 Laura N. Verboon
Laura N. Verboon Hiren C. Patel2,3
Hiren C. Patel2,3 Andrew D. Greenhalgh
Andrew D. Greenhalgh- 1Division of Infection, Immunity and Respiratory Medicine, Faculty of Biology, Medicine and Health, School of Biological Sciences, The University of Manchester, Manchester, United Kingdom
- 2Division of Cardiovascular Sciences, Salford Royal National Health Service Foundation Trust, Faculty of Biology, Medicine and Health, The University of Manchester, Manchester, United Kingdom
- 3Geoffrey Jefferson Brain Research Centre, The Manchester Academic Health Science Centre, Northern Care Alliance National Health Service Group, University of Manchester, Manchester, United Kingdom
- 4Lydia Becker Institute of Immunology and Inflammation, The University of Manchester, Manchester, United Kingdom
Mild traumatic brain injury (mild TBI), often referred to as concussion, is the most common form of TBI and affects millions of people each year. A history of mild TBI increases the risk of developing emotional and neurocognitive disorders later in life that can impact on day to day living. These include anxiety and depression, as well as neurodegenerative conditions such as chronic traumatic encephalopathy (CTE) and Alzheimer's disease (AD). Actions of brain resident or peripherally recruited immune cells are proposed to be key regulators across these diseases and mood disorders. Here, we will assess the impact of mild TBI on brain and patient health, and evaluate the recent evidence for immune cell involvement in its pathogenesis.
Mild Traumatic Brain Injury
Traumatic brain injury (TBI) is a term used to include a spectrum of insults resulting from mechanical injury to the brain. TBI includes injuries that range from severe, with open skull injuries and major parenchymal disruption, to the mildest form of TBI, often termed concussion. Although widely used in everyday language, the term concussion is now less commonly used in medical and scientific terminology, as it lacks diagnostic precision and does not refer to underlying pathological processes (1, 2). Therefore, mild TBI is the preferred term and will be used throughout this review (1, 2). It is estimated that TBI affects 69 million individuals each year world-wide, with the vast majority of cases being mild TBI (3, 4). The main causes of mild TBI are motor accidents, falls, assaults, active-duty of soldiers, and domestic violence (5, 6). Recently, greater attention has been given to this condition due to the high prevalence of mild TBI among young athletes in relation to their involvement in collision sports such as American Football, soccer, and rugby (7).
Mild TBI is a physiological disruption of brain function and occurs due to mechanical distortion of brain tissue, most commonly from a blow to the head, but can also be caused by a blast injury frequently seen in soldiers serving in a war zone (7, 8). Rapid rotational velocity/acceleration (inertial loading) is thought to be a key component of injury (9–11). The underlying pathophysiology of the injury remains poorly understood, as availability of human post-mortem brain tissue for examination from this typically non-lethal injury is limited (12). Although referred to as “mild,” individuals can still experience a variety of physical, emotional and cognitive problems, including sleep disturbance, increased anxiety, and depression (4, 7, 13, 14).
Diagnosing mild TBI and its severity is usually based on the loss of consciousness duration (<30 min), the Glasgow coma scale score (13–15), post-traumatic amnesia duration (momentarily to <24 h), and a lack of intracerebral/subdural/epidural hematoma, cerebral, or hemorrhagic contusion, penetrating TBI (dura penetrated), subarachnoid hemorrhage or brainstem injury (2) (Figure 1). Mild TBI is still a heterogeneous insult, and there can be major variation in the likelihood of significant neuropathology and varying symptoms including blurred vision, confusion, dizziness, focal neurological symptoms, headache, and nausea (2, 7, 8). Whilst most patients recover and return to their normal self, the clinical outcome of concussion is hard to predict. This is because of the heterogeneity of initial trauma, the inability to quantify disease severity and the likely initiation of complex pathogenic pathways (15). Even though men are at greater risk of mild TBI due to greater participation in high-risk activities, studies have shown that females are at greater risk of poor outcomes (16–19) and further research of both sexes is needed to characterize the nature of sex-dependent injury and recovery (20). In addition, pre-existing health conditions, age, genetic background (21), and alcohol or substance abuse also influences recovery and leads to differences in clinical outcome between patients (22).
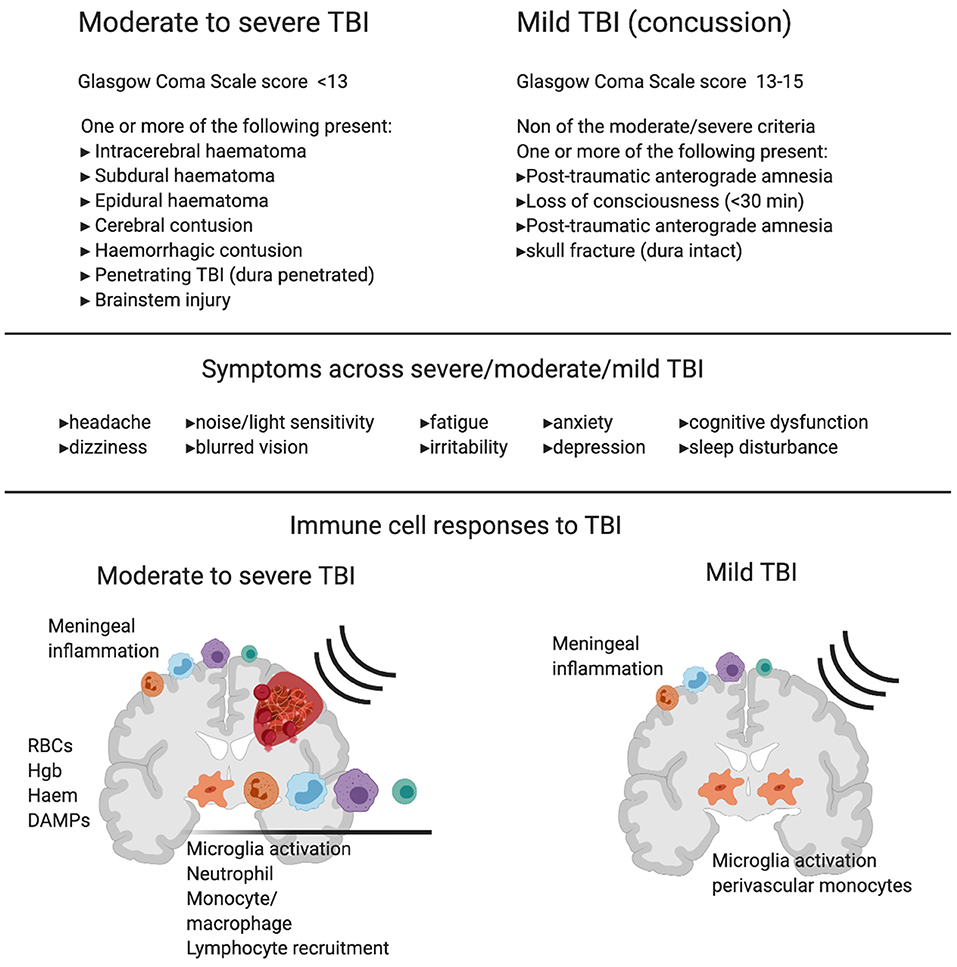
Figure 1. Diagnostic criteria, symptoms and immune cell involvement in moderate to severe traumatic Brain Injury (TBI) in comparison with mild TBI. Commonly used diagnostic criteria in moderate to severe TBI compared to mild TBI shows the major clinical difference between the two reflects hemorrhage or clear contusion in the brain. Symptoms are shared across mild, moderate and severe TBI with increasing likelihood of symptom occurrence and severity with increasing injury. Schematics represent immune response in moderate to severe TBI (left) and mild TBI (right). In moderate to severe TBI in humans and animal models, there is clear evidence for resident microglia activation and recruitment of macrophages, dendritic cells, neutrophils, B cells and T cells, and meningeal inflammation. In addition to active recruitment mechanisms, peripheral immune cells can infiltrate with frank hemorrhage alongside red blood cells (RBCs) and the release of hemoglobin (Hgb), Haem, and other damage associated molecular patterns (DAMPs), which are one set of initiators of the immune response. In contrast, in mild TBI there is little evidence of infiltrating immune cells to the brain tissue in humans or animal models that do not produce hemorrhage or skull opening. In mild TBI, there is evidence of meningeal inflammation, microglial activation, and some monocyte/macrophage recruitment to the cerebrovasculature.
The following review will focus solely on consequences of mild TBI in adults, that would be commonly be referred to as concussion. There is a vast and important literature on more severe TBI that includes evidence of hemorrhage and parenchymal injury and these injuries are defined as moderate or severe TBI. Clinically, moderate or severe TBI is diagnosed by neuroimaging and preclinical modeling of these injuries is much more common than mild TBI, due to the production of frank and measurable tissue damage. As a result, there is a large literature on neuroinflammatory and immune mechanisms that drive both injury and repair in these insults (23–25). Much less is known about the pathology of mild TBI with no overt contusion or hemorrhage in the pathology, and here, a different immune response is likely to occur. The current review will explicitly refer to evidence of the immune response after human mild TBI, in animal models with high translational relevance to mild TBI (without compromising the skull and no evidence of hemorrhage) and studies that investigate patients with a history of head injury through sport.
Evidence for Mild TBI as a Risk Factor for Long-Term Problems
Mild TBI is now recognized as a major public health concern as clinicians and researchers are becoming more aware of the dangers and potential long-term consequences associated with this type of head injury (26). In most mild TBI cases, acutely reported symptoms resolve within 3 months; however, a small proportion of patients continue to suffer life disrupting symptoms (27–29). A range of factors, not necessarily directly reflecting injury severity, are associated with poor outcome following mild TBI, including previous neurological or psychiatric problems and whether the patient had suffered a previous head injury (27, 28). Indeed, patients with a history of mild TBI can experience changes in emotions or behavior, often expressed by increased anxiety and depressive like behaviors (2, 4, 7, 13, 14). Most studies investigating multiple head injuries over a sustained period are derived from participants of contact sports. These patients may differ greatly from those who suffer a mild TBI as a one-off event, not only in the nature of the head injury but also their lifestyle and pre-morbid traits (30). Currently, there is major interest in mild TBI/concussion due to its prevalence in sports such as the National Football League (NFL), rugby, and soccer, where the risk of head injury is high. Single concussive events in these sports can result in the same myriad of symptoms as a one-off mild TBI in the general population, and may trigger that same initial pathological response; however, it is the accumulation of injuries and their long-term effect on mood and neurodegenerative outcomes that is often assessed in these athletes.
Effects of Repeated Mild TBI in Contact Sports
In contact sports, diagnosed mild TBIs/concussions and even head impacts that are frequent but do not cause noticeable immediate injury, such as heading a soccer ball, are now being investigated as risk factors for poor long-term brain health (31). In a population of retired rugby players, the prevalence of major depressive disorder was significantly higher compared to other retired sportsmen (7). Another study investigated professional NFL players and found a link between recurrent concussion and diagnosis of lifetime depression and suggested that the prevalence of depression increases with the number of past mild TBIs (32). Indeed, retired players that reported either one to two, or three or more previous concussions were 1.5 and three times more likely to be diagnosed with depression, respectively, compared to retired players with no history of mild TBIs (32). Regardless of the type of contact sport, diagnostic test scores for major depressive disorder increases with the number of mild TBIs (7).
In addition to emotional disturbances, mTBI is associated with a risk of developing a number of neurodegenerative conditions (33). A series of studies retrospectively investigating a cohort study of former professional footballers [Football's Influence on Lifelong health and Dementia risk (FIELD)] investigated the link between at dementia pathology, mortality and mental health and suicide in ex footballers (soccer players) (34). Mortality from neurodegenerative disease was higher and mortality from other common diseases lower among retired professional soccer players than among matched controls (35). Surprisingly, in these cohorts, hospital admissions for common mental health disorders were lower than population controls, with no difference in suicide, despite evidence of neurodegeneration (36). Outside of professional athletes, a population-based administrative health cohort study, in more than 47,000 cases of mild TBI showed mild TBI was associated with an increased risk of diagnosis of attention-deficit hyperactivity disorder, mood and anxiety disorders, dementia and Parkinson's disease later in life (37).
