 Morgane Bourhis
Morgane Bourhis Juliette Palle1,2
Juliette Palle1,2 Magali Terme
Magali Terme
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 29 March 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.616837
This article is part of the Research TopicThe Impact of Angiogenic Growth Factors and Hypoxia on The Antitumor Immune ResponseView all 8 articles
Vascular endothelial growth factor A is known to play a central role in tumor angiogenesis. Several studies showed that VEGF-A is also an immunosuppressive factor. In tumor-bearing hosts, VEGF-A can modulate immune cells (DC, MDSC, TAM) to induce the accumulation of regulatory T-cells while simultaneously inhibiting T-cell functions. Furthermore, VEGFR-2 expression on activated T-cells and FoxP3high regulatory T-cells also allow a direct effect of VEGF-A. Anti-angiogenic agents targeting VEGF-A/VEGFR contribute to limit tumor-induced immunosuppression. Based on interesting preclinical studies, many clinical trials have been conducted to investigate the efficacy of anti-VEGF-A/VEGFR treatments combined with immune checkpoint blockade leading to the approvement of these associations in different tumor locations. In this review, we focus on the impact of VEGF-A on immune cells especially regulatory and effector T-cells and different therapeutic strategies to restore an antitumor immunity.
Vascular endothelial growth factor A (VEGF-A) is considered to be a critical regulator of angiogenesis, which is the formation of new blood vessels from pre-existing ones, both in physiological and pathological states such as tumor angiogenesis (1). VEGF-A production is regulated by transcriptional factors such as HIF-1 (hypoxia-inducible factor 1) during a hypoxic stress or by oncogenes (2). Its pro-angiogenic activities are mediated by the activation of VEGF receptors (VEGFR-1, VEGFR-2) which can be expressed on endothelial cells, tumor cells and some immune cells (1). However, VEGF-A has a dual function in supporting tumor progression: first, by inducing vessel formation and second, by acting as an immunosuppressive factor (3–5). The immune system has emerged as a pivotal actor in controlling tumor growth. Cytolytic CD8+ T lymphocytes, which have been previously activated by mature dendritic cells presenting tumor antigen-derived peptides, can lyse tumor cells. However, different escape mechanisms are developed by the tumor to evade the immune system such as the development of regulatory T cells (Tregs) or the induction of T cell exhaustion (6). Different works have highlighted a direct or an indirect impact of VEGF-A on this T cell-based immunosuppression. This review will summarize these studies and focus on the immunomodulation induced by anti-angiogenic agents.
Tregs play a critical role in immune homeostasis by regulating effector T cell functions. The proportion of Tregs is enhanced in tumor-bearing mice and in cancer patients and is often associated with a poorer overall survival (7). A correlation between VEGF-A in malignant effusions and the accumulation of Tregs has been observed in cancer patients suggesting a potential role of VEGF-A on Tregs (8). A meta-analysis also revealed that VEGF-A expression is positively associated with intratumoral Tregs in hepatocellular carcinoma (HCC) (9). Thus, VEGF-A has been associated with the induction and maintenance of regulatory T-cells in tumor microenvironment in a direct or indirect-dependent manner.
In cancer, Tregs accumulation could occur through different mechanisms, such as expansion of pre-existing Tregs or conversion of conventional CD4+T cells into Tregs. Immature dendritic cells (DC) can induce Tregs proliferation in a TGF-β-dependent manner in tumor-bearing rodents (10). An initial study showed that tumor cell lines-derived VEGF-A affects the development of hematopoietic progenitor cells (HPCs) at an early state resulting in impaired DC differentiation and maturation (11). Inhibition of DC differentiation is mediated by VEGFR-2 (12). In mouse models, VEGF-A binding to VEGFR-1 on HPCs blocked the activation of nuclear factor κB (NF-κB) thereby blocking DC maturation (13, 14). In cancer patients, an increased VEGF-A plasma level is correlated with the presence of immature DC in the peripheral blood (15). This team also reported that the decrease of mature DC is associated with an increase of myeloid-derived suppressor cell (MDSC) in the peripheral blood of cancer patients (16). MDSC, especially Gr-1+CD11b+CD115+ (monocytic) MDSC, can also generate tumor specific Tregs in tumor-bearing mice and in cancer patients by secreting IL-10 and TGF-β (17, 18) or by arginase activity (19). VEGF-A has also been involved in MDSC increase in a VEGFR-2-dependent manner in mice (13, 20) and in ovarian cancer patients (21). Activation of JAK2 -STAT3 pathway by VEGF-A facilitates circulating MDSC accumulation (22). Accumulation of VEGFR-2+ MDSC in tumors contributes to poor prognosis (21). Thus, VEGF-A can act on both DC maturation and MDSC in tumor-bearing hosts. These myeloid cells producing immunosuppressive factors such as TGF-β or IL-10 could be involved in Tregs accumulation. Furthermore, a correlation has been observed between MDSC decrease and Tregs reduction in metastatic renal cell cancer patients during sunitinib treatment (a tyrosine kinase inhibitor targeting VEGFR) suggesting a link between MDSC and Tregs (23)
Different studies have recently highlighted a population of Tregs expressing VEGFR-2 in tumor-bearing mice and cancer patients (24–26). In a mouse model of colorectal cancer, we observed that a subset of activated/memory Tregs express VEGFR-2 (unlike healthy controls) and that VEGF-A induces Tregs proliferation in a VEGFR-2 dependent manner (25). In humans, Suzuki et al. showed that VEGFR-2 is selectively expressed by human FoxP3high Tregs but not on FoxP3low Tregs and may have stronger suppressive function (24). Effector CD45RA-FoxP3+CD4+ Tregs subset infiltrating the tumors has also been reported to express VEGFR-2 in advanced gastric cancer patients. Furthermore, the ability of VEGF-A to increase Tregs proliferation has been confirmed in this setting (26). VEGFR-2+ Tregs in tumor tissues is also associated with clinical outcome since intratumoral FoxP3+ VEGFR-2+ Tregs, unlike intratumoral FoxP3+ VEGFR2- Tregs are significantly correlated with poor overall survival and disease-free survival. It is an independent factor of recurrence and poor survival in colorectal cancer patients suggesting that VEGFR-2+ Tregs may be a prognostic biomarker in colorectal cancer (27). In some tumor locations, the prognostic role of tumor-infiltrating Tregs is still controversed. Taking VEGFR-2+ Tregs into account and not all Tregs could be more accurate to evaluate patient prognosis. Furthermore, specifically targeting VEGFR-2+ Tregs and not all Tregs could be of interest in cancer patients since it could help to restore an efficient anti-tumor response while limiting autoimmune adverse events.
Disruption of effector T cell infiltration or activation are important mechanisms of tumor-induced immunosuppression. VEGF-A has also been reported to take part in these mechanisms.
