.1997.4721
16. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. (2003) 3:781–90. doi: 10.1038/nri1199
17. Sáez-Borderías A, Gumá M, Angulo A, Bellosillo B, Pende D, López-Botet M. Expression and function of NKG2D in CD4+ T cells specific for human cytomegalovirus. Eur J Immunol. (2006) 36:3198–206. doi: 10.1002/eji.200636682
18. Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. (2002) 17:19–29. doi: 10.1016/S1074-7613(02)00333-3
19. Bauer S, Groh V, Wu J, Steinle a, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. (1999) 285:727–9. doi: 10.1126/science.285.5428.727
20. Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol. (2000) 1:119–26. doi: 10.1038/77793
21. Jin Y, Andersen G, Yorgov D, Ferrara TM, Ben S, Brownson KM, et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat Genet. (2016) 48:1418–24. doi: 10.1038/ng.3680
22. Sahoo A, Lee B, Boniface K, Seneschal J, Sahoo SK, Seki T, et al. MicroRNA-211 regulates oxidative phosphorylation and energy metabolism in human vitiligo. J Invest Dermatol. (2017) 137:1965–74. doi: 10.1016/j.jid.2017.04.025
23. Dwivedi M, Kemp EH, Laddha NC, Mansuri MS, Weetman AP, Begum R. Regulatory T cells in vitiligo: implications for pathogenesis and therapeutics. Autoimmun Rev. (2015) 14:49–56. doi: 10.1016/j.autrev.2014.10.002
24. Klarquist J, Denman CJ, Hernandez C, Wainwright DJ, Strickland FM, Overbeck A, et al. Reduced skin homing by functional Treg in vitiligo. Pigment Cell Melanoma Res. (2010) 23:276–86. doi: 10.1111/j.1755-148X.2010.00688.x
25. O'Malley MA, Mathias CGT, Priddy M, Molina D, Grote AA, Halperin WE. Occupational vitiligo due to unsuspected presence of phenolic antioxidant byproducts in commercial bulk rubber. J Occup Med. (1988) 30:512–6. doi: 10.1097/00043764-198806000-00014
26. Le Poole IC, Wañkowicz-Kaliñska A, Van Den Wijngaard RMJGJ, Nickoloff BJ, Das PK. Autoimmune aspects of depigmentation in vitiligo. J Investig Dermatol Symp Proc. (2004) 9:68–72. doi: 10.1111/j.1087-0024.2004.00825.x
27. Cerwenka A, Bakker ABH, McClanahan T, Wagner J, Wu J, Phillips JH, et al. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. (2000) 12:721–7. doi: 10.1016/S1074-7613(00)80222-8
28. Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci USA. (2001) 98:11521–6. doi: 10.1073/pnas.201238598
29. O'Callaghan CA, Cerwenka A, Willcox BE, Lanier LL, Bjorkman PJ. Molecular competition for NKG2D: H60 and RAE1 compete unequally for NKG2D with dominance of H60. Immunity. (2001) 15:201–11. doi: 10.1016/S1074-7613(01)00187-X
30. Jacquemin C, Martins C, Lucchese F, Thiolat D, Taieb A, Seneschal J, et al. NKG2D defines a subset of skin effector memory CD8 T cells with proinflammatory functions in vitiligo. J Invest Dermatol. (2020) 140:1143–53.e5. doi: 10.1016/j.jid.2019.11.013
31. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. (2005) 436:1186–90. doi: 10.1038/nature03884
32. Cosman D, Müllberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. (2001) 14:123–33. doi: 10.1016/S1074-7613(01)00095-4
33. Zloza A, Kohlhapp FJ, Lyons GE, Schenkel JM, Moore TV, Lacek AT, et al. NKG2D signaling on CD8+ T cells represses T-bet and rescues CD4− unhelped CD8+ T cell memory recall but not effector responses. Nat Med. (2012) 18:422–8. doi: 10.1038/nm.2683
34. Zloza A, Lyons GE, Chlewicki LK, Kohlhapp FJ, O'Sullivan JA, Lacek AT, et al. Engagement of NK receptor NKG2D, but not 2B4, results in self-reactive CD8+ T cells and autoimmune vitiligo. Autoimmunity. (2011) 44:599–606. doi: 10.3109/08916934.2011.593599