The majority of evidence suggests that mild TBI can be detrimental to mental health, but also carries increased risk of developing epilepsy and neurodegenerative disorders, such as Alzheimer's disease, Parkinson's disease, and chronic traumatic encephalopathy (CTE) (2). Significant neurodegeneration observed in retired athletes has been linked to repeated mild TBI at a younger age and mortality from neurodegenerative disease is significantly higher among former professional soccer players than in matched population controls (35). Within this group, Alzheimer's disease as the primary or a contributory cause of death was responsible for the largest increase of deaths (35). In a separate study, there was increased risk and early onset of ALS in professional players from Italian soccer teams (38).
Post-mortem studies on former contact sports athletes show a high prevalence of CTE; a progressive neurodegeneration associated with repetitive head trauma (39, 40). Interestingly, neuropathological severity of CTE seemed to increase in accordance with the level of play and almost all cases had behavioral or mood symptoms or both, cognitive symptoms or signs of dementia (39). Post-mortem brains from U.S. military veterans that have been exposed to blast exposure and/or concussive injury display CTE neuropathology that is similar to the pathology observed in former athletes (41).
These studies add to the growing evidence to suggest that a history of mild TBI is a risk factor for the development of pathological neurodegeneration. They have also brought much needed attention to the dangers of head injury, in general. Research is now focused on understanding the underlying pathology of both single and accumulated mild TBIs, and what can be learned from each individual instance to prevent long-term problems.
Understanding Distinct or Overlapping Mechanisms of a Single, Repeated, or Life-Long History of Mild TBI
The Lancet Commission on dementia prevention recently added TBI as potentially modifiable risk factor for dementia (26). In this important document, a combination of studies relating to severe TBI, those investigating concussion, or a career in professional contact sport, are cited as why traumatic brain injury is considered a risk factor for dementia and neurodegenerative disease (26, 33). As detailed above, severe TBI can be quite different to mild TBI. However, the role of neuroinflammation, propagated by the immune system, is a likely modifiable regulator in both. Even when focusing on mild TBI research alone, there are still many challenges to assessing the role of the immune system. Complicating factors include: heterogeneous pathology and symptoms (2), diffuse injury across brain regions (10, 42) and the overlapping research conclusions between one-off mild TBI and multiple injuries sustained by professional sport participation. However, the investigation of the immune response to discrete injuries will undoubtedly lead to increased understanding of the dangerous cumulative effects of multiple mild TBIs. Indeed, much of our understanding of the mechanisms of mild TBI, and the role of the immune system, is derived from animal models of injury. A combination of clinical and preclinical studies in the acute setting can provide insight into both mild TBI in the general population and those accumulated in professional sport.
The following sections will outline what is known about the immune system's response to CNS injury in general, and why aspects of the immune system have become considered drivers of neurodegenerative disease, of which head injury is now considered a risk factor. This will provide context for the review of the current literature for immune involvement in the pathology of mild TBI.
Positioning of the Immune System Within the CNS
Immune cells are present throughout the adult CNS (43). Microglia are a type of tissue resident macrophage and are the major immune cell type (44). Microglial cell bodies and their processes cover every cubic micrometer of the brain during constant surveillance activities (45, 46). Recent work shows that, of the total number of immune cells in the brain, ~80% are microglia, with the remainder comprised of barrier-associated macrophages and cell types more traditionally associated with the periphery such as neutrophils and T cells (44, 47–49). Microglia are highly plastic and defend the brain against external challenges. Pattern recognition receptors (PRRs) are scattered along their membrane, by which they can recognize pathogen-associated molecular patterns (PAMPs) and host-derived danger-associated molecular patterns (DAMPs), making microglia equipped with the tools to evoke a rapid, fine-tuned inflammatory response to immunological challenges (50). Microglia phenotype and morphology are determined by their local environment and several concepts relating to their function, such as “homeostatic,” “primed,” “trained,” or “tolerant” microglia, have emerged from experimental models (51). Microglial priming is defined as a prolonged and exaggerated immune response resulting from an acute inflammatory event in an ongoing inflammatory environment (52). Innate immune memory is associated to cell reprogramming following a primary immune stimulus that leads to increased (trained) or decreased (tolerant) responses to a secondary inflammatory stimulus (53). These concepts are important in the context of mild TBI as repeated head injuries lead to greater risk of poor outcome (32, 54). Although not defined as immune cells per se, astrocytes, oligodendrocytes, and endothelial cells all perform various functions that are critical to the immune response (55–58), and the important actions specific to these cells in mild TBI are reviewed elsewhere (59–62).
Neuroinflammatory cascades rely on the activation of the inflammasome, a protein complex, consisting of caspase-1, apoptosis-associated speck-like protein (ASC) and nod-like receptor protein (NLRP1 or NLRP3) (63, 64). Common microglial pathways activated upon the detection of a challenge involve NF-κB, which is a pro-inflammatory transcription factor that stimulates cytokine release in conjunction with the inflammasome (50). Metabolic changes within microglia also sustain or restrain inflammation (65). Rapid motility, reactive oxygen species (ROS) and cytokine production require quick energy utilization through glycolysis and fatty acid synthesis (65–67). In contrast, anti-inflammatory microglia require efficient energy production utilizing oxidative phosphorylation for transcription of ATP-dependent tissue repair genes, reduce ROS, perform amino acid and fatty acid oxidation to produce growth factors, including polyamines and prolines, and to support mitochondrial respiration (65–67).
In the event of CNS injury, microglia reduce their ramifications and extend cell processes to the site of injury, helping to maintain the integrity of critical CNS barrier structures such as glial limitans and vasculature (68–70). Moreover, they increase their migration to damaged brain sites and become phagocytic to clear cell debris (71–73). Microglia and other resident immune cells can be joined by their infiltrating counterparts from the circulation, such as neutrophils, monocytes, and lymphocytes, depending on the severity of injury (43, 71, 73–79). These peripheral cells are recruited through a multitude of mechanisms, including endothelial and microglial signaling, and can enter the brain through the compromised blood brain barrier, circumventricular organs or other brain blood interfaces, such as the meninges (80, 81).
In the context of human mild TBI, it is unknown whether circulating immune cells are recruited to the brain in patients, and closed head animal models provide differing results depending on induction and severity of the injury [see below]. Recruitment of immune cells may vastly affect progression of pathology, as the actions of these cells can differ compared to their resident counterparts (43). Furthermore, evidence suggests that infiltrating immune cells influence resident microglia populations, which may have long lasting consequences for injury outcomes (74, 82–85).
Here, it is again important to distinguish between TBI with parenchymal hemorrhage (and associated animal models) and mild TBI, as hemorrhage is likely to create a type injury and immune response that is completely distinct to that of injuries with no bleeding (Figure 1). For example, extravasated red blood cells (RBCs) are a source of multiple immune response triggers and DAMPs (86). RBCs are phagocytosed by immune cells, which drives an inflammatory phenotype in those cells (87), and a portion of which stay within the tissue, die and degranulate, releasing endothelins and oxygen free radicals (88). Extravasated RBCs also releases toxic Hgb which is oxidized to haem and acts as a DAMP to exacerbate the inflammatory response (89). A further metabolite, iron is also implicated in brain injury after hemorrhage (88, 90, 91). Thus, several stages of RBC lysis contribute to a type of brain injury not seen in mild TBI without parenchymal bleeding. For information on the immune response to TBI including hemorrhage, we would like to point readers to the following excellent reviews on the topic (23–25).
In mild TBI, whether immune cells from the periphery are recruited or not (see below), microglia and the other resident immune cells are present and can respond rapidly to changes in the brain (44, 47–49). In the wider field of neuroimmunology, the interest in the microglia-mediated immune response during brain injury and disease has risen exponentially in the past decade, mainly due to genome wide association studies that implicate many microglial genes as risk factors for neurodegenerative disease (92, 93). It is here, in the brain's immune response, where mild TBI and the risk of cognitive decline and neurodegeneration may meet.
The Immune System's Role as a Driver of Neurodegeneration
To understand if the immune response to mild TBI increases the risk of neurodegenerative disease, it is important to understand the known role of the immune system in neurodegeneration. The CNS and innate immune system continuously modulate each other through a sophisticated bidirectional crosstalk (43, 94). Under pathological conditions, disrupted communication may result in an inflammatory response. When the inflammatory response of CNS resident immune cells remains unresolved, this may lead to initiation, propagation, and progression of tissue damage, ultimately resulting in neurodegeneration (50). The immune system may therefore be a driver of neurodegeneration, in general (95–98) and mild TBI's activation of the immune system may be a causative trigger, although this is yet to be formally demonstrated.
Many neurodegenerative disorders display concurrent and chronic alterations in immune function and signaling. However, there is now strong evidence that immune dysregulation can be a direct cause of neurodegenerative disease. Somatic mutation specifically in the erythro-myeloid progenitor lineage from which microglia derive can drive late-onset neurodegeneration in mice (56). More recently, biallelic mutations in NRROS (Negative Regulator Of Reactive Oxygen Species), which is necessary for TGFB-1 signaling in microglia, were found to cause an early onset lethal microgliopathy in humans (99, 100) and NRROS-deficient (Nrros-/-) mice show neurodegeneration (101), defects in motor functions and die before 6 months of age (102). Together, these data show that microglia-specific alterations can cause neurodegenerative disease, confirming that the well-documented immune response to neurodegeneration may not solely be secondary to injury.
Several genes involved in the immune system, and particularly microglia, have been identified as risk factors for the most common form of neurodegenerative disease, Alzheimer's (103–105). The APOE gene, encoding apolipoprotein (Apo)E (106) is mainly expressed in the brain by microglia and astrocytes and is component of Aβ plaques and promotes Aβ aggregation and deposition (107). The three major human isoforms, apoE2, apoE3, and apoE4, are encoded by different alleles and differ in their effects on AD risk and pathology, with one APOE-ε4 allele increasing AD risk 3-fold and two APOE-ε4 alleles increasing AD risk by 12-fold (108). In addition, APOE-ε4 is also implicated in Dementia with Lewy bodies and Frontotemporal dementia (97). Another molecule implicated in AD is the triggering receptor expressed on myeloid cells 2 (Trem2) and is highly expressed in microglia in the brain and important for microglial, phagocytosis, proliferation and environment sensing (109). Single-nucleotide polymorphism (SNP) mutations in Trem2 drastically increase the AD risk (110), estimated to be with a 3.0- to 4.5-fold (96). Moreover, homozygous Trem2 variants have been proposed to be causal for Frontotemporal Dementia (FTD) or linked to increased FTD risk (111). A recent genome-wide meta-analysis identified new loci and functional pathways influencing Alzheimer's disease risk that localized to immune-related tissues and cell types (microglia), highlighting the role of the immune system and its principle brain-resident cell in neurodegenerative disease (92). In sum, the immune system and its dysfunction are now strongly implicated in neurodegenerative disease, for which mild TBI (particularly repeated mild TBI) is proposed as a risk factor. We will now summarize how mild TBI may activate the brain's immune system, potentially linking it to long-term neurodegeneration.
The Immune Response to Mild TBI
To reiterate the focus of this article, we will review the known immune response to injuries that are defined as mild TBI or concussion and their relevant animal models. This would therefore exclude brain injuries with intracerebral, subdural, epidural hematoma, cerebral or hemorrhagic contusion, penetrating TBI (dura penetrated), subarachnoid hemorrhage or brainstem injury (2) (Figure 1). This definition describes a population of patients that have potential pathology too subtle for standard imaging, but represents up to >80% of those that suffer a TBI (3). Animal models representing this “mild” situation are less common as researchers investigating brain injury are in search robust, reliable, modifiable readouts to demonstrate mechanisms and new treatment options; such animal models often employ craniotomy and/or hemorrhage and contusion and would be considered moderate to severe TBI in the clinic. If the immune response to mild TBI is a factor in pathology, it is important to understand how this may be initiated in a brain without bleeding or overt lesion.
Triggers of the Immune Response in Mild TBI
The immune system is ubiquitous in the CNS and is equipped to respond to brain tissue injury after mild TBI. The injury produced after a closed-head impact is the result of physical movements of the brain within the skull, including acceleration and differential inertial loading (10, 11). Physical forces are thought to lead to axonal injury due to excessive regional stretching of axons (112) (Figure 2A). Using head kinematics from athletic events, modeling showed that the brain can be described as a hyperviscoelastic medium and deformation is most sensitive to specific frequency oscillations, particularly in deep brain regions, and is aligned with areas of pathology after mild TBI (10).