As we described above, VEGF-A can block DC maturation and increase MDSC accumulation. Therefore, immature DC are not able to efficiently activate T-cells (11). MDSC are also highly efficient at suppressing effector T cells by different mechanisms: L-arginase depletion (28), NO or ROS production (29, 30) and CD40-CD40L ligation (31). Likewise, tumor-associated macrophages (TAM) express PD-L1 which upon binding with PD-1 inhibits TCR signaling leading to an inactivation of T-cells (32). VEGF-A contributes to TAM recruitment; mainly into poorly vascularized tumor areas, exercising a chemoattractant effect via VEGFR-1 expression on macrophages surface. Nevertheless, VEGF-A alone is not sufficient to their activation which requires other tumor-produced factors such as IL-4 and IL-10 (33, 34). The up-regulation of these pro-inflammatory cytokines seems to be favored by VEGF-A over-expression.
Although tumor angiogenesis driven by pro-angiogenic factors intends to contribute to blood supply to the tumor, the induced vascular network is abnormal. It is characterized by chaotic, immature, disorganized, poorly perfused and permeable blood vessels which are partially mediated by an abnormal level of tumor-secreted VEGF-A and other factors such as TGF-β, PDGF (platelet-derived growth factor) and angiopoietin 2 (35, 36). In many human and mouse solid tumors, the aberrant structure and function of the tumor vasculature generates a barrier to the CD8+ T-cell infiltration and contribute to the maintenance of an immunosuppressive tumor microenvironment (37). Deletion of Rgs5-gene (regulator of G-protein signaling 5), which is responsible for the aberrant morphology of blood vessels, induced a vascular normalization and CD8+ T-cell infiltration in tumor-bearing mice (38). Several in vitro studies have demonstrated that the decrease of T-cell adhesion resulting in a restricted migration is associated with the decrease of intercellular adhesion molecules 1 (ICAM-1) and vascular cell adhesion molecules 1 (VCAM-1) on endothelial cells (39–41). The cooperation of VEGF-A with IL-10 and prostaglandin E2 is also able to induce FasL expression on tumor endothelial cells. In ovarian, colon, bladder, prostate, and renal cancers, FasL+ endothelial cells acquire the ability to kill T-cells while allowing FoxP3+ Tregs accumulation and infiltration (42). Finally, the down-regulation of adhesion molecules and expression of FasL on tumor endothelial cells mainly induced by VEGF-A are responsible for a decrease of tumor infiltration by T-cells.
In tumor producing elevated levels of VEGF-A, studies revealed that this factor and its receptors have important roles in the aberrant hematopoiesis resulting in defects in immunity (20). Mice exposed to recombinant VEGF at similar concentrations to those observed in patients with advanced cancer develop a thymic atrophy with a reduced number of CD4/CD8 thymocytes (43). These results demonstrate that VEGF-A directly interferes with the thymic development of T-cells from HPCs and can contribute to the immune deficiencies associated with tumors. Studies revealed that VEGF-A directly impacts effector T cells. Indeed, in vitro activated T-cells but also tumor-infiltrating T cells express VEGFR-2 (44). In advanced ovarian cancer, VEGF-A directly suppresses T cell proliferation and cytotoxic activity via VEGFR-2 (45, 46). Although Basu et al.’ study reported an enhanced IFN-γ and IL-2 production and migratory responses induced by VEGF-A in human CD45RO+ CD4+ memory T-cells (47), there are growing evidences to support the immunosuppressive role of VEGF-A/VEGFR in T-cells (5) especially on tumor-induced T-cell exhaustion (44, 48). T-cell exhaustion is phenotypically characterized by the co-expression of immune inhibitory receptors called immune checkpoints such as program cell death-1 (PD-1), T-cell immunoglobulin mucin-3 (Tim-3), cytotoxic T-lymphocyte-associated protein (CTLA-4), lymphocyte activation gene 3 (Lag3) in CD8+ T-cells and by a gradual loss of function (49). VEGF-A increases PD-1 expression and other immune checkpoints CTLA-4, Tim-3 and Lag-3 on CD8+ T-cells but also their co-expression which is related to exhaustion. The VEGFR-2- PLCγ- calcineurin- NFAT pathway is involved in this effect (44). These results have been confirmed by others (48, 50). Recently, a study carried out on patients with microsatellite stable colorectal cancer (MSS CRC) resistant to anti-PD-1 therapies has identified that VEGF-A-dependent upregulation of immune checkpoints involved the TOX transcription factor (50).
In conclusion, VEGF-A acts as an immunosuppressive factor in modulating immune cells. Its effects are summarized in Figure 1.
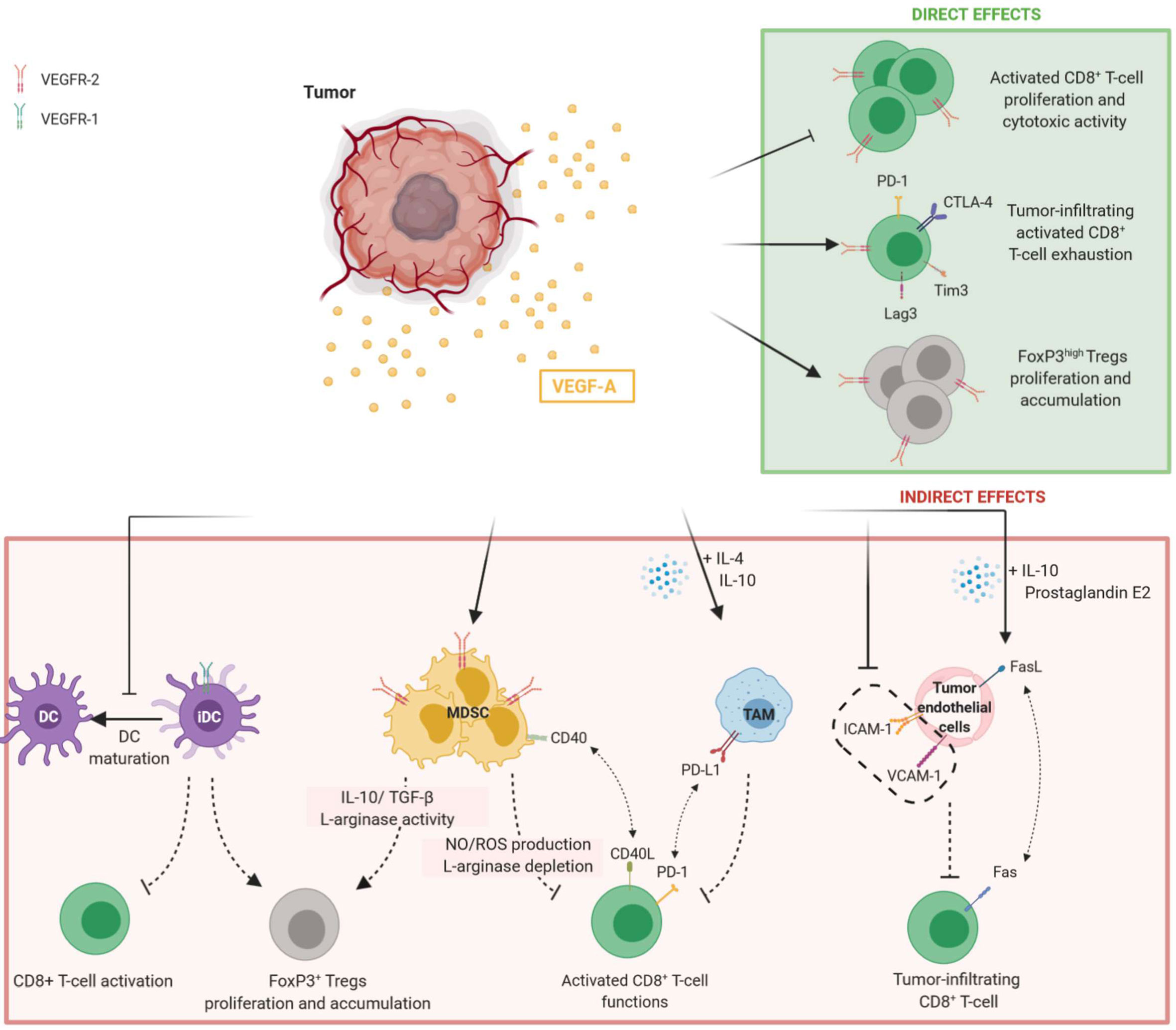
Figure 1 Direct and indirect effects of VEGF-A in promoting immunosuppressive tumor microenvironment. Tumor-secreted VEGF-A induces the inhibition of T-cell functions and proliferation while promoting Tregs accumulation. Both direct and indirect effects of VEGF-A are observed. Direct modulation of T-cell by VEGF-A is mediated by the binding to VEGFR-2 on their surface, whereas the indirect effects of VEGF-A on T-cell results from the modulation of immune cells such as DC, MDSC and TAM expressing VEGFR-1 or VEGFR-2. On tumor endothelial cells, VEGF-A can reduce adhesion molecules expression and induce FasL, preventing tumor-infiltrating CD8+ T-cell. DC, dendritic cells; iDC, immature dendritic cells; MDSC, myeloid derived suppressor cells; TAM, tumor-associated macrophages.
Based on these properties, the immunomodulatory role of anti-angiogenic (AA) agents targeting VEGF-A/VEGFR in antitumor immunity has been investigated in tumor models and cancer patients (51). These effects are summarized in Table 1.
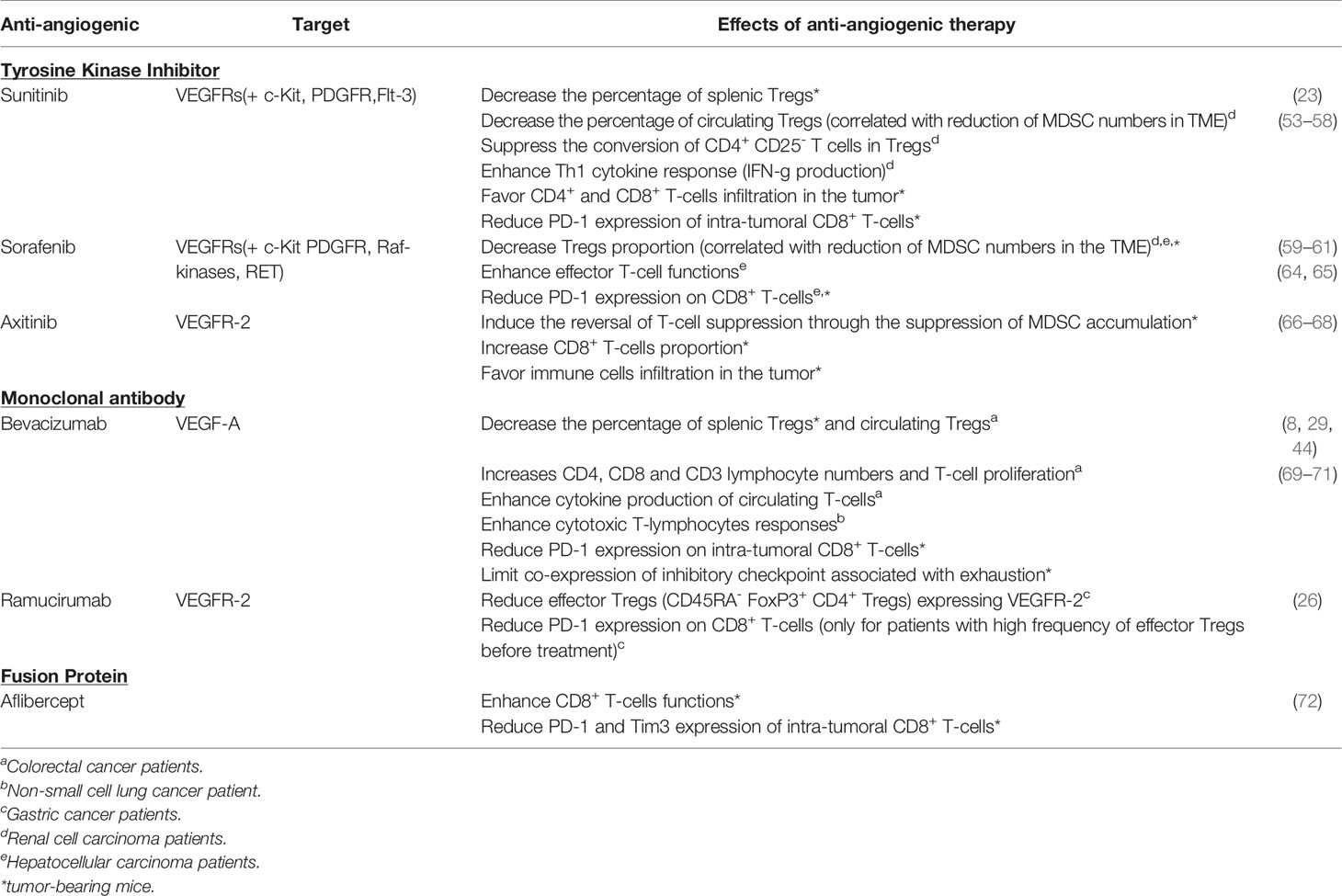
Table 1 Modulation of T-cells by anti-angiogenic therapy.
During the last decade, different AA molecules have been developed and approved to treat cancer patients. They can be classified in three main classes: (i) small tyrosine kinase inhibitors (TKI) such as sunitinib, sorafenib and axitinib (ii) monoclonal antibodies (mAb) such as bevacizumab (anti-VEGF-A) and ramucirumab (anti-VEGFR-2) (iii) aflibercept which is a fusion protein composed of extracellular domains from VEGFR-1 and VEGFR-2 (52). Whereas TKI target VEGFR pathway (but also other receptors), monoclonal antibodies and fusion proteins directly target circulating pro-angiogenic factors or their receptors present on the cell membrane.