35. Perez C, Prajapati K, Burke B, Plaza-Rojas L, Zeleznik-Le NJ, Guevara-Patino JA. NKG2D signaling certifies effector CD8 T cells for memory formation. J Immunother Cancer. (2019) 7:48. doi: 10.1186/s40425-019-0531-2
36. Groh V, Brühl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci USA. (2003) 100:9452–7. doi: 10.1073/pnas.1632807100
37. Andersson AK, Sumariwalla PF, McCann FE, Amjadi P, Chang C, McNamee K, et al. Blockade of NKG2D ameliorates disease in mice with collagen-induced arthritis: a potential pathogenic role in chronic inflammatory arthritis. Arthritis Rheum. (2011) 63:2617–29. doi: 10.1002/art.30460
38. Antonangeli F, Soriani A, Cerboni C, Sciumè G, Santoni A. How mucosal epithelia deal with stress: role of NKG2D/NKG2D ligands during inflammation. Front Immunol. (2017) 8:1583. doi: 10.3389/fimmu.2017.01583
39. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz T, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. (2004) 21:357–66. doi: 10.1016/J.IMMUNI.2004.06.020
40. Ma H, Lu Y, Lowe K, van der Meijden-Erkelens L, Wasserfall C, Atkinson MA, et al. Regulated hAAT expression from a novel rAAV vector and its application in the prevention of type 1 diabetes. J Clin Med. (2019) 8:1321. doi: 10.3390/jcm8091321
41. Qin H, Lee IF, Panagiotopoulos C, Wang X, Chu AD, Utz PJ, et al. Natural killer cells from children with type 1 diabetes have defects in NKG2D-dependent function and signaling. Diabetes. (2011) 60:857–66. doi: 10.2337/db09-1706
42. Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, et al. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity. (2004) 20:757–67. doi: 10.1016/j.immuni.2004.05.008
43. Schallreuter KU, Moore J, Wood JM, Beazley WD, Gaze DC, Tobin DJ, et al. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a UVB-activated pseudocatalase. J Investig Dermatol Symp Proc. (1999) 4:91–6. doi: 10.1038/sj.jidsp.5640189
44. Dell'Anna ML, Urbanelli S, Mastrofrancesco A, Camera E, Iacovelli P, Leone G, et al. Alterations of mitochondria in peripheral blood mononuclear cells of vitiligo patients. Pigment Cell Res. (2003) 16:553–9. doi: 10.1034/j.1600-0749.2003.00087.x
45. Morrone A, Picardo M, De Luca C, Terminali O, Passi S, Ippolito F. Catecholamines and vitiligo. Pigment Cell Res. (1992) 5:65–9. doi: 10.1111/j.1600-0749.1992.tb00003.x
46. Bhattacharyya S, Saha J. Tumour, oxidative stress and host T cell response: cementing the dominance. Scand J Immunol. (2015) 82:477–88. doi: 10.1111/sji.12350
47. Bickers DR, Athar M. Oxidative stress in the pathogenesis of skin disease. J Invest Dermatol. (2006) 126:2565–75. doi: 10.1038/sj.jid.5700340
48. Bossi O, Gartsbein M, Leitges M, Kuroki T, Grossman S, Tennenbaum T. UV irradiation increases ROS production via PKCδ signaling in primary murine fibroblasts. J Cell Biochem. (2008) 105:194–207. doi: 10.1002/jcb.21817
49. Rinnerthaler M, Bischof J, Streubel M, Trost A, Richter K. Oxidative stress in aging human skin. Biomolecules. (2015) 5:545–89. doi: 10.3390/biom5020545
50. Di Dalmazi G, Hirshberg J, Lyle D, Freij JB, Caturegli P. Reactive oxygen species in organ-specific autoimmunity. Autoimmun Highlights. (2016) 7:11. doi: 10.1007/s13317-016-0083-0
51. Nordlund JJ, Abdel-Malek ZA, Boissy RE, Rheins LA. Pigment cell biology: an historical review. J Invest Dermatol. (1989) 92:S53–60. doi: 10.1038/jid.1989.33
52. Hirobe T. Structure and function of melanocytes: microscopic morphology and cell biology of mouse melanocytes in the epidermis and hair follicle. Histol Histopathol. (1995) 10:223–37.