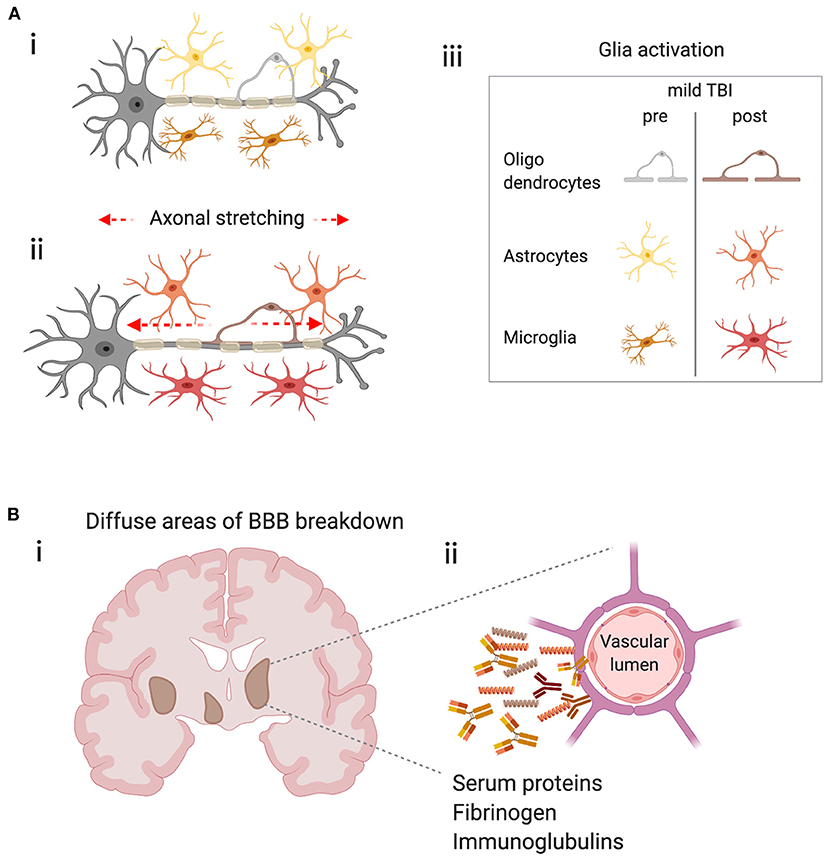
Figure 2. Potential triggers of the immune response in mild TBI. (A) Physical forces lead to stretching of axons, axonal injury and glial activation. (i) Schematic representing a neuron with its myelinated axon surrounded by glia in the healthy brain. (ii) Represents the stretching of axons due to physical forces during mild TBI and the subsequent glial activation (iii) Key–indicates glial subtypes hypothesized to be activated after axonal stretching. (B) Diffuse blood-brain barrier (BBB) in the brain after concussion leads to extravasation of harmful molecules to the parenchyma. (i) Schematic shows a coronal section of a human brain with representations of diffuse BBB breakdown after mild TBI (brown areas). (ii) Enlarged image shows compromised cerebral blood vessel leaking molecules that may trigger inflammation and glial activation.
Stretching of axons is the leading hypothesis as to why white matter appears most sensitive to mild brain injury. However, from the view of the immune system, microglial cells are also sensitive to mechanical signals (113, 114). Viscoelastic testing of individual CNS cells showed that other glial cells, such as astrocytes and Müller glia are twice as soft as neurons and act as compliant structures surrounding the neuronal cells, and are described as “cushioning material” (115). This opens the possibility that glia, in general, are differentially susceptible to forces produced in the brain during mild TBI. Mechanical changes in glial cells may physically protect neurons from initial mechanical damage, but then subsequently become activated and produce factors that drive neuronal pathology (Figure 2A). Alternatively, glia may be bystanders, solely responding to neuronal injury when the force is big enough to affect neurons directly. In either scenario, an aberrant microglial response due to either genetic factors, such as APOE genotype or TREM2 mutations (see above), or environmental influences, such as inflammation due to infection, poor diet or obesity may contribute to concussive injury as a risk factor for neurodegenerative disease (26).
Physical forces may also result in damage to the blood-brain barrier (BBB), a specialized endothelial barrier that tightly regulates molecular and cellular movement into the brain (116). BBB breakdown is long associated with moderate to severe brain injury (Figure 2B) but has recently been shown to be present in clinically relevant models of mild TBI. In awake mice, 24 h after a closed-head impact injury, serum albumin extravasation and evidence of myeloid inflammatory cell infiltration due to BBB breakdown was localized to the lateral surface of the ipsilateral perirhinal cortex adjacent to the impact contact zone (117). In a swine model of head rotational acceleration and in the absence of hemorrhage or other focal pathology, disruption of the BBB was found 6–72 h after injury, by extravasation of serum proteins, fibrinogen and immunoglobulin-G (12), confirming earlier mouse studies (118). BBB disruption was consistent with the biomechanical insult as extravasated serum proteins were observed at interfaces between regions of tissue with differing material properties, including gray–white matter boundaries and periventricular and subpial regions which overlapped with regions of axonal pathology in the white matter (12). This highly relevant model of mild TBI provides insights to subtle, yet significant pathology that is likely to trigger immune responses, yet be undetectable in humans through standard clinical imaging techniques.
Fibrinogen is a central blood coagulation protein that is deposited in the CNS after BBB disruption (62, 119) and is found in brain tissue in models of mild TBI described above (12). Fibrinogen induces encephalitogenic adaptive immune responses and peripheral macrophage recruitment into the CNS leading to demyelination in models of multiple sclerosis (120). Interestingly, fibrinogen deposition in the CNS affects many processes across diseases, including suppression of remyelination through OPC function (121) and induction of microglia-mediated spine elimination, leading to cognitive deficits in a model of AD (122). Both white matter damage and cognitive deficits are described in mild TBI. Currently, a promising advance for treating pathologies in involving fibrinogen deposition, is the generation of a monoclonal antibody 5B8, that selectively inhibits fibrin-induced inflammation and oxidative stress without interfering with clotting and shows efficacy in animal models of MS and AD (123). Whether such strategies will be employed to improve pathology and cognitive and emotional deficits after mild TBI remains to be seen.
More data are required to understand the initial triggers of the immune response in mild TBI, and a combination of neuronal damage through mechanical stretching, direct mechanical damage of glia and endothelial cells and BBB-breakdown are all likely to play a role. Irrespective of the initial driver of brain tissue damage, patient studies indicate there is an active immune response in those individuals exposed to mild TBI injury.
Clinical Evidence of the Immune Response to Mild TBI–Serum Biomarkers
An important area of brain injury and disease research is the search for blood biomarkers that reflect brain processes, to predict disease course and used as readouts to assess utility of interventions to improve outcome for patients. Recently, a range of proteins, including neurofilament light (NfL) polypeptide released from damaged axons, have been proposed as viable biomarkers of mild TBI (60, 124, 125). The brain's immune response to mild TBI is also evident in the blood, as elevated c-reactive protein (CRP) levels at admission are independently associated with the increased risk of persistent psychological problems and cognitive impairment (29). Plasma interleukin (IL)-2 and IL-6 levels are also significantly higher for mild TBI patients compared with orthopedic injury controls, indicating a brain-specific injury initiated an immune response that is present in the periphery (126). Elevated IL-2, 24 h after injury, is associated more severe early post-concussive symptoms, while elevated plasma IL-10 level at 6 months is associated with more severe posttraumatic stress disorder (PTSD) and mood scores (126). Overall plasma levels of IL-1ß, IL-4, IL-6, and IFN-γ are reduced at 6 months compared to acute levels, indicating a subsiding of inflammation caused by the initial injury. Interestingly, plasma levels of the anti-inflammatory cytokine IL-10 have also been shown to be predictive of a mild TBI vs. a moderate-to-severe TBI (as defined by hemorrhage on CT) (127), again highlighting the different immune responses between injury types. Further to immune mediators detected in the plasma, complement pathway proteins (key mediators of the immune response) are elevated in astrocyte-derived exosomes in the plasma within seven days of a mild TBI (128). In the non-acute setting, a study of veterans with a remote history of mild TBIs found an association between concentrations of TNF-α and post-concussive syndrome (PCS) and PTSD symptoms (125). The total number of mild TBIs correlated with exosomal and plasma NfL levels and plasma IL-6 (125). These results indicate a persistent elevated neuronal and neuroinflammatory response many years after mild TBI.
Serum biomarkers of brain injury and the immune response are now being collected in a range of settings, from single injuries to chronic sport related mild TBI in male and female participants (129). Although, the peripheral immune response to more severe TBI is well documented (24, 64, 130), the data specifically in mild TBI highlights that circulating immune mediators are also present and may be involved in symptom progression and resolution.
Clinical Evidence of the Immune Response to Mild TBI–Neuroimaging
The immune response can also be measured indirectly in the brain by clinical neuroimaging. The meninges, a protective layer of membranes surrounding the brain, have gained much attention in the field of neuroimmunology as they are the home of the brain's lymphatic system (131, 132) and a variety of immune cell populations (44, 47, 49) which, in animal models, can regulate brain function in health (133), disease (134, 135), infection (136), and recovery from TBI (137). In patients with mild TBI, enhancement of the meninges on post-contrast images obtained by fluid attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI), show abnormalities that may reflect inflammation in the immune cell rich meningeal membranes (138, 139). Meningeal immune cell-mediated inflammation can influence neuronal populations in the brain parenchyma in mice, resulting behavioral changes that are also associated with symptoms of mild TBI, such as anxiety (133, 137). As yet, it is unknown whether meningeal inflammation in human mild TBI plays a role in emotional symptoms seen after injury.
In the context of athletes with a history of mild TBI, initial neuroimaging studies of former retired NFL players reported cognitive deficits that were correlated with white matter abnormalities and changes in regional cerebral blood flow (140) and that mild TBI is a risk factor for development of mild cognitive impairment (140). Following these findings, work began to show that neuroinflammation in the brain tissue proper was also a key component in those at risk from concussive-symptoms.
It is well-known that brain injury and disease cause a change in the functional state of microglia, the major cell type in the brain responsible for neuroinflammation (141). Neuroinflammation is associated with the de novo expression of the mitochondrial 18 kDa translocator protein (TSPO), a binding site for which many selective high-affinity compounds for PET imaging have been developed (142). During brain injury or disease, TSPO is mainly expressed in microglia but can also be detected in astrocytes (51, 143, 144). Increased regional TSPO expression in the brain typically covaries with disease state and activity and is proposed to be a non-diagnostic biomarker and secondary to disease etiology. As result, many clinical studies use in vivo measurements of TSPO expression as a biomarker of disease progression or therapeutic efficacy (142).
In former NFL players, early studies showed significant increase in binding of the radio-ligand [11C]DPA-713 to TSPO in several brain regions, such as the supramarginal gyrus and right amygdala, compared to age-matched, healthy controls, indicating neuroinflammation in those areas (145). The same former players had varied performance on a test of verbal learning and memory (145). Interestingly, studies performed in much younger, active and recently retired NFL players with a self-reported history of mild TBI also revealed increased [11C]DPA-713 binding to TSPO in eight of the 12 brain regions examined, but did not differ from control participants in regional brain volumes or in neuropsychological performance (146). This opened the possibility of using TSPO binding a biomarker for brain changes in the younger concussed brain, prior to cognitive decline (146). Postmortem studies of former NFL player confirmed that repeated head injury is associated with chronic activation of microglia (147). In addition, the duration of repeated head injury exposure, as defined by the years of football played, predicted greater density of CD68 positive inflammatory microglia in NFL players with and without CTE pathology and that increased neuroinflammation was related to the risk of a subject being diagnosed with dementia, independent of age (147). Together, these studies show that the immune response is detectable in patients before cognitive changes, and persists throughout the lifetime of those exposed to concussive injury.
Other strategies to assess immune cell activation in response to concussion injury include the quantification of protein markers in the CSF, such as soluble TREM2 (sTREM2). TREM2 is a transmembrane receptor of the immunoglobulin superfamily highly expressed in microglia which multiple ligands (148). TREM2 surface expression rapidly declines on activation of microglia which is partly due to sequential cleavage of membrane-bound TREM2 by proteases that release soluble TREM2 (sTREM2) (149). As result, sTREM2 has been hypothesized to be predictive of microglial activation if detected in the CSF. In former NFL players, sTREM2 levels were higher in their CSF compared to controls and were associated with higher t-tau concentrations, which are thought to be a major factor in neurodegeneration (150). However, in patients with TREM2 mutations with AD and FTD, sTREM is reduced in the CSF (151). Other studies have shown increases of CSF TREM2 in AD dementia (152, 153), or increase in MCI-AD but not AD dementia (154). Therefore, the use of sTREM as a direct biomarker for microglia activation is yet unproven, and the biological role sTREM2 in neurodegeneration is unknown.