Sunitinib. Sunitinib is a TKI currently used to treat different types of cancers, in particular metastatic renal cell carcinoma (mRCC) (52). After sunitinib treatment, the percentage of splenic FoxP3+ Tregs in mouse models of renal cancer and circulating Tregs in mRCC patients is decreased (23, 53–55). Sunitinib also reduces MDSC numbers which is correlated with Treg decrease in the tumor microenvironment (23, 56) and favors CD4+ and CD8+ infiltration in the tumor site while reducing PD-1 expression on CD8+ T-cells (44, 53). Moreover, sunitinib suppresses the conversion of CD4+CD25- naïve T cells in Tregs in mouse tumor models (57). In humans, in vitro studies reported a significant improvement of Th1 cytokine response in mRCC patients receiving sunitinib. This effect seems to be linked to a reduction of Tregs (23, 54). In addition, in RCC tumor cells and tumor-associated MDSC, sunitinib inhibits Stat3 activity leading to tumor cell apoptosis and promoting antitumor effect (56).
Sorafenib. As well as sunitinib, sorafenib can reduce Tregs and MDSC proportion in mouse models of liver cancer (58) and in HCC patients (59) or RCC (60). However, sorafenib seems to modulate T-cell functions differently from sunitinib and had no impact on Th1 response (60, 70). An in vitro study examining the effects of sorafenib on the proliferation and activation of human peripheral blood T-cells showed that sorafenib targets LCK phosphorylation implicated in the TCR signaling causing the loss of T-cell immune responses (71). Controversially, studies have revealed that this treatment seems to up-regulate the tumor-specific effector T cell functions while PD-1 expression on CD8+ T-cells is down-regulated (59, 61, 62). Currently, the effects of sorafenib on T-cell functions remain unclear.
Axitinib. Axitinib is a highly selective VEGFR tyrosine kinase inhibitor and has demonstrated its efficacy in the treatment of advanced RCC (aRCC) (63). In tumor bearing mice, axitinib suppressed MDSC accumulation through the inhibition of Stat3 activity and it was correlated with the reversal of T-cell suppression (64, 65). Indeed, the proportion of CD8+ T-cells is increased in a mouse model of renal cancer (64) and an in vivo study in a mouse melanoma model showed an increase of tumor-infiltrating immune cells (65).
Bevacizumab. Bevacizumab, a humanized anti-VEGF-A monoclonal antibody which directly targets VEGF-A, reduced the proportion of Tregs in tumor-bearing mice and in patients with metastatic colorectal cancer (mCRC) (25, 66). This phenomenon is linked to a decrease of Ki67+ expression in Tregs (25). In mCRC patients treated with bevacizumab, Manzoni et al. revealed an increase of CD4, CD8 and CD3 lymphocyte numbers (29), whereas Tsavaris et al. observed a better proliferation and cytokine production of circulating T-cells compared to patients treated with chemotherapy only (67). Moreover, the administration of anti-VEGF-A in tumor-bearing mice decreased PD-1 expression on intratumoral CD8+ T-cells and limited the co-expression of inhibitory checkpoints associated with exhaustion (44). Recently, a study conducted on non-small-cell lung cancer (NSCLC) patients revealed that bevacizumab addition to the chemotherapy doublet based on cisplatin and oral etoposide decreased the plasmatic VEGF-A level and improved cytotoxic T-lymphocytes responses (68) while simultaneously restoring DC functions.
Ramucirumab. Ramucirumab is a monoclonal antibody targeting VEGFR-2. In patients with advanced gastric cancers, CD45RA- FoxP3+ CD4+ effector Tregs cells expressing VEGFR-2 are present in higher frequency in TIL than in PBMC (26). In vitro experiments showed that VEGF-A stimulates their proliferation which can be overcome by ramucirumab. Ramucirumab-containing therapies strongly reduce effector Tregs in tumors of advanced gastric cancer patients. A higher frequency of these effector Tregs in tumors before treatment was associated with an enhanced proportion of partial response and a longer progression-free survival. Since VEGFR-2 is highly expressed by this Tregs subset, it could be envisioned that the proportion of VEGFR-2+ effector Tregs in TIL could be a biomarker of favorable clinical response to ramucirumab therapies (26). A decreased expression of PD-1 on CD8+ T-cells was also reported after ramucirumab treatment (26).
Aflibercept. Aflibercept is a VEGFR fusion protein conjugated to Fc portion of human IgG1, also known as VEGF-Trap. Nowadays the impact of VEGF-Trap treatments on T cells remains poorly described in cancer patients but a recent study highlights the improvement of CD8+ T cell functionality in a mouse model of glioma. Indeed, they observed a decrease of PD-1 and Tim3 expression on infiltrating CD8+ T-cells. Likewise, they found an improvement of DC maturation demonstrated by the increase of co-stimulation molecules expression including CD80, CD86 and MHC II which are required for T-cell activation (69).
An indirect impact of AA treatment on tumor-induced immunosuppression could also be proposed. AA treatments can be responsible of a transient normalization of the vasculature favoring tumor infiltration by immune cells. However, some reports have also indicated a potential enhancement of hypoxia especially during a prolonged AA treatment (72). Hypoxia fuels tumor progression by selecting more malignant cells and also by inducing an immunosuppressive microenvironment. It can lead myeloid cells toward an immunosuppressive phenotype or potentiate regulatory T cell functions (73). Nevertheless, recent studies highlighted that aggravated hypoxia mediated by anti-VEGF-A treatment directly enhances CD8+ T-cell functions in an HIF-1α dependent manner (74, 75). This aspect of anti-angiogenic impact needs further investigation.
To enhance anti-tumor effects, combining AA therapies with immunotherapies such as immune checkpoint blockade (ICB: anti-PD-1, anti-PD-L1, anti-CTLA-4) have raised great interest. In mouse tumor models, VEGF-A/VEGFR-2 and PD-1 blockade induces strong and synergic antitumor responses and limits T-cell exhaustion in VEGF-A-expressing tumor compared to monotherapies in mouse models of MSS colorectal cancer (44, 50). Two studies have been carried out in murine lung cancer models using anti-VEGF/VEGFR combined to anti-PD-L1. They have demonstrated strong anti-tumor effects which are associated with an increase of TIL and T-cell responses (48, 76). The association of anti-VEGFR-2 plus anti-PD-L1 could rescue the PD-1/Tim3 exhaustion T-cell phenotype, while improving overall survival (48). In preclinical murine models, the association of axitinib with ICB (anti-PD-L1 and anti-Tim3 antibodies) resulted in a synergistic therapeutic efficacy (77).