53. Nordlund JJ. The melanocyte and the epidermal melanin unit: an expanded concept. Dermatol Clin. (2007) 25:271–81. doi: 10.1016/j.det.2007.04.001
54. Riley PA. Melanin. Int J Biochem Cell Biol. (1997) 29:1235–9. doi: 10.1016/S1357-2725(97)00013-7
55. D'Mello SAN, Finlay GJ, Baguley BC, Askarian-Amiri ME. Signaling pathways in melanogenesis. Int J Mol Sci. (2016) 17:1144. doi: 10.3390/ijms17071144
56. Slominski A, Tobin DJ, Shibahara S, Wortsman J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. (2004) 84:1155–228. doi: 10.1152/physrev.00044.2003
57. Kobayashi N, Nakagawa A, Muramatsu T, Yamashina Y, Shirai T, Hashimoto MW, et al. Supranuclear melanin caps reduce ultraviolet induced DNA photoproducts in human epidermis. J Invest Dermatol. (1998) 110:806–10. doi: 10.1046/j.1523-1747.1998.00178.x
58. Song X, Mosby N, Yang J, Xu A, Abdel-Malek Z, Kadekaro AL. α-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes. Pigment Cell Melanoma Res. (2009) 22:809–18. doi: 10.1111/j.1755-148X.2009.00615.x
59. Hoogduijn M, Cemeli E, Ross K, Anderson D, Thody A, Wood J. Melanin protects melanocytes and keratinocytes against H2O2-induced DNA strand breaks through its ability to bind Ca2+. Exp Cell Res. (2004) 294:60–7. doi: 10.1016/j.yexcr.2003.11.007
60. Brenner M, Hearing VJ. The protective role of melanin against UV damage in human skin. Photochem Photobiol. (2008) 84:539–49. doi: 10.1111/j.1751-1097.2007.00226.x
61. Koga S, Nakano M, Tero-Kubota S. Generation of superoxide during the enzymatic action of tyrosinase. Arch Biochem Biophys. (1992) 292:570–5. doi: 10.1016/0003-9861(92)90032-R
62. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417:1–13. doi: 10.1042/BJ20081386
63. Lewis DA, Travers JB, Somani AK, Spandau DF. The IGF-1/IGF-1R signaling axis in the skin: a new role for the dermis in aging-associated skin cancer. Oncogene. (2010) 29:1475–85. doi: 10.1038/onc.2009.440
64. Passi S, Grandinetti M, Maggio F, Stancato A, Luca C. Epidermal oxidative stress in vitiligo. Pigment Cell Res. (1998) 11:81–5. doi: 10.1111/j.1600-0749.1998.tb00714.x
65. Luger TA. Epidermal cytokines. Acta Derm Venereol Suppl. (1989) 151:61–76. doi: 10.1007/978-3-642-83360-1_71
66. Swope VB, Abdel-Malek Z, Kassem LM, Nordlund JJ. Interleukins 1α and 6 and tumor necrosis factor-α are paracrine inhibitors of human melanocyte proliferation and melanogenesis. J Invest Dermatol. (1991) 96:180–5. doi: 10.1111/1523-1747.ep12460991
67. Dell'Anna ML, Ottaviani M, Kovacs D, Mirabilii S, Brown DA, Cota C, et al. Energetic mitochondrial failing in vitiligo and possible rescue by cardiolipin. Sci Rep. (2017) 7:6237. doi: 10.1038/s41598-017-13961-5
68. Salem MMAEL, Shalbaf M, Gibbons NCJ, Chavan B, Thornton JM, Schallreuter KU. Enhanced DNA binding capacity on up-regulated epidermal wild-type p53 in vitiligo by H2O2 -mediated oxidation: a possible repair mechanism for DNA damage. FASEB J. (2009) 23:3790–807. doi: 10.1096/fj.09-132621
69. Schallreuter KU, Salem MAEL, Holtz S, Panske A. Basic evidence for epidermal H2H2/ONOO–mediated oxidation/nitration in segmental vitiligo is supported by repigmentation of skin and eyelashes after reduction of epidermal H2H2 with topical NB-UVB-activated pseudocatalase PC-KUS. FASEB J. (2013) 27:3113–22. doi: 10.1096/fj.12-226779
70. Schallreuter KU, Wood JM, Berger J. Low catalase levels in the epidermis of patients with vitiligo. J Invest Dermatol. (1991) 97:1081–5. doi: 10.1111/1523-1747.ep12492612
71. Hasse S, Gibbons NCJ, Rokos H, Marles LK, Schallreuter KU. Perturbed 6-Tetrahydrobiopterin recycling via decreased dihydropteridine reductase in vitiligo: more evidence for H2O2 stress. J Invest Dermatol. (2004) 122:307–13. doi: 10.1046/j.0022-202X.2004.22230.x
72. Zhang Y, Liu L, Jin L, Yi X, Dang E, Yang Y, et al. Oxidative stress-induced calreticulin expression and translocation: new insights into the destruction of melanocytes. J Invest Dermatol. (2014) 134:183–91. doi: 10.1038/jid.2013.268
73. Kang P, Zhang W, Chen X, Yi X, Song P, Chang Y, et al. TRPM2 mediates mitochondria-dependent apoptosis of melanocytes under oxidative stress. Free Radic Biol Med. (2018) 126:259–68. doi: 10.1016/j.freeradbiomed.2018.08.022
74. Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. (2000) 5:415–8. doi: 10.1023/A:1009616228304