Patient studies investigating the microglial response to mild TBI have largely focused on individuals with a known history of head injury, such as professional athletes. Recently, TSPO expression was assessed after a single-event mild TBI in patients without signs of structural damage 1–2 weeks and at 3–4 months after injury (155). Importantly, patients were not included if they showed any intracranial lesion on the initial computed tomography scan or had a history of head trauma with loss of consciousness. Using the single photon emission computed tomography tracer 123I-CLINDE, persistent TSPO upregulation was found at 3–4 months post-injury, even in patients with good clinical recovery (155). This is consistent with the data from active or recently retired NFL players that had increased TSPO binding, but no change in neuropsychological testing (146). Clearly, it is unknown whether a single head injury and subsequent TSPO increases is comparable to a history of head injury and persistent TSPO increases, and whether they have the same long-term consequences, but it is evidence that a sub-chronic immune response occurs in even mild injury with resolved short-term clinical symptoms.
The Immune Response in Animal Models of Mild TBI
Models of traumatic brain injury have provided insight into multiple mechanisms of tissue damage. Even for “mild” TBI, animal models vary widely, from impacts direct to the brain's surface in fixed-head, anesthetized animals to injury with no surgery or anesthesia with forces that allow head rotation (59, 156). Each model (even similar in description), in different laboratory's hands can lead to different etiologies and severities, which should be considered when assessing the literature. Often the least “clinically relevant” model can lead to the most useful mechanistic insight, and those most likely to best recapitulate human concussion should inherently be the most heterogeneous in their outcome. Both extremes are now being used to explore the role of the immune system in mild brain injury. We will not attempt to review the different models of injury here, but we point the reader to excellent reviews in the field (59, 156). As mentioned previously, we will focus our attention on closed head injury models that most closely resemble clinical mild TBI, at least in as much as do not use any type of craniotomy or produce frank tissue damage or hemorrhage. We will assess the only those models that are “closed skull” with no observable bleeding and investigate the immune reaction to injury. These models are relatively few compared to the wider TBI literature but may offer key insights to pathology.
In a pig model of closed-head rotational acceleration, biomechanical loading parameters can be replicated that are thought to underlie neuropathology of mild TBI in humans (157). Pigs have a large gyrencephalic brain and a gray to white matter ratio similar to humans, which is important as diffuse axonal injury in white matter is believed to be the principal pathology of closed-head diffuse brain injury (42, 158). Using these models, changes in microglia morphology around compromised neurons was described as early as 15 min after injury, potentially allowing microglia to influence the evolution of subsequent neuronal damage (159). Evidence from many other models of CNS injury suggest that initial actions of microglia are protective (69, 160–162), though this is unknown in the context of mild TBI. In the same pig model, investigation of later time points after injury, found subtle neuronal changes in the hippocampus (163, 164) and microglia had increased signs of activation up to 1 year after injury (164). Just as acute microglial actions at sites of CNS injury are proposed to be beneficial, long-term microglial-mediated inflammation is thought to be detrimental and may be involved in the long-term complications associated with mild TBI (165–173). Longer-term activation of microglia was also seen in non-surgical, diffuse closed-head injury in mice, in a model characterized by an impact as well as linear and rotational acceleration (174). Thirty days after injury, multifocal, bilateral axonal damage with neuronal death in the hippocampus was detected, microgliosis was prominent and neurobehavioral deficits observed in spatial learning/memory and socialization (174). Indeed, a range of studies in appropriate rodent models show microglial activation across multiple time points, particularly in white matter tracts (165–172) and may be a result of rotational stress or reduced cerebral blood flow (175, 176).
To investigate the causality of inflammation and the immune response on mild TBI deficits, multiple studies have manipulated these pathways and investigated behavioral outcome measures. For example; selective, small molecule inhibition of acute pro-inflammatory cytokines and chemokines, nanopeptides targeting apoE mediated the neuroinflammation and minocycline administration all reduce microglial activation and improve neurological outcome after mild TBI, respectively (177–179). Hippocampal microglia activation is attenuated by inhaled nitrous oxide and correlates with improved performance on memory tasks (180) and statins reduce pro-inflammatory cytokine gene expression in the brain, reduce microglial activation and improve functional neurological deficits after mild TBI (181). Nilvadipine, a tyrosine kinase inhibitor, suppresses inflammation and restores spatial memory deficits (182) and administration of Carnosic acid, and inhibitor of NF-kB, significantly improves motor and cognitive function and reduces microglia activation in white matter tracks in a mouse models of repetitive mild TBI (183). The neuronal expression of pro-inflammatory mediator complement receptor C5a is upregulated after mild TBI and is dependent on TNF (184). Mice lacking a functional alternative pathway of complement activation have reduced neuronal cell death after mild TBI (185) and removal of the protective neuronal-derived complement protein CD59, worsens neurological outcome seven days after mild TBI in mice (186).
These studies show that, like in humans, inflammation and microglial activation are prolonged after injury. Animal models show that the neuroimmune response correlates with cognitive deficits and may be modified to improve outcome, though more evidence of causal contribution of the immune system to behavioral deficits is needed.
Models of mild TBI are continually being refined. Recently, it was shown that a commonly used inhaled anesthetic, isoflurane, inhibits microglial ramification and surveillance in vivo (187), therefore potentially blocking the immediate immune response of the brain to injury when animals and anesthetized. The issue of anesthesia is prominent in almost all models of brain injury, but some have begun to produce concussive injuries in awake, restrained animals. In awake rats, using an injury paradigm with a bespoke helmet, injury produced memory deficits, and microglia activation after impact verses sham (188). Although, these studies remove the issue of anesthetic effects on the immune system, the stress induced by restraint of animals must be considered. Restrain habituation for the above study was performed for 3 days before injury, and stress induced inflammation may still be a contributing factor.
The onset and duration of inflammation driven by immune mechanisms after concussion is likely to play a key role in pathology. Activation of Toll-like receptor 4 (TLR4), which plays a fundamental role in pathogen recognition and activation of innate immunity, following repeated mild TBI is either beneficial or detrimental depending on the timing of activation. Administration of low dose LPS 1 day after injury was associated with a reduction in neuronal injury, a restoration of levels of myelin basic protein (MBP) and PSD-95 and no behavioral changes in locomotion, anxiety, depressive-like behavior or cognition at 3 months post-injury (189). Conversely, when LPS was given at 5 days after injury, it was associated with an acute increase in pro-inflammatory cytokine production, an exacerbation of neuronal damage and increased levels of aggregated and phosphorylated tau which led to a slight exacerbation of cognitive deficits and depressive-like behavior at 3 months (189). Due to the interest in the immune response after mild TBI, a natural avenue for therapeutics are anti-inflammatory treatments. However, modulating the immune response may require a strict, and yet unknown, time course. The strength of immune modulation should also be considered. In a model of AD, it was found that targeting the TLR4 receptor before onset of pathology by inducing either immune tolerance (4xLPS injections) or immune training/priming (1xLPS injection), either alleviates later B-amyloidosis with the former, or makes it worse with the latter (190). Such mechanisms must be considered when approaching inflammation after mild TBI, especially considering mild head injury's convergence with neurodegenerative disease (89).
In a model of lateral closed-head impact injury that uses momentum transfer to induce traumatic head acceleration in unanaesthetised mice, an abrupt onset and rapid resolution of a concussion-like syndrome is characterized by altered arousal, locomotor impairments and neurobehavioral deficits (117). The majority of brains in injured mice (~90%) had no evidence of frank hemorrhage or contusion but BBB breakdown was observed. An increase in the number of microglia and in infiltration of monocytes was observed 3 days after injury, though this was localized to the impact region (117). Uniquely, the study presented post-mortem brains from four teenage athletes in the acute-subacute period after mild closed-head impact injury and found, among other pathology, perivascular neuroinflammation in the form of haemosiderin-laden macrophages surrounding a small blood vessel (117). These data indicate that microglia may not be the only immune cell contributing to mild TBI pathology, as monocyte-derived macrophages from the circulation may enter brain tissue after mild injury, though more data is required to confirm this as an immune response to mild TBI pathology, in general.
Repeated mild TBIs cause more significant neurological damage than a single injury, including longer recovery time and a higher likelihood of subsequent brain injury (32, 54). It is hypothesized that priming of the immune response may play a role. Repeated mild brain injury produces greater microglial activation, anxiety-like behavior and impaired spatial memory compared to a single injury, in mice (191). In rats tested with projectiles and helmets, repeated injury produced a more significant and long lasting inflammatory response associated with microglial activation than a single injury (192). Delivering multiple mild impacts over a shorter inter-injury interval leads to a more significant acute microglial activation and prolonged astrogliosis in select regions of the brain, compared to the same number of administered over a longer time-period (193). The corpus callosum, hippocampus and lateral septum appear particularly vulnerable to injury (191, 193) and these areas may contribute to clinical symptomology, including anxiety and memory problems (191, 193). Frequent mild head injury also promotes trigeminal sensitivity with associated microglial proliferation and increased neuropeptide levels in the trigeminal pain system (194), which is associated with headaches and migraine that accompany post-concussion syndrome. In a mouse model of highly repetitive mild TBI, 30 injuries cause white matter pathology, and microglial proliferation and activation. This pathology is present 60 days after final injury, and is still apparent at 1 year (195).
The Immune Response in Animal Models of Mild TBI and Alzheimer's Disease
As discussed above, epidemiological studies associate increased risk of AD-related clinical symptoms with a history of mild TBI (2, 26, 33, 35). To investigate the link between mild TBI and AD, studies have performed brain injury in genetic mouse models of AD and assessed the neuroinflammatory response driving pathology and clinical symptoms (196–200). In APP/PS1 mice that contain human transgenes for both mutated APP and PSEN1 (201), neuroinflammatory gene expression is increased seven days after injury and microglia activation is greater at 2 months in APP/PS1 compared to WT mild TBI controls (197). Importantly, a small molecule inhibitor, previously described to selectively limit pro-inflammatory cytokine production after mild TBI (202), improved cognitive behavior outcomes in APP/PS1 mice after injury (197). Thus, providing a link between neuroinflammatory responses and altered risk for AD-associated pathology changes after mild TBI (197). Other studies in APP/PS1 mice and mild TBI models have investigated acute neuroinflammatory outcomes in young and old mice, however, the interplay between the immune response and AD-like progression was more complicated as neuroinflammation is increased in aged WT mice but reduced in aged APP/PS1 mice (199, 200). In a repeated mild TBI model (12 injuries in 1 month) and aged APP/PS1 mice, there was no increase in various makers of microglia activation 1 month after final brain injury compared to sham control (203). However, there was an increase in brain insoluble to soluble Aß42 ratio in injured APP/PS1 mice compared with sham and a parallel reduction in phagocytic receptor, TREM2, suggesting pathology mediating microglia changes induced by injury (203). In a transgenic mouse model for human tau (hTau), acute microglia activation after mild TBI is increased in both young and aged animals, however, this response is not as robust in aged animals compared to young, suggesting a diminished acute microglial response to mild TBI in animals with established AD pathology (204) and highlights the complicated nature of priming and tolerance in chronic neurodegenerative disease (53).
To model subthreshold injury be expected in competitive contact sports, a 7 days 42-impact paradigm with helmets in mice was used to simulate frequent head injury (196). This paradigm resulted in chronic gliosis and T-cell infiltration of the superior colliculus and optic tract, with concomitant demyelination of the optic nerve and associated white matter tracts 1 month after injury (196). When injuries were performed in Tau58.4 mice, there was progressive neuroinflammation and neurodegeneration in multiple brain regions compared to WT mice. To investigate the role of T-cells in these specific areas vulnerable to demyelination, T-cell-deficient Rag2/ Il2rg (R2G2) mice were subjected to the same injury paradigm. R2G2 mice had altered myeloid cell gene expression and fewer demyelinated lesions compared to T cell-competent mice. This study suggests that vulnerable regions known to be affected in CTE, such as white matter, may be protected by manipulating the immune system (196).