Based on the interesting results from preclinical studies, many clinical trials have been conducted to evaluate combination therapies in cancer patients (78). In 2014, a phase I clinical trial (NCT00790010) investigated the combination of ipilimumab, an anti-CTLA-4 monoclonal antibody, and bevacizumab in 46 patients with a metastatic melanoma (79). The authors observed an upregulation of VCAM-1 and other adhesion molecules on intratumoral endothelial cells leading to endothelial cell activation. Furthermore, the trafficking of CD8+ T-cells across tumor vasculature was enhanced. When combined to ipilimumab, bevacizumab seems to influence tumor vasculature morphology and immune responses (79). Although antitumor response efficacy has been demonstrated, important immune-related adverse events are induced. Anti-CTLA-4 antibodies are known to generate autoimmune diseases (80). Meta-analysis revealed that about 70% of patients treated with ipilimumab including 25% of high-grade adverse events develop skin rashes, gastrointestinal dysfunctions such as colitis or less frequently endocrine dysfunctions (81, 82). When ipilimumab was combined to bevacizumab, 29.3% of patients experienced grade 3-4 adverse events. Inflammatory events were higher than expected with ipilimumab alone, but remained manageable. Interestingly, the incidence of colitis was not increased (79). High grade adverse events are less common for anti-PD-1/PD-L1 antibodies alone compared to ipilimumab. A study conducted in a small cohort of mRCC patients explored the effects of an anti-PD-L1 (atezolizumab) plus bevacizumab (NCT01633970) (83). Similar findings were highlighted including an improved migration of antigen-specific T-cells and an increase of cytokines and chemokines production in particular CX3CL1 involved in T-cell trafficking (83). Therapies combining AA agents plus ICB have shown their efficiency in phase III clinical trials (84) and have been recently approved in different locations (i) atezolizumab (anti-PD-L1) and bevacizumab with chemotherapy in NSCLC (85) (ii) atezolizumab and bevacizumab in unresectable HCC (86) (iii) pembrolizumab (anti-PD-1) plus lenvatinib (TKI) in advanced endometrial cancer (87) (iv) pembrolizumab (anti-PD-1) plus axitinib in RCC (88) and v) axitinib plus avelumab (an anti-PD-L1) in RCC (89). No major increases of adverse events were reported with these combinations compared to each agent alone. Results of these clinical trials are summarized in Table 2.
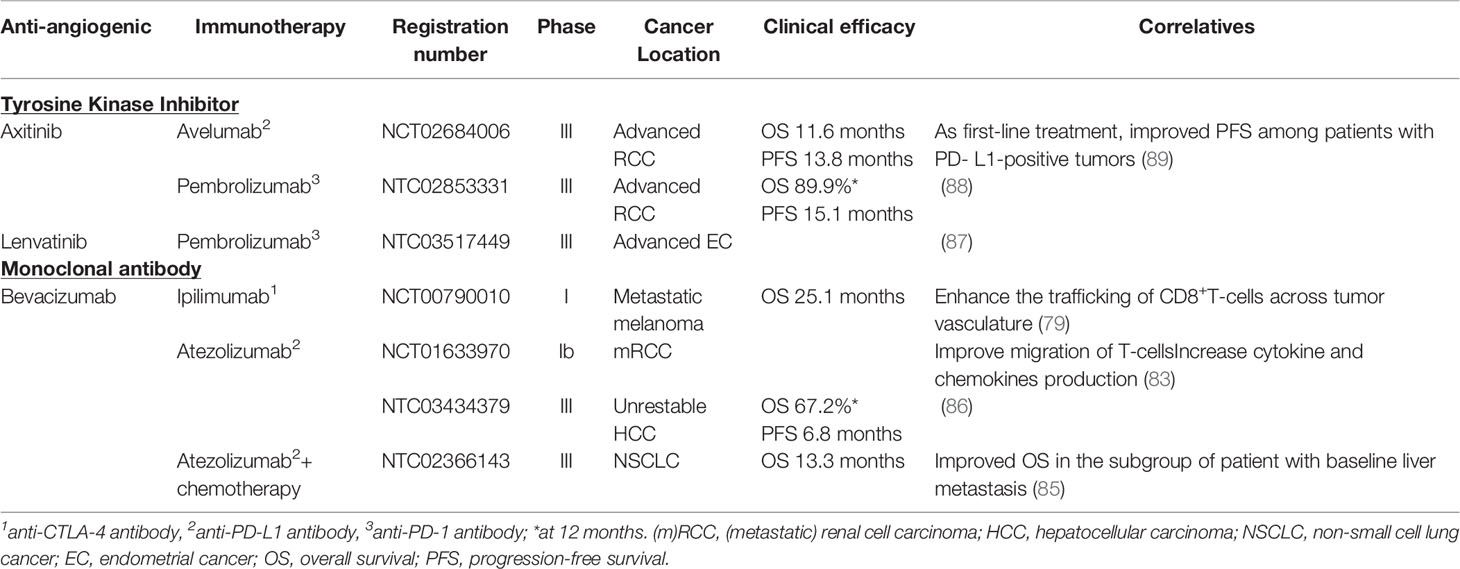
Table 2 Current clinical trials of anti-angiogenic therapies combined with immunotherapies.
The need to find approaches enabling the induction and activation of immune response against cancer remains considerable and in this context, novel approaches are important to consider. A better understanding of the immunomodulatory roles of pro-angiogenic factors produced in tumor-bearing hosts could help to develop new therapeutic strategies or combinations. One of the main issues encountered with AA agents in the development of resistance mechanisms including activation of alternative pro-angiogenic pathways such as Angiopoietin 2 (Ang2)/Tie2, HGF/c-Met or PlGF (90–92). High levels of Ang2 are associated with unfavorable responses to bevacizumab-containing therapies in patients suffering from colorectal cancers and metastatic melanoma treated with ICB (93). Different AA molecules are currently used to treat cancer patients, but the impact of these different treatments on anti-tumor immunity remains unclear. In some tumor locations, combining AA molecules with ICB allowed to improve the outcome of cancer patients, leading to the approval of different anti-angiogenic/ICB combinations by the FDA. However, many questions remain unanswered such as which tumors are sensitive to these associations, or if targeting alternative pro-angiogenic pathways in combination with ICB could also provide interesting anti-tumor effects. Recently, a study has shown in mouse tumor models that the concomitant blocking of VEGF-A and Ang2 with a bi-specific antibody stimulated antitumor immune responses compared with single-agent therapies and could increase the effectiveness of ICB such as anti-PD-1 antibody (94). Currently, multiple clinical trials are being conducted to assess dual inhibition of VEGF-A and Ang2 in patients with cancer and to further investigate mechanisms involved in alternative pro-angiogenic pathways (95).
MB and MT designed and wrote the main manuscript. JP and IG-F critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
JP received financial support from ITMO Cancer AVIESAN (Alliance Nationale pour les Sciences de la Vie et de la Santé, National Alliance for Life Sciences & Health) within the framework of the Cancer Plan. MB was supported by Association pour la Recherche contre le Cancer. This work has been supported by Cancéropôle Ile de France/INCa (2017-1-EMERG-74-INSERM-5-1), SIRIC CARPEM, Labex Immuno-oncology and Ligue contre le Cancer.