75. Liu B, Jian Z, Li Q, Li K, Wang Z, Liu L, et al. Baicalein protects human melanocytes from H2O2-induced apoptosis via inhibiting mitochondria-dependent caspase activation and the p38 MAPK pathway. Free Radic Biol Med. (2012) 53:183–93. doi: 10.1016/j.freeradbiomed.2012.04.015
76. Kroll TM, Bommiasamy H, Boissy RE, Hernandez C, Nickoloff BJ, Mestril R, et al. 4-Tertiary butyl phenol exposure sensitizes human melanocytes to dendritic cell-mediated killing: Relevance to vitiligo. J Invest Dermatol. (2005) 124:798–806. doi: 10.1111/j.0022-202X.2005.23653.x
77. Xie H, Zhou F, Liu L, Zhu G, Li Q, Li C, et al. Vitiligo: how do oxidative stress-induced autoantigens trigger autoimmunity? J Dermatol Sci. (2016) 81:3–9. doi: 10.1016/j.jdermsci.2015.09.003
78. Eagle R, Jafferji I, Barrow A. Beyond stressed self: evidence for NKG2D ligand expression on healthy cells. Curr Immunol Rev. (2009) 5:22–34. doi: 10.2174/157339509787314369
79. Yamamoto K, Fujiyama Y, Andoh A, Bamba T, Okabe H. Oxidative stress increases MICA and MICB gene expression in the human colon carcinoma cell line (CaCo-2). Biochim Biophys Acta - Gen Subj. (2001) 1526:10–12. doi: 10.1016/S0304-4165(01)00099-X
80. Hedlund M, Nagaeva O, Kargl D, Baranov V, Mincheva-Nilsson L. Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PLoS ONE. (2011) 6:16899. doi: 10.1371/journal.pone.0016899
81. Siemens DR, Hu N, Sheikhi AK, Chung E, Frederiksen LJ, Pross H, et al. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res. (2008) 68:4746–53. doi: 10.1158/0008-5472.CAN-08-0054
82. Barsoum IB, Hamilton TK, Li X, Cotechini T, Miles EA, Siemens DR, et al. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: role of nitric oxide. Cancer Res. (2011) 71:7433–41. doi: 10.1158/0008-5472.CAN-11-2104
83. Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter region architecture and transcriptional regulation of the genes for the MHC class i-related chain A and B ligands of NKG2D. J Immunol. (2007) 178:961–9. doi: 10.4049/jimmunol.178.2.961
84. Van Belle TL, von Herrath MG. The role of the activating receptor NKG2D in autoimmunity. Mol Immunol. (2009) 47:8–11. doi: 10.1016/j.molimm.2009.02.023
85. Raulet DH. Interplay of natural killer cells and their receptors with the adaptive immune response. Nat Immunol. (2004) 5:996–1002. doi: 10.1038/ni1114
86. Kraetzel K, Stoelcker B, Eissner G, Multhoff G, Pfeifer M, Holler E, et al. NKG2D-dependent effector function of bronchial epithelium-activated alloreactive T-cells. Eur Respir J. (2008) 32:563–70. doi: 10.1183/09031936.00096407
87. Borchers MT, Harris NL, Wesselkamper SC, Vitucci M, Cosman D. NKG2D ligands are expressed on stressed human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. (2006) 291:L222–31. doi: 10.1152/ajplung.00327.2005
88. Petukhova L, Duvic M, Hordinsky M, Norris D, Price V, Shimomura Y, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. (2010) 466:113–7. doi: 10.1038/nature09114
89. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. (2014) 20:1043–9. doi: 10.1038/nm.3645
90. Dai Z, Xing L, Cerise J, Wang EHC, Jabbari A, de Jong A, et al. CXCR3 blockade inhibits T cell migration into the skin and prevents development of alopecia areata. J Immunol. (2016) 197:1089–99. doi: 10.4049/jimmunol.1501798
91. Markiewicz MA, Wise EL, Buchwald ZS, Pinto AK, Zafirova B, Polic B, et al. RAE1ε ligand expressed on pancreatic islets recruits NKG2D receptor-expressing cytotoxic T cells independent of T cell receptor recognition. Immunity. (2012) 36:132–41. doi: 10.1016/j.immuni.2011.11.014
92. Tang F, Sally B, Lesko K, Discepolo V, Abadie V, Ciszewski C, et al. Cysteinyl leukotrienes mediate lymphokine killer activity induced by NKG2D and IL-15 in cytotoxic T cells during celiac disease. J Exp Med. (2015) 212:1487–95. doi: 10.1084/jem.20150303
93. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. (2009) 155:173–81. doi: 10.1111/j.1365-2249.2008.03860.x
94. Eisenbarth GS. Banting lecture 2009: an unfinished journey: molecular pathogenesis to prevention of type 1A diabetes. Diabetes. (2010) 59:759–74. doi: 10.2337/db09-1855
95. Hüe S, Mention JJ, Monteiro RC, Zhang SL, Cellier C, Schmitz J, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. (2004) 21:367–77. doi: 10.1016/j.immuni.2004.06.018