In summary, animal models of mild TBI are varied, but extremely powerful tools investigate mechanisms of injury. There is now a wealth of evidence linking the immune response to mild TBI and the behavioral and pathological outcomes. This is fueling many investigations into new therapeutics targeting these mechanisms.
Conclusion
Mild TBI, often referred to as concussion, is the most common form of traumatic brain injury and can result in insidious effects including emotional and cognitive dysfunction. In addition, a history of mild TBI is proposed as a risk factor for longer term neurodegenerative disease. Despite this, the underlying pathology is still unclear. Immune activation, and particularly changes to microglia are associated with human mild TBI and a variety of animal models of mild brain injury. In many cases these are descriptive, although evidence suggests immune activation correlates with cognitive and behavioral symptoms. Animal studies are beginning to demonstrate a causative role of the immune system in acute brain dysfunction following mild TBI. The fact that the immune system is readily reactive, and can remain active over a long period of time after injury, leaves open the possibility that an aberrant immune response, driven by factors that have shown to be important for neurodegenerative disease, may contribute to the long-term consequences of mild brain injury.
Author Contributions
LV contributed to writing the manuscript. HP edited the manuscript. AG wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
AG was supported by a University of Manchester Presidential Fellowship. Publishing costs covered by The University of Manchester open access publishing fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Smith DH, Stewart W. “Concussion” is not a true diagnosis. Nat Rev Neurol. (2020) 16:457–8. doi: 10.1038/s41582-020-0382-y
2. Sharp DJ, Jenkins PO. Concussion is confusing us all. Pract Neurol. (2015) 15:172–86. doi: 10.1136/practneurol-2015-001087
3. Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung Y-C, Punchak M, et al. Estimating the global incidence of traumatic brain injury. J Neurosurg. (2018) 1:1–18. doi: 10.3171/2017.10.JNS17352
4. Spira JL, Lathan CE, Bleiberg J, Tsao JW. The impact of multiple concussions on emotional distress, post-concussive symptoms, and neurocognitive functioning in active duty United States marines independent of combat exposure or emotional distress. J Neurotrauma. (2014) 31:1823–34. doi: 10.1089/neu.2014.3363
5. Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. (2006) 21:375–8. doi: 10.1097/00001199-200609000-00001
6. Wagner AK, Sasser HC, Hammond FM, Wiercisiewski D, Alexander J. Intentional traumatic brain injury: epidemiology, risk factors, and associations with injury severity and mortality. J Trauma Acute Care Surg. (2000) 49:404–10. doi: 10.1097/00005373-200009000-00004
7. Decq P, Gault N, Blandeau M, Kerdraon T, Berkal M, ElHelou A, et al. Long-term consequences of recurrent sports concussion. Acta Neurochir. (2016) 158:289–300. doi: 10.1007/s00701-015-2681-4
9. Smith DH, Nonaka M, Miller R, Leoni M, Chen XH, Alsop D, et al. Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. J Neurosurg. (2000) 93:315–22. doi: 10.3171/jns.2000.93.2.0315
10. Laksari K, Kurt M, Babaee H, Kleiven S, Camarillo D. Mechanistic insights into human brain impact dynamics through modal analysis. Phys Rev Lett. (2018) 120:138101. doi: 10.1103/PhysRevLett.120.138101
11. Zhang L, Yang KH, King AI. A proposed injury threshold for mild traumatic brain injury. J Biomech Eng. (2004) 126:226–36. doi: 10.1115/1.1691446
12. Johnson VE, Weber MT, Xiao R, Cullen DK, Meaney DF, Stewart W, et al. Mechanical disruption of the blood–brain barrier following experimental concussion. Acta Neuropathol. (2018) 135:711–26. doi: 10.1007/s00401-018-1824-0
13. Gyoneva S, Ransohoff RM. Inflammatory reaction after traumatic brain injury: therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol Sci. (2015) 36:471–480. doi: 10.1016/j.tips.2015.04.003
14. McInnes K, Friesen CL, MacKenzie DE, Westwood DA, Boe SG. Mild traumatic brain injury (mTBI) and chronic cognitive impairment: a scoping review. PLoS ONE. (2017) 12:e0174847. doi: 10.1371/journal.pone.0174847
15. Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J. Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron. (2017) 95:1246–65. doi: 10.1016/j.neuron.2017.07.010
16. Cassidy JD, Carroll LJ, Peloso PM, Borg J, von Holst H, Holm L, et al. Incidence, risk factors and prevention of mild traumatic brain injury: results of the WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J Rehabil Med. (2004) (43 Suppl):28–60. doi: 10.1080/16501960410023732
17. Berz K, Divine J, Foss KB, Heyl R, Ford KR, Myer GD. Sex-specific differences in the severity of symptoms and recovery rate following sports-related concussion in young athletes. Phys Sportsmed. (2013) 41:58–63. doi: 10.3810/psm.2013.05.2015
18. Bazarian JJ, Blyth B, Mookerjee S, He H, McDermott MP. Sex differences in outcome after mild traumatic brain injury. J Neurotrauma. (2010) 27:527–39. doi: 10.1089/neu.2009.1068
19. Broshek DK, Kaushik T, Freeman JR, Erlanger D, Webbe F, Barth JT. Sex differences in outcome following sports-related concussion. J Neurosurg. (2005) 102:856–63. doi: 10.3171/jns.2005.102.5.0856
20. Rubin TG, Lipton ML. Sex differences in animal models of traumatic brain injury. J Exp Neurosci. (2019) 13:1179069519844020. doi: 10.1177/1179069519844020
21. Mollayeva T, El-Khechen-Richandi G, Colantonio A. Sex & gender considerations in concussion research. Concussion. (2018) 3:CNC51. doi: 10.2217/cnc-2017-0015
22. Xiong Y, Mahmood A, Chopp M. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chin J Traumatol. (2018) 21:137–51. doi: 10.1016/j.cjtee.2018.02.003
23. McKee CA, Lukens JR. Emerging roles for the immune system in traumatic brain injury. Front Immunol. (2016) 7:556. doi: 10.3389/fimmu.2016.00556
24. Simon DW, McGeachy MJ, Bayir H, Clark RSB, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. (2017) 13:572. doi: 10.1038/nrneurol.2017.116
25. Wofford KL, Loane DJ, Cullen DK. Acute drivers of neuroinflammation in traumatic brain injury. Neural Regen Res. (2019) 14:1481–9. doi: 10.4103/1673-5374.255958
26. Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. (2020) 396:413–46. doi: 10.1016/S0140-6736(20)30367-6
27. Ponsford J, Willmott C, Rothwell A, Cameron P, Kelly A-M, Nelms R, et al. Factors influencing outcome following mild traumatic brain injury in adults. J Int Neuropsychol Soc. (2000) 6:568–79. doi: 10.1017/S1355617700655066
28. Bigler ED, Farrer TJ, Pertab JL, James K, Petrie JA, Hedges DW. Reaffirmed limitations of meta-analytic methods in the study of mild traumatic brain injury: a response to Rohling et al. Clin Neuropsychol. (2013) 27:176–214. doi: 10.1080/13854046.2012.693950
29. Su S-H, Xu W, Li M, Zhang L, Wu Y-F, Yu F, et al. Elevated C-reactive protein levels may be a predictor of persistent unfavourable symptoms in patients with mild traumatic brain injury: a preliminary study. Brain Behav Immun. (2014) 38:111–7. doi: 10.1016/j.bbi.2014.01.009
30. Abrahams S, McFie S, Lacerda M, Patricios J, Suter J, September AV, et al. Unravelling the interaction between the DRD2 and DRD4 genes, personality traits and concussion risk. BMJ Open Sport Exerc Med. (2019) 5:e000465. doi: 10.1136/bmjsem-2018-000465
31. Ashton J, Coyles G, Malone JJ, Roberts JW. Effects of an acute bout of repeated soccer heading on cognitive performance. Science and Medicine in Football. (2020) 1–7. doi: 10.1080/24733938.2020.1846769
32. Guskiewicz KM, Marshall SW, Bailes J, McCrea M, Harding HP, Matthews A, et al. Recurrent concussion and risk of depression in retired professional football players. Med Sci Sports Exerc. (2007) 39:903–9. doi: 10.1249/mss.0b013e3180383da5
33. Abner EL, Nelson PT, Schmitt FA, Browning SR, Fardo DW, Wan L, et al. Self-reported head injury and risk of late-life impairment and AD pathology in an AD center cohort. Dement Geriatr Cogn Disord. (2014) 37:294–306. doi: 10.1159/000355478
34. Russell ER, Stewart K, Mackay DF, MacLean J, Pell JP, Stewart W. Football's InfluencE on Lifelong health and Dementia risk (FIELD): protocol for a retrospective cohort study of former professional footballers. BMJ Open. (2019) 9:e028654. doi: 10.1136/bmjopen-2018-028654
35. Mackay DF, Russell ER, Stewart K, MacLean JA, Pell JP, Stewart W. Neurodegenerative disease mortality among former professional soccer players. N Engl J Med. (2019) 381:1801–8. doi: 10.1056/NEJMoa1908483
36. Russell ER, McCabe T, Mackay DF, Stewart K, MacLean JA, Pell JP, et al. Mental health and suicide in former professional soccer players. J Neurol Neurosurg Psychiatry. (2020) 91:1256–60. doi: 10.1136/jnnp-2020-323315
37. Morissette MP, Prior HJ, Tate RB, Wade J, Leiter JRS. Associations between concussion and risk of diagnosis of psychological and neurological disorders: a retrospective population-based cohort study. Fam Med Com Health. (2020) 8:e000390. doi: 10.1136/fmch-2020-000390
38. Pupillo E, Bianchi E, Vanacore N, Montalto C, Ricca G, Robustelli Della Cuna FS, et al. Increased risk and early onset of ALS in professional players from Italian Soccer Teams. Amyotroph Lateral Scler Frontotemporal Degener. (2020) 21:403–9. doi: 10.1080/21678421.2020.1752250
39. Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA. (2017) 318:360–70. doi: 10.1001/jama.2017.8334
40. Lee EB, Kinch K, Johnson VE, Trojanowski JQ, Smith DH, Stewart W. Chronic traumatic encephalopathy is a common co-morbidity, but less frequent primary dementia in former soccer and rugby players. Acta Neuropathol. (2019) 138:389–99. doi: 10.1007/s00401-019-02030-y
41. Goldstein LE, Fisher AM, Tagge CA, Zhang X-L, Velisek L, Sullivan JA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. (2012) 4:134ra60. doi: 10.1126/scitranslmed.3003716
42. Browne KD, Chen X-H, Meaney DF, Smith DH. Mild traumatic brain injury and diffuse axonal injury in swine. J Neurotrauma. (2011) 28:1747–55. doi: 10.1089/neu.2011.1913
43. Greenhalgh AD, David S, Bennett FC. Immune cell regulation of glia during CNS injury and disease. Nat Rev Neurosci. (2020) 21:139–52. doi: 10.1038/s41583-020-0263-9
44. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional ingle-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. (2018) 48:380–395. doi: 10.1016/j.immuni.2018.02.014
45. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
46. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
47. Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. (2019) 22:1021–35. doi: 10.1038/s41593-019-0393-4
48. Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, et al. High-dimensional, single-cell characterization of the brain's immune compartment. Nat Neurosci. (2017) 20:1300. doi: 10.1038/nn.4610
49. Goldmann T, Wieghofer P, Jordao MJC, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. (2016) 17:797–805. doi: 10.1038/ni.3423
50. Scheiblich H, Trombly M, Ramirez A, Heneka MT. Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol. (2020) 41:300–12. doi: 10.1016/j.it.2020.02.002
51. Boche D, Gerhard A, Rodriguez-Vieitez E, MINC Faculty. Prospects and challenges of imaging neuroinflammation beyond TSPO in Alzheimer's disease. Eur J Nucl Med Mol Imaging. (2019) 46:2831–47. doi: 10.1007/s00259-019-04462-w
52. Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. (2014) 10:217–24. doi: 10.1038/nrneurol.2014.38
53. Neher JJ, Cunningham C. Priming microglia for innate immune memory in the brain. Trends Immunol. (2019) 40:358–74. doi: 10.1016/j.it.2019.02.001
54. Guskiewicz KM, McCrea M, Marshall SW, Cantu RC, Randolph C, Barr W, et al. Cumulative effects associated with recurrent concussion in collegiate football players: the NCAA concussion study. JAMA. (2003) 290:2549–55. doi: 10.1001/jama.290.19.2549
55. Damisah EC, Hill RA, Rai A, Chen F, Rothlin CV, Ghosh S, et al. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci Adv. (2020) 6:eaba3239. doi: 10.1126/sciadv.aba3239
56. Zhou T, Zheng Y, Sun L, Badea SR, Jin Y, Liu Y, et al. Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nat Neurosci. (2019) 22:421–35. doi: 10.1038/s41593-018-0324-9
57. Falcão AM, van Bruggen D, Marques S, Meijer M, Jäkel S, Agirre E, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. (2018) 24:1837–44. doi: 10.1038/s41591-018-0236-y
58. Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. (2016) 37:608–20. doi: 10.1016/j.it.2016.06.006
59. Bodnar CN, Roberts KN, Higgins EK, Bachstetter AD. A systematic review of closed head injury models of mild traumatic brain injury in mice and rats. J Neurotrauma. (2019) 36:1683–706. doi: 10.1089/neu.2018.6127
60. Zetterberg H, Blennow K. Fluid biomarkers for mild traumatic brain injury and related conditions. Nat Rev Neurol. (2016) 12:563–74. doi: 10.1038/nrneurol.2016.127
61. Sahyouni R, Gutierrez P, Gold E, Robertson RT, Cummings BJ. Effects of concussion on the blood-brain barrier in humans and rodents. J Concussion. (2017). doi: 10.1177/2059700216684518. [Epub ahead of print].