MT reports personal fees from Roche and Gilead and research grant from Sanofi outside the submitted work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ho QT, Kuo CJ. Vascular Endothelial Growth Factor: Biology and Therapeutic Applications. Int J Biochem Cell Biol (2007) 39:1349. doi: 10.1016/j.biocel.2007.04.010
2. Chouaib S, Messai Y, Couve S, Escudier B, Hasmim M, Noman MZ. Hypoxia Promotes Tumor Growth in Linking Angiogenesis to Immune Escape. Front Immunol (2012) 3:21. doi: 10.3389/fimmu.2012.00021
3. Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev (2008) 222:162–79. doi: 10.1111/j.1600-065X.2008.00602.x
4. Voron T, Marcheteau E, Pernot S, Colussi O, Tartour E, Taieb J, et al. Control of the Immune Response by Pro-Angiogenic Factors. Front Oncol (2014) 4:70. doi: 10.3389/fonc.2014.00070
5. Li Y-L, Zhao H, Ren X-B. Relationship of VEGF/VEGFR with immune and cancer cells: staggering or forward? Cancer Biol Med (2016) 13:206–14. doi: 10.20892/j.issn.2095-3941.2015.0070
6. Lapeyre-Prost A, Terme M, Pernot S, Pointet A-L, Voron T, Tartour E, et al. Chapter Seven - Immunomodulatory Activity of VEGF in Cancer. In: Galluzzi L, editor. International Review of Cell and Molecular Biology, vol. 330. United States: Academic Press (2017). p. 295–342. doi: 10.1016/bs.ircmb.2016.09.007
7. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression — implications for anticancer therapy. Nat Rev Clin Oncol (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
8. Wada J, Suzuki H, Fuchino R, Yamasaki A, Nagai S, Yanai K, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res (2009) 29:881–8.
9. Sun L, Xu G, Liao W, Yang H, Xu H, Du S, et al. Clinicopathologic and prognostic significance of regulatory T cells in patients with hepatocellular carcinoma: a meta-analysis. Oncotarget (2017) 8:39658–72. doi: 10.18632/oncotarget.17340
10. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-β–secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med (2005) 202:919–29. doi: 10.1084/jem.20050463
11. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med (1996) 2:1096–103. doi: 10.1038/nm1096-1096
12. Dikov MM, Ohm JE, Ray N, Tchekneva EE, Burlison J, Moghanaki D, et al. Differential Roles of Vascular Endothelial Growth Factor Receptors 1 and 2 in Dendritic Cell Differentiation. J Immunol (2005) 174:215–22. doi: 10.4049/jimmunol.174.1.215
13. Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood (1998) 92:4150–66. doi: 10.1182/blood.V92.11.4150.423k45_4150_4166
14. Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, et al. Vascular Endothelial Growth Factor Affects Dendritic Cell Maturation Through the Inhibition of Nuclear Factor-κB Activation in Hemopoietic Progenitor Cells. J Immunol (1998) 160:1224–32.
15. Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED, et al. Clinical Significance of Defective Dendritic Cell Differentiation in Cancer. Clin Cancer Res (2000) 6:1755–66.
16. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol (2001) 166:678–89. doi: 10.4049/jimmunol.166.1.678
17. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, et al. A New Population of Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients Induces CD4+CD25+Foxp3+ T Cells. Gastroenterology (2008) 135:234–43. doi: 10.1053/j.gastro.2008.03.020
18. Huang B, Pan P-Y, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299
19. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid derived suppressor cells promote cross-tolerance in B cell lymphoma by expanding regulatory T cells. Cancer Res (2008) 68:5439–49. doi: 10.1158/0008-5472.CAN-07-6621
20. Huang Y, Chen X, Dikov MM, Novitskiy SV, Mosse CA, Yang L, et al. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood (2007) 110:624–31. doi: 10.1182/blood-2007-01-065714
21. Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of Vascular Endothelial Growth Factor in Ovarian Cancer Inhibits Tumor Immunity through the Accumulation of Myeloid-Derived Suppressor Cells. Clin Cancer Res (2017) 23:587–99. doi: 10.1158/1078-0432.CCR-16-0387
22. Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol (2004) 172:464–74. doi: 10.4049/jimmunol.172.1.464
23. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib Mediates Reversal of Myeloid-Derived Suppressor Cell Accumulation in Renal Cell Carcinoma Patients. Clin Cancer Res (2009) 15:2148–57. doi: 10.1158/1078-0432.CCR-08-1332
24. Suzuki H, Onishi H, Wada J, Yamasaki A, Tanaka H, Nakano K, et al. VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. Eur J Immunol (2010) 40:197–203. doi: 10.1002/eji.200939887
25. Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res (2013) 73:539–49. doi: 10.1158/0008-5472.CAN-12-2325
26. Tada Y, Togashi Y, Kotani D, Kuwata T, Sato E, Kawazoe A, et al. Targeting VEGFR2 with Ramucirumab strongly impacts effector/activated regulatory T cells and CD8+ T cells in the tumor microenvironment. J Immunother Cancer (2018) 6:106. doi: 10.1186/s40425-018-0403-1
27. Suzuki H, Onishi H, Morisaki T, Tanaka M, Katano M. Intratumoral FOXP3+VEGFR2+ regulatory T cells are predictive markers for recurrence and survival in patients with colorectal cancer. Clin Immunol (2013) 146:26–33. doi: 10.1016/j.clim.2012.10.007
28. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol (2005) 5:641–54. doi: 10.1038/nri1668
29. Manzoni M, Rovati B, Ronzoni M, Loupakis F, Mariucci S, Ricci V, et al. Immunological Effects of Bevacizumab-Based Treatment in Metastatic Colorectal Cancer. Oncology (2010) 79:187–96. doi: 10.1159/000320609
30. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13:828–35. doi: 10.1038/nm1609
31. Pan P-Y, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune Stimulatory Receptor CD40 Is Required for T-Cell Suppression and T Regulatory Cell Activation Mediated by Myeloid-Derived Suppressor Cells in Cancer. Cancer Res (2010) 70:99. doi: 10.1158/0008-5472.CAN-09-1882
32. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
33. Lewis JS, Landers RJ, Underwood JC, Harris AL, Lewis CE. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J Pathol (2000) 192:150–8. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH687>3.0.CO;2-G
34. Linde N, Lederle W, Depner S, van Rooijen N, Gutschalk CM, Mueller MM. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J Pathol (2012) 227:17–28. doi: 10.1002/path.3989
35. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature (2011) 473:298–307. doi: 10.1038/nature10144
36. Baluk P, Hashizume H, McDonald DM. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev (2005) 15:102–11. doi: 10.1016/j.gde.2004.12.005
37. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis (2018) 9(2):115. doi: 10.1038/s41419-017-0061-0
38. Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature (2008) 453:410–4. doi: 10.1038/nature06868
39. Dirkx AEM, Egbrink MGA, Kuijpers MJE, Van den Niet ST, Heijnen VVT, Steege JCAB, et al. Tumor Angiogenesis Modulates Leukocyte-Vessel Wall Interactions in Vivo by Reducing Endothelial Adhesion Molecule Expression. Cancer Res (2003) 63:2322–9.