96. Allez M, Tieng V, Nakazawa A, Treton X, Pacault V, Dulphy N, et al. CD4+NKG2D+ T cells in Crohn's disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology. (2007) 132:2346–58. doi: 10.1053/j.gastro.2007.03.025
97. Van Belle TL, Ling E, Haase C, Bresson D, Ursø B, Von Herrath MG. NKG2D blockade facilitates diabetes prevention by antigen-specific Tregs in a virus-induced model of diabetes. J Autoimmun. (2013) 40:66–73. doi: 10.1016/j.jaut.2012.08.001
98. Maeda Y, Nishikawa H, Sugiyama D, Ha D, Hamaguchi M, Saito T, et al. Detection of self-reactive CD8+ T cells with an anergic phenotype in healthy individuals. Science. (2014) 346:1536–40. doi: 10.1126/science.aaa1292
99. Richards DM, Ruggiero E, Hofer A-C, Sefrin JP, Schmidt M, von Kalle C, et al. The contained self-reactive peripheral T cell repertoire: size, diversity, and cellular composition. J Immunol. (2015) 195:2067–79. doi: 10.4049/jimmunol.1500880
100. Ruck T, Bittner S, Afzali AM, Göbel K, Glumm S, Kraft P, et al. The NKG2D - IL-15 signaling pathway contributes to T-cell mediated pathology in inflammatory myopathies. Oncotarget. (2015) 6:43230–43. doi: 10.18632/oncotarget.6462
101. Groh V, Smythe K, Dai Z, Spies T. Fas ligand-mediated paracrine T cell regulation by the receptor NKG2D in tumor immunity. Nat Immunol. (2006) 7:755–62. doi: 10.1038/ni1350
102. Saikali P, Antel JP, Pittet CL, Newcombe J, Arbour N. Contribution of astrocyte-derived IL-15 to CD8 t cell effector functions in multiple sclerosis. J Immunol. (2010) 185:5693–703. doi: 10.4049/jimmunol.1002188
103. Beere HM. Death versus survival: Functional interaction between the apoptotic and stress-inducible heat shock protein pathways. J Clin Invest. (2005) 115:2633–9. doi: 10.1172/JCI26471
104. Melcher A, Todryk S, Hardwick N, Ford M, Jacobson M, Vile RG. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat Med. (1998) 4:581–7. doi: 10.1038/nm0598-581
105. Clark PR, Ménoret A. The inducible Hsp70 as a marker of tumor immunogenicity. Cell Stress Chaperones. (2001) 6:121–5. doi: 10.1379/1466-1268(2001)006<0121:TIHAAM>2.0.CO;2
106. Massa C, Guiducci C, Arioli I, Parenza M, Colombo MP, Melani C. Enhanced efficacy of tumor cell vaccines transfected with secretable hsp70. Cancer Res. (2004) 64:1502–8. doi: 10.1158/0008-5472.CAN-03-2936
107. Elsner L, Muppala V, Gehrmann M, Lozano J, Malzahn D, Bickeböller H, et al. The heat shock protein HSP70 promotes mouse NK cell activity against tumors that express inducible NKG2D ligands. J Immunol. (2007) 179:5523–33. doi: 10.4049/jimmunol.179.8.5523
108. Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol. (2002) 2:185–94. doi: 10.1038/nri749
109. Castellino F, Boucher PE, Eichelberg K, Mayhew M, Rothman JE, Houghton AN, et al. Receptor-mediated uptake of antigen/heat shock protein complexes results in major histocompatibility complex class I antigen presentation via two distinct processing pathways. J Exp Med. (2000) 191:1957–64. doi: 10.1084/jem.191.11.1957
110. Qiao Y, Liu B, Li Z. Activation of NK cells by extracellular heat shock protein 70 through induction of NKG2D ligands on dendritic cells. Cancer Immun. (2008) 8:12. Available online at: https://cancerimmunolres.aacrjournals.org/content/canimmarch/8/1/12.short
111. Multhoff G, Mizzen L, Winchester CC, Milner CM, Wenk S, Eissner G, et al. Heat shock protein 70 (Hsp70) stimulates proliferation and cytolytic activity of natural killer cells. Exp Hematol. (1999) 27:1627–36. doi: 10.1016/S0301-472X(99)00104-6
112. Jacquemin C, Rambert J, Guillet S, Thiolat D, Boukhedouni N, Doutre M -S, et al. Heat shock protein 70 potentiates interferon alpha production by plasmacytoid dendritic cells: relevance for cutaneous lupus and vitiligo pathogenesis. Br J Dermatol. (2017) 177:1367–75. doi: 10.1111/bjd.15550
113. Peraldi M-N, Berrou J, Dulphy N, Seidowsky A, Haas P, Boissel N, et al. Oxidative stress mediates a reduced expression of the activating receptor NKG2D in NK cells from end-stage renal disease patients. J Immunol. (2009) 182:1696–705. doi: 10.4049/jimmunol.182.3.1696
114. van den Boorn JG, Melief CJ, Luiten RM. Monobenzone-induced depigmentation: from enzymatic blockade to autoimmunity. Pigment Cell Melanoma Res. (2011) 24:673–9. doi: 10.1111/j.1755-148X.2011.00878.x