62. Sulimai N, Lominadze D. Fibrinogen and neuroinflammation during traumatic brain injury. Mol Neurobiol. (2020) 57:4692–703. doi: 10.1007/s12035-020-02012-2
63. Beamer E, Gölöncsér F, Horváth G, Beko K, Otrokocsi L, Koványi B, et al. Purinergic mechanisms in neuroinflammation: an update from molecules to behavior. Neuropharmacology. (2016) 104:94–104. doi: 10.1016/j.neuropharm.2015.09.019
64. O'Brien WT, Pham L, Symons GF, Monif M, Shultz SR, McDonald SJ. The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J Neuroinflammation. (2020) 17:104. doi: 10.1186/s12974-020-01778-5
65. Ghosh S, Castillo E, Frias ES, Swanson RA. Bioenergetic regulation of microglia. Glia. (2018) 66:1200–12. doi: 10.1002/glia.23271
66. Devanney NA, Stewart AN, Gensel JC. Microglia and macrophage metabolism in CNS injury and disease: the role of immunometabolism in neurodegeneration and neurotrauma. Exp Neurol. (2020) 329:113310. doi: 10.1016/j.expneurol.2020.113310
67. Wang J, Ma MW, Dhandapani KM, Brann DW. Regulatory role of NADPH oxidase 2 in the polarization dynamics and neurotoxicity of microglia/macrophages after traumatic brain injury. Free Radic Biol Med. (2017) 113:119–31. doi: 10.1016/j.freeradbiomed.2017.09.017
68. Calcia MA, Bonsall DR, Bloomfield PS, Selvaraj S, Barichello T, Howes OD. Stress and neuroinflammation: a systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology. (2016) 233:1637–50. doi: 10.1007/s00213-016-4218-9
69. Hines DJ, Hines RM, Mulligan SJ, Macvicar BA. Microglia processes block the spread of damage in the brain and require functional chloride channels. Glia. (2009) 57:1610–8. doi: 10.1002/glia.20874
70. Bernier L-P, York EM, Kamyabi A, Choi HB, Weilinger NL, MacVicar BA. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat Commun. (2020) 11:1559. doi: 10.1038/s41467-020-15267-z
71. David S, Kroner A, Greenhalgh AD, Zarruk JG, López-Vales R. Myeloid cell responses after spinal cord injury. J Neuroimmunol. (2018) 321:97–108. doi: 10.1016/j.jneuroim.2018.06.003
72. Miao W, Zhao Y, Huang Y, Chen D, Luo C, Su W, et al. IL-13 ameliorates neuroinflammation and promotes functional recovery after traumatic brain injury. J Immunol. (2020) 204:1486–98. doi: 10.4049/jimmunol.1900909
73. Ritzel RM, Doran SJ, Glaser EP, Meadows VE, Faden AI, Stoica BA, et al. Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiol Aging. (2019) 77:194–206. doi: 10.1016/j.neurobiolaging.2019.02.010
74. Greenhalgh AD, David S. Differences in the phagocytic response of microglia and peripheral macrophages after spinal cord injury and its effects on cell death. J Neurosci. (2014) 34:6316–22. doi: 10.1523/JNEUROSCI.4912-13.2014
75. Morganti JM, Jopson TD, Liu S, Riparip L-K, Guandique CK, Gupta N. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci. (2015) 35:748–760. doi: 10.1523/JNEUROSCI.2405-14.2015
76. Morganti-Kossmann MC, Semple BD, Hellewell SC, Bye N, Ziebell JM. The complexity of neuroinflammation consequent to traumatic brain injury: from research evidence to potential treatments. Acta Neuropathol. (2019) 137:731–55. doi: 10.1007/s00401-018-1944-6
77. Puntambekar SS, Saber M, Lamb BT, Kokiko-Cochran ON. Cellular players that shape evolving pathology and neurodegeneration following traumatic brain injury. Brain Behav Immun. (2018) 71:9–17. doi: 10.1016/j.bbi.2018.03.033
78. Tapp ZM, Kumar JE, Witcher KG, Atluri RR, Velasquez JA, O'Neil SM, et al. Sleep disruption exacerbates and prolongs the inflammatory response to traumatic brain injury. J Neurotrauma. (2020) 37:1829–43. doi: 10.1089/neu.2020.7010
79. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe C-U, Siler DA. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. (2009) 40:1849–57. doi: 10.1161/STROKEAHA.108.534503
80. Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J. Myeloid cells in the central nervous system. Immunity. (2017) 46:943–56. doi: 10.1016/j.immuni.2017.06.007
81. Benakis C, Llovera G, Liesz A. The meningeal and choroidal infiltration routes for leukocytes in stroke. Ther Adv Neurol Disord. (2018) 11:1756286418783708. doi: 10.1177/1756286418783708
82. Greenhalgh AD, Zarruk JG, Healy LM, Baskar Jesudasan SJ, Jhelum P, Salmon CK, et al. Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PLoS Biol. (2018) 16:e2005264. doi: 10.1371/journal.pbio.2005264
83. Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. (2009) 6:e1000113. doi: 10.1371/journal.pmed.1000113
84. Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. (2013) 38:555–69. doi: 10.1016/j.immuni.2013.02.012
85. Ma S-F, Chen Y-J, Zhang J-X, Shen L, Wang R, Zhou J-S, et al. Adoptive transfer of M2 macrophages promotes locomotor recovery in adult rats after spinal cord injury. BrainBehav Immun. (2015) 45:157–70. doi: 10.1016/j.bbi.2014.11.007
86. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol. (2010) 28:321–42. doi: 10.1146/annurev-immunol-030409-101311
87. Kroner A, Greenhalgh AD, Zarruk JG, Passos dos Santos R, Gaestel M, David S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron. (2014) 83:1098–116. doi: 10.1016/j.neuron.2014.07.027
88. Chaichana KL, Pradilla G, Huang J, Tamargo RJ. Role of inflammation (leukocyte-endothelial cell interactions) in vasospasm after subarachnoid hemorrhage. World Neurosurg. (2010) 73:22–41. doi: 10.1016/j.surneu.2009.05.027
89. Greenhalgh AD, Brough D, Robinson EM, Girard S, Rothwell NJ, Allan SM. Interleukin-1 receptor antagonist is beneficial after subarachnoid haemorrhage in rat by blocking haem-driven inflammatory pathology. Dis Models Mech. (2012) 5:823–33. doi: 10.1242/dmm.008557
90. Lee J-Y, Keep RF, He Y, Sagher O, Hua Y, Xi G. Hemoglobin and iron handling in brain after subarachnoid hemorrhage and the effect of deferoxamine on early brain injury: J Cereb Blood Flow Metab. (2010) 30:1793–803. doi: 10.1038/jcbfm.2010.137
91. Borsody M, Burke A, Coplin W, Miller-Lotan R, Levy A. Haptoglobin and the development of cerebral artery vasospasm after subarachnoid hemorrhage. Neurology. (2006) 66:634–40. doi: 10.1212/01.wnl.0000200781.62172.1d
92. Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. (2019) 51:404–13. doi: 10.1038/s41588-018-0311-9
93. Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, Pocock JM. Microglial genes regulating neuroinflammation in the progression of Alzheimer's disease. Curr Opin Neurobiol. (2016) 36:74–81. doi: 10.1016/j.conb.2015.10.004
94. Jurgens HA, Johnson RW. Dysregulated neuronal-microglial cross-talk during aging, stress and inflammation. Exp Neurol. (2012) 233:40–8. doi: 10.1016/j.expneurol.2010.11.014
95. Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. (2013) 61:71–90. doi: 10.1002/glia.22350
96. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. (2018) 21:1359–69. doi: 10.1038/s41593-018-0242-x
97. Hammond TR, Marsh SE, Stevens B. Immune signaling in neurodegeneration. Immunity. (2019) 50:955–74. doi: 10.1016/j.immuni.2019.03.016
98. Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. (2018) 69:437–49. doi: 10.1146/annurev-med-050715-104343
99. Smith C, McColl BW, Patir A, Barrington J, Armishaw J, Clarke A, et al. Biallelic mutations in NRROS cause an early onset lethal microgliopathy. Acta Neuropathol. (2020) 139:947–51. doi: 10.1007/s00401-020-02137-7
100. Dong X, Tan NB, Howell KB, Barresi S, Freeman JL, Vecchio D, et al. Bi-allelic LoF NRROS variants impairing active TGF-β1 delivery cause a severe infantile-onset neurodegenerative condition with intracranial calcification. Am J Hum Genet. (2020) 106:559–69. doi: 10.1016/j.ajhg.2020.02.014
101. Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J, et al. A milieu molecule for TGF-β required for microglia function in the nervous system. Cell. (2018) 174:156–71.e16. doi: 10.1016/j.cell.2018.05.027
102. Wong K, Noubade R, Manzanillo P, Ota N, Foreman O, Hackney JA, et al. Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nat Immunol. (2017) 18:633–41. doi: 10.1038/ni.3743
103. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. (2015) 16:358–72. doi: 10.1038/nrn3880
104. Spangenberg EE, Green KN. Inflammation in Alzheimer's disease: lessons learned from microglia-depletion models. Brain Behav Immun. (2017) 61:1–11. doi: 10.1016/j.bbi.2016.07.003
105. Newcombe EA, Camats-Perna J, Silva ML, Valmas N, Huat TJ, Medeiros R. Inflammation: the link between comorbidities, genetics, and Alzheimer's disease. J Neuroinflammation. (2018) 15:276. doi: 10.1186/s12974-018-1313-3
106. Ridge PG, Hoyt KB, Boehme K, Mukherjee S, Crane PK, Haines JL, et al. Assessment of the genetic variance of late-onset Alzheimer's disease. Neurobiol Aging. (2016) 41:200.e13–20. doi: 10.1016/j.neurobiolaging.2016.02.024
107. Huynh T-PV, Liao F, Francis CM, Robinson GO, Serrano JR, Jiang H, et al. Age-dependent effects of apoE reduction using antisense oligonucleotides in a model of β-amyloidosis. Neuron. (2017) 96:1013–23.e4. doi: 10.1016/j.neuron.2017.11.014
108. Lambert J-C, Amouyel P. Genetics of Alzheimer's disease: new evidences for an old hypothesis? Curr Opin Genet Dev. (2011) 21:295–301. doi: 10.1016/j.gde.2011.02.002
109. Hickman SE, Kingery ND, Ohsumi T, Borowsky M, Wang L, Means TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neuroscie. (2014) 16:1896–905. doi: 10.1038/nn.3554
110. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. (2013) 368:117–27. doi: 10.1056/NEJMoa1211851
111. Carmona S, Zahs K, Wu E, Dakin K, Bras J, Guerreiro R. The role of TREM2 in Alzheimer's disease and other neurodegenerative disorders. Lancet Neurol. (2018) 17:721–30. doi: 10.1016/S1474-4422(18)30232-1
112. Bazarian JJ, Zhu T, Zhong J, Janigro D, Rozen E, Roberts A, et al. Persistent, long-term cerebral white matter changes after sports-related repetitive head impacts. PLoS ONE. (2014) 9:e94734. doi: 10.1371/journal.pone.0094734
113. Moshayedi P, Ng G, Kwok JCF, Yeo GSH, Bryant CE, Fawcett JW, et al. The relationship between glial cell mechanosensitivity and foreign body reactions in the central nervous system. Biomaterials. (2014) 35:3919–25. doi: 10.1016/j.biomaterials.2014.01.038
114. Bollmann L, Koser DE, Shahapure R, Gautier HOB, Holzapfel GA, Scarcelli G, et al. Microglia mechanics: immune activation alters traction forces and durotaxis. Front Cell Neurosci. (2015) 9:363. doi: 10.3389/fncel.2015.00363
115. Lu Y-B, Franze K, Seifert G, Steinhäuser C, Kirchhoff F, Wolburg H, et al. Viscoelastic properties of individual glial cells and neurons in the CNS. Proc Natl Acad Sci USA. (2006) 103:17759–64. doi: 10.1073/pnas.0606150103
116. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. (2017) 18:123. doi: 10.1038/ni.3666
117. Tagge CA, Fisher AM, Minaeva OV, Gaudreau-Balderrama A, Moncaster JA, Zhang X-L, et al. Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain. (2018) 141:422–58. doi: 10.1093/brain/awx350
118. Lu J, Moochhala S, Kaur C, Ling EA. Cellular inflammatory response associated with breakdown of the blood-brain barrier after closed head injury in rats. J Neurotrauma. (2001) 18:399–408. doi: 10.1089/089771501750170976
119. Davalos D, Kyu Ryu J, Merlini M, Baeten KM, Le Moan N, Petersen MA, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. (2012) 3:1227. doi: 10.1038/ncomms2230
120. Ryu JK, Petersen MA, Murray SG, Baeten KM, Meyer-Franke A, Chan JP, et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun. (2015) 6:8164. doi: 10.1038/ncomms9164
121. Petersen MA, Ryu JK, Chang K-J, Etxeberria A, Bardehle S, Mendiola AS, et al. Fibrinogen activates BMP signaling in oligodendrocyte progenitor cells and inhibits remyelination after vascular damage. Neuron. (2017) 96:1003–12.e7. doi: 10.1016/j.neuron.2017.10.008
122. Merlini M, Rafalski VA, Rios Coronado PE, Gill TM, Ellisman M, Muthukumar G, et al. Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an Alzheimer's disease model. Neuron. (2019) 101:1099–108.e6. doi: 10.1016/j.neuron.2019.01.014
123. Ryu JK, Rafalski VA, Meyer-Franke A, Adams RA, Poda SB, Rios Coronado PE, et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat Immunol. (2018) 19:1212–23. doi: 10.1038/s41590-018-0232-x
124. Shahim P, Politis A, van der Merwe A, Moore B, Ekanayake V, Lippa SM, et al. Time course and diagnostic utility of NfL, tau, GFAP, and UCH-L1 in subacute and chronic TBI. Neurology. (2020) 95:e623–36. doi: 10.1212/WNL.0000000000009985
125. Guedes VA, Kenney K, Shahim P, Qu B-X, Lai C, Devoto C, et al. Exosomal neurofilament light: a prognostic biomarker for remote symptoms after mild traumatic brain injury? Neurology. (2020) 94:e2412–23. doi: 10.1212/WNL.0000000000009577
126. Vedantam A, Brennan J, Levin HS, McCarthy JJ, Dash PK, Redell JB, et al. Early versus late profiles of inflammatory cytokines after mild traumatic brain injury and their association with neuropsychological outcomes. J Neurotrauma. (2020) 38:53–62. doi: 10.1089/neu.2019.6979
127. Lagerstedt L, Egea-Guerrero JJ, Rodríguez-Rodríguez A, Bustamante A, Montaner J, El Rahal A, et al. Early measurement of interleukin-10 predicts the absence of CT scan lesions in mild traumatic brain injury. PLoS ONE. (2018) 13:e0193278. doi: 10.1371/journal.pone.0193278
128. Goetzl EJ, Yaffe K, Peltz CB, Ledreux A, Gorgens K, Davidson B, et al. Traumatic brain injury increases plasma astrocyte-derived exosome levels of neurotoxic complement proteins. FASEB J. (2020) 34:3359–66. doi: 10.1096/fj.201902842R
129. Major BP, McDonald SJ, O'Brien WT, Symons GF, Clough M, Costello D, et al. Serum protein biomarker findings reflective of oxidative stress and vascular abnormalities in male, but not female, collision sport athletes. Front Neurol. (2020) 11:549624. doi: 10.3389/fneur.2020.549624
130. McDonald SJ, Sharkey JM, Sun M, Kaukas LM, Shultz SR, Turner RJ, et al. Beyond the brain: peripheral interactions after traumatic brain injury. J Neurotrauma. (2020) 37:770–81. doi: 10.1089/neu.2019.6885
131. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. (2015) 212:991–9. doi: 10.1084/jem.20142290
132. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523:337–41. doi: 10.1038/nature14432
133. Alves de Lima K, Rustenhoven J, Da Mesquita S, Wall M, Salvador AF, Smirnov I, et al. Meningeal γδ T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat Immunol. (2020) 21:1421–29. doi: 10.1038/s41590-020-0776-4
134. Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci. (2018) 21:1380–91. doi: 10.1038/s41593-018-0227-9
135. Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer's disease. Nature. (2018) 560:185–91. doi: 10.1038/s41586-018-0368-8
136. Fitzpatrick Z, Frazer G, Ferro A, Clare S, Bouladoux N, Ferdinand J, et al. Gut-educated IgA plasma cells defend the meningeal venous sinuses. Nature. (2020) 587:472–6. doi: 10.1038/s41586-020-2886-4
137. Bolte AC, Dutta AB, Hurt ME, Smirnov I, Kovacs MA, McKee CA, et al. Meningeal lymphatic dysfunction exacerbates traumatic brain injury pathogenesis. Nat Commun. (2020) 11:4524. doi: 10.1038/s41467-020-18113-4
138. Russo MV, Latour LL, McGavern DB. Distinct myeloid cell subsets promote meningeal remodeling and vascular repair after mild traumatic brain injury. Nat Immunol. (2018) 19:442–52. doi: 10.1038/s41590-018-0086-2
139. Livingston WS, Gill JM, Cota MR, Olivera A, O'Keefe JL, Martin C, et al. Differential gene expression associated with meningeal injury in acute mild traumatic brain injury. J Neurotrauma. (2017) 34:853–60. doi: 10.1089/neu.2016.4479
140. Hart J Jr, Kraut MA, Womack KB, Strain J, Didehbani N, Bartz E, et al. Neuroimaging of cognitive dysfunction and depression in aging retired National Football League players: a cross-sectional studyNeurobiology of aging NFL players. JAMA Neurol. (2013) 70:326–35. doi: 10.1001/2013.jamaneurol.340
141. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. (2017) 23:1018–27. doi: 10.1038/nm.4397
142. Liu G-J, Middleton RJ, Hatty CR, Kam WW-Y, Chan R, Pham T, et al. The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol. (2014) 24:631–53. doi: 10.1111/bpa.12196
143. Lavisse S, Inoue K, Jan C, Peyronneau MA, Petit F, Goutal S, et al. [18F]DPA-714 PET imaging of translocator protein TSPO (18 kDa) in the normal and excitotoxically-lesioned nonhuman primate brain. Eur J Nucl Med Mol Imaging. (2015) 42:478–94. doi: 10.1007/s00259-014-2962-9
144. Cosenza-Nashat M, Zhao M-L, Suh H-S, Morgan J, Natividad R, Morgello S, et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. (2009) 35:306–28. doi: 10.1111/j.1365-2990.2008.01006.x
145. Coughlin JM, Wang Y, Munro CA, Ma S, Yue C, Chen S, et al. Neuroinflammation and brain atrophy in former NFL players: an in vivo multimodal imaging pilot study. Neurobiol Dis. (2015) 74:58–65. doi: 10.1016/j.nbd.2014.10.019
146. Coughlin JM, Wang Y, Minn I, Bienko N, Ambinder EB, Xu X, et al. Imaging of glial cell activation and white matter integrity in brains of active and recently retired National Football League players. JAMA Neurol. (2017) 74:67–74. doi: 10.1001/jamaneurol.2016.3764
147. Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH, et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun. (2016) 4:112. doi: 10.1186/s40478-016-0382-8
148. Ulland TK, Song WM, Huang SC-C, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 maintains microglial metabolic fitness in Alzheimer's disease. Cell. (2017) 170:649–63.e13. doi: 10.1016/j.cell.2017.07.023
149. Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J Biol Chem. (2013) 288:33027–36. doi: 10.1074/jbc.M113.517540
150. Alosco ML, Tripodis Y, Fritts NG, Heslegrave A, Baugh CM, Conneely S, et al. Cerebrospinal fluid tau, Aβ, and sTREM2 in Former National Football League Players: modeling the relationship between repetitive head impacts, microglial activation, and neurodegeneration. Alzheimers Dement. (2018) 14:1159–70. doi: 10.1016/j.jalz.2018.05.004
151. Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. (2014) 6:243ra86. doi: 10.1126/scitranslmed.3009093
152. Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol Neurodegener. (2016) 11:3. doi: 10.1186/s13024-016-0071-x
153. Piccio L, Deming Y, Del-Águila JL, Ghezzi L, Holtzman DM, Fagan AM, et al. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. (2016) 131:925–33. doi: 10.1007/s00401-016-1533-5
154. Suárez-Calvet M, Kleinberger G, Araque Caballero MÁ, Brendel M, Rominger A, Alcolea D, et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer's disease and associate with neuronal injury markers. EMBO Mol Med. (2016) 8:466–76. doi: 10.15252/emmm.201506123
155. Ebert SE, Jensen P, Ozenne B, Armand S, Svarer C, Stenbaek DS, et al. Molecular imaging of neuroinflammation in patients after mild traumatic brain injury: a longitudinal 123I-CLINDE single photon emission computed tomography study. Eu J Neurol. (2019) 26:1426–32. doi: 10.1111/ene.13971
156. Silverberg ND, Gardner AJ, Brubacher JR, Panenka WJ, Li JJ, Iverson GL. Systematic review of multivariable prognostic models for mild traumatic brain injury. J Neurotrauma. (2015) 32:517–26. doi: 10.1089/neu.2014.3600
157. Cullen DK, Harris JP, Browne KD, Wolf JA, Duda JE, Meaney DF, et al. A porcine model of traumatic brain injury via head rotational acceleration. Methods Mol Biol. (2016) 1462:289–324. doi: 10.1007/978-1-4939-3816-2_17
158. Kinder HA, Baker EW, West FD. The pig as a preclinical traumatic brain injury model: current models, functional outcome measures, and translational detection strategies. Neural Regen Res. (2019) 14:413–24. doi: 10.4103/1673-5374.245334
159. Wofford KL, Harris JP, Browne KD, Brown DP, Grovola MR, Mietus CJ, et al. Rapid neuroinflammatory response localized to injured neurons after diffuse traumatic brain injury in swine. Exp Neurol. (2017) 290:85–94. doi: 10.1016/j.expneurol.2017.01.004
160. Szalay G, Martinecz B, Lenart N, Kornyei Z, Orsolits B, Judak L, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun. (2016) 7:11499. doi: 10.1038/ncomms11499
161. Rice RA, Spangenberg EE, Yamate-Morgan H, Lee RJ, Arora RPS, Hernandez MX, et al. Elimination of microglia improves functional outcomes following extensive neuronal loss in the hippocampus. J Neurosci. (2015) 35:9977–89. doi: 10.1523/JNEUROSCI.0336-15.2015
162. Bellver-Landete V, Bretheau F, Mailhot B, Vallières N, Lessard M, Janelle M-E, et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun. (2019) 10:518. doi: 10.1038/s41467-019-08446-0
163. Wolf JA, Johnson BN, Johnson VE, Putt ME, Browne KD, Mietus CJ, et al. Concussion induces hippocampal circuitry disruption in swine. J Neurotrauma. (2017) 34:2303–14. doi: 10.1089/neu.2016.4848
164. Grovola MR, Paleologos N, Wofford KL, Harris JP, Browne KD, Johnson V, et al. Mossy cell hypertrophy and synaptic changes in the hilus following mild diffuse traumatic brain injury in pigs. J Neuroinflammation. (2020) 17:44. doi: 10.1186/s12974-020-1720-0