40. Bouzin C, Brouet A, De Vriese J, Dewever J, Feron O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J Immunol (2007) 178:1505–11. doi: 10.4049/jimmunol.178.3.1505
41. Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J (2015) 29:227–38. doi: 10.1096/fj.14-250985
42. Motz GT, Santoro SP, Wang L-P, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor Endothelium FasL Establishes a Selective Immune Barrier Promoting Tolerance in Tumors. Nat Med (2014) 20:607–15. doi: 10.1038/nm.3541
43. Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood (2003) 101:4878–86. doi: 10.1182/blood-2002-07-1956
44. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet A-L, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med (2015) 212:139–48. doi: 10.1084/jem.20140559
45. Gavalas NG, Tsiatas M, Tsitsilonis O, Politi E, Ioannou K, Ziogas AC, et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. Br J Cancer (2012) 107:1869–75. doi: 10.1038/bjc.2012.468
46. Ziogas AC, Gavalas NG, Tsiatas M, Tsitsilonis O, Politi E, Terpos E, et al. VEGF directly suppresses activation of T cells from ovarian cancer patients and healthy individuals via VEGF receptor Type 2. Int J Cancer (2012) 130:857–64. doi: 10.1002/ijc.26094
47. Basu A, Hoerning A, Datta D, Edelbauer M, Stack MP, Calzadilla K, et al. Cutting Edge: Vascular Endothelial Growth Factor-Mediated Signaling in Human CD45RO+ CD4+ T Cells Promotes Akt and ERK Activation and Costimulates IFN-γ Production. J Immunol (2010) 184:545–9. doi: 10.4049/jimmunol.0900397
48. Meder L, Schuldt P, Thelen M, Schmitt A, Dietlein F, Klein S, et al. Combined VEGF and PD-L1 Blockade Displays Synergistic Treatment Effects in an Autochthonous Mouse Model of Small Cell Lung Cancer. Cancer Res (2018) 78:4270–81. doi: 10.1158/0008-5472.CAN-17-2176
50. Kim CG, Jang M, Kim Y, Leem G, Kim KH, Lee H, et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci Immunol (2019) 4(41):eaay0555. doi: 10.1126/sciimmunol.aay0555
51. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front Immunol (2018) 9:978. doi: 10.3389/fimmu.2018.00978
52. Qin S, Li A, Yi M, Yu S, Zhang M, Wu K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J Hematol Oncol (2019) 12(1):27. doi: 10.1186/s13045-019-0718-5
53. Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The Novel Role of Tyrosine Kinase Inhibitor in the Reversal of Immune Suppression and Modulation of Tumor Microenvironment for Immune-Based Cancer Therapies. Cancer Res (2009) 69:2514–22. doi: 10.1158/0008-5472.CAN-08-4709
54. Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, et al. Sunitinib Reverses Type-1 Immune Suppression and Decreases T-Regulatory Cells in Renal Cell Carcinoma Patients. Clin Cancer Res (2008) 14:6674–82. doi: 10.1158/1078-0432.CCR-07-5212
55. Adotevi O, Pere H, Ravel P, Haicheur N, Badoual C, Merillon N, et al. A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. J Immunother (2010) 33:991–8. doi: 10.1097/CJI.0b013e3181f4c208
56. Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res (2009) 69:2506–13. doi: 10.1158/0008-5472.CAN-08-4323
57. Kujawski M, Zhang C, Herrmann A, Reckamp K, Scuto A, Jensen M, et al. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res (2010) 70:9599–610. doi: 10.1158/0008-5472.CAN-10-1293
58. Cao M, Xu Y, Youn J, Cabrera R, Zhang X, Gabrilovich D, et al. Kinase Inhibitor Sorafenib Modulates Immunosuppressive Cell Populations in a Murine Liver Cancer Model. Lab Invest (2011) 91:598–608. doi: 10.1038/labinvest.2010.205
59. Kalathil SG, Lugade AA, Miller A, Iyer R, Thanavala Y. PD-1+ and Foxp3+ T cell reduction correlates with survival of HCC patients after sorafenib therapy. JCI Insight 1(11):e86182. doi: 10.1172/jci.insight.86182
60. Busse A, Asemissen AM, Nonnenmacher A, Braun F, Ochsenreither S, Stather D, et al. Immunomodulatory effects of sorafenib on peripheral immune effector cells in metastatic renal cell carcinoma. Eur J Cancer (2011) 47:690–6. doi: 10.1016/j.ejca.2010.11.021
61. Chen M-L, Yan B-S, Lu W-C, Chen M-H, Yu S-L, Yang P-C, et al. Sorafenib relieves cell-intrinsic and cell-extrinsic inhibitions of effector T cells in tumor microenvironment to augment antitumor immunity: Sorafenib Treatment Enhances Antitumor Immunity in Tumor Milieu. Int J Cancer (2014) 134:319–31. doi: 10.1002/ijc.28362
62. Kalathil SG, Hutson A, Barbi J, Iyer R, Thanavala Y. Augmentation of IFN-γ+ CD8+ T cell responses correlates with survival of HCC patients on sorafenib therapy. JCI Insight 4(15):e130116. doi: 10.1172/jci.insight.130116
63. Gross-Goupil M, Françlois L, Quivy A, Ravaud A. Axitinib: A Review of its Safety and Efficacy in the Treatment of Adults with Advanced Renal Cell Carcinoma. Clin Med Insights Oncol (2013) 7:269–77. doi: 10.4137/CMO.S10594
64. Yuan H, Cai P, Li Q, Wang W, Sun Y, Xu Q, et al. Axitinib augments antitumor activity in renal cell carcinoma via STAT3-dependent reversal of myeloid-derived suppressor cell accumulation. BioMed Pharmacother (2014) 68:751–6. doi: 10.1016/j.biopha.2014.07.002
65. Du Four S, Maenhout SK, De Pierre K, Renmans D, Niclou SP, Thielemans K, et al. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology (2015) 4(4):e998107. doi: 10.1080/2162402X.2014.998107
66. Terme M, Tartour E, Taieb J. VEGFA/VEGFR2-targeted therapies prevent the VEGFA-induced proliferation of regulatory T cells in cancer. Oncoimmunology (2013) 2:e25156. doi: 10.4161/onci.25156
67. Tsavaris N, Voutsas IF, Kosmas C, Gritzapis AD, Baxevanis CN. Combined treatment with Bevacizumab and standard chemotherapy restores abnormal immune parameters in advanced colorectal cancer patients. Invest New Drugs (2012) 30:395–402. doi: 10.1007/s10637-010-9533-0
68. Martino E, Misso G, Pastina P, Costantini S, Vanni F, Gandolfo C, et al. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discovery (2016) 2:16025. doi: 10.1038/cddiscovery.2016.25