115. Ewens S, Wulferink M, Goebel C, Gleichmann E. T cell-dependent immune reactions to reactive benzene metabolites in mice. Arch Toxicol. (1999) 73:159–67. doi: 10.1007/s002040050601
116. Gholijani N, Yazdani MR, Dastgheib L. Predominant role of innate pro-inflammatory cytokines in vitiligo disease. Arch Dermatol Res. (2020) 312:123–31. doi: 10.1007/s00403-019-01996-9
117. Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, Turka LA. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-γ for autoreactive CD8+ T-cell accumulation in the skin. J Invest Dermatol. (2012) 132:1869–76. doi: 10.1038/jid.2011.463
118. Engelhorn ME, Guevara-Patiño JA, Noffz G, Hooper AT, Lou O, Gold JS, et al. Autoimmunity and tumor immunity induced by immune responses to mutations in self. Nat Med. (2006) 12:198–206. doi: 10.1038/nm1363
119. Gregg RK, Nichols L, Chen Y, Lu B, Engelhard VH. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice. J Immunol. (2010) 184:1909–17. doi: 10.4049/jimmunol.0902778
120. Harris JE. Cellular stress and innate inflammation in organ-specific autoimmunity: lessons learned from vitiligo. Immunol Rev. (2016) 269:11–25. doi: 10.1111/imr.12369
121. Mosenson JA, Zloza A, Nieland JD, Garrett-Mayer E, Eby JM, Huelsmann EJ, et al. Mutant HSP70 reverses autoimmune depigmentation in vitiligo. Sci Transl Med. (2013) 5:174ra28. doi: 10.1126/scitranslmed.3005127
122. Luiten RM, Van Den Boorn JG, Konijnenberg D, Dellemijn TAM, Van Der Veen JPW, Bos JD, et al. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol. (2009) 129:2220–32. doi: 10.1038/jid.2009.32
123. Richmond JM, Bangari DS, Essien KI, Currimbhoy SD, Groom JR, Pandya AG, et al. Keratinocyte-derived chemokines orchestrate T-cell positioning in the epidermis during vitiligo and may serve as biomarkers of disease. J Invest Dermatol. (2017) 137:350–8. doi: 10.1016/j.jid.2016.09.016
124. Rashighi M, Harris JE. Interfering with the IFN-γ/CXCL10 pathway to develop new targeted treatments for vitiligo. Ann Transl Med. (2015) 3:343. doi: 10.3978/j.issn.2305-5839.2015.11.36
125. Yang L, Wei Y, Sun Y, Shi W, Yang J, Zhu L, et al. Interferon-gamma inhibits melanogenesis and induces apoptosis in melanocytes: a pivotal role of CD8+ cytotoxic T lymphocytes in vitiligo. Acta Derm Venereol. (2015) 95:664–70. doi: 10.2340/00015555-2080
126. Curtsinger JM, Agarwal P, Lins DC, Mescher MF. Autocrine IFN-γ promotes naive CD8 T cell differentiation and synergizes with IFN-α to stimulate strong function. J Immunol. (2012) 189:659–68. doi: 10.4049/jimmunol.1102727
127. Tulic MK, Cavazza E, Cheli Y, Jacquel A, Luci C, Cardot-Leccia N, et al. Innate lymphocyte-induced CXCR3B-mediated melanocyte apoptosis is a potential initiator of T-cell autoreactivity in vitiligo. Nat Commun. (2019) 10:2178. doi: 10.1038/s41467-019-09963-8
128. Morelli JG, Norris DA. Influence of inflammatory mediators and cytokines on human melanocyte function. J Invest Dermatol. (1993) (Suppl. 2):191–5S: doi: 10.1038/jid.1993.75
129. Rath PC, Aggarwal BB. TNF-induced signaling in apoptosis. J Clin Immunol. (1999) 19:350–64. doi: 10.1023/a:1020546615229
130. Englaro W, Bahadoran P, Bertolotto C, Buscà R, Dérijard B, Livolsi A, et al. Tumor necrosis factor alpha-mediated inhibition of melanogenesis is dependent on nuclear factor kappa B activation. Oncogene. (1999) 18:1553–9. doi: 10.1038/sj.onc.1202446
131. Yang X, Yan L, Ha DA, Qu L, Liu L, Tao YU. Changes in sICAM-1 and GM-CSF levels in skin tissue fluid and expression of IL-6, IL-17 and TNF-α in blood of patients with vitiligo. Exp Ther Med. (2019) 17:408–12. doi: 10.3892/etm.2018.6937
132. Sharma CK, Sharma M, Prasad K. Involvement of different genes expressions during immunological and inflammatory responses in vitiligo. Crit Rev Eukaryot Gene Expr. (2017) 27:277–87. doi: 10.1615/CritRevEukaryotGeneExpr.2017019558
133. Richmond JM, Strassner JP, Zapata LZ, Garg M, Riding RL, Refat MA, et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. (2018) 10:7710. doi: 10.1126/scitranslmed.aam7710
134. Burdick KH, Hawk WA. Vitiligo in a case of vaccinia virus-treated melanoma. Cancer. (1964) 17:708–12. doi: 10.1002/1097-0142(196406)17:6<708::AID-CNCR2820170604>3.0.CO;2-3