165. Evanson NK, Guilhaume-Correa F, Herman JP, Goodman MD. Optic tract injury after closed head traumatic brain injury in mice: a model of indirect traumatic optic neuropathy. PLoS ONE. (2018) 13:e0197346. doi: 10.1371/journal.pone.0197346
166. Namjoshi DR, Cheng WH, Bashir A, Wilkinson A, Stukas S, Martens KM, et al. Defining the biomechanical and biological threshold of murine mild traumatic brain injury using CHIMERA (closed head impact model of engineered rotational acceleration). Exp Neurol. (2017) 292:80–91. doi: 10.1016/j.expneurol.2017.03.003
167. Xu L, Nguyen JV, Lehar M, Menon A, Rha E, Arena J, et al. Repetitive mild traumatic brain injury with impact acceleration in the mouse: multifocal axonopathy, neuroinflammation, and neurodegeneration in the visual system. Exp Neurol. (2016) 275:436–49. doi: 10.1016/j.expneurol.2014.11.004
168. Tu T-W, Williams RA, Lescher JD, Jikaria N, Turtzo LC, Frank JA. Radiological-pathological correlation of diffusion tensor and magnetization transfer imaging in a closed head traumatic brain injury model. Ann Neurol. (2016) 79:907–20. doi: 10.1002/ana.24641
169. Collins-Praino LE, Arulsamy A, Katharesan V, Corrigan F. The effect of an acute systemic inflammatory insult on the chronic effects of a single mild traumatic brain injury. Behav Brain Res. (2018) 336:22–31. doi: 10.1016/j.bbr.2017.08.035
170. Gatson JW, Liu M-M, Abdelfattah K, Wigginton JG, Smith S, Wolf S, et al. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J Trauma Acute Care Surg. (2013) 74:470–4. doi: 10.1097/TA.0b013e31827e1f51
171. Chen H, Desai A, Kim H-Y. Repetitive closed-head Impact model ofeEngineered rotational acceleration induces long-term cognitive impairments with persistent astrogliosis and microgliosis in mice. J Neurotrauma. (2017) 34:2291–302. doi: 10.1089/neu.2016.4870
172. Robinson S, Berglass JB, Denson JL, Berkner J, Anstine CV, Winer JL, et al. Microstructural and microglial changes after repetitive mild traumatic brain injury in mice. J Neurosci Res. (2017) 95:1025–35. doi: 10.1002/jnr.23848
173. Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. (2018) 19:622–35. doi: 10.1038/s41583-018-0057-5
174. Sauerbeck AD, Fanizzi C, Kim JH, Gangolli M, Bayly PV, Wellington CL, et al. modCHIMERA: a novel murine closed-head model of moderate traumatic brain injury. Sci Rep. (2018) 8:7677. doi: 10.1038/s41598-018-25737-6
175. Hernandez A, Donovan V, Grinberg YY, Obenaus A, Carson MJ. Differential detection of impact site versus rotational site injury by magnetic resonance imaging and microglial morphology in an unrestrained mild closed head injury model. J Neurochem. (2016) 136(Suppl.1):18–28. doi: 10.1111/jnc.13402
176. Sankar SB, Pybus AF, Liew A, Sanders B, Shah KJ, Wood LB, et al. Low cerebral blood flow is a non-invasive biomarker of neuroinflammation after repetitive mild traumatic brain injury. Neurobiol Dis. (2019) 124:544–54. doi: 10.1016/j.nbd.2018.12.018
177. Lloyd E, Somera-Molina K, Van Eldik LJ, Watterson DM, Wainwright MS. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J Neuroinflammation. (2008) 5:28. doi: 10.1186/1742-2094-5-28
178. Laskowitz DT, Wang H, Chen T, Lubkin DT, Cantillana V, Tu TM, et al. Neuroprotective pentapeptide CN-105 is associated with reduced sterile inflammation and improved functional outcomes in a traumatic brain injury murine model. Sci Rep. (2017) 7:46461. doi: 10.1038/srep46461
179. Sangobowale MA, Grin'kina NM, Whitney K, Nikulina E, St Laurent-Ariot K, Ho JS, et al. Minocycline plus N-Acetylcysteine reduce behavioral deficits and improve histology with a clinically useful time window. J Neurotrauma. (2018) 35:907–17. doi: 10.1089/neu.2017.5348
180. Liu P, Li Y-S, Quartermain D, Boutajangout A, Ji Y. Inhaled nitric oxide improves short term memory and reduces the inflammatory reaction in a mouse model of mild traumatic brain injury. Brain Res. (2013) 1522:67–75. doi: 10.1016/j.brainres.2013.05.032
181. Wang H, Lynch JR, Song P, Yang H-J, Yates RB, Mace B, et al. Simvastatin and atorvastatin improve behavioral outcome, reduce hippocampal degeneration, and improve cerebral blood flow after experimental traumatic brain injury. Exp Neurol. (2007) 206:59–69. doi: 10.1016/j.expneurol.2007.03.031
182. Morin A, Mouzon B, Ferguson S, Paris D, Browning M, Stewart W, et al. Nilvadipine suppresses inflammation via inhibition of P-SYK and restores spatial memory deficits in a mouse model of repetitive mild TBI. Acta Neuropathol Commun. (2020) 8:166. doi: 10.1186/s40478-020-01045-x
183. Maynard ME, Underwood EL, Redell JB, Zhao J, Kobori N, Hood KN, et al. Carnosic acid improves outcome after repetitive mild traumatic brain injury. J Neurotrauma. (2019) 36:2147–52. doi: 10.1089/neu.2018.6155
184. Stahel PF, Kariya K, Shohami E, Barnum SR, Eugster H, Trentz O, et al. Intracerebral complement C5a receptor (CD88) expression is regulated by TNF and lymphotoxin-alpha following closed head injury in mice. J Neuroimmunol. (2000) 109:164–72. doi: 10.1016/S0165-5728(00)00304-0
185. Leinhase I, Holers VM, Thurman JM, Harhausen D, Schmidt OI, Pietzcker M, et al. Reduced neuronal cell death after experimental brain injury in mice lacking a functional alternative pathway of complement activation. BMC Neurosci. (2006) 7:55. doi: 10.1186/1471-2202-7-55
186. Stahel PF, Flierl MA, Morgan BP, Persigehl I, Stoll C, Conrad C, et al. Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. J Neuroinflammation. (2009) 6:2. doi: 10.1186/1742-2094-6-2
187. Madry C, Kyrargyri V, Arancibia-Cárcamo IL, Jolivet R, Kohsaka S, Bryan RM, et al. Microglial ramification, surveillance, and Interleukin-1β release are regulated by the two-pore domain K(+) channel THIK-1. Neuron. (2018) 97:299–312.e6. doi: 10.1016/j.neuron.2017.12.002
188. Pham L, Shultz SR, Kim HA, Brady RD, Wortman RC, Genders SG, et al. Mild closed-head injury in conscious rats causes transient neurobehavioral and glial disturbances: a novel experimental model of concussion. J Neurotrauma. (2019) 36:2260–71. doi: 10.1089/neu.2018.6169
189. Corrigan F, Arulsamy A, Collins-Praino LE, Holmes JL, Vink R. Toll like receptor 4 activation can be either detrimental or beneficial following mild repetitive traumatic brain injury depending on timing of activation. Brain Behav Immun. (2017) 64:124–39. doi: 10.1016/j.bbi.2017.04.006
190. Wendeln A-C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. (2018) 556:332–8. doi: 10.1038/s41586-018-0023-4
191. Broussard JI, Acion L, De Jesús-Cortés H, Yin T, Britt JK, Salas R, et al. Repeated mild traumatic brain injury produces neuroinflammation, anxiety-like behaviour and impaired spatial memory in mice. Brain Inj. (2018) 32:113–22. doi: 10.1080/02699052.2017.1380228
192. Madathil SK, Wilfred BS, Urankar SE, Yang W, Leung LY, Gilsdorf JS, et al. Early microglial activation following closed-head concussive injury is dominated by pro-inflammatory M-1 type. Front Neurol. (2018) 9:964. doi: 10.3389/fneur.2018.00964
193. Acabchuk R, Briggs DI, Angoa-Pérez M, Powers M, Wolferz R, Soloway M, et al. Repeated mild traumatic brain injury causes focal response in lateral septum and hippocampus. Concussion. (2016) 1:CNC13. doi: 10.2217/cnc-2015-0001
194. Tyburski AL, Cheng L, Assari S, Darvish K, Elliott MB. Frequent mild head injury promotes trigeminal sensitivity concomitant with microglial proliferation, astrocytosis, and increased neuropeptide levels in the trigeminal pain system. J Headache Pain. (2017) 18:16. doi: 10.1186/s10194-017-0726-1
195. Winston CN, Noël A, Neustadtl A, Parsadanian M, Barton DJ, Chellappa D, et al. Dendritic spine loss and chronic white matter inflammation in a mouse model of highly repetitive head trauma. Am J Pathol. (2016) 186:552–67. doi: 10.1016/j.ajpath.2015.11.006
196. Cheng H, Deaton LM, Qiu M, Ha S, Pacoma R, Lao J, et al. Tau overexpression exacerbates neuropathology after repeated mild head impacts in male mice. Neurobiol Dis. (2020) 134:104683. doi: 10.1016/j.nbd.2019.104683
197. Webster SJ, Van Eldik LJ, Watterson DM, Bachstetter AD. Closed head injury in an age-related Alzheimer mouse model leads to an altered neuroinflammatory response and persistent cognitive impairment. J Neurosci. (2015) 35:6554–69. doi: 10.1523/JNEUROSCI.0291-15.2015
198. Cheng JS, Craft R, Yu G-Q, Ho K, Wang X, Mohan G, et al. Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS ONE. (2014) 9:e115765. doi: 10.1371/journal.pone.0115765
199. Cheng WH, Stukas S, Martens KM, Namjoshi DR, Button EB, Wilkinson A, et al. Age at injury and genotype modify acute inflammatory and neurofilament-light responses to mild CHIMERA traumatic brain injury in wild-type and APP/PS1 mice. Exp Neurol. (2018) 301:26–38. doi: 10.1016/j.expneurol.2017.12.007
200. Cheng WH, Martens KM, Bashir A, Cheung H, Stukas S, Gibbs E, et al. CHIMERA repetitive mild traumatic brain injury induces chronic behavioural and neuropathological phenotypes in wild-type and APP/PS1 mice. Alzheimers Res Ther. (2019) 11:6. doi: 10.1186/s13195-018-0461-0
201. Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. (2006) 7:940–6. doi: 10.1038/sj.embor.7400784
202. Bachstetter AD, Webster SJ, Goulding DS, Morton JE, Watterson DM, Van Eldik LJ. Attenuation of traumatic brain injury-induced cognitive impairment in mice by targeting increased cytokine levels with a small molecule experimental therapeutic. J Neuroinflammation. (2015) 12:69. doi: 10.1186/s12974-015-0289-5
203. Ojo JO, Leary P, Lungmus C, Algamal M, Mouzon B, Bachmeier C, et al. Subchronic pathobiological response following chronic repetitive mild traumatic brain injury in an aged preclinical model of amyloid pathogenesis. J Neuropathol Exp Neurol. (2018) 77:1144–62. doi: 10.1093/jnen/nly101
Keywords: concussion, neuroimmunology, microglia, neurodegenenerative diseases, inflammation, mild TBI
Citation: Verboon LN, Patel HC and Greenhalgh AD (2021) The Immune System's Role in the Consequences of Mild Traumatic Brain Injury (Concussion). Front. Immunol. 12:620698. doi: 10.3389/fimmu.2021.620698
Received: 23 October 2020; Accepted: 25 January 2021;
Published: 15 February 2021.
Edited by:
Giuseppe Locatelli, University of Bern, SwitzerlandReviewed by:
Fiona Crawford, The Roskamp Institute, United StatesAdam Bachstetter, University of Kentucky, United States
Susanna Rosi, University of California, San Francisco, United States
Copyright © 2021 Verboon, Patel and Greenhalgh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew D. Greenhalgh, YW5kcmV3LmdyZWVuaGFsZ2hAbWFuY2hlc3Rlci5hYy51aw==