69. Malo CS, Khadka RH, Ayasoufi K, Jin F, AbouChehade JE, Hansen MJ, et al. Immunomodulation Mediated by Anti-angiogenic Therapy Improves CD8 T Cell Immunity Against Experimental Glioma. Front Oncol (2018) 8:320. doi: 10.3389/fonc.2018.00320
70. Nagai H, Mukozu T, Matsui D, Kanekawa T, Kanayama M, Wakui N, et al. Sorafenib Prevents Escape from Host Immunity in Liver Cirrhosis Patients with Advanced Hepatocellular Carcinoma. Clin Dev Immunol (2012) 2012:607851. doi: 10.1155/2012/607851
71. Zhao W, Gu YH, Song R, Qu BQ, Xu Q. Sorafenib inhibits activation of human peripheral blood T cells by targeting LCK phosphorylation. Leukemia (2008) 22:1226–33. doi: 10.1038/leu.2008.58
72. Ribatti D, Annese T, Ruggieri S, Tamma R, Crivellato E. Limitations of Anti-Angiogenic Treatment of Tumors. Trans Oncol (2019) 12:981–6. doi: 10.1016/j.tranon.2019.04.022
73. Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: a key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol Cell Physiol (2015) 309:C569–79. doi: 10.1152/ajpcell.00207.2015
74. de Almeida PE, Mak J, Hernandez G, Jesudason R, Herault A, Javinal V, et al. Anti-VEGF Treatment Enhances CD8+ T-cell Antitumor Activity by Amplifying Hypoxia. Cancer Immunol Res (2020) 8:806–18. doi: 10.1158/2326-6066.CIR-19-0360
75. Palazon A, Tyrakis PA, Macias D, Veliça P, Rundqvist H, Fitzpatrick S, et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell (2017) 32:669–83.e5. doi: 10.1016/j.ccell.2017.10.003
76. Zhao S, Jiang T, Li X, Zhou C. OA11.07 Combining Anti-Angiogenesis and Immunotherapy Enhances Antitumor Effect by Promoting Immune Response in Lung Cancer. J Thoracic Oncol (2017) 12:S288. doi: 10.1016/j.jtho.2016.11.293
77. Läubli H, Müller P, D’Amico L, Buchi M, Kashyap AS, Zippelius A. The multi-receptor inhibitor axitinib reverses tumor-induced immunosuppression and potentiates treatment with immune-modulatory antibodies in preclinical murine models. Cancer Immunol Immunother (2018) 67:815–24. doi: 10.1007/s00262-018-2136-x
78. Ciciola P, Cascetta P, Bianco C, Formisano L, Bianco R. Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents. J Clin Med (2020) 9(3):675. doi: 10.3390/jcm9030675
79. Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res (2014) 2:632–42. doi: 10.1158/2326-6066.CIR-14-0053
80. Teply BA, Lipson EJ. Identification and management of toxicities from immune checkpoint-blocking drugs. Oncol (Williston Park) (2014) 28 Suppl 3:30–8.
81. Bertrand A, Kostine M, Barnetche T, Truchetet M-E, Schaeverbeke T. Immune related adverse events associated with anti-CTLA-4 antibodies: systematic review and meta-analysis. BMC Med (2015) 13:211. doi: 10.1186/s12916-015-0455-8
82. Voskens CJ, Goldinger SM, Loquai C, Robert C, Kaehler KC, Berking C, et al. The Price of Tumor Control: An Analysis of Rare Side Effects of Anti-CTLA-4 Therapy in Metastatic Melanoma from the Ipilimumab Network. PloS One (2013) 8:e53745. doi: 10.1371/journal.pone.0053745
83. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun (2016) 7:12624. doi: 10.1038/ncomms12624
84. Yi M, Jiao D, Qin S, Chu Q, Wu K, Li A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer (2019) 18:60. doi: 10.1186/s12943-019-0974-6
85. Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med (2019) 7:387–401. doi: 10.1016/S2213-2600(19)30084-0
86. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. New Engl J Med (2020) 382:1894–905. doi: 10.1056/NEJMoa1915745
87. Makker V, Casado Herraez A, Aghajanian C, Fujiwara K, Pignata S, Penson RT, et al. A phase 3 trial evaluating efficacy and safety of lenvatinib in combination with pembrolizumab in patients with advanced endometrial cancer. JCO (2019) 37(15_supl):TPS5607–TPS5607. doi: 10.1200/JCO.2019.37.15_suppl.TPS5607
88. Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med (2019) 380:1116–27. doi: 10.1056/NEJMoa1816714
89. Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. New Engl J Med (2019) 380:1103–15. doi: 10.1056/NEJMoa1816047
90. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer (2008) 8:592. doi: 10.1038/nrc2442
91. Kopetz S, Hoff PM, Morris JS, Wolff RA, Eng C, Glover KY, et al. Phase II Trial of Infusional Fluorouracil, Irinotecan, and Bevacizumab for Metastatic Colorectal Cancer: Efficacy and Circulating Angiogenic Biomarkers Associated With Therapeutic Resistance. J Clin Oncol (2010) 28:453. doi: 10.1200/JCO.2009.24.8252
92. Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to Anti-Angiogenic Therapy in Cancer—Alterations to Anti-VEGF Pathway. Int J Mol Sci (2018) 19. doi: 10.3390/ijms19041232
93. Goede V, Coutelle O, Neuneier J, Reinacher-Schick A, Schnell R, Koslowsky TC, et al. Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br J Cancer (2010) 103:1407–14. doi: 10.1038/sj.bjc.6605925
94. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassará A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med (2017) 9:eaak9670. doi: 10.1126/scitranslmed.aak9670
Keywords: VEGF-A (vascular endothelial growth factor-A), effector T-cells, regulatory T (Treg) cells, tumor, anti-angiogenic therapy, immunosuppression, immune check point inhibitor (ICI)
Citation: Bourhis M, Palle J, Galy-Fauroux I and Terme M (2021) Direct and Indirect Modulation of T Cells by VEGF-A Counteracted by Anti-Angiogenic Treatment. Front. Immunol. 12:616837. doi: 10.3389/fimmu.2021.616837
Received: 13 October 2020; Accepted: 04 March 2021;
Published: 29 March 2021.
Edited by:
Loredana Albonici, University of Rome Tor Vergata, ItalyReviewed by:
Brian Rini, Vanderbilt University, United StatesCopyright © 2021 Bourhis, Palle, Galy-Fauroux and Terme. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magali Terme, bWFnYWxpLnRlcm1lQGluc2VybS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.