135. Kaercher K. Systematized depigmentation in melanoma treated with x-irradiation. Dermatologica. (1960) 120:255–63.
136. Smith JL, Stehlin JS. Spontaneous regression of primary malignant melanomas with regional metastases. Cancer. (1965) 18:1399–415. doi: 10.1002/1097-0142(196511)18:11<1399::AID-CNCR2820181104>3.0.CO;2-R
137. Nordlund JJ, Kirkwood JM, Forget BM, Milton G, Albert DM, Lerner AB. Vitiligo in patients with metastatic melanoma: a good prognostic sign. J Am Acad Dermatol. (1983) 9:689–96. doi: 10.1016/S0190-9622(83)70182-9
138. Bystryn J, Rigel D, Friedman R, Kopf A. Prognostic significance of hypopigmentation in malignant melanoma. Arch Dermatol. (1987) 123:1053–5.
139. Quaglino P, Marenco F, Osella-Abate S, Cappello N, Ortoncelli M, Salomone B, et al. Vitiligo is an independent favourable prognostic factor in stage III and IV metastatic melanoma patients: results from a single-institution hospital-based observational cohort study. Ann Oncol. (2010) 21:409–14. doi: 10.1093/annonc/mdp325
140. Teulings HE, Tjin EPM, Willemsen KJ, van der Kleij S, Meulen S ter, Kemp EH, et al. Anti-Melanoma immunity and local regression of cutaneous metastases in melanoma patients treated with monobenzone and imiquimod; a phase 2 a trial. Oncoimmunology. (2018) 7:e1419113. doi: 10.1080/2162402X.2017.1419113
141. Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, et al. A Wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity. (2015) 43:1011–21. doi: 10.1016/j.immuni.2015.10.016
142. Tembhre MK, Sharma VK, Sharma A, Chattopadhyay P, Gupta S. T helper and regulatory T cell cytokine profile in active, stable and narrow band ultraviolet B treated generalized vitiligo. Clin Chim Acta. (2013) 424:27–32. doi: 10.1016/j.cca.2013.05.005
143. Dwivedi M, Laddha NC, Shah K, Shah BJ, Begum R. Involvement of interferon-gamma genetic variants and intercellular adhesion molecule-1 in onset and progression of generalized vitiligo. J Interf Cytokine Res. (2013) 33:646–59. doi: 10.1089/jir.2012.0171
144. Basak PY, Adiloglu AK, Ceyhan AM, Tas T, Akkaya VB. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J Am Dermatology. (2009) 60:256–60. doi: 10.1016/j.jaad.2008.09.048
145. Jin Y, Roberts GHL, Ferrara TM, Ben S, van Geel N, Wolkerstorfer A, et al. Early-onset autoimmune vitiligo associated with an enhancer variant haplotype that upregulates class II HLA expression. Nat Commun. (2019) 10:1–10. doi: 10.1038/s41467-019-08337-4
146. Jin Y, Birlea SA, Fain PR, Gowan K, Riccardi SL, Holland PJ, et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N Engl J Med. (2010) 362:1686–97. doi: 10.1056/NEJMoa0908547
147. Jin Y, Birlea SA, Fain PR, Mailloux CM, Riccardi SL, Gowan K, et al. Common variants in FOXP1 are associated with generalized vitiligo. Nat Genet. (2010) 42:576–8. doi: 10.1038/ng.602
148. Touloukian CE, Leitner WW, Robbins PF, Rosenberg SA, Restifo NP. Mining the melanosome for tumor vaccine targets: P.polypeptide is a novel tumor-associated antigen. Cancer Res. (2001) 61:8100–4. Available online at: https://cancerres.aacrjournals.org/content/61/22/8100.article-info
149. Skipper JCA, Hendrickson RC, Gulden PH, Brichard V, Van Pel A, Chen Y, et al. An HLA-A2-restricted tyrosinase antigen on melanoma cells results from posttranslational modification and suggests a novel pathway for processing of membrane proteins. J Exp Med. (1996) 183:527–34. doi: 10.1084/jem.183.2.527
150. Konopacki C, Pritykin Y, Rubtsov Y, Leslie CS, Rudensky AY. Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function. Nat Immunol. (2019) 20:232–42. doi: 10.1038/s41590-018-0291-z
151. Ren J, Han L, Tang J, Liu Y, Deng X, Liu Q, et al. Foxp1 is critical for the maintenance of regulatory T-cell homeostasis and suppressive function. PLoS Biol. (2019) 17:e3000270. doi: 10.1371/journal.pbio.3000270
152. Song P, Wang X-W, Li H-X, Li K, Liu L, Wei C, et al. Association between FOXP3 polymorphisms and vitiligo in a Han Chinese population. Br J Dermatol. (2013) 169:571–8. doi: 10.1111/bjd.12377
153. Jahan P, Cheruvu R, Tippisetty S, Komaravalli PL, Valluri V, Ishaq M. Association of FOXP3 (rs3761548) promoter polymorphism with nondermatomal vitiligo: a study from India. J Am Acad Dermatol. (2013) 69:262–6. doi: 10.1016/j.jaad.2013.01.035
154. Birlea SA, Jin Y, Bennett DC, Herbstman DM, Wallace MR, McCormack WT, et al. Comprehensive association analysis of candidate genes for generalized vitiligo supports XBP1, FOXP3, and TSLP. J Invest Dermatol. (2011) 131:371–81. doi: 10.1038/jid.2010.337
155. Yun JY, Uhm YK, Kim HJ, Lim SH, Chung JH, Shin MK, et al. Transforming growth factor beta receptor II (TGFBR2) polymorphisms and the association with nonsegmental vitiligo in the Korean population. Int J Immunogenet. (2010) 37:289–91. doi: 10.1111/j.1744-313X.2010.00923.x
156. Abanmi A, Al Harthi F, Zouman A, Kudwah A, Al Jamal M, Arfin M, et al. Association of interleukin-10 gene promoter polymorphisms in Saudi patients with vitiligo. Dis Markers. (2008) 24:51–7. doi: 10.1155/2008/210609
157. Zheng SG, Gray JD, Ohtsuka K, Yamagiwa S, Horwitz DA. Generation ex vivo of TGF-β-producing regulatory T cells from CD4+ CD25− precursors. J Immunol. (2002) 169:4183–9. doi: 10.4049/jimmunol.169.8.4183
158. Hsu P, Santner-Nanan B, Hu M, Skarratt K, Lee CH, Stormon M, et al. IL-10 potentiates differentiation of human induced regulatory T cells via STAT3 and Foxo1. J Immunol. (2015) 195:3665–74. doi: 10.4049/jimmunol.1402898
159. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen WJ. A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. (2008) 9:632–40. doi: 10.1038/ni.1607
160. Dey-Rao R, Sinha AA. Vitiligo blood transcriptomics provides new insights into disease mechanisms and identifies potential novel therapeutic targets. BMC Genomics. (2017) 18:109. doi: 10.1186/s12864-017-3510-3
161. Puri N, Mojamdar M, Ramaiah A. In vitro growth characteristics of melanocytes obtained from adult normal and vitiligo subjects. J Invest Dermatol. (1987) 88:434–8. doi: 10.1111/1523-1747.ep12469795
162. Goldstein NB, Koster MI, Hoaglin LG, Spoelstra NS, Kechris KJ, Robinson SE, et al. Narrow band ultraviolet b treatment for human vitiligo is associated with proliferation, migration, and differentiation of melanocyte precursors. J Invest Dermatol. (2015) 135:2068–76. doi: 10.1038/jid.2015.126
163. Mansuri MS, Singh M, Dwivedi M, Laddha NC, Marfatia YS, Begum R. MicroRNA profiling reveals differentially expressed microRNA signatures from the skin of patients with nonsegmental vitiligo. Br J Dermatol. (2014) 171:1263–7. doi: 10.1111/bjd.13109
164. Shi YL, Weiland M, Li J, Hamzavi I, Henderson M, Huggins RH, et al. MicroRNA expression profiling identifies potential serum biomarkers for non-segmental vitiligo. Pigment Cell Melanoma Res. (2013) 26:418–21. doi: 10.1111/pcmr.12086
165. Shi YL, Weiland M, Lim HW, Mi QS, Zhou L. Serum miRNA expression profiles change in autoimmune vitiligo in mice. Exp Dermatol. (2014) 23:140–2. doi: 10.1111/exd.12319
Keywords: vitiligo, NKG2D, T cells, horror autotoxicus, Hsp70, oxidative stress
Citation: Plaza-Rojas L and Guevara-Patiño JA (2021) The Role of NKG2D in Vitiligo. Front. Immunol. 12:624131. doi: 10.3389/fimmu.2021.624131
Received: 30 October 2020; Accepted: 02 February 2021;
Published: 26 February 2021.
Edited by:
Thierry Passeron, Université Côte d'Azur, FranceReviewed by:
Jillian M. Richmond, University of Massachusetts Medical School, United StatesCopyright © 2021 Plaza-Rojas and Guevara-Patiño. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José A. Guevara-Patiño, amFndWV2YXJhQGx1Yy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.